
Cryptococcus neoformans Kin1 protein kinase homologue, identified through a Caenorhabditis elegans...
13
Molecular Microbiology (2004) 54(2), 407–419 doi:10.1111/j.1365-2958.2004.04310.x © 2004 Blackwell Publishing Ltd Blackwell Science, LtdOxford, UKMMIMolecular Microbiology0950-382XBlackwell Publishing Ltd, 2004 ? 2004542407419Original ArticleC. elegans/C. neoformans systemE. Mylonakis et al. Accepted 14 July, 2004. *For correspondence. E-mail [email protected]; Tel. (+1) 617 726 3811; Fax (+1) 617 726 7416. Cryptococcus neoformans Kin1 protein kinase homologue, identified through a Caenorhabditis elegans screen, promotes virulence in mammals Eleftherios Mylonakis, 1 Alexander Idnurm, 2 Roberto Moreno, 1 Joseph El Khoury, 1,3,4 James B. Rottman, 5 Frederick M. Ausubel, 6,7 Joseph Heitman 2,8,9,10 and Stephen B. Calderwood 1,11 * 1 Division of Infectious Diseases, Massachusetts General Hospital, Boston, MA 02114, USA. 2 Department of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, NC 27710, USA. 3 Center for Immunology and Inflammatory Diseases, and 4 Division of Rheumatology, Allergy, and Immunology, Massachusetts General Hospital, Boston, MA 02114, USA. 5 Archemix Corporation, Cambridge, MA 02139, USA. 6 Department of Molecular Biology, Massachusetts General Hospital, Boston, MA 02114, USA. 7 Department of Genetics, Harvard Medical School, Boston, MA 02115, USA. 8 Division of Infectious Diseases, 9 Department of Medicine, and 10 Howard Hughes Medical Institute, Duke University Medical Center, Durham, NC 27710, USA. 11 Department of Microbiology and Molecular Genetics, Harvard Medical School, Boston, MA 02115, USA. Summary Cryptococcal infections are a global cause of signifi- cant morbidity and mortality. Recent studies support the hypothesis that virulence of Cryptococcus neofor- mans may have evolved via survival selection in envi- ronmental hosts, such as amoebae and free-living nematodes. We used killing of the nematode Cae- norhabditis elegans by C. neoformans as an assay to screen a library of random C. neoformans insertion mutants. Of 350 mutants tested, seven were identified with attenuated virulence that persisted after crossing the mutation back into a wild-type strain . Genetic analysis of one strain revealed an insertion in a gene homologous to Saccharomyces cerevisiae KIN1 , which encodes a serine/threonine protein kinase. C. neoformans kin1 mutants exhibited significant defects in virulence in murine inhalation and hae- matogenous infection models and displayed increased binding to alveolar and peritoneal mac- rophages. The kin1 mutant phenotypes were comple- mented by the wild-type KIN1 gene. These findings show that the C. neoformans Kin1 kinase homologue is required for full virulence in disparate hosts and that C. elegans can be used as a substitute host to identify novel factors involved in fungal pathogenesis in mammals. Introduction Cryptococcus neoformans , an emerging human fungal pathogen, exists in the yeast form in its natural habitat as well as in animal and human hosts. Although C. neofor- mans can cause pulmonary or disseminated disease in normal hosts, disease is usually seen in immunocompro- mised hosts. There has been an upsurge in the incidence of cryptococcosis associated with HIV infection, especially in areas with limited access to anti-retroviral therapy, including Africa, Thailand and India (Perfect and Casade- vall, 2002). C. neoformans is also the third most common cause of invasive fungal infection in solid organ transplant recipients (Mueller and Fishman, 2003). In addition to being an important human pathogen, C. neoformans is an excellent model organism to study vir- ulence of human pathogenic fungi. It grows as a haploid yeast and can be readily transformed, allowing gene dis- ruption by homologous recombination. The genomes of three C. neoformans strains are being sequenced (including strain H99 used in our studies). Recently developed congenic mating partners enable genetic anal- ysis of mutations and studies of genetic linkage in C. neoformans serotype A, the most common cause of human cryptococcal disease (Mitchell, 2003; Nielsen et al ., 2003). The study of virulence determinants of C. neoformans in a number of non-mammalian hosts, including amoebae (Steenbergen et al ., 2001), Caenorhabditis elegans (Mylonakis et al ., 2002), Dictyostelium discoideum (Steenbergen et al ., 2003) and Drosophila melanogaster (Apidianakis et al ., 2004), supports the idea that mamma-
Transcript of Cryptococcus neoformans Kin1 protein kinase homologue, identified through a Caenorhabditis elegans...

Molecular Microbiology (2004)
54
(2), 407–419 doi:10.1111/j.1365-2958.2004.04310.x
© 2004 Blackwell Publishing Ltd
Blackwell Science, LtdOxford, UKMMIMolecular Microbiology0950-382XBlackwell Publishing Ltd, 2004
? 2004
54
2407419
Original Article
C. elegans/C. neoformans systemE. Mylonakis
et al.
Accepted 14 July, 2004. *For correspondence. [email protected]; Tel. (+1) 617 726 3811; Fax (+1)617 726 7416.
Cryptococcus neoformans
Kin1 protein kinase homologue, identified through a
Caenorhabditis elegans
screen, promotes virulence in mammals
Eleftherios Mylonakis,
1
Alexander Idnurm,
2
Roberto Moreno,
1
Joseph El Khoury,
1,3,4
James B. Rottman,
5
Frederick M. Ausubel,
6,7
Joseph Heitman
2,8,9,10
and Stephen B. Calderwood
1,11
*
1
Division of Infectious Diseases, Massachusetts General Hospital, Boston, MA 02114, USA.
2
Department of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, NC 27710, USA.
3
Center for Immunology and Inflammatory Diseases, and
4
Division of Rheumatology, Allergy, and Immunology, Massachusetts General Hospital, Boston, MA 02114, USA.
5
Archemix Corporation, Cambridge, MA 02139, USA.
6
Department of Molecular Biology, Massachusetts General Hospital, Boston, MA 02114, USA.
7
Department of Genetics, Harvard Medical School, Boston, MA 02115, USA.
8
Division of Infectious Diseases,
9
Department of Medicine, and
10
Howard Hughes Medical Institute, Duke University Medical Center, Durham, NC 27710, USA.
11
Department of Microbiology and Molecular Genetics, Harvard Medical School, Boston, MA 02115, USA.
Summary
Cryptococcal infections are a global cause of signifi-cant morbidity and mortality. Recent studies supportthe hypothesis that virulence of
Cryptococcus neofor-mans
may have evolved via survival selection in envi-ronmental hosts, such as amoebae and free-livingnematodes. We used killing of the nematode
Cae-norhabditis elegans
by
C. neoformans
as an assay toscreen a library of random
C. neoformans
insertionmutants. Of 350 mutants tested, seven were identifiedwith attenuated virulence that persisted after crossingthe mutation back into a wild-type strain
.
Geneticanalysis of one strain revealed an insertion in a genehomologous to
Saccharomyces cerevisiae KIN1
,which encodes a serine/threonine protein kinase.
C. neoformans kin1
mutants exhibited significantdefects in virulence in murine inhalation and hae-matogenous infection models and displayedincreased binding to alveolar and peritoneal mac-rophages. The
kin1
mutant phenotypes were comple-mented by the wild-type
KIN1
gene. These findingsshow that the
C. neoformans
Kin1 kinase homologueis required for full virulence in disparate hosts andthat
C. elegans
can be used as a substitute host toidentify novel factors involved in fungal pathogenesisin mammals.
Introduction
Cryptococcus neoformans
, an emerging human fungalpathogen, exists in the yeast form in its natural habitat aswell as in animal and human hosts. Although
C. neofor-mans
can cause pulmonary or disseminated disease innormal hosts, disease is usually seen in immunocompro-mised hosts. There has been an upsurge in the incidenceof cryptococcosis associated with HIV infection, especiallyin areas with limited access to anti-retroviral therapy,including Africa, Thailand and India (Perfect and Casade-vall, 2002).
C. neoformans
is also the third most commoncause of invasive fungal infection in solid organ transplantrecipients (Mueller and Fishman, 2003).
In addition to being an important human pathogen,
C.neoformans
is an excellent model organism to study vir-ulence of human pathogenic fungi. It grows as a haploidyeast and can be readily transformed, allowing gene dis-ruption by homologous recombination. The genomes ofthree
C. neoformans
strains are being sequenced(including strain H99 used in our studies). Recentlydeveloped congenic mating partners enable genetic anal-ysis of mutations and studies of genetic linkage in
C.neoformans
serotype A, the most common cause ofhuman cryptococcal disease (Mitchell, 2003; Nielsen
et al
., 2003).The study of virulence determinants of
C. neoformans
in a number of non-mammalian hosts, including amoebae(Steenbergen
et al
., 2001),
Caenorhabditis elegans
(Mylonakis
et al
., 2002),
Dictyostelium discoideum
(Steenbergen
et al
., 2003) and
Drosophila melanogaster
(Apidianakis
et al
., 2004), supports the idea that mamma-

408
E. Mylonakis
et al.
