
miR1 Overexpression Enhances Ca2+ Release and Promotes Cardiac Arrhythmogenesis by Targeting PP2A...
24
miR-1 Overexpression Enhances Ca 2 Release and Promotes Cardiac Arrhythmogenesis by Targeting PP2A Regulatory Subunit B56 and Causing CaMKII-Dependent Hyperphosphorylation of RyR2 Dmitry Terentyev, Andriy E. Belevych, Radmila Terentyeva, Mickey M. Martin, Geraldine E. Malana, Donald E. Kuhn, Maha Abdellatif, David S. Feldman, Terry S. Elton, Sandor Gyorke Abstract—MicroRNAs are small endogenous noncoding RNAs that regulate protein expression by hybridization to imprecise complementary sequences of target mRNAs. Changes in abundance of muscle-specific microRNA, miR-1, have been implicated in cardiac disease, including arrhythmia and heart failure. However, the specific molecular targets and cellular mechanisms involved in the action of miR-1 in the heart are only beginning to emerge. In this study we investigated the effects of increased expression of miR-1 on excitation– contraction coupling and Ca 2 cycling in rat ventricular myocytes using methods of electrophysiology, Ca 2 imaging and quantitative immunoblotting. Adenoviral- mediated overexpression of miR-1 in myocytes resulted in a marked increase in the amplitude of the inward Ca 2 current, flattening of Ca 2 transients voltage dependency, and enhanced frequency of spontaneous Ca 2 sparks while reducing the sarcoplasmic reticulum Ca 2 content as compared with control. In the presence of isoproterenol, rhythmically paced, miR-1– overexpressing myocytes exhibited spontaneous arrhythmogenic oscillations of intracellular Ca 2 , events that occurred rarely in control myocytes under the same conditions. The effects of miR-1 were completely reversed by the CaMKII inhibitor KN93. Although phosphorylation of phospholamban was not altered, miR-1 overexpression increased phosphorylation of the ryanodine receptor (RyR2) at S2814 (CaMKII) but not at S2808 (protein kinase A). Overexpression of miR-1 was accompanied by a selective decrease in expression of the protein phosphatase PP2A regulatory subunit B56 involved in PP2A targeting to specialized subcellular domains. We conclude that miR-1 enhances cardiac excitation– contraction coupling by selectively increasing phosphorylation of the L-type and RyR2 channels via disrupting localization of PP2A activity to these channels. (Circ Res. 2009;104:00-00.) Key Words: ryanodine receptor miR-1 CaMKII PP2A arrhythmia C ardiac contractility relies on release of Ca 2 from the sarcoplasmic reticulum (SR) and alterations in intracel- lular Ca 2 cycling have been implicated in different cardiac diseases, including arrhythmia and heart failure. Normally, SR Ca 2 release is activated by Ca 2 that enters the cell through voltage-dependent Ca 2 channels of the sarcolemma during the plateau phase of the cardiac action potential. This process, known as Ca 2 -induced Ca 2 release (CICR), 1 involves the ryanodine receptor (RyR2) channels on the SR and is essential for activation of contractile filaments during myocardial contraction. 2 Relaxation occurs when Ca 2 re- leased to the cytosol is resequestered to the SR by the phospholamban (PLB)-controlled SR Ca 2 ATPase (SERCA). Cardiac contractility is modulated by reversible phosphorylation of the components of SR Ca 2 release machinery, including the L-type Ca 2 channel (dihydropyri- dine receptor [DHPR]), RyR2, and PLB, by protein kinase (PK)A, 2,3 Ca 2 /calmodulin-dependent protein kinase (CaMKII), 4,5 and phosphatases PP1 and PP2A. 6–8 Although, in general, both PKA and CaMKII potentiate SR Ca 2 release and enhance contractility, 2 the underlying mechanisms of these effects and, in particular, the role of RyR2 phosphory- lation/dephosphorylation remain highly controversial. 3,9 –14 MicroRNAs are recently discovered molecules consisting of 22 nucleotides that regulate gene expression by anneal- ing to target messenger RNAs inhibiting translation or pro- moting mRNA degradation. 15 Of the 600 microRNAs identified in vertebrates, several, including miR-1, are mus- cle-specific. 16 –18 miR-1 has been shown to be involved in cardiac development and apoptosis 17,18 and is reportedly upregulated in hearts from individuals with coronary artery disease 19 and heart failure. 20 Several targets for miR-1 have Original received June 16, 2008; revision received December 8, 2008; accepted December 30, 2008. From the Davis Heart and Lung Research Institute (D.T., A.E.B., R.T., M.M.M., G.E.M., D.E.K., D.S.F., T.S.E., S.G.), Ohio State University, Columbus; and Cardiovascular Research Institute (M.A.), Department of Cell Biology and Molecular Medicine, University of Medicine and Dentistry of New Jersey, Newark. Correspondence to Dmitry Terentyev, PhD, Department of Physiology and Cell Biology, Ohio State University, DHLRI 501, 473 West 12th Ave, Columbus, OH 43210. USA. Phone: 1 614 2923944. Fax:1 614 247 7799. E-mail [email protected] © 2009 American Heart Association, Inc. Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/CIRCRESAHA.108.181651 1 by guest on September 2, 2016 http://circres.ahajournals.org/ Downloaded from by guest on September 2, 2016 http://circres.ahajournals.org/ Downloaded from by guest on September 2, 2016 http://circres.ahajournals.org/ Downloaded from by guest on September 2, 2016 http://circres.ahajournals.org/ Downloaded from by guest on September 2, 2016 http://circres.ahajournals.org/ Downloaded from by guest on September 2, 2016 http://circres.ahajournals.org/ Downloaded from by guest on September 2, 2016 http://circres.ahajournals.org/ Downloaded from by guest on September 2, 2016 http://circres.ahajournals.org/ Downloaded from by guest on September 2, 2016 http://circres.ahajournals.org/ Downloaded from by guest on September 2, 2016 http://circres.ahajournals.org/ Downloaded from by guest on September 2, 2016 http://circres.ahajournals.org/ Downloaded from by guest on September 2, 2016 http://circres.ahajournals.org/ Downloaded from by guest on September 2, 2016 http://circres.ahajournals.org/ Downloaded from by guest on September 2, 2016 http://circres.ahajournals.org/ Downloaded from by guest on September 2, 2016 http://circres.ahajournals.org/ Downloaded from by guest on September 2, 2016 http://circres.ahajournals.org/ Downloaded from by guest on September 2, 2016 http://circres.ahajournals.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of miR1 Overexpression Enhances Ca2+ Release and Promotes Cardiac Arrhythmogenesis by Targeting PP2A...

miR-1 Overexpression Enhances Ca2� Release and PromotesCardiac Arrhythmogenesis by Targeting PP2A Regulatory
Subunit B56� and Causing CaMKII-DependentHyperphosphorylation of RyR2
Dmitry Terentyev, Andriy E. Belevych, Radmila Terentyeva, Mickey M. Martin, Geraldine E. Malana,Donald E. Kuhn, Maha Abdellatif, David S. Feldman, Terry S. Elton, Sandor Gyorke
Abstract—MicroRNAs are small endogenous noncoding RNAs that regulate protein expression by hybridization toimprecise complementary sequences of target mRNAs. Changes in abundance of muscle-specific microRNA, miR-1,have been implicated in cardiac disease, including arrhythmia and heart failure. However, the specific molecular targetsand cellular mechanisms involved in the action of miR-1 in the heart are only beginning to emerge. In this study weinvestigated the effects of increased expression of miR-1 on excitation–contraction coupling and Ca2� cycling in ratventricular myocytes using methods of electrophysiology, Ca2� imaging and quantitative immunoblotting. Adenoviral-mediated overexpression of miR-1 in myocytes resulted in a marked increase in the amplitude of the inward Ca2�
current, flattening of Ca2� transients voltage dependency, and enhanced frequency of spontaneous Ca2� sparks whilereducing the sarcoplasmic reticulum Ca2� content as compared with control. In the presence of isoproterenol,rhythmically paced, miR-1–overexpressing myocytes exhibited spontaneous arrhythmogenic oscillations of intracellularCa2�, events that occurred rarely in control myocytes under the same conditions. The effects of miR-1 were completelyreversed by the CaMKII inhibitor KN93. Although phosphorylation of phospholamban was not altered, miR-1overexpression increased phosphorylation of the ryanodine receptor (RyR2) at S2814 (CaMKII) but not at S2808(protein kinase A). Overexpression of miR-1 was accompanied by a selective decrease in expression of the proteinphosphatase PP2A regulatory subunit B56� involved in PP2A targeting to specialized subcellular domains. Weconclude that miR-1 enhances cardiac excitation–contraction coupling by selectively increasing phosphorylation of theL-type and RyR2 channels via disrupting localization of PP2A activity to these channels. (Circ Res. 2009;104:00-00.)
Key Words: ryanodine receptor � miR-1 CaMKII � PP2A � arrhythmia
Cardiac contractility relies on release of Ca2� from thesarcoplasmic reticulum (SR) and alterations in intracel-
lular Ca2� cycling have been implicated in different cardiacdiseases, including arrhythmia and heart failure. Normally,SR Ca2� release is activated by Ca2� that enters the cellthrough voltage-dependent Ca2� channels of the sarcolemmaduring the plateau phase of the cardiac action potential. Thisprocess, known as Ca2�-induced Ca2� release (CICR),1
involves the ryanodine receptor (RyR2) channels on the SRand is essential for activation of contractile filaments duringmyocardial contraction.2 Relaxation occurs when Ca2� re-leased to the cytosol is resequestered to the SR by thephospholamban (PLB)-controlled SR Ca2� ATPase(SERCA). Cardiac contractility is modulated by reversiblephosphorylation of the components of SR Ca2� releasemachinery, including the L-type Ca2� channel (dihydropyri-
dine receptor [DHPR]), RyR2, and PLB, by protein kinase(PK)A,2,3 Ca2�/calmodulin-dependent protein kinase(CaMKII),4,5 and phosphatases PP1 and PP2A.6–8 Although,in general, both PKA and CaMKII potentiate SR Ca2� releaseand enhance contractility,2 the underlying mechanisms ofthese effects and, in particular, the role of RyR2 phosphory-lation/dephosphorylation remain highly controversial.3,9–14
MicroRNAs are recently discovered molecules consistingof �22 nucleotides that regulate gene expression by anneal-ing to target messenger RNAs inhibiting translation or pro-moting mRNA degradation.15 Of the �600 microRNAsidentified in vertebrates, several, including miR-1, are mus-cle-specific.16–18 miR-1 has been shown to be involved incardiac development and apoptosis17,18 and is reportedlyupregulated in hearts from individuals with coronary arterydisease19 and heart failure.20 Several targets for miR-1 have
Original received June 16, 2008; revision received December 8, 2008; accepted December 30, 2008.From the Davis Heart and Lung Research Institute (D.T., A.E.B., R.T., M.M.M., G.E.M., D.E.K., D.S.F., T.S.E., S.G.), Ohio State University,
Columbus; and Cardiovascular Research Institute (M.A.), Department of Cell Biology and Molecular Medicine, University of Medicine and Dentistryof New Jersey, Newark.
Correspondence to Dmitry Terentyev, PhD, Department of Physiology and Cell Biology, Ohio State University, DHLRI 501, 473 West 12th Ave,Columbus, OH 43210. USA. Phone: �1 614 2923944. Fax:�1 614 247 7799. E-mail [email protected]
© 2009 American Heart Association, Inc.
Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/CIRCRESAHA.108.181651
1
by guest on September 2, 2016
http://circres.ahajournals.org/D
ownloaded from
by guest on Septem
ber 2, 2016http://circres.ahajournals.org/
Dow
nloaded from
by guest on September 2, 2016
http://circres.ahajournals.org/D
ownloaded from
by guest on Septem
ber 2, 2016http://circres.ahajournals.org/
Dow
nloaded from
by guest on September 2, 2016
http://circres.ahajournals.org/D
ownloaded from
by guest on Septem
ber 2, 2016http://circres.ahajournals.org/
Dow
nloaded from
by guest on September 2, 2016
http://circres.ahajournals.org/D
ownloaded from
by guest on Septem
ber 2, 2016http://circres.ahajournals.org/
Dow
nloaded from
by guest on September 2, 2016
http://circres.ahajournals.org/D
ownloaded from
by guest on Septem
ber 2, 2016http://circres.ahajournals.org/
Dow
nloaded from
by guest on September 2, 2016
http://circres.ahajournals.org/D
ownloaded from
by guest on Septem
ber 2, 2016http://circres.ahajournals.org/
Dow
nloaded from
by guest on September 2, 2016
http://circres.ahajournals.org/D
ownloaded from
by guest on Septem
ber 2, 2016http://circres.ahajournals.org/
Dow
nloaded from
by guest on September 2, 2016
http://circres.ahajournals.org/D
ownloaded from
by guest on Septem
ber 2, 2016http://circres.ahajournals.org/
Dow
nloaded from
by guest on September 2, 2016
http://circres.ahajournals.org/D
ownloaded from

