
Activation of myelin basic protein and S6 peptide kinases in phorbol ester- and PAF-treated sheep...
12
Biochimica et Biophysica Acta, 1176 (1993) 287-298 287 © 1993 Elsevier Science Publishers B.V. All rights reserved 0167-4889/93/$06.00 BBAMCR 13368 Activation of myelin basic protein and $6 peptide kinases in phorbol ester- and PAF-treated sheep platelets Mitra Samiei, Jasbinder S. Sanghera and Steven L. Pelech Biomedical Research Centre and The Department of Medicine, Unicersity of British Columbia, Vancouver (Canada) and Kinetek Biotechnology Corporation, Richmond (Canada) (Received 23 July 1992) (Revised manuscript received 9 December 1992) Key words: MAP kinase; $6 kinase; Platelet activating factor; Platelet The involvement of myelin basic protein (MBP) kinases and ribosomal $6 peptide kinases in sheep platelet signal transduction was investigated. Treatment of platelets with 200 nM 12-O-tetradecanoylphorbol-13-acetate (PMA) led to 5-fold stimulations of cytosolic MBP and $6 peptide kinase activities within 1 min. lmmunoblotting analysis of phenyl-Superose-fractionated cytosol from PMA-treated platelets with a panel of mitogen-activated protein (MAP) kinase anti-peptide antibodies revealed that one of the activated MBP kinases was p42 mapk. This MAP kinase isoform was also stimulated to a lesser extent (--2-fold) when platelets were exposed to 200 txM platelet-activating factor (PAF) for 3 min. The pathways of PAF-activation of p42 mapk also involved a protein kinase C-independent route, since the staurosporin analog compound 3 reduced PAF-induced activation by = 30% under conditions in which it inhibited PMA-activation of p42 mapk by = 80%. Another MAP kinase isoform of 44 kDa, most probably p44 erkl, was also detected in platelet cytosol, but it was only marginally modulated in response to PMA or PAF. The predominant PMA- and PAF-activated MBP kinase detected after MonoQ fractionation of platelet cytosol did not appear to correspond to a MAP kinase. MonoQ chromatography of platelet cytosol also resolved two PMA- and PAF-activated $6 peptide kinases, which appeared to coelute on phenyl-Sepharose. Western blotting analysis of the MonoQ fractions with antibodies raised against peptide sequences in the $6 kinases p90 rsk and p70 S6K revealed immunoreactive proteins of -- 75 kDa and = 95 kDa that coincided with the first $6 peptide kinase peak. These proteins probably corresponded to the 502 and 525 amino-acid-length forms of p70 soK. Only the second peak of $6 peptide kinase activity from MonoQ was appreciably stimulated in response to PAF-treatment of platelets, and this was largely abolished by compound 3. It is more likely that the novel MBP and $6 peptide kinases described here, rather than p42 mapk and p70 s6K, play a significant role in PAF signal transduction in the platelet. Introduction Platelets are an important component of the early response to blood vessel injury, and they are exquisitely sensitive to activation by such diverse agents as platelet-activating factor (PAF, 1-O-alkyl-2-acetyl-sn- glycero-3-phosphorylcholine), thrombin, collagen and phorbol ester tumour promoters. These factors trigger a rapid series of events that promote clotting and wound repair, including cytoskeletal rearrangements, aggregation and the release of mediators from various classes of granules [1]. Devoid of a nucleus and chro- mosomes, platelets are terminally differentiated with no capacity to divide. However, they exhibit unusually Correspondence to: S.L. Pelech, The Biomedical Research Centre, 2222 Health Sciences Mall, University of British Columbia, Vancou- ver, BC, V6T 1Z3, Canada. elevated levels of protein-tyrosyl phosphorylation upon activation [2-4], a phenomenon that has generally been linked with stimulated cell proliferation [5]. Inhibition of PAF-induced tyrosyl protein phosphorylation in rab- bit platelets exposed to tyrphostins and other tyrosyl kinase inhibitors leads to loss of platelet responses such as aggregation and secretion [6]. The ability of the protein-tyrosyl phosphatase inhibitors vanadate and molybdate to induce secretion from electropermeabi- lized human platelets also supports a role for tyrosyl phosphorylation in the platelet activation cascade [7]. Platelet tyrosyl protein phosphorylation may reflect in part the exceptionally high level of expression of the tyrosyl kinase p60 src (0.2-0.4% of the total platelet protein) [8,9]. Most of the p60 src resides near the plasma membrane and the surface connected canalicu- lar system [10]. However, while the bulk of p60 src is detergent-soluble in resting platelets, approx. 40% of the total p60 src becomes associated with the cyto-
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Activation of myelin basic protein and S6 peptide kinases in phorbol ester- and PAF-treated sheep...

Biochimica et Biophysica Acta, 1176 (1993) 287-298 287 © 1993 Elsevier Science Publishers B.V. All rights reserved 0167-4889/93/$06.00
BBAMCR 13368
Activation of myelin basic protein and $6 peptide kinases in phorbol ester- and PAF-treated sheep platelets
Mitra Samiei, Jasbinder S. Sanghera and Steven L. Pelech Biomedical Research Centre and The Department of Medicine, Unicersity of British Columbia, Vancouver (Canada)
and Kinetek Biotechnology Corporation, Richmond (Canada)
(Received 23 July 1992) (Revised manuscript received 9 December 1992)
Key words: MAP kinase; $6 kinase; Platelet activating factor; Platelet
The involvement of myelin basic protein (MBP) kinases and ribosomal $6 peptide kinases in sheep platelet signal transduction was investigated. Treatment of platelets with 200 nM 12-O-tetradecanoylphorbol-13-acetate (PMA) led to 5-fold stimulations of cytosolic MBP and $6 peptide kinase activities within 1 min. lmmunoblotting analysis of phenyl-Superose-fractionated cytosol from PMA-treated platelets with a panel of mitogen-activated protein (MAP) kinase anti-peptide antibodies revealed that one of the activated MBP kinases was p42 mapk. This MAP kinase isoform was also stimulated to a lesser extent (--2-fold) when platelets were exposed to 200 txM platelet-activating factor (PAF) for 3 min. The pathways of PAF-activation of p42 mapk also involved a protein kinase C-independent route, since the staurosporin analog compound 3 reduced PAF-induced activation by = 30% under conditions in which it inhibited PMA-activation of p42 mapk by = 80%. Another MAP kinase isoform of 44 kDa, most probably p44 erkl, was also detected in platelet cytosol, but it was only marginally modulated in response to PMA or PAF. The predominant PMA- and PAF-activated MBP kinase detected after MonoQ fractionation of platelet cytosol did not appear to correspond to a MAP kinase. MonoQ chromatography of platelet cytosol also resolved two PMA- and PAF-activated $6 peptide kinases, which appeared to coelute on phenyl-Sepharose. Western blotting analysis of the MonoQ fractions with antibodies raised against peptide sequences in the $6 kinases p90 rsk and p70 S6K revealed immunoreactive proteins of -- 75 kDa and = 95 kDa that coincided with the first $6 peptide kinase peak. These proteins probably corresponded to the 502 and 525 amino-acid-length forms of p70 soK. Only the second peak of $6 peptide kinase activity from MonoQ was appreciably stimulated in response to PAF-treatment of platelets, and this was largely abolished by compound 3. It is more likely that the novel MBP and $6 peptide kinases described here, rather than p42 mapk and p70 s6K, play a significant role in PAF signal transduction in the platelet.
