
Theoretical investigation of CO adsorption on Pd(111) and Pd(111) --- Zn systems
8
Applied Surface Science 258 (2011) 1429–1436 Contents lists available at SciVerse ScienceDirect Applied Surface Science j our nal ho me p age: www.elsevier.com/loc ate/apsusc Theoretical investigation of CO adsorption on TM-doped (MgO) 12 (TM = Ni, Pd, Pt) nanotubes Mingxia Yang a , Yonghong Zhang b , Shiping Huang a,∗ , Hui Liu c , Peng Wang d , Huiping Tian d a Division of Molecule and Materials Simulation, State Key Laboratory of Organic-Inorganic Composites, Beijing University of Chemical Technology, Beijing 100029, China b Dept. of Applied Physics, Tianjin Polytechnic University, Tianjin 300160, China c State Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, China d Research Institute of Petroleum Processing, SINOPEC, Beijing 100083, China a r t i c l e i n f o Article history: Received 14 July 2011 Received in revised form 19 September 2011 Accepted 22 September 2011 Available online 29 September 2011 Keywords: CO adsorption TM-doped (MgO)12 nanotubes Density functional theory Chemisorption Electronic properties Vibrational frequencies a b s t r a c t CO adsorption on TM-doped magnesia nanotubes (TM = Ni, Pd and Pt) have been studied by using density functional theory. Our calculation results show that CO favors adsorption on TM-doped magnesia nan- otubes in the form of C atom bonding with TM atom. Fukui indices analysis clearly exhibits that doping of impurity TM atom allows for a noticeably enhancement of nucleophilic reactivity ability of magnesia nanotube. The adsorption energies demonstrate that CO molecule is more strongly bound on the 3-fold TM atoms than the 4-fold TM atoms. This finding is well confirmed by TM–C bond length, charge transfer and C–O vibrational frequency. The high adsorption energy of 2.55 eV is found when CO adsorbs on 3-fold Pt in Pt-doped magnesia nanotubes, implying the kind of the doping TM atom has a significant influence on the chemical reactivity. © 2011 Elsevier B.V. All rights reserved. 1. Introduction Detection of toxic gas molecules, such as carbon monoxide (CO), is of critical importance in the environment and human beings. In practice, metal oxides, as main gas sensors, are widely used for monitoring this task. Among the metal oxides, MgO plays an important role owing to its simple crystalline structure and easy preparation in experiment [1]. Recently, CO adsorption on MgO sur- faces has been extensively investigated both theoretically [2–11] and experimentally [12–20]. On the theoretical side, Pacchioni et al. [2–4] have treated the MgO surface by a cluster approach. He [8] and Staemmler [11] have reported that none (or at most only one) of the about 20 previous theoretical treatments of this presumably simplest adsorption system came close to the experimental value of the adsorption energy, 0.14 eV [17]. Nygren and Pettersson [5] obtained a CO adsorption energy of 0.08 eV with ab initio model potential (AIMP) embedding and extensive treatment of dynamic electron correlation. Illas and co-workers [7] using the three func- tional of M06-HF family (which contain a nonzero percentage of Hartree-Fock exchange-M06, M06-2x, and M06-HF) studied the adsorption of CO an MgO(0 0 1). It was shown that the results given ∗ Corresponding author. Fax: +86 10 64427616. E-mail address: [email protected] (S. Huang). by both standard and newly developed exchange-correlation func- tional are not completely satisfactory. By adopting a B3LYP+MP2 mixed scheme within a periodic ONIOM-like approach, Ugliengo and Damin [10] have found a binding energy of 0.13 eV and a quite relevant dispersive interactions (0.07 eV) for CO interacting with MgO surface. From the experimental point of view, the adsorption energies of the system of CO/MgO are measured just from 0.09 eV to 0.45 eV, which is a typical physisorption. MgO surfaces cannot detect CO well since the CO adsorbed weakly on the MgO surfaces. The neutral surface of the insulating MgO is no doubt one of the main reasons. Depositing additional materials on the well-defined MgO sur- faces has been shown to be suitable models for heterogeneous environmental catalysis [21–26], where the active additional mate- rials are known to be single atoms, size-selected metal atoms and clusters. Experimentally, small Au aggregates on clean MgO(1 0 0) surface and CO adsorption on this Au/MgO(1 0 0) system have been researched by Freund [22] through scanning tunneling spec- troscopy (STS) experiment. Theoretically, using the hybrid B3LYP functional within DFT calculation, Pacchioni et al. [23] have stud- ied the thermally stable and chemically active gold nanoclusters on MgO surface. Next, his co-works [24] reported the adsorption prop- erties of CO adsorbed on Au clusters supported on MgO/Ag(0 0 1) thin films with GGA-PW91 method in VASP program. They found that CO only binds to the low-coordinated Au atoms and a red 0169-4332/$ – see front matter © 2011 Elsevier B.V. All rights reserved. doi:10.1016/j.apsusc.2011.09.097
Transcript of Theoretical investigation of CO adsorption on Pd(111) and Pd(111) --- Zn systems

Tn
Ma
b
c
d
a
ARR1AA
KCTDCEV
1
iIfipfa[aosoopetHa
0d
Applied Surface Science 258 (2011) 1429– 1436
Contents lists available at SciVerse ScienceDirect
Applied Surface Science
j our nal ho me p age: www.elsev ier .com/ loc ate /apsusc
heoretical investigation of CO adsorption on TM-doped (MgO)12 (TM = Ni, Pd, Pt)anotubes
ingxia Yanga, Yonghong Zhangb, Shiping Huanga,∗, Hui Liuc, Peng Wangd, Huiping Tiand
Division of Molecule and Materials Simulation, State Key Laboratory of Organic-Inorganic Composites, Beijing University of Chemical Technology, Beijing 100029, ChinaDept. of Applied Physics, Tianjin Polytechnic University, Tianjin 300160, ChinaState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, ChinaResearch Institute of Petroleum Processing, SINOPEC, Beijing 100083, China
r t i c l e i n f o
rticle history:eceived 14 July 2011eceived in revised form9 September 2011ccepted 22 September 2011vailable online 29 September 2011
a b s t r a c t
CO adsorption on TM-doped magnesia nanotubes (TM = Ni, Pd and Pt) have been studied by using densityfunctional theory. Our calculation results show that CO favors adsorption on TM-doped magnesia nan-otubes in the form of C atom bonding with TM atom. Fukui indices analysis clearly exhibits that dopingof impurity TM atom allows for a noticeably enhancement of nucleophilic reactivity ability of magnesiananotube. The adsorption energies demonstrate that CO molecule is more strongly bound on the 3-foldTM atoms than the 4-fold TM atoms. This finding is well confirmed by TM–C bond length, charge transferand C–O vibrational frequency. The high adsorption energy of 2.55 eV is found when CO adsorbs on 3-fold
eywords:O adsorptionM-doped (MgO)12 nanotubesensity functional theoryhemisorptionlectronic properties
Pt in Pt-doped magnesia nanotubes, implying the kind of the doping TM atom has a significant influenceon the chemical reactivity.
