
Comparison of nonviral transfection and adeno-associated viral transduction on cardiomyocytes
Upload
independentCategory
view
0download
0
Ronglih LiaoKoh, Douglas B. Sawyer, Jane A. Leopold, Diane E. Handy, Joseph Loscalzo, Carl S. Apstein and Mohit Jain, Daniel A. Brenner, Lei Cui, Chee Chew Lim, Bo Wang, David R. Pimentel, Stanley
Phenotype in Adult CardiomyocytesGlucose-6-Phosphate Dehydrogenase Modulates Cytosolic Redox Status and Contractile
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2003 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/01.RES.0000083489.83704.762003;93:e9-e16; originally published online June 26, 2003;Circ Res.
http://circres.ahajournals.org/content/93/2/e9World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org/content/suppl/2003/07/23/93.2.e9.DC1.htmlData Supplement (unedited) at:
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer process is available in the
click Request Permissions in the middle column of the Web page under Services. Further information about this located,Editorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Research Requests for permissions to reproduce figures, tables, or portions of articles originally published inPermissions:
by guest on October 17, 2013http://circres.ahajournals.org/Downloaded from by guest on October 17, 2013http://circres.ahajournals.org/Downloaded from by guest on October 17, 2013http://circres.ahajournals.org/Downloaded from by guest on October 17, 2013http://circres.ahajournals.org/Downloaded from by guest on October 17, 2013http://circres.ahajournals.org/Downloaded from by guest on October 17, 2013http://circres.ahajournals.org/Downloaded from by guest on October 17, 2013http://circres.ahajournals.org/Downloaded from by guest on October 17, 2013http://circres.ahajournals.org/Downloaded from by guest on October 17, 2013http://circres.ahajournals.org/Downloaded from by guest on October 17, 2013http://circres.ahajournals.org/Downloaded from by guest on October 17, 2013http://circres.ahajournals.org/Downloaded from by guest on October 17, 2013http://circres.ahajournals.org/Downloaded from

Glucose-6-Phosphate Dehydrogenase ModulatesCytosolic Redox Status and Contractile Phenotype in
Adult CardiomyocytesMohit Jain, Daniel A. Brenner, Lei Cui, Chee Chew Lim, Bo Wang, David R. Pimentel,Stanley Koh, Douglas B. Sawyer, Jane A. Leopold, Diane E. Handy, Joseph Loscalzo,
Carl S. Apstein, Ronglih Liao
Abstract—Reactive oxygen species (ROS)–mediated cell injury contributes to the pathophysiology of cardiovasculardisease and myocardial dysfunction. Protection against ROS requires maintenance of endogenous thiol pools, mostimportantly, reduced glutathione (GSH), by NADPH. In cardiomyocytes, GSH resides in two separate cellularcompartments: the mitochondria and cytosol. Although mitochondrial GSH is maintained largely by transhydrogenaseand isocitrate dehydrogenase, the mechanisms responsible for sustaining cytosolic GSH remain unclear. Glucose-6-phosphate dehydrogenase (G6PD) functions as the first and rate-limiting enzyme in the pentose phosphate pathway,responsible for the generation of NADPH in a reaction coupled to the de novo production of cellular ribose. Wehypothesized that G6PD is required to maintain cytosolic GSH levels and protect against ROS injury in cardiomyocytes.We found that in adult cardiomyocytes, G6PD activity is rapidly increased in response to cellular oxidative stress, withtranslocation of G6PD to the cell membrane. Furthermore, inhibition of G6PD depletes cytosolic GSH levels andsubsequently results in cardiomyocyte contractile dysfunction through dysregulation of calcium homeostasis. Cardio-myocyte dysfunction was reversed through treatment with either a thiol-repleting agent (L-2-oxothiazolidine-4-carboxylic acid) or antioxidant treatment (Eukarion-134), but not with exogenous ribose. Finally, in a murine model ofG6PD deficiency, we demonstrate the development of in vivo adverse structural remodeling and impaired contractilefunction over time. We, therefore, conclude that G6PD is a critical cytosolic antioxidant enzyme, essential formaintenance of cytosolic redox status in adult cardiomyocytes. Deficiency of G6PD may contribute to cardiacdysfunction through increased susceptibility to free radical injury and impairment of intracellular calcium transport. Thefull text of this article is available online at http://www.circresaha.org. (Circ Res. 2003;93:e9-e16.)
Key Words: glucose-6-phosphate dehydrogenase � cardiomyocytes � oxidant injury� contractile dysfunction � intracellular calcium
Reactive oxygen species (ROS)–mediated cell injury con-tributes to the pathophysiology of cardiovascular disease
and myocardial dysfunction.1–3 These ROS originate frommultiple sources, including cellular oxidase complexes and asmitochondrial byproducts of aerobic metabolism.3,4 To pro-tect against ROS, cardiomyocytes contain a highly activecellular antioxidant defense. Central to the neutralization ofROS is the endogenous thiol, reduced glutathione (GSH), andglutathione cycling.5,6 GSH provides the reducing equivalentsnecessary for the conversion of hydrogen peroxide and lipidperoxides to water and lipid alcohols, respectively, therebypreventing degradation to highly toxic free radicals, includinghydroxyl and peroxyl radicals.7 GSH also plays an importantrole in protecting against oxidation of protein sulfhydrylgroups.8 In the presence of ROS, the cellular redox state isaltered such that GSH is rapidly converted to its oxidized
form, GSSG. The maintenance of GSH pools, in turn,ultimately requires NADPH, an essential cofactor for regen-eration of GSH from GSSG.6 In cardiomyocytes, GSHresides in two distinct and separate cellular compartments, themitochondria and cytosol.5 Depletion of cytosolic GSH levelsresults in oxidative modification of cytosolic proteins andlipids, as well as polynucleotides. Whereas mitochondrialGSH is maintained largely by NADH interconversion toNADPH via transhydrogenase and by reduction of NADP� toNADPH via isocitrate dehydrogenase (ICD), malic enzyme,and glutamate dehydrogenase, the mechanism responsible forsustaining cytosolic GSH in cardiomyocytes remains unclear.
Glucose-6-phosphate dehydrogenase (G6PD) functions asthe first and rate-limiting enzyme in the pentose phosphatepathway, responsible for the generation of NADPH in areaction coupled to the oxidation of glucose-6-phosphate and
Original received May 23, 2003; revision received June 13, 2003; accepted June 16, 2003.From the Cardiac Muscle Research Laboratory, Whitaker Cardiovascular Institute and Evans Department of Medicine, Boston University School of
Medicine, Boston, Mass.Correspondence to Dr Ronglih Liao, Boston University School of Medicine, 650 Albany St, X-726, Boston, MA 02118. E-mail [email protected]© 2003 American Heart Association, Inc.
Circulation Research is available at http://www.circresaha.org DOI: 10.1161/01.RES.0000083489.83704.76
1
UltraRapid Communication

de novo production of cellular ribose. In nonnucleated mam-malian cells, most notably erythrocytes, G6PD is well-established in the protection against cytosol ROS, as defi-ciency of G6PD is associated with hemolytic disordersstemming from increased susceptibility of erythrocytes tooxidative stress.9,10 The role of G6PD in cardiomyocytes,however, remains highly controversial,11 largely owing to thehigh myocardial concentration of NADP�-dependent ICD, analternative enzyme capable of NADPH generation.12 ThisICD pool, however, remains confined to the mitochondriaand, thus, has little if any effect on cytosolic redox state. Inthis article, we demonstrate that G6PD is a critical cytosolicantioxidant enzyme in adult cardiomyocytes. G6PD activityis rapidly increased in response to cellular oxidative stress,with translocation of G6PD to the cell membrane. Further-more, inhibition of G6PD depletes cytosolic GSH levels andsubsequently results in cardiomyocyte contractile dysfunctionthrough dysregulation of calcium homeostasis. Finally, in amurine model of G6PD deficiency, we demonstrate thedevelopment of in vivo adverse structural remodeling andcontractile dysfunction over time.
