
GPCR Heteromers and their Allosteric Receptor-Receptor Interactions
8
356 Current Medicinal Chemistry, 2012, 19, 356-363 0929-8673/12 $58.00+.00 © 2012 Bentham Science Publishers GPCR Heteromers and their Allosteric Receptor-Receptor Interactions K. Fuxe* ,1 , D.O. Borroto-Escuela 1 , D. Marcellino 1 , W. Romero-Fernández 1 , M. Frankowska 2 , D. Guidolin 3 , M. Filip 2 , L. Ferraro 4 , A.S. Woods 5 , A. Tarakanov 6 , F. Ciruela 7 , L.F. Agnati 8 and S. Tanganelli 4 1 Department of Neuroscience, Karolinska Institutet, Stockholm, Sweden 2 Laboratory of Drug Addiction Pharmacology, Department of Pharmacology, Institute of Pharmacology, Polish Academy of Sciences, Smetna 12, PL 31-343 Kraków, Poland 3 Department of Human Anatomy and Physiology, University of Padova, Italy 4 Department of Clinical and Experimental Medicine, Pharmacology Section and LTTA Centre, University of Ferrara, Ferrara, Italy 5 NIDA-IRP, Structural Biology Unit, MD, USA 6Russian Academy of Sciences, St. Petersburg Institute for Informatics and Automation, Russia 7 Unitat de Farmacologia, Facultat de Medicina, Universitat de Barcelona, Spain 8 IRCCS, Ospedali San Camillo, Venice, Italy Abstract: The concept of intramembrane receptor-receptor interactions and evidence for their existences were introduced in the beginning of the 1980’s, suggesting the existence of receptor heterodimerization. The discovery of GPCR heteromers and the receptor mosaic (higher order oligomers, more than two) has been related to the parallel development and application of a variety of resonance energy transfer techniques such as bioluminescence (BRET), fluorescence (FRET) and sequential energy transfer (SRET). The assembly of interacting GPCRs, heterodimers and receptor mosaic leads to changes in the agonist recognition, signaling, and trafficking of participating receptors via allosteric mechanisms, sometimes involving the appearance of cooperativity. The receptor interface in the GPCR heteromers is beginning to be characterized and the key role of electrostatic epitope-epitope interactions for the formation of the receptor heteromers will be discussed. Furthermore, a "guide-and-clasp" manner of receptor-receptor interactions has been proposed where the "adhesive guides" may be the triplet homologies. These interactions probably represent a general molecular mechanism for receptor-receptor interactions. It is proposed that changes in GPCR function (moonlighting) may develop through the intracellular loops and C-terminii of the GPCR heteromers as a result of dynamic allosteric interactions between different types of G proteins and other receptor interacting proteins in these domains of the receptors. The evidence for the existence of receptor heteromers opens up a new field for a better understanding of neurophysiology and neuropathology. Furthermore, novel therapeutic approaches could be possible based on the use of heteromers as targets for drug development based on their unique pharmacology. Keywords: Receptor mosaic, oligomers, allosteric modulation, GPCR, receptor recognition, receptor signaling, receptor interface, receptor trafficking, receptor G protein coupling, moonlighting, triplet homologies, adenosine receptor, dopamine receptors. INTRODUCTION In 1963-1966 Fuxe, Dahlström and their colleagues discovered and mapped out the central dopamine (DA), noradrenaline (NA) and 5-hydroxytryptamine (5-HT) neurons in the central nervous system (CNS) and here is the first scheme, we prepared of the monoamine pathways [1]. The year was 1965. Later on in the mid and late 1970’s began the mapping out of a large number of peptide neurons. The monoamine-peptide interactions were of great interest to us [2]. How did they in fact interact at the molecular level? One possibility was that the monoamine and peptide signals became integrated through direct peptide receptor-monoamine receptor interactions in the plasma membrane. We began to test this hypothesis in 1980-1981 in membrane preparations of various CNS regions and found that neuropeptides could modulate the binding characteristics especially the affinity of the monoamine receptors in a receptor subtype specific way [3, 4]. Thus, intramembrane receptor-receptor interactions did exist in addition to indirect actions via phosphorylation and changes in membrane potential. However, it took around 10 years before they began to have an impact in the receptor field. But the good news was that the results were in line with earlier findings by Lefkowitz and Limbird and colleagues, 1975, showing negative cooperativity in - adrenergic receptors, which could be explained by the existence of receptor homodimers leading to site-site interactions [5]. It was also clear that adapter proteins can be involved in mediating the receptor-receptor interactions in brain membranes. A *Address correspondence to this author at the Retziusväg 8, 17177 Stockholm, Sweden; Tel/Fax: +46 8 315721; E-mail: [email protected] logical consequence for the indications of direct physical interactions between neuropeptide and monoamine receptors, the term heteromerization was introduced by us in 1993 to describe a specific interaction between different types of GPCRs [6] which sometimes can involve an adapter protein and sometimes require the assistance of scaffolding proteins to allow their interaction to occur. The concept of GPCR heteromer (includes both heterodimer and higher order heteromers) was later confirmed in an excellent way in 1998-99 by studies reporting that two non-functional GPCR monomers, GABAB1 and GABAB2, can assemble in a signaling heterodimer at the cell surface [7].The GABA B receptor belongs to the class C of GPCR with dimerization taking place between the Venus flytrap modules and the C terminal coiled-coil domains. METHODOLOGY FRET and BRET methods gave the evidence needed to demonstrate heteromers among class A GPCRs. Schematic representation of the basic principle of FRET is displayed here (Fig. 1a-b). Two putative proteins bearing a ‘donor’ (CFP or GFP2) or an ‘acceptor’ (YFP) fluorophore are depicted. If these two proteins interact then the donor and acceptor fluorophores are likely in proximity (10 nm or less) and energy transfer between donor and acceptor can occur after donor excitation by demonstration of YFP emission. The bimolecular fluorescence complementation (BiFC) approach is based on the formation of a fluorescent complex when two fragments of a fluorescent protein are brought together by an interaction between receptors fused to the fragments (Fig. 1c). This approach enables the visualization of the subcellular locations of
-
Upload
benthamscience -
Category
Documents
-
view
5 -
download
0
Transcript of GPCR Heteromers and their Allosteric Receptor-Receptor Interactions

356 Current Medicinal Chemistry, 2012, 19, 356-363
0929-8673/12 $58.00+.00 © 2012 Bentham Science Publishers
GPCR Heteromers and their Allosteric Receptor-Receptor Interactions
K. Fuxe*,1, D.O. Borroto-Escuela
1, D. Marcellino
1, W. Romero-Fernández
1, M. Frankowska
2, D. Guidolin
3,
M. Filip2, L. Ferraro
4, A.S. Woods
5, A. Tarakanov
6, F. Ciruela
7, L.F. Agnati
8 and S. Tanganelli
4
1Department of Neuroscience, Karolinska Institutet, Stockholm, Sweden
2Laboratory of Drug Addiction Pharmacology, Department of Pharmacology, Institute of Pharmacology, Polish Academy of
Sciences, Smetna 12, PL 31-343 Kraków, Poland
3Department of Human Anatomy and Physiology, University of Padova, Italy
4Department of Clinical and Experimental Medicine, Pharmacology Section and LTTA Centre, University of Ferrara, Ferrara, Italy
5NIDA-IRP, Structural Biology Unit, MD, USA
6Russian Academy of Sciences, St. Petersburg Institute for Informatics and Automation, Russia
7Unitat de Farmacologia, Facultat de Medicina, Universitat de Barcelona, Spain
8IRCCS, Ospedali San Camillo, Venice, Italy
Abstract: The concept of intramembrane receptor-receptor interactions and evidence for their existences were introduced in the
beginning of the 1980’s, suggesting the existence of receptor heterodimerization. The discovery of GPCR heteromers and the receptor
mosaic (higher order oligomers, more than two) has been related to the parallel development and application of a variety of resonance
energy transfer techniques such as bioluminescence (BRET), fluorescence (FRET) and sequential energy transfer (SRET). The assembly
of interacting GPCRs, heterodimers and receptor mosaic leads to changes in the agonist recognition, signaling, and trafficking of
participating receptors via allosteric mechanisms, sometimes involving the appearance of cooperativity. The receptor interface in the
GPCR heteromers is beginning to be characterized and the key role of electrostatic epitope-epitope interactions for the formation of the
receptor heteromers will be discussed. Furthermore, a "guide-and-clasp" manner of receptor-receptor interactions has been proposed
where the "adhesive guides" may be the triplet homologies. These interactions probably represent a general molecular mechanism for
receptor-receptor interactions. It is proposed that changes in GPCR function (moonlighting) may develop through the intracellular loops
and C-terminii of the GPCR heteromers as a result of dynamic allosteric interactions between different types of G proteins and other
receptor interacting proteins in these domains of the receptors. The evidence for the existence of receptor heteromers opens up a new
field for a better understanding of neurophysiology and neuropathology. Furthermore, novel therapeutic approaches could be possible
based on the use of heteromers as targets for drug development based on their unique pharmacology.
