
The application of DNA microarrays in gene expression analysis
10
Journal of Biotechnology 78 (2000) 271 – 280 The application of DNA microarrays in gene expression analysis Nicole L.W. van Hal a, *, Oscar Vorst b , Ade `le M.M.L. van Houwelingen b , Esther J. Kok a , Ad Peijnenburg a , Asaph Aharoni b , Arjen J. van Tunen b , Jaap Keijer a a RIKILT (State Institute for Quality Control of Agricultural Products), Gene Expression and Detection Centre, PO Box 230, 6700 AE Wageningen, The Netherlands b Plant Research International, Gene Expression and Detection Centre, PO Box 16, 6700 AA Wageningen, The Netherlands Received 12 February 1999; received in revised form 25 June 1999; accepted 6 July 1999 Abstract DNA microarray technology is a new and powerful technology that will substantially increase the speed of molecular biological research. This paper gives a survey of DNA microarray technology and its use in gene expression studies. The technical aspects and their potential improvements are discussed. These comprise array manufacturing and design, array hybridisation, scanning, and data handling. Furthermore, it is discussed how DNA microarrays can be applied in the working fields of: safety, functionality and health of food and gene discovery and pathway engineering in plants. © 2000 Elsevier Science B.V. All rights reserved. Keywords: DNA microarray technology; Gene expression; Food safety; Gene discovery; Pathway engineering; Review www.elsevier.com/locate/jbiotec 1. Introduction Analysis of gene expression is important in many fields of biological research, since changes in the physiology of an organism or a cell will be accompanied by changes in the pattern of gene expression. For example, insight into the func- tional and toxicological characteristics of com- pounds may be derived from their effects on gene expression. Expression studies on clinical samples, normal versus diseased, may lead to the identifica- tion of novel biomarkers. Similarly, gene expres- sion analysis can be used to obtain insight in the physiological consequences of genetic modifica- tion in plants. Several techniques for the analysis of gene ex- pression at the mRNA-level are available, such as Northern blotting (Alwine et al., 1977), differen- tial display (Liang and Pardee, 1992), serial analy- Based on a poster presentation at the ‘First European Symposium on Applied Genome Research’, Brussels, Novem- ber 1998. * Corresponding author. Tel.: +31-317-475561; fax: +31- 317-417717. E-mail address: [email protected] (N.L.W. van Hal) 0168-1656/00/$ - see front matter © 2000 Elsevier Science B.V. All rights reserved. PII:S0168-1656(00)00204-2
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of The application of DNA microarrays in gene expression analysis

Journal of Biotechnology 78 (2000) 271–280
The application of DNA microarrays in gene expressionanalysis�
Nicole L.W. van Hal a,*, Oscar Vorst b, Adele M.M.L. van Houwelingen b,Esther J. Kok a, Ad Peijnenburg a, Asaph Aharoni b, Arjen J. van Tunen b,
Jaap Keijer a
a RIKILT (State Institute for Quality Control of Agricultural Products), Gene Expression and Detection Centre, PO Box 230,6700 AE Wageningen, The Netherlands
b Plant Research International, Gene Expression and Detection Centre, PO Box 16, 6700 AA Wageningen, The Netherlands
Received 12 February 1999; received in revised form 25 June 1999; accepted 6 July 1999
Abstract
DNA microarray technology is a new and powerful technology that will substantially increase the speed ofmolecular biological research. This paper gives a survey of DNA microarray technology and its use in gene expressionstudies. The technical aspects and their potential improvements are discussed. These comprise array manufacturingand design, array hybridisation, scanning, and data handling. Furthermore, it is discussed how DNA microarrays canbe applied in the working fields of: safety, functionality and health of food and gene discovery and pathwayengineering in plants. © 2000 Elsevier Science B.V. All rights reserved.
Keywords: DNA microarray technology; Gene expression; Food safety; Gene discovery; Pathway engineering; Review
www.elsevier.com/locate/jbiotec
1. Introduction
Analysis of gene expression is important inmany fields of biological research, since changesin the physiology of an organism or a cell will beaccompanied by changes in the pattern of gene
expression. For example, insight into the func-tional and toxicological characteristics of com-pounds may be derived from their effects on geneexpression. Expression studies on clinical samples,normal versus diseased, may lead to the identifica-tion of novel biomarkers. Similarly, gene expres-sion analysis can be used to obtain insight in thephysiological consequences of genetic modifica-tion in plants.
Several techniques for the analysis of gene ex-pression at the mRNA-level are available, such asNorthern blotting (Alwine et al., 1977), differen-tial display (Liang and Pardee, 1992), serial analy-
� Based on a poster presentation at the ‘First EuropeanSymposium on Applied Genome Research’, Brussels, Novem-ber 1998.
* Corresponding author. Tel.: +31-317-475561; fax: +31-317-417717.