© 2004 Blackwell Publishing Ltd,
Molecular Microbiology
,
54
, 407–419
lian pathogenesis of
C. neoformans
may be a conse-quence of adaptations that have evolved during theinteraction of
C. neoformans
with environmental preda-tors. Indeed, studies using the free-living nematode
C.elegans
have demonstrated that
C. neoformans
kills
C.elegans
and that the
C. neoformans
polysaccharide cap-sule, as well as several
C. neoformans
genes previouslyshown to be involved in mammalian virulence, are impor-tant for
C. elegans
killing. These genes include thoseassociated with signal transduction pathways (
GPA1
,
PKA1
,
PKR1
and
RAS1
) that control capsule formation,melanin production and mating in response to environ-mental signals, and laccase (
LAC1
), an enzyme catalys-ing melanin biosynthesis (Mylonakis
et al
., 2002).Because
C. neoformans
is an important human patho-gen and an excellent model of fungal pathogenesis,because molecular biology tools are now available for thisorganism and because of significant overlap in the viru-lence determinants required for
C. elegans
killing andmammalian virulence, we exploited the
C. neoformans
/
C. elegans
pathogenesis model to identify novel genesassociated with cryptococcal virulence. Specifically, wedevised a system for screening a pool of random insertionmutants of
C. neoformans
to identify virulence factorsassociated with killing of
C. elegans
. At least one of the
C. neoformans
virulence factors identified in this screen,Kin1, is important both for infection and killing of
C. ele-gans
and for pathogenicity in murine models of haematog-enous and respiratory infection.
Results
Screening a library of random
C. neoformans
mutants in
C. elegans
We tested 350 random insertion mutants of
C. neofor-mans
strain H99 for their ability to kill
C. elegans
. Themutants were generated by biolistic transformation ofstrain H99 with plasmid pCH233, which contains anourseothricin resistance gene (
NAT
) as described in
Experimental procedures
. One-third (118 out of 350) ofthe strains were selected after the first screen. Eighteenof the 118 mutants were selected for further analysis afterthe second screen. Nine of the 18 strains exhibited adifference in killing of
C. elegans
at
P
<
0.05 in a thirdscreen and were selected for further evaluation.
Genetic crosses were used to test whether the reduc-tion in virulence resulted from the insertion of the plasmid,which contains the nourseothricin resistance gene (
NAT
),rather than an unrelated mutation from DNA damage dur-ing biolistic transformation. For seven mutants, analysis ofprogeny (12–20 per isolate) after the cross to wild typeindicated the phenotype was linked to the insertion muta-tion (all NAT-resistant segregates were attenuated and all
NAT-sensitive segregates were as virulent as the wildtype).
Inverse polymerase chain reaction (PCR) was used toobtain the DNA sequence flanking the insertion for five ofthe seven mutant strains (Table 1). Strain 1A3 had aninsertion in the
KIN1
gene, encoding a putative serine/threonine protein kinase and was further evaluated asdescribed below. Two strains (2G2 and 2D4) had inser-tions in genes encoding major facilitator superfamily(MFS) transporters, one closely related to a transporter ofmyoinositol and the other of unknown specificity. Theinsertion in strain 3E3 was in the promoter of a generelated to glyceraldehyde 3-phosphate dehydrogenase.Strain 3E7 had a deletion of 18 kb of DNA that removedthe flavohaemoglobin (
FHB1
) gene and three other pre-dicted genes (Idnurm
et al
., 2004). Two other
fhb1
mutantalleles (insertional mutant 3C10) and a targeted deletionreported in de Jesus-Berrios
et al
. (2003) showed no sig-nificant reduction in virulence for
C. elegans
. Thus, it islikely that one of the other three genes deleted, rather than
FHB1
, is responsible for the observed reduction in viru-lence of 3E7 in
C. elegans
. The remaining two strains hadmultiple copies of the plasmid inserted in tandem in thechromosome (based on Southern blot and PCR analy-ses), thereby complicating amplification of the flankingDNA. Genetic analysis of one of these mutants (1A10)showed that the insertion was linked to the mating typelocus. Progeny from crosses to three independent
MAT
astrains all showed linkage with an average distance of19 cM on the ‘right side’ (centromeric) of the
MAT
locus.
In vitro
evaluation of a
kin1
mutant
We selected mutant 1A3, which has an insertion in thepromoter of a gene homologous to the
Saccharomycescerevisiae KIN1
gene, for further analysis. In
S. cerevi-siae
,
KIN1
encodes a serine/threonine protein kinase andstudies in
Schizosaccharomyces pombe
suggest thatKin1 is a member of the PAR-1/MARK (partitioning-defec-tive 1/microtubule-associated protein/microtubule affinity-regulating kinase) kinase family important in eukaryotic
Table 1.
Mutations in
C. neoformans
yielding reduced virulence for
C. elegans
.
Strain Gene mutated/comments
1A3
KIN1
1A10 Two copies of plasmid inserted; 19 cM from MAT locus; flanking sequence not obtained
2D4 Similar to myoinositol transporter2D5 Two copies of plasmid inserted; flanking sequence not
obtained2G2 Transporter (major facilitator superfamily)3E3 Related to glyceraldehyde 3-phosphate dehydrogenase3E7 18 kb deletion covering
FHB1
and three other predicted genes (Idnurm
et al.
, 2004)

C. elegans/C. neoformans
system
409
© 2004 Blackwell Publishing Ltd,
Molecular Microbiology
,
54
, 407–419
cell polarity and cytoskeletal dynamics (Drewes andNurse, 2003).
We constructed a defined
kin1
deletion mutation in
C.neoformans
strain KN99
a
that replaces all but six aminoacids (the first three and last three of the predicted codingregion) with the NAT cassette, as well as a reconstitutedstrain. The reconstituted strain was designated KN99
a
kin1
+
KIN1
. Because
S. pombe kin1
mutants exhibit mor-phological abnormalities, morphological features of the
kin1
mutant of
C. neoformans
were compared with thewild-type and reconstituted strains. The morphology ofthese three strains was compared under different condi-tions, including during growth, steady state and starvationat 25∞, 30∞ or 37∞C, as well as after exposure to differentcombinations of these conditions.
The KN99a kin1 mutant strain did not exhibit any differ-ences in capsule, melanin production, or growth at 37∞Con rich or minimal media (Fig. 1A and data not shown),compared with the wild-type or reconstituted strains. Also,there were no differences in viability (measured by platingdilutions of these cultures onto yeast peptone dextrose(YPD) plates with appropriate anti-microbials) after expo-sure to b-glucanases, hydrogen peroxide, or the tubulin-binding agent nocodazole (evaluated by comparing colony-forming units after exposure of the kin1 mutant or the wildtype to 0.03 mg ml-1 nocodazole for 1, 3, 6 or 19 h at 30∞C;Woyke et al., 2002). Also, no differences were noted inactin localization during budding (data not shown). Eval-uation of the mutant by electron microscopy demonstratedthat KN99a kin1 did not have an aberrant cell wall (datanot shown). However, in mating experiments, there wasless filament development in the mutant kin1 MATa ¥ kin1MATa compared with the wild-type cross (Fig. 1B). Simi-larly, there was less filament formation in the cross of kin1MATa to kin1 MATa compared with the cross of kin1 MATato KN99a (data not shown). In addition to the differencein filament production observed in the crosses, the mutantdemonstrated the following morphologic differences: underconditions of exponential growth, 60% of the kin1 mutantcells were budding, compared with 35% of wild-type cells.This increased percentage was not retained during sta-tionary growth phase or during starvation. Also, duringearly exponential growth, kin1 cells aggregated comparedwith wild type. These clumps were dispersed with minimalagitation. Finally, after multiple cycles of starvation and re-growth, a small number (<2%) of kin1 cells demonstratedan elongated daughter cell (Fig. 1C); this phenotype wasnot seen with wild-type or reconstituted strains.
kin1 mutation reduces virulence in C. elegans and two murine models
The kin1 mutant exhibited significant defects in its abilityto kill C. elegans. Survival of C. elegans N2 animals feed-
Fig. 1. kin1 mutation does not affect known virulence attributes.A. Two dilutions of yeast cells from wild-type (KN99a), kin1 mutant (kin1) and reconstituted strains (KN99a kin1+KIN1) were plated on YPD medium and grown at 30∞C or 37∞C or onto minimal medium (YNB) and grown at 30∞C for 2 days. Melanin (Mel) and capsule (Caps) production was assessed after 4 days of growth at 22∞C in the presence of L-DOPA or the iron chelation agent EDDHA respec-tively. Capsule was visualized by exclusion of India ink from the yeast cells.B. In mating experiments, there is less filament development in the cross of mutant kin1 MATa ¥ kin1 MATa than in the wild-type cross KN99a ¥ KN99a.C. After starvation and re-growth, ª 2% of the budding cells of the kin1 mutant demonstrate an elongated daughter cell as seen here (arrow).

