
620.pdf - Pakistan Research Repository
249
/ w m IN THE NAME OF ALLAH, THE BENEFICENT, THE MERCIFUL .
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of 620.pdf - Pakistan Research Repository

/w
mIN THE NAME OF ALLAH,
THE BENEFICENT,
THE MERCIFUL .

Dedicated To
My Parents for their love,sacrifice and encouragement

STUDIES ON THECHEMICALCONSTITUENTS OF
PROSOPIS JULIFLORA DC. ANDPLUCHEA ARGUTA BOISS.
THESIS SUBMITTEDFOR THE FULFILMENT OF THE DEGREE
OF DOCTOR OF PHILOSOPHY
/
BY
AZRA SULTANA
H.E.J. RESEARCH INSTITUTE OF CHEMISTRYUNIVERSITY OF KARACHI
KARACHIJULY1990

CONTENT Page No.
1SUMMARY
1 GENERAL INTRODUCTION 7
PARTI
132 BIOSYNTHESIS
2.1 Biosynthesis of Piperidine Alkaloids
2.1.1 Piperidine alkaloids derived from polyacctate
2.1.2 Piperidine alkaloids derivedfrom lysine
14
27
34
3 REFERENCES
4 INTRODUCTION
4.1 The Genus Prosopis
4.1.1 Botanical Classification and Characterization
4.1.2 Use in Folklore and Indigenous system of Medicine
4.13 Pharmacological Importance
4.1.4 Literature Review of Prosopis
5 PRESENT WORK
5.1 N-methyl julifloridine (I) - Isolation and Structure
5.2 Projuline (II) - Isolation and Structure
5.3 Prosopinoline (III) - Isolation and Structure
5.4 Juliprosinene (IV) - Isolation and Structure
5.5 Prosopidione (V) - Isolation and Structure
6 EXPERIMENTAL
6.1 General Notes
6.2 Plant Material
6.3 Procedure for Isolation of Crude Alkaloids
6.4 Alkaloids from Benzene Soluble Fraction F26.4.1 Juliprosopine (Juliflorine) (19)
6.4.2 Julifloricine (29, 30)
35
39
40
40
41
43
45
72
75
80
86
92
97
102
103
105
105
106
108
108

1096.43 Julifloridine (18)
6.4.4 Isolation of N-methyl Julifloridine (I)
6.4.5 Isolation of Projuline (II)
6.4.6 Isolation of Prosopinolinc (III)
6.4.7 Isolation of Juliprosinene (IV)
6.4.8 Isolation of Prosopidionc (V)
BIOLOGICAL ACTIVITY
7.1 Materials and Methods
7.1.1 Acute Toxicity
7.1.2 Neuromuscular Blocking Activity
7.13 Effect on Blood Pressure
7.2 Results and Discussion
7.2.1 Acute Toxicity
7.2.2 Neuromuscular Blocking Activity in IsolatedRat Phrenic Nerve-Hemidiaphragm Preparation
7.23 Effect of blood Pressure
110
112
115
117
119
121
122
122
123
125
126
126
126
128
73 Antimicrobial Activity of Juliprosinene (IV)
REFERENCES
130
132PART 11
BIOSYNTHESIS
1.1 Biosynthesis of Terpenoids
1.1.1 Sesquiterpenes
1.1.2 The eudesmane (selinane)
1.13 Biosynthesis of eudesmane
REFERENCES
INTRODUCTION
3.1 Selection of Plant
3.1.1 Botanical description of Pluchea arguta Boiss.
3.1.2 Medicinal Importance of Pluchea
3.1.3 Literature Review of Pluchea
141
142
144
156
156
162
165
166
166
167
170

2014 PRESENT WORK
4.1 Pluchidione (I) - Isolation and Structure
4.2 Pluchidinol (II) - Isolation and Structure
4.3 ll,12,13-trinor-3,4-diepicuauhtemone (III) -Isolation and structure
5 EXPERIMENTAL
5.1 General Notes
5.2 Plant Material
5.3 Method of Extraction and Isolation
5.3.1 Compounds reported for the first timefrom Pluchca aryuta Boiss.
5.3.2 Compound already reported from Pluchca aryuta Boiss.
5.4 Crude Sesquiterpenic Mixture
5.4.1 Isolation of Pluchidione (I)
5.4.2 Isolation of Pluchidinol (II)
5.4.3 Isolation of 11,12,13-trinor-3,4-diepi-cuauhtemone (III)
204
210
215
220
221
223
223
224
226
226
228
230
233
6 REFERENCES
LIST OF PUBLICATIONS
235
241

ACKNOWLEDGEMENT
I take this opportunity to express my deep sense of
gratitude and ' than ks to my Supervisor, Professor Dr. Viqar
Uddin Ahamd for suggesting the problems, able guidance, keen
and continuous interest, patient and constant encouragement
throughout the course of this investigation.
research work was carried and completed with theThe
excellent facilities made accessible to me by this institute
which is considered to be one of the best and most well
equipped institute of the world. Thanks to the dedication and
Sidd iqui ,
F.R.S., H.I. and Professor Dr. Atta -u r-Rahman , T.I. Director,
H.E.J. Research Institute of Chemistry.
un ti ring efforts of Professor Dr. Salim u z z am an
I would like to thank Miss Nasreen Fatima and Mrs. Tesneem
Marium Sajid ,, Pharmaco log ica1 Section, H.E.J. Research
Institute of Chemistry for carrying out the pharmacological
work. Thanks are also due to Mrs. Attiya Zia-ul-Haq for
determining antimicrobial activity of juliprosinene.
I am indebted to Professor Gunther Snatzke, from West
Germany for useful d iscussion on CD. spectra of pluchidione.
I wish to express here, my sincere appreciation and thanks
to my laboratory colleagues and staff for their constant co¬
opera t ion .
My gratitude is also due to the staff of the spectroscopic

section for their co-operation. I am also thankful to Mr. Ahmed
Ullah for his pains taking efforts in typing out
manu scrip t and Abdul Hafeez for structure drawings.
this
I would specially like to thank Dr. Kaniz Fizza for her
valuable gu id ance and suggestions and Mr. Abdul Qasim for proof
read ing .
I also acknowledge with gratitude the environment of
support and encouragement by my friends particularly , Miss
1f f at Ara , Miss Shaista pa rveen and Miss Teh seen Latif for
their help to persue the work.
My deep gratitude are also to my parents and other family
members for their love and constant encouragement to carryout
the higher studies .
I am verlj much indebted to my husband and my daughter ,
Afshan for letting me use the time that was definitely all
theirs.
I hope all others who have given help and whose names could
not be mentioned here due to limitation of space will accept
this anonymous recognition.
AZRA SULTANA

\
\
SUMMARY

The present thesis is divided into two parts. The first
part entitled "Studies on the Chemical Constituents of prosopis
describes the isolation and structure of fourj u1i f lo ra DC. "
new piperdine alkaloids as well as a novel terpenoidal
compound. These are as follows
Alkaloidal Compounds
1. N-methyl julif loridine (I)
HO-
H3C''*Ss'N'ÿ' >(CH2 '11
CH3 CH2OH
2

Projuline (II)2 .
9 " M
CH _.0HHa 3 8 M " II III
31 0 "2 " 4 " 6 “ 8 " u ii
6 " "2 M II
H3C 1 ” N3 ” 5 ° 7 " 9 *
tj M U j i* n
H
3. Prosopinoline ( III )
HQ4
8 •r M
3 5 1 0M H
2"2 4 ** 6" 8”6 10“ H3H3C Nr 3 " 3" 7 " N-H9"
H
CH2OH
juliprosinene ( IV)4 .
HOvÿ5 1
2 i i t 4 1 • f 6 1 ' 1 8 1 1 1
7 1 1 11 • 3 . . i 5 9> i »i i i
H i i %.1 0
1 H II
8 " "HQ4
3 "3 5 1 M 8 l "o7 M 9 "5 " 2 »* »
7 2 & N2 " 4 ,r6 M M
1 0 " II n3 +
5 " “ 4 " »ÿ
cii
H
3

Terpenoidal Compound
prosopidione (V)1 .
0 071 3
I92 6
8 1 03 5
4
1 1 1 2
There more alkaloids namely juliflorine ( juliprosopine ) ,
julif loricine and julif loridine already reported from this
plant, have also been isolated. The structure of these
compounds have been elucidated on the basis of modern
and NMR ( 1H ,spectroscopic methods including UV, IR, mass
13C and 2D), as well as through chemical studies.
The second part of the thesis entitled "Further studies
on the Chemical Constituents of Pluchea arguta Boiss ( compo-
siteae)1’ describes the isolation of three novel eudesmane type
of sesquiterpenes. These are as follows.
4

Pluchidione (I)1.
10 110OH 7
O.l
62 1 383 5
I 2
Pluchidinol (II)2 .
HÿC CH/ OH 3
/ H-OH
HOiCH
3CH3
H2 CH3H3C
HO H //
H -C OHCH33
11,12,13-trinor-3, 4 -diepicuauhtemone ( III )3 .
1 4
CH3
T.09
6 1 o
l.A. I
2 8
HiH
H3C' OH1 5
5

Moreover a known sesquiterpene, plucheinol (15) was also
isolated. Beside this three triterpenes, $ -sitos terol , taraxa-
sterol acetate, taraxasterol and vanillic acid were also
isolated, and characterized, which have not been reported
previously from this plant. The structure of these compounds
have been studied on the basis of spectroscopic and chemical
studies .
'
6

1. GENERAL fNTRODUCTION
<

The plants and herbs have served as one of man's oldest
resource of useful drugs. If one considers the therapeutic
digitalis, atropine,quinine ,importance of morphine,
it is evident howreserpine, vincristine, vinblastine etc.,
great is the contribution of plant derived drugs to medicine
even today. If one adds the synthetic derivatives of plant
derived products, the role of natural drugs becomes most
partly in view of the plants and herbs as richimpressive ,
and virtually inexhaustible source of old and new potential
drugs and partly in view of ever rising cost of synthetic
medicines.
Medicinal plants are very commonly used for the cure of
various ailments specially in the developing countries.
Approximately one third of all pharmaceuticals are of plant
origin. In fact medicinal plants are the main constituents
of most of the household remedies used by the rural
population. Synthetic chemicals are very expensive to produce.
The phytochemical studies on medicinal plants have served
the dual purpose of bringing up new therapeutic agents and
8

providing useful leads for chemotherapeutic studies directed
the synthesis of drugs modelled on the chemical
structure of natural products. Moreover, they promoted studies
towards
in the correlation of chemical structure and physiological
activity through functional variations in the active
constituents of the plant material.
The history of medicine and pharmacy begins from
the Father of Medicine. In his writings nearlyHippocrates ,
400 samples are named as medicinal substances. Theophrastus
{370-287 B.C.) who received the herb garden of Aristotle as
wrote a book "On the History of Plants" in whicha legacy,
he mentioned 500 drugs and another book "On the classes of
Plants”. His famous treatise on materia medica was first
published in Greek at Venice in 1949 which served as the
pharmacological hand book "Pharmacologic Vade-me-Cum" for
approximately 1,600 years. This book was translated into
Arabic and some European languages.
A great amount of medical knowledge during the early . and
late middle ages, was provided by the Muslim period of science
and medicine. The most famous physician and philosopher of
his time, Ibn Sina (908-1037 A.D) described 760 herbal drugs
9

in his book "Qanun fi al- Tibb" which is known as "Canon of
the West. It was considered to be the mostMedicine" in
authentic materia medica of the era which served as a text
book of medicine in Europe till the 17th century A.D. Another
well known botanist and pharmacist Ibn Al-Baitar (1248 A.D.)
published a list of medicinal plants describing their
therapeutic values in his books "Kitab al-Jami fi al- Adwiya
al- Mufrada" and "Kitab al- Mughni fi al- Adwiya al- Mufrada" .
In the Indo-Pak subcontinent, records of the indigenous
go back to 700 B.C.,system of medicine known as Ayuurveda,
and its systematisation is attributed to Characa and Sushruta,
who cited about 700 medicinal plants. It is not unlikely that
the ancient, Moen Jo Daro civilization of the Indus Valley,
which dates back to -about the same period as Riverine
civilization of Sumer, Egypt and China, had also made sizeable
contributions in the medical field, some of which may have
been absorbed in the Ayurvedic system after the advent of
the Aryans in this region.
During early decades of the current century, the
development of chemotherapy led to an increasing shift towards
synthetic drugs. However, there has been a considerable
10

o
the study of natural products with
the advent of antibiotics and the importance which some of
recovery of interest in
the constituents of medicinal plants have gained in the
treatment of cardio-vascurar diseases, mental disorder and
certain forms of cancer.
Recent scientific studies have continually reported
isolation of active ingredients in many Chinese medicine.
A rough count of 104 new drugs developed in mainland China
since 1949 showed that 60% of the new drugs originated
directly or indirectly from medicinal plants.
Pakistan is very rich in medicinal plant wealth due to
varied climate and terrain. The H.E.J. Research Institute
of Chemistry which has been established at Karachi University,
with the provision of modern sophisticated facilities for
isolation and structure elucidation studies in the
constituents of medicinal plants and their derivatives from
medicinal plants. Taking into account the developments stated
above, work was undertaken at this Institute for the present
doctoral disertation. It relates to the isolation and
structural studies in the chemical constituents of Prosonis
jul if Lora and Pluchea arguta.
11

PART I
STUDIES ON THE CHEMICAL CONSTITUENTS OFPROSOPIS .TULIFLORA DC.

2. BIOSYNTHESIS

2.1 Biosynthesis of Piperidine Alkaloids
A number of piperidine alkalodds are synthesized in the
plant from the amino acid lysine.
The biosynthesis of lysine itself is complex, two distinct
pathways have been established. In bacteria, green algae,
and some higher plants, aspartic acid (1) andsome fungi,
pyruvic acid (2) are involved and a -e -diaminopimelic acid
(3) is a key intermediate. In other algae, fungi and certain
plant lysine (4) is derived from acetate and a-keto glutaric
acid (5) via u -amino adipic acid (6). Scheme 1 and 2 outlined
these pathways respectively [1].
Lysine is incorporated into piperidine alkaloids like
sedamine (7), anabsine (8) and N-methyl pelletierine ( 9 ) [ 2 ] .
Five of the carbon atoms of L-lysine (C-2 to C-6 ) are
incorporated in,to piperidine ring of these bases in a manner
which does not allow carbon 2 and 6 of lysine to become
equivalent i.e. no symmetrical intermediates are possible.
However in the conversion of lysine into the related bases
cernuine (10) lycopodine (11) decodine (12) and lupine
alkaloids C-2 and C-6 become equivalent and at least one
14

C°2H CHO co2o C02HSeveralrCH C =0 tUN— CH2
* Steps
SÿC02H2
h°2' N
*CHNH2*CHNH2 CH3
2 , 3-dihydro-picolinic acid HC
_NH2C°2H C°2H (2)
C°2Hpartic acid ( 1 )
L-a ,e -diaminopimelic acid (3)
CO2H
CH2NH2 H2NCH
-C°2(?H2,3 ( CH )2 3
H-.+ C_
PJHH NH2r
C°2Hlysine ( 4 )
CO„H2
raeso- a, e-diaminopimelic acid
The diaminopimelic acid pathway forthe formation of lysine (4)
neme-1
15

co2H2"\ /0 CO„H2
TCH2COÿHC°2HHOCCO2H CH CH22CH
2
CHCHCH 29H2 2 2
CH CH CH2COCO2H 2 2
CO2H CO2HHoinocitric acid a-Ketoadipic
acid
CO„H2
o-ylutaric: acid (5) a-amine adipicacid ( 6 )+
CH3COCoA
IH N CO„H2 2
rCH
2
CH2CO2H NN CO2H
A-Piperidineb-carboxylic acid
CO-.H
I
CHH 2
2:olic acid
CHO
CH o -aminoadipicacid semaldehyde2
FH2
,CH2
CH2NH2(4 ) lysine
-2 The a-aminoadipic acid pathway for the formation
of lysine
16
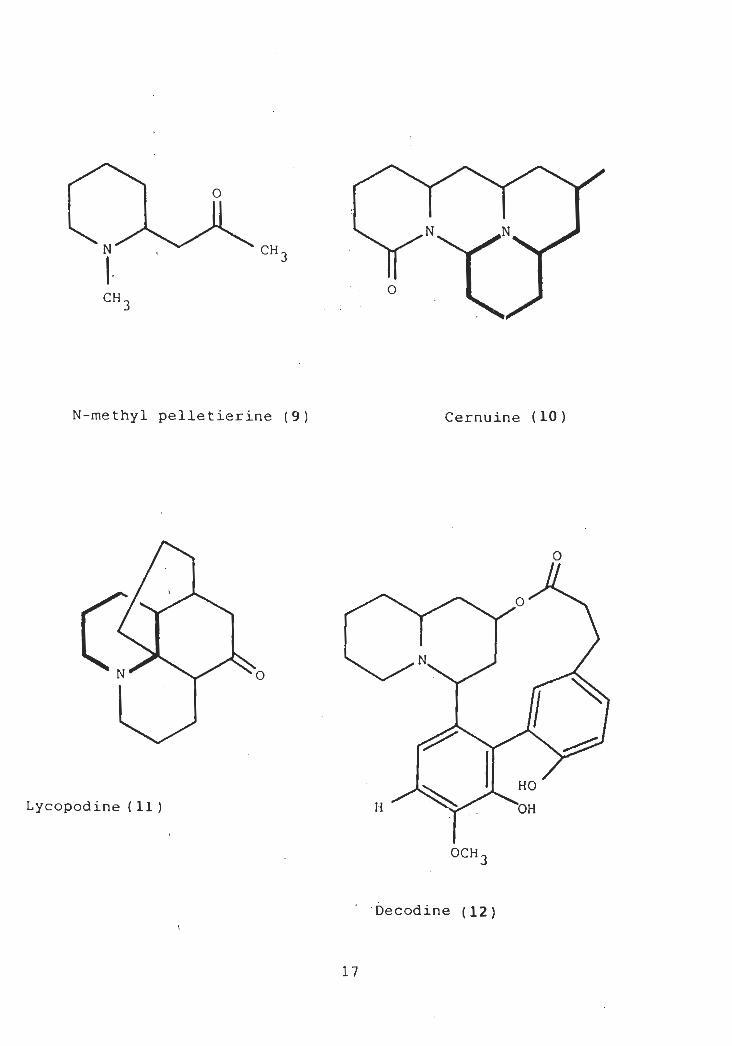
o
N ,NN CH3
OCH3
N-methyl pelletierine (9) Cernuine ( 10 )
0
C;o
N,
%'IHO
Lycopodine ( 11 ) H 'OH
och3
Decodine (12)
17

symmetrical intermediate, must therefore be involved. Such
(13) [Scheme-3] [3].an intermediate could be cadaverine
Cadaverine is a specific precursor of the piperidine ring
from lysine.of sedamine and anabasine, which is formed
Cadaverine may be converted to A-piperidine (14) by oxidative
deamination. It was suggested that the imine (14) condenses
Since Dawson and(15) .with a reduced nicotinic acid
co-workers [4] have shown that nicotinic acid labelled at
C-6 with tritium was less efficiently incorporated than the
other hydrogen-labelled, nicotinic acid. It was suggested
that the active intermediate was 2 , 6-dihydronicotinic acid.
Oxidative decarboxylation of the intermediate (16) affords
anabasine. Adenocarpine (cinnamoyl te trahydroanabas ine ) (17)
is apparently formed via intermediate (18) from A-piperidine
derived from cadaverine. The biogenetic scheme [scheme-4]
showed that lysine undergoes a- transamina tion affording
a -keto-6 -amino carpoic acid which cyclizes to A-piperidine-2-
(19) decarboxylation affords A-piperidinecarboxylic acid
(14) [5-8].
It was generally accepted that many piperidine alkaloids
by a condensation between A -piperidine (14 ) andarose
acetoacetic acid as is shown in scheme-5 [9].
18
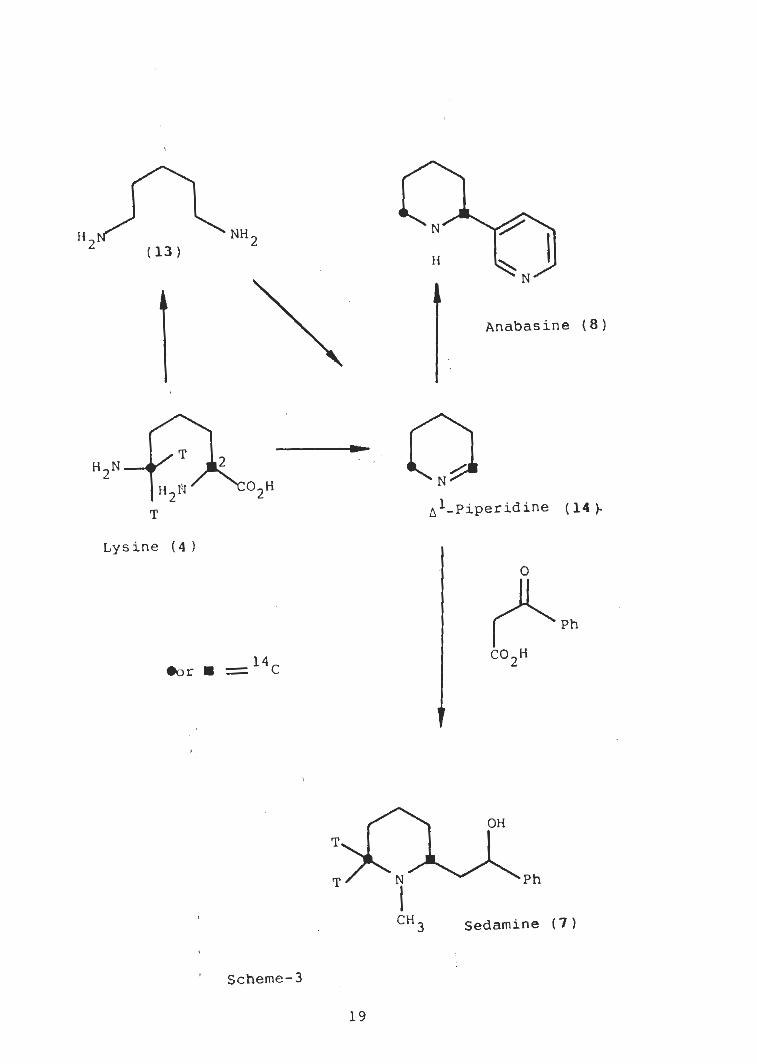
NNHH2 2(13)
H
N
Anabasine (8)
T2H2N
O2HH2W(14).T
Lysine ( 4 )
O
Ph
CO2H=14c•or
OH
AT' N Ph
CH Sedamine (7)3
Scheme-3
19
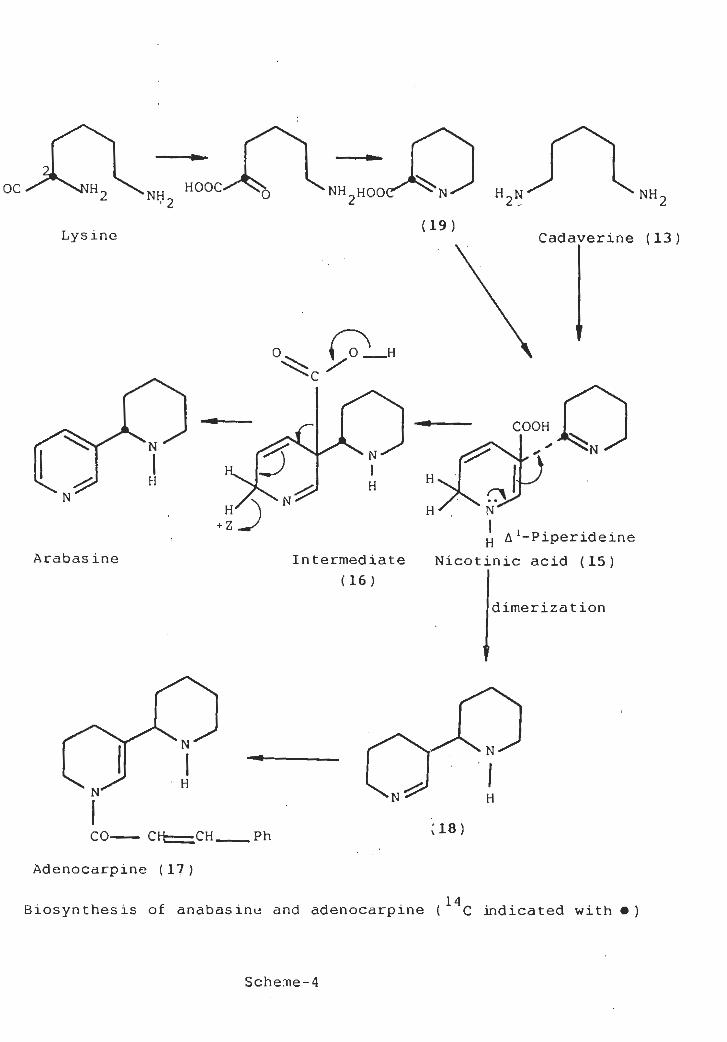
HOOC-ÿsÿ N H2
HOOC'''’*ÿ NOC 'H2 H2N NH2NH
(19)Lysine Cadaverine (13)
(TLH
COOH
N •N
PNP HH H
5Vh/)*Z_/
NH
H A ‘-Piperideine
Nicotinic acid (15)Arabas ine Intermediate
(16 )
dimerization
N N
HN' N H
(18)CO-CH-CH
_Ph
Adenocarpine ( 17 )
14Biosynthesis of anabasine and adenocarpine ( C indicated with •)
Scheme-4

OOH 0 'HN I /
CH3 N'N CH N3 CHl
3HO
H
Isopelie t ierine Sedr id ine
? 1
NNI
CH
T0
3
,>CY H
Y-Pelletierine
O
N N' NCHNH CH „3 32H H
OHCH CH
__COOH
Coniine ( 24 )ConhydrineMimos ine
Hypothetical bigenesis of piperidine alkaloids fromA 1 -piperid ine and acetoacetic acid
Schem- 5

Therefore, any scheme for the biosynthesis of piperidine
alkaloids which does not accomodate cadaverine as a normal
component is unrealistic. A reasonable hypothesis for those
alkaloids derived from lysine without intervention of a
symmetrical intermediate cadaverine formed by decarboxylation
of lysine must remain enzyme-bound, and therefore unsymmetri-
cal. Exogenous cadaverine enters the pathway at this point
by absorption on to the enzyme to give (20). In order to
explain the incorporation of lysine with some alkaloids by
way of symme tr izat ion step, it -is1 necessary only to postulate
equilibration of bound with unbound cadaverine [3]. It was
also noted that L-isomer of lysine was much preferred for
anabasine (8) biosynthesis.
f inalA point arises from the fact that the
stereochemistry at C-2 is not the same in different piperidine
bases. For example sedamine (7) is 2S whereas {-) pelletierine
is 2R . This indicates that addition of the side chain to
A -piperidine , can occur on both re and si faces of the
molecule.
Majority of piperidine alkaloids have been interpreted
in terms of an attractive series of pyridoxal-linked
22

from lysine andintermediates derivable independently
cadaverine. [’Scheme-6] [10].
The transformation of cadaverine into alkaloids(13)
takes place through s terospecif ic removal of one of the
protons attached to C-l (Pro-S hydrogen) and probably
involves stereospecific removal of one of the protons attached
to C-l (pro-S hydrogen). Probably a diamine oxidase takes
part in the reaction. Examination of diamine oxidase-catalysed
oxidation of cadaverine with enzyme from hog kidney and
method, has shown that thisanalysis by an excellent
reaction also involves removal of the 1 -Pro-S proton from
the diamine, pea seedling diamine oxidase has been found to
effect the conversion of benzylamine into benzaldehyde with
similar stereochemistry.
The conversion of lysine into piperidine alkaloids takes-
place with retention of hydrogen isotope at C-2 . The suggested
sequence is given in scheme-6. The catalysis of the reaction
may be attributed to L-lysine decarboxylase. This enzyme,
from the micro-organism Bacillus cadavecis , has been found
to carry out the conversion of L-lysine into cadaverine with
retention of configuration. Decarboxylation of L- [ 2-ÿH ] -lysine
23

