
Oxidized Collagen Stimulates Proliferation of Vascular Smooth Muscle Cells
Upload
independentCategory
view
2download
0
Materials Science and Engineering C 32 (2012) 2583–2588
Contents lists available at SciVerse ScienceDirect
Materials Science and Engineering C
j ourna l homepage: www.e lsev ie r .com/ locate /msec
The effect of point mutations on structure and mechanical properties of collagen-likefibril: A molecular dynamics study
Ashley E. Marlowe, Abhishek Singh, Yaroslava G. Yingling ⁎Department of Materials Science and Engineering, North Carolina State University, 911 Partners Way, Raleigh, NC 27695, USA
⁎ Corresponding author at: Department of Materials SCarolina State University, Campus Box 7907, 911 PartUSA. Tel.: +1 919 513 2624; fax: +1 919 515 7724.
E-mail address: [email protected] (Y.G. Yingli
0928-4931/$ – see front matter © 2012 Elsevier B.V. Allhttp://dx.doi.org/10.1016/j.msec.2012.07.044
a b s t r a c t
a r t i c l e i n f oArticle history:Received 17 April 2012Received in revised form 16 July 2012Accepted 28 July 2012Available online 3 August 2012
Keywords:Collagen fibrilMolecular dynamics simulationsPoint mutationsOsteogenesis ImperfectaMechanical properties
Understanding sequence dependent mechanical and structural properties of collagen fibrils is important forthe development of artificial biomaterials for medical and nanotechnological applications. Moreover, pointmutations are behind many collagen associated diseases, including Osteogenesis Imperfecta (OI). Weconducted a combination of classical and steered atomistic molecular dynamics simulations to examine theeffect of point mutations on structure and mechanical properties of short collagen fibrils which include mu-tations of glycine to alanine, aspartic acid, cysteine, and serine or mutations of hydroxyproline to arginine,asparagine, glutamine, and lysine. We found that all mutations disrupt structure and reduce strength of thecollagen fibrils, which may affect the hierarchical packing of the fibrils. The glycine mutations were more det-rimental to mechanical strength of the fibrils (WT>Ala>Ser>Cys>Asp) than that of hydroxyproline(WT>Arg>Gln>Asn>Lys). The clinical outcome for glycine mutations agrees well with the trend in reduc-tion of fibril's tensile strength predicted by our simulations. Overall, our results suggest that the reduction inmechanical properties of collagen fibrils may be used to predict the clinical outcome of mutations.
© 2012 Elsevier B.V. All rights reserved.
1. Introduction
Collagen is the most abundant protein in mammals and is a keycomponent of the extracellular matrix, supporting and connectivetissues. Collagen, likemany biological materials, has complex hierarchi-cal structure where tropocollagen molecules assemble into a parallel,overlapping staggered array to form collagen fibrils [1]. Diseases associ-ated with collagen often have detrimental consequences, since it is anintegral part of themechanical veracity of themajority of the structuralconstituents in organisms. For example, osteogenesis imperfecta (OI),or brittle bones disease, involves a large deficiency in collagen, resultingin stunted growth, easy bone fracture or breakage, thin skin, and weaktendons, among other symptoms [2]. The disease varies in severityfrommild to lethal, depending on the type and location of themutation,and affects about 1 in 20,000 people worldwide.
Type I collagen is found in the highest quantity in the human bodyand its deficiency is the cause of most collagen diseases. Type I collagenpolypeptides have a Glycine(Gly)-X-Y repeat unit, where glycine isrequired to ensure the formation of the triple helix, and the X and Ypositions are most commonly occupied by proline and hydroxyproline[1,3]. The Gly-Pro-Hyp tripeptide unit is the most stabilizing [4], aswell as the most common repeat sequence in fibrillar type collagens.Amino acid mutations in collagen sequence modify the intermolecular
cience and Engineering, Northners Way, Raleigh, NC 27695,
ng).
rights reserved.
properties of both the tropocollagen molecule and the fibril and leadto formation of a defective connective tissue [5]. It is known that adefect or mutation in the molecular structure of collagen modifies thebehavior and properties of the molecule; however, there is a longstanding debate on the mechanism for the cause of OI itself. Possibleexplanations include the deprivation of molecular stability and correctassociation with other collagen molecules, as well as the lacking ofessential binding sites [6]. A further understanding of the effect ofpoint mutations in type I collagen is necessary to gain perspective inthis disease.
Lethal OI mutations most typically involve single point amino acidmutations [6], which can lead to a creation of a bulge in collagen,affecting molecular mechanics of the collagen fibril and interactionswith other molecules [7]. The majority of OI mutations involve amutation of glycine, which is buried at the center of the triple helix, toa larger amino acid [6,8,9]. Specifically, it has been determined thatmutation of glycine to alanine is 19% lethal, serine is 30%, cysteine is39% and aspartic acid is 68% [9]. The order of destabilization of tropocol-lagen was determined to be AlabSerbCysbArgbValbGlubAspbTrpwhich correlates well with the clinical outcomes [10].