Materials and MethodsAdult Cardiomyocyte Isolation, Culture,and TreatmentAdult ventricular cardiomyocytes were isolated from male Sprague-Dawley rats (weighing 250 to 300 g) obtained from Charles RiverLaboratories (Wilmington, Mass), as previously described.13 Cardi-omyocytes were resuspended in 50 mL Dulbecco’s modified eaglemedium (DMEM) (GIBCO), and layered over a bovine serumalbumin (Sigma Chemical Co) solution to separate ventricularcardiomyocytes from nonmyocytes. For cell physiological or bio-chemical experiments, cells were plated onto laminin-coated cover-slips or culture dishes, respectively. After 1 hour of culture, themedium was changed to ACCT (2 mmol/L L-carnitine, 5 mmol/Lcreatine, 5 mmol/L taurine, bovine serum albumin, 100 IU/mLpenicillin, and 100 �g/mL streptomycin in DMEM).
Cells were treated with H2O2 at the concentrations and timeintervals indicated in Results. For G6PD inhibition experiments,cells were treated with increasing concentrations of dehydroepi-androsterone (DHA), a noncompetitive inhibitor of G6PD,14 in 0.1%dimethylsulfoxide (DMSO), or DMSO vehicle alone (0 �mol/LDHA). DHA significantly affected cell morphometry and survival atconcentrations of 100 �mol/L and higher. Cell physiology experi-ments were, therefore, conducted with DHA concentrations rangingfrom 0 to 30 �mol/L, avoiding the confounding effect of cell deathon experimental results and data interpretation. For ribose treatment,cardiomyocytes were incubated with 5 mmol/L D-ribose for 60minutes before study. This concentration of ribose has previouslybeen shown to augment nucleotide production in cultured cardio-myocytes.15 For G6PD gene transfer, cardiomyocytes were infectedwith recombinant adenovirus containing rat G6PD at an MOI of 10for 24 hours, as previously described.16 For antioxidant experiments,cardiomyocytes were co-treated with either Eukarion-134 (50�mol/L) (EUK), a superoxide-dismutase/catalase mimetic,17 in 0.1%DMSO, or L-2-oxothiazolidine-4-carboxylic acid (5 mmol/L) (OTC),a procysteine glutathione-generating compound,18 in 0.1% EtOH.EUK was generously provided by Eukarion, Inc (Bedford, Mass).All other chemicals were obtained from Sigma Chemical Co.
Immunoblotting and Subcellular FractionationTotal G6PD protein levels were determined by standard Westernblot. Cultured cardiomyocytes were lysed in Triton X-100 lysisbuffer (Cell Signaling) with protease inhibitors (2 �mol/L leupeptin,1 mmol/L PMSF). The lysate was sonicated and centrifuged at
16 000g for 15 minutes at 4°C. The supernatant was collected, andprotein concentration measured according to the method of Lowry.19
Samples were run on 12% Tris-Glycine precast gels (BioWhitaker)and transferred to PVDF membranes. Equal protein loading wasconfirmed by Ponceau staining. After blocking in 5% non-fat milk,PVDF membranes were probed with rabbit anti-rat G6PD IgG(kindly provided by Drs Richard A. Cohen and Reiko Matsui,Boston, Mass) followed by HRP conjugated anti-rabbit secondaryantibody (Pierce Chemical Co). Protein levels were detected bychemiluminescence (Pierce Chemical Co).
Subcellular fractionation of cardiomyocytes was conducted aspreviously described.20 Cardiomyocytes were homogenized in 4°CHES buffer (10 mmol/L HEPES, 250 mmol/L Sucrose, 1.0 mmol/LEDTA, pH 7.4) with protease inhibitors. Homogenates were centri-fuged at 700g for 10 minutes at 4°C. The supernatant was collectedand recentrifuged at 10 000g for 25 minutes. The resulting superna-tant was added to the original pellet and designated the membrane/cytosolic fraction. This fraction was sonicated and again centrifugedat 700g. The supernatant was collected (pellet discarded) andcentrifuged at 100 000g for 60 minutes at 4°C. The pellet wasresuspended in Triton X-100 lysis buffer and designated as themembrane fraction. The supernatant was diluted with Triton X-100lysis buffer and designated as the cytosolic fraction. Protein concen-tration was determined in both membrane and cytosolic fractions andprotein levels of G6PD determined via Western blot, as describedabove.
Measurement of Cardiomyocyte Cell Shorteningand Intracellular CalciumCell shortening and intracellular calcium transients ([Ca2�]i tran-sients) were recorded in cardiomyocytes, as previously de-scribed.13,21,22 Cultured cardiomyocytes were incubated with mem-brane permeant fura-2 (1 �mol/L) (Molecular Probes) andprobenecid (500 �mol/L), to prevent leakage of fura-2 from cells.Cardiomyocytes were perfused with 1.2 mmol/L Ca2� Tyrodesolution at 37°C and electrically paced at 300 beats per minute viaplatinum wires. Cell shortening/relengthening, and [Ca2�]i transientswere measured using video edge detection and fluorescence mea-surements of the Fura-2 ratio, respectively (SoftEdge AcquisitionSystem and IonWizard, IonOptix Inc).13,21,22 Percent cell shortening(% CS) was calculated as diastolic cell length minus systolic celllength normalized to the diastolic cell length. The rate of cardiomyo-cyte contraction (�dL/dt) and rate of cardiomyocyte relaxation(�dL/dt) were calculated by commercially available acquisitionsoftware (IonWizard, IonOptix Inc). The time constant (�), a celllength–independent measure of cardiomyocyte relaxation, was cal-culated as previously described.22 [Ca2�]i was calculated from fura-2florescence as previously described,22 and [Ca2�]i transients wereanalyzed in a similar fashion to cell shortening experiments. Eachvalue presented for the cardiomyocyte contractility or calciumexperiments represents 6 to 10 independent experiments (at eachconcentration of DHA) with 5 to 20 cell measurements perexperiment.
Biochemical AssaysBiochemical assays were performed on mitochondrial-free cardio-myocyte homogenates. Mitochondrial free-lysates were prepared aspreviously described.23 Cardiomyocytes were homogenized in 4°CHES buffer with protease inhibitors (as described above). Homoge-nates were centrifuged at 16 000g for 15 minutes at 4°C. Thesupernatant was collected and used for assays. The absence ofmitochondria in the supernatant was confirmed through Westernblotting for mitochondrial proteins, including cytochrome C andmanganese superoxide dismutase.
Glucose-6-Phosphate Dehydrogenase ActivityGlucose-6-phosphate dehydrogenase activity was determined inmitochondrial-free cardiomyocyte homogenates according to themethods of Tian and colleagues.24 Protein concentration was deter-mined according to the method of Lowry.19
2 Circulation Research July 25, 2003
Glutathione LevelsTotal (GST), reduced (GSH), and oxidized (GSSG) glutathioneconcentrations were determined in mitochondrial-free cardiomyo-cyte homogenates using a commercially available kit (CaymanChemical) based on an enzymatic recycling method described byTietze and coworkers.25 All assays were run in triplicate andaveraged to obtain a mean value per sample.
