
Artigo docking Zn BDC 2012
10
Metal Organic Frameworks for Drug Delivery and Environmental Remediation: A Molecular Docking Approach Marcelo O. Rodrigues, y Marcos V. de Paula, Kaline A. Wanderley, Iane B. Vasconcelos, Severino Alves, Jr., and Thereza A. Soares* Metal organic frameworks (MOFs) are promising materials for the controlled release of drugs. Molecular docking methods have been successfully applied to the fast screening of lead compounds starting from the three- dimensional structure of proteins. We apply molecular docking calculations to MOFs as a fast approach to distinguish between good and poor drug candidates for incorporation. The approach can predict the binding behavior of different guest molecules in agreement to experimental measurements using X-ray powder diffraction, thermogravimetry–differential thermal analysis, and UV–vis spectroscopy techniques. It can also identify the overall binding mode of tested compounds and estimate binding affinity differences above the error of 2 kcal mol 1 or 8.4 kJ mol 1 associated with the empirical scoring function used in the calculations. This exploratory investigation indicates that the molecular docking technique may be useful in the fast screening of drug candidates for adsorption to coordination polymers for controlled drug delivery and/or environmental remediation. V C 2012 Wiley Periodicals, Inc. DOI: 10.1002/qua.24211 Introduction Metal-organic frameworks (MOFs) are a novel class of crystal- line materials constructed by the means of self-assemblage of metal ions or their clusters interconnected by multifunctional organic ligands. [1–6] The wide interest in these materials has been motivated by the remarkable number of potential frame- work constructions as well as promising applications in gas technology, [7,8] catalysis, [9,10] drug delivery, [11–13] luminescent materials, [14] sensors, [15–18] and stationary phase for chroma- tography. [19–21] In particular, MOFs containing Zn 2þ cations and 1,4-benzenedicarboxylate (1,4-BDC 2 ) residues as linkers have been extensively studied through the pioneering contri- butions of Yaghi and coworkers. [22–25] The compound 1,4-BDC plays an important role in the three-dimensional arrangement of MOFs for it bridges metal centers that can adopt different coordination modes to form structures in 1D, 2D, and 3D (Fig. 1). [26–28] Depending on the resulting structure, the MOF skele- ton can be flexible, combining a high cavity volume, regular crystallinity, and the presence of tunable organic groups within the framework. These properties allow an easy modula- tion of the size of the layers, enabling a variety of specific applications. [29] [Zn(BDC)(H 2 O) 2 ] n stands out among 1,4-BDC- derived MOFs due to the fact that its supramolecular structure forms 1-D polymer chains interconnected by p–p stacking and a series of hydrogen bond interactions, whose layer sizes can in principle be modulated on adsorption of other molecules. MOFs exhibit many desired characteristics as drug carriers, including exceptionally high surface areas and large cavity sizes for drug encapsulation, intrinsic biodegradability as a result of relatively labile metal–ligand bonds, and versatile functionality for postsynthetic grafting of drug molecules. [13,30] These materials are made of hydrophilic and hydrophobic enti- ties with manifold possibilities of pore sizes and connectivities that can be adapted to the physical–chemical properties of each drug. Despite its recognition as potential drug car- riers, [11–13,31] little attention has been paid to the characteriza- tion of MOF–drug interactions at the atomic resolution and its impact on controlled drug liberation. In that regard, the ability to predict the binding conformation and binding affinity of a variety of guest molecules for a known framework is highly desirable. Computational simulations can offer a unique insight into the nature of host–guest interactions at the atomic level. [32,33] The molecular docking method allows the prediction of the structure of multicomponent complexes. It has been tradition- ally used to determine the structure of complexes between protein and small molecules, or between two proteins with a wide variety of computational programs publicly or commer- cially available. [34–39] The method has two major components: a search algorithm to sample the configurational and confor- mational degrees of freedom and a scoring function to evalu- ate the binding energies between the parts of the system. In flexible docking, the search algorithm explores different posi- tions for the guest molecule in the host framework using the M. O. Rodrigues, M. V. de Paula, K. A. Wanderley, I. B. Vasconcelos, S. Alves, Jr. and T. A. Soares Departament de Fundamental Chemistry, Universidade Federal de Pernambuco, Cidade Universit aria, Recife 50740-560, Brazil E-mail: [email protected] yPresent address: Instituto de Quı ´mica, Universidade de Brası ´lia, Brası ´lia, DF 70910-900, Brazil. Contract grant sponsor: FACEPE, CNPq, and INCT-INAMI. V C 2012 Wiley Periodicals, Inc. FULL PAPER WWW.Q-CHEM.ORG 3346 International Journal of Quantum Chemistry 2012, 112, 3346–3355 WWW.CHEMISTRYVIEWS.ORG
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Artigo docking Zn BDC 2012

Metal Organic Frameworks for Drug Delivery andEnvironmental Remediation: A Molecular DockingApproach
Marcelo O. Rodrigues,y Marcos V. de Paula, Kaline A. Wanderley, Iane B. Vasconcelos,Severino Alves, Jr., and Thereza A. Soares*
Metal organic frameworks (MOFs) are promising materials
for the controlled release of drugs. Molecular docking
methods have been successfully applied to the fast
screening of lead compounds starting from the three-
dimensional structure of proteins. We apply molecular
docking calculations to MOFs as a fast approach to
distinguish between good and poor drug candidates for
incorporation. The approach can predict the binding
behavior of different guest molecules in agreement to
experimental measurements using X-ray powder diffraction,
thermogravimetry–differential thermal analysis, and UV–vis
spectroscopy techniques. It can also identify the overall
binding mode of tested compounds and estimate binding
affinity differences above the error of 2 kcal mol�1 or 8.4 kJ
mol�1 associated with the empirical scoring function used
in the calculations. This exploratory investigation indicates
that the molecular docking technique may be useful in the
fast screening of drug candidates for adsorption to
coordination polymers for controlled drug delivery and/or
environmental remediation. VC 2012 Wiley Periodicals, Inc.
DOI: 10.1002/qua.24211
Introduction
Metal-organic frameworks (MOFs) are a novel class of crystal-
line materials constructed by the means of self-assemblage of
metal ions or their clusters interconnected by multifunctional
organic ligands.[1–6] The wide interest in these materials has
been motivated by the remarkable number of potential frame-
work constructions as well as promising applications in gas
technology,[7,8] catalysis,[9,10] drug delivery,[11–13] luminescent
materials,[14] sensors,[15–18] and stationary phase for chroma-
tography.[19–21] In particular, MOFs containing Zn2þ cations
and 1,4-benzenedicarboxylate (1,4-BDC2�) residues as linkers
have been extensively studied through the pioneering contri-
butions of Yaghi and coworkers.[22–25] The compound 1,4-BDC
plays an important role in the three-dimensional arrangement
of MOFs for it bridges metal centers that can adopt different
coordination modes to form structures in 1D, 2D, and 3D (Fig.
1).[26–28] Depending on the resulting structure, the MOF skele-
ton can be flexible, combining a high cavity volume, regular
crystallinity, and the presence of tunable organic groups
within the framework. These properties allow an easy modula-
tion of the size of the layers, enabling a variety of specific
applications.[29] [Zn(BDC)(H2O)2]n stands out among 1,4-BDC-
derived MOFs due to the fact that its supramolecular structure
forms 1-D polymer chains interconnected by p–p stacking and
a series of hydrogen bond interactions, whose layer sizes can
in principle be modulated on adsorption of other molecules.