The effect of mutations of proline and hydroxyproline amino acids,which are exposed to the surface, are much less investigated and theclinical effect are less characterized [11]. However, a single substitutionof Hyp to Gln has been associated with a Marfan-like syndrome [12].Persikov et al used a host-guest peptide approach to establish a scaleof inclination for all amino acids to be present in the X or Y positions,putting a single varied “guest triplet” in the center of a tropocollagen[4]. They found that mutations at the X and Y positions to imino acids

2584 A.E. Marlowe et al. / Materials Science and Engineering C 32 (2012) 2583–2588
are stable whilemutations to Gly and aromatic residues are destabilizing[4]. Recent molecular dynamics (MD) simulations of Hyp and Promutations in collagen indicated that mutations change the formation ofinterchain hydrogen bonds, solvent interactions, and puckering ofneighboring imino acids and, thus, the structural stability of the collagen[13]. There has been a considerable research progress in elucidation ofeffect of amino acid mutations on collagen structure; yet the relationbetween structure of collagen and human disease remains unclear.
In this study, MD simulations were used to determine the effect ofpoint mutations on stability and mechanical properties of collagen-likefibril containing the Gly-Pro-Hyp repeat unit. Previous MD simulationsstudies of tropocollagen have been successful in investigating thestructural aspects of the protein and explaining experimental results[6,7,14–18]. For example, multiscale simulations indicated that severityof OI phenotype correlates well with the mechanical properties ofglycine residue mutations [7]. Simulations of mechanical testing oftropocollagen molecule with Gly mutations were successful in correla-tion of the loss of molecular stiffness with the implications of the disease[19]. While previous simulations of collagen mutations were performedon tropocollagen molecules, it is not known to what extent and at whatlevel in a complex hierarchical structure of collagen the mutations influ-ence the properties. Thus, in this study we focus on the effect of muta-tions on the fibril structure. The aim of our simulations is to comparethe effects of Hyp and Gly mutations on fibril mechanical properties.Glycine is located on the interior axis of the tropocollagen helix, wherethere is no room for a larger side group, whereas proline and hydroxy-proline point outward, stabilizing the triple helix. In our first set ofmuta-tions, a glycine residuewasmutated to alanine, aspartic acid, cysteine, or
Fig. 1. (a) The tropocollagen molecule is shown with hydroxyproline highlighted in oranghydroxyproline or glycine. (b and c) End and side view of the initial fibril structure where oof each of the outer tropocollagen molecules to the centroid of the central one (in purple)
serine amino acid. In the second set of mutations, a hydroxyproline wasmutated to arginine, asparagine, glutamine, or lysine amino acid (Fig. 1).
2. Materials and methods
Generally, prediction of protein stability due to mutations can beaccomplished by using methods from four categories: (1) physical orfirst principle methods, (2) empirical potential functions, (3) machinelearning methods, and (4) statistical potential methods. Physicalmethods are the most accurate and the most computationally expen-sive. For our study, we chose physical methods that rely on knowledgeof atomistic models and the use of molecular dynamics, molecularmechanics and/or Monte Carlo simulations. In these methods themutations are usually introduced into a folded protein structureand simulations with carefully parameterized force field are used toassess the changes in protein stability, which provide quantitativeinsights into molecular scale changes associated with mutations[6,13,16,19,20]. The limitations of the physical methods includesampling difficulty of mutated structures, unknown folding pathway,length and size of simulations, and accuracy of the force fields.
In this study, a 15 amino acid homotrimer tropocollagen moleculewas built (Fig. 1) based on the molecule from the Protein Databankwith PDB ID 1QSU [21]. The length of the tropocollagen and fibril usedin our study is comparablewith the length used in experimental studies[4]. The intermolecular and intramolecular bonding patterns of thishomotrimer are analogous to normal type I collagen, making analysisof this structure resonate with true collagen. Single point mutationswere introduced into the wild‐type molecule per specifications listed
e and glycine highlighted in red. Each mutation is shown for substitution with eithernly the central tropocollagen had a point mutation, and the distances from the centroidare shown in Angstroms.

2585A.E. Marlowe et al. / Materials Science and Engineering C 32 (2012) 2583–2588
in Fig. 1. In the first set of simulations, the eighth amino acid, hydroxy-proline, was mutated in the central tropocollagen of the fibril. In thesecond set of simulations, the ninth amino acid, glycine, was mutatedin the center tropocollagen of each fibril. Only single point mutationswere placed in eachmolecule in the same location to better understandthe changes these mutations make. Simulations of a single tropocolla-gen and collagen fibril with and without mutations were performed.
The additional limitation of our study is that the mutations wereintroduced into already assembled and stabilized fibril. It is knownthat certain mutations will not prevent the formation of the tropocol-lagen helix, such as in the case of Ser and Cys mutations [22,23], andsome mutations will lead to folding difficulty, such as in the caseof Arg [24]. However, assessing the time dependent assembly andfolding pathway of mutated collagen molecules is outside of currentcomputational limit for atomistic molecular dynamics studies. Thus,our results provide insights into the effect of local changes in fibrilsassociated with mutations and cannot take into account the effect ofmutations on the fibril's folding pathways and whole hierarchicalstructure of collagen and resultant tissue.
