
Cerivastatin prevents tumor necrosis factor-α-induced downregulation of endothelial nitric oxide...
10
Atherosclerosis 155 (2001) 61 – 70 Cerivastatin prevents tumor necrosis factor-a-induced downregulation of endothelial nitric oxide synthase: role of endothelial cytosolic proteins Fernando Gonza ´lez-Ferna ´ndez, Ana Jime ´nez, Almudena Lo ´ pez-Blaya, Sandra Velasco, Marı ´a M. Arriero, A ´ ngel Celdra ´n, Luis Rico, Jero ´ nimo Farre ´, Santos Casado, Antonio Lo ´ pez-Farre ´* Cardio6ascular Research and Hypertension Laboratory, Fundacio ´n Jime ´nez Dı ´az, A6 Reyes Cato ´licos 2, Madrid 28040, Spain Received 24 January 2000; accepted 28 April 2000 Abstract Cardiovascular disease is accompanied by an impaired endothelium-dependent vasodilatory response. Loss of endothelial nitric oxide synthase (eNOS) expression may contribute to endothelial dysfunction. The aim of the present study was to analyze the effect of cerivastatin, a novel HMG CoA reductase inhibitor, on tumor necrosis factor-a (TNF-a)-induced downregulation of eNOS protein expression in bovine aortic endothelial cells (BAEC). TNF-a (10 ng/ml)- incubated BAEC showed a reduced expression of eNOS protein and decreased eNOS mRNA stabilization. This effect was associated with an increased binding activity of BAEC cytosolic proteins to the 3%-untranslated region (3%UTR) of eNOS mRNA. Cerivastatin prevented TNF-a-in- duced downregulation of eNOS protein expression in a concentration-dependent manner (10 -8 to 10 -5 M). Cerivastatin also prevented the binding of the cytosolic proteins to 3%-UTR of eNOS mRNA and was associated with eNOS mRNA stabilization. The reduced expression of eNOS protein by TNF-a was also prevented by coincubation with cycloheximide. In addition cycloheximide inhibited the binding activity of the cytosolic proteins to 3%-UTR of eNOS mRNA, suggesting the inducible character of the mentioned-cytosolic proteins. TNF-a stimulated the translocation of nuclear factor-kB (NF-kB), an effect that was not modified by cerivastatin. Furthermore, an inhibitor of NF-kB translocation, pyrrolidine dithiocarbamate failed to modify both the downregulation of eNOS expression and the increased binding activity of the cytosolic proteins to 3%-UTR of eNOS mRNA by TNF-a. The effect of cerivastatin on eNOS expression and the binding activity of the cytosolic proteins were reversed by coincubation with L-mevalonate. In conclusion, cerivastatin stabilized eNOS mRNA and upregulated eNOS expression in the endothelium, and this was associated with a decreased binding activity of cytosolic proteins to 3%-UTR of eNOS mRNA. The effect of cerivastatin on the regulation of eNOS expression was independent of NF-kB mobilization by TNF-a. These findings suggest that cerivastatin may have beneficial effects on the endothelial dysfunction associated with cardiovascular diseases beyond its effect on lowering cholesterol. © 2001 Elsevier Science Ireland Ltd. All rights reserved. Keywords: Atherosclerosis; Endothelial cells; Endothelium-derived factors; Nitric oxide synthase; Statins www.elsevier.com/locate/atherosclerosis 1. Introduction Clinical trials with 3-hydroxy-3 methylglutaryl coen- zyme A (HMG-CoA) reductase inhibitors have shown that a reduction in serum cholesterol level is correlated with improved survival in patients with coronary artery disease [1,2]. However, more recent studies have sug- gested that, despite comparable reductions on serum cholesterol achieved with HMG-CoA reductase in- hibitors and other treatment modalities such as partial ileal loop bypass or treatment with cholestyramine, the clinical benefits were higher with HMG-CoA reductase inhibitors [3–5]. One of the earliest recognizable benefits after treatment with HMG-CoA reductase in- hibitors is the restoration of endothelial function [6,7]. The first and second author contributed equally to the article. * Corresponding author. Tel.: +34-91-5504821; fax: +34-91- 5494764. E-mail address: alopeza@fid.es (A. Lo ´ pez-Farre ´). 0021-9150/01/$ - see front matter © 2001 Elsevier Science Ireland Ltd. All rights reserved. PII:S0021-9150(00)00535-9
-
Upload
usantotomas -
Category
Documents
-
view
1 -
download
0
Transcript of Cerivastatin prevents tumor necrosis factor-α-induced downregulation of endothelial nitric oxide...

Atherosclerosis 155 (2001) 61–70
Cerivastatin prevents tumor necrosis factor-a-induceddownregulation of endothelial nitric oxide synthase: role of
endothelial cytosolic proteins�
Fernando Gonzalez-Fernandez, Ana Jimenez, Almudena Lopez-Blaya, Sandra Velasco,Marıa M. Arriero, Angel Celdran, Luis Rico, Jeronimo Farre, Santos Casado,
Antonio Lopez-Farre *Cardio6ascular Research and Hypertension Laboratory, Fundacion Jimenez Dıaz, A6 Reyes Catolicos 2, Madrid 28040, Spain
Received 24 January 2000; accepted 28 April 2000
Abstract
Cardiovascular disease is accompanied by an impaired endothelium-dependent vasodilatory response. Loss of endothelial nitricoxide synthase (eNOS) expression may contribute to endothelial dysfunction. The aim of the present study was to analyze theeffect of cerivastatin, a novel HMG CoA reductase inhibitor, on tumor necrosis factor-a (TNF-a)-induced downregulation ofeNOS protein expression in bovine aortic endothelial cells (BAEC). TNF-a (10 ng/ml)- incubated BAEC showed a reducedexpression of eNOS protein and decreased eNOS mRNA stabilization. This effect was associated with an increased bindingactivity of BAEC cytosolic proteins to the 3%-untranslated region (3%UTR) of eNOS mRNA. Cerivastatin prevented TNF-a-in-duced downregulation of eNOS protein expression in a concentration-dependent manner (10−8 to 10−5 M). Cerivastatin alsoprevented the binding of the cytosolic proteins to 3%-UTR of eNOS mRNA and was associated with eNOS mRNA stabilization.The reduced expression of eNOS protein by TNF-a was also prevented by coincubation with cycloheximide. In additioncycloheximide inhibited the binding activity of the cytosolic proteins to 3%-UTR of eNOS mRNA, suggesting the induciblecharacter of the mentioned-cytosolic proteins. TNF-a stimulated the translocation of nuclear factor-kB (NF-kB), an effect thatwas not modified by cerivastatin. Furthermore, an inhibitor of NF-kB translocation, pyrrolidine dithiocarbamate failed to modifyboth the downregulation of eNOS expression and the increased binding activity of the cytosolic proteins to 3%-UTR of eNOSmRNA by TNF-a. The effect of cerivastatin on eNOS expression and the binding activity of the cytosolic proteins were reversedby coincubation with L-mevalonate. In conclusion, cerivastatin stabilized eNOS mRNA and upregulated eNOS expression in theendothelium, and this was associated with a decreased binding activity of cytosolic proteins to 3%-UTR of eNOS mRNA. Theeffect of cerivastatin on the regulation of eNOS expression was independent of NF-kB mobilization by TNF-a. These findingssuggest that cerivastatin may have beneficial effects on the endothelial dysfunction associated with cardiovascular diseases beyondits effect on lowering cholesterol. © 2001 Elsevier Science Ireland Ltd. All rights reserved.