MOFs exhibit many desired characteristics as drug carriers,
including exceptionally high surface areas and large cavity
sizes for drug encapsulation, intrinsic biodegradability as a
result of relatively labile metal–ligand bonds, and versatile
functionality for postsynthetic grafting of drug molecules.[13,30]
These materials are made of hydrophilic and hydrophobic enti-
ties with manifold possibilities of pore sizes and connectivities
that can be adapted to the physical–chemical properties of
each drug. Despite its recognition as potential drug car-
riers,[11–13,31] little attention has been paid to the characteriza-
tion of MOF–drug interactions at the atomic resolution and its
impact on controlled drug liberation. In that regard, the ability
to predict the binding conformation and binding affinity of a
variety of guest molecules for a known framework is highly
desirable.
Computational simulations can offer a unique insight into
the nature of host–guest interactions at the atomic level.[32,33]
The molecular docking method allows the prediction of the
structure of multicomponent complexes. It has been tradition-
ally used to determine the structure of complexes between
protein and small molecules, or between two proteins with a
wide variety of computational programs publicly or commer-
cially available.[34–39] The method has two major components:
a search algorithm to sample the configurational and confor-
mational degrees of freedom and a scoring function to evalu-
ate the binding energies between the parts of the system. In
flexible docking, the search algorithm explores different posi-
tions for the guest molecule in the host framework using the
M. O. Rodrigues, M. V. de Paula, K. A. Wanderley, I. B. Vasconcelos, S. Alves, Jr.
and T. A. Soares
Departament de Fundamental Chemistry, Universidade Federal de Pernambuco,
Cidade Universit�aria, Recife 50740-560, Brazil
E-mail: [email protected]
yPresent address: Instituto de Quımica, Universidade de Brasılia, Brasılia, DF
70910-900, Brazil.
Contract grant sponsor: FACEPE, CNPq, and INCT-INAMI.
VC 2012 Wiley Periodicals, Inc.
FULL PAPER WWW.Q-CHEM.ORG
3346 International Journal of Quantum Chemistry 2012, 112, 3346–3355 WWW.CHEMISTRYVIEWS.ORG

translational, rotational, and torsional degrees of freedom.
Scores are assigned to the sampled conformations based on
the intermolecular interaction energy, and each conformation
is ranked relative to other poses and other ligands.
A previous molecular docking study suggested that protein-
specific docking programs could predict the binding poses of
small guest molecules into porous crystalline materials based
on either strong interactions or shape-fitting.[40] These findings
imply that the molecular docking technique may be applied
to the virtual screening of potential candidates to immobiliza-
tion in host frameworks. However, some questions need to be
addressed before drug screening for incorporation in crystal-
line frameworks becomes reliably attainable: How dependent
is the performance of actual docking algorithms on the con-
cept of ‘‘molecular complementarity’’, a major feature in protein
recognition, but perhaps less effective for the comparatively
unspecific drug–MOF association? How accurately will scoring
functions designed to model biomolecule–ligand interactions
work? How important is the treatment of one copy versus
multiple copies of the drug for correct prediction of the struc-
ture of MOF–guest complexes? The answers to these questions
will require a systematic evaluation of the method and careful
comparison of calculated structures against the rather scant
crystallographic data for MOF complexes.
In this report, we investigate the potential of the molecular
docking method to distinguish between high- and low-affinity
MOF-binding drugs at different pH conditions. Molecular dock-
ing calculations were carried out to estimate the binding con-
formation and binding affinity of guest molecules (ibuprofen,
methylene blue, amoxicillin, and gentamicin) to the metal-or-
ganic framework [Zn(BDC)(H2O)2]n (Fig. 2). The adsorption (or
not) of these drugs onto the [Zn(BDC)(H2O)2]n framework was
characterized by the means of powder X-ray diffraction and
differential thermal analysis and used to validate the computa-
tional predictions against.
Methodology
Molecular docking calculations
The hybrid search method based on the Lamarckian genetic
algorithm (LGA) implemented in the suite of programs Auto-
Dock v4.0 was used in conjunction with the semiempirical free
energy force field for the molecular docking calculations.[34,41]
This energy function describes the energetics of the binding
process involving two molecules in an aqueous environment
using pair-wise terms to evaluate the interaction between the
two molecules coupled to an empirical method to estimate
the contribution of the surrounding solvent.[41] Hence, the free
energy of binding is given by (i) the difference between the
energy of the guest molecule and the crystalline framework in
a separated unbound state and (ii) the energy of the guest–
host complex. During the conformational search, the intramo-
lecular energetics of the transition from the unbound state to
the bound conformation is evaluated for each of the mole-
cules separately, and subsequently the intermolecular ener-
getics of bringing the two molecules together into the bound
complex is evaluated. The binding energy between two mole-
cules is given by
DG ¼ ðVdrug�drugbound � Vdrug�drug
unbound Þ þ ðVMOF�MOFbound � VMOF�MOF
unbound Þþ ðVdrug�MOF
bound � Vdrug�MOFunbound þ DSconfÞ (1)
Equation (1) includes four terms to describe the intramolec-
ular energies for the bound and unbound states of the guest
molecule and for the bound and unbound states of the host
framework, two terms to describe the change in
Figure 1. Dimensionalities adopted by crystalline framework structures con-
structed from 1,4-BDC and Zn2þ ions. (A) 1-D structure, (B) 2-D structure,
and (C) 3-D structure (MOF-5). The [Zn(BDC)(H2O)2]n in its 1-D structure con-
sists of approximately tetrahedral Zn2þ centers linked by a herringbone pat-
tern of 1,4-BDC molecules aligned approximately along the crystallographic
a/c diagonal. The lamellar structure is kept through offset p–p stacking and
hydrogen bond interactions between adjacent 1-D chains. [Color figure can
be viewed in the online issue, which is available at wileyonlinelibrary.com.]
FULL PAPERWWW.Q-CHEM.ORG
International Journal of Quantum Chemistry 2012, 112, 3346–3355 3347

intermolecular energy between the bound and unbound
states and a one term to represent an estimate of the confor-
mational entropy lost upon binding.[34,41] Pair-wise atomic
interactions account for dispersion/repulsion, hydrogen bond-
ing, electrostatics, and desolvation as described[3]
V ¼ Wwdw
Xij
Aij
r12ij
� Bijr6ij
!þWhbond
Xij
EðtÞ Cijr12ij
� Dij
r10ij
!
