
Prevention of acute/severe hypoglycemia-induced neuron death by lactate administration
Upload
svuniversityCategory
view
0download
0
Seediscussions,stats,andauthorprofilesforthispublicationat:http://www.researchgate.net/publication/260336989
InSilicoinhibitorsforPlasmodiumfalciparumlactatedehydrogenase
ARTICLE·MAY2013
DOWNLOADS
48
VIEWS
31
3AUTHORS,INCLUDING:
MadhuSudhanaSaddala
SriVenkateswaraUniversity
10PUBLICATIONS0CITATIONS
SEEPROFILE
KonidalaKranthiKumar
SriVenkateswaraUniversity
3PUBLICATIONS0CITATIONS
SEEPROFILE
Availablefrom:MadhuSudhanaSaddala
Retrievedon:03July2015

146
MAIN
©1996-2013 All Rights Reserved. Online Journal of Bioinformatics . You may not store these pages in any form except for your own personal use.All other usage or distribution is illegal under international copyright treaties. Permission to use any of these pages in any other way besides thebefore mentioned must be gained in writing from the publisher. This article is exclusively copyrighted in its entirety to OJB publications. Thisarticle may be copied once but may not be, reproduced or re-transmitted without the express permission of the editors. This journal satisfies therefereeing requirements (DEST) for the Higher Education Research Data Collection (Australia). Linking:To link to this page or any pages linking tothis page you must link directly to this page only here rather than put up your own page.
OJBTM
Online Journal of Bioinformatics ©
Volume 14 (2): 146-159, 2013
In Silico inhibitors for Plasmodium falciparum lactate dehydrogenase
Madhu Sudhana Saddala, K. Kranthi Kumar and A. Usha Rani*
Division of Environmental Biology, Department of Zoology, DBT Bioinformatics Center, Sri Venkateswara University, Tirupati A.P., India.
ABSTRACT
Madhu Sudhana Saddala, K. Kranthi Kumar and A. Usha Rani, In Silico Inhibitors for Plasmodiumfalciparum lactate dehydrogenase, Onl J Bioinform., 14 (2): 146-159, 2013 Dysruption of activesites on lactate dehydrogenase from Plasmodium falciparum (PfLDH) could inhibit the malarialparasite. We describe Virtual gossypol-like compounds that might inhibit PfLDH by interactingwith amino acids on its receptors. Multiple sequence alignment of PfLDH with Plasmodium sppwas performed with Clustal W1.83. A phylogenetic tree was constructed with Tree Viewer 3.0.Protein structure was refined by 2ns molecular simulation and energy minimization. Compounds(1997) were then screened Virtually (Autodoc Vina) for similarity with gossypol from The Zincdatabase for binding capacity. Docking showed that sequentially ZINC27313038, ZINC13759138,ZINC13759183, ZINC13759202, ZINC59648667 and ZINC11159075 k.cal/mol had most bindingcapacity with PfLDH compared with gossipol. These compounds bind by hydrogen bonds andhydrophobic interactions and may inhibit PfLDH mediated glycolysis The cavity surrounded byIle31, Gly99, Asn140, Gly32, Thr101, Gly29 Thr97, Asp53, Met30, Phe52 and Glu122 may possiblybe manipulated to inhibit the parasite.
Keywords: PfLDH, NAMD, MDSimulation, Virtual screening, Docking, Zinc database
INTRODUCTION
Malaria is one of the major diseases affecting mankind, which is widely re-emerging in manytropical and subtropical areas of the world, constituting a huge public health problem. It affectsapproximately 216 million individuals throughout the tropical and subtropical areas of developingcountries and causes considerable morbidity and mortality with about 655,000 deaths worldwide