Introduction
Platelets are an important component of the early response to blood vessel injury, and they are exquisitely sensitive to activation by such diverse agents as platelet-activating factor (PAF, 1-O-alkyl-2-acetyl-sn-
glycero-3-phosphorylcholine), thrombin, collagen and phorbol ester tumour promoters. These factors trigger a rapid series of events that promote clotting and wound repair, including cytoskeletal rearrangements, aggregation and the release of mediators from various classes of granules [1]. Devoid of a nucleus and chro- mosomes, platelets are terminally differentiated with no capacity to divide. However, they exhibit unusually
Correspondence to: S.L. Pelech, The Biomedical Research Centre, 2222 Health Sciences Mall, University of British Columbia, Vancou- ver, BC, V6T 1Z3, Canada.
elevated levels of protein-tyrosyl phosphorylation upon activation [2-4], a phenomenon that has generally been linked with stimulated cell proliferation [5]. Inhibition of PAF-induced tyrosyl protein phosphorylation in rab- bit platelets exposed to tyrphostins and other tyrosyl kinase inhibitors leads to loss of platelet responses such as aggregation and secretion [6]. The ability of the protein-tyrosyl phosphatase inhibitors vanadate and molybdate to induce secretion from electropermeabi- lized human platelets also supports a role for tyrosyl phosphorylation in the platelet activation cascade [7].
Platelet tyrosyl protein phosphorylation may reflect in part the exceptionally high level of expression of the tyrosyl kinase p60 src (0.2-0.4% of the total platelet protein) [8,9]. Most of the p60 src resides near the plasma membrane and the surface connected canalicu- lar system [10]. However, while the bulk of p60 src is detergent-soluble in resting platelets, approx. 40% of the total p60 src becomes associated with the cyto-

288
skeletal fraction during platelet aggregation [11]. Such translocation of p60 src could mediate some of the shape changes associated with platelet activation. Other src-related tyrosyl kinases may also participate in sig- nalling from thrombin in platelets, since p54 lyn, p61 fyn and p62 T M (but not p60 src) selectively associate with the GTPase activating protein for p21 ra~ in agonist- stimulated platelets, but not in resting cells [12]. Purifi- cation of a 54-kDa src-like kinase, most likely p54 ty~, revealed that it was expressed as approx. 0.1% of the total protein in human platelets [13].
PAF and thrombin signalling in platelets is also mediated via induction of the phospholipase C-cata- lyzed hydrolysis of polyphosphatidylinositol and the generation of diacylglycerol and inositol 1,4,5-tris- phosphate [14-16]. This latter second messenger trig- gers the release from the endoplasmic reticulum of calcium [17], which together with diacylglycerol acts to stimulate protein kinase C (PKC) [18]. Indeed, PKC is activated in both PAF-and thrombin-treated rabbit platelets [19,20]. Phorbol esters that directly stimulate protein kinase C activity are sufficient to bring about platelet aggregation and secretion, albeit much slower than PAF or thrombin [21]. Since agonist-induced poly- phosphoinositide production and stimulation of PKC activity in rabbit platelets are prevented by tyrphostins [6], it is possible that phospholipase C activation is achieved via tyrosyl phosphorylation.
PKC is but one of more than a dozen seryl/threonyl protein kinases that are activated in agonist-stimulated platelets [22,23]. Ferrell and Martin [22] originally de- tected at least 14 electrophoretically distinct seryl/ threonyl protein kinases from human platelets after renaturation from SDS-polyacrylamide gels, and 10 of these were activated in extracts from thrombin-treated cells. Since a combination of phorbol ester and calcium ionophore was much more effective than either agent alone in the recruitment of these kinases, other cal- cium-dependent protein kinases may also contribute to platelet activation. This may include the calmodulin- dependent myosin light chain kinase, since myosin light chain is a prominent phosphoprotein in thrombin- stimulated human platelets [24]. While the findings of Ferrell and Martin [22] imply that a network of protein kinases modulated by covalent modification mediates platelet activation, few of these kinases have been characterized and their location within the network remains to be mapped.
The remarkably high level of p60 sr~ activity in platelets might reflect the adaption of a cell cycle control protein kinase for alternative functions in a cell type that is incapable of cell division. In this vein, we have previously described the presence and activation of the cell division control (cdc) seryl/threonyl protein kinase p34 cdc2 in PAF- and thrombin-treated sheep platelets [25]. The activation of p60 ~r~ at M-phase has
been attributed to its phosphorylation by p34 cat2 [26- 28]. In transfected cells, introduction of active p60 ....... has also been linked with the indirect stimulations of seryl/threonyl protein kinases that phosphorylate myelin basic protein (MBP) and ribosomal protein $6, referred to as mitogen-activated protein (MAP) kinases [29] and $6 kinases [30], respectively. Two $6 kinases, p70 s6K and p90 rsk, have been shown to be phospho- rylated by MAP kinases in vitro [31-33]. We have recently reported the tyrosyl phosphorylation and acti- vation of a MAP kinase from sea star oocytes, i.e., p44 mpk, with the src-related tyrosyl kinase p56 tck [34]. Furthermore, phorbol ester treatment of a diversity of mammalian cultured cells has been correlated with stimulation of two MAP kinase isoforms, i.e., p42 mapk and p44 erkl, and both $6 kinases (reviewed in Refs. 5, 35 and 36). These observations prompted the present investigation of the MAP kinases and $6 kinases in sheep platelets and their regulation in response to PAF and phorbol esters.
Materials and Methods
Materials Rabbit antipeptides antibodies were generated
against the sea star p44 mp~ sequence GLAYIGE- GAYGMVYKA-GC (mpk-I Ab); the rat erkl se- quences PFEHQTYCQRTLREIQILLGFRHENV1GI- RD1LRAP-GGC (erkl-III Ab) and CGG-PFTFDME- LDDLPKERLKELIFQETARFQPGAPEAP (erkl-CT Ab); the mouse rsk sequence C-LVKGAMAATYSAL- NSSKPTPQLKPIESSILAGRRVRKLPSTI'L (rsk-CT); and the rat $6 kinase sequence AMIVRNAKDTAHT- KAERNILEEVKHP-GGC (S6K-III). These antibodies were immunoaffinity purified on the respective pep- tide-agarose columns and are now commercially avail- able from Upstate Biotechnology, (Lake Placid, NY), along with the 4G10 anti-phosphotyrosine mouse mon- oclonal antibody. The aforementioned peptides and RRRLSSLRA peptide based on two adjacent phosphorylation sites in $6 were kindly provided by Dr. Ian Clark-Lewis (Biomedical Research Centre). Affin- ity-purified rabbit antibody against purified sea star p44 '~pk was prepared as described [37]. PAF, throm- bin, PMA, bovine brain MBP, cAMP-dependent pro- tein kinase inhibitor peptide (TrYADFIASGRTGR- RNAIHD), gelatin, nitro blue tetrazolium, and 5- bromo-4-chloro-3-indolyl phosphate were purchased from Sigma. Goat anti-rabbit IgG conjugated to alka- line phosphatase was bought from Bio-Rad. Complete and Incomplete Freund's Adjuvant were from Gibco Laboratories. [T-3zp]ATP and PY-20 anti-phospho- tyrosine antibody were from ICN Biomedicals.
Preparation of platelet extracts Sheep platelets were isolated as described by
Pinckard et al. [38] with a few modifications. Briefly,

platelet rich plasma (PRP) was obtained by centrifuga- tion of whole blood at 1000 × g for 25 min. PRP was layered over Ficoll, centrifuged at 2000 × g for 25 min, and the platelet bands were washed twice with MTBH buffer (136.9 mM NaCI, 10 mM Hepes (pH 6.5), 2.7 mM KCI, 413 mM NaH2PO4-H20, 1.2 mM NaHCO3, 5.6 mM dextrose, 30.8 mM bovine serum albumin). Isolated sheep platelets were incubated at 37°C for 0-20 min with PMA (200 nM) or PAF (200 /zM) in MTBH buffer (pH 6.5). At the stated concentrations, these agonists optimally induced platelet aggregation in the absence of calcium in the medium. In some experiments, the platelets were preincubated for 20 min with 10 ~M compound 3 prior to treatment with PMA or PAF. In other experiments, when 2 mM CaCI2 was included with the MTBH buffer, 10 nM PAF was sufficient to induce rapid aggregation and was used. Since the presence of CaC12 in the medium in the absence of agonist was sufficient to induce a slow rate of platelet aggregation, it was usually omitted. Follow- ing agonist treatment for various times, the platelets were sedimented (9000 rpm, 5 min) and then sonicated in homogenizing buffer (75 mM /3-glycerophosphate, 20 mM Mops (pH 7.2), 15 mM EGTA, 2 mM EDTA, 1 mM sodium orthovanadate, 1 mM dithiothreitol) for 15 s (Sonics vibra-cell set at 40). The homogenate was centrifuged at 200000Xg for 15 min in a Beckman TL-100 ultracentrifuge, and the supernatant (cytosol) was immediately divided into portions and frozen at -70°C. Protein concentration in cytosolic extracts was estimated by the method of Bradford [39] with bovine serum albumin/'AI% = 6.5) as the standard. ~ " ~ 2 8 0 n m
Column chromatography For most anion-exchange chromatographies, 2 mg of
cytosolic protein were loaded onto a 1-ml MonoQ (Pharmacia) column. The column was eluted with a 10 ml linear 0-0.8 M NaC1 gradient in buffer A (25 mM fl-glycerophosphate, 10 mM Mops (pH 7.2), 5 mM EGTA, 2 mM EDTA, 1 mM sodium orthovanadate and 1 mM dithiothreitol) at a flow rate of 0.8 ml/min, and 250/zl fractions were collected.
