
Sweat gland differentiation in mammary adenoid cystic carcinoma
Upload
independentCategory
view
0download
0
ORIGINAL ARTICLE
Artemin is estrogen regulated and mediates antiestrogen resistance
in mammary carcinoma
J Kang1, PX Qian2, V Pandey1, JK Perry1, LD Miller3, ET Liu4, T Zhu2, DX Liu1 and PE Lobie1,5
1Liggins Institute, University of Auckland, Auckland, New Zealand; 2Hefei National Laboratory for Physical Sciences at Microscaleand School of Life Sciences, University of Science and Technology of China, Hefei, Anhui, PR China; 3Department of CancerBiology, Wake Forest University School of Medicine, Winston-Salem, NC, USA; 4Genome Institute of Singapore, Singaporeand 5Department of Molecular Medicine and Pathology, Faculty of Medical and Health Sciences, University of Auckland,Auckland, New Zealand
We have previously identified an oncogenic role of artemin(ARTN), a member of glial cell derived neurotrophicfactor family of ligands, in mammary carcinoma. We hereinreport that ARTN is an estrogen-inducible gene. Meta-analysis of gene expression data sets showed that ARTNexpression is positively correlated to estrogen receptor(ER) status in human mammary carcinoma. Furthermore,in patients with ER-positive mammary carcinoma treatedwith tamoxifen, high ARTN expression is significantlycorrelated with decreased survival. Forced expression ofARTN in ER-positive human mammary carcinoma cellsincreased ER transcriptional activity, promoted estrogen-independent growth and produced resistance to tamoxifenand fulvestrant in vitro and to tamoxifen in xenograftmodels. ARTN-stimulated resistance to tamoxifen andfulvestrant is mediated by increased BCL-2 expression.Conversely, depletion of endogenous ARTN by small-interfering RNA or functional antagonism of ARTN byantibody enhanced the efficacy of antiestrogens. Tamox-ifen decreased ARTN expression in tamoxifen-sensitivemammary carcinoma cells whereas ARTN expression wasincreased in tamoxifen-resistant cells and not affected bytamoxifen treatment. Antibody inhibition of ARTN intamoxifen-resistant cells improved tamoxifen sensitivity.Functional antagonism of ARTN therefore warrants consi-deration as an adjuvant therapy to enhance antiestrogenefficacy in ER-positive mammary carcinoma.Oncogene (2010) 29, 3228–3240; doi:10.1038/onc.2010.71;published online 22 March 2010
Keywords: artemin; antiestrogen resistance; mammarycarcinoma\
Introduction
The pivotal role of estrogen in the etiology and progres-sion of mammary carcinoma has been well defined (Yager
and Davidson, 2006). The biological effects of estrogenare mediated by two classical estrogen receptors (ERs):ERa and ERb. Estrogen-bound ER translocates to thenucleus and initiates gene transcription by binding toestrogen response element (ERE) in the promoter regionof target genes. Alternatively, ER interacts with othertranscription factors, indirectly modulating gene expres-sion (Marino et al., 2006). In addition to the nuclearaction of ER, plasma membrane and cytoplasm-localized ER activate multiple signaling pathways andexert rapid effects upon estrogen stimulation (Kampaet al., 2008; Levin and Pietras, 2008).
Antiestrogens block the functions of ER and arewidely used as systemic adjuvant therapy for ER-positive breast cancer (Johnston, 2005). Tamoxifencompetes with estrogen for ER binding, and functionsas a selective estrogen receptor modulator with mixedantagonist and agonist activity dependent on tissue type(Osborne, 1998). The pure ER antagonist fulvestrant(ICI 182780) downregulates ER protein, resulting ininhibition of ER signaling without any agonist effects(Dowsett et al., 2005). Although antiestrogens have beenproven to be an effective therapeutic approach forchemoprevention and treatment of mammary carci-noma (Abe et al., 1998), both de novo resistance andacquired resistance to these drugs are major obstaclesto control progression and metastasis (Musgrove andSutherland, 2009). Therefore, an improved understand-ing of the molecular basis of antiestrogen resistance anddevelopment of new strategies to increase the efficacy ofantiestrogens are required.
Artemin (ARTN) belongs to the glial cell derivedneurotrophic factor (GDNF) family of ligands (GFL)that include three other members: GDNF, neurturinand persephin. ARTN signaling is mediated througha multi-component receptor complex including theglycosylphosphatidylinositol-anchored GDNF familyreceptor a3 (GFRa3) or GFRa1 as one ligand bindingcomponent and RET receptor tyrosine kinase as onecommon signaling component (Airaksinen and Saarma,2002; Carmillo et al., 2005). We have recently identifiedan oncogenic role of ARTN in mammary carcinoma(Kang et al., 2009). ARTN protein was detectable in65% of mammary carcinoma and correlated todecreased overall survival (OAS) of breast cancer
Received 30 September 2009; revised 18 January 2010; accepted 29January 2010; published online 22 March 2010
Correspondence: Professor PE Lobie, Liggins Institute, University ofAuckland, Private Bag 92019, 2-6 Park Avenue, Grafton, Auckland,New Zealand.E-mail: [email protected]
Oncogene (2010) 29, 3228–3240& 2010 Macmillan Publishers Limited All rights reserved 0950-9232/10 $32.00
www.nature.com/onc

patients. Forced expression of ARTN in mammarycarcinoma cells promoted anchorage-independentgrowth and enhanced migration and invasion, withresultant increased tumor growth in vivo. In addition,analysis of the cancer microarray database Oncominerevealed a significant association of ARTN expressionwith residual disease after chemotherapy (Kang et al.,2009). Boulay et al. (2008) have recently reported theinduction of RET and GFRa1 expression by estrogentreatment in ER-positive mammary carcinoma cells,indicating that GFL signaling may interact withestrogen signaling and potentially mediate some of thecellular functions of estrogen.
We report herein that ARTN is an estrogen-induciblegene that confers antiestrogen resistance in ER-positivemammary carcinoma cells.
Results
ARTN is an estrogen-inducible gene and is correlatedwith decreased survival in ER-positive breast cancerpatients treated with tamoxifenTo determine the potential effect of estrogen on ARTNexpression, we exposed ER-positive mammary carcino-ma cells MCF-7 and T47D to 10 nM E2 and determinedthe mRNA level of ARTN using quantitative PCR(qPCR). As shown in Figure 1a, E2 stimulation resultedin increased ARTN expression as early as 6 h aftertreatment and the maximal E2-stimulated expressionwas observed after 72 h. An increase of ARTN proteinin both MCF-7 and T47D cells on E2 exposure wasconfirmed by western immunoblotting (Figure 1b).E2-mediated upregulation of ARTN mRNA was
0 6 12 24 72
0
1
2
3
Hours
*
*
Rel
ativ
e ex
pre
ssio
n
MCF-7
0 6 12 24 72
0
1
2
3
Hours
***
Rel
ativ
e ex
pre
ssio
n T47D
0
1
2
3
E2 + - - + + - + - - + +-
*
* *Rel
ativ
e ex
ress
ion
MCF-7
β-ACTIN
ARTN
E2
-
-
28 kDa
43 kDa
- + - +
MCF-711
BT549
T47D3.1±0.2 2.6±0.1
-- -+ -+ -- -+ -+TAM-- +- +- -- +- +-Fulv
ER+, tamoxifen only (DMFS)
Pro
babi
lity
1.0
0.8
0.6
0.4
0.2
0.0
ER+, tamoxifen only (OAS)
Pro
babi
lity
1.0
0.8
0.6
0.4
0.2
0.0
0 2 4 6 8 10 12
low ARTN (n=32)high ARTN (n=36)p=0.02
Distant metastasis-free survival (yrs)
p=0.02low ARTN (n=32)high ARTN (n=36)
0 2 4 6 8 10 12Overall survival (yrs)
Figure 1 Artemin (ARTN) is an estrogen regulated gene and correlated with decreased survival in estrogen receptor (ER)-positivebreast cancer patients treated with tamoxifen. (a) Human ER-positive mammary carcinoma cells MCF-7 and T47D were treated with10 nM E2 in phenol red-free medium containing 10% charcoal stripped-fetal bovine serum and total RNA from a triplicate sample wasisolated at indicated time points. ARTN mRNA level was measured by qPCR. Fold changes were calculated relative to the samplesat 0 time point. *Po0.05; **Po0.01 compared with 0 time point. (b) Western blot analysis of ARTN protein level in MCF-7 andT47D cells treated with either dimethyl sulfoxide (DMSO, vehicle) or 10 nM E2 for 72 h. b-Actin was used as a loading control for celllysate. (c) ER-positive MCF-7 and ER-negative BT549 cells were treated with either DMSO (vehicle), 10 nM E2, 1mM tamoxifen or100 nM fulvestrant for 72 h. ARTN mRNA level was measured by qPCR. *Po0.05 compared with vehicle control. (d) Kaplan–Meieranalysis of the correlation of ARTN mRNA expression to distant metastasis-free survival (DMFS) and overall survival (OAS) intamoxifen treated ER-positive breast cancer patients.