© 2011 Elsevier B.V. All rights reserved.
ibrational frequencies
. Introduction
Detection of toxic gas molecules, such as carbon monoxide (CO),s of critical importance in the environment and human beings.n practice, metal oxides, as main gas sensors, are widely usedor monitoring this task. Among the metal oxides, MgO plays anmportant role owing to its simple crystalline structure and easyreparation in experiment [1]. Recently, CO adsorption on MgO sur-aces has been extensively investigated both theoretically [2–11]nd experimentally [12–20]. On the theoretical side, Pacchioni et al.2–4] have treated the MgO surface by a cluster approach. He [8]nd Staemmler [11] have reported that none (or at most only one)f the about 20 previous theoretical treatments of this presumablyimplest adsorption system came close to the experimental valuef the adsorption energy, 0.14 eV [17]. Nygren and Pettersson [5]btained a CO adsorption energy of 0.08 eV with ab initio modelotential (AIMP) embedding and extensive treatment of dynamiclectron correlation. Illas and co-workers [7] using the three func-
ional of M06-HF family (which contain a nonzero percentage ofartree-Fock exchange-M06, M06-2x, and M06-HF) studied thedsorption of CO an MgO(0 0 1). It was shown that the results given∗ Corresponding author. Fax: +86 10 64427616.E-mail address: [email protected] (S. Huang).
169-4332/$ – see front matter © 2011 Elsevier B.V. All rights reserved.oi:10.1016/j.apsusc.2011.09.097
by both standard and newly developed exchange-correlation func-tional are not completely satisfactory. By adopting a B3LYP+MP2mixed scheme within a periodic ONIOM-like approach, Ugliengoand Damin [10] have found a binding energy of 0.13 eV and a quiterelevant dispersive interactions (0.07 eV) for CO interacting withMgO surface. From the experimental point of view, the adsorptionenergies of the system of CO/MgO are measured just from 0.09 eVto 0.45 eV, which is a typical physisorption. MgO surfaces cannotdetect CO well since the CO adsorbed weakly on the MgO surfaces.The neutral surface of the insulating MgO is no doubt one of themain reasons.
Depositing additional materials on the well-defined MgO sur-faces has been shown to be suitable models for heterogeneousenvironmental catalysis [21–26], where the active additional mate-rials are known to be single atoms, size-selected metal atoms andclusters. Experimentally, small Au aggregates on clean MgO(1 0 0)surface and CO adsorption on this Au/MgO(1 0 0) system havebeen researched by Freund [22] through scanning tunneling spec-troscopy (STS) experiment. Theoretically, using the hybrid B3LYPfunctional within DFT calculation, Pacchioni et al. [23] have stud-ied the thermally stable and chemically active gold nanoclusters on
MgO surface. Next, his co-works [24] reported the adsorption prop-erties of CO adsorbed on Au clusters supported on MgO/Ag(0 0 1)thin films with GGA-PW91 method in VASP program. They foundthat CO only binds to the low-coordinated Au atoms and a red
1 ce Sci
sPMaattabse
otpsctgb
mwMfTtognot
2
2
sFooblrsstaosTfsTCnclawsMcM
430 M. Yang et al. / Applied Surfa
hift of about 50–60 cm−1 occurs. Grönbeck et al. [25] using GGA-BE method studied CO molecule adsorption on the modifiedgO(1 0 0) surface by supported Pd, Ag, Pt, and Au in the form of
toms. They found that there is a strong binding energy between COnd the supported metal atoms, and CO adsorption has increasedhe barrier of diffusion for metal atoms. Ferullo et al. [26] inves-igated the interaction of CO with Au atoms adsorbed on terracend low-coordinates sites (edge and corner) of MgO(1 0 0) surfacey using DFT-B3LYP method. They pointed out that CO adsorbstrongly on atomic Au deposited on anionic (O2−) sites with bindingnergies changing from 0.51 to 0.69 eV.
In this paper, the main aim is to perform a systematic studyn inducing CO in the modified (MgO)12 nanotubes by doping withransition metal atoms (TM) of Ni, Pd and Pt using DFT. For (MgO)12,revious theoretical studies indicated that the tubelike groundtate is about 0.12–0.66 eV higher in energy than the cage andubic isomers [27,28]. In experiment, observations from mass spec-ra [29–31] revealed that small MgO clusters prefer the nanotubeeometry over the bulklike structure. Thus (MgO)12 nanotube cane taken as a good model for the investigation of CO adsorption.
The rest of the paper is organized as follows. In Section 2, theodels and computational methods are introduced. In Section 3,e first focus our attention on the geometries, the stability, theulliken atomic charges, the HOMO–LUMO gaps and the Fukui
unctions of the TM-doped (MgO)12 nanotubes (TM = Ni, Pd and Pt).he detailed results are summarized in Section 3.1. More impor-antly, in Section 3.2, the properties of the CO molecule adsorptionn these TM-doped (MgO)12 nanotubes are systematically investi-ated. The interaction between CO and those TM-doped (MgO)12anotubes is analyzed in terms of adsorption energies, Mayer bondrders, electron transfer, density of states (DOS) and C–O vibra-ional frequency. In Section 4, the conclusion of the study is given.
. Models and computational methods
.1. Models
The pristine (MgO)12 nanotube is a barrel-like structure con-isting of four stacked hexagonal (MgO)3 sub-units as shown inig. S1 in the Supporting Information. It displays a high symmetryf D3h with all ions on the surface. The Mg–O bond lengths in theutmost layers are optimized at 1.915 A, while the average Mg–Oond lengths in the middle layers are about 2.064 A. For the inter-
ayers, the average Mg–O distances are optimized at 1.989 A. Theseesults are in good agreement with previous (MgO)12 nanotubetudies[32,33]. To construct the TM-doped (MgO)12 nanotubes, theubstitutional type is adopted. The substitutional models refer tohe models obtained by replacing a surface magnesium atom with
TM atom. From our calculated results we know that the nan-tube has only two kinds of coordinated sites, edge 3c and planarurface 4c sites (seen in Fig. S1), and all ions are on the surface.hus, only two substitutional sites are investigated: the edge 3-old Mg and the planar surface 4-fold Mg sites. For the sake ofimplicity, the two substitutional models are signified by TM-1 andM-2, the corresponding CO adsorption models are defined as TM1-O and TM2-CO. The models of CO adsorption on pure (MgO)12anotube are named by Mg1-CO and Mg2-CO. Next, we have cal-ulated the main features of the isolated CO molecule. The C–O bondength for an isolated CO molecule is optimized to be 1.142 A, whichgrees well with the experimental value of 1.128 A [34]. Comparedith the experimental value of 2170 cm−1 [34], our calculated C–O
tretching frequency for the isolated CO molecule is 2101 cm−1.eanwhile, the dipole moment of CO is 0.135 D in the present work
ompared to the available experimental data of 0.122 D [34] andullliken atomic charge of the carbon atom is +0.101 |e|.
ence 258 (2011) 1429– 1436
2.2. Computational methods
Full calculations are carried out with the DMol3 software [35],which is based on the density functional theory. The generalized-gradient approximation (GGA) of Perdew, Burke, and Ernzerhof(PBE) exchange-correlation functional [36] is used. Double numer-ical basis sets including polarization functions (DNP) [37,38] areperformed to describe the valence orbitals of all the atoms in ourcalculations. To describe the cores, all-electron relativistic calcu-lations are used for pure magnesia clusters and free CO molecule,which are time-consuming for transition metal atoms. Thus, theDFT-based semi-core pseudopotentials treatment is chosen for theclusters containing TM atom. Full structures are optimized withoutsymmetry constraints. The global cutoff radius of all the clustersis set to be 4.9 A, and the smearing is 0.005 hartree. The conver-gence criteria applied during geometry optimization are set to10−5 hartree for energy. In order to accelerate SCF convergence, weuse Pulay’s direct inversion of iterative subspace (DIIS) technique. A� point of 1 × 1 × 1 has been taken into account when calculate theelectronic density of states of the TM-doped and CO adsorbed mag-nesia nanotubes. In addition, our calculations employ the harmonicvibrational frequency computation to verify that all the structuresare true corresponded to the minima on the potential energy sur-face. Basis set superposition error (BSSE) have been carried out tocorrect the effect of BSSE for the CO adsorption energies using theGaussian03 code [39]. The 6-31G(d) basis set is used for Mg, O andC atoms. The LANL2DZ basis set is adopted for the valence electronsof Ni, Pd and Pt atoms, which core electrons are represented by theLANL2DZ effective core potential (ECP).