For hydrophobic chromatography on phenyl-Super- ose (Pharmacia), cytosolic extract (2 mg) was loaded onto a pre-packed 1-ml column that had been equili- brated with buffer B (12.5 mM Mops (pH 7.2), 12.5 mM /3-glycerophosphate, 0.5 mM EGTA, 7.5 mM MgCI 2, 50 /~M NaF), containing 250 mM NaCI. The column was eluted with a 10 ml linear gradient of 0-3% Brij-35 and 250 mM NaCI to 0 mM NaCI in buffer B, at a flow rate of 0.3 ml/min, and 250 tzl fractions were collected. For phenyl-Sepharose (Pharmacia) chromatography, cytosolic extract (2 mg) was applied to a 2-ml column that had been equili- brated with buffer B. The column was eluted with a 12.5-ml linear gradient of 0-3% Brig-35 in buffer B, at
289
a flow rate of 1 ml/min, and 250 ~1 fractions were collected.
Protein kinase assays The thawed extracts and column fractions were as-
sayed for phosphotransferase activity toward 1 mg/ml MBP or 1 mg/ml RRRLSSLRA $6 peptide with the following components for 10 min at 30°C in a volume of 25/~1:50/zM [y-32p]ATP (1500 cpm/pmol); 25 mM fl-glycerophosphate; 10 mM Mops (pH 7.2); 15 mM MgCI2; 2 mM EGTA; 2 mM EDTA; 1 mM sodium orthovanadate; 1 mM dithiothreitol; and 500 nM TTYADFIASGRTGRRNAIHD peptide. At the end of the reaction period, 20-/xl aliquots were removed and spotted on to 2-cm 2 pieces of Whatman P81 phos- phocellulose paper and 30 s later washed with several changes of 1% (v/v) phosphoric acid. The wet filter papers were transferred into 6-ml plastic scintillation vials that contained 0.5 ml of Ecolume (ICN) scintilla- tion fluid and quantitated for radioactivity in a Wallac 1410 Counter (LKB).
SDS-PAGE and immunoblotting studies SDS-PAGE of the column-fractionated platelet ex-
tracts was performed with 11% separating polyacryl- amide gels as described by Laemmli [40]. Following electrophoresis, the separating gel was soaked in trans- fer buffer (25 mM Tris, 192 mM glycine, 20% methanol) and then placed together with a nitrocellulose mem- brane in a sandwich. Proteins were transferred for 3 h at 75 V. Subsequently, the nitrocellulose membrane was blocked with TBS (Tris-buffered saline) containing 5% skim milk powder for 2 h at 20°C. The membrane was washed twice with TBS containing 0.05% Tween 20 (TTBS) for 5 min before incubation with the pri- mary antibody in 0.05% skim milk overnight at 20°C. The membrane was then washed twice with TTBS and incubated with the secondary antibody (goat anti-rabbit IgG or goat anti-mouse IgG coupled to alkaline phos- phatase in 0.05% skim milk) for 2 h at 20°C. The membrane was rinsed with two washes of T-I'BS, fol- lowed by one wash with TBS, and then incubated with 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetra- zolium colour development solution (a mixture of 3% nitro blue tetrazolium in 1 ml 70% dimethylformamide and 1.5% 5-bromo-4-chloro-3-indolyl phosphate in 1 ml 100% dimethylformamide subsequently added to 100 ml of 0.1 M NaHCO 3, 10 mM MgCI 2 (pH 9.8)). The colour was developed in 1-24 h at 20°C in the dark, and the reaction stopped by rinsing the mem- brane with water. In case of 4G10 antibody, the nitro- cellulose membrane was blocked in 5% bovine serum albumin to minimize the background. In addition, low salt TBS (50 mM NaCI) was used in all the dilutions and washes with 0.05% Nonidet P-40 substituted for Tween 20.

290 Results
Actication of MBP kinases in sheep platelets by PMA and PAF
To assess the p re sence and t ime-course act ivat ion of M B P kinases in sheep p la te le t s , these cells were ex- posed to e i the r 200 n M P M A or 200 /zM P A F in ca lc ium-f ree m e d i u m for var ious t ime intervals up to 20 min. As shown in Fig. 1A, t r e a t m e n t of in tac t p la te le t s with T P A for as l i t t le as 1 min resu l t ed in a 7-fold inc rease in M B P p h o s p h o t r a n s f e r a s e activity as m e a s u r e d in cytosol ic extracts . Exposure of the cells to P A F for 1 min p r o d u c e d only a m o d e s t 50% s t imula- t ion of cytosolic M B P kinase activity, but this ac t ivat ion
was sus ta ined for at leas t 20 min. To cha rac t e r i ze the P M A - a c t i v a t e d M B P kinases in
p l a t e l e t cytosol, these ext rac ts were sub jec ted to an ion -exchange c h r o m a t o g r a p h y on M o n o Q and the co lumn f rac t ions assayed for M B P p h o s p h o t r a n s f e r a s e activity (Fig. 2). S t imula t ion of p l a t e l e t s with 200 n M P M A for 3 min resu l ted in the act ivat ion of at leas t two M B P kinases , the first e lu t ing at = 0.4 M NaC1 and t h e second at = 0.6 M NaCI (Fig. 2A). T h e first M B P kinase was ac t iva ted over 4.5-fold, while the second was s t imu la t ed = 2.5-fold. T h e e lu t ion behav iou r of the first M B P kinase was remin i scen t of two M A P kinase isoforms, namely p42 mapk (also known as p42 erk2) and p44 erkl [32,34].
To inves t iga te the M A P kinase i soform compos i t ion in sheep p la te le ts , the M o n o Q f rac t ions f rom P M A - t r e a t e d p la te le t s shown in Fig. 2 A were ana lyzed on W e s t e r n blots with M A P kinase an t ibod ie s g e n e r a t e d agains t p e p t i d e sequences found in ra t p44 e~kl (Fig. 2B and C) and sea s tar p44 mpk (an t i -mpk- I , da t a not shown) and agains t pur i f i ed p44 mpk p ro t e in (Fig. 2D).
o 700
600
500
'~ 400
0.. 300
200 ¢ :
"A
100
0 - - - | . . . . , . . . . , . . . . , -
0 5 10 15 20
Time (min)
. . . j
a:
t.
800 B
700
60O
5OO
4OO
3OO
20O
1001 ~r-"O-'-"-O- . . . . . . . . . . --O
0 - - . i . . . . i . . . . , . . . . , -
0 5 10 15 20
Time (rain)
Fig. 1. PMA and PAF activation of MBP and $6 peptide kinases. Sheep platelets were exposed to 200 nM PMA ( • ) , 200/zM PAF (o) or no agonist (©) for the indicated time intervals prior to harvesting, and cytosolic extracts were prepared as described under Materials and Methods. The phosphotransferase activity towards MBP (panel A) and $6 peptide (RRRLSSLRA, panel B) in the cytosolic extracts was assayed as described under Materials and Methods. Values are
averages for at least two independent experiments.