Artemin mediates antiestrogen resistanceJ Kang et al
3229
Oncogene

abrogated by tamoxifen and fulvestrant in MCF-7 cells(Figure 1c). In contrast, neither estrogen nor antiestro-gen treatment in ER-negative BT549 cells affectedARTN mRNA level (Figure 1c). The modulation ofARTN expression by E2 and antiestrogens in mammarycarcinoma cells observed by qPCR was also confirmedby semi-quantitative RT–PCR using different ARTN-specific primers (Supplementary Figure S1a and 1b;Supplementary data 1).
Estrogen receptor status in breast cancer is a criticalparameter to determine the response to antiestrogen therapy(Heldring et al., 2007). On the basis of ER status, weevaluated ARTN mRNA expression in 20 breast cancermicroarray data sets available in the cancer microarraydatabase Oncomine (www.oncomine.org). The meta-analysis of these data sets revealed that ARTN expres-sion was positively correlated with ER-positive statusin mammary carcinoma (P¼ 0.029, SupplementaryFigure S2; Supplementary data 2).
We also determined whether ARTN expression inmammary carcinoma would affect the outcome ofER-positive patients treated with antiestrogen. Wetherefore analyzed ARTN mRNA expression in 68patients with ER-positive mammary carcinoma treatedwith tamoxifen (Miller et al., 2005). Kaplan–Meieranalysis showed a highly significant correlation ofincreased ARTN mRNA expression to decreased distantmetastasis-free survival (DMFS; hazard ratio: 2.5,95% confidence interval: 1.1–5.5, P¼ 0.02; Figure 1d).To highlight the significant association of ARTNexpression with poor DMFS outcome, we further splitthe cohort into three groups: lowest 33% (n¼ 22),middle 33% (n¼ 23) and highest 33% (n¼ 23) of theexpression level of ARTN (Supplementary Figure S3;Supplementary data 3). The group with the highestexpression of ARTN had a particularly poor outcome,with 480% recurrence at 10 years. The P-value forthis Kaplan–Meier analysis was 0.001. Increased ARTNexpression was also significantly associated with decreasedOAS (hazard ratio: 2.1, 95% confidence interval: 1.1–4.2,P¼ 0.02; Figure 1d).
ARTN enhanced ER transcriptional activity and functionTo delineate the potential functional interaction be-tween ARTN and ER signaling in mammary carcinomacells, we first determined the effects of modulation ofARTN expression on ER transcriptional activity. Thebasal ER transcriptional activity (obtained in theabsence of E2) in MCF7-ARTN cells was 1.8-foldhigher than MCF7-Vec cells. E2 increased ERE-lucifer-ase activity and forced expression of ARTN increasedER transcriptional activity with E2 in an additivemanner (Figure 2a). The capacity of MCF7-Vec andMCF7-ARTN cells to form colonies in soft agar underestrogen-deprived or estrogen-replete conditions wasexamined. In the absence of estrogen stimulation, forcedexpression of ARTN resulted in a 1.7-fold increase ofcolony formation compared with the vector cells.E2 promoted anchorage-independent growth and forcedexpression of ARTN enhanced the stimulatory effect of
E2 on colony formation in soft agar (Figure 2b). We alsoshowed by qPCR that forced expression of ARTNincreased the expression of estrogen-responsive genesincluding progesterone receptor (PR), TFF1 and TFF3in MCF7-ARTN cells compared with MCF7-Vec cells(Figure 2c).
To examine the effects of depleted ARTN expression,we stably transfected two ARTN-specific small-interferingRNA (siRNA) plasmids into MCF-7 cells. Figure 2dshows that both the mRNA and protein expressionlevels of ARTN were efficiently decreased in the twostable MCF-7 cell lines, MCF7-siARTN A and MCF7-siARTN B, compared with the control MCF7-siCKcells. Depletion of endogenous ARTN by siRNA decreasedERE activity in both the absence and the presence of E2
stimulation (Figure 2e). Furthermore, siRNA-mediateddepletion of ARTN decreased colony formation underestrogen-deprived conditions compared with vector cellsand dramatically abrogated E2-stimulated anchorage-independent growth (Figure 2f).
Forced expression of ARTN reduced antiestrogensensitivity of mammary carcinoma cellsAs our data supported a potential functional interactionbetween ARTN and ER signaling, we assessed whethermodulation of ARTN expression would affect theantiestrogen sensitivity of mammary carcinoma cells.In the absence of antiestrogen treatment, forced expres-sion of ARTN in MCF-7 cells did not affect cell numberin monolayer culture as described previously (Kanget al., 2009; Figure 3a). However, in the presence ofeither tamoxifen or fulvestrant, MCF7-ARTN cellsshowed an increase in cell number compared withMCF7-Vec cells (Figure 3a). As the growth inhibitoryeffect of antiestrogen results from both inhibitionof cell-cycle progression (Doisneau-Sixou et al., 2003)and induction of cell apoptosis (Bursch et al., 1996;Mandlekar and Kong, 2001), we determined S-phaseentry by bromodeoxyuridine (BrdU) incorporation andapoptosis with Hoechst staining. Both tamoxifen andfulvestrant suppressed BrdU incorporation (Figure 3b)and increased apoptosis (Figure 3c) in MCF7-Vec cells.Forced expression of ARTN significantly abrogatedantiestrogen inhibition of cell-cycle progression(Figure 3b) and produced resistance to the apoptoticeffect of antiestrogens (Figure 3c).
We further compared the anchorage-independentgrowth of MCF7-Vec cells and MCF7-ARTN cellstreated with antiestrogens using colony formation insoft agar. Figures 3d and e show that tamoxifen andfulvestrant inhibited the anchorage-independent growthof MCF7-Vec cells in a dose-dependent manner. Again,forced expression of ARTN in MCF-7 cells significantlyabrogated the ability of both tamoxifen and fulvestrantto decrease colony formation in soft agar. A similarprotective effect of ARTN on soft agar colony forma-tion in response to antiestrogen treatment was alsoobserved in T47D cells with forced expression of ARTN(Supplementary Figure S4a and 4b; Supplementarydata 4).
Artemin mediates antiestrogen resistanceJ Kang et al
3230
Oncogene

Cell culture in three-dimensional (3D) Matrigel hasbeen considered as a more relevant model system toanalyze cell behavior (Zhu et al., 2005; Lee et al., 2007).Antiestrogen treatment reduced the number and sizeof colonies formed by MCF7-Vec cells in Matrigel(Figure 3f). An abrogation of the antiestrogen effect oncolony growth in Matrigel was observed in MCF7-ARTN cells, with 67.6 and 70.6% greater colony growth
relative to MCF7-Vec cells following treatment withtamoxifen and fulvestrant, respectively (Figure 3f).
Forced expression of ARTN reduced tamoxifen sensitivityof mammary carcinoma cells in vivoTo determine whether ARTN-induced antiestrogenresistance in vitro correlated with tumor growth
Veh E2
0
1
2
3
4
5 MCF7-VecMCF7-ARTN
*
*
###
##
Rel
ativ
e E
RE
-Lu
cac
tivi
ty
Veh E2
0
100
200
300
400
500 MCF7-VecMCF7-ARTN
**
**
###
###
Co
lon
y fo
rmat
ion
in s
oft
ag
ar (
%)
0
2
4
6 MCF7-Vec VehMCF7-ARTN Veh
MCF7-Vec E2MCF7-ARTN E2
** ***
###
#
Rel
ativ
e ex
pre
ssio
n
# #
##
0.0
0.5
1.0
1.5
** **
siARTN BR
elat
ive
exp
ress
ion
Veh E2
0
1
2
3
4
5 MCF7-siCKMCF7-siARTN A
***
MCF7-siARTN B
**
***
###
### ###
Rel
ativ
e E
RE
-Lu
cac
tivi
ty
Veh E2
0
100
200
300
400
500 MCF7-siCK
MCF7-siARTN B
**
**
MCF7-siARTN A
**
**
##
## ##
Co
lon
y fo
rmat
ion
in s
oft
ag
ar (
%)
siCK
siARTN
B
siARTN
A
β-ACTIN
ARTN -
-
28kDa
44kDaPR TFF1 TFF3
siCK siARTN A
Figure 2 Effect of modulation of artemin (ARTN) expression level on estrogen receptor (ER) transcriptional activity and anchorage-independent growth of MCF-7 cells in the absence and the presence of E2. (a) ERE-luciferase assay: MCF7-Vec and MCF7-ARTNcells were transiently co-transfected with 0.5mg per well of pGL3-2ERE reporter and 0.5 mg per well b- galactosidase (control fortransfection efficiency) in six-well plates in phenol red-free RPMI medium containing 1% charcoal stripped-fetal bovine serum(CS-FBS). After 24 h, the cells were treated with either dimethyl sulfoxide (DMSO, vehicle) or 10 nM E2 for another 24 h before celllysis. The luciferase values were normalized for b-galactosidase activity. ER transcriptional activity is represented as the fold inductionrelative to the level of luciferase activity in vehicle-treated vector control cells. (b) Colony formation in soft agar: MCF7-Vec andMCF7-ARTN cells were cultured in 0.35% agar and treated with DMSO (vehicle) or 10 nM E2 for 10 days. Cell growth is presented asthe percentage of vehicle-treated vector control cells. (c) MCF7-Vec and MCF7-ARTN cells were treated with either DMSO (vehicle)or 10 nM E2 in phenol red-free RPMI medium containing 1% CS-FBS for 24 h before RNA isolation. Progesterone receptor (PR),TFF1 and TFF3 mRNA levels were measured by qPCR. (d) qPCR and western blot showing efficiency of siRNA-mediated depletionof ARTN mRNA and protein. (e) ERE-luciferase assay and (f) colony formation in soft agar for MCF7-siARTN A, MCF7-siARTNB and MCF7-siCK cells were performed as described in (a) and (b), respectively. *Po0.05; **Po0.01; ***Po0.001 compared withvector control group for the same treatment; #Po0.05; ##Po0.01; ###Po0.001 compared with vehicle control.
