
samina nazir - Pakistan Research Repository
228
Combinatorial Design, Synthesis, Bioevaluation and SAR Studies on Some Small Drug‐like Molecular Libraries Islamabad SAMINA NAZIR Department of Chemistry Quaid‐i‐Azam University Islamabad Pakistan 2009
-
Upload
khangminh22 -
Category
Documents
-
view
3 -
download
0
Transcript of samina nazir - Pakistan Research Repository

Combinatorial Design, Synthesis,
Bioevaluation and SAR Studies on Some Small
Drug‐like Molecular Libraries
Islamabad
SAMINA NAZIR
Department of Chemistry
Quaid‐i‐Azam University
Islamabad
Pakistan
2009

Combinatorial Design, Synthesis, Bioevaluation and SAR studies on some small Drug‐like Molecular
Libraries
Islamabad
A Dissertation submitted to the Department of Chemistry,
Quaid‐i‐Azam University, Islamabad, in partial fulfillment
of the requirements for the degree of
Doctor of Philosophy
in
Organic Chemistry
by
Samina Nazir
Department of Chemistry Quaid‐i‐Azam University
Islamabad, Pakistan.
2009

(In the name of Allah, the Beneficent, the most Merciful)

Dedicated
to
my loving parents, my husband Umar
&
dear dayghter Tuba.

CONTENTS
Acknowledgements……………………………………………………………………………. i Abstract…………………………………………………………………………………………………... ii Chapter 1: Introduction………………………………………………………………………………... 1 1.1. Combinatorial library synthesis…………………………………………………………………....... 5 1.1.1. Solution phase library synthesis………………………………………………………………. 7 1.1.2. Solid phase library synthesis …………………………………………………………............. 8 1.1.3. Parallel library synthesis ………………………….………………………………………….. 12 1.1.3.1. Tea bag method………………………………………………………………………………13 1.1.4. Mixture synthesis………………………………………………………………………………... 14 1.1.4.1. Split and mix synthesis………………………………………………………………………15 1.1.4.2. Positional scanning library synthesis…………………………………………………………16 1.2. Deconvolution…………………………….………………………………………………………..... 19 1.3. Drug-like molecules …………………………………………………………………………………. 24 1.3.1. Chalcones and heterocycles …………………………….……………………………………… 25 1.3.2. Peptidyl chalcones and heterocycles…………………………………………………………….. 30 1.4. Plan of work………………………………………………………………………………………… 32 Chapter 2: Results and Discussion…………………………………………………………………….. 34 2.1. Combinatorial synthesis …………………………………………………………………………….. 36 2.1.1. Synthesis of a parallel library of amino chalcones (1-20)………………………………………. 36 2.1.2. Biological screening of a chalcone library……………………………………………………. 39 2.1.2.1. Phosphatase inhibition………………………………………………………………………39 2.1.2.2. Antibacterial activity…………………………………………………………………………42 2.1.2.3. Brine shrimp lethality studies…………………………………………………………………43 2.2. Indexed combinatorial synthesis…………………………………………………………………….. 44 2.2.1. A 120-member chalcone library………………………………………………………………… 45 2.2.1.1. Synthesis………………………………………………………………………………………45 2.2.1.2. Antibacterial studies…………………………………………………………………………51 2.2.1.3. Deconvolution……………………………………………………………………………….52 2.2.1.4. SAR……………………………………………………………………………………………56 2.2.2. 175 Member chalcone library…………………………………………………………………… 59 2.2.2.1. Synthesis………………………………………………………………………………………60 2.2.2.2. Antitumor studies……………………………………………………………………………61 2.2.2.3. Deconvolution……………………………………………………………………………….63 2.2.2.4. SAR………………………………………………………………………………………….71 2.3. Peptidyl chalcones and peptidyl heterocycles………………………………………………………. 72 2.3.1. Synthesis of peptidyl chalcones………………………………………………………………… 73 2.3.1.1. Synthesis of linker reagent…………………………………………………………………..74 2.3.1.2. Protection of triphenylphosphin-PS-support…………………………………………………75 2.3.1.3. Deprotonation leading to Wittig ylide……………………………………………………….76 2.3.1.4. Acylation of Wittig ylide…………………………………………………………………….76 2.3.1.5. Amide coupling leading to peptide synthesis…………………………………………………79 2.3.1.6. N-Terminal acetylation of peptides (92-95)…………………………………………………82 2.3.1.7. Deprotection of phosphorane support……………………………………………………….83 2.3.1.8. Synthesis of peptidyl chalcones (100-107)………………………………………………….84 2.3.2. Synthesis of peptidyl heterocycles……………………………………………………………… 87 2.3.2.1. Synthesis of peptidyl oxazoles………………………………………………………………88 2.3.2.2. Synthesis of peptidyl pyrazolines……………………………………………………………90 2.3.2.3. Synthesis of peptidyl pyrazoles………………………………………………………………91 2.3.2.4. Synthesis of peptidyl benzothiazepines………………………………………………………92 2.3.2.5. Synthesis of peptidyl benzodiazepines………………………………………………………95
Continued…

Chapter 3: Computational Studies.……………………………………………………………………. 98 3.1. Molecular Docking…………………………………………………………………………………… 100 3.2. Structure activity relationship (SAR)………………………………………………………………… 109 3.2.1. In silico guided design and synthesis of cytotoxic chalcones…………………………………………109 3.2.2. SAR of 30 hydroxychalcones for antitumor potency……………………………………………… 122 Chapter 4: Experimental.………………………………………………………………………………. 129 4A. General……………………………………………………………………………………………..... 130 4A.1. Chemicals………………………………………………………………………………………….. 130 4A.2. Purification and drying of solvents………………………………………………………………… 130 4A.3. Analytical methods and instruments……………………………………………………………….. 131 4A.3.1. Thin layer chromatography……………………………………………………………………131 4A.3.2. Green House synthesizer………………………………………………………………………131 4A.3.3. Microwave synthesizers………………………………………………………………………131 4A.3.4. Ika Vibrax orbital shaker………………………………………………………………………131 4A.3.5. Melting point determination…………………………………………………………………132 4A.3.6. FTIR spectroscopy……………………………………………………………………………132 4A.3.7. UV Vis. Spectroscopy……………………………………………………………………….132 4A.3.8. LC MS analysis………………………………………………………………………………132 4A.3.9. HPLC…………………………………………………………………………………………133 4A.3.10. GC MS spectrometry…………………………………………………………………………133 4A.3.11. Mass spectrometry……………………………………………………………………………133 4A.3.12. NMR spectroscopy…………………………………………………………………………134 4A.3.13. Elemental analysis……………………………………………………………………………134 4A.3.14. Kaiser test……………………………………………………………………………………134 4B. Synthesis of chalcone libraries……………………………………………………………………… 135 4B.1. Parallel synthesis of a 20 member library………………………………………………………… 135 4B.1.1. (E)-1-(4′-Aminophenyl)-3-(phenyl)-2-propen-1-one (1)……………………………………135 4B.1.2. (E)-1-(4′-Aminophenyl)-3-(2-hydroxyphenyl)-2-propen-1-one (2)…………………………135 4B.1.3. (E)-1-(4′-Aminophenyl)-3-(3-hydroxyphenyl)-2-propen-1-one (3)…………………………135 4B.1.4. (E)-1-(4′-Aminophenyl)-3-(4-hydroxyphenyl)-2-propen-1-one (4)…………………………136 4B.1.5. (E)-1-(4′-Aminophenyl)-3-(2-methoxyphenyl)-2-propen-1-one (5)…………………………136 4B.1.6. (E)-1-(4′-Aminophenyl)-3-(3-methoxyphenyl)-2-propen-1-one (6)…………………………136 4B.1.7. (E)-1-(4′-Aminophenyl)-3-(4-methoxyphenyl)-2-propen-1-one (7)…………………………137 4B.1.8. (E)-1-(4′-Aminophenyl)-3-(3,4-dimethoxyphenyl)-2-propen-1-one (8)………………………137 4B.1.9. (E)-1-(4′-Aminophenyl)-3-(2-nitrophenyl)-2-propen-1-one (9)……………………………..137 4B.1.10. (E)-1-(4′-Aminophenyl)-3-(3-nitrophenyl)-2-propen-1-one (10)……………………………137 4B.1.11. (E)-1-(4′-Aminophenyl)-3-(4-nitrophenyl)-2-propen-1-one (11)………………………….138 4B.1.12. (E)-1-(4′-Aminophenyl)-3-(2-chlorophenyl)-2-propen-1-one (12)…………………………138 4B.1.13. (E)-1-(4′-Aminophenyl)-3-(3-chlorophenyl)-2-propen-1-one (13)…………………………138 4B.1.14. (E)-1-(4′-Aminophenyl)-3-(4-chlorophenyl)-2-propen-1-one (14)…………………………139 4B.1.15. (E)-1-(4′-Aminophenyl)-2-(4-fluorophenyl)-2-propen-1-one (15)…………………………139 4B.1.16. (E)-1-(4′-Aminophenyl)-3-(2-bromophenyl)-2-propen-1-one (16)…………………………139 4B.1.17. (E)-1-(4′-Aminophenyl)-3-(3-bromophenyl)-2-propen-1-one (17)…………………………139 4B.1.18. (E)-1-(4′-Aminophenyl)-3-(4-methylphenyl)-2-propen-1-one (18)…………………………140 4B.1.19. (E)-1-(4′-Aminophenyl)-3-(3-hydroxy-4-methoxyphenyl)-2-propen-1-one (19) …………140 4B.1.20. (E)-1-(4′-Aminophenyl)-3-(4-N,N-dimethylaminophenyl)-2-propen-1-one (20)……………140 4B.2. Combinatorial synthesis of indexed chalcone library (120 members)……………………………… 141 4B.2.1. Synthesis of sub-libraries of Set 1 (AL1-AL6)…………………………………………………141 4B.2.1.1 Synthesis of AL1……………………………………………………………………141 4B.2.1.2. Synthesis of AL2……………………………………………………………………141 4B.2.1.3. Synthesis of AL3……………………………………………………………………141 4B.2.1.4. Synthesis of AL4……………………………………………………………………141
Continued…

4B.2.1.5. Synthesis of AL5……………………………………………………………………141 4B.2.1.6. Synthesis of AL6……………………………………………………………………141 4B.2.2. Synthesis of sub-libraries of Set 2 (BL1-BL20)………………………………………………142 4B.2.2.1. Synthesis of BL1……………………………………………………………………143 4B.2.2.2. Synthesis of BL2……………………………………………………………………143 4B.2.2.3. Synthesis of BL3……………………………………………………………………143 4B.2.2.4. Synthesis of BL4……………………………………………………………………143 4B.2.2.5. Synthesis of BL5……………………………………………………………………143 4B.2.2.6. Synthesis of BL6……………………………………………………………………143 4B.2.2.7. Synthesis of BL7……………………………………………………………………143 4B.2.2.8. Synthesis of BL8……………………………………………………………………143 4B.2.2.9. Synthesis of BL9……………………………………………………………………143 4B.2.2.10. Synthesis of BL10…………………………………………………………………144 4B.2.2.11. Synthesis of BL11…………………………………………………………………143 4B.2.2.12. Synthesis of BL12…………………………………………………………………143 4B.2.2.13. Synthesis of BL13…………………………………………………………………143 4B.2.2.14. Synthesis of BL14…………………………………………………………………144 4B.2.2.15. Synthesis of BL15…………………………………………………………………144 4B.2.2.16. Synthesis of BL16…………………………………………………………………144 4B.2.2.17. Synthesis of BL17…………………………………………………………………144 4B.2.2.18. Synthesis of BL18…………………………………………………………………144 4B.2.2.19. Synthesis of BL19…………………………………………………………………144 4B.2.2.20. Synthesis of BL20…………………………………………………………………144 4B.2.3. Parallel synthesis of the members of active column (Pool AL1)………………………………144 4B.2.3.1. (E)-1,3-(Diphenyl)-2-propen-1-one, A1B1 (21)……………………………………144 4B.2.3.2. (E)-1-(Phenyl)-3-(2-hydroxyphenyl)-2-propen-1-one, A1B2 (22)…………………144 4B.2.3.3. (E)-1-(Phenyl)-3-(3-hydroxyphenyl)-2-propen-1-one, A1B3 (23)…………………145 4B.2.3.4. (E)-1-(Phenyl)-3-(4-hydroxyphenyl)-2-propen-1-one, A1B4 (24)…………………145 4B.2.3.5. (E)-1-(Phenyl)-3-(2-methoxyphenyl)-2-propen-1-one, A1B5 (25)…………………145 4B.2.3.6. (E)-1-(Phenyl)-3-(3-methoxyphenyl)-2-propen-1-one, A1B6 (26)…………………145 4B.2.3.7. (E)-1-(Phenyl)-3-(4-methoxyphenyl)-2-propen-1-one, A1B7 (27)…………………146 4B.2.3.8. (E)-1-(Phenyl)-3-(3,4-dimethoxyphenyl)-2-propen-1-one, A1B8 (28)……………146 4B.2.3.9. (E)-1-(Phenyl)-3-(2-nitrophenyl)-2-propen-1-one, A1B9 (29)………………………146 4B.2.3.10. (E)-1-(Phenyl)-3-(3-nitrophenyl)-2-propen-1-one, A1B10 (30)……………………146 4B.2.3.11. (E)-1-(Phenyl)-3-(4-nitrophenyl)-2-propen-1-one, A1B11 (31)……………………147 4B.2.3.12. (E)-1-(Phenyl)-3-(2-chlorophenyl)-2-propen-1-one, A1B12 (32)…………………147 4B.2.3.13. (E)-1-(Phenyl)-3-(3-chlorophenyl)-2-propen-1-one, A1B13 (33)…………………147 4B.2.3.14. (E)-1-(Phenyl)-3-(4-chlorophenyl)-2-propen-1-one, A1B14 (34)…………………147 4B.2.3.15. (E)-1-(Phenyl)-3-(4-fluorophenyl)-2-propen-1-one, A1B15 (35)…………………147 4B.2.3.16. (E)-1-(Phenyl)-3-(2-bromophenyl)-2-propen-1-one, A1B16 (36)…………………148 4B.2.3.17. (E)-1-(Phenyl)-3-(3-bromophenyl)-2-propen-1-one, A1B17 (37)…………………148 4B.2.3.18. (E)-1-(Phenyl)-3-(4-methylphenyl)-2-propen-1-one, A1B18 (38)…………………148 4B.2.3.19. (E)-1-(Phenyl)-3-(3-hydroxy-4-methyoxyphenyl)-2-propen-1-one, A1B19 (39)…148 4B.2.3.20. (E)-1-(Phenyl)-3-(4-N,N-dimethylaminophenyl)-2-propen-1-one, A1B20 (40)…148 4B.2.4. Parallel synthesis of members of the active row (Pool BL2)…………………………………149 4B.2.4.1. (E)-1-(4′-Aminophenyl)-3-(2-hydroxyphenyl)-2-propen-1-one, A3B2 (2)…………149 4B.2.4.2. (E)-1-(Phenyl)-3-(2-hydroxyphenyl)-2-propen-1-one, A1B2 (22)…………………149 4B.2.4.3. (E)-1-(2′-Hydroxyphenyl)-3-(2-hydroxyphenyl)-2-propen-1-one, A2B2 (41)……149 4B.2.4.4. (E)-1-(2′,3′,4′-Trimethoxyphenyl)-3-(2-hydroxyphenyl)-2-propen-1-one, A4B2
(42)…………………………………………………………………………………149
4B.2.4.5. (E)-1-(3′,4′,5′-Trimethoxyphenyl)-3-(2-hydroxyphenyl)-2-propen-1-one, A5B2 (43)…………………………………………………………………………………
150
4B.2.4.6. (E)-1-(2′,4′-Dibromophenyl)-3-(2-hydroxyphenyl)-2-propen-1-one, A6B2 (44)…150 Continued…

4B.3. Synthesis of 175 member chalcone library…………………………………………………………. 150 4B.3.1. Synthesis of indexed libraries (AL1-AL7)……………………………………………………150 4B.3.1.1. Synthesis of pool AL1..............................................................................................150 4B.3.1.2. Synthesis of pool AL2………………………………………………………………150 4B.3.1.3. Synthesis of pool AL3………………………………………………………………151 4B.3.1.4. Synthesis of pool AL4………………………………………………………………151 4B.3.1.5. Synthesis of pool AL5………………………………………………………………151 4B.3.1.6. Synthesis of pool AL6..............................................................................................151 4B.3.1.7. Synthesis of pool AL7………………………………………………………………151 4B.3.2. Synthesis of indexed libraries (BL1-BL25)……………………………………………………152 4B.3.2.1. Synthesis of BL1……………………………………………………………………152 4B.3.2.2. Synthesis of BL2……………………………………………………………………152 4B.3.2.3. Synthesis of BL3……………………………………………………………………152 4B.3.2.4. Synthesis of BL4……………………………………………………………………152 4B.3.2.5. Synthesis of BL5……………………………………………………………………152 4B.3.2.6. Synthesis of BL6……………………………………………………………………153 4B.3.2.7. Synthesis of BL7……………………………………………………………………153 4B.3.2.8. Synthesis of BL8……………………………………………………………………153 4B.3.2.9. Synthesis of BL9……………………………………………………………………153 4B.3.2.10. Synthesis of BL10…………………………………………………………………153 4B.3.2.11. Synthesis of BL11…………………………………………………………………153 4B.3.2.12. Synthesis of BL12…………………………………………………………………153 4B.3.2.13. Synthesis of BL13…………………………………………………………………153 4B.3.2.14. Synthesis of BL14…………………………………………………………………153 4B.3.2.15. Synthesis of BL15…………………………………………………………………153 4B.3.2.16. Synthesis of BL16...................................................................................................153 4B.3.2.17. Synthesis of BL17…………………………………………………………………154 4B.3.2.18. Synthesis of BL18…………………………………………………………………154 4B.3.2.19. Synthesis of BL19…………………………………………………………………154 4B.3.2.20. Synthesis of BL20…………………………………………………………………154 4B.3.2.21. Synthesis of BL21…………………………………………………………………154 4B.3.2.22. Synthesis of BL22…………………………………………………………………154 4B.3.2.23. Synthesis of BL23…………………………………………………………………154 4B.3.2.24. Synthesis of BL24…………………………………………………………………154 4B.3.2.25. Synthesis of BL25…………………………………………………………………154 4B.3.3. Synthesis of members of active column AL1 (A1B1-A1B25)…………………………………154 4B.3.3.1. (E)-1-(Phenyl)-3-(3-nitrophenyl)-2-propen-1-one, A1B14 (30)……………………155 4B.3.3.2. (E)-1-(Phenyl)-3-(2-chlorophenyl)-2-propen-1-one, A1B9 (32)……………………155 4B.3.3.3. (E)-1-(Phenyl)-3-(3-chlorophenyl)-2-propen-1-one, A1B10 (33)……………………155 4B.3.3.4. (E)-1-(Phenyl)-3-(4-chlorophenyl)-2-propen-1-one, A1B11 (34)……………………155 4B.3.3.5. (E)-1-(Phenyl)-3-(4-fluorophenyl)-2-propen-1-one, A1B13 (35)……………………155 4B.3.3.6. (E)-1-(Phenyl)-3-(3-bromophenyl)-2-propen-1-one, A1B12 (37)……………………155 4B.3.3.7. (E)-1-(Phenyl)-3-(4-methylphenyl)-2-propen-1-one, A1B17 (38)…………………155 4B.3.3.8. (E)-1-(Phenyl)-3-(3-hydroxy-4-methyoxyphenyl)-2-propen-1-one, A1B15 (39)……155 4B.3.3.9. (E)-1-(Phenyl)-3-(4-N,N-dimethylaminophenyl)-2-propen-1-one, A1B16 (40)……155 4B.3.3.10. (E)-1-(Phenyl)-3-(thiophen-2-yl)-2-propen-1-one, A1B18 (45)……………………155 4B.3.3.11. (E)-1-(Phenyl)-3-(5-methylthiophen-2-yl)-2-propen-1-one, A1B19 (46)…………156 4B.3.3.12. (E)-1-(Phenyl)-3-(5-bromothiophen-2-yl)-2-propen-1-one, A1B20 (47)…………156 4B.3.3.13. (E)-1-(Phenyl)-3-(5-nitrothiophen-2-yl)-2-propen-1-one, A1B21 (48)……………156 4B.3.3.14. (E)-1-(Phenyl)-3-(pyrrol-2-yl)-2-propen-1-one, A1B22 (49)………………………156 4B.3.3.15. (E)-1-(Phenyl)-3-(pyridin-2-yl)-2-propen-1-one, A1B23 (50)……………………157 4B.3.3.16. (E)-1-(Phenyl)-3-(pyridin-3-yl)-2-propen-1-one, A1B24 (51)……………………157 4B.3.3.17. (E)-1-(Phenyl)-3-(pyridin-4-yl)-2-propen-1-one, A1B25 (52)……………………157 Continued…

4B.3.4. Synthesis of members of active column AL3 (A3B1-A3B25)…………………………………157 4B.3.4.1. (E)-1-(3′-Hydroxyphenyl)-3-(phenyl)-2-propen-1-one, A3B1 (53)…………………157 4B.3.4.2. (E)-1-(3′-Hydroxyphenyl)-3-(2-hydroxyphenyl)-2-propen-1-one, A3B2 (54)……157 4B.3.4.3. (E)-1-(3′-Hydroxyphenyl)-3-(3-hydroxyphenyl)-2-propen-1-one, A3B3 (55)……158 4B.3.4.4. (E)-1-(3′-Hydroxyphenyl)-3-(4-hydroxyphenyl)-2-propen-1-one, A3B4(56)………158 4B.3.4.5. (E)-1-(3′-Hydroxyphenyl)-3-(2-methoxyphenyl)-2-propen-1-one, A3B5(57)………158 4B.3.4.6. (E)-1-(3′-Hydroxyphenyl)-3-(3-methoxyphenyl)-2-propen-1-one, A3B6(58)………158 4B.3.4.7. (E)-1-(3′-Hydroxyphenyl)-3-(4-methoxyphenyl)-2-propen-1-one, A3B7 (59)……159 4B.3.4.8. (E)-1-(3′-Hydroxyphenyl)-3-(3,4-dimethoxyphenyl)-2-propen-1-one, A3B8 (60)…159 4B.3.4.9. (E)-1-(3′-Hydroxyphenyl)-3-(2-chlorophenyl)-2-propen-1-one, A3B9 (61)………159 4B.3.4.10. (E)-1-(3′-Hydroxyphenyl)-3-(3-chlorophenyl)-2-propen-1-one, A3B10 (62)……159 4B.3.4.11. (E)-1-(3′-Hydroxyphenyl)-3-(4-chlorophenyl)-2-propen-1-one, A3B11 (63)……159 4B.3.4.12. (E)-1-(3′-Hydroxyphenyl)-2-(3-bromophenyl)-2-propen-1-one, A3B12(64)………160 4B.3.4.13. (E)-1-(3′-Hydroxyphenyl)-2-(4-fluorophenyl)-2-propen-1-one, A3B13 (65)………160 4B.3.4.14. (E)-1-(3′-Hydroxyphenyl)-3-(3-nitrophenyl)-2-propen-1-one, A3B14 (66)………160 4B.3.4.15. (E)-1-(3′-Hydroxyphenyl)-3-(3-hydroxy-4-methoxyphenyl)-2-propen 1-one, A3B15 (67)… ……………………………………………………………………
161
4B.3.4.16. (E)-1-(3′-Hydroxyphenyl)-3-(4-N,N-dimethylaminophenyl)-2-propen 1-one, A3B16 (68)…………………………………………………………………………
161
4B.3.4.17. (E)-1-(3′-Hydroxyphenyl)-3-(4-methylphenyl)-2-propen-1-one, A3B17 (69)……161 4B.3.4.18. (E) -1-(3′-Hydroxyphenyl)-3-(thien-2-yl)-2-propen-1-one, A3B18 (70)…………161 4B.3.4.19. (E)-1-(3′-Hydroxyphenyl)-3-(5-methylthien-2-yl)-2-propen-1-one,A3B19 (71)…162 4B.3.4.20. (E)-1-(3′-Hydroxyphenyl)-3-(5-bromothien-2-yl)-2-propen-1-one,A3B20 (72)…162 4B.3.4.21. (E)-1-(3′-Hydroxyphenyl)-3-(5-nitrothien-2-yl)-2-propen-1-one, A3B21(73)……162 4B.3.4.22. (E)-1-(3′-Hydroxyphenyl)-3-(pyrrol-2-yl)-2-propen-1-one, A3B22 (74)…………162 4B.3.4.23. (E)-1-(3′-Hydroxyphenyl)-3-(pyridine-2-yl)-2-propen-1-one, A3B23 (75)………163 4B.3.4.24. (E)-1-(3′-Hydroxyphenyl)-3-(pyridine-3-yl)-2-propen-1-one, A3B24 (76)………163 4B.3.4.25. (E)-1-(3′-Hydroxyphenyl)-3-(pyridine-4-yl)-2-propen-1-one, A3B25(77)…………163 4B.3.5. Synthesis of members of active row, A1B9-A7B9……………………………………………163 4B.3.5.1. (E)-1-(4′-Aminophenyl)-3-(2-chlorophenyl)-2-propen-1-one, A7B9 (12)…………163 4B.3.5.2. (E)-1-(Phenyl)-3-(2-chlorophenyl)-2-propen-1-one, A1B9 (32)……………………164 4B.3.5.3. (E)-1-(3′-Hydroxyphenyl)-3-(2-chlorophenyl)-2-propen-1-one, A3B9 (61)………164 4B.3.5.4. (E)-1-(2′-Hydroxyphenyl)-3-(2-chlorophenyl)-2-propen-1-one, A2B9 (78)………164 4B.3.5.5. (E)-1-(4′-Hydroxyphenyl)-3-(2-chlorophenyl)-2-propen-1-one, A4B9 (79)………164 4B.3.5.6. (E)-1-(2′-Aminophenyl)-3-(2-chlorophenyl)-2-propen-1-one, A5B9 (80)…………164 4B.3.5.7. (E)-1-(3′-Aminophenyl)-3-(2-chlorophenyl)-2-propen-1-one, A6B9 (81)…………164 4C. Synthesis of peptidyl chalcones (102-109)…………………………………………………………… 165 4C.1. 2-Trimethylsilylethyl-2-bromoacetate (82)………………………………………………………… 165 4C.2. 2-Trimethylsilylethyl-2-diphenylphosphoranylidene salt (83)…………………………………….. 165 4C.3. 2-Trimethylsilylethyl-2-diphenylphosphoranylideneacetate (84)…………………………………. 165 4C.4. 2-Acyl-2-diphenylphosphoranylidene acetates (85-86)…………………………………………… 166 4C.4.1. 2-Trimethylsilylethyl-4-(9H-fluoren-9-yl-methoxycarbonyl)-3-oxo-5-phenyl-2- diphenylphosphoranylidenepentanoate (85)…………………………………………………
166
4C.4.2. 2-Trimethylsilylethyl-4-(9H-fluoren-9-yl-methoxycarbonyl)-5-methyl-3-oxo-2- diphenylphosphoranylidenehexanoate (86)……………………………………………………
166
4C.5. Fmoc protected di- and tri- peptides (87-91)………………………………………………………. 166 4C.5.1. (S)-2-Trimethylsilylethyl 4-[(S)-2-(9H-fluoren-9-yl-methoxycarbonyl)-3- phenylpropanamido]-3-oxo-5-phenyl-2-diphenylphosphoranylidenepentanoate(87)………
166
4C.5.2. (S)-2-Trimethylsilylethyl 4-[(S)-2-(9H-fluoren-9-yl-methoxycarbonyl)-3- phenylpropanamido]-5-methyl-3-oxo-2-diphenylphosphoranylidenehexanoate 88)…………
167
4C.5.3. (S)-2-Trimethylsilylethyl 4-[(S)-2-(9H-fluoren-9-yl-methoxycarbonyl)-4- methylpentanamido]-3-oxo-5-phenyl-2-diphenylphosphoranylidenepentanoate (89)………
167
Continued…

4C.5.4. (S)-2-Trimethylsilylethyl 4-[(S)-2-(9H-fluoren-9-yl-methoxycarbonyl)propanamido]- 3-oxo-5-phenyl-2-diphenylphosphoranylidenepentanoate (90)………….……………………
167
4C.5.5. (S)-2-Trimethylsilylethyl 4-[(S)-2-((S)-2-(9H-fluoren-9-yl-methoxycarbonyl)-3-phenylpro panamido)propanamido]-3-oxo-5-phenyl-2-diphenylphosphoranylidenepentanoate (91)……
167
4C.6. Acetyl di- and tri- peptides (92-95)………………………………………………………………… 167 4C.6.1. (S)-2-Trimethylsilylethyl 4-[(S)-2-acetamido-3-phenylpropanamido]-3-oxo-5-phenyl-2- diphenylphosphoranylidenepentanoate (92)…………………………………………………
168
4C.6.2. (S)-2-Trimethylsilylethyl 4-[(S)-2-acetamido-3-phenylpropanamido]-5-methyl-3-oxo-2- diphenylphosphoranylidenehexanoate (93)……………………………………………………
168
4C.6.3. (S)-2-Trimethylsilylethyl 4-[(S)-2-acetamido-4-methylpentanamido]-3-oxo-5-phenyl-2- diphenylphosphoranylidenepentanoate (94)…………………………………………………
168
4C.6.4. (S)-2-Trimethylsilylethyl 4-[(S)-2-((S)-2-acetamido)-3-phenylpropanamido)propanamido] -3-oxo-5-phenyl-2-diphenylphosphoranylidenepentanoate (95)………………………………
168
4C.7. Peptidyl-3-amino-2-oxo-1-diphenylphosphoranylidenepropanes (96-99)…………………………. 168 4C.7.1. (S)-2-acetamido-N-[(S)-3-oxo-1-phenyl-4-diphenylphosphoranylidenebutan-2-yl]-3- phenylpropanamide (96)………………………………………………………………………
168
4C.7.2. (S)-2-acetamido-N-[(S)-4-methyl-2-oxo-1-diphenylphosphoranylidenepentan-3-yl]-3- phenylpropanamide (97)………………………………………………………………………
168
4C.7.3. (S)-2-acetamido-4-methyl-N-[(S)-3-oxo-1-phenyl-4-diphenylphosphoranylidenebutan-2- yl]pentanamide (98)…………………………………………………………………………
169
4C.7.4. (S)-2-((S)-2-acetamido-3-phenylpropanamido)-N-[(S)-3-oxo-1-phenyl-4- diphenylphosphoranylidenebutan-2-yl]propanamide (99)……………………………………
169
4C.8. Peptidyl chalcones (100-107)………………………………………………………………………. 169 4C.8.1. N-Acetyl-L-phenylalanyl-(S,E)-1-amino-1-benzyl-6-methyl-hept-3-en-2-one (100)…………169 4C.8.2. N-Acetyl-L-phenylalanyl-(S,E)-1-amino-1-benzyl-4-(2-chloro-6-fluorophenyl)-but -3-en-2-one (101)………………………………………………………………………………
169
4C.8.3. N-Acetyl-L-phenylalanyl-(S,E)-1-amino-1-isopropyl-4-(2-chloro-6-fluorophenyl)-but -3-en-2-one (102)……………….……………………………………………………………
170
4C.8.4. N-Acetyl-L-phenylalanyl-(S,E)-1-amino-1-isopropyl-4-thiophen-3-en-2-one (103)…………170 4C.8.5. N-Acetyl-L-phenylalanyl-(S,E)-1-amino-1-isopropyl-(5,5,5-tribromo)-pent-3- en-2-one (104)…………………………………………………………………………………
171
4C.8.6. N-Acetyl-L-leucinyl-(S,E)-1-amino-1-benzyl-4-(2-chloro-6-fluorophenyl)-but-3- en-2-one (105)…………………………………………………………………………………
171
4C.8.7. N-Acetyl-L-phenylalanyl-L-alanyl-(S,S,E)-1-amino-1-benzyl-pent-3-en-2-one (106)...........172 4C.8.8. N-Acetyl-L-phenylalanyl-L-alanyl-(S,S,E)-1-amino-1-benzyl-6-methyl-hept-3-en-2-one (107)……………………………………………………………………………………………
172
4D. Synthesis of peptidyl heterocycles (108-125)……………………………………………………….. 172 4D.1. Synthesis of peptidyl isoxazoles (108-109)………………………………………………………… 173 4D.1.1. N-Acetyl-L-phenylalanyl-(S)-(1S)-benzyl-1-[5-(2-chloro-6-fluorophenyl)isoxazol-3-yl] methylamine (108)……………………………………………………………………………
173
4D.1.2. N-Acetyl-L-phenylalanyl-(S)-(1S)-benzyl-(5-isobutylisoxazol-3-yl)methylamine (109)……173 4D.2. Peptidyl pyrazolines (110-114)……………………………………………………………………. 174 4D.2.1. N-Acetyl-L-phenylalanyl-L-alanyl-(S,S)-(1S)-benzyl-1-(5-methyl-4,5-dihydro-1H- pyrazol-3-yl)methylamine (110)………….…………………………………………………
174
4D.2.2. N-Acetyl-L-phenylalanyl-L-alanyl-(S,S)-(1S)-benzyl-1-(1,5-dimethyl-4,5-dihydro-1H- pyrazol-3-yl)methylamine (111)………………………………………………………………
174
4D.2.3. N-Acetyl-L-phenylalanyl-L-alanyl-(S,S)-(1S)-benzyl-1-(1-phenyl-5-methyl-4,5-dihydro -1H-pyrazol-3-yl)methylamine (112)…………………………………………………………
174
4D.2.4. N-Acetyl-L-phenylalanyl-L-alanyl-(S,S)-(1S)-benzyl-1-(1-methyl-5-isobutyl-4,5-dihydro -1H-pyrazol-3-yl)methylamine (113)......................................................................................
174
4D.2.5. N-Acetyl-L-phenylalanyl-L-alanyl-(S,S)-(1S)-benzyl-1-(5-isobutyl-4,5-dihydro-1H- pyrazol-3-yl)methylamine (114)…………………………………………………………….
174
4D.3. Peptidyl pyrazoles (115-118)………………………………………………………………………. 174 Continued…

4D.3.1. N-Acetyl-L-phenylalanyl-L-alanyl-(S,S)-(1S)-benzyl-1-(5-methyl-1H-pyrazol-3-yl) methylamine (115)……………………………………………………………………………
175
4D.3.2. N-Acetyl-L-phenylalanyl-L-alanyl-(S,S)-(1S)-benzyl-1-(1,5-dimethyl-1H-pyrazol-3-yl) methylamine (116)……………………………………………………………………………
175
4D.3.3. N-Acetyl-L-phenylalanyl-L-alanyl-(S,S)- (1S)-benzyl-1-(1-phenyl-5-methyl-4,5- dihydro-1H-pyrazol-3-yl)methylamine (117)…………………………………………………
175
4D.3.4. N-Acetyl-L-phenylalanyl-L-alanyl-(S,S)-(1S)-benzyl-1-(1-methyl-5-isobutyl-1H-pyrazol -3-yl)methylamine (118)………………………………………………………………………
176
4D.4. Peptidyl benzothiazepines (119-123)…………………………………………………………………176 4D.4.1. N-Acetyl-L-phenylalanyl-(1S)-benzyl-1-(2-(2-chloro-6-fluorophenyl)-2,3-dihydro-1, 5-benzothiazepine-4-yl)methylamine (119)............................................................................
176
4D.4.2. N-Acetyl-L-phenylalanyl-(1S)-benzyl-1-(2-isobutyl-2,3-dihydro-1,5-benzothiazepine-4- yl)methylamine (120).............................................................................................................
177
4D.4.3. N-Acetyl-L-phenylalanyl-(1S)-isopropyl-1-(7-chloro-2-(2-chloro-6-fluorophenyl)-2,3- dihydro-1,5-benzothiazepine-4-yl)methylamine (121)...........................................................
177
4D.4.4. N-Acetyl-L-phenylalanyl-(1S)-isopropyl-1-(7-chloro -2-(thiophen-2-yl)-2,3-dihydro-1,5- benzothiazepine-4-yl)methylamine (122)……………………………………………………
177
4D.4.5. N-Acetyl-L-leucinyl-(1S)-benzyl-1-(7-chloro-2-(2-chloro-6-fluorophenyl)-2,3-dihydro-1, 5-benzothiazepine-4-yl)methylamine (123)............................................................................
178
4D.5. Synthesis of peptidyl benzodiazepines (124-125)……………………………………………………178 4D.5.1. N-Acetyl-L-phenylalanyl-(1S)-isopropyl-1-[2-(2-chloro-6-fluorophenyl)-1H-1,5- benzodiazepine-4-yl)methylamine (124)................................................................................
178
4D.5.2. N-acetyl-L-leucinyl-(1S)-benzyl-1-[2-(2-chloro-6-fluorophenyl)-1H-1,5-benzodiazepine -4-yl)methylamine (125)………………………………………………………………………
179
4E. Bioevaluation…………………………………………………………………………………………. 179 4E.1. Antibacterial assay…………………………………………………………………………………. 179 4E.2. Cytotoxicity studies………………………………………………………………………………… 180 4E.3. Potato disc tumor (PDT) bioassay…………………………………………………………………. 181 4E.4. Phosphatase inhibition……………………………………………………………………………… 181 4E.5. PGM inhibition assay………………………………………………………………………………. 182 References………………………………………………………………………………………………….. 186 Summary…………………………………………………………………………………………………. 201 Future Plan…………………………………………………………………………………………………. 202 List of Publications……………………………………………………………………………………… 203 Appendix………………………………………………………………………………………………… 204 Index of Tables…………………………………………………………………………………….. 205 Index of Figures…………………………………………………………………………………….. 207 Index of Schemes……………………………………………………………………………………. 210 Standard Abbreviations and Acronyms……………………………………………………………. 211

i
Acknowledgements All praises to Almighty Allah, Who puts the sun’s seal on the tablets of the flowing waters and throws clouds to the skies, Who distills the waters of the clouds over the seas to conceive the pearl in the womb of the oyster, Who creates fire in every stone, color in the fire, radiance in the color, Who gives voices to the dust, word to the voices, and life to the word, Who created us as a Muslim and blessed us with knowledge to differentiate between right and wrong. Many many thanks to Him as He blessed us
with the Holy Prophet, Hazrat Muhammad for whom the whole universe is created and who enabled us to worship only one God. He (PBUH) brought us out of darkness and enlightened the way of Heaven. The writing of a PhD dissertation could be a lonely and isolating experience, yet it was obviously not possible without the personal and practical support of numerous people. It reflects the support and care of countless people who influenced my life and this work. Thus, I feel great pleasure in expressing my ineffable thanks to my encouraging and motivating supervisor, Prof. Dr. Farzana Latif Ansari (TI), whose personal interest, thought provoking guidance, valuable suggestions and discussions enabled me to complete this work. My research for this dissertation was made more extensive and proficient by the use of resources at FMP Institute, Berlin, Germany. Thus, I express my gratitude to Prof. Dr. Jörg Rademann and his research group at FMP, especially Mr. Adeeb AlDahshan, with whom I completed a part of my research work. Moreover, I express my thanks to FMP for hiring me as a Guest Scientist for a couple of months. My heartfelt gratitude goes to Higher Education Commission (HEC) Pakistan for providing me Indigenous PhD Fellowship and a fellowship under International Research Support Initiative Program (IRISP) during my PhD. I am thankful to Prof. Dr. Saqib Ali (PoP), the Chairman, Department of Chemistry, Quaid-i-Azam University, Islamabad and Prof. Dr. Nasim Hassan Rama, head of Organic Section, for providing necessary research facilities. Many thanks are due to all the faculty members of Chemistry Department, especially to all teachers of organic section for being a source of inspiration and enlightenment for me. I also owe my recognition to my lab fellows Maj. Mohammad Baseer Khan, Mr. Umer Rasheed, Mr. Ahsan Ullah, Miss. Farukh Jabeen, Mr. Awais Shaukat, Miss. Saima Kalsoom, Miss. Sobia Shaheen, Miss. Aliya Shehzadi, Miss. Zahra Ali, Miss. Shireen Gull and Musfirah Khaliq for their help at crucial times of my research work. I am very obliged to the supporting staff of the department, namely Mr. Sharif Chohan, Mr. Shams, Mr. Tayyab, Mr. Farhan, Mr. Jumma Khan, Mr. Raza, Mr. Rashid, Mr Hanif and Mr. Bilal for their all time help. I feel the sincere gratitude and a feeling of great pleasure in my heart in thanking to my chemistry teachers Ms. Rizwana and Dr. Nagmana Rasheed, whose personal interest and kind attitude inculcated an interest of Chemistry within me at my school and college level.
I have no words to acknowledge the sacrifices, efforts, lot of prayers, guidance, support, encouragement and firm dedication of my loving parents, husband, brother, sisters and my in-laws throughout my academic period. Their endless prayers contributed to the successful completion of this research project. Words become meaningless when I look at them as icons of strength for being what I am today. Had my father not been there as pillar of strength, this building of my achievement would have never stood.
Samina Nazir

ii
Abstract
A combinatorial synthesis of small libraries of a variety of amino chalcones has been
carried out in solution phase under standard Claisen Schmidt conditions. The
compounds were tested for their potential as antibacterial and cytotoxic agents as well
as phosphatase inhibitors and the leads were identified in each bioassay. Chalcone 7
was found to have strongest potential as cytotoxic agent, while chalcone 11 be the
most potent PGM inhibitory agent.
Parallel synthesis of a 120 member chalcone library was carried out as mixture
synthesis following the positional scanning protocol. The identification of lead in this
library was carried out by deconvulution and chalcones 22 and 41 were found to be
the most potential candidates to be developed as antibacterial agents.
Following the same strategy of mixture synthesis, another 175 member chalcones
library was synthesized and most potent anticancer chalcones 31, 61 and 78 were
identified by deconvolution through position scanning protocol.
Peptidyl α,β- unsaturated ketones were synthesized as novel bis- electrophiles
susceptible for 3+2 and 3+4 annulations. As a result, peptidyl oxazoles, pyrazolines,
pyrazoles, benzothiazepines and benzodiazepines were synthesized.
Potent antidiabetic chalcones were docked into the PGM active site and a rationale
was found for greater antidiabetic activity of chalcones over the other. Rational design
and synthesis of some cytotoxic chalcones was based on 3DQSAR studies using
CoMFA as a tool. 3DQSAR studies were also carried out on a library of 30 chalcones
as potential antitumor agents.

1
Synthesis
Drug
Lead Structure
d f
Drug Candidate
Combinatorial Chemistry
Lead Natural Products
Introduction

Introduction The quality of human life is an outcome of the scientific research introducing new
materials and chemical entities every year. In the last century, the orthodox chemical
synthesis practice i.e. making and testing one compound at a time, was replaced by
more efficient combinatorial methods. Combinatorial chemistry is an exciting
approach to chemical synthesis that enables the creation of a collection of large
number of organic compounds called a library. The combinatorial libraries may
contain a chemical mixture, a physical mixture or the individual pure compounds
synthesized by linking chemical blocks in all possible combinations.1 The key to
combinatorial chemistry lies in the concurrent and parallel synthesis of a large number
of analogues using the same reaction conditions and the same reaction vessels.
Building blocks Library
5 5 X 5 = 25
Scheme 1.1. General schematics of a combinatorial library.
Diversity is the key for searching New Chemical Entities (NCE) with better potency
or higher binding affinity in the target active site of an enzyme or receptor. The
incorporation of chemical diversity involves a lot of toil and time consumption when
carried out through conventional synthetic methods, where the number of compounds
is generally proportional to the number of experiments. Diversity is the genuine
feature exploited by Mother Nature for evolutionary experimentation and is
accomplished through combinatorial methods. The implication of combinatorial
chemistry in human beings can be taken as an example. The basis of our immune
system is the combinatorial production of millions of different antibodies synthesized
by recombining segments of a variable region of primary peptide structure (Fig. 1.1).

Chapter 1 ♦Introduction
- 3 -
Human antibody libraries are screened against different antigens for desired
specificity in search of new therapeutic agents.2
Fig. 1.1. Combinatorial production of antibodies in humans.
Furthermore, the deadly south pacific cone snails have been practicing combinatorial
synthesis of mixtures of more than 100 peptide toxins for the past 50 million years.
An example is conotoxin for paralyzing their prey (Fig. 1.2).3
Fig. 1.2. South pacific cone snails.
The goal of combinatorial chemists is to adopt evolutionary concepts of nature in the
laboratory. Although chemists were engaged in mixture synthesis for years, this
mixture synthesis was named as combinatorial synthesis in early 90’s leading to the
birth of a new field named as combinatorial chemistry (CombiChem).4 CombiChem is
used to systematically optimize chemical properties for different blocks of small,
reactive molecules with varying structures. The reactive chemical blocks are

Chapter 1 ♦Introduction
- 4 -
assembled to produce maximum diversity and a wide range of useful chemical
properties, which are expected to produce the desired biological response in a
bioassay. CombiChem has drastically changed the scenario of the drug discovery
process. Through conventional methods, an estimate for the time and cost of bringing
a NCE into the market is eleven years and 690 M $.5 The generation and use of
combinatorial libraries for the identification of novel chemical lead compounds or for
the optimization of promising lead candidates has emerged as a powerful method for
drug discovery.6-10 CombiChem has provided many new leads and drug candidates
and is, therefore, envisioned to decrease the cost and time expense in the drug
discovery process.11-21 The role of CombiChem in the drug discovery process is
illustrated in Fig. 1.3.
Lead Structure
identification
Drug
Drug Candidate
Combinatorial Chemistry
Natural Products Lead
Synthesis
Fig. 1.3. The role of CombiChem in drug discovery.
Formally, CombiChem started with the first report of mixture synthesis on
polypropylene pins in 1986.22 Parallel solid phase synthesis as circles on cellulose
paper and on pins respectively had already been reported.23 Later, the tea-bag method
was introduced that simplified the rapid multiple peptide synthesis on solid support.
Another addition to the repertoire was pool-and-split method introduced for the
mixture synthesis of peptide and oligonucleotide libraries.24 The solid phase peptide
synthesis rapidly evolved in 1980s, when Merrifield25 was awarded the Nobel prize in
chemistry in 1984 for his work on solid phase synthesis. A number of synthetic
strategies were introduced in late nineties to start with the combinatorial chemistry

Chapter 1 ♦Introduction
- 5 -
and till now new are being added to this inventory.26-38 The chemical diversity
methods for the preparation of large compound libraries for biological screening
purposes have become today a significant subject of research. The medicinal chemists
always focus to discover compounds with optimal biological activity and with the
ability to resist endogenous degradative pathways. Biological methods to create
diversity do not offer this capability, though they produce libraries of significantly
greater size than can currently be assessed with chemical methods. The CombiChem
initially evolved with the peptide and oligonucleotide synthesis; however, many
efforts have been exercised to apply combinatorial library synthesis of small non-
peptidic drug-like molecules exploiting the greater diversity and range of useful
properties embodied in small molecular libraries.39-45
The range of combinatorial techniques is highly diverse. The libraries can be
synthesized individually in parallel fashion or in mixtures, using either solution or
solid phase techniques. A wide range of approaches to the generation of chemical
libraries have been disclosed, including split and mix, encoded, indexed or parallel
and spatially addressed synthesis on pins, beads, chips and other solid supports.46
Whatever the technique used, the common denominator is always to amplify
productivity beyond the levels that have been routine for the last hundred years.
The means of identification of active compounds in libraries is called deconvolution.
It includes different approaches for the identification of the most active compound in
the library through further synthesis and screening experiments. Reported
deconvolution methods involve the iterative resynthesis, recursive deconvolution,
radiolabeling, fluorescent labeling, sequencing of peptides themselves, sequencing of
polymer coding for peptide sequences (including DNA or peptides) and sequencing
by organic identifying group.47 A thrilling addition to the repertoire was positional
scanning, a new deconvolution method which was introduced in 1992 and enabled the
identification of active analogue of a particular pharmacophore in a single round of
screening.48
1.1. Combinatorial library synthesis
The combinatorial libraries are synthesized by linking chemical building blocks in all
possible combinations. The number of library members is always equal to the product
of the number of building blocks incorporated at each step.

Chapter 1 ♦Introduction
- 6 -
The power of combinatorial chemistry to produce enormous number of combinations
is well described through a peptide library synthesis. The combinatorial possibilities
for the number of compounds is seen to increase exponentially, that is, the use of N
components is any order in a sequence of M coupling steps leads to the formation of
NM compounds. For example, using 20 natural amino acids for synthesis of a
pentapeptide library would result in a massive of 3,200,000 compounds (20).
Parallel library synthesis is frequently used to produce small libraries (i.e., containing
hundreds to thousands of compounds) in small tubes, microwells or disposable plastic
syringes fitted with Teflon filters. This is most often the case when pre-existing
template information is available and a particular drug or bioactive compound is being
mimicked. The parallel library synthesis involves the reaction of a reactant A with
multiple reactants, B1, B2, B3 … Bn, to produce a compound library of n individual
products AB1, AB2, AB3 … ABn (Fig. 1.4). The parallel synthesis of tens to hundreds
of analogues of a biologically active compounds can be achieved through manual or
automated approaches.49-50 In parallel synthesis, each reaction sequence is performed
separately and simultaneously with every other in separate compartments. The
products are synthesized using reliable coupling and functional group interconversion
chemistry in solution or on solid support.
B1 AB1 A1 B1 A1B1, A2B1, A3B1 ….AnB1 B2 AB2 A2 B2 A1B2, A2B2, A3B2 ….AnB2 A + B3 AB3 A3 B3 A1B3, A2B3, A3B3 ….AnB3 : : : : : : : : : : : : : : : : Bm ABm An Bm A1Bm, A2Bm, A3Bm ….AnBm
Parallel Synthesis Mixture Synthesis
Fig. 1.4. Combinatorial library synthesis.
The main benefit of this technique is that there is no difficulty in determining the
synthetic heritage of the individual compounds; all the members in a particular well is
identical to each other. The pools of synthesized library are subjected to screening
after removal of solvent and volatile by-products or after cleavage from the support.
The library is screened, usually without purification, and with only minimal

Chapter 1 ♦Introduction
- 7 -
characterization of the individual compounds. The active pools identified, thereby,
contain only the LEAD compounds.51
1.1.1. Solution phase library synthesis
Solution phase synthesis is the major contributor in medicinal chemistry projects for
synthesizing drug-like molecules. A strong synthetic experience is always needed for
carrying out successful solution phase chemistry, but the products synthesized are
accessible for analysis; they are usually cheap and easily scalable. Furthermore, no
synthesis development work is needed for solution phase chemistry as compared to
solid phase synthesis. Because of the soluble nature of in-solution synthesized
libraries, they are not only easily accomplished but are also directly accessible for
biological screening of the final products. There are always chances for impure
products in solution phase chemistry unless the by-products are volatile or the
reactions are very clean and high yielding. Therefore, the solution phase synthesis is
only used for making smaller (more focused) libraries involving one to three step
reactions only. New equipment, such as 'personal synthesizers’ and ‘multi vial
apparatus,’ allows for parallel synthesis of many compounds by one chemist in a
simple and quick manner.
Parallel synthesis is the most straightforward method for library preparation owing to
its close resemblance to the traditional synthesis. Individual compounds are
synthesized in different vessels (at the same time) and are directly accessible for
screening only after minimal purification. There is no need of tedious deconvolution
for identifying the synthetic heritage of the active compound. The active hit is easily
identified from its position (X, Y coordinate) in the array, which encodes the reagents
and thus structure of the product.
Mixture-based library synthesis was first accomplished by Hougten.52 Such libraries
are generated through divide couple and recombine (DCR) method in solution, or
attached to soluble polymer supports. These libraries, being soluble, are easily
subjected to routine assays for identification of the LEAD compounds. Many solution
phase mixture based compound libraries have been reported and the active
compounds were identified with potential for developing into a drug candidate.53
Polymer-assisted solution phase synthesis (PASP) is carried out for the in-solution
synthesis of compound libraries (or individual compounds) using polymeric

Chapter 1 ♦Introduction
- 8 -
reagents.54 It employ the polymer reagents or polymeric scavenger reagents and the
library is thus generated using both solution and solid phase methods. Polymer
scavengers are utilized for the purpose of removing by-products by simple filtration.
They can also immobilize the hazardous materials produced during a chemical
reaction. The technique facilitates a complete conversion of reactants into products by
employing excess of reagents. PASP can also be fully automated for rapid generation
of compound libraries.55-56
1.1.2. Solid phase library synthesis.
Solid phase synthesis (SPS) involves the construction of peptides or drug-like
molecules on an appropriate solid support, and is the birthplace of the combinatorial
‘revolution’. In SPS, reactions are carried out on the surface of a solid phase called
resin, which is in heterogeneous contact with the solution. The reactions are, thereby,
completed in a swollen gel system formed by the penetration of solvent and solute
molecules into the polymeric matrix. Three essential requirements for SPS are the
following.
1. A physicochemically and thermally stable solid support
2. A suitable linker reagent
3. A chemical protection strategy
The first requirement is a cross-linked, insoluble polymeric material called resin bead,
which is physicochemically and thermally stable and inert to the reaction conditions
to be carried out onto it. The resins are most commonly polystyrene cross-linked with
1-2% divinylbenzene to make insoluble porous beads a few microns in diameter. The
polystyrene is derivatized, most commonly “chloromethylated” to provide a
functional group to which a small organic molecule may be attached either directly or
via a linker group.
OOH
Wang resin
Linker
Linking functional group
Fig. 1.5. The Wang resin and linker.

Chapter 1 ♦Introduction
- 9 -
Beads must be able to swell in the solvent used. Most reactions occur in the bead
interior. The magnified surface of a polymeric resin bead is shown in Fig. 1.6
showing the linkers attached to polymeric bead as small red lines.
A
s
m
(
t
f
T
resin beadswelling
starting material, reagent and solvent
linkers
Fig. 1.6. The structure of a resin bead
suitable linker reagent is the second requirement, which allows the linking of
ubstrate to the solid support (Fig. 1.7). A linker allows the construction of organic
olecule/peptide on a resin bead and facilitates the projection of resin bound substrate
starting material) into the bulk solvent, generating a reactivity environment closer to
he solution phase chemistry. The quantitative removal of the final products of interest
rom the linker is necessary when the synthesis is complete.
synthetic
resin linker
steps
cleavage
attachment
linker resin molecule
molecule linker resin
molecule
Fig. 1.7. The linker acting as attachment site for construction of organic molecule.
he following are some important characteristics of a linker:
• It is a molecular moiety, which is covalently attached to the solid support and
which contains a reactive functional group.
• It allows the attachment of the first reactant.
• The linker must be stable to the reaction conditions of the synthesis but easily
cleavable to release the final compound.

Chapter 1 ♦Introduction
- 10 -
• Different linkers are available depending on the functional group to be
attached and the desired functional group on the product.
• Resins are named to define the linker e.g. Merrifield, Wang and Rink.
A chemical protection strategy is needed to allow selective protection and
deprotection of reactive groups. Different protecting groups are used for peptide
synthesis. Some of the most common protecting groups are 9-
fluorenylmethyloxycarbonyl (Fmoc), tert-butyloxycarbonyl (Boc), benzyloxy-
carbonyl (Z) and allyloxycarbonyl (alloc) as shown in Fig. 1.8.
Cl
The
reac
filtra
man
desi
end
the N
to th
prev
prim
acid
are
(Fig
synt
spec
auto
Che
OO
O
O
OO
O
O
O
Cl Cl
Fmoc Boc Z alloc
Fig. 1.8. The common protecting groups used in peptide synthesis.
technique of SPS has the advantages of minimal solubility problems since the
tions are performed in a swollen gel system, simplified purification through
tion and washing of the support, improved reaction yield, simplified
ipulation of small molar quantities and easy automation. The synthesis of a
red sequence of polypeptide includes the following essential steps. The carboxyl
of the amino acid is loaded onto the support and every new amino acid is added to
-terminus of the growing peptide chain. When each new amino acid is coupled
e previous one, the amino terminus of the amino acid being added is protected to
ent unnecessary chain growth. After the coupling step, the protection of the
ary amine is removed and the coupling reaction is repeated with the next amino
. Amide coupling, washing, deprotection, washing and then next amide coupling
carried out as a repetitive cycle until the desired peptide sequence is complete
. 1.9). After the synthesis of the peptide of a desired length and sequence,
hesized products are cleaved from the solid support and are purified for routine
tral analysis. This laborious process, therefore, lends itself extremely well to
mation of peptide and oligonucleotide synthesis employing different synthesizers.
mistry required for the synthesis of these biopolymers was quite limited and a

Chapter 1 ♦Introduction
- 11 -
huge research was therefore taken in 1990 to enhance the scope and application of
SPS to the construction of small drug-like molecules. In 1992, Bunin et al. reported
first solid phase organic synthesis of a library of 192 benzodiazepines, which resulted
in an explosion in the area of synthesis of drug-like molecules on solid phase (Fig.
1.10).57-58
P1
Many
techno
gradua
has b
polyet
memb
approa
NH O
P2 H2NO
HN OH
O HN
O
A
NH O
OHN
A
OHNH O
R1OH2N
R2
deprotection
P2
P1
P3
P4
P3
P4activation
A
H2NO
P1
HN
O
A
P3
P4
couplingP4
P3
P1
P4deprotection
NH O
OHN
P4
P3
P1
deprotection
cleavage
P1 Aresin protecting group activator
Fig. 1.9. General scheme for solid phase peptide synthesis.
laboratories and companies started focusing on the development of SPS
logies after this pioneering work and spectacular developments were made
lly regarding different solid supports and chemistry suitable to SPS.59-61 SPS
een performed on a variety of supports including polymeric resin beads,
hylene pins, crowns, tubes, photoresponsive chips, paper, glass, cotton and
ranes. Today, it has spread in every field and is being adapted as an ideal
ch for the synthesis of new drugs, new catalyst, or new natural products.

Chapter 1 ♦Introduction
- 12 -
NH2
O
RB
RA
NH
O
RB
RA
RB
RA
FmocHN CO2H
RC
ORc
NHFmoc
N
HNO
Rc
N
NO
Rc
N
NO
Rc
12 amino acids
RD Hal7 alkylating agents
RB
RA
RD
RB
RA
RD
192 benzodiazepines
Fig. 1.10. Solid phase synthesis of a diazepinone library.
1.1.3. Parallel library synthesis
Both solution and solid phase parallel chemistry can be employed for the construction
of a library of molecules of varying nature. Through manual or automated approaches,
tens to hundreds of analogues of a biologically active substrate are synthesized in
parallel such that a single product is produced in each reaction vessel. Parallel
chemistry has the advantage of time saving in doing all the reactions simultaneously
in different vessels but separate work-up is required for each newly synthesized
product.
Parallel solid phase synthesis can be carried out on any support such as pins or tea
bags and the addition of reactants is made separately using different vessels or wells
of a microtitre plate. A parallel synthesis of nine dipeptides is shown in Fig. 1.11. In
this synthesis, optimization of 12 reactions will provide 9 dipeptides in separate
vessels. Since the library members will be in separate vessels, this technique can be
successfully used for both solution and solid phase synthesis.
Parallel synthesis can be automated through use of automated synthesizers (Fig. 1.12)
containing 6, 24, 42, 96 or 144 reaction vessels or wells. These synthesizers employ
the use of beads or pins as solid support. Reactions and work-ups are carried out

Chapter 1 ♦Introduction
- 13 -
automatically employing the same synthetic route for each vessel. The use of different
reagents in each well results in the synthesis of different products.
diversification reaction divide diversification reaction
Fig. 1.11. A schematic for solid phase parallel synthesis of dipeptides.
Fig. 1.12. Parallel synthesizers, a) Radleys 6-well workstation, b) Process chemistry
workstation, advantage series 3400™.
1.1.3.1. Tea bag method
This methodology is a manual approach to parallel synthesis that was first employed
by Hougten in 1985 for synthesizing a peptide library.62 It has been successfully used
for generating numerous peptidic and heterocyclic libraries. The resin is distributed
into individual polypropylene meshed bags (3 X 4 cm), heat sealed and labeled simply
by using graphite based ink.

Chapter 1 ♦Introduction
- 14 -
bel
Seal
Label
Polypropylene mesh
La
Fig. 1.13. A tea bag
For routine peptide synthesis, sealed tea bags with enclosed resin are distributed into
clean reaction vessels and resin (in each bag) is loaded with the first reactant or
acylated with a different amino acid. After the reaction is complete, the common
washing and deprotection steps are carried out in a single vessel. The reaction with a
further set of activated reagents is made after distributing tea bags to different clean
vessels. The process is continued until desired compound or peptide sequence is
complete. All the resin portions from different bags are cleaved separately and are
analyzed after necessary purification.
1.1.4. Mixture synthesis
This method involves the use of a standard synthetic route to produce a large variety
of different analogues where each reaction vessel or tube contains a mixture of
products.
20 Amino acids (X20)
Hexapeptides
(1,889,568 hexapeptides / vial)
34 Million products
The identities of the structures in each vessel are not known with certainty.
Combinatorial mixture synthesis is called a game of numbers. The number of
compounds synthesized increases exponentially with the number of reaction steps
encountered. Therefore, this method is quite useful for finding a lead compound in a
large molecular library. The synthesis of hexapeptide mixtures from 20 amino acids,
for example, will result into 34 million products. The approach enables the synthesis
of a large number of compounds quickly as mixtures, each of which is tested for

Chapter 1 ♦Introduction
- 15 -
activity. The inactive mixtures are stored in combinatorial libraries, whereas active
mixtures are studied further to identify the active component.
1.1.4.1. Split and mix synthesis. The classical method for the synthesis of all possible
dipeptides using 5 amino acids would involve 25 separate reactions, whereas the
combinatorial procedure would involve five separate steps, following a split and mix
strategy, also known as DCR, as discussed earlier.
This is a simple but amazing methodology for generating chemical diversity. For
example, it has played an implausible role in the development of CombiChem. The
split and mix method was first introduced by Furka63 for the synthesis of a peptide
library as shown in Fig. 1.14. Making a peptide library from three amino acids, for
example glycine (green), alanine (red) and valine (blue), will result into nine
dipeptides in just two steps. Initially, a batch of resin beads is taken and split into
three equal portions followed by the coupling of one amino acid to each of separated
resin portions.
split mix split
diversification reaction diversification reaction
Fig. 1.14. SPS of a library of dipeptides through split and mix strategy.
After washing all the batches thoroughly for removing excess of amino acids and
coupling reagents, they are pooled and mixed thoroughly. The splitting step is
followed by another round of coupling with other amino acid called a diversification
reaction. Following a thorough washing step results in three different dipeptides in
each reaction vessel. Hence, two times splitting and pooling, i.e. two diversification
reactions would result into a library of nine dipeptides.