AJ rN
HÿI H„ N2 H
HCH
HO,CH2O ©CH2O ©H rq
l.oiJ--C02HH H „ C2 H,G /**ÿ N
HH
3 N3IH2 r
H
L-Lys ine (21)Pyridoxalphosphate
HHSs NH
2NH-.Ni 2
>HRHR N'H
CHdaverineA 1 -Piperideine
(13)HO
CH2OPpH.C3 / N
CiH
Alkaloids
(20)
Scheme-6
24

3[IS- H ] -cadaverine. When this
material is converted into alkaloids, e.g. N-methyl-
this enzyme then affordsby
the tritium attached to what becomes C-2 ispellet ierine ,
lost. On the other hand, conversion of lysine into sedamine
(7) in sedum acre results in the retention of the tritium
orginally present at C-2. The simplest explaination is that
protonation of (21) in the micro-organism and plant proceeds
with opposite stereochemistry which differs with the ideas
on the pyridoxal phosphate. Leete et.al. and R.B. Herbert
showed that 1 , 6-dihydronicotinic acid may be an important
intermadiate in the biosynthesis of nicotine anabasine and
of anabat ine [11].
It had been explained that nicotinic acid is a precursor
of the pyridine ring anabasine (8) [12] and nicotine (22)
(13. 14] the carboxyl group being lost at some step in the
biosynthesis [15], It has been established after feeding
experiments that carbon 4, 5 and 6 of nicotinic acid are
derived from a three carbon compound closely related to
glycerol [16-20]. The other carbon atoms are derived from
succinic acid or a closely related compound [21-30]. A
tentative biosynthetic scheme based on these tracer results
is illustrated in Scheme-7 [9]. It was suggested that the
25
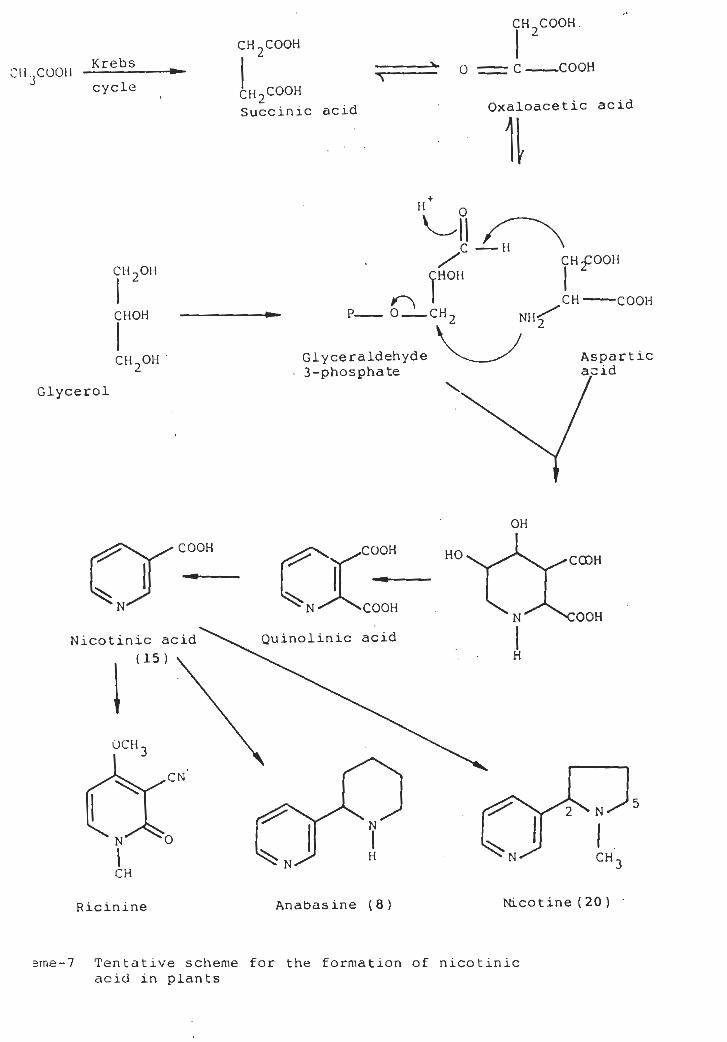
CH COOH.
CH2COOHKrebs V
0-C-COOHCll jCOOII "Vcycle COOHCH2
Succinic acid Oxaloacetic acid
A
r
+H 0
— H/c CH£OOHCH2OH HOH1 CH-COOH£1-CH,CHOH NHÿ
Asparticacid
Glyceraldehyde3-phosphateCH2OH
Glycerol
OH
COOH .COOH HO COOH
COOHOOHN
Quinolinic acidNicotinic acid(15) H
OCH3
CN'
N0N
I H CHN3
CH
Nicotine ( 20 )Anabasine (8)Ricinine
Tentative scheme for the formation of nicotinicacid in plants
sme-7

heterocyclic ring formed during the condensation between
glyceraldehyde-3-phospha te and aspartic acid. The resultant
undergoes dehydration andderivat ive thenpiperidine
dehydrogenation to yield quinolic acid, which was then
decarboxyla ted affording nicotinic acid (15).
Many known piperidine alkaloids which, although struc¬
turally quite similar to the lysine-derived ones, were formed
biosyn thet icaily by different routes. Arecoline (23) is
nicotinic acid (15) coniine (24) isprobably derived from
acetate derived, and probably so are pinidine (25) and
carpaine (26). Skytanthine (27) is a monoterpene alkaloid
[31] .
2.1.1 Piperidine alkaloids derived from polyaceiaie
Pinidine (23) and coniine, (24) and nigrifactine (28)
are the common examples of such type of alkaloids.
2.1.1.1 Biosynthesis of coniine (24)
Connine (24) is the major constituent of the notorious
poison hemlock, Conlum maculatum L. ( Umbellif erae ) .
27

C02CH3
N
CH3
Arecoline (23)
H3C N(CH )
H 2'7H
(CH,)2 '7
Carpaine (26 )
28

H3CCH3
N
CH3
Skytanthine (21)
N (CH=CH)3CH3
Nigrifactine (28)
HOÿ
R
RR
M3C ” NCH
_)
2 10
HC = 0
CH3
Cassine (33)
29

Leete deduced the biosynthesis of coniine (24) after doing
14 C]experiments over many years. Initial experiments with [2-
14C] 1,2-dehyd-14,1 cadaverine (13) and [6-
ropiperidine established that these were not intermediates
lysine ( 4 ) , (1,5—
or precursors of coniine (24). The biosynthetic scheme in
14C] y-coniceine (29)the late stages investigated with [1-
and excellent incorporation into be th coniine (24) and
N-methyl coniine step is reversible, however, the intermediate
steps were elucidated with the aid of labelled fatty acid
derivatives [1-ÿ4C], [7-ÿ4C], and [8-ÿ4C] octanoic acid (30)
were well incorporated into coniine (24), which became
labelled at positions 6, 2 ' , 3 ’ respectively. Even more
14significant however was the incorporation of [6- C)
145-oxooctanoic acid (31) and [6- C] 5-oxooctanal (32) which
are clearly the immediate precursors of Y-coniceine (29).
Whether octanoic acid (30) is a true intermediate remains
to be firmly established. Y-Coniceine reductase is the enzyme
that converts Y-coniceine (29) to coniine (24), depends on
and delivers a hydride from pro-S side of theNADPH
nucleoside 11, 31] [ Scheme-8 ] .
30

< CH3COSCOAHO-.C HOÿC O2
2
(31)Octanoic acid (30)
OHC 0
(32)
\ NN
Y_
Coniceine ( 29 )
H
Coniine ( 24 )Biosynthesis of coniine (24)
Scheme-8

2.1.1.2 Biosynthesis of pinidine (25)
The biosynthetic sequence which leads to pinidine is
similar to that of hemlock alkaloid coniine and is
b iosyn thesized ' by linear combination of five acetate units.
[ Scheme -9 1 [ J2 3h ) .
Alkaloids isolated from the genus Prosopis contain one
or two piperidine rings having generally long aliphatic chains.
In addition there are many alkaloids which occur in other
plant and animal sources [36-37] which contain a piperidine
ring with a long alkyl side chain at the 6-position, and
occasionally a short alkyl chain at 2-position. Their
biosynthesis is unknown. Biogene t ically these alkaloids would
appear to be derived from a polyacetate intermediate in a
manner similar to coniine (24) carpaine (26) and cassine (33)
the most common examples of such alkaloids. Carpaine hadare
been shown to be formed from fourteen acetate units.
32
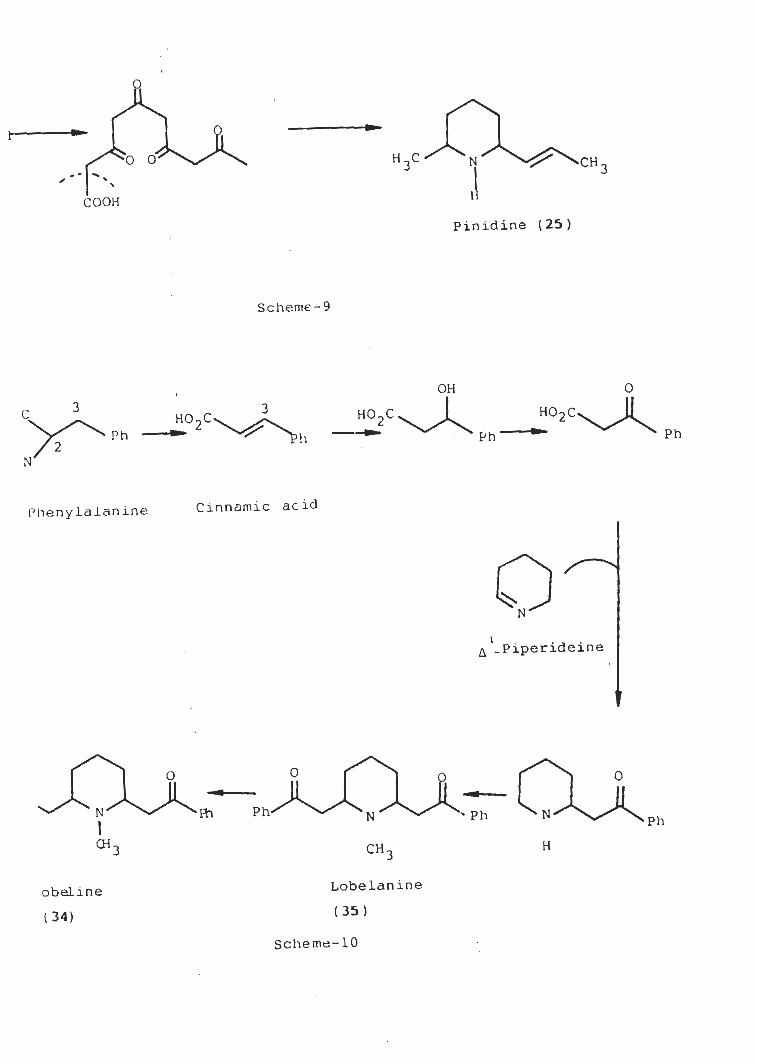
i-
H,C0 0 N
JCH 33
COOH
Pinidine ( 25 )
Scheme-9
OH
3H02Cc HO2C
PhPh Ph2
N
Cinnamic acidPhenylalanine
N
i
A -Piperideine
0 0O O
Ph'N Fh NPhN Ph
CH HCH 33
Lobelanineobeline( 35 )
( 34)
Scheme-10

2.1.2 Piperidine alkaloids derived from lysine
2.1.2.1 Biosynthesis of lobeline (34)
In the biosynthesis of lobeline, the incorporation of
1 2-14 C] lysine into lobeline (34) was found to be symmetrical,
i.o. C-2 and C-6 were equally labelled. Symmetrization of
the label could occur on formation of lobelanine (35), or
at possible earlier intermediate, cadaverine. Cadaverine was
found, however, to be a much less efficient precursor for
lobeline than lysine or Aÿ -piperidine , which strongly sug¬
gests that it is not an obligatory intermediate in lobeline
biosynthesis and thus symmetrization of lysine lable occurs
via lobelanine (38, 39] [ Scheme-10 ) .
3.4

3. REFERENCES

49-52, 206-216G . A. Cordell, Introduction to alkaloids,
(1981) and references cited therein.1.
Chemical and Biolical Perspec-S.W. Pelletier, Alkaloids .t.ives Vol . 1,P. 129 (1982).
J.E. Saxtorv, The Alkaloid. A specialist periodical Report,
The Royal Society of Chemistry, Burlington House, LondonWIV OBN 5 , 5 ( 1975 ) .
2 .
3.
Dawson, D.R. Chirstaman, A.F.D. Adamo, M.L. SoltA . P . Wolf, Chem. Ind., London, 100 (1958), J . Am .
Soc. . 82 . 2628 (1960).
R . F.andChem.
4 .
II. R. Schulte, K.L. Kelling, D. Knofel, and K. Mothes,Phy tochemis try . 3,249( 1964).
3 .
6. T. Griffth, G.D. Griffith, Phytochemistry. 5, 1175 (1966).
E. Leete, J . Am . Chem. Soc. . 78 3520 (1956).7 .
8. E. Leete, G.E. Gros and J.T. Gilbertson, J. Am. Chem.Soc. . 86, 3907 (1964).
11 Edition,Berfeld, Biogenesis of Natural Compounds
Pergamon Press, 961 (1967).9 .
10. M.F. Grundon, The Alkaloids . A Specialist Periodic Report,The Royal Society of Chemistry, Burhington House, London,WIV OBN 10, 9, (1981). references cited therein.
11. E. Leete and Y.Y. Liu, Phytochemistry. 12, 593 (1973).
12. M.L. Solt, F.R. Dawson and D.R. Chirstman, Plant Physio¬
logy , 35, 887 (1960).
13. R.F. Dawson, American Scientists, 48, 321 (1960).
14. R.F. Dawson, D.R. Christman, M.L. Solt and A.P. Wolf,Arch. Biochem. Biophysics, 91, 144 (1960).
15. R.F. Dawson, D.R. Christman, and R.C. Anderson, J. Chem.Soc., 75, 5114 (1953).
36

2, 18216. D.R. Chirstman and R.F. Dawson, Biochemistry,
(1963).
j. Biol, Chem. , 240, 409917. J. Fleeker and R.U. Byerrum,( 1965 ) .
J. Chem., 41, 117 (1963).18. P.F. Juby and L. Marion, Can .
19. T. Griffith, K.P. Heilman and R.U. Byerrum, Biochemistry,
1, 336 (1962).
20. E. Leete and A.R. Friedman, J. Am. Chem. Soc , , 86, 1224( 1964 ) .
21. J.M. Essery, P.F. Juby, L. Marion and Trumbull, J . Am.
Chem. Soc., 84 , 4593 ( 1962 ), Can . J . Chem. , 41, 1142( 1963 ) .
22. A.R. Friedman and E.J. Leete, J . Am. Chem. Soc . , 85, 2141( 1963 ) .
23. T. Griffith and R.U. Byerrum, Biochem. Biophys. Res,
Comm. , 10, 293 (1963).'
24. T. Griffth and R.U. Byerrum, Science, 129, 1485 (1959).
25. T. Griffth, K.P. Heilman and R.U. Byerrum, J. Bio. Chem.235, 800 (1960).
26. T.M. Jackanicz and R.U. Byerrum, J . Biol. Chem. , 241,1296 (1966).
27. S.R. Jhons and L. Marion, Can, J. Chem., 44, 23 (1966).
28. E. Leete, Chem. Ind . , London, 1477 ( 1958).
29. U. Schiedt and H.G.Z. Hoss, Z. Na turf orsch. , 13b, 691(1958), Z. Physiol. Chem., 330, 74 (1962).
30. P.R. Thomas, M.F. Barnes, and I. Marion, Can. J . Chem. ,44, 1997 (1966).
31. J. Mann. Secondary Metabolism 202-207 (1987).
32. E. Leete, J. Amer. Chem. Soc., 85, 3523 (1963).
37

33. E. Leete, J. Amer. Chem. Soc. , 86, 2509 (1964).
34. R.N. Gupta and I.D. Spenser, Phytochemistry , 8, 1937( 1969 ) .
35. E. Leete and K.N. Juneau, J . Am. Chem. Soc. , 91, 5614( 1969 ) .
36. Omnium Chimique Societe, Fr., Patent No. 1.524.395,( 1968) .
37. P. Bourinet and A. Ouevauviller , C.R. Soc. Biol., 162,1138 (1968).
38. D.G. O' Donovan, D.J. Long, E. Forde, and P. Geary, J.C. S. Perkin I. 415 (1975).
39. D.J. O'Donovan and T. Forde, J. Chem. Soc. (C), 2889 (1971).
38

!
4. INTRODUCTION

4.1 The Genus Prosopis
4.1.1 Botanical Classification and Characterization
belongs to the family Leguminoseae
(Mimosaceae ) . The commonly used term "mesquite" includes all
leguminous trees of genus prosopi s • About 43 species of this
i) P rosopis fracta syn.
The genus Prosopis
genus are known, among which,
sr.eph ian iana (M. Bieb) Kunth; Mimosa farcta, ii ) P. chelnesis
P .
stuntz syn. P. spicigera Linn. (11, are common in Pakistan.
In Karachi three species are commonly found, namely P.
cineraria, P. juliflora DC. and P- glandulosa [2].
In Indo-Pak sub-continent P. juliflora is called 'Vilayati
Kikkar' , ’Kabuli Kikkar', 'Vilayati Babul* and 'Vilayati
Khe j ra 1 . The plant is ever-green spiny tree or shrub, with
drooping branches. The leaflets are usually about 1 cm long;
pinnae are smaller with crowded leaflets. The leaves are
bipinnate and flowers are small, yellowish in dense spikes.
The bark is greyish and pods are yellow, 10-25 cm long and
8-15 mm broad, straight or falcate, flat or cylindrical often
with transverse depression between the seeds; 10-30 seeds
are present in a pod and are ovoid, flattened, 7 mm x 3 mm
hard, yellowish brown and shiny [3].
40

nutritional evolutionRecently processing composition,
and utilization of mesquite (Prosopis spp . ) pods as a raw
material for food industry had been reported [4]. The mesquite
gum is used as an adulterant and a substitute for gum arabic.
The gum is also used as a culture medium for bacteria
(arabinose) [5]- P. veluntina has been shown as unexplored
but potential protein source [3]. The seeds of P. glandulosa
furnishes a yellow dye [6].
4.1.2 Use in Folklore and Indigenous system of Medicine
In South Western U.S.A. and in Mexico, it is reported
(mesquite) can be appliedthat the leaves of Prosopis spp.
to irritated conjunctiva. Pima Indians use the extract of
the bark as hot tea for sore throat. Macerated leaf of P .
a f rican a activates harmones and is also considered effective
contains aeroallergensagainst male sterility, P. ju lif lora
causing allergic rhinitis , bronchial asthma and
hypersensitivity pneumonitis. P. ruscif ol ia is used to make
alcoholic beverages. Fruits of P. nigra is used as
carbohydrates source in making beverages for domestic use
[7] .
4 I

The methanolic extract of Pcosopis specigeca has anti-
is also reported toinflammatory activity [8]. P. spicigea
employed against snake bites. The pods and roots of this plant
are stated to possess astringent properties and are used in
dysentery [ 9 ] .
The alkaloids prosopine and prosopinine from prosopis
af ricana are used for the treatment of angina laryngitis,
rhinitis, hermorhoids and as local anaesthetic for dentistry
and minor surgery. An infusion prepared by boiling the leaves
with water is taken internally for the treatment of the
rheumatism and employed externally to treat open sores on
skin. The bark of P. spicigera Linn is used as remedy in
rheumatism and scorpion sting [10].
Antibacterial activity of ethanolic extract of Prosopis
is also reported [8]. P- ru scifolia extracts areg landu losa
active against Staphylococcus [11].
42

4.1.3 Pharmacological Importance
In 1948, P. Shanker-Mur thi and S. Siddiqui reported the
antibiotic activity of leaf extract of Prosopis juliflora
and Escherichia coli 112] .against staphylococcus a u rcu s
The fruits and flowers of P. juliflora contains patulitrin
which showed significant activity against lung113, 14],
carcinoma in vivo [15].
The alkaloids prosopine and prosopinine from P. africana
have been found to act on the central nervous system and have
antibiotic action 116, 17].
An alkaloid mixture extracted from P. sp icigera caused
a decrease in blood pressure and immediate mortality when
given to dogs at 1 mg/kg. After administration to mice, the
histological examination of these animals showed extensive
damage of liver, spleen, kidney, lung and heart [18].
Vaniline, an alkaloid from Prosopis ruscifolia had also
been showed antibacterial activity [19]. The growth of
and Bacillus subtilis was inhibitedChromobacterium v i o1aceum
completely by 2 ug/ml of vinaline of Staphylococcus aureus
43

Sarcina lutea by 10 pg/1 of b anthracis, Mycobacteriumand
by 20 pg/pl of Ustilagophlei and Klebsiella pneumoniae
He lm in t hospo r ium sativum by 40sotrytis cinerea andin a yd i s ,
Escherichia coli,of H . my cod i es ,p g/ pi and 0 . ce ru s ,
Solmonella paratyphi, Heurospora crassa, Candida monosa by
ug/ml Serratia marcescen s , Pseudomones aeruginosa ,80
Absidia glauca, ru sarium moniliCorSaccha romyces cerevisiae,
were inhibited partially. Interavenous injection of 0.25 mg/kg
was lethal for rabbits, 0.01-0.1 mg/15g for rats. Oral dose
of 50 mg/kg had no effect on rabbit.
The presence of cytotoxic principles in Prosopis juliflora
was reported by El-Merzabani et al. [20]. Vitexin, isolated
from P. ruscifolia was reported to be as broad spectrum
antibiotic [21]. More than 90% of the cells exposed to P.
juliflora extract were non-viable after 4 hours incubation.
In vitro-animal test showed that treatment with P. juliflora
extract cured none of tumour-bearing animals although the
mean survival time of mice was significantly increased (T/C
= 1.13) when given in the lowest dose (1.25 g/kg) on days
1 , 5 and 9 .
44

g landu losaOf P rosop is is reportedThe crude extract
to be active against P-338 lymphocytic leukemia (PS) and human
epidermoid carcinoma of the nasopharynx (KB) ED,.Q = 6.8, 7.8,
2.4, 2.6 ug/ml [221 •
Alcoholic extract of the leaves of Prosopis farcta has
shown a dual action of increase and decrease in blood pressure
in vivo and increase in contraction of heart in vitro studies.
Ali. A. Al-jeboory and Wesal. A.H. Al-Husainy in 1984 reported
cardiovascular studies on this plant [23).
4.1.4 Literature Review of Prosopis
The folkoric medicinal use of the genus Prosopis prompted
the investigation of the genus from chemical and biological
point of view which have resulted into the isolation of
several new compounds of medicinal value.
Augusto P. Cerco's isolated the first alkaloid, vinaline
Prosopis ruscifolia in 1951. The antibacterial activityf rom
of the compound was also reported [19] .
45

In 1964 R.C. Sharma et al. reported the isolation of a
flavone glycoside patulitrin (1), 3, 5, 6, 3, 4 pentamethoxy-
7-hydroxy flavone and glucose from flowers of P- spicigera
[14]-
In 1966 G. Rattle et al . isolated prosopine and
P .prosopinine alkaloids from P. at'ricana 124] . From
tj L <ind u Z o no 'the isolation of two new substances prosopol and
prosopenol alongwith g-sitosterol have been reported by
Manzoor-e-Khuda and co-workers in 1968 [25].
P. ru scifolia vitexin (2) was isolated,From the bark of
in 1971 by ,H.R. Juliani and J.C. Oberli [21]. In the same
year M.N. Graziano et al. reported the presence of tyramine
6 -pheny lamines (4) and tryptamine (5) in leaves of P.(3) ,
alba [26] .
Prosopine (6) and prosopinine (7) were isolated from P-
africana (Guill Et Perr) Taub in 1972 by Qui Khoung and Huu
and co-workers [27]. Same authors in the same year reported
the isolation of five new alkaloids from the roots of P .
[28] named as isoprosopinine A (8), isoprosopinine
B (9), prosophylline (10), prosafrine (11) and prosafrinine
africana
46
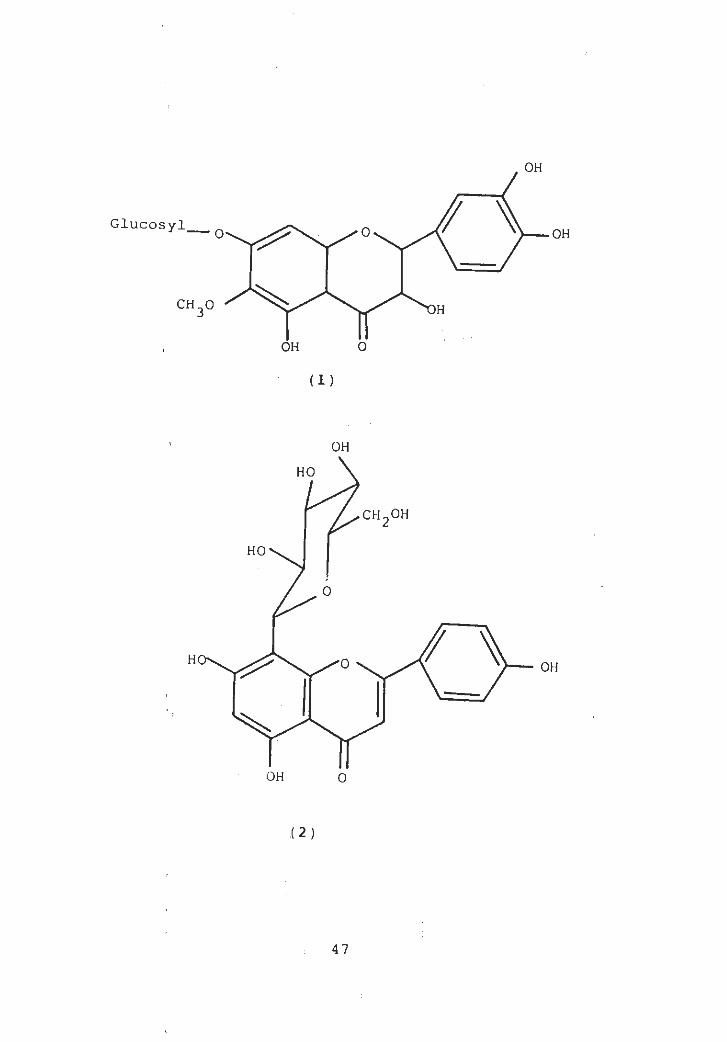
OH
GlucosylO-O OH
CH3° 'H
OH O
(1)
OH
HO
LCH2OH
HO
O
OH
OH O
(2)
4 7

CHÿCI-ÿNHÿ
OH
Tyramine 4- (2-Aminophenyl ) phenol
(3)
Ph.NH2
Phenylamine
(4 )
CH2CH2NH2
SÿN/H
(5)
Tryptamine
40

R = OriginName
Prosopine 6(CH2)10-CHOH-CH3 Prosopis
af ricana0HO,
Prosopinine 7(CH2)9-C-CH2CH3(CHÿ )&-C-(CH2 )4~CH3 a-Isoprosopine 8 „
0HOH2C " RN II(CH ) -C-(CH ) -CH 8-Isoprosopine 9 "
/ 2 J JH
(CH2,)9-CH-CH2-CH3 -Prosafrine 11HO v
0II
ICH2)9-C-CH2-CH3 Prosafrinine 12
H3C'' > RN
H
0II
(CH2)9_C'CH2'CH3 dl Prosophylline 10HO
H0H_C ivRN2 t
H
49