All MD simulations were performed with the AMBER 9.0 softwarepackage [25] using the ff03 Cornell force field [26] and TIP3P watermodel [27]. The method of explicit solvation was used to accuratelymodel the interactions of water and ions with collagen. A conjugatedgradient minimization procedure was first completed for 5000 stepsfor each tropocollagen molecule. Then, using the geometry of pre‐opti-mized tropocollagen, a fibril model was built, which was furtherminimized. Once minimized, collagen fibril was placed in a largeTIP3P water box with periodic boundary conditions. Additional Na+
and Cl− ions were added to create a 0.1 M distribution of salt aroundeach collagen fibril, analogous to biological milieu.
The solvent was carefully equilibrated to produce an accuratemodeling of solvation effects. A simulation setup described in detailsin previous studieswas used [28]. Briefly, eachfibrilwasfirstminimizedin solvent for 10,000 steps, holding the molecule fixed during this timewith a constraint of 20 kcal/mol. The entire system was then heatedgradually to 300 K, still constrained. A 200 ps constant pressure andtemperature (NPT) MD run was completed while holding the fibrilfixed at 200 kcal/mol constraint. Four extra minimization procedureswere completed, each for 1000 steps, while further relaxing the con-straint on the fibril. A second NPT MD run was performed at 25 kcal/mol restraint for 20 ps, before minimizing and reheating the systemto 300 K at constant volume within 40 ps. The production simulationswere performed for 20 ns on each fibril using a 1 fs time step. TheParticle Mesh Ewald (PME) summation method was used to calculatethe electrostatic potential [29] with periodic boundary conditionsapplied in all directions. The non-bonded interactions were cut off at9 Å with a 0.00001 tolerance of Ewald convergence. The temperaturewasmaintained at 300 K using a Berendsen thermostat [30]. All compu-tational simulations were completed on an IBM Linux cluster with dualXeon computing nodes using 8 or 16 Intel Xeon processors per simula-tion. Analysis was completed using AMBER 9.0 tools such as PTRAJ andin-house scripts.
2.1. Determination of mechanical properties
Steeredmolecular dynamics (SMD) simulations can computational-ly emulate the experimental biomacromolecular pulling experiencedwith AFM and optical tweezers [17]. We used SMD to determine howmutations affect the mechanical properties of collagen, as well as todetermine how collagen behaves under normal physiological stress. InSMD simulations harmonic force F(t)=k0[z(t)−z(0)] was applied tothe terminal residues of the collagen, where z(0) is the initial positionand z(t) is the position at time t. The main advantage of SMD is thatthe structural changes and mechanical properties can be determinedand monitored throughout the simulation on an atomistic scale,which are hard to achieve in experimental procedures. However, SMD
does not allow direct comparison with experimental results, as compu-tational predictions are sensitive to the pulling velocities and statisticalensemble of large samplings are required to obtain meaningful result[17].
Two types of SMD simulations were completed in this study forthe wild‐type and for each type of mutation. In the first type we esti-mated the elastic moduli of the tropocollagen by pulling terminalends of all collagens in the fibril in opposite direction with a forceconstant of 40 kcal/mol. Tropocollagen mechanical properties areknown to depend on the pulling rate [17]. We have tested severalpulling rates and chose 1.3 m/s, which is on the upper limit of thepredicted acceptable range for tropocollagen molecule when Youngmodulus is independent of the loading rate [17]. The elastic, or ten-sile, modulus (E) was determined using the following equation:
E ¼ FLoAoΔL
where F is the applied force, Lo is the initial length of the tropocolla-gen molecule, Ao is the initial cross-sectional area, and ΔL is thechange in length of the tropocollagen molecule.
In the second type, we estimated collagen shear strength bypulling the central collagen molecule out at 100 kcal/mol force con-stant to counter intra-strand non‐bonded interactions of the neigh-boring collagens. Because collagen fibrils are anisotropic, theresponse to a shear stress is a measure of the internal strength ofthe collagen fibril, and therefore of the tropocollagen molecule[1,31]. The shear modulus (G) was estimated using the followingequation:
G ¼ FLoAΔx
where F is the applied force, Lo is the initial length of the tropocollagen, Ais the area onwhich the force acts, andΔx is the transverse displacementalong the pulling direction.
The binding energy for each collagen system was computed usingthe Molecular Mechanics-Poisson Boltzmann Surface Area (MM-PBSA)and the MM/GBSA (Generalized Born/Surface Area (LCPO)) approach[32] as implemented inMM‐PBSAmodule for the last 2 ns. The bindingenergies were computed as follows:
ΔE binding = E (fibril)− [E (central tropocollagen)+E (neighboringtropocollagens)];
E=Einternal (bond, angle, dihedral)+Eele+EvdW+EPB+Enp;EPB the electrostatic contribution to the solvation energy calculat-
ed by PB;Eele electrostatic energy as calculated by the MM force field;EvdW van der Waals contribution as calculated by the MM force
field;Enp nonpolar contribution to the solvation energy calculated
with an empirical model.
