
Reversible hydrogen control of antiferromagnetic anisotropy in О

Reversible inactivation of dihydrolipoamide dehydrogenase byAngeli’s salt
Liang-Jun Yan1,*, Li Liu2, and Michael J. Forster1
1Department of Pharmacology and Neuroscience and Institute for Aging and Alzheimer’s DiseaseResearch, University of North Texas Health Science Center, Fort Worth, Texas2The First Affiliated Hospital of Nanjing Medical University, Jiangsu, China
AbstractDihydrolipoamide dehydrogenase (DLDH) is a key component of 3 mitochondrial α-keto aciddehydrogenase complexes including pyruvate dehydrogenase complex, α-ketoglutaratedehydrogenase complex, and branched chain amino acid dehydrogenase complex. It is a pyridine-dependent disulfide oxidoreductase that is very sensitive to oxidative modifications by reactivenitrogen species (RNS) and reactive oxygen species (ROS). The objective of this study was toinvestigate the mechanisms of DLDH modification by RNS derived from Angeli’s salt. Studieswere conducted using isolated rat brain mitochondria that were incubated with varyingconcentrations of Angeli’s salt followed by spectrophotometric enzyme assays, blue native gelanalysis, and 2-dimensional gel-based proteomic approaches. Results show that DLDH could beinactivated by Angeli’s salt in a concentration dependent manner and the inactivation was atargeting rather than a random process as peroxynitrite did not show any detectable inhibitoryeffect on the enzyme’s activity under the same experimental conditions. Since Angeli’s salt canreadily decompose at physiological pH to yield nitroxyl anion (HNO) and nitric oxide, furtherstudies were conducted to determine the actual RNS that was responsible for DLDH inactivation.Results indicate that it was HNO that exerted the effect of Angeli’s salt on DLDH. Finally, two-dimensional Western blot analysis indicates that DLDH inactivation by Angeli’s salt wasaccompanied by formation of protein s-nitrosothiols, suggesting that s-nitrosylation is likely thecause of loss in enzyme’s activity. Taken together, the present study provides insights intomechanisms of DLDH inactivation induced by HNO derived from Angeli’s salt.
KeywordsAngeli’s salt; brain; dihydrolipoamide dehydrogenase; mitochondria; nitroxyl anion (HNO); S-nitrosylation
IntroductionDihydrolipoamide dehydrogenase (DLDH) is an essential component of pyruvatedehydrogenase, α-ketoglutarate dehydrogenase, and branched chain keto aciddehydrogenase complexes [1, 2]. It belongs to the family of flavin-dependent, pyridinedinucleotide disulfide oxidoreductase including glutathione reductase, mercuric reductase,trypanothione reductase, and thioredoxin reductase. It is also the L protein in the glycinecleavage system [3, 4]. DLDH is a stable homodimer, with each monomer owning a tightly
*Corresponding author: Liang-Jun Yan, Ph.D. Department of Pharmacology and Neuroscience, University of North Texas HealthScience Center at Fort Worth, 3500 Camp Bowie Boulevard, Fort Worth, TX 76107, Phone: 817-735-2386, Fax: 817-735-0408, [email protected].
NIH Public AccessAuthor ManuscriptSheng Wu Wu Li Hsueh Bao. Author manuscript; available in PMC 2012 November 06.
Published in final edited form as:Sheng Wu Wu Li Hsueh Bao. 2012 April 20; 28(4): 341–350.
$waterm
ark-text$w
atermark-text
$waterm
ark-text

but non-covalently bound FAD molecule, a transiently bound NAD+ or NADH molecule,and a redox-sensitive center containing two cysteine residues that are directly involved inthiol-disulfide exchanges during catalysis [1, 5–7]. Physiologically, DLDH oxidizes thereoxidation of dihydrolipoamide that is covalently linked to acyltransferase at the expense ofNAD+ [1, 2, 8]. Therefore, the enzymatic reaction also produces NADH that can enter into theelectron transport chain for oxidative phosphorylation and ATP synthesis.
DLDH is redox-sensitive and is a multifunctional oxidoreductase. In vitro, DLDH can serveas a diaphorase [9] that can transfer electrons from NADH to electron acceptors such asubiquinone [10, 11], cytochrome c [12], and artificial electron-accepting dyes including nitro-blue tetrazolium (NBT) [13, 14] and 2,6-dichlorophenolindophenol (DCPIP) [15].Pathophysiologically, DLDH dysfunction can cause maple syrup urine disease due toaccumulation of branched chain amino acids in the body [16], and is involved in arsenitepoisoning due to binding of arsenite to the vicinal thiols of dihydrolipoamide, which thenprevents reoxidation of dihydrolipoamide by DLDH [17].
Interestingly, as a redox-sensitive protein, DLDH can either aggravate or mitigate oxidativedamage depending on experimental conditions. On one hand, DLDH can be a source ofreactive oxygen species [18–22] and can be oxidatively modified leading to potentialimpairment of mitochondrial function [23–28]; on the other hand, DLDH can scavenge nitricoxide [12] and serve as an antioxidant by protecting other proteins against oxidativemodifications [29]. Moreover, when its homodimeric status is perturbed, DLDH can turnitself into a protease [30] and thereby enhances oxidative damage [31].
Our research interest in DLDH is focused on the role of its oxidative modifications in agingand age-related neurodegenerative diseases. Our first stage of studying DLDH oxidativemodifications mainly centered on in vitro oxidative modifications of this protein by RNS orROS. In the present article, we report our findings of in vitro DLDH inactivation byAngeli’s salt. We chose Angeli’s salt as the donor of RNS mainly based on our previousfindings that this chemical, among the RNS donors tested, is the most reactive towardDLDH [32]. Additionally, the use of Angeli’s salt in the present study was also prompted bythe fact that this pharmacological reagent has therapeutic potential [33–37].
Experimental roceduresAnimal and chemicals
Adult male Sprague-Dawley rats were obtained from Harlan (Indianapolis, IN). Use ofanimals was in adherence with the NIH Guidelines for the Care and Use of LaboratoryAnimals and the protocol was approved by the University of North Texas Health ScienceCenter Animal Care and Use Committee. Dihydrolipoamide was prepared using sodiumborohydride reduction of lipoamide as previously described [15, 38]. Rabbit anti-DLDHpolyclonal antibodies (IgG) were from US Biological (Swampscott, MA, USA) and goatanti-rabbit IgG conjugated with horseradish peroxidase was from Zymed (South SanFrancisco, CA, USA). Angeli’s salt and peroxynitrite were purchased from Cayman (AnnArbor, MI) and all other chemicals were purchased from Sigma (St. Louis, MO) unlessotherwise stated.
Preparation of brain mitochondriaThe isolation of mitochondria from whole brain was carried out as previously outlined [39]
with modifications [32]. Brains were removed rapidly and homogenized in 15 ml of ice-cold,mitochondrial isolation buffer containing 0.32 M sucrose, 1 mM EDTA and 10 mM Tris-HCl, pH 7.1. The homogenate was centrifuged at 1,330 g for 10 min and the supernatantwas saved. The pellet was resuspended in 0.5 (7.5 ml) volume of the original isolation buffer
Yan et al. Page 2
Sheng Wu Wu Li Hsueh Bao. Author manuscript; available in PMC 2012 November 06.
$waterm
ark-text$w
atermark-text
$waterm
ark-text

