
Raf1 and ERK2 kinases are required for phorbol 12,13-dibutyrate-stimulated proliferation of rat...
9
2746 C. Lisbona, S. Alemany, V. Calvo and M. Fernandez-Renart Eur. J. Immunol. 1994. 24: 274fj-2754 Carlos Lisbona., Susana Alemany, Victor Calvo and Margarita Fernandez-Renart Dpto de Bioquimica de la Universidad Aut6noma de Madrid, Instituto de Investigaciones Biomkdicas, Madrid Raf-1 and ERK2 kinases are required for phorbol l2,13-dibu tyra t e-stimulat ed proliferation of rat lymphoblasts. ERK2 activation precedes Raf-1 hyperphosphorylation* Rat lymphoblasts are arrested in the G1 phase of the cell cycle and can be promoted to proceed up to the S phase, when they are stimulated by phorbol ester. In this work, we have studied some details of the phorbol12,13-dibutyrate (PBu2)-stimulated proliferation. We show that in response to PBu2 at least four different protein kinase C (PKC) isoforms translocate to the membrane. A specific PKC 5 antibody recognizes two bands of 75 and 82 kDa. These two activities are separated using a Mono Q chromatography and we show that p75 is the classical PKC 5 isoform, while p82 might be a related isoform which is PBu2 sensitive. Our data show that there is a correlation between the ability of PBuz to promote mitogenesis and to activate ERK2 kinase, suggesting that ERK2 kinase might be the limiting step of the process.We also show that ERK kinase activation precedes Raf-1 kinase hyperphosphorylation, suggesting that Raf-1 kinase activation is not required for ERK kinase activation. This idea was checked using a Raf-1 kinase antisense (AS) oligonucleotide. The results obtained with the Raf-1 AS oligonucleotide indicate that this serinekhreonine kinase is dispensable for ERK kinase activation, but needed for the PBu2 mitogenic signaling even as late as 7 h after the delivery of the signal. 1 Introduction T lymphocytes at Go state of the cell cycle can be activated by interactions with a wide range of agonists and their respective cell surface receptors. After stimulation with antigen or other ligands, such as mitogenic plant lectins or antibodies directed against various cell surface determi- nants, they undergo sequential differentiation steps that induce their entry into G1 phase [l-31. Cells in GI are responsive to additional signals, such as the cytokine IL-2, and they can complete the mitotic cycle going from GI to S [4]. Transition from G1 to S can be achieved by means of PKC-dependent (PBu2) or PKC-independent (IL-2) mechanisms. Activation of PKC is a major step in the pathway of T cell activation [5-71. In the Go-GI transition the diacylglycerol (DG) released by phospholipase C activation, enhances PKC activity and phosphorylation of multiple substrates, as [I 133141 ____ ____ * This work was supported by grants from DGYCT, Plan Nacional A predoctoral fellow from the Spanish Ministry for Education and Europharma. and Science. Correspondence: Margarita Fernandez-Renart, Dpto de Bio- quimica de la Universidad Autonoma de Madrid, Instituto de Investigaciones BiomCdicas, CSIC, c/Arzobispo Morcillo, 4, E- 28029 Madrid, Spain (Fax: 34-1 5 85 45 87) Abbreviation: AS: Antisense Key words: Raf-1 / ERK2 / Protein tyrosine kinase well as the activation of secondary signal transducers as p21TaS [8-101 or Raf-1 [ll, 121. For this stage, Siege1 et al. [ 131 have proposed that TcR-mediated activation of Raf-1, in human T cells, is PKC dependent, This PKC dependence of Raf-1 activation by TcR has been also reported in mouse Tcells [ll]. MAP kinase is also activated in this stage [14]. The Ca++ released by inositol trisphosphate (IP3) is also needed for the IL-2 gene expression [15]. The GI-S transition occurs after interaction of the IL-2 cytokine with its high-affinity receptors, which is PKC independent, or by phorbol esters activating different PKC. There is evidence implicating Raf-1 in the progression from GI into S phase, based on studies showing that IL-2 induces Raf-1 hyperphosphorylation, and increases its kinase activ- ity [16, 171. In general, activation of PKC requires the translocation of the enzyme from the cytosol to the membrane [18, 191. After translocation, the enzyme is quickly degraded by Ca++-activated proteases [20]. Activation of theTcells in the Go to GI transition, through the CD3 complex or PMA, promotes translocation of PKC activity from the soluble fraction to the cell membrane [21, 221. At present, the mammalian PKC family consists of 12 different polypeptides indicated by greek symbols a, PI, PII, y, 6, E, 5, L, 0, q, h and p [23-251, most of them encoded by different genes. In mouse or human lymphocytes, or lymphocytic cell lines, protein kinases C a, P, E, 5 and 6 can be detected at the protein level [26], but there is some heterogeneity of PKC isoforms among different cell lines. [27, 281. This suggests that various isoforms perform specific functions. OO14-2980/94/1111-2746$10.00 + .25/0 0 VCH Verlagsgesellschaft mbH, D-69451 Weinheim, 1994
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Raf1 and ERK2 kinases are required for phorbol 12,13-dibutyrate-stimulated proliferation of rat...

2746 C. Lisbona, S. Alemany, V. Calvo and M. Fernandez-Renart Eur. J. Immunol. 1994. 24: 274fj-2754
Carlos Lisbona., Susana Alemany, Victor Calvo and Margarita Fernandez-Renart
Dpto de Bioquimica de la Universidad Aut6noma de Madrid, Instituto de Investigaciones Biomkdicas, Madrid
Raf-1 and ERK2 kinases are required for phorbol l2,13-dibu tyra t e-stimulat ed proliferation of rat lymphoblasts. ERK2 activation precedes Raf-1 hyperphosphorylation*
Rat lymphoblasts are arrested in the G1 phase of the cell cycle and can be promoted to proceed up to the S phase, when they are stimulated by phorbol ester. In this work, we have studied some details of the phorbol12,13-dibutyrate (PBu2)-stimulated proliferation. We show that in response to PBu2 at least four different protein kinase C (PKC) isoforms translocate to the membrane. A specific PKC 5 antibody recognizes two bands of 75 and 82 kDa. These two activities are separated using a Mono Q chromatography and we show that p75 is the classical PKC 5 isoform, while p82 might be a related isoform which is PBu2 sensitive. Our data show that there is a correlation between the ability of PBuz to promote mitogenesis and to activate ERK2 kinase, suggesting that ERK2 kinase might be the limiting step of the process.We also show that ERK kinase activation precedes Raf-1 kinase hyperphosphorylation, suggesting that Raf-1 kinase activation is not required for ERK kinase activation. This idea was checked using a Raf-1 kinase antisense (AS) oligonucleotide. The results obtained with the Raf-1 AS oligonucleotide indicate that this serinekhreonine kinase is dispensable for ERK kinase activation, but needed for the PBu2 mitogenic signaling even as late as 7 h after the delivery of the signal.
1 Introduction
T lymphocytes at Go state of the cell cycle can be activated by interactions with a wide range of agonists and their respective cell surface receptors. After stimulation with antigen or other ligands, such as mitogenic plant lectins or antibodies directed against various cell surface determi- nants, they undergo sequential differentiation steps that induce their entry into G1 phase [l-31. Cells in GI are responsive to additional signals, such as the cytokine IL-2, and they can complete the mitotic cycle going from GI to S [4]. Transition from G1 to S can be achieved by means of PKC-dependent (PBu2) or PKC-independent (IL-2) mechanisms.