3. Results and discussion
The root mean square deviation (RMSD) from the initial structurecan beused as an indicator for stability of afibril (Fig. 2). Themost stablestructure is of fibril without mutations, as expected. Fibrils with thearginine (Arg) and lysine (Lys) mutations in a position of Hyp hadmuch larger deviations from the wild‐type fibril than any other muta-tion. The deviation could be due to the positively charged nature ofthese amino acids combined with their size and polarity. Lysine andarginine are easily twice as large as the hydroxyproline amino acidand are also the two most hydrophilic residues used in this study. Thehydrophobicity index of arginine is −4.5 while that of lysine is −3.9
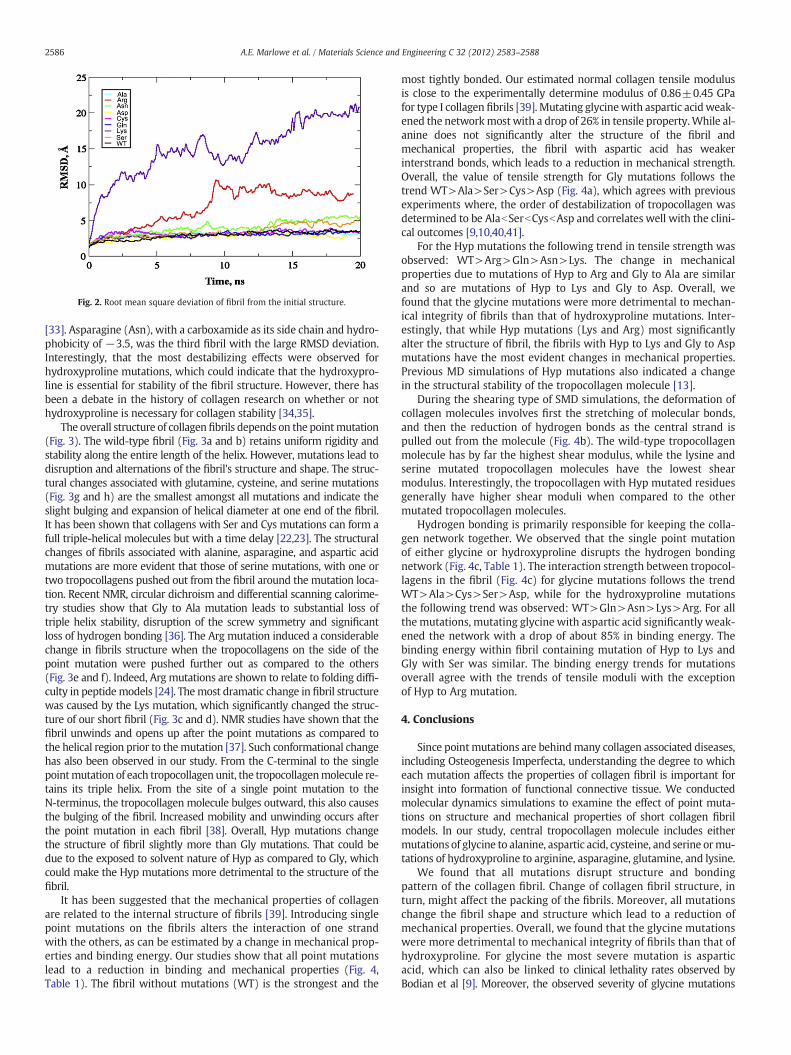
Fig. 2. Root mean square deviation of fibril from the initial structure.
2586 A.E. Marlowe et al. / Materials Science and Engineering C 32 (2012) 2583–2588
[33]. Asparagine (Asn), with a carboxamide as its side chain and hydro-phobicity of −3.5, was the third fibril with the large RMSD deviation.Interestingly, that the most destabilizing effects were observed forhydroxyproline mutations, which could indicate that the hydroxypro-line is essential for stability of the fibril structure. However, there hasbeen a debate in the history of collagen research on whether or nothydroxyproline is necessary for collagen stability [34,35].
