
LTP in hippocampal neurons is associated with a CaMKII-mediated increase in GluA1 surface expression
14
,1 ,1 ,1 ,1 , *MRC Centre for Synaptic Plasticity, School of Physiology and Pharmacology, University of Bristol, Bristol, UK Institut de Pharmacologie Mole ´culaire et Cellulaire, CNRS, Universite ´ de Nice-Sophia Antipolis, Valbonne, France àDepartment of Brain and Cognitive Sciences, College of Natural Sciences, Seoul National University, Gwanak-gu, Seoul, Korea Understanding the molecular basis of long-term potentiation (LTP) is of fundamental importance because it has been implicated in learning and memory as well as other physiological and pathological processes. One form of LTP is dependent on Ca 2+ influx through post-synaptic NMDA receptors (NMDARs), subsequent activation and autophos- phorylation of the Ca 2+ /calmodulin-dependent protein kinase II (CaMKII) and an increase in alpha-amino-3-hydroxy-5- methylisoxazole-4-propionate receptor (AMPAR) insertion at the post-synaptic membrane (Collingridge et al. 2004). Alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate receptors mediate the vast majority of fast excitatory synaptic transmission in the mammalian brain and they are key components of the modifiable synaptic response. AMPARs are composed of four subunits, termed GluA1–4 according to International Union of Basic and Clinical Pharmacology nomenclature. The edited GluA2 subunit is critical for the biophysical properties of AMPARs producing low conduc- tance, non-rectifying, Ca 2+ -impermeable AMPARs, and postnatally the great majority of AMPARs contain edited Received September 1, 2010; revised manuscript received November 29, 2010; accepted December 1, 2010. Address correspondence and reprint requests to Elek Molna ´r, MRC Centre for Synaptic Plasticity, School of Physiology and Pharmacology, University of Bristol, Medical Sciences Building, University Walk, Bristol BS8 1TD, UK. E-mail: [email protected] 1 The present address of V. J. Appleby is the Children’s Brain Tumour Research Centre, Institute of Genetics, University of Nottingham, Queen’s Medical Centre, Nottingham NG7 2UH, UK; S. A. L. Corre ˆa is the Department of Biological Sciences, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK; J. K. Duckworth is Pfizer Regen- erative Medicine, UCB Building, Granta Park, Great Abington, Cam- bridge CB21 6GS, UK; and J. E. Nash is the Centre for Neurobiology of Stress, Department of Biological Sciences, University of Toronto at Scarborough, 1265 Military Trail, Toronto, ON, Canada M1C 1A4. Abbreviations used: AMPAR, alpha-amino-3-hydroxy-5-methylisox- azole-4-propionate receptor; BSA, bovine serum albumin; CaMKII, Ca 2+ /calmodulin-dependent protein kinase II; DIV, days in vitro; GluA1– 4, AMPAR subunits 1–4; HBS, HEPES based saline buffer; LTP, long- term potentiation; mEPSC, miniature excitatory post-synaptic current; NMDAR, NMDA receptor; PBS, phosphate-buffered saline; TTX, tetrodotoxin. Abstract The use of hippocampal dissociated neuronal cultures has enabled the study of molecular changes in endogenous native proteins associated with long-term potentiation. Using immu- nofluorescence labelling of the active (Thr286-phosphory- lated) alpha-Ca 2+ /calmodulin-dependent protein kinase II (CaMKII) we found that CaMKII activity was increased by transient (3 · 1 s) depolarisation in 18- to 21-day-old cultures but not in 9- to 11-day-old cultures. The increase in Thr286 phosphorylation of CaMKII required the activation of NMDA receptors and was greatly attenuated by the CaMKII inhibitor KN-62. We compared the effects of transient depolarisation on the surface expression of GluA1 and GluA2 subunits of the alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate recep- tor and found a preferential recruitment of the GluA1 subunit. CaMKII inhibition prevented this NMDA receptor-dependent delivery of GluA1 to the cell surface. CaMKII activation is therefore an important factor in the activity-dependent recruitment of native GluA1 subunit-containing alpha-amino- 3-hydroxy-5-methyl-4-isoxazole propionate receptors to the cell surface of hippocampal neurons. Keywords: AMPA, CaMKII, GluA1, GluA2, glutamate, LTP, NMDA. J. Neurochem. (2011) 116, 530–543. JOURNAL OF NEUROCHEMISTRY | 2011 | 116 | 530–543 doi: 10.1111/j.1471-4159.2010.07133.x 530 Journal of Neurochemistry Ó 2011 International Society for Neurochemistry, J. Neurochem. (2011) 116, 530–543 Ó 2011 The Authors
-
Upload
landaverde -
Category
Documents
-
view
0 -
download
0
Transcript of LTP in hippocampal neurons is associated with a CaMKII-mediated increase in GluA1 surface expression

,1 ,1 ,1 ,1
,
*MRC Centre for Synaptic Plasticity, School of Physiology and Pharmacology, University of Bristol, Bristol, UK
�Institut de Pharmacologie Moleculaire et Cellulaire, CNRS, Universite de Nice-Sophia Antipolis, Valbonne, France
�Department of Brain and Cognitive Sciences, College of Natural Sciences, Seoul National University, Gwanak-gu, Seoul, Korea
Understanding the molecular basis of long-term potentiation(LTP) is of fundamental importance because it has beenimplicated in learning and memory as well as otherphysiological and pathological processes. One form of LTPis dependent on Ca2+ influx through post-synaptic NMDAreceptors (NMDARs), subsequent activation and autophos-phorylation of the Ca2+/calmodulin-dependent protein kinaseII (CaMKII) and an increase in alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate receptor (AMPAR) insertion atthe post-synaptic membrane (Collingridge et al. 2004).