Chapter 1 ♦Introduction
- 16 -
A diverse library of any drug-like molecular template or polymer can be envisioned
using the concept of “split and mix” synthesis e.g., Elmann reported the first solid
phase synthesis of a 11, 200 member diazepinone library through split-mix method.
The split and mix step is represented by as shown in (Fig. 1.15).64
O
NHBoc
SnMe3O
NH2
O
NH
O
R1R1
O
Cl20 acid chlorides 20 compounds
TFA
F
OR2
NHFmoc
35 amino acids
ONHFmoc
R2
O
R1
20 x 35 = 700 compounds
Pd2dba3
O N
HN
O
R1
R2
16 alkylating agents
Base, R3X TFAHO N
HN
O
R1
R2
20 x 35 x 16 = 11,200 compounds700 compounds
Split-Mix step Pd2dba3 = Tris(dibenzylideneacetone)dipalladium (0)
Fig. 1.15. Synthesis of a diazepinone library through split and mix strategy.
1.1.4.2. Positional scanning library synthesis
In positional scanning library synthesis, all sub-libraries necessary to trace the most
active compound are prepared before screening. This is another type of mixture
synthesis and is a variation to split and mix approach. This technique employs the
synthesis of ‘orthogonal libraries’ called sub-libraries based on the principle of the
creation of two sets of mixtures. The sub-libraries are synthesized by keeping one of
the components of two building blocks of a reaction constant (alternatively) and the
other is used as an equimolar mixture. The same compound is contained in two
different sub-libraries (product mixtures) and the observed activities of the mixtures
in these sub-libraries are used as “indices” to the rows or columns of a two-
dimensional matrix reflecting the activities of individual compound responsible for
that activity.65
The generation of a two-dimensional positional scanning or indexed combinatorial
libraries can be presented in the following fashion:

Chapter 1 ♦Introduction
- 17 -
R−X + nY−R′ ⎯⎯→ mR−R′1−n
m sub-libraries of n compounds
R′−X + mX−R ⎯⎯→ nR′−R1−m
n sub-libraries of m compounds
Each of m molecules (R−X) having one reactive site is reacted with a mixture of n
other molecules (R′−Y) to provide m sub-libraries, each containing n compounds with
variations of only R′ groups. When another type of molecule (R′-Y) is fixed, n
numbers of sub-libraries, each containing m compounds with variations of only R
groups, are obtained. Positional scanning method offers several advantages over solid
phase and solution phase parallel approaches.
The combinatorics in the preparation of such a library can be conceptually presented
as an N-dimensional matrix, wherein this matrix has as many elements as are present
in each set (n). Consider a library of molecules composed of two sets of structures A
and B, each of which has ten structural variants. The number of variants in each
building block set, a and b is 10. They can be envisioned to be composed of a 10 x 10
grid, where each cell contains, for the combination (Ax By), its assay mixture (Fig.
1.16). To examine all as pure compounds would require 100 experiments. Since one
cell possesses the maximum response function in the grid, the task is to find it without
actually preparing them all. Were the contribution of A and B to the response function
completely independent, the best combination would be obtained by choosing any B
for testing with all A’s, and any A for testing with all B’s. When A and B are not
dependent variables in the response function, indexing permits selected combinations
to be tested. By screening the rows and the columns, which are indices to the cells at
their intersections, as indices only 20 reactions/assays are required to find the
maximum response. Each compound is tested twice, once each as a component of an
A mixture and a B mixture (100 compounds x 2 assays = 200 = 20 row/column
reactions x 10 compounds in each). The index to the maximum cell in this example is
its row reaction, composed of one reagent B5 and an equimolar mixture of reagents
A1-A10, and its column reaction, composed of the reagent A4 and an equimolar
mixture of B reagents B1-B10. Because all combinations are tested, an assumption that

Chapter 1 ♦Introduction
- 18 -
parameters do not interact is not required. This example shows a 5 fold improvement
in the synthesis and data collection efficiency (the parallelism advantage) for the
library compared to one -at-a-time processing. This process can be conducted with
more elements in each set and with more sets, leading to higher dimensional arrays
and to higher efficiency in data collection. 1 2 3 4 5 6 7 8 9 1 2 3 4 5 * 6 7 8 9 10
Fig. 1.16. Conceptual matrix for combinatorial synthesis.
The synthesis of positional scanning libraries represents one of the most useful
protocols for mixture synthesis. Not only is it much less time intensive as compared to
parallel synthesis of individual compounds or small mixtures, but also produces
depository libraries for use in multiple screens with immediate deconvolution i.e. the
identification of most active member of the library. Thus, unlike other deconvolution
protocols, positional scanning libraries provide lead identification in a single round of
testing. However, positional scanning protocol requires a significant amount of
synthetic work before the activity of a library is judged. These libraries, being less
demanding to prepare, allow an accurate detection of significant activities, but more
subtle discoveries and less-distinguishable activities are not detected. This is a natural
consequence of testing the larger compound mixtures and the relative insensitivity of
assays to the contribution of any single, uniquely acting compound in the mixture.
Thus, the disadvantages associated with the loss of some information contained within
the library must be balanced against the advantages of the ease of library synthesis
judged in the light of library-screening objectives.
Small mixture libraries can be assembled simultaneously in a simple chemical process
from multiple sub-units and then screened directly. The number of compounds
prepared is much greater than the number of chemical steps required. Therefore, this
approach is fast and relatively inexpensive. Screening two-dimensional matrix

Chapter 1 ♦Introduction
- 19 -
libraries should identify the most active sub-libraries, which can be resynthesized;
thereby simplifying the deconvolution procedure.66
Indexed library method was first introduced by Pirrung.67 Pirrung et al. produced a
series of carbamate and tetrahydroacridine two-dimensional indexed libraries. An
exhaustive review on CombiChem describes the indexed combinatorial method in
detail.68 Smith et al. reported the synthesis of 80 two-dimensional indexed sub-
libraries.69 Nauville et al. produced libraries based on piperazine.70 Kaldor et al.
reported discovery of antirhinoviral leads from two-dimensional indexed
combinatorial libraries.71 Mardar et al. prepared nine indexed sub-libraries by a one-
pot three-step procedure.72 Nielsen et al. reported the generation of phthalhydrazide
indexed libraries,73 while Chung et al. prepared a β-amino alcohol library in solution
phase.74 Dooley et al identified a novel, highly active tetrapeptide agonist for the
opioid receptor was from a positional scanning library of 6.25 million tetrapeptides.75
These types of libraries may be considered more applicable for lead discovery than
the parallel approaches.
1.2. Deconvolution
Deconvolution is the finding of most active compound/sequence in a successfully
synthesized bioactive compound library prepared as a mixture. The lead identification
in a mixture is a tiresome job and several approaches have been introduced for
achieving this goal. The following are some important deconvolution methods:
Iterative re-synthesis.76 Iterative re-synthesis is the process in which sub-libraries are
re-synthesized in order to find the most active compound in the library. After
screening the library as a mixture of compounds, re-synthesis and re-assay of possible
candidates in active pools is carried out. The number of sub-libraries gets smaller and
smaller until the most active compound is identified. At the end, single compounds
are prepared in each pool and tested for their activity. As a consequence, the most
active compound is identified from a mixture of compounds. Consider for example, a
library of 27 compounds (diversity elements A, B and C) is synthesized. In order to
find the most effective compounds from a screen of mixtures, we need to resort to
sub-library synthesis for finding the active compound C-B-A. The iterative re-
synthesis has the problem of interference by unwanted properties of other compounds
e.g. cytotoxicity in each mixture screen. The possible synergistic interactions of

Chapter 1 ♦Introduction
- 20 -
multiple compounds are also possible. Furthermore, sub-library synthesis is
cumbersome for finding the lead structure.
sub-library sub-library sub-library
Recu
the i
1.18
The
whic
this
A-X-X
B-X-X
C-X-X
C-A-X
C-B-X
C-C-X
C-B-A
C-B-B
C-B-C
C-B-A
synthesis
library with C at defined position show
best properties
library with B at defined position show
best properties
most effective compound has structure CBA
screeningscreeningscreening
library with A at defined position show
best properties
synthesis synthesis
Fig. 1.17. Iterative re-synthesis of a tripeptide.
rsive deconvolution.77 This is a modification of iterative re-synthesis in which
ntermediate pools and starting subsets are saved after each coupling step (Fig.
).
Recursive deconvolution
fin
h h
ste
Prepared SavedAnBnC1
AnBnC2
AnBnC3
AnB1
AnB2
AnB3
A1
A2
A3
Pool 1 Pool 2 Pool 3AnBnC1 AnBnC2 AnBnC3
screening
pool 2 was found activeretrieve intermediate poolscoupling with C2
AnB1C2 AnB2C2 AnB3C2
pool 3 was found active
retrieve subset Acoupling with B3, then with C2
A1B3C2 A2B3C2 A3B3C2
screening
screening
pool 2 was found activeA2B3C2 is identified as most active library member
Prepared pools and saved subsets
Fig. 1.18. Recursive deconvolution of a tripeptide.
al pools are tested for their bioactivity and the intermediates of the active pool,
ave been retained before the final step of coupling/synthesis are used to repeat
p (i.e. ‘recursive deconvolution’) and the individual mixture thus obtained are

Chapter 1 ♦Introduction
- 21 -
tested for their potency. Finally, an amino acid or reactant is identified which
constitutes a part of the most active compound. The components of the active mixture
obtained after the final step are re-synthesized individually and tested for finding the
most active compound or sequence.
On-bead screening.78 In this technique, each library component is tested as resin
bound in an assay medium, where a soluble receptor is present and the interaction
between receptor and ligand/inhibitor takes place at the surface of the bead. With the
help of a colorimetric, fluorescent or radiolabeled detection mode in the bioassay, the
active beads are identified. They are removed from the assay medium and analytically
characterized after washing off the receptor. When on-bead screening is carried out
through fluorescent labeling, a fluorophore is attached to the growing peptide.
Enzyme-linked colorimetric assays are used for identifying beads bearing active
fluorogenic peptide sequences. Active beads are discovered by visual inspection and
isolated by micromanipulation. The structures of active peptide isolated thereby are
determined by microsequencing.79,80
Multiple release resins.81 A variety of resins have been introduced, which allow the
release of a certain amount of target molecule from resin bead for its bio-evaluation,
while the rest still attached to bead. Mixtures are synthesized on resin beads, tablets
etc. through split and mix synthesis, so that one particular compound on one bead is
synthesized. The biologically active compound library on beads is divided into
portions, each portion containing many beads/tablets is subjected to cleavage and
cleaved mixtures are bio-evaluated. The one with the desired biological response is
divided, so that each resin entity is separated, cleaved and again tested for desired
bioactivity. The active bead is then subjected to on-bead sequencing using Edman
degradation.
X2
X1
X3
A3AA3A
A3A
A2AA1AFmocA2AA1AFmoc
A2AA1AFmocX2
X1
X3
A3AA3A
A2AA1AFmocA2AA1AFmoc
X2
X1
X3A3AA2AA1AFmocAA3AA2AA1Fmoc OH
AA3AA2AA1Fmoc OH+
+
1% TFA/DCM
95% TFA
Fig. 1.19. Sequential release of peptide from a multiple release resin.

Chapter 1 ♦Introduction
- 22 -
Radiolabelling.82,83 In this technique, radio-frequency microchip tags are used, each
of which are encapsulated in glass. The radiofrequency tags act as a bar-code for
separated portions of resin in vessels like tea bag or glass encapsulated microreactors
employed in the split and mix strategy for library synthesis. The microreactors contain
the regular solid phase synthesis resin (20-50 mg) and a glass encased radiofrequency
tag semiconductor unit capable of receiving, storing and emitting radiofrequency
signals from a distance. The histogram of the synthesis is recorded on each
microreactor (or a macro bead) through remote radiofrequency transmission.
memory chip
inert porous wallresin bead
Fig. 1.20. Schematic representation of a microreactor used in radiofrequency encoded
combinatorial chemistry.
Chemical encoding.84-88 In this technique, a number of chemical tags are incorporated
on the same bead on which the encoded compound is synthesized.
split acylation tagging mix split
Fig. 1.21. Tagging the synthetic heritage of a peptide library.
The tagging process is chosen such that it does not interfere with the synthesis. The
tags should not occupy much of the beads capacity and the synthesized compounds

Chapter 1 ♦Introduction
- 23 -
should be able to cleave selectively from bead in the presence of coding elements
(Fig. 1.22). The decoding processes are selected, so that they are quick and reliable
and the chemical nature of tags permits their rapid determination in small quantities
using conventional analytical techniques. Various chemical tags such as haloaromatic
binary tags, secondary amine binary tags, oligonucleotide tags and peptide tags have
been used for the purpose.
Enc
two
resin
and
stack
colu
diffe
each
x 5
and
small molecule
compound cleavage tag cleavage
Fig. 1.22. Removal of molecular tags for analysis.
oded sheets.89 These sheets are produced by sandwiching resin beads between
sheets of polypropylene. The sheets are fused together to immobilize enclosed
in the form of circles. Each sheet is acylated with a single amino acid, washed
deprotected all in a single vessel (Fig. 1.23). The sheets (for example 3) are then
ed and cut into columns (if are like, as shown in Fig. 1.23, will be cut as five
mns 1-5). Each stack of columns (3 circles) will be acylated separately with
rent amino acid, combined again for common washing and deprotection. Finally,
column will be cut into squares and acylated separately resulting in a library of 3
x 5 = 75 tripeptides. The synthetic origin of each resulting peptide is identified
is recognized through a three letter code assigned to each circle on sheets.
1 2 3 4 5
a
b
c
d
e
Fig. 1.23. A representative encoded sheet.

Chapter 1 ♦Introduction
- 24 -
The tea-bag method and positional scanning of indexed libraries are very effective
and easy-going deconvolution methods, which have already been described in
previous sections, 1.1.3.1. and 1.1.4.2, respectively.
1.3. Drug-like molecules
Drugs are normally low molecular weight chemicals that interact with the
macromolecular targets in the body that are mostly proteins (receptors, enzymes, ion-
channels and transport proteins) or nucleic acids producing desired pharmacological
effects. Drugs have a range of physicochemical features, which result in improved
ADMET (adsorption, distribution, metabolism, excretion and toxicity) properties.90
Lipniski’s rule of five (Ro5).91 Drug-like molecules contain certain drug-like
properties described by certain parameters collected in Lipniski’s rule of five (Ro5)
for orally available drugs. The rules define certain physicochemical constraints, which
are the following:
• M.W < 500
• HBD (H-bond donors) ≤ 5
• HBA (H-bond acceptors) ≤ 10
• log P < 5
Ro5 is associated with 90 % of orally effective drugs that have achieved phase II
clinical status. The rule has been defined through a thorough analysis of the
Comprehensive Medicinal Chemistry (CMC), Derwent Word Drug Index (WDI) and
Modern Drug Data Report (MDDR), which are most commonly utilized drug-like
databases. These parameters ensure acceptable aqueous solubility and intestinal
permeability. If a compound does not obey Ro5, the problem of oral availability is
usually encountered.
Rule of three (Ro3). Certain other findings were made for oral bioavailability of drug
candidates. It was found that number of rotatable bonds (NROT) should not be more
than seven for oral drug delivery.92 Moreover, polar surface area (PSA) is another
essential property for a molecule to be drug-like. Lower bioavailability results for
PSA in a range of 110-140 Å2.93 The term “Lead-Like” has also been introduced
along with the drug-like for molecules identified from High Throughput Screening
(HTS) campaigns.94 As a result, the range of physicochemical properties is narrowed
to “Rule of Three”, which is believed to be helpful in generating fragment libraries

Chapter 1 ♦Introduction
- 25 -
and lead generation.95 Screening experiments indicate that successful hits generally
obey “Rule of Three”, which states that orally bioavailable lead compounds should
have:
• MW < 300
• HBD ≤ 3
• HBA ≤ 3
• log P ≤ 3
• NROT ≤ 3
• PSA ≤ 60
1.3.1. Chalcones and heterocycles
Finding novel biologically active compounds has always been a challenge to
chemists. Since pre-historic times, plants have been the main source of biologically
significant compounds. Chalcones are α,β-unsaturated ketones found as secondary
metabolites in many plant species. Naturally occurring chalcones e.g. 2′,6′-dihydroxy-
4-methoxy chalcone, carthamin and butein are identified from plants such as
cinnamon, red pepper and carthamus flower.96
O
OHHO
O
O
HOOH
OH
HO
O
O
O
HOH
OH
OH
O
HO
HO
HO
OHO
OH
OH
OH
HO
carthamin
butein
In laboratory, their synthesis is accessible by an acid/base catalyzed Claisen-Schmidt
condensation of an aryl aldehyde and a methyl ketone. The reaction can be
accomplished through stirring at room temperature,97 grinding,98 conventional
heating,99 microwave irradiation100 and by sonication101 yielding 1,3-diarylenone in
good yield.
Chalcones mostly are non-toxic, obey Ro5 and are, therefore, expected to be
bioavailable. They, being bioactive, are getting fame in the new era of drug-like

Chapter 1 ♦Introduction
- 26 -
molecules. Chalcones themselves are now getting importance as medicinal agents,
and some chalcones are used as medicines, while others are being investigated as drug
candidates.102 Though structurally simple, various substituted chalcones show an
impressive array of biological activities like anti-inflammatory,103-105 anticancer,106-114
antioxidant,115-116 antiviral,117-118 antibacterial,119-120 antileshmanial,121-123
antimalarial,124-128 antihyperglycemic129 and antituberculotic.130
Some of the activities of chalcones cited in literature are given in Table 1.1.
Table. 1.1. Some recent reports upon bioactive chalcones.
Structure Activity
O
NO2
antitumor131
O
OH
OHH3CO
antibacterial132
OO
OOO
antitrypanosomal133
OH3CO
H3COOCH3
NO2
antileishmanial134
OOH
PtpA inhibitor135
The number of reports on the synthesis and bio-evaluation of chalcones has increased
immensely in the past few decades leading to about 269 manuscripts in 2009 only, as
shown quantitatively in Fig. 1.24.

Chapter 1 ♦Introduction
- 27 -
Fig. 1.24. Pi chart representation of the publications o
searched through ScienceDirect. Some reports on anti-inflammatory chalcone
publishing group. For example, 3′,4′,5′,3,4,5-hex
as an anti-inflammatory agent,136 while 3′-iso
suppress the respiratory burst and degranulation
helps to prevent them from rheumatoid arthritis a
OH3CO
H3COOCH3
OCH3OCH3
OCH3
A
Amino chalcones have been reported with strong
methoxy chalcone (C) was found with quite prom
against a panel of human tumor cell lines, t
multidrug resistant human epidermoid carcino
cancer (IA9).138 3,5-Dimethoxy-4′-amino chalc
potent when tested with some other amino c
potency against murine leukemia (L1210), mu
human T-leukemia (Molt/4 and CEM) and human
O
OCH3
NH2
H2N
C
2009
2006n chalcones dur
s have been
amethoxychalc
propoxychalcon
activity in h
nd asthma.137
OO
B
cytotoxic pot
ising cytotoxic
he most impo
ma (KB-VIN)
one (D) was f
halcones for
rine mammary
cervix carcino
O
D
2008
20042002
2001 20032005
2007
ing 2001-2009 as
published by nature
one (A) was disclosed
e (B) was found to
uman neutrophils and
ential e.g. 2′-amino-3-
potential when tested
rtant of which were
and human ovarian
ound to be the most
their antiproliferative
carcinoma (FM3A),
ma.139
OCH3
OCH3

Chapter 1 ♦Introduction
- 28 -
4′-Hydroxy-4-N,N-dimethylaminochalcone (E) has been reported as a potent
antimalarial agent against chloroquine resistant human malarial parasite Plasmodium
falciparum, but the sulfonamide analogs showed an increased potency with compound
(F) as the most active and effective antimalarial with an IC50 value of 1 µM.140
O
HO
N
O
SO2HN
Cl
Cl
E F
Moreover, many chalcone-based pharmaceutical compositions have also been
patented. An example is the pharmaceutical composition of chalcones and their
derivatives (G-J) patented as antiangiogenic agent141 for treating angiogenic skin
disorders such as psoriasis, venous ulcers, acne, rosacea, warts, eczema, hemangiomas
and lymphangiogenesis, among numerous others. Moreover, internal malignancies
(e.g. colon, cervical, bladder), oral malignancies, cutaneous malignancies, including
basal cell carcinoma, squamous cell carcinoma and inflammation can also be treated
through the same chalcone-based pharmaceutical composition.
O O
H3CO
OCH3
G H
O
O
I J
The pharmaceutical compositions of chalcones and their derivatives were patented as
inhibitors of matrix metalloproteinases (MMP’s), a family of over 20 proteins. This
patent has its pharmacological significance against diseases caused by over expression
of MMP’s, e.g. Alzheimer's disease, asthma, skin aging, arthritis, angiogenesis and
cancer metathesis. The discovered formulations containing chalcone, phloretin, 2-
hydroxy chalcone, 4-hydroxy chalcone, 4-methoxy chalcone, 4′-methoxy chalcone
and 2′,4′-dimethoxy chalcone can be taken as a 1-200 mg /kg dose orally (tablet,

Chapter 1 ♦Introduction
- 29 -
capsule, syrup, sachet, etc.) or topically (cream, lotion, ointment, paste, etc.) or
through injection.142
The pharmaceutical composition containing mixtures of fluorous chalcones (such as
K) as anticancer agents was published in 2007. Analogues of fluorous chalcone were
inducers of caspase cascade and thus activators of apoptosis causing subsequent cell
death in drug-resistant cancer cells. They, being inducers of caspase cascade, were
also claimed as useful therapeutic targets for viral infections as HIV and hepatitis C
virus (HCV).143
OOF
F
F
O
F
FF
K
Amino functionalized chalcones e.g. L and analogues have also been described as
useful drug substances with antiparasitic and bactericidal properties. They were
disclosed to possess enhanced biological effects along with improved metabolic and
physicochemical properties associated with them.144
O
N R
L
Chalcone are not only themselves very important for their biological properties but
they are also important precursors for a large number of heterocyclic systems, not
only in the biota in nature but also in the chemistry laboratory. The important
heterocyclic systems derived from chalcones as precursors are benzothiazepine,
benzodiazepine, benzoxazepine, pyrimidine, pyrazole, oxazole, etc.,145 as shown in
Fig. 1.25. Heterocycles are also important small drug-like molecules with only few
Lipniski’s violations. As a result, they have usually enhanced absorption and
bioavailability and are ideal leads whenever they show potency against any drug
target. Ansari et al. have reported the synthesis of a wide variety of chalcones and
derived benzothiazepines, which are found to be free radical scavengers and enzyme
inhibitors against cholinesterases, urease and α-glucosidase.146 The mixture based

Chapter 1 ♦Introduction
- 30 -
indexed libraries of chalcones have also been synthesized and the leads were
identified through deconvolution in antibacterial and antitumor based screening
experiments.147-148 Some chalcone libraries have also been evaluated for their
antibacterial, cytotoxic and antitumor potencies with the subsequent QSAR and 3D-
QSAR studies.149
O
OO
N O
N NH ONH
N O
N N
Ph
NHNH
O
N S
Fig. 1.25. Important heterocyclic systems based on chalcone precursor.150-156
Furthermore, theoretical and computational studies on the leads identified as AChE
and urease inhibitors have also been reported by the same group.157
1.3.2. Peptidyl chalcones and heterocycles
Protein binders are of primordial importance for the study of functions and structure
of proteins and in many cases can be used as drug molecules.158 Therefore, methods to
develop novel protein-binding molecules are of general interest. In the most direct
approach, proteins themselves are well suited to bind to protein-protein interaction
sites as they can deliver the hydrogen-bonding pattern of proteins and their complete
side chain diversity. α,β-Unsaturated ketone core is the part of chalcones, which in
most cases is responsible for the bioactivity of a chalcone template. The carbonyl part
of the molecule acts as a HBA and is found to co-ordinate with the metalloproteinases
for a wide range of bio-activities.157 The incorporation of a peptidyl chain instead of a
phenyl ring may result in an enhanced diversity and provide an effective hydrogen
bonding pattern, which is characteristic of peptide ligands in chemical biology. The
resulting peptidyl chalcone derivatives are expected to be interesting ligands that may
result in specificity when tested for different bioactivities known to be characteristic
of chalcones.

Chapter 1 ♦Introduction
- 31 -
It is well documented in literature that the use of peptide ligands in chemical biology
and medicinal chemistry is limited severely by several factors.159-162 Many short
length peptides are conformationally flexible and do not possess a rigid solution
structure, and show a decreased binding affinity and biological activity. Moreover,
peptidic ligands are prone to proteolytic degradation under physiological conditions,
possess no significant bioavailability and do not permeate through biological
membranes in most instances.163 For these reasons, small, non-peptidic interfering
molecules can be a powerful complementation to peptides. Heterocycles are
especially suited as drugs presumably due to their limited flexibility and a relatively
lower number of rotating bonds found in these molecules.164 Such combinations have
been especially successful in those cases in which peptides are the native substrates of
ligands of the targeted protein such as in the field of protease inhibitors. Therefore,
novel structural combinations of heterocycles and peptides are highly desirable.165
In this perspective, peptidyl chalcones appear as interesting template for the
construction of a variety of peptidyl heterocycles such as diazepines, thiazepines,
pyrimidines, pyrazoles, oxazoles, etc. Many heterocycles contain HBA as well as
HBD sites for interaction with biological targets. Peptidyl heterocycles are expected
to have high polarizability for efficient interactions with hydrophobic pockets and are
expected to release water, increased membrane permeability and enhanced metabolic
stability. It is expected that this conjugation of peptides and heterocycles would lead
to improvement in the bioavailability of the synthesized drug-like peptidyl
heterocycles. The hybrid structures are expected to widen the scope for developing
more stable short peptide sequences as drug candidates.
Peptidic-heterocyclic conjugates are not new to the field of chemistry or biochemistry.
Microbes employ several catalytic strategies to transform conformationally flexible
peptide chains into heterocyclic rigidified scaffolds that possess antibiotic or toxin
activity. Prominent examples include biosynthesis of lactam antibiotics of the
penicillin and cephalosporin families and the maturation of vancomycin where
distinct structural modifications to the nascent peptide chains confer physiological
function (Fig. 1.26).166

Chapter 1 ♦Introduction
- 32 -
Patellamide A Patellamide C
Fig. 1.26. The ribosomaly derived peptides, Patellamides A and C.
Heterocycles are a recurring motif for altering shape and flexibility of peptides. It is
believed that peptidic heterocycles can be synthesized as artificial toxins for antibiotic
or anticancer applications.167 Such small-molecule fragments may confer stabilized or
unusual 3D structures to these hybrid molecules, leading to improvements in ligand
binding or inhibition, relative to the peptide. Alternatively, these hybrid molecules
may display improved ADMET properties too.168 Peptide isosteres containing a
heterocyclic moiety have also been patented as HIV inhibitors.169 Such endeavors
provide a guideline for the combinatorial biosynthesis of novel variants to optimize
future generations of bioactive molecules.
1.4. Plan of work
Keeping in view the significance of combinatorial synthesis, it was planned to
synthesize a variety of drug-like molecules such as chalcones, heterocycles and their
peptide conjugates employing both solution and solid phase synthetic methodologies.
The synthesis of chalcones and their libraries was planned followed by their screening
as cytotoxic, antibacterial and antitumor agents with a view to identify leads in
different bioassays. It was also planned to design and synthesize large chalcone
libraries through positional scanning protocol and identification of leads through a
simple and much cheep deconvolution procedure.
Moreover, synthesis of a peptidylchalcone library was also planned through solid
phase technique using phosphorane-supported polystyrenedivinyl benzene as solid
support in a multistep process. The resulting free peptidylchalcones obtained after
cleaving off the support were planned to be converted to their respective 5 and 7
membered aza heterocycles e.g. oxazoles, pyrazoles, thiazepines and diazepines
conjugated to different peptide sequences.

Chapter 1 ♦Introduction
- 33 -
Besides carrying out the synthesis of chalcones in the wet laboratory, it was also
planned to conduct some computational studies on these compounds in dry laboratory
with a view to provide a rationale for the greater bioactivity of some compounds over
the others. Molecular docking and structure-activity relationship (SAR) tools of
Molecular Operating Environment (MOE) and SYBYL software would be used to
accomplish the planned work.
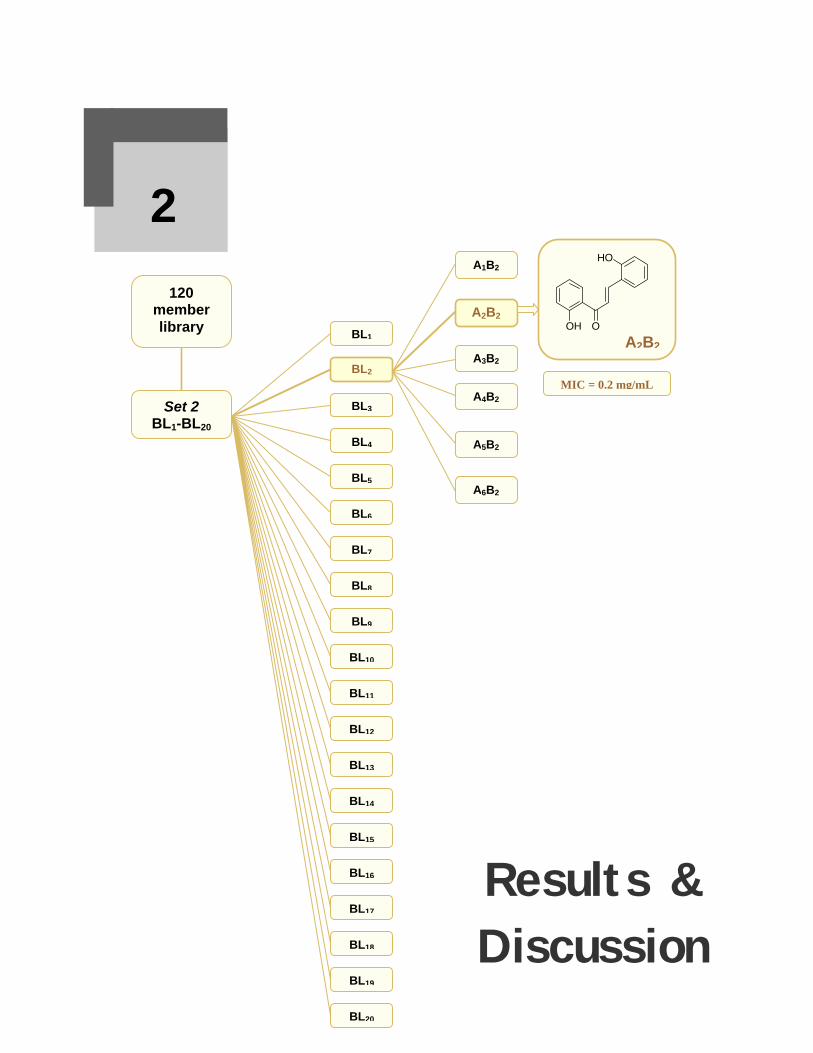
120 member library
A1B2
A2B2
A3B2
A4B2
A5B2
A6B2
Set 2 BL1-BL20
BL1
BL13
BL12
BL11
BL10
BL9
BL8
BL7
BL6
BL5
BL4
BL3
BL2
BL20
BL19
BL18
BL17
BL16
BL15
BL14
Results & Discussion
2
MIC = 0.2 mg/mL
O
HO
OH
A2B2

Results and Discussion The present work describes the design and combinatorial synthesis of small parallel
libraries of chalcones, peptidyl chalcones and peptidyl heterocycles, either in solution
or on solid phase. The chalcone libraries, both parallel and indexed, were synthesized
under Claisen-Schmidt conditions in solution phase. Peptidyl chalcones were
synthesized on solid phase using phosphorane bound resin, while the derived
heterocycles were synthesized in solution phase. The characterization of all the
synthesized compounds was based on their physicochemical and spectroscopic data.
Combinatorial chemistry is an exciting approach to chemical synthesis that enables
the creation of a large number of organic compounds by linking chemical blocks in all
possible combinations. Fig 2.1 shows a comparison of the conventional versus
combinatorial synthesis.
Conventional synthesis
One compound only
Combinatorial synthesis
A library of nine compounds
Fig 2.1. Comparison of conventional versus combinatorial synthesis.
Classical synthesis i.e. the conversion of a reactant A to a final product D usually
involves a multistep sequence, followed by purification and complete characterization
of the products before screening. Guided by the biological activity of the previous
compound, the next analogue is then designed, prepared and screened again. This
process is repeated to optimize both activity and selectivity. On the contrary,
combinatorial synthesis involves the synthesis of a large number of compounds; this
collection may be a chemical mixture, a physical mixture, or individual pure

Chapter 2 ♦Results and Discussion - 36 - compounds. The collection is then tested for biological activity and the active
compound is identified finally by deconvolution and is made in quantity as a single
compound.1
2.1. Combinatorial synthesis
Parallel solution phase synthesis is the most straightforward method for library
preparation due to its close resemblance to traditional synthesis. It involves the
reaction of a single compound A with multiple reactants (B1, B2, …Bn), which gives
rise to a compound library of n individual products AB1, AB2…ABn. The library is
then evaluated, usually without purification and with only minimal characterization of
the individual compounds, by means of a rapid-throughput screening methodology.
Individual compounds are synthesized in different vessels and are available directly
for analysis after purification. Parallel chemistry has the advantage that doing all the
reactions at a time in different vessels saves time. However, the work-up has to be
done separately for each NCE.
2.1.1. Synthesis of a parallel library of amino chalcones (1-20)
A small parallel library of twenty different substituted amino chalcones (1-20) was
synthesized in solution phase under Claisen Schmidt conditions using different
substituted benzaldehydes and 4′-aminoacetophenone (Scheme 2.1). The selection of
substituents on benzaldehyde (B ring) was made on the basis of their size,
lipophilicity and electronic properties and they were changed systematically, one at a
time at 2-, 3- and 4- positions to get a set of regioisomers. The reaction was carried
out at room temperature and the reaction time varied from 3-8 h.146
O
R
O
H
O
R NaOH/ EtOHH2N H2N
1-20
A
B
No. R No. R No. R No. R
1 H 6 3-OCH3 11 4-NO2 16 2-Br
2 2-OH 7 4-OCH3 12 2-Cl 17 3-Br
3 3-OH 8 3,4-(OCH3)2 13 3-Cl 18 4-CH3
4 4-OH 9 2-NO2 14 4-Cl 19 3-OH, 4-OCH3
5 2-OCH3 10 3-NO2 15 4-F 20 4-N(CH3)2
Scheme 2.1. Parallel synthesis of a library of amino chalcones (1-20).

Chapter 2 ♦Results and Discussion - 37 - The synthesized chalcones were either purified by recrystallization or by flash
chromatography using n-hexane and ethyl acetate as eluent. The purity was assessed
through multiple TLC and the Rf values were recorded. Melting points were found to
vary from 55 to 200 oC, while the yield varied from 60-87%. The characterization was
made on the basis of physical parameters and spectral analysis as given in Chapter 4.
In IR, amino chalcones, being the primary amines, gave two bands of medium
intensity in the range of 2847-3474 cm–1 and 2555-3385 cm–1. The C=O and C=C
stretching vibrations appeared in the range of 1600-1690 cm–1 and 1508-1600 cm–1,
respectively. A weak peak of aromatic C–H stretching was often observed at 3041-
3246 cm–1. A broad peak due to –OH group appeared at 3065-3600 cm–1. The
stretching frequencies for different substituents on ring B e.g. C-OCH3, N-CH3, C-Br,
C-Cl, C-F in different chalcones were observed in the range of 3041-3239, 2904, 736-
752, 842-869, 1092 cm–1. The two bands of unsymmetrical and symmetrical
stretching in chalcones 9-11 for C-NO2 appeared at 1404-1514 cm–1. In an inspection
of the IR stretching frequencies of different chalcones, it was observed that electron
withdrawing substituents particularly at ortho and para positions on ring B shifted the
stretching frequency corresponding to an amino group on ring A by a higher value
e.g. the nitro group at ring B was found to shift the stretching frequencies of the 4-
NH2 on ring A by about 10 cm-1 i.e. p-NO2 substituted B ring resulted in the highest
shift of 4-NH2 group; at 3481 cm-1 and 3385 cm-1; o-NO2 resulted in a lower shift;
3472 cm-1, 3382 cm-1, whereas the m-NO2 was found to shift these frequencies to
3469 cm-1 and 3380 cm-1.
The HR-MS spectra of chalcones 1-20 showed the molecular ion peak in each case
confirming the identity of these compounds. Halogen substituted chalcones (12-14
and 16-17) showed isotopic peaks corresponding to molecular ion and respective,
fragments in almost 3:1 ratio for Cl35: Cl37 (12-14) and 1:1 ratio for Br79: Br81 (16-17),
respectively. The mass fragmentation pattern of chalcone 1 is given in Scheme 2.2.

Chapter 2 ♦Results and Discussion - 38 -
-H
+
m/z = 222
O
H2N
O
H2N
+
m/z = 223
-CO+
-H
+
H2N
+
m/z = 92
-C2H2
H2N
+
m/z = 66
O+m/z = 131
CH+
m/z = 103
H2C
m/z = 91
C OH2N+
m/z = 120
-CO
-H
Scheme 2.2. Mass fragmentation pattern of amino chalcone 1.
The 1H NMR data of all compounds 1-20 were recorded and a few generalizations
could be made e.g. the aromatic protons of ring A H-2′, H-6′ and H-3′, H-5′ always
appeared as two distinct doublets with an integration of two protons. The vinylic
protons appeared as two distinct doublets, each with an integration of one proton with
a coupling constant of ~14-16 Hz in all cases. 1H NMR data of chalcone 2 is given in
Table 2.1 as a representative example.
The most characteristic chemical shifts are those of α- and β- olefinic protons (vinylic
protons). α Proton appeared upfield at 6.98 ppm, while β proton appeared at 7.78 ppm
with a coupling constant of 14.8 Hz indicating a trans orientation of the double bond
and hence an E configuration of the compound. The –NH2 protons appeared at 5.98
ppm. The two doublets at 8.01 ppm and 7.31 ppm (J = 8.3 Hz) were assigned to H-2′,
6′ and H-3′, 5′, respectively.

Chapter 2 ♦Results and Discussion - 39 - Table 2.1. 1H NMR data of 1-(4′-aminophenyl)-3-(2-hydroxyphenyl)-2-propen-1-one (2).
O
H2N 1
23
4
5
61'
2'3'
4'
5'6'
HO
α
β
2
δ ppm Multiplicity Integration Coupling constant J (Hz)
Assignment
5.98 bs 2H - NH2
6.98 d 1H 14.8 H−C(α)
7.75 d 1H 14.8 H−C(β)
8.01 d 2H 8.3 H−C(2′,6′)
7.31 d 2H 8.3 H−C(3′,5′)
7.41 d 1H 8.0 H−C(3)
7.04 d 1H 8.1 H−C(6)
6.98 t 1H 8.0 H−C(5)
6.95 t 1H 8.0 H−C(4)
9.88 s 1H - OH
2.1.2. Biological screening of a chalcone library
Amino chalcones have been reported in the literature with potential antitumor, anti-
proliferative, antiparasitic and bactericidal activities.138-139 The synthesized amino
chalcones were tested for their inhibitory potential as phosphatase inhibitors,
antibacterial and cytotoxic agents, as discussed in the following sections.
2.1.2.1. Phosphatase inhibition
A phosphatase is an enzyme that removes a phosphate group from its substrate by
hydrolyzing phosphoric acid monoesters into a phosphate ion and a molecule with a
free hydroxyl group. They act opposite to the kinases/phosphorylases, which add a
phosphate group to proteins by using higher energy molecules such as ATP. The
addition of a phosphate group may activate or deactivate an enzyme (e.g. Kinase
signaling pathways) or enable a protein-protein interaction to occur; therefore,
phosphatases are integral to many signal transduction pathways e.g. in diabetes. Their
role in signal transduction is because they regulate the proteins to which they are

Chapter 2 ♦Results and Discussion - 40 - attached. In order to reverse the regulatory effect, the phosphate group is removed,
which occurs on its own by hydrolysis, or is mediated by protein phosphatases.
Insulin is the major hormone responsible for lowering blood sugar level. The lack of
insulin as a result of the degeneration of special insulin producing cells (called beta
cells) in pancreas,170 or the lack of response of target cells to normal circulating
insulin levels, are the key pathological features of type I and type II diabetes,171
respectively. Insulin binding to its cell surface transmembrane receptor stimulates
autophosphorylation and activation of intrinsic protein tyrosinase kinase (PTK)
activity and subsequent phosphorylation of insulin receptor substrates.
The control of insulin receptor signaling involves the coordinated action of both
positive and negative regulatory proteins. Among the negative regulatory proteins,
protein tyrosinase phosphatases (PTPs) play a prominent role and their inhibition
mimics several actions of insulin including the stimulation of glucose uptake.172-174
The development of drugs capable of inhibiting PTPs may allow the enhanced or
prolonged activation of the insulin receptor and have therapeutic use in the treatment
of type II diabetes.175-177
With the aim of finding some potential antidiabetic compounds, the synthesized
amino chalcones 1-20 were tested against a large variety of fifteen protein
phosphatases, e.g. Mycobacterium tuberculosis inosit phosphatase (Mtb Inosit
Pptase), Mycobacterium tuberculosis phosphoglyceromutase (Mtb PGM),
Mycobacterium tuberculosis aroA (Mtb aroA), Mycobacterium tuberculosis aroK
(Mtb aroK), Mycobacterium tuberculosis protein tyrosinase phosphatase A (Mtb
ptpA), human protein tyrosinase phosphatase 1B (hum ptp1B), human protein
tyrosinase phosphatase N3 (hum ptpN3), human protein tyrosinase phosphatase N5
(hum ptpN5), human protein tyrosinase phosphatase N7 (hum ptpN7), human protein
tyrosinase phosphatase RJ (hum ptpRJ), human protein tyrosinase phosphatase RK6
(hum ptpRK6), human protein tyrosinase phosphatase ROG7 (hum ptpROG7), human
protein tyrosinase phosphatase RR (hum ptpRR), human protein tyrosinase
phosphatase RS (hum ptpRS) and Mycobacterium tuberculosis protein tyrosinase
phosphatase SHP2 (ptp SHP2). All compounds were tested at a concentration of 0.1
mM. They were found weakly active against Mtb Inosit Pptase, but were found to be
very potent when tested against (Mtb PGM). The data of the activity of the tested

Chapter 2 ♦Results and Discussion - 41 - compounds 1-20 represented as % inhibition against Mtb inosit Pptase is given in
Table 2.2. Compounds 1, 3-9 and 13 inhibited the target enzyme only to some extent.
Table 2.2. Mtb inosit Pptase inhibitory activity of chalcones 1-20.
No. % Inh. No. % Inh. No. % Inh. No. % Inh.
1 21 6 4 11 - 16 -
2 - 7 26 12 - 17 -
3 32 8 3 13 7 18 -
4 21 9 14 14 - 19 -
5 8 10 - 15 - 20 -
PGM is the key enzyme for isomerization of 3-phosphoglycerate to 2-
phosphoglycerate in the glycolysis pathway during respiration and PGM inhibitors are
expected to maintain blood sugar levels for routine tasks in ischemic patients.178 The
phosphoglycerate kinase step follows after isomerization step involving PGM and this
results in the production of ATP, synthesis of pyruvic acid for aerobic production of
energy in Krebs cycle and chimiosmosis. PGM is also involved in gluconeogenic
transformation of phosphoenolpyruvate into glycogen and results in weakness after
strenuous exercise. Overacting PGM may result in lower blood sugar levels in
diabetic patients. PGM is found in all species but uses different variants. Chalcones 1-
20 when tested against Mtb PGM, were found to be quite potent. The data is
represented as % inhibition in Table 2.3. Chalcones 8 and 19 showed 60 % inhibition,
while chalcone 3, 13 and 15 did not show any activity. Chalcones 4 and 11 with
substituents 4-OH and 4-NO2 groups were found to be the lead structures of the
library with 96 and 99 % inhibition, respectively.
Table 2.3. Mtb PGM inhibitory activity of chalcones 1-20.
No. % Inh. No. % Inh. No. % Inh. No. % Inh.
1 44 6 43 11 99 16 24
2 17 7 44 12 47 17 17
3 - 8 60 13 - 18 48
4 96 9 45 14 41 19 60
5 15 10 34 15 - 20 41
It may be concluded that the suitable competitive reversible inhibitors of PGM e.g.
the lead structures 4 and 11 are expected to be potential candidates for developing into
antidiabetic drugs. Molecular docking studies on these chalcones in the active site of

Chapter 2 ♦Results and Discussion - 42 - PGM providing further insight into the mechanism of their inhibition have been
carried out as discussed in Chapter 3.
2.1.2.2. Antibacterial activity
Chalcones are well known to have antibacterial properties.140 The members of the
library (1-20) were, therefore, tested for their bactericidal activity against six bacterial
strains, namely Escherichia coli, Bacillus subtilis, Enterobacter aerogenes,
Staphylococcus aureus, Pseudomonas pickitti and Salmonella setubal. Each
compound and the controls were tested at a concentration of 0.1 mg/mL.
Roxithromycin and Cefixim were used as positive control, whereas DMSO was used
as negative control.
Table 2.4. Bactericidal activity of amino chalcones (1-20).
Zone of Inhibition (mm) No. S.aureus B.subtilis E.coli E.aerogens
1 0 0 0 11
2 0 10 0 11
3 0 0 0 11
4 0 0 0 11
5 0 0 0 11
6 0 0 0 11
7 0 0 0 11
8 0 0 0 0
9 0 0 9 0
10 0 0 9.5 0
11 0 0 0 0
12 0 0 0 0
13 0 0 0 11
14 0 10 0 0
15 0 0 9 0
16 0 0 9 0
17 0 0 9 0
18 0 0 9 0
19 11 12 13 0
20 0 0 0 0
Ra 26 26 19 21
Cb 24 26 32 21
a R= Roxithromycin (= Rulid®); b C= Cefixim®.

Chapter 2 ♦Results and Discussion - 43 - It is evident from the data in Table 2.4 that all the tested chalcones did not show any
significant activity against any of the tested bacterial strains. Compounds 1-7 and 13
showed weak activity against E. aerogenes, while 2 and 14 showed weak activity
against B.subtilis. Compounds 9, 10, 15, 16, 17 and 18 showed very weak activities
against E.coli, while chalcone 19 showed moderate activity against E.coli. The same
compound was found weakly active against B.subtilis and S.aureus.
2.1.2.3. Brine shrimp lethality studies
Chalcones, in general, are known to have cytotoxic potential179-180 and amino
chalcones have been reported as cytotoxic agents.138-140 Studies on the cytotoxicity of
chalcones 1-20 was carried out through brine shrimp lethality (BSL) assay, which is a
rapid and economic benchtop assay employing the use of easily available brine
shrimps; Artimisia salina as test animals.
Artimisia salina
The test is a pre-screen of antitumor properties.181-182 BSL testing was performed with
different concentrations (150, 100 & 50 µg/mL) of the test samples prepared in
DMSO and LD50 (lethal dose to kill 50% of test animals) values were recorded in µM
quantities as shown in Table 2.5.
Table 2.5. BSL studies on amino chalcones (1-20).
No. LD50 No. LD50 No. LD50 No. LD50
1 6.68 6 3.79 11 130.56 16 8.34
2 46.50 7 0.24 12 16.22 17 83.71
3 60.54 8 - 13 52.30 18 150.11
4 7.10 9 160.63 14 14.61 19 -
5 2.72 10 557.02 15 614.42 20 2.59
Chalcone 7 with methoxy substituent on ring B was found to be the LEAD structure
with an LD50 value of 0.24 µM, while chalcones 1, 4-6, 16 and 20 showed strong
cytotoxic behavior as reflected from their LD50 values ranging from 2.59-8.34 µM.
Chalcones 2, 3, 12-14 and 17 showed good cytotoxic activity with LD50 < 100.

Chapter 2 ♦Results and Discussion - 44 -
O
H2N
OCH3
7 (LD50 = 0.24 µM)
2.2. Indexed combinatorial synthesis
The synthesis of positional scanning library also called indexed libraries represents
one of the most useful protocols for mixture synthesis. It is much less time intensive
as compared to parallel synthesis of individual compounds and produces depository
libraries for use in multiple screens with immediate deconvolution.183 Deconvolution
is a multistep process where smaller libraries are successively prepared and tested to
identify the individual active members of a combinatorial library. Positional scanning
libraries provide lead identities in a single round of testing and the method is ideally
suited for cellular assays that involve membrane-bound targets.184 The synthesis of
positional scanning libraries represents one of the most useful protocols for mixture
synthesis and can easily be conducted in solution phase but it is not easily adaptable
to solid phase synthesis.185
Indexed combinatorial synthesis is a mixture synthesis, which involves the reaction of
two components (building blocks) at a time such that one component is kept constant,
while the other is used as an equimolar mixture. Alternatively, the synthesis is carried
out through keeping second building block constant and varying the first one. These
efforts result in the synthesis of two daughter libraries (two sets of indexed libraries,
also called sub-libraries or pools). The two libraries (product mixtures) are tested and
their activities are used as “indices” to the rows or columns of a 2D matrix reflecting
the activities of individual compounds. Libraries can be preceded with the third
building block for getting 3D indexed libraries as well and the process may
continue.65
Two indexed combinatorial libraries (120 and 175 member) of chalcones were
synthesized in solution phase and tested for their antibacterial, cytotoxic and
antitumor properties followed by deconvolution, which led to the identification of
potent antibacterial and antitumor chalcones as leads of two libraries. The reliability
and success of the process was assessed by confirming the presence of all component
chalcones in the indexed library (product mixture) through GC-MS analysis.

Chapter 2 ♦Results and Discussion - 45 - 2.2.1. A 120-member chalcone library147
A 120 member chalcone library was designed and synthesized with a view of finding
potential antibacterial chalcones through deconvolution.
2.2.1.1. Synthesis. A 120 member indexed chalcone library was designed by using
six acetophenones (A1-A6) and 20 benzaldehydes (B1-B20) under Claisen Schmidt
conditions (Scheme 2.3).
R'
O
RR'
O
R H
O
NaOH/ EtOH
Acetophenones Benzaldehydes Benzaldehydes
R′ R R
A1 C6H5 B1 Ph B11 4-NO2C6H4
A 2 2′-OHC6H4 B2 2-OHC6H4 B12 2-ClC6H4
A 3 4′-NH2C6H4 B3 3-OHC6H4 B13 3-ClC6H4
A 4 2′,4′,5′-(OCH3)3C6H2 B4 4-OHC6H4 B14 4-ClC6H4
A 5 3′,4′,5′-(OCH3)3C6H2 B5 2-OCH3C6H4 B15 4-FC6H4
A 6 2′,4′-(Br)2C6H3 B6 3-OCH3C6H4 B16 2-BrC6H4
B7 4-OCH3C6H4 B17 3-BrC6H4
B8 3,4-(OCH3)2C6H3 B18 4-CH3C6H4
B9 2-NO2C6H4 B19 3-OH,4-OCH3C6H3
B10 3-NO2C6H4 B20 4-N(CH3)2C6H4
Scheme 2.3. Synthesis of a 120 member chalcone library.
The library was synthesized in the form of two sets of daughter libraries (Set 1 and Set
2). Set 1 contained six daughter libraries, each containing 20 compounds, whereas Set
2 contained twenty daughter libraries, each containing 6 compounds (Fig. 2.2).
Parent indexed library
R'
O
R n = 6 n = 20 n = 120
∑ R´COCH3 + ∑ RCHO ∑
Indexed daughter libraries (Set 1) (6 libraries, each containing 20 compounds) n = 20 n = 20
R´COCH3 + ∑ RCHO ∑
Indexed daughter libraries (Set 2) (20 libraries, each containing 6 compounds) n = 6 n = 6
∑ R´COCH3 + RCHO ∑
R'
O
R
R'
O
R
Fig. 2.2. Synthesis of indexed libraries of Sets 1 and 2.

Chapter 2 ♦Results and Discussion - 46 - The presence of all the library members in each library was confirmed through GC-
MS analysis. The identification of some small molecular libraries through GC-MS has
already been reported.186-187 For indexed library, the daughter libraries were run
through a DB-5ms column containing a stationary phase of 5% phenyl and 95%
dimethylarylenesiloxane with He gas. A 5973 inert MS selective detector was used.
The GC-MS spectrum of AL1 library is given in Fig 2.3, whereas the mass fragments
contained in each peak are given in Fig. 2.4 (a-c) and Fig. 2.5 (d-l). The GC-MS of
the daughter library AL1 is discussed in detail. The library was analyzed using
different GC-MS programs and the best results were found when the temperature was
kept from 120 to 300 °C at a rate of 10°C/min, whereas inlet was kept at 250 °C. The
20 members of the library were found separated as 12 peaks, but the relative peak
areas indicated the presence of all 20 compounds. All the compounds were eluted in
17 minutes. The first peak separated at 11.2 min with M+. peak of 226 indicating the
presence of fluorochalcone. The second peak separated at 11.4 min, which contained
four chalcones as indicated through the relative peak area. The presence of these
chalcones was confirmed through the M+. and fragment ion peaks. The M+. peaks at
208 indicated the presence of unsubstituted chalcone. A hydroxychalcone was
detected through a weak [M + H]+ peak at 226. A strong peak at 207 indicated the
presence of M+. – NO2 from 2- nitrochalcone or 4-nitrochalcone. The third peak
appeared at 12.75 min with a weak M+. peak at 238 and [M – CH3]+ peak at 223
indicating a methoxy chalcone. The peaks at 12.93 min, 13.10 min and 13.28 min
corresponded to chlorochalcones, each giving a molecular ion peak at 242 and M+.+2
peak at 244 with relative abundance of 3:1. The 3-hydroxy-4-methoxychalcone
appeared at 13.71 min giving [M – O]+ peak at 238, [M – OH]+ at 237 and [M –
OCH3]+ at 223. A bromochalcone was eluted at 14.09 min giving M+. and M+.+2
peaks at 286 and 288 in 1:1 ratio. At 14.22 min, a peak eluted with a peak area
indicating the presence of four chalcones. A careful analysis indicated the presence of
two hydroxy- and two methoxy-chalcones. The molecular ion peaks were observed as
M+. at 238 and [M + H]+ at 239 for methoxy chalcones, whereas as M+. at 224 and [M
– H]+ at 223 for hydroxy chalcone. The 238 – 28 and 239 – 28 were the peaks found
at 210 and 211 after CO. removal from methoxy chalcones. Similarly, 224 – CO. and
223 – CO. were observed at 196 and 195 for hydroxy chalcones. At 14.28 min,

Chapter 2 ♦Results and Discussion - 47 -
Fig. 2.3. The GC-MS spectrum of the library AL1 containing 20 compounds.
Fig. 2.4. The mass spectra for each GC peaks (a-c).

Chapter 2 ♦Results and Discussion - 48 -
Fig. 2.5. The mass spectra for each GC peaks (d-l).

Chapter 2 ♦Results and Discussion - 49 - a bromochalcone appeared with M+. and M+. + 2 peaks at 286 and 288. The spectrum
showed very weak peaks at 256 and 258 after CO. removal from M+. and M+. + 2. The
methyl chalcone also appeared in the same peak as indicated by [M + H]+ peak at 223.
Furthermore, a peak at 207 indicated the [M – CH3]+ for methyl chalcone or the M+. –
Br. for bromochalcone. Dimethoxychalcone and a nitrochalcone at 15.79 min were
identified through the M+. peaks at 268 and 253, respectively. The 253 – O. was
observed at 237 whereas 253 – NO. appeared at 223 and 253 – NO2 at 207 confirming
the presence of a nitro chalcone. The last peak appeared at 16.7 min containing the
N,N-dimethylaminochalcone with a molecular ion peak at 251. The M+.– CO.
appeared at 223.
The synthesized indexed library can be represented as a 2D matrix, wherein the x-axis
has six structural variants (acetophenones), and the y-axis has 20 structural variants
(benzaldehydes) leading to a 6 × 20 grid (Table 2.6).
Table. 2.6. Conceptual matrix for an indexed 120 member chalcone library. Acetophenones
Benzaldehydes A1 A2 A3 A4 A5 A6
B1 A1 B1 A2 B1 A3 B1 A4 B1 A5 B1 A6 B1
B2 A1 B2 A2 B2 A3 B2 A4 B2 A5 B2 A6 B2
B3 A1 B3 A2 B3 A3 B3 A4 B3 A5 B3 A6 B3
B4 A1 B4 A2 B4 A3 B4 A4 B4 A5 B4 A6 B4
B5 A1 B5 A2 B5 A3 B5 A4 B5 A5 B5 A6 B5
B6 A1 B6 A2 B6 A3 B6 A4 B6 A5 B6 A6 B6
B7 A1 B7 A2 B7 A3 B7 A4 B7 A5 B7 A6 B7
B8 A1 B8 A2 B8 A3 B8 A4 B8 A5 B8 A6 B8
B9 A1 B9 A2 B9 A3 B9 A4 B9 A5 B9 A6 B9
B10 A1 B10 A2 B10 A3 B10 A4 B10 A5 B10 A6B10
B11 A1 B11 A2 B11 A3 B11 A4 B11 A5 B11 A6B11
B12 A1 B12 A2 B12 A3 B12 A4 B12 A5 B12 A6B12
B13 A1 B13 A2 B13 A3 B13 A4 B13 A5 B13 A6B13
B14 A1 B14 A2 B14 A3 B14 A4 B14 A5 B14 A6B14
B15 A1 B15 A2 B15 A3 B15 A4 B15 A5 B15 A6B15
B16 A1 B16 A2 B16 A3 B16 A4 B16 A5 B16 A6B16
B17 A1 B17 A2 B17 A3B17 A4 B17 A5 B17 A6B17
B18 A1 B18 A2 B18 A3 B18 A4 B18 A5 B18 A6B18
B19 A1 B19 A2 B19 A3 B19 A4 B19 A5 B19 A6B19
B20 A1 B20 A2 B20 A3 B20 A4 B20 A5 B20 A6B20

Chapter 2 ♦Results and Discussion - 50 - The number of the building blocks was chosen such that the library was large enough
to demonstrate the principle but small enough to verify the composition of library by
analytical methods. Besides, the size of the library was kept such that it was soluble
in an assay mixture for its effective screening. The chemistry of the reaction used in
the preparation of library was investigated on a single compound. The choice of the
reagents reflects the chemical diversity in terms of hydrophobic and lipophilic
substituents. All library components share a common α,β-unsaturated carbonyl
functionality.
Synthesis of such a library in indexed fashion resulted in 26 daughter libraries with
six members in Set 1, while 20 members in Set 2. Both sets were subjected to
screening experiments followed by deconvolution and subsequent LEAD
identification. The assay value of each cell is contained in the combination AxBy (x =
1–6, y = 1–20) (Table 2.6). Obviously, the examination of all the pure compounds
would have required 120 experiments. Since only one cell out of 120 may possess the
maximum response function, the next step involves its identification without looking
at all 120 cells. The best way of doing this is to choose any B for testing with all A-
type compounds; conversely, any A for testing with all B-type members.
120 Member Parent
Library
Indexed Libraries
Screening
Set 2. Pools 1-20
1. B1 + A1-62. B2 + A1-6
: 20. B20 + A1-6
LEAD
Set 1: Pools 1-6
1. A1 + B1-202. A2 + B1-20
: 6. A6 + B1-20
Fig. 2.6. A general schematic diagram for library synthesis in search of the LEAD in a bioassay.

Chapter 2 ♦Results and Discussion - 51 - By screening six columns and 20 rows, which are indices to the cells at their
intersections, as mixtures, only 26 reactions (instead of 120) need to be carried out to
find the maximum response. Therefore, a combinatorial synthesis of 26 sub-libraries
was carried out. This method has the advantages over the parallel approach in that it
is fast and relatively inexpensive. However, the number of compounds prepared is
much greater than the number of chemical steps required
2.2.1.2. Antibacterial studies. All 26 indexed libraries were subjected to antibacterial
studies against six bacterial strains; three Gram positive (B. subtillis ATTCC 6633,
M. leuteus and S. aureus ATCC 6538) and three Gram negative (E. coli AATCC
1522, E. aerogenes AATCC 13048 and S. setubal ATCC 19196) using the Agar Well
Diffusion method.188 Each compound was tested twice, once as a component of sub-
libraries of Set 1 and then as a member of sub-libraries of Set 2. Thus, a total of 26
assays were required to reveal the antibacterial activity of 120 compounds, which
corresponded to a factor of 4.6 in terms of time improvement in the synthesis and data
collection efficiency.
The antibacterial activities of the sub-libraries AL1-AL6 and BL1-BL20 are shown in
Tables 2.7 and 2.8, respectively. All libraries were tested at a concentration of 0.1
mg/mL. The libraries were found to be active against S. aureus and B. subtilis, very
weekly active against E. coli, while inactive towards the other three bacterial strains
i.e. M. leuteus, E. aerogenes and S. setubal. Roxithromycin (Rulid®) and Cefixime®
were used as standard positive controls in these studies.
Table 2.7. Antibacterial activities of Set 1 sub-libraries (pools AL1-AL6).
Zone of inhibition(mm)a Zone of inhibition(mm) a
S. aureus B. subtilis E. coli S. aureus B. subtilis E. coli
AL1 15.1±0.15 16.0±0.55 11.5±0.50 AL5 10.7±0.20 10.7±0.16 0 AL2 10.9±0.15 12.0±0.26 0 AL6 17.8±0.20 15.1±0.05 0 AL3 8.8±0.12 10.8±0.11 0 Rb 26.5±1.30 32.1±0.31 11.6±0.10 AL4 0 0 0 Cc 21.3±0.70 12.4±0.18 31.9±0.17
a mean value of three replicates, b R = Roxithromycin, c C =Cefixime®.
After having synthesized the sub-libraries and carrying out their antibacterial
screening, the next step is the synthesis of the library corresponding to the most active
column and the most active row, and to identify the most active library and the lead
structures therein by deconvolution.
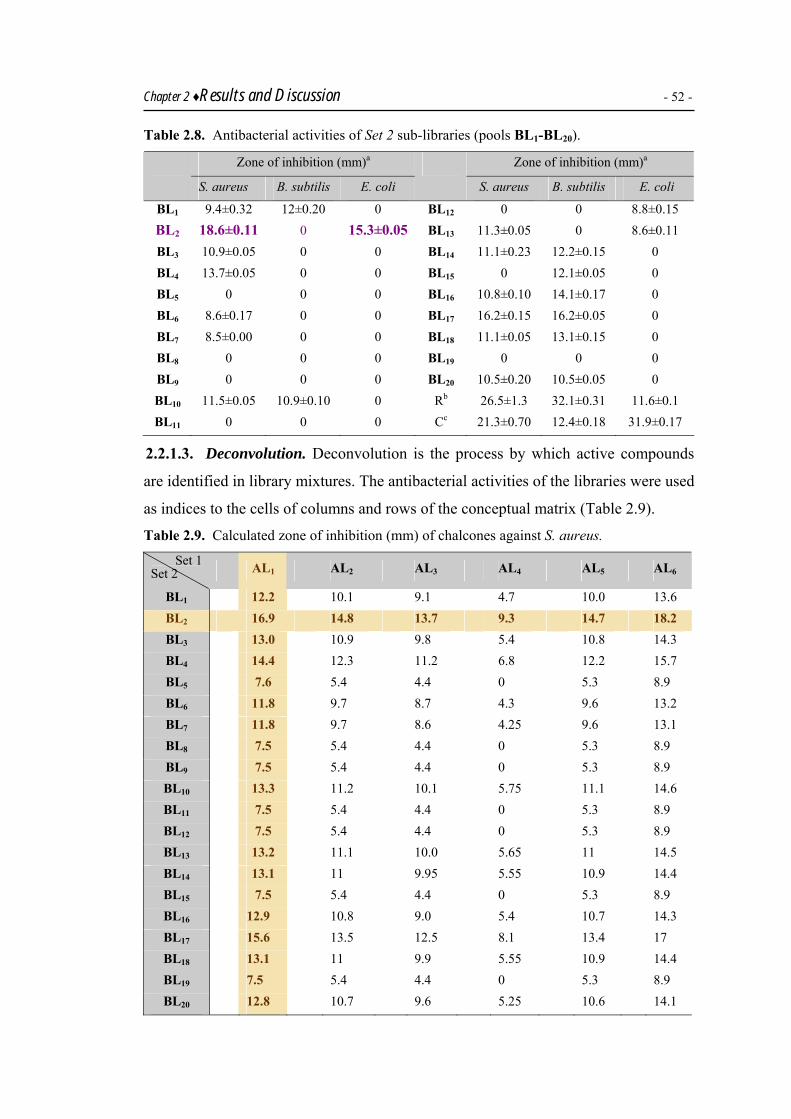
Chapter 2 ♦Results and Discussion - 52 - Table 2.8. Antibacterial activities of Set 2 sub-libraries (pools BL1-BL20).
Zone of inhibition (mm)a Zone of inhibition (mm)a
S. aureus B. subtilis E. coli
S. aureus B. subtilis E. coli
BL1 9.4±0.32 12±0.20 0 BL12 0 0 8.8±0.15 BL2 18.6±0.11 0 15.3±0.05 BL13 11.3±0.05 0 8.6±0.11 BL3 10.9±0.05 0 0 BL14 11.1±0.23 12.2±0.15 0 BL4 13.7±0.05 0 0 BL15 0 12.1±0.05 0 BL5 0 0 0 BL16 10.8±0.10 14.1±0.17 0 BL6 8.6±0.17 0 0 BL17 16.2±0.15 16.2±0.05 0 BL7 8.5±0.00 0 0 BL18 11.1±0.05 13.1±0.15 0 BL8 0 0 0 BL19 0 0 0 BL9 0 0 0 BL20 10.5±0.20 10.5±0.05 0 BL10 11.5±0.05 10.9±0.10 0 Rb 26.5±1.3 32.1±0.31 11.6±0.1 BL11 0 0 0 Cc 21.3±0.70 12.4±0.18 31.9±0.17
2.2.1.3. Deconvolution. Deconvolution is the process by which active compounds
are identified in library mixtures. The antibacterial activities of the libraries were used
as indices to the cells of columns and rows of the conceptual matrix (Table 2.9).
Table 2.9. Calculated zone of inhibition (mm) of chalcones against S. aureus.
Set 1 Set 2 AL1 AL2 AL3 AL4 AL5 AL6
BL1 12.2 10.1 9.1 4.7 10.0 13.6 BL2 16.9 14.8 13.7 9.3 14.7 18.2 BL3 13.0 10.9 9.8 5.4 10.8 14.3 BL4 14.4 12.3 11.2 6.8 12.2 15.7 BL5 7.6 5.4 4.4 0 5.3 8.9 BL6 11.8 9.7 8.7 4.3 9.6 13.2 BL7 11.8 9.7 8.6 4.25 9.6 13.1 BL8 7.5 5.4 4.4 0 5.3 8.9 BL9 7.5 5.4 4.4 0 5.3 8.9 BL10 13.3 11.2 10.1 5.75 11.1 14.6 BL11 7.5 5.4 4.4 0 5.3 8.9 BL12 7.5 5.4 4.4 0 5.3 8.9 BL13 13.2 11.1 10.0 5.65 11 14.5 BL14 13.1 11 9.95 5.55 10.9 14.4 BL15 7.5 5.4 4.4 0 5.3 8.9 BL16 12.9 10.8 9.0 5.4 10.7 14.3 BL17 15.6 13.5 12.5 8.1 13.4 17 BL18 13.1 11 9.9 5.55 10.9 14.4 BL19 7.5 5.4 4.4 0 5.3 8.9 BL20 12.8 10.7 9.6 5.25 10.6 14.1

Chapter 2 ♦Results and Discussion - 53 - The data was expanded on 120 cells of chalcones on the matrix against S. aureus,
since the libraries were found to be the most active against this strain only. This data
expansion was carried out for each cell by taking the average of the activities of the
respective column and row cell, and the data were used as indices to the particular
cell. This resulted in calculated antibacterial activity against S. aureus for all 120
chalcones of the matrix. For the identification of the lead compounds, the individual
members of the most active column and row needed to be identified. A careful
examination of the data revealed that the members of the column AL1 and the row
BL2 showed maximum inhibition. Hence, parallel synthesis of the individual
members of these two sub-libraries was accomplished.
Parallel synthesis of active libraries AL1 and BL2. Parallel solution phase synthesis
of a total of twenty five chalcones belonging to the active libraries AL1 and BL2 was
carried out under standard Claisen-Schmidt conditions using a 24 parallel reactions
workstation, GreenHouse Synthesizer (Scheme 2.4).
H
O
R
OO
NaOH/ EtOHR' R'
R
GreenHouse
Synthesizer
R R R
21 H 28 3,4-(OCH3)2 35 4-F
22 2-OH 29 2-NO2 36 2-Br
23 3-OH 30 3-NO2 37 3-Br
24 4-OH 31 4-NO2 38 4-CH3
25 2-OCH3 32 2-Cl 39 3-OH,4-OCH3
26 3-OCH3 33 3-Cl 40 4-N(CH3) 2
27 4-OCH3 34 4-Cl
O
R
AL1 (A1B1-A1B20)
(21 - 40)
R′ R′ R′
2* 4′-NH2 41 2′-OH 43 3′,4′,5′ -(OCH3)2
22** H 42 2′,3′,4′- (OCH3)2 44 2′,4′-(Br)2O
R'
HO
BL2 (A1B2-A6B2) (2, 22, 41 – 44)
*already mentioned in scheme 2.1.
** already mentioned above.
Scheme 2.4. Synthesis of individual chalcones of active libraries AL1 and BL2.