The overall structure of collagenfibrils depends on the pointmutation(Fig. 3). The wild-type fibril (Fig. 3a and b) retains uniform rigidity andstability along the entire length of the helix. However, mutations lead todisruption and alternations of the fibril's structure and shape. The struc-tural changes associated with glutamine, cysteine, and serine mutations(Fig. 3g and h) are the smallest amongst all mutations and indicate theslight bulging and expansion of helical diameter at one end of the fibril.It has been shown that collagens with Ser and Cys mutations can form afull triple-helical molecules but with a time delay [22,23]. The structuralchanges of fibrils associated with alanine, asparagine, and aspartic acidmutations are more evident that those of serine mutations, with one ortwo tropocollagens pushed out from the fibril around the mutation loca-tion. Recent NMR, circular dichroism and differential scanning calorime-try studies show that Gly to Ala mutation leads to substantial loss oftriple helix stability, disruption of the screw symmetry and significantloss of hydrogen bonding [36]. The Arg mutation induced a considerablechange in fibrils structure when the tropocollagens on the side of thepoint mutation were pushed further out as compared to the others(Fig. 3e and f). Indeed, Arg mutations are shown to relate to folding diffi-culty in peptidemodels [24]. Themost dramatic change in fibril structurewas caused by the Lys mutation, which significantly changed the struc-ture of our short fibril (Fig. 3c and d). NMR studies have shown that thefibril unwinds and opens up after the point mutations as compared tothe helical region prior to themutation [37]. Such conformational changehas also been observed in our study. From the C-terminal to the singlepointmutation of each tropocollagen unit, the tropocollagenmolecule re-tains its triple helix. From the site of a single point mutation to theN-terminus, the tropocollagen molecule bulges outward, this also causesthe bulging of the fibril. Increased mobility and unwinding occurs afterthe point mutation in each fibril [38]. Overall, Hyp mutations changethe structure of fibril slightly more than Gly mutations. That could bedue to the exposed to solvent nature of Hyp as compared to Gly, whichcould make the Hyp mutations more detrimental to the structure of thefibril.
It has been suggested that the mechanical properties of collagenare related to the internal structure of fibrils [39]. Introducing singlepoint mutations on the fibrils alters the interaction of one strandwith the others, as can be estimated by a change in mechanical prop-erties and binding energy. Our studies show that all point mutationslead to a reduction in binding and mechanical properties (Fig. 4,Table 1). The fibril without mutations (WT) is the strongest and the
most tightly bonded. Our estimated normal collagen tensile modulusis close to the experimentally determine modulus of 0.86±0.45 GPafor type I collagen fibrils [39]. Mutating glycinewith aspartic acidweak-ened the networkmostwith a drop of 26% in tensile property.While al-anine does not significantly alter the structure of the fibril andmechanical properties, the fibril with aspartic acid has weakerinterstrand bonds, which leads to a reduction in mechanical strength.Overall, the value of tensile strength for Gly mutations follows thetrend WT>Ala>Ser>Cys>Asp (Fig. 4a), which agrees with previousexperiments where, the order of destabilization of tropocollagen wasdetermined to be AlabSerbCysbAsp and correlates well with the clini-cal outcomes [9,10,40,41].
For the Hyp mutations the following trend in tensile strength wasobserved: WT>Arg>Gln>Asn>Lys. The change in mechanicalproperties due to mutations of Hyp to Arg and Gly to Ala are similarand so are mutations of Hyp to Lys and Gly to Asp. Overall, wefound that the glycine mutations were more detrimental to mechan-ical integrity of fibrils than that of hydroxyproline mutations. Inter-estingly, that while Hyp mutations (Lys and Arg) most significantlyalter the structure of fibril, the fibrils with Hyp to Lys and Gly to Aspmutations have the most evident changes in mechanical properties.Previous MD simulations of Hyp mutations also indicated a changein the structural stability of the tropocollagen molecule [13].
During the shearing type of SMD simulations, the deformation ofcollagen molecules involves first the stretching of molecular bonds,and then the reduction of hydrogen bonds as the central strand ispulled out from the molecule (Fig. 4b). The wild‐type tropocollagenmolecule has by far the highest shear modulus, while the lysine andserine mutated tropocollagen molecules have the lowest shearmodulus. Interestingly, the tropocollagen with Hyp mutated residuesgenerally have higher shear moduli when compared to the othermutated tropocollagen molecules.
Hydrogen bonding is primarily responsible for keeping the colla-gen network together. We observed that the single point mutationof either glycine or hydroxyproline disrupts the hydrogen bondingnetwork (Fig. 4c, Table 1). The interaction strength between tropocol-lagens in the fibril (Fig. 4c) for glycine mutations follows the trendWT>Ala>Cys>Ser>Asp, while for the hydroxyproline mutationsthe following trend was observed: WT>Gln>Asn>Lys>Arg. For allthe mutations, mutating glycine with aspartic acid significantly weak-ened the network with a drop of about 85% in binding energy. Thebinding energy within fibril containing mutation of Hyp to Lys andGly with Ser was similar. The binding energy trends for mutationsoverall agree with the trends of tensile moduli with the exceptionof Hyp to Arg mutation.
4. Conclusions
Since pointmutations are behindmany collagen associated diseases,including Osteogenesis Imperfecta, understanding the degree to whicheach mutation affects the properties of collagen fibril is important forinsight into formation of functional connective tissue. We conductedmolecular dynamics simulations to examine the effect of point muta-tions on structure and mechanical properties of short collagen fibrilmodels. In our study, central tropocollagen molecule includes eithermutations of glycine to alanine, aspartic acid, cysteine, and serine ormu-tations of hydroxyproline to arginine, asparagine, glutamine, and lysine.