Alpha-amino-3-hydroxy-5-methylisoxazole-4-propionatereceptors mediate the vast majority of fast excitatory synaptictransmission in the mammalian brain and they are keycomponents of the modifiable synaptic response. AMPARsare composed of four subunits, termed GluA1–4 according toInternational Union of Basic and Clinical Pharmacologynomenclature. The edited GluA2 subunit is critical for thebiophysical properties of AMPARs producing low conduc-tance, non-rectifying, Ca2+-impermeable AMPARs, andpostnatally the great majority of AMPARs contain edited
Received September 1, 2010; revised manuscript received November 29,2010; accepted December 1, 2010.Address correspondence and reprint requests to Elek Molnar, MRC
Centre for Synaptic Plasticity, School of Physiology and Pharmacology,University of Bristol, Medical Sciences Building, University Walk,Bristol BS8 1TD, UK. E-mail: [email protected] present address of V. J. Appleby is the Children’s Brain TumourResearch Centre, Institute of Genetics, University of Nottingham,Queen’s Medical Centre, Nottingham NG7 2UH, UK; S. A. L. Correa isthe Department of Biological Sciences, University of Warwick, GibbetHill Road, Coventry CV4 7AL, UK; J. K. Duckworth is Pfizer Regen-erative Medicine, UCB Building, Granta Park, Great Abington, Cam-bridge CB21 6GS, UK; and J. E. Nash is the Centre for Neurobiology ofStress, Department of Biological Sciences, University of Toronto atScarborough, 1265 Military Trail, Toronto, ON, Canada M1C 1A4.Abbreviations used: AMPAR, alpha-amino-3-hydroxy-5-methylisox-
azole-4-propionate receptor; BSA, bovine serum albumin; CaMKII,Ca2+/calmodulin-dependent protein kinase II; DIV, days in vitro; GluA1–4, AMPAR subunits 1–4; HBS, HEPES based saline buffer; LTP, long-term potentiation; mEPSC, miniature excitatory post-synaptic current;NMDAR, NMDA receptor; PBS, phosphate-buffered saline; TTX,tetrodotoxin.
Abstract
The use of hippocampal dissociated neuronal cultures has
enabled the study of molecular changes in endogenous native
proteins associated with long-term potentiation. Using immu-
nofluorescence labelling of the active (Thr286-phosphory-
lated) alpha-Ca2+/calmodulin-dependent protein kinase II
(CaMKII) we found that CaMKII activity was increased by
transient (3 · 1 s) depolarisation in 18- to 21-day-old cultures
but not in 9- to 11-day-old cultures. The increase in Thr286
phosphorylation of CaMKII required the activation of NMDA
receptors and was greatly attenuated by the CaMKII inhibitor
KN-62. We compared the effects of transient depolarisation
on the surface expression of GluA1 and GluA2 subunits of the
alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate recep-
tor and found a preferential recruitment of the GluA1 subunit.
CaMKII inhibition prevented this NMDA receptor-dependent
delivery of GluA1 to the cell surface. CaMKII activation is
therefore an important factor in the activity-dependent
recruitment of native GluA1 subunit-containing alpha-amino-
3-hydroxy-5-methyl-4-isoxazole propionate receptors to the
cell surface of hippocampal neurons.
Keywords: AMPA, CaMKII, GluA1, GluA2, glutamate, LTP,
NMDA.
J. Neurochem. (2011) 116, 530–543.
JOURNAL OF NEUROCHEMISTRY | 2011 | 116 | 530–543 doi: 10.1111/j.1471-4159.2010.07133.x
530 Journal of Neurochemistry � 2011 International Society for Neurochemistry, J. Neurochem. (2011) 116, 530–543� 2011 The Authors

GluA2 (Molnar and Isaac 2002; Isaac et al. 2007). Previousstudies indicate that neuronal activity-dependent changes inAMPAR subunit composition can affect the regulation ofsynaptic transmission (Cull-Candy et al. 2006).