Keywords: Atherosclerosis; Endothelial cells; Endothelium-derived factors; Nitric oxide synthase; Statins
www.elsevier.com/locate/atherosclerosis
1. Introduction
Clinical trials with 3-hydroxy-3 methylglutaryl coen-zyme A (HMG-CoA) reductase inhibitors have shownthat a reduction in serum cholesterol level is correlated
with improved survival in patients with coronary arterydisease [1,2]. However, more recent studies have sug-gested that, despite comparable reductions on serumcholesterol achieved with HMG-CoA reductase in-hibitors and other treatment modalities such as partialileal loop bypass or treatment with cholestyramine, theclinical benefits were higher with HMG-CoA reductaseinhibitors [3–5]. One of the earliest recognizablebenefits after treatment with HMG-CoA reductase in-hibitors is the restoration of endothelial function [6,7].
� The first and second author contributed equally to the article.* Corresponding author. Tel.: +34-91-5504821; fax: +34-91-
5494764.E-mail address: [email protected] (A. Lopez-Farre).
0021-9150/01/$ - see front matter © 2001 Elsevier Science Ireland Ltd. All rights reserved.PII: S 0 0 2 1 -9150 (00 )00535 -9

F. Gonzalez-Fernandez et al. / Atherosclerosis 155 (2001) 61–7062
Endothelial dysfunction is an early marker ofatherosclerosis and is often defined as the impairedrelease of nitric oxide (NO) by the endothelium [8].NO is a gas generated in endothelial cells by themetabolic conversion of L-arginine into L-citrulline bythe activity of an enzyme known as endothelial NO-synthase (eNOS) [9,10]. The NO generated by theeNOS activity is responsible for the so-called endothe-lium-dependent vasorelaxation response which hasbeen reported impaired in patients with hypercholes-terolemia and atherosclerosis [11–13]. This loss of en-dothelium-mediated vasodilation is thought to beinvolved in the pathogenesis of myocardial ischemia[14].
Initially the eNOS protein was described as consti-tutive; however, in recent years a number of studieshave demonstrated that several pathological stimuli,such as hypoxia, chronic exercise and the growthstate, upregulate eNOS expression in endothelial cells[15–17]. Furthermore, cytokines and more particu-larly tumor necrosis factor-a (TNF-a) downregulatethe expression of eNOS through destabilization ofeNOS mRNA [18,19]. Interestingly, there is growingevidence that the HMG-CoA reductase inhibitors re-verse the downregulation of eNOS expression inducedby oxidized low density lipoprotein in endothelialcells under cholesterol-clamped conditions [20,21]. Themechanism by which statins prevents eNOS expres-sion in endothelial cells remains to be identified, al-though the stabilization of eNOS mRNA seems toplay a major role [20].
The regulation of mRNA stability has emerged asan important control mechanism which regulates cel-lular mRNA levels. Protein factors control mRNAstability by binding to specific sequences contained ineach mRNA [22,23]. Some of these sequences havebeen identified within the 3%-untranslated region (3%-UTR) of mRNA [22]. In this regard, we recently ob-tained new evidences demonstrating that bovineendothelial cells contain cytosolic proteins thatformed complexes with in vitro transcribed 3%-UTR ofeNOS mRNA [24]. The binding of these cytosolicproteins was increased by cytokines which are com-monly found in atherosclerotic lesions i.e. TNF-a,and was correlated with higher eNOS mRNA destabi-lization [24,25].
Therefore, based on the above described data, theaim of the present study was to study the effect ofcerivastatin, a new HMG CoA reductase inhibitor, onthe TNF-a-dependent downregulation of eNOS inbovine aortic endothelial cells. We further analyzedwhether cerivastatin modified the binding activity ofthe cytosolic proteins that bind to 3%-UTR of eNOSmRNA.
2. Materials and methods
2.1. Cell culture and lysate preparation
Bovine aortic endothelial cells (BAEC) were isolatedfrom thoracic aortas as previously described [16,26].Characterization was based on their typical cobblestoneappearance and biochemically by factor VIII detection.Cells were maintained in RPMI 1640 supplementedwith 10% calf serum, 5 mM glutamine, 100 U/mlpenicillin and 100 mg/m streptomycin in an atmosphereof 95% O2 and 5% CO2. Experiments were performedon confluent monolayers at passages 2–4, madequiescent by serum deprivation. For the TNF-aexperiments, confluent BAEC were incubated withTNF-a (10 ng/ml) in the presence and in the absence ofincreasing doses of cerivastatin. To prepare cytoplasmiclysates, confluent monolayers of BAEC were washedtwice in ice-cold phosphate-buffered saline, gentlyscraped in cold detaching buffer (10 mM Tris–HCl (pH7.6), 100 mM NaCl, 1 mM EDTA), and transferred tomicrocentrifuge tubes. After brief centrifugation, thecells were resuspended in hypotonic buffer (25 mmol/lTris–HCl (pH 7.9), 0.5 mM EDTA, 1 mMphenylmethylsulfonyl fluoride) and lysed by four cyclesof freezing and thawing followed by centrifugation at12 000×g and 4°C for 15 min. The supernatant wasremoved, supplemented with glycerol (10% finalconcentration) and frozen at −70°C until use.
2.2. Western blot analysis
eNOS protein was analyzed by Western blotting aspreviously described [16,27]. Endothelial cells wereincubated in the presence and in the absence of TNF-aand increasing doses of cerivastatin. Endothelial cellswere then homogenated and lysed in Laemmli buffercontaining 2-bmercaptoethanol [28]. Proteins wereseparated on denaturing sodium docecyl sulfate(SDS)-10% polyacrylamide gels. Equal amounts ofprotein (10 mg/lane) estimated by bicinchoninic acidreagent (Pierce, Rockford, IL) were loaded in the gel.To verify that equal amounts of proteins had beenloaded in the gel, a parallel gel with identical sampleswas run and stained with Coomassie Blue to comparethe intensities of the protein bands. The separatedproteins were then blotted into nitrocellulose(Immobilon-P, Millipore Corp., Iberica). Blots wereblocked overnight at 4°C with 5% nonfat dry milk inTBS-T (20 mM of Tris-(hydroxymethyl) aminomethane(Tris–HCl), 137 mM of NaCl, 0.1% Tween-20).Western blot analysis was performed with amonoclonal antibody against eNOS (TransductionLaboratories, Lexington, UK). Blots were incubatedwith the first antibody (1:2500) for 1 h at roomtemperature and, after extensive washing, with the

F. Gonzalez-Fernandez et al. / Atherosclerosis 155 (2001) 61–70 63
second antibody (horseradish peroxidase-conjugatedanti-mouse immunoglobulin antibody) at a dilution of1:1500 for 1 further hour. Specific eNOS protein wasdetected by enhanced chemiluminescence (ECL; Amer-sham Iberica, Madrid, Spain). Prestained protein mark-ers were used for molecular mass determinations(Sigma Chemicals, Spain).
