
The Transcription Factor Aryl Hydrocarbon Receptor Nuclear Translocator Functions as an Estrogen...
13
The Transcription Factor Aryl Hydrocarbon Receptor Nuclear Translocator Functions as an Estrogen Receptor -Selective Coactivator, and Its Recruitment to Alternative Pathways Mediates Antiestrogenic Effects of Dioxin Joe ¨ lle Ru ¨ egg, Elin Swedenborg, David Wahlstro ¨ m, Aurelie Escande, Patrick Balaguer, Katarina Pettersson, and Ingemar Pongratz Karolinska Institute (J.R., E.S., D.W., K.P., I.P.), Department of Biosciences and Nutrition, S-141 57 Huddinge, Sweden; Karolinska Institute (D.W.), Institute of Environmental Medicine, S-171 77 Solna, Sweden; and Institut National de la Sante ´ et de la Recherche Me ´ dicale Unite ´ 824 (A.E., P.B.) Montpellier F-34298, France The biological effects of dioxins are mediated by the aryl hydrocarbon receptor (AhR) and its dimer- ization partner, the AhR nuclear translocator (ARNT), and include interference with hormonal signaling pathways like the response to estrogens. The effects of estrogens are mediated by two es- trogen receptor (ER) isoforms, ER and ER, which belong to the family of nuclear receptors. We have previously shown that ARNT can act as co- activator of the ERs. In this study, we show that recruitment of ARNT to AhR or hypoxia-inducible factor-1 signaling pathways as well as small in- terfering RNA-mediated down-regulation of ARNT levels lead to a reduction in ER transcriptional ac- tivity. Using chromatin immunoprecipitation as- says, we demonstrate that this decrease coincides with reduced recruitment of ARNT to estradiol- regulated promoters. We show further that coac- tivation by ARNT as well as inhibition by dioxin acts stronger on ER than on ER activity. Additionally, we demonstrate that the effects of ARNT are de- pendent on the A/B domain of the ERs with the A/B domain of ER being considerably stronger in medi- ating the coactivating effects of ARNT. Taken to- gether, our studies show that recruitment of ARNT to the AhR after dioxin treatment can account for the antiestrogenic effect of dioxins. Moreover, we show for the first time that the inhibitory effects of dioxin are more pronounced on ER than on ER.(Molec- ular Endocrinology 22: 304–316, 2008) 1 7-ESTRADIOL (E 2 ) is a steroid hormone and reg- ulates several key biological processes such as bone development, reproduction, and brain develop- ment (1). The cellular response to estrogenic com- pounds is mediated by the two estrogen receptor (ER) isoforms, ER (NR3A1) and ER (NR3A2). These re- ceptors belong to the nuclear receptor (NR) superfam- ily, which includes, e.g. the receptors for glucocorti- coids, progestins, and thyroid hormone. The members of the NR family share a conserved structural arrange- ment with a centrally located DNA-binding domain flanked by the N-terminal A/B domain that includes the transcriptional activation function AF-1, and the C- terminal ligand-binding domain (LBD), harboring both the ligand-binding pocket and a second transcrip- tional function known as the AF-2 domain (2). Upon binding of agonists, ER and ER undergo a complex activation process to acquire full tran- scriptional activity. Ligand binding induces a con- formational change, which in turn promotes release of inhibitory repressive factors such as silencing mediator of retinoid and thyroid receptors or nuclear receptor corepressor. Subsequently, positive regu- latory factors are recruited to the receptor, and this complex binds to specific DNA elements located in the regulatory regions of target genes (3). Extensive studies have identified numerous proteins that can interact with and coactivate the transcriptional ac- tivity of the NRs. These factors include the classical coactivators of the p160 family, like steroid receptor coactivator and transcriptional intermediary factor 2, co- modulators such as p300/cAMP response element bind- ing protein-binding protein (CBP)-associated factor, and First Published Online November 8, 2007 Abbreviations: AF, Activation function; AhR, aryl hydrocar- bon receptor; ARNT, AhR nuclear translocator; bHLH basic helix-loop-helix; ChIP, chromatin immunoprecipitation; CBP, cAMP response element binding protein-binding protein; DCC, dextran-coated charcoal; DPN, diarylpropionitrile; E 2 , 17-estradiol; ER, estrogen receptor; ERE, estrogen re- sponse element; FCS, fetal calf serum; HIF-1, hypoxia- inducible factor-1; hsp90, heat-shock protein 90; LBD, li- gand binding domain; 3-MC, 3-methylcholantrene; NR, nuclear receptor; 4-OHT, 4-hydroxy-tamoxifen; PAS, Per- ARNT-Sim; PPT, propyl pyrazole triol; siRNA, small interfer- ing RNA; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin. Molecular Endocrinology is published monthly by The Endocrine Society (http://www.endo-society.org), the foremost professional society serving the endocrine community. 0888-8809/08/$15.00/0 Molecular Endocrinology 22(2):304–316 Printed in U.S.A. Copyright © 2008 by The Endocrine Society doi: 10.1210/me.2007-0128 304
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of The Transcription Factor Aryl Hydrocarbon Receptor Nuclear Translocator Functions as an Estrogen...

The Transcription Factor Aryl HydrocarbonReceptor Nuclear Translocator Functions as anEstrogen Receptor �-Selective Coactivator, and ItsRecruitment to Alternative Pathways MediatesAntiestrogenic Effects of Dioxin
Joelle Ruegg, Elin Swedenborg, David Wahlstrom, Aurelie Escande, Patrick Balaguer,Katarina Pettersson, and Ingemar Pongratz
Karolinska Institute (J.R., E.S., D.W., K.P., I.P.), Department of Biosciences and Nutrition, S-141 57Huddinge, Sweden; Karolinska Institute (D.W.), Institute of Environmental Medicine, S-171 77 Solna,Sweden; and Institut National de la Sante et de la Recherche Medicale Unite 824 (A.E., P.B.)Montpellier F-34298, France
The biological effects of dioxins are mediated bythe aryl hydrocarbon receptor (AhR) and its dimer-ization partner, the AhR nuclear translocator(ARNT), and include interference with hormonalsignaling pathways like the response to estrogens.The effects of estrogens are mediated by two es-trogen receptor (ER) isoforms, ER� and ER�,which belong to the family of nuclear receptors. Wehave previously shown that ARNT can act as co-activator of the ERs. In this study, we show thatrecruitment of ARNT to AhR or hypoxia-induciblefactor-1� signaling pathways as well as small in-terfering RNA-mediated down-regulation of ARNTlevels lead to a reduction in ER transcriptional ac-tivity. Using chromatin immunoprecipitation as-
says, we demonstrate that this decrease coincideswith reduced recruitment of ARNT to estradiol-regulated promoters. We show further that coac-tivation by ARNT as well as inhibition by dioxin actsstronger on ER� than on ER� activity. Additionally,we demonstrate that the effects of ARNT are de-pendent on the A/B domain of the ERs with the A/Bdomain of ER� being considerably stronger in medi-ating the coactivating effects of ARNT. Taken to-gether, our studies show that recruitment of ARNT tothe AhR after dioxin treatment can account for theantiestrogenic effect of dioxins. Moreover, we showfor the first time that the inhibitory effects of dioxinare more pronounced on ER� than on ER�. (Molec-ular Endocrinology 22: 304–316, 2008)
17�-ESTRADIOL (E2) is a steroid hormone and reg-ulates several key biological processes such as
bone development, reproduction, and brain develop-ment (1). The cellular response to estrogenic com-pounds is mediated by the two estrogen receptor (ER)isoforms, ER� (NR3A1) and ER� (NR3A2). These re-ceptors belong to the nuclear receptor (NR) superfam-ily, which includes, e.g. the receptors for glucocorti-coids, progestins, and thyroid hormone. The members
of the NR family share a conserved structural arrange-ment with a centrally located DNA-binding domainflanked by the N-terminal A/B domain that includes thetranscriptional activation function AF-1, and the C-terminal ligand-binding domain (LBD), harboring boththe ligand-binding pocket and a second transcrip-tional function known as the AF-2 domain (2).
Upon binding of agonists, ER� and ER� undergoa complex activation process to acquire full tran-scriptional activity. Ligand binding induces a con-formational change, which in turn promotes releaseof inhibitory repressive factors such as silencingmediator of retinoid and thyroid receptors or nuclearreceptor corepressor. Subsequently, positive regu-latory factors are recruited to the receptor, and thiscomplex binds to specific DNA elements located inthe regulatory regions of target genes (3). Extensivestudies have identified numerous proteins that caninteract with and coactivate the transcriptional ac-tivity of the NRs. These factors include the classicalcoactivators of the p160 family, like steroid receptorcoactivator and transcriptional intermediary factor 2, co-modulators such as p300/cAMP response element bind-ing protein-binding protein (CBP)-associated factor, and
First Published Online November 8, 2007Abbreviations: AF, Activation function; AhR, aryl hydrocar-
bon receptor; ARNT, AhR nuclear translocator; bHLH basichelix-loop-helix; ChIP, chromatin immunoprecipitation; CBP,cAMP response element binding protein-binding protein;DCC, dextran-coated charcoal; DPN, diarylpropionitrile; E2,17�-estradiol; ER, estrogen receptor; ERE, estrogen re-sponse element; FCS, fetal calf serum; HIF-1�, hypoxia-inducible factor-1�; hsp90, heat-shock protein 90; LBD, li-gand binding domain; 3-MC, 3-methylcholantrene; NR,nuclear receptor; 4-OHT, 4-hydroxy-tamoxifen; PAS, Per-ARNT-Sim; PPT, propyl pyrazole triol; siRNA, small interfer-ing RNA; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin.
Molecular Endocrinology is published monthly by TheEndocrine Society (http://www.endo-society.org), theforemost professional society serving the endocrinecommunity.
0888-8809/08/$15.00/0 Molecular Endocrinology 22(2):304–316Printed in U.S.A. Copyright © 2008 by The Endocrine Society
doi: 10.1210/me.2007-0128
304

factors with histone acetyl transferase activity like CBPand p300 (4).
In addition to their endogenous ligands, the activityof ER� and ER� can be modulated by a wide array ofexogenous compounds, such as dietary derived sub-stances like isoflavonoids and chemical pollutants likepolyaromatic hydrocarbons such as dioxins (3). Mod-ulation of receptor activity by chemical contaminantsis commonly referred to as endocrine disruption. Thenegative effect of dioxins and related compounds onestrogen signaling is a well-studied phenomenon atthe organism level (5–7). However, the molecularmechanisms behind the antiestrogenic effect of diox-ins are poorly understood but have been suggested toinvolve increased metabolism of E2, increased ERturnover, and promoter interference (8).