Ca2+/calmodulin-dependent protein kinase II is a serine/threonine kinase and accounts for 1–2% of the total proteinin the brain (Bennett et al. 1983; Kennedy et al. 1983; Kellyet al. 1984; McGuinness et al. 1985). Signals mediated byincreases in intracellular Ca2+ can activate CaMKII, allowingan autophosphorylation event to occur leading to long-lastingactivation (Lou et al. 1986; Miller and Kennedy 1986;Lisman et al. 2002). NMDAR-dependent LTP (NMDAR-LTP) at hippocampal synapses is associated with a persistentactivation of CaMKII (Fukunaga et al. 1993). This activationof CaMKII is accompanied by autophosphorylation ofThr286 and Thr287 on the a and b subunits of CaMKII,respectively (Fukunaga et al. 1995; Barria et al. 1997;Lisman et al. 2002) and can occur in a highly localisedmanner (Inagaki et al. 2000; Lee et al. 2009). LTP isassociated with localised activation of CaMKII at thesynapse and an increase in the synaptic content of CaMKII(Strack et al. 1997; Gardoni et al. 2001; Otmakhov et al.2004b; Skelding and Rostas 2009). The central role forCaMKII in LTP has been confirmed by both loss of functionstudies using pharmacological (Malinow et al. 1989; Itoet al. 1991; Otmakhov et al. 1997; Bortolotto and Colling-ridge 1998) and genetic approaches (Silva et al. 1992; Hindset al. 1998), and gain of function studies utilising post-synaptic application of CaMKII (McGlade-McCulloh et al.1993; Pettit et al. 1994; Lledo et al. 1995).
The molecular mechanisms underlying changes in cellsurface expression and subunit composition of endogenousnative AMPARs that underlie neuronal plasticity are poorlyunderstood. The availability of antibodies that recogniseepitopes of glutamate receptors on living neurons (Richmondet al. 1996; Noel et al. 1999; Pickard et al. 2000) combinedwith models of LTP in cultured, dissociated hippocampalneurons (e.g., Fitzjohn et al. 2001; Liao et al. 2001; Lu et al.2001) has enabled the imaging of changes in native post-synaptic receptor proteins (Liao et al. 2001; Lu et al. 2001;Pickard et al. 2001; Oh et al. 2006). We have developed amethod whereby rapid transient depolarisation, of a fewseconds, induces LTP in cultured dissociated hippocampalneurons. This form of LTP is triggered by the synapticactivation of NMDARs, is dependent on post-synaptic Ca2+
and is expressed by an increase in miniature excitatory post-synaptic currents (mEPSC) frequency, with little or nochange in mEPSC amplitude (Fitzjohn et al. 2001). Itinvolves the insertion of AMPARs into the plasma membraneat sites previously lacking AMPARs (Pickard et al. 2001)and requires protein interactions involving SAP97 and/orMyosin VI (Nash et al. 2010).
In this study, we used this cell culture model of NMDAR-LTP in combination with immunocytochemical and electro-
physiological techniques to investigate the role of CaMKII inthe synaptic activity-induced recruitment of native AMPARsto the plasma membrane. Our results indicate that transientdepolarisation-induced LTP in hippocampal neurons isassociated with a CaMKII mediated increase in GluA1 onthe cell surface, but no change in GluA2 AMPAR subunitdistribution.
Materials and methods
Hippocampal neuronal culturesLow-density primary hippocampal cultures were prepared from
3-day-old Wistar rats (B&K Universal Ltd., Hull, UK) using a
previously described protocol (Pickard et al. 2001; Nash et al.2010) with minor modifications. Briefly, the CA3-CA1 region of the
hippocampus was dissected, and neurons were recovered by
enzymatic digestion with trypsin and mechanical dissociation. Cells
were then plated at a density of � 50 000 per dish onto 22 mm glass
coverslips coated with poly-L-lysine (Sigma, Poole, UK). Neurons
were used for experiments 9–21 days after plating as specified at
individual experiments.
Induction of transient depolarisationTransient depolarisation was induced using three 1 s applications of
90 mM extracellular K+ as described previously (Fitzjohn et al.2001; Pickard et al. 2001; Nash et al. 2010). Briefly, neuronal
cultures were transferred into HBS (HEPES based saline: 119 mM
NaCl, 5 mM KCl, 25 mM HEPES, 33 mM D-glucose, 2 mM CaCl2,
2 mM MgCl2, 1 lM glycine, 0.1 mM picrotoxin (Sigma), 0.5 lMtetrodotoxin (TTX; Tocris Cookson Ltd., Bristol, UK), 300–
310 mOsm, pH 7.4) and then 90 mM K+ in HBS (Na+ adjusted to
maintain osmolarity) was applied for 3 · 1 s periods with 10 s
intervals in HBS. LTP induction was also performed in the presence
of the CaMKII inhibitor KN-62 (3 lM) and NMDAR antagonist
L-689,560 (5 lM; Tocris Cookson Ltd.).