E-mail address: [email protected] (N.L.W. vanHal)
0168-1656/00/$ - see front matter © 2000 Elsevier Science B.V. All rights reserved.
PII: S 0168 -1656 (00 )00204 -2

N.L.W. 6an Hal et al. / Journal of Biotechnology 78 (2000) 271–280272
sis of gene expression (SAGE; Velculescu et al.,1995) and dotblot analysis (Lennon and Lehrach,1991). However, these methods each have theirdisadvantages, which render them unsuitable iflarge numbers of expression products have to beanalysed simultaneously. Northern blot analysisonly allows limited numbers of mRNAs to bestudied at the same time. Differential display doesenable the simultaneous detection of multiple dif-ferences in gene expression. However, only a lim-ited number of different conditions can becompared, the method is not quantitative andscreening is based on differences in mRNA lengthand not identity. SAGE is laborious. It involvescomplex sample preparation procedures, requiresextensive DNA sequencing and is not very sensi-tive. Finally, dotblot analysis requires a relativelylarge amount of material due to the size of thefilters.
Recently, substantial improvement in sensitivityand throughput of expression screening has beenobtained by the introduction of DNA microarraytechnology (reviewed in: Watson et al., 1998;Duggan et al., 1999; Graves, 1999). The study ofgene expression by DNA microarray technology,which is still in development, is based on hybridi-
sation of mRNA to a high-density array of immo-bilised target sequences, each corresponding to aspecific gene. Sample mRNAs are labelled as acomplex mixture, usually by incorporation of afluorescent nucleotide by oligo(dT)-primed reversetranscription. The labelled pool of sample mR-NAs is subsequently hybridised to the array,where each messenger will quantitatively hybridiseto its complementary target sequence. After wash-ing, the fluorescence at each spot on the array is aquantitative measure corresponding to the expres-sion level of the particular gene. The use of twodifferently labelled mRNA samples allows quanti-tative comparison of gene expression in both sam-ples (Fig. 1). The major advance of DNAmicroarray technology, as compared to conven-tional techniques, results from the small size ofthe array, which allows for a higher sensitivity,enables the parallel screening of larger numbers ofgenes and provides the opportunity to use smalleramounts of starting material. Mainly, the intro-duction of fluorescent probes has made miniaturi-sation of arrays possible (Jordan, 1998). The scaleof gene expression analysis is not only extendedby the simultaneous analysis of large numbers ofgenes, but also because microarrays can be pro-duced in series facilitating comparative analysis ofa large number of samples.
In our fields of work, which focus on food andplant research, gene expression analysis withDNA microarrays is a powerful tool. In bothfields, understanding of the pathways and interac-tions that occur in cells and organisms is animportant aspect. DNA microarray technology isexpected to be useful in for example: (i) riskassessment in transgenic agricultural products byanalysis of altered gene expression; (ii) investiga-tion of functional and toxicological effects of foodcomponents; and (iii) unravelling gene functionand metabolic pathways in plants by mutant anal-ysis. Although DNA microarray technology canalso be used for detection and identification ofcomplex samples, this review will focus on its usein gene expression analysis. Next to a number oftechnical aspects, the power and putative im-provements of this technology will be discussedand illustrated by some of the above-mentionedapplications.
Fig. 1. The principle of gene expression analysis by DNAmicroarray technology. mRNA samples are reverse tran-scribed to cDNA while fluorescently labeled nucleotides areincoporated (Cy3 and Cy5 labelled dCTP or dUTP are oftenused for this purpose). The usage of multiple dyes allows thecomparison of multiple RNA samples on one single array.

N.L.W. 6an Hal et al. / Journal of Biotechnology 78 (2000) 271–280 273
Fig. 2. Three approaches for DNA microarray manufacturing.
2. Manufacturing of DNA microarrays
A number of techniques are being developedfor manufacturing DNA microarrays. Two mainapproaches exist. The first approach encompassesdirect synthesis of oligonucleotides on a solidsurface. Best known is synthesis based on pho-tolithography as has been developed by Fodor etal. (1991). Specific areas of a glass surface, deriva-tised with linker molecules carrying a photo-labileprotective group, are selectively illuminated byusing a photomask. Subsequently, the surface isincubated with a solution containing a photopro-tected nucleotide, which will only be coupled tothe light-activated areas. After removal of theexcess nucleotide, a second photomask is used todeprotect other areas on the surface and subse-quently another type of nucleotide is coupled tothese areas. By repeating this procedure, a definedset of oligonucleotides is synthesised on the sur-face (Fig. 2; Chee et al., 1996; McGall et al., 1996;Lipshutz et al., 1999). The method allows themanufacturing of microarrays with very high den-sities (\250 000 oligonucleotide spots per cm2)and facilitates the production of large series ofidentical arrays. Unfortunately, the design of thistype of array, also named a DNA chip, is pro-hibitively expensive, has no flexibility in design
and only a limited number of different arrays ison the market yet.