Chapter 2 ♦Results and Discussion - 54 - All the compounds were recrystallized from EtOH and identified through IR, GCMS
and 1H NMR data. The purity of the compounds was checked through multiple TLC
(solvent system, n-Hex: EtOAc; 9:1, 3:1) and the Rf values were recorded. The yield
of chalcones varied from 61-85 % and the data is given in the Experimental Section.
FT-IR spectral data of chalcones 21-44 is given in the Experimental Section. The
C=O and C=C stretching vibrations appeared in the range of 1631-1690 cm–1 and
1508-1606 cm–1, respectively. A weak peak of Ar-H was observed at 3041-3246 cm–
1. The stretching frequencies for different substituents on the B ring, derived from
benzaldehydes i.e. C-OH, C-OCH3, N-CH3, C-Br, C-Cl, C-F and C-NO2 were also
observed. 1H NMR data of chalcones 21-44 has been reported in Experimental Part in detail.
Generally, protons on ring A appeared as a downfield doublet for H-2′, 6′, then a
triplet for H-4′ followed by a triplet for H-3′, 5′ with a coupling constant in the range
of 7-8 Hz. The α-proton at the vinylic position appeared in the range of 7.39-7.74
ppm, whereas β-proton appeared at 7.73-8.17 ppm, each with a coupling constant of
14-16 Hz. 1H-F coupling was observed in compound 35 (Fig. 2.7). The 1H-F coupling
for ortho position was found in the range of 6-11 Hz, whereas for meta position, it
was 3-9 Hz. In 35, protons 3 and 5 appeared as a doublet of doublets (dd) at 7.98 ppm
(J = 9.0, 7.0 Hz), whereas protons 2 and 6 appeared as an apparent triplet (app t) at
7.30 ppm (J = 8.5 Hz) due to H-F coupling.
Fig. 2.7. 1H NMR of 1-(phenyl)-3-(4′-fluorophenyl)-2-propen-1-one (35).

Chapter 2 ♦Results and Discussion - 55 - Screening of libraries AL1 and BL2. The active libraries AL1 and BL2 were tested
against six bacterial strains; however, they were found active only toward two strains
i.e. S. aureus and B. subtilis.
Table 2.10. Antibacterial activities of individual chalcones of library AL1.
Zone of inhibition (mm)a Zone of inhibition (mm)a
Compound S. aureus B. subtilis Compound S. aureus B. subtilis
A1B1 10.5±0.10 9.2±0.10 A1B12 0 9.5±0.10
A1B2 10.9±0.01 9.7±0.20 A1B13 11.1±0.02 9.2±0.05
A1B3 16.6±0.05 12.5±0.10 A1B14 0 0
A1B4 10.1±0.20 11.7±0.02 A1B15 0 0
A1B5 0 0 A1B16 0 0
A1B6 0 10.7±0.03 A1B17 10.23±0.06 9.5±0.10
A1B7 0 10.4±0.07 A1B18 0 0
A1B8 0 0 A1B19 0 0
A1B9 15.1±0.10 14.9±0.10 A1B20 0 0
A1B10 0 9.5±0.10 Rb 26.5±1.3 32.1±0.31
A1B11 0 0 Cc 21.3±0.70 12.4±0.18
a mean value of three replicates. b R = Roxithromycin (positive control). c C =Cefixime® (positive control).
Table 2.11. Antibacterial activities of individual chalcones of library BL2.
Zone of inhibition (mm)a Zone of inhibition (mm)a
Compound S. aureus B. subtilis Compound S. aureus B. subtilis
A1B2 10.9±0.01 12.5±0.20 A5B2 13.2±0.05 13.4±0.10
A2B2 18.2±0.10 17.6±0.05 A6B2 9.4±0.10 0
A3B2 9.6±0.10 11.5±0.01 Rb 26.5±1.3 32.1±0.39
A4B2 13.1±0.10 13±0.10 Cc 21.3±0.70 12.4±0.18
a mean value of three replicates. b R = Roxithromycin (positive control). c C =Cefixime® (positive control).
The results of the antibacterial studies of chalcones of sub-libraries AL1 and BL2 are
given in Tables 2.10 and 2.11, respectively. The minimum inhibitory concentration
(MIC) values of the members of AL1 and BL2 are given in Table 2.12. The
concentration of each library member and of the control was 0.1 mg/mL in DMSO. It
is evident from the data in Table 2.10 and 2.11 that chalcones A1B3 and A2B2 were
the most active members of the libraries AL1 and BL2. Based on the MIC values,
chalcone A1B2 and A2B2 (MIC = 0.2 mg/mL) may be regarded as the LEAD of the
designed 120-member library.

Chapter 2 ♦Results and Discussion - 56 - Table 2.12. Minimum inhibitory concentration (MICs)a of active chalcones (mg/mL).
Compound S.aureus B.subtilis Compound S.aureus B.subtilis
A1B1 0.5 0.4 A1B9 0.4 0.3
A1B2 0.2 - A1B17 0.5 -
A1B3 0.4 - A2B2 0.2 0.2 A1B4 - 0.5 A4B2 0.3 0.4
A1B6 - 0.6 A5B2 0.4 0.6
A1B7 - 0.6
a MIC = minimum concentration at which no colony was observed after incubation.
O
HO
O
HO
OH
A1B2 (22) A2B2 (41)
LEADS
It is evident that starting from designing of the combinatorial library to a sequential
logical identification of the lead through positional scanning protocol led to a
reduction in the number of steps from 120 to 26, thereby, reducing the cost in terms
of time and resources in the identification of a lead. Hence a positional scanning
protocol can be considered as a cost effective technique for rapid lead discovery. This
identification of the lead from the designed library through deconvolution has been
shown schematically in Figs. 2.8 and 2.9.
2.2.1.4. Structure activity relationship. Based on the antibacterial activities, it is
also possible to deduce some trends in structure activity relationship (SAR). The
following order of decreasing activity was noticed:
A2B2 > A4B2 > A5B2 > A1B2 > A3B2 > A6B2
O
HO
OH O
HO
OO
O
O
HO
O
OO
O
HO
O
HO
O
HO
Br
H2N Br
The activity of the chalcones was found to be dependent on the substitution pattern on
ring A. Chalcones with electron donors at ortho position of A ring were found as the
most active as indicated from 2-OH and 2-OCH3 substituted chalcones A2B2 and

Chapter 2 ♦Results and Discussion - 57 - A5B2. A steric bulk at ortho position of ring A appeared to be detrimental for activity
as indicated from the least potent chalcone A6B2.
A1B18
Set 1 AL1-AL6
AL1
AL2
AL3
AL4
AL5
120 member library
AL6
A1B14
A1B15
A1B16
A1B17
A1B19
A1B20
A1B2
A1B3
A1B4
A1B5
A1B6
A1B7
A1B8
A1B9
A1B10
A1B11
A1B12
A1B13
A1B1
O
HO
A1B2
MIC = 0.2 mg/mL
Fig. 2.8. Identification of lead through deconvolution of a 120 member library (Set 1) against
S. aureus.

Chapter 2 ♦Results and Discussion - 58 -
120 member library
A1B2
A2B2
A3B2
A4B2
A5B2
A6B2
BL1
BL13
BL12
BL11
BL10
BL9
BL8
BL7
BL6
BL5
BL4
BL3
BL2
BL20
BL19
BL18
BL17
BL16
BL15
BL14
MIC = 0.2 mg/mL
O
HO
OH
A2B2
Set 2 BL1-BL20
Fig. 2.9. Identification of lead through deconvolution of a 120 member library (Set 2) against
S. aureus.

Chapter 2 ♦Results and Discussion - 59 - 2.2.1. 175 Member chalcone library148
Chalcones are well known to act as antitumor agents by interacting and
depolymerizing tubulin dimers in living cells.189 Tubulin is a globular protein of
molecular weight 100,000, which exists in dynamic equilibrium with microtubules.
Microtubules are vital components of the cell and are responsible for intracellular
transport, mechanical stabilization and formation of the mitotic spindle during cell
division. Vinblastine, vincristine, epithilone and taxol are known as tubulin binders
that work by depolymerization of microtubules.190 Some chalcones are known to bind
to tubulin dimer and halt mitosis at G2/M phase.191 Using chalcones as antitumor
agents may prove quite successful in cancer chemotherapy, as spindle poisons exert
their influence when mitosis is in the metaphase. Furthermore, a number of α,β-
unsaturated ketones have been reported to demonstrate preferential reactivity towards
thiols in contrast to amino and hydroxy groups.192 The chalcones, therefore, are
expected to be free from problems of mutagenecity and carcinogenecity associated
with a number of alkylating agents used in cancer chemotherapy.193
Based on the well known antitumor properties of a chalcone template,106-114 the
studies were expanded for antitumor evaluation of a diverse set of chalcones.
Therefore, a 175 member chalcone library was designed, synthesized and evaluated
for antitumor potency through potato-disc tumor inhibition (PDT) assay.148
Fig. 2.10. Mechanism of tubulin depolymerization (courtesy from Calbiochem).194

Chapter 2 ♦Results and Discussion - 60 - 2.2.2.1. Synthesis. Having two chalcones as potential candidates to be developed as
antibacterials and two other chalcones as LEADS in BSL bioassay in a 120 member
library, the studies were extended to a still larger library. The building blocks were
chosen with a view to have diversity in terms of hydrophobicity, hydrophilicity and
electronic effects. Seven acetophenones A1-A7 and 25 aldehydes B1-B25 were used to
design this library.
R'
O
R
R'
O
R H
O
NaOH / EtOH
Aldehyde R Aldehyde R B1 C6H5 B14 3-NO2C6H4
Acetophenone R′ B2 2-OHC6H4 B15 3-OH,4-OCH3C6H4
A1 C6H5 B3 3-OHC6H4 B16 4-N(CH3)2C6H4
A2 2΄-OHC6H4 B4 4-OHC6H4 B17 4-CH3C6H4
A3 3΄-OHC6H4 B5 2-OCH3C6H4 B18 2-Thienyl
A4 4΄-OHC6H4 B6 3-OCH3C6H4 B19 5-Methyl-2-thienyl
A5 2΄-NH2C6H4 B7 4-OCH3C6H4 B20 5-Bromo-2-thienyl
A6 3΄-NH2C6H4 B8 3,4-(OCH3)2C6H3 B21 5-Nitro-2-thienyl
A7 4΄-NH2C6H4 B9 2-ClC6H4 B22 2-Pyrrolyl
B10 3-ClC6H4 B23 2-Pyridyl
B11 4-ClC6H4 B24 3-Pyridyl
B12 3-BrC6H4 B25 4-Pyridyl
B13 4-FC6H4
Scheme 2.5. Building blocks for the synthesis of a 175 member library.
The testing of this indexed library has been conceptually represented as a 2-D matrix,
wherein x-axis has 7 structural variants (acetophenones), while y-axis has 25
structural variants (aldehydes) leading to a 7 × 25 grid, as discussed earlier (Fig.
2.11).
The synthesis of these chalcone libraries was carried out using standard Claisen-
Schmidt conditions.148 Each of the 32 row and column reaction was carried out with
one specific acetophenone An and a mixture of 25 aldehydes leading to daughter
libraries of Set 1. The libraries (BL1-BL25) of Set 2 were obtained likewise by the
reaction of an aldehyde Bn with a mixture of seven acetophenones. To eliminate
kinetics effects, all reactions were forced to completion by conducting them with a
stoichiometric quantity of the unitary reagent relative to the total of the mixed

Chapter 2 ♦Results and Discussion - 61 - reagents. The synthesis of library BL7 was carried out independently both under
microwave irradiation and conventional heating. The presence of all the components
of the mixture in BL7 was indicated by means of TLC (thin-layer chromatography).
The composition of the library was then compared in two experiments and found to
be the same under both conventional heating and microwave irradiation. The
successful synthesis of indexed libraries was confirmed through GC-MS analysis.
Set 1 Set 2
AL1 AL2 AL3 AL4 AL5 AL6 AL7
BL1 A1 B1 A2 B1 A3 B1 A4 B1 A5 B1 A6 B1 A7 B1
BL2 A1 B2 A2 B2 A3 B2 A4 B2 A5 B2 A6 B2 A7 B2
BL3 A1 B3 A2 B3 A3 B3 A4 B3 A5 B3 A6 B3 A7 B3
BL4 A1 B4 A2 B4 A3 B4 A4 B4 A5 B4 A6 B4 A7 B4
BL5 A1 B5 A2 B5 A3 B5 A4 B5 A5 B5 A6 B5 A7 B5
BL6 A1 B6 A2 B6 A3 B6 A4 B6 A5 B6 A6 B6 A7 B6
BL7 A1 B7 A2 B7 A3 B7 A4 B7 A5 B7 A6 B7 A7 B7
BL8 A1 B8 A2 B8 A3 B8 A4 B8 A5 B8 A6 B8 A7 B8
BL9 A1 B9 A2 B9 A3 B9 A4 B9 A5 B9 A6 B9 A7 B9
BL10 A1 B10 A2 B10 A3 B10 A4 B10 A5 B10 A6 B10 A7B10
BL11 A1 B11 A2 B11 A3 B11 A4 B11 A5 B11 A6 B11 A7B11
BL12 A1 B12 A2 B12 A3 B12 A4 B12 A5 B12 A6 B12 A7B12
BL13 A1 B13 A2 B13 A3 B13 A4 B13 A5 B13 A6 B13 A7 B13
BL14 A1 B14 A2 B14 A3 B14 A4 B14 A5 B14 A6 B14 A7 B14
BL15 A1 B15 A2 B15 A3 B15 A4 B15 A5 B15 A6 B15 A7 B15
BL16 A1 B16 A2 B16 A3 B16 A4 B16 A5 B16 A6 B16 A7 B16
BL17 A1 B17 A2 B17 A3B17 A4 B17 A5 B17 A6 B17 A7 B17
BL18 A1 B18 A2 B18 A3 B18 A4 B18 A5 B18 A6 B18 A7 B18
BL19 A1 B19 A2 B19 A3 B19 A4 B19 A5 B19 A6 B19 A7 B19
BL20 A1 B20 A2 B20 A3 B20 A4 B20 A5 B20 A6 B20 A7 B20
BL21 A1 B21 A2 B21 A3 B21 A4 B21 A5 B21 A6 B21 A7 B21
BL22 A1 B22 A2 B22 A3 B22 A4 B22 A5 B22 A6 B22 A7 B22
BL23 A1 B23 A2 B23 A3 B23 A4 B23 A5 B23 A6 B23 A7 B23
BL24 A1 B24 A2 B24 A3 B24 A4 B24 A5 B24 A6 B24 A7 B24
BL25 A1 B25 A2 B25 A3 B25 A4 B25 A5 B25 A6 B25 A7 B25
Fig. 2.11. Conceptual matrix for designing a 175 member chalcone library.
2.2.2.2. Antitumor studies. Crown gall is a neoplastic disease of plants, which occurs
in more than 60 families of dicotyledons and many gymnosperms. Crown galls cause

Chapter 2 ♦Results and Discussion - 62 - the bulging of a mass of tissues from stems and roots of woody and herbaceous
plants. These tumors may be spongy or hard, and may or may not have deleterious
effects on the plant. Histologically, crown-gall tumors are similar to those found in
humans and animals. The causative agents of this disease are specific strains of the
Gram-negative Agrobacterium tumefaciens (AT). The inhibition of crown-gall tumor
(induced by A. tumefaciens in potato-disc tissue) is an assay based on antimitotic
activity. This assay is capable of detecting a broad range of known and novel
antitumor effects. During infection of plant material with A. tumefaciens, a tumor-
producing plasmid, found in bacterial DNA, is incorporated in the chromosomal plant
DNA. When plant tissue is wounded, it releases phenols and other compounds that
activate the Ti-plasmid in A. tumefaciens. The Ti-plasmid causes the plant’s cells to
multiply rapidly without going through apoptosis, resulting in tumor formation that
are similar in nucleic acid content and histology to human and animal cancers. The
relevance of the crown-gall-tumor system to the general cancer problem has been
thoroughly reviewed.195-197
The validity of this bioassay is based on the observation that certain tumorigenic
mechanisms are similar in plants and animals. For example, it has been observed that
the inhibition of crown-gall tumor on potato discs and their subsequent growth shows
good correlation with compounds and extracts active in the 3PS leukemic mouse
assay.198 It has also been reported that the potato disc tumor assay is statistically more
predictive in terms of 3PS activity than either the 9KB or the 9PS cytotoxicity
assays199 and can be used as a fairly rapid, inexpensive, and reliable prescreen for
antitumor activity.200
PDT, being a simple bench top assay has been used as a pre-screen for evaluation of
antitumor potential of different compounds obtained either from nature or from
synthetic world. The designed chalcone libraries of Sets 1 and 2 were, therefore,
subjected to this bioassay. The screening of two sets of libraries enabled the testing of
each compound twice, once as a component of the sub-libraries of Set 1, and then as a
member of the sub-libraries of Set 2. Thus, a total of 32 assays were required to find
the antitumor activity of 175 compounds which corresponded to a factor of ~ 5.5 in
terms of time improvement in the synthesis and data collection efficiency.

Chapter 2 ♦Results and Discussion - 63 - The sub-libraries of two sets, AL1-AL7 and BL1-BL25, were screened initially at a
concentration of 1000 ppm and their antitumor activities were determined as shown in
Table 2.13.
Table 2.13. PDT inhibition studies of libraries of Sets 1 and 2 at 1000 ppm concentration.
Library % Inh. Library % Inh. Library % Inh. Library % Inh.
AL1 100 BL3 95 BL12 100 BL21 80
AL2 96 BL4 100 BL13 79 BL22 66
AL3 100 BL5 100 BL14 57 BL23 95
AL4 95 BL6 96 BL15 66 BL24 33
AL5 71 BL7 100 BL16 65 BL25 74
AL6 78 BL8 100 BL17 64
AL7 48 BL9 100 BL18 69
BL1 40 BL10 77 BL19 79
BL2 85 BL11 100 BL20 64
All the libraries showed significant antitumor activity. Libraries AL1, AL3, BL4-BL5,
BL7-BL9 and BL11-BL12 showed 100% tumor inhibition. Further short listing was
done by screening these nine libraries at lower concentrations, e.g. at 100 and 10
ppm, as shown in Table 2.14. As a result, the libraries AL1 and BL9 (corresponding to
column 1 and row 9, respectively) were found to be the most active, both at 10 and
100 ppm.
Table 2.14. PDT inhibition studies on the active libraries at 10 and 100 ppm concentration.
Library % Inh. Library % Inh. Library % Inh.
AL1 64 (88) BL5 33 (54) BL9 73 (95) AL3 60 (84) BL7 15 (29) BL11 14 (35)
BL4 11 (78) BL8 23 (45) BL12 22 (41)
Values in parentheses correspond to inhibition at 100 ppm.
2.2.2.3. Deconvolution. Having synthesized and screened all the sub-libraries, the
identification of the lead(s) was carried out by deconvolution. This required the
calculation of activities of all the members of the library using the experimentally
determined values at 1000 ppm, as indices to the cells of columns and rows of the 2D
matrix. The assay value of each cell is contained in the combination AxBy (x = 1-7
and y = 1-25). Since only one cell out of 175 may possess the maximum response
function (Rxy), deconvolution is the identification of this particular combination

Chapter 2 ♦Results and Discussion - 64 - without looking at all 175 cells. Therefore, each B (aldehyde) was tested with all A’s,
(acetophenones) and each A was tested with all B’s.
The data was expanded in 175 cells of chalcones on the matrix by taking the average
of the activity of the respective column and row, which were used as indices to the
particular cells. This resulted in calculated antitumor activities for all 175 chalcones
in the 2D matrix as shown in Table 2.15.
Table 2.15. Calculated antitumor activities of designed library at a concentration of 1000 ppm.
Set 1 Set 2
AL1 AL2 AL3 AL4 AL5 AL6 AL7
BL1 70 69 70 67 56 59 44 BL2 92 92 92 90 78 82 67 BL3 97 96 97 95. 83 86 71 BL4 100 99 100 97 85 89 74 BL5 100 99 100 97 85 89 74 BL6 98 97 98 96 84 87 72 BL7 100 99 100 97 85 89 74 BL8 100 99 100 97 85 89 74 BL9 100 99 100 97 85 89 74 BL10 88 87 88 86 74 77 62 BL11 100 99 100 97 85 89 74 BL12 100 99 100 97 85 89 74 BL13 89 89 89 87 75 78 64 BL14 78 78 78 76 64 67 53 BL15 83 82 83 80 68 72 57 BL16 82 82 82 80 68 71 57 BL17 82 81 82 79 68 71 56 BL18 84 83 84 82 70 73 58 BL19 89 88 89 87 75 78 63 BL20 82 81 82 79 67 71 56 BL21 90 89 90 87 76 79 64 BL22 83 82 83 80 68 72 57 BL23 97 96 97 95 83 86 71 BL24 66 66 66 64 52 55 41 BL25 87 86 87 84 73 76 61
A number of chalcones showed 100% inhibition at 1000 ppm concentration (Table
2.13). The activities of nine short-listed sub-libraries (inhibiting 100% at 1000 ppm)
were screened at lower concentrations i.e. 100 and 10 ppm values. The inhibitory
potentials at 10 ppm concentrations were used as indices to the cells of the 2D matrix

Chapter 2 ♦Results and Discussion - 65 - comprising two columns and seven rows and the data was spread over 14 cells, as
shown in Table 2.16.
Table 2.16. Calculated antitumor activities of the most active library members at 10 ppm.
Set 1 Set 2
AL1 AL3
BL4 37.5 35.5
BL5 48.5 46.5
BL7 39.5 37.5
BL8 43.5 41.5
BL9 50.5* 48.5
BL11 39.0 37.0
BL12 43.0 41.0 *most active member.
It is evident that both sub-libraries AL1 and BL9 may be predicted to be the most-
active. Thus, for the identification of the most active chalcone, parallel synthesis of
the members of the most active column AL1, and the most active row BL9, was
carried out by means of microwave assisted organic synthesis.
Synthesis of members of active libraries AL1, AL3 and BL9. Members of the active
columns AL1 and AL3 as well as active row BL9 were synthesized in parallel under
microwave irradiation. The structures of the members of AL1 and AL3 are given in
Schemes 2.6, 2.7, whereas those of BL9 are given in Scheme 2.8.
O
R
O
H R
O
NaOH/ EtOH
M.w.
R R R
21 Ph 32 2-ClC6H4 46 5-Methyl-2-thienyl
22 2-OHC6H4 33 3-ClC6H4 47 5-Bromo-2-thienyl
23 3-OHC6H4 34 4-ClC6H4 48 5-Nitro-2-thienyl
24 4-OHC6H4 35 4-FC6H4 49 2-Pyrrolyl
25 2-OCH3C6H4 37 3-BrC6H4 50 2-Pyridyl
26 3-OCH3C6H4 38 4-MeC6H4 51 3-Pyridyl
27 4-OCH3C6H4 39 3-OH,4-OCH3C6H3 52 4-Pyridyl
28 3,4(OCH3)C6H3 40 4-N(CH3)2C6H4
O
R
A1B1-A1B25
(21-30, 32-35,37-40,
45-52)
30 3-NO2C6H4 45 2-Thienyl
Scheme. 2.6. Parallel synthesis of chalcones of active column AL1.

Chapter 2 ♦Results and Discussion - 66 -
R R R
53 C6H5 62 3-ClC6H4 71 5-Methyl-2-thienyl
54 2-OHC6H4 63 4-ClC6H4 72 5-Bromo-2-thienyl
55 3-OHC6H4 64 3-BrC6H4 73 5-Nitro-2-thienyl
56 4-OHC6H4 65 4-FC6H4 74 2-Pyrrolyl
57 2-OCH3C6H4 66 3-NO2C6H4 75 2-Pyridyl
58 3-OCH3C6H4 67 3-OH,4-OCH3C6H3 76 3-Pyridyl
59 4-OCH3C6H4 68 4-N(CH3)2C6H4 77 4-Pyridyl
60 3,4(OCH3)C6H3 69 4-MeC6H4
O
ROH
A3B1-A3B25
(53-77)
61 2-ClC6H4 70 2-Thienyl
Scheme. 2.7. Parallel synthesis of chalcones of active column AL3.
R′ R′
12 4′-NH2C6H4 79 4′-OHC6H4
32 C6H5 80 2′-NH2C6H4
62 3′-OHC6H4 81 3′-NH2C6H4
R'
O
Cl
A1B9-A7B9 (12, 32, 62, 78-81) 78 2′-OHC6H4
Scheme. 2.8. Parallel synthesis of chalcones of active row BL9.
FT-IR spectral analysis of all the chalcones A1B1-A1B25, A3B1-A3B25 and A1B9-A7B9
was carried out. The C=O and C=C stretching vibrations for the prepared compounds
appeared in the range of 1631-1690 cm–1 and 1508-1606 cm–1, respectively. Aryl C-H
stretches are often observed at 3041-3246 cm–1. 1H NMR spectra of the synthesized chalcones were recorded. A detailed spectral
analysis of 1-(phenyl)-3-(5-methyl-2-thienyl)-2-propen-1-one (46) has been given in
Table 2.17. The most characteristic chemical shifts were those of vinylic protons H
(α) and H (β); the former appeared upfield at 7.21 ppm, while the later appeared at
7.88 ppm. The coupling constant was 15.3 Hz indicating a trans orientation of the
double bond. The methyl protons appeared as a singlet at 2.53 ppm. In thienyl ring,
doublet (d) at 6.76 ppm with a coupling constant 3.6 Hz was assigned to H–C(3), a
doublet at 7.18 ppm with 3.6 Hz was assigned to H–C(4). A doublet at 8.01 ppm with
a coupling constant 7.2 Hz was assigned to H–C(2′, 6′). A triplet at 7.51 ppm with
coupling constant 7.2 Hz was assigned to the H–C(3′, 5′), whereas a triplet at 7.59
ppm, again with a coupling constant 7.2 Hz was assigned to H–C(4′).

Chapter 2 ♦Results and Discussion - 67 - Table 2.17. 1H NMR data of 1-(phenyl)-3-(5-methyl-2-thienyl)-2-propen-1-one (46).
O
1
23
4
5
1'
2'3'
4'
5'6'
α
β
SMe
46
δ ppm Multiplicity Integration J (Hz) Assignment
2.53 s 3H - H−C(CH3)
6.76 d 1H 3.6 H−C(3)
7.18 d 1H 3.6 H−C(4)
7.21 d 1H 15.3 H-C(α)
7.51 t 2H 7.2 H−C(3′, 5′)
7.59 t 2H 7.2 H−C(4′)
7.88 d 1H 15.3 H−C(β)
8.01 d 2H 7.2 H−C(2′, 6′)
Antitumor screening. The purified and characterized compounds were assessed for
their antitumor potencies through PDT assay. The individual members of the active
libraries AL1, AL3 and BL9 were tested for their antitumor potential at 10, 100 and
1000 ppm concentrations and the data is shown in Tables 2.18-2.20.
Table 2.18. Antitumor activities of chalcones of library AL1.
Chalcone Inhibitiona Chalcone Inhibitiona Chalcone Inhibitiona
A1B1 17.0, 34.1, 62.1 A1B10 43.1, 47.5, 76.8 A1B19 26.2, 37.7, 45.9
A1B2 35.3, 40.2, 80.5 A1B11 71.9, 75.7, 96.3 A1B20 54.1, 56.4, 78.7
A1B3 53.6, 58.5, 100 A1B12 64.6, 74.4, 78.2 A1B21 44.3, 54.1, 54.1
A1B4 34.1, 56.1, 64.6 A1B13 32.9, 53.6, 93.9 A1B22 40.9, 75.4, 80.3
A1B5 19.5, 52.6, 57.5 A1B14 37.8, 58.5, 69.5 A1B23 67.2, 80.3, 83.6
A1B6 34.1, 48.8, 54.8 A1B15 53.6, 54.9, 74.4 A1B24 63.9, 98.4, 90.2
A1B7 35.4, 47.7, 53.6 A1B16 10.6, 29.5, 31.1 A1B25 42.6, 49.2, 68.8
A1B8 56.1, 68.7, 70.7 A1B17 14.7, 14.7, 36.1
A1B9 100, 100, 100 A1B18 11.5, 32.7, 49.2
a Percent inhibition at 10, 100 and 1000 ppm, respectively. All the chalcones were tested on twelve
potato-discs at each concentration.

Chapter 2 ♦Results and Discussion - 68 - Table 2.19. Antitumor activities of chalcones of library AL3.
Chalcone Inhibitiona Chalcone Inhibitiona Chalcone Inhibitiona
A3B1 72.8, 100, 100 A3B10 51.0, 79.7, 100 A3B19 75.6, 80.2, 100
A3B2 100, 100, 100 A3B11 33.2, 63.6, 100 A3B20 65.4, 66.7, 82.0
A3B3 57.8, 100, 100 A3B12 53.2, 77.9, 100 A3B21 65.4, 70.9, 73.1
A3B4 35.4, 70.5, 100 A3B13 37.1, 76.6, 100 A3B22 73.8, 81.8, 100
A3B5 76.8, 100, 100 A3B14 75.6, 91.0, 96.1 A3B23 57.7, 100, 100
A3B6 25.9, 49.3, 100 A3B15 64.9, 72.7, 100 A3B24 76.9, 77.9, 83.3
A3B7 31.2, 33.8, 100 A3B16 72.3, 80.8, 100 A3B25 80.8, 89.5, 94.9
A3B8 60.2, 83.6, 100 A3B17 78.2, 84.2, 100
A3B9 38.9, 60.2, 100 A3B18 52.5, 100, 100
a Percent inhibition at 10, 100 and 1000 ppm, respectively. All the chalcones were tested on twelve
potato-discs at each concentration.
Table 2.20. Antitumor activities of the chalcones of library BL9.
Chalcone Inhibitiona Chalcone Inhibitiona
A1B9 100, 100, 100 A5B9 49.4, 59.1, 100
A2B9 100, 100, 100 A6B9 55.8, 70.1, 100
A3B9 61.0, 78.6, 98.0 A7B9 52.6, 66.8, 86.4
A4B9 70.7, 77.9, 100
a Percent inhibition at 10, 100 and 1000 ppm, respectively. All the chalcones were tested on twelve
potato-discs at each concentration.
It is evident from the data that the most active members of the library A1B9 and A2B9
and A3B2 (cf. for structures, Scheme 2.5) displayed 100% tumor inhibition at a
concentration of 10 ppm, and thus, can be regarded as leads of the designed library. It
may be recalled that the calculated deconvolution led to the identification of the same
chalcone, A1B9, as lead structure. However, the experimental data showed that A2B9
and A3B2 also exhibited 100% tumor inhibition at 10 ppm and, therefore, are equally
important candidates for developing into highly effective chemotherapeutic agents.
O
Cl
O
OHHO
OOH
Cl
A1B9 (32) A3B2 (61) A2B9 (78)

Chapter 2 ♦Results and Discussion - 69 - The identification of the lead structure in 175 member library through positional
scanning method is shown in Figures. 2.12 and 2.13.
A1B5
AL1
AL2
AL3
AL4
AL5
AL6
175 member library
Set 1 AL1-AL7
AL7
A3B2
A3B15
A3B17
A3B16
A3B21
A3B20
A3B19
A3B18
A3B22
A3B23
A3B25
A3B24
A3B14
A3B13
A3B12
A3B11
A3B10
A3B9
A3B8
A3B7
A3B6
A3B5
A3B4
A3B3
A3B1
A1B1
A1B25
A1B16
A1B17
A1B18
A1B19
A1B20
A1B21
A1B22
A1B23
A1B24
A1B6
A1B7
A1B8
A1B9
A1B10
A1B11
A1B12
A1B13
A1B14
A1B15
A1B4
A1B3
A1B2
O
Cl
A1B9
O
OHHO
A3B2
LEADS
Fig. 2.12. Identification of antitumor leads through deconvolution of 175 member library (Set 1).

Chapter 2 ♦Results and Discussion - 70 -
BL1
BL3
BL4
BL5
BL6
BL7
BL9
BL11
BL12
BL13
BL14
BL2
BL24
BL23
BL22
BL18
BL19
BL20
BL17
BL15
BL8
BL10
BL16
BL21
BL25
BL9
A1B9
A2B9
A3B9
A4B9
A5B9
A6B9
175 member library
Set 2 BL1-BL25
A7B9
OOH
Cl
A2B9
O
Cl
A1B9
LEADS
Fig. 2.13. Identification of antitumor leads through deconvolution of 175 member library (Set 2).

Chapter 2 ♦Results and Discussion - 71 - Therefore, deconvolution by means of positional scanning is a quite cost-effective
protocol capable of identifying a lead compound; however subtle differences in
activity within the library cannot be detected. This disadvantage, however, is more
than compensated by the advantage of the ease of library synthesis.
2.2.2.4. Structure activity relationship. Following the identification of hits of the
designed library by deconvolution, it is essential to highlight the structural features
that contribute to tumor inhibition. Such a correlation may be performed at the library
level as well as at the level of individual compounds. The order of activity within
these seven sub-libraries, in terms of substituent is
H > OH > NH2
or more specifically,
H > 3-OH > 2-OH > 4-OH > 3-NH2 > 2-NH2 > 4-NH2
Evidently, the library derived from unsubstituted acetophenone (AL1) shows 100%
tumor inhibition, while all the three libraries derived from isomeric amino
acetophenones (AL5-AL7) show weak tumor inhibitory potential (Table 2.16).
Moreover, the substituents at para position of ring B lower the activity to a greater
extent as compared to those at ortho or meta positions. Among the libraries of Set 2
(BL1-BL25), seven sub-libraries (BL4, BL5, BL7, BL8, BL9, BL11 and BL12) showed
100% tumor inhibition at a concentration of 1000 ppm. All the sub-libraries of
various analogues of methoxy benzaldehydes (BL5, BL7 and BL8) showed 100%
activity at 1000 ppm, reflecting the significance of a OCH3 substituent at the aromatic
ring. Regarding the tumor inhibitory potential of the individual chalcones, it is
interesting to note that A1B9, A2B9 and A3B2 were found to be the lead structures,
since this further confirms the above conclusion that an unsubstituted A-ring is
important for tumor inhibition, whereas ortho substitution was found to be important
on ring B (o-chloro and o-hydroxy substituents in lead structures). Moreover,
chalcones substituted with an electron donor substituent on ring A, e.g., N(CH3)2 or
3CH3 group were found to be the least active. Varieties of chalcones were derived
from heteroaryl aldehydes, but the pyridyl chalcones A1B23-A1B25 and A3B23-A3B25
were found to be significantly active. Furthermore, A3B19 and A3B22 containing
methyl substituted thienyl and pyrrolyl moieties as ring B resulted in significant
antitumor potencies.

Chapter 2 ♦Results and Discussion - 72 - 2.3. Peptidyl chalcones and peptidyl heterocycles
The standard approach of combining heterocycles and peptides is the fusion of a
small-molecule fragment in the side chain of amino acid building blocks or to
integrate heterocycles via standard acylation of amino groups of the peptide and the
heterocycles to create hybrid molecules.201-206 Only few examples have been reported
where heterocycles are integrated in the peptide backbone itself, as this requires in
most cases specific, C-terminal derivatization of peptides.207-212 Current strategy
describes the synthesis of peptidyl chalcones on solid phase by the use of 2-
phosphoranylideneacetate developed as a linker reagent on polymeric
triphenylphosphine resin. The use of triphenylphosphine resin has been reported in the
Wittig and Mitsunobu reactions.213-214 Polymer-supported C-acylation was reported
for the first time, employing triphenylphosphine resin for the synthesis of polymeric
2-phosphoranylidene acetonitrile as a polymer reagent and an anchoring group
allowing subsequent derivatization of the immobilized product.215-216 Furthermore, C-
acylation of polymer-supported 2-phosphoranylidene acetates was developed as a
0linker reagent with protected amino acids yielding 2-acyl-2-phosphoranylidene
acetates as flexible intermediates for the C-terminal variation of carboxylic acids. As
a result, several novel peptidyl bis- and tris-electrophiles have become accessible,
namely peptidyl diketoesters, peptidylketoaldehydes and peptidyl vinyl ketones (Fig.
2.14).217
POR*
O
Ph Ph
OOR*
O
R″
H
O
OHN
R1
Pep
OHN
R1
PepO
HN
R1
Pep
OHN
R1
Pep
∗∗
∗∗
∗∗∗
Fig. 2.14. Peptidyl bis- and tris- electrophiles obtained from 2-acyl-2-phosphoranylidene
acetate.

Chapter 2 ♦Results and Discussion - 73 - On the basis of above findings, a small library of peptidyl chalcones was designed,
synthesized and then converted to different classes of heterocyles, namely oxazoles,
pyrazolines, pyrazoles, thiazepines and diazepines.218
2.3.1. Synthesis of peptidyl chalcones
The conjugation of peptides with different heterocycles based on C-acylation of
peptides led to peptidyl chalcones, which are also known as peptidyl vinyl ketones.
The peptidyl chalcones were synthesized on phosphorane bound polystyrene
divinylbenzene (PS) support using polymer-supported carbanion equivalents.
Developing carbanion equivalents on solid support is an ideal approach for
establishing complex reaction sequences including C-C coupling steps in polymer-
assisted synthesis. Ideally, the successful method should allow reactions with easily
available building blocks, smooth reaction conditions and further derivatization after
the C-C coupling step. Lithiated dithioacetals have been reported as polymer-
supported carbanion equivalents.219-221 These acetals are strongly basic and, therefore,
are not a convincing approach for the synthesis of sensitive products. The integration
of a C-acylation step into the standard protocol of peptide synthesis would be
especially attractive for the synthesis of peptide mimetics and in the attempts to
reduce the peptide character of inhibitors, which is a common challenge in drug
development programs.
Polymer-supported acyl anion equivalents were first established using phosphorane-
PS support for synthesizing a library of norstatine isosteres, active as aspartic protease
inhibitors.215 Furthermore, C-acylations of polymer-supported 2-phosphoranylidene
acetates (linker reagents) with different protected amino acids yielded 2-acyl-2-
phosphoranylidene acetates, which are flexible intermediates for the C-terminal
variation of carboxylic acids: peptidyl-2,3-diketoesters, peptidyl vinyl ketones,
peptidyl-2-ketoaldehydes and 1,3-diamino-2-hydroxy-propanes.217 Based on the
previous studies, a parallel library of peptidyl chalcones has been synthesized on
phosphorane-PS support and then subsequently converted to a library of peptidic
heterocycles. This integration of heterocycles in the peptide backbone with specific C-
terminal derivatization may lead to novel peptidomimetics.218
The synthetic strategy involved the following main steps:
1. Synthesis of linker reagent.

Chapter 2 ♦Results and Discussion - 74 -
2. Protection of triphenylphosphin-PS support.
3. Deprotonation leading to Wittig ylide.
4. Acylation of Wittig ylide.
5. Amide couplings leading to peptides.
6. N-Acetylation of peptides.
7. Deprotection of phosphorane.
8. Synthesis of peptidyl chalcones.
2.3.1.1. Synthesis of linker reagent
BrOH
O
BrO
OSi
DCC/ DMAP
OHSi
82 As a first step toward polymer-supported C-acylation, linker reagent has been
introduced as a tool in solid phase synthesis combining the functions of a polymer
reagent and those of an anchoring group allowing for subsequent derivatization of the
immobilized product.217 2-Trimethylsilylethyl-2-bromoacetate (82) was used as linker
reagent on phosphorane supported polystyrenedivinylbenzene resin. This ester could
be synthesized by pyridine catalyzed esterification of an acid halide with
trimethylsilyl ethanol or through Steglich esterification. In the second method, the
acid part is activated using dicyclohexylcarbodiimide (DCC) and catalytic amount of
4-N,N-dimethylaminopyridine (DMAP) and then reacted with the desired alcohol.222
This method has the advantage of giving only the desired ester under controlled
conditions. The synthesis was achieved in good yield by activating bromoacetic acid
with DCC/DMAP followed by esterification with 2-trimethylsilylethanol. The crude
product after vacuum distillation gave the desired ester, which was further used as a
facile linker reagent for C-C acylations on phosphorane support. The product was
characterized by 1H NMR and 13C NMR analysis. 1H NMR analysis of 2-trimethylsilylethyl-2-bromoacetate (82) showed a singlet of
nine protons at 0 ppm for trimethylsilyl group, two triplets at 1.10 and 4.21 ppm, each
of two protons for two methylenes attached to Si(CH3)3 and an ester moiety
respectively. The two methylene protons attached to Br appeared as singlet at 3.90
ppm (Fig. 2.15).

Chapter 2 ♦Results and Discussion - 75 -
Fig. 2.15. 1H NMR analysis of 2-trimethylsilylethyl-2-bromoacetate 82.
2.3.1.2. Protection of triphenylphosphin-PS support
Toluene, Mw,15 min
PPh
Ph PhP
Ph
O
O
BrBrO
OSi
Si
83 The linker reagent [2-trimethylsilylethyl-2-bromoacetate 82] was loaded on
triphenylphosphin-PS support for synthesizing a Wittig salt. The commercially
available triphenylphosphin-PS support was alkylated with the synthesized linker
under microwave irradiation at 100 °C for 15 min in toluene (successful alkylation
can also be carried out through stirring at room temperature for 72 h). The alkylation
was confirmed after recording ATR-FT-IR spectrum of the intermediate obtained
(Fig. 2.16).
Absorbance
Wave number cm-1
Fig. 2.16. ATR-FT-IR of triphenylphosphorane support after alkylation with trimethylsilylethyl
bromoacetate.

Chapter 2 ♦Results and Discussion - 76 - The infrared spectrum of alkylated support 83 showed C-H and Ar-H absorbance of
polystyrene at 2851 cm-1 and 3056 cm-1, while a characteristic absorbance at 1735
cm-1 corresponded to the ester carbonyl functionality. The resultant protected
triphenyl phosphin-PS support was accessible for the synthesis of a Wittig ylide
through base catalyzed elimination of an acidic proton at methylene directly attached
to the phosphorous atom.
2.3.1.3. Deprotonation leading to Wittig ylide
84
PhP
Ph
O
O
Br
PhP
Ph
O
O
TEA / DCM
Si Si
The phosphonium salt obtained after alkylation has highly acidic proton and was
easily deprotonated using triethylamine as a base. The resulting Wittig ylide
(polymer-bound phosphoranylidene acetate) enjoys greater stability due to resonance
stabilization with the adjacent carbonyl moiety of the ester group. The product, after
thorough drying, was stored at room temperature. IR analysis of product 84 showed a
peak at 1740 cm-1 that was assigned to ester C=O and this confirmed the identity of
Wittig ylide.
2.3.1.4. Acylation of Wittig ylide
Polymer-bound phosphoranylidene acetate i.e. Wittig ylide can serve as an equivalent
analogue of polymer bound acyl anion. The acylation of the Wittig ylide 84 with
different F-moc protected amino acids serves as the starting point for standard peptide
synthesis. However, polymer-bound phosphoranylidene acetate, being a weak
nucleophile, posed a major challenge in finding efficient acylation conditions. Some
early attempts of acylation remained unsuccessful using different activating agents for
enhancing the electrophilicity of the carbonyl of the amino acid e.g.
diisopropylcarbodiimide (DIC) with catalytic amount of DMAP, N-ethyl-N′-(3-
dimethylaminopropyl)-carbodiimide hydrochloride (EDC) with DMAP and 1-
[bis(dimethylamino)methylene]-1H-1,2,3-triazolo-[4,5-b]pyridinium hexafluorophos
phate-3-oxide (HATU) with diisopropylethylamine (DIPEA). Successful acylations
could be performed with 1-(2-mesitylenesulfonyl)-3-nitro-1H-1,2,4-triazole (MSNT),
whereas usage of fluoro-N,N,N′,N′-bis(tetramethylene)formamidiniumhexafluorophos
phate (TFFH) as acylation agent destroyed labile trimethylsilyl group on linker

Chapter 2 ♦Results and Discussion - 77 - reagent. Therefore, for synthesizing a library of peptidyl chalcones, MSNT and
lutidine (as a base) were used for racemization-free acylation of phosphoranylidene
acetate. MSNT catalysed C-acylation of phosphorane was performed with a broad
choice of carboxylic acids. This reagent tolerated the standard side-chain protecting
groups used in peptide synthesis such as Boc, tert-butyl, and trityl groups.
FmocHN
R1
O
85, R1 =
O
O
PPh
Ph
MSNT, Lutidine, DCM
Fmoc-AA-OH
PhP
Ph
O
O
SiSi
86, R1 =
The mechanism of this C-acylation is shown in Scheme 2.9. Lutidine (as a base)
deprotonates OH of an amino acid and results in a nucleophile. This nucleophile
attacks the sulphone of MSNT. As a result, triazole departs whereas the amino acid
now contains a better leaving group (mesitylenesulfonate). In the following step,
phosphoranylidene acetate (a weak nucleophile) attacks the carbonyl sulphonate ester
resulting in the removal of sulphonate while the amino acid is loaded on 84 through
acylation (Scheme 2.9).
NH
O
O
FmocR1
OSO
O
N
NNS
O
O NO2 NH
O
O
FmocR1
SO
O
PPhPh
OR
O
PPhPh
RO
O
O
R1
NH
Fmoc
N
NN
NO2
Scheme 2.9. MSNT/Lutidine mediated acylation on phosphorane support.
The basicity of the phosphorane reagent did not allow cleavage of detectable amounts
of the Fmoc-protecting group. This was verified by the negative Kaiser test
subsequent to all acylations. The coupling yield of the acylated products 85 and 86

Chapter 2 ♦Results and Discussion - 78 - was determined by spectrophotometric quantification of Fmoc group that was cleaved
off a dried resin sample ~ 5 mg (Table 2.21).
Table 2.21. % Yield of acetylated products obtained on solid support.
Entry Amino acid Conditions Yield (%)a
85
5 equiv Fmoc-Phe, 5 equiv MSNT,
4.9 equiv 2,6-lutidine, DCM, RT.
88
86 FmocHN
OH
O
5 equiv Fmoc-Val, 5 equiv MSNT,
4.9 equiv 2,6-lutidine, DCM, RT.
84
FmocHNOH
O
a Calculated after standard Fmoc cleavage of ~ 5 mg sample.
The IR spectrum of the acylated support 85 showed great similarity to the
corresponding polymeric Wittig salt 83. Important differences were the absorption
due to Si-C bond, which after alkylation could be seen at 837 cm-1, while Fmoc
moiety appeared as a double peak at 742-760 cm-1.
After successful acylation, the protecting group Fmoc a was removed from small
samples ~ 5 mg of resin, using 20% piperidine/DMF and the cleaved adducts c and d
were quantified through measuring absorbance λ1 = 267 nm, λ2 = 289 nm and λ3 =
301 nm, as shown in Scheme 2.10. The concentration of the cleaved Fmoc moiety
was calculated by Beer Lambert Law (Section 4A.3.4.).
H
OO
HN R
N H
OO
HN R
N H
OO
HN R
NH
H2N R
H
N CO2
a b
cd
Scheme 2.10. Elimination of base-labile Fmoc moiety demasking amino functionality.

Chapter 2 ♦Results and Discussion - 79 - 2.3.1.5. Amide coupling leading to peptide synthesis
NH
OR1
OO
PPh
Ph
NFmoc
O
Rnn
Si
n = 1, 2
FmocHNO
R1
OO
PPh
Ph
Si
amide coupling H
87-91 The acylated products 85-86 were subjected to Fmoc deprotection using 20%
piperidine/DMF (2 x 6 min). The resulting free N-terminus amino group was used as
the attachment site for adding amino acid monomers thus yielding peptidyl 4-amino-
3-oxo-2-phosphoranylidene butanoates 87-91. The activating agents were first
developed for racemization-free-amide coupling on phosphorane support through test
reactions. Various coupling methods using different coupling reagents e.g., DIC, N-
[(1H-benzotriazol-1-yloxy)(dimethylamino)methylene]-N-methylmethanaminiumtetra
fluoroborate (TBTU) with DIPEA and DIC with N-hydroxybenzotriazole (HOBT)
were tested for the coupling of Fmoc amino acids. The reaction of DIC with a
carboxylic acid yields a highly reactive O-acyl urea. In order to enhance the
electrophilicity of the carboxylate group, the negatively charged oxygen must first be
"activated" to a better leaving group.
NH
OH
O
FmocN C NR2
NH
O
O
FmocR2
HN
N
H2NOR
O
R1
NH
HN
O
FmocR2
R1
OR
O
NH
O
NH
NH
N
O
FmocR2
O
HN
H
a
c
b
e
f
d
Scheme 2.11. Amide coupling through DIC.
When DIC is used for this purpose, the negatively charged oxygen acts as a
nucleophile, attacking the central carbon in DIC. Nucleophilic attack by an amino
group (of amino acid loaded onto support) to the former C-terminus (carbonyl group
of incoming amino acid now present as an ester b) results into amide coupling
(Scheme 2.11). However, intramolecular rearrangement of b proceeds without H+

Chapter 2 ♦Results and Discussion - 80 - uptake, together with the migration of an amine moiety to carbonyl carbon of ester
functionality resulting into a by product f, which was stuck to resin and was not easily
removed through washings. As a result, the desired product was contaminated with
incomplete peptide sequence and urea derivative f, as indicated through the LC-MS of
the cleaved product obtained in the test reaction.
O-Form
NN
N
NN
NN
N
ON
N
O
O
OR2
HN
Fmoc
O
OR2
HN
Fmoc
O
OR2
HN
Fmoc
N
NN
NN
O
base H BF4
NN
N
O
NN
N
OO
NHFmoc
O
O
ORR1
HN base H
O
ORR1
HN base H
ONH
FmocR2
NN
N
O
N-Form
O-Form N-Form
NHFmocR2
R2
Scheme 2.12. Amide coupling through TBTU/base.
TBTU containing non-nucleophilic tetrafluoroborate anion and DIPEA were tested for
effective amide coupling. TBTU exists in two isomeric forms i.e. O-TBTU and N-
TBTU, existing in dynamic equilibrium under basic conditions. The equilibrium shifts
towards N- Form in strong basic conditions such as in the presence of Et3N of DIPEA.
Under basic reaction conditions, the carboxylic acid part of amino acid deprotonates.
The resulting carboxylate anion initially attacks the α-carbon atom of the immonium
salt (uronium (O-form) or the guanidinium N-oxide (N-form)) to form an unstable
acyloxyimmonium intermediate with the release of benzotriazolyloxy anion (BtO−).

Chapter 2 ♦Results and Discussion - 81 - The BtO− nucleophile, thus generated, attacks the acyloxyimmonium intermediate
generating benzotriazolyl ester, which rearranges into the corresponding N-acylated
isomer. The two forms of the active intermediates reacted with the free amine ending
of phosphorane support resulting in the synthesis of the corresponding amide (Scheme
2.12).223
When the cleaved product was analyzed through LC-MS, the product had low
enantiopurity that was expected to result from intramolecular rearrangement of
acyloxyimmonium intermediate to 4H-5-oxazolone and subsequent epimerization in
the desired product (Scheme 2.13).
O
OHN
NR
OON
BF4
N N
O
NO
O
R'
R H
R' NO
O
R'
H R
NO
O
R'
R
Scheme 2.13. Mechanism of 4H-5-oxazolone mediated epimerization.
Another coupling reagent HOBT was used after pre-activation of Fmoc amino acids
with DIC. HOBT together with DIC was found to be the safest with the phosphorane
support and Fmoc chemistry. The reaction proceeds through nucleophilic attack of the
negatively charged oxygen on the central carbon in DIC (Scheme 2.14). The yields of
the amide coupling reactions using DIC/HOBT are given in Table 2.22.
OH
ONH
R2Fmoc
N C NO
ONH
R2Fmoc N
HN
H
NN
NOH
NN
NO
O
R2 HN
FmocH2NOR
O
R1
NN
NO
NH
NH
O
HN
ONH
R2Fmoc
R1
OR
O
Scheme 2.14. Mechanism of DIC/HOBT mediated amide coupling.
The yields of amide couplings were determined after the Fmoc deprotection of small
resin samples.

Chapter 2 ♦Results and Discussion - 82 - Table 2.22. Yields of the standard amide coupling using DIC/HOBT.
No. Amino acid Product Yield (%)a
87
FmocHNOH
O
86
88
FmocHNOH
O
82
89
FmocHNOH
O
84
90 FmocHNOH
O
NH
O
OO
PPh
Ph
FmocHNO
Si
84
91 FmocHN
OH
O
NH
O
OO
PPh
Ph
HN
O
SiFmocHNO
83
NH
O
OO
PPh
Ph
FmocHNO
Si
NH
O
OO
PPh
Ph
FmocHNO
Si
NH
O
OO
PPh
Ph
FmocHNO
Si
a Calculated after standard Fmoc cleavage of ~ 5 mg sample.
2.3.1.6. N-Terminal acetylation of peptides (92-95)
The amino terminus of the peptides 87-89 and 91 was deprotected from Fmoc moiety
using standard F-moc cleavage conditions. The resulting unprotected peptides were
subjected to N-terminal amine acetylation using acetic anhydride (5 eq) in DMF
resulting in acetylated peptide sequences 92-95. The reaction was repeated two times
to ensure complete acetylation. The complete conversion of N-terminal amino group
to N-acetyl derivatives was confirmed through the Kaiser test (Section 4A.3.11).

Chapter 2 ♦Results and Discussion - 83 -
NH
OR1
OO
PPh
Ph
HN
H
O
Rm
n
Si
n = 1-2
Ac2O/DMF
NH
OR1
OO
PPh
Ph
HN
O
RmO
n
Si
n = 1-2
92, R1 =
93, R1 =
94, R1 =
R2 =
R2 =
R2 =
95, R1 = R2 = R3 =
2.3.1.7. Deprotection of phosphorane support
After having accomplished a successful synthesis of the desired peptide sequence, the
final step was the removal of trimethylsilylethyl group. Tetrabutylammonium fluoride
(TBAF) is well known for TMSE removal.224 However, it was found to destroy
peptide part under basic condition. By controlling the pH in buffer medium, the
removal of tetrabutylammonium ions from the resin could not be achieved even after
repeated washings and always resulted in contaminated products of successive steps.
Furthermore, tris(dimethylamino)sulfonium difluorotrimethylsilicate (TAS-F) was
found as a safe desilylating reagent by removing TMSE without any washing
problems (Fig 2.17).225 TAS-F, a mild neutral agent is an in situ source of fluoride
ion. It successfully removed trimethyl silyl moiety and resulted in subsequent
decarboxylation of supported peptides (Scheme 2.12).
SNN
NSi
F
F
Fig. 2.17. tris(Dimethylamino)sulfonium difluorotrimethylsilicate (TAS-F).
NH
OP
O OSi
Ph PhR1
R2
F
NH
OP
O O
Ph PhR1
R2
H
NH
H
P
O
Ph PhR1
R2CO2C2H4
(CH3)3SiF
Scheme 2.12. Hydrolysis of protected 4-amino-3-oxo-2-phosphoranylidene butanoates with
TAS-F.

Chapter 2 ♦Results and Discussion - 84 - It was, therefore, used for deprotection of all the peptides sequences 92-95.
NH
OR1
OO
PPh
Ph
HN
O
RmO
n
Si
n = 1, 2
TAS-F/DMF
NH
H
R1
O
PPh
Ph
HN
O
RmO
n n = 1, 2
92-95 96-99 The successful removal of TMSE was confirmed through ATR-FT-IR of the resulting
supports 96-99 by the disappearance of Si-C vibration at 837 cm-1 in the resulting
products (Fig 2.18).
Fig. 2.18. ATR FT-IR of 96 after removal of TMSE protection with TAS-F.
2.3.1.8. Synthesis of peptidyl chalcones (100-107)
Peptidyl-3-amino-2-oxo-1-phosphoranylidene propanes 96-99 obtained were
subjected to Wittig cleavage using a variety of aliphatic and aromatic aldehydes that
led to the formation of peptidyl chalcones. All the reactions were performed in THF.
In case of aliphatic aldehydes, the reactions were complete in 4-5 h by stirring at room
temperature, whereas with aromatic aldehydes, the reaction mixture had to be heated
at 50oC for 5-6 h.
100-107
NH
H
R1
O
PPh
Ph
HN
O
RmO
n n = 1, 2
NH
R1
O
HN
O
RmO
n n = 1, 2
R'CHO/THFR'
The resulting peptidyl chalcones were purified through preparative HPLC in some
cases. The yields were calculated for the pure compounds, whereas the purity of the
crude products was assessed through LC analysis (Table 2.23).

Chapter 2 ♦Results and Discussion - 85 - Table 2.23. Synthesis of peptidyl chalcones 100-107.
Entry Product Purity (%)a Yield (%)b
100 HN
NH
OO
O
62 70
101 HN
NH
OO
O
F
Cl
80 68
102 HN
NH
OO
O
F
Cl
61 74
103
HN
NH
OO
O
S
85 65
104
HN
NH
OO
O
BrBr
Br
80 72
105 HN
NH
OO
O
F
Cl
64 78
106 NH
HN
NH
O
O
O
O
90 74
107 NH
HN
NH
O
O
O
O
80 71
a Purity of crude products assessed using LCMS; b isolated yield.

Chapter 2 ♦Results and Discussion - 86 - The structures of the synthesized peptides 100-107 were confirmed through HR-MS, 1H NMR and 13C NMR analysis. In the HR-MS analysis of chalcone 100, the
molecular ion peak (M+H+) was found as 421.2456.
Fig. 2.19. 1H NMR (300 MHz, CDCl3) spectrum of chalcone 100.
Regarding 1H NMR analysis, the methyl protons of the aliphatic chain appeared as a
doublet at 0.91 ppm with a coupling constant of 7.8 Hz and an integration of six
protons, while the methine CH proton appeared as a multiplet at 1.65-1.81 ppm. A
three proton singlet at 1.94 ppm was assigned to protons of the acetyl group. A broad
triplet at 2.07 ppm with a coupling constant of 6.1 Hz was assigned to allylic CH2
protons. Benzylic protons from two phenylalanine units were found as multiplets at
2.91-3.20 ppm with an integration of four protons. α-Protons of phenylalanine units
were found as multiplets at 4.64-4.78 ppm and 4.85-5.05 ppm, each with one proton
integration. The two doublets at 6.15 ppm and 6.56 ppm, each with a coupling
constant of 7.3 Hz were assigned to two NH protons of the amide linkage. The
doublet at 6.15, was assigned to α-vinylic protons. A large coupling constant of 15.9
Hz confirmed the trans configuration of the double bond in the synthesized chalcone.
The β-vinylic proton appeared downfield as a multiplet along with aromatic protons
of phenyl rings at 6.89-7.06 ppm, with an integration of 11 H′s.
13C NMR spectrum further confirmed the identity of the product as shown in Fig.
2.20. Important signals were observed at 21.9, 22.7, 27.3 ppm for methyl, methine
and methylene protons, respectively. Chiral carbons appeared at 53.9 and 56.6 ppm,
respectively, whereas the three carbonyls were found at 169.3, 169.9 and 195.6 ppm.