2.3. Plasmids and in 6itro transcription
Oligonucleotides complementary to endothelial NOScDNA (GenBank accesion number BTNIOXSY) werepurchased from Bio-synthesis Inc (Lewisville, TX).Oligoc1 (5%-GGATCTAGAACGCTATCACGAGG-ACATT-3%) and Oligoc2 (5%-AGGAAGCTTAGTAG-GTCTCCTAACTTCTG-3%) were used to produce byRT-PCR a fragment covering 166 bases of the codingregion and 393 bases of the 3%-UTR of the eNOScDNA (from 3485 to 4012). Amplification productswere purified after agarose gel electrophoresis, sub-jected to restriction endonuclease digestion with XbaIand HindIII and ligated to pGEM4Z (Promega Bio-tech, Madison, MI) to create plasmid pNOS-UTR-L.
To produce single-strand RNA, the plasmids werelinearized with the corresponding restriction enzymeand transcribed with SP6 or T7 RNA polymerase.Radiolabeled RNA was produced according to themanufacturer conditions (Promega Biotech, Madison,WI) with 32P-CTP (Amersham Iberica, Madrid, Spain,109 cpm/mg). For competition experiments, unlabeledRNA was synthesized using the RiboMax Large ScaleRNA System (Promega Biotech, Madison, WI).
2.4. Band shift assays
Cytoplasmic lysates of endothelial cells (10 mg) wereincubated with radiolabeled UTR-L (5–10×104 cpm,approximately 1 ng RNA) in 15 mM HEPES (pH 7.9),10 mM KCl, 5 mM Cl2Mg, 1 mM DTT, 1 mg/ml yeasttRNA, 40 units of RNAsin (Promega Biotech,Madison, WI) and 10% glycerol in a total volume of 15ml for 10 min at 25°C. Afterwards, 20 units of RNAseT1 per reaction (Gibco-BRL, Dieselstrasse, Germany)were added and the reaction mixtures were incubatedfor 30 min at 37°C. Samples were electrophoresed on4% native polyacrilamide gel in 0.25×TBE (Tris–Bo-rate-EDTA) as running buffer, dried and autoradio-graphed with Kodak X-OMAT-S film. For competitionexperiments, unlabeled RNA was preincubated for 10min with cytosolic lysates prior to the addition ofradiolabeled RNA.
2.5. Northern-blot analysis
Total RNA was isolated according to the method ofChomczynski and Sacchi [29]. Twenty micrograms total
RNA were fractionated in 1.3% agarose–formaldehydegels and transferred by capilar to Genescreen™ nylonmembranes (Dupont, Boston, MA). The membraneswere prehybridized at 42°C for 4 h in 50% formamide,1% SDS, 5×SSC, 1×Denhardt’s solution, 100 mg/mldenatured herring sperm DNA and 50 mmol/l phos-phate, pH 6.5, and hybridized for 16–18 h in the samesolution supplemented with dextran sulphate (10% finalconcentration) containing 500 000 cpm/ml of radiola-beled eNOS probe. The cDNA probe used was theEcoRI/BamHI fragment of pNOS-UTR-L (see before).
2.6. Determination of nuclear factor-kB translocation
Nuclear protein extracts were prepared from TNF-a-stimulated BAEC in the presence or in the absence ofcerivastatin by ultrasound disruption of cell membranesfollowed by high salt extraction with Dignam’s buffer Cand diluted with buffer D as described previously [30].Nuclear proteins were mixed with an oligonucleotidecorresponding to a double-stranded NF-kB consensusoligonucleotide (5%-AGTTGAGGGGACTTTCCCAG-G-3%, Promega). The NF-kB oligonucleotide was end-labelled using (g-32P)-ATP and T4 polynucleotide ki-nase. Standard binding reactions were performed byincubating 10 mg of nuclear extracts in 20 ml of 10 mMTris–HCl, pH 7.5, containing 100 mM NaCl, 1 mMdithiothreitol, 1 mM EDTA, 10 mmol/l MgCl2, 20%(v/v) glycerol, and 50 000 dpm of 32P-labelled NF-kBoligonucleotide (approximately 1 pmol) for 20 min atroom temperature. After incubation, the samples wereloaded onto a 4% polyacrylamide gel and run at aconstant current of 100 V in 0.5×Tris–borate-EDTA(TBE) at 4°C. Gels were dried and placed on film at−80°C.
2.7. Statistical methods
Densitometric results of Western and Northern blotswere expressed as mean9SEM. Unless otherwisestated, each study was performed in a minimum of fourdifferent experiments. To determine the statistical sig-nificance of our results, comparisons were performed byANOVA. The Bonferroni correction for multiple com-parisons was used to determine the level of significanceof the P value. A P value B0.05 was consideredstatistically significant.
3. Results
3.1. Effect of ceri6astatin in eNOS protein expressionin endothelial cells
As previously reported, TNF-a (10 ng/ml) downregu-lated eNOS protein expression in BAEC in a time-de-

F. Gonzalez-Fernandez et al. / Atherosclerosis 155 (2001) 61–7064
pendent manner (Fig. 1A and B). The maximal de-crease in eNOS protein expression was observed be-tween 12 and 24 h after TNF-a incubation (Fig. 1A andB). Therefore, the subsequent experiments were per-formed after 18 h of TNF-a incubation.
BAEC were stimulated with TNF-a (10 ng/ml) for 18h in the presence and in the absence of increasing dosesof cerivastatin (10−8–10−5 M). As shown in Fig. 2Aand B, cerivastatin prevented the reduction in eNOSprotein expression induced by TNF-a in BAEC. Theeffect of cerivastatin ocurred in a concentration-depen-
Fig. 3. Western blot that demonstrates the specificity of the mono-clonal antibody used in the experiments to recognize the eNOSprotein. The monoclonal antibody did not recognize the neuronal-type NOS in rat pituitary homogenate and the inducible-type NOS inE. coli LPS-stimulated macrophages but it specifically recognized theeNOS isoform (140 KDa) in homogenates of human umbilical veinendothelial cells (Top). The middle of the figure shows a Western blotthat demonstrates the presence of the neuronal-type NOS (155 KDa)in rat pituitary lisate which is not detectable in homogenates ofhuman endothelial cells. The bottom of the figure shows a Westernblot that demonstrates the presence of the inducible-type NOS (135KDa) in homogenates of E. coli LPS-stimulated macrophages.
