
Calculated rotational and vibrational g factors of LiH X 1Σ+ and evaluation of parameters in radial...
17
Calculated Rotational and Vibrational g Factors of LiH X 1 R þ and Evaluation of Parameters in Radial Functions from Rotational and Vibration-Rotational Spectra S. P. A. SAUER, 1 I. PAIDAROVA ´ , 2 J. ODDERSHEDE, 3 K. L. BAK, 4 J. F. OGILVIE 5,6 1 Department of Chemistry, University of Copenhagen, Universitetsparken 5, DK-2100 Copenhagen, Denmark 2 J. Heyrovsky Institute of Physical Chemistry, Academy of Sciences of the Czech Republic, Dolejvskova 3, 18223 Prague 8, Czech Republic 3 Institute of Physics and Chemistry, University of Southern Denmark, Campusvej 55, DK-5230 Odense M, Denmark 4 Ingeniørhøjskolen i Aarhus, Dalgas Avenue 2, DK-8000 Aarhus C, Denmark 5 Department of Mathematics, Centre for Experimental and Constructive Mathematics, Simon Fraser University, 8888 University Drive, Burnaby, BC, Canada V5A 1S6 6 Escuela de Quimica y CELEQ, Universidad de Costa Rica, San Pedro, San Jose 2060, Costa Rica Received 14 December 2009; accepted 11 February 2010 Published online 12 May 2010 in Wiley Online Library (wileyonlinelibrary.com). DOI 10.1002/qua.22666 ABSTRACT: The vibrational g factor, that is, the nonadiabatic correction to the vibrational reduced mass, of LiH has been calculated for internuclear distances over a wide range. Based on multiconfigurational wave functions with a large complete active space and an extended set of gaussian type basis functions, these calculations yielded Correspondence to: J. F. Ogilvie; e-mail: [email protected] This article is dedicated in the memory of David M. Bishop. Contract grant sponsor: Danish Natural Science Research Council/Danish Councils for Independent Research. Contract grant number: 272-08-0486. Contract grant sponsor: Academy of Sciences of the Czech Republic. Contract grant number: IAA 401870702. Contract grant sponsors: Carlsberg Foundation, Danish Center for Scientific Computing. International Journal of Quantum Chemistry, Vol. 111, 736–752 (2011) V C 2010 Wiley Periodicals, Inc.
Transcript of Calculated rotational and vibrational g factors of LiH X 1Σ+ and evaluation of parameters in radial...

Calculated Rotational and Vibrational gFactors of LiH X 1Rþ and Evaluation ofParameters in Radial Functions fromRotational and Vibration-RotationalSpectra
S. P. A. SAUER,1 I. PAIDAROVA,2 J. ODDERSHEDE,3 K. L. BAK,4
J. F. OGILVIE5,6
1Department of Chemistry, University of Copenhagen, Universitetsparken 5,DK-2100 Copenhagen, Denmark2J. Heyrovsky Institute of Physical Chemistry, Academy of Sciences of the Czech Republic,Dolejvskova 3, 18223 Prague 8, Czech Republic3Institute of Physics and Chemistry, University of Southern Denmark, Campusvej 55,DK-5230 Odense M, Denmark4Ingeniørhøjskolen i Aarhus, Dalgas Avenue 2, DK-8000 Aarhus C, Denmark5Department of Mathematics, Centre for Experimental and Constructive Mathematics,Simon Fraser University, 8888 University Drive, Burnaby, BC, Canada V5A 1S66Escuela de Quimica y CELEQ, Universidad de Costa Rica, San Pedro, San Jose 2060, Costa Rica
Received 14 December 2009; accepted 11 February 2010Published online 12 May 2010 in Wiley Online Library (wileyonlinelibrary.com).DOI 10.1002/qua.22666
ABSTRACT: The vibrational g factor, that is, the nonadiabatic correction to thevibrational reduced mass, of LiH has been calculated for internuclear distances over awide range. Based on multiconfigurational wave functions with a large complete activespace and an extended set of gaussian type basis functions, these calculations yielded
Correspondence to: J. F. Ogilvie; e-mail: [email protected] article is dedicated in the memory of David M. Bishop.Contract grant sponsor: Danish Natural Science Research
Council/Danish Councils for Independent Research.Contract grant number: 272-08-0486.Contract grant sponsor: Academy of Sciences of the Czech
Republic.Contract grant number: IAA 401870702.Contract grant sponsors: Carlsberg Foundation, Danish
Center for Scientific Computing.
International Journal of Quantum Chemistry, Vol. 111, 736–752 (2011)VC 2010 Wiley Periodicals, Inc.

also the rotational g factor, the electric dipolar moment, and its gradient withinternuclear distance for LiH in its electronic ground state X 1Rþ. The vibrational gfactor gv exhibits a pronounced minimum near internuclear distance R ¼ 3.65 � 10�10 m;the derivative of electric dipolar moment and the nonadiabatic matrix element couplingthe electronic ground state to the first electronically excited state exhibit extrema near thesame location that is also near the avoided crossing of the curves for potential energy forthe electronic ground state and excited state A 1Rþ. The irreducible contribution girrr (R) tothe rotational g factor increases monotonically over the calculated domain, whereas theirreducible contribution girrv (R) to the vibrational g factor has a minimum at the samelocation as that of gv itself. From these calculated radial functions, we derived values ofthe rotational g factor and electric dipolar moment for LiH in vibrational states v ¼ 0 and1, and the corresponding rotational dependences, in satisfactory agreement withexperimental values. These calculated data of rotational g factor served as constraints innew fits of 1000 vibration-rotational spectral data of LiH in four isotopic variants, whichyield estimates of adiabatic corrections for comparison with published data and of thevibrational g factor for comparison with our calculated results.VC 2010 Wiley Periodicals,Inc. Int J Quantum Chem 111: 736–752, 2011
Key words: rotational g factor; vibrational g factor; nonadiabatic reduced mass;spectral fitting; LiH
1. Introduction
A research interest of David M. Bishop washighly accurate calculations of properties
of atoms and diatomic molecules. Among the lat-ter, LiH figured in at least five reports [1–5], ofwhich two [1, 2] were concerned with adiabaticpotential energy surfaces for states X 1Rþ and A1Rþ. Our contribution to this memorial issue isthus appropriately a combined theoretical and ex-perimental investigation of nonadiabatic contribu-tions to vibration-rotational spectra of LiH in theelectronic ground state.
According to computational spectrometry [6],we developed a procedure whereby one appliesthe results of quantum-chemical calculations asconstraints within analyses of wavenumber dataof lines in infrared spectra measured at high reso-lution; we applied this approach first to spectra ofLiH [7]. A conventional mechanical model mightserve as a basis of interpreting such spectra interms of the nuclear vibration and rotation of freediatomic molecules in a gaseous sample at smalldensity: Two atomic centers A and B, regarded aspoint masses at distance R apart in a particularmolecule AB, oscillate about the center of massalong an interatomic vector and rotate about thatcenter, apart from translation of the entire mole-cule within an enclosing vessel that produces no
discernible spectral effect of interest here. A corre-sponding primitive Hamiltonian thereby com-prises three terms,
HðRÞ ¼ 1
2
p2
lþ �h2
2lR2JðJ þ 1Þ þ VðRÞ (1)
that pertain to the kinetic energy of relativeatomic motion along the interatomic vector, thekinetic energy of relative motion perpendicular tothat axis, or molecular rotation, and interatomicpotential energy V(R), respectively, all regardedas mechanical effects. For a reduced mass l of aneutral diatomic molecule in terms of two atomicmasses Ma and Mb,
1
l¼ 1
Maþ 1
Mb(2)
p denotes the relative linear momentum conjugateto R; the angular momentum is �h
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiJðJ þ 1Þp
.Because a diatomic molecule contains two atomicnuclei and their associated electrons, rather thanstructureless or spherically symmetric atoms, andbecause electrons follow imperfectly the motionof one or other nucleus in a classical sense, theformer Hamiltonian is inadequate to explaininfrared spectra recorded at the greatest contem-porary spectral resolution; we have thus recourseto an extended effective Hamiltonian [8] for
ROTATIONAL AND VIBRATIONAL g FACTORS OF LiH
VOL. 111, NO. 4 DOI 10.1002/qua INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 737