250
~' ~" 200
~..~ 150
Q.. ~ 100
50
A
o unr..:l ......
• ' • • I • • • • | • • • . I " ' " ' I • • • " I
0 10 20 30 40 50 MonoQ fraction number
u e r k l - I I I
a e r k l - CT
mpk p44
PY-20 Ab
-p44 -p42
-p44 -p42
-p44 -p42
-p44 -p42
1 2 3 4 5 6 7
Lane number
Fig. 2. MonoQ chromatography of MBP kinases from PMA-stimu- lated platelets. Cytosolic protein (2.0 mg) from untreated platelets (©) and those exposed to 200 nM PMA (o) for 3 min were separately fractionated on a MonoQ column as described under Materials and Methods. The column fractions were assayed for phosphotransferase activity towards 1 mg MBP/ml (panel A). Selected fractions desig- nated with outlined numbers in panel A were immunoblotted with anti-erkl-III (panel B), anti-erkl-CT (panel C), anti-p44 mpk (panel D), and PY-20 anti-phosphotyrosine (panel E) antibodies. The upper and lower bounds of each box correspond to the migration positions of the prestained standards ovalbumin (50 kDa) and carbonic anhy- drase (33 kDa), which are shown in lane 7. The positions of p42 ' ' 'pk and p44 erkl are indicated on the right. Similar results were obtained
in three separate experiments.
Al l of these an t ibod ies d e t e c t e d 42-kDa and 44-kDa pro te ins , cons is ten t with p42 map~ and p44 erkl, which co inc ided with the first M B P kinase p e a k in the M o n o Q fract ions. Based upon the in tens i t ies of these immuno-
291
reactive proteins, the amount of p44 erkt appeared to predominate over p42 mapk in the sheep platelet. How- ever, the selective detection of p42 mapk with an anti- phosphotyrosine antibody implied that it was this iso- form that was primarily activated in response to PMA (Fig. 2E) [5,35,36].
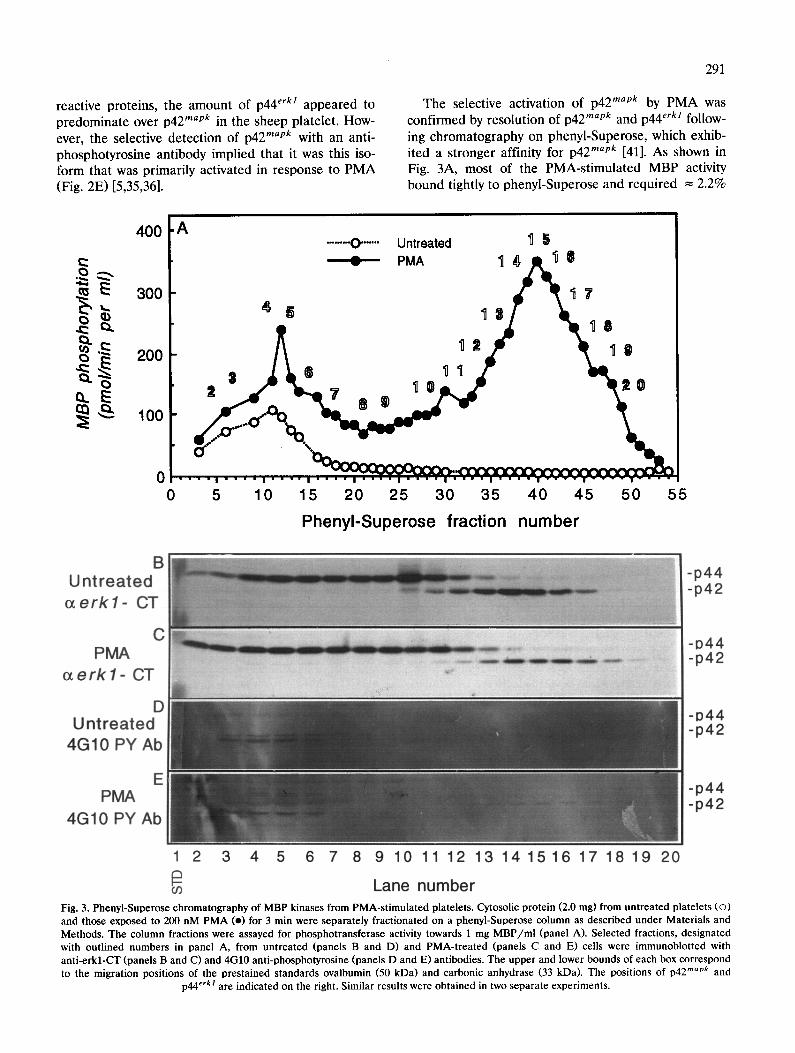
The selective activation of p42 mapk by PMA was confirmed by resolution of p42 rnapk and p44 erkl follow- ing chromatography on phenyl-Superose, which exhib- ited a stronger affinity for p42 mapk [41]. As shown in Fig. 3A, most of the PMA-stimulated MBP activity bound tightly to phenyl-Superose and required = 2.2%
,oo . . ! | ........ O ....... Untreated u ~ I
300 I - d • 1] 7 I
I ,00 °
0 5 10 15 20 25 30 35 40 45 50 55
PhenyI-Superose fraction number
B U nt reated e~erkl- CT
C PMA
o~erkl- CT
D U ntreated
4G 10 PY Ab
E PMA
4G 10 PY Ab
-p44 -p42
-D44 -p42
-D44 -p42
-p44 -p42
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
Lane number Fig. 3. Phenyl-Superose chromatography of MBP kinases from PMA-stimulated platelets. Cytosolic protein (2.0 mg) from untreated platelets (o) and those exposed to 200 nM PMA (o) for 3 min were separately fractionated on a phenyl-Superose column as described under Materials and Methods. The column fractions were assayed for phosphotransferase activity towards 1 mg MBP/ml (panel A). Selected fractions, designated with outlined numbers in panel A, from untreated (panels B and D) and PMA-treated (panels C and E) cells were immunoblotted with anti-erkl-CT (panels B and C) and 4G10 anti-phosphotyrosine (panels D and E) antibodies. The upper and lower bounds of each box correspond to the migration positions of the prestained standards ovalbumin (50 kDa) and carbonic anhydrase (33 kDa). The positions of p42 mapk and
p44 erk! are indicated on the right. Similar results were obtained in two separate experiments.

292
Brij-35 for elution. The MBP phosphotransferase activ- ity of this kinase was elevated = 25-fold by PMA treatment, whereas the MBP kinase activity near the break through fractions from the same column was stimulated by only 2.5-fold. The late eluting MBP kinase activity peak was coincident with a 42-kDa protein that was immunoreactive with an antibody raised against the C-terminus of rat p44 erk/, which can crossreact with p42 mapk (Fig. 3B and C). Immuno- reactivity to p44 erkl was detectable over a broad range in the earlier phenyl-Superose fractions. In fraction- ated extracts from PMA-treated ceils (Fig. 3E) but not untreated platelets (Fig. 3D), this protein was also visualized with a different anti-phosphotyrosine anti- body from that used in Fig. 2E. The tyrosyl phospho- rylation of p42 mapk was associated with a slight retar- dation (= 500 Da) in its mobility on SDS-polyacryl- amide gels, which has also been correlated with activa- tion of MAP kinases [41]. This band shift in p42 mapk was most evident in the Western blots shown in Fig. 2C and D. Collectively, these findings indicated that PMA treatment of platelets resulted in preferential tyrosyl phosphorylation and activation of p42 mapk.