The second approach, DNA micro-dispensing,is more flexible and can be performed in a regularmolecular biology laboratory (Schena et al., 1995full protocol is available at: http://cmgm.stanford.edu/pbrown/protocols.html). Small quantities ofDNA solution, with a minimum volume of ap-proximately 50 pl, are dispensed onto a solidsurface. The number of micro-dispensing robotscommercially available is quickly increasing andthe performance of these machines is continuallyimproving. They can be subdivided into two maincategories: passive dispensers and active dispens-ing units based on ink-jet technology (Fig. 2).Passive dispensers apply the DNA solution witha pin (or multiple pins) that touches the solidsurface. Ink-jet based devices use either piezoe-lectric delivery or delivery by solenoid valves,making direct surface contact redundant. Thedensity of the spots depends on the skills of thedispensing device. However, generally a higherdensity can be reached when using a passive dis-penser.
DNA dispensing is flexible and allows for con-stant update of the array. Oligonucleotides as wellas longer known or unknown DNA sequences canbe deposited on the array in a format that can

N.L.W. 6an Hal et al. / Journal of Biotechnology 78 (2000) 271–280274
vary, if necessary, from one array to the next.Using micro-dispensing it should also be possibleto synthesise oligonucleotides directly on an arrayor to deposit molecules other than nucleic acids,for example proteins. Here, we will only deal withDNA expression arrays.
The DNA molecules can be dispensed ontodifferent kinds of surface (e.g. glass or mem-branes) to which the DNA is then fixed. In thecase of glass-surfaces a positively charged layer(e.g. poly-L-lysine) can be used to bind the nega-tively charged DNA fragments (Schena et al.,1995; Shalon et al., 1996). However, these interac-tions reduce the conformational freedom of thebound DNA and will therefore also decrease theaffinity for complementary molecules in solution.Alternatively, DNA molecules that carry a 5%modification can be covalently bound to a glasssurface that carries reactive groups (Schena et al.,1996; Rogers et al., 1999). Presently surface-modified glass slides (silylated, poly-L-lysinecoated) are most commonly used as a substrate.Besides glass, other materials are being explored
as well, such as gold coated slides (http://www.interactiva.de) or materials which will forma 3-dimensional matrix, e.g. polyacrylamide gelpads, nitrocellulose or nylon membranes, nitrocel-lulose coated slides and other polymers (Proud-nikov et al., 1998). These alternatives aim atbetter signal to noise ratios, higher binding capac-ity, reusability and improved reproducibility.
DNA microarray technology is rapidly evolvingin many aspects. Improvements such as increasedspot density, increased reproducibility, and re-duced production time will certainly be driven bythe needs of especially pharmaceutical companiesfor high throughput screening systems. To analysegene expression in human/animal biopsies, mi-crodissected specimens, or small plant tissues, theoverall sensitivity has to be increased. This willdepend on technical aspects such as the quality ofthe scanners and arrayers, fluorescent dyes withimproved quantum yields and lower background,or target supports with reduced background andmore target sequence binding capacity. Improve-ment may also be found in alternative samplingpreparation, labelling and coupling of DNA tothe array, as well as in the use of peptide nucleicacid (PNA). This is a synthetic polynucleotidethat has an uncharged protein-like backbone,which may improve hybridisation characteristics(Egholm et al., 1993; Wittung et al., 1996; Geigeret al., 1998).
3. Microarray design
Design of a microarray is largely dependent onthe research question that has to be answered.Diagnostic questions will often involve smallerarrays with well defined target sequences, whileelucidation of metabolic pathways or identifica-tion of novel responding genes necessitates the useof large arrays often with undefined target se-quences. Compared to conventional methods,DNA microarrays have the advantage that ex-pression of large sets of genes can be determinedin parallel (Fig. 3; for examples of applicationssee: Spellman et al., 1998; Iyer et al., 1999). If thenumber of genes of an organism is not too largeand all the genes are known, sequences corre-
Fig. 3. Two-color image of a Petunia hybrida cDNA array.The array consists of PCR-amplified random cDNA clonesspotted on glass; spot diameter 500 mm. A sample mixture ofCy3 labelled leaf mRNA and Cy5 labelled flower mRNA washybridised. After scanning for each fluorescent dye separatelyfalse colour images (red for Cy3, green for Cy5) were superim-posed. Red dots correspond to genes higher expressed in leafthan in flower tissue, green dots to genes that are higherexpressed in flower tissue. Yellow dots represent genes thatshow no differential expression. The spot indicated by thearrow corresponds to a photosynthesis-specific rubisco acti-vase gene, showing a higher expression in leaf.