been identified in the heart, including connexin 43 and Kir2.1and miR-1–mediated reductions in expression of these pro-teins has been implicated in arrhythmogenesis.19 However,considering the inherent capacity of miRNAs to target abroad range of proteins, the link between miR-1 and heartfailure and sudden cardiac death is far from being clear andmore miR-1 targets involved in these disease states are likelyremain to be identified. In the present study, we investigatedthe effects of increased expression of miR-1 on excitation–contraction (EC) coupling and Ca2� cycling in rat ventricularmyocytes by using methods of cellular electrophysiology andCa2� imaging. Our results identified a new potentially im-portant target for miR-1 in the heart, namely the PP2Aregulatory subunit B56�. Through translational inhibition ofthis mRNA target, miR-1 causes CaMKII-dependent hyper-phosphorylation of RyR2, enhances RyR2 activity, and pro-motes arrhythmogenic SR Ca2� release.
Materials and MethodsThe cellular and subcellular effects of adenovirally mediated miR-1overexpression were studied in isolated adult rat ventricular myo-cytes maintained in culture for 36 to 48 hours. Cytosolic Ca2�
changes were monitored using confocal microscopy, and whole cellcurrents and membrane potential were recorded with the patch-clamptechnique. Changes in levels of RNA, protein expression and proteinphosphorylation were studied using standard approaches.
An expanded Materials and Methods section can be found in theonline data supplement, available at http://circres.ahajournals.org.
ResultsmiR-1 Stimulates ICa and SR Ca2� Release inCardiac MyocytesMyocytes were infected with either an adenoviral constructfor expression of miR-1 (Ad-miR-1)21 or a construct contain-ing a nontranslatable DNA segment that served as control(Ad-control). As determined by RT PCR, miR-1 abundancewas increased �2-fold in myocytes infected with Ad-miR-1compared with control cells (Figure I in the online datasupplement). First, we investigated the effects of overexpres-sion of miR-1 on EC coupling in voltage clamped cardiacmyocytes. Inward Ca2� currents (ICa) and intracellular Ca2�
transients were simultaneously measured in cardiac myocytesdepolarized to membrane potentials in the range between�40 and 60 mV (Figure 1). Overexpression of miR-1 resultedin a significant increase in the amplitude of ICa at membranepotentials of �40 to 20 mV (Figure 1B). Additionally, thevoltage dependence of ICa (I-V curve) was shifted to the left4 mV in miR-1 myocytes (supplemental Figure II). Despiteincreased ICa, the trigger for SR Ca2� release, maximumCa2� transient amplitude was not changed by miR-1 overex-pression (Figure 1A and 1B). The failure of enhanced ICa toincrease maximum SR Ca2� release is attributable to reducedSR Ca2� content limiting Ca2� release in miR-1 cells (seebelow). At the same time, the voltage dependence of Ca2�
transients was markedly broadened and flattened comparedwith control because of increased Ca2� transient amplitude atsmall and large depolarizations (Figure 1B). Whereas theincrease of Ca2� transient amplitude at small depolarizingpulses can be accounted for by increased ICa, the increase ofCa2� transients at large depolarizing steps occurred without
an increase in ICa and is indicative of enhanced responsive-ness of Ca2� release channels to ICa (ie, enhanced ECcoupling gain).
The effects of miR-1 overexpression on the SR Ca2�
release were further studied by measuring spontaneous localCa2� release events, Ca2� sparks, in intact quiescent myo-cytes loaded with Fluo-3 AM (Figure 2). Spark frequency inmiR-1–overexpressing cells was significantly higher than incontrol cells (4.39�0.54 and 2.88�0.41 100 �m�1 sec�1,respectively; Figure 2A and 2C). We assessed the Ca2�-loading state of the SR in miR-1 versus control myocytes byapplication of caffeine. Judging from the amplitude ofcaffeine-induced Ca2� transients, SR Ca2� content wasreduced to 75% of control in miR-1 myocytes (Figure 2B and2D). The lowered SR Ca2� content is a likely result ofincreased spark-mediated SR Ca2� leak, and it provides anexplanation for the lack of potentiation of maximum Ca2�
Figure 1. miR-1 overexpression stimulates ICa and SR Ca2�
release in cardiac myocytes. A, Representative recordings ofCa2� transients and ICa evoked by depolarizing steps from aholding potential of �50 to �30, 0, and �30 mV in a controlmyocyte and a myocyte overexpressing miR-1. The voltagesteps were applied at intervals of 5 seconds. B and C, Voltagedependencies of ICa (bottom) and Ca2� transient amplitude (top)in control (blue) and miR-1–overexpressing (red) myocytes atbaseline conditions (B) and on application of 100 nmol/L ISO(C). *P�0.05, significantly different vs control (1-way ANOVA);N�5 to 7.
2 Circulation Research February 27, 2009
by guest on September 2, 2016
http://circres.ahajournals.org/D
ownloaded from