When cytosolic extract from sheep platelets treated with 200/xM PAF for 3 min in calcium-free media was subjected to analytical MonoQ chromatography, three distinct peaks of PAF-stimulated MBP kinase activity were resolved (Fig. 4A). Peak I (= 0.4 M NaC1 elution) was activated by = 50%, whereas peaks II (= 0.5 M NaCI elution) and I I I ( = 0.6 M NaC1 elution) were activated by ---3-fold. Peak I MBP kinase coincided with p42 mapk and p44 erkl immunoreactivity with the erkl-C-terminus antibody (Fig. 4B), although these ki- nases were not detectable with an anti-phosphotyrosine antibody (Fig. 4C). Peak III MBP kinase appeared to correspond to the second major PMA-activated MBP kinase, whereas the peak II was selectively stimulated in response to PAF but not PMA. MonoQ peak II and III fractions did not seem to feature any enriched proteins that crossreacted with anti-MAP kinase or anti-phosphotyrosine antibodies (Fig. 4B and C). Al- though = 44-kDa proteins were visualized with the erkl-CT and 4G10 antibodies in the region of the peak MBP kinase III fractions, they were more intensely stained in side fractions.
The 50% activation of the MonoQ MBP kinase peak I by PAF (Fig. 4A) was very modest when compared to the 5-fold stimulation elicited by PMA (Fig. 2A). Phenyl-Superose chromatography of the cytosol from PAF-treated platelets permitted identification of p42 "ap~ as the major PAF-activated MAP kinase (Fig. 5A and B). The phosphorylation of p42 m~pk in re- sponse to PAF was barely visible with anti-phospho- tyrosine antibodies (Fig. 5C), perhaps because the de- gree of phosphorylation was near the limits of detec- tion.
(3 4::
200
E 150
100
E
50
O; 0
A 7 8 . . . .
. . . . . . .
10 20 30 40 50
MonoQ fraction number
B PAF
~ e r k l - CT
C PAF
4G10 PY Ab
-p44 -p42
-p44 -p42
1 2 3 4 5 6 7 8 9 1 0
Lane number Fig. 4. MonoQ chromatography of MBP kinases from PAF-stimu- lated platelets. Cytosolic protein (2.0 mg) from untreated platelets (©) and those exposed to 200 p,M PAF (e) for 3 min were separately fractionated on a MonoQ column as described under Materials and Methods. The column fractions were assayed for phosphotransferase activity towards 1 mg MBP/ml (panel A). For PAF-treated cell extracts, selected MonoQ fractions designated with outlined num- bers in panel A were immunoblotted with anti-erkl-CT (panel B) and 4G10 anti-phosphotyrosine (panel C) antibodies. The upper and lower bounds of each box correspond to the migration positions of the prestained standards ovalbumin (50 kDa) and carbonic anhy- drase (33 kDa), which are shown in lane 10. The positions of p42 mapk and p44 erkl are indicated on the right. Similar results were
obtained in three separate experiments,
Role of PKC in the PAF activation of platelet MBP kinases
PKC is selectively and potently inhibited by the staurosporin analogue compound 3 [42]. After preincu- bation of sheep platelets with 10/xM compound 3 for 20 min, the 200 nM PMA-induced stimulation of MAP kinase activity with MBP (MonoQ peak I) after 3 min was reduced to 21% of the activity measured in the absence of PMA. Compound 3 reduced the MBP ki- nase activity in MonoQ fractions 36-44 (peak III) to near the basal levels found in extracts from untreated platelets (Table I). Furthermore, the presence of com- pound 3 blocked both the PMA-induced band shift of p42 mapk to a higher molecular mass species and its tyrosyl phosphorylation (data not shown). Thus activa- tion of p42 mapk by PMA was primarily mediated by a PKC-dependent pathway.

293
PAF has previously been shown to activate PKC in rabbit platelets albeit not as dramatically as PMA [19,20]. Consequently, the contribution of PKC in the 200 /zM PAF stimulations of the MBP kinases in platelets after 3 min treatment was assessed with com- pound 3. The presence of compound 3 reduced PAF activations of the MBP kinases in MonoQ peaks I, II and III, respectively, to 68, 59 and 54% of the maximal levels seen with PAF in the absence of compound 3 (Table I). Since compound 3 was about 2.5 times more effective in the inhibition of the PMA-induced activa- tion of p42 mapk in MonoQ peak I, it would appear that PAF also stimulated p42 mapk by a PKC-independent pathway. PKC was probably not involved in the PAF activation of MonoQ MBP kinase peak II, since this
kinase was not stimulated in response to PMA (Fig. 2A). Therefore, the compound 3 inhibition of the PAF activation of this MBP kinase was unexpected and implies that this inhibitor may not be selective for PKC. As the PAF- and PMA-stimulations of MonoQ MBP kinase peak III were similarly inhibited by com- pound 3 and reduced to near the basal levels in ex- tracts from untreated platelets, the PAF activation was probably mediated primarily via PKC.
Actiuation of $6 kinases in sheep platelets by PMA and PAF
Previous studies have identified ribosomal $6 pro- tein kinases as in vitro substrates of MAP kinases [31-33]. The primary sites of phosphorylation of $6 by
150 4 ~ ......... 0 ....... Untreated
12°I" 1 3 , 6 I , I
0 0 7 1
60
30
0 0 5 10 15 20 25 30 35 40 45 50 55
PhenyI-Superose fraction number
m Ill m E
1 2 3 4 5 6 7 8 9 10 11 12 13 14 1 5 1 6 17 18 19 20
Lane number Fig. 5. PhenyI-Superose chromatography of MBP kinases from PAF-stimulated platelets. Cytosolic protein (2.0 mg) from untreated platelets (O) and those exposed to 200/~M PAF (e) for 3 min were separately fractionated on a phenyl-Superose column as described under Materials and Methods. The column fractions were assayed for phosphotransferase activity towards 1 mg MBP/ml (panel A). For PAF-treated cell extracts, selected MonoO fractions designated with outlined numbers in panel A were immunoblotted with anti-erkl-CT (panel B) and 4G10 anti-phosphotyrosine (panel C) antibodies. The upper and lower bounds of each box correspond to the migration positions of the prestained standards ovalbumin (50 kDa) and carbonic anhydrase (33 kDa), which are shown in lane 1. The positions of p42 mapk and p44 erkl a r e indicated
on the right. Similar results were obtained in two separate experiments.

294
these $6 kinases are located near the C-terminus of the protein within the sequence Arg-Arg-Arg-Leu-Ser-Ser- Leu-Arg-Ala, and this peptide is a convenient alterna- tive substrate [5]. In cytosolic extracts from platelets, this $6 peptide served as substrate for a protein kinase(s) that was activated 7.5-fold following treat- ment of the cells with 200 nM PMA for 1 min (Fig. 1B). This PMA-induced activation of the $6 peptide kinase was reversed upon further incubation with the
phorbol ester. Treatment of the platelets with 200/zM PAF for 1 to 20 min produced a modest 50% increase in cytosolic $6 peptide phosphotransferase activity that was sustained during this period. Thus, the activation of $6 peptide kinases in platelets by these agonists paralleled the stimulation of MBP kinase activity (Fig. 1A).
Fractionation of cytosol from platelets-treated with 200 nM PMA for 3 rain on MonoQ afforded the
L - -
e-
B
D
A
80 ........ 0 ...... Untreated T ~ ( ~
6 0 '
@
20
0 0 10 20 30 40 50
MonoQ fraction number
S6K-I I I RSK- CT
C
-p75
E
-p75
1 2 3 4 5 6 7 8 9 10 1 2 3 4 5 6 7 8 9 10
Lane number ~ Lane number Fig. 6. MonoQ chromatography of $6 peptide kinases from PMA-stimulated platelets. Cytosolic protein (2.0 mg) from untreated platelets (©) and those exposed to 200 nM PMA (e) for 3 min were separately fractionated on a MonoQ column as described under Materials and Methods. The column fractions were assayed for phosphotransferase activity towards 1 mg/ml RRRLSSLRA peptide (panel A). Selected fractions designated with outlined numbers in panel A from untreated (panels B and C) and PMA-treated (panels D and E) cells were immunoblotted with anti-S6K-III (panels B and D) and anti-RSK-CT (panels C and E) antibodies. Migration positions of the prestained standards phosphorylase (106 kDa), bovine serum albumin (80 kDa) and ovalbumin (50 kDa) are shown in lane 1. The position of a 75 kDa protein is indicated on the
right. Similar results were obtained in three separate experiments.