Fig. 1. (A) Representative Western blot showing eNOS proteinexpression in BAEC. BAEC were stimulated with TNF-a (10 ng/ml)for different periods of time. (B) Bar graph showing the densitometricscanning of the Western blot. Results are represented as mean9SEMof four different experiments. * PB0.05 with respect to control.
dent manner. A slight increase in eNOS protein expres-sion was observed at 10−7 mol/l of cerivastatin. Dosesof cerivastatin lower than 10−7 M failed to affecteNOS protein expression (data not shown). The maxi-mal increase in eNOS protein expression by cerivastatinwas observed at 10−5 M (Fig. 2A and B).
The specificity of the eNOS monoclonal antibodywas further studied. The monoclonal antibody used didnot cross-react with the neuronal-type constitutive iso-form (155 KDa) because the band of nNOS protein wasundetectable in a homogenate of rat pituitary (Fig. 3,top) which was previously positively stained with amononuclear antibody against the neuronal-type NOSisoform (Fig. 3, middle). Moreover, the eNOS bandwas undetectable in a homogenate of E. coli LPS-stimu-lated macrophages (Fig. 3, top) which was previouslypositively stained with a monoclonal antibody againstthe inducible-type NOS (140 KDa, Fig. 3, bottom). Themonoclonal antibody specifically recognized the eNOSisoform (140 KDa) in homogenates of human umbilicalendothelial cells (Fig. 3, top).
Compared with untreated cells, incubation of BAECwith TNF-a decreased eNOS mRNA levels in a time-dependent manner (Fig. 4). A significant decrease ineNOS mRNA expression was observed 6 h after TNF-astimulation of the cells with a marked reduction of thelevel of eNOS mRNA 18 h after TNF-a incubation(Fig. 4).
Fig. 2. (A) Representative Western blot demonstrating the expressionof eNOS protein in TNF-a-stimulated BAEC in the presence ofincreasing concentrations of cerivastatin. (B) Bar graph showing thedensitometric scanning of the Western blot. Results are represented asmean9SEM of six different experiments. * PB0.05 with respect tocontrol. � PB0.05 with respect to TNF-a-incubated BAEC in theabsence of cerivastatin.

F. Gonzalez-Fernandez et al. / Atherosclerosis 155 (2001) 61–70 65
The posttranscriptional regulation of eNOS mRNAwas determined by the presence of the transcriptionalinhibitor 5,6-dichloro-1-b-D-ribofuranosylbenzimida-zole (60 mM DRB, Calbiochem, La Jolla, CA). Thetime-dependent decrease in the levels of eNOS mRNAinduced by TNF-a occurred in the presence of 60mmol/l DRB, indicating that the effect of TNF-a oneNOS mRNA expression was dependent on mRNAdestabilization (Fig. 4). Addition of 10−5 M cerivas-tatin, significantly prevented eNOS mRNA destabiliza-tion induced by TNF-a (Fig. 4).
3.2. Binding of endothelial cell cytosolic proteins to the3 %- UTR of eNOS mRNA
As previously reported [24], addition of endothelialcytoplasmic extracts to a labeled probe containing theentire 3%-UTR of eNOS mRNA, the UTR-L probe,resulted in a gel-shifted band (Fig. 5A, lane 1). Incuba-tion of BAEC with TNF-a for 18 h potentiated thebinding activity of the cytosolic extracts to the labeledUTR-L (Fig. 5A, lane 2). The 18-h incubation time waschosen because we have previously demonstrated thatthe highest binding activity induced by TNF-a occurredafter 12–24 h of incubation [24]. The mRNA-endothe-lial cytoplasmic extract complex was prevented by anexcess (1000 ng) of unlabeled UTR-L (Fig. 5A, lane 3),thus demonstrating the specificity of the reaction.Treatment of cytosolic extracts with proteinase K (87mg/ml) prior to their incubation with the UTR-L abol-ished the complex formation, thus indicating the in-volvement of cytosolic proteins in the eNOSmRNA-endothelial cytosolic extract interactions (Fig.5B).
Fig. 5. (A) Gel mobility-shift assay demonstrating that TNF-a (10ng/ml) stimulated BAEC cytosolic extracts to bind to 3%-UTR ofeNOS mRNA. The complex formation between the BAEC cytosolicextracts and labeled UTR-L was specific since it was prevented by anexcess of unlabeled UTR-L (1000 ng). (B) Treatment of the BAECcytosolic extracts with proteinase K (87 mg/ml) prior to their incuba-tion with the UTR-L probe fully eliminated the gel-shifted band.
Fig. 4. Representative Northern blot analysis of eNOS mRNA ex-pression after incubation of BAEC with TNF-a (10 ng/ml) fordifferent periods of time in the presence and in the absence of 10−5
M cerivastatin. Experiments were performed in the presence of thetranscriptional inhibitor DRB (60 mM). Equal loading of RNA wasconfirmed by the ethidium bromide staining of 28S and 18S RNA.
We then determined whether cerivastatin could affectthe binding activity of endothelial cell cytosolic proteinsto the 3%-UTR of eNOS mRNA. BAEC were stimulatedwith TNF-a (10 ng/ml) for 18 h in the presence and inthe absence of increasing concentrations of cerivastatin.As shown in Fig. 6, cerivastatin reduced the bindingactivity of BAEC cytosolic proteins to labeled UTR-Lstimulated by TNF-a. The effect of cerivastatin oc-curred in a concentration-dependent manner (Fig. 6). Aslight although significant decrease of the endothelialcytosolic protein binding activity was observed at 10−7
M of cerivastatin. Doses of cerivastatin lower than10−7 mol/l failed to consistently affect the bindingactivity of the BAEC cytosolic proteins to eNOS
F. Gonzalez-Fernandez et al. / Atherosclerosis 155 (2001) 61–7066
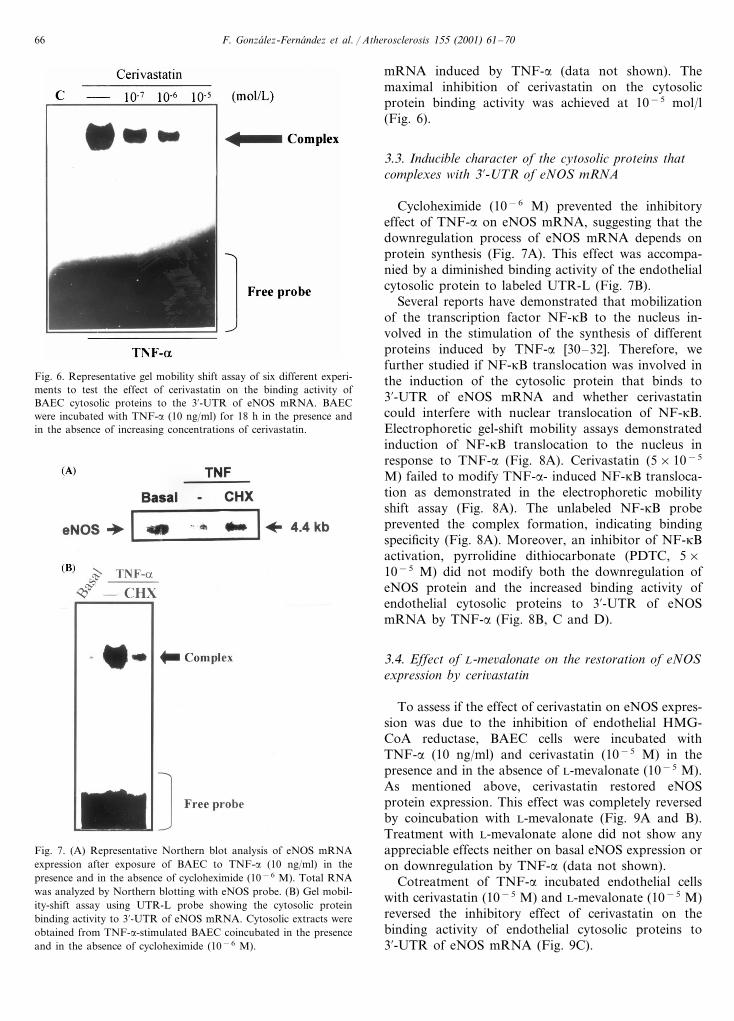
Fig. 6. Representative gel mobility shift assay of six different experi-ments to test the effect of cerivastatin on the binding activity ofBAEC cytosolic proteins to the 3%-UTR of eNOS mRNA. BAECwere incubated with TNF-a (10 ng/ml) for 18 h in the presence andin the absence of increasing concentrations of cerivastatin.