nuclear motion applicable to an electronic state ofsymmetry class 1Rþ as follows.
HeffðRÞ ¼ 1
2lp 1þ gvðRÞme
mp
� �p
þ �h2JðJ þ 1Þ2lR2
1þ grðRÞme
mp
� �þ VðRÞ þ DVðRÞ ð3Þ
This formula contains terms further to those ofEq. (1) in recognition of the existence of extra-me-chanical effects [9] of two types [10]: adiabaticcorrections pertain to theoretical expressions thatinvolve expectation values of quantum-mechani-cal operators within a particular electronic state ofinterest, in practice, the electronic ground state;nonadiabatic effects involve matrix elements ofthe same or other operators connecting that elec-tronic ground state with electronically excitedstates, as follows. A radial function gr(R) for therotational g factor in a term for the kinetic energyof rotation has nuclear and electronic contribu-tions; for a molecule with electronic ground stateof class R, the latter contribution involves matrixelements to electronically excited states of symme-try class P and constitutes the nonadiabatic con-tribution to the rotational reduced mass. Anotherradial function gv(R) for the vibrational g factor inthe term for the kinetic energy of vibration like-wise contains contributions from atomic nucleiand electrons; this contribution involves matrixelements to electronically excited states of thesame class R, and constitutes the nonadiabaticcontribution to the vibrational reduced mass.According to convention, the radial functions forboth rotational and vibrational g factors have asmultiplicand a ratio me/mp of electronic and pro-tonic rest masses. From an experimental point ofview, the splitting of lines in a pure rotationalspectrum on subjection of a gaseous sample to amagnetic field is a direct manifestation of therotational g factor, but gv is experimentally acces-sible through no known magnetic effect of loworder [11]. An additional term DV(R) in the effec-tive Hamiltonian is purely an artifact of an ap-proximate theoretical treatment according towhich electronic and nuclear motions are treatedseparately, which also generates V(R) as the elec-tronic energy at internuclear distance R. This cor-rection term DV(R), which takes partly intoaccount the fact that internuclear potential energyV(R) would otherwise have a small dependenceon nuclear mass, includes both expectation values
of operators in the ground electronic state: thus,adiabatic corrections, with a relative dependenceon ratio me/M of electronic and nuclear masses tothe first power, and matrix elements connectingthe electronic ground state with electronicallyexcited states [8, 10]; thus, further nonadiabaticeffects, but with a relative dependence on a ratiome/M to a power greater than unity. We thusexpress DV(R) as DVad(R) þ DVnad(R), but neglectDVnad(R) because its dependence on ratio me/Mwith exponent 3/2 or larger makes it muchsmaller than retained terms [8]; its effects in ourcollected molecular spectra of LiH would bemuch smaller than the experimental uncertaintyof wavenumber or frequency measurements. Allthese terms due to extra-mechanical effects are ex-pressible as sums of contributions from separateatomic centers A and B [8].
Hence, l 1þ gvðRÞ me
mp
� ��1in Eq. (3) represents
an effective reduced mass for vibration dependenton internuclear separation R, and
l 1þ grðRÞ me
mp
� ��1represents an analogous effective
reduced mass for rotation [11]; thereby gv(R) andgr(R) absorb all effects of the deviation of the distribu-tion of electronic charge from a spherical form cen-tered on an atomic nucleus that would correspond tothe condition of an isolated, electrically neutral atomas part of an electrically neutral diatomic molecule.
For a diatomic molecule of a particular isotopicvariant i in an electronic state of symmetry class1Rþ, we express the spectral terms, or energies di-vided by hc, of this effective Hamiltonian [9],
EivJ ¼
X1k¼0
X1l¼0
Ykl þ Za;vkl þ Za;r
kl þ Zb;vkl þ Zb;r
kl
� �
vþ 1
2
� �k
JðJ þ 1Þ½ �l ð4Þ
in which appear vibrational quantum number vand rotational quantum number J in the absenceof other pertinent contributions to total molecularangular momentum. The principal coefficients Ykl
reflect a presence of terms for mechanical effectsin a primitive Hamiltonian, Eq. (1); auxiliary termcoefficients Za
kl and Zbkl represent additional terms
resulting from extra-mechanical effects in an effec-tive Hamiltonian, Eq. (3), for atomic centers oftypes A and B separately. Following Dunham’salgebraic approach [12] as extended by van Vleck[10] and others [13], we form radial functions aspolynomials in variable z [14, 15]; the latter
SAUER ET AL.
738 INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY DOI 10.1002/qua VOL. 111, NO. 4

quantity denotes a reduced and dimensionless dis-placement of instantaneous internuclear separationR from equilibrium internuclear distance Re,
z ¼ 2R� Re
Rþ Re(5)
As parameters besides Re, we use coefficientsof z in four radial functions [9], with coefficientscj for potential energy independent of mass,
VðRÞ ! VðzÞ ¼ c0z2 1þ
X1j¼1
cjzj
0@
1A (6)
coefficients sj for nonadiabatic vibrational effectsand the associated nuclear contribution that per-tain [16] to the vibrational g factor, gv,
gvðRÞ ! gavðzÞ þ gbvðzÞ ¼ mp
X1j¼0
saj zj
MaþX1j¼0
sbj zj
Mb
24
35 (7)
coefficients tj for nonadiabatic rotational effectsand the associated nuclear contribution that per-tain [17] to the rotational g factor, gr,
grðRÞ ! garðzÞ þ gbrðzÞ ¼ mp
X1j¼0
taj zj
MaþX1j¼0
tbj zj
Mb
24
35 (8)
and coefficients uj for adiabatic corrections [9]:
DVadðRÞ ! me
X1j¼0
uaj zj
MaþX1j¼0
ubj zj
Mb
24
35 (9)
Although extra-mechanical effects taken explic-itly into account in our effective Hamiltonian, Eq.(3), number three, only two contributions to auxil-iary term coefficients are experimentally distin-guishable for atoms of each type A or B sepa-rately through use of isotopic variants; we denotethese as Zv
kl for vibration-rotational terms thatinvolve parameters in DVad(z) and gv(z), and asZrkl for further rotational terms that involve pa-
rameters in gv(z) and gr(z) [9]. The physical basisof this criterion is that these extra-mechanicaleffects are shown to express a dependence onindividual masses of atoms of each type [8], andthe nature of these dependences makes themseparable. It is thus in general impracticable toevaluate multiple parameters of three types s, t,
and u in complete sets up to a particular orderfor atomic centers of types either A or B in a het-eronuclear diatomic molecule from only data offrequencies and wavenumbers of spectral transi-tions for samples of a diatomic molecular com-pound in the absence of an external field.
For that reason [6], in preceding work, we com-bined calculated values of the rotational g factorand electric dipolar moment p as a function ofinternuclear separation to evaluate coefficients oftypes taj and tbj , thereby simulating experimentaldata prospectively derivable from extensive exper-imental application of Zeeman and Stark effects,respectively; we constrained the resulting values ofthose coefficients while adjusting values of param-eters of types cj, s
aj , s
bj , u
aj , and ubj to reproduce satis-
factorily experimental data of frequency and wavenumbers, first for LiH [7] and subsequently forother diatomic molecular species AlH [18] and CO[19]. To assess the significance of these deduced ra-dial functions gv(R) and DVad(R) in the presentwork, we compare them with our calculations forthe vibrational g factor and with adiabatic correc-tions reported by Bishop and Cheung [1]. For therotational g factor, reviewed elsewhere [20], the ex-perimental, theoretical, and computational aspectsare well established, unlike the vibrational g factorthat we here investigate in some detail.
In our original joint work on LiH [7], we fitted 12values of rotational gr(R) factor and electric dipolarmoment p(R) for 7Li1H in a domain R/10�10 m ¼[1.17, 2.18], or |z| < 0.309, to generate, through coef-ficients tLij and tHj in Eq. (8), a radial function gr(z);we applied the latter in fitting 557 wavenumbersand frequencies of vibration-rotational and purerotational spectral transitions. A plot of this functiongr(R) yields a curve varying monotonically withinthat domain, eventually approaching zero asymp-totically at distances for R � Re ¼ 1.5949 � 10�10 m;this behavior is regular, and typical of this functionfor neutral diatomic molecules for which these calcu-lations have been performed [20–24]. In the presentwork in which the fitted spectral data number 1000,we increased the domain of these calculations to R/10�10 m ¼ [1.05, 10.0]; we found a similar behavior,but gr became slightly positive for R > 4 � 10�10 m.In contrast, our present calculations of gv yielded apronounced minimum near R ¼ 3.65 � 10�10 m;because information on the variation of gv with inter-nuclear distance R is available for only a fewmolecu-lar species—specifically, for H2 [25] and HeHþ [26]over a large domain and for NaCl [27] over a smalldomain near Re, we focus attention on this
ROTATIONAL AND VIBRATIONAL g FACTORS OF LiH
VOL. 111, NO. 4 DOI 10.1002/qua INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 739