Intracellular ROSIntracellular ROS was assessed using the ROS-sensitive fluorophore,dichlorofluorescein diacetate (DCF) (Molecular Probes). Oxidation of DCFby intracellular ROS results in the formation of a fluorescent compound.26
Cardiomyocytes were incubated with 20 �mol/L DCF for 30 minutes andDCF fluorescence visualized and quantified using epi-fluorescent micros-copy and video imaging (BIOQUANT, version 2.5).
G6PD-Deficient MiceG6PD-deficient mice (G6PDdef),27 were rederived in our laboratoriesfrom frozen embryos. G6PDdef mice carry a mutation at the 5�untranslated sequence of the X-linked G6PD gene and, through asplicing defect, exhibit decreased G6PD protein expression andenzyme activity.28 Male hemizygous G6PDdef mice and wild-typelittermates (WT) were used in this study. All animal studies strictlyadhered to the regulations of Boston University Animal CareCommittee and the National Society for Medical Research.
Transthoracic EchocardiographyTransthoracic echocardiograms were serially obtained in consciousmice, as previously described,21,29 using an Acuson Sequoia C-256echocardiograph machine and a 15-MHz probe. The heart wasimaged in the two-dimensional parasternal short-axis view, and anM-mode measurement was recorded at the mid-ventricle at the levelof the papillary muscles. Heart rate, septal wall thickness, andend-diastolic and end-systolic dimensions were measured from theM-mode image using analysis software (Acuson, Sequoia). Frac-tional shortening was defined as the end-diastolic dimension minusthe end-systolic dimension normalized for the end-diastolic dimen-sion, and was used as an index of cardiac contractile function.
Statistical AnalysisStatistical significance was evaluated by one-way analysis of variance(ANOVA). A post hoc test of least significant differences was used todetermine differences among groups. All data are expressed asmean�SEM. A value of P�0.05 was considered statistically significant.
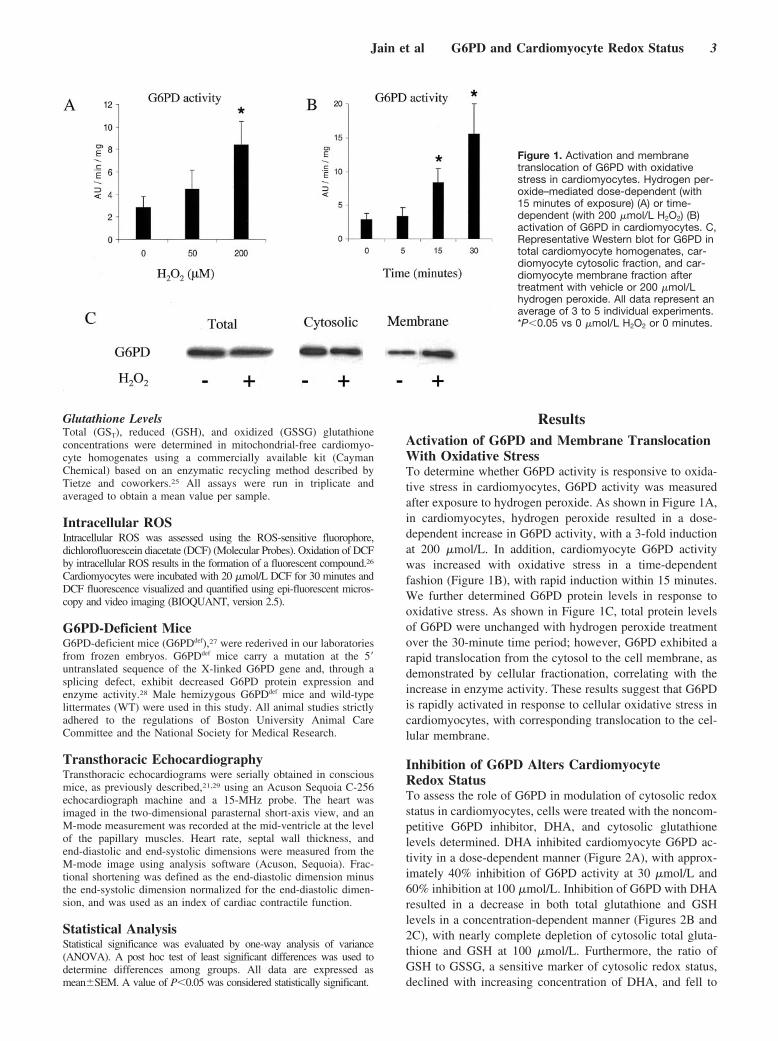
ResultsActivation of G6PD and Membrane TranslocationWith Oxidative StressTo determine whether G6PD activity is responsive to oxida-tive stress in cardiomyocytes, G6PD activity was measuredafter exposure to hydrogen peroxide. As shown in Figure 1A,in cardiomyocytes, hydrogen peroxide resulted in a dose-dependent increase in G6PD activity, with a 3-fold inductionat 200 �mol/L. In addition, cardiomyocyte G6PD activitywas increased with oxidative stress in a time-dependentfashion (Figure 1B), with rapid induction within 15 minutes.We further determined G6PD protein levels in response tooxidative stress. As shown in Figure 1C, total protein levelsof G6PD were unchanged with hydrogen peroxide treatmentover the 30-minute time period; however, G6PD exhibited arapid translocation from the cytosol to the cell membrane, asdemonstrated by cellular fractionation, correlating with theincrease in enzyme activity. These results suggest that G6PDis rapidly activated in response to cellular oxidative stress incardiomyocytes, with corresponding translocation to the cel-lular membrane.
Inhibition of G6PD Alters CardiomyocyteRedox StatusTo assess the role of G6PD in modulation of cytosolic redoxstatus in cardiomyocytes, cells were treated with the noncom-petitive G6PD inhibitor, DHA, and cytosolic glutathionelevels determined. DHA inhibited cardiomyocyte G6PD ac-tivity in a dose-dependent manner (Figure 2A), with approx-imately 40% inhibition of G6PD activity at 30 �mol/L and60% inhibition at 100 �mol/L. Inhibition of G6PD with DHAresulted in a decrease in both total glutathione and GSHlevels in a concentration-dependent manner (Figures 2B and2C), with nearly complete depletion of cytosolic total gluta-thione and GSH at 100 �mol/L. Furthermore, the ratio ofGSH to GSSG, a sensitive marker of cytosolic redox status,declined with increasing concentration of DHA, and fell to
Figure 1. Activation and membranetranslocation of G6PD with oxidativestress in cardiomyocytes. Hydrogen per-oxide–mediated dose-dependent (with15 minutes of exposure) (A) or time-dependent (with 200 �mol/L H2O2) (B)activation of G6PD in cardiomyocytes. C,Representative Western blot for G6PD intotal cardiomyocyte homogenates, car-diomyocyte cytosolic fraction, and car-diomyocyte membrane fraction aftertreatment with vehicle or 200 �mol/Lhydrogen peroxide. All data represent anaverage of 3 to 5 individual experiments.*P�0.05 vs 0 �mol/L H2O2 or 0 minutes.