147
each year (World Health Organization, 2011) and approximately one third of the world’spopulation is at risk for contracting the disease. The World Health Organization has announced anew campaign for global malaria eradication (Wells et al., 2009). The global importance of thisdisease, current limitations of vector control and the absence of an effective vaccine, makes thedevelopment of therapeutic antimalarial drugs the main strategy of malaria control (de Ridder etal., 2008).
The causative agents are protozoan parasites from the genus Plasmodia. In particular the mostvirulent Plasmodium falciparum is mainly responsible for the infection in humans. P. falciparumlactate dehydrogenase (PfLDH) has been suggested as a drug target by several authors (Gomez etal., Menting et al., and Ridley et al., 1997). PfLDH is made up of 316 amino acids (Figure 2), it iscoded by a gene located on chromosome 13 and expressed as a 1.6 kb m-RNA. It is an essentialenzyme for parasite survival as plasmodium species lack a functional Krebs cycle during theirerythrocytic stages and, hence, must generate all their energy from glycolysis coupled withfermentation (Lang-Unnasch et al., 1998). Lactate dehydrogenase catalyses the reduction of theketo group in pyruvate to a hydroxyl (yielding lactate) with the concomitant oxidation of NADH toNAD+. This regenerated NAD+ is essential for the continuation of glycolysis.
The enzyme PfLDH differs from the human isozymes in several important structural and kineticfeatures, among which is the possession of a five-residue insertion in the substrate-specificityloop (Bzik et al., 1993). The dependence of P. falciparum upon glycolysis has inspired a fairamount of research into inhibitors of glycolysis (Brady et al., 2004). Gossypol is a di-sesquiterpenenatural product isolated from seeds of the cotton plant, has been reported to have a wide rangeof biological activities including contraceptive (Rogers et al., 1992), anti-viral (Fidock et al., 1994;Dame et al., 1984) and anti-malarial (Ling et al., 2003; Howard et al., 1998) effects. Gossypol hasbeen reported to non-selectively inhibit the activity of human LDH’s and pfLDH with lowmicromolar inhibitory constant (Ki) (Deck et al., 1998; Yu et al., 2001) binding competitively withNADH. In Silico Virtual inhibitors against PfLDH in Plasmodium spp drug are described herein.
MATERIALS AND METHODSSequence analysis:Lactate dehydrogenase sequence for Plasmodium species were obtained from the publiclyavailable NCBI database (http://www.ncbi.nlm.nih.gov/). GenBank accession numbers of all theLDH are given in Table 1. All the protein sequences were aligned by employing EBI tools ClustalW(http://www.ebi.ac.uk.clustalw) (Higgins et al., 1994) with default parameters. Phylogeneticanalysis of the sequences is done by Molecular Evolutionary Genetic Analysis (MEGA) softwareversion 4 (Tamura et al., 2007), using neighbor-joining method with complete deletion andPoisson correction settings.
Protein preparation and MD simulations:The 3D structure of PfLDH complexed with NADH and the substrate oxamate used in the presentwork was obtained from the Protein Data Bank (PDB) file 1LDG (Bernstein, et al., 1977; Berman etal., 2002). The existing ligands and crystallographic water were removed. It was refined bymolecular dynamics in a solvated layer and equilibration methods using NAMD 2.9 (Nanoscale

148
Molecular Dynamics) software (Kale et al., 1999) using CHARMM27 (Schlick et al., 1999)(Chemistry at Harvard Macromolecular Mechanics) force field for protein in water (Schlenkrich etal., 1996). Protein was energy minimized with 25,000 runs for 500ps and simulation with 1000000steps for 2ns. Spherical periodic boundary conditions were included in this study. Finally, thestructure having the least RMSD of Cα trace was generated by employing the molecular dynamicssimulations which improves the quality of the target protein. The trajectory analysis was analyzedby drawing the graph between Time in Ps on X-axis and RMSD (Å) on Y-axis as shown in Figure.1.The quality structure of protein was used for further analysis.
Figure 1: Root mean square deviation (RMSD) during the molecular simulations of PFLDH. Time(Ps) was taken in X-axis and RMSD was taken in Y-axis.
Active site Identification:Active site of PfLDH was identified using CASTp server (Computer Atlas of Surface Topology ofprotein) (Dundas et al, 2006). A new program, CASTp, for automatically locating and measuringprotein pockets and cavities, is based on precise computational geometry methods, includingalpha shape and discrete flow theory. CASTp identification and measurements of surfaceaccessible pockets as well as interior inaccessible cavities by locating, delineating and measuringconcave surface regions on three-dimensional structure of proteins. The measurement includesthe area and volume of pocket or void by solvent accessible surface model (Richards’ surface) andby molecular surface model (Connolly’s surface), calculated analytically. It can also be used tostudy surface features and functional regions of proteins.
Screening Ligands:Commercially available ligands are listed in public databases, such as ZINC database, that containsmore than 4.6 million compounds in ready to dock and provide 3D formats at the URL

