
Rotational excitation in scattering of hyperthermal NO from Pt(111
13
Rotational excitation in scattering of hyperthermal NO from Pt(111) A. E. Wiskerke, C. A. Taatjes, a) and A. W. Kleyn FOM-Institute for Atomic and Molecular Physics, Kruislaan 407, 1098 SJ Amsterdam, The Netherlands R. J. W. E. Lahaye and S. Stolte Department of Chemistry, Laser Centre of the Free University, De Boelelaan 1083, 1081 HV Amsterdam, The Netherlands D. K. Bronnikov Russian National Scientific Centre ‘‘Kurchatov Institute’’123182, Moscow, Russia B. E. Hayden Department of Chemistry, University of Southampton, Southampton SO9 5NH, United Kingdom ~Received 5 May 1994; accepted 18 November 1994! Rotational excitation of NO scattered from Pt~111! has been measured for incoming energies from 0.3 to 1.6 eV. For an initial energy of 0.3 eVa clear rotational rainbow, which we assign to O-end collisions, is visible at superspecular exit angles. This is very surprising, since NO–Pt~111! is a chemisorption system with a binding energy of 1 eV. Sharp, pronounced rainbows are visible in the range of incoming kinetic energies where the initial sticking coefficient is high. For an initial energy of 0.3 eV the initial sticking coefficient is 0.9, and at this energy the clearest rotational rainbow is observed. In contrast, at an initial energy of 1.6 eV the sticking coefficient is 0.5 and no rotational rainbow is observed. At subspecular exit angles the distributions are indistinguishable from a Boltzmann distribution at all the energies investigated, and show a clear energy dependence. Boltzmann-type distributions at high incident energy indicate a thorough redistribution of the available energy, although they cannot be explained in terms of a simple statistical model. The scattering results are interpreted as indicating a competition between direct scattering from the repulsive wall and indirect scattering via the deep potential well. Most of the direct scattering can be assigned to O-end collisions with the surface, where the binding energy is expected to be much smaller. Indirect scattering becomes more important at higher energies as more molecules are able to escape the well promptly. © 1995 American Institute of Physics. I. INTRODUCTION In recent years the NO–Pt~111! system has become a model for molecular chemisorption dynamics. A large num- ber of experiments have been undertaken to explore the static 1–3 and dynamic 4–12 properties of the interactions in the system. There is a very strong bond ~’1 eV! between NO and the surface, but no dissociation of the molecule takes place. Furthermore, no precursor and no barrier to chemi- sorption have been observed. This makes the NO–Pt~111! system a very simple case of molecular chemisorption, and makes it well suited for the investigation of the formation of a strong, chemical bond. Our investigation focuses on scat- tering dynamics and in particular on rotational excitation arising from scattering in the presence of such a strong at- tractive well. It has been shown that rotational excitation that occurs in a molecule–surface scattering process is very sensitive to the interaction potential. 13–15 If the scattering process is an indi- rect process, such as trapping-desorption, the final rotational populations are thermal distributions with temperatures equal to or lower than the surface temperature. 12 If, on the other hand, the scattering process is direct and the time for accom- modation very short, the final rotational state is sensitive to the initial conditions of the molecule as it approaches the surface. The resultant distributions are nonthermal and can even exhibit rotational rainbows. 15 A rainbow is the manifes- tation of the divergence of a classical differential cross sec- tion. For rotational rainbows the singularity is usually attrib- uted to an extremum in the rotational excitation D J as a function of the initial orientation angle between the molecu- lar axis and the surface normal g ~we choose g50° to cor- respond with the N-end towards the surface!. However, this extremum could also be a maximum of D J as a function of impact parameter. The observation of a rotational rainbow evidences a direct character for the excitation process. Rota- tional rainbows have been observed for a number of systems where little or no attraction is present 16–20 ~see Sec. II be- low!. The rotational excitation in these cases can be modeled by a collision on a purely repulsive potential with an orien- tational anisotropy. Since the molecule spends a relatively long time at the turning point of this potential, the rotational excitation depends mainly on the anisotropy at this point. Such a potential energy surface explains rotational excitation of NO from Ag~111!, and many features of experiments with oriented NO molecules. 16,21 In the case of NO–Pt~111!, how- ever, the molecules have to traverse a deep potential well of 1 eV. During this time period of strong interaction, the ‘‘memory’’ concerning the initial orientation and angle of incidence may be lost, producing a highly-averaged, Boltzmann-type final energy distribution. The physisorption potential in the case of NO–Ag~111! a! Present address: Combustion Research Facility, Sandia National Laborato- ries, P.O. Box 969, Livermore, California 94551-0969. 3835 J. Chem. Phys. 102 (9), 1 March 1995 0021-9606/95/102(9)/3835/13/$6.00 © 1995 American Institute of Physics Downloaded¬29¬Apr¬2005¬to¬130.37.129.78.¬Redistribution¬subject¬to¬AIP¬license¬or¬copyright,¬see¬http://jcp.aip.org/jcp/copyright.jsp
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Rotational excitation in scattering of hyperthermal NO from Pt(111

Rotational excitation in scattering of hyperthermal NO from Pt(111)A. E. Wiskerke, C. A. Taatjes,a) and A. W. KleynFOM-Institute for Atomic and Molecular Physics, Kruislaan 407, 1098 SJ Amsterdam, The Netherlands
R. J. W. E. Lahaye and S. StolteDepartment of Chemistry, Laser Centre of the Free University, De Boelelaan 1083, 1081 HV Amsterdam,The Netherlands
D. K. BronnikovRussian National Scientific Centre ‘‘Kurchatov Institute’’ 123182, Moscow, Russia
B. E. HaydenDepartment of Chemistry, University of Southampton, Southampton SO9 5NH, United Kingdom
~Received 5 May 1994; accepted 18 November 1994!
Rotational excitation of NO scattered from Pt~111! has been measured for incoming energies from0.3 to 1.6 eV. For an initial energy of 0.3 eV a clear rotational rainbow, which we assign to O-endcollisions, is visible at superspecular exit angles. This is very surprising, since NO–Pt~111! is achemisorption system with a binding energy of 1 eV. Sharp, pronounced rainbows are visible in therange of incoming kinetic energies where the initial sticking coefficient is high. For an initial energyof 0.3 eV the initial sticking coefficient is 0.9, and at this energy the clearest rotational rainbow isobserved. In contrast, at an initial energy of 1.6 eV the sticking coefficient is 0.5 and no rotationalrainbow is observed. At subspecular exit angles the distributions are indistinguishable from aBoltzmann distribution at all the energies investigated, and show a clear energy dependence.Boltzmann-type distributions at high incident energy indicate a thorough redistribution of theavailable energy, although they cannot be explained in terms of a simple statistical model. Thescattering results are interpreted as indicating a competition between direct scattering from therepulsive wall and indirect scattering via the deep potential well. Most of the direct scattering canbe assigned to O-end collisions with the surface, where the binding energy is expected to be muchsmaller. Indirect scattering becomes more important at higher energies as more molecules are ableto escape the well promptly. ©1995 American Institute of Physics.
t
ana
n
can
ec-b-
u-
owota-ems
ledn-elyalint.tionth
ll ofefed,
a
I. INTRODUCTION
In recent years the NO–Pt~111! system has become amodel for molecular chemisorption dynamics. A large number of experiments have been undertaken to explorestatic1–3 and dynamic4–12properties of the interactions in thesystem. There is a very strong bond~'1 eV! between NOand the surface, but no dissociation of the molecule takplace. Furthermore, no precursor and no barrier to chemsorption have been observed. This makes the NO–Pt~111!system a very simple case of molecular chemisorption, amakes it well suited for the investigation of the formation oa strong, chemical bond. Our investigation focuses on sctering dynamics and in particular on rotational excitatioarising from scattering in the presence of such a strongtractive well.
It has been shown that rotational excitation that occursa molecule–surface scattering process is very sensitive tointeraction potential.13–15 If the scattering process is an indi-rect process, such as trapping-desorption, the final rotatiopopulations are thermal distributions with temperatures equto or lower than the surface temperature.12 If, on the otherhand, the scattering process is direct and the time for accomodation very short, the final rotational state is sensitivethe initial conditions of the molecule as it approaches th
a!Present address: Combustion Research Facility, Sandia National Laborries, P.O. Box 969, Livermore, California 94551-0969.
J. Chem. Phys. 102 (9), 1 March 1995 0021-9606/95/102(9)/38Downloaded¬29¬Apr¬2005¬to¬130.37.129.78.¬Redistribution¬subject¬
-he
esi-
ndft-
t-
inthe
alal
m-toe
surface. The resultant distributions are nonthermal andeven exhibit rotational rainbows.15A rainbow is the manifes-tation of the divergence of a classical differential cross stion. For rotational rainbows the singularity is usually attriuted to an extremum in the rotational excitationDJ as afunction of the initial orientation angle between the moleclar axis and the surface normalg ~we chooseg50° to cor-respond with the N-end towards the surface!. However, thisextremum could also be a maximum ofDJ as a function ofimpact parameter. The observation of a rotational rainbevidences a direct character for the excitation process. Rtional rainbows have been observed for a number of systwhere little or no attraction is present16–20 ~see Sec. II be-low!. The rotational excitation in these cases can be modeby a collision on a purely repulsive potential with an orietational anisotropy. Since the molecule spends a relativlong time at the turning point of this potential, the rotationexcitation depends mainly on the anisotropy at this poSuch a potential energy surface explains rotational excitaof NO from Ag~111!, and many features of experiments wioriented NO molecules.16,21In the case of NO–Pt~111!, how-ever, the molecules have to traverse a deep potential we1 eV. During this time period of strong interaction, th‘‘memory’’ concerning the initial orientation and angle oincidence may be lost, producing a highly-averagBoltzmann-type final energy distribution.
The physisorption potential in the case of NO–Ag~111!to-
383535/13/$6.00 © 1995 American Institute of Physicsto¬AIP¬license¬or¬copyright,¬see¬http://jcp.aip.org/jcp/copyright.jsp

--tos-l-en-cttic
by–1mr-nt
n-s,
tialtoi-
less
esndbyatly
ed-
ol-
op-
n--tg,
e-be
e-rali-st
u-
3836 Wiskerke et al.: Rotational excitation of NO from Pt
acts to accelerate the molecule towards the surface. Aconsequence, a more violent collision with the repulsive wtakes place than would be expected without the physisotion potential. For NO–Pt~111! the depth of the well is muchlarger and therefore even more rotational excitation mayexpected. However, the final rotational energy is obtainedthe expense of the translational energy of the molecule inwell. The transfer from translational to rotational energy cbe so efficient that the remaining translational energy islow to allow the molecule to escape from the well. Rottional excitation can act in this case as a dynamic precurto chemisorption, which we will refer to as rotationally mediated adsorption~RMA!. This RMA mechanism is analo-gous to rotationally mediated trapping, which has been pposed for the NO–Ag~111! case.22
The initial sticking probability for thermal NO onPt~111! is nearly unity. Brown and Luntz report a stickinprobability S0 which ranges from 0.9 for an initial beamenergyEi of 0.2 eV toS050.2 forEi53.0 eV.10 The wideenergy range over whichS0 changes only gradually suggesa very efficient sticking mechanism. The observedS0 is toohigh to be attributed purely to phonon excitation, whichexpected to remain low due to the large mass mismatchtween NO and Pt. For a single head-on collision withorotational excitation, the binary collision or Baule formu~see e.g. Ref. 23! predicts an energy loss of 46%. For centsite collisions, a higher energy loss can occur, due todisplacement of the surface atoms parallel to the surfaHowever, Lahayeet al. show that this effect occurs only amuch higherEi .