þWcoul
Xij
qiqieðrijÞrij
þWsol
Xij
ðSiVj þ SjViÞeð�r2ij=2r2Þ (2)
where W corresponds to weighting constants optimized to cal-
ibrate the empirical free energy based on a set of experimen-
tally characterized protein complexes.[41] The 6/12 potential
describes dispersion/repulsion interactions with parameters A
and B taken from the Amber force field.[42] The 10/12 potential
is a directional H-bond term whose interaction directionality
E(t) is dependent on the angle t away from ideal bonding ge-
ometry. A screened Coulomb potential is used to describe
electrostatic interactions. The fourth term is a desolvation
potential based on the volume (V) of the atoms surrounding a
given atom, weighted by a solvation parameter (S) and an ex-
ponential term based on the distance weighting r. Rotatable
bonds included all torsional degrees of freedom.[34,41]
All docking simulations were performed using the crystalline
structure of the [Zn(BDC)(H2O)2]n framework [CCSD number
182/1299].[27] During the conformational search, the ligands
were fully flexible concerning its degrees of translation, orien-
tation, and conformation with respect to the [Zn(BDC)(H2O)2]nstructure, which was kept rigid. Each sampled conformation
was evaluated and ranked according to the empirical energy
function [Eq. (2)]. Grid maps with 126 � 126 � 126 points of
dimension and a grid spacing of 0.25 A were calculated using
AutoGrid4.[43] Charges for ligands and the [Zn(BDC)(H2O)2]
framework unite were assigned via a restrained hyperbolic fit
of the electrostatic potential[44] on the nuclei positions of each
atom after geometry optimizations at the HF-6-31G* level
using the NWCHEM software.[45] Formal charges were assigned
to the Zn2þ metal ions. van der
Waals parameters were taken
from the Amber force field
available with the distribution
of AutoDock v4.0.[42] Electro-
static interactions were eval-
uated with a screened Coulomb
potential.[46] A desolvation
potential was used which is
based on the volume of the
atoms surrounding a given
atom, weighted by a solvation
parameter and an exponential
term based on the distance,
with a distance weighting factor
of 3.5 A.[47]
The LGA was used to perform
the conformational search fol-
lowing the same protocol previ-
ously described in Refs. [48,49]. Briefly, LGA parameters were
defined as follows: the initial population of random individuals
had a size of 150 individuals with a maximum number of 2.5
� 106 energy evaluations and a maximum number of genera-
tions equal to 27,000. An elitism rate of 1 was applied to
ensure that the top individual always survives into the next
generation in conjunction with mutation and crossover rates
of 0.02 and 0.08, respectively. A maximum of 300 iterations
per local search was used. The probability of performing a
local search on an individual was 0.06 where the maximum
number of consecutive successes or failures before doubling
or halving the search step was 4. The number of docking sim-
ulations performed for the guest molecules reflected the re-
spective number of degrees of freedom, that is, 100 steps (ibu-
profen and methylene blue) and 200 steps (amoxicillin and
gentamicin). The final lowest energy structures were clustered
based on a RMS positional deviation of 2.0 A.
Experimental Details
The Ibuprofen loading was performed by introducing, under
stirring for 3 days, 100 mg of the dehydrated powder material
(dried overnight at 150�C in an oven) in a 10 mL solution of
ethanol containing 300 mg of Ibuprofen. After drug immobili-
zation, the remaining ethanol was removed at 100�C. Amoxi-
cillin and gentamicin were loaded in the MOF by introducing,
under stirring for 7 days, 100 mg of the [Zn(BDC)(H2O)2]n ma-
terial in 10 mL aqueous solution (Milli-Q water) containing 300
mg of the drug. Adsorption of methylene blue was performed
by stirring 100 mg of the [Zn(BDC)(H2O)2]n material in a 5 mL
aqueous solution (10�3 M) of the drug during 7 days. Subse-
quently, the suspensions were filtered, and the resultant solids
were characterized by X-ray powder diffraction (XRPD) and
thermal analysis Thermogravimetry–differential thermal analy-
sis (TG–DTA) curves were acquired through a SHIMADZU DTG-
60H instrument in the range from 25 to 900�C using an alu-
mina crucible with about 8.0 mg of samples, under dynamic
nitrogen atmosphere (50 mL min�1) and with a heating rate
Figure 2. Chemical structures of ibuprofen, methylene blue, amoxicillin, and gentamicin.
FULL PAPER WWW.Q-CHEM.ORG
3348 International Journal of Quantum Chemistry 2012, 112, 3346–3355 WWW.CHEMISTRYVIEWS.ORG

of 10�C min�1. XRD analyses were performed at room temper-
ature, using a Bruker D8 Advanced with DaVinci design
equipped with a LynxEye Linear position sensitive detector
and a copper (Cu) sealed tube (kka1 ¼ 1.5404 A, kka2 ¼1.5444 A, Ia2/Ia1 ¼ 0.5). Intensity data were collected in step
scanning mode, in the range from 5 to 50� (2y), with a step
size of 0.01�, Soller slit with 2.5� of divergence, 0.5� scattering
slit, and 0.6-mm receiving slit. The Rietveld refinement[50] for
the drug–[Zn(BDC)(H2O)2]n complexes was performed with the
software GSAS/EXPGUI,[51,52] using the X-ray structure of the
uncomplexed MOF as initial atomic coordinates.[27,53] The pref-
erential orientation of the system was corrected using spheri-
cal harmonic model (sixth order) proposed by Jarvinen.[54] The
peak profile was adjusted by the Thompson–Cox–Hastings
function modified by Young and Desai (pV-TCHZ).[55] Surface
roughness correction was refined by the Pitschke function,
and the background was fitted by an eighth-degree shifted
Chebyshev polynomial function. The refined parameters in the
final runs were the scale factor, background and absorption
coefficients, spherical harmonic, unit-cell parameters, and pV-
TCHZ correction for asymmetric parameters.
Results and Discussion
Experimental characterization of the drug–[Zn(BDC)(H2O)2]ncomplexes
The immobilization of ibuprofen on the [Zn(BDC)(H2O)2]n was
ascertained by UV–vis spectroscopy (Fig. 3). This technique
was used to quantify the mass of ibuprofen adsorbed on the
[Zn(BDC)(H2O)2]n after 8 days. The calibration curve used for
that end shows that of 44.5% of ibuprofen was adsorbed on
the [Zn(BDC)(H2O)2]n (Fig. 3). Moreover, the incorporation of
ibuprofen into different 1,4-BDC-derived MOFs has been previ-
ously reported.[11,12,29,31] In the case of methylene blue, amoxi-
cillin, and gentamicin, evidence of drug incorporation was first
assessed by visual inspection of change in the coloration of
the MOF upon incorporation of the drug (Fig. 4). Hence, the
complex amoxicillin–[Zn(BDC)(H2O)2]n acquired a brown color,
whereas the methylene blue–[Zn(BDC)(H2O)2]n became blue.
The complex gentamicin–[Zn(BDC)(H2O)2]n did not exhibit any
alteration of color compared to the pure framework. The color-
less pattern characteristic of the [Zn(BDC)(H2O)2]n framework
remained unaltered during the 7 days, when it was kept
immersed in the gentamicin solution. Overall, these results
attest to the incorporation of amoxicillin and methylene blue
into the framework and suggest that gentamicin is not
Figure 3. Calibration curve obtained from the absorption spectrum in the
ultraviolet–visible (UV–vis) spectral region for the ibuprofen solution. The
red line represents the best linear fit profile.
Figure 4. XRD for (A) pure [Zn(BDC)(H2O)2]n (solid red line) and the amoxicil-
lin–[Zn(BDC)(H2O)2]n complex (black circles), (B) gentamicin–[Zn(BDC)(H2O)2]ncomplex, and (C) methylene blue–[Zn(BDC)(H2O)2]n complex. Rietveld refine-
ment of the XRD data was required for (B) and (C). The difference (green solid
line) and Braggs peaks (blue bars) arising from the Rietveld fit are shown. As
there is an unequivocal structural difference between pure [Zn(BDC)(H2O)2]nand the amoxicillin–[Zn(BDC)(H2O)2]n complex (A), the Rietveld refinement
was not performed for this system. [Color figure can be viewed in the online
issue, which is available at wileyonlinelibrary.com.]