vibrational g function in relation to both theoreticalcalculations and our estimate from spectral data, inour present application of computational spectrome-try. Finally one should note that apart from our pre-vious work on LiH [7] several other authors havealso analysed the infrared spectra of LiH [28–34].
2. Theory
As the pertinent theory is presented elsewhere[8, 16, 17], we here recall some definitions of im-portant terms. The rotational and vibrational gfactors for use with the reduced masses in atomicmasses are expressible as a sum of electronic andnuclear contributions,
gr;v ¼ gelr;v þ gnur;v (10)
of which the nuclear contribution for a diatomicmolecule is common to both factors and inde-pendent of internuclear distance.
gnur;v ¼ mpZaM
2b þ ZbM
2a
MaMbðMa þMbÞ (11)
The electronic contributions contain sums oftransition moments, to electronically excited statesof an appropriate symmetry class weighted withthe reciprocal of the excitation energies, of the op-erator for nuclear linear momentum for gv,
gelv ¼ � 2mp�h2
mel
Xn6¼0
h0j @@R jni
�� ��2EnðRÞ � E0ðRÞ (12)
which hence involve quantity �i�hh0j@=@Rjni as anonadiabatic matrix element coupling the elec-tronic ground state j0i, having associated wavefunction W0({ri}; R), with electronically excited statejni of the same symmetry class, with its associatedwave function Wn({ri}; R); formulae relating thesesums to coefficients sj in Eq. (7) are reported else-where [16]. Related to magnetic dipolar moment,for angular momentum L about the center of massRCM, with Lx ¼ Ly because of cylindrical symmetry,the corresponding formula for gr is
gelr ¼ � 2mp
melR2
Xn6¼0
h0jLxðRCMÞjnij j2EnðRÞ � E0ðRÞ (13)
but here the electronically excited state jni musthave electronic angular momentum K differing by
one unit from that of the electronic ground statej0i. The relation of this sum to coefficients tj inEq. (8) is specified elsewhere [17]. The energies ofwhich differences appear in both latter denomina-tors are electronic energies formally equivalent topotential energy V(R) for nuclear vibration in aparticular electronic state, which for the electronicground state appears in Eq. (3). Because coeffi-cients sj in Eq. (7) and tj in Eq. (8) are definabledirectly [16, 17] at a particular internuclear dis-tance in terms of sums related to the ones in Eqs.(12) and (13), these coefficients are not merely em-pirical fitting parameters but have a sound theo-retical basis.
3. Calculations of MolecularElectronic Structure of LiH
These calculations were performed with a localdevelopment version of the program package Dal-ton [35]. The contributions from sums over statesto the rotational and vibrational g factors, to pa-rameters tLij , t
Hj , s
Lij , and sHj and to the gradient of
electric dipolar moment are efficiently evaluatedas linear response functions [36, 37], whereas theelectric dipolar moment is a simple expectationvalue in the electronic ground state, the imple-mentation is described elsewhere [38]. To calcu-late the rotational g factor, we used rotationalLondon orbitals [39, 40]. We used a multiconfi-gurational self-consistent–field (MCSCF) wavefunction [41, 42] with a large complete activespace [43], that is, no inactive orbitals and 21active orbitals in class A1, 10 in classes B1 and B2,and four in class A2 of the irreducible representa-tion of point group C2v. This active space is thesame as that in our calculation of radial functionsfor the electric polarizability of LiH [44], but ismuch larger than in preceding calculations ofrotational and vibrational g factors [16, 17, 45].The one-electron basis set was taken also from thepreceding work on the polarizability of LiH [44];it comprises 13 s-, 8 p-, 6 d-, and 2 f-type sets ofuncontracted gaussian functions on Li and 12 s-, 8p-, and 5 d-type sets of uncontracted gaussians onH; the exponents were taken from a contractedbasis set of Roos and Sadlej [46]. The basis setthat we used in our previous calculations of rota-tional [7, 17] and vibrational [16] g factors and ofspin-rotation parameters of LiH [47] was basedalso on the basis set of Roos and Sadlej, but wasnot totally decontracted, and lacked functions of
SAUER ET AL.
740 INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY DOI 10.1002/qua VOL. 111, NO. 4

type f on Li. The results of our calculations of var-ious quantities as a function of internuclear dis-tance appear in Table I. Figure 1 shows points ofelectronic energy, relative to zero energy at theminimum and expressed as a wavenumber quan-tity. Our electronic energy, �8.0674 Hartree, atthe equilibrium internuclear distance compareswell with the ‘‘exact’’ Born–Oppenheimer energy,�8.070549 Hartree, estimated from the experimen-tal binding energy De [48]. Figure 2 shows plotsof the rotational and vibrational g factors, electricdipolar moment |p| and its derivative |dp/dR|,and one nonadiabatic coupling matrix element inEq. (12) as a function of internuclear distance.
The quality of our calculated radial functionsof the rotational g factor gr(R) and electric dipolarmoment p(R) can be judged by comparing thevibrational averages of gr(R),
gðv;JÞr ¼ Hv;JðRÞ grðRÞj jHv;JðRÞ
(14)
and analogously of electric dipolar moment p(R)with the available experimental values [49–51].The vibration-rotational wavefunctions Hv,J(R)were obtained by numerical solution of the nu-clear Schrodinger equation
� �h2
2ld2
dR2þ JðJ þ 1Þ
R2
� �þ ZaZbe
2
4pe0Rþ VðRÞ
( )
�Hv;JðRÞ ¼ Ev;JHv;JðRÞ ð15Þ
in which, we applied the calculated MCSCF ener-gies as potential energy V(R) for the nuclearmotion in this equation. The results of these calcu-lations appear in Table II, in which values of rota-tional g factors pertain specifically to 7Li1H ateither the internuclear separation Re or vibration-ally averaged over the vibrational ground stateand first excited state, as indicated; the electricdipolar moment p in the form of a radial functionis independent of isotopic mass for this net elec-trically neutral molecule.
Our value of the rotational g factor at the sepa-ration Re is in perfect agreement with a recentbenchmark CCSD(T) result [52] and our dipolemoment radial function coincides with an earlierCISD [53] and very recent second order perturba-tion theory radial function [54]. Furthermore thesatisfactory agreement between calculated andmeasured quantities, with deviations of less than0.2%, demonstrates the quality of the radial func-tions for the rotational g factor and electric dipo-
lar moment, and lends confidence to their use asconstraints in fitting spectral data. Nevertheless,comparison with experimental values for thevibrational ground state probes mainly our rota-tional g-factor and dipole moment curves in thevicinity of the equilibrium internuclear distanceand not near the dissociation limit. We assign,therefore, no physical significance to the deviationof the calculated rotational g-factor from zero forinternuclear distances larger than 4.0 � 10�10 m,which is certainly due to a decrease in accuracyof our approximated wavefunction for large inter-nuclear distances. These internuclear distances are,however, far outside of the range of internucleardistance relevant for the fitting of spectral data.
4. Analysis of Spectral Data
To convert results in Table I to radial coefficientstj as coefficients of zj, we fitted to polynomials thevalues of tLi(R) and tH(R) from these relations,
tLiðRÞ ¼ lgrðRÞmp
þ 2pðRÞeRMH
� �(16)
tHðRÞ ¼ lgrðRÞmp
� 2pðRÞeRMLi
� �(17)
that enable calculation of the isotopic dependenceof the rotational g factor as a radial function withpolarity þAB� ¼ þLiH�, as indicated by the calcu-lated values and sign of p(R) in Table I. These for-mulae can be derived [17] from Eq. (13) and areconsistent with a general partition of the rota-tional g factor into an irreducible contribution anda contribution containing the electric dipolarmoment, according to this formula that indicatesalso the dependence on the masses of separateatoms A and B [8, 17].
grðRÞ ¼ mpgirrr ðRÞ 1
Maþ 1
Mb
� �þmp
eRpðRÞ 1
Ma� 1
Mb
� �(18)
The corresponding formula for the vibrationalg factor is [8, 16]
gvðRÞ ¼mpgirrv ðRÞ 1
Maþ 1
Mb
� �þmp
e
dpðRÞdR
1
Ma� 1
Mb
� �(19)
From the tabulated values of gr(R) and p(R) ateach value of R in a domain R/10�10 m ¼ [1.05,
ROTATIONAL AND VIBRATIONAL g FACTORS OF LiH
VOL. 111, NO. 4 DOI 10.1002/qua INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 741

TABLE IElectronic energy E0, electric dipolar moment p, vibrational gv, and rotational gr factors at internucleardistance R for 7Li1H in electronic state X 1R1.
R/10�10 m E0/Hartree p/10�30 C m gr gv
1.05 �7.994350052 �16.44303 �1.8031 0.24861.1 �8.011500191 �16.63474 �1.5936 0.19811.15 �8.025414911 �16.84999 �1.4200 0.15101.2 �8.036600616 �17.08190 �1.2746 0.10951.25 �8.045489390 �17.33524 �1.1520 0.06711.3 �8.052435545 �17.60613 �1.0477 0.02971.35 �8.057742373 �17.89172 �0.9584 �0.00451.4 �8.061666611 �18.19025 �0.8814 �0.03611.45 �8.064425535 �18.50019 �0.8147 �0.06511.5 �8.066203195 �18.82008 �0.7564 �0.09171.55 �8.067155663 �19.14853 �0.7053 �0.11631.59491123 �8.067417267 �19.44972 �0.6647 �0.13661.65 �8.067095170 �19.82565 �0.6205 �0.15921.7 �8.066290690 �20.17161 �0.5851 �0.17801.75 �8.065083681 �20.52066 �0.5534 �0.19531.8 �8.063543838 �20.87131 �0.5249 �0.21051.9 �8.059695260 �21.57140 �0.4760 �0.23642.0 �8.055127893 �22.25954 �0.4353 �0.26062.1 �8.050112404 �22.92459 �0.4007 �0.28002.2 �8.044857596 �23.54810 �0.3704 �0.29742.3 �8.039506202 �24.11859 �0.3433 �0.31582.4 �8.034170782 �24.61760 �0.3184 �0.33762.5 �8.028925412 �25.02839 �0.2949 �0.36732.6 �8.023842356 �25.32147 �0.2720 �0.41022.75 �8.016613300 �25.49113 �0.2377 �0.51293.0 �8.005822077 �24.84864 �0.1793 �0.85733.25 �7.996867704 �22.72133 �0.1196 �1.46733.3 �7.995310387 �22.10585 �0.1080 �1.61453.35 �7.993837241 �21.39819 �0.0963 �1.74123.4 �7.992441383 �20.66748 �0.0854 �1.86893.45 �7.991124757 �19.88620 �0.0749 �1.98293.5 �7.989886632 �19.06068 �0.0649 �2.07983.55 �7.988729758 �18.14300 �0.0549 �2.15333.6 �7.987647628 �17.25218 �0.0463 �2.19913.65 �7.986639457 �16.34228 �0.0383 �2.21643.7 �7.985702979 �15.42241 �0.0310 �2.20433.75 �7.984835601 �14.50161 �0.0245 �2.16463.8 �7.984034441 �13.58851 �0.0188 �2.09873.85 �7.983296379 �12.69109 �0.0137 �2.00993.9 �7.982618112 �11.81647 �0.0094 �1.90383.95 �7.981996211 �10.97070 �0.0056 �1.78404.0 �7.981427178 �10.15875 �0.0025 �1.65534.25 �7.979254156 �6.70307 0.0060 �1.01294.5 �7.977915733 �4.27505 0.0075 �0.55454.75 �7.977149024 �2.63273 0.0065 �0.27445.0 �7.976671992 �1.63497 0.0048 �0.14037.5 �7.975938063 �0.00819 0.0000 �0.000210.0 �7.975935907 �0.00020 0.0000 0.0001
SAUER ET AL.
742 INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY DOI 10.1002/qua VOL. 111, NO. 4