Artemin mediates antiestrogen resistanceJ Kang et al
3231
Oncogene

in vivo, we implanted MCF7-Vec or MCF7-ARTNcells into the mammary fat pad of immunodeficientmice, which were randomized to receive estrogensupplement combined with either tamoxifen orvehicle. As shown in Figure 4a, in mice supplementedwith estrogen, tumors formed by MCF7-ARTN cells
were 1.5-fold larger than those formed by MCF7-Vec cells, in confirmation of previously reporteddata (Kang et al., 2009). Tamoxifen inhibited thegrowth of tumors derived from MCF7-Vec cells asexpected (Sarkaria et al., 1993). In contrast, forcedexpression of ARTN reduced tamoxifen efficacy,
Veh TAM Fulv0
50
100
150
200
250 MCF7-VecMCF7-ARTN
*
****
Cel
l gro
wth
in 3
D m
atri
gel
(%
)
0 3 6 90
5
10
15
20
25 MCF7-Vec VehMCF7-ARTN Veh
MCF7-Vec TAMMCF7-ARTN TAM
MCF7-Vec FulvMCF7-ARTN Fulv
*
*
*
Day
To
tal c
ell n
um
ber
(x1
05 )
Veh TAM Fulv0
6
12
18
24
30 MCF7-VecMCF7-ARTN
**
**
Brd
U p
osi
tive
cel
ls (
%)
Veh TAM Fulv0
3
6
9
12
15 MCF7-VecMCF7-ARTN
* **
Ap
op
toti
c ce
lls (
%)
0
50
100
150
200
250 MCF7-VecMCF7-ARTN
TAM(μM)
** ** **
*
Co
lon
y fo
rmat
ion
in s
oft
ag
ar (
%)
0
50
100
150
200
250 MCF7-VecMCF7-ARTN
Fulv(nM)
** **
** **
Co
lon
y fo
rmat
ion
in s
oft
ag
ar (
%)
MC
F7-
Vec
MC
F7-
AR
TN
Veh TAM
510.20 0 1 10 100
Fulv
Figure 3 Forced expression of artemin (ARTN) reduced antiestrogen sensitivity in MCF-7 cells. (a) Total cell number assay: MCF7-Vec and MCF7-ARTN cells were seeded in RPMI medium containing 10% fetal bovine serum (FBS) and treated with either dimethylsulfoxide (DMSO, vehicle), 5 mM tamoxifen or 100 nM fulvestrant for 9 days. At the indicated time points, cells were trypsinized andcounted using Trypan blue. (b) Bromodeoxyuridine (BrdU) incorporation assay: MCF7-Vec and MCF7-ARTN cells were plated inRPMI medium containing 10% FBS. Cells were treated with either DMSO (vehicle), 5mM tamoxifen or 100 nM fulvestrant for 72 h.The percentage of BrdU-positive cell nuclei relative to the total number of cell nuclei was determined. (c) Apoptosis assay: MCF7-Vecand MCF7-ARTN cells were treated with either DMSO (vehicle), 5 mM tamoxifen or 100 nM fulvestrant in the complete medium for 4days and then fixed, stained by Hoechst 33258 dye for assessment of apoptosis. (d, e) Colony formation in soft agar: MCF7-Vec andMCF7-ARTN cells were cultured in 0.35% soft agar and treated with tamoxifen (d) or fulvestrant (e) at different concentration for 10days. The cell growth was measured using alamarBlue and presented as the percentage of vehicle-treated vector control cells. (f) Three-dimensional (3D) Matrigel: MCF7-Vec and MCF7-ARTN cells were cultured in 3DMatrigel and treated with DMSO (vehicle) or 1 mMtamoxifen or 10 nM fulvestrant for 7 days. The cell growth was measured using alamarBlue and presented as the percentage of vehicle-treated vector control cells. Bar¼ 100mm. *Po0.05; **Po0.01 compared with vector control group for the same treatment.
Artemin mediates antiestrogen resistanceJ Kang et al
3232
Oncogene

producing a tumor growth rate comparable with thatobserved in estrogen-stimulated MCF7-Vec tumors.
BrdU incorporation and terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL)assays were performed on tumor sections to quantifyproliferation and apoptosis within the tumor at thetime of termination of the experiment. As previouslydescribed (Kang et al., 2009), forced expression ofARTN in MCF-7 cells generated tumors with increasedproliferation and decreased apoptosis compared withMCF7-Vec cells. Tamoxifen reduced BrdU incorpora-tion (Figure 4b) and increased apoptosis (Figure 4c)in MCF7-Vec tumors. However, there were no signifi-cant changes in BrdU incorporation (Figure 4b)and apoptosis (Figure 4c) in MCF7-ARTN tumorswith tamoxifen treatment, further supporting thein vitro data that ARTN reduces tamoxifen efficacyby preventing cell-cycle arrest and protecting cellsfrom apoptosis.
Forced expression of ARTN reduced ERb expression andincreased BCL-2 expression in mammary carcinoma cellsWe next determined whether ARTN-induced antiestro-gen resistance is due to modification of ER expression.We therefore compared ERa and ERb protein levels inMCF7-Vec and MCF7-ARTN cells. There was nodifference in ERa expression between MCF7-Vec andMCF7-ARTN cells treated with vehicle, E2 or anti-estrogens (Figure 5a), indicating that forced expressionof ARTN did not affect ERa protein level. However,ERb expression was decreased in MCF7-ARTN cellsunder all examined conditions (Figure 5a), indicatingthat ARTN decreased ERb expression in MCF-7 cells.
ARTN has been observed to promote mammarycarcinoma cell survival under serum-reduced conditions
(Kang et al., 2009). Several ARTN-regulated genespossessing anti-apoptotic function have been identifiedby qPCR, including BCL-2 whose expression wasincreased 8.4-fold in MCF7-ARTN cells compared withMCF7-Vec cells (Kang et al., 2009). To elucidate themolecular mechanisms underlying ARTN-enhanced cellsurvival in response to antiestrogen treatment, weinvestigated the effect of forced expression of ARTNon the protein level of BCL-2 following antiestrogentreatment. As observed in Figure 5b, BCL-2 proteinlevel was increased in MCF7-ARTN cells comparedwith MCF7-Vec cells under conditions of estrogendeprivation, estrogen stimulation or estrogen in combi-nation with tamoxifen or fulvestrant. Estrogen increasedBCL-2 expression whereas both tamoxifen and fulves-trant abrogated estrogen-stimulated BCL-2 expressionin vector cells. Forced expression of ARTN preventedinhibition of BCL-2 expression by both tamoxifen andfulvestrant. To determine whether BCL-2 mediatedARTN-stimulated oncogenicity and antiestrogen resis-tance, we examined the anchorage-independent growthof MCF7-Vec and MCF7-ARTN cells in the presence ofthe BCL-2 inhibitor YC137 alone or in combinationwith tamoxifen or fulvestrant. As shown in Figure 5c,colony formation by MCF7-Vec cells was dramaticallyreduced by YC137, supportive of the reported functionof BCL-2 in anchorage-independent growth (Zhanget al., 2003). YC137 also significantly abrogated theARTN enhancement of colony formation in soft agar,suggestive that BCL-2 is required for ARTN-stimulatedanchorage-independent growth. Inhibition of BCL-2also completely eliminated the protective effect ofARTN in response to treatment with tamoxifen andfulvestrant, suggestive that decreased antiestrogen sen-sitivity by forced expression of ARTN is mediated byincreased BCL-2 expression.