410 E. Mylonakis et al.
© 2004 Blackwell Publishing Ltd, Molecular Microbiology, 54, 407–419
ing on C. neoformans kin1 mutants was significantlylonger compared with the parental and reconstitutedstrains (in both cases P < 0.001; Fig. 2A). Of note is thefact that the killing of C. elegans by KN99a and KN99ahad not been previously compared with the isogenic alpha
mating strain H99. Thus in this study, we used strain H99as an additional control. Similar to the findings from mam-malian models (Nielsen et al., 2003), killing of C. elegansby KN99a was similar to that by H99 (Fig. 2A). BothKN99a and KN99a kin1 strains accumulated to a similardegree in the gastrointestinal tract of nematodes (Fig. 3and data not shown).
To determine whether Kin1 plays a role in mammalianvirulence, we compared virulence in two establishedmurine models of cryptococcal infection. The kin1 deletionmutant was hypovirulent in both murine inhalation and tailvein injection models (Fig. 2B and C; P < 0.001 for thekin1 mutant compared with KN99a and KN99akin1+KIN1). The median time to mortality after intranasalinoculation of 2.5 ¥ 105 yeast cells was 21 days and
Fig. 2. Cryptococcus neoformans kin1 mutant is attenuated in C. elegans and mice.A. Survival of C. elegans N2 animals feeding on C. neoformans mutants with disruptions in KIN1. P < 0.001 for the kin1 mutant, compared with the parental strain KN99a, the isogenic alpha mating strain H99 and the reconstituted strain KN99a kin1+KIN1.B. Survival of mice after inhalation of 2.5 ¥ 105 yeast cells of either the kin1 mutant, the parental strain KN99a or the reconstituted strain KN99a kin1+KIN1 (n = 12). P < 0.001 for the kin1 mutant compared with the parental and the reconstituted strains.C. Survival of mice after tail vein injection of 2.2 ¥ 106 yeast cells of either the kin1 mutant (n = 10), the parental strain KN99a (n = 8) or the reconstituted strain KN99a kin1+KIN1 (n = 8). P < 0.001 for the kin1 mutant compared with the parental and reconstituted strains.
A
100
100
P < 0.001
Nematode killing
% S
urv
ival
Time (h)200
KN99a
KN99aKN99a kin1
KN99a kin1
KN99a kin1+KIN1
KN99a kin1+KIN1
H99
H99
75
50
25
00
B
100
500
P < 0.001
Killing in mouse inhalation model
% S
urv
ival
Time (h)1000
KN99a
KN99aKN99a kin1
KN99a kin1
KN99a kin1+KIN1
KN99a kin1+KIN1
75
50
25
00
C
100
100 200 300
P < 0.001
Killing in mouse tail vein injection model
% S
urv
ival
Time (h)
KN99a
KN99aKN99a kin1
KN99a kin1
KN99a kin1+KIN1
KN99a kin1+KIN1
75
50
25
00
Fig. 3. KN99a and KN99a kin1 accumulate in the gastrointestinal tract. Intact yeast cells are present in the distended gastrointestinal tract of C. elegans after feeding 36 h on C. neoformans strains KN99a (A) and KN99a kin1 (B). Black arrows point to the intestinal lumen. The white arrowheads note the pharyngeal grinder organ, which functions to disrupt ingested organisms.
KN99A
KN99 kin1B
C. elegans/C. neoformans system 411
© 2004 Blackwell Publishing Ltd, Molecular Microbiology, 54, 407–419
24.5 days for wild-type and the kin1+KIN1 strain, respec-tively, whereas it was 37 days for the kin1 mutant. Aftertail vein injection of 2.2 ¥ 106 yeast cells, the median timeto mortality was 5.5 days and 6.5 days for wild type andkin1+KIN1, respectively, but was almost twice as long(11.5 days) for the kin1 strain. The kin1 mutant remainedpathogenic in the tail vein injection model even with lowerinocula, but was less virulent than the wild-type strain ateach dose. For example, after tail vein injection of 5 ¥ 104
yeast cells, the median time to mortality in the KN99agroup was 10 days, compared with 14.5 days in the kin1group (P < 0.001). In the same model, after injection of5.5 ¥ 103 yeast cells, the median time to mortality in theKN99a group was 13 days, compared with 19 days in thekin1 group (P < 0.001).
The experiments performed with C. elegans and bothmurine models were repeated using an independentlyconstructed kin1 deletion mutant (kin1 mutant #9) withsimilar results (data not shown). No significant differences
in the killing of nematodes or mice were found betweenthe two kin1 strains (data not shown).
Fungal burden in murine organs
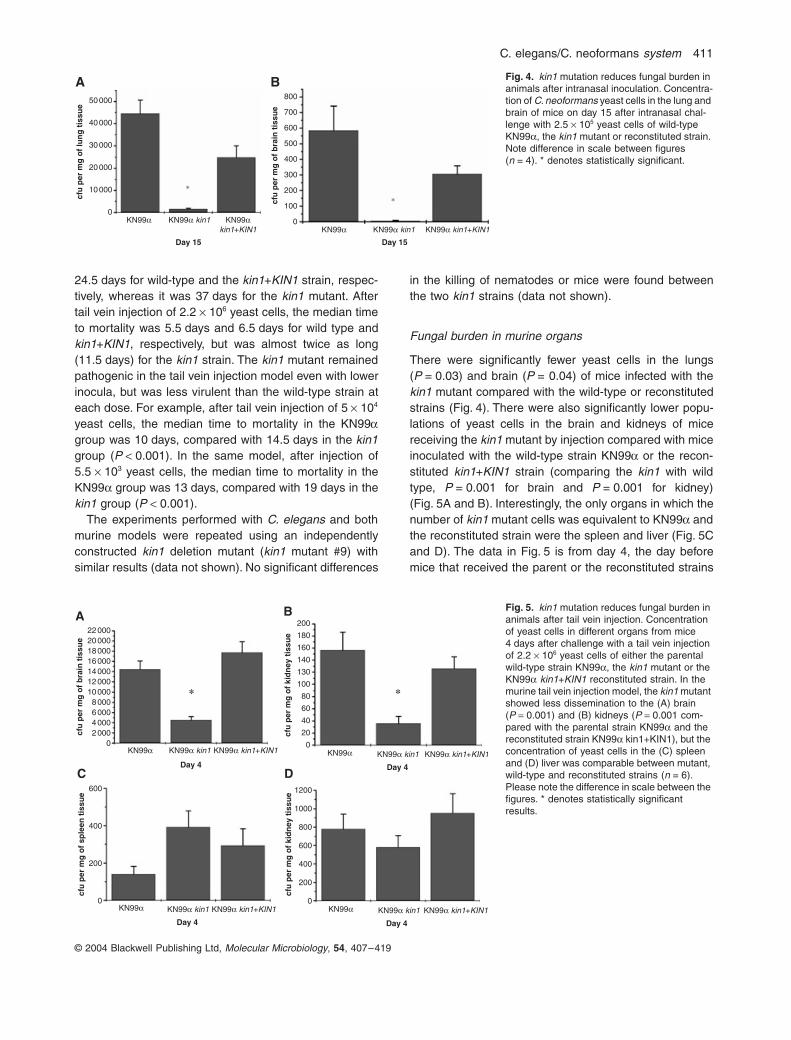
There were significantly fewer yeast cells in the lungs(P = 0.03) and brain (P = 0.04) of mice infected with thekin1 mutant compared with the wild-type or reconstitutedstrains (Fig. 4). There were also significantly lower popu-lations of yeast cells in the brain and kidneys of micereceiving the kin1 mutant by injection compared with miceinoculated with the wild-type strain KN99a or the recon-stituted kin1+KIN1 strain (comparing the kin1 with wildtype, P = 0.001 for brain and P = 0.001 for kidney)(Fig. 5A and B). Interestingly, the only organs in which thenumber of kin1 mutant cells was equivalent to KN99a andthe reconstituted strain were the spleen and liver (Fig. 5Cand D). The data in Fig. 5 is from day 4, the day beforemice that received the parent or the reconstituted strains
*
B
*
A50 000 800
700
600
500
400
300
200
100
0
40 000
30 000
20 000
10 000
0KN99a KN99a kin1
Day 15
cfu
per
mg
of
lun
g t
issu
e
cfu
per
mg
of
bra
in t
issu
e
KN99akin1+KIN1 KN99a KN99a kin1
Day 15
KN99a kin1+KIN1
Fig. 4. kin1 mutation reduces fungal burden in animals after intranasal inoculation. Concentra-tion of C. neoformans yeast cells in the lung and brain of mice on day 15 after intranasal chal-lenge with 2.5 ¥ 105 yeast cells of wild-type KN99a, the kin1 mutant or reconstituted strain. Note difference in scale between figures (n = 4). * denotes statistically significant.