release by increased ICa in miR-1–overexpressing myocytes.Therefore, miR-1 caused profound changes in EC coupling,including increased ICa, enhanced RyR2 channel functionalactivity, and a reduction in the SR Ca2� content.
The Stimulatory Effects of miR-1 in ICa and SRCa2� Release Are Caused by Phosphorylation ofDHPRs and Ryanodine ReceptorsThe effects of miR-1 overexpression on ICa in cardiomyo-cytes, namely the increased ICa amplitude and left-ward shiftof ICa voltage dependence (Figure 1B and 1C and supple-mental Figure II), are similar to the effects of �-adrenergicstimulation. To examine whether the action of miR-1 involvesthe same mechanisms that mediate the response to�-adrenergic stimulation, we investigated the effects of the�-adrenergic agonist isoproterenol (ISO) on ICa and Ca2�
transients in miR-1 versus control myocytes. As shown inFigure 1B and 1C, ISO increased ICa amplitude �2-fold incontrol but was virtually ineffective in miR-1 myocytes.Similarly, whereas ISO resulted in a �2-fold increase inCa2� transient amplitude in control myocytes, ISO failed tocause significant changes in the amplitude and voltagedependence of Ca2� transients in miR-1 myocytes. Theseresults could be explained by miR-1 and ISO acting through
the same intracellular signaling mechanisms resulting inphosphorylation of target proteins including DHPR andRyR2.
RyR2 is phosphorylated at least at 3 different sites: atS2808 by PKA22 and possibly CaMKII23; at S2814 byCaMKII,22 and at S2030 by PKA.11 To test directly thehypothesis that miR-1 overexpression results in increasedphosphorylation of RyR2 at these sites, we quantified RyR2phosphorylation using phospho-specific antibodies. miR-1overexpression caused a marked increase in phosphorylationat the CaMKII site S2814 while leaving phosphorylation ofthe PKA site S2808 unaltered. The involvement of CaMKIIin RyR2 phosphorylation at S2814 was further confirmed bythe ability of KN93, a CaMKII inhibitor, to prevent phos-phorylation at this site in miR-1 myocytes (Figure 3A through3C). Interestingly, ryanodine receptors were not detectiblyphosphorylated at S2030 either in the control or miR-1 groupseven when the myocytes were exposed to 100 nmol/L ISO(supplemental Figure III). miR-1 did not change phosphory-lation of PLB at its PKA or CaMKII sites, S16 and T17,respectively (Figure 3A and 3D), but increased phosphoryla-tion of DHPRs (Figure 3A and 3E).
To test the involvement of CaMKII in the functionaleffects of miR-1 on SR Ca2� release in myocytes, weexamined the impact of KN93 on miR-1–dependent changesin Ca2� cycling. KN93 reversed the effects of miR-1 onfrequency of Ca2� sparks and on the SR Ca2� content ofmyocytes (Figure 2), as well as on Ca2� currents and Ca2�
transients in voltage-clamped cells (supplemental Figure IV).Collectively, these results suggest that miR-1 overexpressionresults in augmented phosphorylation of DHPR and RyR2 byCaMKII, whereas the phosphorylation of PLB is unaltered.
miR-1 Inhibits Expression of the PP2A RegulatorySubunit B56�The phosphorylation-dependent effects of miR-1 on Ca2�
handling point to the possibility that miR-1 targets compo-nents of the phosphorylation–dephosphorylation system inmyocytes. Bioinformatic sequence analysis revealed that thePP2A regulatory subunit B56� is a potential target of miR-1because it harbors 7-bp-long complimentary seed sequence inits 3� untranslated region (UTR) (Figure 4A). Binding ofmiR-1 to B56� encoding RNA was confirmed by a luciferasereporter assay (Figure 4B). Moreover, the miR-1 targeting ofB56� in cardiac myocytes was confirmed by quantitativeimmunoblot analysis using an anti-B56� antibody (Figure 5Band 5C). Importantly, miR-1 overexpression did not appre-ciably change expression levels of a number of relevant Ca2�
and phosphorylation handling proteins, including DHPR,RyR2, SR Ca ATPase (SERCA2a), PLB, sodium/calciumexchanger (NCX1), calsequestrin (CASQ2), catalytic sub-units of PP1 and PP2A, and CaMKII, as well as phosphodi-esterases 3A and 4D. As shown in Figure 5A, expression ofall of these proteins was similar in miR-1 versus controlmyocytes. Of note, miR-1 overexpression resulted in a de-crease in expression of PKA � catalytic subunit, which is alsoa predicted target of miR-1. However, downregulation of thiskinase cannot account for the increase in phosphorylation oftarget proteins by miR-1 in our experiments.
Figure 2. miR-1 overexpression increases Ca2� spark frequencyand decreases SR Ca2� content in intact cardiac myocytes. Aand B, Representative line-scan images of Ca2� sparks (A) andtime-dependent profiles of global Ca2� releases induced byapplication of 10 mmol/L caffeine (B) recorded in a control myo-cyte and a myocyte overexpressing miR-1 at reference condi-tions and after treatment with the CaMKII inhibitor KN93(1 �mol/L). C, Averaged spark frequency for the 3 myocytegroups; n�33, 53, and 45 for control, miR-1, and miR-1 treatedwith KN93, respectively. D, Averaged amplitude of caffeine-induced Ca2� transients; n�14, 14, and 8 for control, miR-1,and miR-1 treated with KN93, respectively. *P�0.05, signifi-cantly different vs control (1-way ANOVA).
Terentyev et al miR-1 Promotes Arrhythmia by Enhancing Ca Release 3
by guest on September 2, 2016
http://circres.ahajournals.org/D
ownloaded from
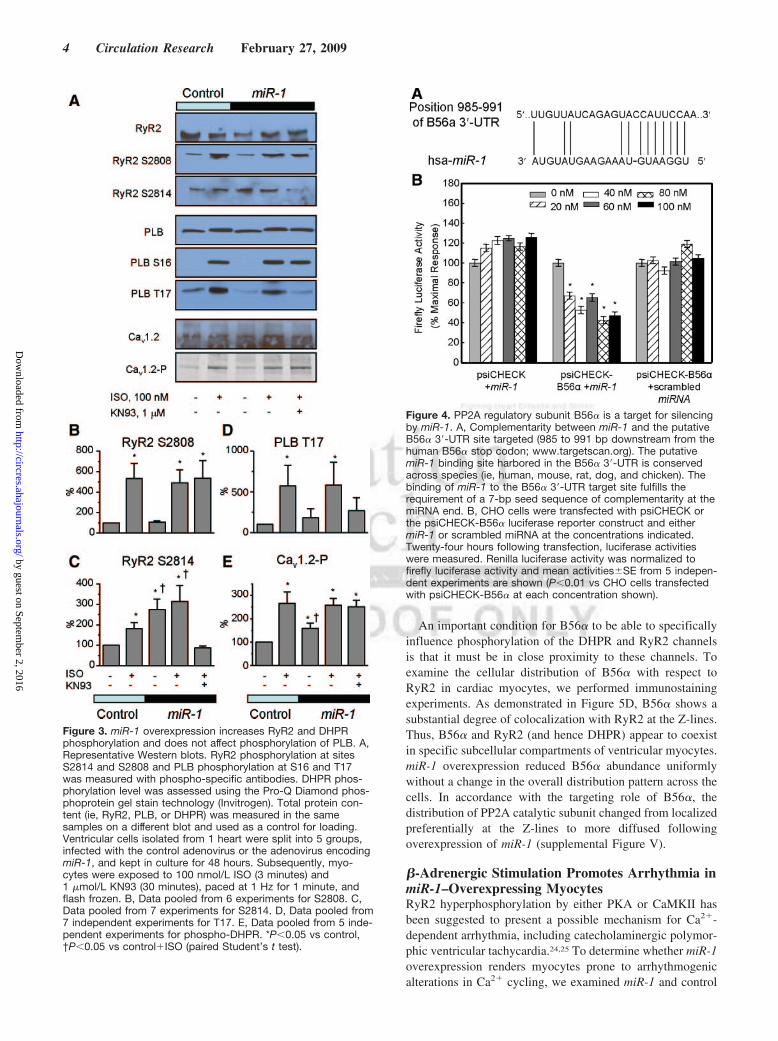
An important condition for B56� to be able to specificallyinfluence phosphorylation of the DHPR and RyR2 channelsis that it must be in close proximity to these channels. Toexamine the cellular distribution of B56� with respect toRyR2 in cardiac myocytes, we performed immunostainingexperiments. As demonstrated in Figure 5D, B56� shows asubstantial degree of colocalization with RyR2 at the Z-lines.Thus, B56� and RyR2 (and hence DHPR) appear to coexistin specific subcellular compartments of ventricular myocytes.miR-1 overexpression reduced B56� abundance uniformlywithout a change in the overall distribution pattern across thecells. In accordance with the targeting role of B56�, thedistribution of PP2A catalytic subunit changed from localizedpreferentially at the Z-lines to more diffused followingoverexpression of miR-1 (supplemental Figure V).
�-Adrenergic Stimulation Promotes Arrhythmia inmiR-1–Overexpressing MyocytesRyR2 hyperphosphorylation by either PKA or CaMKII hasbeen suggested to present a possible mechanism for Ca2�-dependent arrhythmia, including catecholaminergic polymor-phic ventricular tachycardia.24,25 To determine whether miR-1overexpression renders myocytes prone to arrhythmogenicalterations in Ca2� cycling, we examined miR-1 and control
Figure 3. miR-1 overexpression increases RyR2 and DHPRphosphorylation and does not affect phosphorylation of PLB. A,Representative Western blots. RyR2 phosphorylation at sitesS2814 and S2808 and PLB phosphorylation at S16 and T17was measured with phospho-specific antibodies. DHPR phos-phorylation level was assessed using the Pro-Q Diamond phos-phoprotein gel stain technology (Invitrogen). Total protein con-tent (ie, RyR2, PLB, or DHPR) was measured in the samesamples on a different blot and used as a control for loading.Ventricular cells isolated from 1 heart were split into 5 groups,infected with the control adenovirus or the adenovirus encodingmiR-1, and kept in culture for 48 hours. Subsequently, myo-cytes were exposed to 100 nmol/L ISO (3 minutes) and1 �mol/L KN93 (30 minutes), paced at 1 Hz for 1 minute, andflash frozen. B, Data pooled from 6 experiments for S2808. C,Data pooled from 7 experiments for S2814. D, Data pooled from7 independent experiments for T17. E, Data pooled from 5 inde-pendent experiments for phospho-DHPR. *P�0.05 vs control,†P�0.05 vs control�ISO (paired Student’s t test).
Figure 4. PP2A regulatory subunit B56� is a target for silencingby miR-1. A, Complementarity between miR-1 and the putativeB56� 3�-UTR site targeted (985 to 991 bp downstream from thehuman B56� stop codon; www.targetscan.org). The putativemiR-1 binding site harbored in the B56� 3�-UTR is conservedacross species (ie, human, mouse, rat, dog, and chicken). Thebinding of miR-1 to the B56� 3�-UTR target site fulfills therequirement of a 7-bp seed sequence of complementarity at themiRNA end. B, CHO cells were transfected with psiCHECK orthe psiCHECK-B56� luciferase reporter construct and eithermiR-1 or scrambled miRNA at the concentrations indicated.Twenty-four hours following transfection, luciferase activitieswere measured. Renilla luciferase activity was normalized tofirefly luciferase activity and mean activities�SE from 5 indepen-dent experiments are shown (P�0.01 vs CHO cells transfectedwith psiCHECK-B56� at each concentration shown).
4 Circulation Research February 27, 2009
by guest on September 2, 2016
http://circres.ahajournals.org/D
ownloaded from
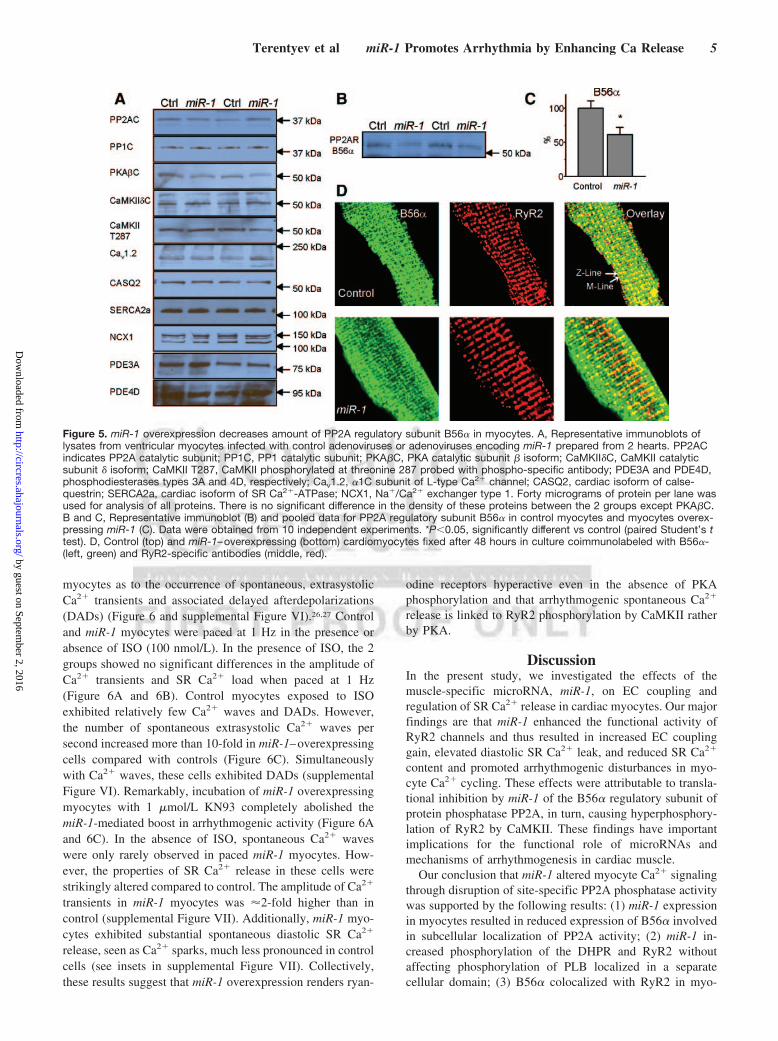
myocytes as to the occurrence of spontaneous, extrasystolicCa2� transients and associated delayed afterdepolarizations(DADs) (Figure 6 and supplemental Figure VI).26,27 Controland miR-1 myocytes were paced at 1 Hz in the presence orabsence of ISO (100 nmol/L). In the presence of ISO, the 2groups showed no significant differences in the amplitude ofCa2� transients and SR Ca2� load when paced at 1 Hz(Figure 6A and 6B). Control myocytes exposed to ISOexhibited relatively few Ca2� waves and DADs. However,the number of spontaneous extrasystolic Ca2� waves persecond increased more than 10-fold in miR-1–overexpressingcells compared with controls (Figure 6C). Simultaneouslywith Ca2� waves, these cells exhibited DADs (supplementalFigure VI). Remarkably, incubation of miR-1 overexpressingmyocytes with 1 �mol/L KN93 completely abolished themiR-1-mediated boost in arrhythmogenic activity (Figure 6Aand 6C). In the absence of ISO, spontaneous Ca2� waveswere only rarely observed in paced miR-1 myocytes. How-ever, the properties of SR Ca2� release in these cells werestrikingly altered compared to control. The amplitude of Ca2�
transients in miR-1 myocytes was �2-fold higher than incontrol (supplemental Figure VII). Additionally, miR-1 myo-cytes exhibited substantial spontaneous diastolic SR Ca2�
release, seen as Ca2� sparks, much less pronounced in controlcells (see insets in supplemental Figure VII). Collectively,these results suggest that miR-1 overexpression renders ryan-
odine receptors hyperactive even in the absence of PKAphosphorylation and that arrhythmogenic spontaneous Ca2�
release is linked to RyR2 phosphorylation by CaMKII ratherby PKA.
DiscussionIn the present study, we investigated the effects of themuscle-specific microRNA, miR-1, on EC coupling andregulation of SR Ca2� release in cardiac myocytes. Our majorfindings are that miR-1 enhanced the functional activity ofRyR2 channels and thus resulted in increased EC couplinggain, elevated diastolic SR Ca2� leak, and reduced SR Ca2�
content and promoted arrhythmogenic disturbances in myo-cyte Ca2� cycling. These effects were attributable to transla-tional inhibition by miR-1 of the B56� regulatory subunit ofprotein phosphatase PP2A, in turn, causing hyperphosphory-lation of RyR2 by CaMKII. These findings have importantimplications for the functional role of microRNAs andmechanisms of arrhythmogenesis in cardiac muscle.
Our conclusion that miR-1 altered myocyte Ca2� signalingthrough disruption of site-specific PP2A phosphatase activitywas supported by the following results: (1) miR-1 expressionin myocytes resulted in reduced expression of B56� involvedin subcellular localization of PP2A activity; (2) miR-1 in-creased phosphorylation of the DHPR and RyR2 withoutaffecting phosphorylation of PLB localized in a separatecellular domain; (3) B56� colocalized with RyR2 in myo-
Figure 5. miR-1 overexpression decreases amount of PP2A regulatory subunit B56� in myocytes. A, Representative immunoblots oflysates from ventricular myocytes infected with control adenoviruses or adenoviruses encoding miR-1 prepared from 2 hearts. PP2ACindicates PP2A catalytic subunit; PP1C, PP1 catalytic subunit; PKA�C, PKA catalytic subunit � isoform; CaMKII�C, CaMKII catalyticsubunit � isoform; CaMKII T287, CaMKII phosphorylated at threonine 287 probed with phospho-specific antibody; PDE3A and PDE4D,phosphodiesterases types 3A and 4D, respectively; Cav1.2, �1C subunit of L-type Ca2� channel; CASQ2, cardiac isoform of calse-questrin; SERCA2a, cardiac isoform of SR Ca2�-ATPase; NCX1, Na�/Ca2� exchanger type 1. Forty micrograms of protein per lane wasused for analysis of all proteins. There is no significant difference in the density of these proteins between the 2 groups except PKA�C.B and C, Representative immunoblot (B) and pooled data for PP2A regulatory subunit B56� in control myocytes and myocytes overex-pressing miR-1 (C). Data were obtained from 10 independent experiments. *P�0.05, significantly different vs control (paired Student’s ttest). D, Control (top) and miR-1–overexpressing (bottom) cardiomyocytes fixed after 48 hours in culture coimmunolabeled with B56�-(left, green) and RyR2-specific antibodies (middle, red).
Terentyev et al miR-1 Promotes Arrhythmia by Enhancing Ca Release 5
by guest on September 2, 2016
http://circres.ahajournals.org/D
ownloaded from