Chapter 2 ♦Results and Discussion - 87 -
Fig. 2.20. 13C NMR (100 MHz, CDCl3) spectrum of chalcone 100.
2.3.2. Synthesis of peptidyl heterocycles
Molecular combinations of peptides and heterocycles have been investigated in order
to blend the desirable properties of peptides with those of heterocycles to obtain
bioactive and bioavailable, metabolically stable and membrane-permeable
molecules.226 Peptidyl chalcone derivatives are accessible for converting into a wide
variety of heterocycles. As mentioned earlier (Chapter 1), chalcones may undergo a
(3+2) annulation with a variety of reagents leading to HC’s of varying size e.g.,
reaction of enone moiety with o-aminothiophenol and o-phenylenediamine leads to 7-
membered heterocycles i.e. 1,5-benzothiazepines and 1,5-benzodiazepines. Likewise,
the reaction of enone with PhNHNH2 or NH2OH led to 5-membered pyrazoles and
oxazoles respectively.218
NH
R2
O
HN
O
RnO
nn = 1, 2
R' N NR
N O
N O
N NR
N S
Fig. 2.21. Variety of heterocycles accessible through α,β-unsaturated ketone template.
Following the same strategy, peptidyl chalcones were reacted with different reagents
to yield different peptidyl heterocycles, as discussed in following sections.

Chapter 2 ♦Results and Discussion - 88 - 2.3.2.1. Synthesis of peptidyl oxazoles
Oxazoles are the rigid scaffolds containing the elements of amide bond.227 They are
therefore astute choice for developing drug-like characteristics in desired peptide
sequences. They are known as adrenergic receptor agonists and bind to the dopamine
receptor.228-229 Peptidyl oxazoles were prepared by reacting peptidyl chalcones with
hydroxylamine (3 eq) in ethanol in the presence of AcOH and NaOAc as catalysts,
whereby the products were separated as white solids.151 The structures of peptidyl
oxazoles were confirmed through HR-MS, 1H NMR and 13C NMR analysis. The
purity and yield of oxazoles 108-109 are shown in Table 2.24.
Table 2.24. Synthesis of peptidyl oxazoles 108-109.
Entry Product Purity (%)a Yield (%)b
108 HN
NH
OO
ON
F
Cl
90 85
109 HN
NH
OO
ON
80 68
a Purity of crude products assessed using LCMS; b isolated yield.
The synthesis of 108, for example, was confirmed through HRMS analysis where the
molecular ion peak (M+H+) was found at 507.1830 as expected (507.1833). In the 1H
NMR spectrum (Fig. 2.22), a singlet at 1.84 ppm with an integration of 3 protons was
assigned to the acetyl group. Three signals, a dd, a multiplet and another dd with an
integration of one, two and one proton, respectively, appeared in the range of 2.45-
3.45 ppm indicating the presence of a CH2 group. These multiplets were assigned to
benzylic protons of phenylalanines. A dd appeared at 2.73 showing geminal coupling
(12 Hz) and a vicinal coupling (9 Hz). Similarly, a dd at 3.14 ppm appeared with the
coupling constants of 15 Hz and 8 Hz, respectively. The two dd’s at 5.39 and 4.52,
each with an integration of one proton were assigned to two α- hydrogens at two

Chapter 2 ♦Results and Discussion - 89 - chiral centers; dd’s showed a vicinal coupling constant of 6 Hz and 15 Hz,
respectively, indicating the cis and trans placement of two vicinal protons with
Fig. 2.22. 1H NMR (300 MHz, CDCl3) spectrum of oxazole 108.
respect to the chiral hydrogen. Two amide NH protons appeared as singlets at 6.70
and 6.90 ppm, whereas a multiplet in the range of 7.03-7.22 ppm with an integration
of 14 protons was assigned to aromatic protons of phenyl and oxazole rings.
Fig. 2.24. 13C NMR (100 MHz, CDCl3) spectrum of 108.
In 13C NMR spectrum of oxazole 108 (Fig. 2.24), peaks for all carbons were
observed. The most upfield signal at 22.58 ppm was assigned to the methyl carbon of
terminal acetyl group. The peaks at 36.98 ppm and 37.64 ppm were assigned to
methylene carbons of two benzyl groups. The methine carbons of the chiral centers
appeared downfield at 43.57 ppm and 53.90 ppm. The other important signals

Chapter 2 ♦Results and Discussion - 90 - corresponded to the carbons of oxazole ring. C-5 attached to nitrogen of oxazole
appeared at 170.90 ppm, whereas the one attached to oxygen (C-3) appeared at 157.56
ppm. The other C-4 was found at 115.4 ppm. The two carbonyl carbons appeared at
169.15 and 171.05 ppm, respectively, while the aromatic carbons appeared in the
range of 115.35 ppm to 159.15 ppm. 13C-F19 couplings were observed in the cases
where peaks were separated. The carbon as a doublet at 157.65 ppm and a 1J of 296
Hz was assigned to one attached to fluorine atom. The ipso carbon of 2-chloro-6-
fluorophenyl ring attached to oxazole ring appeared as a doublet at 139.64 ppm with a 2J coupling as 96 Hz. The carbon attached to chlorine atom appeared at 129.45 with a 3J coupling of ~ 12 Hz.
2.3.2.2. Synthesis of peptidyl pyrazolines
Pyrazolines are important N-containing five-membered heterocyclic compounds used
as antitumor, antibacterial and antituberculotic agents.230 Peptidyl pyrazolines were
prepared by reacting peptidyl chalcone with hydrazines (Table 2.25).153
Table 2.25. Synthesis of peptidyl pyrazolines 110-114.
Entry Product Purity (%)a Yield (%)b
110
NH
HN
NH
O
O
O
N NH
95 65
111
NH
HN
NH
O
O
O
N N
98 61
112 NH
HN
NH
O
O
O
N N
95 66
113
NH
HN
NH
O
O
O
N N
65 53
114
NH
HN
NH
O
O
O
N NH
89 67
a Purity of crude products; b isolated yield.

Chapter 2 ♦Results and Discussion - 91 - A mixture of AcOH and NaOAc was used as catalyst in pyrazoline synthesis. The
synthesized compounds were separated as white solids. The peptidyl pyrazolines 110-
114 were oxidized to the respective pyrazoles without further confirmation.
2.3.2.3. Synthesis of peptidyl pyrazoles
Peptidyl pyrazolines 110-114 were oxidized to the corresponding pyrazoles 115-118
by their reaction with dichlorodicyanoquinone (DDQ). The residue was subjected to
purification by preparative HPLC and pyrazoles were separated as white solids. The
resulting products were characterized through NMR analysis. Yield and purity of the
synthesized products are given in Table 2.26, while the characterization was done on
the basis of LC-MS and 1H NMR data.
Table 2.26. Synthesis of peptidyl pyrazoles 115-118.
Entry Product Purity (%)a Yield (%)b
115 NH
HN
NH
O
O
O
N NH
89 >95
116 NH
HN
NH
O
O
O
N N
84 >95
117 NH
HN
NH
O
O
O
N N
85 >95
118
NH
HN
NH
O
O
O
N N
60 >95
a Purity of crude products; b isolated yield.
In 1H NMR analysis of peptidyl pyrazole 115 (Table 2.27), the most upfield doublet at
1.22 ppm with an integration of 3 Hs and a coupling constant of 7.9 Hz, was assigned
to CH3 group of alanine, whereas the singlet at 1.73 ppm was assigned to methyl of
acetyl group. A singlet of 3 Hs appeared at 1.98 ppm was assigned to methyl group at
C-5 of the pyrazole. Two multiplets at 2.65-2.73 ppm and 2.91-2.95 ppm, each with

Chapter 2 ♦Results and Discussion - 92 - an integration of two protons were assigned to the benzylic protons of two
phenylalanines. The three chiral centers present in the peptide part of the molecule
appeared in the range of 4.20-4.24 ppm, 4.46-4.49 ppm and 4.99-5.05 ppm, each with
a multiplicity of one proton. The aromatic hydrogen of pyrazole ring (H-4) was found
as a singlet at 5.31 ppm, whereas the NH hydrogen of pyrazole appeared as a singlet
at 5.86 ppm. The protons of three amide NH were found at 6.64 ppm, 8.02 ppm and
8.20 ppm as doublets, each with a coupling constant of ~ 8 Hz. The aromatic protons
corresponding to two phenyl rings appeared at 7.12-7.25 ppm as multiplet of ten
protons.
Table 2.27. 1H-NMR analysis (300 MHz, DMSO-d6) of peptidyl pyrazole 115.
δ ppm Multiplicity Integration Coupling constant J (Hz)
Assignment
1.22 d 3H 7.9 H-Cβ(Ala)
1.73 s 3H - H-C(CH3CO)
1.98 s 3H - H-C(CH3 pyrazole)
2.65-2.73 m 2H - H-Cβ1,β1′(Phe, Benzyl)
2.91-2.95 m 2H - H-Cβ2,β2′(Phe, Benzyl)
4.20-4.24 m 1H - H-Cα(Ala)
4.46-4.49 m 1H - H-Cα(Benzyl)
4.99-5.05 m 1H - H-Cα(Phe)
5.31 s 1H - H-C(CH pyrazole)
5.86 s 1H - NH(pyrazole)
6.64 d 1H 8.0 NHI
7.12-7.25 m 10H - H-C(arom.)
8.02 d 1H 8.1 NHII
8.20 d 1H 8.0 NHIII
2.3.2.4. Synthesis of peptidyl benzothiazepines
Benzothiazepines exhibit diverse biological activities. They are known as calcium
antagonist,231 anticonvulsant,232 anticancer,233 antihypertensive234 and
antithrombotic.235 Peptidyl benzothiazepines 119-123 were synthesized by reacting
peptidyl chalcones with o-aminothiophenol at reflux temperatures under an inert N2

Chapter 2 ♦Results and Discussion - 93 - atmosphere.146 The residue was subjected to purification by preparative HPLC to
afford benzothiazepines as off-white to pale yellow solids. The resulting products
were confirmed through 1H NMR, 13C NMR and HRMS analysis. The data of the
purity and yield of peptidyl benzothiazepines is given in Table 2.28.
Table 2.28. Synthesis of peptidyl benzothiazepines 119-123.
Entry Product Purity (%)a Yield (%)b
119 HN
NH
OO
NS
F
Cl
80 52
120 HN
NH
OO
NS
60 63
121
HN
NH
OO
NS
Cl
F
Cl
78 75
122
HN
NH
OO
NS
S
Cl
64 71
123 HN
NH
OO
NS
Cl
F
Cl
50 55
a Purity of crude products assessed through LCMS. b Isolated yield.
Spectroscopic analysis of 119 is discussed as a representative example. Its synthesis
was confirmed through HRMS analysis, wherein molecular ion peak was found as
(M+H+) at 600.1842. In the 1H NMR spectrum (Fig. 2.25), a singlet for acetyl protons
appeared at 1.95 ppm. One of the methylene protons of benzothiazepine ring appeared

Chapter 2 ♦Results and Discussion - 94 - as a multiplet along with β protons of the benzyl rings with an integration of five
protons at 2.84-3.14 ppm. The second methylene proton of benzothiazepine ring
appeared as a dd at 3.55 ppm with an integration of one proton and coupling constant,
9.7 Hz and 6.0 Hz, respectively. The methine proton of benzothiazepine moiety
appeared as a dd at 4.65 ppm with coupling constants of 10.2 Hz and 6.0 Hz. The two
chiral carbons of the peptide part were found as doublet of doublet at 5.04 and 5.39
ppm, each with an integration of one proton and coupling constants of ~ 13 Hz and ~
6 Hz, respectively. Aromatic protons of phenyl rings gave a multiplet of 16 protons in
the range of 7.06-7.58 ppm, whereas one aromatic proton appeared as a doublet at
7.58 ppm with a coupling constant of 7.3 Hz. The two amide hydrogens were found as
singlets at 6.18 and 8.08 ppm, respectively.
Fig. 2.25. 1H NMR (300 MHz, CDCl3) spectrum of 119.
In 13C NMR spectrum (Fig. 2.26), three protons of acetyl group appeared at 22.22
ppm. The important peaks of methane and methylene carbons of benzothiazepine ring
were found at 35.31 ppm and 37.73 ppm, respectively. Furthermore, methylene
protons corresponding to two phenylalanine moieties were found at 45.02 ppm and
48.91ppm. The two peaks for chiral α- carbons appeared at 54.92 and 59.47 ppm. The
characteristic peaks for the two carbonyl carbons were found at 169.53 ppm and
173.23 ppm. The carbon corresponding to C=N part of benzothiazepine ring appeared
at 163.14 ppm, while the other peaks in a range of 114.45 ppm to 158.54 ppm were
assigned to aromatic carbons. 13C-19F coupling was observed for the ipso C attached
to F, resulting in a doublet at 156.9 ppm with a coupling constant of 320 Hz.

Chapter 2 ♦Results and Discussion - 95 -
Fig. 2.26. 13C NMR (100 MHz, CDCl3) spectrum of 119.
The identity of other benzothiazepines 120-123 was also confirmed on the basis of
their spectral data.
2.3.2.5. Synthesis of peptidyl benzodiazepines
Benzodiazepine is a potent scaffold for the treatment of cardiovascular disorders,236
epilepsy237 and HIV.238 Peptidyl benzodiazepines 124-125 were synthesized through a
base catalyzed reaction of peptidyl chalcones with 1,2-phenylenediamine under an
atmosphere of nitrogen. Crude products were subjected to purification by preparative
HPLC. Yield and purity of the isolated peptidyl benzodiazepines are given in Table
2.29.
Table 2.29. Synthesis of peptidyl benzodiazepines 124-125.
Entry Product Purity (%)a Yield (%)b
124
HN
NH
OO
NNH
F
Cl
80 48
125 HN
NH
OO
NNH
F
Cl
62 50
a Purity of crude products assessed through LCMS; b isolated yield

Chapter 2 ♦Results and Discussion - 96 - The identity of synthesized benzodiazepines was confirmed spectroscopically. The
HRMS analysis of the peptidyl benzodiazepine 124 showed a M+H+ peak at
533.2140. In the 1H NMR (Table 2.30), a doublet at 1.15 ppm with an integration of
six protons for two methyl protons of i-Pr group, whereas a multiplet at 1.21 ppm-
1.30 ppm with an integration of one proton was assigned to methine proton of i-Pr
group. A three proton singlet at 1.90 ppm was assigned to N-terminal acetyl group. A
multiplet of two protons at 2.75 ppm-2.80 ppm was assigned to methylene protons of
phenylalanine. A multiplet at 2.95-3.10 ppm with an integration of two protons was
assigned to methylene protons of diazepine ring, whereas a one proton multiplet at
3.40-3.43 ppm was assigned to NH of diazepine ring. The methine proton at the chiral
center of phenylalanine part appeared as a multiplet with one proton integration at
4.17 ppm-4.24 ppm. Another multiplet at 4.75-4.81 ppm with an integration of one
proton was assigned to the chiral carbon attached to isopropyl group. Furthermore, the
methine proton of diazepine ring was assigned to a one proton multiplet at 4.98-5.04
ppm. The aromatic protons of benzene rings and the two amide NHs appeared as a
multiplet at 6.80-7.91 ppm showing an integration of fourteen protons.
Table 2.30. 1H-NMR analysis (300 MHz, CDCl3) of peptidyl benzodiazepine 124.
δ ppm Multiplicity Integration Coupling constant J (Hz)
Assignment
1.15 d 6H 7.9 CH3 isopropyl
1.21-1.30 m 1H - CH isopropyl
1.90 s 3H - H-C(CH3CO)
2.75-2.80 m 2H - H-Cβ1,β2(Phe)
2.95-3.10 m 2H - CH2 (diazepin)
3.40-3.43 m 1H - NH (diazepin)
4.17-4.24 m 1H - H-Cα(Phe)
4.75-4.81 m 1H - H-Cα(CH isopropyl)
4.98-5.04 m 1H - H-C(CH diazepin)
6.8-7.91 m 14H - H-Carom., NHII,III
13C NMR (Table 2.31) of benzodiazepine 124 showed characteristic peaks for all
carbons where 13C-19F coupling was observed for carbon attached to F atom, resulting
in the splitting of the parent peak into two in its appearance as a doublet at 152.3 ppm

Chapter 2 ♦Results and Discussion - 97 - with a JCF of 311 Hz. Similarly, the ipso carbon attached to diazepine ring appeared
as a doublet at 140.7 ppm with 2J as 80 Hz.
Table 2.31. 13C-NMR analysis (100 MHz, CDCl3) of peptidyl benzodiazepine 124.
δ ppm Assignment δ ppm Assignment δ ppm Assignment
18.6 CH3 36.0 CH2 111.6-141.1 aromatic
18.7 CH3 37.1 CH 151.8, 152.7 Aromatic C-F
22.9 CH3 (acetyl) 42.6 CH (chiral) 165.1 C=N
28.6 CH (isopropyl) 54.2 CH (chiral) 172.8, 184.4 C=O
The present work led to establishment of a new strategy for construction of C-
terminal derivatized peptidyl heterocycles using novel bis-electrophiles (peptidyl
chalcones) established after successful C-acylation of phosphorane support. The
results are being compiled in the form of a publication.218 All the synthesized peptidyl
chalcones and derived heterocycles have been submitted for bioevaluation against
different targets.

3
Computational Studies

Computational Studies It is generally recognized that drug discovery and development are very time and
resource consuming processes. There is an ever growing effort to apply computational
power to the combined chemical and biological space in order to streamline drug
discovery, design, development and optimization.
Computational chemistry is a branch of chemistry that uses computers to assist in
solving chemical problems. In biomedical arena, computer-aided or in silico design is
being utilized to expedite and facilitate hit identification, hit-to-lead selection,
optimize the absorption, distribution, metabolism, excretion and toxicity (ADMET)
profile and avoid safety issues.
Pharmaceutical industries are actively involved in the development of computational
tools that will improve effectiveness and efficiency of drug discovery and
development process, decreased use of animals, and increased predictability. With the
advent of fast computers and sophisticated software, computer aided drug design
(CADD)239 is becoming a routine tool in the drug designing and development area and
it is expected that the power of CADD will grow as the technology continues to
evolve. The most commonly used computational approaches in drug designing and
development (DDD) are the following,
o Ligand-based drug design (LBD)
o Structure-based drug design (SBD)
o Enzyme-ligand binding affinity and scoring function (Molecular docking)
o Structure activity relationships (SAR).
This work exploits the use of several computational tools to relate the activities of
chalcones synthesized in the experimental part to their activities. The molecular
docking tool was used for studying the mechanistic interactions of chalcones and
PGM with a view to finding a rationale for increased antidiabetic activity of some
chalcones over the others. In silico design and synthesis of cytotoxic chalcones was
also carried out using comparative molecular field analysis (CoMFA) tool. Moreover,
the QSAR studies on a set of antitumor chalcones revealed useful indicators for
designing new antitumor chalcones.

Chapter 3 ♦Computational Studies - 100 - 3.1 Molecular Docking
Computational chemistry uses the results of theoretical chemistry incorporated into
efficient computer programs to calculate the structure and properties of different
molecules. These programs and docking software are referred to as docking programs
and a large number of different types of software e.g., AutoDock, Dock, FlexX, Gold,
Sybyl and Molecular Operating Environment (MOE) have been developed during the
past few decades for conducting molecular docking studies.
Docking is a method which predicts the preferred orientation between two molecules
when bound to each other to form a stable complex.239 These two macromolecules
may be proteins, nucleic acids, carbohydrates and lipids which play a central role in
signal transduction. The correct relative orientation is studied between a
macromolecule such as a receptor or an enzyme and a small molecule usually called a
ligand.240 The ligand may be of organic or inorganic nature. The first widely used
docking program was Kuntz’s DOCK241 which described the binding site by
intersecting spheres.
The three components of a docking protocol are the representation of the system, the
conformational space search, and the ranking of potential solutions. Solving the
docking problem involves an efficient search procedure and a good scoring function
followed by combining the best matching algorithms and scoring schemes. Docking is
done either by using a matching technique that describes the protein and the ligand as
complementary surface242 or by simulating the actual docking process in which the
ligand-protein pairwise interaction energies are calculated.243 Both these methods
have their advantages as well as some limitations. Molecular docking has a large
number of applications in studying the biological phenomena. For example, binding
interactions between a ligand and an enzyme protein may result in activation or
inhibition of an enzyme, or if the protein is a receptor, ligand binding may result in
agonism or antagonism. Docking is most commonly used in the field of drug design,
lead optimization and bioremediation.244 The present work describes the use of MOE
software for docking of active chalcones into the active site of enzyme e.g.
phosphoglyceromutase (PGM), to provide a rationale for the greater PGM inhibitory
activity of certain chalcones over the others. Moreover, some potent cytotoxic

Chapter 3 ♦Computational Studies - 101 - chalcones were docked into the tubulin protein for predicting their affinity as tubulin
binders.
As discussed earlier in chapter 2, amino chalcones 1-20 were screened against a set of
fifteen different phosphatases namely; Mtb Inosit Pptase, Mtb PGM, Mtb aroA, Mtb
aroK, Mtb ptpA, hum ptp1B, hum ptpN3, hum ptpN5, hum ptpN7, hum ptpRJ, hum
ptpRK6, hum ptpROG7, hum ptpRR, hum ptpRS and ptp ShP2.
The tested chalcones did not show any significant activity as phosphatase inhibitors
against these enzymes except Mtb-PGM. The results of these inhibitory activities
have already been discussed in Chapter 2. However, the same data has been
reproduced in Table 3.1 for a ready reference.
Table 3.1. % inhibiton of 4′-amino chalcones against Mtb PGM.
O
A
BH2N R
No. R % Inh. No. R % Inh. No. R % Inh.
1 H 44 8 3,4-OCH3 60 15 4-F -
2 2-OH 17 9 2-NO2 45 16 2-Br 24
3 3-OH - 10 3-NO2 34 17 3-Br 17
4 4-OH 96 11 4-NO2 99 18 4-CH3 48
5 2-OCH3 15 12 2-Cl 47 19 3-OH,4-OCH3 60
6 3-OCH3 43 13 3-Cl - 20 4-N(CH3) 2 41
7 4-OCH3 44 14 4-Cl 41
It may be seen that the screened chalcones exhibited a varying degree of PGM
inhibition potency ranging from 15-99 %. Chalcones numbered 4 and 11 (4-OH and
4-NO2 groups) were found as the lead structures of the library showing 96 and 99 %
inhibition respectively.
Molecular docking studies on amino chalcones were carried out with PGM to
understand the ligand-enzyme interactions and the relative stability of the ligand-
enzyme complex. MOE (ver. 2008.10 GROMACS) and MOPAC (ver.7) were used to
conduct these studies on a Pentium IV Workstation.245
As a first step, the crystal structure of protein complex of B type PGM was obtained
from the Protein Data Bank (PDB code 1yfk) containing citrate ion (CIT) as a
ligand.246 The X-ray crystallographic structure of 1yfk was used for docking

Chapter 3 ♦Computational Studies - 102 - calculations and the enzyme was prepared for docking studies by carrying out the
following steps,
(i) The ligand molecule was removed from the enzyme active site.
(ii) The crystal structure was edited by removing water molecules and
afterward imported into MOE. All possible hydrogen atoms were added to
the structure with their standard geometry.
(iii) MOE Site Finder was used for the active site search in the enzyme
structure and dummy atoms were created from the obtained alpha spheres.
(iv) The backbone and the residues were fixed and an energy minimization was
performed using MOE.
The produced model was then subjected to systematic conformational search where
all items were set as default with root mean square (RMS) gradient of 0.01 kcal/mol
using the Site Finder tool of the program and the enzyme was searched for its active
site. A conformational search was carried out for each ligand and ten docking
conformations were selected for each ligand from which the best conformation of
each of the enzyme-ligand complex was selected based on energetic grounds. Flexible
docking was performed for finding the receptor and ligand molecules as they appear
in the complex and further energy minimization was used to refine the orientation of
the substrate in the binding site of PGM. The resulting enzyme-ligand complex model
was then used for calculating the energy parameters using MMFF94x force field
energy calculation and the ligand-enzyme interactions at the active site were
predicted. The docked conformations were ranked according to their interaction
affinity.
An inspection of the active site of PGM reveals that it contains Asn17, Phe22, Gly24,
Ser23, Glu89, Arg90, Tyr92, Lys100, Arg116, Arg117, His186, and Asn188,
suggesting that these residues could be involved in the substrate binding or chemical
reaction. The active pocket of PGM is rich in basic residues so that the enzyme can be
inhibited by a wide range of anionic compounds in the millimolar to low micromolar
concentration range. The citrate ligand does not bind to the protein tightly as does the
real substrate because its structure is not as favorite as the substrate to the enzyme.
However, its binding mode indicates the general position and orientation of the
substrate.246

Chapter 3 ♦Computational Studies - 103 - The placement of ligand within active site of PGM in the X-ray crystal structure was
confirmed by the redocking method. The root mean square deviation (RMSD) were
used as a measure of how close the predicted structure of citrate bound to PGM was in
accordance with the citrate found originally in the crystal structure. As expected, the
position and orientation of the placed ligand and the re-docked ligand was well
overlapped (RMSD value computed by SVL program was 1 Å). Fig 3.1 shows a 2D
and 3D view of the citrate ion (ligand) in the active site of PGM. It shows strong
interactions with Asn17, Phe22, Ser23, Gly24, Glu89, Tyr92, Lys100 and Arg116
present within 5 Å of the ligand surface.
It may be seen that amino acids Ser23, Tyr92 and Lys100 show weak hydrogen
bonding but a number of hydrophobic interactions between citrate and amino acid
residues were found as shown in Fig. 3.1.
a. b.
Fig. 3.1 a) Citrate ion docked in the active site of PGM showing interactions with different
amino acids as shown in MOE diagrams a) 2D view b) 3D view.
Having established the interactions of the citrate ion with different amino acids in
PGM, molecular modeling of chalcones was carried out following the same sequence
of steps. A comparison of the molecular docking of citrate and chalcones in PGM
indicates that the same amino acid residues e.g. Asn17, Phe22, Ser23, Gly24, Glu89,
Tyr92, Lys100 and Arg116 were found interacting with chalcones in the active site of
PGM. Table 3.2 shows the hydrophobic interactions of chalcones with different
amino acids in the active site of PGM. Table 3.3 shows the hydrogen bonding as well
as π-π interactions; Arg10 and Arg116, being the key contributors show strong
hydrogen bonding in the most active chalcones e.g. 4 and 11.

Chapter 3 ♦Computational Studies - 104 - Table 3.2. Hydrophobic interactions of amino chalcones in the active site of PGM within
5Å.
Entry Asn 17 Phe 22 Ser 23 Gly 24 Arg 62 Glu 89 Tyr 92 Lys 100
1 + + - + + + + -
2 - + + + + - + +
3 + - + + + - - -
4 + - + + - - + -
5 - + - + + + + -
6 + + + + + - + +
7 + + + + + + + +
8 + + + + + + +
9 - + - + + - + +
10 + + + + + - + +
11 - + - + + - + +
12 + + - + - - + +
13 - + - + + + + +
14 + + + + + - + -
15 + + + + - + + +
16 + - - + - - - +
17 + + - + - - + +
18 + + - + + - - +
19 - + + + + + + +
20 + + - + + + + -
CIT + + + + - + + +
Since hydrogen bonds make important contributions to the interactions between a
ligand and an enzyme, therefore the frequency of residue's occurrence in the
formation of hydrogen bonding reflects the stability of the ligand-enzyme complex. It
may be observed that Arg10 and Arg116 play an important role in stabilizing
chalcones-PGM complex by acting both as hydrogen bond donor and a hydrogen
bond acceptor. The most active chalcones 4 and 11 were found to have strongest H-
bonding interactions with these two amino acids. Moreover, Ser14, Asn 17, Arg21,
Ser23, Lys100, Arg116 and Arg191 also take part in the hydrogen bonding with
relatively high frequency.
Besides hydrogen bonding, weak π-π interactions with Arg10,Asn17, Lys100, Arg116
and Arg191 also help in the stabilization of chalcone-PGM complex.

Chapter 3 ♦Computational Studies - 105 - Table 3.3. Binding interactions of chalcones (1-20) in the active site of PGM.
H bonding Entry % inhibiton Distance Å Score % Amino acids
π-π interactions
1 44 3.05, 1.56 12, 39 Arg10, Asn17 Asn17,Arg10,Arg191
2 17 2.12, 3.02 18, 36 Ser23, Lys100 Arg116
3 - - - - -
4 96 2.44, 1.75, 1.45 73, 21, 73 Arg10, Asn17, Arg116 Arg10, Arg116
5 15 1.45, 2.64 27, 24 Arg21, Ser23 Lys100, Arg191,
6 43 3.05, 3.14, 2.98 23, 15, 16 Ser23, Arg10 Arg191
7 44 2.44, 2.93 44, 29 Lys100, Arg191 Arg10
8 60 2.51, 2.68 60, 76 Lys100, Arg116 Arg10
9 45 2.66, 2.44,2.02 16, 15, 35 Arg10, Ser14, Ser23 Arg10, Arg191
10 34 1.37, 2.52 20, 23 Arg10, Ser23 Arg191
11 99 1.36, 2.26 80, 21 Arg10, Arg116 Arg10
12 47 2.75, 2.46, 2.05 16, 16, 31 Arg10, Ser14, Ser23 Lys100
13 - - - - Lys100
14 41 2.77, 1.53 19, 42 Arg10, Asn17 Arg191
15 - - - - Lys100
16 24 1.83 45 Ser23 Arg10, Arg191
17 17 3.01, 1.56 12, 37 Arg10, Asn17 Arg191
18 48 1.59, 2.71 28, 18 Arg21, Ser23 Lys 100, Arg 191
19 60 2.5, 1.74, 2.83 28,62, 43 Lys100,Arg116,Arg191 Arg10
20 41 2.75, 2.55, 1.47 24, 15, 17 Arg10, Arg191 Arg10, Lys100
CIT - 2.57, 2.86, 2.94 18, 14, 26 Ser23, Tyr92, Lys100 -
Fig. 3.2 and 3.3 show the 2D and 3D views of chalcones 4 and 11 respectively,
showing hydrogen bonding and π-π interactions of the amino acid residues in the
active site of PGM. It may be noted that these interactions were also found in CIT-
PGM complex.
Another feature of these docking studies is that the hydrogen bond interactions found
in the active chalcones were not observed in inactive chalcones e.g. 3, 13, and 15,
however weak π-π interactions were observed in case of 13 and 15 (Fig. 3.4). It may,
therefore, be inferred that the greater activity of compounds 4 and 11 may be
associated to the greater interactions observed in enzyme-ligand complex.

Chapter 3 ♦Computational Studies - 106 -
a. b.
Fig. 3.2. Hydrophilic and hydrophobic interactions of chalcone 4 with amino acids in the
active-site of PGM a) 2D b) 3D.
a. b.
Fig. 3.3. a) 2D b) 3D views of the most active chalcone 11 bound to the active site of PGM.
So far the stability of ligand-enzyme complex was discussed in terms of scoring of
interactions between the two molecules. The strength of the enzyme-ligand complex
can also be evaluated on the basis of binding free energy of the complex formed. The
free energy calculations of the complexes formed as a result of docking of potential
ligands in the PGM active site may complement the binding capacity evaluated on the
basis of scoring functions. Therefore, the binding free energies were calculated using
the MMFF94X free energy calculation method.247

Chapter 3 ♦Computational Studies - 107 -
Fig. 3.4. Ligplot of compound 13, an inactive chalcone
The binding free energy of the inhibitor, Gbind, is obtained by taking the difference
between the free energy of the receptor-ligand complex (Gcomp) and the free energy of
the unbound receptor (Grec) and ligand (Glig) according to the following equation:
∆Gbind = Gcomp- (Grec + Glig)
The binding free energy data of different chalcones and PGM obtained through
docking studies showed direct correlation between docked binding energies and PGM
inhibitory potencies of chalcones as shown in Table 3.4.
Chalcones 4 and 11 are found to have the minimum binding energy (-4.24 and -4.25
Kcal mol-1) resulting in the formation of most stable complex with PGM, while the
least active chalcones e.g. 3, 13 and 15 showed the maximum value (-4.12, -4.10 and -
4.19 respectively). MOE determines a large number of different parameters to study
any correlation of the activity of the compounds with their structures. Besides binding
free energy, electronic energy (Eelec) and Van der Waals energy (Evdw) also help in
predicting the stability of ligand-enzyme complex. These descriptors can also be
calculated using MOE. In the current study, an attempt to find a correlation either
with Eelec or Evdw (Table 3.4) was not successful. These observations are in line with
the fact that the most active chalcones when bound to the active site of PGM through
strong hydrophilic and hydrophobic interactions lead to a greater stability of the
ligand-enzyme complex as reflected in their greater binding free energies. The ligand-
enzyme complex formed by the interaction of the most active chalcone 11 and PGM
given as surface representation shows how well the ligand fits in the active site of
enzyme (Fig. 3.5).

Chapter 3 ♦Computational Studies - 108 - Table 3.4. Binding energies of chalcones (1-20) calculated through molecular docking.
Entry Number of runs Lowest binding energy (kcal/mol) Eele Evdw
1 7 -4.21 -2.18 35.11
2 8 -4.20 -2.47 38.66
3 9 -4.12 -7.31 36.01
4 7 -4.24 -4.93 38.01 5 10 -4.20 -0.97 46.03
6 5 -4.21 -6.46 38.23
7 9 -4.21 -3.34 40.46
8 9 -4.22 -6.49 44.53
9 7 -4.21 -4.21 45.65
10 8 -4.21 1.20 44.83
11 7 -4.25 7.16 41.51 12 10 -4.21 2.08 38.09
13 8 -4.10 -2.55 35.26
14 8 -4.21 -1.22 35.25
15 8 -4.19 -2.91 34.38
16 9 -4.20 1.25 37.84
17 7 -4.20 -2.32 37.73
18 7 -4.21 -2.97 37.45
19 10 -4.22 -8.01 38.71
20 6 -4.21 -9.91 47.99
The binding energy calculations also reflect that compounds 4 and 11 show strong
enzyme-ligand interactions as well as lowest binding free energy rendering them the
most potential PGM inhibitors. It is, therefore, logical to conclude that these
chalcones have strong potential as PGM inhibitors and hence can be developed into
novel antidiabetic agents.
Fig. 3.5. Surface representation of chalcone 11 and PGM complex.

Chapter 3 ♦Computational Studies - 109 - 3.2. Structure activity relationship (SAR)
Computational analysis including 3D-QSAR studies were performed on different
series of active compounds. The chalcones were picked from synthesized libraries and
evaluated for cytotoxic and antitumor properties. Most of the synthesized compounds
were biologically evaluated soon after their purification, and the data were collected.
An understanding of the structural requirements for a particular activity of a
pharmacophore is important in guiding and optimizing the effort of drug design. The
computational studies, therefore, went in parallel to the synthetic assignments. 3D-
QSAR analysis of two series of chalcones was carried out using CoMFA tool and
SAR paradigms were defined as figures indicating favorable electrostatic and steric
contributions towards activity of chalcone template. QSAR studies and some docking
trials of potent chalcone derivatives into binding sites of target proteins revealed
useful indicators for designing new drug candidates of the targets understudy.
3.2.1. In silico guided design and synthesis of cytotoxic chalcones
Rational drug designing also known as in silico drug designing is the process
involving design of small molecules that are complementary in shape and charge to
the biomolecular target to which they interact and bind for a biological response.248
Since computer modeling techniques such as QSAR and molecular docking are used
for in silico drug designing, the process, is also referred to as computer aided drug
designing (CADD).249
Three dimensional quantitative structure-activity relationships (3D-QSAR) enable the
medicinal chemists to perform in silico design of drug molecules based on the ligand
structure. It helps in establishing reliable quantitative structure-activity and structure-
property relationships to derive in silico QSAR models for predicting the activity of
novel compounds prior to their synthesis. This methodology has been successfully
used to generate models for various chemotherapeutic agents.250-251
CoMFA
Comparative molecular field analysis (CoMFA) is one of the most widely used 3D-
QSAR methods that is used for predicting structure activity/property relationship.252 It
is based on the calculated energies of steric and electrostatic interactions between the
compounds and the probe atoms placed at the various intersections of a regular 3D
lattice. Thus, a structure activity relationship is developed by using partial least

Chapter 3 ♦Computational Studies - 110 - squares (PLS) analysis which is a powerful statistical tool. The results are obtained in
the form of steric and electrostatic maps which guide the potential regions of the
molecule in 3D space that should be modified in order to optimize biological activity.
The advantage of CoMFA is its ability to predict the biological activity of molecules
and represent the relationship between steric and electrostatic properties and
biological activity in the form of contour maps. This computational technique has
been used in a wide range of drug designing assignments for different biological
targets.253-255
In order to study and infer a correlation between structure and biological activity,
QSAR studies were carried out using CoMFA based on the results of cytotoxic
behavior of chalcones in the BSL bioassay (Section 2.1.2.3., page 43). The chalcones
with their BSL activities were taken from the synthesized series of amino chalcones
and the individuals synthesized during deconvolution of 120 membered chalcone
library. All the compounds were tested for their BSL activity and their LD50’s were
used to carry out CoMFA analysis. The set of compounds was divided into the
training set and the test set. This model, together with the contour maps derived,
revealed the relevance of the steric and electrostatic fields. Structural variations in the
molecular fields were studied according to the generated 3D-QSAR information to
design some new more potent chalcone analogues. The activities of these newly
designed molecules were predicted using the built CoMFA model based on the
training set and validated on the basis of test set compounds. Furthermore, the
designed analogues were docked into the tubulin protein for finding their potential as
novel antitumors.
Devising a QSAR through CoMFA analysis can be divided into three stages namely;
the data preparation, data analysis, and model validation. The overall process involves
the following steps performed in a molecular modeling Software namely SYBYL.256
• Building 3D structures
• Geometry optimization and calculation of partial atomic charges
• Selection of template molecule and fitting centres for alignment
• Placing the template on a 3D grid
• Alignment of compounds on template molecule and calculation of steric
and electrostatic fields using a probe atom

Chapter 3 ♦Computational Studies - 111 -
• PLS linear regression analysis with q2 > 0.4 and r2 ~ 0.9
• Choosing the training set compounds giving highest q2 with optimum
number of components, leaving others as the test set
• Correlation of activities with magnitudes of electrostatic and steric fields
of training set compounds
• Prediction of activities of test set compounds on the basis of electrostatic
and steric fields given by training set compounds.
Data set for analysis
The data set comprised a total of 35 chalcones, whose cytotoxic potential was
evaluated on the basis of Brine Shrimp Lethality (BSL) assay. The BSL assay is
reported as a quite reliable prescreen for leukemia mouse bioassay.181-182 In vitro
activity data (LD50 µM) of a diverse set of chalcone analogues was used for
developing and validation of CoMFA model. The structures and activities of the
understudy set of chalcones are given in Table 3.5.
Table. 3.5. Brine shrimp cytotoxicity of chalcones.
S.No.* Ring A Ring B LD50 S.No.* Ring A Ring B LD50
121 Ph Ph 0.14 195 4-NH2Ph 2-OCH3Ph 2.72
222 Ph 2-OHPh 84.73 206 4-NH2Ph 3-OCH3Ph 3.79
325 Ph 2-OCH3Ph 0.29 217 4-NH2Ph 4-OCH3Ph 0.24
426 Ph 3-OCH3Ph 2.6 229 4-NH2Ph 2-NO2Ph 160.6
527 Ph 4-OCH3Ph 1.05 2310 4-NH2Ph 3-NO2Ph 557
629 Ph 2-NO2Ph 261.58 2411 4-NH2Ph 4-NO2Ph 130.56
730 Ph 3-NO2Ph 214.23 2515 4-NH2Ph 4-FPh 614.4
831 Ph 4-NO2Ph 364.03 2612 4-NH2Ph 2-ClPh 16.2
932 Ph 2-ClPh 167.85 2713 4-NH2Ph 3-ClPh 52.3
1033 Ph 3-ClPh 0.248 2814 4-NH2 Ph 4-ClPh 14.6
1134 Ph 4-ClPh 4.25 2916 4-NH2Ph 2-BrPh 8.3
1236 Ph 2-BrPh 1.46 3018 4-NH2Ph 4-CH3Ph 150.1
1337 Ph 3-BrPh 0.21 3141 2-OHPh 2-OHPh 0.083
1438 Ph 4-CH3Ph 1.05 322 4-NH2Ph 2-OHPh 46.5
1528 Ph 3,4-(OCH3)2Ph 834.63 3342 2,3,4-(OCH3)3Ph 2-OHPh 0.79
161 4-NH2Ph Ph 6.68 3443 3,4,5-(OCH3)3Ph 2-OHPh 0.34
173 4-NH2 Ph 2-OHPh 60.5 3544 2,4-(Br)2Ph 2-OHPh 54.61
184 4-NH2 Ph 3-OHPh 7.1
*Compound numbers as appear in experimental section.

Chapter 3 ♦Computational Studies - 112 - The data set contained a substituent variety both on A and B rings. The BSL
cytotoxicity data of these chalcones was collected in the form of LD50 values and the
potencies were found within a range of < 1 to ~ 550 µM. The data was randomly
segregated into training and test sets comprising 25 and 10 chalcones respectively.
Molecular conformation and alignment
All molecular modeling studies were performed on a Silicon graphics Workstation
using the molecular modeling package SYBYL 7.0.256 The structures of all 35
compounds were generated by Guassian View and minimized with the Austin Model
1 (AMI) parameterization. Tripos force field, conjugated gradient method,
subsequently Gasteiger-Hückel257 partial charges were calculated and the geometry of
the molecules was optimized. The minimum gradient difference of 0.001 kcal/mol Å
was set as a convergence criterion. The use of a reasonably low energy conformation
in the alignment is a useful starting point for statistical comparison of flexible
structures within the CoMFA model. The lowest energy conformation found for each
molecule in the data set was led to an optimization by means of the HF/3-21g*258
level using Gaussian 94.259 Since the specific molecular target of these compounds in
brine shrimps is unknown, the docking-based alignment method for developing a
QSAR model could not be used. Instead, an active molecule in the training set,
compound 5 (Table 3.5), was chosen as the template and the data set has to be
superposed on the template conformation with the application of the database
alignment method. Structural alignment is perhaps the most subjective, yet critical,
step in CoMFA study, in as much as the resulting 3D QSAR model is often sensitive
to the particular alignment scheme. Each molecule from the training set was
superimposed to the template by minimizing the rms distances between each pair of
corresponding atoms of the template and the compound to be aligned. The common
fitting centers selected for aligning all chalcones are shown by asterisk (Fig. 3.6).
O
OCH3*
**
*
Fig. 3.6. Fitting centers used for overlapping chalcones.

Chapter 3 ♦Computational Studies - 113 - CoMFA interaction energies
The standard Tripos settings were used to carry out the CoMFA analysis. To derive
the CoMFA fields, a 3D cubic lattice was created; the steric and electrostatic
parameters were calculated at each lattice intersection of regularly spaced grid of 2.0
Å in all three dimensions within the defined region. The aligned molecules were
placed in a 3D grid box such that the entire set was included in it. The Van der Waals
potential and the Coulombic term representing the steric and electrostatic fields were
calculated using standard Tripos force fields. An sp3 carbon atom with a positive
charge (+1) was used as a probe atom to generate steric (Lennarde Jones potential)
field energies and electrostatic (Coulombic potential) field energies. A distance
dependent dielectric constant of 1.00 was used. The steric and electrostatic fields were
truncated to +30.00 kcal/mol. The CoMFA fields generated automatically were scaled
by the CoMFA-STD method in SYBYL.
PLS analysis
The CoMFA descriptors were used as independent variables and inverse logarithm of
LD50 values (pLD50) as a dependent variable in PLS regression analysis260 in deducing
the 3D-QSAR model. Normally, cross-validation is employed to check the
predictivity of the derived model. The performance of models was calculated using
the leave-one-out (LOO) cross-validation method. The optimum number of
components used to derive the non-cross-validated model was defined as the number
of components leading to the highest r2 cross-validated and lowest standard error of
prediction (SEP). To speed up the analysis and reduce noise, a minimum filter values
of 2.00 kcal/mol was used. Only those steric and electrostatic energies with values
greater than 2.0 kcal/mol were considered in the PLS linear regression analysis and
the predictive quality of the “best” correlation model was determined. PLS analysis
was used to correlate the biological activity index with CoMFA values that contained
the magnitude of either the steric or electronic potential.261 CoMFA model was
obtained with 25 chalcones in the training set and 10 chalcones in the test set
respectively. The actual, predicted and residual of all the training and test set
compounds are shown in Tables 3.6 and 3.7.

Chapter 3 ♦Computational Studies - 114 - Table 3.6. Observed and predicted pLD50 of the training set compounds.
Chalcones Observed Predicted Residual Chalcones Observed Predicted Residual
1 6.85 7.10 -0.25 18 5.15 5.00 0.15
3 6.54 6.63 -0.09 20 5.42 5.51 -0.09
4 5.59 5.66 -0.07 21 6.62 6.70 -0.08
5 5.98 6.03 -0.05 24 3.88 4.08 -0.20
6 3.58 3.59 -0.01 27 4.28 4.53 -0.25
7 3.67 4.12 -0.45 28 4.84 4.93 -0.09
8 3.44 3.23 0.20 29 5.08 4.59 0.49
10 6.61 6.15 0.46 31 7.08 6.87 0.21
12 5.84 6.14 -0.30 32 4.33 4.39 -0.06
13 6.68 6.26 0.42 33 6.10 6.11 -0.01
14 5.73 5.81 -0.08 34 6.47 6.44 0.03
16 5.18 5.45 -0.28 35 4.26 4.47 -0.21
17 4.22 3.90 0.32
Table 3.7. Observed and predicted pLD50’s of the test set compounds.
Chalcones Observed Predicted Residual Chalcones Observed Predicted Residual
2 4.07 4.22 -0.14 22 3.79 3.43 0.36
9 3.78 4.32 -0.54 23 3.25 2.94 0.31
11 5.37 5.22 0.15 26 4.79 4.51 0.28
15 3.08 2.86 0.22 25 3.21 3.73 -0.52
19 5.57 5.64 -0.08 30 3.82 4.16 -0.34
Cross-validation runs with varying number of groups were also performed to improve
the confidence limits of the derived model. In the best model, when performed with
the optimal number of 5 components and no validation PLS analysis, q2 and r2 values
were obtained as 0.626 and 0.950 respectively, whereas F value was 100.754 and the
Standard Error Estimate (SEE) was found to be 0.335.
The plots of observed versus predicted cytotoxicity of training and test set chalcones
are presented as Figs. 3.7 and 3.8. The CoMFA electrostatic and steric fields based on
PLS analysis are presented as 3D contour plots in Fig. 3.9. The contribution of steric
and electrostatic field is 31.1% and 68.9% respectively.

Chapter 3 ♦Computational Studies - 115 -
R² = 0.95
0
2
4
6
8
0 2 4 6 8
p LC
50(c
alcu
late
d)
pLC50 (experimental)
training set compounds
Fig. 3.7. Calculated vs observed activity of the training set compounds.
R² = 0.826
0
2
4
6
8
0 2 4 6 8
log
LD50
(cal
cula
ted)
log LD50 (experimental)
test set compoundsLinear (test set compounds)
Fig. 3.8. Calculated vs observed activity of the test set compounds.
Fig. 3.9. Stereoview of the CoMFA map for the electronic and steric contributions.

Chapter 3 ♦Computational Studies - 116 - As is evident in Figs. 3.7 and 3.8, there is no outlier in the CoMFA model and the
largest residual found in chalcone 10 is ±0.46 falls within 1 log unit. In CoMFA
method, results are presented as contour maps that correlate the change in biological
activity with the molecular field values. The steric contour maps are represented in
green and yellow colors while the electrostatic contours are depicted in red and blue
colors. Red color indicates areas where more negative density favors activity and blue
color indicates areas where more positive density promotes the activity. Green colour
indicates areas where bulky substituents favor activity and yellow colour indicates
areas where less bulky substituents promote activity. The contour maps are limited to
the regions where various substituents have been modified and have an impact upon
activity of resulting analogues.
Based on the 3D-QSAR analysis, predictions were made for favorable electronic and
steric regions which are expected to enhance activity in the resulting chalcone
derivatives. These results are shown in Fig. 3.10, which give an appropriate
description of the favorable electrostatic and steric substitution pattern.
Negative charge
3-OCH3 & 3-OH
(20) (17) density LD50 3.8, 60.5
Fig. 3.10. Favorable electronic and steric regions for enhanced potency in cytotoxic
chalcones.
It may be generalized that chalcones bearing donors at 3 and 4 positions are expected
to result in increased cytotoxic potential. Furthermore, bulk at 4′ position seems
favorable for activity whereas bulk at 3 and 4 position may result in steric conflict
4-OCH3 & 4-CH3
(21) (30) LD50 0.24 150
Bulk 4-OCH3 (21) & 4-NO2 (24)
A
O
B
Less bulk 4-OCH3 & 3,4-(OCH3)2 (5) (15) LD50 1.05 834.6
Negative charge Density
LD50 0.24, 130.56

Chapter 3 ♦Computational Studies - 117 - leading to decreased potential of the chalcone derivatives. The figure thus presented
provides an insight for designing more chalcone derivatives which may result in
und 1 was expected to be the most potent, whereas compound 4
as the least poten
Table 3.8. Predicted pLC50 of the designed chalcones.
enhanced activity.
On the basis of favorable steric and electrostatic regions obtained from CoMFA
analysis, some new compounds were designed with an expectation of improved
cytotoxic potential. Each of the synthesized compounds retained one or more
electrostatic or steric requirement, predicted as essential for activity in CoMFA
studies. A large red isopleth at 4 position of ring B indicates that electron density at
this position strongly favors the cytotoxic activity. Strong electron donor e.g. -
N(CH3)2 was, therefore, introduced in the designed chalcones. Furthermore, green and
yellow isopleths at position 4 indicated that both bulky and small functional groups
are favorable for activity. Therefore, fluoro group being small in size, having low
polarizability and hence donates electron density to phenyl ring, making position 4 of
ring B as electron rich. This derivatization favors both electrostatic and steric
requirement and is expected to enhance activity. The 3- or 4- methoxy substituted
chalcones resulted in good activities, whereas activity dropped drastically when the
3,4-dimethoxy substituted ring B was present. The position of yellow isopleth
indicated that bulk both at 3 and 4 positions is sterically unfavorable. The one of two
methoxy substituent at 3 and 4 positions of ring B (chalcone 15) was replaced by a
less bulky OH group. Furthermore, an electron donor e.g. -Br was added at 3 position
of ring B, which was expected to result in fair activity. As a result, a total of five
compounds were designed and their pLC50 values were predicted from the derived
QSAR equation based on CoMFA studies (Table 3.8). On the basis of calculated
pLD50 values, compo
t.
S. No. R′ R D50 ca
1 H 4-F 7.20
2 H 3 3
4-N )2
5 4-NH2 4-N(CH3)2 5.46
-OH,4-OCH 6.31
3 H (CH3 5.95
4 4-NH2 3-Br 4.64
pL lc.

Chapter 3 ♦Computational Studies - 118 - Molecular docking
Microtubules (MTs) are cytoskeletal protein polymers and constitute an important
part of the cellular scaffold, providing a network of tracks for intracellular transport
and for separating chromosomes during mitosis.262 Stabilization of microtubule
dynamics is a common mechanism of antimitotic agents resulting in apoptosis of
cancer cells.263 Microtubules and tubulin dimers are dynamic targets for cancer
chemotherapy. Tubulin crystal structures are indispensable to determine the
mechanism of action of different antitumors which are known to target tubulin.
Tubulin to tubulin-binder interactions obtained through X-ray crystal structure data
provide a valuable tool for in silico studies of tubulin-chalcones interactions thus
providing a new arsenal for structure-based drug design.264 In view of the significance
of tubulin as a target in anticancer studies, it was selected for conducting molecular
docking studies on the cytotoxic chalcones, identified as a result of BSL studies.
Chalcones, as already stated are known to bind to tubulin dimer halting mitosis,
resulting in their antimitotic action and subsequently apoptosis in cancer cells.191
For tracing binding interactions and conformations of chalcone as tubulin binders, the
docking studies of all chalcones were performed using MOE software. The crystal
structure of tubulin protein complex was obtained from the Protein data bank (PDB
id: 1z5v) complexed with molecular ligand GSP (Guanosinediphosphatemonothio
phosphate) (Fig. 3.11).264 The X-ray crystallographic structure of complex was used
for the docking calculations after ligand removal and enzyme adjustment. The water
molecules and ligand were removed from the imported protein in MOE and all
possible hydrogen atoms were added to the structure with their standard geometry.
The backbone and the residues were kept fixed and an energy minimization was
performed for all the added hydrogens. The resulting model was subjected to
systematic conformational search where all items were set as default with RMSD
gradient of 0.01 kcal/mol using the Site Finder tool. MOE alpha Site Finder was used
for the active site search in the enzyme structure and dummy atoms were created from
the obtained alpha spheres. The target compounds were built using the builder
interface of MOE program and subjected to energy minimization tool using the
MOPAC 7.0. Chalcone derivatives were docked into the active site and interactions
were studied. Ten docking conformations were selected for each ligand and the best

Chapter 3 ♦Computational Studies - 119 - conformation of each of the ligand-receptor complex was selected based on energetic
grounds. A further energy minimization was used to refine the orientation of substrate
in the binding site of tubulin. The ligand-enzyme complex model obtained henceforth
was then used for calculating the energy parameters using MMFF94x force field
energy calculation and predicting the ligand-enzyme interaction at the active site. The
docked conformations were ranked according to the interaction affinity.
Fig. 3.11. Slab ribbon form of the active site of tubulin protein (PDB id: 1z5v).
Validation of docking reliability
Before using the tubulin crystal structure for docking chalcones, it was necessary to
validate the docking reliability. The known X-ray crystal structure of tubulin obtained
from Protein data bank (PDB id: 1z5v) was found complexed with molecular ligand
GSP and was confirmed by redocking method. The RMSD value was used as a
measure of how close the predicted structure of GSP was bound to receptor. As
expected, the position and orientation of predicted ligand and redocked ligand were
well overlapped (RMSD-tolerance value computed using SVL program was 1 Å).
Therefore, the docking parameters used for GSP were extended for searching the
binding conformation of chalcones to the active site.
Docking results
The designed inhibitors were docked into the binding site of tubulin. The binding free
energies were calculated using the MMFFX94 free energy calculation method.247 The
free energy of inhibitor binding, Gbind, is obtained from the difference between the
free energy of the receptor-ligand complex (Gcpx) and the unbound receptor (Grec) and
ligand (Glig) according to the following equation.
∆Gbind = Gcpx- (Grec + Glig )

Chapter 3 ♦Computational Studies - 120 - The energy scores of the designed chalcones are shown in Table 3.9.
Table 3.9. The energy scores of designed inhibitors.
S. No. Ecomplex Elig Eres G
1 -24.8 -73808 6630 -80
2 -10.6 -72803 6630 -79
3 -12.0 -68374 6630 -75
4 -15.2 -66316 6630 -72
5 -15.9 -67705 6630 -74
The designed chalcone 1, was predicted as the most active from CoMFA model, was
also found as the most potent in docking studies. The descending order of Gibbs free
energy of binding was found to be the same as the descending order of predicted
activities from CoMFA model. In the binding pocket, the compounds 1-5 showed
weak hydrogen bonding and hydrophobic interactions. Chalcone 1 was found to show
50% H-bonding with Ser 140 whereas hydrophobic interactions with Phe 225.
Chalcone 2 showed 36% hydrogen bonding with Asn 207 whereas 3 showed 40% to
the same. Chalcone 4 showed hydrophobic interactions with Phe 225 and 5 showed
15% hydrogen bonding to Asp 180.
Fig. 3.12. Chalcone 1 bound into the binding pocket of tubulin active site.
The designed compounds were already present in the synthesized libraries. These
compounds were evaluated for their cytotoxicity in the BSL bioassay and LD50 values
were recorded (Table 3.10). The observed potencies of compounds are in accordance
with those predicted through QSAR model and docking studies. The observed
potencies of designed chalcones 1-5 decrease in the same order as the predicted
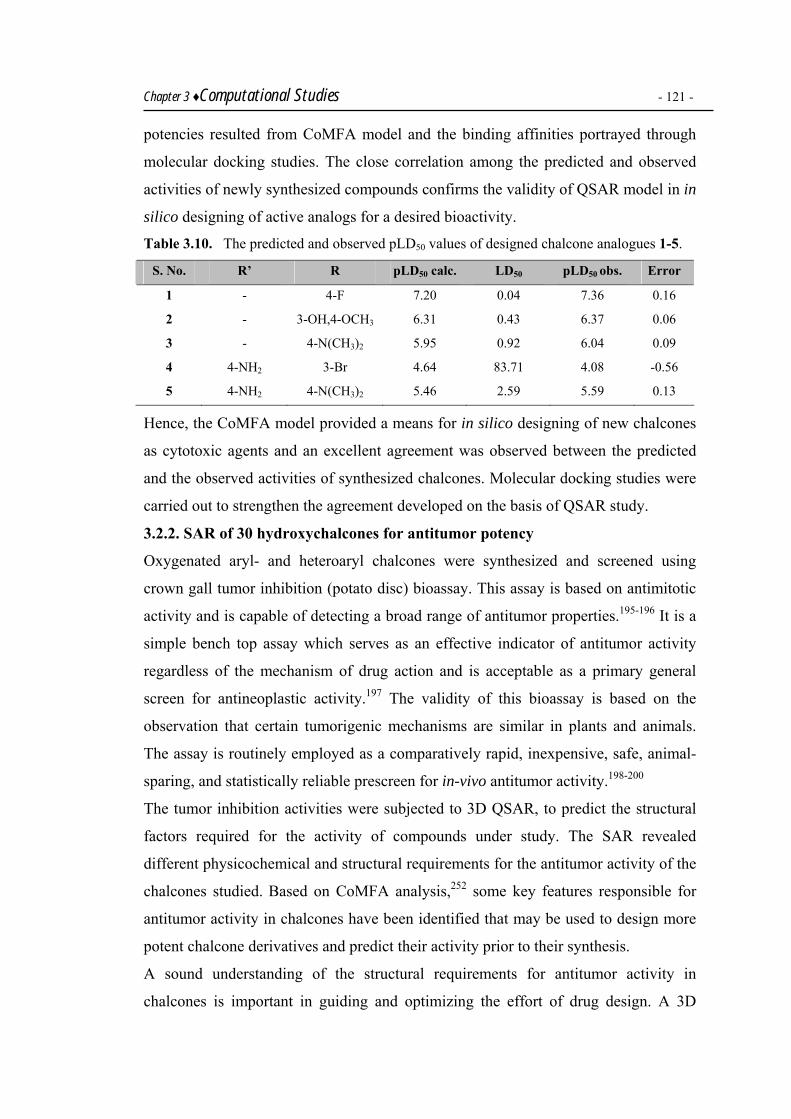
Chapter 3 ♦Computational Studies - 121 - potencies resulted from CoMFA model and the binding affinities portrayed through
molecular docking studies. The close correlation among the predicted and observed
activities of newly synthesized compounds confirms the validity of QSAR model in in
silico designing of active analogs for a desired bioactivity.
Table 3.10. The predicted and observed pLD50 values of designed chalcone analogues 1-5.
S. No. R’ R pLD50 calc. LD50 pLD50 obs. Error
1 - 4-F 7.20 0.04 7.36 0.16
2 - 3-OH,4-OCH3 6.31 0.43 6.37 0.06
3 - 4-N(CH3)2 5.95 0.92 6.04 0.09
4 4-NH2 3-Br 4.64 83.71 4.08 -0.56
5 4-NH2 4-N(CH3)2 5.46 2.59 5.59 0.13
Hence, the CoMFA model provided a means for in silico designing of new chalcones
as cytotoxic agents and an excellent agreement was observed between the predicted
and the observed activities of synthesized chalcones. Molecular docking studies were
carried out to strengthen the agreement developed on the basis of QSAR study.
3.2.2. SAR of 30 hydroxychalcones for antitumor potency
Oxygenated aryl- and heteroaryl chalcones were synthesized and screened using
crown gall tumor inhibition (potato disc) bioassay. This assay is based on antimitotic
activity and is capable of detecting a broad range of antitumor properties.195-196 It is a
simple bench top assay which serves as an effective indicator of antitumor activity
regardless of the mechanism of drug action and is acceptable as a primary general
screen for antineoplastic activity.197 The validity of this bioassay is based on the
observation that certain tumorigenic mechanisms are similar in plants and animals.
The assay is routinely employed as a comparatively rapid, inexpensive, safe, animal-
sparing, and statistically reliable prescreen for in-vivo antitumor activity.198-200
The tumor inhibition activities were subjected to 3D QSAR, to predict the structural
factors required for the activity of compounds under study. The SAR revealed
different physicochemical and structural requirements for the antitumor activity of the
chalcones studied. Based on CoMFA analysis,252 some key features responsible for
antitumor activity in chalcones have been identified that may be used to design more
potent chalcone derivatives and predict their activity prior to their synthesis.
A sound understanding of the structural requirements for antitumor activity in
chalcones is important in guiding and optimizing the effort of drug design. A 3D

Chapter 3 ♦Computational Studies - 122 - QSAR model has been established using CoMFA analysis to illustrate the spatial
orientation of antitumor chalcones and to provide useful indicators for designing new
drug candidates for tumor inhibition.
Data set for analysis
The synthesized set of thirty 3′-hydroxy chalcones contained regioisomeric chalcones
bearing different electron donors and acceptors, both bulky and non bulky substituents
on the ring B as shown in Table 3.11. The activities of synthesized compounds are
given in Table 3.12.
Table 3.11. Structures of synthesized 3′-hydroxy chalcones.
OHO
A
B R
S. No.* R S. No.* R S. No.* R
153 C6H5 1165 4-FC6H4 2175 2-Pyridyl
254 2-OHC6H4 1261 2-ClC6H4 2276 3-Pyridyl
355 3-OHC6H4 1362 3-ClC6H4 2377 4-Pyridyl
456 4-OHC6H4 1463 4-ClC6H4 2474 2-Pyrrolyl
557 2-OCH3C6H4 1564 3-BrC6H4 25** 2-Furyl
658 3-OCH3C6H4 1669 4-MeC6H4 2670 2-Thienyl
759 4-OCH3C6H4 1768 4-N(CH3)2C6H4 2773 5-NO2-2-Thienyl
8** 2-NO2C6H4 1867 3-OH,4-OCH3C6H3 2871 5-Me-2-Thienyl
966 3-NO2C6H4 1960 3,4-OCH3C6H3 2972 5-Br-2-Thienyl
10** 4-NO2C6H4 20** 2-CH3-3,5-OCH3C6H2 30** 3-Indolyl
*Compound numbers as appear in experimental section. **For synthesis, please see reference 149.
The synthesized set of chalcones showed a range of IC50 values varying from 0.004-
132 µM. For the substituted aryl rings at para position, the activities decreased nearly
in the decreasing order of positive inductive effect: CH3 > N(CH3)2 > OH > OCH3 >
F > Cl, with the IC50 values of 0.15, 1.24, 10.96, 28.18, 87.18 and 132.13 respectively.
A chalcone bearing a NO2 group behaved as an exception with an IC50 of 0.93.
Electron withdrawing substituents at the meta position also favored tumor inhibition
resulting in the decreasing order of antitumor activity: NO2 > OH > Cl ~ Br > OCH3.
The chalcones substituted with 3-OH group on ring B was found more active than
expected. Regarding the ortho substitution on ring B, 2-hydroxy substituted 2 was
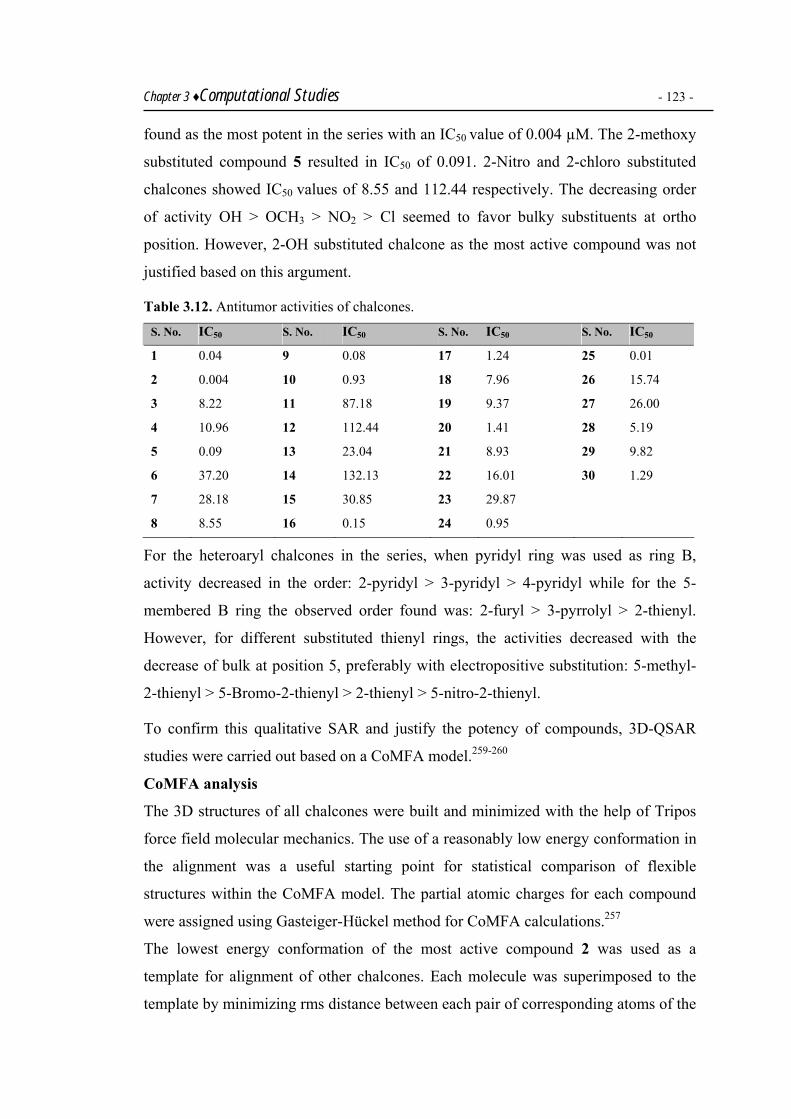
Chapter 3 ♦Computational Studies - 123 - found as the most potent in the series with an IC50 value of 0.004 µM. The 2-methoxy
substituted compound 5 resulted in IC50 of 0.091. 2-Nitro and 2-chloro substituted
chalcones showed IC50 values of 8.55 and 112.44 respectively. The decreasing order
of activity OH > OCH3 > NO2 > Cl seemed to favor bulky substituents at ortho
position. However, 2-OH substituted chalcone as the most active compound was not
justified based on this argument.
Table 3.12. Antitumor activities of chalcones. S. No. IC50 S. No. IC50 S. No. IC50 S. No. IC50
1 0.04 9 0.08 17 1.24 25 0.01
2 0.004 10 0.93 18 7.96 26 15.74
3 8.22 11 87.18 19 9.37 27 26.00
4 10.96 12 112.44 20 1.41 28 5.19
5 0.09 13 23.04 21 8.93 29 9.82
6 37.20 14 132.13 22 16.01 30 1.29
7 28.18 15 30.85 23 29.87
8 8.55 16 0.15 24 0.95
For the heteroaryl chalcones in the series, when pyridyl ring was used as ring B,
activity decreased in the order: 2-pyridyl > 3-pyridyl > 4-pyridyl while for the 5-
membered B ring the observed order found was: 2-furyl > 3-pyrrolyl > 2-thienyl.
However, for different substituted thienyl rings, the activities decreased with the
decrease of bulk at position 5, preferably with electropositive substitution: 5-methyl-
2-thienyl > 5-Bromo-2-thienyl > 2-thienyl > 5-nitro-2-thienyl.
To confirm this qualitative SAR and justify the potency of compounds, 3D-QSAR
studies were carried out based on a CoMFA model.259-260
CoMFA analysis
The 3D structures of all chalcones were built and minimized with the help of Tripos
force field molecular mechanics. The use of a reasonably low energy conformation in
the alignment was a useful starting point for statistical comparison of flexible
structures within the CoMFA model. The partial atomic charges for each compound
were assigned using Gasteiger-Hückel method for CoMFA calculations.257
The lowest energy conformation of the most active compound 2 was used as a
template for alignment of other chalcones. Each molecule was superimposed to the
template by minimizing rms distance between each pair of corresponding atoms of the

Chapter 3 ♦Computational Studies - 124 - template and the compound to be aligned. The structures were fully geometry-
optimized using standard Tripos force field with a distance-dependent dielectric
function until a rms deviation of 0.001 kcal mol-1 Å-1 was achieved.257 The activities
were converted to the corresponding log values. PLS linear regression was used to
correlate the biological activity index with CoMFA values that contained the
magnitude of either the steric or electronic potential. To avoid over fitted 3D QSAR,
the optimum number of components (N) were used for model derivation to give the
highest cross-validated correlation coefficient (q2), quantifying the predictive ability
of the model. The leave-one-out (LOO) procedure of cross-validation was used in
which each compound was successively removed from the model derived and its IC50
value was predicted by using the model built from the remaining compounds. Column
filtering was set at 2.0 kcal/mol so that only those steric and electrostatic energies
with values greater than 2.0 kcal/mol were considered in the PLS analysis and the
predictive quality of the “best” correlation model was determined. The optimal
number of components (N) were selected as three employed for no validation PLS
analysis. With a grid space of 2 Å and F value of 36.344, correlation coefficient (r2)
and LOO (q2) were calculated as 0.997 and 0.643. The results were visualized by
color contour maps displaying both the favorable and unfavorable fields for each
electrostatic and steric interaction surrounding the set of superposed chalcones. The
observed versus predicted activities of the training set and the test sets generated by
CoMFA analysis showed good consistency (Tables 3.13 and 3.14).
Plot of observed versus predicted antitumor activities of chalcones 1-30 including the
members of both the training set and the test set is presented in Fig. 3.13. The
CoMFA electrostatic and steric fields based on PLS analysis are presented as 3D
contour plots in Figs. 3.14 and 3.15 respectively. Red color indicates areas where
more negative density favors activity and blue color indicates areas where more
positive density promotes activity. Green color indicates areas where bulky
substituents favor activity and yellow color indicates areas where less bulky
substituents promote activity.
The compounds containing electronegative atom attached to position 2 in ring B, were
supported for activity as confirmed by a red isopleth in the electrostatic contour map.
The maximum charge concentration on oxygen atom directly attached to the aryl ring

Chapter 3 ♦Computational Studies - 125 - in 2-hydroxy position substituted chalcone was probably the reason for showing the
highest potency (IC50 0.004).
Table 3.13. Observed and predicted log IC50 of training set chalcones.
Chalcones Observed Predicted Residual Chalcones Observed Predicted Residual
1 1.60 1.51 0.09 16 2.18 2.19 0.01
2 0.60 0.62 0.02 17 3.09 3.10 0.01
3 3.92 3.89 -0.03 18 3.90 3.92 0.02
4 4.04 4.03 -0.01 20 3.15 3.18 0.03
5 1.96 1.94 -0.02 21 3.95 3.92 -0.03
6 4.57 4.56 -0.01 22 4.20 4.22 0.02
7 4.45 4.57 0.12 23 4.47 4.52 0.05
8 3.93 3.9 -0.03 25 1.97 1.92 -0.05
10 2.97 3.11 0.14 27 4.41 4.46 0.05
11 4.94 4.91 -0.03 28 3.71 3.78 0.07
12 5.05 5.01 -0.04 29 3.99 3.96 -0.03
13 4.36 4.47 0.11 30 3.11 3.01 0.10
15 4.49 4.54 0.05
Table 3.14. Observed and predicted log IC50 of test set chalcones.
Chalcones Observed Predicted Residual
9 1.91 2.12 0.21
14 5.12 4.9 -0.22
19 3.97 4.12 0.15
24 2.98 2.75 -0.23
26 4.19 3.99 -0.20
The decreasing trend of activity: OH > OCH3 > NO2 > Cl indicated that it was not
actually the electron donor or electron acceptor property of substituent which
supported activity, but only concentration of negative charge on atom directly
attached to ortho position. Similarly, the presence of green isopleth in Fig. 3.15
indicated that the presence of bulk at this position was justified, resulting in more
potency of -OCH3 and -NO2 substituted compounds 5 and 8 (IC50 0.09 and 8.55) than
chloro-substituted compound 12, which showed an IC50 value 112.4.