and centrifuged again under the same conditions. The two supernatants were combined andcentrifuged further at 21,200 g for 10 min. The resulting pellet was resuspended in 12%Percoll solution that was prepared in mitochondrial isolation buffer and centrifuged at 6,900g for 10 min. The resulting supernatant was then carefully removed by vacuum. Theobtained soft pellet was resuspended in 10 ml of the mitochondrial isolation buffer andcentrifuged again at 6,900 g for 10 min. All of the mitochondrial pellets obtained aftercentrifugation were immediately used. All the protein concentrations were determined bybicinchoninic acid assay [40] using a kit obtained from Pierce (Rockford, IL).
In vitro DLDH inactivation by Angeli’s saltDLDH oxidative inactivation in isolated brain mitochondria was performed bysupplementing mitochondria with various concentrations of Angeli’s salt as previouslydescribed [32]. Essentially, mitochondria (0.25 mg/ml) were incubated at 25°C for 30 mininincubation buffer (110 mM mannitol, 10mM KH2PO4, 60 mM Tris, 60 mM KCl, and 0.5mM EGTA, pH 7.4) in the presence or absence of Angeli’s salt. At the end of theincubation, mitochondria were pelleted by centrifugation at 8,000 g for 10 min, andmitochondrial extracts were then prepared as described below. For incubation of themitochondrial extracts with reducing reagents such as DTT, cysteine, and GSH, 10 mM(final concentration) of each of the reducing reagent was added and the sample was furtherincubated for 30 min before gel analysis or spectrophotometric enzyme assay.
Preparation of mitochondrial extracts for spectrophotometric enzyme assay and BN-PAGEanalysis
Whole mitochondrial extracts as the source of DLDH analyses were prepared as previouslydescribed [15, 38]. Briefly, following incubation with Angeli’s salt, mitochondria werepelleted at 8,000 g for 10 min and the pellet was resuspended at a protein concentration ofapproximately 0.5 mg/ml in a hypotonic buffer containing 20 mM potassium phosphate (pH7.4), 1% Triton X-100, 2 mM EDTA, 2 mM EGTA and protease inhibitors. The suspensionwas sonicated in a Branson Sonifier 150, four times for 30 sat 1-min intervals. The sonicatedsample was kept on ice for 30 min and clarified by centrifugation at20,000 g for 30 min. Theclear DLDH-containing supernatant was then used for mitochondrial DLDH assays asdescribed below.
Spectrophotometric measurement of DLDH dehydrogenase activityDLDH dehydrogenase activity was measured by DLDH catalyzed, NAD+-dependentoxidation of dihydrolipoamide [15, 38]. The final volume of reaction was 1 ml, and themixture contained 100 mM potassium phosphate, pH 8.0, 1.5 mM EDTA, 0.6 mg/ml BSA,3.0 mM dihydrolipoamide and 3.0 mM NAD+. A solution containing all the assaycomponents except mitochondrial extracts was used as the blank. The reaction was initiatedby the addition of appropriate amount of mitochondrial extracts (10–20 μg/ml assaysolution) and the change in absorbance at 340 nm was followed at room temperature. Anextinction coefficient of 6.22 mM−1 cm−1 for NADH was used for the calculation of enzymeactivity [15, 38]. One unit of dehydrogenase activity was defined as 1μmol of NAD+ reducedper min.
In-gel DLDH diaphorase activity stainingIn-gel staining of DLDH diaphorase activity by NBT/NADH was performed using bluenative polyacrylamide gel electrophoresis (BN-PAGE) as previously described [32]. The gelbuffer contained 500 mM aminocaproic acid and 50 mM Bis-Tris, pH 7.0. The cathodebuffer contained 50 mM tricine, 15 mM Bis-Tris, pH 7.0, and 0.02% Serva blue G-250,whereas the anode buffer contained 50 mM Bis-Tris, pH 7.0. The sample buffer was 50 mM
Yan et al. Page 3
Sheng Wu Wu Li Hsueh Bao. Author manuscript; available in PMC 2012 November 06.
$waterm
ark-text$w
atermark-text
$waterm
ark-text