0
6
12
18
24 VehTAM
*
Po
siti
ve c
ells
(%
)
BrdU
0
30
60
90
120 VehTAM
*
Po
siti
ve c
ells
(%
)
TUNEL
MCF7-ARTNMCF7-Vec
MCF7-ARTNMCF7-Vec
0
100
200
300
2 3 4 5
400 MCF7-Vec VehMCF7-ARTN VehMCF7-Vec TAMMCF7-ARTN TAM
******
******
******
**
**
*
*
******
Weeks after injection
Vo
lum
e o
f tu
mo
ur
(mm
3 )
Figure 4 Forced expression of artemin (ARTN) attenuated the growth-inhibitory effect of tamoxifen on MCF-7 xenografts. MCF7-Vec and MCF7-ARTN cells in Matrigel were transplanted into the mammary fat pad of immunodeficient mice supplemented with E2,and were randomly assigned to tamoxifen-treated (TAM) or vehicle-treated group (Veh). (a) Tumor growth in the absence and thepresence of tamoxifen. Cell proliferation was assessed by bromodeoxyuridine (BrdU) incorporation (b) and apoptosis was measuredby terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay (c) at the end of experiment. *Po0.05;**Po0.01; ***Po0.001 compared with vector control group for the same treatment.
Artemin mediates antiestrogen resistanceJ Kang et al
3233
Oncogene

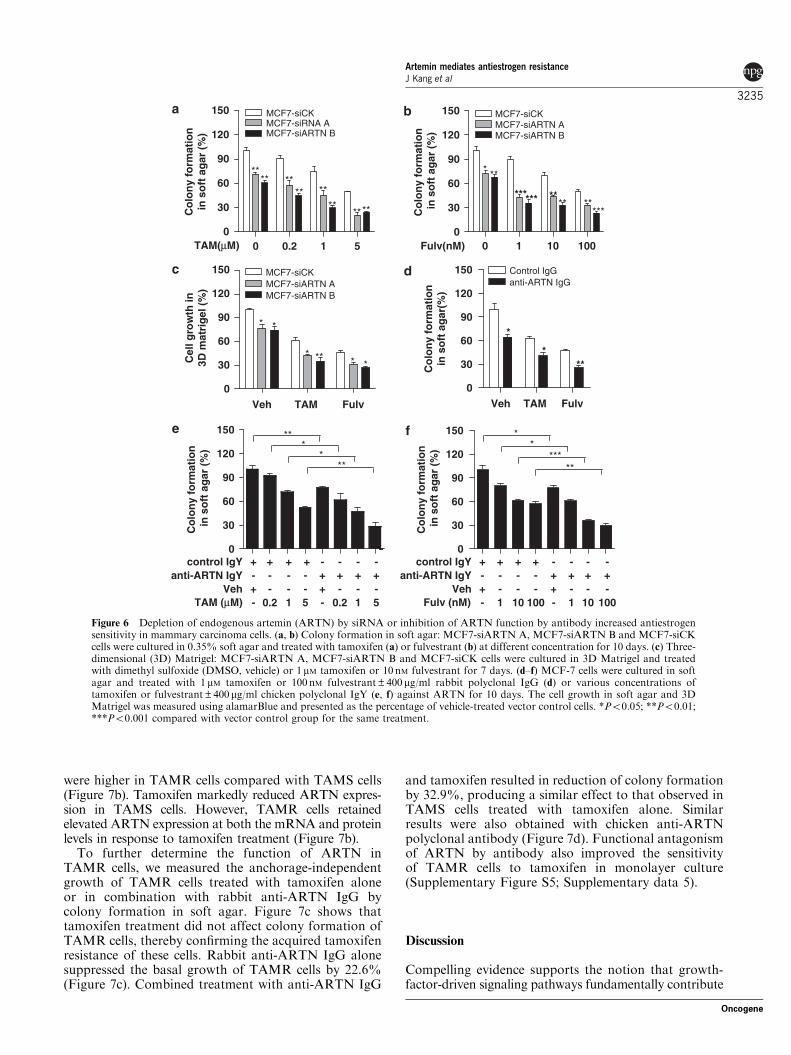
Depletion of endogenous ARTN by siRNA or inhibitionof ARTN function by antibody increased antiestrogensensitivity in mammary carcinoma cellsTamoxifen and fulvestrant inhibited colony formationin soft agar of control cells stably transfected withscrambled siRNA (Figures 6a and b). Similar inhibitoryeffects of tamoxifen and fulvestrant were observed in 3DMatrigel culture (Figure 6c). Depletion of endogenousARTN by two distinct siRNAs dramatically increasedthe sensitivity of MCF-7 cells to both tamoxifen andfulvestrant, resulting in further reduced growth in softagar (Figures 6a and b) and 3D Matrigel (Figure 6c)compared with control siRNA-transfected cells. Simi-larly, depletion of endogenous ARTN in T47D cells byone siRNA (pSilencer-ARTN B) produced a greaterinhibitory effect of tamoxifen and fulvestrant on colonyformation in soft agar (Supplementary Figure S4c–e;Supplementary data 4).
Consistent with the effect of depletion of ARTN bysiRNA, a rabbit polyclonal antibody raised againstARTN enhanced the inhibitory effect of tamoxifen andfulvestrant on colony formation of MCF-7 cells. Rabbitanti-ARTN IgG alone possessed an inhibitory effect onanchorage-independent growth, with 35.6% reductionin colony formation in soft agar (Figure 6d). Tamoxifen
and fulvestrant produced 37.6 and 53.0% growthinhibition, respectively. The combination of anti-ARTNIgG with tamoxifen or fulvestrant produced 59.0and 74.3% reduction, respectively. Similar changeswere also observed after treatment with another poly-clonal antibody against ARTN generated in chicken(Figures 6e and f).
ARTN possesses a functional role in acquired tamoxifenresistanceTo determine whether ARTN contributes to theacquisition of tamoxifen resistance in mammary carci-noma cells, we developed a tamoxifen-resistant MCF-7cell line (designated as TAMR) as described in Materialsand methods. The growth of tamoxifen-sensitive(TAMS) and TAMR cells was measured in estrogen-free medium containing 10 nM E2 or 1 mM tamoxifenover a period of 7 days. TAMS cells retained theresponse to E2 and were sensitive to tamoxifen.In contrast, TAMR cells grew faster and showed lossof response to E2 and loss of tamoxifen sensitivity(Figure 7a). We subsequently compared ARTN expres-sion in TAMS and TAMR cells. In the absence oftamoxifen, both the mRNA and protein levels of ARTN
0
50
100
150
200
250 MCF7-VecMCF7-ARTN
***
*
** *
Co
lon
y fo
rmat
ion
in s
oft
ag
ar (
%)
02468
10
*
MCF7-VecMCF7-ARTN
**
*
Arb
itra
ry u
nit
s
- 43kDa
- 26kDa
0.00.30.60.91.21.5
E2TAMFulv
MCF7-VecMCF7-ARTN
Arb
itra
ry u
nit
s
- 43kDa
- 66kDaERα
β-ACTIN
ERβ
β-ACTIN
0.00.30.60.91.21.5
* **
MCF7-VecMCF7-ARTN
*
Arb
itra
ry u
nit
s
- 43kDa
- 59kDa
-- -- - -
-+ +
++
+
E2TAMFulv
-- -- - -
-+ +
++
+E2TAMFulv
-- -- - -
-+ +
++
+
BCL-2
β-ACTIN
TAM --- 1 - 1 5 1 5-
- - - - -- - + + +
+ + +- - -
FulvYC137 5
Figure 5 Forced expression of artemin (ARTN) reduced estrogen receptor-b (ERb) expression and increased BCL-2 expression inmammary carcinoma cells. (a, b) MCF7-Vec and MCF7-ARTN cells were treated with either dimethyl sulfoxide (DMSO, vehicle),10 nM E2 alone, or in combination with 1 mM tamoxifen or 100 nM fulvestrant in phenol red-free RPMI medium containing 1% charcoalstripped-fetal bovine serum (CS-FBS) for 72 h. Soluble whole cellular extracts were run on a sodium dodecyl sulfate-polyacrylamide gelelectrophoresis and probed for ERa, ERb and BCL-2. b-Actin was used as loading control for whole-cell lysates. Protein level wasdetermined by the Quantity One scanning system and densitometry software (Bio-Rad Laboratories Inc., Hercules, CA, USA), andthen normalized to b-Actin in the same membrane. (c) Colony formation in soft agar: MCF7-Vec and MCF7-ARTN cells werecultured in 0.35% soft agar and treated with BCL-2 inhibitor YC137 at 1 or 5 mM alone or in combination with 1 mM tamoxifen or100 nM fulvestrant for 10 days. The cell growth was measured using alamarBlue and presented as the percentage of vehicle-treatedvector control cells. *Po0.05; **Po0.01; ***Po0.001 compared with vector control group for the same treatment.