Dioxin (also known as TCDD, or 2,3,7,8-tetrachlo-rodibenzo-p-dioxin) is an environmental pollutantformed through incomplete combustion of waste ma-terial or as a side product in certain industrial pro-cesses. The biological responses to dioxin are medi-ated by the aryl hydrocarbon receptor (AhR) (5). TheAhR is a member of the basic helix-loop-helix (bHLH)-Per-ARNT-Sim (PAS) family of proteins that includestranscription factors like the hypoxia-inducible factorshypoxia-inducible factor-1� (HIF-1�) and endothelialPAS domain protein-1 or the circadian regulatory pro-teins Clock and Per. To become transcriptionally ac-tive, bHLH-PAS proteins form heterodimers with theirrespective partner proteins. These partner factors aremembers of the bHLH-PAS subfamily AhR nucleartranslocator (ARNT) (6). Three different ARNT factorshave been identified, ARNT-1, ARNT-2, and ARNT-3,the last also known as bMAL. ARNT-1 and -2 displaya high degree of sequence similarity (9, 10) and func-tion as general dimerization partners for the AhR aswell as the hypoxia-inducible factors HIF-1� and en-dothelial PAS domain protein-1. However, ARNT-1has been described to be more important for the signaltransduction of AhR (11). ARNT-3 is selectively re-cruited by the circadian regulator Clock and does notsupport HIF-1� or AhR function (12, 13).
We have previously shown that ARNT-1 andARNT-2 can interact with and coactivate ER� and ER�transcriptional activities, with the effects being morepotent on the ER� subtype (14). ARNT-3 had no effecton ER transcriptional activity. The C-terminal domainof ARNT was required for its coactivating capacity onthe ERs. On the other hand, the key domains of ARNTin terms of AhR and HIF-1� function, the bHLH andPAS domains, were not needed to support E2 signal-ing, suggesting that the mechanism by which ARNTacts to stimulate ER function is distinct from the doc-umented mechanism (15) of AhR or HIF-1� activation.Furthermore, both the LBD and the A/B domains ofER� and ER� were necessary for functional interactionwith ARNT, suggesting a role of ARNT in AF-1 andAF-2 synergism (14).
The identification of ARNT as a coactivator of theERs (14) is interesting in several regards. It suggests a
novel mechanism for how the antiestrogenic effects ofdioxins are mediated, whereby the AhR and ER com-pete for its common partner ARNT. It can also impli-cate that activation of other signaling pathways usingARNT as cofactor, e.g. activation of HIF-1�, can de-crease ER activity. Furthermore, the preference ofARNT for ER� could indicate a more potent antiestro-genic effect of dioxins on this receptor subtype, whichcould have interesting implications when evaluatingdioxins as endocrine disruptors. Thus, detailed analy-sis of the interplay between ARNT and the ERs isessential for understanding the effects of dioxin on theER system.
In this study, we set out to investigate mechanisticdetails of the coactivating function of ARNT on the ERsand test our hypothesis that competition for ARNTbetween AhR and ER can account at least partly forthe antiestrogenic effect of dioxin. Because ARNT-1has been reported to be more important for AhR sig-naling than ARNT-2 (11), we focused our study on thecross-talk between the ERs and ARNT-1. We reporthere that reducing the available pool of ARNT by eitheractivating the AhR or HIF-1� signaling pathway or bydecreasing ARNT levels using small interfering RNA(siRNA), inhibits ER function, with a more drastic effecton ER�. We show further that ARNT coactivation and,importantly, TCDD inhibition is more pronounced onER� than on ER� and is dependent on the A/B do-mains of the ERs.
RESULTS
Activation of Alternative ARNT-DependentPathways Impairs ER Transcriptional Activity
The mechanism(s) behind the inhibitory cross-talk be-tween the AhR and the ERs are not fully understood, inparticular at the molecular level. We have previouslydemonstrated that the AhR partner protein ARNTfunctions as an ER coactivator (14). It is thereforepossible that sequestering of ARNT to alternative sig-naling pathways, e.g. the AhR pathway, would inter-fere with ER transcriptional activity and therefore ex-plain the inhibitory effect of dioxin on ER signaling.
To test this hypothesis, we coexposed cells to E2
and either dioxin or a hypoxic environment and stud-ied the effects on ER signaling. Hypoxia induces theformation of the HIF-1�/ARNT complex that regulatesexpression of genes involved in the adaptive responseto low oxygen tension in the cell (16). Recruitment ofARNT to the hypoxia signaling network is thereforealso expected to reduce the intracellular levels ofARNT available for the ERs. To activate AhR, we de-cided to use TCDD because this compound is highlyresistant to cellular metabolism, in contrast to otherAhR ligands such as 3-methylcholantrene (3-MC).HC11 cells stably transfected with a 3xERE-TATA-luciferase construct (H-ERE cells) were exposed to 10nM TCDD and/or hypoxia (1% O2) alone or in combi-
Ruegg et al. • ER� Selectivity of ARNT and TCDD Mol Endocrinol, February 2008, 22(2):304–316 305
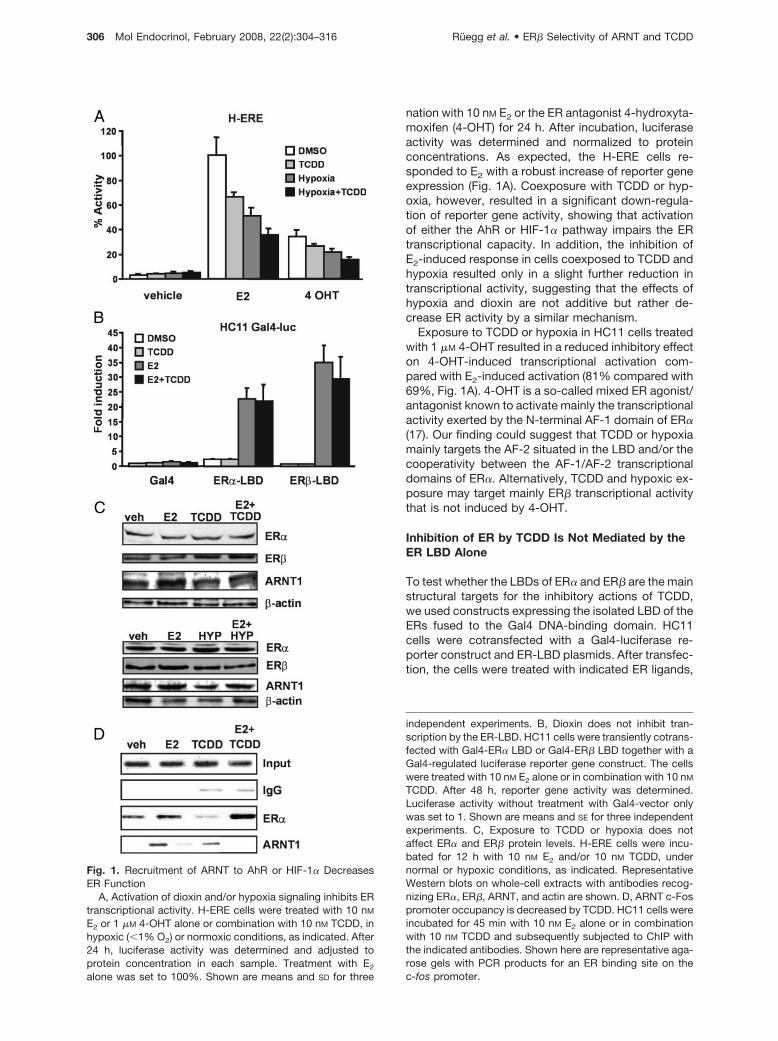
nation with 10 nM E2 or the ER antagonist 4-hydroxyta-moxifen (4-OHT) for 24 h. After incubation, luciferaseactivity was determined and normalized to proteinconcentrations. As expected, the H-ERE cells re-sponded to E2 with a robust increase of reporter geneexpression (Fig. 1A). Coexposure with TCDD or hyp-oxia, however, resulted in a significant down-regula-tion of reporter gene activity, showing that activationof either the AhR or HIF-1� pathway impairs the ERtranscriptional capacity. In addition, the inhibition ofE2-induced response in cells coexposed to TCDD andhypoxia resulted only in a slight further reduction intranscriptional activity, suggesting that the effects ofhypoxia and dioxin are not additive but rather de-crease ER activity by a similar mechanism.
Exposure to TCDD or hypoxia in HC11 cells treatedwith 1 �M 4-OHT resulted in a reduced inhibitory effecton 4-OHT-induced transcriptional activation com-pared with E2-induced activation (81% compared with69%, Fig. 1A). 4-OHT is a so-called mixed ER agonist/antagonist known to activate mainly the transcriptionalactivity exerted by the N-terminal AF-1 domain of ER�(17). Our finding could suggest that TCDD or hypoxiamainly targets the AF-2 situated in the LBD and/or thecooperativity between the AF-1/AF-2 transcriptionaldomains of ER�. Alternatively, TCDD and hypoxic ex-posure may target mainly ER� transcriptional activitythat is not induced by 4-OHT.
Inhibition of ER by TCDD Is Not Mediated by theER LBD Alone
To test whether the LBDs of ER� and ER� are the mainstructural targets for the inhibitory actions of TCDD,we used constructs expressing the isolated LBD of theERs fused to the Gal4 DNA-binding domain. HC11cells were cotransfected with a Gal4-luciferase re-porter construct and ER-LBD plasmids. After transfec-tion, the cells were treated with indicated ER ligands,
Fig. 1. Recruitment of ARNT to AhR or HIF-1� DecreasesER Function
A, Activation of dioxin and/or hypoxia signaling inhibits ERtranscriptional activity. H-ERE cells were treated with 10 nM
E2 or 1 �M 4-OHT alone or combination with 10 nM TCDD, inhypoxic (�1% O2) or normoxic conditions, as indicated. After24 h, luciferase activity was determined and adjusted toprotein concentration in each sample. Treatment with E2
alone was set to 100%. Shown are means and SD for three
independent experiments. B, Dioxin does not inhibit tran-scription by the ER-LBD. HC11 cells were transiently cotrans-fected with Gal4-ER� LBD or Gal4-ER� LBD together with aGal4-regulated luciferase reporter gene construct. The cellswere treated with 10 nM E2 alone or in combination with 10 nM
TCDD. After 48 h, reporter gene activity was determined.Luciferase activity without treatment with Gal4-vector onlywas set to 1. Shown are means and SE for three independentexperiments. C, Exposure to TCDD or hypoxia does notaffect ER� and ER� protein levels. H-ERE cells were incu-bated for 12 h with 10 nM E2 and/or 10 nM TCDD, undernormal or hypoxic conditions, as indicated. RepresentativeWestern blots on whole-cell extracts with antibodies recog-nizing ER�, ER�, ARNT, and actin are shown. D, ARNT c-Fospromoter occupancy is decreased by TCDD. HC11 cells wereincubated for 45 min with 10 nM E2 alone or in combinationwith 10 nM TCDD and subsequently subjected to ChIP withthe indicated antibodies. Shown here are representative aga-rose gels with PCR products for an ER binding site on thec-fos promoter.