(12)- All of these alkaloids are derivatives of piperidine
C-2, a hydroxyl group athaving a hydroxy methyl group at
C-3 and a long alkyl chain bearing hydroxyl and carbonly
groups at C-6 .
in 1972 isolated cassine (13) for theJ . Paren te et al .
time from the bark of P. ruscifolla and P. nigra [29].first
juli flora DC. ( Leguminoseae ) were found toThe fruits of P.
compound (1) [13]. Iches and co-workers in 1973contain the
found that , patulitrin showed significant activity against
the Lewis lung carcinoma in vivo [15].
a piperidine alkaloid wasIn 1974, spicigerine (14 )
P . spicigora [ 30 ,isolated by K. Jewers and co-workers form
31]. It has a methyl group at C-2 of the piperidine ring.
In 1975, G .A . Moro et al. isolated tryptamine, N-acetyl
tryptamine, eleagmine, harman 13 -phenethylamine and tyramine
nigra Gris [32]. This was the firstfrom the leaves of P-
report of the isolation of N-acetyl tryptamine from a natural
source and its occurrence with B-carboline alkaloids isolated
for the first time from the genus P rosopis . Both facts
suggested that as a general rule, N-acetyl tryptamine is a
50

HON.
R
oR SIIH.C " '(CH2)10'C_CH3 N 3
H
(13)
HON.
S.
CH "( CH2 ) 11COOH3 N
( 14 )
ei

direct precursor in the biosynthesis of B-carboline. This
pathway has been previously detected only in genus Passif loca .
3 , 4 -dime thoxy dolbergione (15) and its related quinol
(16) alongwith a phenolic compound were isolated from the
p. Kuncezei by Y. Tomatakan-hexane extract of heartwood of
1975 [33]. Compound (15) and (16) may be skinet al . in
Mesquite leaves also contained 0.8 percent of airritants .
In 1976 K. Jewers and co-workers showed thedark green wax.
presence of lipids, sterol glycoside and sterols. The
hydrocarbon fractions contained hen triacontane , oc tacosan-l-ol
identifiedtriacontan-l-ol. sterolsTheand were as
cholesterol, campesterol, sitosterol and stigmasterol [34].
The stems of P. giandu Iosa were found to contain oleanolic
acid, ursolic acid, a series of higher aliphatic acid sito¬
sterol and D (+) pinitol as reported by K.A. Zirvi and
co-workers in 1977 [35].
D.K. Bhardwaj and co-workers in 1978, isolated a new
prosogerin-C from the seeds of P. spicigeraf lavone , which
has been identified on the basis of colour reactions, spectral
data and degradative studies as 6, 7, 3', 5' pentamethoxy
52

CH3° //
CH3O CFPhCH=CH2/v0
(15)
CH.O OH3
/ %CH.O3 CHPhCH=CH
2
HO
(16)
53

f lavone ( 17c ) [ 36 ] .
The presence of new alkaloids from the leaves of P.
ju 1if lorn was first reported by U.V. Ahmad and co-workers
in 1978. They isolated three new alkaloids, juliflorine,
julifroicine and julif loridine. The structure of julif loridine
(18) was confirmed through spectral data [37]. In 1979 the
same group also published the partial structure of juliflorine
[38]. Afterwards M. Hesse et al. in 1980 published complete
structure of juliprosopine (19) from the leaves of this plant
[39]. On comparison of the published data of juliflorine with
that of juliprosopine it was found that both the compounds
were identical.
Two compounds, prosogerin A (17d) identified as
methoxy-7 -hydroxy-3 ' -4 ' -methylene dioxyf lavone and prosogerin
B (17b) ( 2 ' , 4 ' -dihydroxy-5-methoxy-3, 4 -me thy lenedioxychalk-
one) were isolated from P. spicigura by D.K. Bhardwaj et al.
in 1979 [40] .
I.B. Gianinetto et al. in 1980, reported isolation of
N-methyl cassine from methanolic extract of bark P. nigra ,
P. ru scifolia and P. vin al i1lo [ 41 ] .
34

HO '/ VO
H3C0
O
(17a)
0>)HO iH
V ViHÿCO'
o
(17b)
OCH3
CH.O
f \3 0
OCH3
CH,03
OCH3
0
( 17c)
55

HO
H3g- (CH2 )u-CH OHN
H
( 18)
H0
V<N|3 ' 5 -I 2 ' 6 '
3c2 i » 4 » > i 6' ' « 8 t i i
3" ' i i i1 i i
5" » 7 i i r
H 10' » ' H
1 « 1 1
N
1 i3' ' 3' ' 7,§ •2 1 •1 1
9’ .N
4 1 « I l M2 •• 6 1 1 8 i r4 • l l •
10' ' 5 < i t i
;
H (19)
56

in continuation of theirD.K. Bhardwaj and co-workers
isolated another new flavone from the seeds of the samework ,
(6, 3', 4', 5', tetra methoxy-7-hydroxyplant, prosogerin-D,
flavone ) ( 20 ) in 1980 [42].
the isolation ofFrom the stem bark of p. ju lif lora ,
hexacosan-25-on-l-ol , a new keto -alcohol alongwith ombuin
and a triterpenoid glycoside were reported by R. Vajpeyi and
K. Misra in 1981 [43].
1981 isolated another alkaloidM. Hesse et al. in
juliprosine (21) from the leaves of P. juliflora and proved
the structure [44] . From the roots of P. julif lora a new
glycoside, 3 , 3 1 -di- a-me thyl-ellagic acid 4-o-a-L rhamnopyrano-
side (22) and procyanidine (23) have been characterized by
S. Malhotra and K. Misra in 1981 [45].
A new glycoside ellagic acid 4-0-ct -L-rhamnosylgentiobio-
P . ju liClora byside (24) was identifiea from the pods of
the same group of authors in 1981 [46]. From the stem bark
two flavone glycosides have been characterized as kaempfero.1
4‘ -methyl ether 3-0- B“D-galac topyranos ide (25) by R. Vajpeyi
5 7
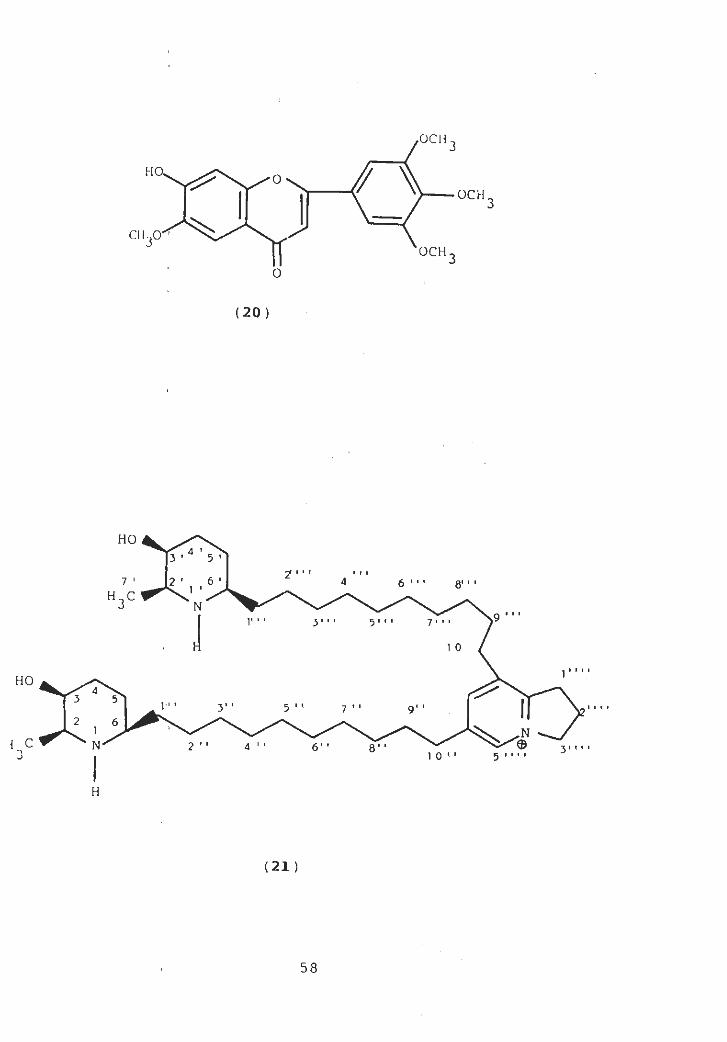
0CM3
$ \HO.
°\ OCH
J 3
CM.XT'3OCH
3O
(20)
HO
3 ’ 5 '2» 1 • f r 1
6 '7 2 1 4 6 1 ' ' 8' 1 '1 'H3C N
J,9 " 'r 1 1 3,
, . v 1 7, , .1 0
* 11HO
43 5
1- 1 r 3* ' 5 ' 1 7 ’ ' 9 ' ' lit*
2 61
i C ©N 2 ' 1 4 1 ' 6' • 1 18 31 t . .1 0 1 1 31111
H
(21 )
58

o
/c0-MeO
HO-/ V-/ ORha
_0C
/OMe
(22)
OH
OH
.OHHO 0
i
OH i
i
>H/ OH
OH
HO
OH
Procyanidine (23)
59

Hi
Rhamnosylgen tiobiosy1-0
OH- 0
/(24 )
Ri 0
OCH3O
OH
OH 0
CH2OHO' H
OH H
OH
H OH(25)
60

nee Shukla et al. in the same year [47].
Work on the ethanolic extract of the roots of P ju liflora
which had already been extracted with acetone was found to
contain three compounds. One of these
glucose 1,3-diester of 3 , 3 ' , 5 , 5 ' - te tra hydroxy-4 , 4 ' -dimethoxy
diphenic acid (26) by colour reaction, degradation and
was characterized as
spectral studies. The isolation of this novel tannin was
reported by S. Malhotra et al. in 1981 [48].
Luteolin, vitexin and isovitexin were isolated from
lu teolin-7-glucos ide and isorhamnrtin-Prosopis vinalillo,
3-ruhtinoside from p. f lexosa , luteolin, luteolin-7-glycoside ,
from P. strombulifera,rhamnosylvi texinquercit in and
by A.M. Gitelliquercetin-3-methylether from P. chilncsis
and co-worker in 1981. They also isolated f lavones from p .
torquata . These were identified as O-rhamnogenco-sylisovite-
xin, isorhamnat in-3- glactoside, quercetin-3-arabinoside and
isorhamnot in . Vitexin and isovitexin and rutin were reported
from P. Kuntizei [49].
D.K. Bhardwaj et al. in 1981 reported the chemical
constituents in the ethanolic extract of the seeds of p-
61

OH
O
HOCH2OCH
3O
OH
OHHO
'H
OO -c OCH3
0
'H
(26)
OCH3
HO '
OCH3
HOCH
3
O
(27)
6 2

Five known polyphenolics , gallic acid, patuletin,
patulitrin and rutin were isolated alongwith a new
Its structure was determined
sp i c i q v i .
luteolin ,
as 6,7-flavone prosoger in-E.
4 ' , 5 ‘ - trime thoxy f lavone (27) [50] -dihydroxy- 3 ' ,
1981, S. Malhotra and K. Misra, identified a newIn
ellagic acid 4-0-rutinoside from the pods of p .ylycos ide ,
juiifloai (51]. From the seeds of the same plant, two
polystyrenes were isolated in 1982 by P. Radha and co-workers,
in the yields of 37.6 and 48.3 mg/lOOg from seed powder.
Respective mol. wts. were determined as 5’725 and 12580 [52].
C .A . Chiale et al . in 1982 isolated cassine, N-methyl cassine
and eleagmine from bark of P. ru scifolia , P . alpataco and
P. sericantha [53],
Figueiredo et al. in 1983, extracted, identified and
characterized the polysaccharides from mesquite beans [54].
Chemical and nutritional studies on mesquite (p, juliflora)
jeans by Del Valle and co-workers in 1983, showed that it
comprised of protein 10-153, fat 2-3%, crude fibre 20-30%,
21% and reducing sugars 2-6%, kernels contain 38%sucrose
3% fat and 9% crude fibres [55], M.M.protein , Ebeid in same
isolated a cholinesterase from mesquite (P. julif lora )/ear ,
63

seedling. The mol. wt . of the isolated enzyme as estimated
by a caliberated column of Sephadex G-200 was>200,000 [56].
isolated from theIn 1983, Si Malhotra and K. Misra,
benzene and ethylacetate soluble fractions of an ethanolic
extract of roots of prosopis ju lif lora two new flavanone
glycosides and • characterized as 3 ' , 4 ' -dihydroxy-5-methoxy-
6-me thyl-f lavanone 7-0-8 -D-glucopyranoside (27a) and 7,4'-
dimethoxy-6 , 8-dime thy If lavanone 5-0-6-D-glactopyranoside ( 27b )
[57] .
In the same year, S.A. Abbas and co-workers isolated
prosopol and it was identified as triacontanol [58]. S.
Malhotra and K. Misra in this year reported from the ethanolic
extract of the green pods of Prosopis juliflora DC., a new
glycoside of ellagic acid (3,3’ di-O-methyl ellagic acid
4-0-( 8-D-glucopyranosyluronic acid) ( l-*4 ) -0- ot-L-arabinopyra-
nosyl-( 1+4 ) -0- a-L-arabinopyranosyl-( !-ÿ 6) -D-glucopyranoside)
(28) which has been isolated alongwith kaempferol, leucocya-
nidin-3-0- 3-D-glucopyranoside and leucodelphinidin-3-O-g -D-
glucopyranosyl (1 4 ) -0- a-L-rhamnopyranos ide [59].
64

'H
CH OH OOH2
OH„C
3OH
0OCII3OH
OH
(27a)
?H3H3G O
\ / CH3I
H 3
0HOCII2 0
H O
OH
(27b)OH
65
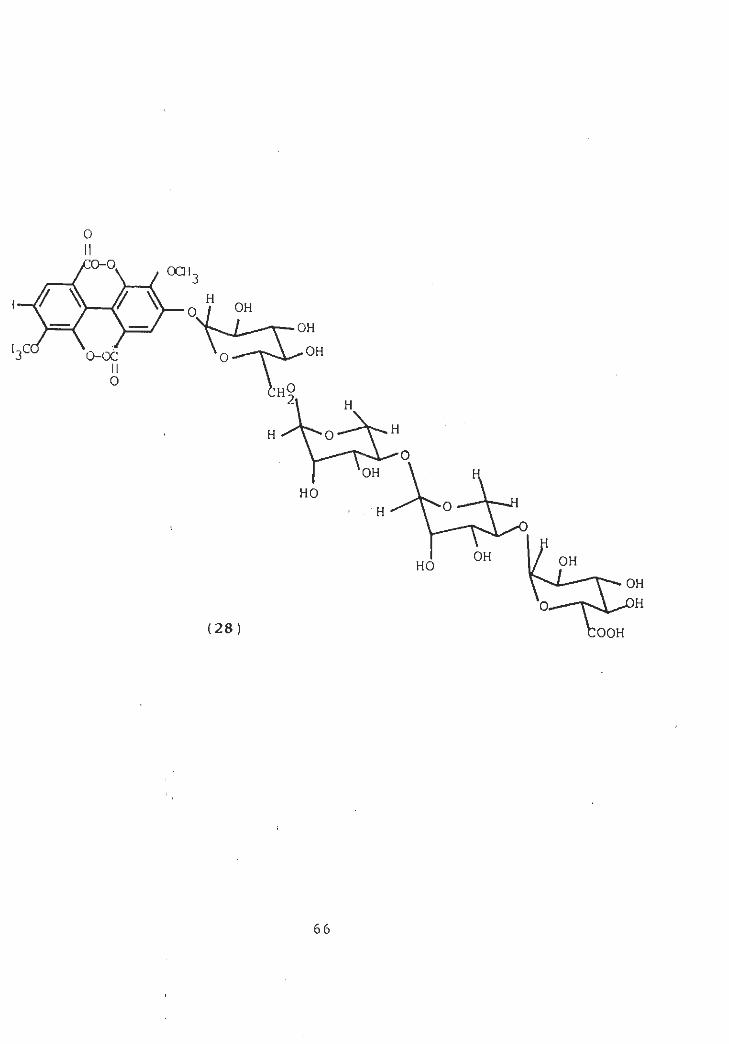
0II:o-o. OCH
3
/ v-rva?\ OH
OH
l3° OHO-OC oII0
H°2i H
HH 0
O
OH
HOOH
OH OHHO
OH
iH0.
(28) OOH
66

From ethanolic extract of p. julit'loca root, the isolation
of a novel tannin was reported by the same authors in 1983.
It was characterized as 4 , 4 1 -dimethoxy-3 , 3 ' , 5 , 5 ' - te trahydroxy
In the same year V.U.diphenic acid 1,3-glucose ester [60].
Ahmad and co-workers reported the absolute configuration of
j ulif loridine '(18) [61] . J.P. Seghal reported that total
tannin contents in Pcosopis cineraria are 2.18% [62].
In 1984, C .A. Chiale et al. reported the flavonoids,
isorhamnetin , quericetin, 6-C-arabinosyl-8-C-glucosylpegenin
and 6 , 8-di-glucosylapigenin from P. humilis From P . repcans,
isorhamnetin, luteolin, vitexin, isovitexin, luteolin 7-O-glu-
coside , 6 , 8-di-C-glucosylapigenin and anthocyanins ,the
petunidin and delphinidin from the corresponding proanthro-
cyanidins were isolated, P. cuizleali contained quercetin,
isorhamnetin, qiiercetin-3-me-ether , isorhamnetin, 3-O-rutino-
side and rutin [63].
In 1984, J.L. Cabrera et al. obtained a product
corresponding to naphtha gasolin or kerosine cycloalkanes,
olefins and aromatic hydrocarbons from the pet. ether extract
)f the leaves of p. ruscifolia which was catalytically cracked
64 ] .
67

The isolation and characterization of allergens of P.
juliflora pollen grains were reported by I.S. Thakur and J.D.
Ahmad et al. suggestedSharma in 1985 165]. In 1985, V.U.
that j ulif loricine , isolated from P. juliflora has one of
two alternative structures (29) and (30) [66]. In the same
Fetuga isolated fatty acids fromyear A.M. Balogun and B.L.
It was shown that low molecularthe seed oil of P. africana.
weight acids (capric and lauric) do not commonly occur in
Leguminoseae . Palmitic acid was the only major saturated acid
present. P. africana was found richer in linoleic acid [67].
Determination of cobalt, molybdenum, selenium, cadmium
and vanadium in leaves and fruits of P. tamarugo Phil was
done by C. Gonzalez and et al. in 1985. Nutritional disorders
may arise from cobalt deficiency rather than mineral toxicity
[68] .
Water soluble vitamins were determined in the fruits of
P. cineraria . The unripe fruits werfe high in pyridoxine folic
acid and ascorbic acid and the ripe fruits were high in
thiamin, riboflavin and nicotinic acid, as reported by M.
Umar and co-worker in 1986 [69].
68

H0V4 '
5 13 •
2 ' ’ 1 r i4 ' 1 ' i i6 86 12 '7
1 r
H3c- N I 1 I I I r1 i i •3 5 7' * ' t • <
9
H1 1 1
1 0HO
H4 1 1 1 r 1
P 5
7-PI I I
' <2’ 4 » 1 6 » 1 8‘ *P * 16 1 02 1 • >1 1 1 » *c 6N 1 « 3r 11 5’ ' t
9' ' :W14
51 t 1 » IIII3
II
v7u1if loricine ( 29 )
HO
4 '
31 5 '
2' 2' 14 ' " 6 1 1 ’6'
8 1 1 *1 1
H3C N1 1 1 11 5'"3 V"
9"H
10 ' «
) H4
1
8 1 » 1 r8,.2' ' 4" 6”2 10' <6 1 t 1 1
1MMN
1 1 6 ’ ' ' '1 53 1 ’ 1 < 7' 1 9' 1 NN.-4 • » * 1
• » 1 \5 1 1 1 »
v7ul if loricine ( 30 )
69

and R.S. PilarIn the sarrÿe year O. Benito, B. Robert
the first limiting amino acidthat threonine isreported ,
for mesquite [70].
The pharmacological activity and toxicity of P. farcca
1986. They alsowere described by A. Kery and. et al. in
isolated proanthocyanidins from P. faccta [71], In the same
year M. Daniel and co-workers described processing, composi¬
tion, nutritional, evaluation and utilization of Prosopis
spp. pods as a raw material for the food industry [72].
Ahmad and co-workers in 1986 reported antimicrobialV . U .
activity of juliflorine from P. julif loca [ 7 3 J . While in same
Nadir and et al. described the effect of differentyear M.T.
methods of extraction on the antimicrobial activity of P.
f arcta [ 74 ] .
In 1986 N. Ahmad and S. Razaq isolated prosopol,
prosopenol, oleanolic acid and 8-sitos terol , alongwith a
0.388% alkaloidal fraction was isolated from the flowers of
[75]. E. Young and et al. in the same yearP. g1and u losa
reported conformation of biphenyl and m-tÿrphenyl analogues
from P. g landulosa (mesquite). They also corrected previous
70

structural assignments for mesquitol [76J.
In 1987, B.V. Bruno and co-workers reported that P.
juliflora ( Sw . ) DC. (mesquite) beans are rich in protein and
J . A. Herrera inlow in lipids, carboydrates and fiber [77).
the same year obtained, vitexin (2), isovitexin isorhamne t in-
3-methyl ether 'and querce t in-4 ' -me thy1 ether from the leaves
of P. fiebrigii and p- hasÿleri. The former three compounds
well isorhamne t in , isorhamnetin-3-rutinoside andas as
myricetin-3 glucoside were also obtained from
[78] .
So n i in 1988, identified monosaccharides ,P. L. some
disaccharides, and natural gums from p. juliflora [79]. V.U.
Ahmad et al. in 1988 reported antimicrobial activity of an
alkaloidal fraction of P. juliflora [80]. In 1989, A. Ahmad,
K.A. Khan, V.U,. Ahmad and S. Qaÿi detected antimicrobial
activity of j ulif loricine isolated from P . juliflora [81]
and also described the antifungal activity of some
hydrosoluble alkaloids of P. juliflora [82].
71

5. PRESENT WORK
i
'
i

The isolation of alkaloids and other chemical constituents
from Prosupis species was generally achieved by the extraction
of dried powdered plant materials and there are wide
variations in the findings reported by various workers. This
is apparently due to the structural changes brought about
by enzymes and aerial oxidation in the drying process. The
alkaloids isolated previously from Prosopis juliflora DC.
were pharmacologically active so this plant was selected for
reinvestigatipn of the chemical constituents. As a result
of the present work on the leaves collected from the grounds
of Karachi University, the following new compounds have been
obtained .
1) N-methyl julif loridine (I)
2) Projuline (II)
3) Prosopinoline (III)
4) Juliprosinene (IV)
5) Prosopidione (V)
Apart from these new compounds, some known alkaloids:
Juliflorine, Julif loricine and julif loridine were also
73

procedure described in theobtained, following up the
experimental.
The residue obtained on removal of the solvent from the
methanolic extract of the leaves of Prosopis juli flora was
partitioned between ethylacetate and water. The aqueous layer
was basified with ammonia (pH-9) and extracted repeatedly
with chloroform. The alkaloid containing chloroform layers
were combined and evaporated at reduced pressure to afford
a gummy residue, this was treated with benzene and benzene
soluble and benzene insoluble portions were obtained. The
benzene soluble portion was selected for investigation and
chromatographed on a (neutral) column and eluted with
increasing polarity of chloroform methanol. Purification
of the compounds was carried out by repeated column
chromatography on (neutral), flash column chromatography
on AlÿOÿ (neutral) and by thin layer chromatographty on sili¬
ca gel.
74

5.1 N-methyl julifloridine (I) - Isolation and Structure
methanolThe elution of column with chloroform
(98:2) furnished a mixture of two alkaloids. The minor
band just above juliflorine (19) was pruified by repeated
column chromatography on (neutral).:
N-methyl julifloridine (1) was Obtained as a gum. It
showed [ M-1 ]+
peak at m/z 312 corresponding to the formula
The predominant ion at m/z 128 in the massC19H38°2 *
spectrum results from the expected cleavage of C-6 side
chain adjacent to the nitrogen in the 2-methyl-3-hydroxy-
6-alkyl piperidine ring. The fromula of this ion (la) is
C7H14N(")' as shown by high resolution mass spectroscopy.
The fragmentation pattern in the mass spectrum resembles
that of cassine [83], carnavaline [83], spicigerine [30,
34 ] . The 2-methyl-3-hydroxy piperidine ring system is
clearly indicated by the presence of peaks at m/z 114,
96, 70. In addition there were important peaks at m/z 298
and 254 . The molecular formula indicated one double bond
equivalent, which was considered to be due to piperidine
ring. Its UV spectrum showed absorption peaks at 203 and
-1283 nm. The absorption peaks at 3500 cm in its IR
75

HO
H3Cÿ" \N/Sÿ(CH2 'll
ICH2OH
N-methyl julif loridine (j)
CH3
HO „
N +
H3C N
CH 3(la)
76

1The H-NMRspectrum indicated the presence of OH group.
= 6.72 Hz) due<5 1.18 ( Jspectrum showed a doublet at
to the methyl, group attached to piperidine ring. Another
2,7
<5 2.51, whichmethyl peak was observed as a singlet at
was assigned to an N-CH2
group. The H-4 and H-5 protons
absorbs as multiplet in the region of 6 1.5 and 1.8
respectively.
In spectaline [84] the H-2 signal is a broad multiplet
at <5 2.90 and the H-3 signal is a broad singlet at <5 3.55
(Wl/2 = 6 Hz). In 6- isocassine , on the other hand, the
H-2 signal is an octet at <53.06 whereas the H-3 singal is
a quintet at<5 3.66 (J = 6.5 Hz, = 3.2 Hz). ThisJ3,43,4
indicates the clear difference in the chemical shift and
coupling constants of the H-2 and H-3 signals between the
two series. In N-methyl julif loridine (I) the H-2 signal
is a distorted octet centered at <S 3.07 which resembles
that of 6-isocassine (31) rather than that of spectaline
(32). It indicated the protons at H-2 and H-3 at a position
to piperidine nitrogen atom. A distorted triplet ( 2H ) at 6
3.63 arises from CHÿOH protons in the side chain, the H-3
signal is masked by this triplet.
77

Two dimensional NMR measurements fully agreed with
proposed structure (I), the coupling interactions were
established by COSY-45 experiment. The methyl doublet at
1.18 was coupled with H-2 , while the octet of H-2 at6
6 3.07 had cross peak with H-3 at 6 3.63. The multiplet
of H-4 is coupled with H-3 and H-5.
13C-NMR spectrum in CDClÿ showed peaks at 6 50.1
{ C-2 ) , 68.48 (C-3) and 49.50 (C-6) which are close to that
The
of 6-isocassine (31 ) series rather than those of
j uliprosopine (19) [39] and spectaline (32). The methyl
carbon signal at 6 14.12 is also nearer to the
corresponding value for 6-isocassine (31) rather than
spectaline (32). It is therefore suggested that
N-methyl julif loridine (I) has the relativessame
configuration as 6-isocassine (31) and julif loridine (18)
[61]. The proposal for a primary alcoholic group in
N-methyl- julif loridine was confirmed through a peak at
6 63.05 (C-12), which is a characteristic for a mthylene
carbon carrying a hydroxyl group. An extra methyl peak
at 6 39.05 is present in spectrum which was
N-CHÿ . Otherwise the
similar to that of julif loridine [61].
assigned to C-NMR spectrum is
78

TABLE-I
nC-NMR chemical shifts for N-methyl julifloridine (I) (CDCI3, 75.43 MHz)
. mm„• ..
ifbon p.v,
; -
___"
___2
9 simm. .. :
50.1 8 39.05
32.868.48 r
26.54 2'-9' 29.55
26.44 10' 25.76
49.50 31.911'
4.12 12’ 63.05
us of each carbon confirmed through DEPT experiment.
79