aminocaproic acid (final concentration) containing 0.3% Serva blue G-250 (w/v, finalconcentration). Following sample loading (typically, 20 μg protein/lane), the gel was run at150 V until the front line had entered into one-third of the gel where the cathode buffer wasreplaced by the one that did not have Serva blue G-250 (50 mM Tricine, 15 mM Bis-Tris,pH 7.0). Gel running was then resumed at 200 V until complete. The gel was incubated in50 mM potassium phosphate buffer (pH 7.0) containing 0.2 mg/ml NBT and 0.1 mg/mlNADH. When an appropriate color of DLDH band appeared, the staining solution wasdiscarded and the gel was scanned immediately for gel documentation.
Labeling of protein s-nitrosothiols by ascorbate reduction/biotin switch assayThe procedure used to label protein s-nitrosothiols was performed as previouslydescribed [41] with modifications. Mitochondrial pellet, immediately following isolation orin vitro treatment with Angeli’s salt, was solubilized in a thiol-group blocking buffercontaining 100 mM sodium acetate (pH 7.0), 20 mM NaCl, 1% SDS and 100 mM N-ethylmaleimide (NEM). The protein mixture was incubated on a rotator at room temperaturefor 2 hrs followed by clarification of the mixture by centrifugation at 13,000 g for 10 min.Excess NEM in the supernatant was removed by gel filtration using PD-10 columns. Thiswas followed by addition of 0.1 mM biotin-maleimide and 5 mM sodium ascorbate (bothfinal concentrations) to the eluate. The sample was further incubated in dark on a rotator atroom temperature for 30 min. Proteins were then precipitated by 10% TCA (finalconcentration) on ice for 10 min followed by centrifugation on a bench top centrifuge at1,000 g for 5 min. The pellet was washed three times with ethyl acetate: ethanol (1:1, v/v).Protein pellet after the third wash was used for 2D Western blot probing of the biotinylated(s-nitrosylated) proteins.
Two-dimensional Western blot detection of sulfenated proteinsTwo-dimensional Western blot was performed as previously described [42] withmodifications. Following biotinylation of the s-nitrosylated cysteine residues, mitochondrialpellets were resuspended in two-dimensional rehydration buffer [8 M urea, 4% CHAPS,0.2% ampholytes (pH 3–10), and 100 mM DTT]. First-dimensional protein separation wasperformed with Bio-Rad Protean IEF Cell. Samples (40 μg/IPG strip) were applied toimmobilized pH gradient strips (7-cm, nonlinear pH 3–10, Bio-Rad) for 1 h at roomtemperature. The strips were then covered with mineral oil overnight, and isoelectricfocusing was performed using the preset rapid voltage ramping method. For the seconddimension, the immobilized pH gradient strips were equilibrated in room temperature for 25min in equilibration buffer (6 M urea, 2% SDS, 0.05 mM Tris-HCl, 20% glycerol) to which2% DTT was added before use. An additional 25 min equilibration period was then usedwith the same equilibration buffer to which 2.5% iodoacetamide, instead of 2% DTT, wasadded. The strips were then embedded in 0.7% agarose on the top of 7.5% Laemmlipolyacrylamide slab gels (no stacking gel) and run by Tricine SDS/PAGE runningbuffer [43]. One of the resulting two-dimensional gels was stained with Coomassie colloidblue as previously described [44], and the other gel underwent electrophoretic transfer toPVDF membrane followed by immunoblotting with HRP-streptavidin. Signals on the PVDFmembrane were visualized with an enhanced chemiluminescence kit. Biotin-conjugatedprotein marker was used for the purpose of both molecular weight ladders and positivecontrols. For 1D Western blot analysis, gel electrophoresis under reducing conditions wasconducted. All images were scanned by an EPSON PERFECTION 1670 scanner.
Data analysisAll experiments were performed independently at least three times using differentmitochondrial preparations. Where applicable, results are expressed as means ± SEM.Statistical analysis was performed with Welch’s t test using Instat software (Graphad
Yan et al. Page 4
Sheng Wu Wu Li Hsueh Bao. Author manuscript; available in PMC 2012 November 06.
$waterm
ark-text$w
atermark-text
$waterm
ark-text

Software, San Diego, CA). A probability value less than 0.05 (p < 0.05) was consideredsignificant.
ResultsAngeli’s salt inactivated DLDH in a concentration-dependent manner
We have previously shown that DLDH is very sensitive to inactivation by Angeli’s salt andAngeli’s salt is more powerful in inhibiting DLDH activity than other RNS donors that weretested [32]. In the present study, we further investigated the mechanisms of DLDHinactivation by Angeli’s salt. Our first experiment was to determine whether DLDHinactivation by Angeli’s salt was concentration dependent. Results in Fig. 1 demonstrate thatwhen analyzed by blue native PAGE, loss of DLDH diaphorase activity was indeed Angeli’ssalt concentration dependent. Loss of the enzyme’s dehydrogenase activity, measuredspectrophotometrically, also exhibited a similar pattern (Fig. 2). Therefore, DLDH activityin whole mitochondria can be impaired by Angeli’s salt in a concentration-dependentfashion.
DLDH in whole mitochondria was specifically targeted by Angeli’s saltTo determine whether DLDH inactivation by Angeli’s salt is a random or targeting process,we compared the effect of Angeli’s salt with that of peroxynitrite. Results in Fig. 3 showthat peroxynitrite, when incubated with whole mitochondria, could not impose its inhibitoryeffect on DLDH activity. In contrast, Angle’s salt, under the same experimental conditions,gave a similar result as shown in Fig. 2. These results indicate that DLDH was specificallytargeted by Angeli’s salt in whole mitochondria.
DLDH inactivation by Angeli’s salt was caused by nitroxyl anion (HNO)It is well-known that at physiological pH, Angeli’s salt can readily decompose to yieldnitroxyl anion (HNO) and nitrite [45, 46]. To determine which of the two decompositionproducts was responsible for the loss of DLDH activity, we first incubated mitochondriawith nitrite. Result in Fig. 4A shows that nitrite did not cause any detectable loss in DLDHactivity, suggesting that HNO is responsible for DLDH inactivation. We then incubatedmitochondria with Angeli’s salt in the presence of ferricyanide that can eliminate HNO byconverting it to nitric oxide [47]. Result in Fig. 4B shows that the ability of Angeli’s salt toinactivate DLDH was completely abolished by ferricyanide. These results indicate that itwas HNO that caused the loss of DLDH activity under our experimental conditions.
DLDH inactivation by Angeli’s salt was largely reversibleTo determine whether DLDH’s inactivation by Angeli’s salt was reversible or not, weevaluated the effects of DTT, cysteine, and glutathione (GSH, reduced form) on enzyme’sactivity following mitochondrial incubation with Angeli’s salt (0.8 mM, in this experiment).At the concentration of 10 mM, each of the three reducing reagents could restore thedehydrogenase activity to greater than 80% of the control value (Fig. 5). Interestingly,DLDH’s diaphorase activity, when analyzed by BN-PAGE, could only be significantlyrestored by cysteine and GSH (10 mM each), but not DTT (Fig. 6). The reason for thisdiscrepancy of DTT’s effect on dehydrogenase and diaphorase activities remains unknown.Nonetheless, these results demonstrate that DLDH inactivation by Angeli’s salt couldlargely be reversed by appropriate reducing reagents.
S-nitrosylation was likely involved in DLDH inactivation by Angeli’s saltAs it is well established that Angeli’s salt can induce protein s-nitrosylation [48] and s-nitrosylation is a reversible process [49, 50], we further determined whether DLDH
Yan et al. Page 5
Sheng Wu Wu Li Hsueh Bao. Author manuscript; available in PMC 2012 November 06.
$waterm
ark-text$w
atermark-text
$waterm
ark-text