306 Mol Endocrinol, February 2008, 22(2):304–316 Ruegg et al. • ER� Selectivity of ARNT and TCDD
alone or in combination with TCDD, for 48 h (Fig. 1B).Interestingly, we note that the negative effects byTCDD on E2-induced activity previously observed inH-ERE cells did not occur using the Gal4 fusion ofeither ER� or ER�. These results therefore suggestthat TCDD does not target the individual transcrip-tional activation domains of the ER isoforms.
Exposure to TCDD and Hypoxia Has No Effect onER Protein Levels in HC11 Cells
Previous studies have suggested that exposure to E2
and TCDD can lead to increased degradation of theER� protein (18, 19). To investigate whether alterationsin protein levels could account for the observed effectsof TCDD and hypoxia, we performed Western blotanalysis of extracts from cells treated with E2, TCDD,or hypoxia, alone or in combination (Fig. 1C). Com-pared with �-actin loading control, we observed no orminimal effects on the protein levels of ER� and ARNTby the different treatments, and the ER� levels wereeven higher after hypoxic treatment. These resultswere confirmed by density measurement of the bandsand normalization to �-actin levels (data not shown).The findings suggest that the decrease in ER-depen-dent transcriptional activity in H-ERE cells cannot beattributed to protein degradation. Somewhat surpris-ingly, ER� levels were not down-regulated by E2 treat-ment. ER� down-regulation after hormone treatmentis a well-documented phenomenon that we observe inother cell lines (data not shown) but not in HC11 cells.In conclusion, these data show that activation of bothAhR and HIF-1� pathways leads to reduced ER tran-scriptional activity, possibly by sequestering of ARNT.
Recruitment of ARNT to E2-Dependent PromotersIs Decreased upon TCDD Exposure
We have previously shown that ARNT is corecruitedtogether with ER on the pS2 promoter in an E2-depen-dent fashion (14). To investigate whether TCDD treat-ment results in a reduced ARNT recruitment to anE2-regulated promoter, we performed chromatin im-munoprecipitation (ChIP) experiments on the c-Fospromoter in HC11 cells. The pS2 gene is not inducedby E2 in these cells, therefore the c-fos promoter waschosen. The cells were treated with E2 alone or incombination with 10 nM TCDD for 45 min. After thistreatment, the cells were cross-linked and harvested,and ChIP assays were performed as described in Ma-terials and Methods. In control experiments, ER� wasrecruited to the c-Fos promoter after treatment with E2
(Fig. 1D). No association of ER� with the c-Fos pro-moter was observed in the presence of TCDD alone.ARNT was also found on the c-Fos promoter in thepresence of E2 but not in the presence of TCDD alone.Interestingly, the levels of ARNT recruited to the c-Fospromoter decreased when the cells were simulta-neously treated with E2 and TCDD. These resultsstrongly suggest that dioxin-activated AhR can se-
quester ARNT away from E2-regulated promoters,thereby reducing ER transcriptional activity.
Reduction of Intracellular Amounts of ARNTInhibits the Transcriptional Activity of the ERs
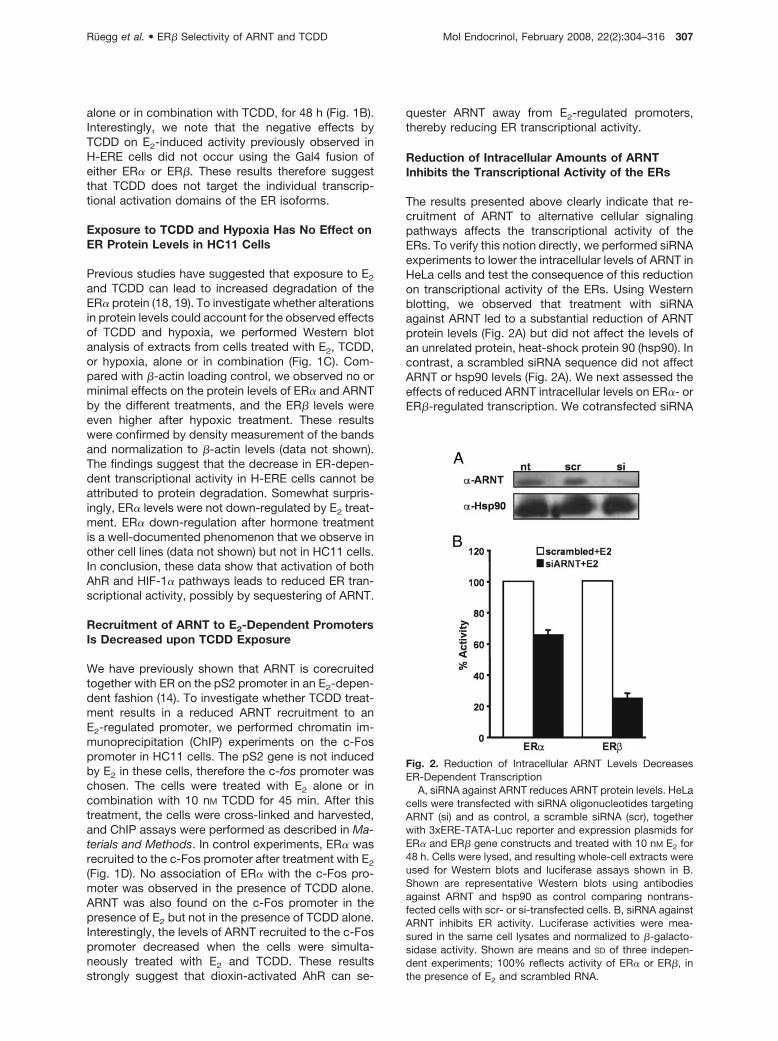
The results presented above clearly indicate that re-cruitment of ARNT to alternative cellular signalingpathways affects the transcriptional activity of theERs. To verify this notion directly, we performed siRNAexperiments to lower the intracellular levels of ARNT inHeLa cells and test the consequence of this reductionon transcriptional activity of the ERs. Using Westernblotting, we observed that treatment with siRNAagainst ARNT led to a substantial reduction of ARNTprotein levels (Fig. 2A) but did not affect the levels ofan unrelated protein, heat-shock protein 90 (hsp90). Incontrast, a scrambled siRNA sequence did not affectARNT or hsp90 levels (Fig. 2A). We next assessed theeffects of reduced ARNT intracellular levels on ER�- orER�-regulated transcription. We cotransfected siRNA
Fig. 2. Reduction of Intracellular ARNT Levels DecreasesER-Dependent Transcription
A, siRNA against ARNT reduces ARNT protein levels. HeLacells were transfected with siRNA oligonucleotides targetingARNT (si) and as control, a scramble siRNA (scr), togetherwith 3xERE-TATA-Luc reporter and expression plasmids forER� and ER� gene constructs and treated with 10 nM E2 for48 h. Cells were lysed, and resulting whole-cell extracts wereused for Western blots and luciferase assays shown in B.Shown are representative Western blots using antibodiesagainst ARNT and hsp90 as control comparing nontrans-fected cells with scr- or si-transfected cells. B, siRNA againstARNT inhibits ER activity. Luciferase activities were mea-sured in the same cell lysates and normalized to �-galacto-sidase activity. Shown are means and SD of three indepen-dent experiments; 100% reflects activity of ER� or ER�, inthe presence of E2 and scrambled RNA.
Ruegg et al. • ER� Selectivity of ARNT and TCDD Mol Endocrinol, February 2008, 22(2):304–316 307
against ARNT or the scrambled sequence togetherwith the 3xERE-TATA-luciferase reporter constructand expression plasmids for either ER� or ER�. Thecells were subsequently incubated with E2 for 24 h,and the transcriptional response was assessed. Asshown in Fig. 2B, siRNA reduction of the intracellularlevels of ARNT resulted in a clear reduction in ER� andER� transcriptional activity. Interestingly, the ER� iso-form was considerably more affected by low ARNTlevels compared with ER�. In fact, whereas the tran-scriptional activity of ER� was reduced by 40% (Fig.2B), the effect of reduced intracellular ARNT levels onER� was substantial with an almost 80% drop in tran-scriptional activity (Fig. 2B).
This experiment demonstrates that reduced avail-ability of ARNT in a cell impairs ER function. Interest-ingly, we observe an ER isoform-specific sensitivity toreduced ARNT levels, which suggests that ARNT ismore critical as a coactivator for ER� than for ER�.