3. Results and discussion
3.1. TM-doped (MgO)12 nanotubes
3.1.1. Geometric structures and stabilitiesIn Fig. 1, the equilibrium geometries of TM-doped (MgO)12 nan-
otubes are presented. The corresponding bond lengths of thesenanotubes are summarized in Fig. S2 in the Supporting Information.For Ni-1 nanotube, there are two shortened Ni-O distance of 1.850 Ain the outermost layer, compared to the pristine Mg–O distance of1.919 A. These relatively shorter Ni–O distances might stem fromthe strong covalent bonding between the Ni and O atoms (theirMayer bond orders are around 0.85 while the Mg–O ones are about0.59). For Pd-1 and Pt-1 nanotubes, the average TM–O bond lengthsare calculated to be 2.065 and 2.284 A, which are larger than thecorresponding average Mg–O bond lengths of 1.933 A. Clearly, thedoping of TM atoms lead to the structural distortion for pristine(MgO)12 nanotube, because the bond distances of TM–O are longerthan that of the pristine Mg–O with the exception of TM = Ni. Insome cases, the TM-2 nanotubes even undergo tremendous distor-tion because the O atoms around substitutional sites are protrudedby TM atoms as can be seen from Fig. S2.
In order to analyze the relative stability of these TM-doped(MgO)12 nanotubes, the averaged binding energies per atom (Eb)are estimated using the following equation:
Eb = (11 × EMg + 12 × EO + ETM − Etotal)24
(1)
where EMg, EO and ETM are the total energy of isolated Mg, Oand TM (TM = Ni, Pd, Pt) atoms, respectively; Etotal is the totalenergy of the corresponding system. The calculated results arelisted in Table 1. One can see that the average binding energy of
the pristine (MgO)12 nanotube is 2.91 eV, which is good consistwith the previous studies [33]. For the stable TM-doped (MgO)12configurations, the calculated average binding energies are in therange of 2.82–2.91 eV. These results indicate that there are rather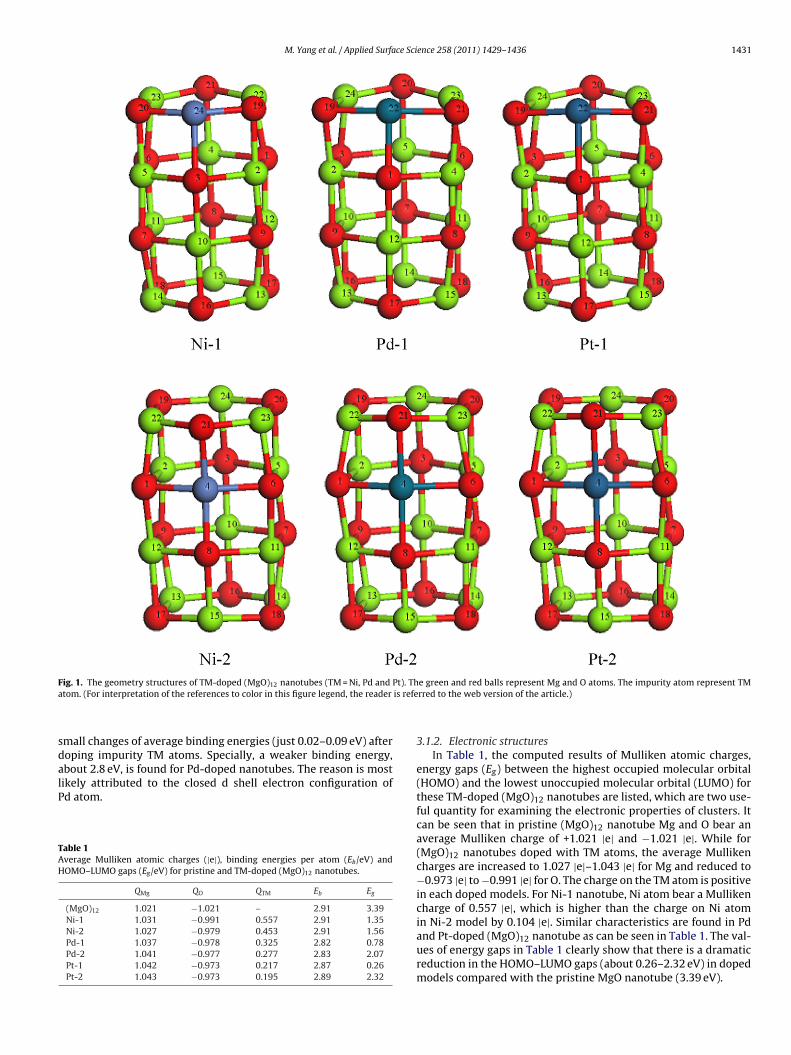
M. Yang et al. / Applied Surface Science 258 (2011) 1429– 1436 1431
Fig. 1. The geometry structures of TM-doped (MgO) nanotubes (TM = Ni, Pd and Pt). The green and red balls represent Mg and O atoms. The impurity atom represent TMa is refe
sdalP
TAH
12
tom. (For interpretation of the references to color in this figure legend, the reader
mall changes of average binding energies (just 0.02–0.09 eV) afteroping impurity TM atoms. Specially, a weaker binding energy,
bout 2.8 eV, is found for Pd-doped nanotubes. The reason is mostikely attributed to the closed d shell electron configuration ofd atom.able 1verage Mulliken atomic charges (|e|), binding energies per atom (Eb/eV) andOMO–LUMO gaps (Eg/eV) for pristine and TM-doped (MgO)12 nanotubes.
QMg QO QTM Eb Eg
(MgO)12 1.021 −1.021 – 2.91 3.39Ni-1 1.031 −0.991 0.557 2.91 1.35Ni-2 1.027 −0.979 0.453 2.91 1.56Pd-1 1.037 −0.978 0.325 2.82 0.78Pd-2 1.041 −0.977 0.277 2.83 2.07Pt-1 1.042 −0.973 0.217 2.87 0.26Pt-2 1.043 −0.973 0.195 2.89 2.32
rred to the web version of the article.)