cytes; and (4) The functional effects of miR-1 on EC couplingand intracellular Ca2� cycling were reversed by the inhibitorof CaMKII KN93. The PP2A catalytic subunit has beenshown to complex with DHPR7 and RyR28 and is critical todephosphorylation of these proteins following their phos-phorylation by PKA and/or CaMKII.7,8 Consistent with therole of B56� in conveying PP2A phosphatase activity toDHPR, B56� has been shown to coimmunoprecipitate withthe � subunit of the cardiac L-type Ca2� channel (Cav1.2�).28
As for RyR2, it has been previously reported that the PP2Acatalytic subunit associates with the RyR2 complex throughthe regulatory subunit PR130, which binds directly to RyR2by a leucine zipper motif.29 However, because PR130 doesnot present a target for miR-1, it is unlikely to play a role inthe effects of miR-1 in our experiments. In support of theinvolvement of B56�, it has been shown to interact with theanchoring protein, ankyrin-B,30 which in turn binds toRyR2,31 thereby linking PP2A activity to the RyR2. Of note,myocytes from genetically altered mice lacking ankyrin-Bexhibit increased propensity for spontaneous Ca2� releaseand DADs when challenged with ISO similar to the effects ofmiR-1 in our study (see below for discussion). Moreover,human mutations in ankyrin-B have been linked to catechol-aminergic polymorphic ventricular tachycardia an arrhythmic
disorder associated with abnormal Ca2� handling andDADs.32,33 Based on our findings, disruption of PP2A activitymay contribute to, or account for, those abnormalities.
The RyR2 is phosphorylated by PKA and CaMKII at anumber of different sites.11,22,23 In our study, the alterations ofRyR2 function and SR Ca release in miR-1–overexpressingmyocytes appear to be caused by CaMKII rather than PKAfor the following reasons. First, the potentiation of RyR2function was associated with increased phosphorylation ofRyR2 at the CaMKII site S2814 but not at the PKA siteS2808, as determined by phospho-specific antibodies directedto these sites. Furthermore, the effects of miR-1 could beprevented by the CaMKII inhibitor KN93. The observedchanges in myocyte Ca2� handling, including increased ECcoupling gain, reduced SR Ca2� content, and increased SRRyR2-mediated Ca2� leak are similar to those observed byothers in cells overexpressing CaMKII25 and are consistentwith miR-1 effects being mediated by CaMKII. Thus, collec-tively, these results suggest that miR-1 influences myocyteCa2� signaling by regulating the spatial distribution ofphosphatase PP2A activity and hence promoting RyR2 phos-phorylation by CaMKII.
Additionally, phosphorylation of the L-type Ca2� channel(�1C and/or � subunits of DHPR) by PKA, PKC, andCaMKII is known to occur at a number of sites and causepotentiation of the L-type Ca2� current.4,7,28,34 At the presenttime, we do not know which of the specific phosphorylationpathways and sites are involved in modulation of ICa bymiR-1 in our study. In principle, by targeting PP2A activity inthe vicinity of the L-type Ca2� channels, miR-1 would beexpected to enhance phosphorylation at all sites. The failureof ISO to further stimulate the enlarged ICa in miR-1 cells(Figure 1) supports the possibility that PKA plays a role inmediating the effects of miR-1 on ICa. At the same time, thefact that CaMKII inhibition attenuates the miR-1-inducedincrease in ICa (supplemental Figure IV) points to the possi-bility that CaMKII might be also involved. Future studiesusing genetic mouse models with targeted mutations of thephosphorylation sites on the L-type Ca2� channel may help toresolve this issue.
An important finding of this work is that miR-1 resulted inprofound disturbances in myocyte Ca2� cycling manifestedas extrasystolic spontaneous Ca2� releases. SpontaneousCa2� releases are arrhythmogenic as they results in DADsand early afterdepolarizations.26,27 DADs and early afterde-polarizations in turn present triggering events for arrhythmia.Interestingly, these abnormal Ca2� releases typically oc-curred after stimulation of the miR-1–overexpressing cellswith ISO and were only rarely observed in ISO-treatedcontrol myocytes or untreated miR-1–overexpressing cells.Correlating these functional effects with the state of RyR2phosphorylation at S2808 and S2814 revealed that disruptedCa2� cycling was associated with maximum phosphorylationat S2814 but not necessarily at S2808, and again thisabnormal Ca2� cycling was completely preventable by inhi-bition of CaMKII. The increased occurrence of global spon-taneous Ca2� waves and DADs in miR-1 cells in the presenceof ISO is likely to be attributable to stimulation of SERCA-mediated SR Ca2� uptake rather than caused by additional
Figure 6. miR-1 overexpression increases arrhythmogenicpotential of myocytes undergoing repetitive stimulation in thepresence of ISO. A, Representative line-scan images and corre-sponding time-dependent profiles of Fluo-3 fluorescence in ratventricular myocytes infected with Ad-control and Ad-miR-1adenoviruses. Ca transients were evoked by electric field stimu-lation at 1 Hz in the presence of 100 nmol/L ISO. miR-1–over-expressing myocytes displayed increased incidence of arrhyth-mogenic spontaneous diastolic Ca2� waves. Incubation of miR-1–overexpressing myocytes with KN93 (1 �mol/L for 30minutes) restored normal rhythmic activity. B, Summarized datafor Ca2� transient amplitude and SR Ca2� content for control,miR-1–overexpressing myocytes and miR-1 myocytes treatedwith KN93 in the presence of 100 nmol/L ISO. The myocyteswere field-stimulated at 0.5 and 1 Hz for 1 minute each thenexposed 10 mmol/L caffeine. C, Averaged number of spontane-ous Ca2� waves per second in field-stimulated myocytes at 1Hz in the presence of 100 nmol/L ISO. *P�0.05, significantlydifferent (1-way ANOVA); number of cells studied, n�8 to 16.
6 Circulation Research February 27, 2009
by guest on September 2, 2016
http://circres.ahajournals.org/D
ownloaded from