295
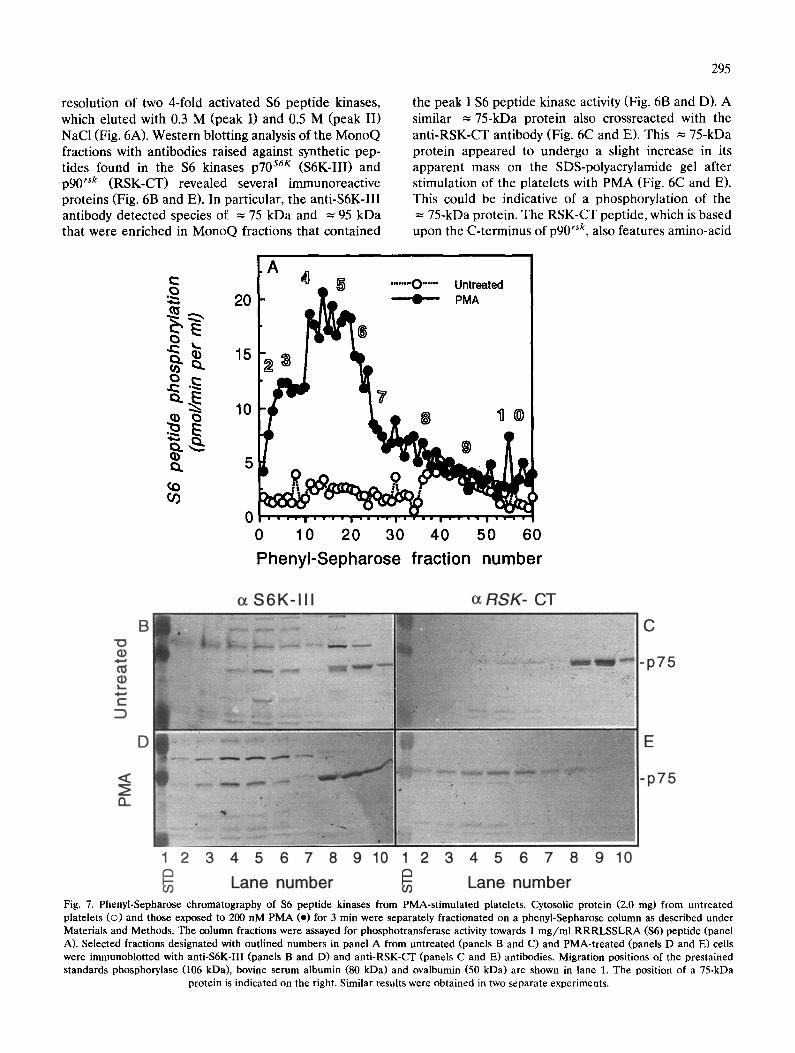
resolution of two 4-fold activated $6 peptide kinases, which eluted with 0.3 M (peak I) and 0.5 M (peak II) NaCI (Fig. 6A). Western blotting analysis of the MonoO fractions with antibodies raised against synthetic pep- tides found in the $6 kinases p70 s6r (S6K-III) and p90 r`k (RSK-CT) revealed several immunoreactive proteins (Fig. 6B and E). In particular, the anti-S6K-III antibody detected species of = 75 kDa and = 95 kDa that were enriched in MonoQ fractions that contained
the peak I $6 peptide kinase activity (Fig. 6B and D). A similar = 75-kDa protein also crossreacted with the anti-RSK-CT antibody (Fig. 6C and E). This = 75-kDa protein appeared to undergo a slight increase in its apparent mass on the SDS-polyacrylamide gel after stimulation of the platelets with PMA (Fig. 6C and E). This could be indicative of a phosphorylation of the = 75-kDa protein. The RSK-CT peptide, which is based upon the C-terminus of p 9 0 rsk, also features amino-acid
¢:::: [ r ~ = ~ ...... o ..... untreated
'°If" q,' I
0 10 20 30 40 50 60 PhenyI-Sepharose fraction number
o~ S6K-II I RSK- CT
"10 I1)
L -
¢..
B C
p75
m 13..
1 2 3 4 5 6 7 8 9 10 1 2 3 4 5 6 7 8 9 10
Lane number ~ Lane number Fig. 7. Phenyl-Sepharose chromatography of $6 peptide kinases from PMA-stimulated platelets. Cytosolic protein (2.0 rag) from untreated platelets (©) and those exposed to 200 nM PMA (e) for 3 min were separately fractionated on a phenyl-Sepharose column as described under Materials and Methods. The column fractions were assayed for phosphotransferase activity towards 1 mg/ml RRRLSSLRA ($6) peptide (panel A). Selected fractions designated with outlined numbers in panel A from untreated (panels B and C) and PMA-treated (panels D and E) cells were immunoblotted with anti-S6K-III (panels B and D) and anti-RSK-CT (panels C and E) antibodies. Migration positions of the prestained standards phosphorylase (106 kDa), bovine serum albumin (80 kDa) and ovalbumin (50 kDa) are shown in lane 1. The position of a 75-kDa
protein is indicated on the right. Similar results were obtained in two separate experiments.

296
TABLE I
Effect of compound 3 on PMA- and PAF-actications of MBP kinases in sheep platelets
Cytosolic protein (2.0 mg) from platelets exposed to 500/~M PAF or 200 nM PMA in the absence or presence of 10 /~M compound 3 were separately fractionated on a MonoQ column, and subsequently the column fractions were assayed for MBP kinase activity as de- scribed under Materials and Methods. When included, the platelets were preincubated for 20 min with 10 /~M compound 3 before the addition of agonist. The percentage inhibition of the MBP kinase activity in each of the three MBP kinase peaks as shown in Fig. 4A in the presence of compound 3 relative to its absence for each agonist is described. Values are the means_+S.E, for three separate experi- ments.
MBP peak Percentage inhibition by compound 3 of:
PMA activation PAF activation
l 79_+8 32+ 5 I1 41 + 7
llI 52_+ 1 46+ 19
sequences found in p70 S6K, and so it is possible that it may also crossreact with p70 s6K. In view of the size of the = 75-kDa immunoreactive protein, its recognition by the S6K-III antibody, its apparent band shift to a higher molecular weight form following PMA treat- ment of platelets, which has been shown to occur to p70 S6K in response to PMA in other mammalian cells [5], and its coincidence with a PMA-stimulated $6 peptide kinase activity on MonoQ (Fig. 6) and phenyl-
Sepharose (Fig. 7), it was probably p70 S6K. It is also feasible that the = 95-kDa immunoreactive protein detected with the anti-S6K-III antibody (Fig. 6B and D, Fig. 7B and D) represented a larger form of p70 S6K that features an additional 23 residues at the N- terminus and migrates as a =90 kDa protein on SDS-polyacrylamide gels [43,44].
The identity of the MonoQ peak II $6 peptide kinase is more equivocal. There were no intensely immunoreactive bands of the expected size for p90 rsk and its phosphorylated forms visualized in the peak fractions of this kinase (Fig. 6C and E). However, the anti-S6K-III antibody weakly crossreacted with a = 60- kDa protein in these MonoQ fractions (Fig. 6B and D). Although the MonoQ peak II $6 kinase was coincident with the MonoQ peak II MBP kinase, these enzymes are likely to be distinct, since this MBP kinase was not significantly activated in response to PMA treatment of platelets.