149
http://ZINC.dock.org . Virtual screening has been emerged as a complementary approach to highthroughput screening and has become an important in silico technique in the pharmaceuticalindustry (Lengauer et al., 2004). The structure based virtual screening begins with theidentification of potential ligand binding sites on the target proteins. Usually, molecules thatmeet the criteria for biological activity fulfill characteristics contained in the Lipinski’s rule of five(Lipinski et al., 1997), or the more relaxed rules revised by Veber et al., 2002. In the present work,we have selected 1997 docked ligands based on structure similarity with query gossypol naturalcompound. The Autodock Vina in PyRx Virtual Screening Tool URL http://pyrx.scripps.edu (Wolf,2009; Trott and Olson, 2010) was used for the screening of selected ligands from Zinc databaseand energy minimization.
Docking studies:For the docking of ligands into target protein binding pockets and to estimate the bindingaffinities of docked ligands, a molecular docking program Autodock Vina in PyRx Virtual screeningtool (1.1.2) were used in this study. Docking studies was performed on refined protein andscreened gossypol like inhibitors. The protein PDB file was transformed into the PDBQT formatfile containing the protein atom coordinates, partial charges and salvation parameters and theligands file (SDF) was transformed into PDBQT format. AutoGrid boxes (x, y, z coordinates 76.54,52.07, 54.65) were fixed around active site of the protein. The search was based on theLamarickian genetic algorithm (LGA) and the obtained dock scores were reported in kcal/mol. Thedocking protocol utilized in the study consisted of 10 independent runs per ligand. The resultswere analyzed on the basis of ranked clusters of compound confirmations.
RESULTS AND DISCUSSIONSequence analysis:All the sequences were aligned by using ClustalW to find out the extent of similarity presentamong the sequences of the same genus, which enjoy a common phylogeny. Alignment ofsequences shows much (99-95%) similarity among the all Plasmodium species of target proteinshown in Figure 2 (next page). For evaluating the phylogenetic relationships of LDHs in differentplasmodium species, an unrooted phylogenetic tree (Figure 3) was constructed by neighbor-joining method keeping bootstrap values higher than 40. P.yoelii, P.berghei, P.chabaudi andP.falciparum are orthologues of P.vivax (bootstrap value 99, 97, 96 and 100), while P. knowlesiseems to be an orthologues of P. cynomolgi (bootstrap value 41; 55% pair -wise similarity).Ideally, drugs directed against this target would be effective against all species of Plasmodium.The crystal structures of the LDH protein from all plasmodial LDHs have been solved and arecompared to the equivalent structure from the P. falciparum enzyme. The active sites andcofactor binding pockets of both enzymes are found to be highly similar with the help ofsequence analysis approach.

150
Figure 2: ClustalW alignment of all the LDH sequences from Plasmodium species. The conservedsignature sequences of the LDHs are highlighted by different colours.
Structural analysis of PfLDH:PfLDH is made up of 316 amino acids (Figure 2), it is coded by a gene located on chromosome 13and expressed as a 1.6 kb m-RNA. PfLDH was also divided into two parts viz. NADH bindingdomain and carboxyl domain. The NADH binding domains contains (18-162a.a) 8 α-helixes and 6β-sheets. This domain key role in catalytic reactions in glycolysis cycle. The structure wasobtained by energy minimization and equilibrium with 25,000 runs for 500ps and simulation with1000000 steps for 2ns. A conserved change of calculated RMSD with time in ps during simulationwas shown in Figure 1 while Figures 7-12 illustrate total, kinetic, total bond, angle, temperatureand total No. of bond energies respectively. The refined protein is shown in Figure 5. The finalrefined protein structure predicted active site using CASTp server and active site regions areshown in Figure 4. Ile31, Gly99, Asn140, Gly32, Thr101, Gly29 Thr97, Asp53, Met30, Phe52 and