Immunocytochemistry and imagingImmunofluorescence labelling of surface expressed ionotropic
glutamate receptor subunits were performed in non-permeabilised
hippocampal neurons as described previously (Pickard et al. 2000,2001; Nash et al. 2010) using extensively characterised primary
antibodies raised against the extracellular epitopes of GluA1 (1 lg/mL; Molnar et al. 1993) and GluA2 (5 lg/mL, Zymed Laboratories,
South San Francisco, CA, USA; Vissavajjhala et al. 1996) AMPAR
subunit proteins. Surface expressed GluN1 NMDAR subunits were
identified using an extracellular N-terminal domain specific guinea
pig polyclonal antibody (1 lg/mL; Molnar et al. 1995; Pickard
et al. 2000). The activated form of aCaMKII was identified using a
rabbit antibody specific for the phosphorylated Thr286 site of
aCaMKII (1 : 20 dilution; Promega, Southampton, UK). Mouse
monoclonal synaptotagmin antibody (1 : 20 dilution; Pharmingen,
Pickard et al. 2000) was used to identify pre-synaptic terminals.
Unless otherwise stated, neuronal cultures on coverslips were
washed with HBS and fixed in 4% paraformaldehyde in phosphate-
buffered saline (PBS, pH 7.4) for 10 min followed by incubation in
PBS supplemented with 10 mM glycine for 10 min to reduce
autofluorescence. Before immunostaining cells were blocked in 5%
� 2011 The AuthorsJournal of Neurochemistry � 2011 International Society for Neurochemistry, J. Neurochem. (2011) 116, 530–543
CaMKII-dependent recruitment of GluA1 in LTP | 531

bovine serum albumin (BSA) in PBS for 15 min at � 20�C. For thelabelling of intracellular epitopes, cells were permeabilised by 0.1%
Triton X-100 in 5% BSA containing PBS. Alternatively, where this
is indicated, neurons were permeabilised by methanol fixation
(Pickard et al. 2000, 2001). Following immunostaining, primary
antibodies were visualized with the appropriate secondary antibod-
ies (Alexa647 or Alexa568 labelled goat anti-mouse, goat anti-rabbit
or anti-guinea pig antibodies, 10 lg/mL; Molecular Probes, Inc.,
Eugene, OR, USA) in 5% BSA containing PBS. Non-specific
labelling and the method specificity of the antibodies were deter-
mined by the use of pre-immune sera, blocking antibody labelling
with the antigenic peptide or fusion protein used for the production
of antibodies (Molnar et al. 1993, 1995; Pickard et al. 2000) and by
performing immunolabelling with the secondary antibodies alone.
Immunofluorescent staining was visualized using a Zeiss
LSM510 Meta confocal microscope (Oberkochen, Germany).
Fluorophores were excited with 488, 543 or 633 nm wavelengths
and emission was detected through 505–530 band-pass, Meta
detector set to detect wavelength between 580–610 nm and 650
long-pass filters.
Quantitative analysis of immunofluorescent receptor clusters was
carried out in 3D using a confocal microscope and Volocity software
(Improvision, Coventry, UK). Twice the level of background
fluorescence was used as the threshold and only clearly identifiable
dendrites were selected for analysis. Puncta (0.3–5 lm3) that were
above threshold intensity were measured. For quantitative compar-
isons, the number of puncta per 100 lm length of dendrite was
expressed for the GluA1 and GluA2 antibodies within a given field.
Quantification of GluA1 and GluA2 average pixel intensities on the
surface of dendrites was carried out using the ImageJ software (NIH,
http://rsbweb.nih.gov/ij) as described previously (Nash et al. 2010).All analysis was performed blind to the experimental manipulation.
A single value was obtained from each independent experiment and
used to construct the mean and standard error. The number of
independent experiments was the number of observations used for
statistical analysis.
Whole-cell recordingsElectrophysiological analysis was carried out as described previ-
ously (Fitzjohn et al. 2001; Nash et al. 2010). Briefly, neurons [21–24 days in vitro (DIV)] were perfused with HBS (containing 0.5 lMTTX at approximately 2 mL/min at 25�C), and voltage-clamped at
)70 mV using an amplifier (Axopatch 200B, Molecular Devices,
Palo Alto, CA, USA). Intracellular solution comprised (mM): Cs
Methane sulphonate (110); HEPES (40); EGTA (0.6); NaCl (10);
Mg-ATP (4); Na2-GTP (0.3); 290 mOsm, pH 7.2. Upon obtaining a
stable baseline (5 min), KCl (90 mM) was perfused directly as a
series of 3 · 1 s pulses, separated by a 10 s interval (Fitzjohn et al.2001). Data were filtered at 5 kHz, collected continuously, digitised
at 10 kHz, and converted to axon binary files using Axoscope
software (Molecular Devices). mEPSCs were identified using
amplitude and area threshold detection using Mini Analysis software
(Synaptosoft, Inc., Decatur, GA, USA). Events were detected by
setting the threshold value for detection at three times the root mean
square of background noise, followed by visual confirmation of
mEPSC detection. The frequency and amplitude of mEPSCs
recorded at least 25 min after depolarisation was compared to
baseline values. Depolarisation induced by each 1 s pulse of K+
typically lasted < 5 s. During and immediately following K+
application mEPSC frequency could not be accurately measured
because of loss of voltage clamp and overlapping mEPSCs.