mRNA induced by TNF-a (data not shown). Themaximal inhibition of cerivastatin on the cytosolicprotein binding activity was achieved at 10−5 mol/l(Fig. 6).
3.3. Inducible character of the cytosolic proteins thatcomplexes with 3 %-UTR of eNOS mRNA
Cycloheximide (10−6 M) prevented the inhibitoryeffect of TNF-a on eNOS mRNA, suggesting that thedownregulation process of eNOS mRNA depends onprotein synthesis (Fig. 7A). This effect was accompa-nied by a diminished binding activity of the endothelialcytosolic protein to labeled UTR-L (Fig. 7B).
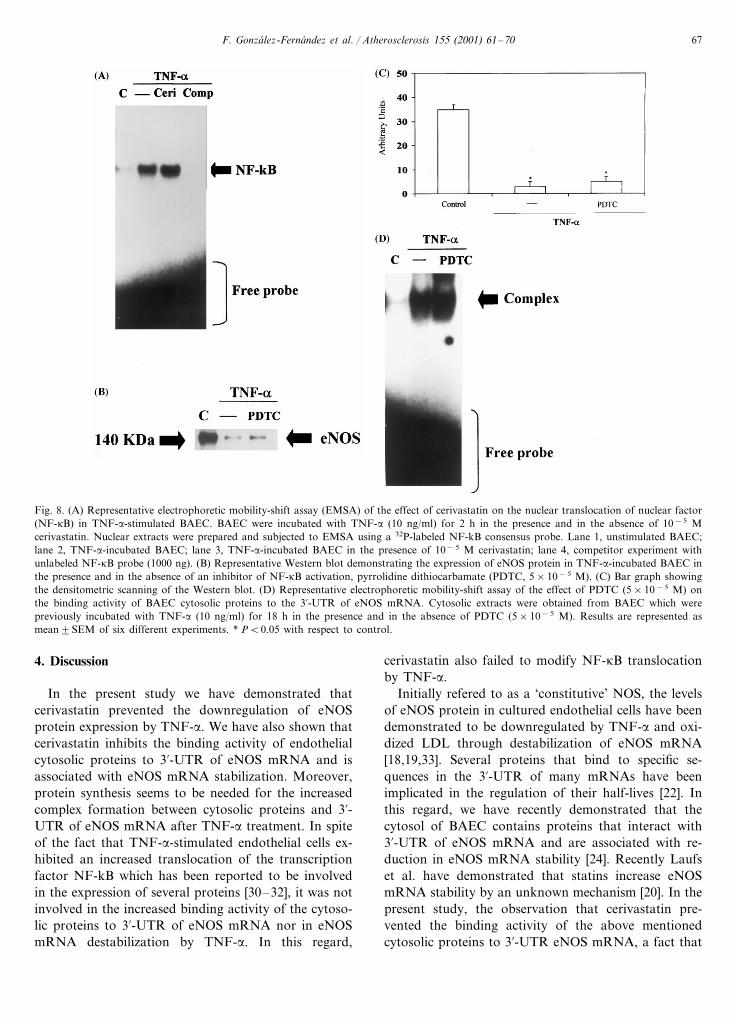
Several reports have demonstrated that mobilizationof the transcription factor NF-kB to the nucleus in-volved in the stimulation of the synthesis of differentproteins induced by TNF-a [30–32]. Therefore, wefurther studied if NF-kB translocation was involved inthe induction of the cytosolic protein that binds to3%-UTR of eNOS mRNA and whether cerivastatincould interfere with nuclear translocation of NF-kB.Electrophoretic gel-shift mobility assays demonstratedinduction of NF-kB translocation to the nucleus inresponse to TNF-a (Fig. 8A). Cerivastatin (5×10−5
M) failed to modify TNF-a- induced NF-kB transloca-tion as demonstrated in the electrophoretic mobilityshift assay (Fig. 8A). The unlabeled NF-kB probeprevented the complex formation, indicating bindingspecificity (Fig. 8A). Moreover, an inhibitor of NF-kBactivation, pyrrolidine dithiocarbonate (PDTC, 5×10−5 M) did not modify both the downregulation ofeNOS protein and the increased binding activity ofendothelial cytosolic proteins to 3%-UTR of eNOSmRNA by TNF-a (Fig. 8B, C and D).