N.L.W. 6an Hal et al. / Journal of Biotechnology 78 (2000) 271–280 275
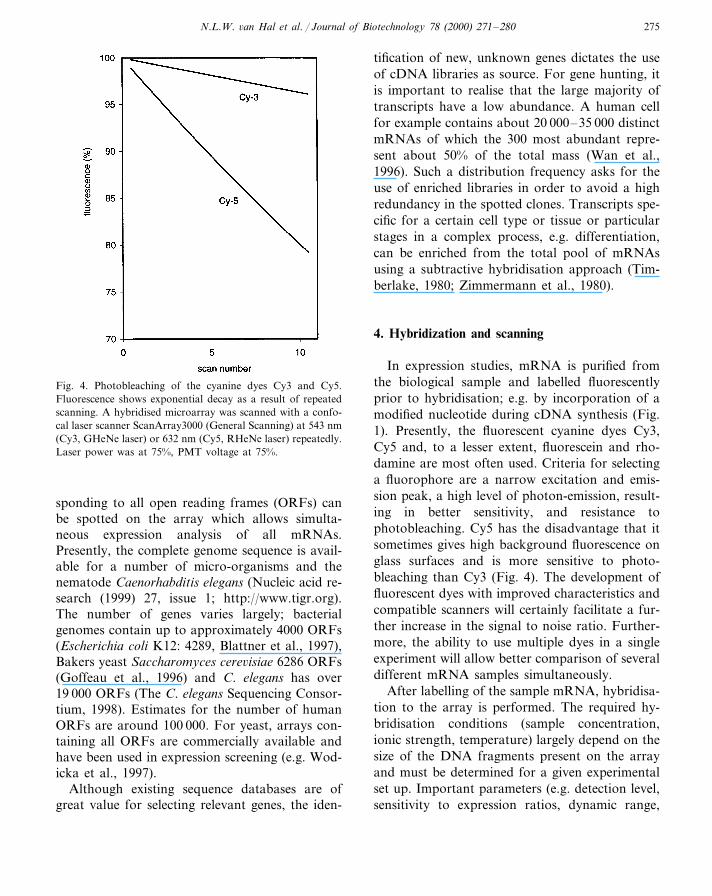
Fig. 4. Photobleaching of the cyanine dyes Cy3 and Cy5.Fluorescence shows exponential decay as a result of repeatedscanning. A hybridised microarray was scanned with a confo-cal laser scanner ScanArray3000 (General Scanning) at 543 nm(Cy3, GHeNe laser) or 632 nm (Cy5, RHeNe laser) repeatedly.Laser power was at 75%, PMT voltage at 75%.
tification of new, unknown genes dictates the useof cDNA libraries as source. For gene hunting, itis important to realise that the large majority oftranscripts have a low abundance. A human cellfor example contains about 20 000–35 000 distinctmRNAs of which the 300 most abundant repre-sent about 50% of the total mass (Wan et al.,1996). Such a distribution frequency asks for theuse of enriched libraries in order to avoid a highredundancy in the spotted clones. Transcripts spe-cific for a certain cell type or tissue or particularstages in a complex process, e.g. differentiation,can be enriched from the total pool of mRNAsusing a subtractive hybridisation approach (Tim-berlake, 1980; Zimmermann et al., 1980).
4. Hybridization and scanning
In expression studies, mRNA is purified fromthe biological sample and labelled fluorescentlyprior to hybridisation; e.g. by incorporation of amodified nucleotide during cDNA synthesis (Fig.1). Presently, the fluorescent cyanine dyes Cy3,Cy5 and, to a lesser extent, fluorescein and rho-damine are most often used. Criteria for selectinga fluorophore are a narrow excitation and emis-sion peak, a high level of photon-emission, result-ing in better sensitivity, and resistance tophotobleaching. Cy5 has the disadvantage that itsometimes gives high background fluorescence onglass surfaces and is more sensitive to photo-bleaching than Cy3 (Fig. 4). The development offluorescent dyes with improved characteristics andcompatible scanners will certainly facilitate a fur-ther increase in the signal to noise ratio. Further-more, the ability to use multiple dyes in a singleexperiment will allow better comparison of severaldifferent mRNA samples simultaneously.
After labelling of the sample mRNA, hybridisa-tion to the array is performed. The required hy-bridisation conditions (sample concentration,ionic strength, temperature) largely depend on thesize of the DNA fragments present on the arrayand must be determined for a given experimentalset up. Important parameters (e.g. detection level,sensitivity to expression ratios, dynamic range,
sponding to all open reading frames (ORFs) canbe spotted on the array which allows simulta-neous expression analysis of all mRNAs.Presently, the complete genome sequence is avail-able for a number of micro-organisms and thenematode Caenorhabditis elegans (Nucleic acid re-search (1999) 27, issue 1; http://www.tigr.org).The number of genes varies largely; bacterialgenomes contain up to approximately 4000 ORFs(Escherichia coli K12: 4289, Blattner et al., 1997),Bakers yeast Saccharomyces cere6isiae 6286 ORFs(Goffeau et al., 1996) and C. elegans has over19 000 ORFs (The C. elegans Sequencing Consor-tium, 1998). Estimates for the number of humanORFs are around 100 000. For yeast, arrays con-taining all ORFs are commercially available andhave been used in expression screening (e.g. Wod-icka et al., 1997).