Chapter 3 ♦Computational Studies 126
R2 = 0.9974
0
1
2
3
4
5
6
0 1 2 3 4 5 6log IC50 (calculated)
log
IC50
(pre
dict
ed)
Training set
Test set
Fig. 3. 13. Calculated vs observed activity for training set and test set compounds.
The presence of blue isopleth at the meta- and para- position of the aryl ring indicated
that the positive charge density at the meta and para positions favored tumor
inhibiting potency. Similarly, the blue isopleth at the substituent attached to the para
position indicated the preference of positive charge density in this area. Electron
withdrawing substituents at the meta position resulted in positive charge density at
this position, and favored tumor inhibition properties. As a result compound 9
containing 3-NO2 was the most potent in the 3-substituted series (IC50 0.08), followed
by OH substituted compound 3 (IC50 8.22). Chalcones 13, 15 and 6 containing 3-Br,
3-Cl and 3-OCH3 showed IC50 values as 30.85, 23.04, 37.20. This observation was
consistent with the presence of small yellow isopleth at the meta substitution,
indicating a disfavor for bulk at this position. At the para position, the blue contour
map surrounded almost the whole of the attached substituent, resulting in increased
favor for an electropositive moiety at this position. This argument was confirmed
when the potencies of para-substituted compounds were evaluated on these grounds.
The methyl group was the most electropositive substituent and therefore the chalcone
16 was found to be the most active (IC50 0.15). Chalcones 4, 7, 11, 14, 16 and 17
showed tumor inhibition in the following decreasing order with a decreasing positive
inductive effect; CH3 > N(CH3)2 > OH > OCH3 > F > Cl. The activity of chalcone 10
bearing a 4-NO2 substituent was supported by the presence of a small red isopleth in
space around 4-position of ring B, as reflected from its small IC50 value of 0.93.

Chapter 3 ♦Computational Studies 127
Fig. 3.14. Stereoview of the CoMFA map for the electronic contribution.
Fig. 3.15. Stereoview of the CoMFA map for the steric contribution.
Compound 19 with 3,4-di OMe groups showed an IC50 value of 9.37 while compound
18 with 3-OH, 4-OMe substitution with a relatively less bulky group at position 3 was
found to have enhanced activity (IC50 7.96). Furthermore, in chalcone 20 carrying a -
CH3 group at position 2 and -OCH3 groups at positions 3 and 5, a decrease in activity
was observed due to the 3-OCH3 group (disfavored by a yellow contour in Fig 3.15).
However, the presence of 2-CH3 group is highly favored for activity as indicated by a
large green isopleth, resulting in an IC50 value of 1.41.
The heteroatoms (N, O, S) on a 5-membered heterocycle in chalcones e.g. 24-26 as
well as nitrogen in chalcones 21 and 22 fall in the dense large green isopleth in steric
contour maps, resulting in high activities with IC50 values of 0.95, 0.01, 15.74, 8.93,
16.01. In the thienyl series as ring B, chalcones 27-29, 28 resulted in an IC50 value of
5.19 with -CH3 at position 5 while in chalcones 29 and 27, the -Br and -NO2 groups at

Chapter 3 ♦Computational Studies 128 position 5 resulted in IC50 values of 9.82 and 26.00. This observation may be
attributed to the fall of these substituents in the dense blue isopleth as indicated in Fig.
3.14. supporting positive electron density on meta- and para- positions of B ring and
electropositive substitution pattern on 5- position of heteroaryl rings.
O
B A
OH
Negative charge
2-OH (2) & 2-Cl (12) 0.004 112.44
2-OCH3 (5) & H (1) density 0.004 0.04 3-Cl (13) & 3-Br (15)
Bulk
Fig. 3.16. Favorable electronic and steric regions for enhanced potency in antitumor
chalcones.
A comprehensive and predictive 3D-QSAR model has been derived using CoMFA
analysis to rationalize the antitumor activity of 30 compounds. A high bootstrapped r2
and small standard deviation indicated that a similar relationship existed in all
compounds. The CoMFA contour maps have successfully rationalized the potencies
of chalcones as discussed on the basis of qualitative SAR. The CoMFA contour maps
showed a good compatibility with the receptor properties even though the
conformations and alignments of ligand were not based on receptor structure. The
structural requirements of the inhibitors identified through the CoMFA contour plots
may help in designing new antitumor agents with enhanced activity.
Positive charge density
Negative charge density
Less bulk 23.04 30.85
3-NO2 (9) & 3-OH (3) 0.08 8.22
Positive charge density
4-NO2 (10) & 4-OH (4) Steric field contribution = 68%, 0.93 10.96
Electrostatic field contribution = 32%

4
120 Member Library
Indexed Libraries
Screening
Set 2. Pools 1-20
1. B1 + A1-62. B2 + A1-6
: 20. B20 + A1-6
LEAD
Set 1: Pools 1-6
1. A1 + B1-202. A2 + B1-20
: 6. A6 + B1-20
Experimental

Experimental 4A. General
4A.1. Chemicals
All acetophenones and benzaldehydes were purchased from Fluka, Sigma-Aldrich or
Merck Biosciences. F-moc protected amino acids were obtained from Novabiochem.
Triphenylphosphine-PS-support and MSNT were acquired from Fluka.
Trimethylsilylethanol, bromoacetic acid, TASF, DCC, DIC, HOBT.H2O, TBTU,
DMAP, lutidine and triethylamine were purchased from Fluka, whereas TFFH and
DIEA were obtained from Aldrich. Other chemicals used were purchased from
Sigma-Aldrich, Merck and BDH. For HPLC, LCMS, Kaiser test and Fmoc
determination, solutions were made in deionized water purified with a Milli-Q
Ultrapure water putification system.
For HPLC and LCMS, acetonitrile and methanol (both of gradient grade) and
trifluoroacetic acid (analytical grade) were obtained from Merck Biosciences.
4A.2. Purification and drying of solvents
Commercial grade solvents were utilized in solution phase synthesis after necessary
purification and drying according to standard methods. A brief account of the
purification procedures follows.
Ethyl acetate was kept over anhydrous calcium chloride for six hours, distilled and
stored over molecular sieves (4Å) under nitrogen.
Chloroform was distilled over calcium hydride and stored over molecular sieves (4Å)
under an atmosphere of nitrogen.
Ethanol was distilled first; the distilled ethanol was refluxed over CaO and redistilled.
The 99% ethanol obtained, thereby, was refluxed over magnesium turnings and
iodine, till the color of iodine disappeared. The distilled dry ethanol was kept over
molecular sieves (4Å).
Anhydrous solvents such as dichloromethane, petroleum ether, DMF and toluene
were obtained from Fluka and stored under nitrogen over molecular sieves for
carrying out solid phase synthesis.

Chapter 4 ♦Experimental - 131 - 4A.3. Analytical methods and instruments
4A.3.1. Thin layer chromatography
All the solution phase reactions were monitored through thin layer chromatography
using precoated silica gel aluminium cards (layer thickness 0.2 mm, HF 254, Riedel-
de-Haen) and precoated silica gel glass plates (layer thickness 0.25 mm, HF 254, E.
Merck). n-Hexane and ethylacetate were used in following different ratios to develop
chromatograms,
(a) (2:1)
(b) (3:1)
(c) (1:1)
Chromatograms were detected under UV light at 254 and 365 nm.
4A.3.2. Green House synthesizer
Radleys Green House synthesizer with 24 well reaction format was used for solution
phase parallel synthesis of chalcone libraries and worked-up parallely using 24 micro-
scale columns provided in parallel purification system with the synthesizer obtained.
4A.3.3. Microwave synthesizers
Solution phase microwave synthesis was performed in 200 mL round-bottom flasks
using a house-hold microwave oven, 900 W, Model No. MS-304A, LG. Water was
used as a heat sink.
Solid phase microwave synthesis was accomplished in a Personal Chemistry Smith
Synthesizer. The permissible sample volume for synthesis was 0.5-5 mL in septa
sealed with stable pressure reaction vessels. A Magneton with a capacity of 15–300 W
generated heat in this synthesizer. The reaction mixture used microwave radiation
with a frequency of ν = 2.45 GHz. The temperature range varied from 60–250 °C at
heating rate of 2 °CS-1. The maximum allowable pressure inside the tubes was 20
bars. During the reaction, temperature inside the reaction vessel was kept constant by
microwave pulses. When the reaction was complete, reaction vessel was brought to
room temperature through rapid cooling under atmospheric pressure.
4A.3.4. Ika Vibrax orbital shaker
Solid phase parallel synthesis was performed in plastic syringes equipped with fritted
Teflon discs and the mixtures were shaken over Ika Vibrax orbital shaker containg 24
positions for stirring reactions in 2 and 5 mL syringes.

Chapter 4 ♦Experimental - 132 - 4A.3.5. Melting point determination
Melting points were recorded on a MEL-Temp. Melting Point Apparatus and were
uncorrected.
4A.3.6. FTIR spectroscopy
IR spectra of the synthesized compounds were recorded on Schimadzu Fourier
Transform Infrared Spectrophotometer Model 270. Solid samples were taken in KBr
pellets and oils were used in NaCl cell. Solid phase reactions were monitored through
FT-ATR-IR spectra of the resins with a Bruker Vector 22/Harrick SplitPea ATR unit.
IR of the solid support was obtained directly by fitting to a Si-crystal setup. Each
spectrum was obtained after 16 scans and was corrected automatically. Carbon
dioxide absorption bands were removed for the purpose of clarity. Absorptions were
reported in wave numbers ν-1 (cm-1).
4A.3.7. UV Vis. spectroscopy
The UV measurements were obtained on a Lambda 2 UV Vis Spectrophotometer
(Perkin-Elmer) equipped with Quartz cell. During SPS of peptidyl chalcones, the
extent of resin loading was determined by photometric determination after Fmoc
cleavage. The loading was estimated by determining the extinction co-efficient after
the separation of Fmoc-protecting group of a defined quantity of resin ~ 5 mg using
20% piperidine/DMF against a reference solution (20% piperidine/DMF). Absorb in
maxima were observed at λ1 = 267 nm, λ2 = 289 nm and λ3 = 301 nm. The extent of
loading (mmol/g) was calculated by Beer-Lambert law using the following equation.
[ ]gmmolm
ELoading /100000••
=λ
λ
ε
where,
Eλ = Eλ 267 , Eλ 289 , Eλ 301
ελ = ελ 267 , ελ 289 , ελ 301
m = mass of support in mg
4A.3.8. LC MS analysis
LC MS of crude peptide derivatives were carried out on an Agilent 1100 series
workstation (Agilent Technologies) equipped with a single Quadrupole Mass
Spectrometer and Electrospray Ionization (ES). Samples were run through analytical
column; 5 µm, 100 Å, 3 mm × 70 mm, Nucleosil 100-5 C18 HD (Macherey-Nagel).

Chapter 4 ♦Experimental - 133 - Injection volume used was 5 µL each time, while flow rate was maintained at 0.5
mL/min. Detections were made on a UV Vis. detector at 214, 254 and 280 nm.
Eluents A (5 mmoL NaCl in water) and B (acetonitrile) were used in a linear gradient;
0–80% B in 0–10 min, isocratic from 10–15 min, 80%–99% B from 15–17 min and
99% B isocratic from 17–20 min.
4A.3.9. HPLC
The purification of crude peptides and the desived heterocycles was carried out by
semi preparative HPLC performed on an Agilent 1100 series instrument (Agilent
Technologies) which consisted of an autosampler, UV-detector (Diode Array Detector
Module 168), Interface (Analog Interface Module 406), High-pressure pump system
(Programmable Solvent Module 126) and PC suite.
Samples were purified on a semi-preparative column; 10 µm, 250 × 20 mm, Grom-
SIL 300 ODS-5 ST RP-C18. Eluents A (0.1% TFA in water) and B (0.1% TFA in
acetonitrile) were used in a linear gradient; 10–100% B in 45 min at a flow rate of 0.3
mL/min of all the peptides. Peptidic heterocycles were subjected to individual
gradients using eluents A (0.1% TFA in water) and B (0.1% TFA in acetonitrile).
Detection was achieved with a UV Vis. detector at wavelengths 220 and 254 nm.
4A.3.10. GCMS spectrometry
GC MS analysis was carried out on a 6890 N Agilent GC workstation coupled with
Agilent 5973 inert Mass Selective Detector. The column used for GC separation was
DB-5ms with He gas supplied at the rate of 5 mL/min. Inlet was kept at 250 °C
whereas temperature programming was set at 120-300 °C at the rate of 10 °C/min.
Gas chromatogram ionization energy was 70 eV and TOF (Time of Flight) was used
as mass detection system. GCMS analysis of the indexed libraries was carried out to
identify all the components of the library. The libraries were analyzed using different
GCMS programs and the best results were found when the temperature program was
kept from 120-300 °C at a rate of 10°C/min whereas inlet was kept at 250 °C.
4A.3.11. Mass spectrometry
Electron impact mass spectrometric analysis (EIMS) was carried out on a Varian
MAT 311A mass spectrometer connected to a Mass Data system, to record the low
resolution mass spectra of chalcones. EIMS was taken at 70 eV through scanning
from m/z 50–300 while maintaining the filter at 1%.

Chapter 4 ♦Experimental - 134 - HREIMS spectra of chalcones were recorded on a JMS 600H mass spectrometer.
Positive EI ionization mode was used at 70 eV. Inlet system used was My-inlet.
Selected isotopes for scanning were C0-100 H0-100 N0-2 O0-12 S0-1. The filter was kept at
Int: 1% and scanning was carried out within a range of m/z 50–800.
HRMS measurements of peptides was carried out on a Ionspec QFT-7 mass
spectrometer, equipped with a Z-Spray Source, spray capillary at 3.8 kV.
4A.3.12. NMR spectroscopy 1H and 13C NMR spectra were recorded on a Bruker AVANCE 400 and 300 MHz
instruments. The signals of the residual protonated solvents (CDCl3, DMSO-d6 and
CD3OD) were used as the reference signal. Chemical shifts were recorded as ppm
whereas coupling constants were measured in Hz. Abbreviations s, d, t, bs and m
have been used for singlet, doublet, triplet, broad singlet and multiplet respectively.
4A.3.13. Elemental analysis
Elemental analysis was performed on a CHN analyzer 932 provided by Leco, USA.
4A.3.14. Kaiser test
Kaiser test was performed to check complete immobilization of free amino groups
obtained through standard amide coupling methods using solutions A, B and C.
Solution A. Ninhydrin (5% w/v) was prepared by dissolving ninhydrin (500mg) in n-
butanol (10mL).
Solution B. Phenol (4:1, w/v) was obtained by dissolving phenol (80 mg) in n-butanol
(20 mL).
Solution C. Potassium cyanide (2%, v/v, 1 mM KCNaq in pyridine) obtained through
mixing 2 mL of 2% KCNaq (33mg/50 mL) to pyridine (98 mL).
The test was carried out by adding 2 drops of soln. A, 1 drop of soln. B and 1 drop of
soln. C to the test sample (usually 1–2 mg of resin, 10–20 resin particles) contained in
a small glass test tube and then heated for 2–5 minutes on a hot plate. A blue
coloration of the resin indicated the presence of free amines on the growing peptide. If
the resin retained its original color, this was a negative test indicating successful
coupling or incomplete deprotection.
The biological evaluation of synthesized compound libraries was carried out on UV
Vis Spectrophotometer (Perkin-Elmer) and Microplate Reader (Tecan Safire) for
detection of absorption at a desired wavelength or within a range.

Chapter 4 ♦Experimental - 135 - 4B. Synthesis of chalcone libraries
Different chalcones and libraries were synthesized in solution phase under Claisen
Schmidt condensation. Some libraries were also synthesized under microwave
irradiation.
4B.1. Parallel synthesis of a 20 member library
General Procedure.146 A round-bottom flask fitted with a mechanical stirrer was
loaded with rectified ethanol (25 mL) and aq. 4M NaOH soln. (30mL). The flask was
cooled in an ice-bath, and acetophenone (5 mmol) was added. To this soln.,
benzaldehyde (5 mmol) was added with stirring. The temp. was kept at ca. 25 oC, and
the mixture was stirred vigorously for 2–3 h, until it became so thick that it could not
be stirred further. The reaction mixture was kept at 0–4 oC overnight. Then, it was
neutralized with aq. HCl. The precipitate separated was recrystallized from aq. EtOH.
The purity was checked by multiple TLC (hexane/EtOAc 2:1, 3:1 and 1:1).
4B.1.1. (E)-1-(4′-Aminophenyl)-3-(phenyl)-2-propen-1-one (1)
1 was synthesized by the general procedure using 4′-aminoacetophenone (0.675g, 5
mmol) and benzaldehyde (0.507 mL, 5 mmol). Yield 68%. M.p. 84-86 ºC. IR (KBr): ν
= 3041, 2847, 2555, 1659, 1597, 1576. 1H NMR (400 MHz, CDCl3): δ = 3.4 (bs, 2H,
H-N(NH2)), 7.02 (d, J = 16.0 Hz, 1H, H-Cα(Vinyl)), 7.06 (d, J = 16.0 Hz, 1H, H-
Cβ(Vinyl)), 7.33-7.34 (m, 4H, H-C(2′,6′,3′,5′)), 7.57-7.61 (m, 5H, H-C(arom.).
HRMS: calcd [(M+) = C15H13NO] 223.0998; found 223.0993 mz (88%).
4B.1.2. (E)-1-(4′-Aminophenyl)-3-(2-hydroxyphenyl)-2-propen-1-one (2)
2 was synthesized by the general procedure using 4′-aminoacetophenone (0.675g, 5
mmol) and 2-hydroxybenzaldehyde (0.388 mL, 5 mmol). Yield 67%. M.p. 104-108
ºC. IR (KBr): ν = 3327, 3259, 3070, 3062, 2380, 1676, 1584, 1574. 1H NMR (400
MHz, CDCl3): δ = 5.98 (bs, 2H, H-N(NH2)), 6.92 (t, J = 8.0 Hz, 1H, H-C(4)), 6.98
(m, 2H, H-Cα(Vinyl), H-C(5)), 7.04 (d, J = 8.1 Hz, 1H, H-C(6)), 7.31 (d, J = 8.3 Hz,
2H, H-C(3′,5′)), 7.41 (d, J = 8.9 Hz, 1H, H-C(3)), 7.75 (d, J = 14.8 Hz, 1H, H-
Cβ(Vinyl)), 8.01 (d, J = 8.3 Hz, 2H, H-C(2′,6′)), 9.88 (s, 1H, H-O(OH)). HRMS:
calcd [(M+) = C15H13NO2] 239.0947; found 239.0939 mz (83.8%).
4B.1.3. (E)-1-(4′-Aminophenyl)-3-(3-hydroxyphenyl)-2-propen-1-one (3)
3 was synthesized by the general procedure using 4′-aminoacetophenone (0.675g, 5
mmol) and 3-hydroxybenzaldehyde (0.610 g, 5 mmol). Yield 65%. M.p. 197 ºC

Chapter 4 ♦Experimental - 136 - (decomp.). IR (KBr): ν = 3380-3120, 3327, 3259, 3062, 1676, 1584, 1573. 1H NMR
(400 MHz, (CDCl3): δ = 4.1 (bs, 2H, H-N(NH2)), 6.68 (d, J = 8.6 Hz, 1H, H-C(6)),
6.80 (d, J = 8.2 Hz, 2H, H-C(3′,5′)), 6.87 (t, J = 8.8 Hz, 8.6 Hz, 1H, H-C(5)), 7.09 (s,
1H, H-C(2)), 7.43 (d, J = 15.4 Hz, 1H, H-Cα(Vinyl)), 7.51 (d, J = 8.8 Hz, 1H, H-
C(4)), 7.73 (d, J = 15.4 Hz, 1H, H-Cβ(Vinyl)), 8.03 (d, J = 8.2 Hz, 2H, H-C(2′,6′)),
9.76 (s, 1H, H-O(OH)). HRMS: calcd [(M+) = C15H13NO2] 239.0936; found 239.0947
mz (80.9%).
4B.1.4. (E)-1-(4′-Aminophenyl)-3-(4-hydroxyphenyl)-2-propen-1-one (4)
4 was synthesized by the general procedure using 4′-aminoacetophenone (0.675g, 5
mmol) and 4-hydroxybenzaldehyde (0.610 g, 5 mmol). Yield 61%. M.p. 183 ºC
(decomp.). IR (KBr): ν = 3372-3065, 3336, 3247, 3064, 1669, 1581, 1572. 1H NMR
(400 MHz, CDCl3): δ = 4.37 (bs, 2H, H-N(NH2)), 6.72 (d, J = 15.2 Hz, 1H, H-
Cα(Vinyl)), 6.92 (bd, J = 8.1 Hz, 4H, H-C(3,5,3′,5′)), 7.52 (bd, J = 8.1 Hz, 4H, H-
C(2,6,2′,6′)), 7.77 (d, J = 15.2 Hz, 1H, H-Cβ(Vinyl)), 9.86 (s, 1H, H-O(OH)). HRMS:
calcd [(M+) = C15H13NO2] 239.0947; found 239.0924 mz (79.2%).
4B.1.5. (E)-1-(4′-Aminophenyl)-3-(2-methoxyphenyl)-2-propen-1-one (5)
5 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5
mmol) and 2-methoxybenzaldehyde (0.604 mL, 5 mmol). Yield 77%. M.p. 105-107
ºC. IR (KBr): ν = 3440, 3349, 3241, 3237, 1639, 1605, 1549. 1H NMR (400 MHz,
CDCl3): δ = 3.46 (s, 3H, H-C(OCH3)), 3.80 (bs, 2H, H-N(NH2)), 6.90 (bd, J = 7.6 Hz,
2H, H-C(3,6)), 7.39 (d, J = 15.6 Hz, 1H, H-Cα(Vinyl)), 7.60 (bt, J = 7.6 Hz, 7.8 Hz,
2H, H-C(4,5)), 7.73 (d, J = 15.6 Hz, 1H, H-Cβ(Vinyl)), 7.90 (d, J = 8.2 Hz, 2H, H-
C(2′,6′)), 8.69 (d, J = 8.2 Hz, 2H, H-C(3′,5′)). HRMS: calcd [(M+) = C16H15NO2]
253.1104; found 253.1091 mz (80.5%).
4B.1.6. (E)-1-(4′-Aminophenyl)-3-(3-methoxyphenyl)-2-propen-1-one (6)
6 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5
mmol) and 3-methoxybenzaldehyde (0.608 mL, 5 mmol). Yield 73%. M.p. 134 ºC. IR
(KBr): ν = 3466, 3329, 3214, 1630, 1600, 1597. 1H NMR (400 MHz, CDCl3): δ =
4.13 (bs, 2H, H-N(NH2)), 6.68 (d, J = 8.0 Hz, H-C(3′,5′)), 6.92 (d, J = 7.4 Hz, 1H, H-
C(6)), 7.22 (d, J = 7.5 Hz, 1H, H-C(4)), 7.23 (s, 1H, H-C(2)), 7.30 (t, J = 7.5 Hz, 1H,
H-C(5)), 7.50 (d, J = 15.4 Hz, 1H, H-Cα(Vinyl)), 7.76 (d, J = 15.4 Hz, 1H, H-

Chapter 4 ♦Experimental - 137 - Cβ(Vinyl)), 7.90 (d, J = 8.0 Hz, 2H, H-C(2′,6′)). HRMS: calcd [(M+) = C16H15NO2]
253.1104; found 253.1089 mz (89.4%).
4B.1.7. (E)-1-(4′-Aminophenyl)-3-(4-methoxyphenyl)-2-propen-1-one (7)
7 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5
mmol) and 4-methoxybenzaldehyde (0.252 mL, 5 mmol). Yield 75%. M.p. 128-130
ºC. IR (KBr): ν = 3449, 3354, 3245, 1646, 1604, 1551. 1H NMR (400 MHz, CDCl3):
δ = 3.83 (s, 3H, H-C(OCH3)), 4.12 (bs, 2H, H-N(NH2)), 6.68 (d, J = 8.2 Hz, 2H, H-
C(3′,5′)), 6.92 (d, J = 7.6 Hz, 2H, H-C(2,6)), 7.23 (d, J = 7.6 Hz, 2H, H-C(3,5)), 7.49
(d, J = 16 Hz, 1H, H-Cα(Vinyl)), 7.72 (d, J = 16 Hz, 1H, H-Cβ(Vinyl)), 7.90 (d, J =
8.2 Hz, 2H, H-C(2′,6′)). HRMS: calcd [(M+) = C16H15NO2] 253.1104; found 253.1093
mz (77.8%).
4B.1.8. (E)-1-(4′-Aminophenyl)-3-(3,4-dimethoxyphenyl)-2-propen-1-one (8)
8 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5
mmol) and 3,4-dimethoxybenzaldehyde (0.83 g, 5 mmol). Yield 85%. M.p. 96-98 ºC.
IR (KBr): ν = 3445, 3351, 3041, 1649, 1606, 1557. 1H NMR (400 MHz, CDCl3): δ =
3.73 (s, 3H, H-C(OCH3)), 3.84 (s, 3H, H-C(OCH3)), 5.27 (bs, 2H, H-N(NH2)), 6.77
(d, J = 8.0 Hz, 2H, H-C(3′,5′)), 7.04 (m, 3H, H-C(2,5,6)), 7.27 (d, J = 15.4 Hz, 1H, H-
Cα(Vinyl)), 7.58 (d, J = 15.4 Hz, 1H, H-Cβ(Vinyl)), 7.84 (d, J = 8.0 Hz, 2H, H-
C(2′,6′)). HRMS: calcd [(M+) = C12H17NO3] 283.1209; found 283.1197 mz (72.1%).
4B.1.9. (E)-1-(4′-Aminophenyl)-3-(2-nitrophenyl)-2-propen-1-one (9)
9 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5
mmol) and 2-nitrobenzaldehyde (0.776 g, 5 mmol). Yield 65%. M.p. 164-166 ºC. IR
(KBr): ν = 3474, 3382, 3227, 1665, 1515, 1482, 1412. 1H NMR (400 MHz, CDCl3): δ
= 3.9 (bs, 2H, H-N(NH2)), 6.79 (d, J = 8.3 Hz, 2H, H-C(3′,5′)), 7.45 (t, J = 7.4 Hz, 7.5
Hz, 1H, H-C(4)), 7.67 (t, J = 7.4 Hz, 8.0 Hz, 1H, H-C(5)), 7.79 (d, J = 7.5 Hz, 1H, H-
C(6)), 7.83 (d, J = 14.0 Hz, 1H, H-Cα(Vinyl)), 8.02 (d, J = 8.3 Hz, 2H, H-C(2′,6′)),
8.13 (d, J = 8.0 Hz, 1H, H-C(3)), 8.22 (d, J = 14.0 Hz, 1H, H-Cβ(Vinyl)). HRMS:
calcd [(M+) = C15H12N2O3] 268.0849; found 268.0847 mz (11.8%).
4B.1.10. (E)-1-(4′-Aminophenyl)-3-(3-nitrophenyl)-2-propen-1-one (10)
10 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5
mmol) and 3-nitrobenzaldehyde (0.776 g, 5 mmol). Yield 83%. M.p. 206-207 ºC. IR
(KBr): ν = 3469, 3380, 3227, 1690, 1595, 1514, 1441. 1H NMR (400 MHz, CDCl3): δ

Chapter 4 ♦Experimental - 138 - = 4.20 (bs, 2H, H-N(NH2)), 6.79 (d, J = 8.0 Hz, 2H, H-C(3′,5′)), 6.90 (d, J = 6.1 Hz,
2H, H-C(6)), 7.60 (bt, J = 8.4 Hz, 6.1 Hz, 1H, H-C(5)), 7.62 (d, J = 16.4 Hz, 1H, H-
Cα(Vinyl)), 7.78 (d, J = 16.4 Hz, 1H, H-Cβ(Vinyl)), 8.03 (d, J = 8.0 Hz, 2H, H-
C(2′,6′)), 8.24 (d, J = 8.4 Hz, 1H, H-C(4)), 8.49 (s, 1H, H-C(2)). HRMS: calcd [(M+)
= C15H12N2O3] 268.0849; found 268.0846 mz (18.8%).
4B.1.11. (E)-1-(4′-Aminophenyl)-3-(4-nitrophenyl)-2-propen-1-one (11)
11 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5
mmol) and 4-nitrobenzaldehyde (0.776 g, 5 mmol). Yield 87%. M.p. 204-205 ºC. IR
(KBr): ν = 3481, 3385, 3228, 1670, 1508, 1478, 1407. 1H NMR (400 MHz, CDCl3): δ
= 4.28 (bs, 2H, H-N(NH2)), 7.62 (d, J = 15.4 Hz, 1H, H-Cα(Vinyl)), 7.76 (d, J = 15.4
Hz, 1H, H-Cβ(Vinyl)), 7.92 (d, J = 8.4 Hz, 2H, H-C(3′,5′)), 8.03 (d, J = 8.4 Hz, 2H,
H-C(2′,6′)), 8.24 (d, J = 8.8 Hz, 2H, H-C(2,6)), 8.28 (d, J = 8.8 Hz, 2H, H-C(3,5)).
HRMS: calcd [(M+) = C15H12N2O3] 268.0849; found 268.0844 mz (12.5%).
4B.1.12. (E)-1-(4′-Aminophenyl)-3-(2-chlorophenyl)-2-propen-1-one (12)
12 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5
mmol) and 2-chlorobenzaldehyde (0.416 mL, 5 mmol). Yield 78%. M.p. 129 ºC. IR
(KBr): ν = 3452, 3351, 3243, 1654, 1582, 1558, 850. 1H NMR (400 MHz, CDCl3): δ
= 4.13 (bs, 2H, H-N(NH2)), 6.68 (d, J = 8.3 Hz, 2H, H-C(3′,5′)), 7.28 (bt, J = 8.5 Hz,
8.7 Hz, 2H, H-C(4,5)), 7.41 (d, J = 8.7 Hz, 1H, H-C(6)), 7.47 (d, J = 15.7 Hz, 2H, H-
Cα(Vinyl)), 7.71 (d, J = 8.5 Hz, 1H, H-C(3)), 7.90 (d, J = 8.3 Hz, 2H, H-C(2′,6′)),
8.11 (d, J = 15.7 Hz, 2H, H-Cβ(Vinyl)). HRMS: calcd [(M+) = C15H12NOCl35]
257.0608; found 257.0595 mz (29.9%), calcd [(M+) = C15H12NOCl37] 259.0567;
found 259.0556 mz (9.9%).
4B.1.13. (E)-1-(4′-Aminophenyl)-3-(3-chlorophenyl)-2-propen-1-one (13)
13 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5
mmol) and 3-chlorobenzaldehyde (0.526 mL, 5 mmol). Yield 81%. M.p. 146 ºC
(decomp.). IR (KBr): ν = 3448, 3354, 3244, 1645, 1583, 1554, 869. 1H NMR (400
MHz, CDCl3): δ = 4.15 (bs, 2H, H-N(NH2)), 6.68 (d, J = 6.8 Hz, 2H, H-C(3′,5′)), 7.32
(t, J = 6.2 Hz, 1H, H-C(5)), 7.46 (bd, J = 5.6 Hz, H-C(4,6)), 7.51 (d, J = 12.5 Hz, H-
Cα(Vinyl)), 7.60 (s, 1H, H-C(2)), 7.68 (d, J = 12.5 Hz, H-Cβ(Vinyl)), 7.91 (d, J = 6.8
Hz, 2H, H-C(2′,6′)). HRMS: calcd [(M+) = C15H12NOCl35] 257.0608; found 257.0595
mz (45.8%), calcd [(M+) = C15H12NOCl37] 259.0567; found 259.0575 mz (15.5%).

Chapter 4 ♦Experimental - 139 - 4B.1.14. (E)-1-(4′-Aminophenyl)-3-(4-chlorophenyl)-2-propen-1-one (14)
14 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5
mmol) and 4-chlorobenzaldehyde (0.652 g, 5 mmol). Yield 84%. M.p. 69-70 ºC. IR
(KBr): ν = 3454, 3350, 3245, 1656, 1581, 1556, 842. 1H NMR (400 MHz, CDCl3): δ
= 4.14 (bs, 2H, H-N(NH2)), 6.68 (d, J = 6.6 Hz, 2H, H-C(3′,5′)), 7.36 (d, J = 6.6 Hz,
1H, H-C(6)), 7.43 (d, J = 6.5 Hz, 1H, H-C(5)), 7.51 (d, J = 15.6 Hz, 1H, H-
Cα(Vinyl)), 7.55 (d, J = 15.6 Hz, 1H, H-Cβ(Vinyl)), 7.81 (d, J = 6.5 Hz, 1H, H-C(3)),
8.02 (d, J = 6.6 Hz, 1H, H-C(2)), 8.12 (d, J = 6.6 Hz, 2H, H-C(2′,6′)). HRMS: calcd
[(M+) = C15H12NOCl35] 257.0608; found 257.0588 mz (100%), calcd [(M+) =
C15H12NOCl37] 259.0567; found 259.0559 mz (32.5%).
4B.1.15. (E)-1-(4′-Aminophenyl)-2-(4-fluorophenyl)-2-propen-1-one (15)
15 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5
mmol) and 4-fluorobenzaldehyde (0.536 mL, 5 mmol). Yield 95%. M.p. 148 ºC. IR
(KBr): ν = 3467, 3343, 3242, 1601, 1575, 1562, 1257. 1H NMR (400 MHz, CDCl3): δ
= 7.12 (bs, 2H, H-N(NH2)), 7.14 (d, J = 16.1 Hz, 1H, H-Cα(Vinyl)), 7.21 (d, J = 16.1
Hz, 1H, H-Cβ(Vinyl)), 7.62(t, J = 7.8 Hz, 2H, H-C(2,6)), 7.90 (dd, J = 9.2 Hz, 7.5 Hz,
2H, H-C(3,5)), 8.11 (d, J = 6.5 Hz, 2H, H-C(3′,5′)), 8.13 (d, J = 6.5 Hz, 2H, H-
C(2′,6′)). HRMS: calcd [(M+) = C15H12NOF] 241.0904; found 241.0885 mz (100%).
4B.1.16. (E)-1-(4′-Aminophenyl)-3-(2-bromophenyl)-2-propen-1-one (16)
16 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5
mmol) and 2-bromobenzaldehyde (0.584 mL, 5 mmol). Yield 83%. M.p. 127-128 ºC.
IR (KBr): ν = 3333, 3228, 1651, 1597, 1584, 752. 1H NMR (400 MHz, CDCl3): δ =
4.76 (bs, 2H, H-N(NH2)), 6.68 (d, J = 8.2 Hz, 2H, H-C(3′,5′)), 7.20 (t, J = 7.1 Hz, 6.8
Hz, 1H, H-C(4)), 7.32 (bt, J = 6.8 Hz, 7.8 Hz, 1H, H-C(5)), 7.41 (d, J = 15.5 Hz, H-
Cα(Vinyl)), 7.62 (d, J = 7.8 Hz, 1H, H-C(6)), 7.69 (d, J = 7.1 Hz, 1H, H-C(3)), 7.90
(d, J = 8.2 Hz, 2H, H-C(2′,6′)), 8.04 (d, J = 15.5 Hz, H-Cβ(Vinyl)). HRMS: calcd
[(M+) = C15H12NOBr79] 301.0103; found 301.0087 mz (40.5%), calcd [(M+) =
C15H12NOBr81] 303.0082; found 303.0072 mz (41.3%).
4B.1.17. (E)-1-(4′-Aminophenyl)-3-(3-bromophenyl)-2-propen-1-one (17)
17 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5
mmol) and 3-bromobenzaldehyde (0.580 mL, 5 mmol). Yield 85%. M.p. 155-157 ºC.
IR (KBr): ν = 3334, 3215, 1649, 1598, 1582, 749. 1H NMR (400 MHz, CDCl3): δ =

Chapter 4 ♦Experimental - 140 - 4.13 (bs, 2H, H-N(NH2)), 6.62 (d, J = 8.2 Hz, 1H, H-C(6)), 6.68 (d, J = 8.3 Hz, 2H,
H-C(3′,5′)), 7.40 (d, J = 15.8 Hz, 1H, H-Cα(Vinyl)), 7.65 (s, 1H, H-C(2)), 7.66 (d, J =
15.8 Hz, 1H, H-Cβ(Vinyl)), 7.91 (d, J = 8.1 Hz, 1H, H-C(4)), 7.94 (d, J = 8.3 Hz, 2H,
H-C(2′,6′)), 8.09 (t, J = 8.2 Hz, 8.1 Hz, 1H, H-C(5)). HRMS: calcd [(M+) =
C15H12NOBr79] 301.0103; found 301.0110 mz (66.1%), calcd [(M+) = C15H12NOBr81]
303.0082; found 303.0079 mz (65.1%).
4B.1.18. (E)-1-(4′-Aminophenyl)-3-(4-methylphenyl)-2-propen-1-one (18)
18 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5
mmol) and 4-methylbenzaldehyde (0.589 mL, 5 mmol). Yield 71%. M.p. 217-220 ºC.
IR (KBr): ν = 2910, 2862, 2575, 1650, 1600, 1597. 1H NMR (400 MHz, CDCl3): δ =
4.51 (bs, 2H, H-N(NH2)), 7.04 (d, J = 8.7 Hz, 2H, H-C(3,5)), 7.21 (d, J = 7.7 Hz, 2H,
H-C(3′,5′)), 7.41 (d, J = 15.6 Hz, 1H, H-Cα(Vinyl)), 7.51 (d, J = 7.7 Hz, 2H, H-
C(2′,6′)), 7.77 (d, J = 15.6 Hz, 1H, H-Cβ(Vinyl)), 7.91 (d, J = 8.7 Hz, 2H, H-C(2,6)).
HRMS: calcd [(M+) = C16H15NO 237.1154]; found 237.1155 mz (100%).
4B.1.19. (E)-1-(4′-Aminophenyl)-3-(3-hydroxy-4-methoxyphenyl)-2-propen-1-one
(19)
19 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5
mmol) and 3-hydroxy-4-methoxybenzaldehyde (0.76 g, 5 mmol). Yield 74%.
M.p.104-106 ºC. IR (KBr): ν = 3450-3445, 3443, 3352, 3030, 1632, 1570, 1265. 1H
NMR (400 MHz, CDCl3): δ = 3.83 (s, 3H, H-C(OCH3)), 5.20 (s, 2H, H-N(NH2)), 6.57
(d, J = 7.9 Hz, 1H, H-C(5)), 6.85 (d, J = 8.0 Hz, 2H, H-C(3′,5′)), 6.91 (s, 1H, H-C(2)),
7.23 (d, J = 7.9 Hz, 1H, H-C(6)), 7.58 (d, J = 15.5 Hz, 1H, H-Cα(Vinyl)), 7.73 (d, J =
15.5 Hz, 1H, H-Cβ(Vinyl)), 8.10 (d, J = 8.0 Hz, 2H, H-C(2′,6′)), 8.97 (s, 1H, H-
O(OH)). HRMS: calcd [(M+) = C16H15NO2 269.1052]; found 269.1054 mz (4.2%).
4B.1.20. (E)-1-(4′-Aminophenyl)-3-(4-N,N-dimethylaminophenyl)-2-propen-1-
one (20)
20 was synthesized by the general procedure using 4′-aminoacetophenone (0.675 g, 5
mmol) and 4-N,N-dimethylaminobenzaldehyde (0.744 g, 5 mmol). Yield 75%. M.p.
140 ºC. IR (KBr): ν = 3332, 3226, 2904, 1657, 1584. 1H NMR (400 MHz, CDCl3): δ
= 2.48 (s, 6H, H-C(N(CH3)2)), 4.09 (s, 2H, H-N(NH2)), 6.62 (bd, J = 8.1 Hz, 4H, H-
C(3,5,3′,5′)), 7.14 (d, J = 13.0 Hz, 1H, H-Cα(Vinyl)), 7.78 (bd, J = 8.1 Hz, 4H, H-

Chapter 4 ♦Experimental - 141 - C(2,6,2′,6′)), 7.90 (d, J = 13.0 Hz, 1H, H-Cβ(Vinyl)). HRMS: calcd [(M+) =
C17H18N2O] 266.1416; found 266.1407 mz (76.3%).
4B.2. Combinatorial synthesis of indexed chalcone library (120 members)146
4B.2.1. Synthesis of sub-libraries of Set 1 (AL1-AL6)
General Procedure: All indexed molecular libraries of chalcones were synthesized by
Claisen Schmidt condensation29 using the following general procedure. An EtOH
soln. (35 mL) of one acetophenone (A1-A6; 10 mmol) and twenty different substituted
benzaldehydes (B1-B20, 0.5 mmol each; 0.5 × 20 = 10 mmol in total) were stirred in a
round bottom flask. A pre-cooled 4M aq. NaOH soln. (60mL) was added and the
mixture was stirred for 5–6 h. The mixture was left at 0–4 °C overnight. The mass
obtained, thereby, was neutralized with ice-cold dil. aq. HCl. The chalcones were then
extracted with AcOEt (5 × 10 mL) and DCM (5 × 10 mL). The combined extracts
were concentrated in vacuo on a rotary evaporator to afford the library as a solid
mass.
4B.2.1.1 Synthesis of AL1. Sub-library AL1 was prepared according to the general
procedure using acetophenone A1 (1.20 mL, 10 mmol) with a mixture of twenty
different benzaldehydes B1-B20, 0.5 mmol each; 0.5 × 20 = 10 mmol in total, using the
quantities given in Table 4.1.
4B.2.1.2. Synthesis of AL2. Pool AL2 was prepared by using 2′-
hydroxyacetophenone A2 (1.20 mL, 10 mmol) and benzaldehydes B1-B20 (Table 4.1).
4B.2.1.3. Synthesis of AL3. Pool AL3 was prepared by using 4′-aminoacetophenone
A3 (1.35 g, 10 mmol) and benzaldehydes B1-B20 (Table 4.1).
4B.2.1.4. Synthesis of AL4. Pool AL4 was prepared by using 2′,3′,4′-
trimethoxyacetophenone A5 (1.35 mL, 10 mmol) and benzaldehydes B1-B20 (Table
4.1).
4B.2.1.5. Synthesis of AL5. Pool AL5 was prepared by using 3′,4′,5′-
trimethoxyacetophenone A4 (2.10 g, 10 mmol) and benzaldehydes B1-B20 (Table 4.1).
4B.2.1.6. Synthesis of AL6. Pool AL6 was prepared by using 2′,4′-
dibromoacetophenone A6 (2.78 g, 10 mmol) and benzaldehydes B1-B20 (Table 4.1).

Chapter 4 ♦Experimental - 142 - Table 4.1. Quantities of benzaldehydes used for the synthesis of AL1-AL6.
O
R
H
Aldehydes R Quantitya Aldehydes R Quantitya
B1 H 0.051 mL B11 4-Nitro 0.076 g
B2 2-Hydroxy 0.039 mL B12 2-Chloro 0.042 g
B3 3-Hydroxy 0.061 g B13 3-Chloro 0.052 mL
B4 4-Hydroxy 0.061 g B14 4-Chloro 0.065 g
B5 2-Methoxy 0.061 mL B15 4-Fluoro 0.054 mL
B6 3-Methoxy 0.060 mL B16 2-Bromo 0.059 mL
B7 4-Methoxy 0.043 mL B17 3-Bromo 0.058 mL
B8 3,4-Dimethoxy 0.083 g B18 4-Methyl 0.059 mL
B9 2-Nitro 0.076 g B19 3-Hydroxy-4-methoxy 0.076 g
B10 3-Nitro 0.076 g B20 4-N,N-dimethylamino 0.075 g
a 0.5 mmol of each of benzaldehydes was used in the synthesis of libraries AL1-AL6.
4B.2.2. Synthesis of sub-libraries of Set 2 (BL1-BL20)
General Procedure: To a stirred soln. of an aldehyde B (1.5 mmol) in EtOH (5 mL)
and six acetophenones A1-A6 (0.25 mmol each; 0.25 × 6 = 1.5 mmol in total) in a
round bottom flask, was added cold 4M aq. soln. of NaOH (8–10 mL). The mixture
was stirred for 5–6 h. When the reaction was complete, the mixture was left at 0–4 °C
overnight. The ususal workup led to the library as a solid mass.
Table. 4.2. Quantities of acetophenones (A1-A6) used for the synthesis of indexed libraries
BL1-BL20.
CH3
O
R Acetophenones R Quantitya Acetophenones R Quantitya
A1 H 0.291 mL A4 2′,3′,4′-Trimethoxy 0.034 mL
A2 2′-Hydroxy 0.030 mL A5 3′,4′,5′-Trimethoxy 0.052 g
A3 4′-Amino 0.034 g A6 2′,4′-Dibromo 0.069 g
a 0.25 mmol of each of acetophenone was used in the synthesis of libraries BL1-BL20.

Chapter 4 ♦Experimental - 143 - 4B.2.2.1. Synthesis of BL1. Pool BL1 was prepared by using benzaldehyde B1 (0.152
mL, 1.5 mmol) and a mixture of six different acetophenones A1-A6 (0.25 mmol each;
0.25 × 6 = 1.5 mmol in total) (Table 4.2).
4B.2.2.2. Synthesis of BL2. Pool BL2 was prepared by using a mixture of
acetophenones A1-A6 (Table 4.2) and 2-hydroxybenzaldehyde B2 (0.116 mL, 1.5
mmol).
4B.2.2.3. Synthesis of BL3. Pool BL3 was prepared by using a mixture of
acetophenones A1-A6 (Table 4.2) and 3-hydroxybenzaldehyde B3 (0.183 g, 1.5 mmol).
4B.2.2.4. Synthesis of BL4. Pool BL4 was prepared by using a mixture of
acetophenones A1-A6 (Table 4.2) and 4-hydroxybenzaldehyde B4 (0.183 g, 1.5 mmol).
4B.2.2.5. Synthesis of BL5. Pool BL5 was prepared by using a mixture of
acetophenones A1-A6 (Table 4.2) and 2-methoxybenzaldehyde B5 (0.181 mL, 1.5
mmol).
4B.2.2.6. Synthesis of BL6. Pool BL6 was prepared by using a mixture of
acetophenones A1-A6 (Table 4.2) and 3-methoxybenzaldehyde B6 (0.183 mL, 1.5
mmol).
4B.2.2.7. Synthesis of BL7. Pool BL7 was prepared by using a mixture of
acetophenones A1-A6 (Table 4.2) and 4-methoxybenzaldehyde B7 (0.130 mL, 1.5
mmol).
4B.2.2.8. Synthesis of BL8. Pool BL8 was prepared by using a mixture of
acetophenones A1-A6 (Table 4.2) and 3,4-dimethoxybenzaldehyde B8 (0.266 g, 1.5
mmol).
4B.2.2.9. Synthesis of BL9. Pool BL9 was prepared by using a mixture of
acetophenones A1-A6 (Table 4.2) and 2-nitrobenzaldehyde B9 (0.234 g, 1.5 mmol).
4B.2.2.10. Synthesis of BL10. Pool BL10 was prepared by using a mixture of
acetophenones A1-A6 (Table 4.2) and 3-nitrobenzaldehyde B10 (0.234 g, 1.5 mmol).
4B.2.2.11. Synthesis of BL11. Pool BL11 was prepared by using a mixture of
acetophenones A1-A6 (Table 4.2) and 4-nitrobenzaldehyde B11 (0.234 g, 1.5 mmol).
4B.2.2.12. Synthesis of BL12. Pool BL12 was prepared by using a mixture of
acetophenones A1-A6 (Table 4.2) and 2-chlorobenzaldehyde B12 (0.125mL, 1.5 mmol).
4B.2.2.13. Synthesis of BL13. Pool BL13 was prepared by using a mixture of
acetophenones A1-A6 (Table 4.2) and 3-chlorobenzaldehyde B13 (0.158 mL, 1.5 mmol).

Chapter 4 ♦Experimental - 144 - 2B.2.2.14. Synthesis of BL14. Pool BL14 was prepared by using a mixture of
acetophenones A1-A6 (Table 4.2) and 4-chlorobenzaldehyde B14 (0.196 g, 1.5 mmol).
4B.2.2.15. Synthesis of BL15. Pool BL15 was prepared by using a mixture of
acetophenones A1-A6 (Table 4.2) and 4-fluorobenzaldehyde B15 (0.161 mL, 1.5 mmol).
4B.2.2.16. Synthesis of BL16. Pool BL16 was prepared by using a mixture of
acetophenones A1-A6 (Table 4.2) and 2-bromobenzaldehyde B16 (0.175 mL, 1.5
mmol).
4B.2.2.17. Synthesis of BL17. Pool BL17 was prepared by using a mixture of
acetophenones A1-A6 (Table 4.2) and 3-bromobenzaldehyde B17 (0.174 mL, 1.5 mmol).
4B.2.2.18. Synthesis of BL18. Pool BL18 was prepared by using a mixture of
acetophenones A1-A6 (Table 4.2) and 4-methylbenzaldehyde B18 (0.177 mL, 1.5
mmol).
4B.2.2.19. Synthesis of BL19. Pool BL19 was prepared by using a mixture of
acetophenones A1-A6 (Table 4.2) and 4-hydroxy-3-methoxy benzaldehyde B19 (0.228
g, 1.5 mmol).
4B.2.2.20. Synthesis of BL20. Pool BL20 was prepared by using a mixture of
acetophenones A1-A6 (Table 4.2) and 4-N,N-dimethylaminobenzaldehyde B20 (0.224
g, 1.5 mmol).
4B.2.3. Parallel synthesis of the members of active column (Pool AL1)
Each of the active column members (A1B1-A1B20, 21-40) was synthesized using
acetophenone; A1 (0.584 mL, 5 mmol) and one of the twenty different benzaldehydes
(B1-B20) (5 mmol) following the general procedure as mentioned in Section 4B.1.
4B.2.3.1. (E)-1,3-(Diphenyl)-2-propen-1-one, A1B1 (21)
21 was synthesized by the general procedure using acetophenone A1 (0.584 mL, 5
mmol) and benzaldehyde B1 (0.507 mL, 5 mmol). Yield 94%. M.p. 56-58 oC. (KBr): ν
= 3076, 1656, 1600, 1598. 1H NMR (300 MHz, DMSO-D6): δ = 7.46 (bt, J = 3 Hz,
3H, H-C(3,4,5)), 7.57 (t, J = 7.33 Hz, 2H, H-C(3′,5′)), 7.67 (t, J = 7.32 Hz, 1H, H-
C(4′)), 7.74 (d, 1H, J = 15.9 Hz, H-Cα(Vinyl)), 7.95 (m, 3H, H-Cβ(Vinyl), H-C(2,6)),
8.15 (d, 1H, J = 7.33 Hz, H-C(2′,6′)).
4B.2.3.2. (E)-1-(Phenyl)-3-(2-hydroxyphenyl)-2-propen-1-one, A1B2 (22)
22 was synthesized by the general procedure using acetophenone A1 (0.584 mL, 5
mmol) and 2-hydroxybenzaldehyde B2 (0.388 mL, 5 mmol). Yield 67%. M.p. 48-50

Chapter 4 ♦Experimental - 145 - oC. IR (KBr): ν = 3600-3100, 1663, 1605, 1575. 1H NMR (300 MHz, DMSO-D6): δ =
7.46 (t, J = 3.2 Hz, 2H, H-C(3,6)), 7.57 (t, J = 7.5 Hz, 2H, H-C(4,5)), 7.67 (t, J = 7.6
Hz, 1H, H-C(4′)), 7.72 (d, J = 15.2 Hz, 1H, H-Cα(Vinyl)), 7.87-7.96 (m, 2H, H-
Cβ(Vinyl), H-C(3′,5′)), 8.15 (d, J = 8.1 Hz, 1H, H-C(2′,6′)).
4B.2.3.3. (E)-1-(Phenyl)-3-(3-hydroxyphenyl)-2-propen-1-one, A1B3 (23)
23 was synthesized by the general procedure using acetophenone A1 (0.584 mL, 5
mmol) and 3-hydroxybenzaldehyde B3 (0.61 g, 5 mmol). Yield 65%. M.p. 148-150 oC. IR (KBr): ν = 3400-3200, 1631, 1584, 1567. 1H NMR (300 MHz, DMSO-D6): δ =
6.87 (d, J = 8.5 Hz, 1H, H-C(4)), 7.21-7.25 (m, 2H, H-C(2,6)), 7.30 (t, J = 7.3 Hz, 1H,
H-C(5)), 7.56 (t, J = 7.3 Hz, 2H, H-C(3′,5′)), 7.61-7.69 (m, 2H, H-Cα(Vinyl), H-
C(4′)), 7.82 (d, J = 15.9 Hz, 1H, H-Cβ(Vinyl)), 8.13 (d, J = 7.3 Hz, 2H, H-C(2′,6′)).
4B.2.3.4. (E)-1-(Phenyl)-3-(4-hydroxyphenyl)-2-propen-1-one, A1B4 (24)
24 was synthesized by the general procedure using acetophenone A1 (0.584 mL, 5
mmol) and 4-hydroxybenzaldehyde B4 (0.610 g, 5 mmol). Yield 61%. M.p. 170-172 oC. IR (KBr): ν = 31-3500, 3042, 1647, 1593, 1506. 1H NMR (300 MHz, DMSO-D6):
δ = 6.84 (d, J = 8.5 Hz, 2H, H-C(3,5)), 7.55 (t, J = 7.3 Hz, 2H, H-C(3′,5′)), 7.64 (t, J =
7.3 Hz, 1H, H-C(4′)), 7.66 (d, J = 15.8 Hz, 1H, H-Cα(Vinyl)), 7.72 (d, J = 14.6 Hz,
1H, H-Cβ(Vinyl)), 7.73 (d, J = 8.5 Hz, 2H, H-C(2,6)), 8.11 (d, J = 7.3 Hz, 2H, H-
C(2′,6′)).
4B.2.3.5. (E)-1-(Phenyl)-3-(2-methoxyphenyl)-2-propen-1-one, A1B5 (25)
25 was synthesized by the general procedure using acetophenone A1 (0.584 mL, 5
mmol) and 2-methoxybenzaldehyde B5 (0.604 mL, 5 mmol). Yield 65%. M.p. 75-77 oC. IR (KBr): ν = 3055, 2940, 1651, 1600, 1595. 1H NMR (300 MHz, DMSO-D6): δ =
3.8 (s, 3H, H-C(OCH3)), 7.01 (d, J = 8.5 Hz, 2H, H-C(3,6)), 7.56 (bt, J = 6.11 Hz, 2H,
H-C(4,5)), 7.62-7.73 (m, 2H, H-Cα(Vinyl), H-C(4′)), 7.80 (d, J = 15.8 Hz, 1H, H-
Cβ(Vinyl)), 7.85 (d, J = 8.5 Hz, 2H, H-C(3′,5′)). 8.13 (d, J = 6.1 Hz, 2H, H-C(2′,6′)).
4B.2.3.6. (E)-1-(Phenyl)-3-(3-methoxyphenyl)-2-propen-1-one, A1B6 (26)
26 was synthesized by the general procedure using acetophenone A1 (0.584 mL, 5
mmol) and 3-methoxybenzaldehyde B6 (0.609 mL, 5 mmol). Yield 65%. M.p. 56-58 oC. IR (KBr): ν = 3034, 2973, 1637, 1587. 1H NMR (400 MHz, CD3OD): δ = 3.83 (s,
3H, H-C(OCH3)), 6.98 (ddd, J = 8.1 Hz, 1.6 Hz, 1.5 Hz, 1H, H-C(4)), 7.24 (dd, J =
1.4 Hz, 1.5 Hz, 1H, H-C(2)), 7.29-7.34 (m, 2H, H-C(5,6)), 7.52 (ddd, J = 7.8 Hz, 7.3

Chapter 4 ♦Experimental - 146 - Hz, 1.4 Hz, 2H, H-C(3′,5′)), 7.61-7.63 (m, 1H, H-C(4′)), 7.66 (d, J = 15.6 Hz, 1H, H-
Cα(Vinyl)), 7.74 (d, J = 15.8 Hz, 1H, H-Cβ(Vinyl)), 8.05 (ddd, J = 7.0 Hz, 1.4 Hz, 1.4
Hz, 2H, H-C(2′,6′)).
4B.2.3.7. (E)-1-(Phenyl)-3-(4-methoxyphenyl)-2-propen-1-one, A1B7 (27)
27 was synthesized by the general procedure using acetophenone A1 (0.584 mL, 5
mmol) and 4-methoxybenzaldehyde B7 (0.432 mL, 5 mmol). Yield 82%. M.p. 74-76 oC. IR (KBr): ν = 3034, 2895, 1651, 1596. 1H NMR (400 MHz, CD3OD): δ = 3.84 (s,
3H, H-C(OCH3)), 6.97 (dd, J = 7.0 Hz, 1.9, 2H, H-C(3,5)), 7.21 (dd, J = 7.0 Hz, 1.9
Hz, 1H, H-C(2,6)), 7.50-7.54 (m, 2H, H-C(3′,5′)), 7.59 (d, J = 15.5 Hz, 1H, H-
Cα(Vinyl)), 7.65-7.68 (m, 1H, H-C(4′)), 7.75 (d, J = 15.5 Hz, 1H, H-Cβ(Vinyl)), 8.04
(ddd, J = 7.3 Hz, 1.9 Hz, 1.4 Hz, 2H, H-C(2′,6′)).
4B.2.3.8. (E)-1-(Phenyl)-3-(3,4-dimethoxyphenyl)-2-propen-1-one, A1B8 (28)
28 was synthesized by the general procedure using acetophenone A1 (0.584 mL, 5
mmol) and 3,4-dimethoxybenzaldehyde B8 (0.830 g, 5 mmol). Yield 73%. M.p. 70-71 oC. IR (KBr): ν = 3041, 2914, 1651, 1600, 1595. 1H NMR (400 MHz, CD3OD): δ =
3.84 (s, 6H, H-C(OCH3)), 6.98 (d, J = 1.5 Hz, 1H, H-C(2)), 7.29 (dd, J = 9.3 Hz, 1.7
Hz, 1H, H-C(6)), 7.36 (d, J = 9.1 Hz, 1H, H-C(5)), 7.52 (ddd, J = 7.7 Hz, 7.3 Hz, 1.5
Hz, 2H, H-C(3′,5′)), 7.61 (d, J = 15.6 Hz, 1H, H-Cα(Vinyl)), 7.73 (d, J = 15.5 Hz, 1H,
H-Cβ(Vinyl)), 7.81 (ddd, J = 7.7 Hz, 1.5 Hz, 1.3 Hz, 2H, H-C(2′,6′)), 7.57-7.59 (m,
1H, H-C(4′)).
4B.2.3.9. (E)-1-(Phenyl)-3-(2-nitrophenyl)-2-propen-1-one, A1B9 (29)
29 was synthesized by the general procedure using acetophenone A1 (0.584 mL, 5
mmol) and 2-nitrobenzaldehyde B9 (0.775 g, 5 mmol). Yield 65%. M.p. 86-88 oC. IR
(KBr): ν = 3072, 1674, 1517, 1472, 1441. Anal. Calculated: C, 71.14%, H, 4.38%, N,
5.53%. Found: C, 71.34%, H, 4.55%, N, 5.65%.
4B.2.3.10. (E)-1-(Phenyl)-3-(3-nitrophenyl)-2-propen-1-one, A1B10 (30)
30 was synthesized by the general procedure using acetophenone A1 (0.584 mL, 5
mmol) and 3-nitrobenzaldehyde B10 (0.775 g, 5 mmol). Yield 83%. M.p. 119-121 ºC.
IR (KBr): ν = 3074, 1690, 1592, 1489, 1441. Anal. Calculated: C, 71.14%, H, 4.38%,
N, 5.53%. Found: C, 71.55%, H, 4.22%, N, 5.86%.