effects on CAMKII-phosphorylated ryanodine receptors. Thestimulatory effects of CaMKII on SR Ca2� release and theinvolvement of CaMKII in the proarrhythmic effects ofmiR-1 are consistent with previous studies which showed thatCaMKII activation is involved in arrhythmia induction invarious pathological settings including cardiac hypertrophyand heart failure.25,35
Previously, we reported that exogenous phosphatases, in-cluding PP2A, elevate cardiac SR Ca2� leak by stimulation ofryanodine receptors.10 This result is in apparent contradictionwith the stimulatory effects of increased RyR2 phosphoryla-tion described in the present study. Recently, we found thatboth phosphorylation and dephosphorylation can stimulateRyR2 activity, resulting in increased SR Ca2� leak incardiomyocytes.36 Although we do not have a definitiveexplanation for these apparently contradicting results, theycan be rationalized by considering that RyR2 is a multimerwith multiple sets of phosphorylation sites. It is possible thatRyR2 activity is lowest at intermediate phosphorylation of acertain set of sites (ie, S2814), whereas both hypo- orhyperphosphorylation of the sites leads to increased activity.It is also possible that the observed stimulatory effects ofkinases and phosphatases are mediated by different sets of sites.Elucidation of the precise mechanisms of action of phosphory-lation and dephosphorylation on RyR2 should await furtherstudies.
miR-1 levels have been shown to increase in human cardiacdiseases, such as infarction and heart failure, and elevatedmiR-1 levels have been implemented in the development ofarrhythmia in these disease settings.19,20 Yang et al19 attrib-uted the proarrhythmic effects of miR-1 to down regulation ofconnexin 43 and the inward rectifier K channel (Kir2.1),which they identified as targets for miR-1. Our study demon-strated that altered Ca2� signaling presents another potentialarrhythmogenic mechanism for miR-1. In vivo, electric re-modeling and alterations of Ca2� signaling are likely to acttogether to induce arrhythmia. For example, the probability ofDADs reaching the threshold for action potential for anygiven magnitude of spontaneous Ca2� release would beexpected to increase on inhibition of Kir2.1 because ofincreased membrane resistance.37
A potential limitation of our study is that the experimentswere performed using primary cultures of adult rat ventricularmyocytes, which are known to change their characteristicsover time in culture. We did not find a significant differencein ICa density either with or without ISO between cellscultured for 48 hours and freshly isolated myocytes (supple-mental Figure VIII). Therefore, the EC coupling machineryof rat cardiomyocytes during the first 48 hours seems to belargely preserved. This result is consistent with the recentstudy by Banyasz et al,38 which showed that the mostprofound changes in morphometric and functional parame-ters, including T-tubular density and ICa, in rat ventricularmyocytes take place after three days in culture.
In conclusion, our results identified a new important targetfor miR-1 in the heart, namely the PP2A regulatory subunitB56�. Through translational inhibition of this mRNA target,miR-1 causes CaMKII-dependent hyperphosphorylation ofRyR2, enhances RyR2 activity, and promotes arrhythmo-
genic SR Ca2� release. This mechanism could contribute toinduction of arrhythmia in disease states accompanied byelevated miR-1.
Sources of FundingThis study was supported by American Heart Association ScientistDevelopment Grants (to D.T. and A.E.B.) and NIH grants (to S.G.,T.S.E., and D.S.F.).
DisclosuresNone.
References1. Fabiato A. Time and calcium dependence of activation and inactivation of
calcium-induced release of calcium from the sarcoplasmic reticulum of askinned canine cardiac Purkinje cell. J Gen Physiol. 1985;85:247–289.
2. Bers DM. Cardiac excitation–contraction coupling. Nature. 2002;415:198–205.
3. Wehrens XH, Marks AR. Novel therapeutic approaches for heart failureby normalizing calcium cycling. Nat Rev Drug Discov. 2004;3:565–573.
4. Maier LS, Bers DM. Role of Ca2�/calmodulin-dependent protein kinase(CaMK) in excitation–contraction coupling in the heart. Cardiovasc Res.2007;73:631–640.
5. Grueter CE, Colbran RJ, Anderson ME. CaMKII, an emerging moleculardriver for calcium homeostasis, arrhythmias, and cardiac dysfunction.J Mol Med. 2007;85:5–14.
6. Steenaart NAE, Ganim JR, DiSalvo J, Kranias EG. The phospholambanphosphatase associated with cardiac sarcoplasmic reticulum is a type 1enzyme. Arch Biochem Biophys. 1992;293:17–24.
7. Davare MA, Horne MC, Hell JW. Protein phosphatase 2A is associatedwith class C L-type calcium channels (Cav1.2) and antagonizes channelphosphorylation by cAMP-dependent protein kinase. J Biol Chem. 2000;275:39710–39717.
8. Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, RosemblitN, Marks AR. PKA phosphorylation dissociates FKBP12.6 from thecalcium release channel (ryanodine receptor): defective regulation infailing hearts. Cell. 2000;101:365–376.
9. Bers DM, Eisner DA, Valdivia HH. Sarcoplasmic reticulum Ca2� andheart failure: roles of diastolic leak and Ca2� transport. Circ Res. 2003;93:487–490.
10. Terentyev D, Viatchenko-Karpinski S, Gyorke I, Terentyeva R, GyorkeS. Protein phosphatases decrease sarcoplasmic reticulum calcium contentby stimulating calcium release in cardiac myocytes. J Physiol. 2003;552:109–118.
11. Xiao B, Jiang MT, Zhao M, Yang D, Sutherland C, Lai FA, Walsh MP,Warltier DC, Cheng H, Chen SR. Characterization of a novel PKAphosphorylation site, serine-2030, reveals no PKA hyperphosphorylationof the cardiac ryanodine receptor in canine heart failure. Circ Res.2005;96:847–855.
12. Benkusky NA, Weber CS, Scherman JA, Farrell EF, Hacker TA, JohnMC, Powers PA, Valdivia HH. Intact beta-adrenergic response andunmodified progression toward heart failure in mice with genetic ablationof a major protein kinase A phosphorylation site in the cardiac ryanodinereceptor. Circ Res. 2007;101:746–749.
13. Yang D, Zhu WZ, Xiao B, Brochet DX, Chen SR, Lakatta EG, Xiao RP,Cheng H. Ca2�/calmodulin kinase II-dependent phosphorylation ofryanodine receptors suppresses Ca2� sparks and Ca2� waves in cardiacmyocytes. Circ Res. 2007;100:399–407.
14. Bridge JHB, Savio-Galimberti E. What are the consequences of phos-phorylation and hyperphosphorylation of ryanodine receptors in normaland failing heart? Circ Res. 2008;102:995–997.
15. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function.Cell. 2004;116:281–297.
16. Zhao Y, Samal E, Srivastava D. Serum response factor regulates amuscle-specific microRNA that targets Hand2 during cardiogenesis.Nature. 2005;436:214–220.
17. Latronico MV, Catalucci D, Condorelli G. Emerging role of microRNAsin cardiovascular biology. Circ Res. 2007;101:1225–1236.
18. Wang Z, Luo X, Lu Y, Yang B. miRNAs at the heart of the matter. J MolMed. 2008.
19. Yang B, Lin H, Xiao J, Lu Y, Luo X, Li B, Zhang Y, Xu C, Bai Y, WangH, Chen G, Wang Z. The muscle-specific microRNA miR-1 regulates
Terentyev et al miR-1 Promotes Arrhythmia by Enhancing Ca Release 7
by guest on September 2, 2016
http://circres.ahajournals.org/D
ownloaded from

cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. NatMed. 2007;13:486–491.
20. Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW,Doevendans PA, Mummery CL, Borlak J, Haverich A, Gross C,Engelhardt S, Ertl G, Bauersachs J. MicroRNAs in the human heart: aclue to fetal gene reprogramming in heart failure. Circulation. 2007;116:258–267.
21. Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M. MicroRNAs playan essential role in the development of cardiac hypertrophy. Circ Res.2007;100:416–424.
22. Wehrens XH, Lehnart SE, Reiken SR, Marks AR. Ca2�/calmodulin-dependent protein kinase II phosphorylation regulates the cardiacryanodine receptor. Circ Res. 2004;94:e61–e70.
23. Rodriguez P, Bhogal MS, Colyer J. Stoichiometric phosphorylation ofcardiac ryanodine receptor on serine 2809 by calmodulin-dependentkinase II and protein kinase A. J Biol Chem. 2003;278:38593–38600.
24. Lehnart SE, Wehrens XH, Laitinen PJ, Reiken SR, Deng SX, Cheng Z,Landry DW, Kontula K, Swan H, Marks AR. Sudden death in familialpolymorphic ventricular tachycardia associated with calcium releasechannel leak. Circulation. 2004;109:3208–3214.
25. Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2�/calmod-ulin-dependent protein kinase modulates cardiac ryanodine receptor phos-phorylation and sarcoplasmic reticulum Ca2� leak in heart failure. CircRes. 2005;97:1314–1422.
26. Kass RS, Tsien RW. Fluctuations in membrane current driven by intra-cellular calcium in cardiac Purkinje fibers. Biophys J. 1982;38:259–269.
27. Venetucci LA, Trafford AW, Eisner DA. Increasing ryanodine receptoropen probability alone does not produce arrhythmogenic calcium waves:threshold sarcoplasmic reticulum calcium content is required. Circ Res.2007;100:105–111.
28. Hall DD, Feekes JA, Arachchige Don AS, Shi M, Hamid J, Chen L,Strack S, Zamponi GW, Horne MC, Hell JW. Binding of protein phos-phatase 2A to the L-type calcium channel Cav1.2 next to Ser1928, itsmain PKA site, is critical for Ser1928 dephosphorylation. Biochemistry.2006;45:3448–3459.
29. Marx SO, Reiken S, Hisamatsu Y, Gaburjakova M, Gaburjakova J, YangYM, Rosemblit N, Marks AR. Phosphorylation-dependent regulation of
ryanodine receptors: a novel role for leucine/isoleucine zippers. J CellBiol. 2001;153:699–708.
30. Bhasin N, Cunha SR, Mudannayake M, Gigena MS, Rogers TB, MohlerPJ. Molecular basis for PP2A regulatory subunit B56alpha targeting incardiomyocytes. Am J Physiol Heart Circ Physiol. 2007;293:H109–H119.
31. Bourguignon LY, Chu A, Jin H, Brandt NR. Ryanodine receptor-ankyrininteraction regulates internal Ca2� release in mouse T-lymphoma cells.J Biol Chem. 1995;270:17917–17922.
32. Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBellWH, Song LS, Haurogne K, Kyndt F, Ali ME, Rogers TB, Lederer WJ,Escande D, Le Marec H, Bennett V. Ankyrin-B mutation causes type 4long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–639.
33. Mohler PJ, Splawski I, Napolitano C, Bottelli G, Sharpe L, Timothy K,Priori SG, Keating MT, Bennett V. A cardiac arrhythmia syndromecaused by loss of ankyrin-B function. Proc Natl Acad Sci U S A. 2004;101:9137–9142.
34. Yang L, Liu G, Zakharov SI, Morrow JP, Rybin VO, Steinberg SF, MarxSO. Ser1928 is a common site for Cav1.2 phosphorylation by proteinkinase C isoforms. J Biol Chem. 2005;280:207–214.
35. Wu Y, Temple J, Zhang R, Dzhura I, Zhang W, Trimble R, Roden DM,Passier R, Olson EN, Colbran RJ, Anderson ME. Calmodulin kinase IIand arrhythmias in a mouse model of cardiac hypertrophy. Circulation.2002;106:1288–1293.
36. Terentyev D, Terentyeva R, Gyorke S. Both phosphorylation and dephos-phorylation of ryanodine receptor enhance sarcoplasmic reticulumcalcium leak in canine ventricular myocytes. Circulation. 2006;114(supplII):II-92.
37. Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmo-genesis and contractile dysfunction in heart failure: roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ Res. 2001;88:1159–1167.
38. Banyasz T, Lozinskiy I, Payne CE, Edelmann S, Norton B, Chen B,Chen-Izu Y, Izu LT, Balke CW. Transformation of adult rat cardiacmyocytes in primary culture. Exp Physiol. 2008;93:370–382.
8 Circulation Research February 27, 2009
by guest on September 2, 2016
http://circres.ahajournals.org/D
ownloaded from

GyorkeMalana, Donald E. Kuhn, Maha Abdellatif, David S. Feldman, Terry S. Elton and Sandor
Dmitry Terentyev, Andriy E. Belevych, Radmila Terentyeva, Mickey M. Martin, Geraldine E.Hyperphosphorylation of RyR2
and Causing CaMKII-Dependentαby Targeting PP2A Regulatory Subunit B56 Release and Promotes Cardiac Arrhythmogenesis2+ Overexpression Enhances CamiR-1
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2009 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
published online January 8, 2009;Circ Res.
http://circres.ahajournals.org/content/early/2009/01/08/CIRCRESAHA.108.181651.citationWorld Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org/content/suppl/2009/01/08/CIRCRESAHA.108.181651.DC1.htmlData Supplement (unedited) at:
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on September 2, 2016
http://circres.ahajournals.org/D
ownloaded from

CIRCRESAHA/2008/181651;R2
1
SUPPLEMENT MATERIAL
MATERIALS AND METHODS
Construction of the MiR-1 Adenovirus. Recombinant adenoviruses were constructed,
propagated, and tittered as previously described by Sayed et al.1. Briefly, pBHGloxΔE1,3Cre
(Microbix), including the ΔE1 adenoviral genome, was cotransfected with the pDC shuttle vector
containing the stem–loop sequence of the mouse miR-1-2 gene, into 293 cells using
Lipofectamine (Invitrogen). Through homologous recombination, the test genes integrate into the
E1-deleted adenoviral genome. The viruses were propagated on 293 cells and purified using
CsCl2 banding, followed by dialysis against 20 mmol/L Tris buffered saline with 2% glycerol.
Tittering was performed on 293 cells overlaid with DMEM plus 5% equine serum and 0.5%
agarose.
Real-time PCR.
Total RNA was isolated from cultured control and miR-1 overexpressing cell samples using Trizol
(Invitrogen). The RNA was subsequently treated with RNase-free DNase I, and mature miR-1
was quantified utilizing the TaqManTM microRNA Assay Kit. The relative miR-1 expression was
calculated from the equation 2-ΔCT , where ΔCT = CTmiR-1-CTinternal control. Fold variations in
expression of miR-1 between RNA samples were calculated after normalization to internal control
18s rRNA 2,3.
Luciferase Reporter Construct. A 1092 bp fragment encompassing the entire human B56α 3′-
untranslated region (3'-UTR) was PCR-amplified utilizing the following sense (5′-
AAAAAAAAGCCTCCCACCTCTGCCGG-3′) and antisense (5′-AGGCAAAATACAAACATAGAC-
3′) primers using standard procedures and a proofreading polymerase (Platinum Pfu, Invitrogen).
Human genomic DNA was used for template. The PCR product was subcloned into the pCR™2.1

CIRCRESAHA/2008/181651;R2
2
vector following the manufacturer’s protocol (Invitrogen) after treating the PCR product 10
minutes with Taq polymerase. Plasmid DNA was subsequently isolated from recombinant
colonies and sequenced to ensure authenticity. The B56α 3′-UTR inserts were removed from the
pCR™2.1 plasmid by EcoRI digestion. The fragments were subsequently gel purified, filled in and
blunt-end ligated into a filled-in XhoI site which is located downstream of the Renilla luciferase (r-
luc) reporter gene (psiCHECK-2™, Promega). The promoter used for Renilla luciferase
expression in the siCHECK vector is the SV40 promoter. Additionally, the psiCHECK-2 vector
possesses a secondary firefly reporter expression cassette which is under the control of the HSV-
TK promoter. This firefly reporter cassette has been specifically designed to be an intraplasmid
transfection normalization reporter; thus when using the psiCHECK-2 vector, the Renilla
luciferase signal is normalized to the firefly luciferase signal. The authenticity and orientation of
the inserts relative to the Renilla luciferase gene were confirmed by sequencing. The resulting
recombinant plasmid was designated, psiCHEK-B56α. Transformed bacterial cultures were
grown and each reporter construct (psiCHECK or psiCHECK-B56α) was purified by using
PureLink™ Hipure Plasmid Maxiprep Kit (Invitrogen).
Transfection and Luciferase Assay. Hsa-miR-1, a partially double-stranded RNA that mimics
endogenous precursor miR-1 and a control scrambled miRNA mimic was purchased from
Dharmacon. Transfection of CHO cells with small RNAs was optimized utilizing Lipofectamine
2000 (Invitrogen) and a fluorescein-labeled double-stranded RNA oligomer designated
BLOCKiTTM fluorescent oligonucleotide (Invitrogen). Once transfection conditions were optimized,
CHO cells were transfected (approaching 100% transfection efficiency) with the reporter
constructs described above and the appropriate miRNA precursor as indicated. After 24 h, cells
were washed and lysed with Passive Lysis Buffer (Promega), and firefly luciferase and Renilla
luciferase activities were determined using the dual-luciferase reporter assay system (Promega)
and a luminometer.