Exposure of platelets to 200 /~M PAF for 3 min failed to produce appreciable activation of the MonoQ peak I $6 peptide kinase (Fig. 8B and data not shown), although there was a modest activation of the peak II $6 peptide kinase. The contribution of PKC in the PAF-induced stimulation of the peak II $6 peptide kinase was evaluated based upon its sensitivity to inhi- bition by compound 3. As shown in Fig. 8B, pretreat- ment with compound 3 blocked most, but not all of the PAF-induced activation of the peak II $6 peptide
o ~ 7O
6o
J:: 50
40
co 20 I1: I1: 10
0 25
.A ..... - o - - Untreated PMA
- ' - ' A ' - " PMA + cmpd 3
7O
6O
5O
4O
30
20
10
0 25
. B ....~... cmpda PAl:
- - I I - PAF + cmpd 3
. . . . I . . . . I " " " ~ I " • . . . . . " I . . . . i " " " " I . . . .
30 35 40 45 30 35 40 45
MonoQ fraction number MonoQ fraction number
Fig. 8. Effect of compound 3 on PMA- and PAF-activations of $6 peptide kinases in platelets. Cytosolic protein (2.0 mg) from untreated platelets (©) and those exposed for 3 min to 200 nM PMA alone (o), 200 nM PMA and 10 tzM compound 3 ( • ) , 10 p.M compound 3 alone (zx), 200/zM PAF alone ([]) and 200 ~M PAF and 10/~ M compound 3 ( • ) , were separately fractionated on a MonoQ column as described under Materials and Methods. When included, the platelets were preincubated for 20 min with 10/zM compound 3 before the addition of agonist. The column fractions were assayed for phosphotransferase activity towards 1 mg/ml RRRLSSLRA ($6) peptide. Similar results were obtained in two
separate experiments.

kinase. Under similar circumstances, compound 3 com- pletely prevented PMA-stimulation of the peak I and II $6 peptide kinases (Fig. 8A).
Discussion
Agonist stimulation of platelets results in recruit- ment of a large continent of protein kinases, the major- ity of which have yet to be identified. Here, we have described the PMA-activation in sheep platelets of three protein kinases, namely p42 mapk, p44 erkl and p70 s6K, which are often 'turned on' in mitogen-treated quiescent mammalian cells and during oocyte matura- tion [5, 45]. These kinases, however, were either poorly activated or not at all in response to PAF treatment of sheep platelets with 200 /zM PAF in the absence of external calcium (Fig. 4A and 8) or 10 nM PAF in the presence of 2 mM CaC12 (data not shown). This was despite the activation by PAF of PKC [19,20], which is the primary target by which PMA exerts its action. This may reflect the very small degree of PKC activation by PAF, which is an order of magnitude poorer than induced by PMA in rabbit platelets [19,46]. However, PMA is a much less potent and slower inducer of sheep platelet aggregation than PAF (data not shown). Consequently, it is questionable whether p42 mapk, p44 erkl and p70 s6r play significant roles in the induc- tion of sheep platelet responses.
PMA caused the activation in sheep platelets of at least two $6 peptide kinases that could be resolved by MonoQ chromatography. While a reasonable case can be made for the early eluting $6 peptide kinase peak containing isoforms of p70 s6r, the identity of the late eluting $6 peptide kinase from MonoQ remains ob- scure. The 90-kDa $6 peptide kinases encoded by the RSK family might have been anticipated to become activated in sheep platelets in response to PMA and PAF. In wide variety of other model systems, PMA and a diversity of agents that stimulate MAP kinases, also elicit p90 rs~ activation [5], p90 rsk is phosphorylated and activated by MAP kinases in vitro [31,32]. How- ever, the RSK-CT antibody did not appear to im- munoreact with any 90- to 95-kDa proteins that coin- cided with either of the PMA-activated $6 peptide kinase peaks on MonoQ (Fig. 6C and E). This antibody strongly immunoblots and immunoprecipitates recom- binant chicken and mouse p90 rsk (J. Blenis, personal communication). Therefore, it seems unlikely that the MonoQ peak II $6 peptide kinase that was activated in response to PMA and PAF by a PKC-dependent path- way was p90 rsk. At this juncture, it appears to be a novel protein kinase.
Although the PAF-induced activation of p42 mapk was small, it was highly reproducible and achieved through PKC-dependent and independent mecha- nisms. Purified PKC does not appear to phosphorylate
297
MAP kinases such as sea star p44 mpk in vitro (data not shown). However, recent studies from several laborato- ries have pointed to -45-kDa protein kinase(s) re- lated to the yeast ste-7- and byr-l-encoded kinases as the major activator(s) of MAP kinases in response to PMA and other agonists [47-50]. This MAP kinase, in turn, can be phosphorylated and stimulated in vitro by active forms of the rafl-encoded protein kinase [51,52]. PMA- and growth factor- activation of p74 rafl such as by insulin has been demonstrated in a range of cul- tured mammalian cells [53,54]. Recent evidence indi- cates that protein kinase C may directly phosphorylate and activate p74 rafl [55]. Reduced mobility of p74 raf~ on SDS-polyacrylamide gels is a commonly observed marker for the activation of this kinase in response to PMA-and mitogenic stimulation of cells. Using three antibodies for three distinct regions in p74 ray1, we detected relatively low levels of a = 75-kDa protein of the appropriate size on SDS-polyacrylamide gels (M. Samiei, C. McNabb and S. Pelech, unpublished results). However, we failed to observe altered mobility of this protein in sheep platelets exposed to PAF or thrombin. These preliminary findings imply that the protein ki- nase C ~ p74 rafl ~ MAP kinase kinase ~ MAP kinase
rsk $6 kinase pathway probably does not play an important role in platelet activation in response to PAF.
Using MBP as an artificial substrate, PAF was shown to lead to the activation of two novel protein kinases (MonoQ peaks II and III) that bound more tightly to MonoQ than p42 mapk (Fig. 4). These kinases did not immunoreact strongly with any of the four MAP kinase antibodies tested on them. MAP kinase antibody im- munoreactivity associated with these kinases appeared to stem from tailing from the MonoQ peak I enzyme. Therefore, they are likely to be distinct from MAP kinases. Peak II MBP kinase was selectively stimulated by PAF but not by PMA. Therefore, this kinase was clearly distinguishable from the PMA-activated $6 pep- tide kinase that coeluted with it on MonoQ. Both PAF and PMA produced comparable activations of the MonoQ peak III MBP kinase (Figs. 2 and 4). There was a fairly high level of basal phosphotransferase activity for MonoQ peak III MBP kinase in extracts from untreated platelets. Since the PAF stimulation of this kinase was abrogated in the presence of compound 3, PKC appears to mediated its activation. Enhance- ment of the MonoQ peak III MBP kinase activity may be a more general response in cytokine-treated hemopoietic cells. We have recently detected the stim- ulation of a similar MBP kinase in various factor-de- pendent hemopoietic cell lines treated with interleukin 3, interleukin 5, granulocyte macrophage-stimulating factor and the steel factor [56, 57]. Current efforts are directed towards the purification and further charac- terization of this novel MBP kinase.

298
Acknowledgements
Al l of the syn the t ic p e p t i d e s u sed as suhs t r a t e s or i m m u n o g e n s were k ind ly p rov ided by Dr . I a n Clark-
Lewis (B.R.C.) . Ms. Faye C h o w a i d ed in the pur i f ica- t ion of the a n t i - p e p t i d e an t ibod ie s . W e t h a n k Dr . M a r i a Gyongyossy - I s sa a n d Dr . D a n a D e v i n e for ass i s tance
a n d use fu l d iscuss ions . M.S. is the r e c i p i en t o f a M e d i - cal R e s e a r c h C o u n c i l ( M . R . C . ) of C a n a d a s t u d e n t s h i p award . S.L.P. is t he r e c i p i e n t o f an M . R . C . of C a n a d a
Schola rsh ip . Th i s work was s u p p o r t e d by o p e r a t i n g g ran t s f rom the B.C. a n d Y u k o n H e a r t a n d S t roke F o u n d a t i o n .
References
1 Seiss, W. (1989) Physiol. Rev. 69, 58-178. 2 Ferrell, J.E. and Martin, G.S. (1988) Mol. Cell Biol. 8, 3603-3610. 3 Golden, A. and Brugge, J.S. (1989) Proc. Natl. Acad. Sci. USA
86, 901-905. 4 Nakamura, S.-I. and Yamamura, H. (1989) J. Biol. Chem. 264,
7089-7091. 5 Pelech, S.L., Sanghera, J.S. and Daya-Makin, M. (1990) Biochem.