151
Glu122 were active site residues. These residues significant role in binding and catalytic activity inthe active site of PfLDH.
Plasmodium yoelii
Plasmodium berghei
Plasmodium chabaudi
Plasmodium falciparum
Plasmodium vivax
lasmodium knowlesi
Plasmodium cynomolgi
Plasmodium malariae
41
100
99
96
97
Figure 3. Unrooted phylogenetic tree of LDHs of different plasmodium species constructed by neighbor-joining method. Bootstrap values are indicated against each branch. Bootstrap similarity is >50% and thetree was built after 1,000 cycles.
Virtual screening:Virtual screening is a proficient approach in discovering inhibitors with novel chemical scaffolds.Two-dimensional structure of gossypol was used as query to search for similar compounds in theZinc database. Then, approximately 1997 compounds were screened, and the all compoundswere saved for further molecular docking. Attempts to screening of gossypol like compounds,that is cytotoxic at high doses, have produced several compounds retaining activity against boththe target enzyme and the parasite but without significantly improving selectivity or parasiticidalactivity.
A BFigure 4: A. Cylindrical image of the structure of PfLDH. It has a Rossmann topology, α-helices (Red) packagainst a central core of β-sheets (Yellow) connecting with loops (Green). B. Binding pocket was predictedthrough CASTp server.

152
Figure 5: After molecular dynamics Simulation of target protein in water content
Docking Studies:Three – dimensional structure information of the target protein was taken from the PDB (ProteinData Bank) entry 1LDG. Processing of the protein included energy minimized and moleculardynamics simulations. The refinement of structure of protein was used for the dock. AutodockVina was used for the docking studies. The docked conformation corresponding to the lowestbinding energy was selected as the most probable binding conformation. The total screened 1997compounds were docked into the active site of PfLDH. The docking energies of all the compoundswere represented in kcal/mol are summarized in supplementary data S. the best six zinccompounds showed low binding energies and significant affinities with target protein of PfLDHthe values are represented in Table 2. Which all the ligands were embedded within the active siteof target protein were observed forming hydrogen bonds with same position as gossypolestablished active site of target protein (Figure 6). The best docked compounds such asZINC27313038, ZINC13759138, ZINC13759183, ZINC13759202, ZINC59648667 and ZINC11159075were found to be shown highest binding energies viz., -11.0, -10.3, -10.3, -10.1, -10.1 and -10.0kcal/mol respectively (Table 2). The lead compounds and their interactions with active site ofresidues are represented in Figure13.

153
Figure 6: All Docked ligands fit into the active site of protein (1LDG)
Figure 7: The Total energy plot of PfLDH structure
Figure 8: The kinetic energy plot of PfLDH structure