Artemin mediates antiestrogen resistanceJ Kang et al
3234
Oncogene
were higher in TAMR cells compared with TAMS cells(Figure 7b). Tamoxifen markedly reduced ARTN expres-sion in TAMS cells. However, TAMR cells retainedelevated ARTN expression at both the mRNA and proteinlevels in response to tamoxifen treatment (Figure 7b).
To further determine the function of ARTN inTAMR cells, we measured the anchorage-independentgrowth of TAMR cells treated with tamoxifen aloneor in combination with rabbit anti-ARTN IgG bycolony formation in soft agar. Figure 7c shows thattamoxifen treatment did not affect colony formation ofTAMR cells, thereby confirming the acquired tamoxifenresistance of these cells. Rabbit anti-ARTN IgG alonesuppressed the basal growth of TAMR cells by 22.6%(Figure 7c). Combined treatment with anti-ARTN IgG
and tamoxifen resulted in reduction of colony formationby 32.9%, producing a similar effect to that observed inTAMS cells treated with tamoxifen alone. Similarresults were also obtained with chicken anti-ARTNpolyclonal antibody (Figure 7d). Functional antagonismof ARTN by antibody also improved the sensitivityof TAMR cells to tamoxifen in monolayer culture(Supplementary Figure S5; Supplementary data 5).
Discussion
Compelling evidence supports the notion that growth-factor-driven signaling pathways fundamentally contribute
Veh TAM Fulv0
30
60
90
120
150 Control IgGanti-ARTN IgG
*
***C
olo
ny
form
atio
nin
so
ft a
gar
(%)
0
30
60
90
120
150 MCF7-siCKMCF7-siRNA A
TAM(μM)
****
**
**
MCF7-siARTN B
**** **
**C
olo
ny
form
atio
nin
so
ft a
gar
(%
)
Veh TAM Fulv0
30
60
90
120
150 MCF7-siCKMCF7-siARTN A
*
* **
MCF7-siARTN B
*
* *Cel
l gro
wth
in3D
mat
rig
el (
%)
0
30
60
90
120
150 MCF7-siCKMCF7-siARTN A
*
*** ****
MCF7-siARTN B
**
*** *****C
olo
ny
form
atio
n in
so
ft a
gar
(%
)
0
30
60
90
120
150
control IgYanti-ARTN IgY
VehTAM (μM)
***
***
Co
lon
y fo
rmat
ion
in s
oft
ag
ar (
%)
0
30
60
90
120
150 **
*****
Co
lon
y fo
rmat
ion
in s
oft
ag
ar (
%)
510.20 1001010Fulv(nM)
+ + + +- -
-- 0.2 1 5
- -- -
-- 0.2 1 5
- -++
+ + +- - - -
+
control IgYanti-ARTN IgY
VehFulv (nM)
+ + + +- -
-- 1 10 100
- -- -
-- 1 10 100
- -++
+ + +- - - -
+
Figure 6 Depletion of endogenous artemin (ARTN) by siRNA or inhibition of ARTN function by antibody increased antiestrogensensitivity in mammary carcinoma cells. (a, b) Colony formation in soft agar: MCF7-siARTN A, MCF7-siARTN B and MCF7-siCKcells were cultured in 0.35% soft agar and treated with tamoxifen (a) or fulvestrant (b) at different concentration for 10 days. (c) Three-dimensional (3D) Matrigel: MCF7-siARTN A, MCF7-siARTN B and MCF7-siCK cells were cultured in 3D Matrigel and treatedwith dimethyl sulfoxide (DMSO, vehicle) or 1mM tamoxifen or 10 nM fulvestrant for 7 days. (d–f) MCF-7 cells were cultured in softagar and treated with 1 mM tamoxifen or 100 nM fulvestrant±400mg/ml rabbit polyclonal IgG (d) or various concentrations oftamoxifen or fulvestrant±400mg/ml chicken polyclonal IgY (e, f) against ARTN for 10 days. The cell growth in soft agar and 3DMatrigel was measured using alamarBlue and presented as the percentage of vehicle-treated vector control cells. *Po0.05; **Po0.01;***Po0.001 compared with vector control group for the same treatment.
Artemin mediates antiestrogen resistanceJ Kang et al
3235
Oncogene

to the development of either de novo or acquiredantiestrogen resistance (Massarweh and Schiff, 2006;Arpino et al., 2008). Elevated expression of epidermalgrowth factor receptor (EGFR) and HER2 and activa-tion of downstream signaling molecules such as p42/44MAPK and phosphatidylinositol 3-kinase (PI3K)/AKThave been shown in antiestrogen-resistant cell linesand tumors (McClelland et al., 2001; Knowlden et al.,2003; Massarweh et al., 2008). Clinical data also suggestthat patients with increased expression of HER2 andEGFR in tumors have a poor outcome when treatedwith tamoxifen (Arpino et al., 2008). Another tyrosinekinase receptor IGF-1R has been also suggested to beinvolved in acquired antiestrogen resistance in vitro,possibly through cross-talk with EGFR and HER2
(Knowlden et al., 2005). In addition, other growthfactors or growth factor receptors such as PC cell-derived growth factor (Tangkeangsirisin et al., 2004)and keratinocyte growth factor receptor (Rotolo et al.,2008) have been reported to confer tamoxifen resistancein human mammary carcinoma cells. In this study,we identified ARTN as a novel growth factor involvedin antiestrogen resistance. As an estrogen-induciblegene, antiestrogen treatment reduced ARTN expressionin ER-positive mammary carcinoma cells. However, inTAMR cells, ARTN expression was substantiallyincreased and was not affected by tamoxifen treatment.The activity of IGF-1R, another estrogen-inducible gene,was also found to be fully restored in tamoxifen-resistant tumors after undergoing initial reduction at
0
50
100
150 TAMSTAMR
**
** **
Co
lon
y fo
rmat
ion
in s
oft
ag
ar (
%)
Veh TAM0
1
2
3
4 TAMSTAMR
** **
##
Rel
ativ
e ex
pre
ssio
n
0
50
100
150
**
**
****
TAMSTAMR
**
Control IgGanti-ARTN IgG
VehTAM
***
Co
lon
y fo
rmat
ion
in s
oft
ag
ar (
%)
TAMR
0 1 3 70
20
40
60 VehE2TAM
Days
To
tal c
ell n
um
ber
(x10
4 )
TAMS
0 1 3 70
10
20
30 VehE2TAM
***
**
Days
To
tal c
ell n
um
ber
(x10
4 )
ARTN
β-ACTIN
1
TAMS
TAM - + +-
- 28 kDa
- 43 kDa
TAMR2.8±0.12.5±0.20.6±0.1
+ ++++
+
+- -
- --
-+-
- + - + - ++- -+ +- -
+ + - - Control IgYanti-ARTN IgY
VehTAM
+ ++++
+
+- -
- --
-+-
- + - + - ++- -+ +- -
+ + - -
d
Figure 7 Artemin (ARTN) possesses a functional role in acquired tamoxifen resistance. (a) TAMS and TAMR cells were cultured inphenol red-free RPMI medium containing 10% charcoal stripped-fetal bovine serum (CS-FBS) for 3 days. Cells were then plated in12-well plates at 20 000 cells per well and treated with either dimethyl sulfoxide (DMSO, vehicle), 10 nM E2 or 1mM tamoxifen over aperiod of 7 days. Cells were trypsinized and counted using Trypan blue. **Po0.01; ***Po0.001 compared with vehicle control.(b) TAMS and TAMR cells were cultured in phenol red-free RPMI medium containing 10% CS-FBS for 3 days and then treated witheither DMSO (vehicle) or 1 mM tamoxifen for 72 h. Left: ARTN mRNA level was measured by qPCR. Right: soluble whole cellularextracts were run on a sodium dodecyl sulfate-polyacrylamide gel electrophoresis and probed for ARTN. b-Actin was used as a loadingcontrol. **Po0.01 compared with TAMS cells for the same treatment. ##Po0.01 compared with vehicle control. (c, d) TAMS andTAMR cells were cultured in soft agar and treated with 1 mM tamoxifen alone or in combination with 400mg/ml rabbit polyclonal IgG(c) or 400mg/ml chicken polyclonal IgY (d) against ARTN for 10 days. Cell growth was presented as the percentage of the group treatedwith vehicle and control IgG (c) or IgY (d). *Po0.05; **Po0.01 compared with the group treated with vehicle and control antibody.
Artemin mediates antiestrogen resistanceJ Kang et al
3236
Oncogene

the tamoxifen-sensitive stage (Massarweh et al., 2008).Interestingly, we found that ARTN expression was alsoincreased upon IGF-1 stimulation in mammary carcinomacells (unpublished observation), indicative of a potentialinteraction between ARTN and IGF-1R signaling.Furthermore, ARTN protein level has also beenobserved to be highly correlated to HER2 expressionin breast tumor tissues (Kang et al., 2009). Thus, ARTNsignaling may integrate with HER2, IGF-1R and othergrowth factor signaling pathways, and contribute to thedevelopment of antiestrogen resistance in mammarycarcinoma.