Keywords: Receptor mosaic, oligomers, allosteric modulation, GPCR, receptor recognition, receptor signaling, receptor interface, receptor trafficking, receptor G protein coupling, moonlighting, triplet homologies, adenosine receptor, dopamine receptors.
INTRODUCTION
In 1963-1966 Fuxe, Dahlström and their colleagues discovered and mapped out the central dopamine (DA), noradrenaline (NA) and 5-hydroxytryptamine (5-HT) neurons in the central nervous system (CNS) and here is the first scheme, we prepared of the monoamine pathways [1]. The year was 1965. Later on in the mid and late 1970’s began the mapping out of a large number of peptide neurons. The monoamine-peptide interactions were of great interest to us [2]. How did they in fact interact at the molecular level? One possibility was that the monoamine and peptide signals became integrated through direct peptide receptor-monoamine receptor interactions in the plasma membrane.
We began to test this hypothesis in 1980-1981 in membrane preparations of various CNS regions and found that neuropeptides could modulate the binding characteristics especially the affinity of the monoamine receptors in a receptor subtype specific way [3, 4]. Thus, intramembrane receptor-receptor interactions did exist in addition to indirect actions via phosphorylation and changes in membrane potential. However, it took around 10 years before they began to have an impact in the receptor field. But the good news was that the results were in line with earlier findings by Lefkowitz and Limbird and colleagues, 1975, showing negative cooperativity in - adrenergic receptors, which could be explained by the existence of receptor homodimers leading to site-site interactions [5]. It was also clear that adapter proteins can be involved in mediating the receptor-receptor interactions in brain membranes. A
*Address correspondence to this author at the Retziusväg 8, 17177 Stockholm,
Sweden; Tel/Fax: +46 8 315721; E-mail: [email protected]
logical consequence for the indications of direct physical interactions between neuropeptide and monoamine receptors, the term heteromerization was introduced by us in 1993 to describe a specific interaction between different types of GPCRs [6] which sometimes can involve an adapter protein and sometimes require the assistance of scaffolding proteins to allow their interaction to occur.
The concept of GPCR heteromer (includes both heterodimer and higher order heteromers) was later confirmed in an excellent way in 1998-99 by studies reporting that two non-functional GPCR monomers, GABAB1 and GABAB2, can assemble in a signaling heterodimer at the cell surface [7].The GABA B receptor belongs to the class C of GPCR with dimerization taking place between the Venus flytrap modules and the C terminal coiled-coil domains.
METHODOLOGY
FRET and BRET methods gave the evidence needed to demonstrate heteromers among class A GPCRs. Schematic representation of the basic principle of FRET is displayed here (Fig. 1a-b). Two putative proteins bearing a ‘donor’ (CFP or GFP2) or an ‘acceptor’ (YFP) fluorophore are depicted. If these two proteins interact then the donor and acceptor fluorophores are likely in proximity (10 nm or less) and energy transfer between donor and acceptor can occur after donor excitation by demonstration of YFP emission. The bimolecular fluorescence complementation (BiFC) approach is based on the formation of a fluorescent complex when two fragments of a fluorescent protein are brought together by an interaction between receptors fused to the fragments (Fig. 1c). This approach enables the visualization of the subcellular locations of

Allosteric Receptor-Receptor Interactions Current Medicinal Chemistry, 2012 Vol. 19, No. 3 357
specific heteromers in the normal cellular environment. Schematic representation of the basic principle of BRET is found in Fig. (1d-
e). Two putative proteins bearing a ‘donor’ (Rluc) or an ‘acceptor’ fluorophore (YFP or GFP2) are depicted. If these proteins are in proximity, e.g. do interact, then a donor–acceptor energy transfer can occur after Rluc substrate (h-coelenterazine) oxidation. The bioluminescence formed can activate YFP and YFP emission develops.
In the case of GPCR trimers for instance, the mGlu5R/A2AR/D2R heteromeric complexes, the detection can be by combined BRET/BiFC assays [8]. Once you get YFP formed from mGluR5-Nterminal-YFP and D2R-Cterminal-YFP you test if BRET can be obtained with A2A-Rluc. Thus, when the three receptors interact the YFP is close enough to the Rluc to be activated by the bioluminescence generated by the oxidation of the substrate h-coelenterazine.
The Ciruela group has also obtained indications for the existence an mGlu5R/A2AR/D2R trimer in the local circuits of the striato-pallidal GABA neurons located extra-synaptically on the dendritic spine which had previously been postulated to exist in previous work [8].
The trimeric RM (receptor mosaic, higher order oligomer, more than two receptors) of GPCRs can also be demonstrated with a combination of BRET and FRET. It is called the sequential BRET-FRET technique or SRET technique (Fig. 1f). In the first variant (SERT2), interacting fusion proteins are composed of GPCRs linked to Rluc, GFP2 and YFP with the emissions representing the light that is emitted following addition of the Rluc substrate DeepBlueC. In SRET1 Rluc, YFP and DsRed acceptor fluorescent proteins emit light after addition of the Rluc substrate coelenterazine h [9].
Atomic Force Microscopy (AFM) has been used to detect clusters of immunogold labeled A2A and D2 receptors. By means of a computer-assisted analysis of the AFM images, it was demonstrated that A2A and D2 receptors are not randomly located within a distance less than 10 nm, instead they are confined in the same micro-domain and likely form heterodimers. In line with this important results another experiment also carried out by means of AFM, suggests the existence of high-order D2 oligomers. It was also observed that at least four dopamine D2 receptors are located in close molecular proximity in living mammalian cells, consistent with their organization as higher order oligomers (possibly formed by eight monomers) at the plasma membrane.
Fig. (1). Schematic representation of the different resonance energy transfer methods used to studied GPCR receptor homo-/heteromers. (a-b) Illustration of
FRETCFP-YFP and FRETYFP-GFP2 characteristics. Color dash circle represent the bandpass of the filters used to measure the emission of the energy donor and
acceptor to calculate FRET efficiency. (c) Application of BiFC to monitor GPCR interactions. The principle of BiFC methods is based in the complementation
of the two N-terminal and C-terminal fragment of a fluorescent protein (e.g, YFP). Upon interaction of the tagged receptors, the protein fragments reconstitute
a functional fluorescent protein interpreted as the results of GPCR homo-/heteromer formation. (d-e) Principle of the detection of GPCR heterodimerization
using the BRET method. In the presence of h-coelenterazine or coelenterazine-400, an energy transfer between Rluc and YFP o GFP2 occurs when the
distance between these proteins is less than 100 Å. (f) To identify heteromeric complexes of more than two neurotransmitter receptors combining BRET and
FRET in the Sequential BRET-FRET (SRET) new technique is employed. In SRET experiments, the oxidation of a Rluc substrate triggers acceptor excitation
by BRET and subsequent energy transfer to a FRET acceptor. Thus, SRET requires the co-expression of three fusion proteins, one coupled to Rluc, another
conjugated with GFP2 or YFP, and the third with YFP or DsRed.