24 Therefore, atEi53 eV efficient stickingof NO to Pt~111! has to involve rotationally mediated adsorption and interactions with several surface atoms as ational mechanisms for losing energy.10 For a deep well, likethat for NO–Pt~111!, such processes are quite likely to occuFor instance, on first impact the normal translational enecan be efficiently transferred into parallel translational arotational energy, and on the second or third impact thisergy can be restored into perpendicular translational eneallowing the molecule to escape the chemisorption weWithout a back-transfer sticking will occur. The effects metioned have been seen in calculations by Harris and Lunt25
Jacobs and Zare,26 and Polanyi and Wolf.27
As a result of these multiple interactions, the initial eergy may be sufficiently scrambled over all degrees of fredom for the final rotational state distributions to have a stistical appearance. Indeed, measurements for thermal NPt~111! by Jacobset al.with normal incident and exit anglesshowed a broad rotational state distribution for the scatteNO with rotational energies exceeding the incident energ5
The high rotational states were attributed to phonon dexcitation. Although phonon de-excitation is not very proable when the initial energy is higher than the surface teperature, the contribution to the directly scattered fractican be significant as a result of the selective removaltrajectories, where phonon excitation occurs leading to stiing. Since the sticking probability at these energies is 90%10
the promptly scattered molecules represent a minority chnel in the total scattering process.
Three classes of scattering processes have been prop
J. Chem. Phys., Vol. 10Downloaded¬29¬Apr¬2005¬to¬130.37.129.78.¬Redistribution¬subjec
s aallrp-
beattheantooa-sor-
ro-
g
ts
isbe-utlaerthece.t
-ddi-
r.rgynden-rgy,ll.n-z,
n-e-ta-O–
redy.e-b-m-onofck-,an-
osed
by Jacobset al. to model their results; direct inelastic scattering, indirect inelastic scattering, and trappingdesorption.26 The promptly scattered molecules are duedirect inelastic or indirect inelastic scattering. Indirect inelatic scattering involves multiple interactions between the moecule and surface, as discussed above. A fairly weak depdence on initial conditions is to be expected for indireinelastic scattering. Due to the deep well, indirect inelasscattering is likely for NO–Pt~111!. However, evidence fordirect inelastic scattering~single molecule surface collisions!is found in several experiments. Recent measurementsHines and Zare showed rotational rainbows for the CONi~111! system, where the chemisorption energy is alsoeV.17 These authors attribute this rainbow to scattering frothe nonbinding O-end, while the C-end collisions are prefeentially stuck to the surface. Measurements of the alignmeof the scattered thermal NO from Pt~111! by Jacobset al.showed no alignment for low rotational states, and preferetially cart wheeling molecules for higher rotational stateuntil the rotational energy equals the initial energy.5 For ro-tational states where the rotational energy exceeds the inienergy, no alignment was observed. Alignment is unlikelysurvive multiple interactions. Experiments scattering orented NO from Pt~111! reveal a large steric effect in thescattered flux at superspecular angles. For high exit angthe scattering is principally due to the fraction of moleculecolliding with their O-ends towards the surface.6,7 Such apronounced dependence on the initial orientation underlinthe direct inelastic character of the scattering process, asuggests that the scattering dynamics may be determinedlarge orientational anisotropy. All these studies suggest thdirect inelastic scattering can occur even for a strongbound system such as NO–Pt~111!.
Large orientational anisotropy has also been observfor NO–Ag~111!. Rotational rainbows observed in this system originate from direct O-end collisions.16 The potentialenergy surface~PES! used to explain the NO–Ag~111! mea-surements has an anisotropy which is smaller for N-end clisions than for O-end collisions.21,28 If only the difference inmass between the O and the N-atom was important, theposite would be expected. For NO–Pt~111! the interaction ismuch stronger and there is a deep well for the N-end orietation. As a result, the rotational excitation for N-end orientations will be larger than for O-end orientations on firsimpact. The high rotational excitation for N-end scatterinhowever, will give rise to RMA or double collisions, andmight not be seen in the scattered distributions. The comptition between RMA and inelastic scattering is expected tostrong for NO–Pt~111! and investigation of this competitionis one of the motivations for the present study.
Recent low energy electron diffraction~LEED! investi-gations show that the NO molecule is tightly bound in thhollow site.3 This means that this potential well is not uniformly deep over the surface but changes with the lateposition and the orientation of the molecule. This nonunform well depth implies variation of the distance of closeapproach with respect to the lateral position~x–y corruga-tion! and with respect tog ~orientational corrugation!. It hasalso been shown that there is an activation energy for diff
2, No. 9, 1 March 1995t¬to¬AIP¬license¬or¬copyright,¬see¬http://jcp.aip.org/jcp/copyright.jsp
ro
t
i
b
a
d
i
wr
n
ua
Pr
cleto
i
be
e
h
an
ee9dey
l-ij
mofsagtoof
an
asl-
3837Wiskerke et al.: Rotational excitation of NO from Pt
sion over the surface of 0.22 eV.8 This barrier is the height ofthe saddle point in the path from one lattice site to anothand yields a lower bound for the lateral variation in bindinenergy. It indicates strong corrugation of the surface. Corgation facilitates the exchange between rotational and bparallel and normal translational energy. The exchangeenergy between the molecular motions and the motion ofsolid in a single collision might be much smaller than foNO–Ag~111! because of the large mass mismatch betweNO and Pt. As a result, energy could be statistically distruted over the molecular degrees of freedom while accommdation to the surface is relatively slow. This is supportedthe sticking measurements of Brown and Luntz,10 whichshow total rather than normal energy scaling. The broadgular distribution of scattered molecules that we recently oserved for a wide range of incoming energies forms adtional evidence for the scrambling of parallel and normtranslational energy.29
The aim of this investigation is to study rotational exctation in inelastic scattering of NO from Pt~111!. By applyinga wide range of initial translational energies, up to 1.6 eV,probe the interaction in more detail than in previous wo~see, e.g., Refs. 5,9!, and sample a larger part of the PESFurthermore, information concerning the orientational dpendence can be used to model the interaction.7 We hope toshed more light on the competition between RMA and ielastic scattering. The rotational state distributions obtainat intermediate initial energies~0.34 eV! show a rainbow forsuperspecular angles and a Boltzmann distribution for sspecular angles. At higher energies the distributionsBoltzmann-type for all final angles. We discuss the resultsthe light of what is already known concerning the NO–interactions and in particular of previous results of expements with oriented molecules.6,7 Finally, implications of thefindings for a model potential are discussed.
II. EXPERIMENT
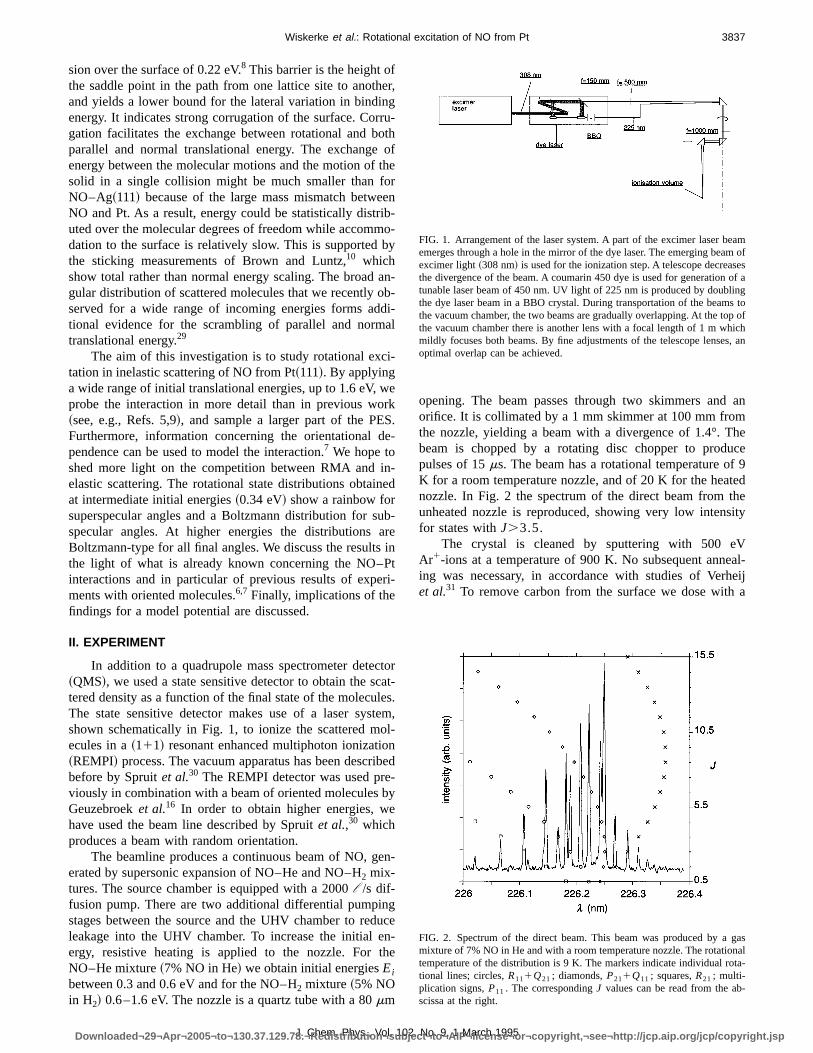
In addition to a quadrupole mass spectrometer detec~QMS!, we used a state sensitive detector to obtain the stered density as a function of the final state of the molecuThe state sensitive detector makes use of a laser sysshown schematically in Fig. 1, to ionize the scattered mecules in a~111! resonant enhanced multiphoton ionizatio~REMPI! process. The vacuum apparatus has been descrbefore by Spruitet al.30 The REMPI detector was used previously in combination with a beam of oriented moleculesGeuzebroeket al.16 In order to obtain higher energies, whave used the beam line described by Spruitet al.,30 whichproduces a beam with random orientation.