Although existing sequence databases are ofgreat value for selecting relevant genes, the iden-

N.L.W. 6an Hal et al. / Journal of Biotechnology 78 (2000) 271–280276
and the correlation between transcript concentra-tion and hybridisation signal) can be determinedby spiking control RNA transcripts in a back-ground of a total messenger population. Work ofothers (Lemieux et al., 1998) has indicated thatthe RNA expression level and hybridisation signalshow a linear correlation providing that the targetDNA is present on the array in at least a ten-foldexcess. The correlation was found to be linear forRNA concentrations ranging from one copy in300 000 till about one copy in 3000 transcripts(Lockhart et al., 1996). The detection limit isexpected to differ per experimental setting. Detec-tion levels of one in 100 000 and one in 300 000have been reported (Lockhart et al., 1996; John-ston, 1998), which implies that it will be possibleto detect about one copy per one to ten cells.Furthermore, it has been estimated by severalgroups that a two to three fold difference in geneexpression will still be detectable (Schena et al.,1996; Ruan et al., 1998). Our own results showthat a four to five fold difference in expressioncan be detected with about 95% probability, whilefor a two to three fold difference the probability isabout 50%.
For imaging, several types of microarray read-ers are on the market. They can be subdividedinto CCD cameras, non-confocal laser scannersand confocal laser scanners. CCD cameras allowfast scanning but their imaging area is rathersmall (max. 1 cm2). Confocal laser scanners havethe advantage that the light collection efficiencyand resolution is usually much higher than for theother two systems. In addition, confocal laserscanners have a small depth of focus that reducesartefacts but also requires more scanning preci-sion. In general, to obtain a reliable quantitativeresult the microarray spot diameter should be atleast five to ten times the pixel size, which is atthis moment about 10 mm for confocal laserscanners.
Hybridisation may be improved by variationsin existing protocols, but also by developing newapproaches, such as the use of electric fields toenhance the hybridisation rate and precisely regu-late the stringency of the hybridisation (Edman etal., 1997; Sosnowski et al., 1997).
5. Data analysis
DNA microarrays produce huge quantities ofdata, which pose a major bottleneck (Ermolaevaet al., 1998). The complex hybridisation data aregenerated within a short time and sophisticatedsoftware is needed to keep a good overview, toassess the quality of the data and to help findstatistical significance and relevant correlationswithin, and between different arrays and experi-ments. Furthermore, integration of informationfrom different sources should be facilitated: link-ing microarray-derived gene expression data toDNA sequence information and experimentaldata available in separate (public) databases. Forexample, one should be able to find genes that arepart of the same metabolic route, compare theirexpression behaviour under all the tested condi-tions and present the data in an orderly and visualway. In the case of clinical specimens, it mustfacilitate access to databases containing all patientinformation and assist in finding significant corre-lations. Finally, more practically, the softwareshould be able to trace back a spot on an array toa clone in the freezer and its sequence in adatabase.
To a limited extent these data handling soft-ware packages are commercially available butlarge improvements are still necessary. Especiallywith regard to spot location, expression patternrecognition, comparison of larger numbers of dif-ferent experimental samples, statistical analysisand visual data presentation. For example, re-cently cluster analysis was applied as a method forthe selection of genes with a similar expressionpattern (Eisen et al., 1998; http://rana.stanford.edu/clustering).
6. Applications
Within our institutes the DNA microarray tech-nology is used for a number of applications. Thefirst application is its use in the safety assessmentof genetic modification of food plants. Thisshould not only be based on the evaluation of thenewly introduced trait, but also on possible unin-tended side effects resulting from the genetic alter-

N.L.W. 6an Hal et al. / Journal of Biotechnology 78 (2000) 271–280 277
ation. Presently, the latter is done by comparisonof a limited number of individual macro- andmicro-nutrients as well as anti-nutritional factors,including natural toxins, between the transformedline and its traditional counterpart(s). However,this approach has its limitations, since it is un-known whether the currently investigated compo-nents are the only and most important factors toscreen with respect to food safety. This type ofanalysis therefore provides only limited informa-tion on the potential effects on human health.DNA microarray technology may be a good alter-native for safety evaluation. It provides the op-portunity to screen for unintended changes inexpression of large numbers of genes in an unbi-ased manner. Presently, we are testing this ap-proach for tomato. For this purpose we haveconstructed tomato cDNA libraries enriched forcDNAs preferentially expressed in either green orred tomato. Arrays containing these sequences aswell as target sequences of known genes will beused for the systematic comparison of control andgenetically modified tomatoes. This will provideinformation on the natural variation in gene ex-pression of tomato at certain stages of ripening aswell as specific changes due to genetic alteration.