3.1.2. Electronic structuresIn Table 1, the computed results of Mulliken atomic charges,
energy gaps (Eg) between the highest occupied molecular orbital(HOMO) and the lowest unoccupied molecular orbital (LUMO) forthese TM-doped (MgO)12 nanotubes are listed, which are two use-ful quantity for examining the electronic properties of clusters. Itcan be seen that in pristine (MgO)12 nanotube Mg and O bear anaverage Mulliken charge of +1.021 |e| and −1.021 |e|. While for(MgO)12 nanotubes doped with TM atoms, the average Mullikencharges are increased to 1.027 |e|–1.043 |e| for Mg and reduced to−0.973 |e| to −0.991 |e| for O. The charge on the TM atom is positivein each doped models. For Ni-1 nanotube, Ni atom bear a Mullikencharge of 0.557 |e|, which is higher than the charge on Ni atomin Ni-2 model by 0.104 |e|. Similar characteristics are found in Pd
and Pt-doped (MgO)12 nanotube as can be seen in Table 1. The val-ues of energy gaps in Table 1 clearly show that there is a dramaticreduction in the HOMO–LUMO gaps (about 0.26–2.32 eV) in dopedmodels compared with the pristine MgO nanotube (3.39 eV).
1 ce Science 258 (2011) 1429– 1436
3
aias[
f
f
weswl(fsf(
nfio(aoadoao(anttTe(tm
3
3
pttf2mprwnwoTPoPv
432 M. Yang et al. / Applied Surfa
.1.3. Fukui indicesSince one of the major objectives of this work is to investigate the
dsorption between CO and TM-doped (MgO)12 nanotubes, Fukuindices [40] are calculated to predict the possible binding site ofn CO molecule on a TM-doped (MgO)12 nanotube. In general, for aystem with N electrons, the two Fukui functions proposed by Yang41] are
+k
= qk(N + 1) − qk(N) (3)
−k
= qk(N) − qk(N − 1) (4)
here qk represents the electronic population of atom k in a consid-red system, and N, N + 1, N − 1 are the number of electrons in theystem, respectively. The two Fukui functions provide a successfulay of measuring the reactivity of regions of clusters [42–44]. The
ocal site with high f +k
value will easily react with electron donorsnucleophiles, such as CO molecule), whereas the local site where−k
is large will well react with electron acceptors (electrophiles,uch as O2 molecule). According to the value of Fukui indices (FI)rom high to low, a part of the calculated results for TM-dopedMgO)12 nanotubes are shown in Table 2.
In the pristine (MgO)12 nanotube, Mg atoms labeled with arabicumeral 13, 14, 15, 22, 23, 24 in Fig. 1 are the most favorable sites
or nucleophilic attack. In the Ni-1 nanotube, FI indicates that thempurity Ni atom (numbered 24) is the most reactive site for nucle-philic guest molecule absorption. On the contrary, the Ni atomnumbered 4) in Ni-2 nanotube is more favored for electrophilicttack. But considering its higher value of f +
k(0.124) than that
f other atoms in Ni-2 nanotube, it is also viable for nucleophilicttack. In the case of Pd-1, the Pd atom (numbered 22) does notisplay obvious differences in selectivity, preferring both nucle-philic and electrophilic attack. The same characteristic is shownt the Pd atom (numbered 4) in Pd-2 nanotube. As for Pt-1 nan-tube, the f +
kand f −
kvalues of Pt atom (numbered 22) is very close
0.248 and 0.243) and higher compared with other atoms, implyingmphiphilic character like Pd-1. In the case of Pt-2, Mg atom withumbered 15 will easily receives nucleophilic molecule absorp-ion, while electrophilic molecule adsorption will likely occur athe Pt atom (numbered 4). Furthermore, it is worth noting fromable 2 that the values of FI in TM atom sites in TM-doped mod-ls are absolutely larger than that of Mg and O atoms in pristineMgO)12 nanotube, implying that there is a noticeably improve inhe nucleophilic reactivity ability with a impurity TM atom in the
agnesia nanotube.
.2. CO adsorption on TM-doped (MgO)12 nanotubes
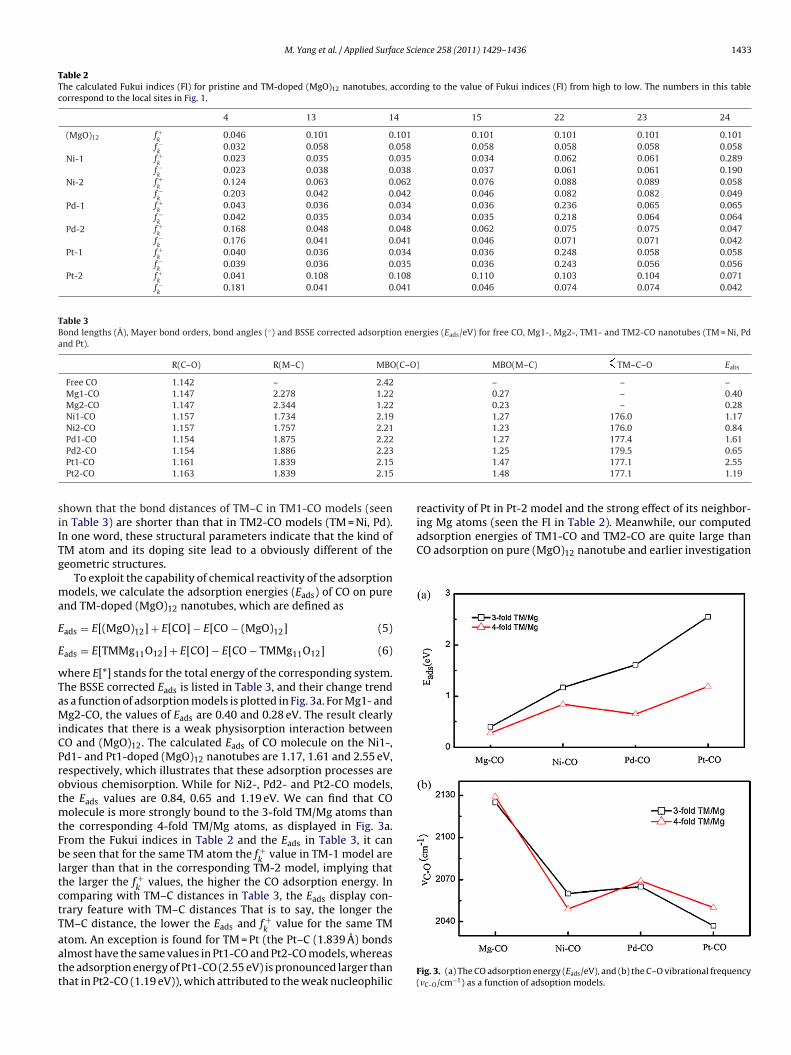
.2.1. Geometric structure and energy analysis of CO adsorptionFig. 2 depicts the most stable structures of CO adsorption on the
ure and TM-doped (MgO)12 nanotubes. The corresponding struc-ural parameters are listed in Table 3. From Fig. 2 we can see thathe C–O bond lengths are slightly shifted from 1.142 to 1.147 Aor Mg1-CO and Mg2-CO, and the Mg–C distances are 2.278 and.344 A. In the TM1- and TM2-CO models, the bond distances of COolecule are in the range of 1.154–1.163 A, which is elongated com-
ared with the bond length of isolated CO molecule, 1.142 A. Theseesults indicate that the C–O bonds in adsorbed models becomeeak by the impurity TM atoms. In addition, for Ni1-CO and Ni2-COanotubes, the Ni–C distances are optimized at 1.734 and 1.757 A,ith a same Ni–C–O angle of 176.0◦. For Pd1-CO and Pd2-CO nan-
tubes, the calculated Pd–C bond distances are 1.875 and 1.886 A.he optimized Pd–C–O angles are 177.4◦ and 179.5◦ in Pd1-CO and
d2-CO adsorption models. In the case of Pt1-CO and Pt2-CO nan-tubes, the Pt–C bond distances are optimized at 1.839 A with at–C–O angle of 177.1◦. These results are good in accord with pre-ious study from Grönbeck et al. [25]. Furthermore, it is obviouslyFig. 2. The geometry structures of Mg1-, Mg2-, TM1- and TM2-CO nanotubes(TM = Ni, Pd and Pt). The C–O and TM/Mg–C bond lengths (A) are noted.