The beamline produces a continuous beam of NO, gerated by supersonic expansion of NO–He and NO–H2 mix-tures. The source chamber is equipped with a 2000l /s dif-fusion pump. There are two additional differential pumpinstages between the source and the UHV chamber to redleakage into the UHV chamber. To increase the initial eergy, resistive heating is applied to the nozzle. For tNO–He mixture~7% NO in He! we obtain initial energiesEi
between 0.3 and 0.6 eV and for the NO–H2 mixture ~5% NOin H2! 0.6–1.6 eV. The nozzle is a quartz tube with a 80mm
J. Chem. Phys., Vol. 102Downloaded¬29¬Apr¬2005¬to¬130.37.129.78.¬Redistribution¬subject
er,gu-thofherenb-o-y
n-b-i-al
-
ek.e-
-ed
b-reinti-
torat-s.em,l-nbed-y
n-
gucen-e
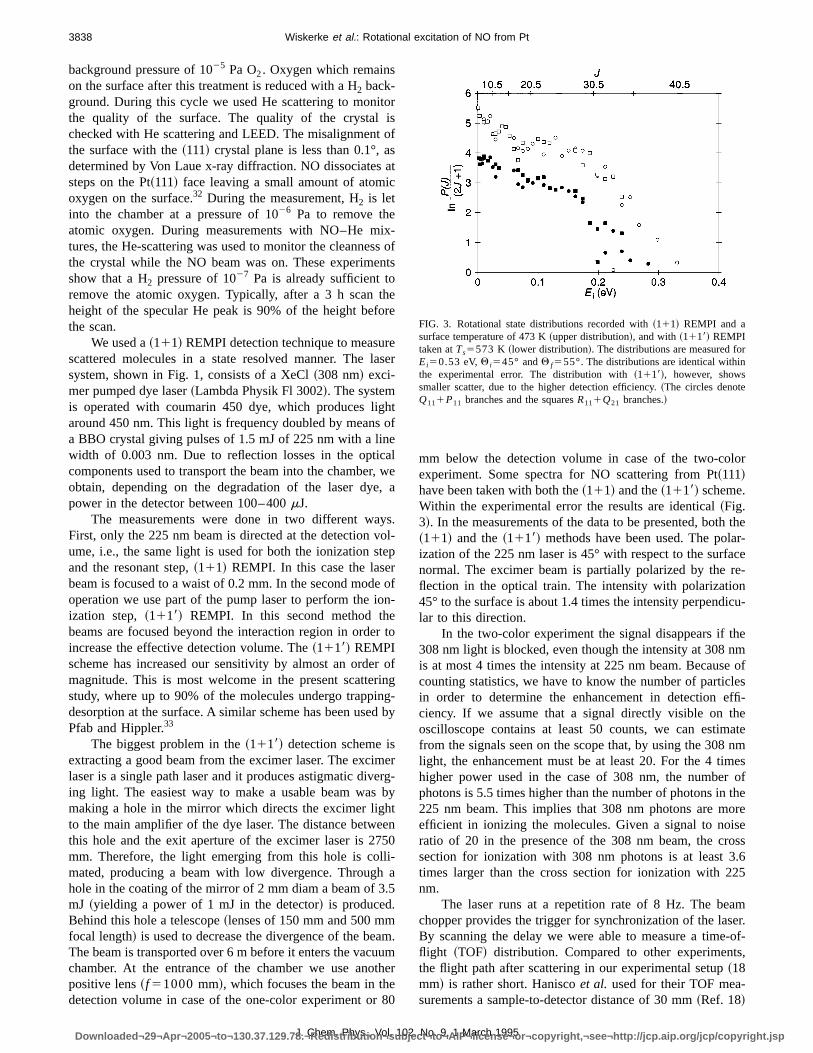
opening. The beam passes through two skimmers andorifice. It is collimated by a 1 mmskimmer at 100 mm fromthe nozzle, yielding a beam with a divergence of 1.4°. Thbeam is chopped by a rotating disc chopper to producpulses of 15ms. The beam has a rotational temperature ofK for a room temperature nozzle, and of 20 K for the heatenozzle. In Fig. 2 the spectrum of the direct beam from thunheated nozzle is reproduced, showing very low intensitfor states withJ.3.5.
The crystal is cleaned by sputtering with 500 eVAr1-ions at a temperature of 900 K. No subsequent anneaing was necessary, in accordance with studies of Verheet al.31 To remove carbon from the surface we dose with a
FIG. 1. Arrangement of the laser system. A part of the excimer laser beaemerges through a hole in the mirror of the dye laser. The emerging beamexcimer light~308 nm! is used for the ionization step. A telescope decreasethe divergence of the beam. A coumarin 450 dye is used for generation oftunable laser beam of 450 nm. UV light of 225 nm is produced by doublinthe dye laser beam in a BBO crystal. During transportation of the beamsthe vacuum chamber, the two beams are gradually overlapping. At the topthe vacuum chamber there is another lens with a focal length of 1 m whichmildly focuses both beams. By fine adjustments of the telescope lenses,optimal overlap can be achieved.
FIG. 2. Spectrum of the direct beam. This beam was produced by a gmixture of 7% NO in He and with a room temperature nozzle. The rotationatemperature of the distribution is 9 K. The markers indicate individual rotational lines; circles,R111Q21 ; diamonds,P211Q11 ; squares,R21 ; multi-plication signs,P11 . The correspondingJ values can be read from the ab-scissa at the right.
, No. 9, 1 March 1995¬to¬AIP¬license¬or¬copyright,¬see¬http://jcp.aip.org/jcp/copyright.jsp
oso
a
s
e
h
e
,
e
n
r
e5-
h
8
or
ther-cee-ncu-
hemof
lesffi-eatenmesoftheoreeoss.625
mer.of-s,
r
3838 Wiskerke et al.: Rotational excitation of NO from Pt
background pressure of 1025 Pa O2. Oxygen which remainson the surface after this treatment is reduced with a H2 back-ground. During this cycle we used He scattering to monitthe quality of the surface. The quality of the crystal ichecked with He scattering and LEED. The misalignmentthe surface with the~111! crystal plane is less than 0.1°, asdetermined by Von Laue x-ray diffraction. NO dissociatessteps on the Pt~111! face leaving a small amount of atomicoxygen on the surface.32 During the measurement, H2 is letinto the chamber at a pressure of 1026 Pa to remove theatomic oxygen. During measurements with NO–He mixtures, the He-scattering was used to monitor the cleannesthe crystal while the NO beam was on. These experimenshow that a H2 pressure of 1027 Pa is already sufficient toremove the atomic oxygen. Typically, after a 3 h scan theheight of the specular He peak is 90% of the height befothe scan.
We used a~111! REMPI detection technique to measurscattered molecules in a state resolved manner. The lasystem, shown in Fig. 1, consists of a XeCl~308 nm! exci-mer pumped dye laser~Lambda Physik Fl 3002!. The systemis operated with coumarin 450 dye, which produces ligaround 450 nm. This light is frequency doubled by meansa BBO crystal giving pulses of 1.5 mJ of 225 nm with a linwidth of 0.003 nm. Due to reflection losses in the opticacomponents used to transport the beam into the chamber,obtain, depending on the degradation of the laser dyepower in the detector between 100–400mJ.
The measurements were done in two different wayFirst, only the 225 nm beam is directed at the detection voume, i.e., the same light is used for both the ionization stand the resonant step,~111! REMPI. In this case the laserbeam is focused to a waist of 0.2 mm. In the second modeoperation we use part of the pump laser to perform the ioization step,~1118! REMPI. In this second method thebeams are focused beyond the interaction region in orderincrease the effective detection volume. The~1118! REMPIscheme has increased our sensitivity by almost an ordermagnitude. This is most welcome in the present scatteristudy, where up to 90% of the molecules undergo trappindesorption at the surface. A similar scheme has been usedPfab and Hippler.33
The biggest problem in the~1118! detection scheme isextracting a good beam from the excimer laser. The excimlaser is a single path laser and it produces astigmatic diveing light. The easiest way to make a usable beam wasmaking a hole in the mirror which directs the excimer lighto the main amplifier of the dye laser. The distance betwethis hole and the exit aperture of the excimer laser is 27mm. Therefore, the light emerging from this hole is collimated, producing a beam with low divergence. Throughhole in the coating of the mirror of 2 mm diam a beam of 3.mJ ~yielding a power of 1 mJ in the detector! is produced.Behind this hole a telescope~lenses of 150 mm and 500 mmfocal length! is used to decrease the divergence of the beaThe beam is transported over 6 m before it enters the vacuumchamber. At the entrance of the chamber we use anotpositive lens~f51000 mm!, which focuses the beam in thedetection volume in case of the one-color experiment or
J. Chem. Phys., Vol. 102Downloaded¬29¬Apr¬2005¬to¬130.37.129.78.¬Redistribution¬subject¬
r
f
t
-ofts
re
ser
tof
lwea
s.l-p
ofn-
to
ofgg-by
erg-bytn0
a5
m.
er
0
mm below the detection volume in case of the two-colexperiment. Some spectra for NO scattering from Pt~111!have been taken with both the~111! and the~1118! scheme.Within the experimental error the results are identical~Fig.3!. In the measurements of the data to be presented, both~111! and the~1118! methods have been used. The polaization of the 225 nm laser is 45° with respect to the surfanormal. The excimer beam is partially polarized by the rflection in the optical train. The intensity with polarizatio45° to the surface is about 1.4 times the intensity perpendilar to this direction.
In the two-color experiment the signal disappears if t308 nm light is blocked, even though the intensity at 308 nis at most 4 times the intensity at 225 nm beam. Becausecounting statistics, we have to know the number of particin order to determine the enhancement in detection eciency. If we assume that a signal directly visible on thoscilloscope contains at least 50 counts, we can estimfrom the signals seen on the scope that, by using the 308light, the enhancement must be at least 20. For the 4 timhigher power used in the case of 308 nm, the numberphotons is 5.5 times higher than the number of photons in225 nm beam. This implies that 308 nm photons are mefficient in ionizing the molecules. Given a signal to noisratio of 20 in the presence of the 308 nm beam, the crsection for ionization with 308 nm photons is at least 3times larger than the cross section for ionization with 2nm.
The laser runs at a repetition rate of 8 Hz. The beachopper provides the trigger for synchronization of the lasBy scanning the delay we were able to measure a time-flight ~TOF! distribution. Compared to other experimentthe flight path after scattering in our experimental setup~18mm! is rather short. Haniscoet al. used for their TOF mea-surements a sample-to-detector distance of 30 mm~Ref. 18!
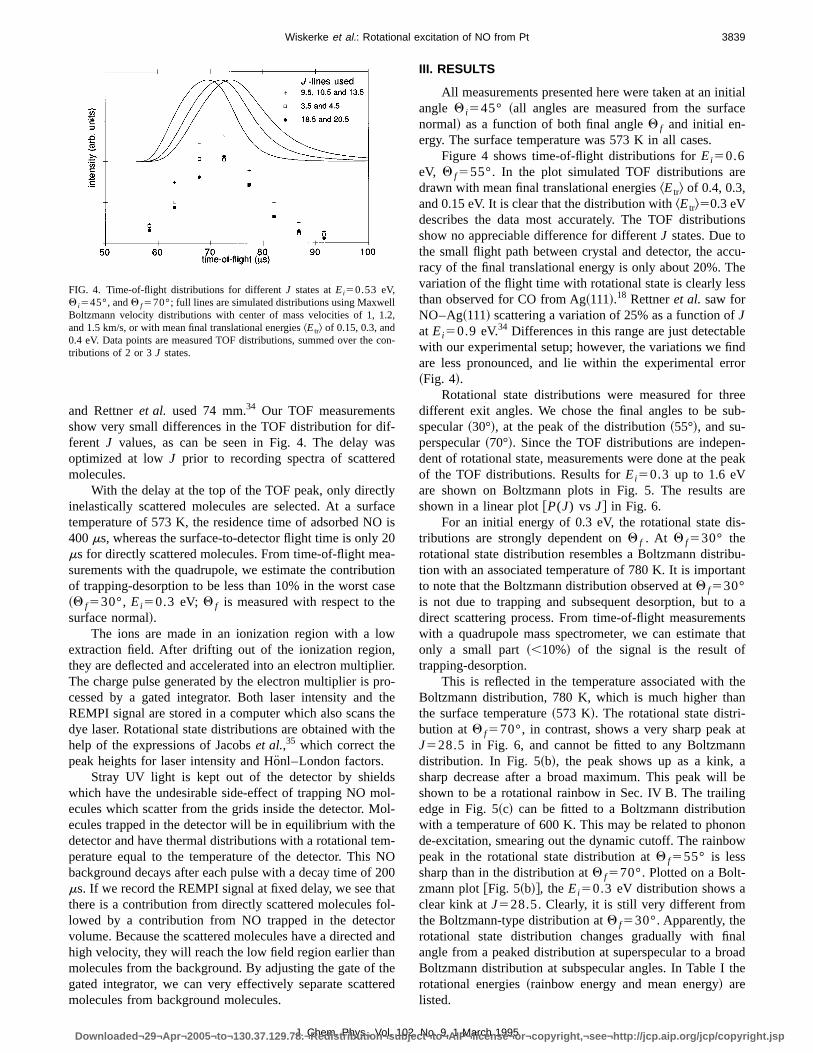
FIG. 3. Rotational state distributions recorded with~111! REMPI and asurface temperature of 473 K~upper distribution!, and with~1118! REMPItaken atTs5573 K ~lower distribution!. The distributions are measured foEi50.53 eV,Q i545° andQ f555°. Thedistributions are identical withinthe experimental error. The distribution with~1118!, however, showssmaller scatter, due to the higher detection efficiency.~The circles denoteQ111P11 branches and the squaresR111Q21 branches.!