* *
A
D
B
C
22 00020 00018 00016 00014 00012 00010 0008 0006 0004 0002 000
0KN99a KN99a kin1
Day 4
cfu
per
mg
of
bra
in t
issu
e
200
600
400
200
0
180
160
140
130
100
80
60
40
20
0
cfu
per
mg
of
kid
ney
tis
sue
KN99a kin1+KIN1 KN99a KN99a kin1
Day 4
KN99a kin1+KIN1
1200
1000
800
600
400
200
0
cfu
per
mg
of
kid
ney
tis
sue
cfu
per
mg
of
sple
en t
issu
e
KN99a KN99a kin1
Day 4
KN99a kin1+KIN1KN99a KN99a kin1
Day 4
KN99a kin1+KIN1
Fig. 5. kin1 mutation reduces fungal burden in animals after tail vein injection. Concentration of yeast cells in different organs from mice 4 days after challenge with a tail vein injection of 2.2 ¥ 106 yeast cells of either the parental wild-type strain KN99a, the kin1 mutant or the KN99a kin1+KIN1 reconstituted strain. In the murine tail vein injection model, the kin1 mutant showed less dissemination to the (A) brain (P = 0.001) and (B) kidneys (P = 0.001 com-pared with the parental strain KN99a and the reconstituted strain KN99a kin1+KIN1), but the concentration of yeast cells in the (C) spleen and (D) liver was comparable between mutant, wild-type and reconstituted strains (n = 6). Please note the difference in scale between the figures. * denotes statistically significant results.

412 E. Mylonakis et al.
© 2004 Blackwell Publishing Ltd, Molecular Microbiology, 54, 407–419
started to die. However, similar trends were noted on days1–3 (data not shown).
Histology studies in murine models of cryptococcosis
Examination of the brain revealed that mice that receivedthe inoculum by tail vein injection had significantly morefungal lesions compared with those that received the inoc-ulum via nasal inhalation. Animals that received theparental C. neoformans strain had increased number andlarger fungal lesions than in the mice that were challengedwith the kin1 mutant (Fig. 6A and B). Besides the numberand the size of the cryptococcal lesions, there were nodifferences in the nature of the lesions between the differ-ent groups. In general, inflammation was not observed inthe brain lesions associated with any of these strains.Rarely, a single neutrophil or minimal haemorrhage wasassociated with the fungal lesions.
Lungs from animals that received intranasal inocula-tion of KN99a contained numerous organisms within res-piratory bronchioles and adjacent alveoli, with largefungal lesions causing expansion/effacement of alveolarseptae. Often, the yeast infiltrate extended into the alve-olar spaces and filled the airway. There was also arobust infiltration/expansion of alveolar septae and bron-
chiolar walls with a mixture of neutrophils and monocyte/macrophages, with less eosinophils. Some air spaceswere filled with alveolar macrophages that containedintracytoplasmic organisms (Fig. 6C). In lungs from ani-mals challenged with kin1, on the other hand, the yeastburden was significantly attenuated (Fig. 6D). Interest-ingly, histopathological evaluation of the spleen frommice that received an inoculum via intravenous injectiondemonstrated a significantly more pronounced hostresponse in the spleen of mice infected with the kin1mutant, compared with mice that received an inoculumwith the parent strain KN99a. Wild-type organismswere surrounded by a few macrophages (Fig. 6E),whereas kin1 organisms were surrounded by numerousmonocytes/macrophages, neutrophils and few eosino-phils forming splenic pyogranulomas around the yeastcells (Fig. 6F).
During histopathology, we identified no difference in thesize of the capsule between the parent strain and themutant. Also, because the cryptococcal capsule may bealtered during processing and staining, the size of thecapsule was evaluated in the frozen samples from thefrontal lobes of the mice, but there was no significantdifference between kin1 and wild-type strains (data notshown).
E F
C D
A BFig. 6. Histopathological comparison of ani-mals infected with wild-type or the kin1 mutant. Brain sections (magnification = 200¥) from mice challenged with wild-type C. neoformans strain KN99a (A) contained fungal lesions (arrows) that were larger than the lesions present in brain sections of animals challenged with kin1 mutants (B). Lungs (magnification = 400¥) from animals chal-lenged intranasally with the wild-type C. neofor-mans KN99 strain contained numerous intra-alveolar fungal lesions which often obliterated the alveolar space and effaced alveolar walls (C). In contrast, animals similarly challenged with the mutant kin1 strain (D) contained fewer intra-alveolar organisms, typically up to one per alveolar space. The pulmonary interstitium was expanded by an inflammatory infiltrate domi-nated by granulocytes (arrowheads). Animals challenged with the KN99a wild-type strain also contained small aggregates of organisms within the splenic white pulp (magnification = 400¥), often colocalizing with B cell follicles, but there was minimal inflammation associated with the organisms (E). By comparison, animals chal-lenged with the kin1 mutant (F) contained scat-tered splenic pyogranulomas characterized by an accumulation of mononuclear cells and granulocytes (arrowheads).

C. elegans/C. neoformans system 413
© 2004 Blackwell Publishing Ltd, Molecular Microbiology, 54, 407–419
The role of KIN1 in binding/phagocytosis
Because of the reduced ability of the kin1 mutant to causepneumonitis and the preferential localization of the kin1mutant strain in organs rich in macrophages, such as thespleen, we studied the binding/phagocytosis of C. neofor-mans KN99a, KN99a kin+KIN1 and kin1 strains by alve-olar and peritoneal macrophages.
After the intranasal administration of an inoculum simi-lar to that used in the murine killing assays, there wasminimal binding of yeast after 2 h to alveolar macroph-ages in all groups (data not shown). However, after 24 h,binding of the kin1 mutant was enhanced compared withwild-type KN99a (P = 0.03) (Fig. 7A). The same increasein binding of the mutant was noted in vitro, when weexamined binding of yeast cells to non-activated residentmacrophages (P = 0.01 kin1 mutant compared withKN99a) or activated resident macrophages (P = 0.01 kin1mutant compared with KN99a). Reintroduction of the wild-type KIN1 gene complemented the macrophage-bindingphenotypes (Fig. 7B and C). The binding index isexpressed as the geometric mean of the number of yeastcells attached or ingested per 100 macrophages. Resultsare expressed as geometric means ± SEs of the bindingindexes obtained for each experiment.
The kin1 mutant shows an increased number of bud-ding yeast in exponential growth. To avoid having thisconfound the binding experiments, we also counted mac-rophages with only a single bound yeast cell and foundthe same increased binding of kin1 mutant compared withwild type.
Discussion
We describe a screening approach for the use of C. ele-gans as a surrogate host. Using this system, we identifiedseven C. neoformans mutant strains with attenuatedpathogenesis in C. elegans and demonstrated that thegene mutated in one of them (KIN1) plays a role in cryp-tococcal infection in mammals and is a key determinantfor the ability of C. neoformans to escape recognition bythe host. This work validates the use of C. elegans as asurrogate host for the identification of C. neoformans vir-ulence factors in mammalian infection.
The screen using C. elegans as a host for the identifi-cation of attenuated mutants of C. neoformans identifiedseven putative mutants out of 350 strains tested. This rateof mutant identification, ª 2%, is comparable to the rateat which hypovirulent mutants were identified in analo-gous screens in C. elegans studying bacterial pathogens.For example, screens of Enterococcus faecalis and Ser-ratia marcescens transposon insertion libraries yielded2% and 1% avirulent mutants respectively (Garsin et al.,2001; Kurz et al., 2003).
Fig. 7. Mutation in kin1 increases binding to alveolar and peritoneal macrophages.A. Binding of C. neoformans KN99a, KN99a kin1 and KN99a kin+KIN1 strains by murine alveolar macrophages 24 h after intrana-sal inoculation. There was minimal binding of the wild-type and recon-stituted strains. However, there was significant binding of kin1 yeast cells to alveolar macrophages (P = 0.03 compared with the wild-type KN99a).B and C. Increased binding of kin1 mutant yeast cells was also seen in vitro using either (B) non-activated (P = 0.01 compared with KN99a) or (C) activated resident peritoneal macrophages (P = 0.01 compared with KN99a). The binding index is expressed as the geo-metric mean of the number of yeast cells attached or ingested per number of macrophages per field.