FULL PAPERWWW.Q-CHEM.ORG
International Journal of Quantum Chemistry 2012, 112, 3346–3355 3349

incorporated into the material (Fig. 4). To establish or clarify
these findings, these samples were characterized by XRPD.
Significant changes of peak positions and relative intensities
were further observed for the complex amoxicillin–
[Zn(BDC)(H2O)2]n compared with their dissociated forms (Fig.
4), indicating that the interaction of amoxicillin onto MOF is
followed by drug degradation. This observation has been fur-
ther verified by the means of mass spectrometry analysis (de
Paula, Soares and Alves, Jr., unpublished results). The molecular
docking calculations indicate that the Zn2þ cations on the
framework surface are the most favorable interaction site for
amoxicillin (Fig. 8). This is also consistent with previous experi-
mental studies reporting on the role of Zn2þ-containing mate-
rials in the degradation of antibiotic molecules (amoxicillin,
ampicillin, penicillin-G, and penicillin-V) in aqueous solution
under natural light exposition.[56,57] However, XRPD patterns
for the methylene blue– and gentamicin–[Zn(BDC)(H2O)2]ncomplexes exhibited a complete overlap of the peaks, prevent-
ing proper determination of the structure (Fig. 4).
Rietveld analyses of the two complexes were carried to allow
a better separation of the peaks. The Rietveld method creates a
virtual separation of these overlapping peaks, thereby allowing
a more accurate determination of the components of the
chemical mixtures (for details about the technique see Refs.
[58,59]). The Rietveld refinement of the XRPD data for methyl-
ene blue–[Zn(BDC)(H2O)2]n and gentamicin–[Zn(BDC)(H2O)2]ncomplexes did not produce the separation of overlapping peaks
as intended. In fact, no significant change in the lattice parame-
ters of the [Zn(BDC)(H2O)2]n crystal or the presence of peaks
corresponding to the drugs after the adsorption experiments
were observed. These observations indicate that
[Zn(BDC)(H2O)2]n has an unique crystalline phase in both sam-
ples. These findings are consistent with the observation that
there is no visual difference between the powder obtained
from the [Zn(BDC)(H2O)2]n pure and in presence of the gentami-
cin solution, further suggesting that gentamicin does not
absorb to the [Zn(BDC)(H2O)2]n framework. However, the color
alteration observed for the methylene blue–[Zn(BDC)(H2O)2]npowder indicates that the complex is formed. As the characteri-
zation by XRPD diffraction has been unsuccessful in directly
identifying the adsorbed methylene blue in the framework ma-
terial possibly due to low quantities and/or positional disorder
of the adsorbed drugs, TGA/DTA analyses were performed to
unambiguously assess the presence of methylene blue in the
[Zn(BDC)(H2O)2]n surface (Fig. 5). These analyses were also
extended for the complex gentamicin–[Zn(BDC)(H2O)2]n to fur-
ther substantiate the XRPD and Rietveld refinement analysis.
The gentamicin–[Zn(BDC)(H2O)2]n and the uncomplexed
[Zn(BDC)(H2O)2]n exhibited similar DTA curves with three well-
defined exothermic peaks between 410 and 550�C, which cor-
respond to the thermal decomposition of the material. The
similar thermal behavior is indicative anew that the adsorption
of gentamicin onto the [Zn(BDC)(H2O)2]n surface did not take
place. In the case of the methylene blue–[Zn(BDC)(H2O)2]ncomplex, an endothermic peak was observed in the same tem-
perature range attributed to desorption of methylene blue or
its fragments concomitant to the degradation of the
[Zn(BDC)(H2O)2]n structure (Fig. 5). These results provide evi-
dence that methylene blue adsorbs onto the [Zn(BDC)(H2O)2]nsurface, in line with the observed color change of the
[Zn(BDC)(H2O)2]n material upon adsorption of the drug (Fig. 4).
The adsorption of drug to the [Zn(BDC)(H2O)2]n framework
The guest molecules used in this study were chosen to repre-
sent drugs of wide medical relevance and different complex-
ities (i.e., degrees of freedom) in the context of the conforma-
tional search algorithm. The trial set of molecules was
comprised of ibuprofen, methylene blue, amoxicillin, and gen-
tamicin. Ibuprofen is a widely used nonsteroidal anti-inflamma-
tory that inhibits the enzyme cyclo-oxygenase. This enzyme
converts arachidonic acid to prostaglandin H2, a precursor of
several other prostaglandins mediators of pain, inflammation,
and fever.[60,61] Methylene blue has been used in many differ-
ent areas of clinical medicine, from dementia to cancer chemo-
therapy.[62] It is also a potent antimalarial chemotherapeutic
that acts via competitive inhibition of the glutathione reduc-
tase of Plasmodium falciparum.[63–66] The photosensitizing
potential of methylene blue has also been recognized ena-
bling its application as antimicrobial agents for blood disinfec-
tion.[62] Amoxicillin is a moderate-spectrum b-lactam antibiotic
that inhibits the synthesis of the bacterial cell wall by a broad
range of Gram-positive and some Gram-negative bacteria.[67]
Amoxicillin has also been successfully used in the eradication
of Helicobacter pylori, a Gram-negative bacterium that can
inhabit different regions of the stomach. Currently, the most
efficient eradication treatment of this bacterium consists of
triple therapy using a proton pump inhibitor or ranitidine bis-
muth citrate, combined with clarithromycin and amoxicil-
lin.[68,69] Gentamicin is an aminoglycoside antibiotic used for
broad-spectrum treatment of various infections.[70,71] It has
Figure 5. TG–DTA curves for the uncomplexed [Zn(BDC)(H2O)2]n (black
line), gentamicin–[Zn(BDC)(H2O)2]n (red line), and methylene blue–
[Zn(BDC)(H2O)2]n (blue line) complexes. Peaks and dips represent exother-
mic and endothermic processes, respectively. The latter is indicated by the
down arrow symbol. The distinctive dip at 500�C observed for the methyl-
ene blue–[Zn(BDC)(H2O)2]n complex (blue line) corresponds to the methyl-
ene blue dissociation event. [Color figure can be viewed in the online
issue, which is available at wileyonlinelibrary.com.]
FULL PAPER WWW.Q-CHEM.ORG
3350 International Journal of Quantum Chemistry 2012, 112, 3346–3355 WWW.CHEMISTRYVIEWS.ORG

been the core of antibacterial therapy for serious Gram-nega-
tive infections. More recently, aminoglycoside antibiotics have
become important tools to study molecular recognition of
ribonucleic acid by exploiting their mechanism of action.[72]
Guest molecules were docked onto the [Zn(BDC)(H2O)2]n struc-
tures, and the lowest energy conformer was selected for each of
the compounds tested (Figs. 5–7). The lowest energy conformer is
defined as the conformation of the ligand with the most favorable
interaction energy as given by an empirical energy function[41] and
selected out of a about 106 sampled conformations.[34,48,49,73] For
the studied drug molecules, the lowest energy conformation cor-
responded also to the most populated cluster of structures based
on a RMSD of 2.0 A. Overall, only one predominant conformation
was identified for the docked molecules, but several clusters of
conformers were formed, because the same conformation could
bind to different regions of the crystalline structure of the frame-
work. The effect of pH on the binding of the guest molecules has
been taken into account through the representation of their pro-
tonation states. Ibuprofen can be unprotonated with a formal
charge of �1 or protonated with a formal charge of 0. Methylene
blue is cationic in nature and only
becomes dicationic at extremely
low pH ¼ 1.[74] Hence, it was rep-
resented as one single protona-
tion state with a formal charge of
þ1. The protonation states of
amoxicillin yielded formal charges
of �1 (protonated, acidic pH), 0
(zwitterionic, neutral pH), and þ1
(unprotonated, basic pH).