M. Yang et al. / Applied Surface Science 258 (2011) 1429– 1436 1433
Table 2The calculated Fukui indices (FI) for pristine and TM-doped (MgO)12 nanotubes, according to the value of Fukui indices (FI) from high to low. The numbers in this tablecorrespond to the local sites in Fig. 1.
4 13 14 15 22 23 24
(MgO)12 f +k
0.046 0.101 0.101 0.101 0.101 0.101 0.101f −k
0.032 0.058 0.058 0.058 0.058 0.058 0.058Ni-1 f +
k0.023 0.035 0.035 0.034 0.062 0.061 0.289
f −k
0.023 0.038 0.038 0.037 0.061 0.061 0.190Ni-2 f +
k0.124 0.063 0.062 0.076 0.088 0.089 0.058
f −k
0.203 0.042 0.042 0.046 0.082 0.082 0.049Pd-1 f +
k0.043 0.036 0.034 0.036 0.236 0.065 0.065
f −k
0.042 0.035 0.034 0.035 0.218 0.064 0.064Pd-2 f +
k0.168 0.048 0.048 0.062 0.075 0.075 0.047
f −k
0.176 0.041 0.041 0.046 0.071 0.071 0.042Pt-1 f +
k0.040 0.036 0.034 0.036 0.248 0.058 0.058
f −k
0.039 0.036 0.035 0.036 0.243 0.056 0.056Pt-2 f +
k0.041 0.108 0.108 0.110 0.103 0.104 0.071
f −k
0.181 0.041 0.041 0.046 0.074 0.074 0.042
Table 3Bond lengths (A), Mayer bond orders, bond angles (◦) and BSSE corrected adsorption energies (Eads/eV) for free CO, Mg1-, Mg2-, TM1- and TM2-CO nanotubes (TM = Ni, Pdand Pt).
R(C–O) R(M–C) MBO(C–O) MBO(M–C) TM–C–O Eabs
Free CO 1.142 – 2.42 – – –Mg1-CO 1.147 2.278 1.22 0.27 – 0.40Mg2-CO 1.147 2.344 1.22 0.23 – 0.28Ni1-CO 1.157 1.734 2.19 1.27 176.0 1.17Ni2-CO 1.157 1.757 2.21 1.23 176.0 0.84Pd1-CO 1.154 1.875 2.22 1.27 177.4 1.61
siITg
ma
E
E
wTaMiCProtmtFbltctTaatt
ing Mg atoms (seen the FI in Table 2). Meanwhile, our computedadsorption energies of TM1-CO and TM2-CO are quite large thanCO adsorption on pure (MgO)12 nanotube and earlier investigation
Pd2-CO 1.154 1.886 2.23Pt1-CO 1.161 1.839 2.15Pt2-CO 1.163 1.839 2.15
hown that the bond distances of TM–C in TM1-CO models (seenn Table 3) are shorter than that in TM2-CO models (TM = Ni, Pd).n one word, these structural parameters indicate that the kind ofM atom and its doping site lead to a obviously different of theeometric structures.
To exploit the capability of chemical reactivity of the adsorptionodels, we calculate the adsorption energies (Eads) of CO on pure
nd TM-doped (MgO)12 nanotubes, which are defined as
ads = E[(MgO)12] + E[CO] − E[CO − (MgO)12] (5)
ads = E[TMMg11O12] + E[CO] − E[CO − TMMg11O12] (6)
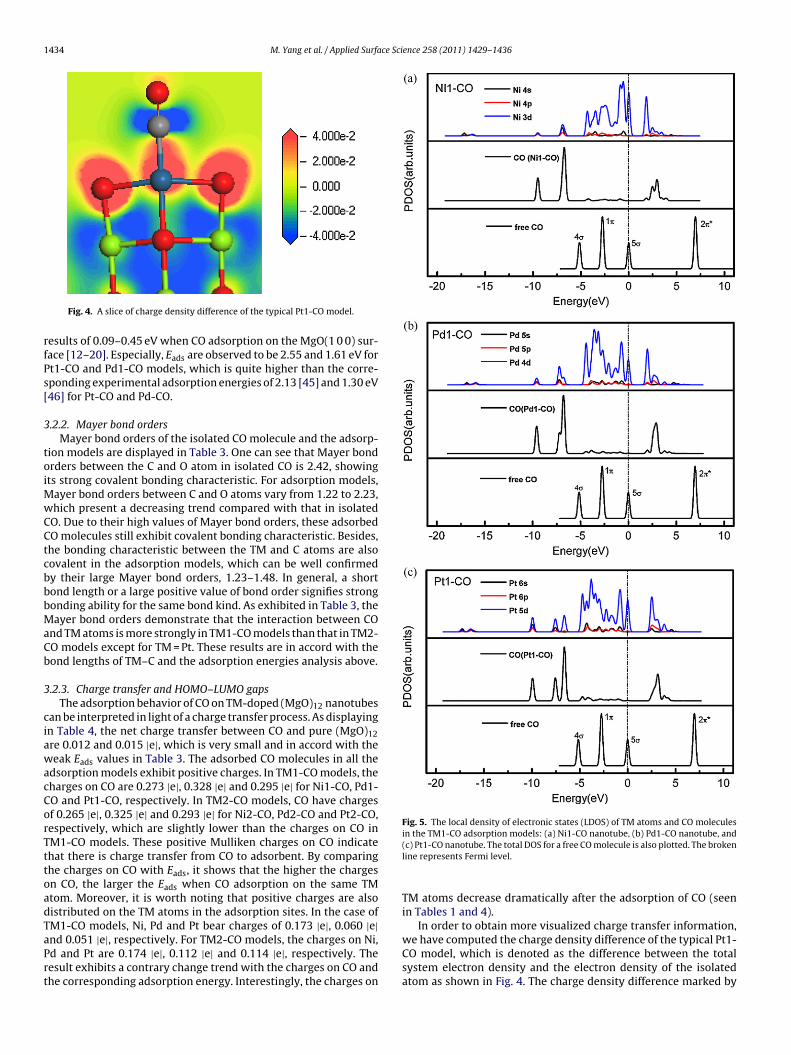
here E[*] stands for the total energy of the corresponding system.he BSSE corrected Eads is listed in Table 3, and their change trends a function of adsorption models is plotted in Fig. 3a. For Mg1- andg2-CO, the values of Eads are 0.40 and 0.28 eV. The result clearly
ndicates that there is a weak physisorption interaction betweenO and (MgO)12. The calculated Eads of CO molecule on the Ni1-,d1- and Pt1-doped (MgO)12 nanotubes are 1.17, 1.61 and 2.55 eV,espectively, which illustrates that these adsorption processes arebvious chemisorption. While for Ni2-, Pd2- and Pt2-CO models,he Eads values are 0.84, 0.65 and 1.19 eV. We can find that CO
olecule is more strongly bound to the 3-fold TM/Mg atoms thanhe corresponding 4-fold TM/Mg atoms, as displayed in Fig. 3a.rom the Fukui indices in Table 2 and the Eads in Table 3, it cane seen that for the same TM atom the f +
kvalue in TM-1 model are
arger than that in the corresponding TM-2 model, implying thathe larger the f +
kvalues, the higher the CO adsorption energy. In
omparing with TM–C distances in Table 3, the Eads display con-rary feature with TM–C distances That is to say, the longer theM–C distance, the lower the Eads and f +
kvalue for the same TM
tom. An exception is found for TM = Pt (the Pt–C (1.839 A) bondslmost have the same values in Pt1-CO and Pt2-CO models, whereashe adsorption energy of Pt1-CO (2.55 eV) is pronounced larger thanhat in Pt2-CO (1.19 eV)), which attributed to the weak nucleophilic
1.25 179.5 0.651.47 177.1 2.551.48 177.1 1.19
reactivity of Pt in Pt-2 model and the strong effect of its neighbor-
Fig. 3. (a) The CO adsorption energy (Eads/eV), and (b) the C–O vibrational frequency(�C–O/cm−1) as a function of adsoption models.