We found that all mutations disrupt structure and bondingpattern of the collagen fibril. Change of collagen fibril structure, inturn, might affect the packing of the fibrils. Moreover, all mutationschange the fibril shape and structure which lead to a reduction ofmechanical properties. Overall, we found that the glycine mutationswere more detrimental to mechanical integrity of fibrils than that ofhydroxyproline. For glycine the most severe mutation is asparticacid, which can also be linked to clinical lethality rates observed byBodian et al [9]. Moreover, the observed severity of glycine mutations
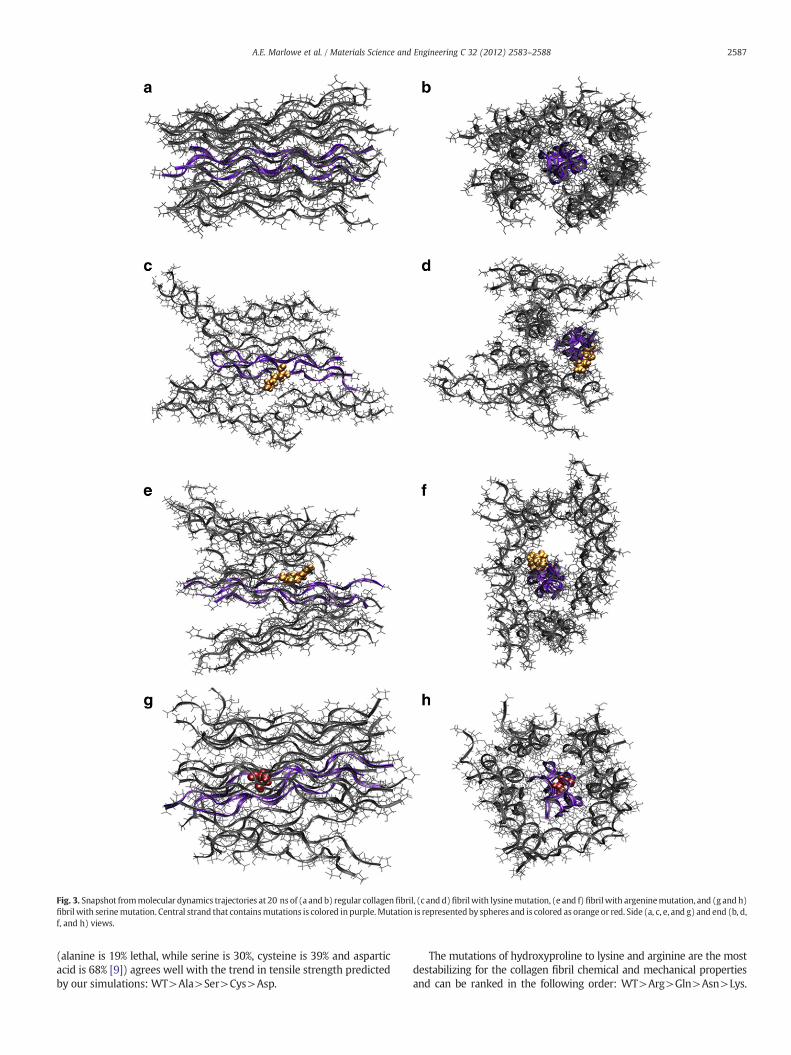
Fig. 3. Snapshot frommolecular dynamics trajectories at 20 ns of (a and b) regular collagenfibril, (c andd)fibrilwith lysinemutation, (e and f) fibrilwith argeninemutation, and (g andh)fibril with serinemutation. Central strand that containsmutations is colored in purple.Mutation is represented by spheres and is colored as orange or red. Side (a, c, e, and g) and end (b, d,f, and h) views.
2587A.E. Marlowe et al. / Materials Science and Engineering C 32 (2012) 2583–2588
(alanine is 19% lethal, while serine is 30%, cysteine is 39% and asparticacid is 68% [9]) agrees well with the trend in tensile strength predictedby our simulations: WT>Ala>Ser>Cys>Asp.
The mutations of hydroxyproline to lysine and arginine are the mostdestabilizing for the collagen fibril chemical and mechanical propertiesand can be ranked in the following order: WT>Arg>Gln>Asn>Lys.