358 Current Medicinal Chemistry, 2012 Vol. 19, No. 3 Fuxe et al.
More recently, evidence has been presented by means of in situ proximity ligation assay (in situ PLA) that in HEK293 cells D2LR can form heteromers with D4.7R and especially with D4.2R and D4.4R. In situ PLA have the potential to enable a fuller understanding of GPCR receptor–receptor and could be highly suited to investigate GPCR heteromers in tissue, providing new insights in basic biological mechanisms, heteroreceptor levels and their locations [10].
ALLOSTERIC RECEPTOR-RECEPTOR INTERACTIONS
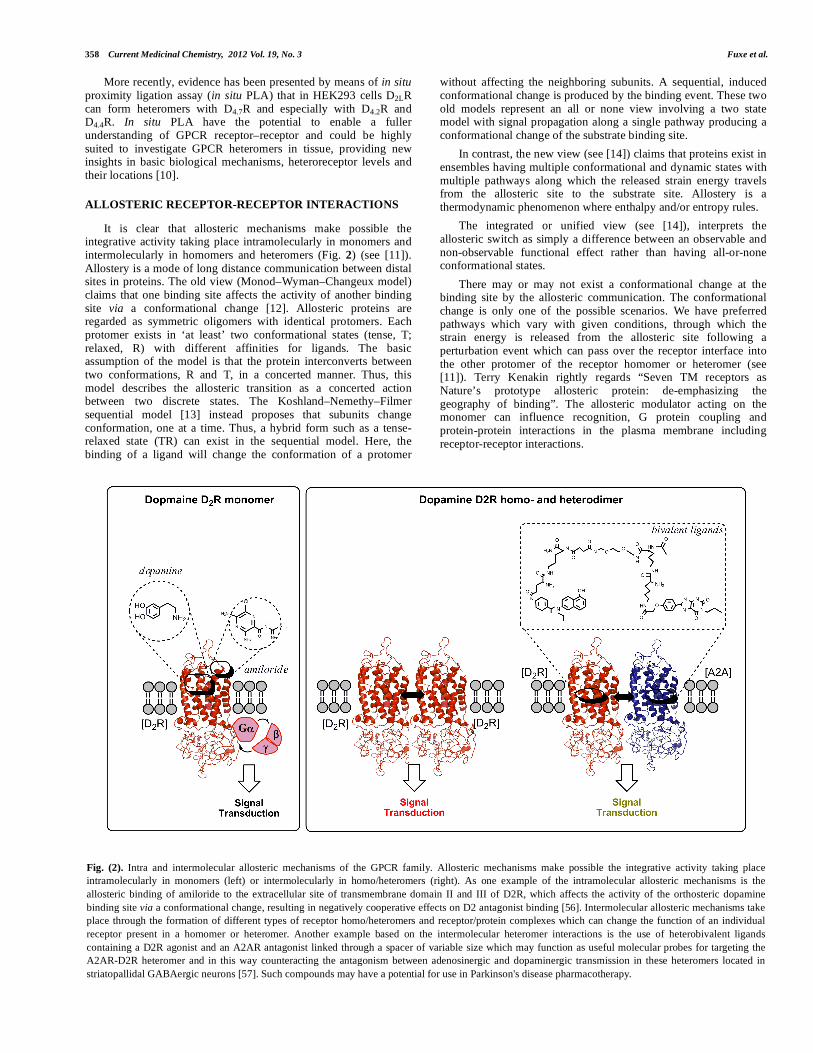
It is clear that allosteric mechanisms make possible the integrative activity taking place intramolecularly in monomers and intermolecularly in homomers and heteromers (Fig. 2) (see [11]). Allostery is a mode of long distance communication between distal sites in proteins. The old view (Monod–Wyman–Changeux model) claims that one binding site affects the activity of another binding site via a conformational change [12]. Allosteric proteins are regarded as symmetric oligomers with identical protomers. Each protomer exists in ‘at least’ two conformational states (tense, T; relaxed, R) with different affinities for ligands. The basic assumption of the model is that the protein interconverts between two conformations, R and T, in a concerted manner. Thus, this model describes the allosteric transition as a concerted action between two discrete states. The Koshland–Nemethy–Filmer sequential model [13] instead proposes that subunits change conformation, one at a time. Thus, a hybrid form such as a tense-relaxed state (TR) can exist in the sequential model. Here, the binding of a ligand will change the conformation of a protomer
without affecting the neighboring subunits. A sequential, induced conformational change is produced by the binding event. These two old models represent an all or none view involving a two state model with signal propagation along a single pathway producing a conformational change of the substrate binding site.
In contrast, the new view (see [14]) claims that proteins exist in ensembles having multiple conformational and dynamic states with multiple pathways along which the released strain energy travels from the allosteric site to the substrate site. Allostery is a thermodynamic phenomenon where enthalpy and/or entropy rules.
The integrated or unified view (see [14]), interprets the allosteric switch as simply a difference between an observable and non-observable functional effect rather than having all-or-none conformational states.
There may or may not exist a conformational change at the binding site by the allosteric communication. The conformational change is only one of the possible scenarios. We have preferred pathways which vary with given conditions, through which the strain energy is released from the allosteric site following a perturbation event which can pass over the receptor interface into the other protomer of the receptor homomer or heteromer (see [11]). Terry Kenakin rightly regards “Seven TM receptors as Nature’s prototype allosteric protein: de-emphasizing the geography of binding”. The allosteric modulator acting on the monomer can influence recognition, G protein coupling and protein-protein interactions in the plasma membrane including receptor-receptor interactions.
Fig. (2). Intra and intermolecular allosteric mechanisms of the GPCR family. Allosteric mechanisms make possible the integrative activity taking place
intramolecularly in monomers (left) or intermolecularly in homo/heteromers (right). As one example of the intramolecular allosteric mechanisms is the
allosteric binding of amiloride to the extracellular site of transmembrane domain II and III of D2R, which affects the activity of the orthosteric dopamine
binding site via a conformational change, resulting in negatively cooperative effects on D2 antagonist binding [56]. Intermolecular allosteric mechanisms take
place through the formation of different types of receptor homo/heteromers and receptor/protein complexes which can change the function of an individual
receptor present in a homomer or heteromer. Another example based on the intermolecular heteromer interactions is the use of heterobivalent ligands
containing a D2R agonist and an A2AR antagonist linked through a spacer of variable size which may function as useful molecular probes for targeting the
A2AR-D2R heteromer and in this way counteracting the antagonism between adenosinergic and dopaminergic transmission in these heteromers located in
striatopallidal GABAergic neurons [57]. Such compounds may have a potential for use in Parkinson's disease pharmacotherapy.

Allosteric Receptor-Receptor Interactions Current Medicinal Chemistry, 2012 Vol. 19, No. 3 359
ON THE RECEPTOR INTERFACE IN GPCR HETERO-
MERS
Let’s now discuss the receptor interface. Two modes of association of GPCRs monomers into dimers have been proposed. One of them is a ‘domain contact’ dimerization, corresponding to the interaction of the molecular surfaces at specific binding sites, without largely changing the conformation of the monomer structure.