5.2 Projuline (II) - Isolation and Structure
(98:2) affordedmethanolElution with chloroform
a fraction containing two alkaloids. The' minor alkaloidal
band just below N-methyljulif loridine (I) was purified
through repeated column chromatography using Ai2°3(neutral) and elution by chloroform methanol (98:2) and
then by preparative TLC on silica gel in chloroform
-diethylamine (90:10).
It afforded a new alkaloid projuline (II) which could
not be crystallized but appeared pure on TLC plate (silica
gel ) . The solvent used for TLC was chloroform
diethylamine (90:10). In order to establish the structure
of this alkaloid its spectra were recorded and studied.
The UV spectrum in chloroform showed absorption band
208 nm and 280 nm. There was no absorption band
at 1640 cm in the IR spectrum indicating that no carbonyl
group was present in the molcule. It also showed the
at X max
-1presnece of OH and NH groups (absorption at 3400 cm )
80

g 'ÿ »r
CH OH4
5 7 I " "
7 p :: "2 " 4 “ 6 "6 8" I o ••1
N 2 »i
6 “ "5 ” 7 "1 Z" 9"' N
" t>Jii iiH
Projuline (II )
81

The high resolution mass measurements on the molecular
ion peak afforded the exact mass to be 420.3750 which was
in agreement with the formula C26H48ÿ2ÿ2‘ Field desorption
mass spectra showed a single peak at m/z 420. A detailed
mass spectroscopic analysis was carried out. The following
major peaks were obtained in its mass spectrum 419 (M+
( M+(C26H47N2°2 5 '
375,
-1 ) -H 2°. 389402 C26H46N20> '
333.3259(*“23ÿ44ÿ2 '
305, 264, 179, 114, 96, 84 and 70. The formulae
(C25H45N2°) '
(C22H41N2 5 '
of all the ions were established by computer monitored
362.35217
high resolution mass measurement and confirmed by carrying
out peak matching experiments on important ions.
Linked scan measurements of metastable transitions
resulting from the fragmentation of the molecular ion at
m/z 420 showed that it fragmented directly to the ions
at m/z 420, 402, 389, 362, 333 , 305, 264 , 221, 179, 114
and 70. From the studies on simple piperidine alkaloids
it had been shown that elimination of the largest alkyl
group was Always preferred and that the base peak of the
spectrum arised from this kind of fragmentations. The base
peak of the mass spectrum of projuline was observed at
m/z 114 having the formula CÿH NO. The strong ion at m/z
82

the base peak by the loss of water96 was derived from
molecule. This confirmed the presence of hydroxyl group
in the piperidine ring.
Projuline acetate was prepared through reaction of
the alkaloid with acetic anhydride and pyridine and the
[M + l]+peak at m/z 547 showed that it was triacetate.
473,Other major peaks in the spectrum were at m/z 502,
443, 400, 368, 324, 313, 264, 207, 156 and m/z 84.
In the 1H-NMR spectrum of projuline recorded in
chloroform, the methyl group appeared as a doublet at 6
1 3C-NMR of projuline was recorded1.08. The broad band
in chloroform. It was noted that the stereochemistry at
iso-6-cassine (31) andC-2 and C-3 same aswas
julif loridine (18) 137] was confirmed by the appearance
of signal at 6 50.4 . It was same in case of iso-6-cassine
(31) for C-2. The signal at 6 63.2 was assigned C-10 which
was due to group. The signal due to C-6 carbon atom
to which alkyl chain was attached appeared at 6 67.6
showing that its stereochemistry was similar to juliflorine
(19) and spectaline (32) [84].
83

m/z 389 forIn the mass spectrum a strong peak at
resembled to that present in juliflorineC25H45N2°( jul iprosopine ) (19). This peak was due to the presence
unit connected to the piperidine ringof a indolizidine
through a long alkyl chain -(CH2)-ÿQ at This clearly
indicated that this part of the molecule had the similar
structure as juliflorine (19). From 13C-NMR a signal at
6 63.2 was clearly seen which may be due to the presence
of CH2OH.this molecule may have the structure (II) i.e. part of
From these observations it was proposed that
the molecule had the same structure as the fragments of
juliflorine with m/z 389 and CH2OH group was present at
C-10 in this moiety of the molecule.
One striking point was that the signals for C(6 f F I )
13Till C-NMR were not seen in projuline. Theseand C(7 ) in
were at 6 136.0 and 123.8 in juliprosopine (juliflorine)
(19 ) . Due to the presence of this alkaloid in very small
amounts as a minor constituent further work to confirm
the structure could not be performed.
84

TABLE-11
"C-NMK chemical shifts for projuline (II) (CDCI3, 75.43 MHz)
IS:
;ppmCarbon ppm Carbon
:: 7. ./ Z.X'Z£::.o
32.1 3"-8" 29,6-28.5
2— 21.5 9" 27.0
3,„, 56.4 10" 35.1
5"" 52.2 2, 50.26
8"" 42.08 3, 67.6
8'"'a 65.5 4, 32.01
gi"i 63.2 5, 26.2
1" 37.0 6, 57.9
2" 25.7 7 15.66
The status of each carbon confirmed through DEPT experiment.
85

5.3 Prosopinoline (III) - Isolation and Structure
The crude alkaloids were isolated from the concentrated
methanolic extract of air dried leaves of P • J u 1i £lora
( neutral ) .repeated column chromatography on A1..0
Prosopinoline (III) was isolated by repeated flash column
by 2 3
chromatography on AlÿO (neutral) using chloroform2 3
methanol (97:3) as solvent for elution. Pure prosopinoline
was obtained as a gum and its purity was checked by thin
layer chromatography on silica gel plates and chloroform
diethylamine (90:10) as the developing solvent. The UV
208spectrum of compound (III) showed absorption at nm
characteristic of piperidine alkaloid [85], The absorption
peaks at 3635 and 3435 cm-1 in its IR spectrum indicated
the presence of OH and NH groups,
The electron impact mass spectrum showed molecular
ion peak at m/z 380. The other significant peaks were
114, 92 and 70. These peaks clearly indicated the presence
of 2-methyl-3-hydroxy piperidine ring. Prosopinoline (III)
appeared to be a monomeric alkaloid as julif loridine (18)
and N-methyl jul if loridine (I).
86

L 8
.
(Ill) 0UTTOUTdOSOJÿ
HOÿHDi 1 1 r 6 H
i. .4 9H-N " . 1 6 >.4 . .4,L
I. . . .1 . .01 ..8 . .9 . . t" . .Z l
44....e
....OkO!

The high resolution mass spectrum of compound (III)
corresponding to380.3401showed molecular ion at m/z
the molecular formula (calcd. 380.3402).
indicated three double bond equivalents.
The
molcular formula
Two of which were considered to be of piperidine rings
and one for the double bond in the ring.
The spectrum of compound (III) bore a distinct
similarity with that of spectaline [84], It showed two
methyl doublets at <S 1.15 (J = 6.5 Hz ) and 6 1.002,7
= 6.36 Hz) which were assigned to the methyl( J8a 1 ' ' ’ 10 '
hydrogens at C-7 and C-10'
» < 1
r i i respectively. The intense
peak corresponding to aliphatic methylene groups appeared
5 1.24 ppm. Two unresolved multiplets were present ata t
<S 2.83 and 6 3.0 which were attributed to the methine
hydrogens at 06 and 02 respectively. The carbinolic
hydrogen at 'C-3 appeared as a broad singlet at 6 3.56 with
a width at half hight of 6 Hz, which is known to be
characteristic ot equatorial hydrogen in thean
3-piperidinol derivatives (86-88) . There was a double
doublet at <5 5.36 (J? = 4.4. •Hz, J6III! ,81111 III* ,7 III!
Hz)’ of C-71111 proton whereas another double doublet was
6 5.30 (J? = 12 Hz,present at J8till fill 1111 , 8a,8
88

1 f I Iascribed to C-8= 6.6 Hz ) proton .
H-NMR spectra (2D, J-resolve,
COSY-450, NOESY) fully agreed with the proposed structure
The two dimensional
(III). The multiplicities of overlapping proton signals
could be determined from the 2D J-resolved spectrum while
the COSY-45 spectrum established the coupling interactions
among the vicinal protons. The C-7 methyl protons 6 1.15
showed cross peaks in the COSY spectrum with the C-2 proton
at 6 3.0. The COSY-45 spectrum also showed coupling
proton at <5 2.1 with the C-10
proton at 6 5.36 and with
interaction of C-6 I I I l I I
proton at 6 1.24, with C-7 till
proton at 6 4.2 whereas NOESY spectrum showed1111C-5
proton at 6 4.1 with C-10interaction of C-9 i r t i i i proton
WHat <5 1.24. These assignments were confirmed by
homonuclear decoupling experiments. Thus, irradiation atl
6 5.30 collapsed the double doublet at 5 5.36 to a doublet
(J = 6.6 Hz) while the irradiation at <S 5.36 collapsed
the double doublet at 6 5.30 to a doublet (J = 4.4 Hz)
and simplified the signal at 6 2.1. Irradiation of H-5 till
signal at 6 4.2 simplified the multiplet at 6 2.1 of
tillH-6 and also the multiplet at 6 4.1 of C-91111
89

13C-NMR spectrum of prosopinoline (III) indicatedThe
23 carbon resonances. The multiplicities of each carbon
using
polarization pulses of 45°, 90° and 135°.These experiments
experimentsdetermined withDEPTatom were
revealed the presence of two methyl, thirteen methylene
and eight pvethine carbons in agreement with structure
(III). Compound (III) displayed signals of methines at
I I f6 130.85 ( C-7 ' ' ' ' ) and 6 128.8 (C-8 1 ) indicated the
presence of one double bond in the ring. The configuration
in piperidine ring is confirmed by comparing the
data with that of reported data [84]. The chemical shifts
of C-2 at 5, 57.16, C-3 at 6 66.23 and of C-6 at 6 55.0
were similar with those of spectaline [84] having 2S, 3S
and 6R configuration. The signal of a methylene at 6 68.18
( C — 9 ’ ' ' ’ ) indicated the presence of an alcoholic group.
There are two methyl signals at6l4.18 of C-10' I I 1 and 6
18.98 of C-7.
90

TABLE-111
I3C-NMR chemical shills for prosopinoline (III) (CDCI3, 75.4 MHz)
"ÿ“'T '
Domp?ra ppCarbon
30.4157.16 9”
66.23 34.1510"
34.13 58.435'”'
24.99 6"" 42.3
55.0 130.85
18.98 128.8
36.94 8""a 52.52
23.8 68.18
-8" 28.9-29.71 10"” 14.18
lus of each carbon confirmed through DEPT experiment.
91

5.4 Juliprosincnc (IV) - Isolation and Structure
methanol mixtureThe fractions eluted from chloroform
(93:7) yielded a new alkaloid j uliprosinene (IV). It was
purified by repeated column chromatography on (neutral)*
The final purification was done by thin layer chromatography
ammonia (70:30:1)on silica gel plates chloroform - methanol
were used as developing solvent.
chloride salt of julipros inene (IV) obtained as aThe
gum showed molecular ion of the cation at m/z 624
corresponding to the formula C40H70N3°2” Tÿe aPPearance of
389 for C25H45N2® indicated that this part
resembled to that of juliflorine (19) [39].
the peak at m/z
of the molecule
Juliprosinene has also indolizidine unit connected through
alkyl chain "(CH2)-1q to the piperidine ring at C-6. The
predominant ion at m/z 114 in the mass spectrum results from
the expected cleavage of the C-6 or C-6' side chain adjacent
to nitrogen in the 2-methyl-3-hydroxy-6-alkyl piperidine ring.
Its UV spectrum showed absorptions at 208 and 285 nm, while
absorption peaks at 3660 and 3350 cm-1 in its IR spectrum
indicated the presence of OH and NH groups. The molecular
formula indicated eight double bond equivalents, two of which
92

'
HO4 *
35 1
2 1
2 • i i 4.*i 0<i.6 •6 r
1 •
3 r , ,1 i i i 5 i » i 7 •
Q l l l
1 o •
1 II II
HO4
8 •I ii
3 5 8 M H
1 " 3" 5M 7 " ” a7" 9 M 2 " >i
2 6 M I*
N2” 4 " 6“ 8"H3
N 4 •< *<1 0M3 M ii +
3" “
H Cl
Julipros inene (IV)
93

were considered to be due to piperidine rings and four to
the double bonds in the rings, while the remaining two
represented the indolizidine system.
1H-NMR spectrum of (IV) showed two three protonThe
doublets at <S 1.13 (J = 6.8 Hz, H-7 ) and 1.05 ( J 2, 7,
6.8 Hz, H-7') indicating they were due to secondary methyl
2,7
<5 2.68 {J = 7.0) which wasgroups. There was triplet at
i Iassigned to H-10 I i I coupled with neighbouringand H-10
methylene protons. There was a broad singlet at <S 3.5 3 due
to H-3 and H-3’ . The doublet at 6 6.85 (J = 10.61111 I I t I, 21III!Hz) of H-l 6 6.82 of H-2 i i iand double doublet at
( J = 10.6 Hz, = 6.2 Hz) showed theJ2lilt , 2 ' t I riii , 3 1 I I I1
presenece of double bonds in the indolizidine ring. The former
value is shifted downfield as compared to juliprosine (21)
(44] due to the presence of another double bond conjugated
with it. The two signal were coupled with each other, which
was confirmed by the cross peaks in the COSY-45 spectrum.
There was a double doublet at 6 6.30 (J = 14 , J3 I I I I till,2gem
proton which appeared downfield= 6.2 Hz), due to the H-3a
as compared to juliprosine because of double bond in
I I l I
juliprosinene' (IV). The singlets at 6 9.35 and 6 7.68 were
1assigned to H-5 I I I I and H-7 ’ ' ' ' . The H-NMR spectrum was
94

similar to that juliprosine (21) (44), the only difference
between juliprosine (21) and juliprosinene (IV) being an
additional double oond in the structure of the latter, the
position of which was confirmed through and
spectrum of juliprosinene also supportedspectra. The
structure (IV), ’.with .nethine signals at 6 129.5 and 115.5
6 68.2 ppm. The latterand a downfield methylene signal at
signal is assignable to C-3 rtii because this carbon has a
double bond on one side and a positively charged nitrogen
on the other side. The relative configuration in the
piperidine ring is confirmed by comparing the C-NMR data
with those reported for spectaline (32) (84), tne chemical
shifts of C-2 and C-2' atS57.2, C-3 and C-3' at667.9 and C-6
and C-6 at 655.85 were similar to those reported for the
corresponding carbons of spectaline (32) (84).
95

TABLE-IV
l3C-NMR chemical shifts for juliprosinene (IV) (CDCI3, 75.43 MHz)
Carbon
129.5 3,3' 67.91”"
32.09115.5 4,4'
3"" 68.2 5,5' 25.84
5 139.49 6,6' 55.85
139.14 7, 7' 18.17, 18.106'”'
!ÿÿÿÿ 143.98 1”, 1'" 36.10,35.86
141.9 2", 2"' 24.98, 25.008"..
8""a 160.73 3"-10" 30.93 - 32.09
57.262,2’ 3’”-10"‘
The status of each carbon confirmed through DEPT experiment.
96

5.5 Prosopidione (V) - Isolation and Structure
Prosopidione was eluted from the AlÿOÿ (neutral) column
methanol (80:20) as mobile phase, but itwith chloroform
was contaminated with some impurity. It was further purified
by repeated column chromatography and its purity was checked
by thin layer chromatography on pre-coated silica gel plate
ammonia (29:10:1) as the develo-using chloroform - methanol
ping solvent.
Prosopidione was isolated as a white amorphous powder,
-19.2° (Methanol, c, 0.052). Themp 202° ( dacomp. ) , ( Q 1 D =UV spectrum displayed maxima at 202 and 228 nm. The IR
spectrum showed bands at 1710 (C=0), 1675 and 1650
(a ,S -unsaturated ketone), 1260. (C-0 stretching), 960 cm
(trans-olefin). The El and FD mass spectra showed a molecular
ion peak at m/z 208. High resolution mass spectrometry gave
the [M]+ peak at m/z 208. 1468 corresponding to the molecular
formula
m/z 165.1270 (calc. for CjiHÿO, 165.1279) was due to the
loss of COCHÿ group from the molecular ion. Another fragment
ion appeared at m/z 140.107,1 having the composition of
and was due to the loss of the side chain ( 2-ketobut-3-enyl )
-1
(calc. 208.1463). An important fragment at
97

o o
1 3
92 6
1 08
3 5
4
1 I \ 2
Prosopidione (V)
98

with proton transfer.
1H-NMR spectrum in CDÿOD displayed signals due to
6 0.81 (d, J = 6.8 Hz, H-l 3 ) , 0.87,
1.10 (2 x s, 2 x Me, H-ll and H-12), 2.26 (s, H-10), the last
The
four methyl groups at
to COCH • There was a multiplet ascribed to H-2 at
S 2.15 and a double doublet at 6 1.7 (J
one due
= 12.56 Hz, J!? .6
= 6.6 Hz, H-5a). The H-3 exhibited multiplet at 6 1.5 (m,
gem
2H-3). The doublets at 6 6.33 ( =16.04 Hz) and 6.88 ( O7_8'
16.04 Hz) are due to trans-olef in ic protons at the C-7 and
C-8 positions, respectively.
Two dimensional NMR measurements fully agreed with the
proposed structure (V). The 2D-J-resolved spectrum determined
the multiplicities of the proton signals, while the coupling
interactions were established by a COSY-45 experiment. The
secondary methyl group showed a doublet at <5 0.81 which was
coupled with H-2 while the multiplet of H-2 at 6 2.15 had
cross peaks with the H-13 and H-3 protons. This technique
indicated coupling of H-5 (6 1.7) with that of H-6 { 6 4.5).
These protons also show reciprocol NOE effects. The doublets
at 6 6.33 and 6.88 interacted with each other which was proved
by NOE difference and homodecoupling technique. The proposed
99

13structure (V) is also confirmed by the C-NMR (75.43 MHz)
spectrum which showed double bonded carbon signals at 6 131.74
(C-7) and 153.96 (C-8). The signal of C-6 appeared downfield
(5 75.76), ,due to the presence of a kc tonic group and double
bond at adjacent positions (C-l and C-7). It also exhibited
signals at 6 200.84 (C-9), 182.64 (C-l) due to the carbonyl
6 48.8groups. Another quaternary carbon signal appeared at
(C-4). The APT spectrum showed the presence of four methyl,
two methylene, four methine and three quaternary carbons.
100

TABLE-V
13C-NMR chemical shifts for prosopidione (V) (CDCI3, 75.43 MHz)
siiSMfiaiai182.64 8 153.96
2 35.34 9 200.84
3 36.97 10 25.81
48.84 11 23.79
5 42.98 12 24.9
6 75.76 13 16.34
7 131.74
The status of each carbon confirmed through DEPT expci iment.
101

6. EXPERIMENTAL
I
1

6.1 General Notes
The column chromatography was perfomed on silica gel
( 70-230neutral 9060 (70-230 mesh, E. Merck) and AÿOÿ
E. Merck), while for thin layer chromatography ( TLC )mesh ,
silica gel pre-coated aluminium cards (Riedel de Haen)
were used. For confirming the purity of the samples, high
performance thin layer chromatography (HPTLC) silica gel
60, precoated glass plates (nano TLC, E. Merck) wereFr2 54
used. The solvent used for different chromatographic
purposes were mostly analytical grade of the firm E. Merck.
Flash column chromatography was performed on Eyela Model
EF-10, using AlÿO-j neutral 90 (70-230 mesh, E. Merck).
The mass spectra were measured or Varian MAT-112 and
spectrometer connected to MAT-188 data system andMAT-312
PDP 11/34 computer system. Optical rotations were measured
on polarotronic-D polarimeter.
All the melting points were determined on a Gallenkamp
melting point apparatus and are uncorrected. The Ultra
103

(UV) spectra were scanned on Shimadzu UV-240Viole t
spec trophoto- meter ahd the Infra Red (IR) spectra were
recorded on JASCO A-302 spectrophotometer. Nuclear Magnetic
Resonance ( NMR ) spectra ( and C) were recorded in CDClÿ
on Bruker Aspect AM 400 and AM 300 spectrometers operating
at 400 and 300 MHz for and 100 and 75.43 MHz for
nuclei respectively. Chemical shifts are reported in ppm
(6 ) .
The purity of alkaloids was checked by TLC on silica
gel precoated plates by spraying with Dragondorf f ' s reagent.
104

6.2 Plant Material
Leaves qf Prosopis julif lora were collected from the
Karachi University and identified by the Department of
Botany, University of Karachi. A voucher specimen has been
deposited in the Herbarium of the Botany Department,
Univeristy of Karachi.
63 Procedure for Isolation of Crude Alkaloids
The air dried leaves of Prosopis julif lora were finely
crushed with an Ultraturrax homogeniser and then extracted
in ethanol. The ethaholic extracts were filtered and
concentrated to gum under vacuum. The residue was
partitioned between pet. ether and water. The aqueous layer
was separated and extracted three times with pet. ether.
The green pet. ether extract contained steroids, triterpe¬
noids, waxes and other neutral and acidic compounds. The
aqueous layer contained alkaloids, amino acids and sugars
etc. The aqueous layer wa£ acidified with 10% acetic acid
and extracted with chloroform. The residue from chloroform
extract contained some non-alkaloidal compounds.
105

layer was basified with ammonia and
extracted exhaustively with large quantity of chloroform.
The aqueous
The chloroform extract was dried over anhydrous sodium
sulphate and concentrated to the crude alkaloidal gum. The
residue was extracted with chloroform and the alkaloids were
separated into chloroform-soluble and chloroform- insoluble
portions [Scheme-11. The chloroform-insoluble material was
and the chloroform- soluble(Fi>taken up in methanol
fraction was evaporated and then extracted with benzene
(F3)yielding benzene-soluble ( F3 ) and benzene-insoluble
alkaloids.
The present work was carried out on fraction F2 i.e.
on benzene-soluble alkaloids.
6.4 Alkaloids from Benzene Soluble Fraction F2
TLC of the fraction on silica gel in solvent system
chloroform - diethylamine (90:10) showed two major alkaloids
alongwith traces of a number of minor alkaloids. These were
first separated into different fractions by column
chromatography on AlÿOÿ
chloroform -methanoL in order of increasing polarity. Purity
(neutral) (Type E), using solvents
106

Scheme-1
TOTAL ETHANOLIC EXTRACT OF LEAVES OF PROSOPIS JULI FLORA DC.
Water and Ethylacetate
1EthylacetateWater
1 evaporatesolvent
Basify with Ammoniato pH 9.0 and extractwith chloroform
Residue
Chromatography
t I Neutral compounds
oroformolublealqids
jTaken up
Ain Methanol.
Chloroform solublealkaloids taken upin benzene
thanollublekaloids
roma tographicjara t ion
Benzene solublechromatographicseparation
Benzene insoluble
alkaloids
F2 F3
107

checked on HPTLC plates using the same solvent system.was
A12°3These alkaloids were purified by repeated column
(neutral, Activity 1).
6.4.1 Juliprosopine (Juliflorine) (19)
Elution with chloroform- methanol (98:2) solvent system
afforded a fraction containing two alkaloids. The major one
purified by repeated column chromatography on (neutral).
The pure compound was obtained as a gum. The purity was
checked by thin layer chromatography on silica gel using
chloroform diethylamine as developing solvent (90:10).
It was also obtained in crystalline form as juliprosopine
60°. The spectra obtained were comparedhydrochloride m.p.
with the reported values in literature [38, 39}.
6.4.2 Julifloricine (29, 30)
The alkaloid mixture eluted by chloroform, methanol
(95:5) yielded another known major alkaloid. It was further
purified by repeated flash column chromatography technique
(neutral) with chloroform- methanol (95:5). It couldon
not be crystallized and was obtained as colourless oily
108

compound- The purity of this compound was checked on silica
gel plates for which chloroform
was used as developing solvents. Julif loricine (29, 30) was
diethylamine (90:10)
identified by spectroscopic means as well as by comparison
with an authentic sample [66].
6.43 Julifloridine (18)
The fractions obtained by chloroform- methanol (90:10)
contained mixture of alkaloids, julif loricine and another
polar compound which purif ied by flashmore was
chromatography on (neutral) column and eluted with
chloroform- methanol mixture. The alkaloid was crystallized
from methanol and was identified as julifloridine m.p.
82-8 3°C. Its spectral data were also compared with published
data and also with an authentic sample [37, 61 ].
109

6.4.4 Isolation of N-methyl Julifloridine (I)
This alkaloid was eluted from the column as a mixture
methanol (98:2) onof alkaloids chloroformby
(neutral) column. The fractions were evaporated and the
mixture thus obtained was rechromatographed on AÿO,
(neutral activity 1) repeatedly and it yielded a new
alkaloid.
N-methyl julifloridine (I) was isolated as a yellowish
gum (9 mg). Its purity was checked on TLC plates and HPTLC
plates of silica gel (E. Merck, Art No. 5556) and solvent
system for this was chloroform - diethylamine (90:10). The
molecular formula of compound was cÿgH37°2'
6.4.4.1 Characterization of N-Methyl julifloridine (l)
UV (MeOH): x 203 and 283 nm.;max
3500 ( O-H ) , 2940 ( C-H ) and 1220 (C-N) cm .IR (CHC13): v :max
talD = -30 ( c= 0 .1, CHC13) .
110

( M-H ]+ ,312.2849 (calcd. for C19H38N02 '
298.2745 ) ,
m/ z ,HRMS:
C18H36N02 '
254 . 2483 ) and 128.1059 (calcd.
(calcd. for298 .2687312.2902 ) ,
254.2465 (calcd. for CÿHÿNO,
NO, 128.1075).for C? H14
EIMS: m/z, ( rel . int. % ) ;ÿ 312 ( 1.2), 298 ( 1.8), 254 ( 2 ),
120 ( 100 ) and 114 ( 10 ) .
1H-NMR : (300 MH2 , CDC13 ) ; 6 1.18 ( 3H , d, J = 6.72 Hz, H-7),
1.30 (d, CH2 of side chain), 1.50 (2H, m, H-4 ) , 1.80 (2H,
m, H-5 ) , 2.51 (3H, s, H-8), 3.07 ( 1H , oct distorted, H-2 ) and
3.63 (t, J = 6.57 Hz, 2H-12 masked by the H-3 signal).
13C-NMR: Table-1
111

6.4.5 Isolation of Projuline (II)
Projuline was obtained with chloroform- methanol (98:2).
The fractions obtained were impure and contained two
alkaloids. Projuline (II) was a minor alkaloid just below
juliprosopine (19) on TLC plates. This alkaloid was purified
by repeated column chromatography using A1_0 Neutral) and2 3
elution with chloroform- methanol mixture (80:20).
Finally 'projuline (II) was purified through thin layer
chromatography on silica gel in chloroform d ie thylamine
( 90:10 ) . Its pruity was checked on HPTLC plate of silica
gel in the above mentioned system. The molecular formula
of the compound ws found to be C_ -H . nN_,0-, .2 b 4 8 2 2
6.4.5.1 Characterization of Projuline (\ I)
UV (MeOH): x 208 and 280' nm.;max
vmax; 3400 1640 <c = c) H50 (C-O) cm"1.IR (CHC13)S
I a )D
B +16.66 ( c=0.06 , CHC13).
112