ARNT Coactivates ER�-ER� Heterodimers to aSimilar Extent As ER�
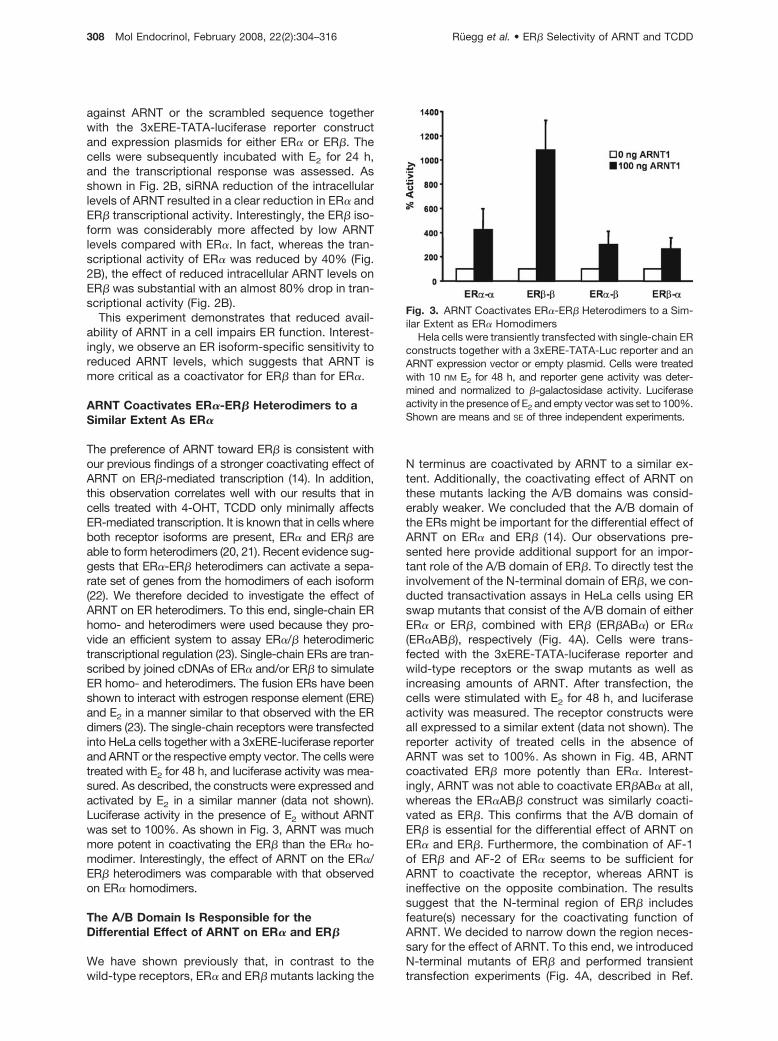
The preference of ARNT toward ER� is consistent withour previous findings of a stronger coactivating effect ofARNT on ER�-mediated transcription (14). In addition,this observation correlates well with our results that incells treated with 4-OHT, TCDD only minimally affectsER-mediated transcription. It is known that in cells whereboth receptor isoforms are present, ER� and ER� areable to form heterodimers (20, 21). Recent evidence sug-gests that ER�-ER� heterodimers can activate a sepa-rate set of genes from the homodimers of each isoform(22). We therefore decided to investigate the effect ofARNT on ER heterodimers. To this end, single-chain ERhomo- and heterodimers were used because they pro-vide an efficient system to assay ER�/� heterodimerictranscriptional regulation (23). Single-chain ERs are tran-scribed by joined cDNAs of ER� and/or ER� to simulateER homo- and heterodimers. The fusion ERs have beenshown to interact with estrogen response element (ERE)and E2 in a manner similar to that observed with the ERdimers (23). The single-chain receptors were transfectedinto HeLa cells together with a 3xERE-luciferase reporterand ARNT or the respective empty vector. The cells weretreated with E2 for 48 h, and luciferase activity was mea-sured. As described, the constructs were expressed andactivated by E2 in a similar manner (data not shown).Luciferase activity in the presence of E2 without ARNTwas set to 100%. As shown in Fig. 3, ARNT was muchmore potent in coactivating the ER� than the ER� ho-modimer. Interestingly, the effect of ARNT on the ER�/ER� heterodimers was comparable with that observedon ER� homodimers.
The A/B Domain Is Responsible for theDifferential Effect of ARNT on ER� and ER�
We have shown previously that, in contrast to thewild-type receptors, ER� and ER� mutants lacking the
N terminus are coactivated by ARNT to a similar ex-tent. Additionally, the coactivating effect of ARNT onthese mutants lacking the A/B domains was consid-erably weaker. We concluded that the A/B domain ofthe ERs might be important for the differential effect ofARNT on ER� and ER� (14). Our observations pre-sented here provide additional support for an impor-tant role of the A/B domain of ER�. To directly test theinvolvement of the N-terminal domain of ER�, we con-ducted transactivation assays in HeLa cells using ERswap mutants that consist of the A/B domain of eitherER� or ER�, combined with ER� (ER�AB�) or ER�(ER�AB�), respectively (Fig. 4A). Cells were trans-fected with the 3xERE-TATA-luciferase reporter andwild-type receptors or the swap mutants as well asincreasing amounts of ARNT. After transfection, thecells were stimulated with E2 for 48 h, and luciferaseactivity was measured. The receptor constructs wereall expressed to a similar extent (data not shown). Thereporter activity of treated cells in the absence ofARNT was set to 100%. As shown in Fig. 4B, ARNTcoactivated ER� more potently than ER�. Interest-ingly, ARNT was not able to coactivate ER�AB� at all,whereas the ER�AB� construct was similarly coacti-vated as ER�. This confirms that the A/B domain ofER� is essential for the differential effect of ARNT onER� and ER�. Furthermore, the combination of AF-1of ER� and AF-2 of ER� seems to be sufficient forARNT to coactivate the receptor, whereas ARNT isineffective on the opposite combination. The resultssuggest that the N-terminal region of ER� includesfeature(s) necessary for the coactivating function ofARNT. We decided to narrow down the region neces-sary for the effect of ARNT. To this end, we introducedN-terminal mutants of ER� and performed transienttransfection experiments (Fig. 4A, described in Ref.
Fig. 3. ARNT Coactivates ER�-ER� Heterodimers to a Sim-ilar Extent as ER� Homodimers
Hela cells were transiently transfected with single-chain ERconstructs together with a 3xERE-TATA-Luc reporter and anARNT expression vector or empty plasmid. Cells were treatedwith 10 nM E2 for 48 h, and reporter gene activity was deter-mined and normalized to �-galactosidase activity. Luciferaseactivity in the presence of E2 and empty vector was set to 100%.Shown are means and SE of three independent experiments.
308 Mol Endocrinol, February 2008, 22(2):304–316 Ruegg et al. • ER� Selectivity of ARNT and TCDD
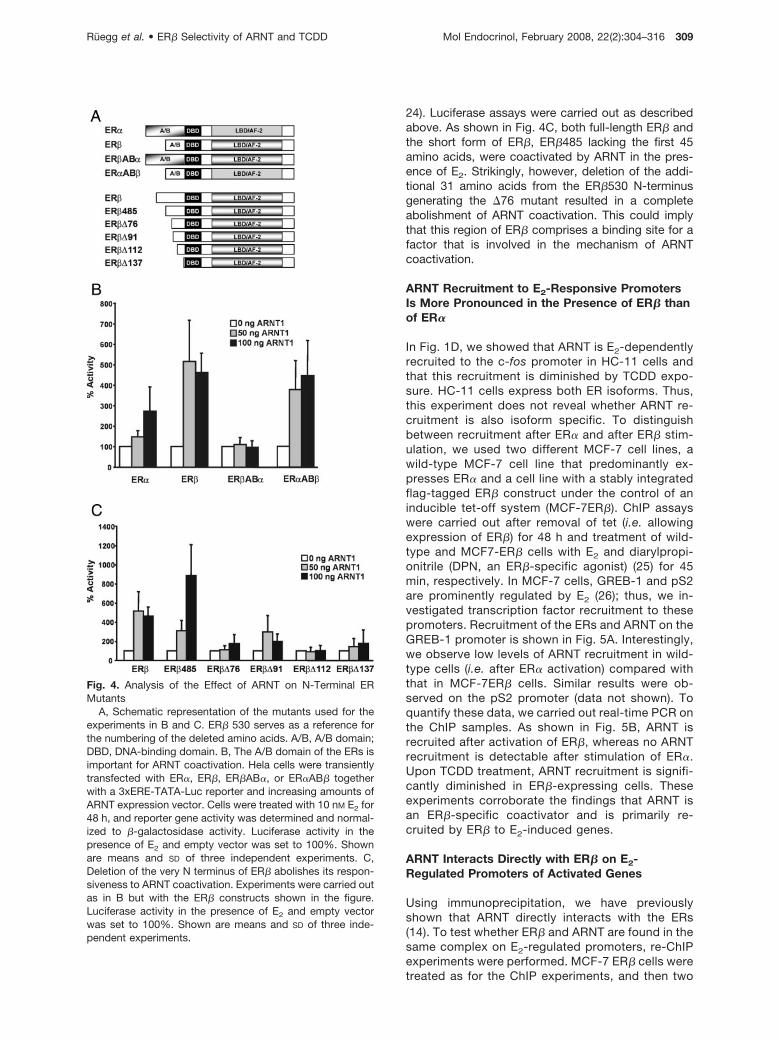
24). Luciferase assays were carried out as describedabove. As shown in Fig. 4C, both full-length ER� andthe short form of ER�, ER�485 lacking the first 45amino acids, were coactivated by ARNT in the pres-ence of E2. Strikingly, however, deletion of the addi-tional 31 amino acids from the ER�530 N-terminusgenerating the �76 mutant resulted in a completeabolishment of ARNT coactivation. This could implythat this region of ER� comprises a binding site for afactor that is involved in the mechanism of ARNTcoactivation.
ARNT Recruitment to E2-Responsive PromotersIs More Pronounced in the Presence of ER� thanof ER�
In Fig. 1D, we showed that ARNT is E2-dependentlyrecruited to the c-fos promoter in HC-11 cells andthat this recruitment is diminished by TCDD expo-sure. HC-11 cells express both ER isoforms. Thus,this experiment does not reveal whether ARNT re-cruitment is also isoform specific. To distinguishbetween recruitment after ER� and after ER� stim-ulation, we used two different MCF-7 cell lines, awild-type MCF-7 cell line that predominantly ex-presses ER� and a cell line with a stably integratedflag-tagged ER� construct under the control of aninducible tet-off system (MCF-7ER�). ChIP assayswere carried out after removal of tet (i.e. allowingexpression of ER�) for 48 h and treatment of wild-type and MCF7-ER� cells with E2 and diarylpropi-onitrile (DPN, an ER�-specific agonist) (25) for 45min, respectively. In MCF-7 cells, GREB-1 and pS2are prominently regulated by E2 (26); thus, we in-vestigated transcription factor recruitment to thesepromoters. Recruitment of the ERs and ARNT on theGREB-1 promoter is shown in Fig. 5A. Interestingly,we observe low levels of ARNT recruitment in wild-type cells (i.e. after ER� activation) compared withthat in MCF-7ER� cells. Similar results were ob-served on the pS2 promoter (data not shown). Toquantify these data, we carried out real-time PCR onthe ChIP samples. As shown in Fig. 5B, ARNT isrecruited after activation of ER�, whereas no ARNTrecruitment is detectable after stimulation of ER�.Upon TCDD treatment, ARNT recruitment is signifi-cantly diminished in ER�-expressing cells. Theseexperiments corroborate the findings that ARNT isan ER�-specific coactivator and is primarily re-cruited by ER� to E2-induced genes.
ARNT Interacts Directly with ER� on E2-Regulated Promoters of Activated Genes
Using immunoprecipitation, we have previouslyshown that ARNT directly interacts with the ERs(14). To test whether ER� and ARNT are found in thesame complex on E2-regulated promoters, re-ChIPexperiments were performed. MCF-7 ER� cells weretreated as for the ChIP experiments, and then two
Fig. 4. Analysis of the Effect of ARNT on N-Terminal ERMutants
A, Schematic representation of the mutants used for theexperiments in B and C. ER� 530 serves as a reference forthe numbering of the deleted amino acids. A/B, A/B domain;DBD, DNA-binding domain. B, The A/B domain of the ERs isimportant for ARNT coactivation. Hela cells were transientlytransfected with ER�, ER�, ER�AB�, or ER�AB� togetherwith a 3xERE-TATA-Luc reporter and increasing amounts ofARNT expression vector. Cells were treated with 10 nM E2 for48 h, and reporter gene activity was determined and normal-ized to �-galactosidase activity. Luciferase activity in thepresence of E2 and empty vector was set to 100%. Shownare means and SD of three independent experiments. C,Deletion of the very N terminus of ER� abolishes its respon-siveness to ARNT coactivation. Experiments were carried outas in B but with the ER� constructs shown in the figure.Luciferase activity in the presence of E2 and empty vectorwas set to 100%. Shown are means and SD of three inde-pendent experiments.