The other one, termed ‘domain swapping’ dimerization, is a mechanism in which a substructure (or domain) of a monomer is exchanged with the corresponding substructure (or domain) of the other monomer. Thus, a large conformational change of a monomer structure is required for this mechanism (see [15] for a detailed review). The two mechanisms for GPCR dimerization are still under debate.
Prediction of receptor interfaces may be made with intrinsic disorder analysis showing flexible and malleable regions [16]. Extracellular, intracellular and transmembrane domains of 14 GPCRs were considered and the propensity of each of these domains for a structured or unstructured conformation has been evaluated by means of ad hoc computer programs. The N- and C-terminals as well as intracellular loop 3 were shown to have a high propensity towards an unstructured conformation. Thus, they are potentially very plastic domains, which interact particularly with other protein domains. The disorder located especially in the intra-cellular loop 3 (ICL3) and in the C-terminus domain has been shown to be strongly linked to receptor-receptor interactions.
Predicted interaction interfaces of ordered structures of the A2A adenosine receptor according to Guidolin, Agnati et al. [17], have been obtained by mapping A2A on the 3D structure of bovine rhodopsin (used as a template for class A GPCRs). Potential interfaces in the trans-membrane helices can be estimated using the G-protein coupled receptor interaction partners (GRIP) method.
Recently, a high energy strength double arginine–phosphate electrostatic interaction has been found in the A2A–D2 heteromer by Amina Woods and colleagues which possess a covalent-like stability as demonstrated with mass spectrometry in combination with collision induced dissociation experiments and confirmed by pull-down techniques [18, 19]. A mass spectrum was produced from a mixture of A2AR and D2R epitopes. The mixture of the negatively charged A2A serine-phosphate containing motif from the C terminal A2A and the positively charged D2 VLRRRRKRVN) arginine rich motif from the IC loop 3 resulted in the formation of a noncovalent complex seen at m/z 3854.5 (Fig. 3).
Based on a mathematical approach, we have deduced, based on 48 pairs of receptors that form or not form heterodimers, a set of triplet amino acid homologies that may be responsible for receptor–receptor interactions [20, 21]. We show how such triplets of amino acid residues and their 'teams' may be utilized to construct a kind of code that determines (and/or predicts) which receptors should or should not form heterodimers. We propose a 'guide-and clasp’ manner for receptor–receptor interactions where ‘adhesive guides' may be the triplet homologies (Fig. 3).
Woods, Ciruela and colleagues showed an amazing stability of epitope–epitope electrostatic interaction based on arginine-phosphate [18, 22]. Thus, we should note that two main players contain R (Arg): IDR and AAR. Others have distinct relationships to negatively and positively charged amino acids.
It is postulated that in the receptor contact interface, these homologous Arg containing triplets can form electrostatic bonds with homologous domains of the other receptor through a matching phosphorylated serine and threonine. In this way a hot spot of two close by arginine-phosphate bonds may develop in the receptor interface that may be dynamically regulated by kinases and
phosphatases increasing and reducing the stability of the heterodimer formed. The arginine-rich basic motif epitope exists in the third intracellular loop (IL3) also of DA D3 and D4 receptors implying the likely existence of also A2A–D3 and A2A–D4 receptor heteromers in the CNS [23]. We have in fact demonstrated A2A-D3 heteromers in cell lines [24].
We have now validated the importance of these electrostatic interactions between A2A and D2 by a point mutation of serine 374 to alanine in the A2A C-terminal tail which reduced the A2AR ability to interact with D2R as seen from the reductions of BRET signals and thus of heteromerization especially in combination with mutation of the two aspartates (Fig. 3). BRET
1 studies were
performed on A2AR and D2LR heteromerization in HEK293T cells. BRET
1 saturation curves were obtained for the D2LR-A2AR
hetero-oligomers and compared to D2LR-A2AR mutant hetero-oligomers which showed markedly reduced BRETmax values after serine 374 point mutation especially in combination with mutation of the two aspartates in the A2A C-terminal [25, 26]. Also, this point mutation abolished the A2AR-mediated inhibition of both the D2R high affinity agonist binding and signaling as studied for example with forskolin-induced CRE-luciferase reporter gene assay. The forskolin-induced increase of luciferase activity by a direct activation of AC is significantly reduced by quinpirole, an action which is fully counteracted by the A2A agonist and by the D2R like antagonist raclopride. However, after the single point mutation of serine 374 in the A2AR, it is no longer possible for the A2A agonist (50 nM) to counteract the quinpirole induced reduction of luciferase activity, while raclopride is still able to counteract it. These results validate a key role of serine 374 in the A2AR–D2R interface in making the allosteric modulation of the D2 receptor possible. Serine 374 is also evolutionary conserved.
It was also demonstrated that D2R TM domains IV and V play a major role in the A2AR-D2R interface since the incubation of co-transfected cells with peptides corresponding to these TM domains significantly reduced the ability of A2AR and D2R to heteromerize [25, 26]. The incubation with either TM-IV or TM-V blocked the allosteric communication between A2AR and D2R in which A2AR agonist-induced counteraction of quinpirole-induced inhibition of CRE activity could no longer be observed. In addition, results from molecular dynamic modeling showed that the A2AR (TM-IV/V)-D2R (TM-IV/V) and A2AR (TM-I/VII)-D2R (TM-IV/V) interaction pattern probably is the most favorable model from the energy point of view. Molecular modeling analysis also indicate the position of the pro-triplet homologies found via bioinformatic analysis (Fig. 3) and are compatible with the theory [20] that the pro-triplets AAR, AQE, and VLS through guide-clasp interactions play an important role in the A2AR-D2LR heteromer interface.
In the case of M2-M3 heteromers also covalent interactions exist namely a disulfide crosslink between extracellular loops besides the non-covalent interaction name represented by electrostatic and hydrophobic bonds [27].
MOONLIGHTING CHARACTERISTICS OF GPCRs AND
ION CHANNELS RECEPTORS IN HETEROMERS
As we have repeatedly discussed over the years the allosteric mechanisms in receptor heteromers make possible a marked rise of the repertoire of GPCR recognition and signaling (Fig. 4) [11, 28]. This is achieved through modulation of the orthosteric and allosteric binding sites of the adjacent protomer, its G protein activation, its G protein selectivity, and of its signaling cascades among others switching from G proteins to beta-arrestin2 and through appearance of novel allosteric sites that may alter for instance G protein coupling and selectivity (Fig. 4).
The term moonlighting protein is used to describe multifunctional proteins in which several functions can be found in

360 Current Medicinal Chemistry, 2012 Vol. 19, No. 3 Fuxe et al.
a single strand of amino acids unrelated to splicing, posttranslational changes, etc [29]. The schematic representation depicts some of the principal, non-exclusive, molecular mechanisms by which moonlighting of GPCR heteromers produces novel functions. This is brought about by the allosteric receptor-receptor interactions altering the function of the protomers of the heteromer [30]. Some examples are given below.
In the D1R/D2R heteromer [31], the moonlighting switches the G protein coupling to Gq from Gs (D1R) and Gi (D2R) upon coactivation of D1R and D2R [26] (Fig. 4D-E). The formation of the D1R-D2R heteromer also promotes the formation of a new binding pocket, generating new recognition specificity. Thus, the dopamine D1R agonist SKF83959 can now fully activate the D1R orthosteric site and partially also the D2R orthosteric site resulting in the activation of Gq which requires the activation of both receptors. In the A2AR-D2R heteromer, moonlighting develops
upon coactivation of the A2AR and D2R favoring the coupling of D2R to b-arrestin2 instead of Gi/o [32] (Fig. 4A). In the 5-HT2AR-D2R heteromer moonlighting will block upon coactivation of the two receptors the D2R-Gi/o coupling and enhance the 5-HT2AR-Gq coupling [33] (Fig. 4B).