Estrogen receptor has been considered as the primarydeterminant of the clinical response to antiestrogentherapy (Heldring et al., 2007). Forced expression ofARTN in MCF-7 cells did not affect ERa expression.Instead, decreased expression of ERb was observed incells with forced expression of ARTN. Several reportshave described loss of ERb during mammary tumor-igenesis, supporting the potential role of ERb as a tumorsuppressor (Park et al., 2003; Shaaban et al., 2003;Bardin et al., 2004; Rody et al., 2005). ERb has beenreported to modulate ERa activity and cellular responseto estrogen (Hall and McDonnel, 1999; Pettersson et al.,2000). Moreover, loss of ERb was observed to beassociated with tamoxifen resistance whereas increase ofERb expression in MCF-7 cells enhanced the antipro-liferative and apoptotic effect of antiestrogens (Hodges-Gallagher et al., 2008). However, data regarding theassociation of ERb expression with clinical pathologicalparameters and antiestrogen responsiveness are stillmixed (Speirs et al., 1999; O’Neill et al., 2004; Murphyand Watson, 2006).
Although the mechanisms by which growth factorsignaling pathways promote antiestrogen resistancehave not been fully determined, cumulative experimen-tal data suggest that activated growth factor signalingcross-talks with ER signaling resulting in ligand-independent or tamoxifen-induced ER activation withconsequent promotion of tamoxifen-resistant tumor cellgrowth (Riggins et al., 2007; Arpino et al., 2008). Ourdata also supported a potential functional interactionbetween ARTN and ER signaling through which ERactivation upregulates ARTN expression and ARTNstimulation increases ER activity. As a secreted growthfactor, ARTN presumably exerts its biological effects bybinding to the receptor complex on the cell surface withconsequent activation of multiple signal transductionpathways. Several growth factor receptor signalingpathways, such as HER2, EGFR and IGF-1R, havebeen reported to induce estrogen-independent ERaactivation by phosphorylation of ER and increase ERtransactivation domain AF-1 activity through MAPKand PI3K/AKT signaling pathways (Kato et al., 1995;Campbell et al., 2001; Fagan and Yee, 2008). Indeed,ARTN stimulates AKT activity in endometrial carci-noma cells (Pandey et al., 2010). We have also observedincreased AKT phosphorylation in mammary carci-noma cells with forced expression of ARTN (paper inpreparation). As discussed above, a potential functionalinteraction of ARTN with IGF-1R, HER2 signaling and
other kinase signaling pathways may also contribute toARTN-stimulated ER activity. However, the precisemechanisms by which ARTN promotes ER activationneed to be determined.
Forced expression of ARTN also promotes tumorcell growth that is resistant to the pure antiestrogenfulvestrant, which exerts its effect by inducing ERdegradation and abrogation of ER transcription (Dowsettet al., 2005), implicating that ER-independent mecha-nisms for ARTN-stimulated oncogenicity may exist.Indeed, activated growth factor-initiated signaling path-ways not only promote ER activation, but also functionin an ER-independent manner (Nicholson et al., 2007).For example, in the fulvestrant-resistant cell model,increased MAPK, AKT and NF-kB activities supportthe important role of growth factor signaling in theresistance to pure antiestrogens (Nicholson et al., 2005;Riggins et al., 2005). We have also previously reportedthat ARTN stimulates oncogenicity of ER-negativemammary carcinoma cells (Kang et al., 2009).
Moreover, activated growth factor signaling canprovide survival signals to protect from tamoxifen-and fulvestrant-induced apoptosis (Kumar et al., 1996;Chung et al., 2002; Siddiqa et al., 2008). We showherein that ARTN-stimulated resistance to antiestrogenis mediated by upregulation of BCL-2 protein expres-sion. The estrogen-dependent expression of BCL-2 hasbeen reported to contribute to the prosurvival effectof estrogen (Teixeira et al., 1995; Perillo et al., 2000).Downregulation of BCL-2 is involved in antiestrogen-induced apoptosis and depletion of BCL-2 expressionenhances sensitivity of breast cancer cells to tamoxifen(Diel et al., 1999; Zhang et al., 1999; Kim et al., 2005).BCL-2 is positively associated with ERa expression(Elledge et al., 1997) whereas decreased BCL-2 expres-sion has been observed after induction of ERb expres-sion in T47D mammary carcinoma cells (Williams et al.,2008), concordant with our observation that ERb isdecreased and BCL-2 expression is increased by forcedexpression of ARTN.
In summary, this study has identified ARTN as anovel estrogen-regulated gene and showed that ARTNreduced the efficacy of antiestrogens in ER-positivemammary carcinoma cells. This effect may be mediated,at least in part, through reduced ERb expression level,enhanced ER transcriptional activity and increasedexpression of the anti-apoptotic protein BCL-2. Combi-nation of antiestrogens and anti-ARTN antibodyenhanced the efficacy of antiestrogens in mammarycarcinoma cells and improved tamoxifen sensitivity intamoxifen-resistant cells. Therefore, functional antagon-ism of ARTN warrants consideration as a noveladjuvant therapeutic approach for the treatment ofER-positive mammary carcinoma.
Materials and methods
Cell cultureThe mammary carcinoma cell lines MCF-7 and T47D wereobtained from the American Type Culture Collection. We
Artemin mediates antiestrogen resistanceJ Kang et al
3237
Oncogene

have previously generated the stable cell lines MCF7-ARTNand T47D-ARTN with forced expression of ARTN and theircontrol cell lines MCF7-Vec and T47D-Vec, respectively.A detailed description of characterization of these cell lines hasbeen reported previously (Kang et al., 2009). To generatesiRNA oligonucleotides targeting all five transcript variants ofARTN, we selected the DNA sequence 50-AACAGCACCTGGAGAACCG-30 and 50-AACTGGCCTGTACTCACTCAT-30
to construct siARTN A and siARTN B plasmids, respectively,using the pSilencer 2.1-U6 hygro vector (Ambion, Austin, TX,USA) according to the manufacturer’s protocol. For estrogen-free cell culture, MCF-7 cells were cultured in phenol red-freeRPMI medium supplemented with 10% dextran-coatedcharcoal-stripped fetal bovine serum (CS-FBS; Invitrogen,Carlsbad, CA, USA) for 3 days before estrogen or antiestrogentreatment.The tamoxifen-resistant (TAMR) cell line was established as
described by Shaw et al. (2006). Briefly, the parental MCF-7cells were continuously exposed for 6 months to 1mMtamoxifen in phenol red-free RPMI medium containing 10%CS-FBS until the cells developed resistance to the growth-inhibitory effect of tamoxifen. MCF-7 cells sensitive totamoxifen, cultured in the same medium without tamoxifen,were designated as tamoxifen-sensitive (TAMS) cells.
Reagentsb-Estradiol (E2), tamoxifen and fulvestrant were purchasedfrom Sigma-Aldrich (St Louis, MO, USA). Bcl-2 inhibitorYC137 was purchased from Calbiochem (San Diego, CA,USA). The chicken and rabbit anti-ARTN polyclonal anti-bodies were generated as described (Kang et al., 2009).
Quantitative PCR and semiquantitative RT–PCRqPCR and semi-quantitative RT–PCR were performed asdescribed previously (Kang et al., 2009). ARTN primers usedfor qPCR were forward: 50-ATGAACACTACAGTGGCTGAGG-30 and reverse: 50-AGCTCCCATGAGTGAGTACAGG-30.
Western blot analysisWestern blot analysis was performed as described previously(Liu and Lobie, 2007) using the following antibodies: goatanti-ARTN polyclonal antibody (R&D Systems, Minneapolis,MN, USA), rabbit anti-ERa, anti-ERb monoclonal antibodies(DakoCytomation, Glostrup, Denmark), mouse anti-BCL-2monoclonal antibody (BD Pharmingen, San Diego, CA, USA)and mouse anti-b-ACTIN monoclonal antibody (Sigma,St Louis, MO, USA).
Luciferase assayCells were plated in six-well plates and cultured in phenolred-free RPMI supplemented with 10% CS-FBS for 72 hbefore transfection. Total DNA (1 mg) per well containing0.5 mg pGL2-ERE-luciferase plasmid and 0.5 mg pcDNA3b-galactosidase reporter plasmid was transfected usingFuGENE 6 reagent (Roche, Mannheim, Germany). Cellswere then treated with either dimethyl sulfoxide (DMSO;vehicle) or 10 nM E2 for 24 h. The cell lysate was used toquantify luciferase and b-galactosidase activities using the kitfollowing the manufacturer’s protocols (Promega, Madison,WI, USA).