3], we calculated values of tLi(R) and tH(R), andthen fitted them as a polynomial in z, taking suffi-cient terms to fit the input data essentially withintheir precision. The resulting values of coefficientstLij and tHj are listed in Table III. An alternativeprocedure involves fitting of the directly calcu-lated values of tLi,H(R)[17] to yield essentially thesame polynomials.
To achieve a global fit of spectral data of LiH,we applied procedure Radiatom [9]: in the origi-nal version (in Fortran) with precision 32 decimaldigits, expressions for Ykl [7] were derived sym-bolically (with program Reduce) through a JBKWtreatment [55]; in a new version (entirely in soft-ware Maple) with precision 24 decimal digits, allexpressions were freshly evaluated symbolicallyaccording to hypervirial perturbation theory [56,57]. We verified that algebraic expressions of termcoefficients Ykl derived by either method are iden-tical, but the latter method [56, 57] is computa-tionally more efficient to produce those expres-sions than the former. Numerical values ofparameters derived through Radiatom in eitherversion are not quite identical—because the algo-rithm for nonlinear regression [9] according to theapproach of Levenberg and Marquardt hasslightly different implementations, but differencesin values are within estimated single standarderrors as stated in Table III from the Fortran ver-sion. Initial estimates of all unconstrained param-eters were set to zero except rough estimates of
U1,0 and U0,1, and generic values of c1 and c2 [13];during progress toward convergence through �20iterations, the weighted value of v2 decreasedfrom �1013 to �103. The duration of a given fitwith Maple, for which code is partially inter-preted, is about 30 times that with the fully com-piled Fortran procedure on computers with com-parable speeds of processors. Other fitting of datawas affected with Maple procedures.
FIGURE 1. Energy/hc of LiH as a function of internu-clear separation R. The small squares indicate points ofelectronic energy E0(R) from calculations of molecularelectronic structure relative to zero energy at Re; thecurve represents potential energy V(R) from analysis ofmolecular spectra, and horizontal lines denote energiesof vibrational states of 7Li1H with v ¼ 0 … 6 in ascend-ing order.
FIGURE 2. (a) Rotational and vibrational g factors,gr(R) and gv(R), as a function of internuclear distance,from quantum-chemical calculations; the black curvedenotes the result from spectral analysis over its domainof validity, (b) electric dipolar moment p(R) and its gradi-ent dp(R)/dR as a function of internuclear distance R,from quantum-chemical calculations, and (c) squarednonadiabatic coupling matrix element |h0|q/qR|1i|2 con-necting electronic ground state 0 ¼ X 1Rþ and electroni-cally excited state 1 ¼ A 1Rþ as a function ofinternuclear separation R. [Color figure can be viewed inthe online issue, which is available atwileyonlinelibrary.com.]
ROTATIONAL AND VIBRATIONAL g FACTORS OF LiH
VOL. 111, NO. 4 DOI 10.1002/qua INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 743

Because our previous analysis of data on LiH[7], new measurements of wave numbers of purerotational and vibration-rotational transitions ofLiH in four isotopic variants—6Li1H, 6Li2H, 7Li1H,and 7Li2H—were reported [58] up to v ¼ 6 forspecies involving 7Li, 1H, and 2H, but up to onlyv ¼ 3 for species involving 6Li. We used thesedata instead of earlier vibration-rotational data of
smaller extent and poorer precision, and com-bined them with frequency data for pure rota-tional transitions [59–62] in a global fit of 1000data that yielded significant values of only 20adjusted parameters: c1 – c9, U0,1 ¼ Y0,1/l,U1;0 ¼ Y1;0=l
12, sLi0 , s
H0 , s
H1 , u
Li1 , u
Li2 , u
H1 , u
H2 , u
H3 , and
uH4 , of which the values appear in Table III with afew other derived values. The weight of each
TABLE IIRotational g factor and electric dipolar moment/10230 C m of 7Li1H at Re 5 1.59491123 3 10210 m and forvibrational states v 5 0 and 1 and their rotational dependences.
Property Calculation Experiment
gr Re �0.6647v ¼ 0 �0.6572 þ 1.52 10�4 J (J þ 1) (�0.65842 6 0.00017)a þ (1.2 6 0.6) 10�4 J (J þ 1)b
v ¼ 1 �0.6439 þ 1.51 10�4 J (J þ 1)|p| Re 19.4493 19.439c
v ¼ 0 19.6376 þ 1.25 10�3 J (J þ 1) 19.620 6 0.001c
v ¼ 1 20.0004 þ 1.29 10�3 J (J þ 1) 19.982 6 0.001c
aRef. [49].b Ref. [50].c Ref. [51].
TABLE IIICoefficients of radial functions and associated parameters of LiH X 1R1a.
c0/m�1 6572379.3 6 5.6 sLi0 0.91260 6 0181
c1 �0.8970678 6 0.0000057c2 0.348233 6 0.000040 sH0 �0.2612 6 0.0029c3 �0.093085 6 0.000196 sH1 �0.3733 6 0.0195c4 �0.044426 6 0.00087c5 0.0765 6 0.0027 uLi0 /10
6 m�1 [168.411 6 0.002]c6 �0.1143 6 0.0078 uLi1 /10
6 m�1 �5.53900 6 0.0082c7 �0.2078 6 0.024 uLi2 /10
6 m�1 6.357 6 0.134c8 0.6024 6 0.049 uH0 /10
6 m�1 [12.829 6 0.002]c9 �0.7538 6 0.115 uH1 /10
6 m�1 �5.15624 6 0.00096U0,1/m
�1 u 662.708918 6 0.000082 uH2 /106 m�1 4.923 6 0.021
U1,0/m�1 u
1=2 131993.551 6 0.064 uH3 /106 m�1 �3.340 6 0.051
Re/10�10 m 1.59491242 6 0.00000020 uH4 /10
6 m�1 4.828 6 0.158ke/N m�1 102.649202 6 0.000100tLi0 [0.749508 6 0.000142] tH0 [�0.772779 6 0.000051]tLi1 [0.60714 6 0.00115] tH1 [1.28086 6 0.00047]tLi2 [�1.2181 6 0.0022] tH2 [�1.7927 6 0.00191]tLi3 [1.077 6 0.022] tH3 [2.0040 6 0.0088]tLi4 [�1.710 6 0.106] tH4 [�1.6797 6 0.0198]tLi5 [3.546 6 0.160] tH5 [2.375 6 0.033]tLi6 [�2.586 6 0.34] tH6 [�0.5685 6 0.079]tLi7 [�6.825 6 0.65] tH7 [�0.8264 6 0.042]tLi8 [6.37 6 0.31]
a In this table, apart from coefficients in radial functions defined through formulae above, appear U1,0 and U0,1, which correspond
to Y1,0 and Y0,1, respectively, in equation (4) with their dependence on mass eliminated, and equilibrium force coefficient ke. Val-ues enclosed within brackets are fitted from results of quantum-chemical calculations to serve, except uLi0 and uH0 , as constraintsin fits of spectral data. Stated uncertainties represent single standard deviations in the fitting, and uncertainties of ke and Re
include uncertainties of the pertinent physical constants.
SAUER ET AL.
744 INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY DOI 10.1002/qua VOL. 111, NO. 4