CIRCRESAHA/2008/181651;R2
3
Ca2+ measurements and Analysis. Isolation of rat ventricular myocytes was carried out as
previously described4 and approved by The Ohio State University Institutional Animal Care and
Use Committee. Isolated myocytes were plated on laminin-coated glass coverslips in serum-free
medium 199 supplemented with (in mmol/L): 25 NaHCO3, 5 creatine, 5 taurine, 10 U/mL
penicillin, 10 µg/mL streptomycin, and 10 µg/mL gentamycin (pH 7.3). Cells were cultured at 37°C
in a humidified atmosphere with 95% air/5% CO2. After 2 hours, unattached cells were removed
and myocytes were infected with adenoviruses at multiplicity of infection (MOI) 100 and cultured
for 36-48 hours before analysis. At such conditions culture-dependent changes in myocyte
structure and function are minimal5 (Online Figure VIII).
Whole-cell patch clamp recordings of transmembrane Ca2+ currents and membrane potential
were performed with an Axopatch 200B amplifier (Axon Instruments, MDS, Sunnyvale, CA). The
external solution contained (in mmol/L): 140 NaCl, 5.4 KCl, 1.0 CaCl2, 0.5 MgCl2, 10 Hepes, and
5.6 glucose (pH 7.3). Patch pipettes for voltage clamp experiments were filled with a solution that
contained (in mmol/L): 123.4 CsCl, 20 TEACl, 5 MgATP, 5 NaCl, 1 MgCl2, 0.1 Tris GTP, 10
Hepes, and 0.1 Fluo-3 K-salt (pH 7.2). In current clamp experiments patch pipettes were filled
with a solution that contained (in mmol/L): 90 K-aspartate, 50 KCl, 5 MgATP, 5 NaCl, 1 MgCl2,
0.1 Tris GTP, 10 HEPES, and 0.1 Fluo-3 K-salt (pH 7.2). Experiments on intact myocytes were
carried out using the following external solution (in mmol/L): 140 NaCl, 5.4 KCl, 1.0 CaCl2, 0.5
MgCl2, 10 Hepes, and 5.6 glucose (pH 7.3). Intracellular Ca2+ imaging was performed using
Olympus Fluoview 1000 confocal microscope equipped with 60X1.4 N.A. oil objective in line-scan
mode at a rate 2 or 5 ms per line. Intact myocytes were loaded with 10 µmol/L Fluo-3 AM Ca2+
indicator for 30 min at room temperature. Fluo-3 was excited by 488-nm line of argon-ion laser,
and fluorescence was acquired at wavelengths of 500–530. Ca sparks were analyzed with IDL
software4. Statistical analysis was performed using Origin 7.0 (OriginLab Corp., MA).
Western Blot Analysis and Immunochystochemistry. For western blot analysis RIPA buffer
supplemented with phosphatase, calpain and protease inhibitors (Sigma) was directly added to

CIRCRESAHA/2008/181651;R2
4
the cells after treatment and the samples were instantly frozen in liquid N2. Cell lysate proteins
were subjected to 4% to 20% SDS-PAGE and blotted onto nitrocellulose membranes (Bio-Rad
Labs, CA). Primary antibodies used were: anti-PLB and anti-SERCA2a (both a gift from Dr. M.
Periasamy, The Ohio State Univ., OH); anti-phospho-PLB- T17, anti-phospho-PLB-S16 (both
from Badrilla, UK); anti-phospho-RyR2-S2808, anti-phospho-RyR2-S2814 (both a gift from Dr.
A.R. Marks, Columbia Univ., NY); anti-phospho-RyR2-S2030(a gift from Dr. H.H. Valdivia, Univ.
of Wisconsin, WI); anti-RyR2, anti-CASQ2, anti-CAMKII Tr-287 and anti-NCX1 (all from ABR,
CO); anti-PP2A B56α, anti-PP1 (both from Upstate, NY); anti-PP2AC (from Calbiochem); anti-
CaMKII (from Santa Cruz Biotech., CA); anti-Cav1.2 (from Alomone, Israel). Protein bands were
visualized using the Super Signal West Pico kit (PIERCE, IL). Phosphorylation analysis for Cav1.2
(α1C) was carried out using Pro-Q Diamond stain technology. In brief, protein gel was fixed
overnight and stained using the Pro-Q Diamond phosphoprotein basic staining protocol
(Invitrogen Corp., CA). The gel was imaged in a Bio-Rad Pharos FX Plus molecular imager using
an excitation wavelength of 532 nm. Scans were quantified with Image J (NIH).
For immunocytochemistry, myocytes were fixed on glass coverslips with 4% paraformaldehyde
and permeabilized with 0.2% Triton X-100/PBS (pH 7.2) containing 1% BSA. Specimens were
incubated with anti-PP2A B56α (1:200) or anti-PP2AC (1:20) and anti-RyR2 (1:1,000) primary
antibodies followed by fluorophore-conjugated, highly cross-adsorbed IgGs (Alexa Fluor-488 and
Alexa Fluor-633; 1:200, Invitrogen Corp., CA). Images were acquired using Olympus Fluoview
1000 confocal system equipped with 60X1.4 N.A. oil objective.
Statistical Analysis. Results are presented as Mean±SEM. Statistical significance was
evaluated either by Student's t-test for paired observations or by One Way ANOVA as
appropriate. P<0.05 was considered significant.

CIRCRESAHA/2008/181651;R2
5
Online Figure Legends.
Online Figure I.
Relative expression of miR-1 in myocytes infected with control and miR-1 adenoviruses. Cells
from the same heart preparation were split onto two samples, infected with control or miR-1
adenoviruses at MOI 100 and kept in culture at 370C for 48 hours. Total RNA was isolated from
myocytes using standard procedures and mature miR-1 miRNas were quantified utilizing RT-PCR
as previously described2, 3. Gene expression was calculated relative to 18S rRNA and CT values
were normalized to “1” for control samples to simplify data presentation. The error bars represent
the average±SE of 6 independent experiments. (*Significantly different vs. control at p<0.05).
Online Figure II.
MiR-1 overexpression induces a leftward shift in the voltage-dependence of ICa activation.
Voltage-dependence curves for ICa activation (GCa/GCa,max) fitted with Boltzmann equations in
control and miR-1-overexpressing myocytes. The membrane potential at which activation was
half-maximal (V0.5) was -19.1±0.7 and -22.8±1.0 for control and miR-1 respectively.
Online Figure III.
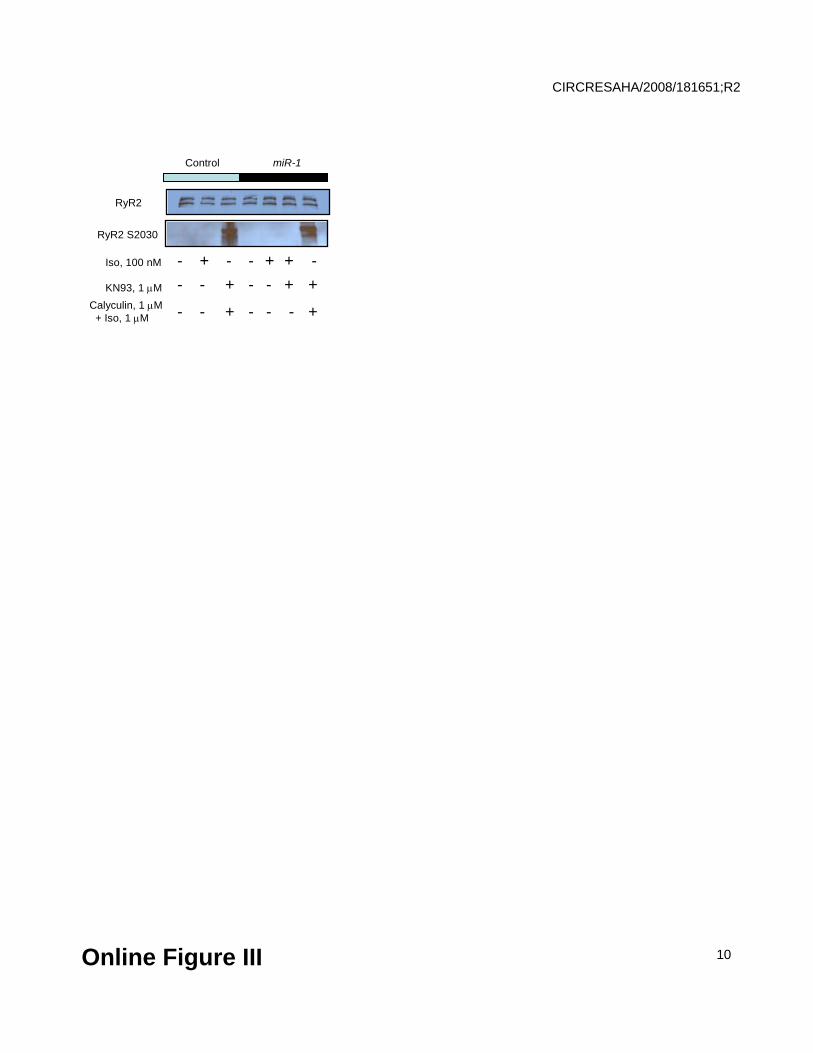
Lack of RyR2 phosphorylation at S2030 in control and miR-1 overexpressing myocytes in the
presence and absence of ISO. RyR2 phosphorylation at site S2030 was measured with phospho-
specific antibodies. Total RyR2 was measured in the same samples on a different blot and used
as a control for loading. Ventricular cells isolated from one heart were split into 7 groups, infected
with the control adenovirus or the adenovirus encoding miR-1 and kept in culture for 48 hrs.
Subsequently, myocytes were exposed to 100 nmol/L ISO (3 min) and 1 μmol/L KN93 (30 min),
paced at 1 Hz for 1 min and flash frozen. To attain maximal phosphorylation cells were incubated
for 30 min with 1 μmol/L ISO and 5 μmol/L Calyculin A. Phosphorylation of S2030 was
undetectable in experimental conditions comparable to the conditions used in Ca cycling studies
for both control and miR-1 groups. Similar results were obtained in 5 independent experiments.