The [Zn(BDC)(H2O)2]n consists
of approximately tetrahedral Zn2þ
centers linked by a herringbone
pattern of 1,4-BDC spacers
aligned approximately along the
crystallographic a/c diagonal.[27]
The herringbone packing pro-
duces a molecular surface com-
posed of V-shaped clefts, so that
a guest molecule can bind either
inside the cleft or across it, some-
times bridging the its boundary-
setting crests. Throughout the
docking procedure, the host
framework atomic coordinates
were kept fixed, whereas the
guest conformation was fully flex-
ible. In this study, guest molecules
were chosen to represent either
simple or complex test cases from
the perspective of conformational
search and suitability of the scor-
ing function in predicting host–
guest interactions in MOFs. Sim-
pler molecules have a low num-
ber of degrees of freedom and
interact through a well-defined
pattern of molecular interactions. That is the case of compounds
such as ibuprofen and methylene blue. In contrast, complex mol-
ecules have a higher number of degrees of freedom and interact
through a less-defined pattern of molecular interactions. This
class is represented in our calculations by the b-lactam antibiotic
amoxicillin and the aminoglycoside antibiotic gentamicin.
Ibuprofen binds along one of the sides of the V-shaped cleft
in the [Zn(BDC)(H2O)2]n surface via electrostatic and p–p inter-
actions (Fig. 6). It binds in parallel to cleft wall, spanning its
full bottom-up length. Electrostatic interactions occur between
the ibuprofen carboxylic group and Zn2þ cation in the MOF
surface, whereas p–p stacking takes place between the ben-
zene rings of host and guest (Fig. 6). Protonated and unproto-
nated states of the ibuprofen molecule led to the same bind-
ing conformation. However, the estimated binding affinity (Kd)
of the unprotonated ibuprofen for the [Zn(BDC)(H2O)2]n sur-
face is in the femtomolar (10�15 M) range whereas the proto-
nated form is the micromolar (10�6 M). It has been previously
shown that ibuprofen is incorporated with remarkably high
load and slow release to MOFs made of 1,4-BDC and iron or
Figure 6. Lowest energy conformers of ibuprofen in the protonated (blue) and unprotonated (green) states.
The respective lowest energy conformers are representative of the most populated conformational cluster. A
total of 4.05 � 106 conformations were sampled for the each of the protonated and deprotonated states of ibu-
profen. (A) Longitudinal and (B) top views of the complex predicted structure. [Color figure can be viewed in
the online issue, which is available at wileyonlinelibrary.com.]
FULL PAPERWWW.Q-CHEM.ORG
International Journal of Quantum Chemistry 2012, 112, 3346–3355 3351

chromium metals.[12,29] It was also asserted that the amount of
adsorbed ibuprofen depended on the relative affinity between
the drug and the porous surface.[29]
Methylene blue was expected to bind to [Zn(BDC)(H2O)2]n in an
analogous manner to ibuprofen, that is, through p–p interactions
between its conjugated ring and the benzene ring of the BDC
molecule in the framework. However, this molecule has a formal
charge of þ1, resulting in a strong electrostatic repulsion when in
contact with the Zn2þ populated surface of the framework. As a
proof of concept, docking calculations were performed for the col-
orless reduced form of the methylene blue (leucomethylene blue),
whose formal charge is 0. The leucomethylene blue binds along
the longitudinal axis of the V-shaped cleft in the [Zn(BDC)(H2O)2]nsurface through p–p interactions between its conjugated ring and
the benzene ring of the BDC molecule of the host framework (Fig.
7). It binds to the [Zn(BDC)(H2O)2]n surface with a binding affinity
of the order of millimolar (10�3 M). In contrast with the docking
calculations, the experimental characterization of the complex
shows that the colorless crystal of the [Zn(BDC)(H2O)2]n becomes
blue upon the adsorption of the methylene blue (Fig. 4). The
amount of adsorbed drug
increases with increasing pH,
indicating that electrostatic
interactions govern the binding
of methylene blue and
[Zn(BDC)(H2O)2]n. A similar pat-
tern has been previously
observed for the adsorption of
methylene blue to the posi-
tively charged iron-containing
MOF253.[75] As for [Zn(BDC)
(H2O)2]n, the adsorbed amount
of methylene blue into MOF253
increased with increasing pH.
However, in the pH range of
the our measurements, that is,
pH 5–12, the protonation state
of the methylene blue does not
change, because it becomes
dicationic only at pH 1.[74] There-
fore, the pH-dependent increase
of methylene blue adsorption is
likely to result from a decrease
of the concentration of Hþ in
the aqueous milieu and not from
changes in the protonation state
of the drug. In such cases, the
molecular docking approach is
intrinsically unsuitable due to its
implicit treatment of the solvent.
Due to its larger molecular
size, gentamicin and amoxicil-
lin do not fit entirely inside
the V-shaped cleft. The dock-
ing calculations show that
gentamicin does not bind to
[Zn(BDC)(H2O)2]n framework
due to strong electrostatic repulsion. This finding corroborates
the XRPD and TGA/DTA analyses (Figs. 4 and 5). Amoxicillin
binds to the [Zn(BDC)(H2O)2]n surface through electrostatic
interactions involving oxygen atoms (Fig. 8). The binding affin-
ity of amoxicillin for the [Zn(BDC)(H2O)2]n surface increases as
more deprotonated oxygen sites becomes available, that is,
with increasing pH. In acidic conditions, amoxicillin binds to a
single Zn2þ cation through the lactam carbonyl and the car-
boxyl groups (Fig. 8A), whereas in neutral conditions, it inter-
acts with two Zn2þ cations through the amide carbonyl and
carboxyl groups (Fig. 8B). In basic conditions, the lowest
energy conformer binds to three Zn2þ cations via the carboxyl,
amide carbonyl, and the deprotonated hydroxyl groups (Fig.
8C). However, the predominant conformer in basic conditions
interacts with two Zn2þ cations through the carboxyl and the
deprotonated hydroxyl groups and has a binding energy of
about 2.51 kJ mol�1 less favorably that the lowest energy con-
former. It is not possible distinguish energetically between
these two conformers given that the error associated with the
potential energy function is ca. 8.4 kJ mol�1.[41] It is likely that
Figure 7. Lowest energy conformer of leucomethylene blue, the colorless reduced form of the methylene blue.