frequency or wavenumber item in the fit was gen-erally a reciprocal of its squared uncertainty.There is considerable scatter of residuals of thepure rotational data [59–62], relative to a standarddeviation of a global fit dominated by infrareddata [58]; seven data from Matsushima et al. [61]had to be accorded reduced weights. Even so, thereduced standard deviation of the global fit is1.25, significantly larger than unity, implying thatuncertainties assigned to some data by their origi-nal authors seem too conservative or that tensionremains between measurements of the same tran-sitions in separate laboratories. Further parame-ters yielded neither statistically significant valuesnor a significantly improved fit.
5. Discussion
According to the values, in Table III, of Re
derived from fitted U0,1, of c0 derived from fittedU1,0 and U0,1, and of fitted coefficients cj, 1 � j � 9,and assuming zero values for all further cj, we eval-uated formula (6) for internuclear potential energyV(z) of LiH independent of atomic mass; becausethe latter experimentally derived quantity fromwhich all mass dependence has been eliminated,which is formally equivalent to electronic energyE0 from Table I relative to E0 at Re, we plot in Figure1 this curve applicable to the electronic groundstate of LiH, X 1Rþ, for comparison with our resultsfrom quantum-chemical calculations; the vibra-tional terms for states 0 � v � 6 of 7Li1H are indi-cated also in Figure 1 to illustrate the range ofenergy sampled by the included spectral data. Therange of energy in which this curve is valid is thusapproximately limited by the energy of that state v¼ 6; the corresponding domain of internuclear dis-tance is R/10�10 m ¼ [1.05, 2.47]. In succeedingplots, the region of validity of a formula for a dis-played curve in terms of its interval of internucleardistance is indicated approximately by its corre-sponding projection on the abscissal axes.
We consider next the rotational and vibrationalg factors, according to the results of our calcula-tions. Both gr(R) and gv(R) are expected to havezero values for internuclear distances R ¼ 0, forthe united atom, and R ! 1, for the separateatoms, and to exhibit at least one extremumbetween these limits that reflects molecular bind-ing. Comparison of the computed points or thecorresponding curves in Figure 2(a) reveals thatvalues of gr(R) increase monotonically from �1.8
at 1.05 � 10�10 m and appear to approach zeroasymptotically as R increases within the domainof calculation, whereas gv decreases from 0.25 atR ¼ 1.05 � 10�10 m to a pronounced minimumwith gv ¼ �2.22 at R ¼ 3.65 � 10�10 m beforetending to zero for R � Re. We observe that ourcalculated g-factors do not yet approach theunited atom limiting values at an internuclear dis-tance 1.05 � 10�10 m. Additional calculations forsmaller internuclear distances were computation-ally impracticable due to convergence problemsand linear dependences in the used basis set.Futhermore, these internuclear distances are notrelevant for the fitting of available spectral datadespite their importance in the study of the limit-ing behavior of the g-factors.
We inquire whether these behaviors might beattributed to either electric dipolar moment p(R)for gr(R) and its derivative dp(R)/dR for gv(R), asdepicted in Figure 2(c), or the irreducible contri-butions, girrr (R) and girrv (R), according to Eqs. (16)and (17), respectively. There is no evident relationbetween features of curves of p(R) and gr(R). Fig-ure 3 shows that girrr (R) increases from �0.85 at1.05 � 10�10 m to a maximum about 0.23 at 3 �10�10 m and then decreases gradually towardzero with increasing internuclear distance. girrv (R)decreases from 0.38 at 1.05 � 10�10 m to a deepminimum, �2.8, at 3.65 � 10�10 m beforeapproaching zero for R � Re; that minimum coin-cides with both the minimum of gv(R) and themaximum of dp(R)/dR. The latter calculated mini-mum in gv(R) lies beyond the domain of internu-clear distance to which the available spectral dataare sensitive, according to Figure 1. Another inter-pretation involves recognition of each g factor as
FIGURE 3. Irreducible components, girrr (R) and girr
v (R),of rotational and vibrational g functions as a function ofinternuclear distance, from quantum-chemical calcula-tions. [Color figure can be viewed in the online issue,which is available at wileyonlinelibrary.com.]
ROTATIONAL AND VIBRATIONAL g FACTORS OF LiH
VOL. 111, NO. 4 DOI 10.1002/qua INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 745

a sum of contributions from the two atomic cen-ters, as indicated in Eqs. (7) and (8) consistentwith Eqs. (16) and (17) [8, 16, 17]; explicitly, gv(R)becomes the vibrational g factor of a molecule inwhich Ma is infinite so that only atomic center Bvibrates, and analogously for gr(R) [16, 17]. A fur-ther interpretation of girrv (R) is that this contribu-tion to gv(R) is inexpressible in terms of expecta-tion values of the wave function for the electronicground state, unlike the other term involving p(R)[16, 17]. In either case, these two interpretationsprovide no explanation of a deep minimum ofgv(R) at a particular internuclear separation. Todiscover the source of this minimum, we calcu-lated the nonadiabatic coupling matrix elementfor transitions between the electronic groundstate, denoted X 1Rþ, and the first electronicallyexcited state, A 1Rþ, of hence the same symmetryclass; the plotted points from this calculationappear in Figure 2(c) for the applicable region. Amaximum of the squared nonadiabatic matrix ele-ment h0j @
@R j1i�� ��2 that couples electronic ground
state X 1Rþ of LiH to electronically excited state A1Rþ appears at the same internuclear separation3.65 � 10�10 m. The difference of energies En(R)� E0(R) between electronically excited state A 1Rþ
and electronic ground state X 1Rþ must be a posi-tive quantity at all internuclear separationsbecause the corresponding curves for potentialenergy must not intersect. As Eq. (12) indicates,the maximum in the squared matrix elementinvolving the first electronically excited state A1Rþ, hence produces a minimum of gv(R) at R ¼3.65 � 10�10 m. For H2, we concluded also thatsuch matrix elements in the numerator of Eq. (12),rather than the energy difference in the corre-sponding denominator, were responsible for theproduction of a minimum in that vibrational gfactor [25]; in particular, the matrix element cou-pling the first electronically excited state of class1Rþ
g of H2 exhibited a maximum near the sameinternuclear distance at which appeared a mini-mum of gv(R), but the exact position of the latterminimum was influenced by the excitation energyand by the coupling to more highly excited states.Likewise for HeHþ [26], the extrema in the calcu-lated curve of gv(R) correspond to extrema in thefirst-order matrix element coupling the electronicground state and first excited state.
Regarding the adiabatic contributions to thevibration-rotational energies of LiH in its isotopicvariants, from values of uLij in Table III fittedfrom spectra plus a constant, uLi0 , derived from
published numerical values at several internucleardistances [1], we derived the following formula torepresent a radial function for an adiabatic correc-tion associated with the lithium atomic center in-dependent of atomic mass,
DVLiad=u m�1 ¼ 92385:19
þ ð�3086:86 8:2Þzþ ð35976 73Þz2 ð20Þ
which is plotted as a curve in Figure 4(a), withcalculated points [1] for comparison; for an addi-tional comparison, an exact polynomial represen-tation of those calculated points [1] yields thisformula:
DVLiad=u m�1 ¼ 92385:19� 3169:90 zþ 4896:87 z2
þ 1987:27 z3 � 11905:13 z4 � 37933:27 z5
þ 71718:27 z6 þ 74959:08 z7 ð21Þ
From fitted values of uHj in Table III, we deriveanalogously a corresponding formula to representa radial function for an adiabatic correction asso-ciated with the hydrogen atomic center,
DVHad=u m�1 ¼ 7035þ ð�2828:4860:52Þz
þ ð2688:2611:5Þz2 þ ð�1405659Þz3 þ ð13316187Þz4(22)
which is plotted as a curve in Figure 4(b), withthe calculated points [1].
Because data for isotopic species 6Li are avail-able to only v ¼ 3 and because the relative massdifference between masses of 6Li and 7Li issmaller than between 1H and 2H, the formula forDVH
ad(z) in Eq. (9) is truncated at a quadratic term,whereas the quartic polynomial for DVLi
ad(z) in Eq.(9) reflects data to v ¼ 6. A corresponding exactfit to calculated points [1] yields
DVHad=u m�1 ¼ 7035:2� 3160:75 zþ 5584:07 z2
þ 13854:46 z3 � 100067:32 z4
þ 13622:76 z5 þ 502328:20 z6 � 523003:44 z7 ð23Þ
In Figure 4(c), we compare the curve for thetotal adiabatic correction for 6Li1H from a sum ofcorrections of separate atomic centers in Eqs. (20)and (22) divided by their masses,
DVad ¼ DVLiad
MLiþ DVH
ad
MH(24)
SAUER ET AL.
746 INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY DOI 10.1002/qua VOL. 111, NO. 4