Cumulative probability plots were constructed by analysing all
mEPSCs in a 5 min baseline period immediately prior to KCl
application and all mEPSCs in a 5 min period at least 25 min post-
KCl. Access resistance was monitored throughout using LTP
software (Anderson and Collingridge 2001) and recordings disre-
garded if this varied more than 20% throughout the experiment.
StatisticsAll data are presented as mean ± SEM (standard error of the mean).
The ‘n’ number refers to the number of independent experiments
carried out. Statistical analysis was carried out using an unpaired
Student’s t-test. For non-equal ‘n’ numbers, a two sample equal
variance t-test was performed. An ANOVA (SPSS, IBM Corporation,
Armonk, NY, USA) was used for comparison of multiple data sets.
For the electrophysiological experiments, ANOVA with Tukey
multiple comparisons test post hoc was performed. Statistically
significant difference was judged to be p < 0.05.
Results
Activation of CaMKII at synapses following transientdepolarisationThe increase in post-synaptic Ca2+ leads to persistentactivation of CaMKII and the autophosphorylation ofthreonine 286 residue (Thr286) of aCaMKII (Lisman et al.2002). Therefore, the activation of the endogenous CaMKIIenzyme can be monitored in hippocampal neurons using anantibody specific for the Thr286 phosphorylation site. Firstwe tested the effects of transient depolarisation (3 · 1 spulses of 90 mM KCl, applied at 10 s intervals) on CaMKII-Thr286 autophosphorylation and subcellular distribution inhippocampal neuronal cultures at 9–11 and 18–21 DIV.Sham treated and KCl depolarised cells were fixed andpermeabilised in methanol 30 min after the induction processand stained for phospho-Thr286 aCaMKII (P-CaMKII) andthe synaptic protein synaptotagmin (Fig. 1). At 9–11 DIV,most of the P-CaMKII labelling was identified in the cellbodies and only few P-CaMKII immunopositive puncta wereidentified in sham treated neurons (6.7 ± 1.9 puncta/100 lmof dendrite, n = 6) or KCl treated cells (6.3 ± 1.2 puncta/100 lm of dendrite, n = 6, p > 0.05) indicating that CaMKIIis not activated significantly in response to depolarisation inrelatively immature neurons (Fig. 1a). In more maturehippocampal neuronal cultures at 18–21 DIV, the identicalKCl treatment produced a marked increase in P-CaMKIIimmunoreactivities in the soma, dendritic shafts and espe-cially at synaptotagmin positive sites of dendrites (Fig. 1b;sham treated neurons: 2.1 ± 0.6 P-CaMKII puncta/100 lmof dendrite, n = 6, KCl treated neurons: 40.5 ± 1.9 P-CaMKII puncta/100 lm of dendrite, n = 6, p < 0.001).These results indicate that CaMKII is activated upon KCl
Journal of Neurochemistry � 2011 International Society for Neurochemistry, J. Neurochem. (2011) 116, 530–543� 2011 The Authors
532 | V. J. Appleby et al.

treatment in primary hippocampal neuronal cultures at 18–21DIV but not in less mature neurons at 9–11 DIV and that itsactivity is especially high at synapses. Previous studiesestablished that transient KCl depolarisation increasedCaMKII-Thr286 autophosphorylation without detectablechanges in total CaMKII levels up to 2 h post-treatment(Lengyel et al. 2004; Migues et al. 2006; Zhou et al. 2007).Therefore it is very unlikely that total CaMKII levelschanged under our experimental conditions.
We also investigated whether the essential GluN1NMDAR subunit co-localises with P-CaMKII and synapto-tagmin in hippocampal neurons at 18–21 DIV (Figure S1).This immunolabelling revealed that the majority (> 85%) of
the P-CaMKII puncta co-localised to synapses that containedNMDARs.