HRMS: m/z 420.3750 [M ] * , ( colcd. for C26H48N2°2 420.3715).
E1MS: m/z, (rel. int. %); 420 (1), 419 ( 4), 418 (8 ), 402
(5), 389 (36), 362 (19), 333 (46), 305 (4 ), 264 (4), 236
207 (2), 165 (22 ), 114 (100 ), 96 ( 55 ), 84 (98) and.70(3) ,
(84).
1H-NMR: (300 MHz, CDC13); 6 1.08 ( 3H , d, J = 7.1 Hz, H-7),
1.25 (br. s, CH2 of side chain), 4.0-4. 3 ( m ) and 5.3 (1H, m
H-7 ' 1 1
13C-NMR: Table-11
Acety 1at ion of (IX)
6 mg of (II) was taken for preparation of the acetate.
The compound was taken in pyridine and acetic anhydride
(1:5) and the mixture was allowed to stand overnight at room
temperature. The reaction mixture was worked up in the usual
manner. The residue from CHCI3 extract was collected.
UV (MeOH): X 203 and 285 nm.1735 (C=0), 1300 (C-N) and 1050 (C-O) cm-1.
max
IR ( CHC13) : V
max
113

= +1.66 ( c=0 .006 , CHC1 T )[ a]D 3
E IMS: m/z 547 [M + 1 ]+, 502, 473, 400, 324 , 313, 264 , 207 ,
156 and 84.
114

6.4.6 Isolation of Prosopinoline (III)
Prosopinoline was obtained with chloroform - methanol
obtained were impure and contained(97:3). The fractions
two alkaloids. Prosopinoline (III) was a minor alkaloid
which was just above the julif loricine on TLC plate. It was
further purified by repeated flash chromatography using
AlÿO-j (neutral)'-nd eluted with chloroform- methanol mixture
(97:3).
The purity of compound was checked by thin layer
chromatography on HPTLC plate of silica gel and the
developing solvents were chloroform -die thylamine (90:10).
Prosopinoline (III) was isolated as gum (10 mg) which
was soluble in chloroform and having molecular formula
C23H44N2°2 ‘
6.4.6.1 Characterization of Prosopinoline (III)
UV (MeOH): A 208 and 280 nm.max '
IR (CHC13)S vmgx; 3635 (O-H), 3435 (N-H) and 1550 (N-H) cm-1.
115

( a 1 D = +22.72 ( c = 0 . 22 , CHCI3 ) .
HRMS: m/z 380.3401 [ M ] * baled, for C23H44N2°2' 380-3402)-
E IMS s m/z, ( rel . int. %), 380 ( 2 ), 114 (100), 92 (12) and 70
(96).
1H-NMR: (400 MHz, CDCl-j ) ; 6 1.00
= 6.36 Hz, H-10
( 3H , d, 19 i i i tJ8a 1 f I I 9
= 6.5 Hz, H-7 ) ,lift ), 1.15 ( 3H , d, J2,7-10'1), 2.007 ( 2H , m, H-5), 2.1 (1H,1. 24 ( 20H , 1 IH-1s,
m, H-6 t l I ), 2.30 (2H. m. H-4 ) , 2.83 (1H, m, H-6), 3.0 (1H,
m, H-2), 3.56 (1H, bs , H-3), 3.7 (1H, m, H-8 ), 4.1 ( 2H ,a‘ • ’ •tillH-9 ), 4.2 (1H, m, H-5 till ), 5.30 (1H, dd,m,
J7 12 Hz, = 6.6 Hz, H-8 I » » I I ) andJ8fill fill,8 lift , 8a
= 4.4 Hz, H-7
I I I I
5.36 [ 1H, dd, I f t I ) •J6 I I 1 1 VI I I I .7
13C-NMR: Table-Ill
116

6.4.7 Isolation of Juliprosinene (IV)
The fractions eluted by chloroform - methanol mixture
fractionscontained mixture of alkaloids-. The(93:7)
major quantitycontaining this alkaloid
chromatographed repeatedly
in were
AI2O3 (neutral Activity 1)
column. Juliprosinene (IV) were further purified by thin
on
layer chromatography using silica gel as stationary phase
ammonia '70:30:1) as mobileand chloroform- methanol
phase.
The purity was checked by thin layer chromatography on
silica gel preparative plates. The solvent for thin layer
chromatography was chloroform - methanol ' diethylamine
(90:10:10).
19.5 mg of the chloride salt of juliprosinine (IV)
obtained as a gum showed the molecular ion peak of the
cation, corresponding to the formula c4oH7oN3°2’
6.4.7.1Characterization of juliprosinene (IV)
UV (MeOH ) : . X 208 and 285 nm.;max
117

3660 (O-H), 3350 (N-H), 2935 ( C-H ) , 1625IR (CHC13): v
(C=C), 1503 (N-H), 965 (C-H) and 877 (C-H) cm-1.;max
D = + 9.5° ( c = 0 .04 , CHC13).[or 1
[M+1 ]+
,625.5532m/z ,HRMS: (calcd. for
625.5531) and 389.3550 (calcd. for C25H45N20, 389.3510).
EIMS: m/z, (rel. int. %) (M]+ 624 (3.63), 608 ( 3.67 ), 389
(21.76), 114 (100), 92 (12.30) and 70 (96.68).
XH-NMR: (300 MHz, CDC13); 6 1.05, 1.13 ( 3H , 3H, 2d, J = 6.8
CH2 of side chain), 2.65-2.55
7.0 Hz. H-10
), 2.75-2.97 ( 2H , m, H-2 , H-2 ’ ) . 3.53 (2H, br s, H-3,
Hz, H -7 1 , H-7 ) , 1.25 (32H, s ,
(2H, m, H-6 , H-6 1 ) , 2.68 (4H, t, J I I
H-10 I I I
H-3’ ) , 6.25 ( 1H, dd, J3b = 6.2 Hz, J = 14 Hz ,
Jgem10.6 Hz,
lilt I f I I-2 gemI I I IH. 3 ), 6.30 ( 2H , dd, J
3
), 6.82 (1H, dd,
= 14= 6.2 Hz ,i i i i r i i,2b
Hz , Ha"3 VIII
J1 I t I I .2 I I I I
= 6.2 Hz, H-2 I I I I ), 6.85 (1H, d.J2 J1till I I I.3 I I I I ,2 lilt
= 10.6 Hz, H-l lit* ). 7.68 (1H, I I I IH-7 ) and 9.35 ( 1H ,s ,
tills, H-5 ) •
13C-NMR: Table-IV
118

6.4.8 Isolation of Prosopidione (V)
The fractions obtained on elution with 20% methanol in
chloroform afforded a non-alkaloidal compound with some
impurity. An interesting new compound was isolated by
subjecting the impure fraction of compound to column
chromatography on Al-,0 (neutral, type-E) as stationary
phase and chloroform - methanol (80:20) as mobile phase.
2 3
The fractions rich in this new terpenoid compound were
collected and rechromatographed. Purity of the compound was
checked by thin layer chromatography in chloroform
methanol - diethylamine (80:10:10) on silica gel.
12.2 mg , of the new terpenoid p.'osopidone (V) was
obtained as a white amorphous powder. The molecular formula
of the compound was CI3H2Q02‘
6.4.8.1 Characterization of Prosopidione (V)
UV ( MeOH ) : X 202 and 228 nm .;max
LR ( KBr ) : v 1710 (C=0), 1675, 1650 ( e , 8-unsaturated ke-max
119

tone), 1260 (C-O) and 960 (C-li stretching for C=C ) .
M.p. 202° {decomp).
= -19.2° ( c = 0 .052 , MeOH).ta]D
HRMS: m/z, 208.1468 [M]+, ( caicd . for 208.1463),
165.1270 (caicd. for •C11H1?0, 165.1279), 140.1073 (caicd.
C9H16° '
125.1330) .
for 125.1307 (caicd. for CÿH140.1201) and 9 17
1H-NMR: ( 300 MHz, CD,,OD): 5 0.81 ( 3H , d, J = 6.8 Hz, H-13),
H-11 ) , 1.10 { 3H , s, H-12 ) , 1.5 ( 2H , m, H-3),0.87 ( 3H , s ,
1.7 (1H, dd, = 6.6 Hz, H-5 ), 1.8Jgem'
12.52 Hz,
= 12.56 Hz, J5 ,6
= 4.84 Hz, H-5 ), 2.15 (1H,(1H, dd, J5 ,6Jgem'H-2 ) , 2.26 (s, COMe), 4.5 (1H, m, h-6 ) , 6.33 (1H, d, J7,8m ,
= 16.04 Hz) .= 16.04 Hz, H-7 ) and 6.88 (1H, d, J7 , 3
13C-NMR : Taf>le-V
120

;
7. BIOLOGICAL ACTIVITY

Screening of isolated constituents of a plant for
biological activity is gradually gaining importance. The
discovery of useful biological activity is essentially the
objective of phytochemical research. Prosopis juli flora
alkaloids juliflorine and julif loricine were tested for the
following:
a) Acute toxicity in mice.
b) Neuromuscular blocking activity in isolated rat phrenic
nerve hemidiaphragm preparation.
c) effect on blood pressure in anesthetised rats.
Antimicrobial testing of juliprosinene , a new alkaloid
from Prosopis ju lif lora was conducted
7.1 Materials and Methods
7.1.1 Acute Toxicity
LiD50 or the dose which kills 50% of
189) .the animals was
determined in mice Juliflorine givenwas
122

intraperitonially to groups, each consisting of four mice
of either sex, in doses 10 mg/kg, 21.5 mg/kg, 46.4 mg/kg and
100 mg/kg body weight. Mortality was observed for 24 hours.
Behavioural changes were also recorded continuously for 2
hours on a work sheet standarized by Malone and Robichaud
[90], intermittently for a further 4 hours. LDÿQ
limits was calculated.
with fiducial
7 1.2 Neuromuscular Blocking Activity
The isolated rat phrenic nerve - hemidiaphragm preparation
for recording neurally evoked twitch was based on the method
described by Bulbring [91].
Rats were killed by stunning followed by exsanguinat ion .The thorax was cut open and the left phrenic nerve located.
Triangular section of the left hemidiaphragm was dissected
out along with its accompanying phrenic nerve. The preparation
was then mounted in a double- jacke ted organ bath (100 ml)
(Harvard) containing oxygenated Krebs so..ution (NaCl 6.9 g/1;
NaHC03 2.1 g/1; Glucose 2.0 g/1; KC1 10% 3.5 ml/1; MgSOÿ .
7H20 10% 2.9 ml/1; KH2PC>4 10% 1.6 ml/1; CaCl2 1M 2.5 ml/1),
the temperature of which was maintained at 30°C. A thread
123

to the apex of the fan-shaped preparation was connectedt ied
FTO-j (Grass Instrumentto a force-displacement transducer
Co., Quincy, Mass., USA). The phrenic nerve was passed between
the loops of a bipolar platinium electrode connected to a
stimulator (Harvard). Nerve stimulation was elicited by
rectangular pulses of supramaximal voltage of 0.5 msec
duration at a frequency of 0.2 Hz.
The resting tension was maintained at 2g and isometric
contractions were recorded on a polygraph (Grass Instrument
Co., Quincy, Mass., USA). Thirty minutes equilibration time
was allowed before starting the experiment.
Muscle twitch in response to single repeated stimulation
was recorded before and after adding the drug into the tissue
bath. The response was observed for 10 minutes after drug
'
administration .
The amplitude of neurally evoked twitch without the effect
of any drug was taken as control. The twitch depression
produced by the tested drug was expressed as percent of the
control according to the following equation:
124

T X 100%% twitch depression = C
C
where C - the twitch amplitude of the control
T = the twitch amplitude measured at 10 minutes after
drug administration
calculatedThe response was expressed as mean +_ S-.E.M.
from at least 3 observations. Student's t-cest was applied
to determine the significance of difference between mean
P 0.05 was accepted as statistically significant.
7.1.3 Effect on Blood Pressure
The effect of juliflorine on blood pressure was observed
in anesthetised (Pentothal sodium, 40 mg/kg body weight) rats
of either sex, weighing 200-250 g [92).
The trachea and jugular vein were cannulated. The tested
alkaloid was injected via the jugular vein. Blood pressure
was recorded from the common carotid artery with a strain
(P23BC Gould Statham) connected
to a polygraph (Grass Instrument Co., Quincy, Mass., USA).
gauge pressure transducer
125

7.2 Results and Discussion
7.2.1 Acute Toxicity
The 24 hours LD in mice was found to be 21.5 mg/kg body50
weight with confidence limits 8.88 52.3. This is considered
to be highly • toxic according to Ghosh [931. The animals
exhibited depression at lower doses while at higher doses,
clonic convulsions preceded death [Table-A] .
7.2.2 Neuromuscular Blocking Activity in Isolated Rat Phrenic Nerve-Hemidiaphragm
Preparation
Prosopis juliflora alkaloids blocked neurally-evoked
muscle twitch, but not in a dose-dependent manner. Juliflorine
in doses 50 mg/ml and 75mg/ml produced no significant change,
whereas at 100 mg/ml dose 79.06 7.28% (P 0.01, n= 7 )+
depression of the muscle twitch was observed. Julif loricine
also behaved similarly at the above dose, producing 67.77
i 15.40% (P 0.05, n=3) depression.
Pharmacodynamic studies reveal that the block induced
by these alkaloids preceded muscle fasiculation and
126
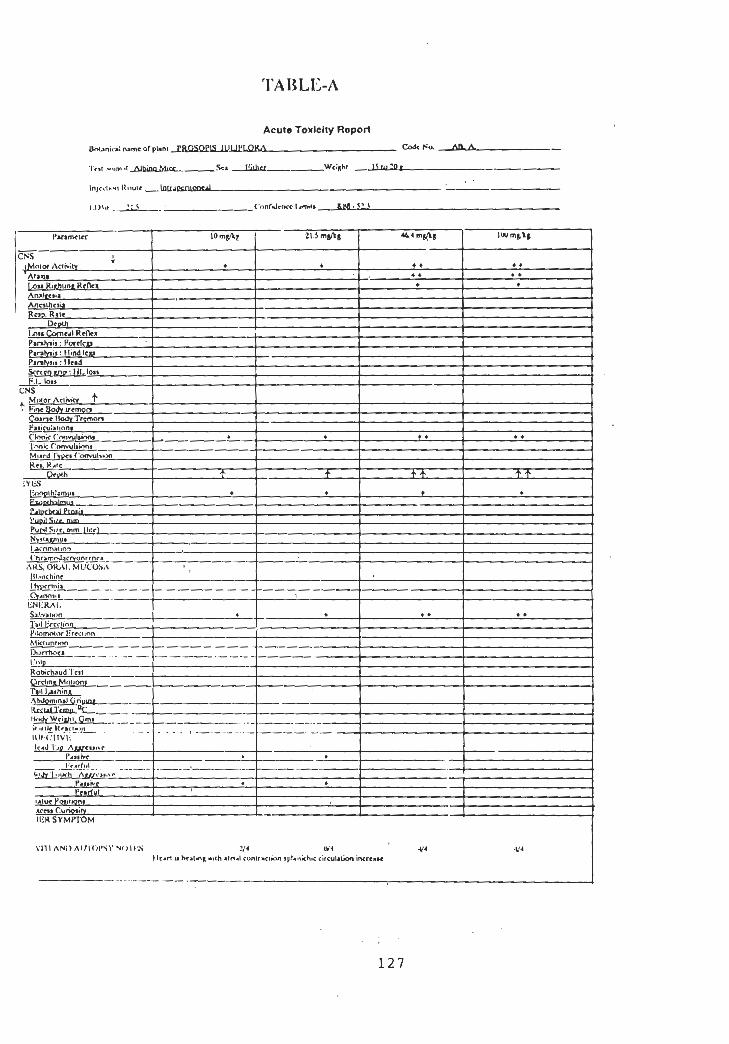
1ABLI>A
Acute Toxicity Report
Cod< No. Aik, ADoomoi rv>mc of pl«m PROSOPIS JULll'LQKA
.WeJjhi I S in ?01S«i IvidiffTol AJbino Mit
Inject* «n Route_
Intt jpcriioocJ
<'(>nfHlcn<-c OOMB1UI .!>*«
2l.5mgAs 444 m?A*10 m$/*?Parameter
CNSiMoiof Activity
fAiw>
I4 4
\jytt Kj'uMmf Reflex
Anctihcii*KCIJ. R<1«
Depthtÿn Comral ReflexPiraViii ; PofftewPiralym : Hind leg*
Pinhÿn : Hc>dS<TC£fJ_£C£ÿHÿlM»F-L IOM
__CNS
M<*or Activity
t Fir>e Efoft iremonCe»r»e Ikxty TrymorB
F«<icu!>iionfQpfvg ronvyiltioni 4 f 4 4 44
1Apic ronvy|>k>o<
MucJ Typet Convulvop
Ket K<»cA fDepth tT
•YiiSlv>ppihba)u> 4 4 4
Fti h n I mi .' i
gjlucbcai PtQ« j.Pupi! Si / r mm
Pupil S»/c. mm Ilitr )
Nyiujÿuit_I xrimaiion( PfÿrroJjcryoorrnf 4
ArtS, ORAL MUCOSAHi.mchineI lypermia
_OfÿfVlVI
i£NI:RALSjjgation_
1
4 4 4 4 4
Tail IxecliooP'toirwxof iTect.onMUrtuniton_IXirrhoci
Hotwctwu<j I cilCircling MoiJQn*
TeM l,aihing
Abdominal Cripinyrtectal Temp. °CIkwiy Weighs Cm»k.irfj£ lineIN MI
_iviM:iTvir“lc J«ij Tap. AgJ/CHiVf
PÿMIVf » 4
Psft'J'MKh Al!//\-llAfPUBIVf 44
Ff rfpt
mtue Ptttil*QnB
Jif Cl» Curiptily
ICR SYMPTOM
vm AND AUTOPSY NOD.S V* IH -Y4 A!A) le*rt I* br*tÿg «i(h »tn*l contraction ip*4»khrc circulation increase
127

augmentation of the maximal twitch. This action is similar
to that of succinyl choline, resulting from the repetitive
firing of the muscle fibres. Moreover, the fact that
physos t igmine , a cholinesterase inhibitor fails to reverse
the block produced by these alkaloids, provides evidence that
the blockade is probably of depolarising nature [Fig-1].
this result does not preclude blockage ofHowever ,
neuromuscular transmission other mechanisms such asby
prejunctional interference with the synthesis or release of
acetyl choline. The high toxicity of the alkaloid may also
be associated with its blocking activity at the neuromuscular
junction .
7.23 Effect of blood Pressure
No significant effect of juliflorine was observed on the
blood pressure of anesthetised rats in doses of 50 mg/kg,
250 mg/kg, 1000 mg/kg and 2000 mg/kg body weight, while 5000
mg/kg was lethal.
128

The effect of physos t igmine (10 yg/ml) on depression ofneural ly-evoked twitch produced Dy A.d- tubocurarineB-succinyl choline C- julif lorine
19rÿin‘
Fig-1:
IIIA
* *physostignn.ne10 yg/nil.
d- tubocurarine1.5 yg/ml .
B
7physostigmine
10 yg/ml.succinyl choline
15 yg/ml.
C
mjulif lorine
100 yg/ml.physos t iqmine
10 yg /ml .
129

7.3 Antimicrobial Activity of Juliprosincnc (IV)
Inocula were prepared by incubating one loopful of
10 ml of nutrient broth for 24 hours atstock culture in
37°. Nutrient agar plates were swabbad with 0.1 ml of the
overnight inocula. Wells of 0.6 cm were dug with a sterile
borer in the inoculated agar. Into the well was placed 0.1
ml of the test solution. A control was maintained by using
0.1 ml of DMSO. The plates were incuoated at 37° for 24
hours. All tests were done in duplicate.
130

TABLE-1
Results of Antibacterial activity of Juliprosinenc
Activity3No. Bactetialttrata
Wm istsi&si v j
Staphylococcus aureus B-8271. + +
Sterptococcus faecalis 8413 Her 10442. +
3. E. Coli 11303 HER 1024 + +
E. Coli K-125 HER 10374. + +
Klebsiella pneumoiae C3HER 11115. + +
6. Shigella sonnei YER HER 1048 + +
7. S. dysenteriae a. SH HER 1031 +
8. Pseudamonas aeruginosa PA 01 HER 1018 + +
9. Bacillus subtilis - ATCC 6051
10. Salmonella typhimurium HER - 1038
ution of 0.1 mg Juliprosinene in 0.1 ml of DMSO (see experimental).
or activity
tivity (No test organism inhibited or killed)
/e (Less than 50% of test organism inhibited or killed)
ghly active (More than 75% of test organism inhibited or killed)

'
8. REFERENCES o
o

1. E. Nasir 'and S.I. Ali, Flora of West Pakistan, FakhriPrinting Press, Karachi, Pakistan, 383 (1972).
Jafri, Flora of Karachi, The Book CorporationPakistan, 149 (1966).
2 . S.M.H .Karachi .
3. A. Krishnamurthi , Wealth of India, Council of Scientificand Industrial Research, New Delhi, VIII, 245 (1969).
4. D. Meyer, R. Becker, M. R. Gumbmann, P. Vohra, H. Neukomand R.M. Saunders, J. Aqric. Food Chem. , 34(5), 914( 1986 ) .
5 . Hoppe-Drogen Kunde, Band 1 Angiospeim 8, Auflage, Waterde Gruyter, Berlin, New York, 874 (1975).
6 . K.M. Nadkarni, Indian Materia Medica, Bombay popularprakashan, 1, 1011 (1976).
7. W.H.Wiley 45 , 69 , 224 ,
Lewis and M.P.F. Elvin Lewis, Medical Botany, John302, 329, 362, 434 (1977).
8. M.S. Attia, S. Ahmad, S.A.H. Zaidi and Z. Ahmad, Pak.j . Sci. and Ind . Res. , 15(3), 199 (1972).
9. K.R. Kitikar and B.D. Basu, Indian Medicinal Plants,Indian Press, Allahabad, 473 (1918).
10. R.N. Chopra, S.L. Nayar and I.C. Chopra, Glossary ofIndian Medicinal Plants. CSIR, New Delhi, 204 (1956).
11. R.R. Cruse, Economic Botany, 13, Nr. 3 (1959).
12. P.S. Murthi and S. Siddiqui, J. Sci. and Ind. Res.,7B( 11 ) , 188 ( 1948 ) . '
3. G.M. Wassel, A.M. Rizk and E.F.Mater. Veq., XXII, 1, 119 (1972).
Abdel-Bary, Qual. Plant:
4. R.C. Sharma, A. Zaman ana A.R. Kidwai, Indian J. Chem.,2(2), 83 (1964).
133

15. G.R. Iches, H.S. Fona, P.L. Schiff Jr., R.E. Perdue
Jr. and W.R. Farnsworth, J Pharm. Sci . , 62, 1009 (1973).
Patent No.l. 524.395,16. Omnium Chimique Societe, Fr.,( 1968 ) .
17. P. Bourinet and A. Quevauviller , C R . Soc. Biol . , 162,1138(1968).
18. D. Shankaranarayan, C. Gopalkishnan , S.K. Nazimudeen,L. Kameswaran and 3. Arumugam, Me j iscope , 22(2), 37( 1979 ) .
Augusto Cerco's, Rev. Argentina Argon, 18, 200-9,19. P.( 1951 ) .
20. M.M. At tia ,El-Duweini and Ghazal, Planta Medica, 36, 150 (1979).
El-Merzaban.i , El-Aaser , A.K.A . A. M. A.
21. J.C. Oberti and H.R. Juliani, An. Asoc. Quim. Argent.,59(2), 101 (1971).
22. M. Ikram, Fitoterpia , 123 (1983).
23. Ali A. Al-Jeboory and Wesal. A.J. Al-Husainy, Fitoterpia,LV, (3) 137 (1984).
24. G. Rattle, X. Monseur, B.C. Das, J. Yassi, Q. Khuong-Huuand R. Goutarel, Bull. Soc. Chim. France, 2945 (1966).
25. M. Manzoor-e-Khuda , S.A. Abbas and W.A. Zaidi, Pak.J. Sci and. Ind. Res. , 11(1), 1 (1968).
26. M.N. Cranziano, G.E. Ferraro and J.D. Coussio, Lloydia,34(4 ), 453 ( 1971 ) .
27. Q. Khuong Huu, G. Rattle and X. Monseur, Bull. Soc.Chim. Belg. , 81, (7-8), 425 (1972).
28. Q. Khuong Huu, G. Rattle, X. Monseur and G. Robert,Bull. Soc. Chim. Belg., 81, (7-8), 443 (1972).
29. J. Parente, J. Herrera, J. Oberti and H.R.An . Quim. Arg . , 60(6), 527 (1972).
Jaliani ,
134

F. Amir and F.H.30. K. jewers, M.J. Nagler, K.A. Zirvi,Cottee, Pahalvi Med, j., 5(1), 1 (1974).
31. K. Jewers, M.J. Nagler, K.A. Zirvi, F. Amir and F.H.Cottee, Phytochemistry, 15, 238 (1976).
32. G.A. Moro, M.N. Graziano and J.D. Coussio, Phytochemistry ,14, 827 (1975).
33. Y. Tomataka, M. Tetsuya, M. Kyoji, Mokuzai , 21(12),
686 (1975).
34. K. Jewers* M.J. Nagler, K.A. Zirvi and F. Amir,Phytochemistry , 15, 238 (1976).
32,35. K.A. Zirvi, K. Jewers, M.J. Nagler, Planta Medica,
244 ( 1977 ) .36. D.K. Bhardwaj , R.K. Jain, G.C. Shjarma and C.K. Mehta,
Ind. J. of Chem. Sec. B, 1133 (1978).
37. V.U. Ahmad, A. Basha and W. Haque, Z. Naturforsch, 33b347 (1978).
38. V.U. Ahmad, Z.G. Mohammad, J. Chem. Soc. Pak. , 1, 2(1979).
39. R. Ott. Longoni, N. Viswanathan and M. Hesse, Helv.Chim. Acta, 63, 2119 (1980).
40. D.K. Bhardwaj, M.S. Brisht, C.K. Mehta and G.C. Sharma,Phytochemistry, 19, 355 (1979).
11. I.B. Gianinetto, J.L. Cabrera, H.R. Juliani, J. Nat.Prod. , 43(5), 632 (1980).
2. D.K. Bhardwaj, M.S. Brisht, C.K. Mehta and G.C. Sharma,Phytochemistry, 19, 1269 (1980).
3. R. Vajpeyi and K. Misra, Ind. J. Chem., Sect. 20B, 348( 1981 ) .
4. V. P. Datwyler, R. Ott. Longoni, E. Schopp and M. Hesse,Helv. Chim. Acta, 64, Fase 6, Nr. 188, 1959 (1981).
135

©
45. S. Malhot;:a and K. Misra, Phytochemistry, 20( 8), 2043(1981).
46. S. Malhotra and K. , Misra, Phytochemistry , 20(4), 860( 1981 ) .
47. R. Vajpeyi, N. Shukla and K. Misra, Phytochemistry,
20, 339 (1981).
48. S. Malhotra and K. Misra, Int. Conf. Chem. Biotechnol.Biol. Act. Nat. Prod. [Proc.), 1st, 3(1), 129 ( 1981 ).
Edited by B. Atanasova B., Bulg. Acad. Sci. Sofia, Bulg.
49. A.M. Gitelli, I.B. Gianinello, J.L. Cabrera and H.R.Juliani, Ar. Asoc. Quim Argent, 69( 1 ), 33 (1981).
50. D.K. Bhardwa j , A.K. Gupta, R.K. Jain and G.C. Sharma ,J. Nat. Prod., 44(6), 656 (1981).
51. S. Malhotra and K. Misra, Phy tochemis try , 20( 10), 2439( 1981 ) .
Curr. Sci., 51(23),52. P. Radha, Bishnov and1124 (1982).
P. Lata,
53. C.A . Chiale, I.B. Gianinelto, J.L. Cabrera and H.R.Juliani, An Asoc. Quim. Aargent., 70(2), 3379 (1982).
54. Figueiredo, Antonio de A, Cienc. Tecnol. Aliment, 3(1),
82 (1983).
55. F.R. Del. Valle, M. Escobedo, M.J. Munoz, R. Ortegaand H.J. Bourges, J. Food Sci. , 48(3), 914 (1983).
56. M.M. Ebeid, Fitoterapia, 54(4), 179 (1983).
57. S. Malhotra and K. Misra, Planta Medica, 47(1), 46 (1983).
58. S.A. Abbas and P. Mison, Pak. J. Sci. Ind. Res., 26(3),
140 (1983).
59. S. Malhotra and K. Misra, Ind. J. of Chem., 22B , 936( 1983) .
60. S. Malhotra and K. Misra, Curr. Sci., 52(12), 583 (1983).
136