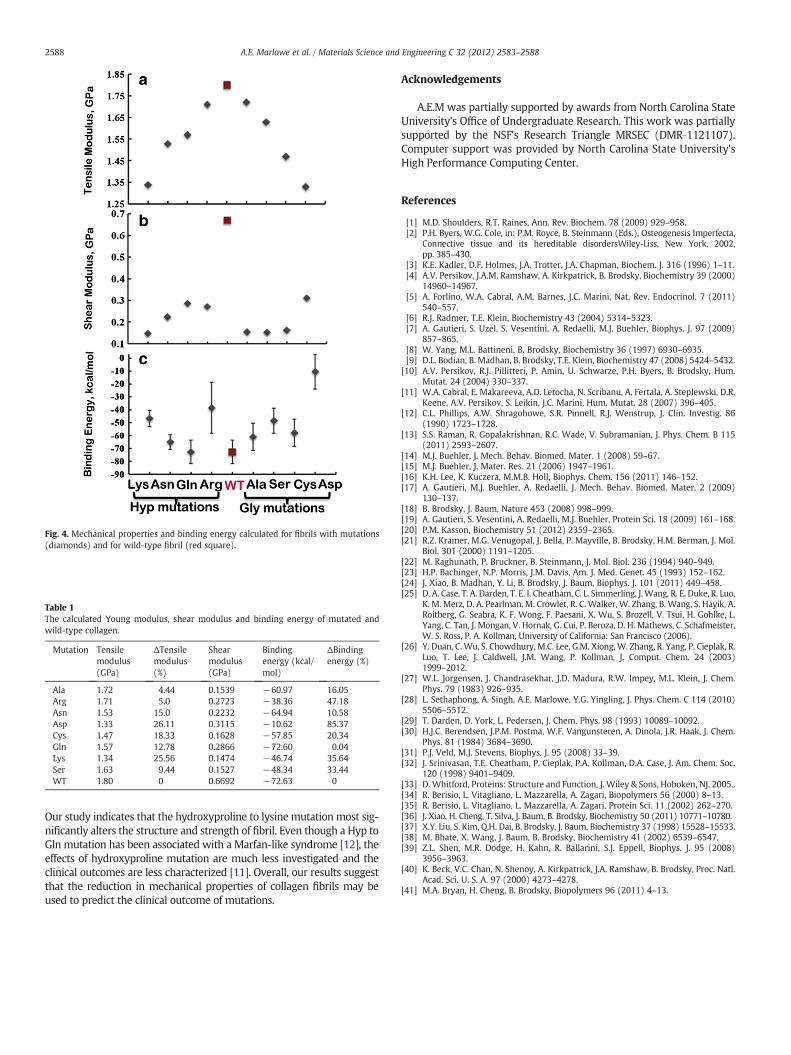
Table 1The calculated Young modulus, shear modulus and binding energy of mutated andwild-type collagen.
Mutation Tensilemodulus(GPa)
ΔTensilemodulus(%)
Shearmodulus(GPa)
Bindingenergy (kcal/mol)
ΔBindingenergy (%)
Ala 1.72 4.44 0.1539 −60.97 16.05Arg 1.71 5.0 0.2723 −38.36 47.18Asn 1.53 15.0 0.2232 −64.94 10.58Asp 1.33 26.11 0.3115 −10.62 85.37Cys 1.47 18.33 0.1628 −57.85 20.34Gln 1.57 12.78 0.2866 −72.60 0.04Lys 1.34 25.56 0.1474 −46.74 35.64Ser 1.63 9.44 0.1527 −48.34 33.44WT 1.80 0 0.6692 −72.63 0
Fig. 4. Mechanical properties and binding energy calculated for fibrils with mutations(diamonds) and for wild-type fibril (red square).
2588 A.E. Marlowe et al. / Materials Science and Engineering C 32 (2012) 2583–2588
Our study indicates that the hydroxyproline to lysine mutation most sig-nificantly alters the structure and strength of fibril. Even though a Hyp toGln mutation has been associated with a Marfan-like syndrome [12], theeffects of hydroxyproline mutation are much less investigated and theclinical outcomes are less characterized [11]. Overall, our results suggestthat the reduction in mechanical properties of collagen fibrils may beused to predict the clinical outcome of mutations.
Acknowledgements
A.E.M was partially supported by awards from North Carolina StateUniversity's Office of Undergraduate Research. This work was partiallysupported by the NSF's Research Triangle MRSEC (DMR‐1121107).Computer support was provided by North Carolina State University'sHigh Performance Computing Center.
References
[1] M.D. Shoulders, R.T. Raines, Ann. Rev. Biochem. 78 (2009) 929–958.[2] P.H. Byers, W.G. Cole, in: P.M. Royce, B. Steinmann (Eds.), Osteogenesis Imperfecta,
Connective tissue and its hereditable disordersWiley-Liss, New York, 2002,pp. 385–430.
[3] K.E. Kadler, D.F. Holmes, J.A. Trotter, J.A. Chapman, Biochem. J. 316 (1996) 1–11.[4] A.V. Persikov, J.A.M. Ramshaw, A. Kirkpatrick, B. Brodsky, Biochemistry 39 (2000)
14960–14967.[5] A. Forlino, W.A. Cabral, A.M. Barnes, J.C. Marini, Nat. Rev. Endocrinol. 7 (2011)
540–557.[6] R.J. Radmer, T.E. Klein, Biochemistry 43 (2004) 5314–5323.[7] A. Gautieri, S. Uzel, S. Vesentini, A. Redaelli, M.J. Buehler, Biophys. J. 97 (2009)
857–865.[8] W. Yang, M.L. Battineni, B. Brodsky, Biochemistry 36 (1997) 6930–6935.[9] D.L. Bodian, B. Madhan, B. Brodsky, T.E. Klein, Biochemistry 47 (2008) 5424–5432.
[10] A.V. Persikov, R.J. Pillitteri, P. Amin, U. Schwarze, P.H. Byers, B. Brodsky, Hum.Mutat. 24 (2004) 330–337.
[11] W.A. Cabral, E. Makareeva, A.D. Letocha, N. Scribanu, A. Fertala, A. Steplewski, D.R.Keene, A.V. Persikov, S. Leikin, J.C. Marini, Hum. Mutat. 28 (2007) 396–405.