3.4. Effect of L-me6alonate on the restoration of eNOSexpression by ceri6astatin
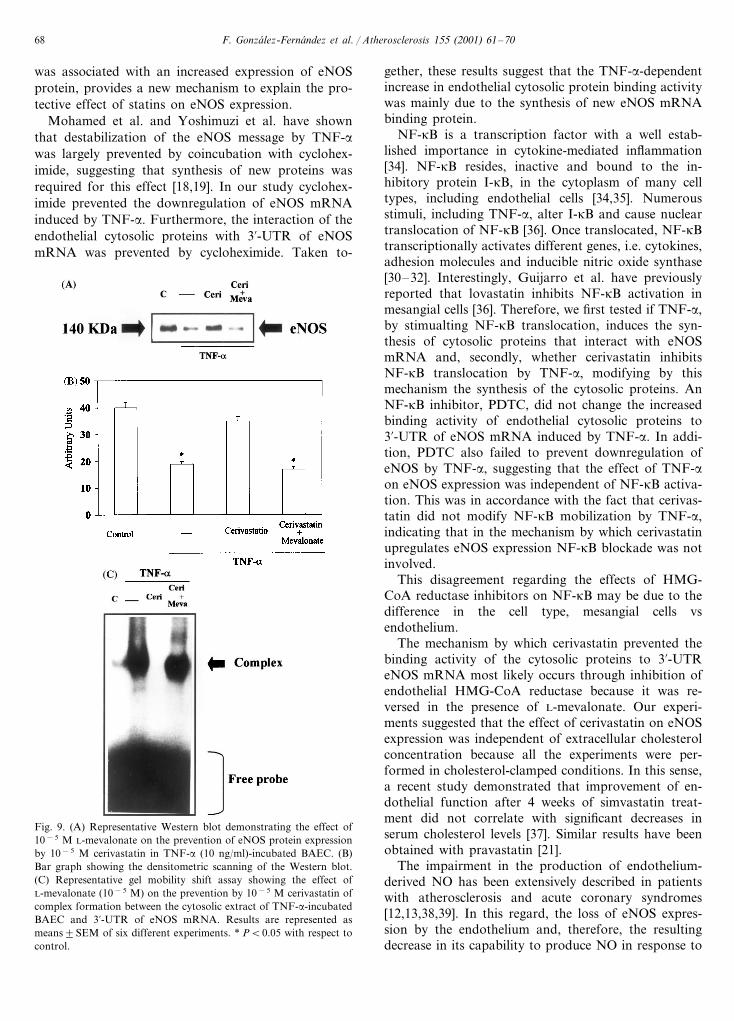
To assess if the effect of cerivastatin on eNOS expres-sion was due to the inhibition of endothelial HMG-CoA reductase, BAEC cells were incubated withTNF-a (10 ng/ml) and cerivastatin (10−5 M) in thepresence and in the absence of L-mevalonate (10−5 M).As mentioned above, cerivastatin restored eNOSprotein expression. This effect was completely reversedby coincubation with L-mevalonate (Fig. 9A and B).Treatment with L-mevalonate alone did not show anyappreciable effects neither on basal eNOS expression oron downregulation by TNF-a (data not shown).
Cotreatment of TNF-a incubated endothelial cellswith cerivastatin (10−5 M) and L-mevalonate (10−5 M)reversed the inhibitory effect of cerivastatin on thebinding activity of endothelial cytosolic proteins to3%-UTR of eNOS mRNA (Fig. 9C).
Fig. 7. (A) Representative Northern blot analysis of eNOS mRNAexpression after exposure of BAEC to TNF-a (10 ng/ml) in thepresence and in the absence of cycloheximide (10−6 M). Total RNAwas analyzed by Northern blotting with eNOS probe. (B) Gel mobil-ity-shift assay using UTR-L probe showing the cytosolic proteinbinding activity to 3%-UTR of eNOS mRNA. Cytosolic extracts wereobtained from TNF-a-stimulated BAEC coincubated in the presenceand in the absence of cycloheximide (10−6 M).
F. Gonzalez-Fernandez et al. / Atherosclerosis 155 (2001) 61–70 67
Fig. 8. (A) Representative electrophoretic mobility-shift assay (EMSA) of the effect of cerivastatin on the nuclear translocation of nuclear factor(NF-kB) in TNF-a-stimulated BAEC. BAEC were incubated with TNF-a (10 ng/ml) for 2 h in the presence and in the absence of 10−5 Mcerivastatin. Nuclear extracts were prepared and subjected to EMSA using a 32P-labeled NF-kB consensus probe. Lane 1, unstimulated BAEC;lane 2, TNF-a-incubated BAEC; lane 3, TNF-a-incubated BAEC in the presence of 10−5 M cerivastatin; lane 4, competitor experiment withunlabeled NF-kB probe (1000 ng). (B) Representative Western blot demonstrating the expression of eNOS protein in TNF-a-incubated BAEC inthe presence and in the absence of an inhibitor of NF-kB activation, pyrrolidine dithiocarbamate (PDTC, 5×10−5 M). (C) Bar graph showingthe densitometric scanning of the Western blot. (D) Representative electrophoretic mobility-shift assay of the effect of PDTC (5×10−5 M) onthe binding activity of BAEC cytosolic proteins to the 3%-UTR of eNOS mRNA. Cytosolic extracts were obtained from BAEC which werepreviously incubated with TNF-a (10 ng/ml) for 18 h in the presence and in the absence of PDTC (5×10−5 M). Results are represented asmean9SEM of six different experiments. * PB0.05 with respect to control.
4. Discussion
In the present study we have demonstrated thatcerivastatin prevented the downregulation of eNOSprotein expression by TNF-a. We have also shown thatcerivastatin inhibits the binding activity of endothelialcytosolic proteins to 3%-UTR of eNOS mRNA and isassociated with eNOS mRNA stabilization. Moreover,protein synthesis seems to be needed for the increasedcomplex formation between cytosolic proteins and 3%-UTR of eNOS mRNA after TNF-a treatment. In spiteof the fact that TNF-a-stimulated endothelial cells ex-hibited an increased translocation of the transcriptionfactor NF-kB which has been reported to be involvedin the expression of several proteins [30–32], it was notinvolved in the increased binding activity of the cytoso-lic proteins to 3%-UTR of eNOS mRNA nor in eNOSmRNA destabilization by TNF-a. In this regard,
cerivastatin also failed to modify NF-kB translocationby TNF-a.
Initially refered to as a ‘constitutive’ NOS, the levelsof eNOS protein in cultured endothelial cells have beendemonstrated to be downregulated by TNF-a and oxi-dized LDL through destabilization of eNOS mRNA[18,19,33]. Several proteins that bind to specific se-quences in the 3%-UTR of many mRNAs have beenimplicated in the regulation of their half-lives [22]. Inthis regard, we have recently demonstrated that thecytosol of BAEC contains proteins that interact with3%-UTR of eNOS mRNA and are associated with re-duction in eNOS mRNA stability [24]. Recently Laufset al. have demonstrated that statins increase eNOSmRNA stability by an unknown mechanism [20]. In thepresent study, the observation that cerivastatin pre-vented the binding activity of the above mentionedcytosolic proteins to 3%-UTR eNOS mRNA, a fact that
F. Gonzalez-Fernandez et al. / Atherosclerosis 155 (2001) 61–7068
was associated with an increased expression of eNOSprotein, provides a new mechanism to explain the pro-tective effect of statins on eNOS expression.
Mohamed et al. and Yoshimuzi et al. have shownthat destabilization of the eNOS message by TNF-awas largely prevented by coincubation with cyclohex-imide, suggesting that synthesis of new proteins wasrequired for this effect [18,19]. In our study cyclohex-imide prevented the downregulation of eNOS mRNAinduced by TNF-a. Furthermore, the interaction of theendothelial cytosolic proteins with 3%-UTR of eNOSmRNA was prevented by cycloheximide. Taken to-
gether, these results suggest that the TNF-a-dependentincrease in endothelial cytosolic protein binding activitywas mainly due to the synthesis of new eNOS mRNAbinding protein.