Z. Naturforsch , 38b, 660 (1983).6 l . V.U. Ahmad and S. Qazi,
62. J.P. Sehgal, Indian J. Anim. Sci., 54 ( i ) , 126 (1984).
63. C.A. Chiale, J.L. Cabrera and J.R. Juliani, An Assoc.Quim. Arg. , 72(5), 501 (1984).
64. J.L. Cabrera, II. R. Juliani, O.A. Orio and R.E. Herreo,An. Asoc. Quim. Arg., 72(3), 269 (1984).
65. I.S. Thakur and J.D. Sharma, Biochemistry International,11(6), 903 (1985).
66. V.U. Ahamad and S. Qazi, J. Chem. Soc. Pak., 7(4), 347( 1985 ) .
67. A.M. Balogun and B.L. Fetuga, Food Chem., 17(3), 175(1985).
68. C. Gonzalez, M. Baez and M. Lachica, Agrochimica, 29(5-6),478-89 (1985).
69. M.D. Umar, A.N. Memon and S.A. Ali, J. Pharm. , (Univ.Karachi), 5(1), 25-7 (1986).
70. D. Lumen, .0. Benito, B. Robert and R.S. Pilar J. Agric.Food Chem. ,, 34(2), 361-4 (1986).
71. A. Kery, H.A. Twaij and A. A. Al-Jeboory, Study, org.Chem., 23 ( Flavanoids and Bis f lavanoids , 1985), 171-81(1986).
72. M. Daniel, B. Robert, M.R.and S.M. Robin, J. Agric.( 1986 ) .
Gumbmann, V. Pran, H. NeukomFood Chem., 34(5), 914-19
73. A. Ahmad, K.A. Khan, V.U. AhmadMedica, 247 ' ( 1986 ) .
and S. Qazi, Planta
74. M.T. Nadir, A.J. Baqi, S.M.Fitoterapia, 57(5), 359-63 (1986).
Al-Sarraj and W.A. Hussein
75. N. Ahmad and S. Razaq, Fitoterapia , 57(6), 457 (1986).
137

E.V. Brandt, D .A. Young, D. Ferreira and D.G.J. Chem. Soc. Perkin Trans I, 10, 1937-49 (1986).
7 6. E. young ,
Roux ,
77. B.V. Bruno, G.J. Carlos, S.A. Renato and C. Renato,
Arq. Biol. Tecnol. , 30(2), 275-86 (1987).
78. J.A. Herrera, l.B. Gianinetto, J.L. Cabrera and H.R.Juliani, An. Asoc. Quim. Argent, 75(4), 379-80 (1987).
79. P.L. Soni,. S.S. Bisen, Indian For, 114(1), 26-8, 4 plates( 1988 ) .
80. A. Ahmad, K.A. Khan, V.U. Ahmad and S. Qazi, Fitoterapia ,59(6), 481 ( 1988 ) .
81. A. Ahmad, K.A. Khan, V.U. Ahmad and S. Qazi, Ar2neimit tel-Forschung (Drug Research, 39(1), No. 6, 652 (1989).
82. A. Ahmad, K.A. Khan, S. Qazi and V.U. Ahmad, Fitoterapia,60(1), 85 (1989).
83. D. Lythgoe and M.J. Vernengo, Tetrahedron Lett., 1133( 1967 ) .
84. I. Chris tofidis , A. Welter and J. Jadot, Tetrahedron,33, 977 (1977).
85. A. I. Scott,17 ( 1964 ) .
Ultraviolet Spectra c£ Natural Product,
86. E. Brown and A. Bonte, Tetrahedron Lett., 33, 2881 (1975).
87. E. Brown, R. Dhal and P.F.5607 (1972).
28,Casals, Tetrahedron ,
88. R. Lyle, DChem., 31, 4164 (1966).
McMamon , W. Krueger and C. Specer, J. Qrg.
89. P.R. Dua, "The use of Pharmacological Techniques forthe Evaulation oC Natural Products”, B.N. Dhawan andR.C. Srimal (Eds.) UNESCO Publication 25 (1984).
90. M.H. Malone and R.C. Robichoud, Lloydia, 25, No. 4, 320( 1962 ) .
138

91. E. Bulbring, Brit J . Pharmacol. , 1,38 (1946).
92. S. Shibata, K. Karakashi and M. Kachii, J. Pharmacol.Exp. Ther., 185, 406 (1973).
93. M.N. Ghosh, Fundamentals o£ Experimental Pharmacology,Scientific Book Agency, Calcutta, India, 157, (1984).
139

PART II
FURTHER STUDIES ON THE CHEMICALCONSTITUENTS OF PLUCHEA ARGUTA BOISS.

I
1. BIOSYNTHESIS

1.1 Biosynthesis of Terpenoids
is the building block for terpene(1)I soprene
condensation of successive isoprene units in
tail fashion would produce compounds of formula
biosynthesis ,
a head to
This is called as the "Isoprene rule" and hence<C5H8Vthe term isoprenoids .
Earlier chemists hypothesised a direct participation of
isoprene in vivo by the possible formation of dipentene from
two isoprene units by a simple Dieis-Alder process. [Eq-1]
An objection to this early postulate was that isoprene
itself did not appear to be present in nature and could only
be obtained by pyrolysis of certain monoterpenes. In 1959
Cornforth in his work on biosynthesis of steroidsJ .W.
characterized two active forms of isoprene, isopentenyl
pyrophosphate (IPP) (2) and dimethyl allyl pyrophosphate
( DMAPP ) (3).
The intermediates, IPP and DMAPP, arise from ( + ) mevalonic
acid ( MVA ) (4) and an enzyme complex which effects this
conversion in high yields.
142

2 methyl buta-1, 3-dieneIsoprene (1 )
In Vivo
Pyrolysis
Ov
Dipentene (d,l)
Eq-1
C
H3 \
C=CHH3Cÿ / iH-X3CH2-0©(£) CH2-O®®
IPP (2 ) DMAPP (3)
CH2-COOH
H3 OHCtii
CH2-CH2OHMV A (4)
143

Acetic acid or its derivative acetyl CoA, is the only
of theof mevalonic acid and, hence,carbon source
intermediates .IPP and DMAPP. It had been observed that only
the R form of mevalonic acid is utilized by organisms for
producing terpenes while S form is metabolically inert.
tail mannerA DMAPP molecule can condense in a head to
with IPP to produce geranyl pyrophosphate. DMAPP thus acts
as the foundation stone upon which are added the building
bricks of IPP units. [1]
The terpenoid end products can be considered as product
of pathways branching from intermediates of a common central
pathway Fig.1 [ 2 ] .
1.1.1 Sesquiterpenes '
The carbon atom skeleton of almost all the known
sesqui terpenoids derived trans-f arnesy1frombecan
pyrophosphate (6) and the cis isomer (5), through appropriate
cyclisation and rearrangemen ts . Cis-FPP should derive from
trans FPP via (reversible) mechanisms very similar to the
ones connecting GPP to NPP. The schemes for sesquiterpenes
144
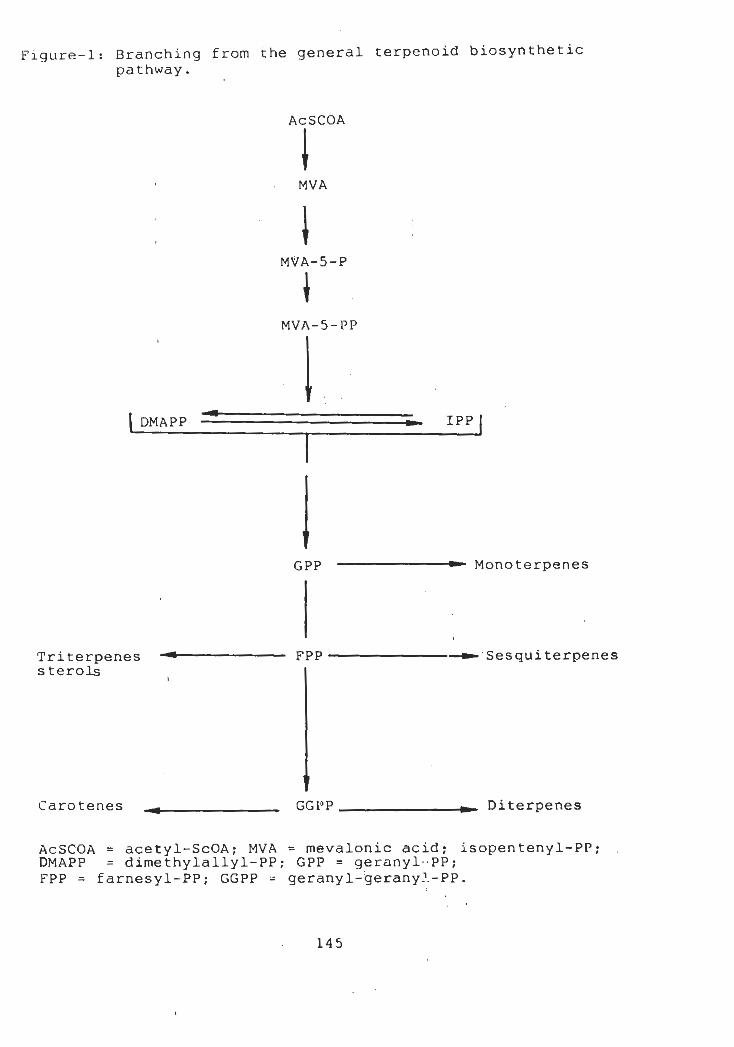
Figure-1: Branching from the general terpenoid biosynthetic
pathway.
AcSCOA
tMVA
IMVA-5-P
*MVA-5-PP
\ DMAPP IPP
Mono terpenesGPP
SesquiterpenesFPPTri terpeness terols
DiterpenesCarotenes GGPP
AcSCOA = acetyl-ScOA; MVA = mevalonic acid; isopentenyl-PP ;DMAPP = dime thylallyl-PP; GPP = geranyl-PP;FPP = farnesyl-PP; GGPP - geranyl-geranyl-PP.
145

biogenesis has great similarity to those for the cyclohexane
monoterpenes.
The departure of the pyrophosphate anion from (5) and
(6) leads the six possible monocyclic cations (10-15) through
(7) and (9) [Scheme-1]. Thethe non classical cations
stabilization of such classical carbocat ions , either througn
loss of a proton from the adjacent carbon atoms, or attack
of a hydroxide ion, produces compounds referred to as primary
skeletal sesquiterpenes [1].
Many total synthesis of sesquiterpenes have been modelled
on proposed pathways (i.e., biomimetic synthesis), and often
proceed via intermediates which contain incipient cationic
sites. Some resonable pathways are shown in Scheme-1 [3].
A small number of bicyclic sesquiterpenes are derived
from cyclisation shown in Scheme-2. The departure of the
pyrophosphate anion is the starting point for all the
cycl isat ions , an incipient carbocation being formed at
tail position of the FPP chain. In forming the bicyclof arnesyl
skeleton [Scheme-2]. The cyclisation takes place after an
the
3lec trophilic attack, e.g. by a proton, at the double bond
146

i)\o©©O
0©©(5) (6)
ppÿ/ cppi
O'1~ I
t •»I.©ÿI
___/
I
(8)(7)
©©
( 11 )(10)(9)
© © © EZ
(14 )(13)(12)
Biosynthesis of sesquiterpenes
©E
Scheme-1
(15)

00©
1
000
+
//
\' o/( CHO
0
CHO
JO <•'
HHOH2C' !
HIres in Scheme-2 Polygodial
148

at the head of, FPP, or on-to the corresponding epoxide. The
relative positions of the double bonds (of E- configuration)
in the conformation assumed by the farnesyl chain at the
moment of electrophilic attack determines the structure and
stereochemistry of the final products. This mode of
cyclisation, is uncommon for the sesquiterpenes. [1)
The known naturally occuring sesquiterpenoids isolated
from plants, can, be classified as follows (4]
1 ) The eudesmane
2) The guaiane
3 ) The germacrane
4 ) The drimane
The formation of the compounds of type (1-3) may be ratio¬
nalized by assuming a cyclization process that is initiated
)y ionization of all trans-f arnesyl pyrophosphate [5, 6).
>lthough this scheme has not yet been documented experimen-
ally the laboratory conversions of some germacrane to
udesmane [7-10], and of a germacrane to guaiane [11] offer
upport for step e and d of Scheme-3. The direct conversion
E a farnesol derivative to a germacrane [step a], eudesmane
149
r>

092 di 8
k3 8 54 3 7
66 7 4
Germacranes
cae
-30P„0
2 6
trans-Fernesyl
pyrophosphate b
92 1 0 86
35. 7
4 6
Eudesmanes
Scheme-3
150

or guaians [step e] has not yet 'Deen realized[step b ] ,
[ Scheme- 3 ] [ 1 ] .
Germacradiene structures are not so important for their
range and wide occurence but because they play a fundamental
role as intermediate for a variety of bicyclic structural
types. ’Ihe two double bonds are held in an appropriate
position and configuration in the ten membered ring to allow
intramolecular electrophilic cyclisations , to yield bicyclic
products. The final structure of the products depends on the
mtimate structure of initial conformation adopted by the
lacrocyclic ring, taking into account the orientation of the
.sopropyl group and the s tereoelectronic features of the
:yclisation reaction [Scheme-4].
Scheme-4 depicts some possible cyclisations of the
ermacradiene intermediates in various conformations, the
ouble bonds are always in the same positions and with the
- configuration typical of primary cation (14). Electrophilic
idition follows the Markownikoff rule for both double bonds.
ten on oxidative ( OH )+
attack is required, it appears to
jllow the pattern set by opening the corresponding "epoxide"
itermediates themselves originated by attack of an oxidase
151

1 +
1
f( Germacradiene Intermediates)
+x+
*r\ R,R
c HO'•OH
X HOX
R Hi
©\i
HO IH
R
.'Hi
OHHOH
3 -Eudesmoi( Eudesmane or Seiinane)
=H+ of a different electrophile ( e . g. OH+)
=-*-OH ,
Occidentalol
etc.COOH
Scheme-4

on the double bond [1],
The fungal metabolite avocettin (17) nay be derived from
(16) by three plausibleantipodol y-cadinene rou tes
[Scheme-5]. It was suggested that in the present instance
(18) could be the initial precursor in a scheme that bypasses
formation of the 2-cis-isomer [Scheme-6). The key idea was
;hat enough conformational flexibility occured in the diene
:ontaining the CÿQ ring to permit the conformational change
19, 20) necessary to allow cyclization. This had been
roposed by others (1973) to accomodate the formation of
endrobine from 2-trans-FPP via an intermediate with a
ermacrane- type ring without the prior interconversion of
-cis-FPP [ 12 ] .
On the other hand, the drimanes, a few examples of which
re known, may arise by the electrophile catalyzed cyclisation
f f ernesol pyrophosphate typical of di- and triterpenes
Scheme-7] [4]. A laboratory analogy is known [13].
153

OPP
1,3-hydride shift
)f
1, 2 -hydride.q h i f tfr 1.2
++
HHo:. »o
HCOÿ
(17)(16)
Scheme-5
154

OPP
HRHsHs( 18 )
H
cH •
I,
GH+H
:fo, Hs(16)
(19)Scheme-6
OP20‘3
Fernesyl pyrophosphate Dr imane
Scheme-'/

1.1.2 The eudcsmane (selinane)
The logical precursor of this group [14] is the cation
[Scheme-1] which can undergo a favourable cyclization
decline system, examplified by cryp tomeridiol
[15]. Originally this group was characterized by the formation
(15)
to the trans
3f eudalene on dehydrogenation.
1.1.3 Biosynthesis of eudesmane
According to biosynthetic point of view the eudesmane
iroup of sesqui terpenoids is derived by cyclization of a
f arnesylermacrane derivative formed from trans, trans
yrophosphate (6) [ Scheme-1] [16].
Thermal cyclization of elemol (21) produces a mixture
f hedrocaryol (22) and starting material which undergoes
ilver ion-catalyzed rearrangement to a-eudesmane (23) and
-eudesmane (24). Elemol (25) and the eudemols co-occur in
he citronella oil (Ceylone variety) and this may indicate
nat a biosynthetic relationship exists between them [Scheme-8
id 9 ] [ 17 1 .
156

Germacrane
**
*
trans, trans- OPPFernesy1pyrophosphate
Eudesmane
I1
OPP
Scheme-8
157

I
H OH OH
(22)(21 )
+ 1Ag
+
i
H
(23) (24 )
Scheme-9
158

The biosynthesis of eremophilone [Scheme-10] requires
the prior migration of one intermediate, forming the isomer
(26) with both double bonds in the ( E ) -conf igura t ion .
Subsequent cyclisation, induced by the usual electrophilic
addition, then takes place ( Markownikof f type on both double
bonds ) [ 1 ] .
The biosynthesis of rishitin (28) a member of the group,
involves a remarkable second rearrangement of the vetispirane,
hydroxy lubimin (27) [Scheme -11] [ 18 ] .
159

12 )
/7>
1 r*R
R
ii
OH
Elemol( Elemane ) (26)
t
r\'OH
/ :0H
±1X
H7 for X=OHV
\ I
OHH R+ i
H
I0H
i
l 0
R\Scheme-10\ \
\
R = -4 OH , H" * v \' ' COOI1 e tC+
of a different 'electrophile (e.gOH )X=H
9CH3C02H
3MVA
1HO
CHO0X
X
+
II
IHO,
HO, „CHOX
*
H HO
IIi
(28)
(27 )
Scheme-11
161

2. REFERENCES
r
f

1. P. Manito, "Biosynthesis of Natural Products" Ellis HowardLimited, Chichester, 213-253 (1981); and references citedthere in.
2. A.W . Charlesd, W.D. Marks and T.D. Micheal, Recentadvances in phyto-chemistry. Topics in Biochemistry ofnatural Products 13, 163 (1978).
3 . J. Mann, Secondary Metabolism, 117-128, (1987).
4. W. Herz, Recent advances in Phytochemistry, 1. 229 (1968).
5. J.H. Richards and J.B.Steroids, Terpenes and(1964-) cited Ref. 7.
Hendrickson, The Biosynthesis, ofAcetoaenins . Flew York , Benjamn
6. J.B. Hendrickson, Tetrahedron , 7, 82 (1959).
7. D.H.R. Barton and P. de Mayo, J.Chem. Soc . , 150, Quart.
Rev . Biolÿ 11 189 ( 1957 ) .
8. G.ll. Kulkarrni, G.R. Kelkar and S.C.Tetrahedron , 20, 2639 (1964).
Bha t tacharyya ,
9 . A.S. Rao, G.R.9. 274 (1960).
Kelkar and S.C. Bhattacharyya , Te trahedron ,
10. M. Suchy, V. Herout and F. Sorm,Commun. . 24, 1548 (1959).
Collection Czech. Chem.
11. T.R. Govindachari , B.S.21, 1509 (1965).
Joshi and V.N. Kamat, Tetrahedron
12. J.R. Hanson, Terpenoids and Steroids, Special PeriodicalReports, The chemical society, 7, 187-188, (1977).
13. E.E. Van Tamelen, A. Storni, F.J[. Hessler and M. Schwartz,J. Amer. Chem. Soc., 85, 3295 (1963).
14 . W . Cocker and T.B.H. Me. Murry, Te trahedron , 8, 181( 1960 ) .
5. M. Sumimoto, H. I to, H.(London), 780 (1963).
Hirai and K. Wadd , Chem. Ind.
163

16. T.W. Sam, and J.K. Sutherland, Chem. Comm., 970, (1971).
17. T.C. Jan and J.E. Me ClosVey, Tethrahedron Lett., 5139( 1972 ) .
18. A. Stoessi and J.B. Stothers, Can. J . Chem. . 64 , 1,( 1986 ) .
O
164

1
'
:
3. INTRODUCTION
;
;

3.1 Selection of Plant
"A large number of plants which were not used as medicinal
purposes have been found to contain pharmacologically
interesting compounds", this was observed by Dr. N.J. de Souza
for"Screening of Indian plantsreview ,in his
pharmacologically active substanes" [1], The above observation
of N.J. de Souza was further justified, when we see the
phytochemical report of "Prosopis Juliflora" the plant which
is not used for medicinal purposes, but the isolated alkaloids
were pharmacologically active [2-6]. In the light of above
Pluchea acguta for phyto¬
chemical studies, although no medicinal value of this plant
observations, we selected a plant
is known.
3.1.1 Botanical description of Pluchea arguta Boiss.
Pluchea argucaiÿyn. Conyza odonotophylla Boiss.) is a
member of family. Compositae, about 1000 genera of this family
are known in the world. The plants of this family are easily
identified by their characteristic capitulum inf lorecense ,
and hairy papus [7]. This genus belongs to tribe Inuleae,
and contains 50 species- distributed in the tropical and
166

subtropical regions of the world (8). Out of these, only 7
species are found in Pakistan [9] Pluchea plants are usually
shrub or under shrubs, rarely herbs, often woody below,
pubescent or tomentose.
is commonly found as shrub on thePluchea arguta Boiss,
sandy plains of Pakistan [9]. It has many branches, is
glandular, pubescent, with offensive smell and upto about
50 cm tall. The leaves are obovate-oblcng or oblanceolate ,
actually serrate or dentate, 2.5-5 cm long, 2.5-22 m broad.
The violaceous or pinkish heads are 8-12 m.m. in diameter
pappus hairs are shortly barbellate [7].
3.1.2 Medicinal Importance of Pluchea
The leaves and roots extract of Pluchea indica Less., have
been reported to possess astringent and antipyretic
and are given in decoction as a diaphoretic inproperties ,
fevers [10, 11 j. The leave juice of this plant is used ‘for
the treatment of dysentery, and the leaves are used as
constituents in slimming tea. In Indo-China the leaves and
young shoots are crushed, mixed with alcohol, and applied
to the back in cases of lumbago and also are used for
rheumatic pains and in baths to treat scabies [12]. Leaves
167

of p. indica Less, is also a remedy of leucorrhoea [10]. Fresh
leaves are used in the forms of poultices, against atonic and
gangrenous ulcers [13] . Cigarettes prepared from the chopped
Less, smoked to relieve the painP . indicastem bark of
of sinusitis [ 14 ] .
The air dried whole plant of Pluchea lanceo laca Olive.
and Hiern is used as drug. This drug had 2 primary actions.
The first one is the acetylcholine-like action the second
is the relaxant spasmolytic action on different muscles
preparations. The drug potentiated the barbiturate hyponsis
15] .of the central nervous system [10,
Olive and Hiern, areThe leaves >of Pluchea lanceo la ta
used as substitute or adulterant for Senna [10, 15, 16]. This
plant is also used in Ayurvedic system of medicine in various
chemical conditions. It is used as a bitter, laxative, an
analgesic, an antipyretic and a nerve tonic. It is used for
the treatment of rheumatism, dyspepsia and bronchitis [10].
A decoction of the plant has been reported to prevent the
swelling of joints in experimental arthritis [15].
168

Pluchea sagittalis (Lam.) Cabrera isIn folk medicine,
used due to its tonic, sour and digestive properties. Infusion
(Lam.) Cabrera have been described asof P . sag i11a 1i s
colagogue and choleretia when given to patients with liver
and gall bladder problem [17]. It is also used for the
preparation of a digestive liquor.
Pluchea odorata (L.) Cass, is known as "Shiuapata" which
'Sihua' and 'Pate') [18]. Inmeans "Woman illness" (from
central America and Caribbean, the reported emmenagogue [19-
21] and abort if acien t [22, 23] activities of P. odorata (L.)
Cass are known.
3,6 Dimethoxyf lavones isolated from Plu.chea chingogo DC.
vitro inhibitory activity against human[2] exhibited in
carcinoma cells, with 5 , 7 , 4 trihydroxy , 3 , 6-dimethoxy flavone
being the most active of the anticancer agents [24],
The whole plant of Pluchea pinnat i f id a Hook, rubbed on
to the inflammed or wounded places while stems employed as
cure against pain in bowels, anorexia and debility after fever
[11 1 .
169

3.1.3 Literature Review of Pluchea
In 1961, J,. Cardosa dp Vale, A. Fernandes Costa, M.A.
Maiae Vale. stated the phytochemical investigation of the
genus, Pluchea: They isolated catechuic acid, d-catchin and
1-epicatechin , with different metals like Fe , Al, Ca , Mg,
Mn , K and Na [ 25 ] .
Preliminary phytochemical and pharmacological studies
of Pluchea lanceolate Olive, were reported by D.N. Prasad,
K.D. Gode, P.S. Sinha and P.K. Das [26] in 1965, they also
reported the antiinflammatory activity [27] of the same genus
in 1966.
E. Dasgupta has reported the isolation of g -sitos terol ,
acetylcholine chloride and a quaternary base plucheine from
P. lanceolata Olive and Hiern [28].
F. Bohlman ' and M. Grenz isolated small amount of
sesquiterpene lactone 1 a -anageloxy-3 , 4 -epoxy-a -hydroxy-a -
cyclocos tunolide (1) and thio-phenace tylene compound (2) from
P. dioscocidis DC. [29].
170
H CH\ I
c=cocoh3C /
HHOHo''CH3H,, o"
H
'fitin «>»H
=I=0 HH‘
CH3 0
1H
(1 )0
H3ÿ ~ C = C — CH
S
(2 j
171

reported [30] cr-pinane, camphene, cincol,R. Manzi et al.
p-cymens , linalol, 1-camphor, a-tripencol, borneol, caryophy-
llene and humulene from P. sagi t ta1is ( Lam. ) in 1969.
A . X . Domingues and A. Zamudio in 1972 proved the presence
of 3-amyrin acetate and campesterol in p. odorata [31]. The
isolation and structure of cuauhtemone (3) from P. odorata
Cass was confirmed through single crystal x-ray( L. )
diffraction techniques, which showed that the compound has
trans-fused cyclohexane rings with a,B -unsaturated ketone
moeity imparting biological activity [32], K. Nakanishi et
al. isolated cuauhtemone (3) and its acetate, epoxyangelate
derivative (4) from P. odorata (L.) Cass [33]. F. Bohlmann and
C. Zedero investigated the roots of F. odorata { L. ) Cass.
and isolated, in addition to ' thiophenace ty lene compounds,
a 5- ( angeloyloxy ) carvatogeton (5), thymoherochinon- dimethyl-
other (6) and 3, 5-dimethoxy phatalsciure dimethylester , while
the aerial parts of p. odorata ( L. ) Cass, contain besides
the known cuauhtemone (3), 3 (2',3'epoxy- 2 ’ , 3 ' dihydroangeli-
cate)-4a acetoxy cuauhetemone (4), cuauhtemone- 3a- angelicate
(7) and cuauh temone- 3 a- ( 2 ' 3 ' - epoxy-2'- 3'- dihydroangelicate )
(8) [34].
172
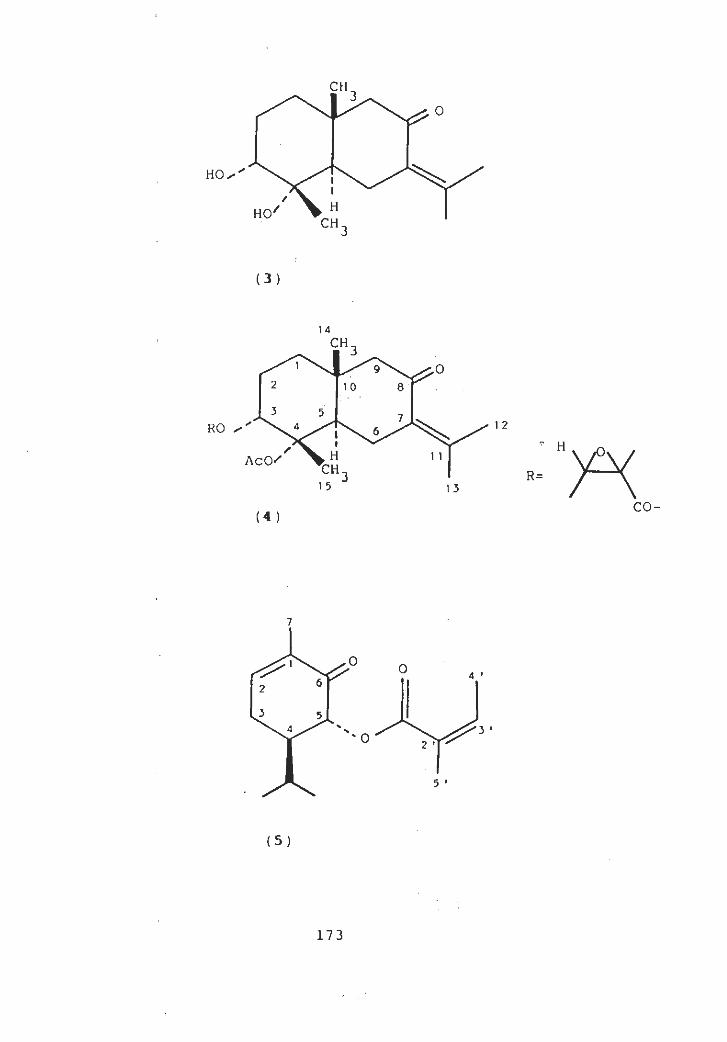
0
HO.*"l
/HHO/
CH3
(3)
14
CH3
1 09
2 1 0 8
3 5 71 2RO 4 61
t r HH 1 1AcO'
CH3 R =1 5 13
co-(4)
7
01 o4
62
3 54 3 'N
' 02 ’
5 1
(5)
173