Chapter 4 ♦Experimental - 147 - 4B.2.3.11. (E)-1-(Phenyl)-3-(4-nitrophenyl)-2-propen-1-one, A1B11 (31)
31 was synthesized by the general procedure using acetophenone A1 (0.584 mL, 5
mmol) and 4-nitrobenzaldehyde B11 (0.775 g, 5 mmol). Yield 60%. M.p. 145 oC. IR
(KBr): ν = 3065, 1670, 1508, 1467, 1331. Anal. Calculated: C, 71.14%, H, 4.38%, N,
5.53%. Found: C, 71.37%, H, 4.98%, N, 5.22%.
4B.2.3.12. (E)-1-(Phenyl)-3-(2-chlorophenyl)-2-propen-1-one, A1B12 (32)
32 was synthesized by the general procedure using acetophenone A1 (0.584 mL, 5
mmol) and 2-chlorobenzaldehyde B12 (0.417 mL, 5 mmol). Yield 78%. M.p. 45-46
ºC. IR (KBr): ν = 3055, 1652, 1600, 1770. 1H NMR (400 MHz, CD3OD): δ = 7.08 (dt,
J = 7.5 Hz, 7.5 Hz, 1.5 Hz, 1H, H-C(4)), 7.10-7.12 (m, 1H, H-C(5)), 7.22 (dd, J = 7.4
Hz, 1.4 Hz, 1H, H-C(3)), 7.24 (dd, J = 7.5 Hz, 1.7 Hz, 1H, H-C(6)), 7.39 (d, J = 14.8
Hz, H-Cα(Vinyl)), 7.36-7.53 (m, 5H, H-C(arom.)), 8.17 (d, J = 14.9 Hz, 1H, H-
Cβ(Vinyl)). Anal. Calculated: C, 74.23%, H, 4.57%. Found: C, 73.93%, H, 4.58%.
4B.2.3.13. (E)-1-(Phenyl)-3-(3-chlorophenyl)-2-propen-1-one, A1B13 (33)
33 was synthesized by the general procedure using acetophenone A1 (0.584 mL, 5
mmol) and 3-chlorobenzaldehyde B13 (0.506 mL, 5 mmol). Yield 81%. M.p. 68-70
ºC. IR (KBr): ν = 1645, 1583, 1554, 689. Anal. Calculated: C, 74.23%, H, 4.57%.
Found: C, 74.18%, H, 4.57%.
4B.2.3.14. (E)-1-(Phenyl)-3-(4-chlorophenyl)-2-propen-1-one, A1B14 (34)
34 was synthesized by the general procedure using acetophenone A1 (0.584 mL, 5
mmol) and 4-chlorobenzaldehyde B14 (0.652 g, 5 mmol). Yield 82%. M.p. 114-116 oC. IR (KBr): ν = 3086, 1655, 1600, 761. Anal. Calculated: C, 74.23%, H, 4.57%.
Found: C, 74.07%, H, 4.70%.
4B.2.3.15. (E)-1-(Phenyl)-3-(4-fluorophenyl)-2-propen-1-one, A1B15 (35)
35 was synthesized by the general procedure using acetophenone A1 (0.584 mL, 5
mmol) and 4-fluorobenzaldehyde B15 (0.536 mL, 5 mmol). Yield 70%. M.p. 78-80 oC. IR (KBr): ν = 3043, 1658, 1594, 1157. 1H NMR (300 MHz, DMSO-D6): δ = 7.30
(t, J = 8.5 Hz, 2H, H-C(2)), 7.57 (t, J = 7.3 Hz, 2H, H-C(3′,5′)), 7.67 (t, J = 7.3 Hz,
1H, H-C(4′)), 7.74 (d, J = 14.6 Hz, 1H, H-Cα(Vinyl)), 7.91 (d, J = 15.9 Hz, 1H, H-
Cβ(Vinyl)), 7.98 (dd, J = 9.0 Hz, 7.0 Hz, 2H, H-C(3,5)), 8.15 (d, J = 7.3 Hz, 2H, H-
C(2′,6′)).

Chapter 4 ♦Experimental - 148 - 4B.2.3.16. (E)-1-(Phenyl)-3-(2-bromophenyl)-2-propen-1-one, A1B16 (36)
36 was synthesized by the general procedure using acetophenone A1 (0.584 mL, 5
mmol) and 2-bromobenzaldehyde B16 (0.584 mL, 5 mmol). Yield 83%. M.p. 98-100 oC. IR (KBr): ν = 3059, 1683, 1593, 1567, 751. Anal. Calculated: C, 62.74%, H,
3.86%. Found: C, 62.51%, H, 3.83%.
4B.2.3.17. (E)-1-(Phenyl)-3-(3-bromophenyl)-2-propen-1-one, A1B17 (37)
37 was synthesized by the general procedure using acetophenone A1 (0.584 mL, 5
mmol) and 3-bromobenzaldehyde B17 (0.580 mL, 5 mmol). Yield 85%. M.p. 82-84 oC. IR (KBr): ν = 3061, 1658, 1601, 1556, 769. Anal. Calculated: C, 62.74%, H,
3.86%. Found: C, 62.24%, H, 3.94%.
4B.2.3.18. (E)-1-(Phenyl)-3-(4-methylphenyl)-2-propen-1-one, A1B18 (38)
38 was synthesized by the general procedure using acetophenone A1 (0.584 mL, 5
mmol) and 4-methylbenzaldehyde B18 (0.587 mL, 5 mmol). Yield 79%. M.p. 92-94 oC. IR (KBr): ν = 3079, 2710, 1650, 1600, 1595. 1H NMR (400 MHz, CD3OD): δ =
2.35 (s, 3H, H-C(CH3)), 7.01 (dd, J = 7.9 Hz, 1.6 Hz, 2H, H-C(3,5)), 7.18 (dd, J = 7.7
Hz, 1.5 Hz, 1H, H-C(2,6)), 7.47 (dt, J = 8.0 Hz, 1.4 Hz, 2H, H-C(3′,5′)), 7.60 (m, 1H,
H-C(4′)), 7.67 (d, J = 15.7 Hz, 1H, H-Cα(Vinyl)), 7.80 (dd, J = 8.1 Hz, 1.4 Hz, 2H, H-
C(2′,6′)), 7.87 (d, J = 15.7 Hz, 1H, H-Cβ(Vinyl)). Anal. Calculated: C, 86.45%, H,
6.35%. Found: C, 86.66%, H, 6.61%.
4B.2.3.19. (E)-1-(Phenyl)-3-(3-hydroxy-4-methyoxyphenyl)-2-propen-1-one,
A1B19 (39)
39 was synthesized by the general procedure using acetophenone A1 (0.584 mL, 5
mmol) and isovanillin B19 (0.760 g, 5 mmol). Yield 78%. M.p. 108-110 oC. IR (KBr):
ν = 3400-3200, 3057, 2913, 1632, 1604. 1H NMR (300 MHz, CDCl3): δ = 3.98 (s, 3H,
H-C(OCH3)), 6.97 (d, J = 8.1 Hz, 1H, H-C(5)), 7.15 (d, J = 1.8 Hz, 1H, H-C(2)), 7.23
(dd, J = 8.1 Hz, 1.8 Hz, 1H, H-C(6)), 7.40 (d, J = 15.9 Hz, 1H, H-Cα(Vinyl)), 7.52 (t,
J = 7.8 Hz, 2H, H-C(3′,5′)), 7.60 (t, J = 7.2 Hz, 1H, H-C(4′)), 7.77 (d, J = 15.3 Hz,
1H, H-Cβ(Vinyl)), 8.02 (d, J = 8.4 Hz, 2H, H-C(2′,6′)).
4B.2.3.20. (E)-1-(Phenyl)-3-(4-N,N-dimethylaminophenyl)-2-propen-1-one, A1B20
(40)
40 was synthesized by the general procedure using acetophenone A1 (0.584 mL, 5
mmol) and 4-N,N-dimethylaminobenzaldehyde B20 (0.745 g, 5 mmol). Yield 65%.

Chapter 4 ♦Experimental - 149 - M.p. 105-106 oC. IR (KBr): ν = 2904, 1657, 1584. 1H NMR (400 MHz, CD3OD): δ =
3.03 (s, 6H, H-C(N(CH3)2)), 6.75 (dd, J = 7.3 Hz, 1.7 Hz, 2H, H-C(3,5)), 7.21 (dd, J =
7.8 Hz, 1.6 Hz, 2H, H-C(2,6)), 7.46 (d, J = 15.5 Hz, 1H, H-Cα(Vinyl)), 7.52 (ddd, J =
8.4 Hz, 7.8 Hz, 1.4 Hz, 2H, H-C(3′,5′)), 7.55-7.56 (m, 2H, H-C(2′,6′)), 7.74 (d, J =
15.4 Hz, 1H, H-Cβ(Vinyl)), 8.01-8.02 (m, 1H, H-C(4′)). Anal. Calculated: C, 81.24%,
H, 6.82%, N, 5.57%. Found: C, 81.48%, H, 6.98%, N, 5.59%.
4B.2.4. Parallel synthesis of members of active row (Pool BL2)
Each of the active row members (A1B2-A6B2, 2, 22, 41-44) was synthesized using 2-
hydroxybenzaldehyde; B2 (0.388 mL, 5 mmol) and one of the six different substituted
acetophenones (A1-A6) (5 mmol) following the general procedure as mentioned in
section 4B.1.
4B.2.4.1. (E)-1-(4′-Aminophenyl)-3-(2-hydroxyphenyl)-2-propen-1-one, A3B2 (2)
For the synthesis and analysis of 2, see section 4B.1.2.
4B.2.4.2. (E)-1-(Phenyl)-3-(2-hydroxyphenyl)-2-propen-1-one, A1B2 (22)
For the synthesis and analysis of 22, see section 4B.2.3.2.
4B.2.4.3. (E)-1-(2′-Hydroxyphenyl)-3-(2-hydroxyphenyl)-2-propen-1-one, A2B2
(41)
41 was synthesized by the general procedure using 2′-hydroxyacetophenone (0.601
mL, 5 mmol). Yield 78%. M.p. 160-162 °C. IR (KBr): ν = 3445-3450, 3060, 2915,
1632, 1570, 1265. Anal. Calculated: C, 74.99%, H, 5.03%. Found: C, 74.87%, H,
5.32%.
4B.2.4.4. (E)-1-(2′,3′,4′-Trimethoxyphenyl)-3-(2-hydroxyphenyl)-2-propen-1-one,
A4B2 (42)
42 was synthesized by the general procedure using 2′,3′,4′-trimethoxyacetophenone
(0.677 mL, 5 mmol). Yield 81%. M.p. 114-116 ºC. IR (KBr): ν = 3230-3450, 2986,
2967, 2874, 1634, 1595. 1H NMR (300 MHz, CDCl3): δ = 3.88, 3.92, 3.94 (s, 3H, H-
C(OCH3)), 6.73 (d, J = 9.0 Hz, 1H, H-C(5′)), 6.77 (d, J = 7.8 Hz, 1H, H-C(3)), 6.91
(d, J = 7.8 Hz, 1H, H-C(5)), 6.98 (t, J = 8.4 Hz, 1H, H-C(4)), 7.25 (d, J = 16.5 Hz,
1H, H-Cα(Vinyl)), 7.58 (m, 2H, H-C(6,6′)), 8.07 (d, J = 16.4 Hz, 1H, H-Cβ(Vinyl)).
Anal. Calculated: C, 68.78%, H, 5.77%. Found: C, 68.75%, H, 5.65%.

Chapter 4 ♦Experimental - 150 - 4B.2.4.5. (E)-1-(3′,4′,5′-Trimethoxyphenyl)-3-(2-hydroxyphenyl)-2-propen-1-one,
A5B2 (43)
43 was synthesized by the general procedure using 3′,4′,5′-trimethoxyacetophenone
(1.05 g, 5 mmol). Yield 75%. Oil. IR (KBr): ν = 3400-3200, 2949, 2987, 2838, 1631,
1604. 1H NMR (300 MHz, CDCl3): δ = 3.91-3.92 (bs, 9H, H-C(OCH3)), 6.93 (t, J =
8.7 Hz, 2H, H-C(3,6)), 7.15(t, J = 7.2 Hz, 1H, H-C(4)), 7.19-7.28 (m, 3H, H-
C(2′,6′,5)), 7.39 (d, J = 15.3 Hz, 1H, H-Cα(Vinyl)), 7.83 (d, J = 15.4 Hz, 1H, H-
Cβ(Vinyl)). Anal. Calculated: C, 68.78%, H, 5.77%. Found: C, 68.72%, H, 5.52%.
4B.2.4.6. (E)-1-(2′,4′-Dibromophenyl)-3-(2-hydroxyphenyl)-2-propen-1-one, A6B2
(44)
44 was synthesized by the general procedure using 2′,4′-dibromoacetophenone (1.39g,
5 mmol). Yield 61%. Oil. IR (KBr): ν = 3220-3430, 1659, 1562, 796. Anal.
Calculated: C, 47.16%, H, 2.64%. Found: C, 47.60%, H, 2.40%. 4B.3. Synthesis of 175 member chalcone library147
A 175 member chalcones library was designed and synthesized as the following two
sets of indexed libraries.
4B.3.1. Synthesis of indexed libraries (AL1-AL7)
General Procedure. Microwave assisted synthesis of chalcone libraries was carried
out according to a modified literature procedure.ref To an EtOH soln. (40 mL) of one
acetophenone (from A1–A7; 25 mmol) and a mixture of the 25 aldehydes (B1-B25, 1
mmol each; 1 × 25 = 25 mmol in total) was added a 4M soln. of aq. NaOH (30 mL).
The mixture was exposed to microwave irradiation for 3 min, and then left at 0–4 °C
overnight. The mass obtained was cooled and neutralized with cold dil. aq. HCl. The
chalcones were successively extracted with AcOEt (5 × 10 mL) and CH2Cl2 (5 × 10
mL). All org. phases were combined and concentrated in vacuo, which led to a dark-
brown solid mass.
4B.3.1.1. Synthesis of Pool AL1. AL1 was prepared according to the general
procedure by using acetophenone A1 (2.92 mL, 25 mmol) with a mixture of twenty
five substituted aldehydes B1-B25 (Table 4.3).
4B.3.1.2. Synthesis of pool AL2. AL2 was prepared by using 2′-
hydroxyacetophenone A2 (3.01 mL, 25 mmol) and aldehydes B1-B25 (25 mmol total)
(Table 4.3).

Chapter 4 ♦Experimental - 151 - Table. 4.3. Quantities of aldehydes used for the synthesis of AL1-AL7.
O
R
H
O
R H
B1-B17 B18-B25
Aldehydes R Quantity Aldehydes R Quantity
B1 H 0.101 mL B14 3-Nitro 0.155 g
B2 2-Hydroxy 0.078 mL B15 3-Hydroxy-4-methoxy 0.152 g
B3 3-Hydroxy 0.122 g B16 4-N,N-dimethylamino 0.149 g
B4 4-Hydroxy 0.122 g B17 4-Methyl 0.118 mL
B5 2-Methoxy 0.121 mL B18 2-Thienyl 0.092 mL
B6 3-Methoxy 0.122 mL B19 5-Methyl-2-thienyl 0.108 mL
B7 4-Methoxy 0.086 mL B20 5-Bromo-2-thienyl 0.119 mL
B8 3,4-Dimethoxy 0.166 g B21 5-Nitro-2-thienyl 0.156 g
B9 2-Chloro 0.083 mL B22 2-Pyrrolyl 0.095 g
B10 3-Chloro 0.105 mL B23 2-Pyridyl 0.095 mL
B11 4-Chloro 0.130 g B24 3-Pyridyl 0.094 mL
B12 3-Bromo 0.116 mL B25 4-Pyridyl 0.095 mL
B13 4-Fluoro 0.107 mL
a 1 mmol of each of aldehydes was used in the synthesis of libraries AL1-AL7.
2B.3.1.3. Synthesis of pool AL3. AL3 was prepared by using 3′-
hydroxyacetophenone A3 (3.40 g, 25 mmol) and aldehydes B1-B25 (25 mmol total)
(Table 4.3).
4B.3.1.4. Synthesis of pool AL4. AL4 was prepared by using 4′-
hydroxyacetophenone A4 (3.40 g, 25 mmol) and aldehydes B1-B25 (25 mmol total)
(Table 4.3).
4B.3.1.5. Synthesis of pool AL5. AL5 was prepared by using 2′-aminoacetophenone
A5 (3.38 g, 25 mmol) and aldehydes B1-B25 (25 mmol total) (Table 4.3).
4B.3.1.6. Synthesis of pool AL6. AL6 was prepared by using 3′-aminoacetophenone
A6 (3.38 g, 25 mmol) and aldehydes B1-B25 (25 mmol total) (Table 4.3).
4B.3.1.7. Synthesis of pool AL7. Pool AL7 was prepared by using 4′-
aminoacetophenone A7 (3.38 g, 25 mmol) and aldehydes B1-B25 (25 mmol total)
(Table 4.3).

Chapter 4 ♦Experimental - 152 - 4B.3.2. Synthesis of indexed libraries (BL1-BL25)
All the libraries of Set 2 were synthesized according to the general procedure
described for Set 1 (Section 4B.3.1.), by reacting an EtOH soln. (10mL) of the
corresponding aldehyde (one of B1-B25; 7 mmol) with a mixture of seven
acetophenones (all of A1-A7, 1 mmol each; 1 × 7 = 7 mmol in total) in the presence of
4M aq. NaOH soln. (10mL).
Table. 4.4. Quantities of acetophenones used for the synthesis of indexed libraries BL1-BL25.
CH3
O
R
Acetophenones R Quantitya Acetophenones R Quantitya
A1 H 0.117 mL A5 2′-Amino 0.135 g
A2 2′-Hydroxy 0.120 mL A6 3′-Amino 0.135 g
A3 3′-Hydroxy 0.136 g A7 4′-Amino 0.135 g
A4 4′-Hydroxy 0.136 g
a 1 mmol of each of acetophenone was used in the synthesis of libraries BL1-BL25.
4B.3.2.1. Synthesis of BL1. Pool BL1 was prepared following the general procedure
using equimolar quantities of benzaldehyde B1 (0.710 mL, 7 mmol) with a mixture of
acetophenones A1-A7 (Table 4.4).
4B.3.2.2. Synthesis of BL2. Pool BL2 was prepared by using 2-hydroxybenzaldehyde
B2 (0.543 mL, 7 mmol) and a mixture of seven acetophenones A1-A7 (7 mmol total)
(Table 4.4).
4B.3.2.3. Synthesis of BL3. Pool BL3 was prepared by using 3-hydroxybenzaldehyde
B3 (0.854 g, 7 mmol) and a mixture of acetophenones A1-A7 (7 mmol total) (Table
4.4).
4B.3.2.4. Synthesis of BL4. Pool BL4 was prepared by using 4-hydroxybenzaldehyde
B4 (0.854 g, 7 mmol) and a mixture of acetophenones A1-A7 (7 mmol total) (Table
4.4).
4B.3.2.5. Synthesis of BL5. Pool BL5 was prepared by using 2-methoxybenzaldehyde
B5 (0.845 mL, 7 mmol) and a mixture of acetophenones A1-A7 (7 mmol total) (Table
4.4).

Chapter 4 ♦Experimental - 153 - 4B.3.2.6. Synthesis of BL6. Pool BL6 was prepared by using 3-methoxybenzaldehyde
B6 (0.852 mL, 7 mmol) and a mixture of acetophenones A1-A7 (7 mmol total) (Table
4.4).
4B.3.2.7. Synthesis of BL7. Pool BL7 was prepared by using 4-methoxybenzaldehyde
B7 (0.605 mL, 7 mmol) and a mixture of acetophenones A1-A7 (7 mmol total) (Table
4.4).
4B.3.2.8. Synthesis of BL8. Pool BL8 was prepared by using 3,4-
dimethoxybenzaldehyde B8 (1.162 g, 7 mmol) and a mixture of acetophenones A1-A7
(7 mmol total) (Table 4.4).
4B.3.2.9. Synthesis of BL9. Pool BL9 was prepared by using 2-chlorobenzaldehyde
B9 (0.583 mL, 7 mmol) and a mixture of acetophenones A1-A7 (7 mmol total) (Table
4.4).
4B.3.2.10. Synthesis of BL10. Pool BL10 was prepared by using 3-
chlorobenzaldehyde B10 (0.736 mL, 7 mmol) and a mixture of acetophenones A1-A7
(7 mmol total) (Table 4.4).
4B.3.2.11. Synthesis of BL11. Pool BL11 was prepared by using 4-
chlorobenzaldehyde B11 (0.914 g, 7 mmol) and a mixture of acetophenones A1-A7 (7
mmol total) (Table 4.4).
4B.3.2.12. Synthesis of BL12. Pool BL12 was prepared by using 3-
bromobenzaldehyde B12 (0.812 mL, 7 mmol) and a mixture of acetophenones A1-A7
(7 mmol total) (Table 4.4).
4B.3.2.13. Synthesis of BL13. Pool BL13 was prepared by using 4-fluorobenzaldehyde
B13 (0.750 mL, 7 mmol) and a mixture of acetophenones A1-A7 (7 mmol total) (Table
4.4).
4B.3.2.14. Synthesis of BL14. Pool BL14 was prepared by using 3-nitrobenzaldehyde
B14 (1.085 g, 7 mmol) and a mixture of acetophenones A1-A7 (7 mmol total) (Table
4.4).
4B.3.2.15. Synthesis of BL15. Pool BL15 was prepared by using 4-hydroxy-3-
methoxybenzaldehyde B15 (1.064 g, 7 mmol) and a mixture of acetophenones A1-A7
(7 mmol total) (Table 4.4).
4B.3.2.16. Synthesis of BL16. Pool BL16 was prepared by using 4-N,N-
dimethylaminobenzaldehyde B16 (1.043 g, 7 mmol) and a mixture of acetophenones
A1-A7 (7 mmol total) (Table 4.4).

Chapter 4 ♦Experimental - 154 - 4B.3.2.17. Synthesis of BL17. Pool BL17 was prepared by using 4-
methylbenzaldehyde B17 (0.824 mL, 7 mmol) and a mixture of acetophenones A1-A7
(7 mmol total) (Table 4.4).
4B.3.2.18. Synthesis of BL18. Pool BL18 was prepared by using thiophene-2-
carboxaldehyde B18 (0.644 mL, 7 mmol) and a mixture of acetophenones A1-A7 (7
mmol total) (Table 4.4).
4B.3.2.19. Synthesis of BL19. Pool BL19 was prepared by using 5-methylthiophene-2-
carboxaldehyde B19 (0.756 mL, 7 mmol) and a mixture of acetophenones A1-A7 (7
mmol total) (Table 4.4).
4B.3.2.20. Synthesis of BL20. Pool BL20 was prepared by using 5-bromothiophene-2-
carboxaldehyde B20 (0.840 mL, 7 mmol) and a mixture of acetophenones A1-A7 (7
mmol total) (Table 4.4).
4B.3.2.21. Synthesis of BL21. Pool BL21 was prepared by using 5-nitrothiophene-2-
carboxaldehyde B21 (1.092 g, 7 mmol) and a mixture of acetophenones A1-A7 (7
mmol total) (Table 4.4).
4B.3.2.22. Synthesis of BL22. Pool BL22 was prepared by using pyrrole-2-
carboxaldehyde B22 (0.672 g, 7 mmol) and a mixture of acetophenones A1-A7 (7
mmol total) (Table 4.4).
4B.3.2.23. Synthesis of BL23. Pool BL23 was prepared by using pyridine-2-
carboxaldehyde B23 (0.672 mL, 7 mmol) and a mixture of acetophenones A1-A7 (7
mmol total) (Table 4.4).
4B.3.2.24. Synthesis of BL24. Pool BL24 was prepared by using pyridine-3-
carboxaldehyde B24 (0.658 mL, 7 mmol) and a mixture of acetophenones A1-A7 (7
mmol total) (Table 4.4).
4B.3.2.25. Synthesis of BL25. Pool BL25 was prepared by using pyridine-4-
carboxaldehyde B25 (0.672 mL, 7 mmol) and a mixture of acetophenones A1-A7 (7
mmol total) (Table 4.4).
4B.3.3. Synthesis of members of active column AL1 (A1B1-A1B25)
General Procedure: To an ethanolic solution (20 mL) of acetophenone A1 (1.168 mL,
10 mmol) and an aldehyde B (10 mmol) was added a 4.0 M aq. NaOH soln. (60 mL),
and the mixture was irradiated in a microwave oven till the reaction was found to be
complete. The mixture was kept at 0–4°C overnight, neutralized with ice-cold dil. aq.
HCl. The resulting precipitate was filtered off and purified either by recrystallization

Chapter 4 ♦Experimental - 155 - from EtOH or by flash chromatography (SiO2; hexane/AcOEt). The purity of the
compounds was checked by multiple TLC (SiO2; hexane/AcOEt 3:1, 2:1 and 1:1).
The synthesis of A1B1-A1B8 (21-28) has already been discussed in Sections 4B.2.3.1-
4B.2.3.8.
4B.3.3.1. (E)-1-(Phenyl)-3-(3-nitrophenyl)-2-propen-1-one, A1B14 (30)
For details, see Section 4B.2.3.10.
4B.3.3.2. (E)-1-(Phenyl)-3-(2-chlorophenyl)-2-propen-1-one, A1B9 (32)
For details, see Section 4B.2.3.12.
4B.3.3.3. (E)-1-(Phenyl)-3-(3-chlorophenyl)-2-propen-1-one, A1B10 (33)
For details, see Section 4B.2.3.13.
4B.3.3.4. (E)-1-(Phenyl)-3-(4-chlorophenyl)-2-propen-1-one, A1B11 (34)
For details, see Section 4B.2.3.14.
4B.3.3.5. (E)-1-(Phenyl)-3-(4-fluorophenyl)-2-propen-1-one, A1B13 (35)
For details, see Section 4B.2.3.15.
4B.3.3.6. (E)-1-(Phenyl)-3-(3-bromophenyl)-2-propen-1-one, A1B12 (37)
For details, see Section 4B.2.3.17.
4B.3.3.7. (E)-1-(Phenyl)-3-(4-methylphenyl)-2-propen-1-one, A1B17 (38)
For details, see Section 4B.2.3.18.
4B.3.3.8. (E)-1-(Phenyl)-3-(3-hydroxy-4-methoxyphenyl)-2-propen-1-one,
A1B15 (39)
For details, see Section 4B.2.3.19.
4B.3.3.9. (E)-1-(Phenyl)-3-(4-N,N-dimethylaminophenyl)-2-propen-1-one, A1B16
(40).
For details, see Section 4B.2.3.20.
4B.3.3.10. (E)-1-(Phenyl)-3-(thiophen-2-yl)-2-propen-1-one, A1B18 (45)
45 was prepared according to the general procedure using acetophenone A1 (1.168
mL, 10 mmol) and thiophen-2-carboxaldehyde B18 (0.920 mL, 10 mmol). Yield 79%.
M.p. 62-64 ºC. IR (KBr): ν = 1656, 1587. 1H NMR (300 MHz, CDCl3): δ = 7.12 (dd,
J = 4.9, 3.6 Hz, 1H, H-C(4)), 7.32 (d, J = 15.9 Hz, 1H, H-Cα(Vinyl)), 7.38 (bs, 1H, H-
C(3)), 7.44 (d, J = 4.8 Hz, 1H, H-C(5)), 7.52 (t, J = 7.5 Hz, 1H, H-C(3′,5′)), 7.61 (t, J
= 7.2 Hz, 1H, H-C(4′)), 7.96 (d, J = 15.3 Hz, 1H, H-Cβ(Vinyl)), 8.03 (d, J = 7.2 Hz,

Chapter 4 ♦Experimental - 156 - 1H, H-C(2′,6′)). Anal. Calculated: C, 72.87%, H, 4.70%, S, 14.96%. Found: C,
72.47%, H, 4.24%, S, 14.54%.
4B.3.3.11. (E)-1-(Phenyl)-3-(5-methylthiophen-2-yl)-2-propen-1-one, A1B19 (46)
46 was prepared according to the general procedure using acetophenone A1 (1.168
mL, 10 mmol) and 5-methylthiophen-2-carboxaldehyde B19 (1.080 mL, 10 mmol).
Yield 80%. M.p. 78-80 ºC. IR (KBr): ν = 1656, 1588. 1H NMR (300 MHz, CDCl3): δ
= 2.53 (s, 3H, H-C(CH3), 6.76 (d, J = 3.6 Hz, 1H, H-C(4)), 7.18 (d, J = 3.6 Hz, 1H, H-
C(3)), 7.21 (d, J = 15.3 Hz, 1H, H-Cα(Vinyl)), 7.51 (t, J = 7.2 Hz, 1H, H-C(3′,5′)),
7.59 (t, J = 7.2 Hz, 1H, H-C(4′)), 7.88 (d, J = 15.3 Hz, 1H, H-Cβ(Vinyl)), 8.01 (d, J =
7.2 Hz, 1H, H-C(2′,6′)). Anal. Calculated: C, 73.65%, H, 5.30%, S, 14.04%. Found:
C, 73.50%, H, 5.39%, S, 14.09%.
4B.3.3.12. (E)-1-(Phenyl)-3-(5-bromothiophen-2-yl)-2-propen-1-one, A1B20 (47)
47 was prepared according to the general procedure using acetophenone A1 (1.168
mL, 10 mmol) and 5-bromothiophen-2-carboxaldehyde B20 (1.200 mL, 10 mmol).
Yield 77%. M.p. 100-101 ºC. IR (KBr): ν = 1657, 1591. 1H NMR (300 MHz, CDCl3):
δ = 7.01 (d, J = 3.9 Hz, 1H, H-C(4)), 7.11 (d, J = 3.9 Hz, 1H, H-C(3)), 7.25 (d, J =
15.3 Hz, 1H, H-Cα(Vinyl)), 7.52 (t, J = 7.5 Hz, 2H, H-C(3′,5′)), 7.61 (t, J = 7.2 Hz,
1H, H-C(4′)), 7.82 (d, J = 15.3 Hz, 1H, H-Cβ(Vinyl)), 8.00 (d, J = 7.5 Hz, 2H, H-
C(2′,6′)). Anal. Calculated: C, 53.26%, H, 3.09%, S, 10.94%. Found: C, 53.45%, H,
2.93%, S, 10.53%.
4B.3.3.13. (E)-1-(Phenyl)-3-(5-nitrothiophen-2-yl)-2-propen-1-one, A1B21 (48)
48 was prepared according to the general procedure using acetophenone A1 (1.168
mL, 10 mmol) and 5-nitrothiophen-2-carboxaldehyde B21 (1.560 g, 10 mmol). Yield
83%. M.p. 200 ºC (decomp). IR (KBr): ν = 1670, 1595. Anal. Calculated: C, 60.22%,
H, 3.50%, N, 5.40%, S, 12.37%. Found: C, 60.15%, H, 3.45%, N, 5.49%, S, 12.50%.
4B.3.3.14. (E)-1-(Phenyl)-3-(pyrrol-2-yl)-2-propen-1-one, A1B22 (49)
49 was prepared according to the general procedure using acetophenone A1 (1.168
mL, 10 mmol) and pyrrole-2-carboxaldehyde B22 (0.960 g, 10 mmol). Yield 85%.
M.p. 133-134 ºC. IR (KBr): ν = 1652, 1586. 1H NMR (300 MHz, CDCl3): δ 7.14 (dd,
J = 5.1, 6.0 Hz, 1H, H-C(4)), 7.35 (d, J = 15.1 Hz, 1H, H-Cα(Vinyl)), 7.50 (t, J = 7.8
Hz, 2H, H-C(3′,5′)), 7.60-7.68 (m, 2H, H-C(4′,3)), 7.94 (d, J = 7.2 Hz, 2H, H-

Chapter 4 ♦Experimental - 157 - C(2′,6′)), 8.39 (d, J = 15.0 Hz, 1H, H-Cβ(Vinyl)). Anal. Calculated: C, 79.16%, H,
5.62%, N, 7.10%. Found: C, 79.57%, H, 5.73%, N, 7.14%.
4B.3.3.15. (E)-1-(Phenyl)-3-(pyridin-2-yl)-2-propen-1-one, A1B23 (50)
50 was prepared according to the general procedure using acetophenone A1 (1.168
mL, 10 mmol) and pyridine-2-carboxaldehyde B23 (0.960 mL, 10 mmol). Yield 76%.
M.p. 119-120 ºC. IR (KBr): ν = 1677, 1589. Anal. Calculated: C, 80.36%, H, 5.30%,
N, 6.69%. Found: C, 80.99%, H, 5.98%, N, 6.35%.
4B.3.3.16. (E)-1-(Phenyl)-3-(pyridin-3-yl)-2-propen-1-one, A1B24 (51)
51 was prepared according to the general procedure using acetophenone A1 (1.168
mL, 10 mmol) and pyridine-3-carboxaldehyde B24 (0.940 mL, 10 mmol). Yield 87%.
M.p. 88-112 ºC. IR (KBr) ν (cm-1): 1675, 1591. Anal. Calculated: C, 80.36%, H,
5.30%, N, 6.69%. Found: C, 80.67%, H, 5.72%, N, 6.26%.
4B.3.3.17. (E)-1-(Phenyl)-3-(pyridin-4-yl)-2-propen-1-one, A1B25 (52)
52 was prepared according to the general procedure using acetophenone A1 (1.168
mL, 10 mmol) and pyridine-4-carboxaldehyde B25 (0.960 mL, 10 mmol). Yield 75%.
M.p. 92-96 ºC (decomp). IR (KBr): ν = 1682, 1597. Anal. Calculated: C, 80.36%, H,
5.30%, N, 6.69%. Found: C, 80.82%, H, 5.56%, N, 6.52%.
4B.3.4. Synthesis of members of active column AL3 (A3B1-A3B25)
Each of A3B1-A3B25 was synthesized and worked up following the general procedure
(section 4B.3.3.) using an EtOH soln. (20 mL) of 3′-hydroxyacetophenone A3 (0.68 g,
5 mmol) and one of twentyfive aldehydes (B1-B25) (5 mmol) added to 4M soln. of aq.
NaOH (30 mL).
4B.3.4.1. (E)-1-(3′-Hydroxyphenyl)-3-(phenyl)-2-propen-1-one, A3B1 (53)
53 was prepared by using 3′-hydroxyacetophenone A3 (0.68 g, 5 mmol) and
benzaldehyde B1 (0.507 mL, 5 mmol). Yield 88%. M.p. 127-130 ºC. IR (KBr): ν =
3248, 1644, 1593. 1H NMR (300 MHz, CDCl3): δ = 6.46-7.42 (m, 9H, H-C(arom.)),
7.60 (d, 1H, J = 15.9 Hz, H-Cα(Vinyl)), 7.83 (d, J = 15.9 Hz, 1H, H-Cβ(Vinyl)), 10.0
(s, 1H, H-O(OH)).
4B.3.4.2. (E)-1-(3′-Hydroxyphenyl)-3-(2-hydroxyphenyl)-2-propen-1-one, A3B2
(54)
54 was prepared by using 3′-hydroxyacetophenone A3 (0.68 g, 5 mmol) and 2-
hydroxybenzaldehyde B2 (0.388 mL, 5 mmol). Yield 75%. M.p 152 ºC. IR (KBr): ν =

Chapter 4 ♦Experimental - 158 - 3364, 1652, 1606. 1H NMR (300 MHz, CDCl3): δ = 6.63-7.48 (m, 8H, H-C(arom.)),
7.50 (d, J = 15.9 Hz, 1H, H-Cα(Vinyl)), 7.83 (d, J = 15.9 Hz, 1H, H-Cβ(Vinyl)), 9.90
(bs, 1H, H-O(OH)).
4B.3.4.3. (E)-1-(3′-Hydroxyphenyl)-3-(3-hydroxyphenyl)-2-propen-1-one, A3B3
(55)
55 was prepared by using 3′-hydroxyacetophenone A3 (0.68 g, 5 mmol) and 3-
hydroxybenzaldehyde B3 (0.61 g, 5 mmol). Yield 80%. M.p. 182 ºC. IR (KBr): ν =
3409, 1644, 1585. 1H NMR (300 MHz, CDCl3): δ = 6.86-7.59 (m, 8H, H-C(arom.)),
7.59 (d, J = 15.6 Hz, 1H, H-Cα(Vinyl)), 7.71 (d, J = 15.6 Hz, 1H, H-Cβ(Vinyl)), 9.68
(s, 1H, H-O(OH)), 9.85 (s, 1H, H-O(OH)).
4B.3.4.4. (E)-1-(3′-Hydroxyphenyl)-3-(4-hydroxyphenyl)-2-propen-1-one, A3B4
(56)
56 was prepared by using 3′-hydroxyacetophenone A3 (0.68 g, 5 mmol) and 4-
hydroxybenzaldehyde B4 (0.61 g, 5 mmol). Yield 78%. M.p. 194-195 ºC. IR (KBr): ν
= 3339, 1653, 1561. 1H NMR (300 MHz, CDCl3): δ = 7.16-7.50 (m, 8H, H-C(arom.)),
7.56 (d, J = 15.0 Hz, 1H, H-Cα(Vinyl)), 7.88 (d, J = 14.8 Hz, 1H, H-Cβ(Vinyl)), 9.73
(s, 1H, H-O(OH)), 10.06 (s, 1H, H-O(OH)).
4B.3.4.5. (E)-1-(3′-Hydroxyphenyl)-3-(2-methoxyphenyl)-2-propen-1-one, A3B5
(57)
57 was prepared by using 3′-hydroxyacetophenone A3 (0.68 g, 5 mmol) and 2-
methoxybenzaldehyde B5 (0.60 mL, 5 mmol). Yield 73%. M.p. 128 ºC. IR (KBr): ν =
3347, 1651, 1555. 1H NMR (300 MHz, CDCl3): δ = 3.84 (s, 3H, H-C(OCH3)), 6.66-
7.56 (m, 8H, H-C(arom.)), 7.42 (d, J = 15.3 Hz, 1H, H-Cα(Vinyl)), 8.16 (d, J = 15.6
Hz, 1H, H-Cβ(Vinyl)), 9.00 (bs, 1H, H-O(OH)).
4B.3.4.6. (E)-1-(3′-Hydroxyphenyl)-3-(3-methoxyphenyl)-2-propen-1-one, A3B6
(58)
58 was prepared by using 3′-hydroxyacetophenone A3 (0.68 g, 5 mmol) and 3-
methoxybenzaldehyde B6 (0.608 mL, 5 mmol). Yield 82%. M.p. 143 ºC. IR (KBr): ν
= 3361, 1660, 1576. 1H NMR (300 MHz, CDCl3): δ = 4.59 (s, 3H, H-C(OCH3)), 6.76-
7.58 (m, 8H, H-C(arom.)), 7.67 (d, J = 15.8 Hz, 1H, H-Cα(Vinyl)), 7.83 (d, J = 15.6
Hz, 1H, H-Cβ(Vinyl)), 9.87 (bs, 1H, H-O(OH)).

Chapter 4 ♦Experimental - 159 - 4B.3.4.7. (E)-1-(3′-Hydroxyphenyl)-3-(4-methoxyphenyl)-2-propen-1-one, A3B7
(59)
59 was prepared by using 3′-hydroxyacetophenone A3 (0.68 g, 5 mmol) and 4-
methoxybenzaldehyde B7 (0.252 mL, 5 mmol). Yield 81%. M.p. 129-130 ºC. IR
(KBr): ν = 3350, 1651, 1546. 1H NMR (300 MHz, CDCl3): δ = 3.41 (s, 3H, H-
C(OCH3)), 6.60-7.43 (m, 8H, H-C(arom.)), 7.58 (d, J = 15.6 Hz, 1H, H-Cα(Vinyl)),
7.85 (d, J = 15.6 Hz, 1H, H-Cβ(Vinyl)), 8.96 (bs, 1H, H-O(OH)).
4B.3.4.8. (E)-1-(3′-Hydroxyphenyl)-3-(3,4-dimethoxyphenyl)-2-propen-1-one,
A3B8 (60)
60 was prepared by using 3′-hydroxyacetophenone A3 (0.68 g, 5 mmol) and 3,4-
dimethoxybenzaldehyde B8 (0.83 g, 5 mmol). Yield 90%. M.p. 135 ºC (decomp). IR
(KBr): ν = 3447, 1648, 1567. 1H NMR (300 MHz, CDCl3): δ = 3.45, 3.47 (s, 6H, H-
C(OCH3)), 6.63-7.59 (m, 7H, H-C(arom.)), 7.55 (d, J = 15.6 Hz, 1H, H-Cα(Vinyl)),
8.00 (d, J = 15.6 Hz, 1H, H-Cβ(Vinyl)), 8.42 (bs, 1H, H-O(OH)).
4B.3.4.9. (E)-1-(3′-Hydroxyphenyl)-3-(2-chlorophenyl)-2-propen-1-one, A3B9
(61)
61 was prepared by using 3′-hydroxyacetophenone A3 (0.68 g, 5 mmol) and 2-
chlorobenzaldehyde B9 (0.416 mL, 5 mmol). Yield 77%. M.p. 150 ºC (decomp). IR
(KBr): ν = 3327, 1652, 1585, 786. 1H-NMR (300 MHz, CDCl3): δ = 7.03-7.61 (m,
8H, H-C(arom.)), 7.64 (d, J = 15.9 Hz, 1H, H-Cα(Vinyl)), 7.83 (d, J = 15.9 Hz, 1H,
H-Cβ(Vinyl)), 10.10 (bs, 1H, H-O(OH)).
4B.3.4.10. (E)-1-(3′-Hydroxyphenyl)-3-(3-chlorophenyl)-2-propen-1-one, A3B10
(62)
62 was prepared by using 3′-hydroxyacetophenone A3 (0.68 g, 5 mmol) and 3-
chlorobenzaldehyde B10 (0.526 mL, 5 mmol). Yield 82%. M.p. 127-128 ºC. IR (KBr):
ν = 3404, 1661, 1582, 784. 1H NMR (300 MHz, CDCl3): δ = 7.04-7.93 (m, 8H, H-
C(arom.)), 7.62 (d, J = 15.9 Hz, 1H, H-Cα(Vinyl)), 8.10 (d, J = 15.6 Hz, 1H, H-
Cβ(Vinyl)), 8.76 (bs, 1H, H-O(OH)).
4B.3.4.11. (E)-1-(3′-Hydroxyphenyl)-3-(4-chlorophenyl)-2-propen-1-one, A3B11
(63)
63 was prepared by using 3′-hydroxyacetophenone A3 (0.68 g, 5 mmol) and 4-
chlorobenzaldehyde B11 (0.652 g, 5 mmol). Yield 84%. M.p. 155 ºC. IR (KBr): ν =

Chapter 4 ♦Experimental - 160 - 3317, 1646, 1546, 780. 1H NMR (300 MHz, CDCl3): δ = 7.06-8.05 (m, 8H, H-
C(arom.)), 7.63 (d, J = 15.4 Hz, 1H, H-Cα(Vinyl)), 8.10 (d, J = 15.6 Hz, 1H, H-
Cβ(Vinyl)), 10.03 (bs, 1H, H-O(OH)). Anal. Calculated: C, 69.64%, H, 4.29%. Found:
C, 69.44%, H, 4.61%.
4B.3.4.12. (E)-1-(3′-Hydroxyphenyl)-2-(3-bromophenyl)-2-propen-1-one, A3B12
(64)
64 was prepared by using 3′-hydroxyacetophenone A3 (0.68 g, 5 mmol) and 3-
bromobenzaldehyde B12 (0.580 mL, 5 mmol). Yield 76%. M.p. 182 ºC. IR (KBr): ν =
3409, 1661, 1584. 1H NMR (300 MHz, CDCl3): δ = 7.01-7.63 (m, 8H, H-C(arom.)),
7.51 (d, J = 15.6 Hz, 1H, H-Cα(Vinyl)), 8.13 (d, J = 15.6 Hz, 1H, H-Cβ(Vinyl)), 8.79
(bs, 1H, H-O(OH)).
4B.3.4.13. (E)-1-(3′-Hydroxyphenyl)-2-(4-fluorophenyl)-2-propen-1-one, A3B13
(65)
65 was prepared by using 3′-hydroxyacetophenone A3 (0.68 g, 5 mmol) and 4-
fluorobenzaldehyde B13 (0.536 mL, 5 mmol). Yield 84%. M.p. 136 ºC. IR (KBr): ν =
3297, 1647, 1568. 1H NMR (300 MHz, CDCl3): δ = 7.07 (dd, J = 7.8 Hz, 2.4 Hz, 1H,
H-C(4′)), 7.28 (t, J = 7.8 Hz, 2H, H-C(2,6)), 7.37 (t, J = 8.1 Hz, 1H, H-C(5′)), 7.49 (s,
1H, H-C(2′)), 7.63 (d, J = 8.1 Hz, 1H, H-C(6′)), 7.70 ( d, J = 15.9 Hz, 1H, H-
Cα(Vinyl)), 7.85 (d, J = 15.9 Hz, 1H, H-Cβ(Vinyl)), 7.95 (dd, J = 9.6 Hz, 7.4 Hz, 2H,
H-C(3,5)), 9.90 (s, 1H, H-O(OH)).
4B.3.4.14. (E)-1-(3′-Hydroxyphenyl)-3-(3-nitrophenyl)-2-propen-1-one, A3B14
(66)
66 was prepared by using 3′-hydroxyacetophenone A3 (0.68 g, 5 mmol) and 3-
nitrobenzaldehyde B14 (0.776 g, 5 mmol). Yield 88%. M.p. 207-209 ºC (decomp). IR
(KBr): ν = 3400, 1658, 1574. 1H NMR (300 MHz, DMSO-d6): δ = 7.08 (d, J = 8.1 Hz,
1H, H-C(4′)), 7.34 (d, J = 7.8 Hz, 1H, H-C(6′)), 7.38 (t, J = 7.8 Hz, 1H, H-C(5′)), 7.51
(s, 1H, H-C(2′)), 7.72 (t, J = 8.1 Hz, 1H, H-C(5)), 7.70 (d, J = 8.7 Hz, 1H, H-C(6)),
7.82 (d, J = 15.6 Hz, 1H, H-Cα(Vinyl)), 8.01 (d, J = 15.6 Hz, 1H, H-Cβ(Vinyl)), 8.23
(d, J = 8.1 Hz, 1H, H-C(4)), 8.29 (s, 1H, H-C(2)).

Chapter 4 ♦Experimental - 161 - 4B.3.4.15. (E)-1-(3′-Hydroxyphenyl)-3-(3-hydroxy-4-methoxyphenyl)-2-propen
1-one, A3B15 (67)
67 was prepared by using 3′-hydroxyacetophenone A3 (0.68 g, 5 mmol) and 3-
hydroxy-4-methoxybenzaldehyde B15 (0.76 g, 5 mmol). Yield 65%. M.p. 210 ºC
(decomp). IR (KBr): ν = 3462, 1653, 1535. 1H NMR (300 MHz, CDCl3): δ = 2.33 (s,
3H, H-C(CH3)), 7.01-7.57 (m, 7H, H-C(arom.)), 7.61 (d, J = 15.6 Hz, 1H, H-
Cα(Vinyl)), 7.86 (d, J = 16.5 Hz, 1H, H-Cβ(Vinyl)), 8.89 (bs, 1H, H-O(OH)).
4B.3.4.16. (E)-1-(3′-Hydroxyphenyl)-3-(4-N,N-dimethylaminophenyl)-2-propen
1-one, A3B16 (68)
68 was prepared by using 3′-hydroxyacetophenone A3 (0.68 g, 5 mmol) and 4-N,N-
dimethylaminobenzaldehyde B16 (0.744 g, 5 mmol). Yield 70%. M.p. 169-171 ºC. IR
(KBr): ν = 3447, 1648, 1567. 1H NMR (300 MHz, DMSO-d6): δ = 3.0 (s, 6H, H-C(N-
CH3)), 6.73 (d, J = 8.7 Hz, 2H, H-C(3,5)), 7.02 (d, J = 7.8 Hz, 1H, H-C(4′)), 7.34 (t, J
= 7.5 Hz, 1H, H-C(5′)), 7.43 (s, 1H, H-C(2′)), 7.50 (bd, J = 5.7 Hz, 1H, H-C(6′)), 7.55
(d, J = 15.3 Hz, 1H, H-Cα(Vinyl)), 7.66 (d, J = 15.5 Hz, 1H, H-Cβ(Vinyl)), 7.68 (d, J
= 8.1 Hz, 2H, H-C(2,6)), 8.80 (bs, 1H, H-O(OH)).
4B.3.4.17. (E)-1-(3′-Hydroxyphenyl)-3-(4-methylphenyl)-2-propen-1-one, A3B17
(69)
69 was prepared by using 3′-hydroxyacetophenone A3 (0.68 g, 5 mmol) and 4-
methylbenzaldehyde B17 (0.589 mL, 5 mmol). Yield 77%. M.p. 119-120 ºC. IR
(KBr): ν = 3412, 1653, 1566. 1H NMR (300 MHz, DMSO-d6): δ = 2.33 (s, 3H, H-
C(CH3)), 7.05 (d, J = 7.2 Hz, 2H, H-C(3,5)), 7.35 (t, J = 7.8 Hz, 1H, H-C(5′)), 7.49 (s,
1H, H-C(2′)), 7.60 (d, J = 7.5 Hz, 1H, H-C(4′)), 7.70 (d, J = 8.4 Hz, 1H, H-C(6′)),
7.72 (d, J = 15.6 Hz, 1H, H-Cα(Vinyl)), 7.81 (d, J = 16.5 Hz, 1H, H-Cβ(Vinyl)), 7.84
(d, J = 7.8 Hz, 2H, H-C(2,6)), 8.81 (bs, 1H, H-O(OH)).
4B.3.4.18. (E) -1-(3′-Hydroxyphenyl)-3-(thien-2-yl)-2-propen-1-one, A3B18 (70)
70 was prepared by using 3′-hydroxyacetophenone A3 (0.68 g, 5 mmol) and thiophen-
2-carboxaldehyde B18 (0.46 mL, 5 mmol). Yield 76%. M.p. 123 ºC. IR (KBr): ν =
3387, 1638, 1531. 1H NMR (300 MHz, DMSO-d6): δ = 7.05 (dd, J = 8.1 Hz, 2.4 Hz,
1H, H-C(4′)), 7.20 (t, J = 4.4 Hz, 1H, H-C(4)), 7.36 (t, J = 7.5 Hz, 1H, H-C(5′)), 7.41
(s, 1H, H-C(2′)), 7.46 (d, J = 15.0 Hz, 1H, H-Cα(Vinyl)), 7.54 (d, J = 8.1 Hz, 1H, H-

Chapter 4 ♦Experimental - 162 - C(6′)). 7.67 (d, J = 3.0 Hz, 1H, H-C(3)), 7.78 (d, J = 5.4 Hz, 1H, H-C(5)), 7.89 (d, J =
15.0 Hz, 1H, H-Cβ(Vinyl)).
4B.3.4.19. (E)-1-(3′-Hydroxyphenyl)-3-(5-methylthien-2-yl)-2-propen-1-one,
A3B19 (71)
71 was prepared by using 3′-hydroxyacetophenone A3 (0.68 g, 5 mmol) and 5-
methylthiophen-2-carboxaldehyde B19 (0.54 mL, 5 mmol). Yield 78%. M.p. 114-115
ºC. IR (KBr): ν = 3372, 1654, 1576. 1H NMR (300 MHz, DMSO-d6): δ = 2.50 (s, 3H,
H-C(CH3)), 6.90 (d, J = 3.6 Hz, 1H, H-C(4)), 7.04 (dd, J = 7.8 Hz, 8.1 Hz, 1H, H-
C(5′)), 7.30 (d, J = 15.3 Hz, 1H, H-Cα(Vinyl)), 7.32 (s, 1H, H-C(2′)), 7.36 (d, J = 8.1
Hz, 1H, H-C(4′)), 7.46 (d, J = 3.3 Hz, 1H, H-C(3)), 7.49 (d, J = 7.5 Hz, 1H, H-C(6′)),
7.8 (d, J = 15.3 Hz, 1H, H-Cβ(Vinyl)).
4B.3.4.20. (E)-1-(3′-Hydroxyphenyl)-3-(5-bromothien-2-yl)-2-propen-1-one,
A3B20 (72)
72 was prepared by using 3′-hydroxyacetophenone A3 (0.68 g, 5 mmol) and 5-
bromothiophen-2-carboxaldehyde B20 (0.60 mL, 5 mmol). Yield 74%. M.p. 133-136
ºC. IR (KBr): ν = 3291, 1667, 1593. 1H NMR (300 MHz, DMSO-d6): δ = 6.92 (d, J =
7.8 Hz, 1H, H-C(4′)), 7.17 (d, J = 7.5 Hz, 1H, H-C(6′)), 7.21 (s, 1H, H-C(2′)), 7.24 (t,
J = 7.5 Hz, 1H, H-C(5′)), 7.28 (d, J = 3.9 Hz, 1H, H-C(4)), 7.36 (d, J = 15.3 Hz, 1H,
H-Cα(Vinyl)), 7.45 (d, J = 3.9 Hz, 1H, H-C(3)), 7.74 (d, J = 15.3 Hz, 1H, H-
Cβ(Vinyl)).
4B.3.4.21. (E)-1-(3′-Hydroxyphenyl)-3-(5-nitrothien-2-yl)-2-propen-1-one, A3B21
(73)
73 was prepared by using 3′-hydroxyacetophenone A3 (0.68 g, 5 mmol) and 5-
nitrothiophen-2-carboxaldehyde B21 (0.78 g, 5 mmol). Yield 85%. M.p. 170 ºC
(decomp). IR (KBr): ν = 3074, 1705, 1597. 1H NMR (300 MHz, DMSO-d6): δ = 6.0-
7.2 (m, 6H, H-C(arom.)); 7.55 ( d, J=15.3 Hz, 1H, H-C (α)), 7.66 ( d, J=15.5 Hz, 1H,
H-C (β)), 10.00 (bs, 1H, H-O).
4B.3.4.22. (E)-1-(3′-Hydroxyphenyl)-3-(pyrrol-2-yl)-2-propen-1-one, A3B22 (74)
74 was prepared by using 3′-hydroxyacetophenone A3 (0.68 g, 5 mmol) and pyrrole-
2-carboxaldehyde B22 (0.48 g, 5 mmol). Yield 80%. M.p. 197 ºC. IR (KBr): ν = 3304,
1645, 1561. 1H NMR (300 MHz, (CD3)2CO): δ = 6.12 (m, 2H, H-C(3,4)), 6.58 (d, J =
2.1 Hz, 1H, H-C(5)), 6.90 (s, 1H, H-N(NH)), 6.95 (m, 2H, H-C(2′,4′)), 7.20 (t, J = 7.8

Chapter 4 ♦Experimental - 163 - Hz, 1H, H-C(5′)), 7.31 (d, J = 15.3 Hz, 1H, H-Cα(Vinyl)), 7.40 (dt, J = 7.5 Hz, 1.5
Hz, 1H, H-C(6′)), 7.52 (d, J = 15.3 Hz, 1H, H-Cβ(Vinyl)), 8.87 (bs, 1H, H-O(OH)).
4B.3.4.23. (E)-1-(3′-Hydroxyphenyl)-3-(pyridin-2-yl)-2-propen-1-one, A3B23
(75)
75 was prepared by using 3′-hydroxyacetophenone A3 (0.68 g, 5 mmol) and pyridine-
2-carboxaldehyde B23 (0.48 mL, 5 mmol). Yield 66%. M.p. 146 ºC. IR (KBr): ν =
3318, 1659, 1591. 1H NMR (300 MHz, CDCl3): δ = 6.62-7.24 (m, 4H, H-C(arom.)),
7.36-8.49 (m, 4H, H-C(pyridyl)), 8.05 (d, J = 15.0 Hz, 1H, H-Cα(Vinyl)), 8.20 (d, J =
15.31 Hz, 1H, H-Cβ(Vinyl)), 8.96 (bs, 1H, H-O(OH)).
4B.3.4.24. (E)-1-(3′-Hydroxyphenyl)-3-(pyridin-3-yl)-2-propen-1-one, A3B24
(76)
76 was prepared by using 3′-hydroxyacetophenone A3 (0.68 g, 5 mmol) and pyridine-
3-carboxaldehyde B24 (0.47 mL, 5 mmol). Yield 64%. M.p. 188-190 ºC. IR (KBr): ν =
3301(broad), 1675, 1582. 1H NMR (300 MHz, CDCl3): δ = 6.65-7.46 (m, 4H, H-
C(arom.)), 7.49 (d, J = 15.5 Hz, IH, H-Cα(Vinyl)), 7.90 (d, J = 15.7 Hz, IH, H-
Cβ(Vinyl)), 8.29-8.50 (m, 4H, H-C(pyridyl)), 9.10 (bs, 1H, H-O(OH)).
4B.3.4.25. (E)-1-(3′-Hydroxyphenyl)-3-(pyridin-4-yl)-2-propen-1-one, A3B25
(77)
77 was prepared by using 3′-hydroxyacetophenone A3 (0.68 g, 5 mmol) and pyridine-
4-carboxaldehyde B25 (0.48 mL, 5 mmol). Yield 67%. M.p. 175-177 ºC. IR (KBr): ν =
3129, 1677, 1606. 1H NMR (300 MHz, DMSO): δ = 7.07 (d, J = 7.8 Hz, 1H, H-
C(4′)), 7.29 (dd, J = 7.5 Hz, 3.4, 1H, H-C(5′)), 7.36 (d, J = 6.6 Hz, 1H, H-C(6′)), 7.37
(s, 1H, H-C(2′)), 7.42 (d, J = 8.4 Hz, 2H, H-C(2,6)), 8.06 (d, J = 14.7 Hz, 1H, H-
Cα(Vinyl)), 8.13 (d, J = 14.7 Hz, 1H, H-Cβ(Vinyl)), 8.4 (d, J = 6.0 Hz, 2H, H-C(3,5)),
9.21 (bs, 1H, H-O(OH)).
4B.3.5. Synthesis of members of active row, A1B9-A7B9
Active row members; A1B9-A7B9 were synthesized using an ethanolic solution (20
mL) of acetophenone A (10 mmol) and aldehyde B (10 mmol) following the general
procedure as mentioned in section 4B.3.3.
4B.3.5.1. (E)-1-(4′-Aminophenyl)-3-(2-chlorophenyl)-2-propen-1-one, A7B9 (12)
For details, see Section 4B.1.12.