Ruegg et al. • ER� Selectivity of ARNT and TCDD Mol Endocrinol, February 2008, 22(2):304–316 309
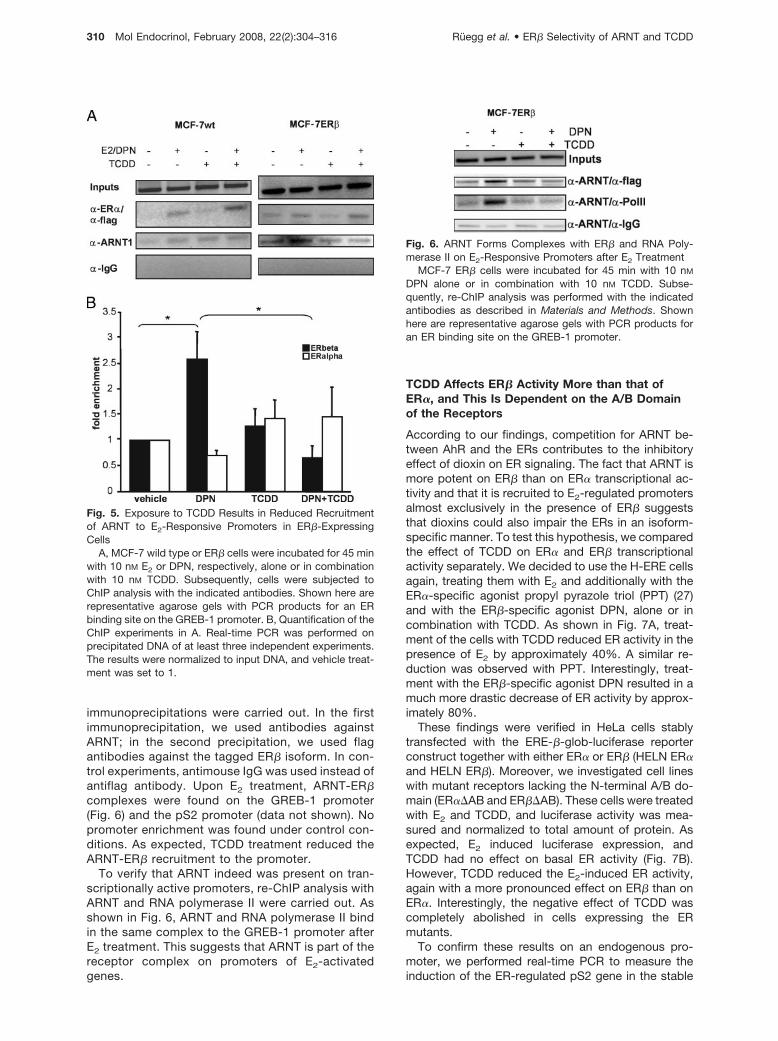
immunoprecipitations were carried out. In the firstimmunoprecipitation, we used antibodies againstARNT; in the second precipitation, we used flagantibodies against the tagged ER� isoform. In con-trol experiments, antimouse IgG was used instead ofantiflag antibody. Upon E2 treatment, ARNT-ER�complexes were found on the GREB-1 promoter(Fig. 6) and the pS2 promoter (data not shown). Nopromoter enrichment was found under control con-ditions. As expected, TCDD treatment reduced theARNT-ER� recruitment to the promoter.
To verify that ARNT indeed was present on tran-scriptionally active promoters, re-ChIP analysis withARNT and RNA polymerase II were carried out. Asshown in Fig. 6, ARNT and RNA polymerase II bindin the same complex to the GREB-1 promoter afterE2 treatment. This suggests that ARNT is part of thereceptor complex on promoters of E2-activatedgenes.
TCDD Affects ER� Activity More than that ofER�, and This Is Dependent on the A/B Domainof the Receptors
According to our findings, competition for ARNT be-tween AhR and the ERs contributes to the inhibitoryeffect of dioxin on ER signaling. The fact that ARNT ismore potent on ER� than on ER� transcriptional ac-tivity and that it is recruited to E2-regulated promotersalmost exclusively in the presence of ER� suggeststhat dioxins could also impair the ERs in an isoform-specific manner. To test this hypothesis, we comparedthe effect of TCDD on ER� and ER� transcriptionalactivity separately. We decided to use the H-ERE cellsagain, treating them with E2 and additionally with theER�-specific agonist propyl pyrazole triol (PPT) (27)and with the ER�-specific agonist DPN, alone or incombination with TCDD. As shown in Fig. 7A, treat-ment of the cells with TCDD reduced ER activity in thepresence of E2 by approximately 40%. A similar re-duction was observed with PPT. Interestingly, treat-ment with the ER�-specific agonist DPN resulted in amuch more drastic decrease of ER activity by approx-imately 80%.
These findings were verified in HeLa cells stablytransfected with the ERE-�-glob-luciferase reporterconstruct together with either ER� or ER� (HELN ER�and HELN ER�). Moreover, we investigated cell lineswith mutant receptors lacking the N-terminal A/B do-main (ER��AB and ER��AB). These cells were treatedwith E2 and TCDD, and luciferase activity was mea-sured and normalized to total amount of protein. Asexpected, E2 induced luciferase expression, andTCDD had no effect on basal ER activity (Fig. 7B).However, TCDD reduced the E2-induced ER activity,again with a more pronounced effect on ER� than onER�. Interestingly, the negative effect of TCDD wascompletely abolished in cells expressing the ERmutants.
To confirm these results on an endogenous pro-moter, we performed real-time PCR to measure theinduction of the ER-regulated pS2 gene in the stable
Fig. 5. Exposure to TCDD Results in Reduced Recruitmentof ARNT to E2-Responsive Promoters in ER�-ExpressingCells
A, MCF-7 wild type or ER� cells were incubated for 45 minwith 10 nM E2 or DPN, respectively, alone or in combinationwith 10 nM TCDD. Subsequently, cells were subjected toChIP analysis with the indicated antibodies. Shown here arerepresentative agarose gels with PCR products for an ERbinding site on the GREB-1 promoter. B, Quantification of theChIP experiments in A. Real-time PCR was performed onprecipitated DNA of at least three independent experiments.The results were normalized to input DNA, and vehicle treat-ment was set to 1.
Fig. 6. ARNT Forms Complexes with ER� and RNA Poly-merase II on E2-Responsive Promoters after E2 Treatment
MCF-7 ER� cells were incubated for 45 min with 10 nM
DPN alone or in combination with 10 nM TCDD. Subse-quently, re-ChIP analysis was performed with the indicatedantibodies as described in Materials and Methods. Shownhere are representative agarose gels with PCR products foran ER binding site on the GREB-1 promoter.
310 Mol Endocrinol, February 2008, 22(2):304–316 Ruegg et al. • ER� Selectivity of ARNT and TCDD

HeLa cells. After treatment with E2, pS2 transcriptionwas induced in all four cell lines (Fig. 7C). TCDD alonehad no effect on pS2 induction, whereas treatmentwith TCDD decreased E2-induced pS2 transcriptionfor ER� by about 40%, and particularly for ER� with arobust reduction of 80%. ER��AB activity was onlymarginally affected by TCDD treatment, whereas ac-tivity of ER��AB was somewhat increased in the pres-ence of TCDD.
In conclusion, TCDD decreased ER� and ER� ac-tivity in different cell lines and on different promoters.In addition, our experiments reveal a remarkably stron-ger sensitivity of the ER� isoform to the inhibitoryactions of TCDD. Moreover, our results suggest thatthe inhibitory effect of TCDD requires the ER A/B do-main because the �AB mutants were only marginallyor not at all affected upon exposure to dioxin.
DISCUSSION
Competition between AhR and ER for ARNT CanAccount for the Antiestrogenic Effect of TCDD
The ability of industrial chemicals and environmentalpollutants to interfere with hormonal signaling path-ways, a phenomenon known as endocrine disruption,has caused considerable attention and concern fordecades. The potency of dioxins as endocrine disrup-tors is well documented, in particular with regard totheir negative effects on E2 signaling pathways (28).The biological effects of dioxins are mediated by theAhR, a ligand-dependent transcription factor. The ac-tivation process of the AhR has been thoroughly stud-ied and involves the recruitment of its dimerizationpartner ARNT (6). We have previously shown thatARNT can act as coactivator of the ERs (14). Theability of ARNT to coregulate E2 signaling pathwaysprompted us to suggest that competition for the intra-cellular pool of ARNT can be an important regulatorycomponent that may explain, at least in part, the dis-ruptive effects of dioxins such as TCDD on estrogensignaling. In this study, our experiments show thatactivation of alternative ARNT-dependent signalingpathways such as exposure to TCDD or hypoxic con-ditions, which leads to the formation of AhR/ARNT orHIF-1�/ARNT complexes, respectively, results in a de-crease in ER transcriptional activity (Fig. 8). In addition,exposure of cells to TCDD and the subsequent forma-tion of the AhR/ARNT complex resulted in reducedoccupation of ARNT on ER-regulated promoters in
with 10 nM TCDD. After 6 h, cells were harvested, total RNAextracted, and mRNA transcribed into cDNA. Amounts ofpS2 cDNA was measured by real-time PCR and normalizedto amount of 18S RNA. Treatment with E2 alone was set to100%. Shown are means and SD of four independent exper-iments.