NF-kB is a transcription factor with a well estab-lished importance in cytokine-mediated inflammation[34]. NF-kB resides, inactive and bound to the in-hibitory protein I-kB, in the cytoplasm of many celltypes, including endothelial cells [34,35]. Numerousstimuli, including TNF-a, alter I-kB and cause nucleartranslocation of NF-kB [36]. Once translocated, NF-kBtranscriptionally activates different genes, i.e. cytokines,adhesion molecules and inducible nitric oxide synthase[30–32]. Interestingly, Guijarro et al. have previouslyreported that lovastatin inhibits NF-kB activation inmesangial cells [36]. Therefore, we first tested if TNF-a,by stimualting NF-kB translocation, induces the syn-thesis of cytosolic proteins that interact with eNOSmRNA and, secondly, whether cerivastatin inhibitsNF-kB translocation by TNF-a, modifying by thismechanism the synthesis of the cytosolic proteins. AnNF-kB inhibitor, PDTC, did not change the increasedbinding activity of endothelial cytosolic proteins to3%-UTR of eNOS mRNA induced by TNF-a. In addi-tion, PDTC also failed to prevent downregulation ofeNOS by TNF-a, suggesting that the effect of TNF-aon eNOS expression was independent of NF-kB activa-tion. This was in accordance with the fact that cerivas-tatin did not modify NF-kB mobilization by TNF-a,indicating that in the mechanism by which cerivastatinupregulates eNOS expression NF-kB blockade was notinvolved.
This disagreement regarding the effects of HMG-CoA reductase inhibitors on NF-kB may be due to thedifference in the cell type, mesangial cells vsendothelium.
The mechanism by which cerivastatin prevented thebinding activity of the cytosolic proteins to 3%-UTReNOS mRNA most likely occurs through inhibition ofendothelial HMG-CoA reductase because it was re-versed in the presence of L-mevalonate. Our experi-ments suggested that the effect of cerivastatin on eNOSexpression was independent of extracellular cholesterolconcentration because all the experiments were per-formed in cholesterol-clamped conditions. In this sense,a recent study demonstrated that improvement of en-dothelial function after 4 weeks of simvastatin treat-ment did not correlate with significant decreases inserum cholesterol levels [37]. Similar results have beenobtained with pravastatin [21].
The impairment in the production of endothelium-derived NO has been extensively described in patientswith atherosclerosis and acute coronary syndromes[12,13,38,39]. In this regard, the loss of eNOS expres-sion by the endothelium and, therefore, the resultingdecrease in its capability to produce NO in response to
Fig. 9. (A) Representative Western blot demonstrating the effect of10−5 M L-mevalonate on the prevention of eNOS protein expressionby 10−5 M cerivastatin in TNF-a (10 ng/ml)-incubated BAEC. (B)Bar graph showing the densitometric scanning of the Western blot.(C) Representative gel mobility shift assay showing the effect ofL-mevalonate (10−5 M) on the prevention by 10−5 M cerivastatin ofcomplex formation between the cytosolic extract of TNF-a-incubatedBAEC and 3%-UTR of eNOS mRNA. Results are represented asmeans9SEM of six different experiments. * PB0.05 with respect tocontrol.

F. Gonzalez-Fernandez et al. / Atherosclerosis 155 (2001) 61–70 69
physiological stimuli could compromise the ability ofthese cells to protect against thrombosis and leukocyteadhesion and favor the impaired endothelium-depen-dent hypotensive response described in atherosclerosisand hypercholesterolemia [9,12,13]. It has also beenreported that cerivastain attenuates the infiltration ofmacrophages in early hypercholesterolemic rabbits [40],in which the upregulation of eNOS expression by theendothelium reported here could be involved. In sum-mary, cerivastatin upregulates eNOS expression inTNF-a-stimulated BAEC and stabilizes eNOS mRNA.Moreover, cerivastatin prevents the binding activity ofendothelial cytosolic proteins to 3%-UTR eNOS mRNA.There is no direct evidence demonstrating the involve-ment of these cytosolic proteins in the regulation ofeNOS mRNA stability. However, the fact that cerivas-tatin reduced the binding activity of the cytosolicproteins to eNOS mRNA and favored eNOS expressionin the cultured endothelial cells suggests at least acausal relationship between the presence of the cytoso-lic proteins and eNOS expression. Therefore, thesecytosolic proteins could represent suitable therapeutictargets to protect endothelial function.
Acknowledgements
This work was supported by grants from FondoInvestigaciones Sanitarias de la Seguridad Social (FISS99/0117) and Laboratorios Bayer. A. Jimenez, M.M.Arriero, A. Lopez-Blaya and S. Velasco are fellows ofFundacion Conchita Rabago de Jimenez Dıaz. F. Gon-zalez-Fernandez is a postdoctoral fellows of Cardiovas-cular Research and Hypertension Laboratory. Theauthors thank Marıa Begona Ibarra for secretarialassistance.
References
[1] Scandinavian Simvastatin Survival Study Group. Randomizedtrial of cholesterol lowering in 4444 patients with coronary heartdisease: the scandinavian simvastatin survival study (4S). Lancet1994;344:1383–9.
[2] Shepherd J, Cobbe SM, Ford I, et al. Prevention of coronaryheart disease with provastatin in men with hypercholesterolemia:West of Scotland Coronary Prevention Study Group. New EnglJ Med 1995;333:1301–7.
[3] Byington RP, Jukima JW, Salomon JT, et al. Reduction incardiovascular events during pravastatin therapy: pooled analy-sis of clinical events of the pravastatin atherosclerosis interven-tion program. Circulation 1995;92:2419–25.
[4] Buchwald H, Barco R, Matts JP. Effect of partial ileal bypasssurgery on mortability and morbidity from coronary heart dis-ease in patients with hypercholesterolemia. Report of the Pro-gram on the Surgical Control of Hyper lipidemias (POSCH).New Engl J Med 1990;323:946–55.
[5] Brom BG, Zhao XQ, Sacco DE, Albers JJ. Lipid lowering andplaque regression: new insights into prevention of plaque disrup-
tion and clinical events in coronary disease. Circulation1993;87:1781–91.
[6] Egashira K, Hirooka Y, Kai H, Sugimachi M, Suzuki S, ImuoT, Takeshita A. Reduction on serum cholesterol with pravastatinimproves endothelium dependent coronary vasomotion in pa-tients with hypercholesterolemia. Circulation 1999;89:2519–24.
[7] Treasure CB, Klein JL, Weintranb WS, et al. Beneficial effects ofcholesterol-lowering therapy on the coronary endothelium inpatients with coronary artery disease. New Engl J Med1995;332:481–7.
[8] Harrison DG. From isolated cells to the catheterization labora-tory: studies of endothelial function in the coronary circulationof humans. Circulation 1989;80:703–6.
[9] Moncada S, Higgs A. L-arginine nitric oxide pathway. New EnglJ Med 1993;329:2002–12.