with the corresponding points for the total calcu-lated adiabatic correction [1]. According to thesethree plots in Figure 4, the calculated points andcurves agree satisfactorily within the appropriatedomain; that the values of corresponding coeffi-cients in the Eqs. (20) and (21), or (22) and (23),appear more discordant results in part from theuncertainty in deriving small corrections DV(R)from spectral data prone to error of measurementin the presence of the dominant influence V(R) ofwavenumbers and frequencies of spectral transi-tions, and in part because of the disparate ordersof polynomials.
In Figure 5(a), we plot points calculated forsLi(R) from values of gv in Table I with a linereflecting the constant term sLi0 in Table III, and inFigure 5(b) the corresponding points for sH(R)and a curve for sLi0 þ sLi1 z with values of fittedcoefficients in Table III. From those three deducedvalues of sLi and sH, we generate a formula forthe radial dependence of the vibrational g factorof 7Li1H,
gv ¼ ð�0:129160:0039Þ þ ð�0:373360:0195Þz (25)
truncated at a linear term because only sLi;H0 andsLi1 are evaluated significantly with available spec-tral data; this curve is plotted in Figure 2(a) withcalculated points according to Table I. A fit ofthose calculated data to a polynomial in z, in adomain R/10�10 m ¼ [1.05, 3.0] that exceeds thedomain of internuclear distance sampled in spec-tral measurements, requires an eighth order foran essentially exact fit,
gv¼ð�0:13655360:000056Þ�ð0:6906460:00073Þzþð0:638160:0038Þz2þð0:043560:027Þz3
�ð0:38860:056Þz4�ð0:15560:29Þz5þð1:40260:35Þz6�ð6:28560:93Þz7�ð17:9661:14Þz8 ð26Þ
in which the stated uncertainties represent onlythe standard errors of the coefficients in the fit.The constant terms agree within two larger stand-ard errors, but the coefficient of z from spectraldata has a magnitude too small despite the correctsign; this disparity reflects in part the lack of sLi1 ,in part the much greater order of the polynomialin the latter formula for the calculated data, andin part the experimental error in measurements oftransition wavenumbers.
A fit of the data for gr(z) in Table III requires apolynomial of degree 10 to reproduce all data in a
FIGURE 4. (a) Adiabatic correction for atomic centerLi as a function of internuclear separation R, (b)adiabatic correction for atomic center H as a functionof internuclear separation R, and (c) total adiabaticcorrection for 6Li1H as a function of internucleardistance R; in all cases, the curve within the domain ofits validity is deduced from the spectral analysis with afixed constant term, and the calculated points arisefrom theoretical calculations [1].
ROTATIONAL AND VIBRATIONAL g FACTORS OF LiH
VOL. 111, NO. 4 DOI 10.1002/qua INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 747
domain R/10�10 m ¼ [1.05, 3.0] within the preci-sion of the calculation,
gr ¼ ð�0:66467360:000016Þ þ ð1:3658860:00024Þz� ð1:987660:0018Þz2 þ ð2:196260:014Þz3� ð1:69160:044Þz4 þ ð2:87660:26Þz5
� ð2:54860:38Þz6 � ð5:32161:93Þz7 þ ð8:27361:73Þz8þ ð14:9565:0Þz9 � ð21:7465:4Þz10 ð27Þ
in which the stated uncertainties represent onlythe standard errors of the coefficients in the fit.The full data are plotted in Figure 2(a). Note that
gr is not equal to zero at Re, as assumed or set foran equivalent quantity by Watson [63].
The curves pertaining to both adiabatic correc-tions and the vibrational g factor generally con-form to points from the quantum-chemical calcu-lations, as a comparison of pertinent formulaealso shows. Because the domain of molecularenergies sampled in spectral data [58] for isotopicspecies of LiH containing 6Li, up to only v ¼ 3, isless than that for species containing 2H, up to v ¼6, the corresponding domains of radial functionsfor atomic center Li are less wide than for H. Theslopes and curvatures of curves for both DVLi
ad
and DVHad near Re conform satisfactorily to the
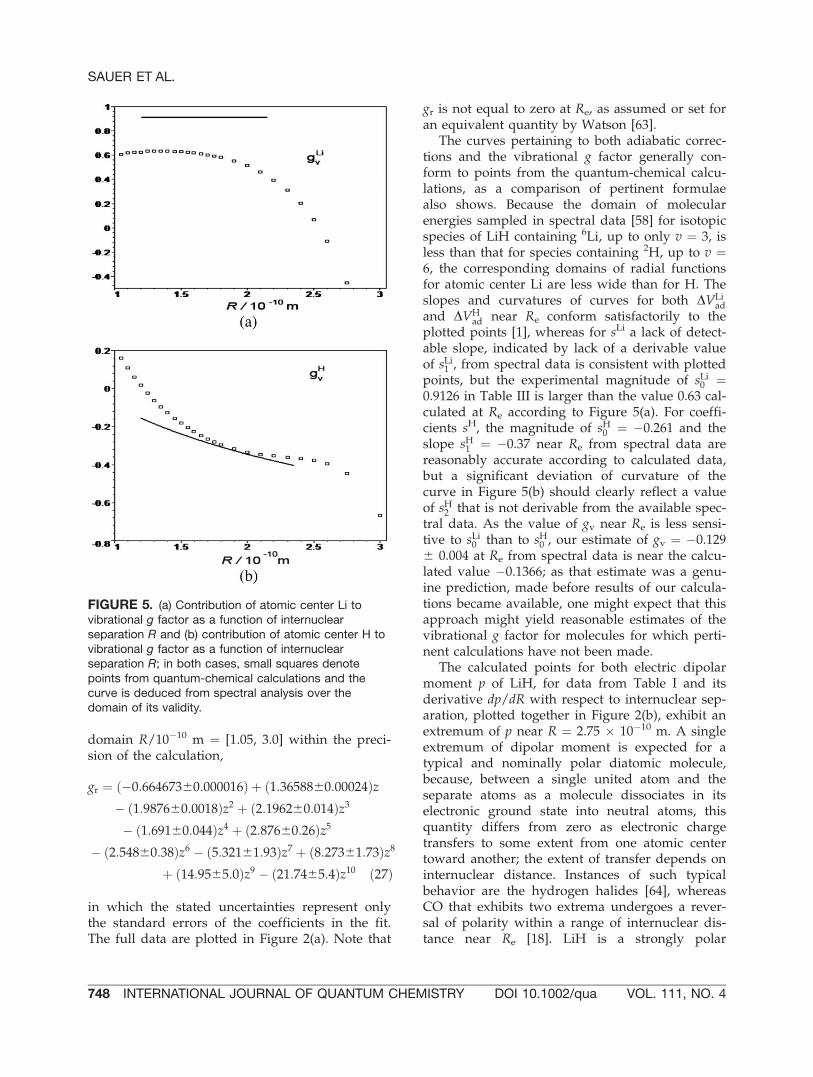
plotted points [1], whereas for sLi a lack of detect-able slope, indicated by lack of a derivable valueof sLi1 , from spectral data is consistent with plottedpoints, but the experimental magnitude of sLi0 ¼0.9126 in Table III is larger than the value 0.63 cal-culated at Re according to Figure 5(a). For coeffi-cients sH, the magnitude of sH0 ¼ �0.261 and theslope sH1 ¼ �0.37 near Re from spectral data arereasonably accurate according to calculated data,but a significant deviation of curvature of thecurve in Figure 5(b) should clearly reflect a valueof sH2 that is not derivable from the available spec-tral data. As the value of gv near Re is less sensi-tive to sLi0 than to sH0 , our estimate of gv ¼ �0.1296 0.004 at Re from spectral data is near the calcu-lated value �0.1366; as that estimate was a genu-ine prediction, made before results of our calcula-tions became available, one might expect that thisapproach might yield reasonable estimates of thevibrational g factor for molecules for which perti-nent calculations have not been made.
The calculated points for both electric dipolarmoment p of LiH, for data from Table I and itsderivative dp/dR with respect to internuclear sep-aration, plotted together in Figure 2(b), exhibit anextremum of p near R ¼ 2.75 � 10�10 m. A singleextremum of dipolar moment is expected for atypical and nominally polar diatomic molecule,because, between a single united atom and theseparate atoms as a molecule dissociates in itselectronic ground state into neutral atoms, thisquantity differs from zero as electronic chargetransfers to some extent from one atomic centertoward another; the extent of transfer depends oninternuclear distance. Instances of such typicalbehavior are the hydrogen halides [64], whereasCO that exhibits two extrema undergoes a rever-sal of polarity within a range of internuclear dis-tance near Re [18]. LiH is a strongly polar
FIGURE 5. (a) Contribution of atomic center Li tovibrational g factor as a function of internuclearseparation R and (b) contribution of atomic center H tovibrational g factor as a function of internuclearseparation R; in both cases, small squares denotepoints from quantum-chemical calculations and thecurve is deduced from spectral analysis over thedomain of its validity.
SAUER ET AL.
748 INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY DOI 10.1002/qua VOL. 111, NO. 4