A second application is the assessment of thefunctionality of food components. For many foodcomponents the biological effect on humans iscurrently unknown. In other cases, more than onefunction has been described, but their relevanceremains unclear. The phytoestrogen genestein, forexample, has estrogenic activity, anti-oxidantproperties, and is a potent tyrosine kinase in-hibitor. One way to identify relevant biologicalfunctions is by large scale expression analysis,since a large number of characters can be tested inparallel and a large number of different exposurescan be compared. We are focussing on effects offood components on the human intestine. Tostudy these effects, CaCo-2 cells, which are widelyused as an human model intestine cell line, areexposed to food components and mRNA expres-sion profiles are determined at different timepoints and at different concentrations. For thispurpose, DNA microarrays containing unknownintestine specific genes as well as known genes willbe constructed. Genes with a known function,
selected from public databases, serve as controls(e.g. intestine specificity) and as indicators for theinvolvement of different physiological pathways(e.g. apoptosis). Unknown, intestine specificgenes, obtained by the construction of enrichedcDNA libraries from differentiated and undiffer-entiated CaCo-2 cells as well as from humanintestine biopsies, serve to find new functionalindicators. Differentially expressed genes will becharacterised by sequencing and further expres-sion analysis in a number of different intestinalcell lines as well as in vivo.
The third application is the use of DNA mi-croarrays to unravel gene functions and metabolicpathways in plants by exploiting mutations. Aclassical approach for elucidating the function ofa gene is by mutant analysis. Mutant organismscan be either mutated in the native alleles or mayresult from the introduction of additional copiesof the gene leading to co-suppression or over-ex-pression of the gene in question. Phenotypic anal-ysis of such mutants can then point to thepossible function of the gene. This can be acumbersome and complicated task. Moreover,many mutations do not result in an altered phe-notype. Especially in flexible organisms, such asplants, a mutation is often overcome by physio-logical responses as a result of the altered expres-sion of other genes. DNA-microarray-assistedgene expression analysis offers a powerful tool toidentify the genes that are affected in a certainmutant. Those genes will be related, either directlyor indirectly, to the nature of the mutation andmay give an important clue in elucidating thefunction of the mutated gene. This new approachmight take away the strong bias associated withexpression analysis of only a small number ofgenes.
7. Concluding remarks
DNA microarray technology is a new and pow-erful technology that will substantially increasethe speed at which differential gene expression canbe analysed and gene functions are elucidated.Genome sequencing programs (genomics) havealready produced large amounts of sequence data,

N.L.W. 6an Hal et al. / Journal of Biotechnology 78 (2000) 271–280278
Fig. 5. Factors for consideration for further improvement of DNA microarray technology with respect to reproducibility, speed, costand sensitivity.
and will continue to do so. DNA microarraytechnology might help to answer the questionwhat the functions of these sequences are (func-tional genomics). Implementation of the tech-nique in applications like risk assesment oftransgenic plants, toxicological analysis of foodcomponents or pathway engineering is expected toaccelerate research and improve knowledge inthese fields. To fully fulfil these expectations, fur-ther improvement of the technology with respectto reproducibility, speed, cost and sensitivity willbe needed (Fig. 5). Consequently, sufficient atten-tion should be paid to the development of goodbiological model systems, which will facilitate fur-ther optimisation that is asked for in its variousapplications.
DNA microarray technology, however, limitsexpression studies to the mRNA level. Ideally, itshould be accompanied by analyses at the proteinlevel. Proteomics, large-scale parallel analysis ofthe proteins that are present in a cell, is develop-ing rapidly (Celis et al., 1998; Blackstock andWeir, 1999). Both technologies are complemen-
tary while focussing on different steps of the sameprocess: the expression of genetic information intofunctional molecules, cells and organisms. More-over, proteomics allows the detection of induciblemodifications (phosphorylation and glycosylation)of the gene products, which are not regulated atthe transcriptional level. Information obtainedfrom genomics, large-scale expression analysis,proteomics and metabolite profiling will be in-valuable to identify gene functions, pathways andinteractive cellular physiology. Ultimately, thiswill provide an interactive atlas of the biologicalprocesses that take place in a cell. Such an atlaswill help to better select pharmaceutical targets, topredict toxicological effects, or to fully employ theplant cell as a factory.
Acknowledgements
The authors would like to thank Robert Halland Harry Kuiper for critically reading themanuscript and Henk Aarts, Lies van der Wal-

N.L.W. 6an Hal et al. / Journal of Biotechnology 78 (2000) 271–280 279
Winnubst and Evelien Kramer for stimulatingdiscussions.