Activation of PKC is a major step in the pathway of T cell activation [5-71. In the Go-GI transition the diacylglycerol (DG) released by phospholipase C activation, enhances PKC activity and phosphorylation of multiple substrates, as
[I 133141 ____ ____
* This work was supported by grants from DGYCT, Plan Nacional
A predoctoral fellow from the Spanish Ministry for Education and Europharma.
and Science.
Correspondence: Margarita Fernandez-Renart, Dpto de Bio- quimica de la Universidad Autonoma de Madrid, Instituto de Investigaciones BiomCdicas, CSIC, c/Arzobispo Morcillo, 4, E- 28029 Madrid, Spain (Fax: 34-1 5 85 45 87)
Abbreviation: AS: Antisense
Key words: Raf-1 / ERK2 / Protein tyrosine kinase
well as the activation of secondary signal transducers as p21TaS [8-101 or Raf-1 [ l l , 121. For this stage, Siege1 et al. [ 131 have proposed that TcR-mediated activation of Raf-1, in human T cells, is PKC dependent, This PKC dependence of Raf-1 activation by TcR has been also reported in mouse Tcells [ l l ] . MAP kinase is also activated in this stage [14]. The Ca++ released by inositol trisphosphate (IP3) is also needed for the IL-2 gene expression [15].
The GI-S transition occurs after interaction of the IL-2 cytokine with its high-affinity receptors, which is PKC independent, or by phorbol esters activating different PKC. There is evidence implicating Raf-1 in the progression from GI into S phase, based on studies showing that IL-2 induces Raf-1 hyperphosphorylation, and increases its kinase activ- ity [16, 171.
In general, activation of PKC requires the translocation of the enzyme from the cytosol to the membrane [18, 191. After translocation, the enzyme is quickly degraded by Ca++-activated proteases [20].
Activation of theTcells in the Go to GI transition, through the CD3 complex or PMA, promotes translocation of PKC activity from the soluble fraction to the cell membrane [21, 221.
At present, the mammalian PKC family consists of 12 different polypeptides indicated by greek symbols a, PI, PII, y , 6, E, 5 , L, 0, q, h and p [23-251, most of them encoded by different genes. In mouse or human lymphocytes, or lymphocytic cell lines, protein kinases C a, P, E , 5 and 6 can be detected at the protein level [26], but there is some heterogeneity of PKC isoforms among different cell lines. [27, 281. This suggests that various isoforms perform specific functions.
OO14-2980/94/1111-2746$10.00 + .25/0 0 VCH Verlagsgesellschaft mbH, D-69451 Weinheim, 1994

Eur. J. Immunol. 1994. 24: 2746-2754 Raf-1 kinase activation is not required for ERK2 kinase activation 2747
MAP kinase and Raf-1 kinase are serinehhreonine kinases that become activated in response to numerous stimuli in multiple cell types [29-341, their activity being needed in order to reach the S phase.
From signals starting at tyrosine kinases receptors, or mitogenic receptors, coupled to src-like tyrosine kinases, it is becoming clear that Raf-1 is an upstream activator of MEK and MAP kinase [30, 31, 351. This idea comes from reports on Raf- and Ras-transformed cells, in which MAP kinases are constitutively active [36, 371. In the case of Raf-transformed cells, the MAP constitutive activity is due to the ability of Raf to phosphorylate and activate MEK directly [38]. The use of a dominant Raf mutant has established that Ras protein is located upstream in this signaling pathway [39]. By the use of the yeast two-hybrid system and by in vitro binding assays, an in vivo protein- protein interaction between Raf and Ras has been shown [40]. It is not clear at the moment how Raf is activated; it has been proposed that Raf binding to activated Ras may serve as a mechanism to translocate Raf to the membrane [41]. Then, the pathway starting at PTK would be PTK- Ras-Raf-MEK-MAP kinase.
It has been proposed that another means to activate Raf is through PKC. Siege1 et al. have presented evidence that PKC is required for Raf activation after stimulation of the Tcell receptor [13], but on the other hand, the ability of PKC-phosphorylated Raf to activate MEK, and conse- quently MAP kinase, has been recently questioned [42].
Sozeri et al. [43], measuring Raf-1 autophosphorylation as indicative of activation, have proposed that PKC a, p and y were able to activate Raf-1 kinase, but on the contrary, Macdonald et al. [42], using an in vitro assay, have reported that these isoforms phosphorylate Raf-1, but do not activate Raf-1 to phosphorylate MEK.
In this report we have analyzed the different PKC isoforms activated, in GI to S transition, by PBu2 stimulation of rat lymphoblasts, studying the membrane translocation of the different isoenzymes. We also present data showing that MAP kinase activation and Raf kinase activation, are PKC-dependent events, and Raf-1 activation is dispensable for MAP kinase activation. MAP kinase activation is a limiting step in the mitogenic process. Using a Raf-1 antisense (AS) oligodeoxyribonucleotide, we show that Raf-1 kinase is required to reach the S phase and is needed late after the proliferative signal delivered by PBu2.
2 Materials and methods
2.1 Antibodies
Rabbit peptide-specific antibodies (IgG fraction) that recognize PKC ‘s and E were purchased from Boehringer and 6 from Amersham. mAb specific to PKC M and p were purchased from Seikagaku Kogyo, the mAb specific to MAP kinase was obtained from Zymed (San Francisco, CA), peptide-specific antibody for Raf-1 was kindly pro- vided by Dr. Tom Roberts, and anti phosphotyrosine antibody was the 4G10 from Amersham. The specificity of the PKC antibodies were determined by Western blot analysis, in which immunogenic peptides were used to
compete with the epitope of the correct molecular size immobilized on the membrane.
2.2 Oligonucleotides
Raf-1 AS (5’-TCCCTGTATGTGCTCCAT-3’) and sense (5’-ATGGAGCACATACAGGGA-3’) oligodeoxiribonu- cleotides corresponding to codons 1-6 of murine c-Raf were from Scandinavian Gene Synthesis (Koping , Swed- en).
2.3 Obtaining T lymphoblasts
Quiescent rat spleen lymphocytes were obtained as in [44]. Lymphoblasts were obtained by stimulation of spleen lymphocytes at a concentration of 5 x lo6 celYml for 48 h, with 2 pg/ml Con A (Aldrich), in basal medium (RPMI, Gibco), 60 pg/ml gentamycin (Llorente), 0.25 pg/ml fungi- zone (Sigma), 2 mM glutamine (Flow Laboratories), 5% (v/v) heat-inactivated fetal calf serum (FCS, Gibco), 5 x
M 2-mercaptoethanol (2-ME, Merck)). After this time, cells were treated with buffered 0.14 ammonium chloride and washed twice with gentamycin, fungizone, and 2% (v/v) FCS in RPMI, and resuspended in basal medium for 24 h, to let the cells return to the GI phase.
2.4 Cell proliferation
Cell proliferation was measured as the incorporation of [3H] thymidine. Lymphoblasts (20 000 cells/100 pl) were resuspended in basal medium, and incubated in 96-well plates. IL-2 (Boehringer), or PBu2 (Sigma), were added at the indicated concentration, and DNA synthesis was measured 24 h after stimulation, for the last 4 h (20-24 h) cells were pulse-labeled with 0.5 pCi/well (methyl [3H])- thymidine (Amersham). Cells were harvested and radioac- tivity was measured using standard scintillation counting.