1434 M. Yang et al. / Applied Surface Science 258 (2011) 1429– 1436
rfPs[
3
toiMwCCtcbbbMaCb
3
ciawacCorTttoadTaPrt
Fig. 5. The local density of electronic states (LDOS) of TM atoms and CO moleculesin the TM1-CO adsorption models: (a) Ni1-CO nanotube, (b) Pd1-CO nanotube, and
Fig. 4. A slice of charge density difference of the typical Pt1-CO model.
esults of 0.09–0.45 eV when CO adsorption on the MgO(1 0 0) sur-ace [12–20]. Especially, Eads are observed to be 2.55 and 1.61 eV fort1-CO and Pd1-CO models, which is quite higher than the corre-ponding experimental adsorption energies of 2.13 [45] and 1.30 eV46] for Pt-CO and Pd-CO.
.2.2. Mayer bond ordersMayer bond orders of the isolated CO molecule and the adsorp-
ion models are displayed in Table 3. One can see that Mayer bondrders between the C and O atom in isolated CO is 2.42, showingts strong covalent bonding characteristic. For adsorption models,
ayer bond orders between C and O atoms vary from 1.22 to 2.23,hich present a decreasing trend compared with that in isolatedO. Due to their high values of Mayer bond orders, these adsorbedO molecules still exhibit covalent bonding characteristic. Besides,he bonding characteristic between the TM and C atoms are alsoovalent in the adsorption models, which can be well confirmedy their large Mayer bond orders, 1.23–1.48. In general, a shortond length or a large positive value of bond order signifies strongonding ability for the same bond kind. As exhibited in Table 3, theayer bond orders demonstrate that the interaction between CO
nd TM atoms is more strongly in TM1-CO models than that in TM2-O models except for TM = Pt. These results are in accord with theond lengths of TM–C and the adsorption energies analysis above.
.2.3. Charge transfer and HOMO–LUMO gapsThe adsorption behavior of CO on TM-doped (MgO)12 nanotubes
an be interpreted in light of a charge transfer process. As displayingn Table 4, the net charge transfer between CO and pure (MgO)12re 0.012 and 0.015 |e|, which is very small and in accord with theeak Eads values in Table 3. The adsorbed CO molecules in all the
dsorption models exhibit positive charges. In TM1-CO models, theharges on CO are 0.273 |e|, 0.328 |e| and 0.295 |e| for Ni1-CO, Pd1-O and Pt1-CO, respectively. In TM2-CO models, CO have chargesf 0.265 |e|, 0.325 |e| and 0.293 |e| for Ni2-CO, Pd2-CO and Pt2-CO,espectively, which are slightly lower than the charges on CO inM1-CO models. These positive Mulliken charges on CO indicatehat there is charge transfer from CO to adsorbent. By comparinghe charges on CO with Eads, it shows that the higher the chargesn CO, the larger the Eads when CO adsorption on the same TMtom. Moreover, it is worth noting that positive charges are alsoistributed on the TM atoms in the adsorption sites. In the case ofM1-CO models, Ni, Pd and Pt bear charges of 0.173 |e|, 0.060 |e|
nd 0.051 |e|, respectively. For TM2-CO models, the charges on Ni,d and Pt are 0.174 |e|, 0.112 |e| and 0.114 |e|, respectively. Theesult exhibits a contrary change trend with the charges on CO andhe corresponding adsorption energy. Interestingly, the charges on(c) Pt1-CO nanotube. The total DOS for a free CO molecule is also plotted. The brokenline represents Fermi level.
TM atoms decrease dramatically after the adsorption of CO (seenin Tables 1 and 4).
In order to obtain more visualized charge transfer information,we have computed the charge density difference of the typical Pt1-
CO model, which is denoted as the difference between the totalsystem electron density and the electron density of the isolatedatom as shown in Fig. 4. The charge density difference marked by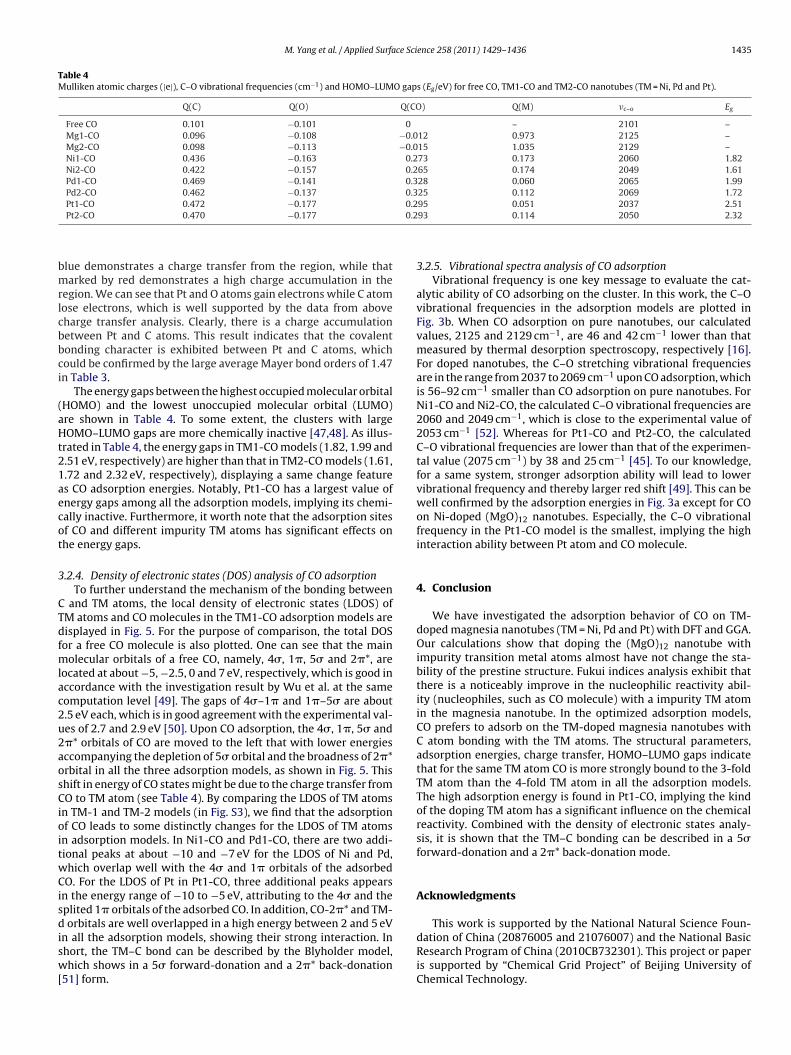
M. Yang et al. / Applied Surface Science 258 (2011) 1429– 1436 1435
Table 4Mulliken atomic charges (|e|), C–O vibrational frequencies (cm−1) and HOMO–LUMO gaps (Eg/eV) for free CO, TM1-CO and TM2-CO nanotubes (TM = Ni, Pd and Pt).