The lowest energy conformer is representative of the most populated conformational cluster. A total of 4.05 � 106
conformations were sampled. (A) Longitudinal and (B) top views of the predicted structure of the complex. [Color
figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
FULL PAPER WWW.Q-CHEM.ORG
3352 International Journal of Quantum Chemistry 2012, 112, 3346–3355 WWW.CHEMISTRYVIEWS.ORG
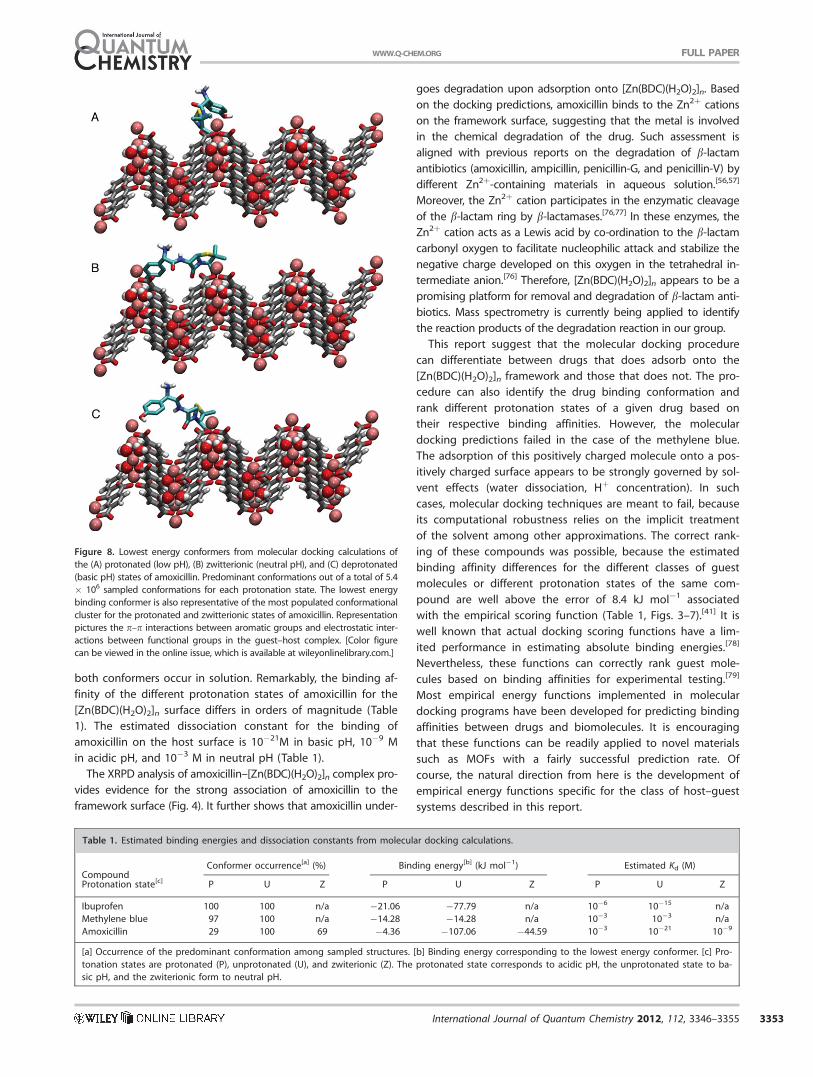
both conformers occur in solution. Remarkably, the binding af-
finity of the different protonation states of amoxicillin for the
[Zn(BDC)(H2O)2]n surface differs in orders of magnitude (Table
1). The estimated dissociation constant for the binding of
amoxicillin on the host surface is 10�21M in basic pH, 10�9 M
in acidic pH, and 10�3 M in neutral pH (Table 1).
The XRPD analysis of amoxicillin–[Zn(BDC)(H2O)2]n complex pro-
vides evidence for the strong association of amoxicillin to the
framework surface (Fig. 4). It further shows that amoxicillin under-
goes degradation upon adsorption onto [Zn(BDC)(H2O)2]n. Based
on the docking predictions, amoxicillin binds to the Zn2þ cations
on the framework surface, suggesting that the metal is involved
in the chemical degradation of the drug. Such assessment is
aligned with previous reports on the degradation of b-lactam
antibiotics (amoxicillin, ampicillin, penicillin-G, and penicillin-V) by
different Zn2þ-containing materials in aqueous solution.[56,57]
Moreover, the Zn2þ cation participates in the enzymatic cleavage
of the b-lactam ring by b-lactamases.[76,77] In these enzymes, the
Zn2þ cation acts as a Lewis acid by co-ordination to the b-lactam
carbonyl oxygen to facilitate nucleophilic attack and stabilize the
negative charge developed on this oxygen in the tetrahedral in-
termediate anion.[76] Therefore, [Zn(BDC)(H2O)2]n appears to be a
promising platform for removal and degradation of b-lactam anti-
biotics. Mass spectrometry is currently being applied to identify
the reaction products of the degradation reaction in our group.
This report suggest that the molecular docking procedure
can differentiate between drugs that does adsorb onto the
[Zn(BDC)(H2O)2]n framework and those that does not. The pro-
cedure can also identify the drug binding conformation and
rank different protonation states of a given drug based on
their respective binding affinities. However, the molecular
docking predictions failed in the case of the methylene blue.
The adsorption of this positively charged molecule onto a pos-
itively charged surface appears to be strongly governed by sol-
vent effects (water dissociation, Hþ concentration). In such
cases, molecular docking techniques are meant to fail, because
its computational robustness relies on the implicit treatment
of the solvent among other approximations. The correct rank-
ing of these compounds was possible, because the estimated
binding affinity differences for the different classes of guest
molecules or different protonation states of the same com-
pound are well above the error of 8.4 kJ mol�1 associated
with the empirical scoring function (Table 1, Figs. 3–7).[41] It is
well known that actual docking scoring functions have a lim-
ited performance in estimating absolute binding energies.[78]
Nevertheless, these functions can correctly rank guest mole-
cules based on binding affinities for experimental testing.[79]
Most empirical energy functions implemented in molecular
docking programs have been developed for predicting binding
affinities between drugs and biomolecules. It is encouraging
that these functions can be readily applied to novel materials
such as MOFs with a fairly successful prediction rate. Of
course, the natural direction from here is the development of
empirical energy functions specific for the class of host–guest
systems described in this report.
Figure 8. Lowest energy conformers from molecular docking calculations of
the (A) protonated (low pH), (B) zwitterionic (neutral pH), and (C) deprotonated
(basic pH) states of amoxicillin. Predominant conformations out of a total of 5.4
� 106 sampled conformations for each protonation state. The lowest energy
binding conformer is also representative of the most populated conformational
cluster for the protonated and zwitterionic states of amoxicillin. Representation
pictures the p–p interactions between aromatic groups and electrostatic inter-
actions between functional groups in the guest–host complex. [Color figure
can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Table 1. Estimated binding energies and dissociation constants from molecular docking calculations.
CompoundConformer occurrence[a] (%) Binding energy[b] (kJ mol�1) Estimated Kd (M)
Protonation state[c] P U Z P U Z P U Z
Ibuprofen 100 100 n/a �21.06 �77.79 n/a 10�6 10�15 n/a
Methylene blue 97 100 n/a �14.28 �14.28 n/a 10�3 10�3 n/a
Amoxicillin 29 100 69 �4.36 �107.06 �44.59 10�3 10�21 10�9
[a] Occurrence of the predominant conformation among sampled structures. [b] Binding energy corresponding to the lowest energy conformer. [c] Pro-
tonation states are protonated (P), unprotonated (U), and zwiterionic (Z). The protonated state corresponds to acidic pH, the unprotonated state to ba-
sic pH, and the zwiterionic form to neutral pH.