KN-62 prevents CaMKII activationKN-62 is an inhibitor of aCaMKII and acts by competing forthe binding of Ca2+/calmodulin complex to the CaMKIIenzyme (Tokumitsu et al. 1990). To investigate the involve-ment of Ca2+/calmodulin-dependent activation of CaMKII,we performed KCl depolarisation of hippocampal neuronswith and without KN-62 (3 lM) followed by anti-P-CaMKIIand anti-synaptotagmin immunolabelling 30 min after theKCl treatment (Fig. 2a). The obtained P-CaMKII immuno-reactivity was significantly lower (by � 80%) in cells
P-CaMKII Synaptotagmin P-CaMKII Synaptotagmin
P-CaMKII Synaptotagmin P-CaMKII Synaptotagmin
3 × 1 s K+ –
21 DIV
9 DIV(a)
(b)
+
3 × 1 s K+ – +
Fig. 1 Co-localisation of autophosphorylat-
ed CaMKII (P-CaMKII) and synaptotagmin
in sham treated and K+ treated hippocam-
pal neuronal cultures at 9 (a) and 21 (b)
days in vitro (DIV). Postnatal hippocampal
neuronal cultures were either sham treated
with HBS buffer (3 · 1 s K+)) or depolar-
ised with brief exposures to 90 mM KCl in
HBS (3 · 1 s K++) to induce LTP. After
30 min, the cultures were fixed and per-
meabilised with methanol and then immu-
nolabelled to visualise active (Thr286
autophosphorylated) CaMKII (P-CaMKII,
green) and the synaptic marker synapto-
tagmin (red). Representative staining of P-
CaMKII and synaptotagmin from the inset
windows in each low magnification image is
shown underneath the image. Co-localisa-
tion of P-CaMKII with synaptotagmin is
shown in yellow. While at 9 DIV (a) 3 · 1 s
K+ treatment produced no detectable
changes in P-CaMKII immunoreactivity, at
21 DIV (b) the same treatment dramatically
increased P-CaMKII immunostaining at
synapses (yellow puncta). Scale bars:
20 lm (top panels), 10 lm (enlarged re-
gions).
� 2011 The AuthorsJournal of Neurochemistry � 2011 International Society for Neurochemistry, J. Neurochem. (2011) 116, 530–543
CaMKII-dependent recruitment of GluA1 in LTP | 533

depolarised in the presence of KN-62 (8.4 ± 2.4 P-CaMKIIpuncta/100 lm of dendrite, n = 6, p < 0.001) compared tocells treated with KCl without KN-62 (40.5 ± 1.9 P-CaMKIIpuncta/100 lm of dendrite, n = 6, Fig. 2a and c). No effectof KN-62 was seen at 9–11 DIV (Fig. 2c), which isconsistent with no change in P-CaMKII levels at thisdevelopmental stage of hippocampal neurons (Fig. 1a) andrules out potential non-specific effects of KN-62.
An NMDAR antagonist prevents CaMKII activationActivation of NMDARs is critical for the induction of LTP inhippocampal neuronal cultures (Fitzjohn et al. 2001; Lu
et al. 2001; Pickard et al. 2001). We have used a highlyselective NMDAR glycine site antagonist, L-689,560(5 lM), to investigate the role of NMDARs in CaMKIIactivation in hippocampal neuronal cultures following tran-sient depolarisation. In the presence of L-689,560, KCltreatment produced a significantly lower level of P-CaMKIIimmunolabelling (3.7 ± 1.5 puncta/100 lm dendrite, n = 6,p < 0.001) compared to KCl induction performed in theabsence of the glycine site antagonist (40.5 ± 1.9 puncta/100 lm dendrite, n = 6; Fig. 2b and c). The P-CaMKIIlabelling obtained with L-689,560 and KCl treated neurons(3.7 ± 1.5 puncta/100 lm dendrite, n = 6) was not signifi-
KN-62
3 × 1 s K+ ––
+–
++
L-689,560
3 × 1 s K+ ––
+–
++
P-CaMKII Synaptotagmin P-CaMKII Synaptotagmin P-CaMKII Synaptotagmin
P-CaMKII Synaptotagmin P-CaMKII Synaptotagmin P-CaMKII Synaptotagmin
(a)
(b)
(c)
0
5
10
15
20
25
30
35
40
45
9–11 DIV 18–21 DIV
% o
f syn
apse
s co
ntai
ning
P-C
aMK
II ** ****
3 × 1 s K+
KN-62L-689,560
– + +– – +– – –
– + +– – +– – –
+–+
Fig. 2 Immunocytochemical analysis of the
effects of the CaMKII inhibitor KN-62 (a)
and NMDAR antagonist L-689,560 (b) on
transient depolarisation induced autophos-
phorylation of CaMKII in hippocampal neu-
rons. Co-localisation of autophosphorylated
CaMKII (P-CaMKII, green) and synapto-
tagmin (red) in sham treated (left panels) K+
treated (90 mM KCl, 3 · 1 s, middle pan-
els) and 3 lM KN-62 (a) or 5 lM L-689,560
(b) treated (right panels) hippocampal neu-
rons in 21 DIV cultures. Scale bars: 20 lm
(top panels), 10 lm (enlarged regions).