Cell function assaysTotal cell number, apoptosis, BrdU incorporation and 3Dmorphogenesis assays were performed as described (Kang
et al., 2009). Soft agar colony formation assay in 96-well plateswas performed as described (Ke et al., 2004) with minormodification. Briefly, 5� 103 cells were seeded into 100ml0.35% agarose onto the 0.5% agarose base layer. Serum-supplemented medium (100ml) containing drug or antibodywas added on the top and changed every 3 days. After 10 days,alamarBlue was added (1:10 volume reagent; Invitrogen) andincubated at 37 1C for 4 h followed by measurement offluorescence with excitation wavelength at 530 nm and emis-sion wavelength at 590 nm.
Tumor xenograft in nude miceMCF7-Vec or MCF7-ARTN cells (3� 106 cells per site) wereinjected into the mammary fat pad of 3- to 4-week-old BALBcnu/nu mice (Shanghai Slaccas Co., Shanghai, China). E2 pellets(1.7mg; 60-day release) were implanted into the back 1 daybefore cell inoculation. The animals were then randomizedto implant with either tamoxifen base pellets (5mg; 60-dayrelease) or placebo pellets (n¼ 8 for each group). BrdU andTUNEL immunostainings were preformed as described(Pandey et al., 2008).
StatisticsAll experiments were repeated 3–4 times. All numerical dataare expressed as mean±s.e.m. (standard error of the mean)from a representative experiment performed in triplicate, andstatistical analyses were assessed by Student’s t-test usingMicrosoft Excel XP.
Abbreviations
ARTN, artemin; BrdU, bromodeoxyuridine; CS-FBS, char-coal stripped-fetal bovine serum; DMFS, distant metastasis-free survival; ER, estrogen receptor; ERE, estrogen responseelement; GDNF, glial cell derived neurotrophic factor; GFL,GDNF family of ligand; GFRa, GDNF family receptor-a;OAS, overall survival; PR, progesterone receptor; qPCR,quantitative PCR; siRNA, small-interfering RNA; TUNEL,terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling.
Conflict of interest
JK, VP, JKP, DXL and PEL have equity interests in SaratanTherapeutics Ltd. DXL and PEL are inventors on PCTapplication PCT/NZ2008/000152 and US provisional applica-tions 61/234902. PEL is an inventor on US provisionalapplication 61/252513. TZ and PEL are consultants forSaratan Therapeutics Ltd.
Acknowledgements
We thank Carol Chelimo (MPH) for the meta-analysis ofbreast cancer microarray data. This work was funded by theBreast Cancer Research Trust (NZ); the Foundation forResearch, Science and Technology of New Zealand; theHundred-Talent Scheme of Chinese Academy of Sciences,National Natural Science Foundation of China (30571030)and National Basic Research Program of China(2007CB914503).
Artemin mediates antiestrogen resistanceJ Kang et al
3238
Oncogene

References
Abe O, Abe R, Enomoto K, Kikuchi K, Koyama H, Nomura Y et al.(1998). Tamoxifen for early breast cancer: an overview of therandomised trials. Lancet 351: 1451–1467.
Airaksinen MS, Saarma M. (2002). The GDNF family: signalling,biological functions and therapeutic value. Nat Rev Neurosci 3:383–394.
Arpino G, Wiechmann L, Osborne CK, Schiff R. (2008). Crosstalkbetween the estrogen receptor and the HER tyrosine kinase receptorfamily: molecular mechanism and clinical implications for endocrinetherapy resistance. Endocr Rev 29: 217–233.
Baloh RH, Tansey MG, Lampe PA, Fahrner TJ, Enomoto H,Simburger KS et al. (1998). Artemin, a novel member of theGDNF ligand family, supports peripheral and central neurons andsignals through the GFRa3-RET receptor complex. Neuron 21:1291–1302.
Bardin A, Boulle N, Lazennec G, Vignon F, Pujol P. (2004). Loss ofER beta expression as a common step in estrogen-dependent tumorprogression. Endocr Relat Cancer 11: 537–551.
Boulay A, Breuleux M, Stephan C, Fux C, Brisken C, Fiche M et al.(2008). The ret receptor tyrosine kinase pathway functionallyinteracts with the ERalpha pathway in breast cancer. Cancer Res
68: 3743–3751.Bursch W, Ellinger A, Kienzl H, Tarak L, Pandey S, Sikorska M
et al. (1996). Active cell death induced by the anti-estrogenstamoxifen and ICI 164 384 in human mammary carcinoma cells(MCF-7) in culture: the role of autophagy. Carcinogenesis 17:1595–1607.
Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S,Nakshatri H. (2001). Phosphatidylinositol 3-kinase/AKT-mediatedactivation of estrogen receptor alpha: a new model for anti-estrogenresistance. J Biol Chem 276: 9817–9824.
Carmillo P, Dag� L, Day ES, Worley DS, Rossomando A, Walus Let al. (2005). Glial cell line-derived neurotrophic factor (GDNF)receptor a-1 (GFRa1) is highly selective for GDNF versus artemin.J Biol Chem 44: 2545–2554.
Chung YL, Sheu ML, Yang SC, Lin CH, Yen SH. (2002). Resistanceto tamoxifen-induced apoptosis is associated with direct interactionbetween Her2/neu and cell membrane estrogen receptor in breastcancer. Int J Cancer 97: 306–312.
Diel P, Smolnikar K, Michna H. (1999). The pure antiestrogen ICI182780 is more effective in the induction of apoptosis and downregulation of BCL-2 than tamoxifen in MCF-7 cells. Breast Cancer
Res Treat 58: 87–97.Doisneau-Sixou SF, Sergio CM, Carroll JS, Hui R, Musgrove EA,
Sutherland RL. (2003). Estrogen and antiestrogen regulation of cellcycle progression in breast cancer cells. Endocr Relat Cancer 10:179–186.
Dowsett M, Nicholson RI, Pietras RJ. (2005). Biological character-istics of the pure antiestrogen fulvestrant: overcoming endocrineresistance. Breast Cancer Res Treat 93: S11–S18.
Elledge RM, Green S, Howes L, Clark GM, Berardo M, Allred DCet al. (1997). bcl-2, p53, and response to tamoxifen in estrogenreceptor-positive metastatic breast cancer: A Southwest OncologyGroup Study. J Clin Oncol 15: 1916–1922.
Fagan DH, Yee D. (2008). Crosstalk between IGF1R and estrogenreceptor signaling in breast cancer. J Mammary Gland Biol
Neoplasia 13: 423–429.Hall JM, McDonnel DP. (1999). The estrogen receptor beta-isoform
(ERbeta) of the human estrogen receptor modulates ERalphatranscriptional activity and is a key regulator of the cellular responseto estrogens and antiestrogens. Endocrinology 140: 5566–5578.
Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman Jet al. (2007). Estrogen receptors: how do they signal and what aretheir targets. Physiol Rev 87: 905–931.
Hodges-Gallagher L, Valentine CD, Bader SE, Kushner PJ. (2008).Estrogen receptor beta increases the efficacy of antiestrogens byeffects on apoptosis and cell cycling in breast cancer cells. Breast
Cancer Res Treat 109: 241–250.
Johnston SRD. (2005). Selective oestrogen receptor modulators anddownregulators for breast cancer—have they lost their way? Breast
Cancer Res 7: 119–130.Kampa M, Pelekanou V, Castanas E. (2008). Membrane-initiated
steroid action in breast and prostate cancer. Steroids 73: 953–960.Kang J, Perry JK, Pandey V, Fielder GC, Mei B, Qian PX et al. (2009).
Artemin is oncogenic for human mammary carcinoma cells.Oncogene 28: 2034–2045.
Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki Het al. (1995). Activation of the estrogen receptor throughphosphorylation by mitogen-activated protein kinase. Science 270:1491–1494.
Ke N, Albers A, Claassen G, Yu DH, Chatterton JE, Hu X et al.(2004). One-week 96-well soft agar growth assay for cancer targetvalidation. Biotechniques 36: 826–833.
Kim R, Tanabe K, Emi M, Uchida Y, Toge T. (2005). Modulation oftamoxifen sensitivity by antisense Bcl-2 and trastuzumab in breastcarcinoma cells. Cancer 103: 2199–2207.
Knowlden JM, Hutcheson IR, Barrow D, Gee JMW, Nicholson RI.(2005). Insulin-like growth factor-I receptor signaling in tamoxifen-resistant breast cancer: a supporting role to the epidermal growthfactor receptor. Endocrinology 146: 4609–4618.
Knowlden JM, Hutcheson IR, Jones HE, Madden T, Gee JMW,Harper ME et al. (2003). Elevated levels of epidermal growth factorreceptor/c-erbB2 heterodimers mediate an autocrine growth reg-ulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology
144: 1032–1044.Kumar R, Mandal M, Lipton A, Harvey H, Thompson CB. (1996).