, No. 9, 1 March 1995to¬AIP¬license¬or¬copyright,¬see¬http://jcp.aip.org/jcp/copyright.jsp
al
s
u-e
dr
e-
ak
e
-t
atsat
e
t
e
n
lde
3839Wiskerke et al.: Rotational excitation of NO from Pt
and Rettneret al. used 74 mm.34 Our TOF measurementsshow very small differences in the TOF distribution for different J values, as can be seen in Fig. 4. The delay woptimized at lowJ prior to recording spectra of scatteredmolecules.
With the delay at the top of the TOF peak, only directlinelastically scattered molecules are selected. At a surfatemperature of 573 K, the residence time of adsorbed NO400ms, whereas the surface-to-detector flight time is only 2ms for directly scattered molecules. From time-of-flight measurements with the quadrupole, we estimate the contributiof trapping-desorption to be less than 10% in the worst ca~Q f530°, Ei50.3 eV;Q f is measured with respect to thesurface normal!.
The ions are made in an ionization region with a lowextraction field. After drifting out of the ionization region,they are deflected and accelerated into an electron multiplThe charge pulse generated by the electron multiplier is pcessed by a gated integrator. Both laser intensity andREMPI signal are stored in a computer which also scans tdye laser. Rotational state distributions are obtained with thelp of the expressions of Jacobset al.,35 which correct thepeak heights for laser intensity and Ho¨nl–London factors.
Stray UV light is kept out of the detector by shieldswhich have the undesirable side-effect of trapping NO moecules which scatter from the grids inside the detector. Moecules trapped in the detector will be in equilibrium with thdetector and have thermal distributions with a rotational temperature equal to the temperature of the detector. This Nbackground decays after each pulse with a decay time of 2ms. If we record the REMPI signal at fixed delay, we see ththere is a contribution from directly scattered molecules folowed by a contribution from NO trapped in the detectovolume. Because the scattered molecules have a directedhigh velocity, they will reach the low field region earlier thanmolecules from the background. By adjusting the gate of tgated integrator, we can very effectively separate scattemolecules from background molecules.
FIG. 4. Time-of-flight distributions for differentJ states atEi50.53 eV,Q i545°, andQ f570°; full lines are simulated distributions using MaxwellBoltzmann velocity distributions with center of mass velocities of 1, 1.2and 1.5 km/s, or with mean final translational energies^Etr& of 0.15, 0.3, and0.4 eV. Data points are measured TOF distributions, summed over the ctributions of 2 or 3J states.
J. Chem. Phys., Vol. 102Downloaded¬29¬Apr¬2005¬to¬130.37.129.78.¬Redistribution¬subject
-as
yceis0-onse
ier.ro-thehehe
l-l-e-O00atl-rand
hered
III. RESULTS
All measurements presented here were taken at an initiangleQ i545° ~all angles are measured from the surfacenormal! as a function of both final angleQ f and initial en-ergy. The surface temperature was 573 K in all cases.
Figure 4 shows time-of-flight distributions forEi50.6eV, Q f555°. In the plot simulated TOF distributions aredrawn with mean final translational energies^Etr& of 0.4, 0.3,and 0.15 eV. It is clear that the distribution with^Etr&50.3 eVdescribes the data most accurately. The TOF distributionshow no appreciable difference for differentJ states. Due tothe small flight path between crystal and detector, the accracy of the final translational energy is only about 20%. Thvariation of the flight time with rotational state is clearly lessthan observed for CO from Ag~111!.18 Rettneret al. saw forNO–Ag~111! scattering a variation of 25% as a function ofJatEi50.9 eV.34 Differences in this range are just detectablewith our experimental setup; however, the variations we finare less pronounced, and lie within the experimental erro~Fig. 4!.
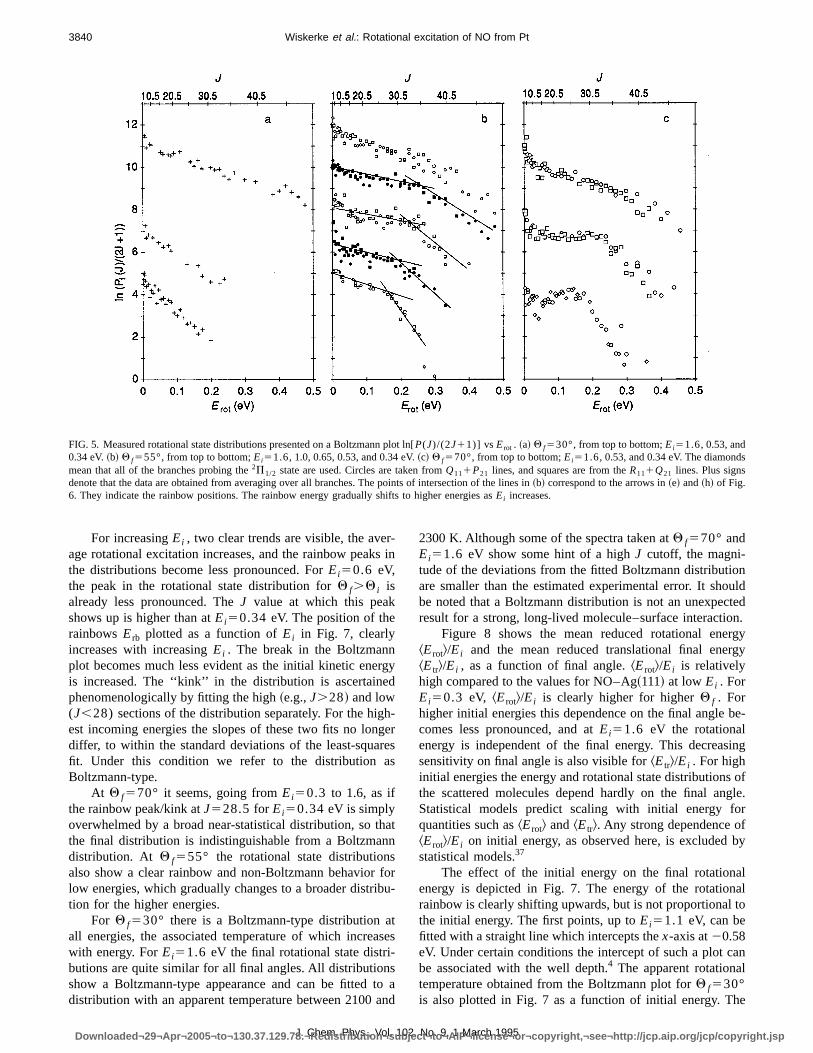
Rotational state distributions were measured for thredifferent exit angles. We chose the final angles to be subspecular~30°!, at the peak of the distribution~55°!, and su-perspecular~70°!. Since the TOF distributions are indepen-dent of rotational state, measurements were done at the peof the TOF distributions. Results forEi50.3 up to 1.6 eVare shown on Boltzmann plots in Fig. 5. The results arshown in a linear plot@P(J) vs J# in Fig. 6.
For an initial energy of 0.3 eV, the rotational state dis-tributions are strongly dependent onQ f . At Q f530° therotational state distribution resembles a Boltzmann distribution with an associated temperature of 780 K. It is importanto note that the Boltzmann distribution observed atQ f530°is not due to trapping and subsequent desorption, but todirect scattering process. From time-of-flight measuremenwith a quadrupole mass spectrometer, we can estimate thonly a small part~,10%! of the signal is the result oftrapping-desorption.
This is reflected in the temperature associated with thBoltzmann distribution, 780 K, which is much higher thanthe surface temperature~573 K!. The rotational state distri-bution atQ f570°, in contrast, shows a very sharp peak aJ528.5 in Fig. 6, and cannot be fitted to any Boltzmanndistribution. In Fig. 5~b!, the peak shows up as a kink, asharp decrease after a broad maximum. This peak will bshown to be a rotational rainbow in Sec. IV B. The trailingedge in Fig. 5~c! can be fitted to a Boltzmann distributionwith a temperature of 600 K. This may be related to phonode-excitation, smearing out the dynamic cutoff. The rainbowpeak in the rotational state distribution atQ f555° is lesssharp than in the distribution atQ f570°. Plotted on a Bolt-zmann plot@Fig. 5~b!#, theEi50.3 eV distribution shows aclear kink atJ528.5.Clearly, it is still very different fromthe Boltzmann-type distribution atQ f530°.Apparently, therotational state distribution changes gradually with finaangle from a peaked distribution at superspecular to a broaBoltzmann distribution at subspecular angles. In Table I throtational energies~rainbow energy and mean energy! arelisted.
,
on-
, No. 9, 1 March 1995¬to¬AIP¬license¬or¬copyright,¬see¬http://jcp.aip.org/jcp/copyright.jsp
s
3840 Wiskerke et al.: Rotational excitation of NO from Pt
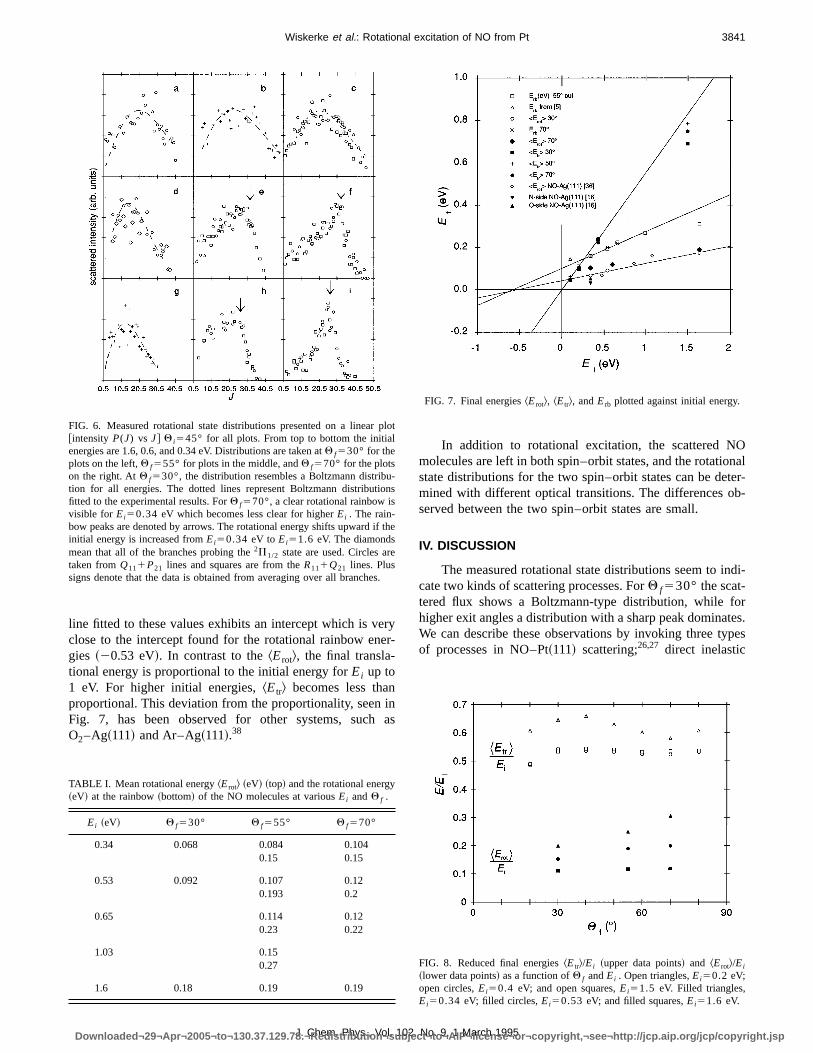
FIG. 5. Measured rotational state distributions presented on a Boltzmann plot ln[P(J)/(2J11)] vsErot . ~a! Q f530°, from top to bottom;Ei51.6,0.53, and0.34 eV.~b! Q f555°, from top to bottom;Ei51.6,1.0, 0.65, 0.53, and 0.34 eV.~c! Q f570°, from top to bottom;Ei51.6,0.53, and 0.34 eV. The diamondmean that all of the branches probing the2P1/2 state are used. Circles are taken fromQ111P21 lines, and squares are from theR111Q21 lines. Plus signsdenote that the data are obtained from averaging over all branches. The points of intersection of the lines in~b! correspond to the arrows in~e! and~h! of Fig.6. They indicate the rainbow positions. The rainbow energy gradually shifts to higher energies asEi increases.