154
Figure 9: The Total bond energy plot of PfLDH structure
Figure 10: The Total angle energy plot of PfLDH structure
Figure 11: The Total temperature plot of PfLDH structure
Figure 12: The Total No. of bonds plot of PfLDH structure
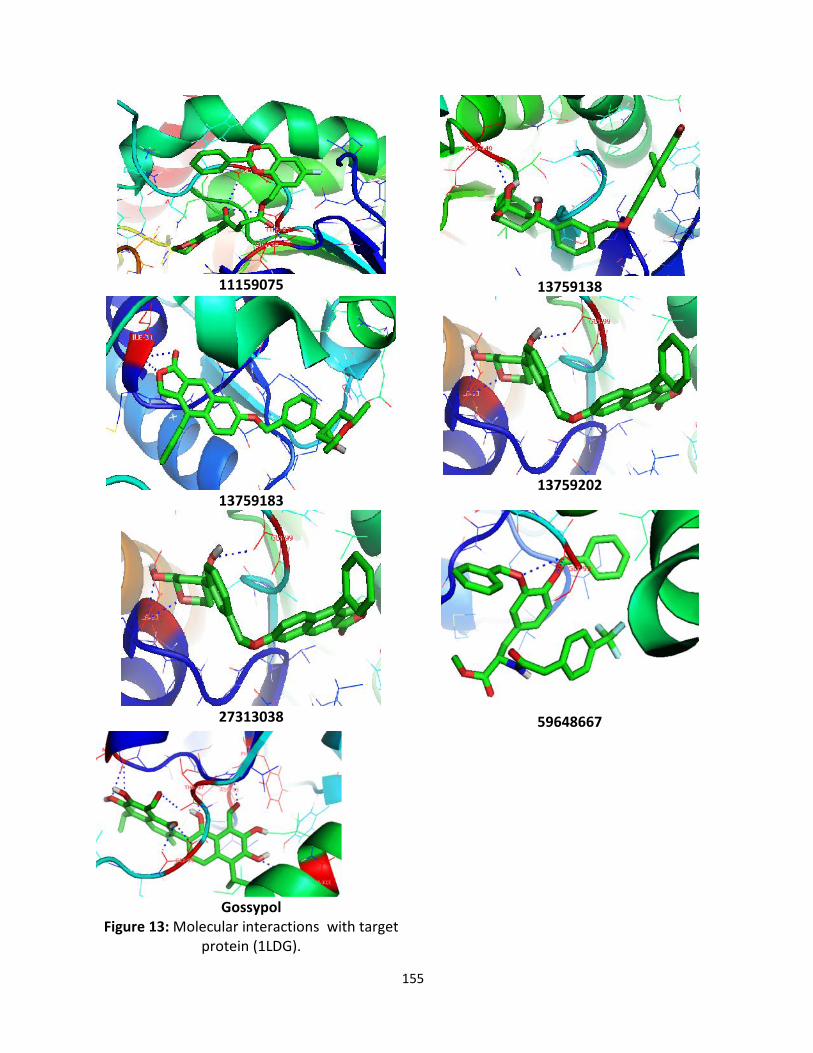
155
11159075 13759138
1375918313759202
27313038 59648667
GossypolFigure 13: Molecular interactions with target
protein (1LDG).

156
Hydrogen bonds play a role in stabilizing the protein-ligand complex (Gao et al., 2005). The Zincdatabase compounds also exhibit several hydrogen bonding moieties. The compoundZINC27313038 (O26, O29, O31) was bound with the binding affinity -11.0 by the formation of threehydrogen bonds to Ile31 and Gly99 of loop connecting NADH binding domain of protein.ZINC13759138 (O29) was bound with the binding affinity -10.3 by the formation of one hydrogenbond with Asn140 in NADH binding domain of protein. ZINC13759183 (O12, O14) was bound withthe binding affinity -10.3 by the formation of two hydrogen bonds with Ile31 and Gly99 in NADHbinding domain of protein. ZINC13759202 (O12, O14, O14, N33) was bound with the binding affinity-10.1 by the formation of four hydrogen bonds with Ile31, Ile31, Gly32 and Thr101 in NADHbinding domain of protein. ZINC59648667 (O36, O28, ) was bound with the binding affinity -10.1 bythe formation of two hydrogen bonds with Gly99 and Gly99 in NADH binding domain of protein.ZINC11159075 (O21, O24, O24) was bound with the binding affinity -10.0 by the formation of fourhydrogen bonds with Gly29, Gly99 and Thr97 in NADH binding domain of protein. The querycompound gossypol (O27, O29, O37, O34, O23, O23, O26 and O43) was bound with the binding affinity -9.3 by the formation of eight hydrogen bonds with Met30, Met30, Phe52, Asp53, Gly99, Gly99,Thr97 and Glu122 in NADH binding domain of protein.
The lead hit compounds satisfied the Lipinski’s rule of five with zero violations and also theoctanol/water partition coefficient (miLogp), a useful parameter for predicting the drug transportproperties like absorption, bioavailability, permeability and penetration. As well as topologicalmolecular polar surface area (TPSA), number of atoms, their molecular weight (MW), number ofhydrogen donors and number of hydrogen acceptors.
A topological parameter is number of rotatable bonds and it describes the molecular flexibility ofthese compounds. This parameter was calculated for the six lead molecules that satisfy the ‘rule-of-5’ and it is found that all the molecules have rotatable bonds in the range of 3-5 exceptZINC27313038 and ZINC59648667. All the above mentioned ‘rule –of-5’ parameters listed inTable.3A and 3B. ZINC13759138 has been satisfied all the parameters with clogP 4.67, solubility-7.22, molecular weight 482.0, Hbond donors 2, H bond acceptors 6, Drug likeness 0.54, Drugscore 0.28, and no adverse effects. The compounds ZINC13759202 and ZINC11159075 also havesatisfied the above parameters. In addition to that, ZINC13759183, ZINC59648667 have alsosatisfied the Lipinski rule but do not satisfied drug transport properties. ZINC11159075 andGossypol have do not satisfied the Lipinski rule and drug transport properties. Our investigationrevealed that the selected compounds have exhibited significant binding affinity with in theactive site of 1LDG protein, when compare to query compound gossypol. Based upon this study,these compounds may be used as leads for developing effective antimalarial drugs.
ACKNOWLEDGMENTS
Authors are grateful to Department of Biotechnology (DBT), Ministry of Science andTechnology, Government of India for their financial assistance. Madhu Sudhana Saddalaespecially grateful to University Grants Commission, New Delhi for their financial assistance with