Jain et al G6PD and Cardiomyocyte Redox Status 3
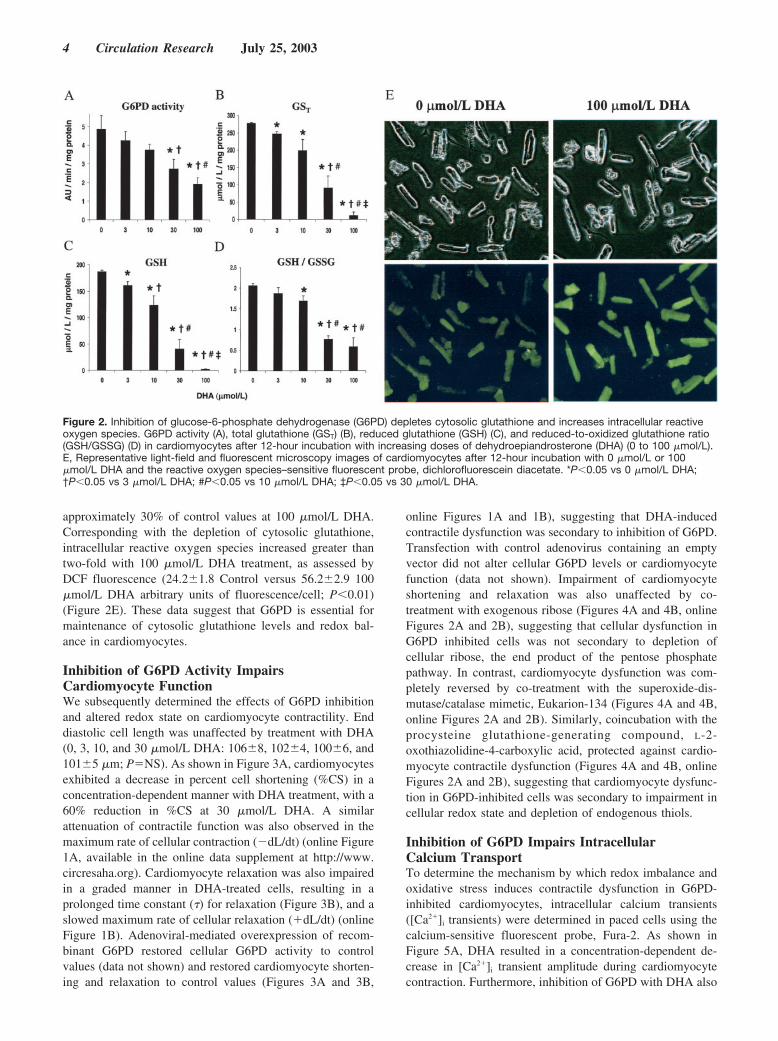
approximately 30% of control values at 100 �mol/L DHA.Corresponding with the depletion of cytosolic glutathione,intracellular reactive oxygen species increased greater thantwo-fold with 100 �mol/L DHA treatment, as assessed byDCF fluorescence (24.2�1.8 Control versus 56.2�2.9 100�mol/L DHA arbitrary units of fluorescence/cell; P�0.01)(Figure 2E). These data suggest that G6PD is essential formaintenance of cytosolic glutathione levels and redox bal-ance in cardiomyocytes.
Inhibition of G6PD Activity ImpairsCardiomyocyte FunctionWe subsequently determined the effects of G6PD inhibitionand altered redox state on cardiomyocyte contractility. Enddiastolic cell length was unaffected by treatment with DHA(0, 3, 10, and 30 �mol/L DHA: 106�8, 102�4, 100�6, and101�5 �m; P�NS). As shown in Figure 3A, cardiomyocytesexhibited a decrease in percent cell shortening (%CS) in aconcentration-dependent manner with DHA treatment, with a60% reduction in %CS at 30 �mol/L DHA. A similarattenuation of contractile function was also observed in themaximum rate of cellular contraction (�dL/dt) (online Figure1A, available in the online data supplement at http://www.circresaha.org). Cardiomyocyte relaxation was also impairedin a graded manner in DHA-treated cells, resulting in aprolonged time constant (�) for relaxation (Figure 3B), and aslowed maximum rate of cellular relaxation (�dL/dt) (onlineFigure 1B). Adenoviral-mediated overexpression of recom-binant G6PD restored cellular G6PD activity to controlvalues (data not shown) and restored cardiomyocyte shorten-ing and relaxation to control values (Figures 3A and 3B,
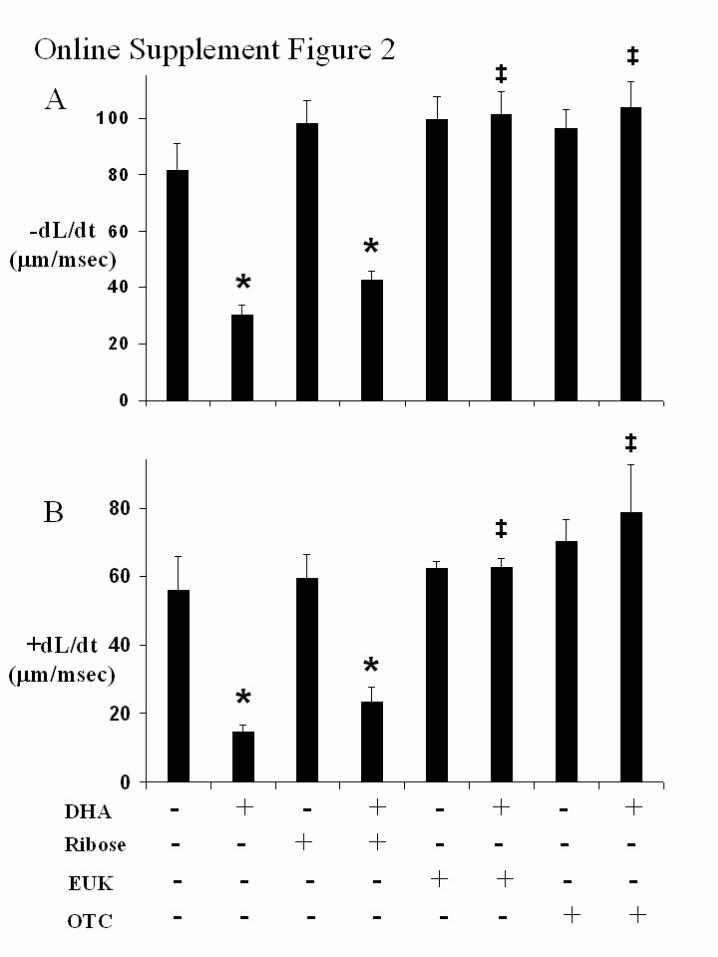
online Figures 1A and 1B), suggesting that DHA-inducedcontractile dysfunction was secondary to inhibition of G6PD.Transfection with control adenovirus containing an emptyvector did not alter cellular G6PD levels or cardiomyocytefunction (data not shown). Impairment of cardiomyocyteshortening and relaxation was also unaffected by co-treatment with exogenous ribose (Figures 4A and 4B, onlineFigures 2A and 2B), suggesting that cellular dysfunction inG6PD inhibited cells was not secondary to depletion ofcellular ribose, the end product of the pentose phosphatepathway. In contrast, cardiomyocyte dysfunction was com-pletely reversed by co-treatment with the superoxide-dis-mutase/catalase mimetic, Eukarion-134 (Figures 4A and 4B,online Figures 2A and 2B). Similarly, coincubation with theprocysteine glutathione-generating compound, L-2-oxothiazolidine-4-carboxylic acid, protected against cardio-myocyte contractile dysfunction (Figures 4A and 4B, onlineFigures 2A and 2B), suggesting that cardiomyocyte dysfunc-tion in G6PD-inhibited cells was secondary to impairment incellular redox state and depletion of endogenous thiols.