*
C Thio-macrophages
B Resident macrophages*
A Alveolar macrophages
*
0
5
10
15
20
25
30
35
40
KN99a KN99a kin1
Nu
mb
ers
of
org
anis
ms
per
100
alve
ola
r m
acro
ph
ages
KN99a kin1+KIN1
0
50
100
150
200
250
KN99a KN99a kin1
Nu
mb
ers
of
org
anis
ms
per
100
mac
rop
hag
es
KN99a kin1+KIN1
0
50
100
150
200
250
KN99a KN99a kin1
Nu
mb
ers
of
org
anis
ms
per
100
mac
rop
hag
es
KN99a kin1+KIN1

414 E. Mylonakis et al.
© 2004 Blackwell Publishing Ltd, Molecular Microbiology, 54, 407–419
One of the challenges in screening large collections ofinsertion mutants, including studies in C. elegans, is toeliminate those in which the phenotype is caused by ageneralized debilitation of the pathogen or an effect ongrowth, rather than a specific effect on virulence. Forexample, in a C. elegans screen of 3400 transposonmutants of the bacterium Burkholderia pseudomallei,workers identified 39 mutants that exhibited reducedlethality towards C. elegans, but only five exhibited growthrates comparable to the wild-type parental strain (Ganet al., 2002). This concern may be even more relevant inscreening C. neoformans libraries, because of possibleDNA damage associated with biolistic transformation andrearrangement of multiple transposons in the genome. Inour screen, we specifically excluded mutants with dimin-ished growth rate and we evaluated linkage of the pheno-type to the insertion by screening progeny after crossingthe insertional mutation to a pathogenic wild-type strain.
The Kin1 protein kinase of the fission yeast S. pombeis a member of the PAR-1/MARK family important ineukaryotic cell polarity and cytoskeletal dynamics. Proteinkinases of the PAR-1/MARK family play crucial roles in theestablishment of cell polarity in organisms as diverse asC. elegans (Guo and Kemphues, 1995; 1996), D. melan-ogaster (Brunner and Nurse, 2000; Tomancak et al.,2000; Cox et al., 2001) and cultured mammalian cells(Bohm et al., 1997; Brown et al., 1999; Biernat et al.,2002). In S. cerevisiae, kin1 mutations result in morphol-ogy and growth defects (Levin and Bishop, 1990) and inS. pombe, kin1 mutations result in delayed cytokinesis(Drewes and Nurse, 2003). Also, differential sensitivity ofS. pombe kin1-disrupted cells to lysis by treatment witha- and b-glucanases suggests an alteration in either thecomposition or organization of the cell wall (Levin andBishop, 1990; Tibbetts et al., 1994). Similar to our obser-vations in C. neoformans, a higher proportion of kin1 cellsof S. pombe undergo septation under conditions of expo-nential growth compared with wild type. In contrast to ourfindings in C. neoformans, in S. pombe this percentage isretained in the stationary growth phase upon re-growthand these septated cells do not complete cytokinesis butform cells with two or three septa (Drewes and Nurse,2003).
Similar to what has been previously reported in S. cer-evisiae, there were no dramatic differences in C. neofor-mans between the kin1 mutant and the parent strainobservable by light microscopy (even after multiple cyclesof starvation and re-growth). We found no difference inactin organization of the kin1 mutant or the morphologyof the kin1 cells by histological examination (includingfrozen sections). Moreover, organisms that were boundto or phagocytosed by murine alveolar or macrophages(in the ex vivo and in vitro experiments) were indistin-guishable from that of the KN99a cells (Fig. 6 and data
not shown). The similarities in actin localization, theresponse to the tubulin-binding agent nocodazole andpreliminary results using the anti-tubulin antibody TAT1 (amonoclonal antibody developed by Woods et al., 1989that has been used for the study of microtubular cytosk-eleton in C. neoformans; protocol detailed in Kopeckaet al., 2001) in yeast cells growing in YPD suggest thatmicrotubule differences (if any) in the KN99a kin1 mutantcompared with the wild-type strain will be minor. Aftercycles of starvation and re-growth, a small number (<2%)of kin1 cells exhibited an elongated daughter cell(Fig. 1C). This phenotype was not seen in the wild-typeor the reconstituted strains but it appears to be toorare to explain the virulence defect of the mutant.
The kin1 mutant did demonstrate some defects in fila-mentation associated with mating (Fig. 1B) and a differ-ence in budding during growth that suggests that KIN1may play a role in cytokinesis. The difference in bindingto macrophages by the kin1 mutant, the tendency of kin1yeast cells to form clumps during growth and the morpho-logic differences suggest that Kin1 may also affect the cellwall or polarity. The similarities in actin formation as wellas the similarities on electron microscopy between thekin1 mutant and wild type suggest that KIN1 may mainlyact in an actin-independent pathway(s).
In human disease, C. neoformans enters through therespiratory tract and first causes a pneumonitis (oftensubclinical) and alveolar macrophages represent one ofthe cell types that inhibit and kill C. neoformans (Casade-vall and Perfect, 1998; Luberto et al., 2003). From thelung, C. neoformans reaches the central nervous systemand causes an often fatal form of meningoencephalitis.This process has been divided into five stages: initiation,dormancy, reactivation, dissemination and proliferation(Casadevall and Perfect, 1998; Feldmesser et al., 2001).Our data support the conclusion that KIN1 plays a signif-icant role at different steps in this process. Specifically, inour ex vivo studies, we found that there is significantlyhigher binding of the kin1 mutant to alveolar macrophages(Fig. 7A), suggesting a defect in the ability of the kin1mutant to escape recognition by the host during initiationof infection. The significantly lower burden of cryptococciin the lungs of mice inoculated with the kin1 mutant (>15-fold lower; Fig. 4) suggests that macrophages adhere bet-ter to kin1 cells and may more efficiently clear kin1 yeastcells. The concentration of kin1 mutant cells in the spleenand the liver, and the presence of splenic pyogranulomasassociated with the kin1 mutant, suggest that the host ismore successful at containing the kin1 mutant than wild-type cells.
Of note is that none of the mice was able to clear thekin1 inoculum and all mice in this group eventually died.Moreover, eosinophils were part of the host response tothe kin1 strain and the presence of eosinophils has been

C. elegans/C. neoformans system 415
© 2004 Blackwell Publishing Ltd, Molecular Microbiology, 54, 407–419
associated with the inability to clear infection (Huffnagleet al., 1998). Taken together, we conclude that kin1mutants are attenuated but not avirulent. Further studiesare needed to further elucidate the interaction betweenthe kin1 mutant and host tissues.
Previous work has suggested that bacteria (Greub andRaoult, 2004) and C. neoformans (Steenbergen et al.,2001; Casadevall et al., 2003) use mechanisms to survivewithin amoebae that are similar to those used for survivalwithin macrophages, and Kin1 may be one such factor inC. neoformans. Because C. elegans has no macroph-ages, Kin1 may be more generally involved in the survivalof C. neoformans in stressful environments, such as afteringestion by C. elegans.
In addition to Kin1, six other mutants were identified inour screen (Table 1). For example, mutant 2D4 has aninsertion in a gene that is probably involved in myoinositoltransport. Inositol metabolism is important for growth anddevelopment of C. neoformans and has been involved inthe ability of C. neoformans to survive as both asaprophyte in soil and a parasite in humans (Molina et al.,1999). The insertion in strain 3E3 is in the promoterof a gene whose product is related to glyceraldehyde3-phosphate dehydrogenase, a key glycolytic enzyme thatis highly and constitutively expressed in yeast andaccounts for 2–5% of total mRNA (Varma and Kwon-Chung, 1999). Altered metabolism in these mutants maycontribute to attenuation in C. elegans.
It should be noted, however, that not all the genesinvolved in cryptococcal pathogenesis in mammals play arole in killing of C. elegans. For example, our screenmissed at least one gene (FHB1 encoding flavohaemo-globin denitrosylase, an enzyme that consumes nitricoxide) that has been shown to be involved in mammalianvirulence. Neither mutant 3C10 nor a targeted deletion ofFHB1 reported in de Jesus-Berrios et al. (2003) showeda significant reduction in virulence for C. elegans. Thismay relate to the apparent lack of a gene for nitric oxidesynthase in C. elegans (Morton et al., 1999), illustratingthat not all traits associated with C. neoformans pathogen-esis in mammals can be identified in the C. elegansmodel.
Although some features of the C. elegans/C. neofor-mans interaction may not be directly relevant to mamma-
lian pathogenesis, we show here that C. elegans can beused as a surrogate host to identify traits associated withfungal pathogenesis in mammals. Our findings provideevidence that the KIN1 gene is associated with C. neofor-mans virulence and binding to macrophages; this sup-ports the use of C. elegans as a screen to identify factorsinvolved in C. neoformans pathogenesis. New approachesare needed for the more rapid identification of fungal vir-ulence factors. The C. elegans/C. neoformans interactionappears to provide a novel system for the rapid and facilestudy of fungal pathogenesis.
Experimental procedures
Strains and media
The C. neoformans strains used in these experiments aresummarized in Table 2 or described in the text. Yeast cultureswere maintained as frozen stocks or on YPD (Difco) agarplates, and grown in YPD. Yeast cultures were grown at 30∞Cunless otherwise specified. The standard C. elegans strainN2 Bristol was maintained at 15∞C and propagated onEscherichia coli strain OP50 using established procedures(Brenner, 1974; Garsin et al., 2001).