Quantitative analysis (c) indicates that at
9–11 DIV K+ induction produced no
detectable changes in P-CaMKII immuno-
reactivity (p > 0.05; n = 6). In contrast, at
18–21 DIV K+ induction (3 · 1 s) signifi-
cantly increased P-CaMKII immunoreac-
tivity at synapses (**p < 0.001; n = 6)
compared to sham treated sister cultures.
K+ treatment induced increase in P-CaMKII
immunoreactivity at 18–21 DIV was signifi-
cantly reduced in the presence of KN-62
(**p < 0.001; n = 6) or L-689,560 (**p <
0.001; n = 4).
Journal of Neurochemistry � 2011 International Society for Neurochemistry, J. Neurochem. (2011) 116, 530–543� 2011 The Authors
534 | V. J. Appleby et al.

cantly different from non-depolarised sham treated cells(2.1 ± 0.6 puncta/100 lm dendrite, n = 6, p > 0.05), indi-cating that blockade of NMDARs reduces CaMKII activationto basal levels. Therefore the synaptic activation of CaMKIIin hippocampal neuronal cultures at 18–21 DIV is mediatedby NMDARs.
The CaMKII inhibitor KN-62 blocks the activity-inducedincrease in mEPSC frequencyIn cultured hippocampal neurons, transient depolarisation byKCl resulted in a large initial increase followed by a smaller
(� 50%) sustained increase in the frequency of mEPSCs(Fig. 3a and b) as reported previously (Fitzjohn et al. 2001).In the presence of KN-62 (3 lM), there was still a short-termincrease in mEPSC frequency, that lasted 5–10 min, but thesustained increase was prevented (Fig. 3c and d; p < 0.05,)KN-62 n = 10; +KN-62 n = 6). In contrast to mEPSCfrequency (Fig. 3e and g), there was no change in meanmEPSC amplitude following KCl treatment either in theabsence or the presence of KN-62 (Fig. 3f and h). Inaddition, mEPSCs had the same amplitude distributionsbefore and after the induction of LTP (Fig. 3f). These
mE
PS
C fr
eque
ncy
0
5
10
15
20
25
30
Time (min)0 5 10 15 20 25 30
Time (min)0 5 10 15 20 25 30
mE
PS
C fr
eque
ncy
05
10152025303540
(a)
(c)
(g) (h)
*
(e) (f)
Amplitude (pA)0 50 100
0.0
0.5
1.0
Interval (ms)0 200 400C
umul
ativ
e pr
obab
ility
0.0
0.5
1.0
Cum
ulat
ive
prob
abili
ty
Amplitude (pA)0 20 40
0.0
0.5
1.0
Interval (ms)0 1000 2000
0.0
0.5
1.0
KN-62
mE
PS
C fr
eque
ncy
(Hz)
0
4
8
12
KN-62
mE
PS
C a
mpl
itude
(pA
)
0
10
20
30
KN-62
+ KN-62
– + – +
– + KN-62 – +
(b)
(d)
– KN-62
Pre 3 × 1 s K+
Post 3 × 1 s K+
Pre 3 × 1 s K+
Post 3 × 1 s K+
*
Pre 3 × 1 s K+
Post 3 × 1 s K+Pre 3 × 1 s K+
Post 3 × 1 s K+
Pre 3 × 1 s K+
Post 3 × 1 s K+
Pre 3 × 1 s K+
Post 3 × 1 s K+
3 × 1 s K+
3 × 1 s K+
Fig. 3 KN-62 inhibits LTP in cultured hip-
pocampal neurons at 21 days in vitro. (a)
mEPSC frequency-time plot from an
example experiment (performed without
KN-62) showing an increase in frequency
following KCl application (arrow). Each
point represents 1 min bins. (b) Example
traces from the experiment shown in (a)
taken from before (top) and 25 min follow-
ing (bottom) KCl application. Downward
deflections are mEPSCs. Scale bar: 15 pA,
1 s. (c, d) Example experiment showing a
block of LTP induction in the presence of
KN-62. Scale bar: 10 pA, 1 s. (e) Cumula-
tive probability plots for example experi-
ments shown in (a–d) for mEPSCs
recorded before (solid line) and after (da-
shed line) KCl treatment. Note the shift in
inter-event interval to shorter intervals post-
KCl (e, )KN-62) but overlapping amplitude
curves (f). (g) Pooled data showing an in-
crease in mEPSC frequency post-KCl in
cells without KN-62 treatment (baseline
white bar, post-KCl black bar; n = 10;
p < 0.05) but not when KCl was applied in
the presence of KN-62 (*p < 0.05, n = 6).
(h) mEPSC amplitude was not changed by
KCl application with or without KN-62
treatment.
� 2011 The AuthorsJournal of Neurochemistry � 2011 International Society for Neurochemistry, J. Neurochem. (2011) 116, 530–543
CaMKII-dependent recruitment of GluA1 in LTP | 535

experiments demonstrate that in hippocampal neuronalcultures, the LTP that is observed as a sustained increase inmEPSC frequency, following brief transient depolarisationwith KCl, is dependent upon the activation of CaMKII.