Fig. 7. TCDD Shows Selectivity for the ER� Isoform andActs on the A/B Domain on the ERs
A, Differential effects of TCDD on ER� and ER� transcrip-tional activity in HC11 cells. H-ERE cells were treated with 10nM E2, 10 nM PPT, or 10 nM DPN alone or combination with 10nM TCDD. After 24 h, luciferase activity was determined andadjusted to protein concentration in each sample. Treatmentwith the respective agonist alone was set to 100%. Shownare means and SD of three independent experiments. B, Ef-fect of TCDD in HELN cell lines measured by the stablyintegrated ERE-� glob-luciferase activity. HELN cells weretreated with 10 nM E2 alone or combination with 10 nM TCDD.After 24 h, luciferase activity was determined and adjusted toprotein concentration in each sample. Treatment with E2
alone was set to 100%. Shown are means and SD of threeindependent experiments. C, Effect of TCDD in HELN celllines measured by induction of the endogenous pS2 gene.HELN cells were treated with 10 nM E2 alone or combination
Ruegg et al. • ER� Selectivity of ARNT and TCDD Mol Endocrinol, February 2008, 22(2):304–316 311

ChIP assays. ARNT recruitment was observed on thec-fos promoter in HC11 cells and the GREB-1 and pS2promoters in MCF-7 cells; all of them are part of E2-regulated genes. This suggests that ARNT could be ageneral coactivator for ER� and not only recruited tospecific genes. However, this hypothesis has to bestudied more closely, for example by using ChIP-on-chip technology. Reducing the cellular levels of ARNTby siRNA significantly attenuated the transcriptionalresponse of ER� and in particular ER�, supporting ournotion and demonstrating an important role of ARNT inER-mediated transcription. The siRNA was directedagainst a sequence that is homologous betweenARNT-1 and ARNT-2. We therefore cannot concludeat that point whether it is sufficient to knock down oneform or whether both have to be down-regulated tosee an effect on ER transcription. Obviously, an evenmore conclusive experiment would be to investigateER activity in ARNT knockout animals. However, un-conditional knockout of ARNT-1 as well as ARNT-2 islethal in mice (29). Knockout mice have been usedwhere ARNT-1 is disrupted in liver (30), in T cells (31),in endothelial cells (32), and in mammary epithelium(33). Using the last, the authors reported that disrup-tion of ARNT has no implication for mammary glanddevelopment, which would suggest that ARNT is dis-pensable for ER function (33). This observation is inline with the results presented in this study, becausethe main ER isoform in the breast is ER�. Yet ourexperiments suggest that ER� rather than ER� is af-fected by ARNT-1 down-regulation. In addition, it ispossible that ARNT-2 still interacts and coactivates ERin the breast-specific ARNT-1 knockout mouse. OnlyARNT1-ARNT2 double-knockout mice can give infor-mation about the necessity of ARNT in ER signaling invivo.
We show here that ARNT is a much more potentcoactivator for ER� than for ER�. Interestingly, ER�activity is also affected to a much greater extent byTCDD than that of ER�. Furthermore, we demonstratethat both coactivation by ARNT and inhibition byTCDD are dependent on the presence of the N-termi-nal A/B domain of both ER� and ER�. This parallelbehavior between coactivation and inhibition by ARNTand TCDD, respectively, substantially corroboratesour hypothesis that recruitment of ARNT to AhR is atleast partly responsible for the antiestrogenic effect ofdioxins.
A recent publication showed that in the presence ofthe AhR agonist 3-MC, the AhR/ARNT complex acti-vated the ERs in the absence of E2, suggesting aproestrogenic function of the activated AhR/ARNTcomplex (34). This study is inconsistent with numerousepidemiological studies and experimental data dem-onstrating an antiestrogenic effect of AhR ligands (7,35–38). We speculate that this inconsistency can beexplained by the choice of AhR ligand. AlthoughTCDD, the most potent dioxin in the environment,remains biochemically stable, metabolism of polycy-clic aromatic hydrocarbons such as 3-MC potentiallyleads to the formation of metabolites with the ability toactivate alternative signaling pathways. This has beenshown to occur for instance with benzo-(a)-pyrene, acompound related to 3-MC. For this study, we choseto use TCDD and could not observe a proestrogeniceffect in any of our experimental settings.
Our results suggest that TCDD inhibits ER activityand that this antiestrogenic action involves competi-tion for limited intracellular levels of ARNT (Fig. 8). Thismakes ARNT an interesting factor in the context ofendocrine disruption and puts forward the importanceof studying the mechanistic details of the interplaybetween ARNT and the ERs.
The N-Terminal Region of ER Plays an ImportantRole for ARNT and TCDD Action on the Receptor
We have shown previously that, in contrast to thewild-type receptors, ARNT coactivated deletion mu-tants of ER� and ER� lacking the N terminus to asimilar extent. Additionally, the coactivating effect ofARNT on these mutants was weaker. We concludedthat the A/B domain of the ERs might be responsiblefor the differential effect of ARNT on ER� and ER� (14).Here, by swapping the A/B domains of the two iso-forms, we show that the A/B domain of ER� is respon-sible for the preference of ARNT toward ER�. Addi-tionally, the combination between ER� AF-1 and ER�AF-2 was not responsive to ARNT coactivation at all.These findings were corroborated by the fact thatTCDD was less potent in inhibiting the function of A/Bdomain deletion mutants of the receptors (and con-structs exhibiting only the ER LBD). In addition, weinvestigated the effect of ARNT on ER heterodimersusing single-chain ER constructs. We show that theeffect of ARNT coactivation on heterodimers is com-
Fig. 8. Model for the Proposed Mechanism of the Antiestro-genic Effects of TCDD and Hypoxia
Transcriptional activity of the ERs, particularly of ER�, isenhanced by the presence of ARNT. Activation of the AhR orHIF-1� signaling pathway leads to reduction of ARNT avail-able for ER and thus to decreased ER function.
312 Mol Endocrinol, February 2008, 22(2):304–316 Ruegg et al. • ER� Selectivity of ARNT and TCDD

parable with that on ER�. We can only speculate aboutthe reason for this finding. It is possible, for instance,that the presence of one ER� receptor is sufficient forthe dimer to adopt a conformation that resembles anER� homodimer. Previous reports have shown that ERheterodimers emulate ER� properties with respect totheir potency on different E2 response elements andDNA-independent pathways (23) as well as the acti-vation of AF-1 by 4-OHT and their recruitment of co-activators to AF-2 and AF-1 (23, 39). It is thereforepossible that recruitment of additional coactivatorsbesides ARNT is involved in the activation process ofthe ER�/� heterodimer function.
The role of ER� AF-1 remains to be fully solved.Several publications suggest that the N terminus ofER� has an inhibitory effect on ER� AF-2 (24, 40, 41).The mechanism behind this finding is unclear; it hasbeen suggested, however, that steric hindrance couldaccount for less efficient recruitment of coactivators toAF-2 (41). It is possible that binding of ARNT to ER�AF-1 overcomes the interference between the twoactivation functions and hence leads to enhanced re-cruitment of coactivators to the AF-2. Alternatively,ARNT could enhance recruitment of other coactivatorsto ER� AF-1. Several coactivators are reported tointeract not only with AF-2 but also with AF-1 of theERs, e.g. p300/CBP (42). Interestingly, p300 has beenshown to bind to amino acids 62–72 of ER� (42). Wefound here that this region is crucial for the coactivat-ing effect of ARNT-1 on ER�. P300 has also beenshown to interact with the C-terminal domain of ARNT.This coincides with our previous finding that the Cterminus of ARNT is essential for its coactivating func-tion on the ERs (14). In addition, CBP/p300 has beendemonstrated to mediate the interaction betweenARNT and HIF-1� (43). Thus, our results together withprevious findings could put forward a role of p300 inmediating the coactivating effect of ARNT on ER�.
TCDD Is Considerably More Potent in DisruptingActivity of ER� than of ER�
In accordance with our previous findings, we showhere that ARNT exhibits a remarkable preference forER� compared with ER�. Coactivation by ARNT over-expression as well as inhibition by ARNT down-regu-lation using siRNA was more pronounced for ER�.This prompted us to investigate whether TCDD showsa similar preference for ER�. Indeed, we could show intwo different cell lines and on stably transfected re-porters as well as on an endogenous E2-regulatedgene that the inhibitory effect of TCDD is much morepronounced on the activity of ER� than of ER�.
ER� and ER� are known to be expressed in differenttissues and to have distinct functions and targetgenes. In cells expressing both receptors, stimulationof ER� induces proliferation and cell growth, whereasactivation of ER� inhibits cell growth and leads toincreased apoptosis (44). This is of particular interestbecause the literature about the endocrine-disruptive
effects of TCDD is controversial and focused on ER�-containing cells and tissues. For example, its anties-trogenic effect has led to the development of relateddrugs to treat estrogen-dependent breast cancer (45).On the other hand, studies in nonhuman primates haveshown that TCDD is associated with an increasedprevalence and severity of endometriosis (46). Further-more, follow-up studies of the TCDD-exposed popu-lation in Seveso, Italy, indicate that both all-cancer andlung cancer incidences tended to be higher than ex-pected (47). Thus, TCDD seems to be both carcino-genic and antiproliferative, depending on the tissuetype and the receptors expressed. Studies investigat-ing the effect of TCDD on the estrogen system mostlyrefer to classical ER�-expressing tissues like breastand uterus and rarely investigated organs predomi-nantly expressing ER�. Our findings may be criticalwith regard to the sensitivity of different tissues to thebiological effects of dioxin-like pollutants. Future stud-ies should evaluate the impact of dioxins on tissuespredominantly expressing ER�.
Taken together, we present evidence that the re-cruitment of the ER coactivator ARNT to the AhRsignaling pathway after dioxin exposure can accountfor the antiestrogenic action of dioxins. The effects ofboth ARNT and dioxin are considerably more pro-nounced on ER� than on ER�. More studies have tobe carried out to unravel the exact mechanism behindthe ER-ARNT cross-talk and to evaluate the physio-logical importance of the selectivity of TCDD for ER�.
MATERIALS AND METHODS
Plasmids and Reagents
TCDD, 3-MC, and benzo(a)pyrene were purchased from Ac-cuStandard (New Haven, CT); E2 and 4-OHT from SigmaChemical Co. (St. Louis, MO); ICI 182,780 from AstraZeneca(Sodertalje, Sweden); and DPN and PPT from Tocris Bio-science (Bristol, UK).
The plasmids encoding for single-chain ERs were a kindgift from Dr. Mesut Muyan (23). The plasmids pSG5-hER�,pSG5-hER�, 3xERE-TATA-Luc, pCMV-ARNT1, Gal4-Luc,pCMV-�Gal, Gal4-ER�-LBD, and Gal4-ER�-LBD have beendescribed elsewhere. ER� deletion mutants have been de-scribed previously (24). In the original publication, deletionswere made on ER� 485. Here, we refer to ER� 530 as full-length receptor; the nomenclature is therefore different, e.g.�31 becomes �76. Additional details can be obtained fromthe authors upon request.