Q(C) Q(O) Q(CO) Q(M) �c–o Eg
Free CO 0.101 −0.101 0 – 2101 –Mg1-CO 0.096 −0.108 −0.012 0.973 2125 –Mg2-CO 0.098 −0.113 −0.015 1.035 2129 –Ni1-CO 0.436 −0.163 0.273 0.173 2060 1.82Ni2-CO 0.422 −0.157 0.265 0.174 2049 1.61Pd1-CO 0.469 −0.141 0.328 0.060 2065 1.99Pd2-CO 0.462 −0.137 0.325 0.112 2069 1.72Pt1-CO 0.472 −0.177 0.295 0.051 2037 2.51
0.2
bmrlcbbci
(aHt21aecot
3
CTdfmlac2u2aosCioitwCisdisw[
Pt2-CO 0.470 −0.177
lue demonstrates a charge transfer from the region, while thatarked by red demonstrates a high charge accumulation in the
egion. We can see that Pt and O atoms gain electrons while C atomose electrons, which is well supported by the data from aboveharge transfer analysis. Clearly, there is a charge accumulationetween Pt and C atoms. This result indicates that the covalentonding character is exhibited between Pt and C atoms, whichould be confirmed by the large average Mayer bond orders of 1.47n Table 3.
The energy gaps between the highest occupied molecular orbitalHOMO) and the lowest unoccupied molecular orbital (LUMO)re shown in Table 4. To some extent, the clusters with largeOMO–LUMO gaps are more chemically inactive [47,48]. As illus-
rated in Table 4, the energy gaps in TM1-CO models (1.82, 1.99 and.51 eV, respectively) are higher than that in TM2-CO models (1.61,.72 and 2.32 eV, respectively), displaying a same change features CO adsorption energies. Notably, Pt1-CO has a largest value ofnergy gaps among all the adsorption models, implying its chemi-ally inactive. Furthermore, it worth note that the adsorption sitesf CO and different impurity TM atoms has significant effects onhe energy gaps.
.2.4. Density of electronic states (DOS) analysis of CO adsorptionTo further understand the mechanism of the bonding between
and TM atoms, the local density of electronic states (LDOS) ofM atoms and CO molecules in the TM1-CO adsorption models areisplayed in Fig. 5. For the purpose of comparison, the total DOSor a free CO molecule is also plotted. One can see that the main
olecular orbitals of a free CO, namely, 4�, 1�, 5� and 2�*, areocated at about −5, −2.5, 0 and 7 eV, respectively, which is good inccordance with the investigation result by Wu et al. at the sameomputation level [49]. The gaps of 4�–1� and 1�–5� are about.5 eV each, which is in good agreement with the experimental val-es of 2.7 and 2.9 eV [50]. Upon CO adsorption, the 4�, 1�, 5� and�* orbitals of CO are moved to the left that with lower energiesccompanying the depletion of 5� orbital and the broadness of 2�*rbital in all the three adsorption models, as shown in Fig. 5. Thishift in energy of CO states might be due to the charge transfer fromO to TM atom (see Table 4). By comparing the LDOS of TM atoms
n TM-1 and TM-2 models (in Fig. S3), we find that the adsorptionf CO leads to some distinctly changes for the LDOS of TM atomsn adsorption models. In Ni1-CO and Pd1-CO, there are two addi-ional peaks at about −10 and −7 eV for the LDOS of Ni and Pd,hich overlap well with the 4� and 1� orbitals of the adsorbedO. For the LDOS of Pt in Pt1-CO, three additional peaks appears
n the energy range of −10 to −5 eV, attributing to the 4� and theplited 1� orbitals of the adsorbed CO. In addition, CO-2�* and TM-
orbitals are well overlapped in a high energy between 2 and 5 eV
n all the adsorption models, showing their strong interaction. Inhort, the TM–C bond can be described by the Blyholder model,hich shows in a 5� forward-donation and a 2�* back-donation51] form.
93 0.114 2050 2.32
3.2.5. Vibrational spectra analysis of CO adsorptionVibrational frequency is one key message to evaluate the cat-
alytic ability of CO adsorbing on the cluster. In this work, the C–Ovibrational frequencies in the adsorption models are plotted inFig. 3b. When CO adsorption on pure nanotubes, our calculatedvalues, 2125 and 2129 cm−1, are 46 and 42 cm−1 lower than thatmeasured by thermal desorption spectroscopy, respectively [16].For doped nanotubes, the C–O stretching vibrational frequenciesare in the range from 2037 to 2069 cm−1 upon CO adsorption, whichis 56–92 cm−1 smaller than CO adsorption on pure nanotubes. ForNi1-CO and Ni2-CO, the calculated C–O vibrational frequencies are2060 and 2049 cm−1, which is close to the experimental value of2053 cm−1 [52]. Whereas for Pt1-CO and Pt2-CO, the calculatedC–O vibrational frequencies are lower than that of the experimen-tal value (2075 cm−1) by 38 and 25 cm−1 [45]. To our knowledge,for a same system, stronger adsorption ability will lead to lowervibrational frequency and thereby larger red shift [49]. This can bewell confirmed by the adsorption energies in Fig. 3a except for COon Ni-doped (MgO)12 nanotubes. Especially, the C–O vibrationalfrequency in the Pt1-CO model is the smallest, implying the highinteraction ability between Pt atom and CO molecule.
4. Conclusion
We have investigated the adsorption behavior of CO on TM-doped magnesia nanotubes (TM = Ni, Pd and Pt) with DFT and GGA.Our calculations show that doping the (MgO)12 nanotube withimpurity transition metal atoms almost have not change the sta-bility of the prestine structure. Fukui indices analysis exhibit thatthere is a noticeably improve in the nucleophilic reactivity abil-ity (nucleophiles, such as CO molecule) with a impurity TM atomin the magnesia nanotube. In the optimized adsorption models,CO prefers to adsorb on the TM-doped magnesia nanotubes withC atom bonding with the TM atoms. The structural parameters,adsorption energies, charge transfer, HOMO–LUMO gaps indicatethat for the same TM atom CO is more strongly bound to the 3-foldTM atom than the 4-fold TM atom in all the adsorption models.The high adsorption energy is found in Pt1-CO, implying the kindof the doping TM atom has a significant influence on the chemicalreactivity. Combined with the density of electronic states analy-sis, it is shown that the TM–C bonding can be described in a 5�forward-donation and a 2�* back-donation mode.