Inhibition of G6PD Impairs IntracellularCalcium TransportTo determine the mechanism by which redox imbalance andoxidative stress induces contractile dysfunction in G6PD-inhibited cardiomyocytes, intracellular calcium transients([Ca2�]i transients) were determined in paced cells using thecalcium-sensitive fluorescent probe, Fura-2. As shown inFigure 5A, DHA resulted in a concentration-dependent de-crease in [Ca2�]i transient amplitude during cardiomyocytecontraction. Furthermore, inhibition of G6PD with DHA also
Figure 2. Inhibition of glucose-6-phosphate dehydrogenase (G6PD) depletes cytosolic glutathione and increases intracellular reactiveoxygen species. G6PD activity (A), total glutathione (GST) (B), reduced glutathione (GSH) (C), and reduced-to-oxidized glutathione ratio(GSH/GSSG) (D) in cardiomyocytes after 12-hour incubation with increasing doses of dehydroepiandrosterone (DHA) (0 to 100 �mol/L).E, Representative light-field and fluorescent microscopy images of cardiomyocytes after 12-hour incubation with 0 �mol/L or 100�mol/L DHA and the reactive oxygen species–sensitive fluorescent probe, dichlorofluorescein diacetate. *P�0.05 vs 0 �mol/L DHA;†P�0.05 vs 3 �mol/L DHA; #P�0.05 vs 10 �mol/L DHA; ‡P�0.05 vs 30 �mol/L DHA.
4 Circulation Research July 25, 2003

impaired removal of [Ca2�]i during cellular relaxation (Figure5B). Importantly, impairment of both calcium release andcalcium reuptake in cardiomyocytes closely correlated withcontractile dysfunction (Figures 3A and 3B) and were re-stored to control values with normalization of G6PD activitythrough G6PD gene transfer (Figures 5A and 5B), but wereunaffected by transfection with empty viral vector controls(data not shown). In addition, dysfunctional calcium ho-meostasis, as observed with cardiomyocyte contractile func-tion, was unaffected through treatment with ribose, butreturned to control values with antioxidant treatment andrestoration of cellular thiols (Figures 6A and 6B). Represen-tative tracings of cardiomyocyte shortening and correspond-ing [Ca2�]i transients are demonstrated in Figure 7. Theseresults suggest that dysregulation of calcium homeostasismay be accountable for contractile dysfunction in G6PDinhibited cardiomyocytes.
Adverse Structural Remodeling and ContractileDysfunction in G6PD-Deficient MiceAlthough our results suggest that inhibition of G6PD mayalter redox homeostasis and impair contractile function inisolated cardiomyocytes, it is uncertain whether G6PD defi-ciency results in the development of contractile dysfunctionin vivo. In vivo cardiac structure and function were, therefore,
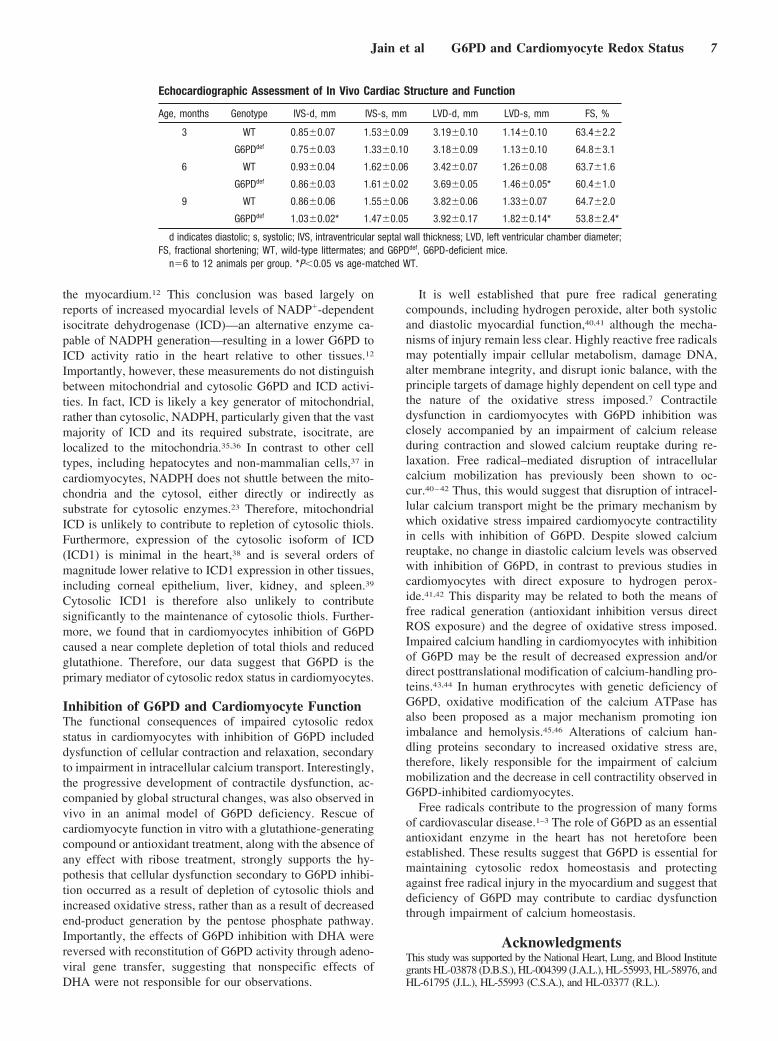
serially evaluated in male hemizygous mutant mice (G6PDdef)and wild-type (WT) littermate counterparts. Hearts fromG6PDdef mice exhibited approximately 20% of WT G6PDactivity (6.2�1.5 G6PDdef versus 30.8�1.8 WT AU/mgprotein; P�0.01). Young WT and G6PDdef mice (3 months)exhibited similar cardiac structural and contractile pheno-types (Table). G6PDdef mice, however, exhibited progressiveadverse structural remodeling and cardiac dysfunction overtime, marked by increased ventricular septal wall thickeningand end-systolic chamber dimensions, as well as a significantdecrease in fractional shortening relative to age-matched WTcontrols at 9 months of age. These results suggest that G6PDdeficiency may modulate cardiac structure and contractilefunction in vivo.
DiscussionThe exquisite sensitivity of myocardium to even ambientlevels of oxidative injury is well established.30,31 This articledemonstrates the necessity of G6PD as a critical componentof the cellular antioxidant system in adult cardiomyocytes,required for maintaining cytosolic redox homeostasis andcontractile function in adult cardiomyocytes, both in vitro andin vivo.
Figure 3. Inhibition of G6PD impairs contractile function in car-diomyocytes. Percent cell shortening (%CS) (A) and time con-stant for cell relengthening (�) (B) in cardiomyocytes paced at300 bpm after 12-hour incubation with dehydroepiandrosterone(DHA) (0 to 30 �mol/L) and/or 24-hour incubation withadenovirus-containing recombinant rat G6PD (Ad-G6PD) at anMOI of 10. *P�0.05 vs 0 �mol/L DHA; †P�0.05 vs 3 �mol/LDHA; #P�0.05 vs 10 �mol/L DHA; ‡P�0.05 vs 30 �mol/L DHA.