[12] C.L. Phillips, A.W. Shragohowe, S.R. Pinnell, R.J. Wenstrup, J. Clin. Investig. 86(1990) 1723–1728.
[13] S.S. Raman, R. Gopalakrishnan, R.C. Wade, V. Subramanian, J. Phys. Chem. B 115(2011) 2593–2607.
[14] M.J. Buehler, J. Mech. Behav. Biomed. Mater. 1 (2008) 59–67.[15] M.J. Buehler, J. Mater. Res. 21 (2006) 1947–1961.[16] K.H. Lee, K. Kuczera, M.M.B. Holl, Biophys. Chem. 156 (2011) 146–152.[17] A. Gautieri, M.J. Buehler, A. Redaelli, J. Mech. Behav. Biomed. Mater. 2 (2009)
130–137.[18] B. Brodsky, J. Baum, Nature 453 (2008) 998–999.[19] A. Gautieri, S. Vesentini, A. Redaelli, M.J. Buehler, Protein Sci. 18 (2009) 161–168.[20] P.M. Kasson, Biochemistry 51 (2012) 2359–2365.[21] R.Z. Kramer, M.G. Venugopal, J. Bella, P. Mayville, B. Brodsky, H.M. Berman, J. Mol.
Biol. 301 (2000) 1191–1205.[22] M. Raghunath, P. Bruckner, B. Steinmann, J. Mol. Biol. 236 (1994) 940–949.[23] H.P. Bachinger, N.P. Morris, J.M. Davis, Am. J. Med. Genet. 45 (1993) 152–162.[24] J. Xiao, B. Madhan, Y. Li, B. Brodsky, J. Baum, Biophys. J. 101 (2011) 449–458.[25] D. A. Case, T. A. Darden, T. E. I. Cheatham, C. L. Simmerling, J.Wang, R. E. Duke, R. Luo,
K. M. Merz, D. A. Pearlman, M. Crowlet, R. C. Walker, W. Zhang, B. Wang, S. Hayik, A.Roitberg, G. Seabra, K. F. Wong, F. Paesani, X. Wu, S. Brozell, V. Tsui, H. Gohlke, L.Yang, C. Tan, J. Mongan, V. Hornak, G. Cui, P. Beroza, D. H. Mathews, C. Schafmeister,W. S. Ross, P. A. Kollman, University of California: San Francisco (2006).
[26] Y. Duan, C.Wu, S. Chowdhury, M.C. Lee, G.M. Xiong, W. Zhang, R. Yang, P. Cieplak, R.Luo, T. Lee, J. Caldwell, J.M. Wang, P. Kollman, J. Comput. Chem. 24 (2003)1999–2012.
[27] W.L. Jorgensen, J. Chandrasekhar, J.D. Madura, R.W. Impey, M.L. Klein, J. Chem.Phys. 79 (1983) 926–935.
[28] L. Sethaphong, A. Singh, A.E. Marlowe, Y.G. Yingling, J. Phys. Chem. C 114 (2010)5506–5512.
[29] T. Darden, D. York, L. Pedersen, J. Chem. Phys. 98 (1993) 10089–10092.[30] H.J.C. Berendsen, J.P.M. Postma, W.F. Vangunsteren, A. Dinola, J.R. Haak, J. Chem.
Phys. 81 (1984) 3684–3690.[31] P.J. Veld, M.J. Stevens, Biophys. J. 95 (2008) 33–39.[32] J. Srinivasan, T.E. Cheatham, P. Cieplak, P.A. Kollman, D.A. Case, J. Am. Chem. Soc.
120 (1998) 9401–9409.[33] D. Whitford, Proteins: Structure and Function, J. Wiley & Sons, Hoboken, NJ, 2005..[34] R. Berisio, L. Vitagliano, L. Mazzarella, A. Zagari, Biopolymers 56 (2000) 8–13.[35] R. Berisio, L. Vitagliano, L. Mazzarella, A. Zagari, Protein Sci. 11 (2002) 262–270.[36] J. Xiao, H. Cheng, T. Silva, J. Baum, B. Brodsky, Biochemistry 50 (2011) 10771–10780.[37] X.Y. Liu, S. Kim, Q.H. Dai, B. Brodsky, J. Baum, Biochemistry 37 (1998) 15528–15533.[38] M. Bhate, X. Wang, J. Baum, B. Brodsky, Biochemistry 41 (2002) 6539–6547.[39] Z.L. Shen, M.R. Dodge, H. Kahn, R. Ballarini, S.J. Eppell, Biophys. J. 95 (2008)
3956–3963.[40] K. Beck, V.C. Chan, N. Shenoy, A. Kirkpatrick, J.A. Ramshaw, B. Brodsky, Proc. Natl.
Acad. Sci. U. S. A. 97 (2000) 4273–4278.[41] M.A. Bryan, H. Cheng, B. Brodsky, Biopolymers 96 (2011) 4–13.
Copyright © 2022 FDOKUMEN




