molecule: the permanent electric dipolar momentof 7Li1H, which is 19.4393 � 10�30 C m at Re fit-ted from experiment [51], with which our calcula-tion according to Table II agrees satisfactorily, isabout three quarters of the value that wouldapply to a simple cation and anion with charge ofmagnitude e at that separation. As internucleardistance R increases from Re for such a stronglypolar species, the molecule initially tends towarddissociation into ions—Liþ and H�, each with onenet electronic charge. Because, for all neutral dia-tomic molecules in their electronic ground states,dissociation into neutral atoms requires lessenergy than dissociation into ions, at some pointfor R > Re the path towards that ionic limit altersinstead to the neutral limit for atoms Li and H. Interms of curves for potential energy, one (adia-batic) curve, similar to that depicted in Figure 1,for the electronic ground state with that neutrallimit approaches, but fails to cross, another curvefor an electronically excited state that correlateswith an ionic limit; the point Rx of nearestapproach between these curves is called anavoided crossing, which for LiH occurs near 3.5 �10�10 m [65]. To define adequately various prop-erties in the vicinity of this avoided crossing, wehave calculated points for R in small increments,as presented in Table I and appearing in the perti-nent plots. Already before a point marking thisavoided crossing, the electric dipolar moment ofthe electronic ground state begins to decay to-ward zero, but the derivative of dipolar momentwith respect to distance has an extremum nearthis point. The vibrational g factor, which accord-ing to Eq. (17) has a contribution from that deriv-ative of dipolar moment, likewise exhibits anextremum near the same point. These features areperceptible in Figures 2(a) and (b). Nonadiabaticvibrational effects, which the electronic contribu-tion to gv(R) reflects, involve squares of matrixelements connecting these same two electronicstates of the same symmetry class 1Rþ; these ma-trix elements that produce the values of gv in Ta-ble I have accordingly a large magnitude near Rx,as shown in Figure 2(c). In sum, unlike the caseof H2 for which avoided crossings occur betweenelectronically excited states in two separate pairsare not associated with the prominent minimumin the curve of gv(R), for LiH an avoided crossinginvolving electronic ground state X 1Rþ and firstelectronically excited states A 1Rþ is associatedwith the prominent minimum in the curve ofgv(R).
As a result of our combining data from experi-ment and theoretical calculations [6], we haveidentified and separated quantitatively the contri-butions from adiabatic corrections and nonadia-batic rotational and vibrational effects on theenergies of molecular vibration-rotational statesthrough auxiliary term coefficients ZLi;H
kl in Eq. (4).As explained in detail elsewhere [13] and men-tioned in the introduction above, these term coef-ficients Zkl can be divided in turn into two com-ponents for atoms of each type, Zv
kl for purelyvibrational effects with an associated centrifugalterm, and Zr
kl for further rotational effects. Thesecomponents reflect the fact that, through isotopicvariants, one can in principle deduce informationabout extra-mechanical effects of only two kindsfor each atomic type, beyond the function forpotential energy derived from purely coefficientsYkl when coefficients Zkl are present to absorb theextra-mechanical effects. Adiabatic correctionscontribute only to Zv
kl and the rotational g factoronly to Zr
kl, but vibrational g factor contributes toboth [9]. Through separate calculation, one canestimate numerically not only each such contribu-tion but also, and more meaningfully, a ratio ofthat contribution to the corresponding principalterm coefficient Ykl. As that ratio Zkl/Ykl isexpected to have a value of order a ratio me/M ofelectronic and atomic mass, further division bythe latter ratio might yield values of order unity.In Table IV, we present such values, each withfour significant digits, of that ratio Zkl/Ykl/(me/M) for k and l of small values for four specifiedcontributions to Zkl and with MH or MLi as appro-priate; for this purpose, we use parameters Re
and cj, 0 � j � 9, of potential energy derived fromspectra but, in Zkl, parameters sj, tj, and uj ofextra-mechanical effects from quantum-chemicalcalculations, because the latter are more abundantthan those from experiment.
As noted above, there is no contribution of grthrough coefficients tj to purely vibrational termcoefficients Zk,0; there is likewise no net contribu-tion of gv through coefficients sj to rotational termcoefficients Z0,l because separate contributions toZr and Zv cancel identically. The fact that thereare thus only two contributions to Z0,1 in total,with no possible interference from gv, permit inprinciple an estimate of gr and electric dipolarmoment p at Re from spectral data in absence ofapplied external fields, from spectra of adequatequality and quantity of transitions; such an esti-mate by this method of gr of GaH [66] was
ROTATIONAL AND VIBRATIONAL g FACTORS OF LiH
VOL. 111, NO. 4 DOI 10.1002/qua INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 749

subsequently confirmed by quantum-chemical cal-culations [67]. For LiH, the available spectral datalack sufficient quality and quantity for its isotopicvariants for such a purpose. Trends evident fromTable IV indicate that, for given k and increasingl, values of this adjusted ratio might have magni-tudes much greater than unity, but that magni-tudes of contributions from gv are generallysmaller than those from gr and adiabatic correc-tions. Whether the latter trend is general remainsto be tested with other molecules, but for both H2
[25] and LiH the magnitudes of gv at Re are mark-edly smaller than of those of gr there. AlthoughFigure 2(b) demonstrates that values of both gvand gr cover comparable ranges, the effect of an
avoided crossing might contribute atypically to themagnitude of gv for R > Re, whereas gr is unaf-fected by this particular perturbation from an elec-tronically excited state because of separate symme-try classes. Some magnitudes of the adjusted ratioZkl/Ykl in Table IV are even comparable with a ratioM/me, which is �1,837 for 1H, but the effect onthose energies remains small because for corre-sponding values of k and l the magnitude of Ykl isalso much smaller than for Y1,0 or Y0,1 that are prin-cipal contributors to molecular vibration-rotationalenergies. A common chemical perception of atomicstructure within a molecule, as implemented in atraditional separation of electronic and nuclearmotions, hence remains a useful approximation,within definable limits. On subtracting the nuclearcontribution to the rotational and vibrational g fac-tors form the total values at each internuclear dis-tance, one can calculate the adiabatic and nonadia-batic contributions to coefficients Zkl, and hence,with Eq. (4), these contributions to the energy orspectral term of vibration-rotational state.
Our results here demonstrate that the agreementbetween radial functions deduced from spectraand resulting from quantum-chemical calculationsis satisfactory, within experimental error that isreasonably small for quantities pertaining to, ornear, equilibrium internuclear separation Re.Through calculation of these radial functions, ofwhich that for V(R) is most critical, one can hencecalculate a vibration-rotational spectrum essen-tially within the error of experimental measure-ment [6]. This approach is, therefore, a viable alter-native to much more complicated calculations ofmolecular electronic structure undertaken withoutinvocation of separate treatment of electronic andnuclear motions [68]. The latter approach requiresa full calculation for each vibration-rotational stateand has so far been achieved for states of H2 withzero total angular momentum of electrons andnuclei [68], thus for only vibrational states, and forLiH [69] in vibrational states v ¼ 0 and v ¼ 1.
6. Conclusions
We present here the first quantum-chemicalcalculation of the vibrational g factor for a neutralheteroatomic diatomic molecule for internucleardistance over a broad range, with correspondingresults for the rotational g factor, the electric dipo-lar moment and its gradient. Using the latter dataas constraints of available spectral data of
TABLE IVSeparate contributions to auxiliary term coefficientsZkl from adiabatic corrections (ad) and vibrationaland rotational g factors as a ratio withcorresponding Ykl divided by a ratio of atomic andelectronic masses, so Zkl/Ykl /(me/M).
Li in 7Li1H
Zr Zv
k l gr gv ad gv0 1 0.7495 0 �0.8788 00 2 0.8914 0.6188 �1.932 �0.61880 3 �0.3613 1.852 �1.992 �1.8520 4 �2.432 3.033 �0.4592 �3.0331 0 0 0 �0.3522 0.30951 1 1.124 �0.0697 �2.632 0.44781 2 4.041 0.3809 �11.78 �0.30412 0 0 0 �1.031 0.81352 1 1.888 �0.0362 �56.27 �0.42622 2 �0.5970 �4.144 51.64 5.9473 0 0 0 �47.67 �0.3677
H in 7Li1H
Zr Zv
k l gr gv ad gv0 1 �0.7727 0 �0.8764 00 2 �2.827 �0.2255 �2.122 0.22550 3 �7.020 �0.3482 0.7656 0.34820 4 �12.74 �0.1741 18.76 0.17411 0 0 0 �0.2537 �0.11281 1 �0.2714 �0.1087 �6.588 0.23921 2 2.472 �1.033 �90.85 2.1132 0 0 0 5.432 �0.09902 1 1.677 �0.4588 179.7 1.8542 2 14.20 �3.287 2353 8.9343 0 0 0 16.21 0.6157
SAUER ET AL.
750 INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY DOI 10.1002/qua VOL. 111, NO. 4