inactivation by Angeli’s salt involved s-nitrosylation. To this end, we used a biotin-switchmethod [41] followed by localization of s-nitrosylated proteins by 2D Western blot probedwith streptavidin linked with horse radish peroxidase. DLDH was localized by 2D Westernblots probed with anti-DLDH antibodies. Results are shown in Fig. 7. Fig. 7A shows a 2DWestern blot map in the absence of ascorbate that was used to reduce protein nitrosothiolsback to free thiols for biotinylation and detection. As can be seen in Fig. 7B, morenitrosylated proteins could be visualized by the use of ascorbate and the arrow-indicatedspot was that of DLDH. The identity was further confirmed by blotting the same membranewith anti-DLDH antibodies (Fig. 7C) as spot in Fig. 7C overlapped with that indicated inFig. 7B. Moreover, when the same spot in Fig. 7B was excised and analyzed by 1D Westernblot probed with anti-DLDH antibodies, a positive immunostaining could also be visualized(Fig. 7D), which further confirmed the spot’s identity in Fig. 7B as that of DLDH. Theseresults indicate that DLDH inactivation by Angeli’s salt likely involved s-nitrosylation.
DiscussionThe main findings of the present study are as the following: 1) mitochondrial DLDH couldbe reversibly inactivated by Angeli’s salt; 2) the inactivation was a targeting rather than arandom process as peroxynitrite, at similar concentrations, did not show any inhibitoryeffect on DLDH activity; 3) the RNS that actually inactivated DLDH was HNO derivedfrom Angeli’s salt; and 4) the inactivation likely involved formation of s-nitrosothiols onDLDH.
It is noteworthy that when incubated with whole mitochondria, Angeli’s salt seemed totarget DLDH. In contrast, peroxynitrite lacked this targeting ability in the presence of othermitochondrial proteins (Fig. 3); although peroxynitrite, when incubated with purifiedDLDH, could indeed inactivate the enzyme (unpublished data, this laboratory). Suchobservations suggest that peroxynitrite is consumed by other mitochondrial proteins before itcan get to DLDH when whole mitochondria are treated by peroxynitrite. Alternatively,peroxynitrite, at the same concentration as that of Angeli’s salt, may not be as effective asAngeli’s salt in inactivating DLDH. Therefore, no inhibitory effect of peroxynitrite could bedetected under our experimental conditions. Regardless, it is suffice to say that inactivationof DLDH by Angeli’s salt is a targeting process.
Angeli’s salt is a unique donor of RNS in that it can decompose readily at physiological pHwith the production of two reactive nitrogen species, i.e., HNO and nitrite. Nevertheless,most of Angeli’s salt actions on proteins have been attributed to HNO [51]. In the presentstudy, we also found that it was indeed HNO, but not nitrite, that actually inactivated DLDH(Fig. 3). This result is therefore in consistent with those by others that HNO is the actualspecies that imposes the biochemical and pharmacological effects of Angeli’s salt [36, 37]. Itshould be noted that nitric oxide can also be produced when Angeli’s salt is incubated withferricyanide [52]. Nonetheless, nitric oxide generated this way apparently had no detectableinhibitory effect on DLDH activity (Fig. 3).
While we determined that DLDH inactivation by Angeli’s salt likely involved the formationof s-nitrosothiols, we were unable to determine which of DLDH’s redox-sensitive cysteineresidues that were actually nitrosylated and led to decrease in DLDH activity. Our attempt toanalyze modified cysteine residues by mass spectrometry techniques failed mainly becausethe peptide containing the two cysteine residues could not be recovered following HPLC andmass spectrometry analysis. Therefore, the redox cysteine residue(s) that was nitrosylatedcould only be speculated. DLDH has two cysteine residues at its active center that areengaged in catalysis, namely, cys45 and cys50 [53]. During catalysis, cys45 is the substrate-binding residue while cys50 is the FAD interacting residue. Studies by others have indicated
Yan et al. Page 6
Sheng Wu Wu Li Hsueh Bao. Author manuscript; available in PMC 2012 November 06.
$waterm
ark-text$w
atermark-text
$waterm
ark-text

that cys45 is more reactive than cys50 toward thiol-reactive reagents [6]. In glutathionereductase whose structure is similar to that of DLDH, the substrate binding cysteine residuecould be nitrosylated and sulfenated upon treatment with nitric oxide donors [54]. Suchstudies suggest that the nitrosylated cysteine residue on DLDH is likely that of cys45following treatment with Angeli’s salt.
It is well-established that protein s-nitrosylation is a reversible process and is involved notonly in oxidative damage [55], but also in regulation and expansion of protein function [56].For example, s-nitrosylation can contribute to protein misfolding and neuronal synapticdamage [55]; s-nitrosylation of mitochondrial proteins can also be involved in ischemicpreconditioning that protects hearts against subsequent, severe ischemic injury [57]. Whilethe physiological and pathophysiological significance of DLDH nitrosylation remains to bedetermined, such modification will certainly impair the enzyme’s function that may furtherperturb mitochondrial oxidative phosphorylation. It should be noted that while it has beenpreviously reported that DLDH underwent s-nitrosylation [58, 59], none of these earlierstudies examined the relationship between s-nitrosylation and loss of enzyme activity. Incontrast, our present data suggest that nitrosylation was likely responsible, at least in part,for the loss of DLDH activity.
It should be pointed out that the ascorbate/biotin switch method may yield a false-positivesignal in detection of protein S-nitrosylation [60]. Therefore, it might be argued that ourobservation of DLDH nitrosylation may have nothing to do with the observed reversibleinactivation of DLDH function. This is the reason why we have only cautiously concludedthat S-nitrosylation is probably the cause of DLDH reversible inactivation. A definitiveconclusion can only be reached when both control and treated samples undergo theprocedures of ascorbate reduction and biotin switch, whereby no positive or less intensivenitrosylation signals will be detected in the control samples. On the other hand, it is alsopossible that formation of protein sulfenic acids (S-sulfenation) could be partiallyresponsible for the reversible inactivation of DLDH function observed in the present studyas protein sulfenic acids can be readily derived from protein S-nitrosothiols [61]. If this isindeed the case, DLDH nitrosylation induced by Angeli’s salt would be only a transientform of DLDH modification and may only be considered partially responsible for the loss inDLDH activity. In other words, both S-nitrosylation and S-sulfenation may be involved inloss of DLDH activity induced by Angeli’s salt. Regardless, whether Angeli’s salt caninduce DLDH sulfenic acid formation under our experimental conditions will need to befurther investigated.
In summary, our study has indicated that brain mitochondrial DLDH could be inactivated ina dose-dependent manner by Angeli’s salt and the inactivation was caused by HNO. Ourresults have also revealed that DLDH inactivation by HNO was a targeting process and wasaccompanied by HNO-induced S-nitrosylation. Overall, the present study provides insightsinto the mechanisms of DLDH inactivation by Angeli’s salt, albeit that further studies areneeded to determine which cysteine residues are nitrosylated.
AcknowledgmentsThis work was supported in part by the National Institutes of Health (AG022550).
References1. Williams, CHJ. Chemistry and Biochemistry of Flavoenzymes. Muller, F., editor. CRC Press; Boca
Raton: 1992. p. 121-212.
2. Patel MS, Roche TE. Molecular biology and biochemistry of pyruvate dehydrogenase complexes.FASEB J. 1990; 4(14):3224–3233. [PubMed: 2227213]
Yan et al. Page 7
Sheng Wu Wu Li Hsueh Bao. Author manuscript; available in PMC 2012 November 06.
$waterm
ark-text$w
atermark-text
$waterm
ark-text