2.5 Cell lysis and Western blot conditions
Cells (10 x 106-20 x 106cells/ml) were harvested,washed in cold PBS containing 5 mM EDTA, 10 mM NaF, 0.4 mM sodium orthovanadate and 10 mM sodium pyrophosphate and spun down to cell pellets. For Western blot analysis the cell pellets were resuspended in 100-200 pl of buffer A (50mM Tris-HC1 pH7.5, 0.5% Triton X-100, 3 0 0 m ~ NaCl, 5 mM EDTA, 10 mM NaF, 10 mM sodium pyrophos- phate, 1 mM sodium orthovanadate, 10 mM iodoacetic acid, 125 pg/ml aprotinin, 10 pg/ml leupeptin, and 4 mM PMSF). After 20 s of vortexing, cells were kept on ice for 15 min and centrifuged in a microcentrifuge for 10 min at 4 “C. Supernatants were collected and protein concentra- tion was determined with the Bio-Rad protein assay system. Loading buffer (187.5 mM Tris-HC1, pH 6.8, 15% 2-ME, 30% glycerol, 6.9% SDS, 0.003% bromophenol blue) was added to extracts containing equal amounts of proteins, and subjected to SDS-PAGE on an 8% or 10% gel or on a 10-20% gradient gel. Gels were transferred to Immobilon-P membranes (Millipore), in transfer buffer (25 mM Tris-HC1 pH 8.8, 200 mM glycine, and 10% (v/v) methanol), at 2 mA/cm2 for 1 h 30 min. After transfer,
2748 C. Lisbona, S. Alemany,V. Calvo and M. Fernandez-Renart Eur. J. Immunol. 1994. 24: 2746-2754
membranes were incubated with the blocking solution (3% (w/v skim milk inTBS-T (50 mM Tris-HCI pH 7.5, 150 mM NaCl and 0.05% Tween-20)), for 1 h at room temperature and, after three washes in TBS-T, membranes were incu- bated with the corresponding antibody. Dilution of each particular antibody was used as recommended by the manufacturer. After three washes with TBS-T, membranes were incubated with peroxidase-labeled goat anti-rabbit IgG antibody (ICN) , or peroxidase-labeled goat anti- mouse IgG antibody (ICN), in TBS-T for 1 h at room temperature. After three washes with TBS-Tand one with TBS, membrane bound antibody was visualized by use of chloronaftol and H202 or ECL detection reagent (Amer- sham) and luminiscent detection film (Konica).
2.6 Membrane and cytosol preparation
Membrane and cytosol fractions of the cells were prepared following the procedure of Isakov and Altman [45]. Briefly, harvested cells (50 x lo6) were washed once with ice-cold PBS, resuspended in 2 ml ice-cold buffer B (20 mM Tris- HC1, pH 7.5,0.33 M sucrose, 2 mM EDTA, 0.5 mM EGTA, 2 mM PMSF and 10 pg/ml leupeptin) and were sonicated on ice with three strokes 10 s each at 4 microns of amplitude. The extracts were centrifuged at 100 000 x g for 20 min at 4°C. The supernatants were saved, and the pellet resus- pended in 2 ml of buffer B containing 1% Triton X-100, and kept on ice for 1 h at 4 "C.The suspensions were centrifuged at 100000 x g for 20 min at 4"C, and the supernatants containing the detergent-solubilized particulate fraction were collected and stored with loading buffer in aliquots at -70 "C.
2.7 Mono Q chromatography
Aliquots of 125 x 106 cells were harvested, washed in PBS and resuspended in buffer C (50 mM Tris-HCI, pH 7.5, 2 mM EDTA, 10 mM EGTA, 1 mM DTT, 0.25 M sucrose, 5 pglml pepstatin, 40 pg/ml PMSF, 10 mM benzamidine, 8 pg/ml antipain, 25 pg/ml aprotinin, 0.5 mg/ml leupeptin and 0.1 mg/ml ovoalbumin), and sonicated, as indicated above, on ice. Extracts were centrifuged at 100000 X g for 30min at 4°C. Supernatants (6-8 mg of protein) were removed and applied to a Mono Q-Sepharose column, driven by an FPLC system, equilibrated in buffer contain- ing 50 mM Tris-HC1, pH 7.5, 1 mM EDTA, 0.5 mM EGTA and 1 mM DTT. Bound proteins were eluted with a 15 ml gradient from 0 mM to 400 mM NaCl in equilibration buffer. Fractions were assayed for PKC activity as described in Sect. 2.8.
2.8 PKC assay
The activity of PKC was determined by measuring the incorporation of 32P from ATP into myelin basic protein (MBP). The reaction mixture contained in a volume of 25 p1: 50 mM Tris-HC1, pH 7.5, 10 mM MgC12, 0.5 mg/ml MBP, 40 VM ATP, and [ Y - ~ ~ P ] ATP (2500 cprdpmol) with or without 1 mM calcium chloride, 500 pg/ml phosphatidylser- ine (PS), and 100 ng/ml PBu2. PS was dried by evaporating the organic solvent, under a stream of argon and was dispersed in 50 mM Tris-HC1, pH 7.5, by two to tree 10 s
burst of sonication. Reactions were initiated byte addition of AT€! After 10 min of incubation at 30 "C reactions were terminated by spotting the 25 pl on p-81 paper. Papers were washed three times on 75 mM phosphoric acid to remove unincorporated ATP and dried. Bound radioactivity was determined by scintillation counting.
2.9 Statistics
For statistics, Kolmogorov-Smirnov test was used for normality, and Spearman test for correlation.
3 Results
3.1 The p82 polypeptide detected with the PKC 5 antiserum is not the characterized PKC 5 isoform
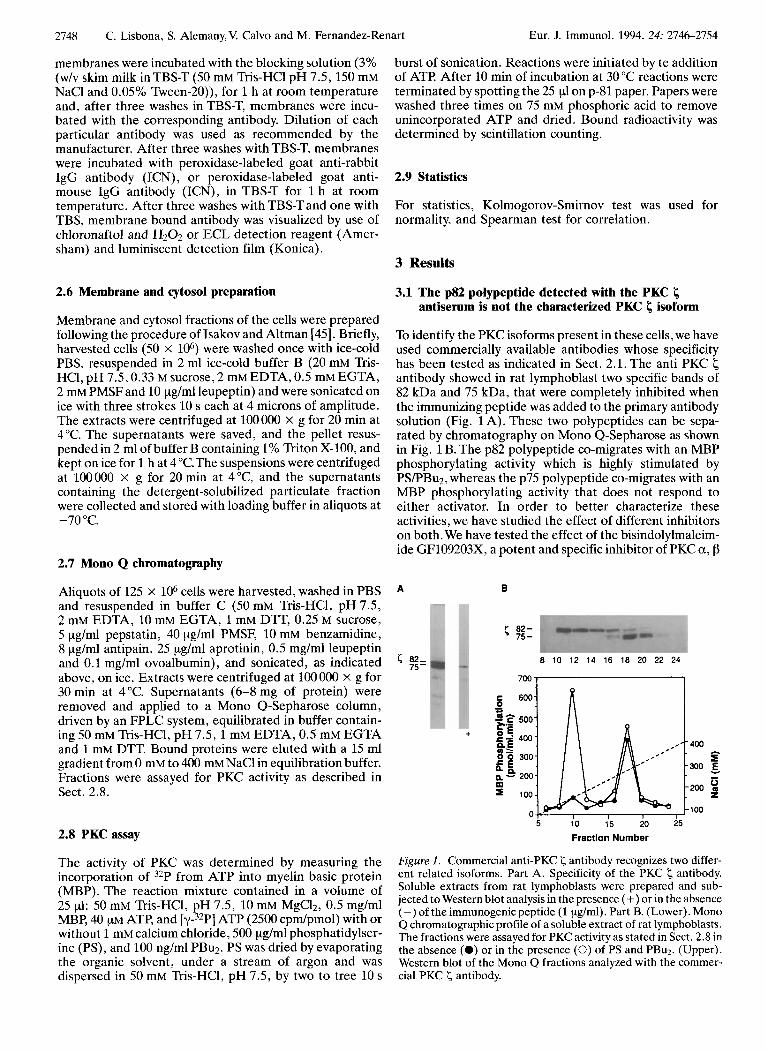
To identify the PKC isoforms present in these cells, we have used commercially available antibodies whose specificity has been tested as indicated in Sect. 2.1. The anti PKC antibody showed in rat lymphoblast two specific bands of 82 kDa and 75 kDa, that were completely inhibited when the immunizing peptide was added to the primary antibody solution (Fig. 1A). These two polypeptides can be sepa- rated by chromatography on Mono Q-Sepharose as shown in Fig. 1 B. The p82 polypeptide co-migrates with an MBP phosphorylating activity which is highly stimulated by PS/PBu2, whereas the p75 polypeptide co-migrates with an MBP phosphorylating activity that does not respond to either activator. In order to better characterize these activities, we have studied the effect of different inhibitors on both. We have tested the effect of the bisindolylmaleim- ide GF109203X7 a potent and specific inhibitor of PKC a, p
A B
< 02- 75 -
< 82- 8 10 12 14 16 18 20 22 24 75-
I
+
Fraction Number
Figure 1. Commercial anti-PKC < antibody recognizes two differ- ent related isoforms. Part A. Specificity of the PKC < antibody. Soluble extracts from rat lymphoblasts were prepared and sub- jected to Western blot analysis in the presence (+) or in the absence (-) of the immunogenic peptide (1 pg/ml). Part B. (Lower). Mono Q chromatographic profile of a soluble extract of rat lymphoblasts. The fractions were assayed for PKC activity as stated in Sect. 2.8 in the absence (0) or in the presence (0) of PS and PBu2. (Upper). Western blot of the Mono Q fractions analyzed with the commer- cial PKC < antibody.