OCH3
H3CO
Thymohydrochinondime thyle ther
(6)
0
o'RO ''' /
5HHO'* CH 3 3’
C0-(7)
SH3
o'' IRO o'zH
HOCH
3
R(8) CO-
174

The occurence of tannins, steroids, phytosterols, flavo-
noids, saponins and essential oil, pyrocatechol , resorcinol,
(Lam.) Cabrera [35] wasand pyrogallol in P. sagittalis
reported by E..C.J. Talenti in 1974. The same workers also
reported the isolation of quercitrin and quercetin in addition
to above mentioned compounds from p. sagittalis (Lam.) Cabrera
in 1976 [36]. S.V. Martino et al. reported the isolation of
a 5, 7, 3', 4 ' - te trahydroxy-3-6 , 8- trime thoxy flavone (9) from
p. sagittalis (Lam.) Cabrera and this was the first report
of this compound as a natural product. [37].
The isolation of four anticancer 3 , 6-dimethoxyf lavones
which were characterized as centaureidin (10), 5,7,4’-trihyd¬
roxy-3, 6-di-methoxy flavone (11), axilla’rin (12) and 7,4-
dihydroxy-3, 6-dimethoxy flavone (13) from the leaves and stem
of p. Chingoyo DC. was reported [38]. Among these 5,7,4’ -tri¬
hydroxy-3 , 6-dimethoxy flavone (ID was the most active
anticancer agent.
isolation ofThe eudesmanone , 4a-acetoxy-3or-( 2 1 -
methyl-2' , 3 ' epoxy-butyryloxy )-11-hydroxy-6 , 7 -dehydroxudesman-
8-one (14) together with known cuauhtemone derivatives (3,
4, 7, 8) and alkylnylthiopenes were reported by F. Bohlmann
175

OHOCH
3
H O
OH
H3CO'OCH
3
OOH
(9)
OH
HO.O
OCH3
H3C0 OCH3
OH O
Centaureidin
(10)
176

HO. 0OH
H3CO. OCH3
OH 0
(ID
•H
HO
H3CO ‘OCH 3
0OH
Axillarine
(12)
HO
OH
HO OCH 3
0
7 , 4 ' -Dhydroxy-3 , 6-dimethoxy flavone
(13)
177

and P.K. Mahanta from p- feotida (L.) DC. [39).
In 1979, -the isolation and structure elucidation of
plucheinol (15) and cuauhtemone (3), has been reported from
p. chingoyo DC., by M.T. Chiang, M. Bittner, M. Silva, W.H.
[40]. Continuing their studies theWatson and P.G. Sammes
same workers also reported the isolation pluchein (16), from
DC. 141). Isochlorogenic acidstem and leaves of p. chingoyo
which is a mixture of 3,4,-; 4,5,-; and 3 , 5-dicaf feoylquinic
isolated from P . sagittal is , (Lam.) Cabrera and thisacid was
the first report of 3 , 4 -dicaf feoylquinic acid as a singleis
produc t [42].
F. Bohlmann, J. Ziesche, M. Robert King and H. Robinson,
isolated the cuauhtemones (17-19) and eudesmanes (20, 21)
[43] .from P. suaveolens (Veil.) O. Kuntze,
A . X . Domingues , R. France, G.C. Romelia Villarreal, B.
Maringanti and F. Bohlmann reported the investigation of the
aerial parts of P. roses Godfrey, which afforded, in addition
to 3-a ( epoxyangeloxy ) -4-a -acetoxy cuauhtemone (4) three more
whicheudesmanolides 12, lac tone , 8,8, 9are
dehydroix thixoch'ilin (22) 8, 9 dehydroixthixochilin-4-O-
178
0
o'I
/ H OHAcO/
(14)
O
HO"
H</ OH
(15)
CH3
0
HO'
HOHH /
CH3
(16 )
179

:
CH3
AngO»
H/
AcO /
(17 )
CH3
0
AngOI
H/
HO /
CH3:
(18)
CH30
HO»
H/
HO /
CH3
(19)
180

CH3 o
AngOi
/ H OHAcO /
CH3
(20)
CH3
AngO’i
H/ OHHO' ‘CH3
(21)
CH30o
iO
0 "I0, / H
RO ' CH3
R=H ( 22 )
R= Ac ( 23 )
181

acetate (24)8-hydroxyixhit ixochlin-4-0-(23) andacetate
[44] .
The chemical investigation of p. sericea by V.D. Romo,
R. Alfanso and his co-workers afforded the new sesquiterpenes
(11s), -11, 13-di-hydrotessanic acid (25) in addition to
tessaric acid (26) , quercetin-3-3 ' -di-dimethyl ether (27),
and taraxas teryl-ace tate (28) [45].
In 1983, F. Bohlmann et al. reported the isolation of
lg-angeloxyeudesmanolide ,andthiopheneacetylene a
9 -a -hydroxy-a -cyclocos tunl ide (29) from the aerial parts of
146] .Pluchc'ii dioscord.ios (L.) DC.
From the leaves of P. odocata (L.) Cass. F.J. Arriaga-
Giner et al. reported the isolation of five 3a ,4a , 11
(30-33) together withtrihydroxy-6 , 7 -dehydroeudesman-8-one
the known flavones, artemetin (34) and herbacetin (35) [47].
M. Sibabrata et al. reported the isolation of 3a(2',3’-di-
from P. indicaacetoxy-2 ' -me thylbutyryl ) -cuauhtemone (36)
Less [ 48 ] .
S.V. Martino et al. reported the isolation of 5, 7, 3',4',
182

0 „' 0
0
oI0 /ÿ
HAcO "
(24 )
O
COOH
CH3(25)
0
COOH
Tessaric acid (26)
183

OCH3
OH
HO O'
OCH 3
0OH
Quercetin-3 , 3 ' -dimethylether
(27)
X
I
ss
Taraxas terylace tate
(28)
CH3 O
0OH
CH3 I
H i
b
(29) 0
184

0
AngO -/ H OHAcer
o(30)
OH/ 3
Ang =H C3
0
HMCBO-' "i
/HAcO/ OH
0
CH(31) 3
HMCB = H,C3 Cl
OH
0
EMBO-"''I
/ HH<yOH
CH3(32 )
EMB = H3C' \0185

o
HMCBO "i
/ H/
HO
,CH3(33)
HMCB = H3COH Cl
OCH_I J
OCH3OH
H3C0 o
H,CO
OCH3OH O
(34 )
H
OHOH
H3C0 O
iCHÿ
OH 0
(3b)
186

6 ,te trahydroxy-3, 6 , 8- tr ime thoxyf lavone 5,7,4’ trihydroxy-3 ,
3’ . 4 trihydroxy 3,6,7-tri-trime thoxy Elavone and 5,4 ’ ,
methoxyf lavone , isochlorogenic acid, chlorogenic acid and
cafferic acid from P. sagittalis (Lam.) Cabrera 149).
In 1983 M. Sibabrata, C.A. Geoffery R. Nijsir, R.
Supreeya, T. Payom and P.J. Hylands, isolated eudesmanolide
(37-41) alongwith thiopheneace ty lene and thiopheneacetylene
derivatives (42) from the aerial parts of P dioscoridis (L.)
DC. [48-50]. M.A . Lopez et al. reported the isolation of
a-amyrin, taraxas terol-acetate and stigmasterol from p.
odorala (L. ) Cass [51] .
E.W. Wollen et al. studied the leave resin of the above
plant collected from Tamaulipas, Maxico. It mostly consists
tciterpenes, phytosterols, and eudesmane type ofof
sesquiterpene and very small amounts of ten different
6-methoxy flavonols. A comparison with the P. odoraca
Cass from Elsalvador shows that the latter contains, a lesser
(L. )
number of triterpenes and flavonoids but more sesquiterpenes
152) .
In 1985 F.J. Arriaae and J. Borges-del-Cas t illo were
187

.0
R0
A/
HO /
\\ 1ÿ „— c — C — CH3(36) R =
OAcOAc
OH OHli
I
CH2I
H llO
0
(37)
OHi»
'
Xi
H J
IO
0(38)
X=Me.H
188

OH OAng
i i
l iO,'
H i
I0.
(39)
OH Oivali
ii
I
iiOs' H i
I0
o(40)
OH OCOCH9CH(CH,)C-H_I z J Z 0
i i
I
Io H
b
0
(41 )
189

isolated two eudesmane derivatives, plucheinol (15) and its
3ct-( ( 2' -3' - dihydroxy-2 '-methyl-derivative ,mono-ester
butryloxy ) 4a -ll-dihydroxy-6 , 7-dehydro- eudesman-8-one (43)
from P. odorata(L.) Cass. [53].
V. Martino et al. investigated the chloroform extracts
of P . sag i tt a li s (Lam.) Cabrera and isolated two methylated
flavonols, 3, 5, 7- trihydroxy-3, 4 ' , 6-trimethoxy flavone
( centaureidin ) 3', 4', 5-trihydroxy-3, 6, 7-trimethoxy flavone
( chrysosplenol D) [54].
15-hydroxy-isocos tic acid (44), the corresponding aldehyde
(45) and the eudesmanolide (46a, b) alongwith the known
compounds (29,39,40,47) including thiophenacetylene were
(L.) DC. by A. A. Darwar and M.obtained from P. discoridis
Metwally [55].
F. Bohlman et al. isolated two pairs of similar
cuauhtemone (3,7,17) and eudesmane compounds (48-52) from
P. symphy t i £ol i a (Miller) Gills. They also reported already
known, thiophenace tylenes and 5-angeloyloxy carvetogetone
(5) [56].
190
/ \3c[c—C]2 — CCHCH?! fsH OH
Thiophene acetylene derivative
(42 )
0
s
RO"/ OH
HO7
(43) 4 'O
3'
OH5'R =
COOH
CH-.OH2
(44 )
191

COOH
CHO
(45)
OH: R!I
.0
HiO
o(46)
a: R=OHb: R = OH
OH
0
(47 ) 0
192

y>
AngO T
IH
HO// OH
AngO=COC(CH )=CHCHÿ ( Z )J 3
(48)
.0
Angi
HO '' HOOH
(49 )
.0
AngiI
fHHO 7 OOH
(50)
193

0
EpangO
HAcO/ OOH
Epango=COC ( CH ) -CHCH' \ 3
(51) 0
0
AngO
H OOHAcOx
(52)
0
HO"" CH3H
HO ' OHCH3CH3
4-Epi-piucheinol (53)
194

Fizza [57] and T.A. Farooqui [58] reportedV . U . Ahmad , K .
the isolation of several eudesmane and cuauhtemone (53-67]
derivatives, alongwith known terpenes, from hexane and ether
extract of p. arguta Boiss.
195

CH3
.0
HO •CH' 3
P
HOOHz'HO CH
3
CH3Arguticin (54)
CH3
.0
H CH„ 0
I IIH3C— c c — c —O'' I CH 3hs
H3C'0 OHO
\ CH3C
\CH
(55)
CH3
PH
CH OI 3 II
H3C— C — C c_
Os' TI
V CH3r0/
H3C' COH0
\ <ÿ° CH3C
\CH
3
(56)
196
CH- 03
H
c — c __oc —
3c/1
/ OOHHH3C- CH
Pluchecin ( 57 )
CH3
CH_ 0
I3
IIH
11-jC — C — C-C — O Cll3
IH !H H/
“OOHHO
7CH
3CH3
Odont icin ( 58 )
CH3
0
H
H3C — C — C — C — O' CH3i
H//
CHOH 0
C
3CH
30= C
H3CCHÿ
Argut in ( 59 )
197

CM3
0
H CHT 01 3 1 1
C-c-C-0H3C 3f
I
V /
HO' CH h3
Pluchecinin (60)
CH3
0
, OOH
HO CH3
i/
HHO/CH
3 CH3
Odontin ( 6 J )
CH
llH CHÿ O
I I3
IIH -,C-C
3 I CH3i
/Cl
H3C'OHOH0
\c OCH
3
\CH 3
3-Ch loro-2 1 -hydroxy-argu t i cin in ( 62 )

CH3
0
CH3 °H
H3C c —c —C —o- - CH3I
IH /
H3OH OH HO 'CH
3
Ui-acetoxyargutin (63)
O
CH
i3 pH CH7 0
I IIH3( C-C-C-O- -
CH3IIH / H
H3C 'Cl OH OHOH
CH3De-acetoxy-3' -chloro-2 1 -hydroxy-argut icinin ( 64 )
CH7o
H, CH 0
ila_c.H3C-C
CH3I
i/
Oil HO / OHH30 0
CH3/HCH3
Odont inin ( 65 )
CH3
•O
HO-_
3IH / OH •OH
OH
CH3
5-a -hydroxy plucheinol (66)
CH3
OH CH O
I I 3 IIH3C-c —c —c-0 — CH 3i
iH /
H •OHOH OH H3CA OH
CH 3
Odonticinin (67)
200

;
4. PRESENT WORK

of Pluchea arguca Bioss • was colectedThe plant material
from various areas in Karachi and extracted with hexane. After
getting the hexane extract, the crushed plant material was
soaked in ethanol. The ethanol soluble fraction of Pluchea
Bioss. was extracted with ether. The ether insolublearguca
Theportion was partitioned with ethylacetate and water.
ethylacetate layer was separated and aqueous phase was further
extracted with ethylacetate. The ethylacetate portion was
evaporated in a rotary evaporator. The gummy residue was
subjected to silica gel column chromatography as described in
detail in experimental section. The column chromatography with
chloroform, ethylacetate mixtures furnished some fractions
movingcon taihedwhich closely sesquiterpenes. These
sesqui terpenic fractions were purified on high performance
liquid chromatography (HPLC) using RP-8 semi-preparative column
with methanol, water as mobile phase.
The structures of three new isolated sesquiterpenes were
proposed with the help of modern spectroscopic methods, thus
structure elucidation of individual sesquiterpene was discussed
separately in this section of thesis.
202

taraxas terol andB -Si tos terol , taraxas terol-acetate,
vanillic acid were isolated for the first time from this plant,
while plucheinol was already reported from this plant.
203

4.1 Pluchidione (I) - Isolation and Structure
The sesquiterpenes containing fraction was isolated by
silica gel column chroma tography with chlorof orm-ethylacetate
from the ethylacetate soluble portion of the crude(75:25),
ethanolic extract of Pluchea arguta Boiss. This fraction was
subjected to thin layer chromatography for final purification.
obtained as yellow gum and its purityPluchidione (I) was
was checked on HPTLC plates (Merck cat. no. 5719).
The UV spectrum showed an absorption at 236 nm indicating
the presence of a ,3 -unsaturated ketone in the molecule [59].
The IR spectrum showed absorptions at 3400 (OH), 1710 (C=0),
-11650 and 1660 ( a , 0-unsaturated ketone). The FDMScm
indicated a molecular ion peak at m/z 222, whereas HRMS
revealed that the exact mass of the molecular ion was
222.1263 consistent with the formula C The formula13H18°3‘
indicates five double bond equivalents in the molecule, two
of which are accounted for by carbonyl groups, two by the
double bonds and one by the ring of the skeleton.
The terpenoidal nature of (I) was indicated by the presence
of four methyl singlets in the '*’H-NMR spectrum, each
204

I
I 1I 0o\
OH\ 7
o 19
2 6 1 38
3 5
41 2
Pluchidione (I )
205

5 1.09, 1.10, 1.80 and 2.28. These areintegrating for 3H at
C-10, C-11 , C-12 and C-13 respectively.due to protons at
The chemical shift of the last methyl mdicated the presence
6 1.80The methyl group which resonated atof a COCH3 group.
= 0.9 Hz) was attached to an unsaturated carbon. A( J12,4
double doublet at 6 2.34 (J = 17 Hz, = 1-14 Hz) is
due to H-38 and another double doublet ascribed to H-3a was
gem
= 1.04 Hz ) . The6 2.46 ( J = 17 Hz, J3a ,4
= 14.7 Hz) are due to
observed at gem
6 6.40 and 6 6.80 (Jdoublets at 7 , 8
protons at C-8 and C-7 respectively coupled to each other;
their trans olefinic coupling constant also clearly indicated
the presence of the double bond outside the ring.
The multiplicities of the proton signals were determined
through a 2D- J -resolved spectrum and vicinal coupling was
confirmed by COSY-45. A strong cross peak was observed at
6 2.34 and 6 5.90. Beside, this another cross peak at 5 2.46
6 5.90 showing the coupling interaction of H-3 a, H-3$
protons with that of C-4 proton. A corresponding cross peak
and
of H-3 a and H-3 8 protons with each other was also observed
in the COSY-45 spectrum, which also showed a cross peak at
6 6.40 and 6 6.80. They are due to vicinal protons in the
skeleton of (I) The NOESY spectrum also confirmed structure
206

(I) showing spatial connectivities of H-ll with H-7, H-3 ,
CH-j-13 with H-8 , CH3-12 with H-4 .
The CD curve in ethanol shows a cotton effect, with a
positive maximum around 243 nm and a negative maximum at 210
nm. due to the n -IT transitions of the a, 8-unsaturated ketone.
In addition, there are weaker negative maxima at 318 ascribed
*to the unconjugated ketone (g-n ) and 340 nm due to the enone
( n— TT ). The positive maximum at 242.8 ( Ae 14.6) corresponds
in the position and intensity to that of +a - ionone [60] which
has R configuration at C-6. It is therefore concluded that the
pluchid ione (I ) has the same i.e. R configuration at C-6.
1 3C-NMR spectrum (CDC13, 100 MHz) r showed the presence
of thirteen carbon atoms in the molecule. The multiplicity
The
the pulse sequence with the
last polarization pulse angle 0 = 45°, 90° and 135°. It showed
assignments were made by DEPT.,
6 18.69, 22.97, 24.37 and 28.37presence of four methyls at
CH T -13which were assigned to CH3-10, CH3-11, CH3~12 and
respectively. There was only one methylene at 6 49.61 due to
3
207

127.80, 130.38 andC-3. Three methine carbon signals at 6
144.98 are due to C-4 , C-8 and C-7 respectively which also
there must be two double bonds in the skeletonsuggested that
of ( I) . Five quaternary carbon signals at 5 41.47, 79.30,
160.31, 196.90 and 197.30 are ascribed to C-l, C-6 , C-5 , C-2
and C-9. The last two values indicated the presence of a
carbonyl function while the value of C-6 indicated an oxygen
C-NMR chemical shifts
1 13two dimensional H- C chemicl
13bearing carbon atom. The assignments of
were also confirmed through a
which showedshift experimentcorrela t ion ( Heterocosy )
connectivities of C-ll ( 6 22.97) with H-ll ( <5 1.10), C-12 ( 6
24.37) with H -12 (6 1.80), C-4 ( 6 127.80 ) with H-4 ( 6 5.90),
C-3 ( 6 49.61 ) with H-3a ( 6 2.46) and H-3 8 ( 6 2.34), C-10 (6
18.69) with H-10 (6 1.09), C-8 (6 130.38) with H-8 ( <5 6.40),
C-7 (6 144.98) with H-7 ( 6 6.80) and C-13 (6 28.37) with H-13
( <5 2.28 ).
208

TABLE-1
’•'C-NMR chemical shifts for pluchidionc (I) (CDCI3, 100 MHz)
w ;iiSiillS
41.47 7 144.981
197.3 8 130.382
49.6 93 196.9
4 127.8 10 18.69
5 160.3 22.97
79.36 12 24.37
13 28.37
The status of each carbon confirmed through DEPT experiment.
209

4.2 Pluchidinol (II) - Isolation and Structure
The compound pluchidinol (II) was isolated from the
sesquiterpenic fraction, obtained from silica gel column,
when the mobile phase was chloroform-e thy lace ta te (75:25).
Purification was carried out by repeated flash chromatography
on silica gel. . The pure compound (II) was collected in the
form of colourless gum.
UV spectrum showed an absorption maximum at 233 nm.Its
which indicated the presence of a, B-unsaturated ketone (59).
The 1R spectrum contains peaks at 3200-3600 (OH) and 1674
-1 ( a , B -unsa t ura ted ketone). The fast atom1645and cm
bombardment (FAB) mass spectrum (positive ion mode) of (II)
a ( M+H )+
contains peak at m/z 549 corresponding to the
In the El mass spectrum, the M+molecular formula C
31'*48°8 *
peak was not observed, the peak at m/z 268 which appeared
after the breaking of the dimer into the monomeric form. The
rest of the important peaks at m/z 235 (base peak) 217, 193,
149 and 123 which are identical to those reported earlier175 ,
40 ) .
spectrum (400 MHz)The very similar to thatwas
210

H,C C,H3/OH7
H OH
»HO
H3CH
T.oi
CH2 CH3
H3CS.
OHiHO, H /
H3C'CH OH3
Pluchid.ionol (II)
211

[61]. The compound (II) showed methylof 4 -epiplucheinol
6H , at, 6 0.94 (C-14 andsinglets, each intergrating for
6 1.18 (methyls at C-4 and . C-4 ' ) , 6 1.42 ( C-12 andC-14 * ) ,
<51.44 (C-13 and C-131 ). There is a narrow triplet
at <5 3.68 (J = 2.70 Hz) characteristic of the protons geminal
C-1 2 1 ) and
to hydroxy group at C-3 and C-3'. A doublet at 67.03 (J 5,.6
= 2.30 Hz) is assigned to the olefinic protons at C-6& 3 ' 6 ’
and C-6'. There was an AB quartet centered at 6 2.30 ppm (
- 18.5 Hz due to H-9'). A multiplet at 615.7 Hz, 5 A-6 B
1.80 was assigned to C-2 and C-2' protons. Besides these
signals , there was a multiplet at 6 1.25 (2H, m, H-16) which
was not present in 4-epiplucheinol (53) [61].
Two dimensional NMR measurements were carried out to
/erify the 1H-NMR assignment. The coupling interactions were
istablished through correlated spectroscopy (COSY-45) while
:he multiplicity of the overlapping proton signals was
letermined from the 2D-J-resolved spectrum. The assignment
:or C-2 and C-21’ protons at 6 1.80 could thus be confirmed
its COSY-45 spectrum, which showed a strong cross peak>Y
6 1.25 (H-16), 1.80 (H-2, H-2‘), 3.68 (H-3, H-3').it
imilarly the assignments of H-5, H-5' at 6 2.68 and H-6,
-6’ at 6 7.03 were also confirmed by COSY-45 spectrum since
212

they showed interaction with each other.
13C-NMR spectra of (II) were veryThe broad band and DEFT
useful in elucidating the structure of the compound, which
showed the presence of eight methyls, seven methylene, six
methine and ten quaternary carbon in the structure of (II).
C-2 ' isThe chemical shift and multiplicity of C-2 and
effected because in pluchidinol (II) they were methines at
6 50.86 (in the case of 4 -epiplucheinol (53) C-2 was a
methylene) attached to the C-16 methylene which linked the
two monomers of the same stereochemistry, whereas it was
absent in 4-epiplucheinol (53). This extra methylene i.e.
C-16 appeared at <5 29.73.
Heterocosy experiments were carried out to identify the
relationship between the carbons and their respective protons.
6 29.73 showed a cross peak with protonsThe C-16 signal at
at 6 1.25. Similarly the C-2 and C-2’ at 6 50.86 showed a
6 1.80.cross peak with It also confirmed the assignments
of other protons with their respective carbon atoms. The
structure of (II) was also confirmed by the acetylation of
the compound (II). The E1MS of the acetylated derivative
showed peak at m/z 799 [M -1)+,
absorption peaks were at 1735, 1300, 1050 cm~ÿ confirmes (II)
where as in IR spectra,
as a dimeric compound.
213

TABLE-II
13C-NMR chemical shifts for pluchidinol (II) (CDCI3, 100 MIIz)
Carbon
9,9’ 57.831.981, r
50.86 10, 10’ 39.252,2’
3,3’ 73.52 11, 11’ 71.99
4,4’ 72.1 12, 12’ 29.36
5,5' 18.9 13, 13’ 28.90
143.26, 6’ 14, 14’ 17.77
7,7' 145.0 15, 15’ 22.70
8,8’ 201.3 16 29.73
The status of each carbon confirmed through DEPT experiment.
214

4.3 ll,12,13-trinor-3,4-diepicuauhtenione (III) - Isolation and structure
of the crude ethanolicThe ethylacoiale soluble portion
subjected to column chromatography on silica gelextract was
and the mobile phase at the time of elution was chloroform-
ethylacetate (30:70) furnished the sesquiterpenic mixture.
The sesquiterpenic fraction was rechromatographed on HPLC
using RP8 semi-preparative column with methanol-water (70:30)
as mobile phase. Pure 11, 12, 13-trinor-3, 4-diepicuauhtemone
(III) was obtained in crystalline form. The purity of the
compound (III) was checked on HPT'LC plate and the developing
solvent was for chloroform-methanol (90:10).
Its UV spectrum showed only end absorption at 208 nm.
indicating the absence of an a , 8 -unsaturated ketone or, a, 8.8 ~
trisubs ti tuted ketonic group in the molecule.
In 1R spectrum important peaks were observed at 3400 cm *
showing the presence of hydroxyl group, whereas the presence
of ketonic group was confirmed by the absorption peak at 1700
. Besides, the IR spectrum also indicated that (III) was-1cm
different from other reported eudesmane [33, 34, 39, 40, 43,
47, 48, 53, 56] compounds with respect to the absence of
215