r-s
rgd
hgres
hnsfob
ts-soa
nuldtedn.rgygy
be-
ing
ofgle.rfby
lalto
an
e
For increasingEi , two clear trends are visible, the aveage rotational excitation increases, and the rainbow peakthe distributions become less pronounced. ForEi50.6 eV,the peak in the rotational state distribution forQ f.Q i isalready less pronounced. TheJ value at which this peakshows up is higher than atEi50.34 eV. Theposition of therainbowsErb plotted as a function ofEi in Fig. 7, clearlyincreases with increasingEi . The break in the Boltzmannplot becomes much less evident as the initial kinetic eneis increased. The ‘‘kink’’ in the distribution is ascertainephenomenologically by fitting the high~e.g.,J.28! and low(J,28) sections of the distribution separately. For the higest incoming energies the slopes of these two fits no londiffer, to within the standard deviations of the least-squafit. Under this condition we refer to the distribution aBoltzmann-type.
At Q f570° it seems, going fromEi50.3 to 1.6, as ifthe rainbow peak/kink atJ528.5 forEi50.34 eV issimplyoverwhelmed by a broad near-statistical distribution, so tthe final distribution is indistinguishable from a Boltzmandistribution. At Q f555° the rotational state distributionalso show a clear rainbow and non-Boltzmann behaviorlow energies, which gradually changes to a broader distrition for the higher energies.
For Q f530° there is a Boltzmann-type distribution aall energies, the associated temperature of which increawith energy. ForEi51.6 eV the final rotational state distributions are quite similar for all final angles. All distributionshow a Boltzmann-type appearance and can be fitted tdistribution with an apparent temperature between 2100
J. Chem. Phys., Vol. 102Downloaded¬29¬Apr¬2005¬to¬130.37.129.78.¬Redistribution¬subject¬
in
y
-ers
at
ru-
es
and
2300 K. Although some of the spectra taken atQ f570° andEi51.6 eV show some hint of a highJ cutoff, the magni-tude of the deviations from the fitted Boltzmann distributioare smaller than the estimated experimental error. It shobe noted that a Boltzmann distribution is not an unexpecresult for a strong, long-lived molecule–surface interactio
Figure 8 shows the mean reduced rotational ene^Erot&/Ei and the mean reduced translational final ener^Etr&/Ei , as a function of final angle.Erot&/Ei is relativelyhigh compared to the values for NO–Ag~111! at lowEi . ForEi50.3 eV, ^Erot&/Ei is clearly higher for higherQ f . Forhigher initial energies this dependence on the final anglecomes less pronounced, and atEi51.6 eV the rotationalenergy is independent of the final energy. This decreassensitivity on final angle is also visible for^Etr&/Ei . For highinitial energies the energy and rotational state distributionsthe scattered molecules depend hardly on the final anStatistical models predict scaling with initial energy foquantities such asErot& and^Etr&. Any strong dependence o^Erot&/Ei on initial energy, as observed here, is excludedstatistical models.37
The effect of the initial energy on the final rotationaenergy is depicted in Fig. 7. The energy of the rotationrainbow is clearly shifting upwards, but is not proportionalthe initial energy. The first points, up toEi51.1 eV, can befitted with a straight line which intercepts thex-axis at20.58eV. Under certain conditions the intercept of such a plot cbe associated with the well depth.4 The apparent rotationaltemperature obtained from the Boltzmann plot forQ f530°is also plotted in Fig. 7 as a function of initial energy. Th
, No. 9, 1 March 1995to¬AIP¬license¬or¬copyright,¬see¬http://jcp.aip.org/jcp/copyright.jsp
r
n
alr--
i-
rs.es
n
t
.
3841Wiskerke et al.: Rotational excitation of NO from Pt
line fitted to these values exhibits an intercept which is veclose to the intercept found for the rotational rainbow enegies ~20.53 eV!. In contrast to theErot&, the final transla-tional energy is proportional to the initial energy forEi up to1 eV. For higher initial energies,Etr& becomes less thanproportional. This deviation from the proportionality, seen iFig. 7, has been observed for other systems, suchO2–Ag~111! and Ar–Ag~111!.38
FIG. 6. Measured rotational state distributions presented on a linear p@intensityP(J) vs J# Q i545° for all plots. From top to bottom the initialenergies are 1.6, 0.6, and 0.34 eV. Distributions are taken atQ f530° for theplots on the left,Q f555° for plots in the middle, andQ f570° for the plotson the right. AtQ f530°, thedistribution resembles a Boltzmann distribu-tion for all energies. The dotted lines represent Boltzmann distributiofitted to the experimental results. ForQ f570°, a clear rotational rainbow isvisible for Ei50.34 eVwhich becomes less clear for higherEi . The rain-bow peaks are denoted by arrows. The rotational energy shifts upward ifinitial energy is increased fromEi50.34 eV toEi51.6 eV. The diamondsmean that all of the branches probing the2P1/2 state are used. Circles aretaken fromQ111P21 lines and squares are from theR111Q21 lines. Plussigns denote that the data is obtained from averaging over all branches
TABLE I. Mean rotational energyErot& ~eV! ~top! and the rotational energy~eV! at the rainbow~bottom! of the NO molecules at variousEi andQ f .
Ei ~eV! Q f530° Q f555° Q f570°
0.34 0.068 0.084 0.1040.15 0.15
0.53 0.092 0.107 0.120.193 0.2
0.65 0.114 0.120.23 0.22
1.03 0.150.27
1.6 0.18 0.19 0.19
J. Chem. Phys., Vol. 102Downloaded¬29¬Apr¬2005¬to¬130.37.129.78.¬Redistribution¬subject¬
yr-
as
In addition to rotational excitation, the scattered NOmolecules are left in both spin–orbit states, and the rotationstate distributions for the two spin–orbit states can be detemined with different optical transitions. The differences observed between the two spin–orbit states are small.
IV. DISCUSSION
The measured rotational state distributions seem to indcate two kinds of scattering processes. ForQ f530° the scat-tered flux shows a Boltzmann-type distribution, while fohigher exit angles a distribution with a sharp peak dominateWe can describe these observations by invoking three typof processes in NO–Pt~111! scattering;26,27 direct inelastic
lot
s
he
FIG. 7. Final energiesErot&, ^Etr&, andErb plotted against initial energy.
FIG. 8. Reduced final energies^Etr&/Ei ~upper data points! and ^Erot&/Ei
~lower data points! as a function ofQ f andEi . Open triangles,Ei50.2 eV;open circles,Ei50.4 eV; and open squares,Ei51.5 eV. Filled triangles,Ei50.34 eV;filled circles,Ei50.53 eV; andfilled squares,Ei51.6 eV.
, No. 9, 1 March 1995to¬AIP¬license¬or¬copyright,¬see¬http://jcp.aip.org/jcp/copyright.jsp

or
dr
e
r
od
segu
l
reaf-
nstp
reorr
dictumr
not
–toall
ibleiontoul-na-toper-ld
er-
ob-
ss,in-
rgy,tot,thec-c-heelineatlas-nal-
onalta-lllthe
a-ef-forlcti-b-nal
3842 Wiskerke et al.: Rotational excitation of NO from Pt
scattering, indirect inelastic scattering, trapping-desorptiA distinct microscopic origin can be presumed for each pcess, and each type of scattering will yield a qualitativedifferent final rotational distribution. While at lower energiethe data show clear signs of the three mechanisms, a gramerging of the scattering mechanisms seems to occuhigher energies. In the present experiments we discriminagainst trapping-desorption contributions, so we can conctrate on the competition between direct and indirect inelasscattering.
A. Surface corrugation
Measurements of inelastic NO–Pt~111! scattering showa broad angular distribution and a final translational ene~see Fig. 8! which hardly depends onQ f .
4,29 For direct scat-tering, the angular distribution is related to the corrugationthe surface. Parallel momentum conservation~as would beobserved in a completely smooth surface! is not observed inNO–Pt~111!, since in most casesEf /Ei does not obey therelation predicted by cube models@Etr/Ei5~sinQ i /sinQ f!
2#.Only at high Q f does the final energy converge t~sinQ i /sinQ f)
2Ei . Since the angular distribution is peakenear the specular angle, the other extreme case, scattefrom individual surface atoms is not seen either. In additiothe energy transfer for such single-atom collisions underemates the amount observed.29 The present results can only bexplained by an intermediate case; we presume scatterintake place from a corrugated repulsive potential, with mtiple scattering playing an important role.
The large corrugation is apparent from the dramaticalarge angular width for NO–Pt~111! scattering of 40°, ascompared to that of O2–Pt~111!, where an angular width of20° is observed for 0.4,Ei,1.6 eV.39 Both the molecularmass and the moment of inertia for O2 and NO are compa-rable, indicating an extra source of broadening in the caseNO–Pt~111! scattering, which might be additional corrugation due to a deeper potential well. If there is a large corgation, the observed large angular width can be explainHowever, the final translational energy should then increwith final scattering angle as observed in calculationscorrugated surfaces.38,40 The experimental angular distributions seem to be in contradiction with the^Etr&/Ei depen-dence onQ f . The chemisorption well for the NO–Pt~111!interaction makes the scattering process more complicathan a single collision with a corrugated wall.