Eur. J. Immunol. 1994. 24: 2746-2754 Raf-1 kinase activation is not required for ERK2 kinase activation 2749
and y isoforms [46], on these two PKC activities. Both of them were inhibited with an IC50 of 30 nM, both activities were also inhibited with the same ICso (2 pM) by a pseudosubstrate designed according to the amino terminal of the I; isoenzyme.The p75 PKC I; behaved as the classical PKC I; whereas p82 seemed to be a t;-related isoform.
3.2 PBuz down-regulates most of the PKC isoforms
The intracellular targets for PBuz are the different PKC isoforms susceptible to activation by the phorbol ester, which includes most except PKC I; which does not bind phorbol esters [47].
PBu2 binds to the regulatory site of most PKC, and promotes their translocation to membrane; the translo- cated enzyme is activated by the membrane lipids and rapidly degraded [48]. We have studied the susceptibility of the different isoforms to PBu2, by looking at their down- regulation at different phorbol ester concentrations, using Western blot analysis. Fig. 2 shows that the isoforms a, p, 6, E and p82 I; are down-regulated after overnight incuba- tion with PBu2. There is a difference in the susceptibility of the different isoforms to down-regulation, a, p and E are more susceptible than the other isoforms to the phorbol ester.
The down-regulation experiment implied that isoforms a, p, 6, E and p82 PKC I; are susceptible to activation by PBu2. We investigated the translocation of the different isoen- zymes from cytosol to cell membrane, soon after phorbol ester activation (80 ng/ml), using isotype-specific antisera.
a 80 -
P 80 -
6
80 -
E 106-
80 -
5 80 -
0 0 5 10 30 80 150
[PDBu] (ng/ml)
Figure 2 . Rat lymphoblast PKC isoforms have a different sensitiv- ity to PBu2 down-regulation. Rat lymphoblasts were incubated with the indicated concentrations (ng/ml) of PBu2 for 14 h, and the cellular extracts were subjected to Western blot analysis with the different PKC isoform antibodies. First lane, control unstimulated cells harvested at the same time as the stimulus. Second lane, control unstimulated cells harvested 14 h after the stimulus. Bar and number in the left indicate molecular mass (kDa).
Figure 3. PBuz induces translocation of PKC a, fJ, 6 and < p82 but not PKC E or p75. Rat lymphoblasts were stimulated with 80 ng/ml PBuz (PDBu) or 20 U/ml interleukin 2 (IL-2) and (5 x lo7) cells were harvested at the indicated time points (min). Membrane and cytosol fractions prepared as stated in Sect. 2.6, were subjected to Western blot analysis. Cy, cytosol fraction. M, membrane fraction. Bar and number in the left indicate molecular mass (kDa). The Western blots are representative of three experiments.
As shown in Fig. 3, PKC a, p, 6 and the p82 kDa polypeptide, detected with the PKC < antisera, are mainly cytosolic in unstimulated cells, and were translocated to the membrane in response to the phorbol ester; p75 PKC I;, which is also mainly cytosolic in these cells, does not change. IL-2 does not promote any PKC translocation of the different isoforms, tested under the same conditions (Fig. 3). A large proportion of PKC E is located in the membrane in unstimulated cells, and upon PBuz stimula- tion no further enzyme seems to be recruited to the membrane. This isoenzyme is especially prone to proteoly- sis because a couple of rounds of freezing and thawing in Laemmly buffer results in complete degradation (data not shown). As shown by the translocation exeriments, at least the PKC a, p, 6 and p82 I; isoenzymes can be activated by PBuz.
We next investigated whether the proliferative signal started by PBuz, which activates the PKC isoforms a , p, 6 and p82 I;, needs the Raf-1 kinase activity to activate ERK kinase located further down in the pathway.