1 4
CH3I 9 .0
2 1 0 8
73 5
4 6. HOI
H/
H3.15 OH
11 , 12 ,13-Trinor -3 , 4 -diepicuauhtemone (III)
216

a, S-unsa t ura t ion or an a, a, &-trisubs tituted ketone group.
The HRMS showed ( M )+
at m/z 212.141 corresponding to the
molecular formula C12H20C->, and suggesting a trinor-sesquiter-
spectrum (CDClÿ,
400 MHz) showed only two methyl signals instead of four. They
appeared at' 3 0.87 and 1.12 and correspond to Me-14 and Me-15
pene character for the molecule. The
13C-NMR DEPT experiment also showed therespectively. A
signals for two methyl groups instead of four. These findings
confirmed the trinor- sesquiterpene skeleton of the molecule.
6 2.20A multiplet, centred at was due to the methylene
protons of C-9. A doublet of doublets resonated at 5 2.0 (J
= 3.5, 9.3 Hz) was assigned to H-5a . H-3a appeared as a
5 3.56 (J = 3.04, 12 Hz) suggestingdoublet of doublets at
the J -orientation of the OH at C-3. The chemical shift of
15-Me at high field (1.12) suggested the cr -orien tation of
15-Me at C-4 and B-orientat ion of the OH group [61).
The multiplicities of the proton signals were determined
2D-J-resolvedthrough while the couplingspectrum,
interactions were established by COSY-45 spectrum. A strong
6 1.82 and 3.56, showing thecross peak was observed at
217

13c_Thecoupling interactions between C-2 and C-3 protons.
NMR spectrum (CDCl-ÿ, 100 MHz) indicated the presence of two
6 19.01 and 21.63 which were assigned to
Me-14 and Me-15 respectively. The signals at
methyl signals at
5 59.63, 47.50
and 39.50 corresponded to C-9, C-5 and C-10 respectively on
the basis of their chemical shifts and multiplicities, while
the assignments of the remaining methylene groups at <5 34.53
( C — 1 ) , 26.88 (C-2) and 22.9 ( C-6 ) were made by comparison with
data reported for the same carbons in cuauhtemone [33]. There
is one more signal for a methylene carbon in the DEPT spectrum
at 6 42.58 which was assigned to C-7 . The absence of a C-ll
signal
1tr inor-eudesmane character of the molecule. The
in broad bandthe confirmed thespectrum
C chemical
shift assignments were confirmed by hetero-.cosy spectrum which
showed connectivities of C-l (6 34.53) with H-l (6 1.84),
C-2 (6 26.88) with H-2 (5 1.82), C-3 .6 73.87 ) with H-3 (6
3.56), C-5 ( 6 47.50) with H-5 (5 2.0), C-6 (6 22.99 ) with
H-6 ( 5 2.30), C-7 ( 6 42.58 ) with H-7 ( 6 2.4), C-9 (6 59.63 )
with H-9 (6 2.2), C-14 (6 19.01) with H-14 ( 6 0.87) and C-
15 ( 6 21.63) With H-15 (6 1.12).
218

TABLE-III
’ÿ'C-NMR chemical shifls for II, 12, 13-trinor-3-4-diepcuauhlemone (III)(CDCI3, 100 MHz)
Carbon; mppm
1 34.53 7 42.58
2 26.88 8 214.32
3 75.16 9 59.63
4 73.87 10 39.50
5 47.50 14 19.01
6 22.99 15 21.63
status of each carbon confirmed through DEPT experiment.
219

'
5. EXPERIMENTAL
i

5.1 General Notes
The column chromatography was perfomed on silica gel 60
(70-230 mesh, E. Merck), while for thin layer chromato-
grapghy ( TLC ) silica gel F pre-coated aluminium cards254
(Riedel de Haen) was used. For confirming the purity of the
samples, high performance thin layer chromatography ( HPTLC )
precoated glass plates used forsilica gel 60, F254chromatographic purposes were mostly analytical grade of
the firm E.. Merck.
Flash column chromatography was performed on Eyela Model
EF-10, using, Silica gel 60, 230-400 mesh size (E. Merck).
Analytical arid preparative HPLC were carried out on JASCO
RP-8 column, with the help ofMode 1 havingVI,-b 1 4
UV1DEC100-11 detector.
The mass spectra were measured on Varian MAT-112 and
MAT-312 spectrometer connected to MAT-188 data system and
POP 11/34 computer system. Optical rotations were measured
on polartronic-D polarimeter.
221

All the melting points were determined on a Gallenkamp
melting point apparatus and are uncorrected. The Ultra Violet
(UV) spectra were scanned on shimadzu UV-240 spectro¬
photometer and the Infra Red (IR) spectra were recorded on
JASCO A-302 spec trophotome terr . Nuclear Magnetic Resonance
spectra ( 1H and 13C) were recorded in CDClÿ on Bruker Aspect
AM 400 and AM 300 spectrometers operating at 400 and 300 MHz
for H and 100 and 75.43 MHz for C nuclei respectively.
Chemical shifts are reported in ppm (6;.
The sesquiterpenes were detected on TLC plate with the
help of UV lamp (254 nm. ) , and by spraying Ehrlich reagent.
When TLC plate was heated at 100°C most of them gave purplish
pink and violet colour.
Ehrlich reagent 25 ml 36% HC1 +ÿ 75 ml, MeOH + one gm of
4 - ( dime thy1amino ) benzaldehyde [63].
222

5.2 Plant Material
The fresh plant material of Pluchea arguta Boiss. was
collected from, Karachi region in the month of August. The
identification and authentication of plant material was
carried out by taxonomist of Botany Department, University
of Karachi.
5.3 Method of Extraction and Isolation
The fresh plant material of Pluchea arguta Boiss (20kg)
soaked in hexane and homogenized with an Ultra-was crushed,
Turrax homogenizer and kept at room temperature for about
15 days. After, removal of hexane extract (tw_t.ce) from the
homogenized material of P arguta, the plant was soaked
in distilled ethanol and kept for 15 days at room temperature.
The ethanolic extract was taken and the solvent was removed
in vacuo. The residue obtained was extracted with ether.
The hexane and ether fractions were already investigated
by Miss Kaniz Fizza and Mr. Tanveer Ahmed Farooqui [57,
58). The ether insoluble portion was partitioned with ethyl-
acetate and water. The ethylacetate layer was separated
and aqueous phase was further extracted with ethylacetate.
223

The ethylacetate soluble portion was evaporated in the rotary
chromatographed( 2 0 gm )evaporator. The gummy residue was
on a large silica gel column and eluted with hexane, hexane-
chli/.jform mixtures, ethyl - acetate, ethylacetate-methanol
mixtures and finally with pure methanol. The non polar
fractions contained mostly known triterpenes and sesqui¬
terpenes some’ of which were already reported in literature.
5.3.1 Compounds reported for the first time from Pluchea arguta Boiss.
5-3.1.1 /S-Sitosterol
The fractions obtained from hexane-chloroform (98:2)
containing 8-sitosterol were collected, concentrated in
vacuum, taken in methanol, and kept at room temperature.
The colourless micro-crystals were separated out, melted
at 137°C and identified as 8-sitosterol through comparison
of its physical and spectral data with those of the authentic
sample (63, 64 ] .
224

5-3.1.2 Taraxasterol,-acetate
(96:4) andSubsequent elution with hexane-chloroform
crystallization with methanol gave colourless crystals,
m.p. 247°, which were characterized as taraxas terol-acetate
through comparison of its spectral data and mixed melting
point with authentic sample (65-67 ].
5-3.1-3 Taraxasterol
The hexane-chlorof orni ( 16:84 ) eluted fractions were
combined, concentrated and taken in methanol. When kept
overnight at room temperature, afforded pure crystalline
taraxasterol melted at 219°C which was identified through
direct comparison with an authentic sample and also through
comparison of sepctral data [67].
5.3.1.4 Vanillic acid
It was also isolated for the first time from this source.
"he fractions eluted from silica gel column, with chloroform-
ithylacetate ( 80:20 )were further purified through repeated
column chromatography. Vanillic acid was obtained form these
225

210°C. It identified byfractions and melted at was
69] as well as by direct comparisonspectroscopic means [68,
with an aunthentic sample.
53.2 Compound already reported from Pluchca arguta Boiss.
53.2.1 Plucheinol (15)
The sesquiterpenic mixture obtained from ethylacetate
soluble portion was chromatographed by chloroform-ethylacetate
on silica gel column, impure plucheinol (15) was( 90:10 )
collected. Final purification was done by repeated flash
chromatography by using chloroform-methanol (95:5) as mobile
phase. Plucheinol was crystallized from methanol (m.p.
87-88°). In order to identify the plucheinol (15) its spectral
data were compared with the reported data [40, 53]. The
compound was also identified by direct comparison with an
authentic sample.
5.4 Crude Sesquiterpenic Mixture
fractions, which were obtained with the mixtureThe
of chloroform-ethylacetate, gave positive test for sesquiter-
226

pene by Ehrlich's reagent. These fractions were separately
chromatographed by flash column chromatography on silica
gel, HPLC using RP-8 Reversed Phase Column, and by thin layer
chromatography on silica gel. The isolation process of
individual sesquiterpene is mentioned separately on next
pages .
227

5.4.1 Isolation of Pluchidione (I)
Silica gel column chromatography of ethylacetate soluble
(75:25) afforded aportion with chlorof orm-ethylacetate
mixture of closely moving sesquiterpenes. The sesquiterpenic
mixture was subjected to flash column chromatography using
chloroform-methanol (96:4) as eluant furnished pluchidione
with some impurities which was further purified through
preprative thin layer chromatography on silica gel plates.
Pure pluchidione (I) obtained as light yellow gum, which
was soluble in chloroform as well as in methanol. The purity
of pluchidione was confirmed on HPTLC plate (chloroform-
methanol, 85:15). Field desorption mass spectrum showed
molecular ion at m/z 222. The molecular formula attributed
for the compound was £ÿ3ÿ18ÿ3
5.4.1.1 Characterization of Pluchidione (\)
UV ( MeOH ) : X 236 nm.;max
3400 (OH), 1720 (C=0), 1680 (C=0), 1360IR (CHCI3):
(C-O) cm--*-.v max '
[ a ] D= +23.25 ( c = 0 .04 3 , CHCl-j).
228

HRMS: m/z 222.1263 [M] +, <Ci3Hi803,
189.0918 (ci2H13°2' caÿ-ccl* f°r 189.0915 ) and 124.0575 (C-yHgCÿ*
calcd. for 124.0524).
for 222.1255),calcd.
FDMS: m/z 222.
CD: (0.03 gm/1 EtOH), [0 1 340-1918 , [0)3i8 -2730, [9]
-44400 .242.8
+ 48174, 10 1 2 25 ~° and
H-NMR : ( 400-MHz , CDC13), 6 1.09 ( 3H ,
J12 ,4
2101
H-10 ) , 1.10 ( 3H ,s ,
= 0.9 Hz, H-12), 2.28 (3H,H-l1 ) , 1.80 (3H, d,s ,
H-l 3 ) , 2.34 ( 1H, dd, J = 1.14 Hz H-30 ) ,= 17 Hz, J3 8' 4
= 1.04 Hz, H-3 a ) , 5.90 (lH,m,
s ,gem
' J3a,4
,8 =
2.46 (1H, dd, = 17JgemH-6 ) , 6.40 (1H, d, J?
= 14.7 Hz, H-7 ) .14.7 Hz, H-8 ) and 6.80 (1H, d,
J7 , 8
C-NMR: Table-I13
229

5.4.2 Isolation of Pluchidinol (II)
The crude ethylacetate soluble portion, after evaporation
in vacuo, subjected to silica gel column chromatography,
using solvent gradients of increasing polarity as mentioned
in method Of extraction and isolation. Chlorof orm-e thyl-
acetate (75:25) eluent afforded a fraction of sesquiterpenic
mixture. The mixture was subjected to repeated flash column
chromatography with chloroform-methanol (96:4) as eluent,
the last few fractions were furnished pluchidinol (II).
The purity of the compound was checked on silica gel
plates and ' HPTLC plate using chloroform-methanol (95:5)
as mobile phase. The pure pluchidinol was isolated as a
colourless gum and was soluble in chloroform as well as
in methanol. 1 The fast atom bombardment mass spectrum showed
[ M+H ]f
at m/z 549 corresponding the molecular formula
C31H4 7°8’
5.4.2.1 Characterization of Pluchidinol (II)
UV (MeOH): X 204 and 233 nm.max
3200-3600 (OH), 1710 (C =0), 1674 and 1645IR (CHC13): vmax
230

(a,8 unsaturated ketone) cm .+ 74.07° ( c= 0 .0 0 6 7 , CHC13 ) •[a ]D=
FAB + ve: m/z 549 [M+H]+
EIMS: m/z ( rel. int. %), 268 (10), 253 (12 ), 235 ( 100),
217 (12), 193 (14), 175 (10), 149 (74), 132 (12), 123 (16),
109 (22), 95 (14), 83 (14), 77 (12) and 55 (12).
1H-NMR: (400 MHz, CDC13) 6 0.94 (6H, s, H-14, 14'), 1.18
( 6H , s, H-15, 15'). 1-25 (2H, m, H-16), 1.42 (6H, s, H-12,
12’). 1-44 ( 6H , s, H-13 , 13'), 1.80 (2H, m, H-2, 2’), 2.30
= 18.5 Hz, H-9, 9' ) , 2.68 (2H,3 = 15.7 Hz, 6 a_6 Q( 4 H , ABq'
d, 3.68 ( 2H , t, J = 2.7J5 , 6 &5 1 ,6 ’
Hz, H-3 , 3’) and 7.03 (2H, d,
= 2.10 Hz, H-5 , 5’ ) ,
= 2.30 Hz, H-6 ,J5,6S.5' ,6'
6’ ) .13C-NMR: Table-11
Acetylation of (II)
A solution of (II) ( 8mg ) in dry chloroform (3.5 ml)
was cooled. Ace tylchloride ( li.il ) and pyridine (0.75ml) was
added to it and the mixture was allowed to stand overnight
at 0°. Ice was added and the reaction mixture was worked
up in the usual manner. The residue from chloroform extract
was collected [70].
231

UV (MeOH): 254 and 204 nm.
IR ( CHC13 ) : v
D = +76.2 ( c=0 .013, CHCJ3) .
E IMS: m/z, 799 [M-l]+, 537, 269 and 253.
1735 (C=0), 1300 and 1050 (-C0-0) cm-1.max
232

5.4.3 Isolation of 11,12,13-trinor-3,4-diepicuauhtemone (III)
from chloroform-ethylacetatefractions obtainedThe
(30:70) were' combined, and rechromatographed on a small
column (silica gel) using chloroform-ethylacetate mixtures
withru order of increasing polarity as eluent. Elution
whichchloroform-ethylacetate , furnished sample PAE-1 ,
indicated the major sesquiterpene with some streaking
impurities on silica gel plate. The 11, 12, 13- trinor-3, 4-
with RP-8purif ied HPLCdiep i.cuauhtemo.pe onwas
(70:30) assemi-preparative column using methanol-water
mobile phase. The isolated 11, 12, 13-trinor-3, 4-diepicuauhte-
mone (III) was soluble in chloroform as well as in methanol.
The purity of the compound (III) was ch.ecked on HPTLC
plate using chloroform-methanol (9o:10) as the developing
solvent. This compound was obtained as colourless crystals
and molecular formula of the compound was established by
HRMS at rn/z 212.1410 c12H20O3-
5.4.3.1 Characterization of 11,12,13-trinor-3,4-diepicuauhtemone fill)
UV ( MeOH ) : X 208 nm.max '
233

I
3400 (OH) and 1700 (C=0) cm"1.
14 2.85 ( c = 0 .014 , CHCl-j ) -HRMS: m/z (rel. int.%) 212.1410 (M]+
IR ( CHC13 ) : Amax ’
1 0 1 D '
( C12H20°3 'calcd .
(3), 194.130 (C12H1802, calcd. for 194.130 (14),212.1410)
167 .107 ( C10H15°2 'calcd. for 167.107) ( 14.1) , 153.091
153.099) (70) and 111.081 (C7H11O,calcd. for( CgH 4O2 >
calcd for 111.080 (20).
FDMS: m/z 212.
LH-NMR: (400 MHz, CDCl-j), 6 0.87 (3H,
H-15), 1.82 (2H, m, H-2 ) , 1.84 (2H,
H-14 ) , 1.12 (3H,s ,
H-l ) , 2.0 (1H,m,i ,
Id, J5 a, 6 a =
-6), 2.4 ( 2H ,
3.5 Hz, 9.3 Hz, H-5a ) , 2.30 (2H,J5a,68 =
H-7 ) and 3.56 (1H,
m,
= 3.04dd, J2B,3am,
J3 a, 2a = 12 Hz , H-3a ) .z ,
3C-NMR: Table-Ill
r%!
11t.
234

6. REFERENCES

de Souza, Manager and Head, Chemistry ResearchDr .Center, Hoechst Pharmaceuticals Ltd., Bombay 400-080.
N. J.1.
2. K . A. Khan, V.U. Ahmad, A. Faruqi, S. Qazi, S.A. Rasuland T.S. Harocn . Arznemittel - Forsching (Drug Research)
36(1), 17 ( 1986 ) . '
3. A. Ahmed, K.A. Khan, V.U. Ahmad and S. Qazi, Planta Med.,247, (1986).
A. Ahmed, K.A. Khan, V.U. Ahmad and S. Qazi, Fitoterapia,59(6), 481 (1988).
4 .
5 . A. Ahmad, K.A. Khan, V.U. Ahmad and S. Qazi, Arzneimettel- Forchung (Drug Research), 39(1) No. 6, 652 (1989).
A. Ahmad, K.A. Khan, s. Qazi and V.U. Ahmad, Fitoterapia,60(1), 85 (1989).
6.
7 . S.M.H. Jafri, Flora of Karachi, 335 (1966).
8. N. Ruangrungs , P. Tantivatana, R.P. Borris and G.A.Cordel. J. 5ci. Soc. Thailand, 1, 123 (1981).
9. R.R. Stewart, “Flora of West Pakistan". (E. Nasir andS.I. Ali, Editors), Fakhri Printing Press Karachi, p.768 (1972).
10. R.N. Chopra, I.C. Chopra, K.K.Indigenous drugs of India, 2nd End. U.N.Ltd. Calcutta, India, p.520 (1958). K.R.B.D. Basu, Indian Medicinal Plants (L.M. Basu, Allahabad,India), 1, 679 (1918).
Handa and L.D. Kapur,Dhur and SonsKirtikar and
11. R.N. Chopra, S.L. Nayar and I.C. Chopra,Indian Medicinal PlantsJ , 197, (1956).
Glossary of
12. L.M. Perry, Medicinal Plants of East and South East AsiaMIT Press, Cambridge, M.A. P.96 , ( 1980).
13. B.M. Sa’stri, The Wealth of India, A Dictionary of IndianRaw Materials and Industrial Products, VIII, CSIR, Delhi,India (1948).
236

14. S. Pongboonrod, Maiter Meang Thai, Kasembunakij Press,Bangkok, 117 (1971).
15. S.A. Krishnamurthi , The Wealth of India, Publicationsand Information Directorate CSIR. New Delhi, editorVol. VIII, 161 (1969).
I (> . W. Dymock, Pharmucographia Indica, 255, (1972).
17. C.B. Udaondo, G.P. Gonalons, A.R. Basile, H. Zunino, andJ.J. Lacour, Bol. Acad. Nac. Med. Buenos Aires, 20, 441(1937).
18. F.J. Arriaga, J.B. Castilo, M.T. Manersa, P. Vazquez andS.V. Iraheta, Phytochemistry , 23(5), 1767 (1983).
19. N. f Farama Copea Maya, Imprenta "GamboaSouza-Novelo ,Guzman" , Medica Yucatan , Mexico ( 194U ) .
20. I. Rivera, An. Inst. Biol. Mex , 14, 38 (1943).
21. J. Eerhault, Flor Illustree de Senegal 11 Govt. Sengal,Ministry of Rural Development, Water and Forest division,Dakar (1974).
22. E.S. Ayensu, Medicinal Plants of West Indies, unpublishedmanuscript. Ref. 18-21 were cited in ref. 46.
23. M. steggerda and B. Korsch, Bull. Hist. Med., 13, 54(1943).
24. M.T. Chiang and M. Silva, Rev. Latinoam Quim, 9(2), 102(1978).
25. J. Cardosa do Vale, A. Fernandes Costa, M.A. Maiae Vale,Garcia Orta, 9(3), 491-9 (1961).
26. D.N. Prasad, K.D. Gode, P.S. Sinha and P.K. Das, IndianJ . Med. Res . , 53(11), 1062-8 (1965). (Eng).
27. D.N. Prasad, S.K. Bhattacharya and P.K. Das, Indian J.Med. Res. 54(6) 582-90 (1966). (Eng).
28. B. Dasgupta, Experientia, 23(12), 989 (1967).
237

Lett.. 58 , 511 ;29. F. Bohlmann and M. Grenz, Tetrahedron( 1969).
30. R. Manzi, F.A. Tedon, E. Arinogoli, and R.A. Yunes, Rev.Fac. ing. Quim., Univ. Nac. Litoral, 38, 251 (1969).
11, 117931. A.X. Dominguez and A. Zamudio, Phytochemistry,( 1972) . \
932. R. Ivie and W.H. Watson, Acta Crystallogr. Sect B, B30
(12), 2891 ( 1974 ) .33. K. Nakanishi, R. Crouch, 1. Miura, X. Dominguez, A.
Zamudio, and R. Villarreal. J. Amer. Chem. Soc. , 96(2),
609 (1974).
34. F. Bohlmann and C. Zedro. Chemi. Ber . , 109. 2653 (1976).:
Int. Congr. Essential Oils [pap],35. E.C.J.6, 89, 22 (1974) (Span).
Talent i ,
I
46(1), 40i 6 . E.C.J. Talenti ,( 1976 ) .
Essenze Deriv. Agrum ,
17. S.V. Martino, E.G. Ferraro and D.J. Coussio, Phytochemis¬try, 15(6), 1086 (1976).
8. M.T. Chiang and M. Silva, Rev. Latinoam Quim, 9(2), 102( 1978 ) .
9. F. Bohlmann and P.K. Mahanto, Phytochemistry, 17(7), 1189(1978).
0. M.T. Chiang, M. Bittner, M. Silva, W.H. Watson and P.G.Sammes , Phytochemistry , 18(12), 2033 (1979).
L. M.T. Chiang, M. Bittner, M. Silva and W.H. Watson, Rev.Latinoam Quim, 10(2), 95 (1979).
'. S.V. Martino, L,.S. Debenedetti and D.J.Phytochemis try, 18(12), 2052 (1979).
Coussio
F. Bohlmann, J. Ziiesche, M.R. King and S. Robinson,Phytochemis try , 19(5), 696 ( 1980).
238

44. A. X. Dominguez, R. Franco, G. Cano, R. Viuorrel, M. Bapuji
and F. .Bohlmann, Phytochemistry, 20(9), 2297 (1981).
45. V.D. Romo, R. Alfanso, D. Benito, S. Guillermo and 0.Elmer, Chem. Lett.. (7), 957 (1982).
46. A. A. Omar, M.T. Sarg , M.S. Khafagy, E.Y. Ibrahim, C.Zdergo and F. Bohlmann, Phytochemistry. 22(3), 779 (1983).
47. F.J. Arriagag-Giner , J. Borges-Del-Castillo , M.T. Manresa-Ferrero, P. Vazques-Bueno , F. Radriguez and S.v. Iraheta,
Phy.tochemis t r_y , 22(8), 1767 (1983).
48. M. Sibabrata, C.A. Geoffery, R. Nijsir, R. Supreeya, T.Payom, and P.J. Hylands, J . Nat. Products, 46(5), 671( 1983) ..
49. S.V. Martino, E.G. Ferraro, L.S. Debendett, and D.J.Coussio, Acta Farm. Bonaerense, 3(2), 141 (1984).
Jakupovic ,50. F. Bohlmann , Metwally andM.A.Phytochemis try , 23(9), 1975 (1984).
J .
51. M.A. Lopez, F.J. Arriage-Giner , J. Boges-del-Cas tillo,and P. Vazquez-Bueno , Fitoterapia, 56(2), 123 (1985).
52. E.W. Wollen, K. Mann, F.J. Arriaga, G.E. Yatski,Naturforsch. C. Boi. Sci. . 40C (56), 321 (1985).
53. F.J. Arriaga and J. Borges-del-Cas tillo, Planta Med. (3),290 (1985).
54. S.V. Martino, F.G. Ferraro, L.S. Debendetti and D.J.Coussio, An. Asoc. Quim, Argent, 73(4), 401 (1985 (Span).
55. A. A. Dawar and M.M. Wally, Chem. Pharm. Bull., 33(11),5068 (1985).
56. J. Jakupovic, L.N. Misso, T.V. Chau, Thi, F. Bohlmannand V. Castro, Phytochemistry, 24(12), 3053 (1985).
57. K. Fizza, "Studies on Chemical Constituents of PlucheaArguta", H . E. J . Research Institute of Chemistry Universityof Karachi April (1987).
239

studies on the sesquiterpenesH.L.J. Research Institute of
58. T.A. Farooqui, "Furtherof Piuchea Arguta Boiss" ,
Chemistry, University of Karachi (1988).
59. A.l. Scott', The Ultraviolet Spectra of Natural Products,
7, 56 (1964)
60. O. Gunther, 0. Eberhard, R. Valentin and S. Sunther, Helv .Chem. Acta, 56, Fase 6, 1874 (1973).
61. V.U. Ahmad’’ ad K. Fizza, Phytochemistry, 25(4), 949,( 1986 ) .
62. E. Stahl, "Thin laver chromatography”, { Spr inger-Var lagNew York) , 868 ( 1969 ) .
63. T.C. Jain and C.M. Banks, Can. J . Chem. , 46, 2325 (1968).
64. I. Rubinstein, L.J. Goad, A . D.H . Clague and L.J. Mulheim,Phytochemistry, 15, 195 (1976).
65. R.E. Chandler, S.N. Hooper, D.L. Hooper, W.D. Jamiesonand E. Lewis, Lipids , 17(2), 102 (1982).
i
66. A. Patra, A.K. Mukhopadhyay and A .K. Mitra, Org. Magn.Resonance , 17, 166 (1981).
67. S.K. Talapa'tra, M. Bhattacharya and B. Talapatra, IndianJ. Chem., 11B , 977 (1973).
68. M. wmdholz. The Merck Index, 9, 9590 (1976).
69. E. Bnemair, G. Hass and W. Voelter, Atlas of Carbon - 13NMR Data, 2, 1909 (1979).
;
70. B. Singh and R.P. Rastogi, Phytochemistry, 11,1797 (1972).
240

LIST OF PUBLICATIONS
Viqar Uddin Ahmad and Azra Sultana, A diketone from theleaves of Prosopis j ulif lora , Phytochemistry , 28(1), 278(1989)
1 .
2. Viqar Uddin Ahmad, Azra Sultana and Viqar Sultana,"Constituents of B lumea obliqua" , Fi to terapia , 60(3), 282( 1989 ) .
3. Viqar Uddin Ahmad, Azra Sultana and Sabiha Qazi,
"Alkaloids from the leaves of Prosopis juliflora", j. Nat.Prod , 52(3), 497 (1989).
4. Viqar Uddin Ahmad, Kaniz Fizza and Azra Sultana,"Isolation of two sesquiterpenes from Pluchea arguta,Boiss." , Phytochemistry, 28(11), 3081 ( 1909).
Viqar Uddin Ahmad, Azra Sultana and Kaniz fizza, "Two newterpenoids from Pluchea arguta Boiss.", 7, . Natur f orsch . ,45b, 385
7
( 1990 ) .
5 .
6 . Viqar Uddin Ahmad and Azra Sultana, "A new alkaloid fromProsopis juliflora" , Sci. Pharm . , (accepted).
7 . Viqar Uddin Ahmad and Azra Sultana, "A new piperidinealkaloid . from Prosopis juliflora" , Phytochemistry .( submit ted ) .
8. Leslie B. de Silva, M. Wimal Herath, Chandani Liyanage,Vijaya Kumar, Viqar Uddin Ahmad and Azra Sultana," Dime thylacroves tone from Acronychia pedunculata fruits",Phytochemistry, (submitted).
241




