The final rotational state distributions provide more isight into the role of corrugation in the scattering proceFor high exit angles the absolute momentum change ofmolecule is small. If the scattering process were a simbinary collision, these ‘‘soft collisions’’ would give rise to asmall energy transfer. Nevertheless, the observed^Etr&/Ei isnot significantly higher for superspecular exit angles. Fhigh exit angles more translational energy is transferredrotation, which compensates for the lower energy transfethe surface. Apparently, the rotational excitation is obtainmainly at the cost of normal translational energy. FQ f530° the final rotational energy is lower, leaving moenergy for the translational degree of freedom. For habody scattering, the rotational excitationDJ is directly re-
J. Chem. Phys., Vol. 102Downloaded¬29¬Apr¬2005¬to¬130.37.129.78.¬Redistribution¬subject
n.o-lysualat
aten-tic
gy
of
ringn,ti-
tol-
ly
of-u-d.seor
ted
-s.hele
ortotodred-
lated to the change in momentumDp. Indeed, models ofhard ellipsoids scattered from corrugated surfaces prethis correlation between rotational excitation and momentchange.41 This would predict larger rotational excitation folarger total scattering angles~i.e., smallerQ f!, in contrast tothe experimental observations. Thus, these models doreproduce the NO–Pt~111! scattering results either.
While these models cannot completely explain the NOPt~111! distributions, simple considerations are sufficientdetermine that corrugation is required in the repulsive w~to obtain the observed angular distributions!, but that thiscorrugated repulsive surface is not completely responsfor the scattering dynamics. It is possible that the deviatfrom the behavior predicted by simple models is relatedthe occurrence of different scattering processes which simtaneously contribute to the scattered distribution. A combition of a distribution with low rotational excitation peakedsubspecular directions and a distribution peaked to suspecular directions showing high rotational excitation wouexplain our observations.
B. Rotational rainbows
The rotational state distributions observed for supspecular scattering at lower incoming energies~especiallyQ f570°, Ei50.34 eV! show very sharp peaks~Fig. 6!.Such pronounced rainbow peaks have previously beenserved for weakly interacting systems,18,19 where only asimple single collision takes place. Such a direct procedominated by repulsive interactions, is indicated by a learly increasing~though not necessarily proportional! rela-tionship between the rainbow energy~Erb! andEi . If energyconservation alone determines the highest rotational enean energetic cutoff will be observed which is proportionalthe initial energy. If the cutoff is due to a dynamical effecthe rotational excitation can be caused predominantly byorientational anisotropy in the repulsive wall or in the attrative part of the potential. If it is only anisotropy in the attrative part of the potential that is important, increasing tinitial energy results in a shorter time for the molecule to fethe torque in the well. This, in turn, results in a decreasethe rotational excitation for higher initial energies. If, on thother hand, it is only the anisotropy in the repulsive wall this important, the rotational excitation takes place at the csical turning point on the potential. In that case, rotatioexcitation increases with initial energy.42 Because the position of the peak is not proportional toEi , we believe that thepeak observed under superspecular conditions is a rotatirainbow, and not an effect due to an energetic cutoff or stistical partitioning.37,43,44 Furthermore, since the rotationaenergy increases withEi , the anisotropy in the repulsive wais of greater importance for the rotational excitation thananisotropy in the attractive potential.
An alternative explanation for the high rotational excittion could be an output channel effect. Output channelfects in the translational degree of freedom are observedH2 desorbing from various metals.45 In these cases, high finatranslational energy is observed, which is caused by an avation barrier for desorption. A similar effect might be oserved for rotational excitation. In this case, the rotatio
, No. 9, 1 March 1995¬to¬AIP¬license¬or¬copyright,¬see¬http://jcp.aip.org/jcp/copyright.jsp
3843Wiskerke et al.: Rotational excitation of NO from Pt
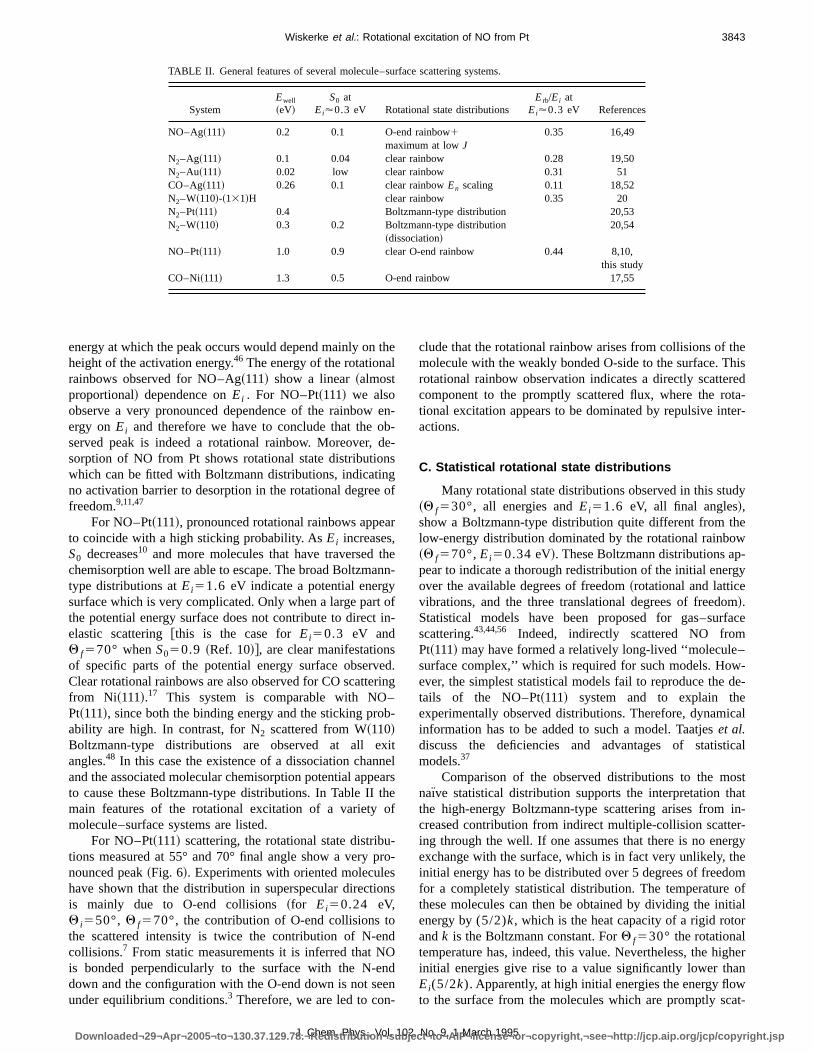
TABLE II. General features of several molecule–surface scattering systems.
SystemEwell
~eV!S0 at
Ei'0.3 eV Rotational state distributionsErb/Ei at
Ei'0.3 eV References
NO–Ag~111! 0.2 0.1 O-end rainbow1maximum at lowJ
0.35 16,49
N2–Ag~111! 0.1 0.04 clear rainbow 0.28 19,50N2–Au~111! 0.02 low clear rainbow 0.31 51CO–Ag~111! 0.26 0.1 clear rainbowEn scaling 0.11 18,52N2–W~110!-~131!H clear rainbow 0.35 20N2–Pt~111! 0.4 Boltzmann-type distribution 20,53N2–W~110! 0.3 0.2 Boltzmann-type distribution
~dissociation!20,54
NO–Pt~111! 1.0 0.9 clear O-end rainbow 0.44 8,10,this study
CO–Ni~111! 1.3 0.5 O-end rainbow 17,55
eiseda-r-
y
ew
y
ce
-e-
al
cal
statn-r-gyemofial
hern
t-
energy at which the peak occurs would depend mainly onheight of the activation energy.46 The energy of the rotationarainbows observed for NO–Ag~111! show a linear~almostproportional! dependence onEi . For NO–Pt~111! we alsoobserve a very pronounced dependence of the rainbowergy onEi and therefore we have to conclude that the oserved peak is indeed a rotational rainbow. Moreover,sorption of NO from Pt shows rotational state distributiowhich can be fitted with Boltzmann distributions, indicatinno activation barrier to desorption in the rotational degreefreedom.9,11,47
For NO–Pt~111!, pronounced rotational rainbows appeto coincide with a high sticking probability. AsEi increases,S0 decreases
10 and more molecules that have traversed tchemisorption well are able to escape. The broad Boltzmatype distributions atEi51.6 eV indicate a potential energsurface which is very complicated. Only when a large partthe potential energy surface does not contribute to directelastic scattering@this is the case forEi50.3 eV andQ f570° whenS050.9 ~Ref. 10!#, are clear manifestationsof specific parts of the potential energy surface observClear rotational rainbows are also observed for CO scatterfrom Ni~111!.17 This system is comparable with NO–Pt~111!, since both the binding energy and the sticking proability are high. In contrast, for N2 scattered from W~110!Boltzmann-type distributions are observed at all eangles.48 In this case the existence of a dissociation chanand the associated molecular chemisorption potential appto cause these Boltzmann-type distributions. In Table II tmain features of the rotational excitation of a varietymolecule–surface systems are listed.
For NO–Pt~111! scattering, the rotational state distributions measured at 55° and 70° final angle show a very pnounced peak~Fig. 6!. Experiments with oriented moleculehave shown that the distribution in superspecular directiois mainly due to O-end collisions~for Ei50.24 eV,Q i550°, Q f570°, thecontribution of O-end collisions tothe scattered intensity is twice the contribution of N-encollisions.7 From static measurements it is inferred that Nis bonded perpendicularly to the surface with the N-edown and the configuration with the O-end down is not seunder equilibrium conditions.3 Therefore, we are led to con
J. Chem. Phys., Vol. 10Downloaded¬29¬Apr¬2005¬to¬130.37.129.78.¬Redistribution¬subjec
thel
en-b-de-nsgof
ar
henn-yofin-
ed.ing
b-
xitnelearsheof
-ro-sns
dOnden-
clude that the rotational rainbow arises from collisions of thmolecule with the weakly bonded O-side to the surface. Throtational rainbow observation indicates a directly scattercomponent to the promptly scattered flux, where the rottional excitation appears to be dominated by repulsive inteactions.
C. Statistical rotational state distributions
Many rotational state distributions observed in this stud~Q f530°, all energies andEi51.6 eV, all final angles!,show a Boltzmann-type distribution quite different from thlow-energy distribution dominated by the rotational rainbo~Q f570°,Ei50.34 eV!. These Boltzmann distributions ap-pear to indicate a thorough redistribution of the initial energover the available degrees of freedom~rotational and latticevibrations, and the three translational degrees of freedom!.Statistical models have been proposed for gas–surfascattering.43,44,56 Indeed, indirectly scattered NO fromPt~111! may have formed a relatively long-lived ‘‘molecule–surface complex,’’ which is required for such models. However, the simplest statistical models fail to reproduce the dtails of the NO–Pt~111! system and to explain theexperimentally observed distributions. Therefore, dynamicinformation has to be added to such a model. Taatjeset al.discuss the deficiencies and advantages of statistimodels.37
Comparison of the observed distributions to the monaıve statistical distribution supports the interpretation ththe high-energy Boltzmann-type scattering arises from icreased contribution from indirect multiple-collision scatteing through the well. If one assumes that there is no enerexchange with the surface, which is in fact very unlikely, thinitial energy has to be distributed over 5 degrees of freedofor a completely statistical distribution. The temperaturethese molecules can then be obtained by dividing the initenergy by(5/2)k, which is the heat capacity of a rigid rotorandk is the Boltzmann constant. ForQ f530° the rotationaltemperature has, indeed, this value. Nevertheless, the higinitial energies give rise to a value significantly lower thaEi(5/2k). Apparently, at high initial energies the energy flowto the surface from the molecules which are promptly sca
2, No. 9, 1 March 1995t¬to¬AIP¬license¬or¬copyright,¬see¬http://jcp.aip.org/jcp/copyright.jsp

h
onoainenh
eo
t
h
i
i
unb
eho
-
r
b-
n
n-o-asi-
to
n-nal
eal.hell
-a--In
ter-
ci-enonlel,ssi-
ita-eat-tial
tileleedrals arsthelltice
3844 Wiskerke et al.: Rotational excitation of NO from Pt
tered is relatively higher. This effect can be related to tsticking probabilityS0 , which decreases for increasing initiaenergies. That is, for higher initial energies, a higher proption of molecules attains the required velocity for escapithe well. The resulting trajectories, which lead to high phnon excitation, increasingly contribute to the promptly sctered flux as the initial energy increases. This observatmay also indicate energy redistribution by an increasinumber of multicollisional events for molecules scatterwith higher initial energy. If the redistribution process is coceptually divided into separate sequential collisions with tsurface, this observation suggests that a larger average nber of encounters occurs for scattering at higher initial eergy.