3.3 Bisindolylmaleimide is a specific PKC inhibitor
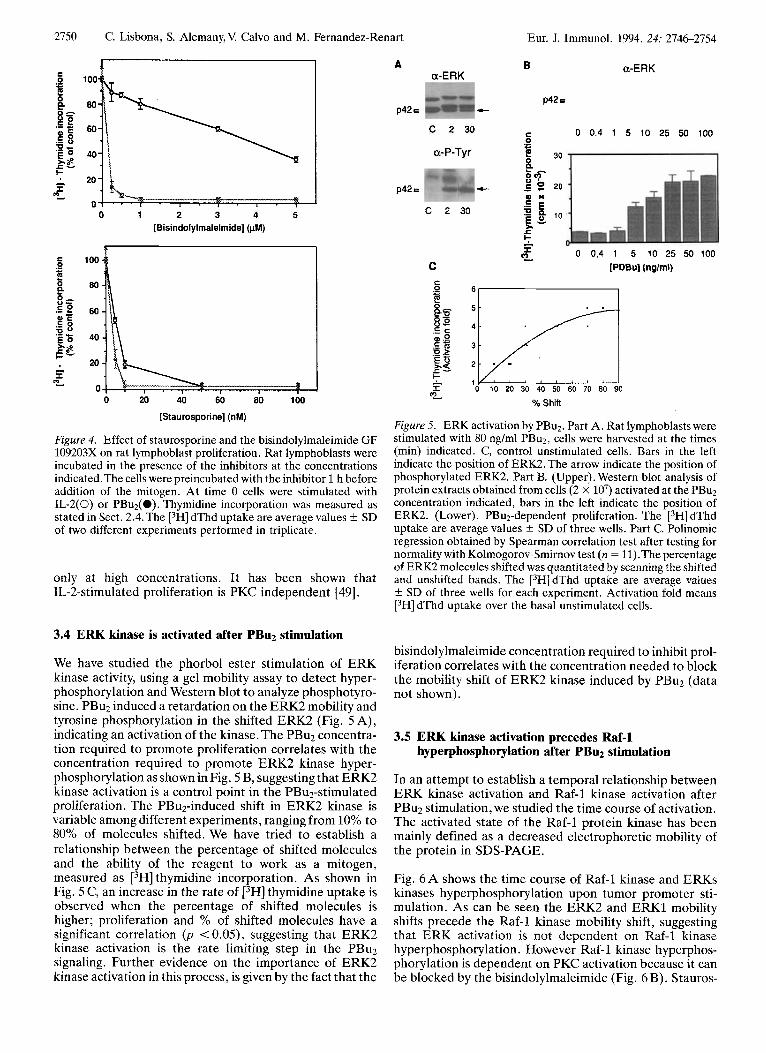
We have checked the specificity of G109203X by comparing its ability to interfere with the PBu2, or IL-2 mediated proliferation. As can be seen in Fig. 4, whereas a more nonspecific protein kinase C inhibitor as staurosporine blocks the proliferation elicited by both mitogens, the bisindolylmaleimide GF 109203X only inhibits the PBu2- induced proliferation, affecting IL-2-induced proliferation
C. Lisbona, S. Alemany,V. Calvo and M. Fernandez-Renart Eur. J. Immunol. 1994. 24: 2746-2754
20
0 0 1 2 3 4 5
[Bisindolylmaleimide] (pM)
0 20 40 60 80 100
[Staurosporine] (nM)
Figure 4. Effect of staurosporine and the bisindolylmaleimide GF 109203X on rat lymphoblast proliferation. Rat lymphoblasts were incubated in the presence of the inhibitors at the concentrations indicated.The cells were preincubated with the inhibitor 1 h before addition of the mitogen. At time 0 cells were stimulated with IL-2(0) or PBu2(0). Thymidine incorporation was measured as stated in Sect. 2.4. The [3H] dThd uptake are average values f SD of two different experiments performed in triplicate.
only at high concentrations. It has been shown that IL-2-stimulated proliferation is PKC independent [49].
3.4 ERK kinase is activated after PBuz stimulation
We have studied the phorbol ester stimulation of ERK kinase activity, using a gel mobility assay to detect hyper- phosphorylation and Western blot to analyze phosphotyro- sine. PBuz induced a retardation on the ERK2 mobility and tyrosine phosphorylation in the shifted ERK2 (Fig. 5 A), indicating an activation of the kinase. The PBuz concentra- tion required to promote proliferation correlates with the concentration required to promote ERK2 kinase hyper- phosphorylation as shown in Fig. 5 B, suggesting that ERK2 kinase activation is a control point in the PBu2-stimulated proliferation. The PBu2-induced shift in ERK2 kinase is variable among different experiments, ranging from 10% to 80% of molecules shifted. We have tried to establish a relationship between the percentage of shifted molecules and the ability of the reagent to work as a mitogen, measured as [3H] thymidine incorporation. As shown in Fig. 5 C, an increase in the rate of [3H] thymidine uptake is observed when the percentage of shifted molecules is higher; proliferation and % of shifted molecules have a significant correlation (p < 0.05), suggesting that ERK2 kinase activation is the rate limiting step in the PBuz signaling. Further evidence on the importance of ERK2 kinase activation in this process, is given by the fact that the
A a-ERK
B a-ERK
p42 E
p42 c c
0 0.4 1 5 10 25 50 100 5
C 0 0,4 1 5 10 25 50 100 F z
[PDBu] (nglml) C 0 .-
.- c 0 .o .c z 3
* i? 0 10 20 30 40 50 60 70 80 90 ., I
%Shill
Figure 5. ERK activation by PBu2. Part A. Rat lymphoblasts were stimulated with 80 ng/ml PBu2, cells were harvested at the times (min) indicated. C, control unstimulated cells. Bars in the left indicate the position of ERK2. The arrow indicate the position of phosphorylated ERK2. Part B. (Upper). Western blot analysis of protein extracts obtained from cells (2 X lo7) activated at the PBu2 concentration indicated, bars in the left indicate the position of ERK2. (Lower). PBu2-dependent proliferation. The [3H] dThd uptake are average values k SD of three wells. Part C. Polinomic regression obtained by Spearman correlation test after testing for normality with Kolmogorov-Smirnov test (n = 11).The percentage of ERK2 molecules shifted was quantitated by scanning the shifted and unshifted bands. The [3H] dThd uptake are average values k SD of three wells for each experiment. Activation fold means [3H] dThd uptake over the basal unstimulated cells.
bisindolylmaleimide concentration required to inhibit prol- iferation correlates with the concentration needed to block the mobility shift of ERK2 kinase induced by PBu2 (data not shown).
3.5 ERK kinase activation precedes Raf-1 hyperphosphorylation after PBuz stimulation
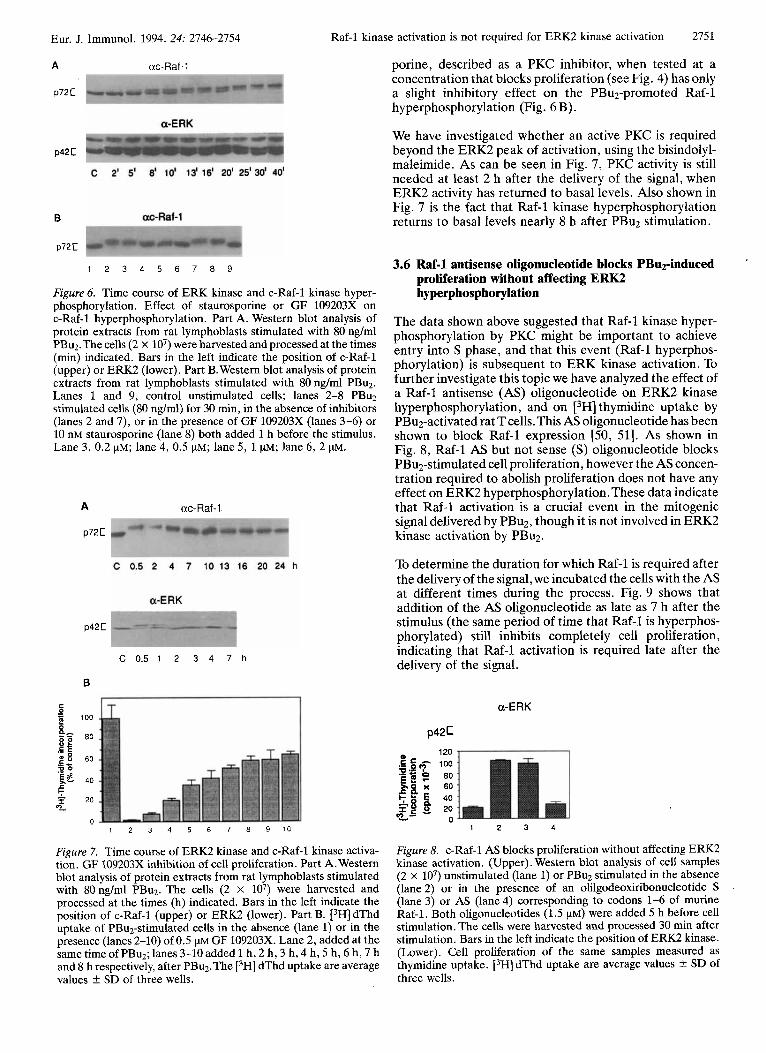
In an attempt to establish a temporal relationship between ERK kinase activation and Raf-1 kinase activation after PBuz stimulation, we studied the time course of activation. The activated state of the Raf-1 protein kinase has been mainly defined as a decreased electrophoretic mobility of the protein in SDS-PAGE.