FULL PAPERWWW.Q-CHEM.ORG
International Journal of Quantum Chemistry 2012, 112, 3346–3355 3353

Conclusions
This work reported on the use of the molecular docking meth-
odology for the identification of candidate drug molecules
with high affinities for the [Zn(BDC)(H2O)2]n framework. If suc-
cessful, such approach can offer a view of host–guest interac-
tions at the atomistic level not easily accessible by experimen-
tal means. It is also a very robust approach for the screening
of large databanks containing thousands of compounds or
biomedical relevance. Evaluated systems were comprised of
the [Zn(BDC)(H2O)2]n framework and four guest molecules ibu-
profen, methylene blue, amoxicillin, and gentamicin. These
compounds were chosen to represent drugs of wide medical
use and different complexities in terms of their number of
degrees of freedom. To validate the theoretical estimates, the
experimental characterization of the host–guest complexes
was performed by the means of XRD and differential thermal
analysis. The proposed molecular docking procedure was able
to identify the overall binding mode of three out of four
tested compounds as well as to estimate binding affinity dif-
ferences for the different classes of guest molecules which are
well above the error of 8.4 kJ mol�1 associated with the em-
pirical scoring function.[41] Mostly notably, the procedure was
also able to distinguish between bad (gentamicin) and good
(ibuprofen, methylene blue, and amoxicillin) drug candidates
for immobilization in the [Zn(BDC)(H2O)2]n framework as vali-
dated via experimental measurements. Hence, the proposed
approach has the potential to offer a fast and affordable alter-
native prior to experiments for identification of the binding
preferences of a host system for a set of guest molecules. We
have taken an initial step toward the evaluation of the
approach applied to metal-organic frameworks.
Acknowledgments
Experimental measurements were performed by M.V. P., K.A.W., and
I.B.V. Interpretation of measurements were performed by M.V. P. and
S.A.-Jr. Computational simulations and related analyses were per-
formed by T.A.S.
Keywords: host–guest systems � crystalline nanosurfaces �AutoDock � molecular modeling � b-lactam � Zn21-catalysed
degradation � environmental remediation
How to cite this article: MO. Rodrigues, MV. de Paula, KA.
Wanderley, IB. Vasconcelos, S. Alves, Jr., TA. Soares, Int. J.
Quantum Chem. 2012, 112, 3346–3355. DOI: 10.1002/qua.24211
Additional Supporting Information may be found in the
online version of this article.
[1] C. Janiak, Dalton Trans. 2003, 2781, doi: 10.1039/B305705B.
[2] S. Kitagawa, R. Kitaura, S. Noro, Angew. Chem. Int. Ed. Engl. 2004, 43,
2334.
[3] U. Mueller, M. Schubert, F. Teich, H. Puetter, K. Schierle-Arndt, J. Pastre,
J. Mat. Chem. 2006, 16, 626.
[4] A. Ramanan, M. S. Whittingham, Cryst. Growth Des. 2006, 6, 2419.
[5] J. L. C. Rowsell, O. M. Yaghi, Microporous Mesoporous Mater. 2004,
73, 3.
[6] O. M. Yaghi, M. O’Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi, J.
Kim, Nature 2003, 423, 705.
[7] M. Hartmann, S. Kunz, D. Himsl, O. Tangermann, S. Ernst, A. Wagener,
Langmuir 2008, 24, 8634.
[8] Z. Zhao, Z. Li, Y. S. Lin, Ind. Eng. Chem. Res. 2009, 48, 10015.
[9] Y. Liu, W. Xuan, Y. Cui, Adv. Mater. 2010, 22, 4112.
[10] A. M. Shultz, O. K. Farha, J. T. Hupp, Nguyen, S. T. J. Am. Chem. Soc.
2009, 131, 4204.
[11] P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F.
Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J. S. Chang, Y. K. Hwang, V.
Marsaud, P. N. Bories, L. Cynober, S. Gil, G. Ferey, P. Couvreur, R. Gref,
Nature Mat. 2010, 9, 172.
[12] P. Horcajada, C. Serre, M. Vallet-Regi, M. Sebban, F. Taulelle, G. Ferey,
Angew. Chem. Int. Ed. Engl. 2006, 45, 5974.
[13] R. C. Huxford, J. Della Rocca, W. Lin, Curr. Opin. Chem. Biol. 2010, 14,
262.
[14] M. O. Rodrigues, F. A. Paz, R. O. Freire, G. F. de Sa, A. Galembeck, M. C.
Montenegro, A. N. Araujo, S. Alves, J. Phys. Chem. B 2009, 113, 12181.
[15] B. Chen, X. Zhao, A. Putkham, K. Hong, E. B. Lobkovsky, E. J. Hurtado,
A. J. Fletcher, K. M. Thomas, J. Am. Chem. Soc. 2008, 130, 6411.
[16] B. L. Chen, Y. Yang, F. Zapata, G. N. Lin, G. D. Qian, E. B. Lobkovsky,
Adv. Mater. 2007, 19, 1693.
[17] Y. F. Chen, R. Babarao, S. I. Sandler, J. W. Jiang, Langmuir 2010, 26,
8743.
[18] L. E. Kreno, J. T. Hupp, R. P. Van Duyne, Anal. Chem. 2010, 82, 8042.
[19] A. Aquino, K. A. Wanderley, O. Paiva-Santos, G. F. de Sa, R. Alexandre,
S. A. Junior, S. Navickiene, Talanta 2010, 83, 631.
[20] A. S. Barreto, R. L. da Silva, S. C. Dos Santos Silva, M. O. Rodrigues, C.
A. de Simone, G. F. de Sa, S. A. Junior, S. Navickiene, M. E. de Mes-
quita, J. Sep. Sci. 2010, 33, 3811.
[21] P. H. de Carvalho, A. S. Barreto, M. O. Rodrigues, M. Prata, P. B. Alves,
M. E. de Mesquita, S.Alves, Jr., S. Navickiene, J. Sep. Sci. 2009, 32, 2132.
[22] D. Britt, D. Tranchemontagne, O. M. Yaghi, Proc. Natl. Acad. Sci. U.S.A.
2008, 105, 11623.
[23] M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O’Keeffe, O. M.
Yaghi, Science 2002, 295, 469.
[24] M. Eddaoudi, H. L. Li, O. M. Yaghi, J. Am. Chem. Soc. 2000, 122, 1391.
[25] D. J. Tranchemontagne, J. R. Hunt, O. M. Yaghi, Tetrahedron 2008, 64,
8553.
[26] M. Edgar, R. Mitchell, A. M. Z. Slawin, P. Lightfoot, P. A. Wright, Chem.
Eur. J. 2001, 7, 5168.
[27] G. Guilera, J. W. Steed, Chem. Comm. 1999, 16, 1563.
[28] H. Li, M. Eddaoudi, M. O’Keeffe, O. M. Yaghi, Nature 1999, 402, 276.
[29] P. Horcajada, C. Serre, G. Maurin, N. A. Ramsahye, F. Balas, M. Vallet-
Regi, M. Sebban, F. Taulelle, G. Ferey, J. Am. Chem. Soc. 2008, 130,
6774.
[30] S. M. Cohen, Curr. Opin. Chem. Biol. 2007, 11, 115.
[31] P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G.
Ferey, R. E. Morris, C. Serre, Chem. Rev. 2012, 112, 1232.
[32] W. van Gunsteren, D. Bakowies, R. Burgi, I. Chandrhhasekhar, M. Chris-
ten, X. Daura, P. Gee, A. Glattli, T. Hansson, C. Oostenbrink, C. Peter, J.
Pitera, L. Schuler, T. Soares, H. B. Yu, Chimia 2001, 55, 856.
[33] W. F. van Gunsteren, J. Dolenc, A. E. Mark, Curr. Opin. Struct. Biol. 2008,
18, 149.