V. REQUIREMENTS FOR A PES
Any model for the NO–Pt~111! interaction has to ex-plain the results of both dynamic and static experiments. Dspite the large amount of information which is available othis system, it will be hard to come up with a unique modbecause many degrees of freedom are involved. Furthermin it must be taken into consideration that the rainbow oserved at superspecular scattering angles represents nothalf of the promptly scattered flux. This flux corresponds10% of the incident flux at low energies, which means ththe probability of rainbow scattering is less than 5%. Tscattered distributions may therefore reflect extremely locized features of the PES. To understand the general scattebehavior, the main features may be represented by a simoscillator description of the solid and an adiabatic potentbetween the anisotropic molecule and the surface oscillarepresenting that part of the PES that gives rise to scatterThe requirements for such a potential energy surface willdiscussed in this section.
A. Orientation dependence of the potential
The orientation dependence of the potential can be stied with beams of oriented NO molecules. Total scatteriexperiments with oriented NO have been carried outKuipers et al. and Tenneret al.,6,7 who observed a largesteric effect for NO–Pt~111! scattering at superspeculaangles. The steric effect is defined as the difference betwthe O-end intensity and the N-end intensity divided by tintensity obtained for an nonoriented beam. The hexapstate selection used for these experiments provides NO mecules in the upperL-doublet of the2P1/2 ~V51,S521/2!state. In the orientation field, which is needed to measuresteric effect, an orientational distributionW(g)5~16cosg!is obtained. The measured steric effect for scattered mecules can be interpreted as 2^cosg&, where the average cosine of the initial orientation angleg is weighted by thecontribution ofg values to the scattered flux.
ForQ f530°, thecontribution of O-end collisions to thescattered flux is approximately equal to the contributionN-end collisions.6,7 Molecules scattered in the latter orientation may have been in the chemisorption well and therefoshow a statistical~Boltzmann-type! distribution. Yet, manyO-end collisions contribute to this distribution atQ f530°.Only molecules within a more restricted orientational dist
J. Chem. Phys., Vol. 102Downloaded¬29¬Apr¬2005¬to¬130.37.129.78.¬Redistribution¬subject
elr-g-t-ongd-eum-n-
e-nl,re,b-evenoateal-ringplealtorng.be
d-gy
reneleol-
the
ol-
of-re
i-
bution appear to contribute to the rotational rainbow oserved atQ f570°. For Q f570°, 2 cosg&50.64, whichcorresponds tog&571°. A correction has to be applied to thesteric effect because of imperfections in the orientatioscheme.16 This yields a value ofg& in the range 52° to 65°.This suggests that the rainbow arises from molecular orietations close to upright, with the O-end down. We have prposed a PES showing this high directionality, which also hthe capability of steering the molecule away from the chemsorption well located on the N-end.42 In this potential therepulsive wall has to be flattened on the O-side, in contrastthe PES for NO–Ag~111!, which exhibits a minimum turningpoint distance for molecules lying flat.28
B. Dependence on initial energy
The dependence of the rainbow energy on the initial eergy provides explicit information concerning the interactiopotential. This dependence is plotted in Fig. 7. If rotationexcitation is mainly due to the anisotropy of the attraction,57
the amount of rotational excitation will depend on the timthe molecule spends in the attractive region of the potentiIn that case, increasing the initial energy will decrease ttime of interaction. Therefore, the rotational excitation widecrease and~]Erot/]Ei! can become negative.
42 The positiveslope ofEr vsEi suggests that the molecules which contribute to the promptly scattered flux experience little reorienttion in the well. Instead, the rotational excitation mainly occurs on the repulsive wall of the potential energy surface.a simple square well picture~showing first attraction andthen repulsion!, the intercept will be equal to the bindingenergy of the system. For physisorption systems, these incepts have indeed the same value as the well depth.51 Thismodel predicts a common intercept for the^Erot& andErb onEi . The correspondence of these intercepts might be coindental, since a single well depth across the unit cell has beassumed. However, measurements of the mobility of NOthe Pt~111! surface indicate a minimum variation in the weldepth of at least 0.2 eV. Moreover, using a simple modTanaka and Sugano showed an intercept different and pobly larger than the well depth of the model potential.58 Theintercept seen in the energy dependence of rotational exction in Fig. 7 is about 0.6 eV, which is clearly less than thwell depth determined by other means. Apparently, the sctered molecules have not sampled regions of the potenenergy surface where the well is deepest.
C. Sticking coefficient and multiple collisions
The mismatch between surface mass and the projecmass prohibits the loss of all the initial energy in a singcollision. Therefore, the translational energy has to be storin rotational energy before it can be dissipated in sevebounces to the lattice. Even if the surface is described asimple cube model without dissipation, multiple encountecan be observed; however, since no energy is dissipatedmolecules will eventually make a collision which returns aavailable energy into translational energy. In a more realismodel, these molecules will have lost their energy to thsurface in a limited number of bounces. AtEi50.34 eV,
, No. 9, 1 March 1995¬to¬AIP¬license¬or¬copyright,¬see¬http://jcp.aip.org/jcp/copyright.jsp

-wi-
ee
easdn,rteal
re-n
rt ofwthes a
toisill
sa-s
3845Wiskerke et al.: Rotational excitation of NO from Pt
only 30% of the kinetic energy in the well has to be disspated, which may be accomplished in just one collision. Fhigher energies, more bounces are needed for the molecuaccommodate to the surface, which causesS0 to be lower.
The polarized rotational state distribution for desorptioprovides additional information on the shape of the PESimulations by Doren and Tully59 show that the rotationalalignment in desorption is strongly dependent on the chopotential. Jacobs and Zare observed a preferential helicoping distribution in desorption.26 This means that the moleculehas to convert cartwheel motion into translational motiondesorb, causing rotational cooling mainly for cartwheel mtion. This mechanism reduces the number of molecules wa high cart wheeling angular momentum, and induces poization for high J values. This implies that for adsorptiothere is a dependence of the sticking probability on the initalignment of the molecules; helicoptering molecules will bunable to orient the molecular axis parallel to the surfanormal, thereby avoiding the binding orientations. Therefoit is less probable that they will stick.
D. Schematic PES
From the discussion above it is clear that the potenenergy surface describing the NO–Pt~111! interaction will bevery complex and has to depend on several degrees of fdom. Because the well is nonuniform and strong corrugathas been inferred, there will be both a lateral dependencean orientation dependence. The vibrational degree of frdom will be of less relevance, because dissociation doesoccur,10 and minimal vibrational excitation is observed in thscattering.9 The question remains whether the complex hpersurface can, for purposes of discussion, be meaningfreduced to a one or two dimensional representation. Thare two arguments that this is the case. First, Doren and Tconstructed a full hypersurface for the similar CO–Ni~111!system and concluded that its lateral dependence allowdiscussion of the dynamics in terms of a two-dimensionhypersurface. Second, the rainbows seen in Fig. 6 are vclear, which suggests that the potential energy surface habe stationary in the lateral coordinates around the imppoint where the rotational rainbow occurs. Because thetensity of the rainbow represents only a small fraction of ttotal incident flux, rotational rainbow scattering may not bpossible at all impact points in the unit cell. We will discusthree types of two-dimensional potential energy surfaces,pending on molecule–surface distancez and molecular ori-entationg, that describe the interaction around the impapoints leading to rotational rainbow scattering, where tscattering experiments provide information. These thschematic forms for an NO–Pt~111! potential energy surfaceare depicted in Fig. 9. The suggested potentials are basepreliminary calculations using a Morse-type potential.42 Theprecise details of the PES and trajectory calculations willdescribed more fully in the next chapter, but here we wishhighlight general features of the interaction which canestablished by simple arguments.
In the potential shown in Fig. 9~a!, rotational excitationis caused by the asymmetry of the repulsive part of thetential. This potential energy surface will indeed give rise
J. Chem. Phys., Vol. 10Downloaded¬29¬Apr¬2005¬to¬130.37.129.78.¬Redistribution¬subjec
i-orle to
nS.
senter-
too-ithlar-nialecere,
tial
ree-ionandee-notey-ullyereully
s aalerys toactin-heesde-
ctheree
d on
betobe
po-to
a rotational rainbow for side-on collisions. As a consequence, there will be no steric effect for rotational rainboscattering, which is strongly contradicted by our experments.
To obtain a steric effect for the rotational rainbow, thinflection point in the anisotropy must be moved towards thO-end of the molecule, as shown in Fig. 9~b!. This potentialwould give a rotational rainbow for O-end collisions in thabsence of an attractive potential at the N-end. However,depicted in Fig. 9~b!, a possible rainbow trajectory is steereinto the attractive well and might undergo a second collisioeliminating the rainbow. If the potential well has a very shorange, indirect scattering may occur if the velocity after thfirst impact is high enough to avoid the attractive potenti
FIG. 9. Three types of potential energy surfaces. Schematic contour repsentations of the potential as a function of the molecule surface separatioz~vertical! and molecular orientationg ~horizontal!. Dashed lines indicatenegative energy contours. The shaded area represents the repulsive pathe potential.~a! This PES represents a potential for molecules which shobinding on one side of the molecule. This model can be rejected becauserotational rainbow appears for flat lying molecules. The experiment showclear O-end rotational rainbow.~b! Modification of potential~a! with theinflection point of the repulsive wall on the O-end. Trajectories leadinghigh rotational excitation may be attracted to the surface when the N-endturned to the surface as indicated by the trajectory. A next bounce wdestroy the potential rainbow.~c! This potential energy surface steers themolecule from the N-end-down orientation. There exist initial orientationfor which the molecule does not feel the chemisorption well along the trjectory. One such trajectory is indicated. In this potential rotational rainboware likely to survive the scrambling effect of the potential well.
2, No. 9, 1 March 1995t¬to¬AIP¬license¬or¬copyright,¬see¬http://jcp.aip.org/jcp/copyright.jsp

e
i-
n
,
.
.
.
.
,
.
,
.
3846 Wiskerke et al.: Rotational excitation of NO from Pt
well, i.e., scatter before the N-side faces the surface.‘‘short range’’ energy well, however, cannot account for thvery large values ofS0 , which indicate that the majority ofimpact trajectories result in experiencing the attractive weRotationally mediated adsorption requires a large rotationsteering which would be less probable with a ‘‘sharply decaying’’ attractive potential. Therefore, the topology of thpotential in Fig. 9~b! might be incorrect.
A PES which gives rise to a rotational rainbow for O-encollisions and RMA for N-end collisions has to steer thmolecule away from the N-end orientations for initial O-enapproaches.