Chapter 4 ♦Experimental - 164 - 4B.3.5.2. (E)-1-(Phenyl)-3-(2-chlorophenyl)-2-propen-1-one, A1B9 (32)
For details, see Section 4B.2.3.12.
2B.3.5.3. (E)-1-(3′-Hydroxyphenyl)-3-(2-chlorophenyl)-2-propen-1-one, A3B9 (61)
For details, see Section 4B.3.4.9.
4B.3.5.4. (E)-1-(2′-Hydroxyphenyl)-3-(2-chlorophenyl)-2-propen-1-one, A2B9 (78)
78 was prepared by the general procedure using 2′-hydroxyacetophenone (1.203 mL,
10 mmol) and 2-chlorobenzaldehyde (0.832 mL, 10 mmol). Yield 85%. Anal.
Calculated: C, 69.91%, H, 4.69%. Found: C, 69.57%, H, 4.38%.
4B.3.5.5. (E)-1-(4′-Hydroxyphenyl)-3-(2-chlorophenyl)-2-propen-1-one, A4B9
(79)
79 was prepared by the general procedure using 4′-hydroxyacetophenone (1.360 g, 10
mmol) and 2-chlorobenzaldehyde (0.832 mL, 10 mmol). Yield 80%. Anal.
Calculated: C, 69.91%, H, 4.69%. Found: C, 69.66%, H, 4.35%.
4B.3.5.6. (E)-1-(2′-Aminophenyl)-3-(2-chlorophenyl)-2-propen-1-one, A5B9 (80)
80 was prepared by the general procedure using 2′-aminoacetophenone (1.350 g, 10
mmol) and 2-chlorobenzaldehyde (0.832 mL, 10 mmol). Yield 76%. M.p. 64-65 ºC.
Anal. Calculated: C, 69.91%, H, 4.69%. Found: C, 69.57%, H, 4.65%.
4B.3.5.7. (E)-1-(3′-Aminophenyl)-3-(2-chlorophenyl)-2-propen-1-one, A6B9 (81)
81 was prepared by the general procedure using 3′-aminoacetophenone (1.350 g, 10
mmol) and 2-chlorobenzaldehyde (0.832 mL, 10 mmol). Yield 94%. Anal.
Calculated: C, 69.91%, H, 4.69%. Found: C, 69.78%, H, 4.87%.

Chapter 4 ♦Experimental - 165 - 4C. Synthesis of peptidyl chalcones (102-109)
Synthesis of a variety of peptidyl chalcones was carried out on solid phase using
phosphorane supported polyethyldivinyl benzene following a multistep strategy.
4C.1. 2-Trimethylsilylethyl-2-bromoacetate (82)217
To a solution of 2-bromoacetic acid (5.70 g, 41 mmol) in dry DCM (30 mL) at 0 °C
was added, 2-trimethylsilylethanol (2.37 g, 20 mmol) and DMAP (200 mg). To this
stirred solution, carefully in small portions was added DCC (9.5 g, 42 mmol) over a
short period of 5 min followed by stirring at 0°C for 1 h. The reaction mixture was
filtered and the remaining filter cake was washed with a mixture of n-hexane/ether
(100 mL, 4:1 v:v) and aq. KHCO3. The organic layer was dried over anhydrous
sodium sulphate and the solvent was removed in a rotary evaporator. The crude
product, thus obtained, was distilled under vacuo. The residue was characterized by
NMR. Yield 83%. 1H NMR (300 MHz, CDCl3): δ = 0.08 (s, 9H, H-C(CH3)), 1.10 (m,
2H, H-C(CH2Si)), 3.90 (s, 2H, H-C(BrCH2CO)), 4.21 (m, 2H, H-C(COCH2)). 13C
NMR (100 MHz, CDCl3): δ = 0.0, 18.7, 27.6, 66.3, 168.8.
4C.2. 2-Trimethylsilylethyl-2-diphenylphosphoranylidene salt (83)217
Wittig salt 83 was synthesized through C-C alkylation of triphenylphosphine-bound
polystyrene support. Triphenylphosphine polystyrene resin (0.5 g, 1.6 mmol/g, 0.8
mmol, 1% divinyl benzene, 100–200 mesh) was weighed into a microwave vial and
suspended in dry toluene (4 mL). After addition of 2-trimethylsilylethyl-2-
bromoacetate (82) (991 mg, 4 mmol, 5 eq), the vial was sealed and heated at 100 °C
for 15 min in a microwave synthesizer. The vial was cooled to room temperature
before opening; the resin was filtered and washed successively with dry toluene,
DCM and ether.
4C.3. 2-Trimethylsilylethyl-2-diphenylphosphoranylideneacetate (84)217
The polymer-bound Wittig salt (83) (0.68 g, 1.16 mmol/g, 0.79 mmol) was suspended
in dry DCM (5 mL) and TEA (550 µL, 3.95 mmol, 5 eq) was added. After shaking for
2 h at rt, the resin turned yellow indicating the deprotonation of Wittig salt. The
yellow-colored resin 84 was filtered, washed and dried in vacuo. The identity of
Wittig Ylide was confirmed through IR spectroscopy of the dried resin sample. IR
(ATR): ν-1 = 3056, 3025, 2923, 2852, 1725, 1600, 1493, 1452, 1437, 1380, 1310,
1249, 1183, 1116, 835, 751, 698, 542.

Chapter 4 ♦Experimental - 166 - 4C.4. 2-Acyl-2-diphenylphosphoranylidene acetates (85-86)
General procedure.217 The resin-bound ester 84 (200 mg, 1.28 mmol/g, 0.26 mmol)
was pre-swollen in dry DCM. The respective Fmoc-amino acids (1.27 mmol, 5 eq)
were suspended in dry DCM (4 mL) and dissolved with addition of 2,6-lutidine
(145.6 µL, 1.25 mmol, 4.9 eq). The clear solutions obtained after addition of MSNT
(370.37 mg, 1.25 mmol, 5 eq) were directly mixed with the resin suspensions and
shaken overnight at room temperature. The resins were washed after the coupling step
and dried in vacuo. The yield of the acylated products was determined after Fmoc
cleavage of small samples.
4C.4.1. 2-Trimethylsilylethyl-4-(9H-fluoren-9-yl-methoxycarbonyl)-3-oxo-5-
phenyl-2-diphenylphosphoranylidenepentanoate (85)
The ester 85 was synthesized using Fmoc-Phe (492.04 mg, 1.27 mmol, 5 eq) as
acylation agent leading to a pale yellow resin. Yield 88%.
4C.4.2. 2-Trimethylsilylethyl-4-(9H-fluoren-9-yl-methoxycarbonyl)-5-methyl-3-
oxo-2-diphenylphosphoranylidenehexanoate (86)
The ester 86 was synthesized using Fmoc-Val (431.03 mg, 1.27 mmol, 5 eq) as
acylation agent leading to pale yellow resin. Yield 84%.
4C.5. Fmoc protected di- and tri- peptides (87-91)
General procedure.217 Fmoc-protected 2-acyl-2-phosphoranylidene acetates (85-86)
were cleaved by the reaction of 20% piperidine/DMF (v/v, 2 × 6 min) with acylated
supports. The deprotected resins (200mg, ~0.22 mmol) were washed and suspended in
dry DMF. Fmoc protected amino acids (1.10 mmol, 5 eq) and HOBt·H2O (1.10 mmol,
5 eq) were dissolved in dry DMF (4 mL), activated by addition of DIC (1.10 mmol, 5
eq), and added to the respective resins. The mixture was shaken for 3 h at rt, filtered,
and the resins was washed. Quantitative coupling was verified by the Kaiser test.
Yield was calculated after Fmoc cleavage of the dried resin sample (~ 5 mg).
4C.5.1. (S)-2-Trimethylsilylethyl 4-[(S)-2-(9H-fluoren-9-yl-methoxycarbonyl)-3-
phenylpropanamido]-3-oxo-5-phenyl-2-diphenylphosphoranylidene
pentanoate (87)
Fmoc deprotected ester 85 (200 mg, 1.08 mmol/g, 0.22 mmol) suspended in dry DMF
was subjected to amide coupling with Fmoc-Phe (416.37 mg, 1.08 mmol, 5 eq)
according to the general procedure. Yield 86%.

Chapter 4 ♦Experimental - 167 - 4C.5.2. (S)-2-Trimethylsilylethyl 4-[(S)-2-(9H-fluoren-9-yl-methoxycarbonyl)-3-
phenylpropanamido]-5-methyl-3-oxo-2-diphenylphosphoranylidenehexa
noate (88)
Fmoc deprotected ester 86 (200 mg, 1.33 mmol/g, 0.23 mmol) suspended in dry DMF
was subjected to amide coupling with Fmoc-Val (385.2mg, 1.14 mmol, 5 eq)
according to the general procedure. The reaction was complete in 4 hours as verified
by the negative Kaiser test. The resin was filtered, washed and dried in vacou. Yield
82%.
4C.5.3. (S)-2-Trimethylsilylethyl 4-[(S)-2-(9H-fluoren-9-yl-methoxycarbonyl)-4-
methylpentanamido]-3-oxo-5-phenyl-2-diphenylphosphoranylidenepenta
noate (89)
Fmoc deprotected ester 85 (200 mg, 1.08 mmol/g, 0.22 mmol) in dry DMF was
subjected to amide coupling with Fmoc-Leu (370.02 mg, 1.08 mmol, 5 eq) according
to the general procedure. Yield 84%.
4C.5.4. (S)-2-Trimethylsilylethyl 4-[(S)-2-(9H-fluoren-9-yl-methoxycarbonyl)pro
panamido]-3-oxo-5-phenyl-2-diphenylphosphoranylidenepentanoate (90)
Fmoc deprotected ester 85 (200 mg, 1.08 mmol/g, 0.22 mmol) in dry DMF was
subjected to amide coupling with Fmoc-Ala (325.98 mg, 1.08 mmol, 5 eq) according
to the general procedure. Yield 84%.
4C.5.5. (S)-2-Trimethylsilylethyl 4-[(S)-2-((S)-2-(9H-fluoren-9-yl-methoxycarbon
yl)-3-phenylpropanamido)propanamido]-3-oxo-5-phenyl-2-diphenylphos
phoranylidenepentanoate (91)
Fmoc deprotected ester 90 (200 mg, 1.0 mmol/g, 0.20 mmol) in dry DMF was
subjected to amide coupling with Fmoc-Phe (386.85 mg, 1.00 mmol, 5 eq) according
to the general procedure. Yield 83%.
4C.6. Acetyl di- and tri- peptides (92-95)
The synthesized peptides 87-89 and 91 were subjected to Fmoc deprotection using
20% piperidine/DMF (v/v, 2 × 6 min) leading to resin-bound Fmoc deprotected
peptides. The deprotected petide sequences were washed with DCM, ether and DMF.
The free amino terminus of peptides was acetylated by stirring the resin samples with
acetic anhydride in DMF (5 eq) (2 × 30 min), which resulted into acetylated peptides
92-95. The acetylated products were again washed and dried in vacou.

Chapter 4 ♦Experimental - 168 - 4C.6.1. (S)-2-Trimethylsilylethyl 4-[(S)-2-acetamido-3-phenylpropanamido]-3-
oxo-5-phenyl-2-diphenylphosphoranylidenepentanoate (92)
The Fmoc deprotected ester 87 (200 mg, 0.93 mmol/g, 0.184 mmol) was acetylated
by stirring with acetic anhydride (98.7 µL, 0.92 mmol, 5 eq) in DMF (2 × 30 min).
4C.6.2. (S)-2-Trimethylsilylethyl 4-[(S)-2-acetamido-3-phenylpropanamido]-5-
methyl-3-oxo-2-diphenylphosphoranylidenehexanoate (93)
The Fmoc deprotected ester 88 (200 mg, 1.02 mmol/g, 0.204 mmol) was acetylated
by stirring with acetic anhydride (109.5 µL, 1.02 mmol, 5 eq) in DMF (2 × 30 min).
4C.6.3. (S)-2-Trimethylsilylethyl 4-[(S)-2-acetamido-4-methylpentanamido]-3-
oxo-5-phenyl-2-diphenylphosphoranylidenepentanoate (94)
The Fmoc deprotected ester 89 (200 mg, 0.96 mmol/g, 0.192 mmol) was acetylated
by stirring with acetic anhydride (103.1 µL, 0.96 mmol, 5 eq) in DMF (2 × 30 min).
4C.6.4. (S)-2-Trimethylsilylethyl 4-[(S)-2-((S)-2-acetamido)-3-phenylpropanami
do)propanamido]-3-oxo-5-phenyl-2-diphenylphosphoranylide
nepentanoate (95)
The Fmoc deprotected ester 91 (200 mg, 0.87 mmol/g, 0.20 mmol) was acetylated by
stirring it with acetic anhydride (107.4 µL, 1.0 mmol, 5 eq) in DMF (2 × 30 min).
4C.7. Peptidyl-3-amino-2-oxo-1-diphenylphosphoranylidenepropanes (96-99)
The N-acylated esters 92-95 were converted to their corresponding amides 96-99 by
the following general procedure.
General procedure. To a suspension of an ester 92-95 (0.254 mmol) in dry DMF was
added TAS-F (3 eq) and mixture was stirred for 3 h at rt. The resulting resin was
filtered, washed and dried in vacuo. IR (ATR) for 96: ν = 3059, 3025, 2926, 2851,
2149, 1731, 1599, 1492, 1451, 1437, 1256, 1185, 1111, 751, 698, 569, 517, 512.
4C.7.1. (S)-2-acetamido-N-[(S)-3-oxo-1-phenyl-4-diphenylphosphoranylidene
butan-2-yl]-3-phenylpropanamide (96)
96 was synthesized according to the general procedure using ester 92 (200 mg, 0.893
mmol/g, 0.179 mmol) and TASF (161.49 mg, 0.537 mmol, 3 eq).
4C.7.2. (S)-2-acetamido-N-[(S)-4-methyl-2-oxo-1-diphenylphosphoranylidenepen
tan-3-yl]-3-phenylpropanamide (97)
97 was synthesized according to the general procedure using ester 93 (200 mg, 0.977
mmol/g, 0.195 mmol) and TASF (161.43 mg, 0.586 mmol, 3 eq).

Chapter 4 ♦Experimental - 169 - 4C.7.3. (S)-2-acetamido-4-methyl-N-[(S)-3-oxo-1-phenyl-4-diphenylphosphorany
lidenebutan-2-yl]pentanamide (98)
98 was synthesized according to the general procedure using ester 94 (200 mg, 0.921
mmol/g, 0.184 mmol) and TASF (152.23 mg, 0.553 mmol, 3 eq).
4C.7.4. (S)-2-((S)-2-acetamido-3-phenylpropanamido)-N-[(S)-3-oxo-1-phenyl-4-
diphenylphosphoranylidenebutan-2-yl]propanamide (99)
99 was synthesized according to the general procedure using ester 95 (200 mg, 0.871
mmol/g, 0.174 mmol) and TASF (144.08 mg, 0.523 mmol, 3 eq).
4C.8. Peptidyl chalcones (100-107)
The synthesized peptides 96-99 were converted to peptidyl vinyl ketones (100-107)
generally described as peptidyl chalcones by the following general procedure.
General procedure. To a suspension of peptides 96-99 (0.250 mmol) in dry THF, the
aldehyde (3 eq, 0.75 mmol) was added and the mixture was stirred for 12 h at rt. THF
and the excess of aldehyde was removed in vacuo. The yellow-white solid thus
obtained were characterized by LC-MS and NMR. The crude products were purified
through HPLC before NMR analysis.
4C.8.1. N-Acetyl-L-phenylalanyl-(S,E)-1-amino-1-benzyl-6-methyl-hept-3-en-2-
one (100)
100 was prepared by the reaction of peptide 96 (180 mg, 1.026 mmol/g, 0.185 mmol)
with isovaleraldehyde (59.42 µL, 0.554 mmol, 3 eq). Yield 70 %. 1H NMR (300
MHz, CDCl3): δ = 0.91 (d, J = 7.8 Hz, 6H , H-C(Methyl)), 1.65-1.81 (m, 1H, H-C(CH
Allyl)), 1.94 (s, 3H, H-C(CH3CO)), 2.07 (bt, J = 6.1 Hz, 2H, H-C(CH2 Vinyl)), 2.91-
3.20 (m, 4H, H-Cβ1,β1′,β2,β2′ (Phe, Benzyl)), 4.64-4.78 (m, 1H, H-Cα(Benzyl)), 4.85-
5.05 (m, 1H, H-Cα(Phe)), 6.05 (d, J = 15.9 Hz, 1H, H-Cα(Vinyl)), 6.15 (d, J = 7.3 Hz,
1H, NHI), 6.56 (d, J = 7.3 Hz, 1H, NHII), 6.89-7.06 (m, 11H, H-C(arom.), H-
Cβ(Vinyl)). 13C NMR (100 MHz, CDCl3): δ = 21.9, 22.7, 27.3, 37.6, 37.8, 41.4, 53.9,
56.6, 126.5, 126.5, 127.6, 127.9, 128.1, 128.7, 128.9, 135.3, 135.9, 148.9, 169.3,
169.9, 195.6. HRMS: calcd [(M+H+) = [C26H33N2O3] 421.2481; found 421.2456 m/z.
4C.8.2. N-Acetyl-L-phenylalanyl-(S,E)-1-amino-1-benzyl-4-(2-chloro-6-fluorophe
nyl)-but-3-en-2-one (101)
101 was prepared by the reaction of peptide 96 (180 mg, 1.026 mmol/g, 0.185 mmol)
with 2-chloro-6-fluorobenzaldehyde (87.84 mg, 0.554 mmol, 3 eq). Yield 68 %. 1H

Chapter 4 ♦Experimental - 170 - NMR (300 MHz, CDCl3): δ = 1.95 (s, 3H, H-C(CH3CO)), 2.98-3.19 (m, 4H, H-
Cβ1,β1′,β2,β2′(Phe, Benzyl)), 4.65-4.74 (m, 1H, H-Cα(Phe)), 4.99-5.06 (m, 1H, H-
Cα(Benzyl)), 6.16 (d, J = 15.4 Hz, 1H, H-Cα(Vinyl )), 6.57 (d, J = 15.4 Hz, 1H, H-
Cβ(Vinyl)), 6.95-7.33 (m, 13H, H-C(arom.)), 7.80-7.81 (d, J = 7.8, 1H, NHI), 7.85-
7.86 (d, J = 7.8, 1H, NHII). 13C NMR (100 MHz, CDCl3): δ = 22.7, 37.1, 37.9, 53.9,
57.9, 114.3, 114.7, 120.9, 121.1, 125.7, 126.7, 128.0, 128.1, 128.2, 128.3, 128.5,
128.6, 128.0, 128.9, 130.9, 133.9, 135.0, 135.9, 135.9, 136.2, 136.3, 169.5, 170.0,
195.7. HRMS: calcd [(M+H+) = [C28H27ClFN2O3]+Et3N 595.2926; found 595.2939
m/z.
4C.8.3. N-Acetyl-L-phenylalanyl-(S,E)-1-amino-1-isopropyl-4-(2-chloro-6-fluoro
phenyl)-but-3-en-2-one (102)
102 was prepared by the reaction of peptide 97 (180 mg, 1.139 mmol/g, 0.205 mmol)
with 2-chloro-6-fluorobenzaldehyde (97.35 mg, 0.614 mmol, 3 eq). Yield 74 %. 1H
NMR (300 MHz, CDCl3): δ = 0.82 (d, J = 7.3 Hz, 3H, H-Cγ1(CH3 isopropyl)), 0.89 (d,
J = 7.3 Hz, 3H, H-Cγ2(CH3 isopropyl)), 1.27 (m, 1H, H-Cβ(CH isopropyl)), 1.74 (s,
3H, H-C(CH3CO)), 2.71 (dd, J = 14.6 Hz, 9.7 Hz, 1H, H-Cβ1(Phe)), 2.95 (dd, J = 13.4
Hz, 4.9 Hz, 1H, H-Cβ2(Phe)), 4.37 (t, J = 7.3 Hz, 1H, H-Cα(isopropyl)), 4.64 (ddd, J =
9.8 Hz, 9.8 Hz, 4.9 Hz, 1H, H-Cα(Phe)), 7.07-7.24 (m, 5H, H-Carom.(Phe, 2-chloro-6-
fluorophenyl)), 7.32 (dd, J = 7.3 Hz, 2.4 Hz, 1H, H-C(2-chloro-6-fluorophenyl)), 7.40
(d, J = 16.1 Hz, 1H, H-Cα(Vinyl)), 7.45-7.51 (m, 2H, H-C(Phe)), 7.65 (d, J = 15.9 Hz,
1H, H-Cβ(Vinyl)), 8.14 (d, J = 8.5 Hz, 1H, NHI), 8.39 (d, J = 7.3 Hz, 1H, NHII). 13C
NMR (100 MHz, CDCl3): δ = 18.1, 19.4, 22.4, 29.1, 37.5, 36.0, 53.4, 60.8, 115.4,
115.7, 120.9, 121.1, 126.1, 126.3, 126.4, 127.9, 129.1, 129.5, 129.7, 131.6, 131.7,
132.1, 132.3, 135.1, 135.2, 137.8, 159.6, 162.9, 169.0, 172.0, 197.2. HRMS: calcd
[(M+) = [C24H27ClFN2O3]+Et3N 546.2893; found 546.2912 m/z.
4C.8.4. N-Acetyl-L-phenylalanyl-(S,E)-1-amino-1-isopropyl-4-thiophen-3-en-2-
one (103)
103 was prepared by the reaction of peptide 97 (180 mg, 1.139 mmol/g, 0.205 mmol)
with 2-thiophenecarboxaldehyde (57.38 µL, 0.614 mmol, 3 eq). Yield 65 %. 1H NMR
(300 MHz, CDCl3): δ = 0.78 (d, J = 7.5 Hz, 3H, H-Cγ1(isopropyl)), 0.96 (d, J = 7.5
Hz, 3H, H-Cγ2(isopropyl)), 2.76 (s, 3H, H-C(CH3CO), 3.08-3.11 (m, 3H, H-
Cβ(isopropyl), H-Cβ,β′(Phe)), 4.64 (m, 1H, H-Cα(Phe)), 4.74 (m, 1H, H-

Chapter 4 ♦Experimental - 171 - Cα(isopropyl)), 7.08-7.55 (m, 10H, H-C(arom)., H-Cα,β(Vinyl), H-C(thiophene)). 13C
NMR (100 MHz, CDCl3): δ = 13.0, 23.3, 32.0, 34.2, 50.1, 56.4, 121.3, 126.5, 127.6,
128.1, 128.5, 128.7, 130.4, 132.4, 135.6, 143.5. HRMS: calcd [(M+H+) =
[C22H27N2O3S]+Et3N 500.2959; found 500.2967 m/z.
4C.8.5. N-Acetyl-L-phenylalanyl-(S,E)-1-amino-1-isopropyl-(5,5,5-tribromo)-
pent -3-en-2-one (104)
104 was prepared by the reaction of peptide 97 (180 mg, 1.139 mmol/g, 0.205 mmol)
with tribromoacetaldehyde (64.68 µL, 0.614 mmol, 3 eq). Yield 72 %. 1H NMR (300
MHz, CDCl3): δ = 0.83 (d, 3H, J = 7.8 Hz, H-Cγ1(CH3 isopropyl)), 0.86 (d, J = 6.7
Hz, 3H, H-Cγ2(CH3 isopropyl)), 2.52-2.56 (m, 1H, H-Cβ(CH isopropyl)), 2.72 (m, 2H,
H-Cβ1,β2(Phe)), 2.73 (s, 3H, H-C(CH3CO)), 4.3 (m, 1H, H-Cα(Phe)), 4.60-4.62 (m,
1H, H-Cα(CH isopropyl)), 6.90-7.24 (m, 7H, H-C(arom.), H-Cα,β(Vinyl)), 8.12 (d, J =
7.8 Hz, 1H, NHI). 8.93 (d, J = 7.8 Hz, 1H, NHII). 13C NMR (100 MHz, CDCl3): δ =
17.6, 19.2, 22.4, 34.1, 36.2, 58.4, 93.8, 125.8, 126.5, 128.2, 136.3, 140.1, 167.2,
171.0, 178.4, 196.1.
4C.8.6. N-Acetyl-L-leucinyl-(S,E)-1-amino-1-benzyl-4-(2-chloro-6-fluorophenyl)-
but-3-en-2-one (105)
105 was prepared by the reaction of peptide 98 (180 mg, 1.063 mmol/g, 0.213 mmol)
with 2-chloro-6-fluorobenzaldehyde (101.16 mg, 0.638 mmol, 3 eq). Yield 78 %. 1H
NMR (300 MHz, CDCl3): δ = 0.97 (d, J = 7.3 Hz, 3H, H-Cδ1(Leu)), 0.98 (d, J = 6.1
Hz, 3H, H-Cδ2(Leu)), 1.14-1.28 (m, 2H, H-Cβ1,β2(Leu)), 1.32-1.50 (m, 1H, H-
Cγ(Leu)), 1.76 (s, 3H, H-C(CH3CO)), 2.85 (dd, J = 15 Hz, 9.7 Hz, 1H, H-
Cβ1(Benzyl)), 3.15 (dd, J = 15 Hz, 7.5 Hz, 1H, H-Cβ2(Benzyl)), 4.27 (m, 1H, H-
Cα(Benzyl)), 4.71 (m, 1H, H-Cα(Leu)), 7.13 (d, J = 15.8 Hz, 1H, H-Cα(Vinyl)), 7.15-
7.50 (m, 7H, H-C(Benzyl, 2-chloro-6-fluorophenyl)), 7.60 (d, J = 14.7 Hz, 1H, H-
Cβ(Vinyl)), 7.68 (dd, J = 8.5 Hz, 5.9 Hz, 1H, H-C(2-chloro-6-fluorophenyl)), 7.95 (d,
J = 8.6 Hz, 1H, NHI), 8.54 (d, J = 8.6 Hz, 1H, NHII). 13C NMR (100 MHz, CDCl3): δ
= 21.8, 22.3, 22.6, 23.9, 37.9, 40.9, 50.8, 58.6, 115.9, 116.2, 121.1, 121.5, 126.2, 126,
127.9, 128, 129.2, 129.3, 136.1, 136.2, 137.5, 137.7, 160.6, 168.7, 168.8, 172.3,
172.5, 178.2. HRMS: calcd [(M+H+) = [C25H29ClFN2O3]+Et3N 561.3082; found
561.3110 m/z.

Chapter 4 ♦Experimental - 172 - 4C.8.7. N-Acetyl-L-phenylalanyl-L-alanyl-(S,S,E)-1-amino-1-benzyl-pent-3-en-2-
one (106)
106 was prepared by the reaction of peptide 99 (180 mg, 0.957 mmol/g, 0.172 mmol)
with acetaldehyde (29 µL, 0.517 mmol, 3 eq). Yield 74 %. 1H NMR (300 MHz,
DMSO-d6): δ = 1.15 (d, J = 7.4 Hz, 3H, H-Cβ(CH3 Ala)), 1.71 (dd, J = 6.7 Hz, 3.2 Hz,
3H, H-C (CH3 Vinyl)), 1.84 (s, 3H, H-C(CH3CO)), 2.66-2.77 (m, 2H, H-Cβ1,β1′(Phe)),
2.94-3.06 (m, 2H, H-Cβ2,β2′(Benzyl)), 4.21-4.26 (m, 1H, H-Cα(Benzyl)), 4.44-4.47 (m,
1H, H-Cα(Phe)), 4.66-4.68 (m, 1H, H-Cα(Ala)), 6.33 (d, J = 15.3 Hz, 1H H-
Cα(Vinyl)), 6.81-6.89 (m, 1H, H-Cβ(Vinyl)), 7.1-7.25 (m, 10H, H-C(arom.)), 8.15 (d,
J = 7.8 Hz, 1H, NHI), 8.26 (d, J = 7.7 Hz, 1H, NHII), 8.35 (d, J = 7.7 Hz, 1H, NHIII). 13C NMR (100 MHz, DMSO-d6): δ = 18.1, 22.4, 30.4, 34.3, 48.3, 57.4, 124.8, 126.0,
126.2, 128.0, 127.9, 129.0, 129.1, 137.5, 137.6, 137.1, 138.1, 138.0, 139.1, 143.8,
169.1, 171.1, 172.1, 196.7. HRMS: calcd [(M+H+) = [C26H32N3O4] 450.2350; found
450.2347 m/z.
4C.8.8. N-Acetyl-L-phenylalanyl-L-alanyl-(S,S,E)-1-amino-1-benzyl-6-methyl-
hept-3-en-2-one (107)
107 was prepared by the reaction of peptide 99 (180 mg, 0.957 mmol/g, 0.172 mmol)
with isovaleraldehyde (55.45 µL, 0.517 mmol, 3 eq). Yield 71 %. 1H NMR (300
MHz, DMSO-d6): δ = 0.83 (d, J = 7.6 Hz, 6H, H-C(CH3 iBu)), 1.14 (d, J = 7.6 Hz,
3H, H-Cβ(Ala)), 1.71 (s, 3H, H-C(CH3CO)), 1.88 (m, 1H, H-Cδ(Vinyl)), 2.03-2.17 (m,
2H, H-Cγ (Vinyl)), 2.65-3.55 (m, 4H, H-Cβ1,β1′β2,β2′(Phe)), 4.21-4.26 (m, 1H, H-
Cα(Benzyl)), 4.44-4.50 (m, 1H, H-Cα(Phe)), 4.67-4.70 (m, 1H, H-Cα(Ala)), 6.29 (d, J
= 14.5 Hz, 1H, H-Cα(Vinyl)), 6.70-6.86 (m, 1H, H-Cβ(Vinyl)), 7.10-7.28 (m, 10H, H-
C(arom.)), 8.23 (d, J = 7.8 Hz, 1H, NHI), 8.49 (d, J = 7.8 Hz, 1H, NHII), 8.64 (d, J =
7.8 Hz, 1H, NHIII). 13C NMR (100 MHz, DMSO-d6): δ = 22.1, 22.4, 22.8, 25.6, 27.2,
30.4, 34.4, 46.4, 53.9, 124.9, 126.0, 126.2, 127.9, 128.0, 128.1, 129.1, 137.6, 138.2,
139.2, 169.1, 171.1, 172.2, 196.9. HRMS: calcd [(M+H+) = [C29H38N3O4] 492.2818;
found 492.2830 m/z.
4D. Synthesis of peptidyl heterocycles (108-125)
The peptidyl chalcones were converted to 5- and 7- membered azaheterocycles e.g.
isoxazoles, pyrazolines, pyrazoles, benzothiazepines and benzodiazepines. These
reactions were carried out in solution phase.

Chapter 4 ♦Experimental - 173 - 4D.1. Peptidyl isoxazoles (108-109)
General Procedure.151 Hydroxylamine hydrochloride (3 eq, 0.072 mmol) was added
to a stirred solution of peptidyl chalcone (1 eq., 0.024 mmol) in EtOH with catalytic
amount of acetic acid (5 mol%) and a few crystals of sodium acetate under N2. The
reaction mixture was stirred at 80 °C overnight. The solvent was removed under
reduced pressure on a rotary evaporator. The residue was subjected to purification by
preparative HPLC to afford the isoxazole as a white solid. The resulting compounds
were characterized by LC-MS and NMR.
4D.1.1. N-Acetyl-L-phenylalanyl-(S)-(1S)-benzyl-1-[5-(2-chloro-6-fluorophenyl)
isoxazol-3-yl]methylamine (108)
108 was prepared by the reaction of peptidyl chalcoone 101 (20 mg, 0.04 mmol) with
hydroxylamine hydrochloride (8.34 mg, 0.12 mmol, 3 eq). Yield 85%. 1H NMR (300
MHz, CDCl3): δ = 1.84 (s, 3H, H-C(CH3CO)), 2.68-2.76 (dd, J = 12.0 Hz, 6.0 Hz,
1H, H-Cβ(Phe)), 2.93-3.04 (m, 2H, H-Cβ(Phe, Benzyl)), 3.12-3.19 (dd, J = 18.0 Hz,
6.0 Hz, 2H, H-Cβ(Benzyl)), 5.38 (dd, J = 15.0 Hz, 6.0 Hz, 1H, H-Cα(Benzyl)), 5.52
(dd, J = 15.2 Hz, 6.0 Hz, 1H, H-Cα(Phe)), 7.03-7.22 (m, 14H, H-C(arom.)), 6.70 (s,
1H, NHI), 6.90 (s, 1H, NHII).13C NMR (100 MHz, CDCl3): δ = 22.6, 38.9, 37.6, 43.9,
53.9, 115.4, 115.7, 125.9, 126.1, 126.3, 128.0, 128.0, 128.1, 128.4, 128.5, 129.0,
129.2, 129.2, 129.5, 138.1, 138.2, 156.2, 157.4, 159.2, 169.2, 170.9, 171.0. HRMS:
calcd [(M+H+) = [C28H28ClFN3O3] 507.1830; found 507.1833 m/z.
4D.1.2. N-Acetyl-L-phenylalanyl-(S)-(1S)-benzyl-(5-isobutylisoxazol-3-yl)methyl
amine (109)
109 was prepared by the reaction of peptidyl chalcoone 100 (20 mg, 0.05 mmol) with
hydroxylamine hydrochloride (3.31 mg, 0.12 mmol, 3 eq). Yield 68%. 1H NMR (300
MHz, CDCl3): δ = 0.90 (d, J = 7.3 Hz, 3H, H-C(CH3 isobutyl)), 0.95 (d, J = 7.3 Hz,
3H, H-C(CH3 isobutyl)), 1.40-1.56 (m, 3H, CH-(CH3)2, CH2-CH-(CH3)2), 2.16 (s, 3H,
H-C(CH3CO)), 2.9-3.1 (m, 4H, H-Cβ1,β1’,β2,β2’(Benzyl, Phe)), 4.1 (dd, J = 9.8 Hz, 3.7
Hz, 1H, H-Cα(Phe)), 4.09 (dd, J = 14.6 Hz, 4.9 Hz, 1H, H-Cα(Benzyl)), 7.11-7.39 (m,
11H, H-C(arom.)), 6.31 (s, 1H, NHI), 7.60 (s, 1H, NHII). 13C NMR (100 MHz,
CDCl3): δ = 22.0, 22.1, 28.9, 35.9, 37.2, 48.6, 60.1, 85.7, 126.2, 127.9, 128.0, 128.1,
129.0, 129.1, 135.9, 137.0, 164.9, 173.3, 173.6. HRMS: calcd [(M+H+) =
[C26H34N3O3]+Et3N 433.2831; found 433.2832 m/z.

Chapter 4 ♦Experimental - 174 - 4D.2. Peptidyl pyrazolines (110-114)
General Procedure.153 Hydrazine in THF (3 eq) was added to a stirred solution of
peptidyl chalcone (1 eq) in THF under N2. The reaction mixture was stirred at reflux
temperature for 8 h. The solvent was removed under reduced pressure on a rotary
evaporator. The resulting crude products were subjected to purification by preparative
HPLC to afford the pyrazoline as a white solid. The obtained pyrazolines were
oxidized to the respective pyrazoles without characterization.
4D.2.1. N-Acetyl-L-phenylalanyl-L-alanyl-(S,S)-(1S)-benzyl-1-(5-methyl-4,5-dihy
dro-1H-pyrazol-3-yl)methylamine (110)
110 was prepared by the reaction of peptidyl chalcone 106 (20 mg, 0.044 mmol) with
hydrazine (1.38 µL, 0.133 mmol, 3 eq) in THF. Yield 65%.
4D.2.2. N-Acetyl-L-phenylalanyl-L-alanyl-(S,S)-(1S)-benzyl-1-(1,5-dimethyl-4,5-
dihydro-1H-pyrazol-3-yl)methylamine (111)
111 was prepared by the reaction of peptidyl chalcone 106 (20 mg, 0.044 mmol) with
methylhydrazine (2.34 µL, 0.133 mmol, 3 eq) in THF. Yield 61%.
4D.2.3. N-Acetyl-L-phenylalanyl-L-alanyl-(S,S)-(1S)-benzyl-1-(1-phenyl-5-methyl
-4,5-dihydro-1H-pyrazol-3-yl)methylamine (112)
114 was prepared by the reaction of peptidyl chalcone 106 (20 mg, 0.044 mmol) with
phenyldrazine (4.33 µL, 0.133 mmol, 3 eq) in THF. Yield 66%.
4D.2.4. N-Acetyl-L-phenylalanyl-L-alanyl-(S,S)-(1S)-benzyl-1-(1-methyl-5-isobut
yl-4,5-dihydro-1H-pyrazol-3-yl)methylamine (113)
113 was prepared by the reaction of peptidyl chalcone 107 (20 mg, 0.041 mmol) with
methyl hydrazine(2.18 µL, 0.122 mmol, 3 eq) in THF. Yield 53%.
4D.2.5. N-Acetyl-L-phenylalanyl-L-alanyl-(S,S)-(1S)-benzyl-1-(5-isobutyl-4,5-dih
ydro-1H-pyrazol-3-yl)methylamine (114)
114 was prepared by the reaction of peptidyl chalcone 107 (20 mg, 0.041 mmol) with
hydrazine (1.28 µL, 0.122 mmol, 3 eq) in THF. Yield 67%.
4D.3. Peptidyl pyrazoles (115-118)
General Procedure. DDQ (1.2 eq) in toluene was added to a stirred solution of
peptidyl pyrazoline (1 eq) in toluene. The reaction mixture was stirred at room
temperature for 12 h. The mixture was filtered through a plug of celite with diethyl
ether. The filtrate was concentrated in vacuo. The residue was subjected to

Chapter 4 ♦Experimental - 175 - purification by preparative HPLC to afford the pyrazole as a white solid. The purified
compounds were characterized by LC-MS and NMR.
4D.3.1. N-Acetyl-L-phenylalanyl-L-alanyl-(S,S)-(1S)-benzyl-1-(5-methyl-1H-
pyrazol-3-yl)methylamine (115)
115 was prepared by the reaction of pyrazoline 110 (20 mg, 0.043 mmol) with DDQ
(11.8 mg, 0.052 mmol, 1.2 eq). Yield 95%.1H NMR (300 MHz, DMSO-d6): δ = 1.22
(d, J = 7.7 Hz, 3H, H-Cβ(Ala)), 1.73 (s, 3H, H-C(CH3CO)), 1.98 (s, 3H, H-C(CH3
pyrazole)), 2.65-2.73 (m, 4H, H-Cβ1,β1′(Phe, Benzyl)), 2.91-2.95 (m, 4H, H-
Cβ2,β2′(Phe, Benzyl)), 4.20-4.24 (m, 1H, H-Cα(Ala)), 4.46-5.49 (m, 1H, H-
Cα(Benzyl)), 4.99-5.05 (m, 1H, H-Cα(Phe)), 5.31 (s, 1H, H-C(CH pyrazole)), 5.86 (s,
1H, NH(pyrazole)), 6.64 (d, J = 7.9 Hz, 1H, NHI), 7.12-7.25 (m, 10H, H-C(arom.)),
8.02 (d, J = 7.8 Hz, 1H, NHII), 8.20 (d, J = 7.8 Hz, 1H, NHIII).
4D.3.2. N-Acetyl-L-phenylalanyl-L-alanyl-(S,S)-(1S)-benzyl-1-(1,5-dimethyl-1H-
pyrazol-3-yl)methylamine (116)
116 was prepared by the reaction of pyrazoline 111 (20 mg, 0.042 mmol) with DDQ
(11.35 mg, 0.050 mmol, 1.2 eq). Yield 96%. 1H NMR (300 MHz, DMSO-d6): δ =
1.12-1.13 (d, J = 7.8 Hz, 3H, H-Cβ(Ala)), 1.72 (s, 3H, H-C(CH3CO)), 2.16 (s, 3H, H-
C(CH3 pyrazole)), 2.64-2.68 (m, 2H, H-Cβ1,β1′(Phe, Benzyl)), 2.92-2.94 (m, 2H, H-
Cβ2,β2′(Phe, Benzyl)), 3.60 (s, 3H, H-C(CH3-N-pyrazole)), 4.20-4.26 (m, 1H, H-
Cα(Ala)), 4.47-4.49 (m, 1H, H-Cα(Phe)), 4.92-4.98 (m, 1H, H-Cα(Benzyl)), 5.87 (s,
1H, H-C(CH pyrazole)), 7.12-7.22 (m, 11H, H-C(arom., NHI), 7.96-7.97 (d, J = 7.8
Hz, 1H, NHII), 8.04-8.08 (d, J = 7.8 Hz, 1H, NHIII).
4D.3.3. N-Acetyl-L-phenylalanyl-L-alanyl-(S,S)- (1S)-benzyl-1-(1-phenyl-5-
methyl-4,5-dihydro-1H-pyrazol-3-yl)methylamine (117)
117 was prepared by the reaction of pyrazoline 112 (20 mg, 0.037 mmol) with DDQ
(9.99 mg, 0.044 mmol, 1.2 eq). Yield 95%. 1H NMR (300 MHz, DMSO-d6): δ = 1.17
(d, J = 6.9 Hz, 3H, H-Cβ(Ala)), 1.74 (s, 3H, H-C(CH3CO)), 2.28 (s, 3H, H-C(CH3
pyrazole)), 2.62-2.71 (m, 2H, H-Cβ1,β1′(Phe, Benzyl)), 2.91-3.04, 3.12-3.21 (m, 2H, H-
Cβ2,β2′(Phe, Benzyl)), 4.21-4.28 (m, 1H, H-Cα(Ala)), 4.43-4.52 (m, 1H, H-Cα(Phe)),
5.05-5.13 (m, 1H, H-Cα(Benzyl)), 6.16 (s, 1H, H-C(CH pyrazole)), 7.14-7.25 and
7.36-7.50 (m, 16H, H-C(arom., NHI), 8.04 (d, J = 8.0 Hz, 1H, NHII), 8.12 (d, J = 8.0
Hz, 1H, NHIII).

Chapter 4 ♦Experimental - 176 - 4D.3.4. N-Acetyl-L-phenylalanyl-L-alanyl-(S,S)-(1S)-benzyl-1-(1-methyl-5-
isobutyl-1H-pyrazol-3-yl)methylamine (118)
118 was prepared by the reaction of pyrazoline 113 (20 mg, 0.040 mmol) with DDQ
(10.67 mg, 0.047 mmol, 1.2 eq). Yield 97%. 1H NMR (300 MHz, DMSO-d6): δ =
0.85-0.87 (d, J = 6.7 Hz, 6H, H-C(CH3 iBu)), 1.13 (d, J = 7.1 Hz, 3H, H-Cβ(Ala)),
1.72 (s, 3H, H-C(CH3CO)), 1.80-1.95 (m, 1H, H-C(CH iBu)), 2.40 (d, J = 6.7 Hz, 2H,
H-C(CH2 iBu)), 2.61-2.71 (m, 2H, H-Cβ1,β1′(Phe, Benzyl)), 2.89-2.97 (m, 2H, H-
Cβ2,β2′(Phe, Benzyl)), 3.60 (s, 3H, H-C(CH3-N-pyrazole)), 4.17-4.28 (m, 1H, H-
Cα(Ala)), 4.42-4.50 (m, 1H, H-Cα(Phe)), 4.92-5.02 (m, 1H, H-Cα(Benzyl)), 5.84 (s,
1H, H-C(CH pyrazole)), 7.09-7.23 (m, 10H, H-C(arom.)), 7.93 (d, J = 7.7 Hz, 1H,
NHI), 7.96 (d, J = 7.8 Hz, 1H, NHII), 8.04 (d, 1H, J = 7.7 Hz, NHIll).
4D.4. Peptidyl benzothiazepines (119-123)
General Procedure.146 1,2-Aminothiophenol (3 eq) was added to a stirred solution of
peptidyl chalcone (1 eq) in EtOH with catalytic amount of acetic acid (5 mol%) under
N2. The reaction mixture was stirred at 80 °C for 12h. The solvent was removed under
reduced pressure on a rotary evaporator. The residue was subjected to purification by
preparative HPLC to afford benzothiazepines as off-white to pale yellow solids.
4D.4.1. N-Acetyl-L-phenylalanyl-(1S)-benzyl-1-(2-(2-chloro-6-fluorophenyl)-2,3-
dihydro-1,5-benzothiazepine-4-yl)methylamine (119)
119 was prepared by the condensation of peptidyl chalcone 101 (20 mg, 0.040 mmol)
with 1,2-aminothiophenol (12.84 µL, 0.120 mmol, 3 eq). Yield 52%. 1H NMR (300
MHz, CDCl3): δ = 1.95 (s, 3H, H-C(CH3CO)), 2.84-3.14 (m, 5H, Cβ1,β2H2 Benzyl,
Cβ1,β2H2 Phe, CH2CH-S), 3.55 (dd, J = 9.7 Hz, 6.0 Hz, 1H, CH2CH-S) 4.65 (dd, J =
10.2 Hz, 6.0 Hz, 1H, CH-S), 5.04 (dd, J = 13.4 Hz, 6.3 Hz, 1H, CαH Phe), 5.39 (dd, J
= 12.6 Hz, 6.2 Hz, 1H,CαH Benzyl), 6.18 (bs, 1H, NHI), 7.06-7.58 (m, 16H, H-
C(arom.)), 7.85 (d, J = 7.3 Hz, 1H, H-C(arom.)), 8.08 (s, 1H, NHII).13C NMR (100
MHz, CDCl3): δ = 22.2, 35.3, 37.7, 45.0, 48.9, 55.0, 59.5, 114.4, 114.8, 118.2, 121.2,
122.1, 125.8, 126.6, 126.9, 127.0, 128.4, 128.4, 128.7, 128.7, 128.8, 128.9, 129.8,
129.9, 136.4, 136.4, 151.4, 155.3, 158.5, 163.2, 169.5, 173.3. HRMS: calcd [(M+H+)
= [C34H32ClFN3O2S] 600.1845; found 600.1852 m/z.

Chapter 4 ♦Experimental - 177 - 4D.4.2. N-Acetyl-L-phenylalanyl-(1S)-benzyl-1-(2-isobutyl-2,3-dihydro-1,5-benzo
thiazepine-4-yl)methylamine (120)
120 was prepared by the condensation of peptidyl chalcone 100 (20 mg, 0.05 mmol)
with 1,2-aminothiophenol (15.3 µL, 0.143 mmol, 3 eq). Yield 63%. 1H NMR (300
MHz, CDCl3): δ = 0.87 (d, 3H, J = 7.3 Hz, H-C(CH3 isobutyl)), 0.93 (d, 3H, J = 7.3
Hz, H-C(CH3 isobutyl)), 1.42-1.60 (m, 1H, H-C(CH2 isobutyl)), 1.81-2.20 (m, 5H,
CH(CH3)2, CH2CH(CH3)2, CH3CO), 2.82-3.18 (m, 6H, H-Cβ1,β2Benzyl, H-Cβ1,β2Phe,
CH2CH-S), 4.03 (dd, J = 14.6 Hz, 6.1 Hz, 1H, H-Cα(Phe)), 4.70 (m, 1H, CH-S), 5.60
(dd, J = 15.9 Hz, 7.3 Hz, 1H, H-Cα(Benzyl)), 6.16 (bs, 1H, NHI), 7.04-7.37 (m, 13H,
H-C(arom.)), 7.82 (d, J = 7.3 Hz, 1H, H-C(arom.)), 8.32 (bs, 1H, NHII). HRMS: calcd
[(M-H+) = [C32H36N3O2S]+Et3N 624.2409; found 624.2421 m/z.
4D.4.3. N-Acetyl-L-phenylalanyl-(1S)-isopropyl-1-(7-chloro-2-(2-chloro-6-fluoro
phenyl)-2,3-dihydro-1,5-benzothiazepine-4-yl)methylamine (121)
121 was prepared by the condensation of peptidyl chalcone 102 (20 mg, 0.045 mmol)
with 4-chloro-1,2-aminothiophenol (21.54 mg, 0.135 mmol, 3 eq). Yield 75%. 1H
NMR (300 MHz, CDCl3): δ = 0.75 (d, J = 6.7 Hz, 3H, H-Cγ1(CH3 isopropyl)), 0.91 (d,
J = 6.7 Hz, 3H, H-Cγ2(CH3 isopropyl)), 1.97 (s, 3H, H-C(CH3CO)), 1.98-2.07 (m, 1H,
H-Cβ(CH isopropyl)), 2.43-2.60 (m, 2H, H-Cβ1,β2(Phe)), 2.95-3.16 (m, 2H, CH2CH-
S), 4.39-4.50 (m, 1H, H-Cα(Phe)), 4.62-4.70 (m, 1H, CH-S), 4.75-4.90 (m, 1H, H-
Cα(CH isopropyl)), 6.50-7.50 (m, 13H, H-C(arom.), NHI,II).13C NMR (100 MHz,
CDCl3): δ = 18.5, 19.3, 22.8, 25.2, 30.4, 30.9, 35.9, 36.9, 38.2, 53.4, 54.7, 62.4, 114.9,
115.2, 122.2, 122.3, 122.8, 126.0, 126.3, 129.2, 129.3, 130.1, 132.7, 135.5, 135.9,
136.0, 154.2, 162.3, 163.5, 169.2, 171.3. HRMS: calcd [(M+H+) =
[C30H31Cl2FN3O2S] 587.1524; found 587.1532 m/z.
4D.4.4. N-Acetyl-L-phenylalanyl-(1S)-isopropyl-1-(7-chloro -2-(thiophen-2-yl)-
2,3-dihydro-1,5-benzothiazepine-4-yl)methylamine (122)
122 was prepared by the condensation of peptidyl chalcone 103 (20 mg, 0.050 mmol)
with 4-chloro-1,2-aminothiophenol (29.94 mg, 0.150 mmol, 3 eq). Yield 71%. 1H
NMR (300 MHz, CDCl3): δ = 0.78 (d, J = 6.9 Hz, 3H, H-Cγ1(CH3 isopropyl)), 0.94 (d,
J = 6.9 Hz, 3H, H-Cγ2(CH3 isopropyl)), 1.98 (s, 3H, H-C(CH3CO)), 1.99-2.03 (m, 1H,
H-Cβ(CH isopropyl)), 2.60-2.82 (m, 2H, H-Cβ1,β2(Phe)), 3.02-3.14 (m, 2H, CH2CH-
S), 5.07-5.16 (dd, J = 11.6 Hz, 4.8 Hz, 1H, CH-S), 5.26-5.35 (m, 1H, H-Cα(CH

Chapter 4 ♦Experimental - 178 - isopropyl)), 6.5 (d, J = 3.2 Hz, 1H, H-C(thiophen)), 6.69 (t, 1H, H-C(thiophen)), 6.90-
7.30 (m, 10H, H-Carom., NHI), 7.5 (bs, NHII). HRMS: calcd [(M+H+) =
[C28H31ClN3O2S2] 541.1571; found 541.1589 m/z.
4D.4.5. N-Acetyl-L-leucinyl-(1S)-benzyl-1-(7-chloro-2-(2-chloro-6-fluorophenyl)
-2,3-dihydro-1,5-benzothiazepine-4-yl)methylamine (123)
123 was prepared by the condensation of peptidyl chalcone 105 (20 mg, 0.044 mmol)
with 4-chloro-1,2-aminothiophenol (20.9 mg, 0.131 mmol, 3 eq). Yield 55%. 1H
NMR (300 MHz, CDCl3): δ = 0.81, 0.92 (d, J = 6.5 Hz, 6H, H-Cδ1,δ2(Leu)), 1.22-1.35
(m, 2H, H-Cβ1,β2(Leu)), 1.48-1.65 (m, 1H, H-Cγ(Leu)), 1.73 (s, 3H, H-C(CH3CO)),
2.64-2.74 (m, 2H, H-Cβ1,β2(Benzyl)), 3.06-3.36 (m, 2H, CH2CH-S), 3.97 (m, 1H, H-
Cα(Leu)), 5.05-5.16 (m, 2H, H-Cα(Benzyl), CH-S), 5.94 (bs, NHI), 6.54-7.47 (m,
12H, H-C(arom.), NHII). 13C NMR (100 MHz, CDCl3): δ = 22.9, 24.8, 38.7, 40.9, 43.1, 44.7, 52.1, 58.6, 60.8,
114.7, 114.8, 119.3, 122.2, 123.0, 123.7, 126.1, 126.3, 126.9, 128.1, 128.5, 128.9,
130.1, 131.6, 132.8, 133.3, 137.1, 153.8, 154.7, 160.5, 167.5, 182.5. HRMS: calcd
[(M+H+) = [C31H33Cl2FN3O2S] 601.1681; found 601.1692 m/z.
4D.5. Synthesis of peptidyl benzodiazepines (124-125)
General Procedure. 1,2-Phenylenediamine (3 eq) was added to a stirred solution of
peptidyl vinyl ketone (1 eq) in EtOH with catalytic amount of Et3N (10 mol%) under
N2. The reaction mixture was stirred at 80 °C overnight and then brought to room
temperature. The solvent was removed under reduced pressure on a rotary evaporator.
The residue was subjected to purification by preparative HPLC to afford the
benzodiazepines.
4D.5.1. N-Acetyl-L-phenylalanyl-(1S)-isopropyl-1-[2-(2-chloro-6-fluorophenyl)-
1H-1,5-benzodiazepine-4-yl)methylamine (124)
124 was synthesized by the reaction of peptidyl chalcone 102 (20 mg, 0.050 mmol)
with 1,2-phenylenediamine (16.22 mg, 0.150 mmol, 3 eq). Yield 48%. 1H NMR (300
MHz, CDCl3): δ = 1.15 (d, J = 6.8 Hz, 6H, H-Cγ1,γ2(CH3 isopropyl)), 1.21-1.30 (m,
1H, H-Cβ(CH isopropyl)), 1.9 (s, 3H, H-C(CH3CO)), 2.75-2.80 (m, 2H, H-
Cβ1,β2(Phe)), 2.95-3.10 (m, 2H, H-C(CH2 diazepin)), 3.42 (m, 1H, NHI), 4.17-4.24
(m, 1H, H-Cα(Phe)), 4.75 (m, 1H, H-Cα(CH isopropyl)), 4.98-5.04 (m, 1H, H-C(CH
diazepin)), 6.8-7.9 (m, 14H, H-C(arom., NHII,III). 13C NMR (100 MHz, CDCl3): δ =

Chapter 4 ♦Experimental - 179 - 18.6, 18.7, 22.9, 28.6, 36.0, 37.1, 42.6, 54.8, 62.7, 111.6, 113.7, 120.3, 126.1, 127.3,
127.6, 128.8, 129.1, 129.2, 131.4, 135.2, 135.7, 136.3, 139.9, 140.3, 141.1, 151.8,
152.7, 165.1, 172.8, 184.4. HRMS: calcd [(M+H+) = [C30H33ClFN4O2] 533.2133;
found 533.2140 m/z.
4D.5.2. N-acetyl-L-leucinyl-(1S)-benzyl-1-[2-(2-chloro-6-fluorophenyl)-1H-1,5-
benzodiazepine-4-yl)methylamine (125)
125 was prepared by the condensation of peptidyl chalcone 105 (20 mg, 0.044 mmol)
with 1,2-phenylenediamine (14.17 mg, 0.131 mmol, 3 eq). Yield 50%. 1H NMR (300
MHz, DMSO-d6): δ = 0.66 (d, J = 6.6 Hz, 6H, H-Cδ1,δ2(Leu)), 1.68-1.72 (m, 2H, H-
Cβ(Leu)), 1.96 (s, 3H, H-C(CH3CO)), 2.12-2.24 (m, 2H, H-Cβ1,β2(Benzyl)), 2.86-2.96
(m, 2H, H-C(CH2 diazepin)), 3.68 (m, 1H, NHI), 4.21-4.29 (m, 1H, H-Cα(Leu)), 5.26-
5.32 (m, 1H, H-Cα(Benzyl)), 6.21-6.29 (m, 1H, H-C(CH diazepin)), 6.70-7.60 (m,
14H, H-C(arom., NHII,III). 13C NMR (100 MHz, DMSO-d6): δ = 21.1, 22.2, 30.7,
43.1, 43.3, 46.3, 47.1, 62.6, 78.5, 78.9, 79.4, 104.3, 122.4, 128.3, 129.1, 129.5, 130.1,
139.7, 147.1, 147.7, 152.3. HRMS: calcd [(M+H+) = [C31H35ClFN4O2] 550.2459;
found 550.2451 m/z.
4E. Bioevaluation
The libraries synthesized were screened for their antibacterial, phosphatase inhibition,
cytotoxic and antitumor properties.
4E.1. Antibacterial assay
Compounds were tested against three Gram-positive bacterial strains (Bacillus
subtillis ATTCC 6633, Micrococcus leuteus and Staphylococcus aureus ATCC 6538)
and three Gram-negative ones (Escherichia coli AATCC 1522, Enterobacter
aerogenes AATCC 13048 and Salmonella setubal ATCC 19196). The agar Well-
Diffusion method188 was used for the determination of inhibition zones and minimum
inhibitory concentration (MIC). Briefly, 0.75 mL of the broth culture containing ca.
106 colony-forming units (CFU) per mL of test strain was added to 75 mL of nutrient
agar medium at 45 oC, mixed well, and then poured into a 14-cm sterile metallic Petri
plate. The medium was allowed to solidify, and 8 mm wells were dug with a sterile
metallic borer. Then, a DMSO soln. of test sample (100 uL) as 1mg/mL was added to
the respective wells. DMSO served as neg. control, and the standard antibacterial
drugs roxithromycin (1 mg/mL) and Cefixime® (1 mg/mL) were used as positive

Chapter 4 ♦Experimental - 180 - controls. Triplicate plates of each bacterial strain were prepared. The plates were
incubated aerobically at 37 oC for 24 h. Activity was determined by precisely
measuring the diameter of zones showing complete inhibition (mm) with the aid of a
Vernier caliper (precision ± 0.1 mm). The growth inhibition was calculated with
reference to positive controls.
Determining of Minimum-Inhibitory Concentrations. For the indivisual chalcones that
showed inhibition zones > 10 mm, MIC values were determined by means of agar
well diffusion method for concentrations of 1.0, 0.8, 0.6, 0.5, 0.4, 0.2 and 0.1 mg/mL
in DMSO. The tests were performed in triplicate, and the results were averaged.
4E.2. Cytotoxicity studies
Brine shrimp was used as the test animal for the investigation of cytotoxicity
according to the standard reported procedure.188 An artificial saline water was
prepared with 30-35 ppm saline (3.8g sea salt/L) and placed in a small unequally
divided tank. About one teaspoon of brine shrimp eggs were added to the larger
compartment covered with aluminum foil. Eggs were maintained under strong
aeration at room temperature for two days so that napulii were mature enough to test.
The illuminated compartment attracted phototropic napulii through perforations in the
dam.
Different concentrations (1000, 100 & 10 µg/mL) of the test samples were prepared in
DMSO. These test samples were taken into the separated test tubes, so that each test
tube contained not more than 50 µL of DMSO. Then ten brine shrimps were
transferred to each test tube using micro pipettes. All tests were performed in
replicates at each concentration. The vials were maintained under strong illumination.
After 24 hours had elapsed, the number of survived napulii were counted and
recorded with the aid of a 3x magnifying glass. The percentage of lethality of brine
shrimp nauplii was counted at each concentration for each sample. An approximate
linear correlation was observed when logarithm of concentration was plotted against
percentage of mortality and the values of LD50 (95% confidence intervals) were
calculated using a simple PC program; Probid analysis. DMSO was used as a negative
control.

Chapter 4 ♦Experimental - 181 - 4E.3. Potato disc tumor (PDT) bioassay
This assay was performed according to the standard reported procedure.200 Potato
discs (0.5 cm thickness) were obtained from surface sterilized potatoes by using a
metallic cork borer and special cutter under completely aseptic conditions. These
potato discs were then transferred to petri plates each containing 25 mL of 1.5% agar
solution. Then 500, 50 and 5 µL of stock (10 mg/mL) of the test sample was added to
2000 µL, 2450 µL and 2495 µL of broth culture of Agrobacterium tumefaciens (a 48 h
culture containing 5 × 109 cells/mL) and 2.5mL of autoclaved distilled water was
added to make 1000 µg/mL, 100 µg/mL, 10 µg/mL concentration. 50µL of these
cultures were poured on each potato disk. The Petri dishes were incubated at 28 ºC.
After 21 days incubation, the number of tumors was counted with the aid of a
dissecting microscope after staining with Lugol’s solution (5% I2, 10%). Then
%inhibition of tumors was calculated by the formula, 100–(NS/NC×100) where NS
are the number of tumors for sample and NC are the number of tumors for negative
control.
IC50 values were calculated after plotting percent activity against the logarithmic
transformation of the corresponding inhibitor with the help of SigmaPlot (SSPS).
4E.4. Phosphatase inhibition265
Phosphatase inhibition assays were performed using 6,8-difluoro-4-
methylumbelliferyl phosphate (DiFMUP) as a substrate obtained from Invitrogen
(D22065) whereas Ptps were obtained as 10mg/mL stock solutions in 10% glycerol
from Oxford. Buffer was made by mixing 10µM NaCl, 0.03% Tween20, 2mM DTT
added to 50mM BIS-TRIS at a pH of 6.5. The reaction buffer was made by mixing
buffer, 0.00078 ng/µL PTP, 10µM DiFMUP and 33.3µg/ml BSA.
Procedure. 15µL of 500µM BIS-TRIS buffer pH 6.5 was dispensed to each well of
MTP. With the robot, 5µL from 2mM compound stock solutions were dispensed into
15µL buffer. Plates were sealed with aluminium foil and stored at -20 °C as pre-
dilution plate A. From this solution 10µL/well reaction buffer was added with
dispenser to another new MTP plate B 0.6 µL of compound stock from the pre-
dilution plate A was pipetted into the 10µL buffer/well in plate B with the robot. This
plate was sealed carefully with foil until the subsequent treatment and stored at -20

Chapter 4 ♦Experimental - 182 - °C. From MTP B, 10µL of the substrate solution was dispensed to rows 1-24 in a new
MTPC. Stored plate in the dark.
The enzyme dilution was prepared for the plate (0.00078 ng/µL PTP). Added
33.3µg/mL BSA in the enzyme solution in a ratio of 1:100, so that the enzyme would
remain stable. Stored this MTP D on ice. From MTPD, 10µL enzyme solution was
dispensed in rows 1-23 of MTP C. Centrifuged briefly and placed into the plate reader
(Tecan Safire). From 20mL reaction buffer containg 10µM DIFMUP and
0.00078ng/µl PTP, 28.5µL/well was transfered with a multi-channel pipette to the
wells of C and 1.5 µL of 50mM buffer was added quickly after the enzyme addition.
The plate was centrifuged and stirred on the shaker at 500 rpm for 15 sec.
Fluorescence was measured in the plate reader (Tecan Safire). Fluorescence was
measured for the DiFMU at 360nm (ε = 17mM-1 cm-1) released as a result of
enzymatic reaction from DiFMUP.
In the next step, 5mL reaction buffer BIS-TRIS pH 6.5 was prepared with an enzyme
concentration of 0.00078ng/µL. 60µL of the enzyme-buffer solution are transferred to
the wells of the MTP. Into the wells, pipetted 60µL 200µM DiFMUP and diluted
rapidly with the multi-channel pipette in a ratio of 1:2. The plate is again briefly
centrifuged and then measured in the platereader (Tecan Safire).
Inhibition of ptp by a particular compound was calculated as a percent activity of the
uninhibited phosphatase. With the tested compound, the absorbance (initially taken as
100%) was decreased to a certain value, and, therefore, % inhibition values for
ligands were calculated by substracting the % absorbance from 100.
4E.5. PGM inhibition assay
A different protocol was followed for carrying out PGM inhibition activity. The assay
was performed by continuous spectrophotometric rate determination at 25 °C, and a
pH of 7.6. The following reagents were used in the enzyme assay.
A. 100 mM triethanolamine buffer, pH 7.6 at 25 °C
(Prepared 100 ml in deionized water using triethanolamine hydrochloride,
Sigma Prod. No. T-1502. Adjusted to pH 7.6 at 25 °C with 1 M NaOH.)
B. 200 mM 3-Phosphoglyceric acid solution (3-PGA)
(Prepared 1 mL in deionized water using D(-)3-phosphoglyceric acid,
trisodium salt, Sigma Prod. No. P-0769.)