frequencies and wave numbers of pure rotationaland vibration-rotational transitions of LiH in fourisotopic variants, we have estimated the separatecontributions of adiabatic corrections and vibra-tional g factor to the eigenenergies of any heteroa-tomic diatomic species; our analysis of spectraldata is based on a comprehensive molecularHamiltonian [8] and algebraic expressions forterm coefficients Ykl [55, 70] and Zkl [55]. The val-ues of gv at Re for LiH from these separate theo-retical and experimental approaches agreeroughly within error propagated from measure-ment of frequency data. Previous estimates of gvat Re for several diatomic molecules were implic-itly achieved, whenever values of both sa;b0 weresignificantly evaluated, but such estimates of gvappear to be more sensitive to the quality andquantity of spectral data available for a given dia-tomic molecular species than estimates of adia-batic corrections. Intervals in which these radialfunctions are defined from experimental data aremuch smaller than the range for which theoreticalcalculations are practicable, but this conditionreflects the quality and quantity of available spec-tral data. Spectral data of LiH have relative preci-sion �2 � 10�7 or worse, whereas the best avail-able measurements of transitions in the midinfrared region have precision �2 � 10�10 [71].When measurements approaching the latter preci-sion become more generally available, in datareduction to coefficients of applicable radial func-tions one must clearly take into account the con-tributions of DVnad(R) in DV(R) in the effectiveHamiltonian, Eq. (3), that we neglect here; apartfrom that additional term, with its implied de-pendence on a greater ratio of electronic to atomicmasses than we include explicitly, the presentapproach to analysis of spectral data continues tobe applicable. For the present data of LiH, 20 val-ues of fitted coefficients pertaining to specified ra-dial functions and likely about 10 values of con-strained parameters, listed in Table III, suffice toreproduce not only at least 1000 measured dataalmost within their uncertainties but also hun-dreds of further measured and unmeasured tran-sitions within the same range of vibration-rota-tional energy. Although only the radial functionfor the rotational g factor has a truly direct experi-mental basis, the generally satisfactory agreementbetween theoretically calculated points and exper-imentally derived curves for potential energy, adi-abatic corrections and the vibrational g factor,within the context of an effective Hamiltonian in
Eq. (3), implies that even these artifacts of separa-tion of electronic and nuclear motions can assumealmost a physical and chemical significance. Thefact that the ratios of magnitudes of the most im-portant term coefficients Zkl for extra-mechanicaleffects relative to corresponding mechanical coef-ficients Ykl are of order me/M, denoting a ratio ofelectronic and atomic masses, likewise impliesthat, to an approximation satisfactory for manychemical purposes, a molecule might be consid-ered to comprise two or more atomic centers,more or less distinct, rather than formally justelectrons and atomic nuclei.
References
1. Bishop, D. M.; Cheung, L. M. J Chem Phys 1983, 78, 1396.
2. Bishop, D. M.; Cheung, L. M. J Chem Phys 1983, 78, 7265.
3. Bishop, D. M.; Cheung, L. M. J Chem Phys 1983, 79, 2945.
4. Bishop, D. M.; Lam, B. Chem Phys Lett 1985, 120, 69.
5. Maroulis, G.; Bishop, D. M. Theor Chim Acta 1986, 69, 161.
6. Ogilvie, J. F.; Oddershede, J. Adv Quantum Chem 2005, 48,253.
7. Ogilvie, J. F.; Oddershede, J.; Sauer, S. P. A. Chem PhysLett 1994, 228, 183.
8. Herman, R. M.; Ogilvie, J. F. Adv Chem Phys 1998, 103, 287.
9. Ogilvie, J. F. J Phys B 1994, 27, 47.
10. van Vleck, J. H. J Chem Phys 1936, 4, 327.
11. Herman, R. M.; Asgharian, A. J Mol Spectrosc 1966, 19,305.
12. Dunham, J. L. Phys Rev 1932, 41, 721.
13. Ogilvie, J. F. The Vibrational and Rotational Spectrometryof Diatomic Molecules; Academic: London, UK, 1998.
14. Ogilvie, J. F. Proc R Soc London A 1981, 378, 287.
15. Ogilvie, J. F. Proc R Soc London A 1982, 381, 479.
16. Kjaer, H.; Sauer, S. P. A. Theor Chem Acc 2009, 122, 137.
17. Sauer, S. P. A. Chem Phys Lett 1998, 297, 475.
18. Sauer, S. P. A.; Ogilvie, J. F. J Phys Chem 1994, 98, 8617.
19. Ogilvie, J. F.; Cheah, S.-L.; Lee, Y.-P.; Sauer, S. P. A. TheorChem Acc 2002, 108, 85.
20. Ogilvie, J. F.; Oddershede, J.; Sauer, S. P. A. Adv ChemPhys 2000, 111, 475.
21. Sauer, S. P. A.; Oddershede, J.; Geertsen, J. Mol Phys 1992,76, 445.
22. Sauer, S. P. A.; Enevoldsen, T.; Oddershede, J. J ChemPhys 1993, 98, 9748.
23. Enevoldsen, T.; Rasmussen, T.; Sauer, S. P. A. J Chem Phys2001, 114, 84.
24. Sauer, S. P. A. Adv Quantum Chem 2005, 48, 319.
25. Bak, K. L.; Sauer, S. P. A.; Oddershede, J.; Ogilvie, J. F.Phys Chem Chem Phys 2005, 7, 1747.
26. Sauer, S. P. A.; Jensen, H. J. A.; Ogilvie, J. F. Adv QuantumChem 2005, 48, 319.
ROTATIONAL AND VIBRATIONAL g FACTORS OF LiH
VOL. 111, NO. 4 DOI 10.1002/qua INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 751

27. Ogilvie, J. F.; Jensen, H. J. A.; Sauer, S. P. A. J Chin ChemSoc 2005, 52, 631.
28. Coxon, J. A. J Mol Spectrosc 1992, 152, 274.
29. Uehara, H. J Mol Spectrosc 1997, 182, 57.
30. Molski, M. J Mol Spectrosc 1997, 185, 256.
31. Molski, M. Acta Phys Pol A 1997, 92, 1117.
32. Uehara, H. J Mol Spectrosc 1998, 188, 215.
33. Molski, M. J Phys Chem A 1999, 103, 5269.
34. Watson, J. K. G. J Mol Spectrosc 2004, 223, 39.
35. Dalton, a program for molecular electronic structure,release 2.0; Available at: http://www.daltonprogram.org,2005. Accessed on March 2010.
36. Linderberg, J.; Ohrn, Y. Propagators in Quantum Chemis-try; Academic Press: London, UK, 1973.
37. Sauer, S. P. A.; Packer, M. J. In Computational MolecularSpectroscopy; Bunker, P. R., Jensen, P., Eds.; Wiley: Lon-don, UK, 2000; Chapter 7, p 221.
38. Helgaker, T. U.; Jensen, H. J. A.; Jørgensen, P. J Chem Phys1986, 84, 6280, and references therein.
39. Helgaker, T.; Jørgensen, P. J Chem Phys 1991, 95, 2595.
40. Gauss, J.; Ruud, K.; Helgaker, T. J Chem Phys 1996, 105,2804.
41. Olsen, J.; Jørgensen, P. J Chem Phys 1985, 82, 3235.
42. Helgaker, T.; Jørgensen, P.; Olsen, J. Molecular ElectronicStructure Theory; Wiley: Chichester, UK, 2000.
43. Roos, B. O. Adv Chem Phys 1987, 69, 399.
44. Paidarova, I.; Sauer, S. P. A. Adv Quantum Chem 2005, 48,185.
45. Ruud, K.; Helgaker, T.; Jørgensen, P. J Chem Phys 1997,107, 10599.
46. Roos, B. O.; Sadlej, A. J Chem Phys 1985, 94, 43.
47. Sauer, S. P. A.; Paidarova, I. Chem Phys 1995, 201, 405.
48. Cencek, W.; Rychlewski, J. Chem Phys Lett 2000, 320, 549.
49. Freeman, R. R.; Jacobson, A. R.; Johnson, D. W.; Ramsey,N. F. J Chem Phys 1975, 63, 2597.
50. Lawrence, T. R.; Anderson, C. H.; Ramsey, N. F. Phys Rev1963, 130, 1865.
51. Rothstein, E. J Chem Phys 1969, 50, 1899.
52. Lutnæs, O. B.; Teale, A. M.; Helgaker, T.; Tozer, D. J.;Ruud, K.; Gauss, J. J Chem Phys 2009, 131, 144104.
53. Partridge, H.; Langhoff, S. R. J Chem Phys 1981, 74, 2361.
54. Theis, D.; Khait, Y. G.; Pal, S.; Hoffmann, M. R. Chem PhysLett 2010, 487, 116.
55. Ogilvie, J. F.; Tipping, R. H. Int Rev Phys Chem 1983, 3, 3.
56. Fernandez, F. M.; Ogilvie, J. F. Chin J Phys 1992, 30, 177.
57. Fernandez, F. M.; Ogilvie, J. F. Chin J Phys 1992, 30, 599.
58. Dulick, M.; Zhang, K.-Q.; Guo, B.; Bernath, P. F. J MolSpectrosc 1998, 188, 14.
59. Pearson, E. F.; Gordy, W. Phys Rev 1969, 177, 59.
60. Plummer, G. M.; Herbst, E.; de Lucia, F. C. J Chem Phys1984, 81, 4893.
61. Matsushima, F.; Odashima, H.; Wang, D. B.; Tsunekawa,S.; Takagi, K. Jpn J Appl Phys 1994, 33, 315.
62. Bellini, M.; de Natale, P.; Inguscio, M.; Varberg, T. D.;Brown, J. M. Phys Rev A 1995, 52, 1954.
63. Watson, J. K. G. J Mol Spectrosc 1980, 80, 411.
64. Ogilvie, J. F.; Rodwell, W. R.; Tipping, R. H. J Chem Phys1980, 73, 5221.
65. Yang, S. C.; Stwalley, W. C. In Metal Bonding and Interac-tions in High-Temperature Systems; Gole, J. L., Stwalley,W. C., Eds.; American Chemical Society: Washington, DC,USA, 1982; p 241.
66. Ogilvie, J. F.; Liao, S. C. Chem Phys Lett 1994, 226, 281.
67. Sauer, S. P. A. Chem Phys Lett 1996, 260, 271.
68. Bubin, S.; Adamowicz, L. J Chem Phys 2003, 118, 3079.
69. Bubin, S.; Adamowicz, L.; Molski, M. J Chem Phys 2005,123, 134310.
70. Ogilvie, J. F. Comput Phys Commun 1983, 30, 101.
71. Saupe, S.; Meyer, B.; Wappelhorst, M. H.; Urban, W.; Maki,A. G. J Mol Spectrosc 1996, 179, 13.
SAUER ET AL.
752 INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY DOI 10.1002/qua VOL. 111, NO. 4




