References
Alwine, J.C., Kemp, D.J., Stark, G.R., 1977. Method fordetection of specific RNAs in agarose gels by transfer todiazobenzyloxymethyl-paper and hybridization with DNAprobes. Proc. Natl. Acad. Sci. USA 74, 5350–5354.
Blackstock, W.P., Weir, M.P., 1999. Proteomics: quantitativeand physical mapping of cellular proteins. Trends Biotech-nol. 17, 121–127.
Blattner, F.R., Plunkett, G., Bloch, C.A., Perna, N.T., Bur-land, V., Riley, M., ColladoVides, J., Glasner, J.D., Rode,C.K., Mayhew, G.F., Gregor, J., Davis, N.W., Kirk-patrick, H.A., Goeden, M.A., Rose, D.J., Mau, B., Shao,Y., 1997. The complete genome sequence of Escherichiacoli K-12. Science 277, 1453–1474.
Celis, J.E., Østergaard, M., Jensen, N.A., Gromova, I., Ras-mussen, H.H., Gromov, P., 1998. Human and mouseproteomic databases: novel resources in the protein uni-verse. FEBS Lett. 430, 64–72.
Chee, M., Yang, R., Hubbell, E., Berno, A., Huang, X.C.,Stern, D., Winkler, J., Lockhart, D.J., Morris, M.S.,Fodor, S.P.A., 1996. Accessing genetic information withhigh-density DNA arrays. Science 274, 610–614.
Duggan, D.J., Bittner, M., Chen, Y., Meltzer, P., Trent, J.M.,1999. Expression profiling using cDNA microarrays. Nat.Genet. 21, S10–S14.
Edman, C.F., Raymond, D.E., Wu, D.J., Tu, E., Sosnowski,R.G., Butler, W.F., Nerenberg, M., Heller, M.J, 1997.Electric field directed nucleic acid hybridization on mi-crochips. Nucl. Acids Res. 25, 4907–4914.
Egholm, M., Buchardt, O., Christensen, L., Behrens, C.,Freier, S.M., Driver, D.A., Berg, R.H., Kim, S.K., Nor-den, B., Nielsen, P.E., 1993. PNA hybridizes to comple-mentary oligonucleotides obeying the Watson–Crickhydrogen-bonding rules. Nature 365, 566–568.
Eisen, M.B., Spellman, P.T., Brown, P.O., Botstein, D., 1998.Cluster analysis and display of genome-wide expressionpatterns. Proc. Natl. Acad. Sci. USA 95, 14863–14868.
Ermolaeva, O., Rastogi, M., Pruitt, K.D., Schuler, G.D.,Bittner, M.L., Chen, Y., Simon, R., Meltzer, P., Trent,J.M., Boguski, M.S., 1998. Data management and analysisfor gene expression arrays. Nat. Genet. 20, 19–23.
Fodor, S.P.A., Read, J.L., Pirrung, M.C., Stryer, L., Lu, A.T.,Solas, D., 1991. Light-directed, spatially addressable paral-lel chemical synthesis. Science 251, 767–773.
Geiger, A., Lester, A., Kleiber, J., Ørum, H., 1998. PNA arraytechnology in molecular diagnostics. Nucleosides Nucle-otides 17, 1717–1724.
Goffeau, A., Barrell, B.G., Bussey, H., Davis, R.W., Dujon,B., Feldmann, H., Galibert, F., Hoheisel, J.D., Jacq, C.,Johnston, M., Louis, E.J., Mewes, H.W., Murakami, Y.,Philippsen, P., Tettelin, H., Oliver, S.G., 1996. Life with6000 genes. Science 274, 546–552.
Graves, D.J., 1999. Powerful tools for genetic analysis come ofage. Trends Biotechnol. 17, 127–134.
Iyer, V.R., Eisen, M.B., Ross, D.T., Schuler, G., Moore, T.,Lee, J.C.F., Trent, J.M., Staudt, L.M., Hudson, J., Bo-guski, M.S., Lashkari, D., Shalon, D., Botstein, D.,Brown, P.O., 1999. The transcriptional program in theresponse of human fibroblasts to serum. Science 283, 83–87.
Johnston, M., 1998. Gene chips: array of hope for understand-ing gene regulation. Curr. Biol. 8, R171–R174.
Jordan, B.R., 1998. Large-scale expression measurement byhybridization methods: from high-density membranes to‘DNA chips’. J. Biochem. 124, 251–258.
Lemieux, B., Aharoni, A., Schena, M., 1998. Overview ofDNA chip technology. Mol. Breed. 4, 277–289.
Lennon, G.G., Lehrach, H., 1991. Hybridization analyses ofarrayed cDNA libraries. Trends Genet. 7, 314–317.
Liang, P., Pardee, A.B., 1992. Differential display of eukary-otic messenger RNA by means of the polymerase chainreaction. Science 257, 967–971.