3. Neuburger M, Polidori AM, Pietre E, Faure M, Jourdain A, Bourguignon J, Pucci B, Douce R.Interaction between the lipoamide-containing H-protein and the lipoamide dehydrogenase (L-protein) of the glycine decarboxylase multienzyme system. 1 Biochemical studies. Eur J Biochem.2000; 267(10):2882–2889. [PubMed: 10806385]
4. Faure M, Bourguignon J, Neuburger M, MacHerel D, Sieker L, Ober R, Kahn R, Cohen-Addad C,Douce R. Interaction between the lipoamide-containing H-protein and the lipoamide dehydrogenase(L-protein) of the glycine decarboxylase multienzyme system 2. Crystal structures of H- and L-proteins. Eur J Biochem. 2000; 267(10):2890–2898. [PubMed: 10806386]
5. Ciszak EM, Makal A, Hong YS, Vettaikkorumakankauv AK, Korotchkina LG, Patel MS. Howdihydrolipoamide dehydrogenase-binding protein binds dihydrolipoamide dehydrogenase in thehuman pyruvate dehydrogenase complex. J Biol Chem. 2006; 281(1):648–655. [PubMed:16263718]
6. Thorpe C, Williams CH Jr. Differential reactivity of the two active site cysteine residues generatedon reduction of pig heart lipoamide dehydrogenase. J Biol Chem. 1976; 251(12):3553–3557.[PubMed: 6457]
7. Brautigam CA, Chuang JL, Tomchick DR, Machius M, Chuang DT. Crystal structure of humandihydrolipoamide dehydrogenase: NAD+/NADH binding and the structural basis of disease-causingmutations. J Mol Biol. 2005; 350(3):543–552. [PubMed: 15946682]
8. Vettakkorumakankav NN, Patel MS. Dihydrolipoamide dehydrogenase: structural and mechanisticaspects. Indian J Biochem Biophys. 1996; 33(3):168–176. [PubMed: 8828286]
9. Massey V, Gibson QH, Veeger C. Intermediates in the catalytic action of lipoyl dehydrogenase(diaphorase). Biochem J. 1960; 77:341–351. [PubMed: 13767908]
10. Olsson JM, Xia L, Eriksson LC, Bjornstedt M. Ubiquinone is reduced by lipoamide dehydrogenaseand this reaction is potently stimulated by zinc. FEBS Lett. 1999; 448(1):190–192. [PubMed:10217438]
11. Xia L, Bjornstedt M, Nordman T, Eriksson LC, Olsson JM. Reduction of ubiquinone by lipoamidedehydrogenase. An antioxidant regenerating pathway. Eur J Biochem. 2001; 268(5):1486–1490.[PubMed: 11231302]
12. Igamberdiev AU, Bykova NV, Ens W, Hill RD. Dihydrolipoamide dehydrogenase from porcineheart catalyzes NADH-dependent scavenging of nitric oxide. FEBS Lett. 2004; 568(1–3):146–150.[PubMed: 15196936]
13. Scouten WH, McManus IR. Microbial lipoamide dehydrogenase. Purification and somecharacteristics of the enzyme derived from selected microorganisms. Biochim Biophys Acta.1971; 227(2):248–263. [PubMed: 4323856]
14. Sokatch JR, McCully V, Gebrosky J, Sokatch DJ. Isolation of a specific lipoamide dehydrogenasefor a branched-chain keto acid dehydrogenase from Pseudomonas putida. J Bacteriol. 1981;148(2):639–646. [PubMed: 6895373]
15. Patel MS, Vettakkorumakankav NN, Liu TC. Dihydrolipoamide dehydrogenase: activity assays.Methods Enzymol. 1995; 252:186–195. [PubMed: 7476353]
16. Hengeveld AF, de Kok A. Structural basis of the dysfunctioning of human 2-oxo aciddehydrogenase complexes. Curr Med Chem. 2002; 9(4):499–520. [PubMed: 11945122]
17. Lai MW, Boyer EW, Kleinman ME, Rodig NM, Ewald MB. Acute arsenic poisoning in twosiblings. Pediatrics. 2005; 116(1):249–257. [PubMed: 15995066]
18. Sreider CM, Grinblat L, Stoppani AO. Catalysis of nitrofuran redox-cycling and superoxide anionproduction by heart lipoamide dehydrogenase. Biochem Pharmacol. 1990; 40(8):1849–1857.[PubMed: 2173592]
19. Bando Y, Aki K. Mechanisms of generation of oxygen radicals and reductive mobilization offerritin iron by lipoamide dehydrogenase. J Biochem (Tokyo). 1991; 109(3):450–454. [PubMed:1652585]
20. Gazaryan IG, Krasnikov BF, Ashby GA, Thorneley RN, Kristal BS, Brown AM. Zinc is a potentinhibitor of thiol oxidoreductase activity and stimulates reactive oxygen species production bylipoamide dehydrogenase. J Biol Chem. 2002; 277(12):10064–10072. [PubMed: 11744691]
Yan et al. Page 8
Sheng Wu Wu Li Hsueh Bao. Author manuscript; available in PMC 2012 November 06.
$waterm
ark-text$w
atermark-text
$waterm
ark-text