Chapter 4 ♦Experimental - 183 -
C. 21 mM adenosine 5'-diphosphate solution (ADP)
(Prepared 1 mL in deionized water using adenosine 5'-diphosphate, sodium
salt, sigma Prod. No. A-6521. Prepared fresh)
D. 40 mM 2,3-diphosphoglyceric acid solution (DPGA)
(Prepared 1 mL in deionized water using 2,3-diphospho-D-glyceric acid,
pentacyclohexylammonium salt, Sigma Prod. No. D-9134. Prepared fresh)
E. 6.4 mM ß-nicotinamide adenine dinucleotide, reduced form solution (ß-
NADH) (dissolved the contents of one 5 mg vial of ß-nicotinamide adenine
dinucleotide, reduced form, disodium salt, Sigma stock No. 340-105 in the
appropriate volume of reagent A.)
F. 50 mM magnesium sulfate solution (MgSO4) (prepared 2 mL in deionized
water using magnesium sulfate, heptahydrate, Sigma Prod. No. M-1880.)
G. 2 M potassium chloride solution (KCl)
(prepare 2 mL in deionized water using potassium chloride, Sigma Prod. No.
P-4504.)
H. PK/LDH enzymes (PK/LDH) (Used PK/LDH enzymes suspension, Sigma
Stock No. 40-7.) Removed glycerol before reaction to avoid its inhibitory
activity in PGA.
I. Enolase enzyme solution (enolase)
(prepared a solution containing 100 units/mL of enolase in cold deionized
water immediately before use, Sigma Prod No. E-0379.)
J. Phosphoglycerate mutase enzyme solution (PGM)
(prepared a solution containing 0.3-0.6 unit/mL of PGM in cold reagent A
immediately before use.)
Procedure.
Prepared a reaction cocktail by pipetting 22.0 µL of 100 mM triethanolamine buffer at
pH 7.6, 1.0 µL of 200 mM 3-PGA, 1.0 µL of 21 mM ADP, 1.0 µL of 40 mM DPGA,
0.70 µL of 6.4 mM ß-NADH, 1.50 µL of 50 mM MgSO4, 1.50 µL of 2 M KCl
solutions. Mixed by swirling and adjusted to pH 7.6 at 25 °C with either 1 M NaOH
or 1 M HCl, if necessary.
Pipetted out 949 µL of reaction cocktail, added 7 µL of PK/LDH enzymes and 10 µL
of enolase enzyme solution into the 1mL cuvette. Mixed by inversion and equilibrated

Chapter 4 ♦Experimental - 184 - to 25°C. Monitored the absorbance at 340 nm at UV vis. spectrophotometer. This
reading served as a negative control.. After that added 34 µL of PGM solution into the
same cuvette, immediately mixed by inversion and recorded the decrease in
absorbance at 340 nm for approximately 5 minutes.
Ligand mixture was prepared by mixing 272 µL of the ligand (5 mM stock soln.) into
3728 µL of the 100 mM Tris buffer at pH 7.6). PGM % inhibition studies of ligand
were performed on a 384 well plate. For performing each reaction as 20 µL total
reaction volume, added 9.49 µL reaction cocktail, 0.7 µL of PK/LDH, 0.1 µL
enolase. Monitored the absorbance at 340 nm. Added in 34 µL PGM soln. to make a
total of 10 µL of reaction mixture. Added rapidly 10 µL of the ligand soln. and mixed
well. The decrease in absorbance was recorded at 340 nm for approximately 5
minutes using a plate reader (Tecan).
Inhibition of PGM by a particular compound was calculated as a percent activity of
the uninhibited phosphatase. The % inhibition values for ligands were calculated by
substracting the % absorbance from 100.

References

References 1. Wilson, S. R.; Czarnik, A. W. Combintorial Synthesis and Application; John
Wiley & Sons: New York, 1997, p 2.
2. Marasco, W. A.; Sui, J. Nat. Biotech. 2007, 25, 1421.
3. Olivera, B. M.; Hillyard, D. R.; Marsh, M.; Yoshikami, D. Trends Biotechnol.
1995, 13, 422.
4. Fassina, G.; Lebl, M.; Chaiken, I. Collect. Czech. Chem. Commun. 1988, 53,
2627.
5. Peakman, T.; Bonduelle, Y. Drug Discov. Today, 2000, 5, 337.
6. Gallop, M. A.; Barrett, R. W.; Dower, W. J.; Fodor, S. P. A.; Gordon, E. M. J.
Med. Chem. 1994, 37, 1233.
7. Pavia, M. R.; Sawyer, T. K.; Moos, W. H. Bioorg. Med. Chem. Lett. 1993, 3,
387.
8. Pinilla, C.; Appel, J. R.; Borras, E.; Houghten, R. A. Nat. Med. 2003, 9, 118.
9. Boger, D. L.; Desharnais, J.; Capps, K. Angew. Chem. Int. Ed. 2003, 42, 4138.
10. Terrett, N. K.; Gardner, M.; Gordon, D. W.; Kobylecki, R. J.; Steele, J.
Tetrahedron, 1995, 51, 8135.
11. Venturini, S.; Allicotti, G.; Zhao, Y.; Simon, R.; Burton, D. R.; Pinilla, C.;
Poignard, P. Eur. J. Immunol. 2006, 36, 27.
12. Lustgarten, J.; Dominguez, A. L.; Pinilla, C. J. Immunol. 2006, 176, 1796.
13. Sospedra, M.; Muraro, P. A.; Stefanova, I.; Zhao, Y.; Chung, K.; Li, Y.;
Giulianotti, M.; Simon, R.; Mariuzza, R. A.; Pinilla, C.; Martin, R. ibid, 2006,
176, 1951.
14. Sospedra, M.; Zhao, Y.; Zur Hausen, H.; Muraro, P. A.; Hamashin, C.; de
Villiers, E. M.; Pinilla, C.; Martin, R. PLoS. Pathog. 2005, 1, 41.
15. Raghavan, S.; Yang, Z.; Mosley, R. T.; Schleif, W. A.; Gabryelski, L.; Olsen,
D. B.; Stahlhut, M.; Kuo, L. C.; Emini, E. A.; Chapman, K. T.; Tata, J. R.
Bioorg. Med. Chem. Lett. 2002, 12, 2855.
16. Choe, Y.; Leonetti, F.; Greenbaum, D. C.; Lecaille, F.; Bogyo, M.; Bromme,
D.; Ellman, J. A.; Craik, C. S. J. Biol. Chem. 2006, 281, 12824.
17. Lee, Y.; Kang, D. K.; Chang, S. I.; Han, M. H.; Kang, I. C. J. Biomol. Screen.
2004, 9, 687.

References
- 187 -
18. Fugere, M.; Appel, J.; Houghten, R. A.; Lindberg, I.; Day, R. Mol. Pharmacol.
2007, 71, 323.
19. Kacprzak, M. M.; Peinado, J. R.; Than, M. E.; Appel, J.; Henrich, S.; Lipkind,
G.; Houghten, R. A.; Bode, W.; Lindberg, I. J. Biol. Chem. 2004, 279, 36788.
20. Fujimoto, D. F.; Pinilla, C.; Segall, A. M. J. Mol. Biol. 2006, 363, 891.
21. Fujii, K.; Zhu, G.; Liu, Y.; Hallam, J.; Chen, L.; Herrero, J.; Shaw, S. Proc.
Natl. Acad. Sci. U.S.A. 2004, 101, 13744.
22. Szostak, J. W. Chem. Rev. 1997, 97, 347.
23. Lebl, M. J. Comb. Chem. 1999, 1, 3.
24. Thompson, L. A.; Ellman, J. A. Chem. Rev. 1996, 96, 555.
25. Merrifield, R. B. J. Am. Chem. Soc. 1963, 85, 2149.
26. Gordon, E. M.; Gallop, M. A.; Patel, D. V. Acc. Chem. Res. 1996, 29, 144.
27. Miranda, L. P.; Alewood, P. F. Proc. Natl. Acad. Sci. USA, 1999, 96, 1181.
28. Haoyun An, P.; Dan Cook, Chem. Rev. 2000, 100, 3311.
29. Boyle, N. A.; Janda, K. D. Curr. Opin. Chem. Biol. 2002, 6, 339.
30. Ley, S. V.; Baxendale, I. R.; Brusotti, G.; Caldarelli, M.; Massi, A.; Nesi, M.;
II Farmaco, 2002, 57, 321.
31. Cin, M. D.; Davalli, S.; Marchioro, C.; Passarini, M.; Perini, O.; Provera, S.;
Zaramella, A. II Farmaco, 2002, 57, 497.
32. Yurek, D. A.; Branch, D. L.; Kuo, M. -S. J. Comb. Chem. 2002, 4, 138.
33. Gelens, E.; Koot, W. J.; Menge, W. M. P. B.; Ottenheijm, H. C. J.;
Timmerman, H. Comb. Chem. High Throughput Screening, 2003, 6, 79.
34. Terret, N. Comb. Chem. -an online Journal, 2003, 5, 1. b) Terret, N. K. ibid,
2003, 5, 5.
35. Albericio, F. Curr. Opin. Chem. Biol. 2004, 8, 211.
36. Fergus, S.; Bender, A.; Spring, D. R. ibid. 2005, 9, 304.
37. Bauer, J.; Rademann, J. J. Am. Chem. Soc. 2005, 127, 7296.
38. Ahsanullah, Schmieder, P.; Kuhne, R.; Rademann, J. Angew. Chem. Int. Ed.
2009, 48, 5042.
39. Powers, D. G.; Casebier, D. S.; Fokas, D.; Ryan, W. J.; Troth, J. R.; Coffen.
D. L. Tetrahedron, 1998, 54, 4085.
40. Lee, H. -K.; Chui, W. -K. Bioorg. Med. Chem. 1999, 7, 1255.
41. Vicent, M. J.; -Paya, E. P.; Orzáez. M. Curr. Topics. Med. Chem. 2007, 7, 83.

References
- 188 -
42. Cerezo, V.; Amblard, M.; Martinez, J.; Verdié, P.; Planas, M.; Feliu. L.
Tetrahedron, 2008, 64, 10538.
43. Lim, H, -J.; Myung, D.; Lee, I. Y. C.; Jung. M. H. J. Comb. Chem. 2008, 10,
501.
44. Cheng, Y.; Rano, T. A.; Huening, T. T.; Zhang, F.; Lu, Z.; Schleif, W. A.;
Gabryelski, L.; Olsen, D. B.; Stahlhut, M.; Kuo, L. C.; Lin, J. H.; Xu, X.; Jin,
L.; Olah, T. V.; McLoughlin, D. A.; King, R. C.; Chapman, K. T.; Tata, J. R.
Bioorg. Med. Chem. Lett. 2002, 12, 529.
45. Lindell, S. D.; Pattenden, L. C.; Shanoon. J. Bioorg. Med. Chem. 2009, 17,
4035.
46. Nefzi, A.; Ostresh, J. M.; Houghten, R. A. Chem. Rev. 1997, 97, 449.
47. Breaker, R. R. ibid. 1997, 97, 371.
48. Pirrung, M. C. ibid. 1997, 97, 473.
49. Fodor, S. P. A.; Read, J. L.; Pirrung, M. C.; Stryer, L.; Lu, A. T.; Solas, D.
Science, 1991, 251, 767.
50. Watts, P.; Wiles, C.; Haswell, S. J.; Pombo-Villar, E. Tetrahedron, 2002, 58,
5427.
51. Geysen, H. M.; Rodda, S. J.; Mason, T. J. Mol. Immunol. 1986, 23, 709.
52. Houghten, R. A.; Pinilla, C.; Blondelle, S. E.; Appel, J. R.; Dooley, C. T.;
Cuervo, J. H. Nature, 1991, 354, 84.
53. Gravert, D. J.; Janda, K. D. Chem. Rev. 1997, 97, 489.
54. Senten, K.; Veken, P. V.; Bal, G.; Haemers, A.; Augustyns, K. Tetrahedron
Lett. 2001, 42, 9135.
55. Ley , S. V.; Baxendale, I. R.; Bream, R. M.; Jackson, P. S.; Leach, A. G.;
Longbottom, D. A.; Nesi, M.; Scott, J. S.; Storer, R. I.; Taylor, S. J. J. Chem.
Soc. Perkin Trans. 1, 2000, 23, 3815.
56. Kirschning, A.; Monenschein, H.; Wittenberg, R. Angew. Chem. 2001, 113,
670.
57. Bunin, B. A.; Elmann, J. A. J. Am. Chem. Soc, 1992, 114, 10997.
58. Bunin, B. A.; Plunkett, M.J.; Ellman, J. A. Proc. Natl. Acad. Sci. USA, 1994,
91, 4708.
59. Lindsley, C. W.; Hodges, J. C.; Filzen, G. F.; Watson, B. M.; Geyer, A. G. J.
Comb. Chem. 2000, 2, 550.

References
- 189 -
60. Haag, R.; Hebel, A.; Stumbé, J. Chap 3. Solid-phase and Soluble polymers for
Combinatorial Synthesis; Editors: Nicolaou, K. C.; Hanko, R.; Hartwing. W.
Handbook of Combinatorial Chemistry, Wiley-VCH Verlag GmbH:
Weinheim, Germany, 2002.
61. Nagata, K.; Fakunaga, T.; Kato, S.; Jyo, A. React. Funct. Polym., 2008, 68,
1127.
62. Houghten, R. A. Proc. Natl. Acad. Sci. U.S.A. 1985, 82, 5131.
63. Furka, A.; Sebestye´n, F.; Asgedom, M.; Dibo´, G. Int. J. Peptide Prot. Res.
1991, 37, 487.
64. Plunkett, M. J.; Ellman, J. A. J. Am. Chem. Soc. 1995, 117, 3306.
65. Pirrung, M. C.; Chen, J. J. Am. Chem. Soc. 1995, 117, 1240.
66. Pirrung, M. C.; Chau, J. H. L.; Chen, J. In Combinatorial Chemistry,
Synthesis, and Application; John Wiley and Sons: New York, 1997, p191.
67. Pirrung, M. C.; Chau, J. H. L.; Chen, J. J. Chem. Biol. 1995, 2, 61.
68. An, H.; Cook, P. D. Chem. Rev. 2000, 100, 3311.
69. Smith, P. W.; Lai, J. Y. Q.; Whittington, A. R.; Cox, B.; Houstol, J. G.; Stylli,
C. H.; Banks, M. N.; Tiller, P. R. Bioorg. Med. Chem. Lett. 1994, 4, 2821.
70. Neuville, L.; Zhu, J. Tetrahedron Lett. 1997, 38, 4091.
71. Kaldor, S. W.; Fritz, J. E.; Tang, J.; McKinney, E. O. Bioorg. Med. Chem.
Lett. 1996, 6, 3041.
72. Marder, M.; Viola, H.; Bacigaluppu, J. A.; Colombu, M. I.; Wasowski, C.;
Wolfman, C.; Medina, J. H.; Ruveda, E. A.; Paladini, A. C. Biochem. Biophys.
Res. Commun. 1998, 249, 481.
73. Nielsan, J; Rasmussen, P. H. Tetrahedron Lett. 1996, 37, 3351.
74. Chung, B. L; Ganesan. A. Bioorg. Med. Chem. Lett. 1997, 7, 1511.
75. Dooley, C. T.; Ny, P.; Bidlack, J. M.; Houghten, R. A. J. Biol. Chem. 1998,
273, 18848.
76. Wilson-Lingardo, L.; Davis, P. W.; Ecker, D. J.; He´bert, N.; Acevedo, O.;
Sprankle, K.; Brennan, T.; Schwarcz, L.; Freier, S. M.; Wyatt, J. R. J. Med.
Chem. 1996, 39, 2720.
77. Erb, E.; Janda, K. D.; Brenner, S. Proc. Natl. Acad. Sci. USA, 1994, 91,
11422.

References
- 190 -
78. Lam, K. S.; Salmon, S. E.; Hersh, E. M.; Hruby, V. J.; Kazmierski, W. M.;
Knapp, R. J. Nature, 1991, 354, 82.
79. Chersi, A.; Sezzi, M. L.; Romano, T. F.; Evangelista, M.; Nista, A. Biochem.
Biophys. Acta, 1990, 1034, 333.
80. Medal, M.; Svendsen, I. J. Chem. Soc. Perkin Trans. 1, 1995, 1, 1591.
81. Cardno, M.; Bradley, M. Tetrahedron Lett. 1996, 37, 135.
82. Moran, E. J.; Sarshar, S.; Cargill, J. F.; Shabaz, M. M., Lio, A.; Mjalli, A. M.
M.; Armstrong, R. W. J. Am. Chem. Soc. 1995, 117, 10787.
83. Czarnik, T.; Nova, M. Chem. Br. 1997, 33, 39.
84. Brenner, S.; Lerner, R. A. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 5381.
85. Kerr, J. M.; Banville, S. C.; Zuckermann, R. N. J. Am. Chem. Soc. 1993, 115,
2529.
86. Still, W.C. Acc. Chem. Res. 1996, 29, 155.
87. Ni, Z. –J.; Maclean, D.; Holmes, C. P.; Murphy, M. M.; Ruhland, B.; Jacobs,
J. W.; Gordon, E. M.; Gallop, M. M. J. Med. Chem. 1996, 39, 1601.
88. Barnes, C.; Balasubramanian. S. Curr. Opin. Chem. Biol. 2000, 4, 346.
89. Smith, H. J.; Williams, H. Smith and Williams Introduction to the Drug
Design and Action; 4th Edit, Edit. Smith, H. J.; CRC Press, Taylor & Francis
Group: FL, USA, 2006, p-365.
90. Lipniski, C. A.; Lombardo, F.; Dominy, B. W. Adv. Drug Delivery Rev. 1997,
23, 3.
91. Lipniski, C. A. Drug Discovery Today: Technol. 2004, 1, 337.
92. Veber, D. F.; Johnson, S. R.; Cheng, H. -Y.; Smith, B. R.; Ward, K. W.;
Kopple, K. D. J. Med. Chem. 2002, 45, 2615.
93. Clark, D. E.; Picket. S. D. Drug Discovery Today, 2000, 5, 49.
94. Oprea. T. I. J. Chem. Inf. Comput. Sci. 2001, 41, 1308.
95. Teague, S. J.; Davis, A. M.; Leeson, P. D.; Oprea, T. Angwe. Chem. Int. Ed.
Engl. 1999, 38, 3743.
96. Daniel, M. Medicinal Plants: Chemistry and Properties, Science Publishers:
Enfield, NH, USA, 2005, p-157.
97. Prasad, Y. R.; Rao, A. L. Rambabu, R. E- J. Chem. 2008, 5, 461.
98. Palleros, D. R. J. Chem. Educ. 2004, 81, 1345.
99. Narender, T.; Reddy, K. P. Tetrahedron Lett. 2007, 48, 3177.

References
- 191 -
100. a) Cabrera, M.; Simoens, M.; Falchi, G.; Lavaggi, M. L.; Piro, O. E.;
Castellano, E.; Vidal, A.; Azqueta, A.; Monge, A.; Deceráin, A. L.; Sagrera,
G.; Seoane, G.; Cerertto, H.; Gonzalez, M. Bioorg. Med. Chem. 2007, 15,
3356. b) Go, M. L.; Wu, X.; Liu, X. L. Curr. Med. Chem. 2005, 12, 483.
101. a) Calvino, V.; Picallo, M.; Lopez-Peinado, A. J.; Martín-Aranda, R. M.;
Durán-Valle, C. J. Appl. Surf. Sci. 2006, 252, 6071. b) Perozo-Randón, E.;
Martin-Aranda, R. M.; Casal, B.; Durán-Valle, C. J.; Lau, W. N.; Zhang, F.;
Yeung, K. L. Catal. Today, 2006, 114, 183.
102. Luchooman, J.; Sedgwick, J. B.; Sundell, C. L.; Meng, C. Q. 99th
International Conference of the American Thoracic Society, Seattle, May
16-21, 2003, Poster C31.
103. Furusawa, J.; Funakoshi-Tago, M.; Mashino, T.; Tago, K.; Inoue, H.;
Sonoda, Y.; Kasahara, T. Int. J. Immunopharmacol. 2009, 9, 499.
104. Yang, H. -M.; Shin, H. -R.; Cho, S. -H.; Bang, S. -C.; Song, G. -Y.; Ju, J. -
H.; Kim, M. -K.; Lee, S. -H.; Ryu, J. -C.; Kim, Y.; Jung, S. -H. Bioorg. Med.
Chem. 2007, 15, 104.
105. Won, S. -J.; Liu, C. -T.; Tsao, L. -T.; Weng, J. -R.; Ko, H. -H.; Wung, J. -P.;
Lin, C. -N. Eur. J. Med. Chem. 2005, 40, 103.
106. Zhou. J.; Geng. G.; Batist. G.; Wu. J. H. Bioorg. Med. Chem. Lett. 2009, 19,
1183.
107. Lee, Y. M.; Lim, D. Y.; Choi, H. J.; Jung, J. I.; Chung, W.; Park, J. H. Y. J.
Med. Food, 2009, 12, 8.
108. Ye, L.; Gho, W. M.; Chan, F. L.; Chen, S.; Leung, L. K. Int. J. Cancer,
2009, 124,1028.
109. Lee, S.; Ryu, H. W.; Kim, Y. M.; Choi, S.; Lee, M. J.; Kwak, T. K.; Kim, H.
J.; Cho, M.; Park, K. H.; Lee, J. W. Hepatology, 2009, 49, 1316.
110. Na, Y.; Cha, J.; Yoon, H.; Kwon, Y.; Chem. Pharm. Bull. 2009, 57, 607.
111. Ducki, S. Anticancer Agents Med. Chem. 2009, 9, 336.
112. Hsu. Y. L.; Kuo. P. L.; Tzeng. W. S.; Lin. C. C. Food Chem. Toxicol. 2006,
44, 704.
113. Modzelewska. A.; Pettit. C.; Achanta. G.; Davidson. N. E.; Huang. P.; Khan.
S. R. Bioorg. Med. Chem. 2006, 14, 3491.
114. Klein. C.; Vassilev. L. T. Br. J. Cancer. 2004, 91, 1415.

References
- 192 -
115. Ghosh, A.; Mandal, S.; Banerji, A.; Kar, M.; Banerji, J. Nat. Prod. Commun.
2009, 4, 209.
116. Yayli, N.; ScFncF, O.; Aydin, E.; Gçk, Y.; Yas¸ar, A.; Baltasi, C.; Yildirim,
N.; KFÅFk, M. J. Photochem. Photobiol. A, 2005, 169, 229.
117. Deng, J.; Sanchez, T.; Al-Mawsawi, L. Q.; Dayam, R.; Yunes, R.A.;
Garofalo, A.; Bolger, M. B.; Neamati, N. Bioorg. Med. Chem. 2007, 15,
4985.
118. Kiat, T. S.; Pippen, R.; Yusof, R.; Ibrahim, H.; Khalid, N.; Rahman, N. A.
Bioorg. Med. Chem. Lett. 2006, 16, 3337.
119. Sivakumar, P. M.; Priya, S.; Doble, M. Chem. Biol. Drug. Dis. 2009, 73,
403.
120. Sivakumar, P. M.; Seenivasan, S. P.; Kumar, V.; Doble, M. Bioorg. Med.
Chem. Lett. 2007, 17, 1695.
121. Boeck. P.; Falcào. C. A. B.; Leal. P. C.; Yunes. R. A.; Filho. V. C.;-Santos,
E. C. T., -Bergmann. B. R. Bioorg. Med. Chem. 2006, 14, 1538.
122. Liu, M.; Wilairat, P.; Croft, S. L.; Tan, A. L. -C.; Go, M. -L. ibid. 2003, 11,
2729.
123. Kayser, O.; Kiderlen, A. F. Phytother. Res. 2001, 15, 148.
124. Geyer, J. A.; Keenan, S. M.; Woodard, C. L.; Thompson, P. A.; Gerena, L.;
Nichols, D. A.; Gutteridge, C. E.; Waters, N. C. Bioorg. Med. Chem. Lett.
2009, 19, 1982.
125. Dominguez, J. N.; Leon, C.; Rodrigues, J.; de Dominguez, N. G.; Gut, J.;
Rosenthal, P. J. J. Med. Chem. 2005, 48, 3654.
126. Dominguez, J. N.; León, C.; Rodrigues, J.; Dominguez, N. G.; Gut, J.;
Rosenthal, P. J. II Farmaco, 2005, 60, 307.
127. Xue, C. X.; Cui, S. Y.; Liu, M. C.; Hu, Z. D.; Fan, B. T. Eur. J. Med. Chem.
2004, 39, 745.
128. Liu, M.; Wilarat, P.; Go, M. L. J. Med. Chem. 2001, 44, 4443.
129. Shukla, P.; Singh, A. B.; Srivastava, A. K.; Pratap. R. Bioorg. Med. Chem.
Lett. 2007, 17, 799.

References
- 193 -
130. Sivakumar, P. M.; Geetha, S. K. B.; Mukesh, D. Chem. Pharm. Bull. 2007,
55, 44.
131. Via, L. D.; Gia, O.; Chiarelotto, G. M.; Ferlin, G. Eur. J. Med. Chem. 2009,
2854.
132. Ávila, H. P.; Smânia, E. A.; Monache, F. D.; Júnior, A. S. Bioorg. Med.
Chem. 2008, 16, 9790.
133. Aponte, J. C.; Verastegui, M.; Malaga, E.; Zimic, M.; Quiliano, M.;
Viasberg, A. J.; Gilman, R. H.; Hammond, G. B. J. Med. Chem. 2008, 51,
6230.
134. Quintin, J.; Desrivot, J.; Thoret, S.; Le Menez, P.; Cresteil, T.; Lewin, G.
Bioorg. Med. Chem. 2009, 19, 167.
135. Chiaradia, L. D.; Mascarello. A.; Purificação, M.; Vernal, J.; Cordeiro, M.
N. S.; Zenteno, M. E.; Villarino, A.; Nunes, R. J.; Yunes, R. A.; Terenzi. H.
Bioorg. Med. Chem. Lett. 2008, 18, 6227.
136. Alcaraz, M. J.; Vicente, A. M.; Araico, A.; Dominguez, J. N.; Terencio, M.
C.; Ferra´ndiz, M. L. Br. J. Pharmacol. 2004, 142, 1191.
137. Hwang, T. -L.; Yeh, S. -H.; Leu, Y. -L.; Chern, C. -Y.; Hsu, H. -C. Br. J.
Pharmacol. 2006, 148, 78.
138. Xia, Y.; Yang, Z. -Y.; Xia, P.; Bastow, K. F.; Nakanishi, Y.; Lee, K. -H.
Bioorg. Med. Chem. Lett. 2000, 10, 699.
139. Ramagnoli, R.; Baraldi, P. G.; Carrion, M. D.; Cruz-Lopez, O.; Cara, C. L.;
Balzarini, J.; Hamel, E.; Canella, A.; Fabbri, E.; Gambari, R.; Basso, G.;
Viola. G. ibid. 2009, 19, 2022.
140. Nowakowska, Z. Eur. J. Med. Chem. 2007, 42, 125.
141. US Patent pub. No. 6906105 (2005).
142. Patent pub. No. WO/2003/037315 (2003).
143. US Patent pub. No. 7256219 (2007).
144. Patent pub. No. WO/2003/097575 (2003).
145. Brown, T.; Jr., H. H.; Lee, M. Top. Heterocycl. Chem. 2006, 2, 1.

References
- 194 -
146. Ansari, F. L.; Umbreen, S.; Hussain, L.; Makhmoor, T.; Nawaz, S. A.;
Lodhi, M. A.; Khan, S. N.; Shaheen, F.; Choudhary, M. I.; Atta-ur-Rahman,
Chem. Biodiv. 2005, 2, 487.
147. Ansari, F. L.; Nazir, S.; Naureen, H.; Mirza, B. Chem. Biodiv. 2005, 2, 1656.
148. Ahsanullah, Ansari, F. L.; Nazir, S.; Haq, E.; Mirza, B. ibid. 2007, 4, 203.
149. Ansari, F. L.; Baseer, M.; Iftikhar, F.; Kulsoom, S.; Ahsanullah, Nazir, S.;
Shaukat, A.; Ihsan-ul-Haq, Mirza, B. ARKIVOC, 2009, (x), 318-332. ISSN
1551-7012.
150. Bhat, B. A.; Puri, S. C.; Qurishi, M. A.; Dhar, K. I.; Quzi, G. N. Synth.
Commun. 2005, 35, 1135.
151. Xu. J.; Ma, L.; Jiao, P. Chem. Commun. 2004, 14, 1616.
152. Selvam, C.; Jachak, SM.; Thilagavathi, R.; Chakraborti, A. K. Bioorg. Med.
Chem. Lett. 2005, 15, 1793.
153. Moustafa, O. S. J. Chin. Chem. Soc. 2003, 50, 1205.
154. Kidwai, M.; Misra, P. Synth. Commun. 1999, 29, 3237.
155. Nagaraj, N.; Reddy, C. S. J. Iran. Chem. Soc. 2008, 5, 262.
156. Micheli, F.; Degiorgis, F.; Feriani, A.; Paio, A.; Pozzan, A.; Zarantonello,
P.; Seneci, P. J. Comb. Chem. 2001, 3, 224.
157. Ansari, F. L.; Wadood, A.; Ahsanullah.; Iftikhar, F.; Zaheer-ul-Haq. J.
Enzym. Inhib. Med. Chem. 2009, 24, 151.
158. Hutton, C. A.; Bartlett, P. A. J. Org. Chem. 2007, 72, 6865.
159. Garvilyuk, J. I.; Ghotas, E.; Batey, R. A. J. Comb. Chem. 2006, 8, 237.
160. Souers, A. J.; Ellman, J. A. Tetrahedron, 2001, 57, 7431.
161. Freidunger, R. M. J. Med. Chem. 2003, 46, 5553.
162. Abell, A. D. Lett. Pept. Sci. 2002, 8, 267.
163. Park, C.-M.; Sun, C.; Olejniczak, E. T.; Wilson, A. E.; Meadows, R. P.;
Betz, S. F.; Elmore, S. W.; Fesik, S. W. Bioorg. Med. Chem. Lett. 2005, 15,
771.
164. Yu, Y.; Ostresh, J. M.; Houghten, R. A. J. Comb. Chem. 2002, 4, 484.
165. Franzen, R. G.; Ostresh, J. M.; Houghten, R. A. ibid. 2000, 2, 195.
166. Walsh, C. T.; Nolan, E. M. Proc. Natl. Acad. Sci. USA, 2008, 105, 5655.
167. Mitchell, D. A.; Lee, S. W.; Pence, M. A.; Markley, A. L.; Limm, J. D.;
Nizet, V.; Dixon, J. E. J. Biol. Chem. 2009, 284, 13004.

References
- 195 -
168. Gavrilyuk, J. I.; Evindar, G.; Chen, J. Y.; Batey, R. A. J. Comb. Chem. 2007,
9, 644.
169. Patent pub. No. WO/1993/005026 (1993).
170. http://www.nlm.nih.gov/medlineplus/ency/article/000305.htm, Medline Plus
Medical Encyclopedia.
171. Salmeen, A.; Andersen, J. N.; Myers, M. P.; Tonks, N. K.; Barford, D. Mol.
Cell, 2000, 6, 1401.
172. Tiganis, T.; Bennett, A. M. Biochem. J. 2007, 402, 1.
173. Boduła, A.; Wdowczyk, M.; Adamiec, R.; Postepy. Hig. Med. Dosw. 2005,
59, 203.
174. Lam, M. H.; Fodero-Tavoletti, T. M.; Michell, B.; Kemp, B. E.; Tonks, N.
K.; Tiganis, T. J. Biol. Chem. 2001, 276, 37700.
175. Galic, S.; Hauser, C.; Kahn, B. B.; Haj, F. G.; Neel, B. G.; Tonks, N. K.;
Tiganis, T. Mol. Cell Biol. 2005, 25, 819.
176. Meng, T.C.; Buckley D.A.; Galic, S.A.; Tiganis, T.; Tonks, N.K. J. Biol.
Chem. 2004, 279, 37716.
177. Galic, S.; Klingler-Hoffmann, M.; Fodero-Tavoletti, T. M.; Puryer, M. A.;
Meng, T. -C.; Tonks, N. K.; Tiganis, T. Mol. Cell. Biol. 2003, 23, 2096.
178. Vogt, A. M.; Poolman, M.; Ackermann, C.; Yildiz, M.; Schoels, W.; Fell, D.
A.; Kubler, W. J. Biolog. Chem. 2002, 277, 24411.
179. Hadfield. J. A.; Ducki. S.; Hirst. N.; McGown. A. T. Prog. Cell R. 2003, 5,
309.
180. Dimmock, J. R.; Jha, A.; Zello, G. A.; Allen, T. M.; Santos, C. L.; Balzarini,
J.; Clercq, E. De.; Manavathu, E. K.; Stables. J. P. Pharmazie, 2003, 58,
227.
181. Manilal, A.; Sujith, S.; Kiran, S.; Selvin, J.; Shakir, C. Global J. Pharmacol.
2009, 3, 90.
182. Anderson, J. E.; Goetz, C. M.; McLaughlin, J. L.; Suffness, M. Phytochem.
Analysis, 1991, 2, 107.
183. Houghten, R. A.; Pinilla, C.; Giulianotti, M. A.; Appel, J. R.; Dooley, C. T.;
Nefzi, A.; Ostresh, J. M.; Yu, Y.; Maggiora, G. M.; Medina-Franco, J. L.;
Brunner, D.; Schneider, J. J. Comb. Chem. 2008, 10, 3.

References
- 196 -
184. Andrus, M. B.; Turner, T. M.; Sauna, Z. E.; Ambudkar, S. V. J. Org. Chem.
2000, 65, 4973.
185. Boger, D. L.; Dechantsreiter, A. M.; Ishii, T.; Frink, B. E.; Hedrick, M. P.
Bioorg. Med. Chem. 2000, 8, 2049.
186. Bhatia, N. M.; Mahadik, K. Sci. Pharm. 2008, 76, 259.
187. Maciejewicz, W.; Daniewski, M.; Bal. K.; Markowski, W.
Chromatographia, 2001, 53, 343.
188. Atta-ur-Rehman; Choudhary, M. I; Thomsen, W. J. ‘Bioassay Techniques
for Drug Development’, Harwood Academic Publishers, 2001, p 9 and 16.
189. Lawrence, N. J.; Patterson, R. P.; Ooi, L. -L.; Cook, D.; Ducki, S. Bioorg.
Med. Chem. Lett. 2006, 16, 5844.
190. Bacher, G.; Beckers, T.; Emig, P.; Klenner, T.; Kutscher, B.; Nickel, B.
Pure Appl. Chem. 2001, 73, 1459.
191. Liu, X.; Go, M, -L. Bioorg. Med. Chem. 2006, 14, 153.
192. Baluja, G.; Municio, A. M.; Vega, S. Chem. Ind. 1964, 2053. b) Dimmock,
J. R.; Raghavan, S. K.; Logan, B. M.; Bigam, G. E. Eur. J. Med. Chem.
1983, 18, 248.
193. Benvenuto, J. A.; Conner, T. H.; Monteith, D. K.; Laidlaw, J. L.; Adams, S.
C.; Matney, T. S.; Theiss, J. C. J. Pharm. Sci. 1993, 82, 988.
194. Tubulin Polymerization and Depolymerization: Expoliting dynamic
instability of microtubules. Calbiochem. Biologics 30.2, 2004, p3.
195. Kahl, G.; Schell, J. S. ‘Molecular Biology of Plant Tumors’, Academic
Press, New York, 1982, p 269-298.
196. Agrios, G. N. ‘in Plant Pathology’, 4th edn., Ed. Agrios, G. N. Academic
Press, New York, 1997, p 457-470.
197. McLaughlin J. L.; Rogers, L. Drug Inf. J. 1998, 32, 513.
198. Galasky, A. G.; Kozimer, R.; Piotroski, D.; Powell, R. G. J. Natl. Cancer
Inst. 1981, 67, 689.
199. Ferrigni, N. R.; Putnam, J. E.; Jacobsen, L. B.; Nichols, D. E.; McLaughlin,
J. L.; Powell, P. G.; Smith, C. R. J. Nat. Prod. 1982, 45, 679.
200. McLaughlin, J. L. ‘Methods in Plant Biochemistry’, Academic Press,
London, 1991, p 1-31.

References
- 197 -
201. N-terminus Heterocyclic peptides, Garvilyuk, J. I.; Ghotas, E.; Chen, Y. J.;
Batey, R. A. J. Comb. Chem. 2007, 9, 644.
202. Gavrilyuk, J. I.; Evindar, G.; Batey, R. A. ibid. 2006, 8, 237.
203. Combs, A. P.; Yue, E. W.; Bower, M.; Ala, P. J.; Wayland, B.; Douty, B.;
Takvorian, A.; Polam, P.; Wasserman, Z.; Zhu, W.; Crawley, M. L.; Pruitt,
J.; Sparks, R.; Glass, B.; Modi, D.; McLaughlin, E.; Bostrom, L.; Li, M.;
Galya, L.; Blom, K.; Hillman, M.; Gonneville, L.; Reid, B. G.; Wei, M.; -
Pasha, M. B.; Klabe, R.; Huber, R.; Li, Y.; Hollis, G.; Burn, T. C.; Wynn,
R.; Liu, P.; Metcalf, B. J. Med. Chem. 2005, 48, 6544.
204. Johannesson, P.; Erdelyi, M.; Lindeberg, G.; Frändberg, P.-A.; Nyberg, F.;
Karlen, A.; Hallberg, A. ibid. 2004, 47, 6009.
205. Tornoe, W. C.; Sanderson, J.; Mottram, J. C.; Coombs, G. H.; Meldal, M. J.
Comb. Chem. 2004, 6, 312.
206. McComsey, D. F.; Hawkins, M. J.; Andrade-Gordon, P.; Addo, M. F.;
Oksenberg, D.; Maryanoff, B. E. Bioorg. Med. Chem. Lett. 1999, 9, 1423.
207. Biron, E.; Chatterjee, J.; Kessler, H. Org. Lett. 2006, 8, 2417.
208. Costanzo, M. J.; Almond Jr., H. R.; Hecker, L. R.; Schott, M. R.; Yabut, S.
C.; Zhang, H.-C.; -Gordon, P. A.; Corcoran, T. W.; Giardino, E. C.;
Kauffman, J. A.; Lewis, J. M.; de Garavilla, L.; Haertlein, B. J.; Maryanoff,
B. E. J. Med. Chem. 2005, 48, 1984.
209. Costanzo, M. J.; Yabut, S. C.; Almond, H. R.; -Gordon, P. A.; Corcoran, T.
W.; De Garavilla, L.; Kauffman, J. A.; Abraham, W. M.; Recacha, R.;
Chattopadhyay, D.; Maryanoff. B. E. ibid. 2003, 46, 3865.
210. Akamatsu, H.; Fukase, K.; Kusumoto, S. J. Comb. Chem. 2002, 4, 475.
211. Tamura, S. Y.; Shamblin, B. M.; Brunck, T. K.; Ripka, W. C. Bioorg. Med.
Chem. Lett. 1997, 7, 1359.
212. Raddatz, P.; Janczyk, A.; Minck, K.-O.; Rippmann, F.; Schittenhelm, C.;
Schmitges, C. J. J. Med. Chem. 1992, 35, 3525.
213. Heitz, W.; Michels, R.; Liebigs, J. Ann. Chem. 1973, 227.
214. Bolli, M. H.; Ley, S. V. J. Chem. Soc. Perkin Trans. 1, 1998, 15, 2243.
215. Weik, S.; Rademann, J. Angew. Chem. 2003, 115, 2595.
216. S. Weik, J. Rademann, Angew. Chem. Int. Ed. 2003, 42, 2491.
217. El-Dahshan, A.; Weik, S.; Rademann, J. Org. Lett. 2007, 9, 949.

References
- 198 -
218. El-Dahshan, A.; Nazir, S.; Ahsanullah, Rademann, J. Manuscript in
preparation.
219. Bertini, V.; Lucchesini , F.; Pocci, M.; DeMunno, A. Tetrahedron Lett.
1998, 39, 9263.
220. Bertini, V.; Lucchesini, F.; Pocci, M.; De Munno, A. J. Org. Chem. 2000,
65, 4839.
221. Lucchesini , F.; Bertini, V.; Pocci, M.; Micali, E.; De Munno, A. Eur. ibid.
2002, 1546.
222. Neises, B.; Steglich, W. Angew. Chem. Int. Ed., 1978, 17, 522.
223. Li, P.; Xu, J. C. J. Chem. Soc. Perkin. Trans. 2, 2001, 2001, 113.
224. Scheidt, K. A.; Chen, H.; Follows, B. C.; Chemler, S. R.; Coffey, D. S.;
Roush, W. R. J. Org. Chem. 1998, 63, 6436.
225. Lynas, J.; Martin, S.; Walker, B. J. Pharm. Pharmacol. 2001, 53, 473.
226. Abell, A. D. Lett. Pep. Sci. 2002, 8, 267.
227. There are over 1000 oxazoles represented in the MDDR database (MDL
version of Drug Data Reports) b) Revesz, L.; Blum, E.; Di Padova, F. E.;
Buhl, T.; Feifel, R.; Gram, H.; Hiestand, P.; Manning, U.; Rucklin. G.
Bioorg. Med. Chem. Lett, 2004, 14, 3595. c) Patent pub. No. USRE40795
(2009).
228. Ok, H. O.; Reigle, L. B.; Candelore, M. R.; Cascieri, M. A.; Colwell, L. F.;
Deng, L. W.; Feeny, W. P.; Forrest, M. J.; Hom, G. J.; MacIntyre, D. E.;
Strader, C. D.; Tota, L.; Wang, P.; Wyvratt, M. J.; Fisher, M. H.; Weber, A.
E. Bioorg. Med. Chem. Lett. 2000, 10, 1531.
229. Einsedeil, J.; Thomas, C.; Hubner, H.; Gmeiner, P. Bioorg. ibid. 2000, 10,
2041.
230. Bhat, B. A.; Dhar, K. L.; Puri, S. C.; Saxena, A. K.; Shanmugavel, M.;
Qazi, G. N. ibid. 2005, 15, 3177.
231. Chaffman, M.; Brogden, R. N. Drugs, 1985, 29, 387.
232. Bock, M. G.; Dipardo, R. M.; Evans, B. E.; Rittle, K. E.; Whittner, W. L.;
Veber, D. F.; Anderson, P. S.; Friedinger, R. M. J. Med. Chem. 1989, 32, 13.
233. Nahed, K. Eur. Pat. Appl. EP 430,036, 1991 (Chem Abstr. 1992, 116, 717c).
234. Hirano, K.; Kamaide, H.; Abe, S.; Nakamusra, M. Br. J. Pharmacol. 1990,
101, 173.

References
- 199 -
235. Myers, A. K.; Formon, G.; Duarte, A. P. T.; Penhos, J.; Ramwell, P. Proc.
Soc. Exp. Biol. Med. 1986, 183, 86.
236. US Patent pub. No. 4677102 (1987)
237. Markand, O. N. J. Clin. Neurophysiol. 2003, 20, 426.
238. US Patent pub. No. 5663333 (1997)
239. Kapetanovic, M. Chem. Biol. Interact. 2008, 171, 156. Lengauer, T.; Rarey,
M. Curr. Opin. Struct. Biol. 1996, 6, 402.
240. Halperin, I.; Ma, B.; Wolfson, H.; Nussinov, R. Proteins, 2002, 47, 409.
241. Kuntz, I.; Blaney, J.; Oatley, S.; Langridge, R.; Ferrin, T. J. Mol. Biol. 1982,
161, 269.
242. Meng, E. C.; Shoichet, B. K.; Kuntz, L. D. J. Comput. Chem. 2004, 13, 505.
243. Morris, G. M.; Goodsell, D.S.; Halliday, R .S.; Huey, R.; Hart, W.E.; Belew,
R. K.; Olson, A. J. ibid. 1998, 19, 1639.
244. Feig, M.; Onufriev, A.; Lee, M. S.; Im, W.; Case, D. A.; Brooks, C. L. ibid.
2004, 25, 265.
245. Molecular Operating Environment (MOE ver. 2008.10) Chemical
Computing Group Inc., 1010 Sherbrooke Street West, Guite 91, Monsteal,
H3A 2R7, Canada.
246. Wang, Y.; Wei, Z.; Liu, L.; Cheng, Z.; Lin, Y.; Ji, F.; Gong, W. Biochem.
Biophys. Res. Commun. 2005, 331, 1207.
247. Basili, S.; Bergen, A.; Dall’Acqua, F.; Faccio, A.; Granzhan, A.; Ihmels, H.;
Moro, S.; Viola, G. Biochemistry-US, 2007, 46, 12721.
248. Schneider, G.; Fechner, U. Nat. Rev. Drug Discov. 2005, 4, 649.
249. Jorgensen, WL. Science, 2004, 303, 1813.
250. Gokhale, V. M.; Kulkarni, V. M. Bioorg. Med. Chem. 2000, 8, 2487.
251. Karki, R. G.; Kulkarni, V. M. ibid. 2001, 9, 3153.
252. Cramer, R. D.; Patterson, D. E.; Bunce, J. D. J. J. Am. Chem. Soc. 1988, 110,
5959.
253. Pramod, C. N.; Sobhia, M. E. J. Mol. Graphics Modell. 2007, 26, 117.
254. Gueto, C.; Ruiz, J. L.; Torres, J. E.; Me´ndez, J.; Vivas-Reyes, R. Bioorg.
Med. Chem. 2008, 16, 2439.
255. Lee, J. H.; Kang, N. S.; Yoo, S. Bioorg. Med. Chem. Lett. 2008, 18, 2479.

References
- 200 -
256. SYBYL, Ligand-Based Design Manual, version 7.0, Tripos Inc., St. Louis,
MO.
257. Gasteiger, J.; Marsili, M. Tetrahedron, 1980, 36, 3219.
258. Dobbs, K.; Hehre, W. J. Comput. Chem. 1987, 8, 880.
259. Guassian 94, Revision B.02, Guassian, Inc., Pittsburgh, PA, 1995.
260. Cramer, R. D.; Bunce, J. D.; Patterson, D. E. Quant. Struct. -Act. Rel. 1988,
7, 18.
261. Clark, M.; Cramer III, R. D.; Jones, D. M.; Patterson, D. E.; Simeroth, P. E.
Tetrahedron Comput. Methodol. 1990, 3, 47.
262. Mozziconacci1, J.; Sandblad, L.; Wachsmuth, M.; Brunner, D.; Karsenti, E.
PLoS ONE, 2008, 3, e3821.
263. Jordan, M. A.; Wilson, L. Curr. Opion. Cell. Biol. 1998, 10, 123.
264. Aldaz, H.; Rice, L. M.; Stearns, T.; Agard, D. A. Nature, 2005, 435, 523.
265. Montalibet, J.; Skorey, K. I.; Kennedy, B. P. Methods, 2005, 35, 2.

Summary

- 201 -
Summary
• Parallel synthesis of a library of 20 amino chalcones was carried out. The
synthesized chalcones were subjected to antidiabetic, antibacterial and brine
shrimp cytotoxicity studies. The LEADS were identified with promising
activities.
• Combinatorial synthesis and antibacterial screening of a 120-member chalcone
library was carried out and leads were identified by deconvolution through
Positional Scanning protocol.
• Combinatorial synthesis and antitumor screening of another 175 member
chalcone library was carried out and identification of leads was carried out
through the same procedure.
• A solid phase synthetic route for peptidyl α,β-unsaturated ketones and peptidic
heterocycles was established. As a result twenty-three different peptidyl
chalcones and heterocycles i.e. peptidyl oxazoles, pyrazolines, pyrazoles,
benzothiazepines and benzodiazepines were synthesized.
• All synthesized compounds were confirmed through LC MS, 1H NMR, 13C
NMR and HR MS analysis.
• Computational studies were carried out on the active compounds. Mechanistic
interactions of chalcones and PGM were studied using molecular docking tool
and a rationale was found for greater antidiabetic activity of some chalcones
over the others.
• In silico design and synthesis of cytotoxic chalcones was carried out through
3DQSAR using CoMFA model. This study provided useful indicators for
designing new antitumor chalcones.

Future Plan

- 202 -
Future Plan
• During present work, a small library of twenty chalcones was subjected to
antidiabetic screening against different phosphatases involved in insulin
signaling pathway. This study would be extended over to other synthesized
chalcone derivatives and peptide conjugates in order to find other leads with
higher antidiabetic potency.
• Chalcones identified as antitumor agents in a 175 member library have been
found to have a great potential for developing into antitumor agents.
Therefore, it is planned to test these compounds against human tumor cell
lines.
• The leads identified during PGM inhibition studies will be subjected to further
biological evaluation against these targets for kinetic studies and
determination of their IC50 values.
• Phosphorane chemistry will be exploited for its scope with new linkers and
deprotection strategies for synthesizing novel pharmacologically important
peptide mimics.

- 203 -
List of Publications
• Samina Nazir, Farzana Latif Ansari, AhsanUllah, Humaira Noureen, Bushra
Mirza, ‘Combinatorial Synthesis and Antibacterial Studies on an Indexed
Chalcone Library’, Chem. Biodiv. 2005, 2, 11656.
• Ahsan Ullah, Farzana Latif Ansari, Ihsan-ul-Haq, Samina Nazir, Bushra
Mirz, ‘Combinatorial Synthesis, Lead Identification, and Antitumor Study of a
Chalcone-Based Positional-Scanning Library’, Chem. Biodiv. 2007, 4, 203-
214.
• Farzana Latif Ansari, Muhammad Baseer, Fatima Iftikhar, Saima Kulsoom,
AhsanUllah, Samina Nazir, Awais Shaukat, Ihsan-ul-Haq, Bushra Mirza,
‘Microwave assisted synthesis, antibacterial activity against Bordetella
bronchiseptica of a library of 3΄-hydroxyaryl and heteroaryl chalcones and
molecular descriptors-based SAR’, ARKIVOC, 2009, (x), 318-332. ISSN
1551-7012.
• Adeeb El-Dahshan, Samina Nazir, Ahsanullah, Jörg Rademann, ‘Peptide-
heterocycle-chimera: new classes of drug-like molecules by ligations of
peptide bis-electrophiles with bis-nucleophiles’. Manuscript submitted.

Appendix

- 205 -
Index of Tables
1.1 Some recent reports upon bioactive chalcones. 26 2.1. 1H NMR data of 1-(4′-Aminophenyl)-3-(2-hydroxyphenyl)-2-propen-1-one (2). 39 2.2. Mtb inosit Pptase inhibitory activity of chalcones 1-20. 41 2.3. Mtb PGM inhibitory activity of chalcones 1-20. 41 2.4. Bactericidal activity of amino chalcones (1-20). 42 2.5. BSL studies on amino chalcones (1-20). 43 2.6. Conceptual matrix for an indexed 120 member chalcone library. 49 2.7. Antibacterial activities of Set 1 sub-libraries (pools AL1-AL6). 51 2.8. Antibacterial activities of Set 2 sub-libraries (pools BL1-BL20). 52 2.9. Calculated zone of inhibition (mm) of chalcones against S. aureus. 52 2.10. Antibacterial activities of individual chalcones of library AL1. 55 2.11. Antibacterial activities of individual chalcones of library BL2. 55 2.12. Minimum inhibitory concentration (MIC) of active chalcones (mg/mL). 56 2.13. PDT inhibition studies of libraries of Sets 1 and 2 at 1000 ppm concentration. 63 2.14. PDT inhibition studies on the active libraries at 10 and 100 ppm concentration. 63 2.15. Calculated antitumor activities of designed library at a concentration of 1000 ppm. 64 2.16. Calculated antitumor activities of the most active library members at 10 ppm. 65 2.17. 1H NMR data of 1-(phenyl)-3-(5-methyl-2-thienyl)-2-propen-1-one (46). 67
2.18. Antitumor activities of chalcones of library AL1. 67
2.19. Antitumor activities of chalcones of library AL3. 68 2.20. Antitumor activities of chalcones of library BL9. 68 2.21. % Yield of acetylated products obtained on solid support. 78 2.22. Yields of the standard amide coupling using DIC/HOBT. 82 2.23 Synthesis of peptidyl chalcones 100-107. 85 2.24. Synthesis of peptidyl oxazoles 108-109. 88 2.25. Synthesis of peptidyl pyrazolines 110-114. 90 2.26. Synthesis of peptidyl pyrazoles 115-118. 91 2.27. 1H-NMR analysis (300 MHz, DMSO-d6) of peptidyl pyrazole 115. 90 2.28. Synthesis of peptidyl benzothiazepines 119-123. 93 2.29. Synthesis of peptidyl benzodiazepines 124-125. 95 2.30. 1H-NMR analysis (300 MHz, CDCl3) of peptidyl benzodiazepine 124. 96 2.31. 13C-NMR analysis (100 MHz, CDCl3) of peptidyl benzodiazepine 124. 97 3.1 % inhibiton of chalcones 1-20 against Mtb PGM. 101 3.2 Hydrophobic interactions of chalcones (1-20) in the active site of PGM within 5Å. 104 3.3 Binding interactions of chalcones (1-20) in the active site of PGM. 105 Contiue..

- 206 -
3.4 Binding energies of chalcones (1-20) calculated through molecular docking. 108 3.5 Brineshrimp cytotoxicity of chalcones. 111 3.6 Observed and predicted pLC50’s of training set compounds. 114 3.7 Observed and predicted pLC50’s of test set compounds. 114 3.8 Predicted pLC50 of the designed chalcones. 117 3.9 The energy scores of designed inhibitors. 120 3.10 The predicted and observed pLC50 values of designed chalcone analogues 1-5. 121 3.11 Structures of synthesized 3´-hydroxychalcones (1-30). 122
3.12 Antitumor activities of chalcones (1-30). 123
3.13 Observed and predicted log IC50 of training set compounds. 125 3.14 Observed and predicted log IC50 of test set. 125 4.1 Quantities of benzaldehydes used for the synthesis of AL1-AL6. 142
4.2 Quantities of acetophenones (A1-A6) used for the synthesis of indexed libraries
BL1-BL20.
142
4.3 Quantities of aldehydes used for the synthesis of AL1-AL7. 151 4.4 Quantities of acetophenones used for the synthesis of indexed libraries BL1-BL25. 152

- 207 -
Index of Figures
1.1 Combinatorial production of antibodies in humans. 3 1.2 South pacific cone snails. 3 1.3 The role of CombiChem in drug discovery. 4 1.4 Combinatorial library synthesis. 5 1.5 The Wang resin and linker. 8 1.6 The structure of a resin bead. 9 1.7 The linker acting as attachment site of construction of organic molecule. 9 1.8 The common protecting groups used in peptide synthesis. 10 1.9 General scheme for solid phase peptide synthesis. 11 1.10 Solid phase synthesis of a diazepinone library. 12 1.11 A schematic for solid phase parallel synthesis of dipeptides. 13
1.12 Parallel synthesizers, a) Radleys 6-well workstation, b) Process Chemistry
Workstation, Advantage series 3400™.
13
1.13 A tea bag 14 1.14 SPS of a library of dipeptides through split and mix strategy. 15 1.15 Synthesis of a diazepinone library through split and mix strategy. 16 1.16 Conceptual matrix for combinatorial synthesis. 18 1.17 Iterative re-synthesis of a tripeptide. 20 1.18 Recursive deconvolution of a tripeptide. 20
1.19 Sequential release of peptide from a multiple release resin. 21
1.20 Schematic representation of a microreactor used in radiofrequency encoded
combinatorial chemistry.
22
1.21 Tagging the synthetic heritage of a peptide library. 22 1.22 Removal of molecular tags for analysis. 23 1.23 A representative encoded sheet. 23
1.24 Pi chart representation of the publications on chalcones during 2000-2008 as
searched through ScienceDirect.
27
1.25 Important heterocyclic systems based on chalcone precursor. 30 1.26 The ribosomaly derived peptides Patellamides A and C. 32 2.1 Comparison of conventional versus combinatorial synthesis. 35 2.2 Synthesis of indexed libraries of Sets 1 and 2. 45 2.3 The GC-MS spectrum of the library AL1 containing 20 compounds. 47 2.4 The mass spectra for each GC peaks (a-c). 47 2.5 The mass spectra for each GC peaks (d-l). 48 Contiue…

- 208 -
2.6 A general schematic diagram for library synthesis in search of the LEAD in a bioassay.
50
2.7 1H NMR of 1-(phenyl)-3-(4′-fluorophenyl)-2-propen-1-one (35). 54
2.8 Identification of lead through deconvolution of a 120 member library (Set 1) against S. aureus.
57
2.9 Identification of lead through deconvolution of a 120 member library (Set 2) against S. aureus.
58
2.10 Mechanism of tubulin depolymerization with courtesy from Calbiochem. 59 2.11 Conceptual matrix for designing a 175 member chalcone library. 61
2.12 Identification of antitumor leads through deconvolution of 175 member library
(Set 1).
69
2.13 Identification of antitumor leads through deconvolution of 175 member library
(Set 2).
70
2.14 Peptidyl bis- and tris- electrophiles obtained from 2-acyl-2-phosphoranylidene acetate.
72
2.15 1H NMR analysis of 2-trimethylsilylethyl-2-bromoacetate 82. 75
2.16 ATR-FT-IR of triphenylphosphorane support after alkylation with trimethylsilyl ethylbromoacetate.
75
2.17 tris(Dimethylamino)sulfonium difluorotrimethylsilicate (TAS-F). 83
2.18 ATR FT-IR of 96 after removal of TMSE protection with TAS-F. 84 2.19 1H NMR (300 MHz, CDCl3) spectrum of chalcone 100. 86 2.20 13C NMR (100 MHz, CDCl3) spectrum of chalcone 100. 87 2.21 Variety of heterocycles accessible through α,β-unsaturated ketone template. 87 2.22 1H NMR (300 MHz, CDCl3) spectrum of oxazole 108. 89 2.23 13C NMR (100 MHz, CDCl3) spectrum of oxazole 108. 89 2.24 1H NMR (300 MHz, CDCl3) spectrum of benzodiazepine 119. 94 2.25 13C NMR (100 MHz, CDCl3) spectrum of benzodiazepine 119. 95
3.1 Citrate ion docked in the active site of PGM showing interactions with different amino acids as shown in MOE diagrams a) 2D view b) 3D view.
103
3.2 Hydrophilic and hydrophobic interactions of chalcone 4 with amino acids in the active-site of PGM a) 2D b) 3D view
106
3.3 2D and 3D views of the most active chalcone 11 bound to the active site of PGM. 106 3.4 Ligplot of compound 13, an inactive chalcone. 107 3.5 Surface representation of chalcone 11 and PGM complex. 108 3.6 Fitting centers used for overlapping chalcones. 112
Contiue..

- 209 -
3.7 Calculated vs observed activity of the training set compounds. 115 3.8 Calculated vs observed activity of the test set compounds. 115 3.9 Stereoview of the CoMFA map for the electronic and steric contributions. 116
3.10 Favorable electronic and steric regions for enhanced potency in cytotoxic chalcones.
117
3.11 Slab ribbon form of active site of tubulin protein (1z5v). 119 3.12 Chalcone 1 bound into the binding pocket of tubulin active site. 120
3.13 Calculated vs observed activity for training set and test set compounds. 126
3.14 Stereoview of the CoMFA map for the electronic contribution. 127 3.15 Stereoview of the CoMFA map for the steric contribution. 127
3.16 Favorable electronic and steric regions for enhanced potency in cytotoxic chalcones.
128

- 210 -
Index of Schemes
1.1 General schematics of a combinatorial library. 2 2.1 Parallel synthesis of a library of amino chalcones (1-21). 36 2.2 Mass fragmentation pattern of amino chalcone 1. 38 2.3 Synthesis of a 120 member chalcone library. 45 2.4 Synthesis of individual chalcones of active libraries AL1 and BL2. 53 2.5 Building blocks for the synthesis of a 175 member library. 60 2.6 Parallel synthesis of chalcones of active column AL1. 65 2.7 Parallel synthesis of chalcones of active column AL3. 66 2.8 Parallel synthesis of chalcones of active row BL9. 66 2.9 MSNT/Lutidine mediated acylation on phosphorane support 77 2.10 Elimination of base-labile Fmoc moiety demasking amino functionality. 78 2.11 Amide coupling through DIC. 79 2.12 Amide coupling through TBTU/base. 80
2.13 Mechanism of 4H-5-oxazolone mediated epimerization. 81
2.14 Mechanism of DIC/HOBT mediated amide coupling. 81

- 211 -
Standard Abbreviations and Acronyms AcOH Acetic acid Ac2O Acetic anhydride ACN Acetonitrile Ac Acetyl ADME adsorption, distribution, metabolism and excretion ADMET adsorption, distribution, metabolism, excretion and toxicology AA-OH Amino acid Ala Alanine Boc tert.-Butyloxycarbonyl bs Broad singlet BTFFH Fluoro-N,N,N′,N′-bis(Tetramethyl)formamidiniumhexafluorophosphate CIT Citrate ion δ Chemical shift CombiChem Combinatorial chemistry CMC Comprehensive Medicinal Chemistry CADD Computer aided drug design DWDI Derwent Word Drug Index d Doublet DCC Dicyclohexylcarbodiimide DCM Dichlormethane Et2O Diethylether DCR Divide-Couple-Recombine DDD Drug design and development DIC N,N′-Diisopropylcarbodiimide DIEA N-Ethyldiisopropylamine DIPEA Diisopropylethylamine DMAP 4-Dimethylaminopyridine DMSO Dimethylsulfoxide DMF Dimethylformamide EDC N-ethyl-N′-(3-dimethylaminopropyl)-carbodiimide hydrochloride eq. Equivalent EI MS Electron impact Mass Spectrometry EtOH Ethanol Fmoc Fluorenylmethoxycarbonyl- Continued…

- 212 -
FT-ATR-IR Fourier transform attenuated total reflection infrared spectroscopy
EtOH Ethanol
Fmoc Fluorenylmethoxycarbonyl-
FT-ATR-IR Fourier transform attenuated total reflection infrared spectroscopy
GSP Guanosinediphosphatemonothiophosphate
HATU 1-[bis(Dimethylamino)methylene]-1H-1,2,3-triazolo-[4,5-b]pyridiniumhexafluo
rophosphate 3-oxide
HTS High Throughput Screening
HBA H-bond acceptor HBD H-bond donor HOAT 1-Hydroxy-7-azabenzotriazole HOBt 1-Hydroxybenzotriazol HPLC High performance liquid chromatography HR MS High resolution mass spectrometry IR Infrared spectroscopy LBDD Ligand-based drug design LC MS Liquid chromatography - mass spectrometry Leu Leucine LOO Leave-one-out m Multiplet MAOS Microwave assisted organic synthesis MDDR Modern Drug Data Report MMP’s Matrix metalloproteinases MOE Molecular Operating Environment MS Mass spectrometry MSNT 1-(2-mesitylenesulfonyl)-3-nitro-1H-1,2,4-triazole Mtb PGM Micobacterium tuberculosis phosphoglycerate mutase NROT Number of rotatable bonds NCE New Chemical Entities NRPS Nonribosomal protein synthesis PSA Polar surface area PASP Polymer-assisted solution phase synthesis PDB Protein Data bank Phe Phenylalanine Continued…

- 213 -
PLS Partial least squares ppm Parts per million PS Polystyrene QSAR Qualitative Structure Activity Relationships Ro3 Rule of three Ro5 Lipniski’s rule of five RMS Root mean square RMSD Root mean square deviation RT Room temperature s Singlet SAR Structure activity relationship SBDD Structure-based drug design SPS Solid phase synthesis SPOS Solid phase organic synthesis TAS-F tris(Dimethylamino)sulphonium difluorotrimethylsilicate
TBTU N-[(1H-Benzotriazol-1-yloxy)dimethylaminomethylene]-N-(methylmethanamin iumtetrafluoroborate)
TFA Trifluoroacetic acid TFFH Fluoro-N,N,N′,N′-bis(tetramethylene)formamidiniumhexafluorophosphate THF Tetrahydrofuran t Triplet Val Valine UV Ultraviolet




