Moonlighting in the A2AR-D2R heteromer may determine the preference of the binding of the Arg rich motif in the D2R-ICL3 to A2AR, Gi/o or calmodulin involving two alternative conformations of the same motif, or two different but overlapping motifs. CaM is a small but highly conserved intracellular acidic calcium binding protein with negatively charged amino acids and a major transducer of calcium information in the cell. A Ca2+/CaM-dependent modulation of MAPK signaling takes place upon agonist activation of A2A–D2 receptor heteromers demonstrating how proteins in this case Ca-calmodulin can modulate the integrative activity of the A2A-D2 heteromer by direct interactions [34].
Fig. (3). GPCR homo/heteromer receptor interface. Three-dimensional molecular models of the seven TM regions of A2AR and D2LR were built based on its
crystal structure (PDB code 3EML [58]) and the crystal structure of rhodopsin (PDB codes 2Z73 [59]), respectively, as structural templates, by means of the
homology modeling program Accelrys Discovery Studio 2.5 (San Diego, CA, USA) to show that dimerization of GPCR can result from either covalent
(disulfide crosslink) and non-covalent unions (electrostatic and hydrophobic union) between receptor protomers. (Helix-helix interaction) Seen from a lateral
view, representation of the D2LR (TM-IV/)-A2AR (TM-V) interaction in the A2AR-D2LR heterodimer is shown. (Electrostatic interactions) Illustration of
positively charged arginine-rich epitope 215VLRRRRKRVN224 in the N-terminal part of the third intracellular loop of D2R electrostatically interacting with
the negatively charged C-terminal epitopes of the A2AR (370SAQEpSQGNT378
). The most important residues in the A2AR appear to be the phosphorylated
serine in the 370SAQEpSQGNT378 in the C-terminal epitope of the A2AR. These electrostatic interactions represent important hot spots in the receptor interface
of the A2AR-D2R, A2AR-D3R and A2AR-D4R heteromers. (Pro-triplet homology interactions) In silico analysis and sequence alignment have been made of
A2AR and D2R receptor homologies containing the triplet homologies AAR, AQE, VLS, and VYI in the receptor interface. The triplet homology VYI is found
in the N-terminal of A2AR and in the fifth transmembrane -helix (TM-V) of D2R; the triplet homology VLS is found in the TM-IV of both A2AR and D2R; the
triplet homology AAR is found in the third intracellular loop (IL3, between TM-V and TM-VI) of both A2AR and D2R and the triplet homology AQE is found
in the IL3 of D2R and in the C-tail of A2AR. Note that the triplet AQE is adjacent to S374 which seems to play a key role in the A2AR-D2R receptor interface.
The following amino acid residues are marked by a color code as the basic elements of leucine-rich motifs. Red bold L is leucine (Leu). Orange bold I and V
are isoleucine (Ile) and valine (Val) that may also occupy a position of Leu in leucine-rich motifs. Green N and C are asparagine (Asn) and cysteine (Cys).
Black bold S and T are serine (Ser) and threonine (Thr) where agonist-regulated phosphorylation may occur. White letters are charged amino acids: negatively
(dark blue background) E (Glu), D (Asp) or positively (dark red background) R (Arg), K (Lys), H (His) [25].

Allosteric Receptor-Receptor Interactions Current Medicinal Chemistry, 2012 Vol. 19, No. 3 361
There is another impressive moon-lighting example from Martin Lohse, Vilardaga et al. [35, 36]. They have been able to measure a millisecond activation switch in GPCRs and beautifully demonstrated cross-conformational switches between alpha2A and u-opioid receptors leading to direct inhibition of G protein signaling. For instance, morphine binding to the μ-opioid receptor triggered a rapid conformational change in the norepinephrine (NE)-bound 2A adrenergic receptor that proceeds with a t1/2 < 400 ms, which is faster than the kinetics for G protein activation. This fast trans-conformational switch between receptor decreases both activation of inhibitory G protein (Gi) signaling and stimulation of MAP kinase phosphorylation.
This work is in line with our early work on reciprocal antagonistic NPY receptor-alpha2 adrenoceptor interactions in the nucleus tractus solitarius as well as in central vasodepressor responses [37, 38]. We have also demonstrated antagonistic NPY Y1-alpha2 receptor interactions at the membrane and cardiovascular level in the nuctractus solitarius (NTS) counteracting the vasodepressor responses to microinjections of adrenaline in the NTS with increased potency in hypertension [39].
Interaction among members of divergent families of receptors represents an additional layer of complexity when considering the mechanisms of receptor signal transduction. Some reports provide
compelling biochemical and functional information about direct interaction between GPCRs and ion channels receptors [40, 41]. For instance, the Fang Liu group demonstrated that D1 receptors were able to regulate NMDA receptor functions through direct protein-protein interactions involving the carboxyl terminals of D1 receptors and NMDA receptor NR1a and NR2A subunits, respectively [42, 43]. Electrophysiological experiments showed that the activation of D1 receptor up-regulates NMDA receptor-mediated long-term potentiation and working memory in a CaMKII-dependent manner. Furthermore, D1 receptor agonist stimulation promoted the NR1-CaMKII coupling and enhanced the CaMKII activity; effects that was blocked by the application of a TM-II peptide of the D1 receptor [44]. Understanding this moonlighting protein complex may enable a more rational approach to the treatment of schizophrenia, drug addiction, and other psychiatric disorders.
G PROTEIN COUPLING OF RECEPTOR HETEROMERS
When discussing receptor coupling to G proteins (fig), we should be aware that a single active GPCR seems to be sufficient for G-protein binding and activation and that the presence of a second receptor in a homodimer impedes signaling [45]. These
Fig. (4). Moonlighting GPCR heteromers. The schematic representation depicts some of the principal, non-exclusive, molecular mechanisms by which moonlighting of GPCR heteromers produces novel functions. (A) In the A2AR-D2R heteromer moonlighting develops upon coactivation of the A2AR and D2R favoring the coupling of D2R to -arrestin instead of Gi/o. Thus, moonlighting in the A2AR-D2R heteromer may determine the preference of the binding of the Arg rich motif in the D2R-ICL3 to A2AR, Gi/o or calmodulin involving two alternative conformations of the same motif, or two different but overlapping motifs. (B) In the 5-HT2AR-D2R heteromer moonlighting will upon coactivation of the two receptors block the D2R-Gi/o coupling and enhance the 5-HT2AR-Gq coupling. (C) Moonlighting in A2A/CB1/D2R receptor mosaic can result in a switch from Gi/o to Gs/olf of the CB1 receptor contributing to increasing the AC signal. (D) In the D1R/D2R heteromer the moonlighting switches the G protein coupling to Gq from Gs (D1R) and Gi (D2R) upon coactivation of D1R and D2R. (E) The formation of the D1R-D2R heteromer promotes the formation of a new binding pocket, generating new recognition specificity. Thus, the dopamine D1R agonist SKF83959 can now fully activate the D1R orthosteric site and partially also the D2R orthosteric site resulting in the activation of Gq which requires the activation of both receptors. (F) Direct interaction of dopamine D1R with NMDA receptor lead to an enhancement of CaMKII activity at the NMDA receptor with a concomitant increase in the NMDA receptor-dependent long term potentiation and working memory via the activation of CaMKII.

362 Current Medicinal Chemistry, 2012 Vol. 19, No. 3 Fuxe et al.
results lend strong support to the traditional one-to-one model as the minimal signaling unit in the homomer: one helical domain in active conformation couples to a G protein, whereas the neighbor of a homodimer simply gets in the way, more so if activated. There exists a model in which one G protein simultaneously docks to both receptors in a homodimer but it was made on the basis of crystal structures of an inactive receptor and an inactive (in a complex with GDP) heterotrimeric G protein, which do not interact [46].