Antibodies
Monoclonal anti-ER� Ab10 was from NeoMarkers (Fremont,CA) and ER� GTX14021 from GeneTex (San Antonio, TX),and monoclonal anti-flag M2 (Sigma), polyclonal anti-ER�H-184, polyclonal anti-ARNT1 H-172, polyclonal anti-polII,monoclonal anti-hsp90 F-8, and actin antibody sc-8432 wereall from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
Cell Culture and Transient Transfection Assays
HC11 cells and stably transfected 3xERE HC11 cells (H-ERE)(previously described in Ref. 48) were maintained in RPMI
Ruegg et al. • ER� Selectivity of ARNT and TCDD Mol Endocrinol, February 2008, 22(2):304–316 313

1640 medium (Invitrogen, Carlsbad, CA) supplemented with8% fetal calf serum (FCS; Invitrogen), L-glutamine (GIBCO/BRL, Carlsbad, CA), 50 �g/ml gentamycin (GIBCO/BRL), 10ng/ml epidermal growth factor (Sigma), and 5 �g/ml insulin(Sigma). HeLa and MCF-7 cells were routinely maintained inDMEM supplemented with 10% FCS and penicillin (100 U/ml)and streptomycin (100 �g/ml). HELN ER� and HELN ER�cells have been already described (49) and were kept inDMEM supplemented with 5% dextran-coated charcoal(DCC)-treated FCS (Hyclone, Logan, UT), penicillin (100U/ml), streptomycin (100 �g/ml), 1 mg/ml G418, and 0.5�g/ml puromycin. MCF7-ER� cells were kindly provided byDr. Anders Strom and were generated as follows: MCF-7cells were first transfected with pTet-tTAk (GIBCO/BRL)modified to contain puromycin resistance by using Lipofectinaccording to the manufacturer’s instructions (GIBCO/BRL).Selection was performed with 0.5 �g/ml puromycin in thepresence of 1 �g/ml tetracycline. A clone showing high levelsof induction upon tetracycline withdrawal and low basal ac-tivity was selected by using the pUHC13-3 control plasmid(GIBCO/BRL). The short form of ER� encoding 485 aminoacids (ER� 485) was fused to the flag tag and cloned intoPBI-EGFP (Clontech, Palo Alto, CA). This construct was thentransfected into the above-described inducible clone to-gether with a neomycin resistance plasmid, and selectionwas performed with 700 �g/ml G418 (Calbiochem, La Jolla,CA). The cells were maintained in medium containing 0.5�g/ml puromycin and 2 mg/ml tetracycline.
Cells were transfected as described earlier (14). Typically,cells were seeded in 12- or 24-well plates in phenol red-freemedium 24 h before transfection. Cells were transfected us-ing Lipofectamine reagent (Invitrogen) according to the man-ufacturer’s recommendations. We used 100 ng of the appro-priate reporter plasmid (3xERE-TATA-Luc or Gal4-Luc)together with 20 ng pCMV5-�Gal as internal transfectioncontrol and 5 ng receptor plasmids. After transfection, themedium was exchanged with phenol red-free medium sup-plemented with 5% DCC-treated FCS, and the cells wereallowed to grow for an additional 24–48 h. At this point, cellswere harvested, and luciferase and �-galactosidase activitieswere determined. Each graph represents the mean of at leastthree independent transfections performed in duplicate ortriplicate. Stably transfected cells (H-ERE) were grown,treated, and lysed similarly before luciferase activity wasdetermined and protein concentrations measured by theBradford method. The luciferase values are correlated to theprotein concentration of each sample.
Western Blot Analysis
HC11 cells were grown on 10-cm plates in phenol red-freemedium for 24 h before they were treated with 10 nM E2, 10nM TCDD, 10 �M 3-MC, and/or 10 nM TCDD for another 6 h.For the hypoxia treatment, cells were placed in a hypoxicchamber (O2 � 1%). Whole-cell extracts were prepared bylysing the cells on ice in RIPA buffer [20 mM Tris (pH 7.5), 150mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 1mM EDTA, 0.1% SDS] supplemented with protease inhibitorcocktail (Complete Protease Inhibitor; Roche Diagnostics,Basel, Switzerland). Protein concentrations were measuredby the Bradford method. The protein extracts were separatedon a 7.5% SDS-PAGE gel, and the proteins were detectedwith the respective antibodies. Western blots measuringsiRNA efficiency were carried out directly on the whole-cellextracts used for luciferase assays.
ChIP and Re-ChIP Assays
ChIP assays for HC11 cells were performed as described(50). Assays with MCF-7 cells were performed as describedpreviously (51) with slight modifications. Briefly, MCF-7 cellswere grown on 15-cm plates to 80–90% confluency in phenol
red-free DMEM supplemented with 5% DCC-stripped FCSfor 48 h. After treating with 10 nM E2/DPN and/or 10 nM TCDDfor 45 min, cells were washed with PBS and chromatin wascross-linked for 15 min with 1.5% formaldehyde. Cells wereharvested and nuclear extracts produced. Chromatin wasthen sonicated 3 times for 10 sec with a Branston sonifier250, duty cycle 90%, output 2.5, and a fraction of the solublechromatin was put aside as input material. ChIP was per-formed overnight with various antibodies and protein Sepha-rose A or G (Pharmacia, Uppsala, Sweden). After washing,the Sepharose beads were eluted three times with 50 �lelution buffer (0.1 M NaHCO3/1% SDS), and bound chromatinwas reverse cross-linked overnight at 65 C. For re-ChIPassays, beads were washed and eluted with 10 mM dithio-threitol after the first immunoprecipitation. Eluates were di-luted, and the second antibody was added overnight. Thenthe samples were processed as for normal ChIP assays.Eluted DNA fragments were isolated and purified using QIA-quick Spin Kit (QIAGEN, Valencia, CA), and the PCR-amplifiedfragments were analyzed on 2% agarose gels and by real-timePCR as described below. Specific primers used for the 3�-flankingregion of the mouse c-Fos gene were 5�-GGCAGTTGTAAAC-TAGC-3� and 5�-GGAACTTGGAGAAACC-3�; the 5�-flanking re-gion of GREB-1, 5�-AGCAGTGAAAAAAAGTGTGGCAACT-GGG-3� and 5�-CGACCCACAGAAATGAAAAGGCAGCAAACT-3�; and the pS2 5�-flanking region, 5�-CCGGCCATCTCTCACTA-TGAA-3� and 5�-CCTCCCGCCAGGGTAAATAC-3�.
siRNA-Mediated Down-Regulation of ARNT Levels
HeLa or MCF-7ER� cells were seeded in six-well plates 24 hbefore transfection. Cells were transfected with 500 pmolsiRNA (5�-CCAUCUUACGCAUGGCAGUTT-3�) (publishedsequence in Ref. 22), 200 ng 3xERE-TATA-Luc, 40 ng �-gal,and 30 ng pSG5-ER� or pSG5-ER� per well, using Lipo-fectamine (Invitrogen) according to the manufacturer’s rec-ommendations. After transfection, cells were treated withvehicle or 10 nM E2 or DPN for 48 h. Luciferase activity wasmeasured as described above.
Detection of pS2 Transcription by Real-Time PCR
HELN cells were seeded out into six-well plates and grown inphenol red-free medium with 5% DCC-treated FCS for 48 h.After treatment with E2 and/or TCDD for 6 h, RNA was iso-lated using Trizol (Invitrogen) according to the manufacturer’srecommendations. One microgram of total RNA was treatedwith DNase I and reverse transcribed using random hexamerprimers (Invitrogen). One microliter of the resulting cDNA wasthen used for real-time PCR with SYBR green (Invitrogen).The pS2 primer sequences were 5�-CCTCCCAGTGTG-CAAATAAGG-3� and 5�-TGGAGGGACGTCGATGGTAT-3�.Gene transcripts were normalized to the 18S rRNA contentand to the untreated samples.
Acknowledgments
We thank Dr. Malin Hedengran-Faulds and Dr. AndersStrom for kindly providing the H-ERE and MCF-7ER� celllines, respectively. We also thank Mesut Muyan for kindlymaking available the single-chain ER constructs.
Received March 7, 2007. Accepted October 30, 2007.Address all correspondence and requests for reprints to:
Ingemar Pongratz, Karolinska Institute, Department of Bio-sciences and Nutrition, S-141 57 Huddinge, Sweden. E-mail:[email protected].
This work was supported by the German Research Foun-dation (DFG), the Swiss National Science Foundation (SNF),
314 Mol Endocrinol, February 2008, 22(2):304–316 Ruegg et al. • ER� Selectivity of ARNT and TCDD

the European Union (EU)-funded CASCADE Network of Ex-cellence, the EU-funded CRESCENDO project, the SwedishCancer Foundation, and the Swedish Research Council.
Disclosure Statement: The authors have nothing todisclose.