[10] Sessa WC. The nitric oxide synthase family of proteins. J VascRes 1994;31:131–43.
[11] Busse R, Mulsch A, Flemming I, Hecker M. Mechanisms ofnitric oxide release from the vascular endothelium. Circulation1993;87:18–25.
[12] Zeiher AM, Drexler H, Saurbier B, Just H. Endothelium-medi-ated coronary blood flow modulation in human: effects of age,atherosclerosis, hypercholesterolemia and hypertension. J ClinInvest 1993;92:652–62.
[13] Lumer PL, Selwyn AP, Shook TL, Wayne RR, Mudge GH,Alexander RW, Ganz P. Paradoxical vasoconstriction inducedby acetylcholine in atherosclerotic coronary arteries. New Engl JMed 1986;315:1046–51.
[14] Osborne JA, Lento PH, Siefgried MR, Stahl GL, Fusman B,Lefer AM. Cardiovascular effects of acute hypercholesterolemiain rabbits: reversal with lovastatin treatment. J Clin Invest1989;83:465–73.
[15] Le Cras TD, Xue C, Rengasamy A, Johns RA. Chronic hypoxiaupregulates endothelial and inducible NO synthase gene andprotein expression in rat lung. Am J Physiol 1996;270:L164–70.
[16] Lopez-Farre A, Sanchez de Miguel L, Caramelo C, et al. Role ofnitric oxide in the autocrine control of growth and apoptosis ofendothelial cells. Am J Physiol 1997;272:H760–8.
[17] Sessa WC, Pritchard K, Seyedi N, Wang J, Hintze TH. Chronicexercise increases coronary vascular nitric oxide production andendothelial nitric oxide synthase gene expression. Circ Res1994;74:349–53.
[18] Yoshizumi M, Perella MA, Burnett JC, Lee ME. Tumor necrosisfactor downregulates and endothelial nitric oxide synthasemRNA by shortening its half-life. Circ Res 1993;73:205–9.
[19] Mohamed F, Mongue JC, Gordon A, Cernacek P, Blais D,Stewart DJ. Lack of role for nitric oxide (NO) in the selectivedestabilization of endothelial NO synthase mRNA by tumornecrosis factor-a. Arterioscler Thromb Vasc Biol 1995;15:52–7.
[20] Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation ofendothelial nitric oxide synthase by HMG CoA reductase in-hibitors. Circulation 1998;97:1129–35.
[21] Kaesemeyer WH, Caldwell RB, Huang J, Caldwell RW. Pravas-tatin sodium activates endothelial nitric oxide synthase indepen-dent of its choelsterol-lowering actions. J Am Coll Cardiol1999;33:234–41.
[22] Jackson RJ. Cytoplasmic regulation of mRNA function: theimportance of the 3%-untranslated region. Cell 1993;74:9–14.
[23] Zhon B, Malter JS. Regulation of mRNA stability and itsrelevance to disease. Lab Invest 1991;65:610–21.
[24] Alonso J, Sanchez de Miguel L, Monton M, Casado S, Lopez-Farre A. Endothelial cytosolic proteins bind to the 3%-untrans-lated region of endothelial nitric oxide synthase mRNA:regulation by tumor necrosis factor alpha. Mol Cell Biol1997;17:5719–26.
[25] Rus HG, Niculescu F, Vlaim R. Tumor necrosis factor-alpha inhuman arterial wall with atherosclerosis. Atherosclerosis1991;89:247–54.

F. Gonzalez-Fernandez et al. / Atherosclerosis 155 (2001) 61–7070
[26] de Frutos T, Sanchez de Miguel L, Garcıa-Duran M, et al.Nitric oxide generated by smooth muscle cells decreases nitricoxide synthase expression in coincubated endothelial cells: in-volvement of TNF-a. Am J Physiol 1999;277:H1317–25.
[27] Cernadas MR, Sanchez de Miguel L, Garcıa-Duran M, et al.Expression of constitutive and inducible nitric oxide synthases inthe vascular wall of young and aging rats. Circ Res 1998;83:279–86.
[28] Laemmli NK. Change of structural proteins during the assemblyof the head of bacteriophage Tk. Nature 1970;227:680–5.
[29] Chomazynski P, Sacchi N. Single-step method of RNA isolationby acid guaridin thiocyanate–phenol–chloroform extraction.Anal Biochem 1987;162:9940–5.
[30] Sanchez de Miguel L, de Frutos T, Gonzalez-Fernandez F.Aspirin inhibits inducible nitric oxide synthase expression andtumour necrosis factor-a release by cultured smooth muscle cells.Eur J Clin Invest 1999;29:93–9.
[31] Weber C, Erl W, Pietsch A, Weber PC. Aspirin inhibits nuclearfactor-kB mobilization and monocyte adhesion in stimulatedhuman endothelial cells. Circulation 1995;91:1914–7.
[32] Weber C, Erl W, Pietsch A, Strobel M, Ziegler-Heitbrock HWL,Weber PC. Antioxidants inhibit monocyte adhesion by suppress-ing nuclear factor-kB mobilization and induction of vascular celladhesion molecule-1 in endothelial cells stimulated to generateradicals. Arterioscler Thromb 1994;14:1665–73.
[33] Liao JK, Shin WS, Lee WY, Clark SL. Oxidized low-densitylipoprotein decreases the expression of endothelial nitric oxidesynthase. J Biol Chem 1995;270:319–24.
[34] Barnes PJ, Karin M. Mechanisms of disease: nuclear factor-kB:a pivotal transcription factor in chronic inflammatory diseases.New Engl J Med 1997;336:1041–5.
[35] Beg AA, Baldwin AS. The IKB proteins: multifunctional regula-tors of Rel/NF-kB transcription factors. Genes Dev1993;7:2064–70.
[36] Guijarro C, Kim Y, Schooner CM, Massy ZA. Lovastatininhibits lipopolysaccharide-induced NF-kB activation in humanmesangial cells. Nephrol Dial Trans 1996;11:990–6.
[37] O’Driscoll G, Green D, Taylor RR. Simvastatin, an HMG-coen-zyme A reductase inhibitor, improves endothelial function within1 month. Circulation 1997;95:1126–31.
[38] Okumura K, Yassue H, Matsuyama K, Ogawa H, Morikemi Y,Obata K, Sakaino N. Effect of acetylcholine on the highlystenotic coronary artery: difference between the constrictor re-sponse of the infarct-related coronary and that of the noninfarct-related artery. J Am Coll Cardiol 1992;19:753–8.
[39] Bogaty P, Hackett D, Daries G, Maseri A. Vasoreactivity of theculprit lesion in unstable angina. Circulation 1994;90:5–11.
[40] Shiomi M, Ito T. Effect of cerivastatin sodium, a new inhibitorof HMG-CoA reductase, on plasma lipid levels, progressium ofatherosclerosis, and the lesional composition in the plaques ofWHHL rabbits. Br J Pharmacol 1999;126:961–8.
.




