RECEPTOR HETEROMERS AS TARGETS FOR DRUGS
There exist novel strategies for drug treatment based on targeting the heteromers in view of the likely existence of disease and tissue specific heteromers. Such drugs will have reduced collateral effects by targeting the heterodimers vs the monomers and homodimers [47-50].
A2A-D2 Receptor Mosaics in Parkinson’s Disease: Advantages
of A2A Antagonists
The demonstrated anti-parkinsonian effect of A2A antagonists in clinical studies has given proof of concept that intramembrane receptor-receptor interactions in receptor mosaics can lead to the development of novel therapies [47, 51].The A2A receptor in the A2A/D2 heteromer in the dorsal striato-pallidal GABA neurons with antagonistic A2A/D2 receptor-receptor interactions is the major target for A2A antagonists when used in treatment of PD. A2A antagonists may have not only anti-parkinson properties but also neuroprotective and anti-dyskinetic properties A2A antagonists therefore clearly offer a realistic opportunity to improve PD treatment [51-53].
A2A-D2 Heteromers in Schizophrenia: A2A Agonists as
Atypical Antipsychotic Drugs
A2A agonists by targeting A2A-D2 heterodimers/RM in the ventral striato-pallidal GABA may represent atypical antipsychotic drugs [11, 54]. This target offers a novel strategy for treatment of schizophrenia by reducing D2 like mediated transmission in the meso-limbic DA neurons. In this way, the reduction of NMDA mediated glutamate transmission in the prefrontal cortex in schizophrenia may be counteracted [54].
CONCLUSIONS
There exists an evidence for the existence of direct physical interactions between different types of GPCRs leading to assemblies of receptors like heterodimers and heteromeric receptor mosaics for example A2A-D2 ,A2A-D2-CB1 and A2A-D2-mGlu5 trimeric RMs [8, 9, 23, 40, 55].Within these receptor assemblies, there exist direct allosteric receptor-receptor interactions with one orthosteric site (neurotransmitter binding site) causing changes in recognition, G protein coupling and selection and trafficking of at least one of the other receptors through allosteric interactions sometimes involving cooperative interactions when homomers are involved. These allosteric mechanisms give special moonlighting characteristics of GPCRs in receptor heteromers. In addition, allosteric modulatory sites also exist in these assemblies likely interacting also with orthosteric and allosteric modulatory sites of other protomers of the receptor heteromer.
Heterodimers and heteromeric receptor mosaics are integrators of receptor signaling in the CNS at the plasma mebrane and targets for novel drug development. A2A receptor antagonists introduced in PD targeting the A2A-D2 heterodimer/RM based on the antagonistic allosteric A2A-D2 receptor -receptor interactions present in these heteromers in the dorsal striato-pallidal GABA pathway.
ACKNOWLEDGEMENTS
This work was supported by: grants from the Swedish Research Council (04X-715), Hjärnfonden, Torsten and Ragnar Söderberg, Telethon TV3's La Marató Foundation 2008 and M.M. Wallenberg Foundation to KF, Karolinska Institutets Forskningsstiftelser 2010 and 2011 to D.O.B-E.; grants SAF2008-01462 and Consolider-Ingenio CSD2008-00005 from Ministerio de Ciencia e Innovación and ICREA Academia-2010 from the Catalan Institution for Research and Advanced Studies to FC. A.O.T. has not received any support for this work; a grant from the Ministry of Science and Higher Education (no. N N401 019635), Warszawa, Poland.
REFERENCES
[1] Fuxe, K.; Dahlstrom, A.; Hoistad, M.; Marcellino, D.; Jansson, A.; Rivera, A.; Diaz-Cabiale, Z.; Jacobsen, K.; Tinner-Staines, B.; Hagman, B.; Leo, G.;
Staines, W.; Guidolin, D.; Kehr, J.; Genedani, S.; Belluardo, N.; Agnati, L.F.
From the Golgi-Cajal mapping to the transmitter-based characterization of the neuronal networks leading to two modes of brain communication: wiring
and volume transmission. Brain. Res. Rev., 2007, 55 (1), 17-54. [2] Fuxe, K.; Andersson, K.; Hokfelt, T.; Mutt, V.; Ferland, L.; Agnati, L.F.;
Ganten, D.; Said, S.; Eneroth, P.; Gustafsson, J.A. Localization and possible function of peptidergic neurons and their interactions with central
catecholamine neurons, and the central actions of gut hormones. Fed. Proc.,
1979, 38 (9), 2333-2340.
[3] Agnati, L.F.; Fuxe, K.; Zini, I.; Lenzi, P.; Hokfelt, T. Aspects on receptor regulation and isoreceptor identification. Med. Biol., 1980, 58 (4), 182-187.
[4] Fuxe, K.; Agnati, L.F.; Benfenati, F.; Cimmino, M.; Algeri, S.; Hokfelt, T.; Mutt, V. Modulation by cholecystokinins of 3H-spiroperidol binding in rat
striatum: evidence for increased affinity and reduction in the number of
binding sites. Acta. Physiol. Scand., 1981, 113 (4), 567-569. [5] Limbird, L.E.; Meyts, P.D.; Lefkowitz, R.J. Beta-adrenergic receptors:
evidence for negative cooperativity. Biochem. Biophys. Res. Commun., 1975,
64 (4), 1160-1168.
[6] Zoli, M.; Agnati, L.F.; Hedlund, P.B.; Li, X.M.; Ferre, S.; Fuxe, K. Receptor-receptor interactions as an integrative mechanism in nerve cells.
Mol. Neurobiol., 1993, 7 (3-4), 293-334. [7] Marshall, F.H.; White, J.; Main, M.; Green, A.; Wise, A. GABA(B) receptors
function as heterodimers. Biochem. Soc. Trans., 1999, 27 (4), 530-535. [8] Cabello, N.; Gandia, J.; Bertarelli, D.C.; Watanabe, M.; Lluis, C.; Franco, R.;
Ferre, S.; Lujan, R.; Ciruela, F. Metabotropic glutamate type 5, dopamine D2 and adenosine A2a receptors form higher-order oligomers in living cells. J.
Neurochem., 2009, 109 (5), 1497-1507.
[9] Carriba, P.; Navarro, G.; Ciruela, F.; Ferre, S.; Casado, V.; Agnati, L.; Cortes, A.; Mallol, J.; Fuxe, K.; Canela, E.I.; Lluis, C.; Franco, R. Detection
of heteromerization of more than two proteins by sequential BRET-FRET. Nat. Methods., 2008, 5 (8), 727-733.
[10] Borroto-Escuela, D.O.; Van Craenenbroeck, K.; Romero-Fernandez, W.; Guidolin, D.; Woods, A.S.; Rivera, A.; Haegeman, G.; Agnati, L.F.;
Tarakanov, A.O.; Fuxe, K. Dopamine D2 and D4 receptor heteromerization and its allosteric receptor-receptor interactions. Biochem. Biophys. Res.
Commun., 2011, 404 (4), 928-934. [11] Fuxe, K.; Marcellino, D.; Leo, G.; Agnati, L.F. Molecular integration via
allosteric interactions in receptor heteromers. A working hypothesis. Curr.
Opin. Pharmacol., 2010, 10 (1), 14-22.
[12] Monod, J.; Wyman, J.; Changeux, J.P. On the Nature of Allosteric
Transitions: A Plausible Model. J. Mol. Biol., 1965, 12 88-118. [13] Koshland, D.E., Jr.; Nemethy, G.; Filmer, D. Comparison of experimental
binding data and theoretical models in proteins containing subunits. Biochemistry, 1966, 5 (1), 365-385.