Fig. 6 A shows the time course of Raf-1 kinase and ERKs kinases hyperphosphorylation upon tumor promoter sti- mulation. As can be seen the ERK2 and ERKl mobility shifts precede the Raf-1 kinase mobility shift, suggesting that ERK activation is not dependent on Raf-1 kinase hyperphosphorylation. However Raf-1 kinase hyperphos- phorylation is dependent on PKC activation because it can be blocked by the bisindolylmaleimide (Fig. 6B). Stauros-
Eur. J. Immunol. 1994. 24: 2746-2754
A uc-Raf-1
p72 C
p42 C
B
Raf-1 kinase activation is not required for ERK2 kinase activation 2751
porine, described as a PKC inhibitor, when tested at a concentration that blocks proliferation (see Fig. 4) has only a slight inhibitory effect on the PBu2-promoted Raf-1 hyperphosphorylation (Fig. 6 B).
We have investigated whether an active PKC is required beyond the ERK2 peak of activation, using the bisindolyl- maleimide. As can be seen in Fig. 7, PKC activity is still needed at least 2 h after the delivery of the signal, when ERK2 activity has returned to basal levels. Also shown in Fig. 7 is the fact that Raf-1 kinase hyperphosphorylation returns to basal levels nearly 8 h after PBu2 stimulation.
p72E
1 2 3 4 5 6 7 8 9
Figure 6. Time course of ERK kinase and c-Raf-1 kinase hyper- phosphorylation. Effect of staurosporine or GF 109203X on c-Raf-1 hyperphosphorylation. Part A. Western blot analysis of protein extracts from rat lymphoblasts stimulated with 80 ng/ml PBu2.The cells (2 x lo7) were harvested and processed at the times (min) indicated. Bars in the left indicate the position of c-Raf-1 (upper) or ERK2 (lower). Part B. Western blot analysis of protein extracts from rat lymphoblasts stimulated with 80 ng/ml PBu2. Lanes 1 and 9, control unstimulated cells; lanes 2-8 PBuz stimulated cells (80 nglml) for 30 min, in the absence of inhibitors (lanes 2 and 7), or in the presence of GF 109203X (lanes 3-6) or 10 nM staurosporine (lane 8) both added 1 h before the stimulus. Lane 3, 0.2 pM; lane 4, 0.5 pM; lane 5 , 1 pM; lane 6, 2 pM.
A uc-Raf - 1
p72 C
p42 C
C 0 . 5 1 2 3 4 7 h
E
'5 100
80 .- c Iy 8 60 .E - D O
'Eg 40
e b
6 f 20 0-
0 1 2 3 4 5 6 7 8 9 10
Figure 7. Time course of ERK2 kinase and c-Raf-1 kinase activa- tion. GF 109203X inhibition of cell proliferation. Part A. Western blot analysis of protein extracts from rat lymphoblasts stimulated with 80nglml PBuz. The cells (2 X lo7) were harvested and processed at the times (h) indicated. Bars in the left indicate the position of c-Raf-1 (upper) or ERK2 (lower). Part B. [3H] dThd uptake of PBuz-stimulated cells in the absence (lane 1) or in the presence (lanes 2-10) of 0.5 p~ GF 109203X. Lane 2, added at the same time of PBu2; lanes 3-10 added 1 h, 2 h, 3 h, 4 h, 5 h, 6 h, 7 h
3.6 Raf-1 antisense oligonucleotide blocks PBu2-induced proliferation without affecting ERK2 hyperphosphorylation
The data shown above suggested that Raf-1 kinase hyper- phosphorylation by PKC might be important to achieve entry into S phase, and that this event (Raf-1 hyperphos- phorylation) is subsequent to ERK kinase activation. To further investigate this topic we have analyzed the effect of a Raf-1 antisense (AS) oligonucleotide on ERK2 kinase hyperphosphorylation, and on t3H] thymidine uptake by PBuz-activated rat T cells.This AS oligonucleotide has been shown to block Raf-1 expression [50, 511. As shown in Fig. 8, Raf-1 AS but not sense (S) oligonucleotide blocks PBu2-stimulated cell proliferation, however the AS concen- tration required to abolish proliferation does not have any effect on ERK2 hyperphosphorylation. These data indicate that Raf-1 activation is a crucial event in the mitogenic signal delivered by PBuz, though it is not involved in ERK2 kinase activation by PBu2.
To determine the duration for which Raf-1 is required after the delivery of the signal, we incubated the cells with the AS at different times during the process. Fig. 9 shows that addition of the AS oligonucleotide as late as 7 h after the stimulus (the same period of time that Raf-1 is hyperphos- phorylated) still inhibits completely cell proliferation, indicating that Raf-1 activation is required late after the delivery of the signal.
a-ERK
p421
1 2 3 4
Figure 8. c-Raf-1 AS blocks proliferation without affecting ERK2 kinase activation. (Upper). Western blot analysis of cell samples (2 X lo7) unstimulated (lane 1) or PBu2 stimulated in the absence (lane 2) or in the presence of an olilgodeoxiribonucleotide S (lane 3) or AS (lane 4) corresponding to codons 1-6 of murine Raf-1. Both oligonucleotides (1.5 pM) were added 5 h before cell stimulation. The cells were harvested and processed 30 min after stimulation. Bars in the left indicate the position of ERK2 kinase. (Lower). Cell proliferation of the same samples measured as thymidine uptake. i3HJ dThd uptake are average values k SD of and 8 h respectively, after PBuz.The t3H] dThd uptake are average
values f SD of three wells. three wells.

2752 C. Lisbona, S. Alemany,V. Calvo and M. Fernandez-Renart Eur. J. Immunol. 1994. 24: 2746-2754
-5 0 5 10 15
Time (h)
Figure 9. c-Raf-1 AS blocks proliferation 7 h after the delivery of the signal. Cells (2 x 10’) were PBu2 stimulated in the presence of c-Raf-1 oligonucleotide AS (0) or S (0). The oligonucleotides were added at the times indicated in the figure.Time 0 indicates the PBuz addition. [3H] dThd uptake are average values iz SD of three wells.
4 Discussion
The primary aim of this work was to investigate the protein kinase C isoforms activated by PBu2, and whether the serine threonine kinases Raf-1 and the ERK2 are involved in the mitogenic pathway started by PBuz in rat T 1ymphoblasts.We wanted to know how many PKC isoforms were activated in response to PBu2. All the isoforms but PKC E were mainly soluble and down-regulated by PBuz treatment, although there are some differences in their sensitivity to the phorbol ester. PKC a, fI and E are more sensitive than p82 <, the isoform 6 being less sensitive.
PKC 6 is not detectable in murine thymocytes [52] but is apparent in rat spleen lymphoblasts at a level comparable to the other isoenzymes, suggesting a role for this enzyme in this subset of cells. In some circumstances, a large fraction of PKC is found membrane associated in unstimulated cells. It has been hypothesized that only the membrane- bound enzyme is activated upon stimulation, the soluble enzyme being a reservoir of functional enzyme that may be mobilized to replace proteolysed membrane PKC [53]. The 6 isoform, which is mainly cytosolic in our rat lymphoblasts, is found membrane associated in unstimulated Jurkat cells, and in these cells no translocation could be detected after stimulation [22]. The E isoform is found in to a large extent membrane associated in our rat lymphoblasts, as it is the case of U937 cells [54], rat fibroblasts [55] or Jurkat cells P21.
