
The histone deacetylase inhibitor AN9 has selective toxicity to acute leukemia and drug-resistant...
28
doi:10.1182/blood-2002-02-0567 Prepublished online July 12, 2002; Nudelman and John Yu Ayse Batova, Li-en Shao, Mitchell B Diccianni, Alice L Yu, Tetsuya Tanaka, Ada Rephaeli, Abraham Leukemia and Drug-Resistant Primary Leukemia and Cancer Cell Lines The Histone Deacetylase Inhibitor AN-9 has Selective Toxicity to Acute (4217 articles) Neoplasia Articles on similar topics can be found in the following Blood collections http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#repub_requests Information about reproducing this article in parts or in its entirety may be found online at: http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#reprints Information about ordering reprints may be found online at: http://bloodjournal.hematologylibrary.org/site/subscriptions/index.xhtml Information about subscriptions and ASH membership may be found online at: digital object identifier (DOIs) and date of initial publication. the indexed by PubMed from initial publication. Citations to Advance online articles must include final publication). Advance online articles are citable and establish publication priority; they are appeared in the paper journal (edited, typeset versions may be posted when available prior to Advance online articles have been peer reviewed and accepted for publication but have not yet Copyright 2011 by The American Society of Hematology; all rights reserved. 20036. the American Society of Hematology, 2021 L St, NW, Suite 900, Washington DC Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.org From
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of The histone deacetylase inhibitor AN9 has selective toxicity to acute leukemia and drug-resistant...

doi:10.1182/blood-2002-02-0567Prepublished online July 12, 2002;
Nudelman and John YuAyse Batova, Li-en Shao, Mitchell B Diccianni, Alice L Yu, Tetsuya Tanaka, Ada Rephaeli, Abraham Leukemia and Drug-Resistant Primary Leukemia and Cancer Cell LinesThe Histone Deacetylase Inhibitor AN-9 has Selective Toxicity to Acute
(4217 articles)Neoplasia �Articles on similar topics can be found in the following Blood collections
http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#reprintsInformation about ordering reprints may be found online at:
http://bloodjournal.hematologylibrary.org/site/subscriptions/index.xhtmlInformation about subscriptions and ASH membership may be found online at:
digital object identifier (DOIs) and date of initial publication. theindexed by PubMed from initial publication. Citations to Advance online articles must include
final publication). Advance online articles are citable and establish publication priority; they areappeared in the paper journal (edited, typeset versions may be posted when available prior to Advance online articles have been peer reviewed and accepted for publication but have not yet
Copyright 2011 by The American Society of Hematology; all rights reserved.20036.the American Society of Hematology, 2021 L St, NW, Suite 900, Washington DC Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom

2
The Histone Deacetylase Inhibitor AN-9 has Selective Toxicity to Acute Leukemia and Drug-Resistant Primary Leukemia and Cancer Cell Lines
Running title: Selective Toxicity of AN-9 to Acute Leukemia
Batova, Ayse,*, Shao, Li-en.,”, Diccianni, Mitchell B.,*, Yu, Alice L.,*, ‡,Tanaka, Tetsuya,”, Rephaeli, Ada,#, Nudelman, A., §, and Yu, John,”
* Department of Pediatrics, Division of Pediatric Hematology/Oncology, University of California San Diego, 200 West Arbor Drive, San Diego, California 92103-8447
" The Scripps Research Institute, Department of Immunology, 10666 North Torrey Pines Road, La Jolla, CA 92037
# Felsenstein Medical Research Center, Sackler School of Medicine, Tel Aviv University, Beilinson Campus, Petach Tikva, 49100, Israel
§ Chemistry Department, Bar Ilan University, Ramat Gan, 52900, Israel
This work was supported by grants from the Leukemia & Lymphoma Society 6125 (to A.L.Y.) and 6226 (to J.Y.), CA79951 (to J.Y.), The Cindy Matters Fund (to A.L.Y.) and in part by Grants MO1 RR00827 from the General Clinical Research Center program. In addition, it is supported by grant 542/0 (to A.R.) from the Israel Science Foundation.
‡ To whom requests for reprints should be addressed at UCSD Medical Center, Division of Pediatric Hematology/Oncology, 200 West Arbor Drive, San Diego, CA 92103-8447;
Key Words: AN-9, histone deacetylase inhibitor, acute leukemia
Abstract 240 words
Neoplasia
Copyright 2002 American Society of Hematology
Blood First Edition Paper, prepublished online July 12, 2002; DOI 10.1182/blood-2002-02-0567 For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom

3
Abstract
The novel pro-drug of butyric acid, pivaloyloxymethyl butyrate (AN-9), a histone deacetylase
inhibitor, shows great promise as an effective and relatively non-toxic anti-cancer agent.for solid
malignancies. However, little is known about its effects on hematopoietic malignancies. In this
study, we show that 21 primary samples of acute leukemia are sensitive to the anti- proliferative
effects of AN-9 with an IC50 of 45.8 ± 4.1 µM. In colony forming assays, primary T-ALL cells
were 3-fold more sensitive than the normal hematopoietic progenitors, BFU-E and CFU- GM, to
AN-9. AN-9 induced apoptosis in the T-ALL cell line CEM. A common problem with cancer is
chemo-resistance which is often typical of relapsed cancers. Remarkably, a T-ALL sample at
diagnosis and an AML sample at relapse that were resistant to Adriamycin in vitro, were
sensitive to AN-9, with IC50 of 50 µM for both samples. More strikingly, samples from 2 infants
with t(4;11) ALL obtained at diagnosis and relapse each were the most sensitive to AN-9, with
IC50 of 25 µM and 17 µM, respectively. Furthermore, an Adriamycin-resistant clone of HL60,
HL60/ADR, obtained by the transfection of the MDR-1 gene, was equally sensitive to AN-9
cytotoxicity as the parental cells. AN-9 induced expression of p21 in an infant leukemia sample
with 11 q23 rearrangement, but not in T- or B-precursor ALL. Collectively, our results suggest
that AN-9 is a selective agent for hematopoietic malignancies which can circumvent mechanisms
of chemo-resistance limiting most conventional chemotherapy.
Introduction
The leukemias account for the largest number of cases of childhood cancer. Acute lymphoblastic
leukemia (ALL) represents approximately three fourths while the acute myeloid leukemia
(AML) represents one sixth of the cases. Despite recent advances in the treatment of childhood
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom

4
cancers, leukemias remain the primary cause of cancer related mortality of children in the United
States. Although ALL and AML are potentially curable diseases with a 5 year survival of 70%
for ALL and 40% for AML, once leukemia recurs, the outcome is dismal. Therefore, there is a
need for novel anti- leukemic agents, especially those which are effective for relapsed leukemias
resistant to existing chemotherapeutic agents.
Histone acetylation plays a key role in the regulation of transcription by modulating
chromatin structure (1-3). In general, histone acetylation is associated with activation of
transcription, whereas histone deacetylation is associated with transcriptional repression. It has
Figure 1. Structure of AN-9 and the products released upon metabolic hydrolysis.
recently been established that many malignancies, particularly leukemias, are associated with the
aberrant recruitment of histone deacetylases (HDAC)s or with mutations in histone acetyl
transferases (HAT)s such as EP300 (4-6). For example, several investigators reported that the
oncoprotein PML/RARα, generated as a result of a translocation in acute promyelocytic
leukemia, suppressed transcription by recruiting HDACs and thus interfering with normal cell
growth and differentiation (7-9). Furthermore, resistance to the differentiating actions of all-trans
retinoic acid in APL patient-derived cells could be overcome by co-treatment with the HDAC
inhibitor, sodium phenyl butyrate (10). There is in fact an increasing body of evidence that
HDAC inhibitors are effective therapeutic agents for a variety of cancers that are refractory to
conventional anti-cancer agents (11, 12).
Pivaloyloxymethyl butyrate (AN-9), a butyric acid prodrug, is a relatively new member
of an established family of acyloxyalkyl ester prodrugs of carboxylic acids that undergo rapid
hydrolysis. Upon hydrolysis, the resulting products are butyric acid, pivalic acids, and
formaldehyde (fig. 1). The anti-cancer effect of AN-9 is assumed to stem primarily from the
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom

5
release of butyric acid, a HDAC inhibitor (13). It is important to note, however, that AN-9 is 10-
fold more potent than butyric acid in vitro, and has anti-cancer effects in vivo not observed with
butyric acid even at 10-fold higher concentrations (14). The increased potency of AN-9 over
butyric acid is most likely due to its increased permeability across cell membranes allowing for
efficient delivery of butyric acid to sub-cellular targets (15). AN-9 inhibits cell proliferation and
soft agar colony formation of a variety of cancer cell lines and primary human solid tumor cells
including those of colon, breast, ovarian, lung, renal and bladder (13, 16, 17). In addition to
being an anti-proliferative agent, AN-9 has also been shown to induce differentiation and
apoptosis in HL-60 cells (18). Earlier studies in mouse tumor models have demonstrated that
AN-9 is an effective anti-tumor agent and displays low toxicity (19). Currently, AN-9 is in a
phase II clinical trial for non-small cell lung cancer. To date, all in vitro and in vivo studies of
AN-9 have been confined to solid tumors with the exception of a murine monocytic leukemia
and the HL60 cell line. In this study, we investigated the in vitro therapeutic efficacy of AN-9 in
primary human acute leukemias including doxorubicin-resistant and/or clinically refractory acute
leukemias. Our findings demonstrate the selective toxicity of AN-9 to acute leukemias including
drug-resistant relapsed leukemias and thus provide the rationale for the initiation of clinical trials
of AN-9 in relapsed acute leukemias.
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom

6
OO
OO
Me CMe3
O
Me OH
O
CMe3HO
O
H H++
pivaloyloxymethyl butyrate (AN-9)
hydrolysis
butyric acid formaldehyde 2,2-dimethylpropanoic acid
Figure 1. Structure of AN-9 and the products released upon metabolic hydrolysis.
Materials and methods
Antibodies and reagents
AN-9 was prepared as previously described and determined to be 99 % pure by NMR
spectroscopy (13). Doxorubicin was obtained from Bedford Laboratories (Bedford, OH ). The
p21 and p27 antibodies were purchased from Pharmingen (San Diego, CA), and the Β-actin
antibody was from Sigma (St Louis, MO). Ficoll-Hypaque was purchased from Pharmacia
(Piscataway, NJ).
Cell lines
The leukemia cell lines HL-60 and HL60/ADR, derived by transfection of HL-60 with the mdr1
gene, were generous gifts from Dr. Michael Kelner (20, 21). The T-ALL cell line CEM was
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom

7
obtained from ATTC (Rockville, MD). The neuroblastoma cell lines Be2c and Be2c/ADR,
derived by selection in doxorubicin containing media, were generous gifts from J. Biedler (22).
Patient Population and Isolation of Primary Leukemia Cells
Heparinized bone marrow or peripheral blood samples were obtained at diagnosis and relapse
from patients with AML or B-precursor ALL at University of California in San Diego. T-ALL
samples were obtained from patients enrolled in POG protocols 9900 (ALL Biology Study) and
9673 (Relapsed T-ALL Study), and were shipped overnight to UCSD for biology studies. Bone
marrow cells obtained from patients in complete remission who undergo diagnostic bone marrow
examination were used as normal controls. Only excess samples obtained for clinical purposes
were analyzed. These samples were collected under an Institutional Review Board-approved
protocol. Mononuclear cells were isolated by density gradient centrifugation through Ficoll-
Hypaque (specific gravity 1.077 g/ml) at 400 g for 30 min, followed by two washes in RPMI
1640. The content of lymphoblasts, as determined by Wright’s stain, was generally ≥ 80 %.
Leukemia cells were cultured in RPMI 1640 supplemented with 10 % fetal calf serum, 2 mM
glutamine, and 1 % penicillin/streptomycin (complete media)
3H-Thymidne Incorporation Assay
Primary leukemia and normal bone marrow cells were plated in complete RPMI at 75 x 103
cells/well in a 96-well plate. Leukemia and neuroblastoma cell lines were plated at 10 and 5 x
103 cells /well, respectively. Cells were treated with increasing concentrations of AN-9 or with
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom

8
0.1 % DMSO (control) for 3 days. Cells were then pulsed with 3H-thymidine (1.6 µCi/well) for 6
hrs. Incorporation of 3H-thymidine was determined in a scintillation counter (Beckman Coulter
Inc., Fullerton, CA) after cells were washed and deposited onto glass microfiber filters using a
cell harvester M-24 (Brandel, Gaithersbur, MD).
Colony Formation Assays
Primary T-ALL and normal bone marrow cells were plated at 5 x 105/35 mm dish in
methylcellulose medium and increasing concentrations of AN-9 for 14 days. An erythroid burst-
forming unit (BFU-E) was identified as a large aggregate of more than 64 hemoglobinized cells
or as clusters of 3 or more subcolonies consisting of 8 or more hemoglobinized cells per
subcolony. The granulocyte/monocyte colony-forming unit (CFU-GM) was enumerated as a
group of more than 50 granulocytic/monocytic translucent cells. T-ALL colonies were identified
as clusters of greater than 50 cells.
Cytotoxicity Assays
Primary leukemia and CEM cells were plated in complete RPMI at 75 x 103 cells/well and 10 x
103 cells/well (96-well plate), respectively. Cells were treated with increasing concentrations of
AN-9 or 0.1 % DMSO for 3 days. The number of viable cells was determined by Trypan-blue
exclusion.
Apoptosis Assays
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom

9
CEM cells were plated at 1.2 x106 cells/well (6-well plate) in complete RPMI and treated with
0.1 % DMSO, or 60 and 75µM AN-9 for 20 and 45 h. Cells were then visualized and
photographed using a Nikon Eclipse TE-300 inverted microscope and the SPOT camera (SPOT
Diagnostic Instruments, Sterling Heights, MI). Alternatively, cells were stained with Alexa Fluor
488 annexin V and propidium iodide using the Vybrant apoptosis assay kit (Molecular Probes
Inc., Eugene, OR) and then analyzed by flow cytometry using a FACScan flow cytometer and
CellQuest 3.2.1 software (Becton Dickinson).
Western Blot Analysis
Primary leukemia cells (10-20 x106) were treated with 10, 25, 50, and 100 µM AN-9 for 24 and
48h. As a control, cells were treated with 0.1% DMSO. Protein was extracted with 30 µl of SDS
lysis buffer (50 mM Tris-HCl pH 6.8, 2 % SDS, 0.1 % bromophenol blue, 10 % glycerol, 5 mM
DTT). Samples were heated in boiling water for 10 min and then the lysate was passed through a
28G needle to shear chromosomal DNA. Supernatants were then collected after spinning in a
microfuge at 13,000 rpm for 10 min at 4oC. Proteins were resolved on an 8 % SDS-
polyacrylamide gel and transferred to Immobilon-P nylon membranes (Millipore Corp., Bedford,
MA). Membranes were probed with p21 and p27 antibodies at 0.5 µg/ml. Membranes were also
probed with a Β-actin antibody at 2 µg/ml as a control for protein loading. Antibody-bound
proteins were detected by chemifluorescence and analyzed using the Storm 840 Phoshorimager
(Molecular Dynamics).
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom

10
Results
Primary ALL cells are more sensitive than normal bone marrow cells to AN-9
The effect of AN-9 on DNA synthesis in both primary ALL and normal bone marrow was
examined by 3H-thymidine incorporation assays. The results demonstrated that ALL cells were
more sensitive than normal bone marrow cells to inhibition of 3H-thymidine uptake by AN-9
(fig. 2).
Figure 2. Inhibition of 3H-thymidine incorporation by AN-9 in primary ALL cells and normal bone marrow cells. Normal bone marrow cells and primary ALL cells were treatedwith increasing concentrations of AN-9 for 3 days. Cells were then pulsed with 3H-thymidine for 6h. �, T-ALL; �, B-precursor ALL; �, relapse infant ALL; �, diagnosis infant ALL ▲, normal bone marrow. All experimental conditions were performed in triplicate. Error bars represent standard error of the mean for data obtained with normal bone marrow (N=6) and primary T-ALL (N=16). For data obtained with infant ALL (N=1) and for a representative B-precursor ALL (N=1), error bars represent standard deviation.
The mean IC50 of AN-9 in 6 normal bone marrow cells was 87 ± 5 µM. In contrast, the mean
AN-9 IC50 in 15 primary T-ALL was 50 ± 5 µM, and that in two B-precursor ALL was 44 µM
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom

11
and 54 µM (data not shown). The finding that both T- and B- ALL have similar sensitivity to
AN-9 suggests that the effects of AN-9 are not lineage specific. Remarkably, however, primary
leukemias obtained from 2 infants with an 11q23 rearrangement were the most sensitive to AN-
9. One of these samples was obtained from a patient who suffered from leukemia relapse shortly
after bone marrow transplantation and became refractory to reinduction chemotherapy. On two
separate occasions along the course of his relapse, the IC50 of AN-9 was only 17 µM (fig. 2) and
13 µM (data not shown), respectively. The second infant ALL sample, collected at the time of
diagnosis had an AN-9 IC50 of 25 µM (fig. 2). To confirm the results obtained by 3H-thymidine
incorporation on the selectivity of AN-9 for leukemia, colony formation assays were performed
using normal bone marrow cells and primary T-ALL cells. The mean IC50 of AN-9 in 6 primary
T-ALL was 46.6 ± 1.1 µM and that in 5 normal hematopoietic progenitors BFU-E and CFU-GM
was 159.7 ± 2.7 µM and 159.5 ± 2.9 µM, respectively (fig. 3). The difference in AN-9 IC50
between T-ALL and normal hematopoietic progenitors is highly significant with P < 0.001.
Thus, the results of colony formation assays confirm the selectivity of AN-9 for ALL over
normal bone marrow cells.
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom

12
Figure 3. Inhibition of Colony Formation by AN-9 of Primary T-ALL and Normal Hematopoietic Progenitors. Primary T-ALL (N=6) and normal bone marrow cells (N=5) were cultured in methylcellulose medium and increasing concentrations of AN-9 for 14 days. The erythroid burst-forming unit (BFU-E) was identified as a large aggregate of more than 64 hemoglobinized cells, or as clusters of 3 or more subcolonies consisting of 8 or more hemoglobinized cells per subcolony. The granulocyte/monocyte colony-forming unit (CFU-GM) was enumerated as a group of more than 50 granulocytic/monocytic translucent cells. �, T-ALL; ▲, CFU-GM; �, BFU-E. Error bars represent standard error of the mean.
AN-9 is cytotoxic to ALL
To distinguish between cytostatic and cytotoxic effects of AN-9, cell viability assays were
performed using both a T-ALL cell line, CEM, as well as primary T-ALL cells. The results of
trypan-blue exclusion assays demonstrated that AN-9 was cytotoxic to CEM and primary T-ALL
in a dose-dependent manner (fig. 4).
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom

13
Figure 4. Cytotoxicity of AN-9 in CEM and Primary T-ALL cells. CEM and T-ALL cells were treated with increasing concentrations of AN-9 for 3 and 5 days, respectively. Viable cells were then determined by trypan blue exclusion. �, Primary T-ALL; ▲, CEM. Results obtained with primary T-ALL is representative of two independent experiments with 2 different patient samples. Data obtained with CEM cells is an average of two independent experiments. All experimental conditions were performed in triplicate. Error bars represent standard deviation.
The IC50 of AN-9 was 47 µM in CEM and 41 µM in a primary T-ALL, after 3 and 5 days of AN-
9 treatment, respectively (fig. 4). At 75 µM AN-9, cytotoxic effects on CEM were evident as
early as after 24h of incubation (data not shown).
Induction of apoptosis by AN-9
In order to determine whether cytotoxicity of AN-9 involved the induction of apoptosis, CEM
cells were treated with 0.1 % DMSO (negative control), 60, 75 µM AN-9, or with 5 % ethanol
(positive control) for 20 and 45 h, and then examined by flow cytometry after staining with
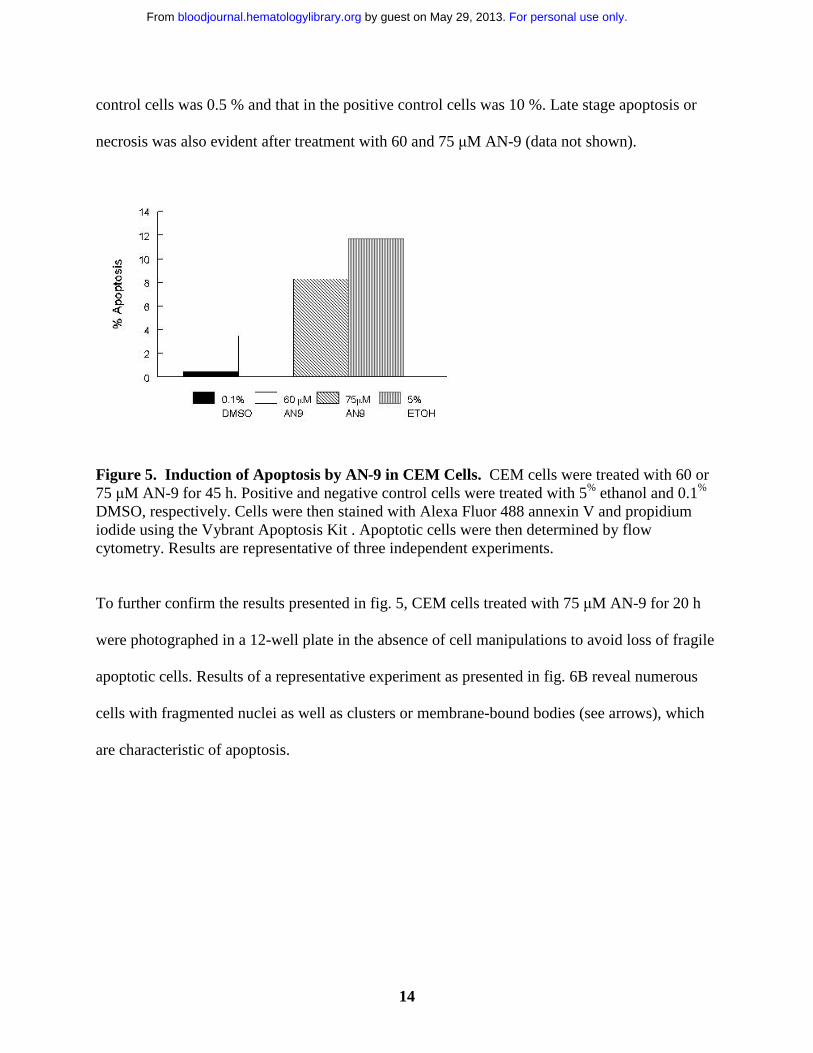
Alexa Fluor 488 annexin V and propidium iodide. As shown in fig. 5, AN-9 induced apoptosis in
CEM cells in a dose-dependent manner. For instance, treatment with 60 µM and 75 µM AN-9 for
45 h resulted in 3.5 % and 8.3 % apoptotic cells, respectively. Percent apoptosis in negative
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom
14
control cells was 0.5 % and that in the positive control cells was 10 %. Late stage apoptosis or
necrosis was also evident after treatment with 60 and 75 µM AN-9 (data not shown).
Figure 5. Induction of Apoptosis by AN-9 in CEM Cells. CEM cells were treated with 60 or 75 µM AN-9 for 45 h. Positive and negative control cells were treated with 5% ethanol and 0.1%
DMSO, respectively. Cells were then stained with Alexa Fluor 488 annexin V and propidium iodide using the Vybrant Apoptosis Kit . Apoptotic cells were then determined by flow cytometry. Results are representative of three independent experiments.
To further confirm the results presented in fig. 5, CEM cells treated with 75 µM AN-9 for 20 h
were photographed in a 12-well plate in the absence of cell manipulations to avoid loss of fragile
apoptotic cells. Results of a representative experiment as presented in fig. 6B reveal numerous
cells with fragmented nuclei as well as clusters or membrane-bound bodies (see arrows), which
are characteristic of apoptosis.
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom

15
Figure 6. Morphology of CEM cells treated with AN-9. CEM cells were treated with either 75 µM AN-9 or with 0.1 % DMSO (control) for 20 h and then cells were photographed using the SPOT camera. (A) control; (B) 75 µM AN-9.
These findings demonstrate that apoptosis occurs within 20 h and appears to be more extensive
than the flow cytometry results indicate since no apoptosis was evident by flow cytometry at
20 h (data not shown). This result is most likely due to the loss of apoptotic cells during the
staining procedure prior to flow cytometry.
AN-9 induces p21 in infant ALL but not in T-ALL or B-precursor ALL
Previous studies have reported the induction of p21 by butyrate and its correlation with cell cycle
arrest and apoptosis (23, 24). In order to determine whether p21 is involved in AN-9 action, we
examined the expression of p21 in response to AN-9 by Western Blot analysis. As shown in
fig.7A, p21 is induced in a primary infant ALL within 24 h of AN-9 treatment, and the levels
declined by 48 h indicating a transient induction. In contrast, as presented in a representative
experiment (fig. 7), induction of p21 by AN-9 was not observed in any of the 2 B-precursor ALL
(fig. 7B) or the 4 primary T-ALL (fig. 7C, data shown for 1 patient) cells examined even at AN-
9 concentrations that effectively arrest DNA synthesis in ALL (fig. 2). The effects of AN-9 on
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom

16
the levels of p27, a homolog of p21, were also examined. The levels of p27 were not induced by
AN-9 in B-precursor ALL or T-ALL (data not shown). These results suggest that the effects of
AN-9 on T-ALL is independent of p21 or p27.
Figure 7. Western Blot Analysis of p21 in primary ALL. Protein was extracted by theSDS/boiling method from (A) infant ALL; (B) B-precusor ALL; and (C) T-ALL cells treated with AN-9 as indicated. SJ SA-1 osteosarcoma cells known to express p21 were used as the positive control. Western blot analysis was performed using a p21 antibody at 0.5 µg/ml. Equal amounts of protein in each lane was verified by probing the blot with a Β-actin antibody at 2 µg/ml. U, untreated (0.1 % DMSO); C, positive control for p21; pt, patient. AN-9 IC50 in the infant ALL was 13 µΜ in a 3H-thymidine uptake assay.
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom
17
Drug-resistant primary leukemia and cancer cell lines are sensitive to AN-9
The finding that clinically refractory infant ALL cells are sensitive to AN-9 prompted us to
determine whether AN-9 is cytotoxic to other drug resistant malignant cells. The effect of AN-9
was examined in a doxorubicin-resistant clone of the myeloid leukemia cell line HL60,
HL60/ADR, obtained by the transfection of the MDR-1 gene, and a doxorubicin-resistant
neuroblastoma cell line Be2c-ADR, induced by repeated exposure of Be2C to doxorubicin. The
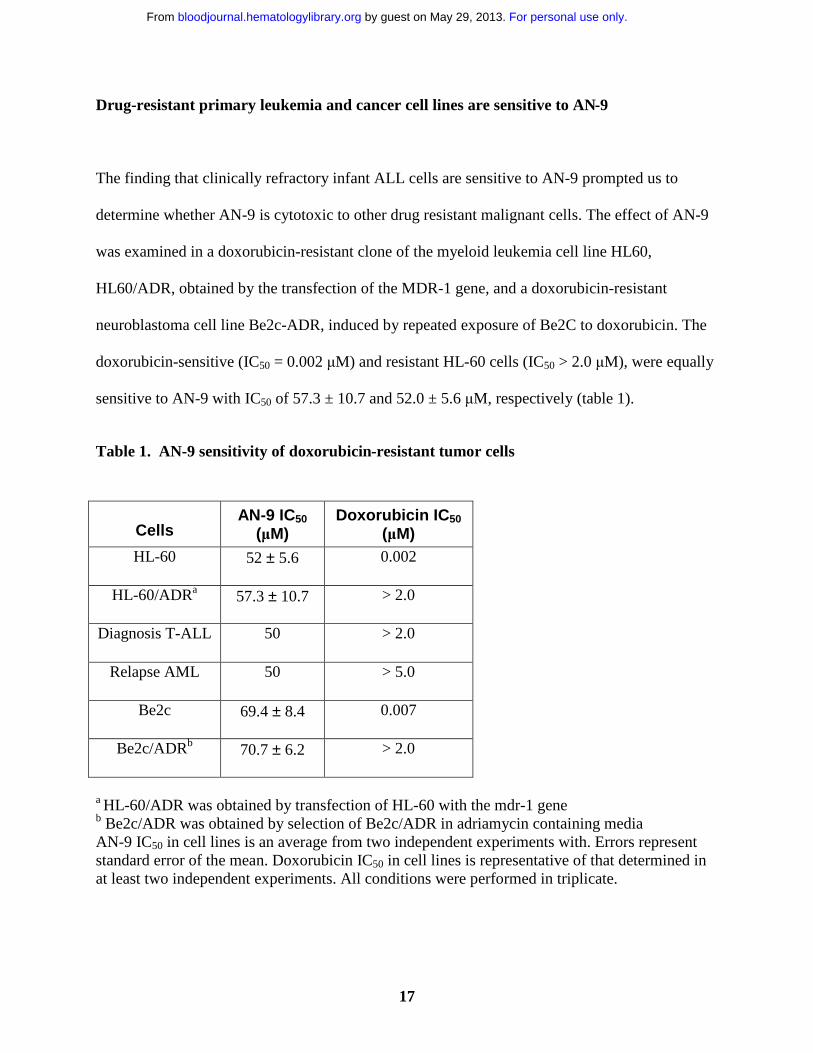
doxorubicin-sensitive (IC50 = 0.002 µM) and resistant HL-60 cells (IC50 > 2.0 µM), were equally
sensitive to AN-9 with IC50 of 57.3 ± 10.7 and 52.0 ± 5.6 µM, respectively (table 1).
Table 1. AN-9 sensitivity of doxorubicin-resistant tumor cells
CellsAN-9 IC50
(µM)Doxorubicin IC50
(µM)HL-60 52 ± 5.6 0.002
HL-60/ADRa 57.3 ± 10.7 > 2.0
Diagnosis T-ALL 50 > 2.0
Relapse AML 50 > 5.0
Be2c 69.4 ± 8.4 0.007
Be2c/ADRb 70.7 ± 6.2 > 2.0
a HL-60/ADR was obtained by transfection of HL-60 with the mdr-1 geneb Be2c/ADR was obtained by selection of Be2c/ADR in adriamycin containing mediaAN-9 IC50 in cell lines is an average from two independent experiments with. Errors represent standard error of the mean. Doxorubicin IC50 in cell lines is representative of that determined in at least two independent experiments. All conditions were performed in triplicate.
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom

18
Similarly, the doxorubicin-sensitive (IC50 = 0.007 µM) and resistant neuroblastoma cell lines
(IC50 > 2.0 µM) were equally sensitive to AN-9 with IC50 of 70.7 ± 6.2 and 69.4 ± 8.4 µM,
respectively (table 1). More importantly, a doxorubicin-resistant primary T-ALL and a relapsed
AML who was refractory to chemotherapy, were sensitive to the cytotoxicity of AN-9, each with
an IC50 of 50 µM (table 1), an IC50 similar to that obtained in doxorubicin-sensitive primary ALL
(fig. 2). Taken together, these findings in addition to the finding of hightened sensitivity to AN-9
of relapsed infant leukemia (fig. 2), suggest that AN-9 may be a promising agent for acute
leukemias, even in cases of relapsed/refractory leukemias.
Discussion
In the present study of 21 primary samples of acute leukemias, we demonstrated that AN-9 has
anti-proliferative and cytotoxic effects on leukemic cells. AN-9 arrested DNA synthesis in both
T-ALL and B-precursor ALL at similar concentrations indicating that its effects on leukemia
cells were not lineage-specific. More importantly, leukemic cells were 2- and 3-fold more
sensitive than normal hematopoietic progenitors to the anti-proliferative effects of AN-9 in 3H-
thymidine incorporation and colony formation assays, respectively. These findings indicate that
AN-9 is less toxic to normal hematopoietic progenitors and thus has selectivity for leukemic
cells. In line with these results, earlier mouse tumor model studies as well a phase I clinical trial
of AN-9 conducted in adults with solid tumors have shown that AN-9 has efficacy and is
relatively non-toxic (19, 25). Notably, in vitro data from solid tumors (13, 16, 17 and table 1)
indicate that IC50 of AN-9 is about 2-fold higher than that observed by us in leukemia,
suggesting that leukemia cells are particularly sensitive to AN-9 cytotoxicity. Collectively, our
results indicate that AN-9 is a selective anti-cancer agent and may have an advantage over
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom

19
standard chemotherapeutic agents such as daunorubicin and adriamycin which are equally toxic
to leukemic cells and hematopoietic progenitors in vitro (26-28).
The anti-cancer effects of several HDAC inhibitors including butyrate, were found to be
dependent on their ability to inhibit HDACs, and correlated with their capability to modulate cell
cycle and apoptosis-regulatory genes (12, 23, 24). A recent study reported that induction of p21
expression by butyrate was required for its effect in arresting cell growth in colon carcinoma
cells (23). However, another study reported that butyrate arrested cell growth in 3T3 fibroblasts
derived from p21 knockout mice, indicating that p21 was not required for the anti-proliferative
effects of butyrate (29). Interestingly, p21 induction by AN-9 was observed only in an infant
ALL sample that was the most sensitive to AN-9 cytotoxicity. The levels of p21were not induced
by AN-9 in any of the primary T-ALL or B- precursor ALL cells examined, even at AN-9
concentrations that completely inhibited DNA synthesis. Thus, it appears that AN-9 may exert its
anti-proliferative effects through different pathways depending on the cell type. Our results
demonstrate that in addition to being an anti-proliferative agent, AN-9 also induces apoptosis, as
observed in CEM cells. Zimra et al. (18) previously reported that AN- 9 induced apoptosis in HL-
60 cells and that this was accompanied by the reduction of Bcl-2 expression. However, another
study reported that butyrate activated caspase 3 and induced apoptosis in lymphoid and
colorectal cancer cells that was dependent on the inhibition of HDACs but independent of
changes in the levels of Bcl-2 and Bax (30). As these authors suggested, it is possible that other
members of the Bcl-2/Bax family may influence butyrate-induced apoptosis. Alternatively,
apoptosis could be induced through the death receptors including CD95, TNF, and the receptor
for the TNF-related apoptosis inducing ligand. Although the hybrid polar HDAC inhibitor, M-
carboxycinnamic acid bishydroxamide, was found to induce CD95/CD95 ligand expression,
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom

20
butyrate did not alter the expression of either the ligand or receptor, but did alter cell sensitivity
to CD95-mediated apoptosis (31). The repression of Bcl-2 (18) and the induction of Bax
expression (Rephaeli, unpublished observations) by AN-9 suggest that the intrinsic pathway of
apoptosis that does not involve the death receptors may be the pathway responsible for AN-9
induced apoptosis. As the precise mechanisms underlying the effects of HDAC inhibitors still
remains unknown, future work is aimed at further examining the pathways and key components
involved in AN-9-induced growth arrest and apoptosis in ALL.
Unfortunately, as with many cancers, relapses are common in ALL and are associated
with multi-drug resistance. In this study, AN-9 was effective in inhibiting the growth of HL-60
myeloid and neuroblastoma clones that harbor multiple genetic alterations (32) and display
multidrug resistant phenotype (20-22). Moreover, AN-9 was equally effective in arresting
proliferation of all the primary leukemia cells tested including, a doxorubicin-resistant T-ALL, a
clinically refractory relapsed AML, as well as relapsed infant ALL characterized by an 11q23
rearrangement and a very poor prognosis. Thus, our finding that AN-9 arrested growth of these
different cancer cell types indicates that AN-9 is a unique agent with potential advantage over
standard chemotherapeutic agents. Similar observations have been reported using other HDAC
inhibitors (10-12). For instance, the HDAC inhibitor, MS-27-275, strongly inhibited the growth
of solid tumor implants in nude mice that did not respond to the standard chemotherapeutic agent
5-fluorouracil (11). Furthermore, the more familiar HDAC inhibitor, trichostatin A (TSA), was
shown to inhibit proliferation and induce apoptosis in gastric and oral carcinoma cell lines
resistant to 9-cis-retinoic acid- and INF-β (12). Another advantage of AN-9 is its ability to
synergize with anthracyclins such as Daunomycin, commonly used in the treatment of leukemia
patients (19, 33). The molecular basis for the synergistic actions of doxorubicin and AN-9 was
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom

21
reported to be due to the ability of formaldehyde, released upon cellular hydrolysis of AN-9, to
facilitate the formation of DNA adducts with Adriamycin (34, 35). Concurrently, butyrate, also
released upon cellular hydrolysis, inhibits HDACs resulting in histone hyperacetylation and
relaxation of the chromatin structure, allowing greater accessibility of DNA for the formation of
adriamycin-DNA adducts. The synergistic action of AN-9 with doxorubicin would allow the use
of significantly lower doses of these agents in treatment and consequently greatly improve the
therapeutic index. Thus, AN-9, a HDAC inhibitor with low toxicity and selectivity toward cancer
cells, may provide a novel therapeutic strategy for cancers refractory to traditional anti-tumor
agents. The present study provides strong evidence for the warrant of clinical trials in relapse
ALL using AN-9, as a single agent, or in combination with standard chemotherapeutic agents.
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom

22
Acknowledgements
The authors gratefully acknowledge investigators from the Pediatric Oncology Group for
providing T-ALL samples. We also thank Louis Bridgeman, Ruby Gribi, and, Dr. Sigrun
Gebauer for technical assistance. We are very grateful to Dr. Denis Sasaki for assistance with
flow cytometry.
References
1. Grunstein M. Histone Acetylation in chromatin structure and transcription. Nature.
1997;389: 349-352.
2. Allfrey VG, Faulknere R,Mirsky AE. Acetylation and methylation of histones and their
possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci., USA , 1964;61:
786-794.
3. Pazin MJ, Kadonaga JT. What’s up and down with histone deacetylation and
transcription? Cell 1997;89: 325-328.
4. Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: Inducers of
differentiation or apoptosis of transformed cells. Nat. Cancer Inst. 2000;92: 1210-1216.
5. Brehm A, Nielsen SJ, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T.
The E7 oncoprotein associates with Mi2 and histone deaceytlasse activity to promote cell
growth. EMBO 1999;18: 2449-2458.
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom

23
6. Gayther SA, Batley SJ, Linger L, Bannister A, Thorpe K, Chin SF, Daigo Y, Russel P,
Wilson A, Sowter HM, Delhanty JD, Ponder BA, Kouzarides T, Caldas C. Mutations
truncating the EP300 acetylase in human cancers. Nat. Genet. 2000;24: 300-303.
7. Grignani F, De Matteis S, Nervi C, Tomassoni L, Gelmetti V, Cioce M. Fusion proteins
of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukemia.
Nature 1998;391: 815-818.
8. Lin RJ, Nagy L, Inoue S, Shao W, Miller WH. Jr., Evans RM. Role of the histone
deacetylase complex in acute promyelocytic leukemia. Nature 1998;391: 811-814.
9. He LZ, Guidez F, Tribioli C, Peruzzi D, Ruthardt M, Zelent A. Distinct interactions of
PML-RARalpha and PLZF- RARalpha with corepressors determine differential responses
to RA in APL. Nat. Genet. 1998;18: 126-135.
10. Warrel RP, He L-Z, Richon V, Calleja E, Pandolfi PP. Therapeutic targeting of
transcription in acute promyelocytic leukemia by use of an inhibitor of histone
deacetylase. J. Nat. Cancer Inst. 1998;90: 1621-1625.
11. Saito A, Yamashita T, Mariko Y, Nosaka Y, Tsuchiya K, Ando T, Suzuki T, Tsuruo T,
Nakanishi O. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in
vivo anti-tumor activity against human tumors. Proc. Natl. Acad. Sci. 1999;96: 4592-
4597.
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom

24
12. Suzuki T, Yokozaki H, Kuniyasu H, Hayashi K, Naka K, Ono S, Ishikawa T, Tahara E,
Yasui W. Effect of trichostatin A on cell growth and expression of cell cycle and
apoptosis-related molecules in human gastric and oral carcinoma cell lines. Int. J. Cancer.
2000;88: 992-997.
13. Nudelman A, Ruse M, Aviram A, Rabizadeh E, Shaklai M, Zimra Y, Rephaeli A. Novel
anticancer prodrugs of butyric acid. J. Med. Chem. 1992;35: 687-694.
14. Aviram A, Rephaeli A, Shaklai M, Nudelman A, Ben-Dror I, Maron L, Rabizadeh E.
Effect of butyric acid pro-drug, pivaloyloxymethyl butyrate, on the tumorigenicity of
cancer cells. J. Cancer Res. Clin. Oncol. 1997;123: 267-271.
15. Zimra Y, Maron L, Shaklai M, Nudelman A, Rephaeli A. Pivaloyloxymethyl butyrate
(AN-9) exhibits higher anticancer activity than butyric acid (BA): It penetrates faster than
the acid and induces apoptosis in HL-60 cells. Blood. 1994;84: 579a.
16. Rephaeli A, Rabizadeh E, Aviram A, Shaklai M, Ruse M, Nudelman A. Derivatives of
butyric acid as potential anti-neoplastic agents. Int. J. Cancer. ;49: 66-72.
17. Siu LL, Von Hoff DD, Rephaeli A, Izbicka E, Cerna C, Gomez L, Rowinsky EK,
Eckhardt SG. Activity of pivaloyloxymethyl butyrate, a novel anticancer agent, on
primary human tumor colony-forming units. Invest. New Drugs. 1998;16: 113-119.
18. Zimra Y, Wasserman L, Maron L, Shaklai M, Rephaeli A, Nudelman A. Butyric acid and
pivaloyloxymethyl butyrate, AN-9, a novel butyric acid derivative, induce apoptosis in
HL-60 cells. J. Cancer Res. Clin. Oncol. 1997;123: 152-160.
19. Kasukabe T, Rephaeli A, Honma Y. An anti-cancer derivative of butyric acid
(pivaloyloxymethyl butyrate) and daunorubicin cooperatively prolong survival of mice
inoculated with monocytic leukemia cells. Brit. J. Cancer. 1997;75: 850-854.
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom

25
20. Kelner MJ.,McMorris TC., Montoya MA, Estes L, Rutherford M, Samson KM, Taetle R.
Characterization of cellular accumulation and toxicity of illudin S in sensitive and
nonsensitive tumor cells. Cancer Chemoth. Pharm. 1997;40: 65-71.
21. Marsh W, Center MS. Adriamycin resistance in HL60 cell and accompanying
modification of a surface membrane protein contained in drug-sensitive cells. Cancer
Res. 1987;47: 5080-5086.
22. LaQuaglia MP, Kopp EB, Spengler BA, Meyers MB, Biedler JL. Multidrug resistance in
human neuroblastoma cells. J. Pediatr. Surg. 1991;26: 1107-1112.
23. Archer SY, Meng S, Shei A, Hodin RA. p21waf1 is required for butyrate-mediated growth
inhibition of human colon cancer cells. Proc. Natl. Acad. Sci. USA 1998;95: 6791-6796.
24. Chai F, Evdokiou A, Young GP, Zalewski PD. Involvement of p21(Waf1/Cip1) and its
cleavage by DEVD-caspase during apoptosis of colorectal cancer cells induced by
butyrate. Carcinogenesis 2000;21: 7-14.
25. Eckhardt SG, Villalona-Calero MA, Hammond L, Moczygemba J, Kraynak M, de Moor
C, Von Hoff DD, Rowinsky EK. A phase I study of AN-9 (pivaloyloxymethyl butyrate,
Pivanex), a lipophilic butyric acid analog, in patients with advanced solid tumors. Proc.
Amer. Ass. Cancer Res. 1998;39: 507.
26. Singer CRJ, Linch DC. Comparison of the sensitivity of normal and leukaemic myeloid
progenitors to in-vitro incubation with cytotoxic drugs:a study of pharmacological
purging. Leukemia Research 1987;11: 953-959.
27. Spiro TE, Mattelaer M-A, Efira A, Stryckmans P. Sensitivity of myeloid progenitor cells
in healthy subjects and patients with chronic myeloid leukemia to chemotherapeutic
agents. JNCI. 1981;66: 1053-1059.
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom

26
28. Linssen P, Brons P, Knops G, Wessels H, de Witte T. Plasma and cellular
pharmacokinetics of m-AMSA related to in vitro toxicity towards normal and leukemic
clonogenic bone marrow cells (CFU-GM, CFU-L). Eur J Haematol. 1993;50: 149-154.
29. Vaziri C, Stice L, Faller DV. Butyrate-induced G1 arrest results from p21-independent
disruption of retinoblastoma protein-mediated signals. Cell Growth Differ. 1998;9: 465-
474.
30. Medina V, Edmonds B, Young GP, James R, Appleton S, Zalewski PD. Induction of
caspase-3 protease activity and apoptosis by butyrate and trichostatin A (Inhibitors of
histone deacetylase) Dependence on protein synthesis and synergy with a
mitochondrial/cytochrome c-dependent pathway. Cancer Res. 1997;57: 3697-3707.
31. Bonnotte B, Favre N, Reveneau S, Micheau O, Droin N, Garrido C, Fontana A, Chauffert
B, Solary E, Martin F. Cancer cell sensitization to Fas-mediated apoptosis by sodium
butyrate. Cell Death Differ. 1998;5: 480-487.
32. Diccianni MB, Omura-Minamisawa M, Batova A, Le T, Bridgeman L, Yu AL. Frequent
deregulation of p16 and the p16/G1 Cell cycle-regulatory pathway in neuroblastoma. Int.
J. Cancer 1999;80: 145-154.
33. Niitsu N, Kasukabe T, Yokoyama A, Okabe-Kado J, Yamamoto-Yamaguchi Y, Umeda
M, Honma Y. Anticancer derivative of butyric acid (pivalyloxymethyl butyrate)
specifically potentiates the cytotoxicity of doxorubicin and daunorubicin through the
suppression of microsomal glycosidic activity. Molecular Pharmacol. 2000;58: 27-36.
34. Cutts SM, Rephaeli A, Nudelman A, Hmelnitsky I, Phillips DR. Molecular basis for the
synergistic interaction of adriamycin with the formaldehyde-releasing prodrug
pivaloyloxymethyl butyrate (AN-9). Cancer Res. 2001;61: 8194-8202.
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom

27
35. Zeman SM, Phillips DR, Crothers DM. Characterization of covalent Adriamycin-DNA
adducts. Proc. Natl. Acad. Sci. 1998;95: 11561-11565.
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom

28
For personal use only. by guest on May 29, 2013. bloodjournal.hematologylibrary.orgFrom




