[14] Tsai, C.J.; Del Sol, A.; Nussinov, R. Protein allostery, signal transmission and dynamics: a classification scheme of allosteric mechanisms. Mol.
Biosyst., 2009, 5 (3), 207-216. [15] Gouldson, P.R.; Higgs, C.; Smith, R.E.; Dean, M.K.; Gkoutos, G.V.;
Reynolds, C.A. Dimerization and domain swapping in G-protein-coupled receptors: a computational study. Neuropsychopharmacology, 2000, 23 (4
Suppl), S60-77. [16] Guo, W.; Urizar, E.; Kralikova, M.; Mobarec, J.C.; Shi, L.; Filizola, M.;
Javitch, J.A. Dopamine D2 receptors form higher order oligomers at
physiological expression levels. EMBO. J., 2008, 27 (17), 2293-2304. [17] Guidolin, D.; Ciruela, F.; Genedani, S.; Guescini, M.; Tortorella, C.;
Albertin, G.; Fuxe, K.; Agnati, L.F. Bioinformatics and mathematical modelling in the study of receptor-receptor interactions and receptor
oligomerization: focus on adenosine receptors. Biochim. Biophys. Acta.,
2011, 1808 (5), 1267-1283.
[18] Ciruela, F.; Burgueno, J.; Casado, V.; Canals, M.; Marcellino, D.; Goldberg, S.R.; Bader, M.; Fuxe, K.; Agnati, L.F.; Lluis, C.; Franco, R.; Ferre, S.;
Woods, A.S. Combining mass spectrometry and pull-down techniques for the study of receptor heteromerization. Direct epitope-epitope electrostatic

Allosteric Receptor-Receptor Interactions Current Medicinal Chemistry, 2012 Vol. 19, No. 3 363
interactions between adenosine A2A and dopamine D2 receptors. Anal.
Chem., 2004, 76 (18), 5354-5363. [19] Woods, A.S.; Ciruela, F.; Fuxe, K.; Agnati, L.F.; Lluis, C.; Franco, R.; Ferre,
S. Role of electrostatic interaction in receptor-receptor heteromerization. J.
Mol. Neurosci., 2005, 26 (2-3), 125-132.
[20] Tarakanov, A.O.; Fuxe, K.G. Triplet puzzle: homologies of receptor heteromers. J. Mol. Neurosci., 2010, 41 (2), 294-303.
[21] Tarakanov, A.O.; Fuxe, K.G. The triplet puzzle of homologies in receptor
heteromers exists also in other types of protein-protein interactions. J. Mol.
Neurosci., 2011, 44 (3), 173-177.
[22] Woods, A.S.; Ferre, S. Amazing stability of the arginine-phosphate electrostatic interaction. J. Proteome. Res., 2005, 4 (4), 1397-1402.
[23] Fuxe, K.; Ferre, S.; Canals, M.; Torvinen, M.; Terasmaa, A.; Marcellino, D.; Goldberg, S.R.; Staines, W.; Jacobsen, K.X.; Lluis, C.; Woods, A.S.; Agnati,
L.F.; Franco, R. Adenosine A2A and dopamine D2 heteromeric receptor complexes and their function. J. Mol. Neurosci., 2005, 26 (2-3), 209-220.
[24] Torvinen, M.; Marcellino, D.; Canals, M.; Agnati, L.F.; Lluis, C.; Franco, R.; Fuxe, K. Adenosine A2A receptor and dopamine D3 receptor interactions:
evidence of functional A2A/D3 heteromeric complexes. Mol. Pharmacol.,
2005, 67 (2), 400-407.
[25] Borroto-Escuela, D.O.; Marcellino, D.; Narvaez, M.; Flajolet, M.; Heintz, N.;
Agnati, L.; Ciruela, F.; Fuxe, K. A serine point mutation in the adenosine A2AR C-terminal tail reduces receptor heteromerization and allosteric
modulation of the dopamine D2R. Biochem. Biophys. Res. Commun., 2010,
394 (1), 222-227.
[26] Borroto-Escuela, D.O.; Romero-Fernandez, W.; Tarakanov, A.O.; Gomez-Soler, M.; Corrales, F.; Marcellino, D.; Narvaez, M.; Frankowska, M.;
Flajolet, M.; Heintz, N.; Agnati, L.F.; Ciruela, F.; Fuxe, K. Characterization of the A2AR-D2R interface: focus on the role of the C-terminal tail and the
transmembrane helices. Biochem. Biophys. Res. Commun., 2010, 402 (4), 801-807.
[27] Borroto-Escuela, D.O.; Correia, P.A.; Romero-Fernandez, W.; Narvaez, M.; Fuxe, K.; Ciruela, F.; Garriga, P. Muscarinic receptor family interacting
proteins: role in receptor function. J. Neurosci. Methods., 2011, 195 (2), 161-
169. [28] Kenakin, T.; Agnati, L.F.; Caron, M.; Fredholm, B.; Guidoli, D.; Kobilka,
B.; Lefkowitz, R.W.; Lohse, M.; Woods, A.; Fuxe, K. International Workshop at the Nobel Forum, Karolinska Institutet on G protein-coupled
receptors: finding the words to describe monomers, oligomers, and their molecular mechanisms and defining their meaning. Can a consensus be
reached? J. Recept. Signal. Transduct. Res., 2011, 30 (5), 284-286. [29] Jeffery, C.J. Moonlighting proteins--an update. Mol. Biosyst., 2009, 5 (4),
345-350. [30] Borroto-Escuela, D.O.; Tarakanov, A.O.; Guidolin, D.; Ciruela, F.; Agnati,
L.F.; Fuxe, K. Moonlighting characteristics of G protein-coupled receptors: Focus on receptor heteromers and relevance for neurodegeneration. IUBMB.
Life., 2011, 63 (7), 463-472.
[31] Rashid, A.J.; So, C.H.; Kong, M.M.; Furtak, T.; El-Ghundi, M.; Cheng, R.; O'Dowd, B.F.; George, S.R. D1-D2 dopamine receptor heterooligomers with
unique pharmacology are coupled to rapid activation of Gq/11 in the striatum. Proc. Natl. Acad. Sci. U S A., 2007, 104 (2), 654-659.
[32] Borroto-Escuela, D.O.; Romero-Fernandez, W.; Tarakanov, A.O.; Ciruela, F.; Agnati, L.F.; Fuxe, K. On the existence of a possible A2A-D2-beta-
Arrestin2 complex: A2A agonist modulation of D2 agonist-induced beta-arrestin2 recruitment. J. Mol. Biol., 2011, 406 (5), 687-699.
[33] Borroto-Escuela, D.O.; Romero-Fernandez, W.; Tarakanov, A.O.; Marcellino, D.; Ciruela, F.; Agnati, L.F.; Fuxe, K. Dopamine D2 and 5-
hydroxytryptamine 5-HT(A) receptors assemble into functionally interacting
heteromers. Biochem. Biophys. Res. Commun., 2010, 401 (4), 605-610. [34] Woods, A.S.; Marcellino, D.; Jackson, S.N.; Franco, R.; Ferre, S.; Agnati,
L.F.; Fuxe, K. How calmodulin interacts with the adenosine A(2A) and the dopamine D(2) receptors. J. Proteome. Res., 2008, 7 (8), 3428-3434.
[35] Vilardaga, J.P.; Nikolaev, V.O.; Lorenz, K.; Ferrandon, S.; Zhuang, Z.; Lohse, M.J. Conformational cross-talk between alpha2A-adrenergic and mu-
opioid receptors controls cell signaling. Nat. Chem. Biol., 2008, 4 (2), 126-131.