Cell Biol. 68, 1297-1330. 6 Salari, H., Duronio, V., Howard, S.L., Demos, M., Jones, K.,
Reany, A., Hudson, A.T. and Pelech, S.L. (1990) FEBS Lett. 263, 104-108.
7 Lerea, K.M., Tonks, N.K., Krebs, E.G., Fischer, E.H. and GIom- set, J.A. (1989) Biochemistry 28, 9286-9292.
8 Golden, A., Nemeth, S.P. and Brugge, J.S. (1986) Proc. Natl. Acad. Sci. USA 83, 852-856.
9 Feder, D. and Bishop, J.M. (1990)J. Biol. Chem. 265, 8205-8211. 10 Ferrell, J.E., Noble, J.A., Martin, G.S., Jacques, Y.V. and Bain-
ton, D.F. (1990) Oncogene 5, 1033-1036. 11 Horvath, A.R., Muszbek, L. and Kellie, S. (1992) EMBO J. 11,
855-861 12 Cichowski, K., McCormick, F. and Brugge, J.S. (1992) J. Biol.
Chem. 267, 5025-5028. 13 Presek, P., Reuter, C., Findik, D. and Bene, P. (1988) Biochim.
Biophys. Acta 969, 271-280. 14 Ieyasu, H., Takai, Y., Kaibuchi, K., Sawamura, M. and Nishizuka,
Y. (1982) Biochem. Biophys. Res. Commun. 108, 1701-1708. 15 Watson, S.P., Reep, B., McConnell, R.T. and Lapetina, E.G.
(1985) Biochem. J. 226, 831-837. 16 Shulka, S.D., Franklin, C.C. and Carter, M.G. (1987) Biochim.
Biophys. Acta 929, 134-141. 17 Berridge, M.J. (1987) Biochim. Biophys. Acta 907, 33-45. 18 Nishizuka, Y. (1988) Nature 334, 661-665. 19 Salari, H., Duronio, V., Howard, S., Demos, M. and Pelech, S.L.
(1990) Biochem. J. 267689-696. 20 Pelech, S.L., Charest, D.L., Howard, S.L., Paddon, H.B. and
Salari, H. (1990) Biochim. Biophys. Acta 1051, 100-107. 21 Watson, S.P. and Hambleton, S. (1989) Biochem. J. 258, 479-485. 22 Ferrell, J.E. and Martin, G.S. (1989) J. Biol. Chem. 264, 20723-
20729. 23 Kocher, M. and Clemetson, K.J. (1991) FEBS Lett. 291,363-366. 24 Rendu, F., Marche, P., Viret, J., MacLouf, J., Lebret, M., Tenza,
D., Caen, J. and Levy-Toledano, S. (1987) Biochemie 69, 305-313. 25 Samiei, M., Daya-Makin, M., Clark-Lewis, I. and Pelech, S.L.
(1991) J. Biol. Chem. 266, 14889-14892. 26 Tamura, T., Friis, F.R. and Bauer, H. (1984) FEBS Lett. 177,
151-156.
27 Morgan, D.O., Kaplan, J.M., Bishop, J.M. and Varmus, H.E. (1989) Cell 57, 775-786.
28 Shenoy, S., Choi, J.-K., Bagrodia, S., Copeland, T.D., Mailer, J.L. and Shalloway, D. (1989) Cell 57, 763-774.
29 Gupta, S.K., Gallego, C., Johnson, G.L. and Heasley, L.E. (1992) J. Biol. Chem. 267, 7987-7990,
30 Vik, T.A., Sweet, L.J. and Erikson, R.L. (1990) Proc. Natl. Acad. Sci. USA 87, 2685-2689.
31 Sturgill, T.W., Ray, L.B., Erikson, E. and Mailer, J.L. (1988) Nature 334, 715-718.
32 Chung, J., Pelech, S.L. and Blenis, J. (1991) Proc. Natl. Acad. Sci. USA 88, 4981-4985.
33 Mukhopadhyay, N.K., Price, D.J., Kyriakis, J.M., Pelech, S., Sanghera, J. and Avruch, J. (1992) J. Biol. Chem. 267, 3325-3335.
34 Ettehadieh, E., Sanghera, J.S., Pelech, S.L., Hess-Bienz, D., Watts, J., Shastri, N. and Aebersold, R. (1992) Science 255, 853-855.
35 Sturgill, T.W. and Wu, J. (1991) Biochim. Biophys. Acta 1092, 350-357.
36 Cobb, M.H., Boulton, T.G. and Robbins, D.J. (1991) Cell Regul. 2, 965-978.
37 Sanghera, J.S., Paddon, H.B. and Pelech, S.L. (1991) J. Biol. Chem. 266, 6700-6707.
38 Pinckard, R.N., Farr, R.S. and Hanahan, D.G. (1984) J. Im- munol. 123, 1847-1853.
39 Bradford, M.M. (1976) Anal. Biochem. 72, 420-428. 40 Laemmli, U.K. (1970) Nature 227, 680-684. 41 Rossomando, A.J., Sanghera, J.S., Marsden, L.A., Weber, M.J.,
Pelech, S.L. and Sturgill, T.W. (1991) J. Biol. Chem. 266, 20270- 20275.
42 Davis, P.D., Hill, C.H., Keech, E., Lawton, G., Nixon, J.S., Sedgwick, A.D., Wadsworth, J., Westmacott, D. and Wilkinson, S.E. (1989) FEBS Lett. 259, 61-63.
43 Grove, J.R., Banerjee, P., Balasubramanyam, Coffer, P.J., Price, D.J., Avruch, J. and Woodgett, J.R. (1991) Mol. Cell. Biol. 11, 5541-5550.
44 Reinhard, C., Thomas, G. and Kozma, S.C. (1992) Proc. Natl. Acad. Sci. USA 89, 4052-4056.
45 Pelech, S.L. and Sanghera, J.S. (1992) Trends Biochem. Sci. 17, 233-238.
46 Pelech, S.L., Samiei, M., Charest, D.L., Howard, S.L. and Salari, H. (1991) J. Biol. Chem. 266, 8696-8705.
47 Wu, J., Michel, H., Rossomando, A., Haystead, T.A.J., Sha- banowitz, J., Hunt, D.F. and Sturgill, T.W. (1992) Biochem. J. 285, 701-705.
48 Crews, C., Alessandrini, A. and Erikson, R.L. (1992) Science 258, 478-480.
49 Kosako, H., Gotoh, Y., Matsuda, S., Ishikawa, M. and Nishida, E. (1992) EMBO J. 11, 2903-2908.
50 Nakielny, S., Campbell, D.G. and Cohen, P. (1992) FEBS Lett. 308, 183-189.
51 Kyriakis, J.M., App, H., Zhang, X., Banerjee, P., Brautigan, D.L., Rapp, U.R. and Avruch, J. (1992) Nature 358, 417-420.
52 Dent, P., Haystead, T.A.J., Haser, W., Vincent, L.A., Roberts, T.M. and Sturgill, T.W. (1992) Science 257, 1404-1407.
53 Li, P., Wood, K., Maroon, H., Haser, W. and Roberts, T. (1991) Cell 64, 479-482.
54 Rapp, U.R. (1991) Oncogene 6, 495-500. 55 Sozeri, O., Vollmer, K., Liyanage, M., Frith, D., Kour, G., Mark
IlI, G.E., Stabel, S. (1992) Oncogene 7, 2259-2262. 56 Welham, M., Duronio, V., Sanghera, J.S., Pelech, S.L and
Schrader, J.W. (1992) J. Immunol. 149, 1683-1693. 57 Okuda, K., Sanghera, J.S., Pelcch, S.L., Kanakura, Y., Hallek,
M., Griffin, J.D. and Druker, BJ. (1992) Blood 79, 2880-2887.




