REFERENCES
1. Nilsson S, Gustafsson JA 2002 Biological role of estro-gen and estrogen receptors. Crit Rev Biochem Mol Biol37:1–28
2. Gronemeyer H, Gustafsson JA, Laudet V 2004 Principlesfor modulation of the nuclear receptor superfamily. NatRev Drug Discov 3:950–964
3. Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J,Andersson G, Enmark E, Pettersson K, Warner M,Gustafsson JA 2001 Mechanisms of estrogen action.Physiol Rev 81:1535–1565
4. Pettersson K, Gustafsson JA 2001 Role of estrogen re-ceptor � in estrogen action. Annu Rev Physiol 63:165–192
5. Poland A, Knutson JC 1982 2,3,7,8-Tetrachlorod-ibenzo-p-dioxin and related halogenated aromatichydrocarbons: examination of the mechanism of tox-icity. Annu Rev Pharmacol Toxicol 22:517–554
6. Gu YZ, Hogenesch JB, Bradfield CA 2000 The PASsuperfamily: sensors of environmental and developmen-tal signals. Annu Rev Pharmacol Toxicol 40:519–561
7. Safe S 2001 Molecular biology of the Ah receptor and itsrole in carcinogenesis. Toxicol Lett 120:1–7
8. Wormke M, Stoner M, Saville B, Safe S 2000 Crosstalkbetween estrogen receptor � and the aryl hydrocarbonreceptor in breast cancer cells involves unidirectionalactivation of proteasomes. FEBS Lett 478:109–112
9. Drutel G, Kathmann M, Heron A, Schwartz JC, Arrang JM1996 Cloning and selective expression in brain and kid-ney of ARNT2 homologous to the Ah receptor nucleartranslocator (ARNT). Biochem Biophys Res Commun225:333–339
10. Hirose K, Morita M, Ema M, Mimura J, Hamada H, Fujii H,Saijo Y, Gotoh O, Sogawa K, Fujii-Kuriyama Y 1996cDNA cloning and tissue-specific expression of a novelbasic helix-loop-helix/PAS factor (Arnt2) with close se-quence similarity to the aryl hydrocarbon receptor nu-clear translocator (Arnt). Mol Cell Biol 16:1706–1713
11. Sekine H, Mimura J, Yamamoto M, Fujii-Kuriyama Y2006 Unique and overlapping transcriptional roles ofarylhydrocarbon receptor nuclear translocator (Arnt) andArnt2 in xenobiotic and hypoxic responses. J Biol Chem281:37507–37516
12. Gekakis N, Staknis D, Nguyen HB, Davis FC, WilsbacherLD, King DP, Takahashi JS, Weitz CJ 1998 Role of theCLOCK protein in the mammalian circadian mechanism.Science 280:1564–1569
13. Takahata S, Sogawa K, Kobayashi A, Ema M, Mimura J,Ozaki N, Fujii-Kuriyama Y 1998 Transcriptionally activeheterodimer formation of an Arnt-like PAS protein, Arnt3,with HIF-1a, HLF, and clock. Biochem Biophys ResCommun 248:789–794
14. Brunnberg S, Pettersson K, Rydin E, Matthews J, Han-berg A, Pongratz I 2003 The basic helix-loop-helix-PASprotein ARNT functions as a potent coactivator of estro-gen receptor-dependent transcription. Proc Natl AcadSci USA 100:6517–6522
15. Reisz-Porszasz S, Probst MR, Fukunaga BN, HankinsonO 1994 Identification of functional domains of the arylhydrocarbon receptor nuclear translocator protein(ARNT). Mol Cell Biol 14:6075–6086
16. Jiang BH, Rue E, Wang GL, Roe R, Semenza GL 1996Dimerization, DNA binding, and transactivation proper-
ties of hypoxia-inducible factor 1. J Biol Chem 271:17771–17778
17. Metzger D, Berry M, Ali S, Chambon P 1995 Effect ofantagonists on DNA binding properties of the humanestrogen receptor in vitro and in vivo. Mol Endocrinol9:579–591
18. Wormke M, Castro-Rivera E, Chen I, Safe S 2000 Estro-gen and aryl hydrocarbon receptor expression andcrosstalk in human Ishikawa endometrial cancer cells. JSteroid Biochem Mol Biol 72:197–207
19. Wormke M, Stoner M, Saville B, Walker K, Abdelrahim M,Burghardt R, Safe S 2003 The aryl hydrocarbon receptormediates degradation of estrogen receptor � throughactivation of proteasomes. Mol Cell Biol 23:1843–1855
20. Cowley SM, Hoare S, Mosselman S, Parker MG 1997Estrogen receptors � and � form heterodimers on DNA.J Biol Chem 272:19858–19862
21. Pettersson K, Grandien K, Kuiper GG, Gustafsson JA1997 Mouse estrogen receptor � forms estrogen re-sponse element-binding heterodimers with estrogen re-ceptor �. Mol Endocrinol 11:1486–1496
22. Monroe DG, Secreto FJ, Subramaniam M, Getz BJ, Kho-sla S, Spelsberg TC 2005 Estrogen receptor � and �heterodimers exert unique effects on estrogen- and ta-moxifen-dependent gene expression in human U2OSosteosarcoma cells. Mol Endocrinol 19:1555–1568
23. Li X, Huang J, Yi P, Bambara RA, Hilf R, Muyan M 2004Single-chain estrogen receptors (ERs) reveal that theER�/� heterodimer emulates functions of the ER� dimerin genomic estrogen signaling pathways. Mol Cell Biol24:7681–7694
24. Delaunay F, Pettersson K, Tujague M, Gustafsson JA2000 Functional differences between the amino-terminaldomains of estrogen receptors � and �. Mol Pharmacol58:584–590
25. Meyers MJ, Sun J, Carlson KE, Marriner GA, Katzenel-lenbogen BS, Katzenellenbogen JA 2001 Estrogen re-ceptor-� potency-selective ligands: structure-activity re-lationship studies of diarylpropionitriles and theiracetylene and polar analogues. J Med Chem 44:4230–4251
26. Lin CY, Strom A, Vega VB, Kong SL, Yeo AL, ThomsenJS, Chan WC, Doray B, Bangarusamy DK, Ramasamy A,Vergara LA, Tang S, Chong A, Bajic VB, Miller LD,Gustafsson JA, Liu ET 2004 Discovery of estrogen re-ceptor � target genes and response elements in breasttumor cells. Genome Biol 5:R66
27. Stauffer SR, Coletta CJ, Tedesco R, Nishiguchi G, Carl-son K, Sun J, Katzenellenbogen BS, KatzenellenbogenJA 2000 Pyrazole ligands: structure-affinity/activity rela-tionships and estrogen receptor-�-selective agonists.J Med Chem 43:4934–4947
28. Safe SH 1995 Modulation of gene expression and endo-crine response pathways by 2,3,7,8-tetrachlorodibenzo-p-dioxin and related compounds. Pharmacol Ther 67:247–281
29. Kozak KR, Abbott B, Hankinson O 1997 ARNT-deficientmice and placental differentiation. Dev Biol 191:297–305
30. Tomita S, Sinal CJ, Yim SH, Gonzalez FJ 2000 Condi-tional disruption of the aryl hydrocarbon receptor nucleartranslocator (Arnt) gene leads to loss of target gene in-duction by the aryl hydrocarbon receptor and hypoxia-inducible factor 1�. Mol Endocrinol 14:1674–1681
31. Tomita S, Jiang HB, Ueno T, Takagi S, Tohi K, MaekawaS, Miyatake A, Furukawa A, Gonzalez FJ, Takeda J,Ichikawa Y, Takahama Y 2003 T cell-specific disruptionof arylhydrocarbon receptor nuclear translocator (Arnt)gene causes resistance to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced thymic involution. J Immunol 171:4113–4120
32. Yim SH, Shah Y, Tomita S, Morris HD, Gavrilova O,Lambert G, Ward JM, Gonzalez FJ 2006 Disruption of theArnt gene in endothelial cells causes hepatic vascular
Ruegg et al. • ER� Selectivity of ARNT and TCDD Mol Endocrinol, February 2008, 22(2):304–316 315

defects and partial embryonic lethality in mice. Hepatol-ogy 44:550–560
33. Le Provost F, Riedlinger G, Hee Yim S, Benedict J,Gonzalez FJ, Flaws J, Hennighausen L 2002 The arylhydrocarbon receptor (AhR) and its nuclear translocator(Arnt) are dispensable for normal mammary gland devel-opment but are required for fertility. Genesis 32:231–239
34. Ohtake F, Takeyama K, Matsumoto T, Kitagawa H,Yamamoto Y, Nohara K, Tohyama C, Krust A, Mimura J,Chambon P, Yanagisawa J, Fujii-Kuriyama Y, Kato S2003 Modulation of oestrogen receptor signalling by as-sociation with the activated dioxin receptor. Nature 423:545–550
35. Zacharewski TR, Bondy KL, McDonell P, Wu ZF 1994Antiestrogenic effect of 2,3,7,8-tetrachlorodibenzo-p-di-oxin on 17�-estradiol-induced pS2 expression. CancerRes 54:2707–2713
36. Kharat I, Saatcioglu F 1996 Antiestrogenic effects of2,3,7,8-tetrachlorodibenzo-p-dioxin are mediated bydirect transcriptional interference with the ligandedestrogen receptor. Cross-talk between aryl hydrocar-bon- and estrogen-mediated signaling. J Biol Chem 271:10533–10537
37. Safe S, Wormke M, Samudio I 2000 Mechanisms ofinhibitory aryl hydrocarbon receptor-estrogen receptorcrosstalk in human breast cancer cells. J MammaryGland Biol Neoplasia 5:295–306
38. Chen I, Hsieh T, Thomas T, Safe S 2001 Identification ofestrogen-induced genes downregulated by AhR agonistsin MCF-7 breast cancer cells using suppression subtrac-tive hybridization. Gene 262:207–214
39. Tremblay GB, Tremblay A, Labrie F, Giguere V 1998Ligand-independent activation of the estrogen receptors� and � by mutations of a conserved tyrosine can beabolished by antiestrogens. Cancer Res 58:877–881
40. Hall JM, McDonnell DP 1999 The estrogen receptor�-isoform (ER�) of the human estrogen receptor modu-lates ER� transcriptional activity and is a key regulator ofthe cellular response to estrogens and antiestrogens.Endocrinology 140:5566–5578
41. Huang J, Li X, Maguire CA, Hilf R, Bambara RA, MuyanM 2005 Binding of estrogen receptor � to estrogen re-sponse element in situ is independent of estradiol and
impaired by its amino terminus. Mol Endocrinol 19:2696–2712
42. Kobayashi Y, Kitamoto T, Masuhiro Y, Watanabe M,Kase T, Metzger D, Yanagisawa J, Kato S 2000 p300mediates functional synergism between AF-1 and AF-2of estrogen receptor � and � by interacting directly withthe N-terminal A/B domains. J Biol Chem 275:15645–15651
43. Kobayashi A, Numayama-Tsuruta K, Sogawa K, Fujii-Kuriyama Y 1997 CBP/p300 functions as a possible tran-scriptional coactivator of Ah receptor nuclear transloca-tor (Arnt). J Biochem (Tokyo) 122:703–710
44. Helguero LA, Faulds MH, Gustafsson JA, Haldosen LA2005 Estrogen receptors � (ER�) and � (ER�) differen-tially regulate proliferation and apoptosis of the normalmurine mammary epithelial cell line HC11. Oncogene24:6605–6616
45. Chen I, McDougal A, Wang F, Safe S 1998 Aryl hydro-carbon receptor-mediated antiestrogenic and antitu-morigenic activity of diindolylmethane. Carcinogenesis19:1631–1639
46. Rier S, Foster WG 2003 Environmental dioxins and en-dometriosis. Semin Reprod Med 21:145–154
47. Mandal PK 2005 Dioxin: a review of its environmentaleffects and its aryl hydrocarbon receptor biology.J Comp Physiol [B] 175:221–230
48. Faulds MH, Olsen H, Helguero LA, Gustafsson JA, Hal-dosen LA 2004 Estrogen receptor functional activitychanges during differentiation of mammary epithelialcells. Mol Endocrinol 18:412–421
49. Escande A, Pillon A, Servant N, Cravedi JP, Larrea F,Muhn P, Nicolas JC, Cavailles V, Balaguer P 2006 Eval-uation of ligand selectivity using reporter cell lines stablyexpressing estrogen receptor � or �. Biochem Pharma-col 71:1459–1469
50. Burakov D, Crofts LA, Chang CP, Freedman LP 2002Reciprocal recruitment of DRIP/mediator and p160 co-activator complexes in vivo by estrogen receptor. J BiolChem 277:14359–14362
51. Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M 2000Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 103:843–852
Molecular Endocrinology is published monthly by The Endocrine Society (http://www.endo-society.org), the foremostprofessional society serving the endocrine community.
316 Mol Endocrinol, February 2008, 22(2):304–316 Ruegg et al. • ER� Selectivity of ARNT and TCDD




