Figure 4. Rescue of contractile function in G6PD inhibited car-diomyocytes with glutathione-generating or antioxidant treat-ment. Percent cell shortening (%CS) (A) and time constant forcell relengthening (�) (B) in cardiomyocytes paced at 300 bpmafter treatment with vehicle or 30 �mol/L dehydroepiandros-terone (DHA), 5 mmol/L D-ribose (ribose), 50 �mol/L Eukarion-134 (EUK), or 5 mmol/L L-2-oxothiazolidine-4-carboxylic acid(OTC). *P�0.05 vs 0 �mol/L DHA; ‡P�0.05 vs 30 �mol/L DHA.
Jain et al G6PD and Cardiomyocyte Redox Status 5

G6PD and Cardiomyocyte Redox StateCardiomyocyte defense against oxidative stress is mediatedprimarily through neutralization of deleterious ROS viareducing equivalents in the form of intracellular thiols.7 Incardiomyocytes, we demonstrate that acute exposure to theoxidant hydrogen peroxide resulted in rapid upregulation ofG6PD activity. Importantly, this increase in G6PD activitywas not translationally regulated, as has been suggested tooccur with more chronic stress response.32 Rather, G6PDactivation was associated with translocation from its cytosolicposition to its active site on the cellular membrane. Thistranslocation allows for entering glucose to shunt rapidly tothe pentose phosphate pathway, thereby providing substratefor G6PD for NADPH generation. In fact, previous reportshave demonstrated an increase in pentose phosphate pathwayflux after exposure to oxidative stressors.33 Similar regulationof G6PD activity through cellular translocation has beenshown to occur in neutrophils, in which acute increases inG6PD activity regulate cytosolic NADPH-dependent oxidasecomplexes.34 Furthermore, in cardiomyocytes, inhibition ofG6PD resulted in loss of redox homeostasis and an increasein intracellular oxidative stress, evidenced by a decrease intotal glutathione, reduced glutathione, and the ratio ofreduced-to-oxidized glutathione, as well as a marked increasein cytosolic ROS. Importantly, G6PD activity correlated
directly with levels of cytosolic reduced glutathione. Takentogether, these data suggest that G6PD is essential formaintenance of cytosolic glutathione stores and subsequentprotection against cellular ROS.
These data differ significantly from previous reports,which have suggested an almost nonexistent role for G6PD in
Figure 5. Inhibition of G6PD impairs intracellular calcium([Ca2�]i) transport in cardiomyocytes. [Ca2�]i transient amplitude(A) and time constant for calcium removal (�) (B) in cardiomyo-cytes paced at 300 bpm after 12-hour incubation with dehydro-epiandrosterone (DHA) (0 to 30 �mol/L) and/or 24-hour incuba-tion with adenovirus-containing recombinant rat G6PD (Ad-G6PD) at an MOI of 10. *P�0.05 vs 0 �mol/L DHA; †P�0.05 vs3 �mol/L DHA; ‡P�0.05 vs 30 �mol/L DHA.
Figure 6. Rescue of intracellular calcium ([Ca2�]i) transport inG6PD inhibited cardiomyocytes with glutathione-generating orantioxidant treatment. [Ca2�]i transient amplitude (A) and timeconstant for calcium removal (�) (B) in cardiomyocytes paced at300 bpm after treatment with vehicle or 30 �mol/L dehydroepi-androsterone (DHA), 5 mmol/L D-ribose (ribose), 50 �mol/LEukarion-134 (EUK), or 5 mmol/L L-2-oxothiazolidine-4-carboxylic acid (OTC). *P�0.05 vs 0 �mol/L DHA; ‡P�0.05 vs30 �mol/L DHA.
Figure 7. Representative tracings of cell length and intracellularcalcium ([Ca2�]i) transients in cardiomyocytes paced at 300 bpmafter treatment with vehicle (control), 30 �mol/L dehydroepi-androsterone (DHA), or 30 �mol/L DHA plus 5 mmol/L L-2-oxothiazolidine-4-carboxylic acid (OTC).
6 Circulation Research July 25, 2003
the myocardium.12 This conclusion was based largely onreports of increased myocardial levels of NADP�-dependentisocitrate dehydrogenase (ICD)—an alternative enzyme ca-pable of NADPH generation—resulting in a lower G6PD toICD activity ratio in the heart relative to other tissues.12
Importantly, however, these measurements do not distinguishbetween mitochondrial and cytosolic G6PD and ICD activi-ties. In fact, ICD is likely a key generator of mitochondrial,rather than cytosolic, NADPH, particularly given that the vastmajority of ICD and its required substrate, isocitrate, arelocalized to the mitochondria.35,36 In contrast to other celltypes, including hepatocytes and non-mammalian cells,37 incardiomyocytes, NADPH does not shuttle between the mito-chondria and the cytosol, either directly or indirectly assubstrate for cytosolic enzymes.23 Therefore, mitochondrialICD is unlikely to contribute to repletion of cytosolic thiols.Furthermore, expression of the cytosolic isoform of ICD(ICD1) is minimal in the heart,38 and is several orders ofmagnitude lower relative to ICD1 expression in other tissues,including corneal epithelium, liver, kidney, and spleen.39
Cytosolic ICD1 is therefore also unlikely to contributesignificantly to the maintenance of cytosolic thiols. Further-more, we found that in cardiomyocytes inhibition of G6PDcaused a near complete depletion of total thiols and reducedglutathione. Therefore, our data suggest that G6PD is theprimary mediator of cytosolic redox status in cardiomyocytes.
Inhibition of G6PD and Cardiomyocyte FunctionThe functional consequences of impaired cytosolic redoxstatus in cardiomyocytes with inhibition of G6PD includeddysfunction of cellular contraction and relaxation, secondaryto impairment in intracellular calcium transport. Interestingly,the progressive development of contractile dysfunction, ac-companied by global structural changes, was also observed invivo in an animal model of G6PD deficiency. Rescue ofcardiomyocyte function in vitro with a glutathione-generatingcompound or antioxidant treatment, along with the absence ofany effect with ribose treatment, strongly supports the hy-pothesis that cellular dysfunction secondary to G6PD inhibi-tion occurred as a result of depletion of cytosolic thiols andincreased oxidative stress, rather than as a result of decreasedend-product generation by the pentose phosphate pathway.Importantly, the effects of G6PD inhibition with DHA werereversed with reconstitution of G6PD activity through adeno-viral gene transfer, suggesting that nonspecific effects ofDHA were not responsible for our observations.
It is well established that pure free radical generatingcompounds, including hydrogen peroxide, alter both systolicand diastolic myocardial function,40,41 although the mecha-nisms of injury remain less clear. Highly reactive free radicalsmay potentially impair cellular metabolism, damage DNA,alter membrane integrity, and disrupt ionic balance, with theprinciple targets of damage highly dependent on cell type andthe nature of the oxidative stress imposed.7 Contractiledysfunction in cardiomyocytes with G6PD inhibition wasclosely accompanied by an impairment of calcium releaseduring contraction and slowed calcium reuptake during re-laxation. Free radical–mediated disruption of intracellularcalcium mobilization has previously been shown to oc-cur.40–42 Thus, this would suggest that disruption of intracel-lular calcium transport might be the primary mechanism bywhich oxidative stress impaired cardiomyocyte contractilityin cells with inhibition of G6PD. Despite slowed calciumreuptake, no change in diastolic calcium levels was observedwith inhibition of G6PD, in contrast to previous studies incardiomyocytes with direct exposure to hydrogen perox-ide.41,42 This disparity may be related to both the means offree radical generation (antioxidant inhibition versus directROS exposure) and the degree of oxidative stress imposed.Impaired calcium handling in cardiomyocytes with inhibitionof G6PD may be the result of decreased expression and/ordirect posttranslational modification of calcium-handling pro-teins.43,44 In human erythrocytes with genetic deficiency ofG6PD, oxidative modification of the calcium ATPase hasalso been proposed as a major mechanism promoting ionimbalance and hemolysis.45,46 Alterations of calcium han-dling proteins secondary to increased oxidative stress are,therefore, likely responsible for the impairment of calciummobilization and the decrease in cell contractility observed inG6PD-inhibited cardiomyocytes.