[36] Vilardaga, J.P.; Bunemann, M.; Krasel, C.; Castro, M.; Lohse, M.J. Measurement of the millisecond activation switch of G protein-coupled
receptors in living cells. Nat. Biotechnol., 2003, 21 (7), 807-812. [37] Harfstrand, A.; Fuxe, K.; Melander, T.; Hokfelt, T.; Agnati, L.F. Evidence
for a cardiovascular role of central galanin neurons: focus on interactions
with alpha 2-adrenergic and neuropeptide Y mechanisms. J. Cardiovasc.
Pharmacol., 1987, 10 Suppl 12 S199-204.
[38] Harfstrand, A.; Fuxe, K.; Agnati, L.; Fredholm, B. Reciprocal interactions between alpha 2-adrenoceptor agonist and neuropeptide Y binding sites in
the nucleus tractus solitarius of the rat. A biochemic and autoradiographic analysis. J. Neural. Transm., 1989, 75 (2), 83-99.
[39] Yang, S.N.; Fior, D.R.; Hedlund, P.B.; Narvaez, J.A.; Agnati, L.F.; Fuxe, K.
Coinjections of NPY(1-36) or [Leu31,Pro34]NPY with adrenaline in the nucleus tractus solitarius of the rat counteract the vasodepressor responses to
adrenaline. Neurosci. Lett., 1994, 171 (1-2), 27-31. [40] Liu, F.; Wan, Q.; Pristupa, Z.B.; Yu, X.M.; Wang, Y.T.; Niznik, H.B. Direct
protein-protein coupling enables cross-talk between dopamine D5 and gamma-aminobutyric acid A receptors. Nature, 2000, 403 (6767), 274-280.
[41] Salter, M.W. D1 and NMDA receptors hook up: expanding on an emerging
theme. Trends. Neurosci., 2003, 26 (5), 235-237. [42] Lee, F.J.; Xue, S.; Pei, L.; Vukusic, B.; Chery, N.; Wang, Y.; Wang, Y.T.;
Niznik, H.B.; Yu, X.M.; Liu, F. Dual regulation of NMDA receptor functions by direct protein-protein interactions with the dopamine D1
receptor. Cell, 2002, 111 (2), 219-230. [43] Li, Y.C.; Liu, G.; Hu, J.L.; Gao, W.J.; Huang, Y.Q. Dopamine D(1) receptor-
mediated enhancement of NMDA receptor trafficking requires rapid PKC-dependent synaptic insertion in the prefrontal neurons. J. Neurochem., 2010,
114 (1), 62-73.
[44] Nai, Q.; Li, S.; Wang, S.H.; Liu, J.; Lee, F.J.; Frankland, P.W.; Liu, F.
Uncoupling the D1-N-methyl-D-aspartate (NMDA) receptor complex promotes NMDA-dependent long-term potentiation and working memory.
Biol. Psychiatry., 2010, 67 (3), 246-254.
[45] Gurevich, V.V.; Gurevich, E.V. GPCR monomers and oligomers: it takes all kinds. Trends. Neurosci., 2008, 31 (2), 74-81.
[46] Gurevich, V.V.; Gurevich, E.V. How and why do GPCRs dimerize? Trends
Pharmacol. Sci., 2008, 29 (5), 234-240.
[47] Fuxe, K.; Marcellino, D.; Borroto-Escuela, D.O.; Guescini, M.; Fernandez-Duenas, V.; Tanganelli, S.; Rivera, A.; Ciruela, F.; Agnati, L.F. Adenosine-
dopamine interactions in the pathophysiology and treatment of CNS disorders. CNS. Neurosci. Ther., 2010, 16 (3), e18-42.
[48] Agnati, L.F.; Guidolin, D.; Vilardaga, J.P.; Ciruela, F.; Fuxe, K. On the expanding terminology in the GPCR field: the meaning of receptor mosaics
and receptor heteromers. J. Recept. Signal. Transduct. Res., 2010, 30 (5), 287-303.
[49] Fuxe, K.; Marcellino, D.; Borroto-Escuela, D.O.; Frankowska, M.; Ferraro,
L.; Guidolin, D.; Ciruela, F.; Agnati, L.F. The changing world of G protein-coupled receptors: from monomers to dimers and receptor mosaics with
allosteric receptor-receptor interactions. J. Recept. Signal. Transduct. Res.,
2010, 30 (5), 272-283.
[50] Fuxe, K.; Marcellino, D.; Rivera, A.; Diaz-Cabiale, Z.; Filip, M.; Gago, B.; Roberts, D.C.; Langel, U.; Genedani, S.; Ferraro, L.; de la Calle, A.;
Narvaez, J.; Tanganelli, S.; Woods, A.; Agnati, L.F. Receptor-receptor interactions within receptor mosaics. Impact on neuropsychopharmacology.
Brain. Res. Rev., 2008, 58 (2), 415-452. [51] Fuxe, K.; Marcellino, D.; Genedani, S.; Agnati, L. Adenosine A(2A)
receptors, dopamine D(2) receptors and their interactions in Parkinson's disease. Mov. Disord., 2007, 22 (14), 1990-2017.
[52] Antonelli, T.; Fuxe, K.; Agnati, L.; Mazzoni, E.; Tanganelli, S.; Tomasini,
M.C.; Ferraro, L. Experimental studies and theoretical aspects on A2A/D2 receptor interactions in a model of Parkinson's disease. Relevance for L-dopa
induced dyskinesias. J. Neurol. Sci., 2006, 248 (1-2), 16-22. [53] Schwarzschild, M.A.; Agnati, L.; Fuxe, K.; Chen, J.F.; Morelli, M. Targeting
adenosine A2A receptors in Parkinson's disease. Trends. Neurosci., 2006, 29 (11), 647-654.
[54] Fuxe, K.; Marcellino, D.; Woods, A.S.; Giuseppina, L.; Antonelli, T.; Ferraro, L.; Tanganelli, S.; Agnati, L.F. Integrated signaling in heterodimers
and receptor mosaics of different types of GPCRs of the forebrain: relevance for schizophrenia. J. Neural. Transm., 2009, 116 (8), 923-939.
[55] Ciruela, F.; Gomez-Soler, M.; Guidolin, D.; Borroto-Escuela, D.O.; Agnati,
L.F.; Fuxe, K.; Fernandez-Duenas, V. Adenosine receptor containing oligomers: their role in the control of dopamine and glutamate
neurotransmission in the brain. Biochim. Biophys. Acta., 2011, 1808 (5), 1245-1255.
[56] Hoare, S.R.; Strange, P.G. Regulation of D2 dopamine receptors by amiloride and amiloride analogs. Mol. Pharmacol., 1996, 50 (5), 1295-1308.
[57] Soriano, A.; Ventura, R.; Molero. A.; Hoen, R.; Casadó, V.; Cortés, A.; Fanelli, F.; Albericio, F.; Lluís, C.; Franco, R.; Royo, M. Adenosine A2A
receptor-antagonist/dopamine D2 receptor-agonist bivalent ligands as pharmacological tools to detect A2A-D2 receptor heteromers. J.Med.Chem.,
2009, 52(18), 5590-602. [58] Jaakola, V.P.; Griffith, M.T.; Hanson, M.A.; Cherezov, V.; Chien, E.Y.;
Lane, J.R.; Ijzerman, A.P.; Stevens, R.C. The 2.6 angstrom crystal structure
of a human A2A adenosine receptor bound to an antagonist. Science, 2008,
322 (5905), 1211-1217.
[59] Li, J.; Edwards, P.C.; Burghammer, M.; Villa, C.; Schertler, G.F. Structure of bovine rhodopsin in a trigonal crystal form. J. Mol. Biol., 2004, 343 (5),
1409-1438.
Received: August 01, 2011 Revised: November 01, 2011 Accepted: November 03, 2011




