The bottom picture@Fig. 9~c!# shows a PES which givesrise to a rotational rainbow with the same steric effect aobserved in the experiments. In this case the trajectoavoids the deep potential well, because the collision causthe molecule to orient O-end down. For this potential, thturning point for molecules approaching with the O-endown~having the symmetry axis along the surface normal! isfurther out than for molecules oriented along the surfacThis potential describes a ‘‘disklike’’ molecule rather than a‘‘egglike’’ molecule. In the energy range of the present experiments, the repulsive part of the potential is being pushback at the N-end, due to the attractive interaction. The ptential which has been proposed for this system by Dorand Tully59 shows a similar curve in the contour lines foV50.2 eV. Our measurements indicate higher potential eergy contours to be disk shaped as well. A secondary mimum for the O-end-down configuration may be a result ofweaker surface–molecule attraction. Calculations with sua potential describe the data qualitatively.42 Recently takenHREELS data indicate that energy minima for both thN-end and O-end orientations exist at the NO–Ru~001!system.60 In addition, the orientational barrier betweenbound N-end and nonbound O-end configurations is constent with the static binding of NO on Pt~111!.
VI. CONCLUSIONS
Despite strong chemisorption, clear rotational rainboware observed for NO–Pt~111! scattering. The most pro-nounced rotational rainbow is observed forEi50.3 eV,Q f570°. It is mainly under these conditions that scatterinvia the less reactive O-end contributes to the signal. Shapronounced rainbows are visible, due to selective removaltrajectories which lead to trapping. AtEi51.6 eV, the stick-ing coefficient is reduced to 0.5, allowing a higher proportioof these molecules to escape the potential well after a coplicated interaction. These molecules undergo complicatinteractions, leading to a Boltzmann-type rotational state dtribution. These trajectories overwhelm the rotational rainbow scattering at higher initial energies. The potential goerning NO–Pt~111! is likely to exhibit repulsive anisotropywhich causes rotational excitation; a second, weaker attrtive well associated with the O-end-down configuratiowhich causes the O-end rainbow.
J. Chem. Phys., Vol. 102Downloaded¬29¬Apr¬2005¬to¬130.37.129.78.¬Redistribution¬subject
Ae
ll.al-e
ded
sryesed
e.n-edo-enrn-ni-ach
e
is-
s
grp,of
nm-edis--v-
ac-n
ACKNOWLEDGMENTS
These results are part of the Research Program of thStichting voor Fundamenteel Onderzoek der Materie~Foun-dation for Fundamental Research on Matter! and was madepossible by financial support from the Nederlandse Organsatie voor Wetenschappelijk Onderzoek~Netherlands Orga-nization for the Advancement of Research!. The participationof B. E. H. was made possible by a grant from the EuropeaScience Foundation.
1C. M. Comrie, W. H. Weinberg, and R. M. Lambert, Surf. Sci.57, 619~1976!.
2B. E. Hayden, Surf. Sci.131, 419 ~1983!.3N. Materer, A. Barbieri, D. Gardin, U. Starke, J. D. Batteas, M. A. vanHove, and G. A. Somorjai, Phys. Rev. B48, 2859~1993!.
4A. E. Wiskerke, C. A. Taatjes, A. W. Kleyn, R. J. W. E. Lahaye, S. StolteD. K. Bronnikov, and B. E. Hayden, Chem. Phys. Lett.216, 93 ~1993!.
5D. C. Jacobs, K. W. Kolasinski, S. F. Shane, and R. N. Zare, J. ChemPhys.91, 3182~1989!.
6E. W. Kuipers, M. G. Tenner, A. W. Kleyn, and S. Stolte, Phys. Rev. Lett62, 2152~1989!.
7M. G. Tenner, E. W. Kuipers, A. W. Kleyn, and S. Stolte, J. Chem. Phys94, 5197~1991!.
8J. A. Serri, M. J. Cardillo, and G. E. Becker, J. Chem. Phys.77, 2175~1982!; J. A. Serri, J. C. Tully, and M. J. Cardillo,ibid. 79, 1530~1983!.
9M. Asscher, W. L. Guthrie, T. -H. Lin, and G. A. Somorjai, J. Chem. Phys78, 6992~1983!.
10J. K. Brown and A. C. Luntz Chem. Phys. Lett.204, 451 ~1993!.11J. Segner, H. Robota, W. Vielhaber, G. Ertl, F. Frenkel, J. Ha¨ger, W.Krieger, and H. Walther, Surf. Sci.131, 273 ~1983!.
12D. A. Mantell, R. R. Cavanagh, and D. S. King, J. Chem. Phys.84, 5131~1986!.
13J. A. Barker and D. J. Auerbach, Surf. Sci. Rep.4, 1 ~1984!.14J. Hager and H. Walther, Annu. Rev. Mater. Sci.19, 265 ~1989!.15A. W. Kleyn, and T. C. M. Horn, Phys. Rep.199, 191 ~1991!.16F. H. Geuzebroek, A. E. Wiskerke, M. G. Tenner, A. W. Kleyn, S. Stolteand A. Namiki, J. Phys. Chem.95, 8409 ~1991!; M. G. Tenner, F. H.Geuzebroek, E. W. Kuipers, A. E. Wiskerke, A. W. Kleyn, S. Stolte, and ANamiki, Chem. Phys. Lett.168, 45 ~1990!.
17M. A. Hines and R. N. Zare, J. Chem. Phys.98, 9134~1993!.18T. F. Hanisco, C. Yan, and A. C. Kummel, J. Chem. Phys.97, 1484~1992!.19G. O. Sitz, A. C. Kummel, and R. N. Zare, J. Chem. Phys.89, 2558
~1988!.20T. F. Hanisco and A. C. Kummel, J. Chem. Phys.99, 7076~1993!.21M. G. Tenner, E. W. Kuipers, A. W. Kleyn, and S. Stolte, Surf. Sci.242,376 ~1991!.
22E. W. Kuipers, M. G. Tenner, A. W. Kleyn, and S. Stolte, Surf. Sci.211/212, 819 ~1989!; Chem. Phys.138, 451 ~1989!.
23J. Harris, inDynamics of gas–surface interactions, edited by C. T. Rettnerand M. N. R. Ashfold~The Royal Society of Chemistry, Cambridge,1991!, p. 1.
24R. J. W. E. Lahaye, S. Stolte, A. W. Kleyn, R. J. Smith, and S. HollowaySurf. Sci.309, 187 ~1994!.
25J. Harris and A. C. Luntz, J. Chem. Phys.91, 6421~1989!.26D. C. Jacobs, and R. N. Zare, J. Chem. Phys.91, 3196~1989!.27J. C. Polanyi and R. J. Wolf, Ber. Bunsenges. Phys. Chem.86, 356~1982!;J. Chem. Phys.82, 1555~1985!.
28H. Voges and R. Schinke, Chem. Phys. Lett.100, 245 ~1983!.29A. E. Wiskerke and A. W. Kleyn~to be published!.30M. E. M. Spruit, E. W. Kuipers, M. G. Tenner, J. Kimman, and A. W.Kleyn, J. Vac. Sci. Technol. A5, 496 ~1987!.
31L. K. Verheij, M. B. Hugenschmidt, B. Poelsema, and G. Comsa, ChemPhys. Lett.174, 449 ~1990!.
32C. T. Campbell, G. Ertl, and J. Segner, Surf. Sci.115, 309 ~1982!.33J. Pfab and M. Hippler~1993!, Comment on Ref. 42, in Faraday Discuss.96, 354 ~1993!.
34C. T. Rettner, J. Kimman, and D. J. Auerbach, J. Chem. Phys.94, 734~1991!.
, No. 9, 1 March 1995¬to¬AIP¬license¬or¬copyright,¬see¬http://jcp.aip.org/jcp/copyright.jsp

r
m
te
3847Wiskerke et al.: Rotational excitation of NO from Pt
35D. C. Jacobs, R. J. Madix, and R. N. Zare, J. Chem. Phys.85, 5469~1986!.
36A. W. Kleyn, A. C. Luntz, and D. J. Auerbach, Phys. Rev. Lett.47, 1169~1981!; Surf. Sci.117, 33 ~1982!.
37C. A. Taatjes, A. E. Wiskerke, and A. W. Kleyn J. Chem. Phys.102, 3848~1995!.
38M. E. M. Spruit, P. J. van den Hoek, E. W. Kuipers, F. H. Geuzebroek, aA. W. Kleyn, Surf. Sci.214, 591 ~1989!.
39A. E. Wiskerke, F. H. Geuzebroek, A. W. Kleyn, and B. E. Hayden, SuSci. 272, 256 ~1992!.
40J. A. Barker, C. T. Rettner, and D. S. Bethune, Chem. Phys. Lett.188, 471~1992!; J. A. Barker and C. T. Rettner, J. Chem. Phys.97, 5844~1992!.
41U. van Slooten, D. R. Andersson, A. W. Kleyn, and E. A. Gislason, ChePhys. Lett.185, 440 ~1991!; Surf. Sci.274, 1 ~1992!.
42A. E. Wiskerke, C. A. Taatjes, A. W. Kleyn, R. J. W. E. Lahaye, S. StolD. K. Bronnikov, and B. E. Hayden, Faraday Discuss.96, 297 ~1993!.
43G. Nyman, L. Holmlid, and J. B. C. Petterson, J. Chem. Phys.93, 845~1990!.
44J. B. C. Pettersson, J. Chem. Phys.100, 2359~1994!.45G. Comsa and R. David, Surf. Sci. Rep.5, 145 ~1985!.46E. Hasselbrink, Chem. Phys. Lett.170, 329 ~1990!.
J. Chem. Phys., Vol. 102Downloaded¬29¬Apr¬2005¬to¬130.37.129.78.¬Redistribution¬subject
nd
f.
.
,
47K. Fukutani, A. Peremans, K. Mase, and Y. Murata, Surf. Sci.283, 158~1993!.
48T. F. Hanisco and A. C. Kummel, J. Vac. Sci. Technol. A11, 1907~1993!.49R. J. Behm and C. R. Brundle, J. Vac. Sci. Technol. A2, 1040~1984!.50C. W. Muhlhausen, J. A. Serri, J. C. Tully, G. E. Becker, and M. J. Car-dillo, Isr. J. Chem.22, 315 ~1982!.
51K. R. Lykke and B. D. Kay, J. Phys. Condens. Matter3, S65~1991!.52G. McElhiney, H. Papp, and J. Pritchard, Surf. Sci.54, 617 ~1976!.53R. A. Shigeishi and D. A. King, Surf. Sci.62, 379 ~1977!.54J. C. Lin, N. Shamir, Y. B. Zhao, and R. Gomer, Surf. Sci.231, 333
~1990!.55S. L. Tang, J. D. Beckerle, M. B. Lee, and S. T. Ceyer, J. Chem. Phys.84,6488 ~1986!.
56E. Zamir and R. D. Levine, Chem. Phys. Lett.104, 143 ~1984!.57C. Haug, W. Brenig, and T. Brunner, Surf. Sci.265, 56 ~1992!.58S. Tanaka and S. Sugano, Surf. Sci.136, 488 ~1984!.59D. J. Doren and J. C. Tully, Langmuir4, 256 ~1988!; J. Chem. Phys.94,8428 ~1991!.
60K. M. Neyman, N. Ro¨sch, K. L. Kostov, P. Jacob, and D. Menzel, J.Chem. Phys.100, 2310~1994!.
, No. 9, 1 March 1995¬to¬AIP¬license¬or¬copyright,¬see¬http://jcp.aip.org/jcp/copyright.jsp




