In our rat lymphoblasts, PKC a, p, 6 and p82 < isoforms translocate to membrane immediately after PBu2 stimula- tion; proteolysis seems to be involved, since the amount of protein recovered in the membrane is less than the protein lost from the soluble fraction. Two distinct anti-PKC < immunoreactive bands were detected with commercially available antibody, as has been previously reported for lymphocytic cell lines [22]. PKC < 75 represents the already described isoform insensitive to PBu2 [47]. We show that PKC f 82 represents an isoform different to p75 <, and we propose that p82 might represent a PKC <-related isoform, since it is inhibited by the < pseudosubstrate with the same ICs0 as p75 PKC 5. On the other hand, p82 does not seem to be related to a or fI isoforms because the activity co-
migrating with p82 in the mono Q is not activated by ca++ , and the concentration dependence of PBu2 for down- regulation is different from the a and f3 isoforms.
It is known that MAP kinase is activated in primary T cells upon phorbol ester stimulation [14, 561. The data we present in this work show that MAP kinse activation alone is not sufficient to trigger PBu2 stimulated cell prolifera- tion, but seems to be the rate limiting step of the process. Perkins et al. [57] have shown using IL-3 dependent cells, expressing IL-2 receptors, that ERK2 stimulation is neces- sary but not sufficient for IL-Zmediated proliferation of those cells.
There is evidence implicating Raf-1 in the progression from GI in S phase, based on studies showing that IL-2 induces Raf-1 hyperphosphorylation and increases its kinase activ- ity [ 16,171. Raf-1 may also play an important role mediating the Go to GI transition as reported by Siegel et al. in human and mouse T cells [ 131. We show a Raf-1 hyperphosphory- lation promoted by PBuz stimulation; this result agrees with the data obtained by Zmuidzinas et al. I171 and by Siegel et al. in human Tcells [13], but the Raf-1 kinase hyperphosphorylation is posterior to the MAP kinase activation upon stimulation.
Since Raf-1 hyperphosphorylation might not necessarily mean activation as reported by Samuels et al. [58], we decided to address whether Raf-1 activity was or was not required to activate MAP kinase by using a Raf-1 AS oligonucleotide. Using a Raf-1 AS oligodeoxiribonucleo- tide, we were able to block completely PBu2-induced proliferation without affecting MAP kinase hyperphospho- rylation, indicating that Raf-1 kinase activation is not required to activate MAP kinase in rat lymphoblasts. A blockage of the IL-2 mitogenic response has been reported by Riedel et al. 1591 with the same AS oligonucleotide corresponding to codons 1-6 of murine Raf-1. Tornkvist et al. [50] reported an abolishment of insulin-stimulated DNA synthesis using the same Raf-1 AS oligonucleotide. Tropp- mair et al. [60] proposed that Raf-1 kinase is an obligatory entry point into the signaling cascade of the several mitogens tested included PBu2, leading to the activation of MAP kinase; on the contrary Chao et al. [61] present data showing that Raf-1 is dispensable for MAP kinase activa- tion in the signaling pathway of EGF and PBu2. Both used transfection assays with a different dominant-negative mutant of Raf-1. The opposite results might reflect uncon- trolled effects of the Raf mutants or the existence of cell-specific signaling pathways.
It is worth mentioning that Kizaka-Kondoh et al. [62] using rat fibroblast cell lines have shown that Raf-1 was not the major activator of ERK in vivo; they proposed that, as in the case of yeast [63], a protein kinse similar to STE 11 may be acting on MAP kinase kinase (MEK). A protein kinase homologous to STEll has been identified recently as an activator of MEK in mice [64]. It would be interesting to find out if a MEK kinase could be involved in the PBu2-mediated activation of ERK kinase in rat lympho- blasts.
The different specificities of staurosporine and bisindolyl- maleimide GF 109203X are evident in two different experiments. Staurosporine blocks at nanomolar concen-

Eur. J. Immunol. 1994. 24: 27462754 Raf-1 kinase activation is not required for ERK2 kinase activation 2753
tration the proliferation induced by either PBu2 or IL-2 (it is known, that the cytokine signaling is PKC independent). The concentration of staurosporine which inhibits prolifer- ation has only a slight effect on Raf-1 hyperphosphoryla- tion. Nevertheless, the bisindolylmaleimide behaves in both events as expected for a specific PKC inhibitor.
Since the proliferation blockage effect observed in rat lymphoblasts occurs when Raf-1 AS is added to the cells as late as 7 h after PBuz stimulation, we suggest that Raf-1 kinase activity must be required late in the G1 to S transition. Zmuidzinas et al. [17] reported a mRNA increase of Raf-1 transcripts through GI, reaching 8-10 times greater levels within the 10-12 h just prior to the onset of DNA synthesis. It is unknown whether the Raf-1 kinase activity is also required early upon the stimulation to
20 21
22
23
24
25
26
27
28
Suzuki, K., Trends. Biol. Sci. 1987. 12: 103. Kranta, A., Jondal, M. and Fredholm, B. B., FEBS Lett. 1991. 283: 321. Tsutsumi, A., Kubo, M., Fujii, H., Freire-Moar, J., Turck, Ch. W. and Ransom, J. T., J. Immunol. 1993. 150: 1746. Knopf, J. L., Lee, M. H., Sultzman, L. A., Kriz, R. N. Loomis, C. R., Hewick, R. M. and Bell, R. M., Cell 1986. 46: 491. Ono, Y., Fuji, T., Ogita, K., Kikkawa, U., Igarashi, K. and Nishizuka,Y., J. Biol. Chem. 1988. 263: 6927. Dekker, L. and Parker, F! J., Trends Biochem. Sci. 1994. 19: 73. Beyers, A. D., Hanekom, C., Rheeder, A., Strachan, A. F., Wooten, M. W. and Nel, A. E., J. Immunol. 1988. 141: 3463. Koretzky, G. A., Wahi, M., Newton, M. E. and Weiss, A., J. Immunol. 1989. 143: 1692. Lucas, S., Marais, R., Graves, J. D., Alexander, D., Parker, F! and Cantrell. D.. FEBS Lett. 1990. 260: 53.
phosphorylate another Raf-1 substrate.
We have also shown, that an active PKC is still required even when most ERK2 molecules have returned to basal levels, indicating that other PKC substrates, besides ERK2, have to be phosphorylated in order to reach the s phase.
29 Carroll, M. F!, Clark-Lewis, I . , Rapp, U. R. and May, W. S.,
30 Morrison, D. K., Kaplan, D. R., Escobedo, J. A,, Rapp, U. R.,
31 ApP, H., Hazan, R., Zilberstein, A., Ullrich, A., Schlessinger,
32 Hoshi, M., Nishida, E. and Sakai, H., J. Biol. Chem. 1988.263:
J. Biol. Chem. 1990. 265: 19812.
Roberts, T. M. and Williams, M. T., Cell 1989. 58: 649.
J. and Rapp, U., Mol. Cell. Biol. 1991. 11: 913.
5396.
We are grateful to Carmen Dominguez for technical assistance. We thank Dr. Lisardo Bosca for helpful discussions. Thanks to Dr. Tom Roberts for providing the Raf-1 antibody.
Received May 20, 1994; accepted August 8, 1994.
5 References
1 Imboden, J. and Stobo, J. D., J. Exp. Med. 1985. 161: 446. 2 Kroczek, R. A., Gunter, K. C., Seligmann, B. and Shevach, E.