Free radicals contribute to the progression of many formsof cardiovascular disease.1–3 The role of G6PD as an essentialantioxidant enzyme in the heart has not heretofore beenestablished. These results suggest that G6PD is essential formaintaining cytosolic redox homeostasis and protectingagainst free radical injury in the myocardium and suggest thatdeficiency of G6PD may contribute to cardiac dysfunctionthrough impairment of calcium homeostasis.
AcknowledgmentsThis study was supported by the National Heart, Lung, and Blood Institutegrants HL-03878 (D.B.S.), HL-004399 (J.A.L.), HL-55993, HL-58976, andHL-61795 (J.L.), HL-55993 (C.S.A.), and HL-03377 (R.L.).
Echocardiographic Assessment of In Vivo Cardiac Structure and Function
Age, months Genotype IVS-d, mm IVS-s, mm LVD-d, mm LVD-s, mm FS, %
3 WT 0.85�0.07 1.53�0.09 3.19�0.10 1.14�0.10 63.4�2.2
G6PDdef 0.75�0.03 1.33�0.10 3.18�0.09 1.13�0.10 64.8�3.1
6 WT 0.93�0.04 1.62�0.06 3.42�0.07 1.26�0.08 63.7�1.6
G6PDdef 0.86�0.03 1.61�0.02 3.69�0.05 1.46�0.05* 60.4�1.0
9 WT 0.86�0.06 1.55�0.06 3.82�0.06 1.33�0.07 64.7�2.0
G6PDdef 1.03�0.02* 1.47�0.05 3.92�0.17 1.82�0.14* 53.8�2.4*
d indicates diastolic; s, systolic; IVS, intraventricular septal wall thickness; LVD, left ventricular chamber diameter;FS, fractional shortening; WT, wild-type littermates; and G6PDdef, G6PD-deficient mice.
n�6 to 12 animals per group. *P�0.05 vs age-matched WT.
Jain et al G6PD and Cardiomyocyte Redox Status 7

References1. Halliwell B, Gutteridge JM. Role of free radicals and catalytic metal ions
in human disease: an overview. Methods Enzymol. 1990;186:1–85.2. Das DK. Redox regulation of cardiomyocyte survival and death. Antioxid
Redox Signal. 2001;3:23–37.3. Sawyer DB, Siwik DA, Xiao L, Pimentel DR, Singh K, Colucci WS. Role
of oxidative stress in myocardial hypertrophy and failure. J Mol CellCardiol. 2002;34:379–388.
4. Hess ML, Manson NH. Molecular oxygen: friend and foe: the role of theoxygen free radical system in the calcium paradox, the oxygen paradoxand ischemia/reperfusion injury. J Mol Cell Cardiol. 1984;16:969–985.
5. Kehrer JP, Lund LG. Cellular reducing equivalents and oxidative stress.Free Radic Biol Med. 1994;17:65–75.
6. Deneke SM, Fanburg BL. Regulation of cellular glutathione. Am JPhysiol. 1989;257:L163–L173.
7. Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. 3rded. New York, NY: Oxford University Press; 1999.
8. Yoshida T, Maulik N, Engelman RM, Ho YS, Magnenat JL, Rousou JA,Flack JE III, Deaton D, Das DK. Glutathione peroxidase knockout miceare susceptible to myocardial ischemia reperfusion injury. Circulation.1997;96:II-216–II-220.
9. Luzzatto L, Mehta A. Glucose-6-phosphate dehydrogenase deficiency. In:Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic andMolecular Basis of Inherited Disease. Toronto: McGraw Hill; 1995.
10. Beutler E. Study of glucose-6-phosphate dehydrogenase: history andmolecular biology. Am J Hematol. 1993;42:53–58.
11. Goodwin GW, Cohen DM, Taegtmeyer H. [5-3H]Glucose overestimatesglycolytic flux in isolated working rat heart: role of the pentose phosphatepathway. Am J Physiol Endocrinol Metab. 2001;280:E502–E508.
12. Andres A, Satrustegui J, Machado A. Development of NADPH-producing pathways in rat heart. Biochem J. 1980;186:799–803.
13. Nagata K, Communal C, Lim CC, Jain M, Suter TM, Eberli FR, Satoh N,Colucci WS, Apstein CS, Liao R. Altered �-adrenergic signal trans-duction in nonfailing hypertrophied myocytes from Dahl salt-sensitiverats. Am J Physiol Heart Circ Physiol. 2000;279:H2502–H2508.
14. Gordon G, Mackow MC, Levy HR. On the mechanism of interaction ofsteroids with human glucose 6-phosphate dehydrogenase. Arch BiochemBiophys. 1995;318:25–29.
15. Geisbuhler TP, Schwager TL. Ribose-enhanced synthesis of UTP, CTP,and GTP from parent nucleosides in cardiac myocytes. J Mol CellCardiol. 1998;30:879–887.
16. Leopold JA, Zhang YY, Scribner AW, Stanton RC, Loscalzo J. Glucose-6-phosphate dehydrogenase overexpression decreases endothelial celloxidant stress and increases bioavailable nitric oxide. ArteriosclerThromb Vasc Biol. 2003;23:411–417.
17. Melov S, Ravenscroft J, Malik S, Gill MS, Walker DW, Clayton PE,Wallace DC, Malfroy B, Doctrow SR, Lithgow GJ. Extension of life-spanwith superoxide dismutase/catalase mimetics. Science. 2000;289:1567–1569.
18. Williamson JM, Meister A. Stimulation of hepatic glutathione formationby administration of L-2-oxothiazolidine-4-carboxylate, a 5-oxo-L-prolinase substrate. Proc Natl Acad Sci U S A. 1981;78:936–939.
19. Lowry OJ, Rosebrough NJ, Farr AL, Randall RJ. Protein concentrationdetermination. J Biol Chem. 1951;193:265–275.
20. Frey RS, Rahman A, Kefer JC, Minshall RD, Malik AB. PKC� regulatesTNF-�–induced activation of NADPH oxidase in endothelial cells. CircRes. 2002;90:1012–1019.
21. Jain M, Lim CC, Nagata K, Davis VM, Milstone DS, Liao R, MortensenRM. Targeted inactivation of G�(i) does not alter cardiac function or�-adrenergic sensitivity. Am J Physiol Heart Circ Physiol. 2001;280:H569–H575.
22. Lim CC, Apstein CS, Colucci WS, Liao R. Impaired cell shortening andrelengthening with increased pacing frequency are intrinsic to thesenescent mouse cardiomyocyte. J Mol Cell Cardiol. 2000;32:2075–2082.