[34] G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S.
Goodsell, A. J. Olson, J. Comp. Chem. 2009, 30, 2785.
[35] R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T.
Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P.
Francis, P. S. Shenkin, J. Med. Chem. 2004, 47, 1739.
[36] T. A. Halgren, R. B. Murphy, R. A. Friesner, H. S. Beard, L. L. Frye, W. T.
Pollard, J. L. Banks, J. Med. Chem. 2004, 47, 1750.
[37] A. N. Jain, J. Comput. Aided Mol. Des. 2007, 21, 281.
[38] B. Kramer, M. Rarey, T. Lengauer, Proteins 1999, 37, 228.
[39] B. K. Shoichet, D. L. Bodian, I. D. Kuntz, J. Comp. Chem. 1992, 13, 380.
[40] O. Korb, P. A. Wood, Chem. Commun. 46, 3318.
[41] R. Huey, G. M. Morris, A. J. Olson, D. S. Goodsell, J. Comp. Chem. 2007,
28, 1145.
[42] S. J. Weiner, P. A. Kollman, D. A. Case, U. C. Singh, C. Ghio, G. Alagona,
S. Profeta, P. K. Weiner, J. Am. Chem. Soc. 1984, 106, 765.
FULL PAPER WWW.Q-CHEM.ORG
3354 International Journal of Quantum Chemistry 2012, 112, 3346–3355 WWW.CHEMISTRYVIEWS.ORG

[43] P. J. Goodford, J. Med. Chem. 1985, 28, 849.
[44] W. D. Cornell, P. Cieplak, C. I. Bayly, P. A. Kollman, J. Am. Chem. Soc.
1993, 115, 9620.
[45] M. Valiev, E. J. Bylaska, N. Govind, K. Kowalski, T. P. Straatsma, H. J. J.
van Dam, D. Wang, J. Nieplocha, E. Apra, T. L. Windus, W. A. de Jong,
Comput. Phys. Commun. 2010, 181, 1477.
[46] E. L. Mehler, T. Solmajer, Protein Eng. Des. Sel. 1991, 4, 903910.
[47] P. F. W. Stouten, C. Fr€ommel, H. Nakamura, C. Sander, Mol. Simulat.
1993, 10, 97.
[48] T. A. Soares, D. S. Goodsell, J. M. Briggs, R. Ferreira, A. J. Olson, Biopoly-
mers 1999, 50, 319.
[49] T. Soares, D. Goodsell, R. Ferreira, A. J. Olson, J. M. Briggs, J. Mol.
Recogn. 2000, 13, 146.
[50] H. M. Rietveld, J. Appl. Crystallogr. 1969, 2, 65.
[51] A. C. Larson, R. B. Von Dreele, General Structure Analysis System
(GSAS). Los Alamos National Laboratory: Los Alamos, 2004, p.224.
[52] B. H. Toby, J. Appl. Crystallogr. 2001, 34, 210.
[53] L. R. Hoffman, D. A. D’Argenio, M. J. MacCoss, Z. Zhang, R. A. Jones, S.
I. Miller, Nature 2005, 436, 1171.
[54] M. Jarvinen, J. Appl. Crystallogr. 1993, 26, 525.
[55] R. A. Young, P. Desai, Archiwun Nauki o Materialach 1989, 10, 71.
[56] E. S. Elmolla, M. Chaudhuri, J. Harzard. Mater. 2010, 173, 445.
[57] R. Nosrati, A. Olad, R. Maramifar, Environ. Sci. Poll. Res. Intl. 2012. [Epub
ahead of print].
[58] R. E. Dinnebier, S. J. Billinge, Powder Diffraction; Royal Society of
Chemistry: Cambridge, 2008.
[59] L. B. McCusker, R. B. Von Dreele, D. E. Cox, D. Loueur, P. Scardi, J. Appl.
Crystallogr. 1999, 32, 36.
[60] M. Busson, J. Int. Med. Res. 1986, 14, 53.
[61] E. R. Southey, K. Soares-Weiser, J. Kleijnen, Curr. Med. Res. Opin. 2009,
25, 2207.
[62] M. Wainwright, K. B. Crossley, J. Chemother. 2002, 14, 431.
[63] M. Bountogo, A. Zoungrana, B. Coulibaly, C. Klose, U. Mansmann, F. P.
Mockenhaupt, J. Burhenne, G. Mikus, I. Walter-Sack, R. H. Schirmer, A.
Sie, P. Meissner, O. Muller, Trop. Med. Int. Health. 2010, 15, 713.
[64] F. Gut, W. Schiek, W. E. Haefeli, I. Walter-Sack, J. Burhenne, Eur. J.
Pharm. Biopharm. 2008, 69, 582.
[65] R. Pastrana-Mena, R. R. Dinglasan, B. Franke-Fayard, J. Vega-Rodriguez,
M. Fuentes-Caraballo, A. Baerga-Ortiz, I. Coppens, M. Jacobs-Lorena, C.
J. Janse, A. E. Serrano, J. Biol. Chem. 2010, 285, 27045.
[66] A. Zoungrana, B. Coulibaly, A. Sie, I. Walter-Sack, F. P. Mockenhaupt, B.
Kouyate, R. H. Schirmer, C. Klose, U. Mansmann, P. Meissner, O. Muller,
PLoS One 2008, 3, e1630.
[67] S. M. Drawz, R. A. Bonomo, Clin. Microbiol. Rev. 2010, 23, 160.
[68] P. Malfertheiner, F. Megraud, C. O’Morain, F. Bazzoli, E. El-Omar, D. Gra-
ham, R. Hunt, T. Rokkas, N. Vakil, E. J. Kuipers, Gut 2007, 56, 772.
[69] P. Malfertheiner, F. Megraud, C. O’Morain, A. P. Hungin, R. Jones, A.
Axon, D. Y. Graham, G. Tytgat, Aliment Pharmacol. Ther. 2002, 16, 167.
[70] A. Forge, J. Schacht, Audiol. Neurootol. 2000, 5, 3.
[71] C. G. Kumar, M. Himabindu, A. Jetty, Crit. Rev. Biotechnol. 2008, 28,
173.
[72] T. Hermann, Cell. Mol. Life Sci. 2007, 64, 1841.
[73] T. A. Soares, R. D. Lins, T. P. Straatsma, J. M. Briggs, Biopolymers 2002,
65, 313.
[74] M. Wainwright, L. Amaral, Trop. Med. Int. Health. 2005, 10, 501.
[75] E. Haque, J. W. Jun, S. H. Jhung, J. Hazardous Mater. 2011, 185, 507.
[76] M. I. Page, A. Badarau, Bioinorg. Chem. Appl. 2008, 576297, doi:
10.1155/2008/576297.
[77] Z. Wang, W. Fast, A. M. Valentine, S. J. Benkovic, Curr. Opin. Chem. Biol.
1999, 3, 614.
[78] D. L. Mobley, K. A. Dill, Structure 2009, 17, 489.
[79] N. Brooijmans, I. D. Kuntz, Annu. Rev. Biophys. Biomol. Struct. 2003, 32,
335.
Received: 18 February 2012Revised: 14 May 2012Accepted: 18 February 2012Published online on 8 June 2012
FULL PAPERWWW.Q-CHEM.ORG
International Journal of Quantum Chemistry 2012, 112, 3346–3355 3355




