CIRCRESAHA/2008/181651;R2
6
Online Figure IV.
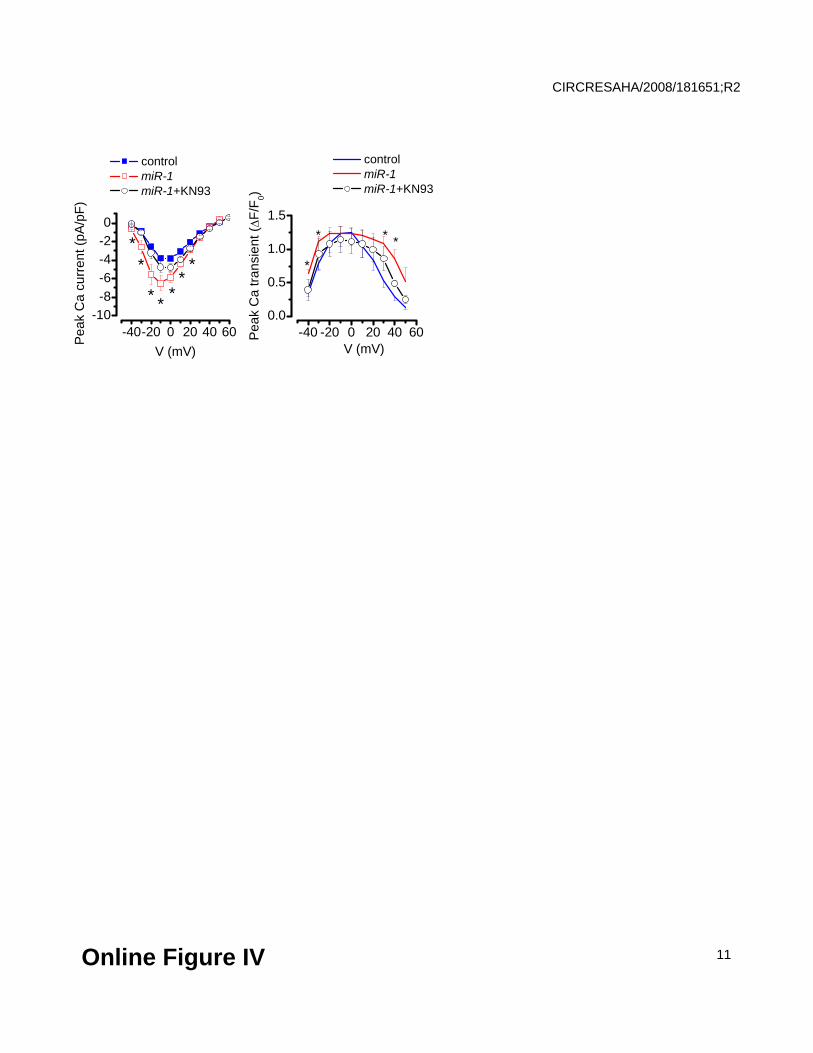
Inhibition of CaMKII abolishes the effects of miR-1 overexpression on Ca2+ currents and Ca2+
transients. Voltage-dependencies of ICa (left) and Ca2+ transient amplitude (right) in control (blue)
and miR-1-overexpressing myocytes at baseline conditions (red) and after 15 min incubation with
1 μmol/L KN93 (black). *Significantly different vs. control at P<0.05, One Way ANOVA (N=5-7).
Online Figure V.
Control (top) and miR-1-overexpressing (bottom) cardiomyocytes fixed after 48 hours in culture
coimmunolabeled with PP2A catalytic subunit- (left, green) and RyR2-specific antibodies (middle,
red).
Online Figure VI.
Arrhythmogenic disturbances in Ca2+ cycling in myocytes overexpressing miR-1. Current clamp
recordings of membrane potential (top traces) along with line-scan images (middle panel) and
averaged temporal profiles (bottom traces) of Fluo-3 fluorescence in a control myocyte and a
myocyte overexpressing miR-1. Myocytes were stimulated at 1 Hz in the presence of 100 nmol/L
ISO. Similar results with DADs and underlying spontaneous Ca2+ releases were obtained in 3 out
of 4 miR-1 overexpressing cells.
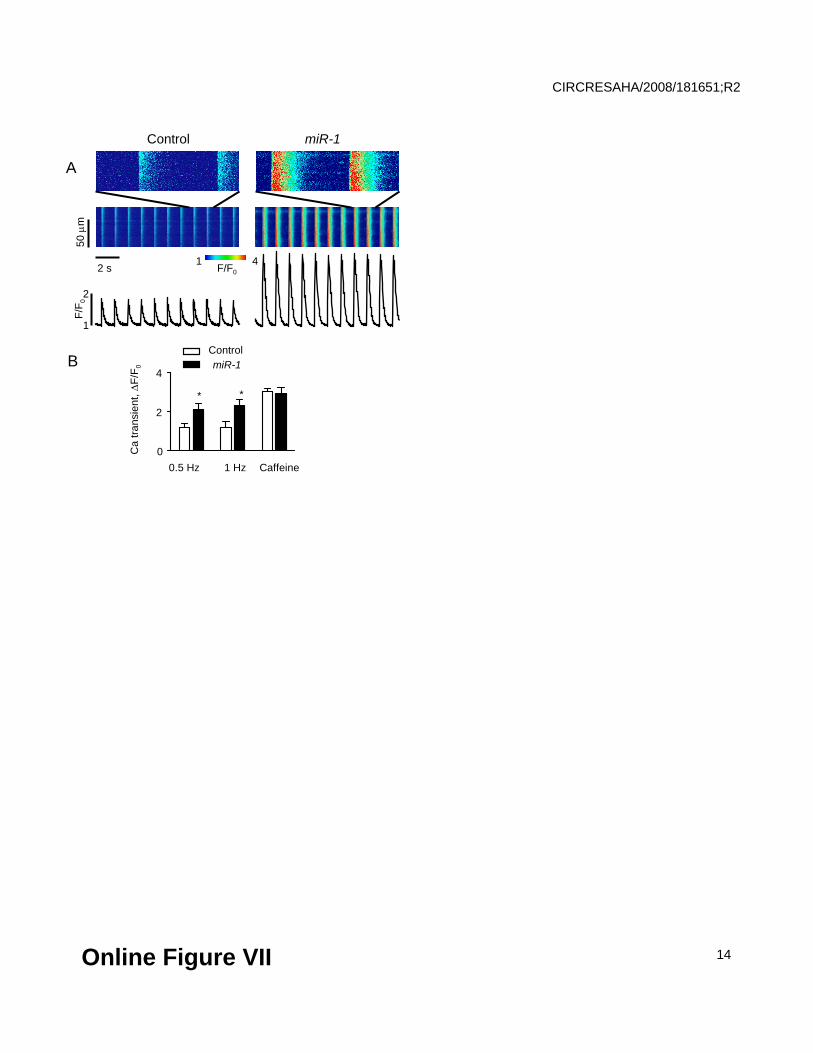
Online Figure VII.
Altered Ca2+ cycling in paced miR-1 overexpressing myocytes in the absence of beta-adrenergic
stimulation. A, Representative line scan images and averaged temporal profiles of Fluo-3
fluorescence in a control myocyte and a myocyte overexpressing miR-1. Insets show images at
expanded time scale. B, Pooled data for Ca2+ transients amplitudes recorded after 1 min of
pacing at 0.5 Hz, 1 Hz and upon application of 10 mmol/L caffeine after 1 min of pacing at 1 Hz
to assess the SR Ca2+ load for two groups of cells. N=10-14, *Significantly different vs. controls at
p<0.05.

CIRCRESAHA/2008/181651;R2
7
Online Figure VIII.
Culturing of myocytes for 48 hours does not affect ICa density and response to beta-adrenergic
stimulation. A, Pooled data for density of ICa measured at 0 mV in fresh isolated myocytes and
cells maintained in culture for 2 days at basal conditions and in the presence of 100 nmol/L ISO
(not significant at p<0.05, n=6). B, Representative traces of ICa evoked by depolarization to 0 mV
recorded in fresh isolated myocyte before (black) and after application of 100 nmol/L ISO (red).
Cell capacitance was 238 pF.
REFERENCES:
1. Sayed D, Hong C, Chen I, Lypowy J, Abdellatif M. MicroRNAs Play an Essential Role in the
Development of Cardiac Hypertrophy. Circ Res. 2007;100:416-424.
2. Schmittgen TD, Lee EJ, Jiang J, Sarkar A, Yang L, Elton TS, Chen C. Real-time PCR
quantification of precursor and mature microRNA. Methods. 2008;44:31-38.
3. Kuhn DE, Nuovo GJ, Martin MM, Malana GE, Pleister AP, Jiang J, Schmittgen TD, Terry AV
Jr, Gardiner K, Head E, Feldman DS, Elton TS. Human chromosome 21-derived miRNAs are
overexpressed in down syndrome brains and hearts. Biochem Biophys Res Commun.
2008;370:473-477.
4. Terentyev D, Viatchenko-Karpinski S, Gyorke I, Terentyeva R, Gyorke S. Protein
phosphatases decrease sarcoplasmic reticulum calcium content by stimulating calcium
release in cardiac myocytes. J Physiol. 2003;552:109-118.
5. Banyasz T, Lozinskiy I, Payne CE, Edelmann S, Norton B, Chen B, Chen-Izu Y, Izu LT, Balke
CW. Transformation of adult rat cardiac myocytes in primary culture. Exp Physiol.
2008;93:370-382.

Online Figure I
CIRCRESAHA/2008/181651;R2
8
Control miR-1
*
0
1
2
Rel
ativ
e E
xpre
ssio
n

-50 -40 -30 -20 -10 0 100.0
0.5
1.0
GC
a/GC
a, max
Control
miR-1
V, mV
Online Figure II
CIRCRESAHA/2008/181651;R2
9
Control miR-1
Iso, 100 nM
KN93, 1 μM
- + - - +
RyR2 S2030
RyR2
+ -- - + - +- +
Calyculin, 1 μM+ Iso, 1 μM - - + - -- +
Online Figure III
CIRCRESAHA/2008/181651;R2
10
-40 -20 0 20 40 600.0
0.5
1.0
1.5
-40-20 0 20 40 60-10-8-6-4-20
V (mV)
**
***
*
control miR-1 miR-1+KN93
Pea
k C
a cu
rren
t (pA
/pF)
V (mV)
*
control miR-1 miR-1+KN93
***
Pea
k C
a tra
nsie
nt (Δ
F/F 0)
*
Online Figure IV
CIRCRESAHA/2008/181651;R2
11

PP2Ac RyR2 Overlay
Control
miR-1
Online Figure V
CIRCRESAHA/2008/181651;R2
12

Online Figure VI
30 μ
m80
mV
3
F/F0
1
1 F/F04
Control miR-1
CIRCRESAHA/2008/181651;R2
13
ControlmiR-1
Ca
trans
ient
, ΔF/
F 0
0.5 Hz 1 Hz
4
0
2
Caffeine
Control miR-1
50 μ
m
1 F/F042 s
F/F 0
1
2
**
A
B
Online Figure VII
CIRCRESAHA/2008/181651;R2
14

Online Figure VIII
0
5
10
Ca
curr
ent d
ensi
ty (p
A/p
F)
ControlISO, 100 nmol/L
0 2Days in culture
A
0 2
B
0
2
1
3
I Ca,
nA
ControlISO, 100 nmol/L
100 ms
CIRCRESAHA/2008/181651;R2
15




