21. Tahara EB, Barros MH, Oliveira GA, Netto LE, Kowaltowski AJ. Dihydrolipoyl dehydrogenase asa source of reactive oxygen species inhibited by caloric restriction and involved in Saccharomycescerevisiae aging. Faseb J. 2007; 21(1):274–283. [PubMed: 17110466]
22. Ambrus A, Torocsik B, Tretter L, Ozohanics O, Adam-Vizi V. Stimulation of reactive oxygenspecies generation by disease-causing mutations of lipoamide dehydrogenase. Hum Mol Genet.2011; 20(15):2984–2995. [PubMed: 21558426]
23. Hussain SN, Matar G, Barreiro E, Florian M, Divangahi M, Vassilakopoulos T. Modifications ofproteins by 4-hydroxy-2-nonenal in the ventilatory muscles of rats. Am J Physiol Lung Cell MolPhysiol. 2006; 290(5):L996–1003. [PubMed: 16603597]
24. Lee HM, Reed J, Greeley GH Jr, Englander EW. Impaired mitochondrial respiration and proteinnitration in the rat hippocampus after acute inhalation of combustion smoke. Toxicol ApplPharmacol. 2009; 235(2):208–215. [PubMed: 19133281]
25. Pankotai E, Lacza Z, Muranyi M, Szabo C. Intra-mitochondrial poly(ADP-ribosyl)ation: potentialrole for alpha-ketoglutarate dehydrogenase. Mitochondrion. 2009; 9(2):159–164. [PubMed:19460292]
26. Kehr S, Jortzik E, Delahunty C, Yates JR 3rd, Rahlfs S, Becker K. Protein s-glutathionylation inmalaria parasites. Antioxid Redox Signal. 2011; 15(11):2855–2865. [PubMed: 21595565]
27. Tyther R, Ahmeda A, Johns E, Sheehan D. Protein carbonylation in kidney medulla of thespontaneously hypertensive rat. Proteomics Clin Appl. 2009; (3):338–346.
28. Tyther R, Ahmeda A, Johns E, Sheehan D. Proteomic identification of tyrosine nitration targets inkidney of spontaneously hypertensive rats. Proteomics. 2007; 7(24):4555–4564. [PubMed:18072209]
29. Korotchkina LG, Yang H, Tirosh O, Packer L, Patel MS. Protection by thiols of the mitochondrialcomplexes from 4-hydroxy-2-nonenal. Free Radic Biol Med. 2001; 30(9):992–999. [PubMed:11316579]
30. Babady NE, Pang YP, Elpeleg O, Isaya G. Cryptic proteolytic activity of dihydrolipoamidedehydrogenase. Proc Natl Acad Sci U S A. 2007; 104(15):6158–6163. [PubMed: 17404228]
31. Vaubel RA, Rustin P, Isaya G. Mutations in the Dimer Interface of DihydrolipoamideDehydrogenase Promote Site-specific Oxidative Damages in Yeast and Human Cells. J BiolChem. 2011; 286(46):40232–40245. [PubMed: 21930696]
32. Yan LJ, Yang SH, Shu H, Prokai L, Forster MJ. Histochemical staining and quantification ofdihydrolipoamide dehydrogenase diaphorase activity using blue native PAGE. Electrophoresis.2007; 28(7):1036–1045. [PubMed: 17315258]
33. Favaloro JL, Kemp-Harper BK. The nitroxyl anion (HNO) is a potent dilator of rat coronaryvasculature. Cardiovasc Res. 2007; 73(3):587–596. [PubMed: 17189622]
34. Irvine JC, Favaloro JL, Widdop RE, Kemp-Harper BK. Nitroxyl anion donor, Angeli’s salt, doesnot develop tolerance in rat isolated aortae. Hypertension. 2007; 49(4):885–892. [PubMed:17309955]
35. Yuill KH, Yarova P, Kemp-Harper BK, Garland CJ, Dora KA. A novel role for HNO in local andspreading vasodilatation in rat mesenteric resistance arteries. Antioxid Redox Signal. 2011; 14(9):1625–1635. [PubMed: 20615121]
36. Fukuto JM, Switzer CH, Miranda KM, Wink DA. Nitroxyl (HNO): chemistry, biochemistry,pharmacology. Annu Rev Pharmacol Toxicol. 2005; 45:335–355. [PubMed: 15822180]
37. Flores-Santana W, Salmon DJ, Donzelli S, Switzer CH, Basudhar D, Ridnour L, Cheng R, GlynnSA, Paolocci N, Fukuto JM, Miranda KM, Wink DA. The specificity of nitroxyl chemistry isunique among nitrogen oxides in biological systems. Antioxid Redox Signal. 2011; 14(9):1659–1674. [PubMed: 21235346]
38. Patel, MS.; Hong, YS. Free Radical and Antioxidant Protocols. Armstrong, D., editor. HumanaPress; Totowa, NJ: 1998. p. 337-346.
39. Sims, NR. Methods in Toxicology: Mitochondrial Dysfunction. Vol. 2. Academic Press; SanDiego: 1993.
40. Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK,Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. AnalBiochem. 1985; 150(1):76–85. [PubMed: 3843705]
Yan et al. Page 9
Sheng Wu Wu Li Hsueh Bao. Author manuscript; available in PMC 2012 November 06.
$waterm
ark-text$w
atermark-text
$waterm
ark-text

41. Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. SciSTKE. 2001; 2001(86):pl1. [PubMed: 11752655]
42. Yan LJ. Analysis of oxidative modification of proteins. Curr Protoc Protein Sci. 2009; Chapter14(Unit14):4.
43. Khalkhali-Ellis Z. An improved SDS-polyacrylamide gel electrophoresis for resolution of peptidesin the range of 3.5–200kDa, Prep Biochem, 1995; 25(1–2): 1–9 5–200kDa. Prep Biochem. 1995;25(1–2):1–9. [PubMed: 7603968]
44. Yan LJ, Forster MJ. Resolving mitochondrial protein complexes using nongradient blue nativepolyacrylamide gel electrophoresis. Anal Biochem. 2009; 389(2):143–149. [PubMed: 19348780]
45. Miranda KM, Dutton AS, Ridnour LA, Foreman CA, Ford E, Paolocci N, Katori T, Tocchetti CG,Mancardi D, Thomas DD, Espey MG, Houk KN, Fukuto JM, Wink DA. Mechanism of aerobicdecomposition of Angeli’s salt (sodium trioxodinitrate) at physiological pH. J Am Chem Soc.2005; 127(2):722–731. [PubMed: 15643898]
46. Amatore C, Arbault S, Ducrocq C, Hu S, Tapsoba I. Angeli’s salt (Na2N2O3) is a precursor ofHNO and NO: a voltammetric study of the reactive intermediates released by Angeli’s saltdecomposition. ChemMedChem. 2007; 2(6):898–903. [PubMed: 17436261]
47. Ma XL, Gao F, Liu GL, Lopez BL, Christopher TA, Fukuto JM, Wink DA, Feelisch M. Oppositeeffects of nitric oxide and nitroxyl on postischemic myocardial injury. Proc Natl Acad Sci U S A.1999; 96(25):14617–14622. [PubMed: 10588754]
48. Liu M, Hou J, Huang L, Huang X, Heibeck TH, Zhao R, Pasa-Tolic L, Smith RD, Li Y, Fu K,Zhang Z, Hinrichs SH, Ding SJ. Site-specific proteomics approach for study protein S-nitrosylation. Anal Chem. 2010; 82(17):7160–7168. [PubMed: 20687582]
49. Huang B, Chen C. Detection of protein S-nitrosation using irreversible biotinylation procedures(IBP). Free Radic Biol Med. 2010; 49(3):447–456. [PubMed: 20466056]
50. Ghezzi P. Oxidoreduction of protein thiols in redox regulation. Biochem Soc Trans. 2005; 33(Pt6):1378–1381. [PubMed: 16246123]
51. Flores-Santana W, Switzer C, Ridnour LA, Basudhar D, Mancardi D, Donzelli S, Thomas DD,Miranda KM, Fukuto JM, Wink DA. Comparing the chemical biology of NO and HNO. ArchPharm Res. 2009; 32(8):1139–1153. [PubMed: 19727606]
52. Mitchell JB, DeGraff W, Kim S, Cook JA, Gamson J, Christodoulou D, Feelisch M, Wink DA.Redox generation of nitric oxide to radiosensitize hypoxic cells. Int J Radiat Oncol Biol Phys.1998; 42(4):95–0798.
53. Yan LJ, Thangthaeng N, Forster MJ. Changes in dihydrolipoamide dehydrogenase expression andactivity during postnatal development and aging in the rat brain. Mech Ageing Dev. 2008;129:282–290. [PubMed: 18316113]
54. Becker K, Savvides SN, Keese M, Schirmer RH, Karplus PA. Enzyme inactivation throughsulfhydryl oxidation by physiologic NO-carriers. Nat Struct Biol. 1998; 5(4):267–271. [PubMed:9546215]
55. Nakamura T, Lipton SA. Redox modulation by S-nitrosylation contributes to protein misfolding,mitochondrial dynamics, and neuronal synaptic damage in neurodegenerative diseases. Cell DeathDiffer. 2011; 18(9):1478–1486. [PubMed: 21597461]
56. Lima B, Forrester MT, Hess DT, Stamler JS. S-nitrosylation in cardiovascular signaling. Circ Res.2010; 106(4):633–646. [PubMed: 20203313]
57. Sun J, Kohr MJ, Nguyen T, Aponte AM, Connelly PS, Esfahani SG, Gucek M, Daniels MP,Steenbergen C, Murphy E. Disruption of caveolae blocks ischemic preconditioning-mediated S-nitrosylation of mitochondrial proteins. Antioxid Redox Signal. 2012; 16(1):45–56. [PubMed:21834687]
58. Foster MW, Stamler JS. New insights into protein S-nitrosylation. Mitochondria as a modelsystem. J Biol Chem. 2004; 279(24):25891–25897. [PubMed: 15069080]
59. Rhee KY, Erdjument-Bromage H, Tempst P, Nathan CF. S-nitroso proteome of Mycobacteriumtuberculosis: Enzymes of intermediary metabolism and antioxidant defense. Proc Natl Acad Sci US A. 2005; 102(2):467–472. [PubMed: 15626759]
60. Huang B, Chen C. An ascorbate-dependent artifact that interferes with the interpretation of thebiotin switch assay. Free Radic Biol Med. 2006; 41(4):562–567. [PubMed: 16863989]
Yan et al. Page 10
Sheng Wu Wu Li Hsueh Bao. Author manuscript; available in PMC 2012 November 06.
$waterm
ark-text$w
atermark-text
$waterm
ark-text

61. Poole LB, Karplus PA, Claiborne A. Protein sulfenic acids in redox signaling. Annu RevPharmacol Toxicol. 2004; 44:325–347. [PubMed: 14744249]
Yan et al. Page 11
Sheng Wu Wu Li Hsueh Bao. Author manuscript; available in PMC 2012 November 06.
$waterm
ark-text$w
atermark-text
$waterm
ark-text

Fig. 1.Effect of Angeli’s salt on DLDH diaphorase activity. Mitochondria were incubated withvarying concentrations of Angeli’s salt at room temperature for 30 min followed bymitochondrial extract preparation in an hypotonic buffer containing 20 mM potassiumphosphate (pH 7.4), 1% Triton X-100, 2 mM EDTA, 2 mM EGTA. DLDH was resolved byblue native PAGE followed by diaphorase activity staining using the NBT/NADH system asdescribed in the text. A shows the gel image of activity staining; B shows quantification ofrelative diaphorase activity derived by scanning the band intensity in A. Values (mean ±SEM) are expressed as percentage of control that contained no Angeli’s salt; N=3.
Yan et al. Page 12
Sheng Wu Wu Li Hsueh Bao. Author manuscript; available in PMC 2012 November 06.
$waterm
ark-text$w
atermark-text
$waterm
ark-text