Overexpression of HER2 modulates bcl-2, Bcl-X(L) and tamoxifeninduced apoptosis in human MCF-7 breast cancer cells. Clin CancerRes 2: 1215–1219.
Lee GY, Kenny PA, Lee EH, Bissell MJ. (2007). Three-dimensionalculture models of normal and malignant breast epithelial cells. Nat
Methods 4: 359–365.Levin ER, Pietras RJ. (2008). Estrogen receptors outside the nucleus in
breast cancer. Breast Cancer Res Treat 108: 351–361.Liu DX, Lobie PE. (2007). Transcriptional activation of p53 by Pitx1.
Cell Death Differ 14: 1893–1907.Mandlekar S, Kong ANT. (2001). Mechanisms of tamoxifen-induced
apoptosis. Apoptosis 6: 469–477.Marino M, Galluzzo P, Ascenzi P. (2006). Estrogen signaling
multiple pathways to impact gene transcription. Curr Genomics 7:497–508.
Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A,Huang S et al. (2008). Tamoxifen resistance in breast tumorsis driven by growth factor receptor signaling with repressionof classic estrogen receptor genomic function. Cancer Res 68:826–833.
Massarweh S, Schiff R. (2006). Resistance to endocrine therapy inbreast cancer: exploiting estrogen receptor/growth factor signalingcrosstalk. Endocr Relat Cancer 13: S15–S24.
McClelland RA, Barrow D, Madden TA, Dutkowski CM, Pamment J,Knowlden JM et al. (2001). Enhanced epidermal growth factorreceptor signaling in MCF7 breast cancer cells after long-termculture in the presence of the pure antiestrogen ICI 182,780(Faslodex). Endocrinology 142: 2776–2788.
Miller LD, Smeds J, George J, Vega VB, Vergara L, Ploner A et al.(2005). An expression signature for p53 status in human breastcancer predicts mutation status, transcriptional effects, and patientsurvival. Proc Natl Acad Sci USA 102: 13550–13555.
Murphy LC, Watson PH. (2006). Is oestrogen receptor-beta apredictor of endocrine therapy responsiveness in human breastcancer? Endocr Relat Cancer 13: 327–334.
Musgrove EA, Sutherland RL. (2009). Biological determinants ofendocrine resistance in breast cancer. Nat Rev Cancer 9: 631–643.
Nicholson RI, Hutcheson IR, Hiscox SE, Knowlden JM, Giles M,Barrow D et al. (2005). Growth factor signalling and resistance toselective oestrogen receptor modulators and pure anti-oestrogens:
Artemin mediates antiestrogen resistanceJ Kang et al
3239
Oncogene

the use of anti-growth factor therapies to treat or delay endocrineresistance in breast cancer. Endocr Relat Cancer 12: S29–S36.
Nicholson RI, Hutcheson IR, Jones HE, Hiscox SE, Giles M, TaylorKM et al. (2007). Growth factor signalling in endocrine and anti-growth factor resistant breast cancer. Rev Endocr Metabol Disord 8:241–253.
O’Neill PA, Davies MPA, Shaaban AM, Innes H, Torevell A, SibsonDR et al. (2004). Wild-type oestrogen receptor beta mRNA andprotein expression in tamoxifen-treated post-menopausal breastcancers. Br J Cancer 91: 1694–1702.
Osborne CK. (1998). Tamoxifen in the treatment of breast cancer. N
Engl J Med 339: 1609–1618.Pandey V, Perry JK, Mohankumar KM, Kong XJ, Liu SM, Wu ZS
et al. (2008). Autocrine human growth hormone stimulatesoncogenicity of endometrial carcinoma cells. Endocrinology 149:3909–3919.
Pandey V, Qian PX, Kang J, Perry JK, Mitchell MD, Yin ZNet al. (2010). Artemin stimulates oncogenicity and invasivenessof endometrial carcinoma cells. Endocrinology 151: 909–920.
Park BW, Kim KS, Heo MK, Ko SS, Hong SW, Yang WI et al.(2003). Expression of estrogen receptor-beta in normal mammaryand tumor tissues: is it protective in breast carcinogenesis? Breast
Cancer Res Treat 80: 79–85.Perillo B, Sasso A, Abbondanza C, Palumbo G. (2000). 17B-estradiol
inhibits apoptosis in MCF-7 cells, inducing bcl-2 expression via twoestrogen-responsive elements present in the coding sequence. Mol
Cell Biol 20: 2890–2901.Pettersson K, Delaunay F, Gustafsson JA. (2000). Estrogen receptor
beta acts as a dominant regulator of estrogen signaling. Oncogene
19: 4970–4978.Riggins RB, Schrecengost RS, Guerrero MS, Bouton AH. (2007).
Pathways to tamoxifen resistance. Cancer Lett 256: 1–24.Riggins RB, Zwart A, Nehra R, Clarke R. (2005). The nuclear factor
kappa B inhibitor parthenolide restores ICI 182,780 (Faslodex;fulvestrant)-induced apoptosis in antiestrogen-resistant breast can-cer cells. Mol Cancer Ther 4: 33–41.
Rody A, Holtrich U, Solbach C, Kourtis K, Von Minckwitz G, EngelsK et al. (2005). Methylation of estrogen receptor beta promotercorrelates with loss of ER-beta expression in mammary carcinomaand is an early indication marker in premalignant lesions. Endocr
Relat Cancer 12: 903–916.Rotolo S, Ceccarelli S, Romano F, Frati L, Marchese C, Angeloni A.
(2008). Silencing of keratinocyte growth factor receptor restores 5-fluorouracil and tamoxifen efficacy on responsive cancer cells. PLoS
ONE 3: e2528.
Sarkaria JN, Gibson DFC, Jordan VC, Fowler JF, Lindstrom MJ,Mulcahy RT. (1993). Tamoxifen-induced increase in thepotential doubling time of MCF-7 xenografts as determined bybromodeoxyuridine labeling and flow cytometry. Cancer Res 53:4413–4417.
Shaaban AM, O’Neill PA, Davies MPA, Sibson R, West CR, SmithPH et al. (2003). Declining estrogen receptor-beta expression definesmalignant progression of human breast neoplasia. Am J Surg Pathol
27: 1502–1512.Shaw LE, Sadler AJ, Pugazhendhi D, Darbre PD. (2006). Changes in
oestrogen receptor-alpha and -beta during progression to acquiredresistance to tamoxifen and fulvestrant (Faslodex, ICI 182,780) inMCF7 human breast cancer cells. J Steroid Biochem Mol Biol 99: 19–32.
Siddiqa A, Long LM, Li L, Marciniak RA, Kazhdan I. (2008).Expression of HER-2 in MCF-7 breast cancer cells modulates anti-apoptotic proteins Survivin and Bcl-2 via the extracellular signal-related kinase (ERK) and phosphoinositide-3 kinase (PI3K)signalling pathways. BMC Cancer 8: 129.
Speirs V, Malone C, Walton DS, Kerin MJ, Atkin SL. (1999).Increased expression of estrogen receptor beta mRNA in tamoxifen-resistant breast cancer patients. Cancer Res 59: 5421–5424.
Tangkeangsirisin W, Hayashi J, Serrero G. (2004). PC cell-derivedgrowth factor mediates tamoxifen resistance and promotes tumorgrowth of human breast cancer cells. Cancer Res 64: 1737–1743.
Teixeira C, Reed JC, Pratt MAC. (1995). Estrogen promoteschemotherapeutic drug resistance by a mechanism involving Bcl-2proto-oncogene expression in human breast cancer cells. Cancer Res
55: 3902–3907.Williams C, Edvardsson K, Lewandowski SA, Stram A, Gustafsson
JA. (2008). A genome-wide study of the repressive effects ofestrogen receptor beta on estrogen receptor alpha signaling in breastcancer cells. Oncogene 27: 1019–1032.
Yager JD, Davidson NE. (2006). Estrogen carcinogenesis in breastcancer. N Engl J Med 354: 270–282.
Zhang GJ, Kimijima I, Onda M, Kanno M, Sato H, Watanabe T et al.(1999). Tamoxifen-induced apoptosis in breast cancer cells relates todown-regulation of bcl-2, but not bax and bcl-X(L), withoutalteration of p53 protein levels. Clin Cancer Res 5: 2971–2977.
Zhang X, Zhu T, Chen Y, Mertani HC, Lee KO, Lobie PE. (2003).Human growth hormone-regulated HOXA1 is a human mammaryepithelial oncogene. J Biol Chem 278: 7580–7590.
Zhu T, Emerald BS, Zhang X, Lee KO, Gluckman PD, Mertani HCet al. (2005). Oncogenic transformation of human mammaryepithelial cells by autocrine human growth hormone. Cancer Res
65: 317–324.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
Artemin mediates antiestrogen resistanceJ Kang et al
3240
Oncogene
Copyright © 2022 FDOKUMEN




