Lipshutz, R.J., Fodor, S.P.A., Gingeras, T.R., Lockhart, D.J.,1999. High density synthetic oligonucleotide arrays. Nat.Genet. 21, S20–S24.
Lockhart, D.J., Dong, H., Byrne, M.C., Follettie, M.T., Gallo,M.V., Chee, M.S., Mittmann, M., Wang, C., Kobayashi,M., Horton, H., Brown, E.L., 1996. Expression monitoringby hybridization to high-density oligonucleotide arrays.Nat. Biotechnol. 14, 1675–1680.
McGall, G., Labadie, J., Brock, P., Wallraff, G., Nguyen, T.,Hinsberg, W., 1996. Light-directed synthesis of high-den-sity oligonucleotide arrays using semiconductor photore-sists. Proc. Natl. Acad. Sci. USA 93, 13555–13560.
Proudnikov, D., Timofeev, E., Mirzabekov, A., 1998. Immobi-lization of DNA in polyacrylamide gel for the manufactureof DNA and DNA-oligonucleotide microchips. Anal.Biochem. 259, 34–41.
Rogers, Y.H., Jiang-Baucom, P., Huang, Z.J., Bogdanov, V.,Anderson, S., Boyce-Jacino, M.T., 1999. Immobilization ofoligonucleotides onto a glass support via disulfide bonds: amethod for preparation of DNA microarrays. Anal.Biochem. 266, 23–30.
Ruan, Y., Gilmore, J., Conner, T., 1998. Towards Arabidopsisgenome analysis: monitoring expression profiles of 1400genes using cDNA microarrays. Plant J. 15, 821–833.
Schena, M., Shalon, D., Davis, R.W., Brown, P.O., 1995.Quantitative monitoring of gene expression patterns with acomplementary DNA microarray. Science 270, 467–470.
Schena, M., Shalon, D., Heller, R., Chai, A., Brown, P.O.,Davis, R.W., 1996. Parallel human genome analysis: mi-croarray-based expression monitoring of 1000 genes. Proc.Natl. Acad. Sci. USA 93, 10614–10619.
Shalon, D., Smith, S.J., Brown, P.O., 1996. A DNA microar-ray system for analyzing complex DNA samples usingtwo-color fluorescent probe hybridization. Genome Res. 6,639–645.
Sosnowski, R.G., Tu, E., Butler, W.F., O’Connell, J.P., Heller,M.J., 1997. Rapid determination of single base mismatch

N.L.W. 6an Hal et al. / Journal of Biotechnology 78 (2000) 271–280280
mutations in DNA hybrids by direct electric field control.Proc. Natl. Acad. Sci. USA 94, 1119–1123.
The C. elegans Sequencing Consortium, 1998. Genome se-quence of the nematode C. elegans : a platform for investi-gating biology. Science 282, 2012–2018.
Spellman, P.T., Sherlock, G., Zhang, M.Q., Iyer, V.R., An-ders, K., Eisen, M.B., Brown, P.O., Botstein, D., Futcher,B., 1998. Comprehensive identification of cell cycle-regu-lated genes of the yeast Saccharomyces cere6isiae by mi-croarray hybridization. Mol. Biol. Cell. 9, 3273–3297.
Timberlake, W.E., 1980. Developmental gene regulation inAspergillus nidulans. Dev. Biol. 78, 497–510.
Velculescu, V.E., Zhang, L., Vogelstein, B., Kinzler, K.W.,1995. Serial analysis of gene expression. Science 270, 484–487.
Wan, J.S., Sharp, S.J., Poirier, G.M., Wagaman, P.C., Cham-bers, J., Pyati, J., Hom, Y.L., Galindo, J.E., Huvar, A.,
Peterson, P.A., Jackson, M.R., Erlander, M.G., 1996.Cloning differentially expressed mRNAs. Nat. Biotechnol.14, 1685–1691.
Watson, A., Mazumder, A., Stewart, M., Balasubramanian,S., 1998. Technology for microarray analysis of gene ex-pression. Curr. Opin. Biotechnol. 9, 609–614.
Wittung, P., Nielsen, P., Norden, B., 1996. Direct observationof strand invasion by peptide nucleic acid (PNA) intodouble-stranded DNA. J. Am. Chem. Soc. 118, 7049–7054.
Wodicka, L., Dong, H., Mittman, M., Ho, M.H., Lockhart,D.J., 1997. Genome-wide expression monitoring in Saccha-romyces cere6isiae. Nat. Biotechnol. 15, 1359–1367.
Zimmermann, C.R., Orr, W.C., Leclerc, R.F., Barnard, E.C.,Timberlake, W.E., 1980. Molecular cloning and selectionof genes regulated in Aspergillus development. Cell 21,709–715.
..




