Screen of mutant library in C. elegans
The development and in vitro testing of a insertional mutationlibrary in C. neoformans strain H99 is described elsewhere(Idnurm et al., 2004). C. neoformans strain H99 is a virulentclinical isolate that is widely used in pathogenesis studies(Heitman et al., 1999). H99 mutants were generated bybiolistic introduction into the fungal genome of BamHI or XhoIlinearized plasmid pCH233, which contains the nourseothri-cin resistance gene (NAT) flanked by the cryptococcal actinpromoter and TRP1 terminator.
Testing for loss or reduction of virulence using a C. eleganskilling assay was performed as previously described(Mylonakis et al., 2002), with minor modifications. In brief, C.neoformans strains were inoculated into 2 ml of YPD andgrown at 30∞C for 24 h; 10 ml of the culture was spread on35 mm tissue-culture plates (Falcon) containing brain–heartinfusion (BHI) agar (Difco). The plates were incubated at30∞C for 48 h. Ampicillin was added to the medium to preventgrowth of E. coli that might have been carried over on transferof worms to the yeast-containing plates. C. elegans animalsat the L4 developmental stage were transferred from a lawn
Table 2. Cryptococcus neoformans strains used in this study.
Strain Relevant characteristics or phenotype Reference
H99 ATCC #208821 Serotype A; clinical isolate; genome sequence determinedKN99a Congenic H99 mating parent, MATa Nielsen et al. (2003)KN99a Congenic H99 mating parent, MATa Nielsen et al. (2003)KN99-5 Fifth backcross in creation of KN99a and KN99a; MATa Nielsen et al. (2003)KN99a kin1 KN99a+kin1::NAT This reportKN99a kin1 MATa product of kin1 ¥ KN99a This reportKN99a kin1+KIN1 MATa product of kin1+KIN1-NEO This report

416 E. Mylonakis et al.
© 2004 Blackwell Publishing Ltd, Molecular Microbiology, 54, 407–419
of E. coli OP50 on nematode growth medium to a lawn of theyeast to be tested on BHI medium, incubated at 25∞C, andexamined for viability at 24 h intervals with a Nikon SMZ645dissecting microscope. Worms were considered dead whenthey failed to respond to touch with a platinum wire pick. Ofnote is that nematodes exposed to C. neoformans are unableto grow and/or produce progeny; thus there are no progenyto confound counts of viable worms.
For the initial screen, 25 nematodes were used to analyseeach C. neoformans mutant tested. In the second screen,and in the evaluation of the progeny from crosses of theinsertional mutant to a pathogenic wild-type strain, 60 nem-atodes were used for each mutant strain. In the third screenand in all other killing assays, 120–150 nematodes were usedfor each mutant strain, divided across three plates. After thefirst screen, one-third of the strains that demonstrated thehighest reduction of virulence compared with the parentstrain H99 were selected. After the second screen, allmutants that demonstrated a P-value < 0.10 compared withthe parent strain H99 were selected for the third screen.In the third screen and in all other killing assays,P-values < 0.05 were considered statistically significant.
Identification of disrupted genes in mutant strains
Strains which exhibited reduced killing of C. elegans werecrossed to the congenic MATa strains KN99a or KN99-5(Nielsen et al., 2003). Basidiospores were isolated by micro-manipulation. Progeny (12–20 strains per insertional mutant)were tested for their ability to kill C. elegans and nourseothri-cin resistance, as well as mating type as an independentmarker for segregation analysis. DNA fragments flanking theinserted NAT plasmid were obtained by inverse PCR andsequenced as described previously (Idnurm et al., 2004).Sequences were compared with the H99 genome databaseat Duke University, and genes predicted in these regions byFGENESH software (http://www.softberry.com) were com-pared with the GenBank database. Subsequent analysesused the annotated database from TIGR.
Disruption and reconstitution of the KIN1 gene
The KIN1 coding region was identified by FGENESH soft-ware and the partial cDNA sequence of a C. neoformans EST(University of Oklahoma). A disruption allele was constructedby overlap PCR to replace most of the coding regionwith a cassette conferring resistance to nourseothricin(Davidson et al., 2002; Idnurm et al., 2004). HomologousDNA fragments were amplified from H99 genomic DNA withprimers (5¢-3¢) JOHE9656 CGAAGGATCAACAAAGATGGand JOHE9657 AGCTCACATCCCGCAGCAGGTAGTCATTATCACGG, and JOHE9659 TAGTTTCTACATCTCTTCATGAGATTATAGTCGGGC and JOHE9661 GTTTCGACTTCAGCAGAGTG. The NAT cassette was amplified with primersJOHE9658 CCGTGATAATGACTACCTGCTGCGAGGATGTGAGCTG and JOHE9660 GCCCGACTATAATCTCATGAAGAGATGTAGAAACTAG. The three products were mixed inequimolar amounts and a single contiguous DNA fragmentamplified with primers JOHE9656 and JOHE9661. This frag-ment was transformed into strain KN99a (MATa) and
mutated strains were identified by PCR and Southern blotanalysis. The kin1 mutation was crossed to strain KN99a andbasidiospore progeny was selected to identify a kin1 MATastrain. Disruption of KIN1 with the NAT cassette was con-firmed using PCR and Southern blot hybridization. Onlystrains in which a single insertion had occurred were furtherstudied.
A wild-type copy of the KIN1 gene was reintroduced into akin1 mutant as follows. A PCR product amplified from H99genomic DNA with primers JOHE9656 and JOHE9661 wascloned into a plasmid conferring resistance to neomycin(Fraser et al., 2003). The plasmid was linearized and trans-formed into the kin1 mutant strain using a biolistic apparatus.Transformants were selected on medium containingneomycin (MediaTech) to identify strains in which the KIN1-containing fragment had integrated ectopically into thechromosome by non-homologous recombination. Strainswith equal growth on medium with or without neomycin wereconsidered to have stable integration of the wild-type copy ofKIN1.
Microscopy
Cryptococcus neoformans cells were fixed for 50 min in 2%paraformaldehyde at room temperature. Calcoflour white wasadded to live or fixed cells for 15–20 min at a final concen-tration of 0.005% (w/v). To visualize F-actin, aliquots of fixedcells were incubated with rhodamine-conjugated phalloidin(Waugh et al., 2002). Light and fluorescence microscopywere performed on an Olympus BX51 microscope andimages were captured and processed using the digital cam-era Qimaging Retiga (Burnaby) and Olympus MicrosuiteSoftware. Vectrashield mounting medium was used for fluo-rescence microscopy (Vector Laboratories).
In vitro evaluation of fungal strains
Melanin production in wild-type KN99a, KN99a kin1 andKN99a kin1+KIN1 strains was examined on niger seed agarmedium or medium containing the diphenolic molecule L-DOPA. Production of the anti-phagocytic polysaccharide cap-sule was examined in strains grown in low-iron medium (con-taining 20 mg l-1 EDDHA to chelate iron) for 4 days at 30∞C.Capsule production was observed by the exclusion of Indiaink from the yeast cells. The ability to mate was testedbetween strains on V8 pH 5 medium grown for 7–10 days atroom temperature in the dark (Alspaugh et al., 1997).Crosses were also set up between strains to examine theeffects of mutation of kin1 in a single or both parents in thecross.
Murine models of virulence
Murine C. neoformans pathogenesis models were asdescribed elsewhere (Casadevall and Perfect, 1998). Wild-type 8-week-old female ICR/CD1 mice (Charles River Labo-ratories) were used in all studies. In brief, C. neoformansstrains were grown at 30∞C with shaking overnight in YPD tolate-log phase. The yeast cells were centrifuged, washed inPBS and resuspended to a concentration of 107 cells per ml

C. elegans/C. neoformans system 417
© 2004 Blackwell Publishing Ltd, Molecular Microbiology, 54, 407–419
(measured with a haemocytometer). For the tail vein injectionmodel, 250 ml of this suspension was inoculated into the tailvein. For the inhalation model, mice were anaesthetized byintraperitoneal injection of tribromoethyl alcohol (Aldrich;400 mg kg-1) and suspended by the incisors on a silk thread.The inoculum was prepared as detailed above for the tail veininjection and 50 ml volumes of the inoculum were slowlypipetted into the nares with continued suspension for 10 min.In all mammalian experiments, the concentration of yeastcells in the inoculum was confirmed by plating serial dilutionsand enumerating colony-forming units 48–72 h later. In bothmurine models, survival was monitored at least twice daily,and moribund animals or those in pain were sacrificed byCO2 asphyxiation. The murine protocols were approvedby the Massachusetts General Hospital Committee onResearch, Subcommittee on Research Animal Care, andspecial attention was given to minimize suffering of the mice.