3 Coutinho, A., Moller, G., Andersson, J. and Bullock, W. W.,
4 Smith, K. A., Science 1988. 240: 1169. 5 Imboden, J. B. and Stobo, J. D., J. Exp. Med. 1985. 161:
6 Samelson, L. E., Patel, M. D.,Weissman, A. M., Harford, J. B.
7 Patel, M. D., Samelson, L. E. and Klausner, R. D., J. Biol.
8 Graves, J. , Downward, J. , Izquierdo, M., Rayter, S.,Warne, F!
9 Satoh,T., Nakafuku, M., Miyasuna, A. and Kaziro,Y., Proc.
10 Izquierdo, M., Leevers, S. J., Marshall, C. J. and Cantrell, D.,
11 Siegel, J. N., Klausner, R. D., Rapp, U. R. and Samelson, L.
12 Siegel, J. N., Klausner, R. D., Rapp, U. R. and Samelson, L.
13 Siegel, J. N., June, C. H., Yamada, H., Rapp, U. R. and
14 Whitehurst, C. E., Boulton,T. G., Cobb, M. H. and Geppert,T.
15 Gardner, F!, Cell 1989. 59: 15. 16 Turner, B., Rapp, U., App, H., Greene, M., Dobaski, K. and
17 Zmuidznas, A., Mamon, H. J., Roberts, T. M. and Smith, K.
18 Nishizuka, Y., Nature 1988. 334: 661. 19 Nishizuka, Y., Nature 1984. 308: 693.
M., J. Immunol. 1986. 136: 4379.
Eur. J. Immunol. 1973. 3: 299.
446.
and Klausner, R. D., Cell 1986. 46: 1083.
Chem. 1987.262: 5831.
and Cantrell, D., J. Immunol. 1992. 148: 2417.
Natl. Acad. Sci. USA 1991. 88: 3314.
J. Exp. Med. 1993. 178: 1199.
E., J. Biol. Chem. 1990. 265: 18472.
E., J. Biol. Chem. 1992. 265: 18472.
Samelson, L. E., J. lmrnunol. 1993. 151: 4116.
D., J. Imrnunol. 1992. 148: 3230.
Reed, J., Proc. Natl. Acad. Sci. USA 1991. 88: 1227.
A., Mol. Cell. Biol. 1991. 11: 2194.
33 Ray, L. B. and Sturgill,T.W., Proc. Natl. Acad. Sci. USA 1987. 84: 1502.
34 Boulton,T., Nye, S. H., Robbins, D. J., Ip, N.Y., Radzieejews- ka, E., Morgenbesser, S. D., DePinho, R. A., Panayotatos, N., Cobb, M. and Yancopoulos, G. D., Cell 1991. 65: 663.
35 Gupta, S. K., Gallego, C., Jhonson, G. L. and Heasley, L. E., J. Biol. Chem. 1992. 267: 7987.
36 Leevers, S. J. and Marshall, C. J. , EMBO J. 1992. 11: 569. 37 Dent, F!, Haser,W., Haystead,T. A. J.,Vincent, L. A., Roberts,
T. M. and Sturgill, T. W., Science 1992. 257: 1404. 38 Kyriakis, J. M., App, H., Zang, X. Z . , Banerjee, I?, Brautigan,
D. L., Rapp, U. R. and Avruch, J., Nature 1992. 358: 417. 39 Shaap, D., Howe, L. R., Marshall, C. J. and Van Blillerswijk,W.
J., J. Biol. Chern. 1993. 268: 20232. 40 Vojtek, A. B., Gollenburg, J. M. and Cooper, J. A., Cell 1993.
74: 205. 41 Fabian, J. R., Daar, I. 0. andMorrison, D. K., Mol. Cell. Biol.
1993. 13: 7170. 42 Macdonald, S. G., Crews, C. M.,Wu, L., Driller, J., Clark, R.,
Erikson, R. L. and McCormick, F., Mol. Cell Biol. 1993. 13: 6615.
43 Sozeri, O., Vollmer, K., Liyanage, M., Frith, D., Kour, G., Mark 111, G. E. and Stabel, S., Oncogene 1992. 7: 2259.
44 Perez-Castillo, A., Pipaon, C., Garcia, I . and Alemany, S., J. Biol. Chem. 1993. 268: 19445.
45 Isakov, N. and Altman, A., J. Immunol. 1987. 138: 3100. 46 Toullec, D., Pianetti, P., Coste, H., Bellevergue, P., Grand-
Perret, T., Ajakane, M., Baudet,V., Boissins, F! , Boursier, E., Loirolle, F., Duhamel, L., Charon, D. and Kirilovsky, J., J. Biol. Chem. 1991. 266: 15771.
47 Ono, Y., Fujii, T., Ogita, K., Kikkawa, U., Igarashi, K. and Nishizuka,Y., Proc. Natl. Acad. Sci. USA 1989. 86: 3099.
48 Young, S., Parker, F! J., Ullrich, A. and Stabel, S., Biochem. J. 1987. 244: 775.
49 Redondo, J. M., Lopez-Rivas, A.,Vila,V., Cragoe, E. J. and Fresno, M . , J. Biol. Chem. 1988. 263: 17467.
50 Tornkvist, A, , Parpal, S., Gustavsson, J. and Stralfors, F!, J. Biol. Chem. 1994. 269: 1.
51 Carroll, M., Spivak, J., McMahon, M.,Weich, N.. Rapp, U. R. and May, W. S., J. Biol. Chem. 1991. 266: 14964.
52 Freire-Moar, J., Cherwinski, H., Hwang, F., Ransom, J.T. and Webb, D., J. Immunol. 1991.147: 405.
53 Diaz-Guerra, M. J. and B o d , L., Biochem. J. 1990. 269: 163.
54 Ways, D. K., Messer, B. R., Garris,T. O., Quin,W., Cook, P. P. and Parker, F! J., Cancer Res. 1992. 52: 5604.

2754
55 Borner, C., Nichols-Guadagno, S . , Fabbro, D. and Weinsten, I. B., J. Biol. Chem. 1992. 267: 12892.
56 Nel, A. N., Pollack, S., Landreth, G., Ledbetter, J. A., Hultin, L.,Williams, K., Katz, R. and Akerley, B., J. Zmmunol. 1990. 145: 971.
57 Perkins, G. R., Marvel, J. and Collins, M. K. L., J. Exp. Med. 1993. 178: 1429.
58 Samuels, M. L. ,Weber, M. J., Bishop, J. M. and McMahon, M., Mol. Cell. Biol. 1994. 13 No 10: 6241.
5Y Riedel, D., Brennscheidt, U., Kiehntopf, M., Brach, M. and Herrmann, F., Eur. J. Immunol. 1993. 23: 3146.
C. Lisbona, S. Alemany,V. Calvo and M. Fernandez-Re1 nart Eur. J. Immunol. 1994.24: 2746-2754
60 Troppmair, J., Bruder, J.T., Muiioz, H., Lloyd, F? A., Kyriakis, J., Banerjee, I?, Avruch, J. and Rapp, U. R., J. Biol. Chem. 1994. 269: 7030.
61 Chao,T.-S. O., Foster, D. A., Rapp, U. R. and Rosner, M. R., J. Biol. Chem. 1994. 269: 7337.
62 Kizaka-Kondoh, S . and Okayama, H., FEBS Lett. 1993.336: 255.
63 Nishida, E. and Gotho, Y., Trends Bzochem. Sci. 1993. 18: 128.
64 Lange-Carte, C. A., Pleiman, C. M., Gardner, A. M., Blumer, K. J. and Johnson, G. L., Science 1993.260: 315.




