157
BSR fellowship. This work was carried out in DBT-Bioinformatics Infrastructure Facility (BIF),Department of Zoology, Sri Venkateswara University, Tirupati (BT/BI/25/001/2006).
REFERENCES
1. Berman, H., Battistuz, T., Bhat, T., Bluhm, W., Bourne, P., et al., (2002). The Protein Data Bank.Acta.Crystallogr. D. Biol. Crystallogr. 58: 899–907.
2. Bernstein, F., Koetzle, T., Williams, G., Meyer, E.J., Brice, M., et al., (1977). The Protein Data Bank: acomputer-based archival file for macromolecular structures. J. Mol. Biol. 112: 535–542.
3. Bzik, D.J., Fox, B.A., Gonyer, K., (1993). Expression of Plasmodium falciparum lactate dehydrogenase inEscherichia coli. Mol. Biochem .Parasitol. 59: 155–166.
4. Cameron, A., Read, J., Tranter, R., Winter, V. J., Sessions, R. B., Brady, R. L., Vivas, L., Easton, A.,Kendrick, H., Croft, S. L., Barros, D., Lavandera, J. L., Martin, J. J., Risco, F., Garcia-Ochoa, S., Gamo, F.J., Sanz, L., Leon, L., Ruiz, J. R., Gabarro, R., Mallo, A., and Gomez de las Heras, F., (2004). Identificationand activity of a series of azole-based compounds with lactate dehydrogenasedirected anti-malarialactivity. J. Biol. Chem. 279: 31429-31439.
5. Dame, J.B., Williams, J.L., McCutchan, T.F., et al., (1984). Structure of the gene encoding theimmunodominant surface antigen on the sporozoite of human malaria parasite Plasmodiumfalciparam. Science. 225: 593-9.
6. De Ridder, S., van der Kooy, F., Verpoorte, R., (2008). Artemisia annua as a self-reliant treatment formalaria in developing countries. J. Ethnopharmacol. 120: 302–314.
7. Dundas, J., Ouyan, Z., Tseng, J., Binkowski, A., Turpaz, Y., Liang, J., (2006). CASTp: computed atlas ofsurface topography of proteins with structural and topographical mapping of functionally annotatedresidues. Nucleic acids Res. 34: W116-W118.
8. Fidock, D.A., Bottius, E., Brahimi, K.A., et al., (1994). Cloning and characterization of a novelPlasmodium falciparam sporozoite surface antigen STARP. Mol. Biochem Parasitol. 64: 219-31.
9. Gao, F., Bren, N., Burghardt, T.P., Hansen, S., Henchman, R.H., Taylor, P., McCammon, J.A., Sine, S.M.,(2005). Agonist-mediated conformational changes in acetylcholine-binding protein revealed bysimulation and intrinsic tryptophan fluorescence. J. Biol. Chem. 280: 8443–8451.
10. Gomez, M.S., Granchi Simone Bertini, Marco Macchia, Filippo Minutolo. Piper, R.C., Hunsaker, L.A.,Royer, R.E., Deck, L.M., Makler, M.T., Vander Jagt, D., (1997). Mol. Biochem. Parasitol. 90: 235–246.
11. Higgins, D., et al., (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequencealignment through weighing, position-specific gap penalties and weith matrix choice. Nucleic AcidRes.22: 4673-4680.
12. Howard, R.F., Narum, D.L., Blackman, M., Thurman, J., (1998). Analysis of the process of Plasmodiumfalciparam rhoptry-associated protein1 and localization of Pr86 to schzont rhoptries and p67 to freemerozoites. Mol. Biochem. Parasitol. 92:111-22.
13. Irwin, J.J., Shoichet, B.K., (2005). ZINC--a free database of commercially available compounds forvirtual screening. J. Chem. Inf. Model. 45: 177–182.
14. Kale, L., Skeel, R., Bhandarkar, M., Brunner, R., Gursoy, A., Krawetz, N., Phillips, J., Shinozaki, A.,Varadarajan, K., Schulten, K., (1999). NAMD2: greater scalability for parallel molecular dynamics. J.Comput .Phy. 15: 283–312.
15. Lang-Unnasch, N., Murphy, A. D., (1998). Metabolic changes of the malaria parasite during thetransition from the human to the mosquito host. Annu. ReV. Microbiol. 52: 561-590.
16. Lengauer, T., Lemmen, C., Rarey, M., Zimmermann, M., (2004). Novel technologies for virtualscreening. Drug Discovery Today. 9: 27-34.