Evaluation of tissue burden and histology studies in murine models of cryptococcosis
In the tail vein injection model, cryptococcal burden wasdetermined by sacrificing two to four mice from each groupon days 1–3 and six mice from each group on day 4 afterinoculation. In the inhalation model, tissue burden was deter-mined by sacrificing four mice from each group on day 15after infection. After euthanasia, in the tail vein injectionmodel, the brain, spleen, kidneys and liver were harvested.In addition to these organs, the lungs were also harvestedfrom mice that received the inoculum by nasal inhalation.
Subsequently, the right lobe of the brain, one-third of thespleen and half of the lower right lung were fixed in 4%paraformaldehyde, and then embedded in paraffin for histo-pathology. Sections were stained with periodic acid–Schiff(PAS) to identify fungal cell wall, and with haematoxylin andeosin to examine the host inflammatory response. Sectionswere examined by light microscopy (Olympus CH stereomi-croscope). Also, the frontal lobes of the left brains werefrozen at -80∞C (used for evaluation of capsule in vivo).
The remaining organs were weighed and homogenized in10 ml of sterile PBS using a Tissue Tearor (Model #398,Biospec Products). Quantitative cultures were performedby plating dilutions of the tissue homogenates on YPDmedium containing ampicillin (100 mg ml-1) and streptomycin(100 mg ml-1). Plates were incubated at 30∞C for 72 h, andyeast colonies were counted.
Ex vivo isolation of alveolar macrophages
Nasal inoculation with 2 ¥ 105 cells of either KN99a, KN99akin1 and KN99a kin1+KIN1 in 8-week-old female ICR/CD1mice (Charles River Laboratories) was performed asdescribed above. Twenty-four hours later, animals were euth-anized by CO2 inhalation. This procedure does not affect thephagocytic ability of alveolar macrophages (Hu et al., 2000;Luberto et al., 2003). The trachea was then exposed andbronchoalveolar lavage (BAL) was performed. Briefly, a 20-gauge Luer stub adapter attached to a 1 ml syringe wasinserted into the trachea. Warm Hanks’ balanced salt solution(HBSS) with 1 mM EDTA and 100 U ml-1 ampicillin was
injected into the lung and recovered. The lavage wasrepeated three times (1 ml was used per lavage for a total of3 ml per mouse) and recovered fluid was pooled for assess-ment of cellular content (c. 0.8 ml was recovered every 1 mladministered). From each mouse, 3–5 ¥ 105 alveolar mac-rophages were obtained. The recovered cells were washedtwice with PBS and an aliquot of viable cells were set asideto study cellular phenotype and for fungal binding studies.The percentage of macrophages was determined by count-ing the number of cells that endocytosed DiI-AcLDL, a fluo-rescent ligand for macrophage scavenger receptors asdescribed (El Khoury et al., 2003). Using this method, wefound that >95% of the cells were macrophages. The remain-ing cells were fixed in 2% paraformaldehyde for 30 min.
For evaluation of binding, 10 000 cells were allowed tosediment onto each spot of a multispot slide. The bindingindex was determined as described above using a Nikonmicroscope equipped with a water immersion lens, as previ-ously described (Luberto et al., 2003). For each experiment,at least 10 fields per well were observed in three differentwells per strain or treatment condition. The binding index isexpressed as the geometric mean of the number of yeastcells attached or ingested per 100 macrophages. Results areexpressed as geometric means ± SEs of the binding indexesobtained for each experiment.
Isolation of resident peritoneal macrophages
C57BL/6 mice (Charles River Laboratories) were sacrificedby CO2 asphyxiation and peritoneal lavage was performed byintroducing 10 ml of ice-cold PBS into the peritoneal cavityand then aspirated back into the syringe. Using this method,1–2 ¥ 106 cells per mouse were obtained. The cells were thenallowed to adhere onto eight-well chamber slides for 2 h inDMEM plus 10% fetal bovine serum. Non-adherent cellswere removed by washing with 0.5 ml of warm medium perwell. The remaining cells were confirmed to be macrophages(>95%) as described above for alveolar macrophages(Greenberg et al., 1991).
Isolation of stimulated peritoneal macrophages
C57BL/6 mice (Charles River Laboratories) were injectedintraperitoneally with 1 ml of thioglycollate medium. After96 h, the mice were sacrificed and the peritoneum wasinjected with 10 ml of ice-cold PBS. The medium wasremoved and the cells were centrifuged (1400 r.p.m., 10 min,4∞C) and resuspended (106 cells per ml) in RPMI 1640 with10% fetal bovine serum supplemented with L-glutamine andpenicillin/streptomycin. The cells were then plated onto mul-tispot slides (Shandon-Lipshaw) 25 000 cells per spot andallowed to adhere for 30 min. The cells were then washedthree times with warm medium to remove non-adherent cells.Using this method, we routinely obtained >98% macroph-ages (Thomas et al., 2000).
Statistical analysis
Each murine experiment was repeated at least twice andeach experiment independently gave statistically significant

418 E. Mylonakis et al.
© 2004 Blackwell Publishing Ltd, Molecular Microbiology, 54, 407–419
results. The figures and the P-values provided herein areeach from one representative experiment. Murine and C.elegans killing curves were plotted and estimation of differ-ences (log-rank and Wilcoxon tests) in survival was analysedwith the Kaplan-Meier method performed using STATA 6 sta-tistical software (Stata). The same software program wasused for the statistical analysis of the colony-forming unitsfrom murine organs (Mann–Whitney and Kruskal–Wallistests). Plotting of colony-forming unit data and plotting andstatistical evaluation of binding studies were performed usingMicrocal Origin (OriginLab Corporation). A P-value of lessthan 0.05 was considered to be significant.
Acknowledgements
Financial support was provided by Howard Hughes MedicalInstitute; the Pfizer Fellowship in Medical Mycology from theInfectious Diseases Society of America; the MassachusettsGeneral Hospital Center for the Study of Inflammatory BowelDisease; Harvard Medical School Center for AIDS Research;New Scholar Award in Global Infectious Diseases of theEllison Medical Foundation; Charles E. Culpeper BiomedicalPilot Initiative to E.M.; by a grant from Aventis, SA to F.M.A.and S.B.C. and by NIH Grants GM48707 and AI44975awarded to F.M.A. and J.H.
References
Alspaugh, J.A., Perfect, J.R., and Heitman, J. (1997) Crypto-coccus neoformans mating and virulence are regulated bythe G-protein alpha subunit GPA1 and cAMP. Genes Dev11: 3206–3217.
Apidianakis, Y., Rahme, L.G., Heitman, J., Ausubel, F.M.,Calderwood, S.B., and Mylonakis, E. (2004) Challenge ofDrosophila melanogaster with Cryptococcus neoformansand role of the innate immune response. Eukaryot Cell 3:413–419.
Biernat, J., Wu, Y.Z., Timm, T., Zheng-Fischhofer, Q., Man-delkow, E., Meijer, L., and Mandelkow, E.M. (2002) Proteinkinase MARK/PAR-1 is required for neurite outgrowth andestablishment of neuronal polarity. Mol Biol Cell 13: 4013–4028.
Bohm, H., Brinkmann, V., Drab, M., Henske, A., andKurzchalia, T.V. (1997) Mammalian homologues of C.elegans PAR-1 are asymmetrically localized in epithelialcells and may influence their polarity. Curr Biol 7: 603–606.
Brenner, S. (1974) The genetics of Caenorhabditis elegans.Genetics 77: 71–94.
Brown, A.J., Hutchings, C., Burke, J.F., and Mayne, L.V.(1999) Application of a rapid method (targeted display) forthe identification of differentially expressed mRNAs follow-ing NGF-induced neuronal differentiation in PC12 cells.Mol Cell Neurosci 13: 119–130.
Brunner, D., and Nurse, P. (2000) New concepts in fissionyeast morphogenesis. Philos Trans R Soc Lond B Biol Sci355: 873–877.
Casadevall, A., and Perfect, J.R. (1998) Cryptococcus Neo-formans. Washington, DC: ASM Press.
Casadevall, A., Steenbergen, J.N., and Nosanchuk, J.D.
(2003) ‘Ready made’ virulence and ‘dual use’ virulencefactors in pathogenic environmental fungi – the Cryptococ-cus neoformans paradigm. Curr Opin Microbiol 6: 332–337.
Cox, D.N., Lu, B., Sun, T.Q., Williams, L.T., and Jan, Y.N.(2001) Drosophila par-1 is required for oocyte differentia-tion and microtubule organization. Curr Biol 11: 75–87.
Davidson, R.C., Blankenship, J.R., Kraus, P.R., de JesusBerrios, M., Hull, C.M., D’Souza, C., et al. (2002) A PCR-based strategy to generate integrative targeting alleleswith large regions of homology. Microbiology 148: 2607–2615.