Fig. 2.Effect of Angeli’s salt on DLDH dehydrogenase activity measured spectrophotometrically.Mitochondria incubation with Angeli’s salt and preparation of mitochondrial extracts wereperformed as described in Fig. 1. The spectrophotometric measurement was performed bymonitoring the increase of NADH absorbance at 340 nm upon DLDH-mediated, NAD+-dependent oxidation of dihydrolipoamide. A solution containing all assay componentsexcept mitochondrial sample was used as the blank. Data are mean ± SEM of threeindependent experiments.
Yan et al. Page 13
Sheng Wu Wu Li Hsueh Bao. Author manuscript; available in PMC 2012 November 06.
$waterm
ark-text$w
atermark-text
$waterm
ark-text
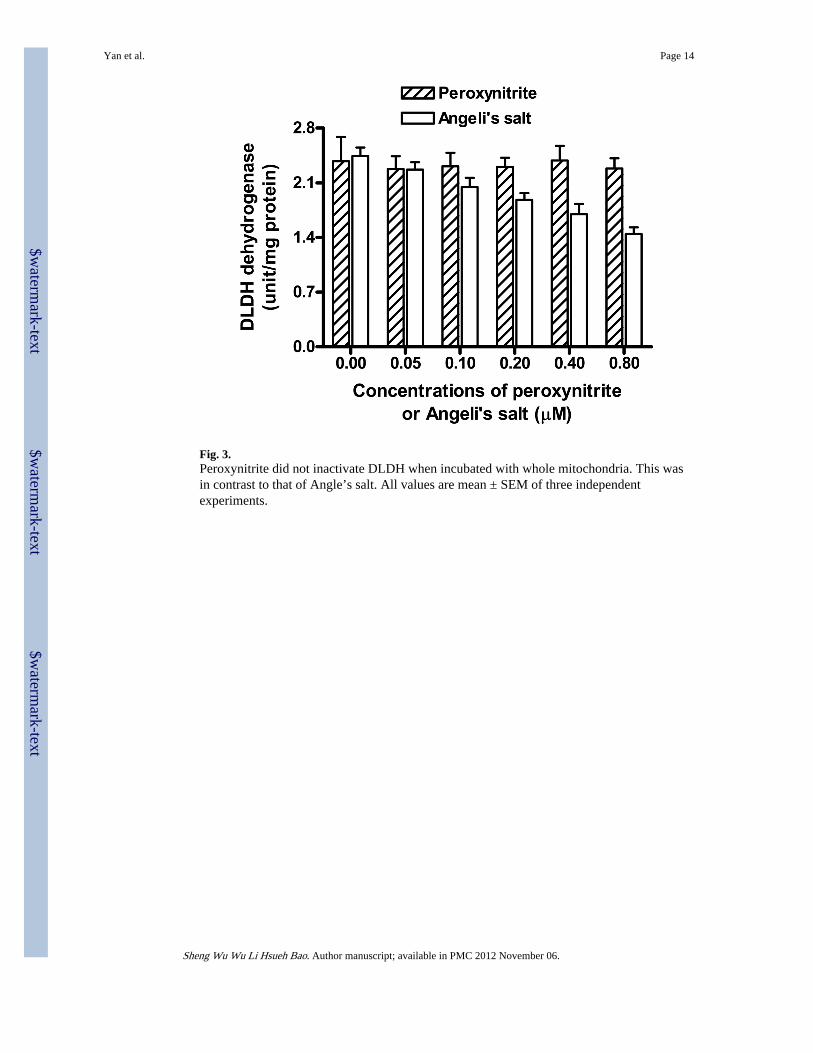
Fig. 3.Peroxynitrite did not inactivate DLDH when incubated with whole mitochondria. This wasin contrast to that of Angle’s salt. All values are mean ± SEM of three independentexperiments.
Yan et al. Page 14
Sheng Wu Wu Li Hsueh Bao. Author manuscript; available in PMC 2012 November 06.
$waterm
ark-text$w
atermark-text
$waterm
ark-text

Fig. 4.No changes in dehydrogenase activity could be observed after mitochondria were incubatedwith (A) nitrite or (B) Angeli’s salt + ferricyanide. Mitochondria incubation with theindicated chemicals, mitochondrial extract preparation, and dehydrogenase activitymeasurement were performed as described in Fig. 2. In (B), equal concentrations of Angeli’ssalt and ferricyanide were present in the same incubation. Data are mean ± SEM of threeindependent experiments.
Yan et al. Page 15
Sheng Wu Wu Li Hsueh Bao. Author manuscript; available in PMC 2012 November 06.
$waterm
ark-text$w
atermark-text
$waterm
ark-text

Fig. 5.Loss of dehydrogenase activity induced by Angeli’s salt could be restored by reducingreagents such as DTT, cysteine, and GSH (10 mM each). After mitochondria incubationwith Angeli’s salt, mitochondrial extracts were prepared in the presence of the respectivereducing reagent. This was followed by spectrophotometric measurement of dehydrogenaseactivity. Values, expressed as percentage of control, are mean ± SEM of three independentexperiments. *p<0.05 compared with control; *<0.05 compared with AS. Welch’s t test wasperformed.
Yan et al. Page 16
Sheng Wu Wu Li Hsueh Bao. Author manuscript; available in PMC 2012 November 06.
$waterm
ark-text$w
atermark-text
$waterm
ark-text

Fig. 6.Loss of diaphorase activity induced by Angle’s salt could be restored by cysteine and GSH,but not by DTT. (A) BN-PAGE measurement of DLDH diaphorase activity. (B)Densitometric values derived from (A). Values, expressed as percentage of control in eachcondition, are mean ± SEM of three independent experiments. *p<0.05 compared withcontrol; *<0.05 compared with AS. Welch’s t test was performed.
Yan et al. Page 17
Sheng Wu Wu Li Hsueh Bao. Author manuscript; available in PMC 2012 November 06.
$waterm
ark-text$w
atermark-text
$waterm
ark-text

Fig. 7.Two dimensional Western blot detection of DLDH nitrosylation by Angeli’s salt. Sampleswere biotinylated in the absence (A) or presence (B) of ascorbate, a specific reducingreagent for protein s-nitrosothiols. In (B) the spot indicated by the arrow was that of DLDH.(C) Confirmation of the spot as that of DLDH by anti-DLDH Western blot assay. The samemembrane as used in B was used. (D) When the arrow-pointed spot in B was excised andanalyzed by 1D Western blot probed with anti-DLDH antibodies, DLDH was also detected,further confirming the arrow-pointed spot in B as that of DLDH
Yan et al. Page 18
Sheng Wu Wu Li Hsueh Bao. Author manuscript; available in PMC 2012 November 06.
$waterm
ark-text$w
atermark-text
$waterm
ark-text
Copyright © 2022 FDOKUMEN




