Acknowledgments
This work is supported by the National Natural Science Foun-
dation of China (20876005 and 21076007) and the National BasicResearch Program of China (2010CB732301). This project or paperis supported by “Chemical Grid Project” of Beijing University ofChemical Technology.
1 ce Sci
A
t
R
[[[[
[[
[
[
[
[
[
[[[
[[[[[[[
[
[[
[
[[[[[
[[[
[
[[[[
436 M. Yang et al. / Applied Surfa
ppendix A. Supplementary data
Supplementary data associated with this article can be found, inhe online version, at doi:10.1016/j.apsusc.2011.09.097.
eferences
[1] C.T. Campbell, Surf. Sci. Rep. 27 (1997) 1.[2] G. Pacchioni, G. Cogliandro, P.S. Bagus, Surf. Sci. 255 (1991) 344.[3] C. Pacchioni, P.S. Bagus, in: H.-J. Freund, E. Umbach (Eds.), Adsorption on
Ordered Surfaces of Ionic Solids and Thin Films, vol. 33, Springer Series Surf.Sci., Springer-Verlag, Berlin, 1993, p. 180.
[4] G. Pacchioni, K.M. Neyman, N. Rösch, J. Electron Spectrosc. Relat. Phenom. 69(1994) 13.
[5] M.A. Nygren, L.G.M. Pettersson, J. Chem. Phys. 105 (1996) 9339.[6] F. Illas, G. Pacchinoi, A. Pelmenschikov, L.G.M. Pettersson, R. Dovesi, C. Pisani,
K. Neyman, N. Rösch, Chem. Phys. Lett. 306 (1999) 202.[7] R. Valero, J.R.B. Gomers, D.G. Truhlar, F. Illas, J. Chem. Phys. 129 (2008) 124710.[8] G. Pacchioni, Surf. Rev. Lett. 7 (2000) 277.[9] A. Damin, R. Dovesi, A. Zecchina, P. Ugliengo, Surf. Sci. 479 (2001) 255.10] P. Ugliengo, A. Damin, Chem. Phys. Lett. 366 (2002) 683.11] V. Staemmler, Top. Organomet. Chem. 12 (2005) 219.12] S. Furuyama, H. Fujii, M. Kawamura, T. Morimoto, J. Phys. Chem. 82 (1978) 1028.13] E.A. Paukshtis, R.I. Soltanov, E.N. Yurchenko, React. Kinet. Catal. Lett. 16 (1981)
93.14] C.R. Henry, C. Chapon, C. Duriez, J. Chem. Phys. 95 (1991) 700.15] J. He, C.A. Estrada, J.S. Corneille, M. Wu, D.W. Goodman, Surf. Sci. 261 (1992)
164.16] R. Wichtendahl, M. Rodriguez-Rodrigo, U. Härtel, H. Kuhlenbeck, H.J. Freund,
Surf. Sci. 423 (1999) 90.17] R. Wichtendahl, M. Rodriguez-Rodrigo, U. Härtel, H. Kuhlenbeck, H.J. Freund,
Phys. Status Solidi A 173 (1999) 93.18] Z. Dohnálek, G.A. Kimmel, S.A. Joyce, P. Ayotte, R.S. Smith, B.D. Kay, J. Phys.
Chem. B 105 (2001) 3747.19] G. Spoto, E. Gribov, A. Damin, G. Ricchiardi, A. Zecchina, Surf. Sci. 540 (2003)
L605.20] J.A. Rodriguez, T. Jirsak, M. Pérez, L. González, A. Maiti, J. Chem. Phys. 114 (2001)
4186.
[[[[[
ence 258 (2011) 1429– 1436
21] H.J. Freund, Surf. Sci. 500 (2002) 271.22] H.J. Freund, Catal. Today 117 (2006) 6.23] G. Pacchioni, S. Sicolo, C. Di Valentin, M. Chiesa, E. Giamello, J. Am. Chem. Soc.
130 (2008) 8690.24] S. Sicolo, L. Giordano, G. Pacchioni, J. Phys. Chem. C 113 (2009) 10256.25] H. Grönbeck, P. Broqvist, J. Phys. Chem. B 107 (2003) 12239.26] R.M. Ferullo, S.A. Fuente, P.G. Belelli, N.J. Castellani, Surf. Sci. 603 (2009) 1262.27] J. Carrasco, F. Illas, S.T. Bromley, Phys. Rev. Lett. 99 (2007) 235502.28] R.B. Dong, X.S. Chen, X.F. Wang, W. Lu, J. Chem. Phys. 129 (2008) 044705.29] G.W. Wang, H. Hattori, J. Chem. Soc., Faraday Trans. 1 (80) (1984) 1039.30] W.A. Saunders, Phys. Rev. B 37 (1988) 6583;
W.A. Saunders, Z. Phys. D 12 (1989) 601.31] D. van Heijnsbergen, G. von Helden, G. Meijer, M.A. Duncan, J. Chem. Phys. 116
(2002) 2400–2406, and references therein.32] R. Kakkar, P.N. Kapoor, K.J. Klabunde, J. Phys. Chem. B 110 (2006) 5941.33] L. Chen, C. Xu, X.F. Zhang, C. Cheng, T. Zhou, Int. J. Quantum Chem. 109 (2009)
349.34] K.P. Huber, G. Herzberg, Molecular Spectra and Molecular Structure. IV. Con-
stants of Diatomic Molecules, Van Nostrand Reinhold, New York, 1979, p. 166.35] B. Delley, J. Chem. Phys. 92 (1990) 508.36] J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865.37] B. Delley, J. Phys. Chem. 100 (1996) 6104.38] B. Delley, J. Chem. Phys. 113 (2000) 7756.39] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman,
et al., Gaussian 03, Revision C.02, Gaussian, Inc., Wallingford, CT, 2004.40] R.G. Parr, W. Yang, J. Am. Chem. Soc. 106 (1984) 4049.41] W. Yang, W. Mortier, J. Am. Chem. Soc. 108 (1986) 5708.42] E. Florez, W. Tiznado, F. Mondragón, P. Fuentealba, J. Phys. Chem. A 109 (2005)
7815.43] A. Poater, M. Duran, P. Toro-Labbé, A. Jaque, M. Solà, J. Phys. Chem. B 110 (2006)
6526.44] H. Sekhar De, S. Krishnamurty, S. Pal, J. Phys. Chem. C 114 (2010) 6690.45] A. Bourane, O. Dulaurent, D. Bianchi, J. Catal. 196 (2000) 115.46] J. Szanyi, W.K. Kuhn, D.W. Goodman, J. Vac. Sci. Technol. A 11 (1993) 1969.47] D. Die, X.Y. Kuang, J.J. Guo, B.X. Zheng, J. Phys. Chem. Solids 71 (2010) 770.
48] Y. Li, Y.J. Liu, D. Wu, Z.R. Li, Phys. Chem. Chem. Phys. 11 (2009) 5703.49] G.F. Wu, J.L. Wang, Y.M. Lu, M.L. Yang, J. Chem. Phys. 128 (2008) 224315.50] G. Pacchioni, A.M. Ferrari, P.S. Bagus, Surf. Sci. 350 (1996) 159.51] G. Blyholder, J. Phys. Chem. 68 (1964) 2772.52] S. Derrouiche, D. Bianchi, Appl. Catal. A: Gen. 313 (2006) 208.



