Drewes, G., and Nurse, P. (2003) The protein kinase kin1,the fission yeast orthologue of mammalian MARK/PAR-1,localises to new cell ends after mitosis and is important forbipolar growth. FEBS Lett 554: 45–49.
El Khoury, J.B., Moore, K.J., Means, T.K., Leung, J., Terada,K., Toft, M., et al. (2003) CD36 mediates the innate hostresponse to beta-amyloid. J Exp Med 197: 1657–1666.
Feldmesser, M., Tucker, S., and Casadevall, A. (2001) Intra-cellular parasitism of macrophages by Cryptococcus neo-formans. Trends Microbiol 9: 273–278.
Fraser, J.A., Subaran, R.L., Nichols, C.B., and Heitman, J.(2003) Recapitulation of the sexual cycle of the primaryfungal pathogen Cryptococcus neoformans var. gattii:implications for an outbreak on Vancouver Island, Canada.Eukaryot Cell 2: 1036–1045.
Gan, Y.H., Chua, K.L., Chua, H.H., Liu, B., Hii, C.S., Chong,H.L., and Tan, P. (2002) Characterization of Burkholderiapseudomallei infection and identification of novel virulencefactors using a Caenorhabditis elegans host system. MolMicrobiol 44: 1185–1197.
Garsin, D.A., Sifri, C.D., Mylonakis, E., Qin, X., Singh, K.V.,Murray, B.E., et al. (2001) A simple model host for identi-fying Gram-positive virulence factors. Proc Natl Acad SciUSA 98: 10892–10897.
Greenberg, S., El Khoury, J., di Virgilio, F., Kaplan, E.M., andSilverstein, S.C. (1991) Ca(2+)-independent F-actinassembly and disassembly during Fc receptor-mediatedphagocytosis in mouse macrophages. J Cell Biol 113:757–767.
Greub, G., and Raoult, D. (2004) Microorganisms resistantto free-living amoebae. Clin Microbiol Rev 17: 413–433.
Guo, S., and Kemphues, K.J. (1995) par-1, a gene requiredfor establishing polarity in C. elegans embryos, encodes aputative Ser/Thr kinase that is asymmetrically distributed.Cell 81: 611–620.
Guo, S., and Kemphues, K.J. (1996) Molecular genetics ofasymmetric cleavage in the early Caenorhabditis elegansembryo. Curr Opin Genet Dev 6: 408–415.
Heitman, J., Casadevall, A., Lodge, J.K., and Perfect, J.R.(1999) The Cryptococcus neoformans genome sequenc-ing project. Mycopathologia 148: 1–7.
Hu, B., Sonstein, J., Christensen, P.J., Punturieri, A., andCurtis, J.L. (2000) Deficient in vitro and in vivo phagocyto-sis of apoptotic T cells by resident murine alveolar mac-rophages. J Immunol 165: 2124–2133.
Huffnagle, G.B., Boyd, M.B., Street, N.E., and Lipscomb,M.F. (1998) IL-5 is required for eosinophil recruitment,crystal deposition, and mononuclear cell recruitment dur-ing a pulmonary Cryptococcus neoformans infection in

C. elegans/C. neoformans system 419
© 2004 Blackwell Publishing Ltd, Molecular Microbiology, 54, 407–419
genetically susceptible mice (C57BL/6). J Immunol 160:2393–2400.
Idnurm, A., Reedy, J.L., Nussbaum, J.C., and Heitman, J.(2004) Cryptococcus neoformans virulence gene discov-ery through insertional mutagenesis. Eukaryot Cell 3: 420–429.
de Jesus-Berrios, M., Liu, L., Nussbaum, J.C., Cox, G.M.,Stamler, J.S., and Heitman, J. (2003) Enzymes that coun-teract nitrosative stress promote fungal virulence. Curr Biol13: 1963–1968.
Kopecka, M., Gabriel, M., Takeo, K., Yamaguchi, M., Svo-boda, A., Ohkusu, M., et al. (2001) Microtubules and actincytoskeleton in Cryptococcus neoformans compared withascomycetous budding and fission yeasts. Eur J Cell Biol80: 303–311.
Kurz, C.L., Chauvet, S., Andres, E., Aurouze, M., Vallet, I.,Michel, G.P., et al. (2003) Virulence factors of the humanopportunistic pathogen Serratia marcescens identified byin vivo screening. EMBO J 22: 1451–1460.
Levin, D.E., and Bishop, B.J. (1990) A putative protein kinasegene (kin1+) is important for growth polarity in Schizosac-charomyces pombe. Proc Natl Acad Sci USA 87: 8272–8276.
Luberto, C., Martinez-Marino, B., Taraskiewicz, D., Bol-anos, B., Chitano, P., Toffaletti, D.L., et al. (2003) Identifi-cation of App1 as a regulator of phagocytosis andvirulence of Cryptococcus neoformans. J Clin Invest 112:1080–1094.
Mitchell, A.P. (2003) Updated view of Cryptococcus neofor-mans mating type and virulence. Infect Immun 71: 4829–4830.
Molina, Y., Ramos, S.E., Douglass, T., and Klig, L.S. (1999)Inositol synthesis and catabolism in Cryptococcus neofor-mans. Yeast 15: 1657–1667.
Morton, D.B., Hudson, M.L., Waters, E., and O’Shea, M.(1999) Soluble guanylyl cyclases in Caenorhabditis ele-gans: NO is not the answer. Curr Biol 9: R546–R547.
Mueller, N.J., and Fishman, J.A. (2003) Asymptomatic pul-monary cryptococcosis in solid organ transplantation:report of four cases and review of the literature. TransplInfect Dis 5: 140–143.
Mylonakis, E., Ausubel, F.M., Perfect, J.R., Heitman, J., andCalderwood, S.B. (2002) Killing of Caenorhabditis elegansby Cryptococcus neoformans as a model of yeast patho-genesis. Proc Natl Acad Sci USA 99: 15675–15680.
Nielsen, K., Cox, G.M., Wang, P., Toffaletti, D.L., Perfect,J.R., and Heitman, J. (2003) Sexual cycle of Cryptococcus
neoformans var. grubii and virulence of congenic a andalpha isolates. Infect Immun 71: 4831–4841.
Perfect, J.R., and Casadevall, A. (2002) Cryptococcosis.Infect Dis Clin North Am 16: 837–874, v–vi.
Steenbergen, J.N., Shuman, H.A., and Casadevall, A. (2001)Cryptococcus neoformans interactions with amoebae sug-gest an explanation for its virulence and intracellular patho-genic strategy in macrophages. Proc Natl Acad Sci USA98: 15245–15250.
Steenbergen, J.N., Nosanchuk, J.D., Malliaris, S.D., andCasadevall, A. (2003) Cryptococcus neoformans viru-lence is enhanced after growth in the genetically mallea-ble host Dictyostelium discoideum. Infect Immun 71:4862–4872.
Thomas, C.A., Li, Y., Kodama, T., Suzuki, H., Silverstein,S.C., and El Khoury, J. (2000) Protection from lethal gram-positive infection by macrophage scavenger receptor-dependent phagocytosis. J Exp Med 191: 147–156.
Tibbetts, M., Donovan, M., Roe, S., Stiltner, A.M., and Ham-mond, C.I. (1994) KIN1 and KIN2 protein kinases localizeto the cytoplasmic face of the yeast plasma membrane.Exp Cell Res 213: 93–99.
Tomancak, P., Piano, F., Riechmann, V., Gunsalus, K.C.,Kemphues, K.J., and Ephrussi, A. (2000) A Drosophilamelanogaster homologue of Caenorhabditis elegans par-1 acts at an early step in embryonic-axis formation. NatCell Biol 2: 458–460.
Varma, A., and Kwon-Chung, K.J. (1999) Characterization ofthe glyceraldehyde-3-phosphate dehydrogenase gene[correction of glyceraldehyde-3-phosphate gene] and theuse of its promoter for heterologous expression in Crypto-coccus neoformans, a human pathogen. Gene 232: 155–163.
Waugh, M.S., Nichols, C.B., DeCesare, C.M., Cox, G.M.,Heitman, J., and Alspaugh, J.A. (2002) Ras1 and Ras2contribute shared and unique roles in physiology andvirulence of Cryptococcus neoformans. Microbiology 148:191–201.
Woods, A., Sherwin, T., Sasse, R., MacRae, T.H., Baines,A.J., and Gull, K. (1989) Definition of individual compo-nents within the cytoskeleton of Trypanosoma brucei by alibrary of monoclonal antibodies. J Cell Sci 93: 491–500.
Woyke, T., Roberson, R.W., Pettit, G.R., Winkelmann, G.,and Pettit, R.K. (2002) Effect of auristatin PHE on micro-tubule integrity and nuclear localization in Cryptococcusneoformans. Antimicrob Agents Chemother 46: 3802–3808.




