23. Robertson JD, Starnes JW, Kehrer JP. Cosubstrates involved in thereduction of cytosolic glutathione disulfide in rat heart. Toxicology. 1997;124:11–19.
24. Tian WN, Braunstein LD, Pang J, Stuhlmeier KM, Xi QC, Tian X,Stanton RC. Importance of glucose-6-phosphate dehydrogenase activityfor cell growth. J Biol Chem. 1998;273:10609–10617.
25. Tietze F. Enzymic method for quantitative determination of nanogramamounts of total and oxidized glutathione: applications to mammalianblood and other tissues. Anal Biochem. 1969;27:502–522.
26. Rothe G, Valet G. Flow cytometric analysis of respiratory burst activityin phagocytes with hydroethidine and 2�,7�-dichlorofluorescin. J LeukocBiol. 1990;47:440–448.
27. Pretsch W, Charles DJ, Merkle S. X-linked glucose-6-phosphate dehy-drogenase deficiency in Mus musculus. Biochem Genet. 1988;26:89–103.
28. Sanders S, Smith DP, Thomas GA, Williams ED. A glucose-6-phosphatedehydrogenase (G6PD) splice site consensus sequence mutation asso-ciated with G6PD enzyme deficiency. Mutat Res. 1997;374:79–87.
29. Liao R, Jain M, Cui L, D’Agostino J, Aiello F, Luptak I, Ngoy S,Mortensen RM, Tian R. Cardiac-specific overexpression of GLUT1prevents the development of heart failure attributable to pressure overloadin mice. Circulation. 2002;106:2125–2131.
30. Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ,Yoshimura MP, Berger C, Chan PH, et al. Dilated cardiomyopathy andneonatal lethality in mutant mice lacking manganese superoxide dis-mutase. Nat Genet. 1995;11:376–381.
31. Lebovitz RM, Zhang H, Vogel H, Cartwright J Jr, Dionne L, Lu N, HuangS, Matzuk MM. Neurodegeneration, myocardial injury, and perinataldeath in mitochondrial superoxide dismutase-deficient mice. Proc NatlAcad Sci U S A. 1996;93:9782–9787.
32. Ursini MV, Parrella A, Rosa G, Salzano S, Martini G. Enhancedexpression of glucose-6-phosphate dehydrogenase in human cells sus-taining oxidative stress. Biochem J. 1997;323:801–806.
33. Zimmer HG, Bunger R, Koschine H, Steinkopff G. Rapid stimulation onthe hexose monophosphate shunt in the isolated perfused rat heart:possible involvement of oxidized glutathione. J Mol Cell Cardiol. 1981;13:531–535.
34. Kindzelskii AL, Huang JB, Chaiworapongsa T, Fahmy RM, Kim YM,Romero R, Petty HR. Pregnancy alters glucose-6-phosphate dehydro-genase trafficking, cell metabolism, and oxidant release of maternalneutrophils. J Clin Invest. 2002;110:1801–1811.
35. Jo SH, Son MK, Koh HJ, Lee SM, Song IH, Kim YO, Lee YS, Jeong KS,Kim WB, Park JW, Song BJ, Huh TL, Huhe TL. Control of mitochondrialredox balance and cellular defense against oxidative damage by mito-chondrial NADP�-dependent isocitrate dehydrogenase. J Biol Chem.2001;276:16168–16176.
36. Minaga T, Yasumi M, Nakamura K, Kimura I, Kizu A, Ijichi H. Apossible mechanism of adriamycin cardiotoxicity. Inhibition of NADP-linked isocitrate dehydrogenase. Adv Myocardiol. 1983;4:247–253.
37. Yamamoto K, Farber JL. Metabolism of pyridine nucleotides in culturedrat hepatocytes intoxicated with tert-butyl hydroperoxide. BiochemPharmacol. 1992;43:1119–1126.
38. Sun L, Sun TT, Lavker RM. Identification of a cytosolic NADP�-dependent isocitrate dehydrogenase that is preferentially expressed inbovine corneal epithelium: a corneal epithelial crystallin. J Biol Chem.1999;274:17334–17341.
39. Henderson NS. Isozymes of isocitrate dehydrogenase: subunit structureand intracellular location. J Exp Zool. 1965;158:263–273.
40. Nakamura TY, Goda K, Okamoto T, Kishi T, Nakamura T, Goshima K.Contractile and morphological impairment of cultured fetal mousemyocytes induced by oxygen radicals and oxidants: correlation withintracellular Ca2� concentration. Circ Res. 1993;73:758–770.
41. Goldhaber JI, Liu E. Excitation-contraction coupling in single guinea-pigventricular myocytes exposed to hydrogen peroxide. J Physiol. 1994;477:135–147.
42. Josephson RA, Silverman HS, Lakatta EG, Stern MD, Zweier JL. Studyof the mechanisms of hydrogen peroxide and hydroxyl free radical–in-duced cellular injury and calcium overload in cardiac myocytes. J BiolChem. 1991;266:2354–2361.
43. Xu KY, Zweier JL, Becker LC. Hydroxyl radical inhibits sarcoplasmicreticulum Ca2�-ATPase function by direct attack on the ATP binding site.Circ Res. 1997;80:76–81.
44. Viner RI, Williams TD, Schoneich C. Peroxynitrite modification ofprotein thiols: oxidation, nitrosylation, and S-glutathiolation of func-tionally important cysteine residue(s) in the sarcoplasmic reticulumCa-ATPase. Biochemistry. 1999;38:12408–12415.
45. Turrini F, Naitana A, Mannuzzu L, Pescarmona G, Arese P. Increased redcell calcium, decreased calcium adenosine triphosphatase, and alteredmembrane proteins during fava bean hemolysis in glucose-6-phosphatedehydrogenase-deficient (Mediterranean variant) individuals. Blood.1985;66:302–305.
46. De Flora A, Benatti U, Guida L, Forteleoni G, Meloni T. Favism:disordered erythrocyte calcium homeostasis. Blood. 1985;66:294–297.
8 Circulation Research July 25, 2003

Online Supplement Figure Legend
Online Figure 1: Inhibition of G6PD impairs contractile function in cardiomyocytes.
Maximum rate of cell shortening (-dL/dt) (Figure 1A) and maximum rate of cell
relengthening (+dL/dt) (Figure 1B) in cardiomyocytes paced at 300 bpm following 12-
hour incubation with dehydroepiandrosterone (DHA) (0-30 µmol/L), and/or 24 hour
incubation with adenovirus containing recombinant rat G6PD (Ad-G6PD) at an MOI of
10. *: p<0.05 vs 0 µmol/L DHA; †: p<0.05 vs 3 µmol/L DHA; ‡: p<0.05 vs 30 µmol/L
DHA.
Online Figure 2: Rescue of contractile function in G6PD inhibited cardiomyocytes with
glutathione-generating or antioxidant treatment. Maximum rate of cell shortening (-dL/dt)
(Figure 2A) and maximum rate of cell relengthening (+dL/dt) (Figure 2B) in
cardiomyocytes paced at 300 bpm following treatment with vehicle or 30 µmol/L
dehydroepiandrosterone (DHA), 5 mmol/L D-ribose (ribose), 50 µmol/L Eukarion-134
(EUK), or 5 mmol/L L-2-oxothiazolidine-4-carboxylic acid (OTC). *: p<0.05 vs 0
µmol/L DHA; ‡: p<0.05 vs 30 µmol/L DHA.

Copyright © 2022 FDOKUMEN



