158
17. Ling, I.T, Kaneko, O., Narum, D.L., et al., (2003). Characterisation of the rhoph2 gene of Plasmodiumfalciparam and Plasmodium yoelii. Mol. Biochem .Parasitol. 127: 47-57.
18. Lipinski, C.A., Lombardo, F., Dominy, B.W., Feeny, P.J., (1997). Experimental and computationalapproaches to estimate solubility and permeability in drug discovery and development settings. Adv.Drug Delivery Rev. 23: 3–25.
19. Menting, J. G., Tilley, L., Deady, L. W., Ng, K., Simpson, R. J., Cowman, A. F., and Foley, M., (1997). Mol.Biochem. Parasitol. 88: 215–224.
20. Ridley, R. G., (1997). Exp. Parasitol. 87: 293–304.21. Rogers, W.O., Malik, A., Mellouk, S., et al., (1992). Characterization of Plasmodium falciparam
sporozoite surface protein 2. Poc. Natl. Acad. Sci .USA. 89: 9176-80.22. Schlenkrich, M., Brickmann, J., MacKerell, A.D., Jr Karplus, M., (1996). Empirical potential energy
function for phospholipids: criteria for parameter optimization and applications. In: Merz KM, Roux B(eds) Biological membranes: a molecular perspective from computation and experiment. Birkhauser,Boston, MA, pp 31–81.
23. Schlick, T., Skeel, R., Brunger, A., Kale, L., Board, J.A., Jr Hermans, J., Schulten, K., (1999). Algorithmicchallenges in computational molecular biophysics. J. Comput. Phys. 151: 9–48.
24. Sousa, S.F., Fernandes, P.A., Ramos, M.J., 2006. Protein-ligand docking: current status and futurechallenges. Proteins. 65: 15–26.
25. Tamura, K., et al., (2007). MEGA 4.5: Molecular Evolutionary Genetics Analysis (MEGA) softwareversion 4.5. Mol. Bio. Evol. 24, 1596-1599.
26. Trott, O., Olson, A.J., (2010). AutoDock Vina: improving the speed and accuracy of docking with a newscoring function, efficient optimization, and multithreading. J. Comput. Chem. 31: 455–461.
27. Veber, D.F., Johnson, S.R., Cheng, H.Y., Smith, B.R., Ward, K.W., Kopple, K.D., Molecular Wells, T.N.,Alonso, P.L., and Gutteridge, W.E., (2009). New medicines to improve control and contribute to theeradication of malaria. Nat. Rev. Drug Discov. 8: 879–891.
28. WHO World Malaria Report., 2011. Available at: http://www.who.int/malaria/world_malaria_report_2011/worldmalariareport2011.pdf. Accessed: March/ 2013.
29. Wolf, L.K., (2009). PyRx. C&EN. 87: 31.30. Yu, Y., deck, J.A, Hunsaker, L.A., et al., (2001). Selective active site inhibitors of human lactate
dehydrogenase A4, B4, and C4. Biochem Pharmacol. 62: 81-9.

159
Copyright © 2022 FDOKUMEN




