LTP is associated with differential insertion of GluA1 andGluA2 subunitsThe transient depolarisation method that we have developedfor the investigation of LTP in primary hippocampal neuronalcultures recapitulates one aspect of the LTP repertoire,namely the insertion of AMPARs into the plasma membraneat sites initially lacking detectable AMPARs (i.e., anatom-ically ‘silent synapses’; Fitzjohn et al. 2001; Pickard et al.2001; Nash et al. 2010). Using sequential immunostaining(before and after LTP induction) we have shown thatAMPARs are inserted into the plasma membrane fromintracellular stores (Pickard et al. 2001). However, thesubunit composition of the newly recruited endogenousreceptors is unknown. Given the substantial amount ofevidence implicating CaMKII in LTP in general (Lismanet al. 2002), we set out to explore the role of this enzyme inthe activity-dependent recruitment of endogenous GluA1 andGluA2 subunit containing AMPARs to the cell surface. Toinvestigate this, we used extensively characterised GluA1and GluA2 subunit-specific antibodies to detect changes insurface expression of AMPAR subunits (Pickard et al. 2000,2001; Nash et al. 2010) at 21 DIV. Because the NMDARantagonist L-689,560 (5 lM) blocks KCl-induced LTP andthe associated AMPAR insertion in neuronal cultures (Fitz-john et al. 2001; Pickard et al. 2001; Nash et al. 2010), wecompared the effects of depolarisation in the presence andabsence of L-689,560. First, however, we confirmed theeffectiveness of the NMDAR inhibition by comparing thenumber of GluA1 puncta in depolarised, L-689,560 treatedneurons with non-depolarised neurons and observed nosignificant difference (Figure S2).
Five and thirty minutes after transient depolarisation ofneurons with brief pulses of high K+, cultures were labelledsimultaneously for surface GluA1 and GluA2, using anti-bodies directed at their extracellular N-terminal domains(Fig. 4a and b). The density of surface GluA1 and GluA2puncta along dendrites was compared in sister culturesexposed to high K+ with or without L-689,560 (Fig. 4). Incultures depolarised in the presence of L-689,560, there wereless GluA1 surface puncta (11.5 ± 1.2 puncta/100 lm den-drite, n = 6; Fig. 4c) compared to GluA2 positive surfacepuncta (17.4 ± 1.3 puncta/100 lm dendrite, n = 6; Fig. 4d).Five minutes after transient depolarisation (without L-689,560) there was a selective increase in the number ofsurface GluA1 puncta (19.9 ± 1.0 puncta/100 lm dendrite,p < 0.001, n = 6; Fig. 4c) with no significant change inGluA2 puncta number (19.0 ± 1.0 puncta/100 lm dendrite,p > 0.05, n = 6; Fig. 4d). Thirty minutes after KCl treatmentthere were similar increases in the number of GluA1 surface
puncta (19.1 ± 1.3 puncta/100 lm dendrite, versus 12.8 ±0.9 puncta/100 lm dendrite in interleaved L-689,560-treatedneuronal cultures, p < 0.001, n = 6; Fig. 4c), whereas GluA2puncta were again unaffected (LTP: 18.4 ± 1.2 puncta/100 lm dendrite versus L-689,560: 16.6 ± 1.2 puncta/100 lm dendrite, p > 0.05, n = 6; Fig. 4d). In addition tothe analysis of GluA1 and GluA2 immunopositive puncta,we also assessed overall changes in total GluA1 and GluA2fluorescence on the surface of dendrites (Fig. 4e and f).Quantification of pixel intensities in dendrites revealed a 2.0and 1.8-fold increase in surface GluA1 immunoreactivity 5and 30 min after KCl treatment, respectively (Fig. 4e),without detectable changes in GluA2 labelling (Fig. 4f).
Insertion of GluA1 is dependent on the activation CaMKIITo investigate the role of CaMKII in the plasma membranerecruitment of native AMPARs we focussed on the 30 mintime-point. In these experiments, we interleaved experimentswith either L-689,560, KN-62, or vehicle (LTP) (Fig. 5).Consistent with the previous data set, obtained with adifferent combination of secondary antibodies (Fig. 4), therewas a significant increase in the number of GluA1 positivepuncta in the LTP group relative to the other two groups(LTP: 18.5 ± 2.3 puncta/100 lm dendrite, L-689,560:9.7 ± 2.1 puncta/100 lm dendrite, KN-62: 10.6 ± 2.3 punc-ta/100 lm dendrite, n = 7, p < 0.002; Fig. 5b). Once again,the number or GluA2 positive puncta remained the same30 min following transient depolarisation in all three groups(LTP: 25.8 ± 2.5 puncta/100 lm dendrite, L-689,560: 23.3 ±3.5 puncta/100 lm dendrite, KN-62: 23.6 ± 2.1 puncta/100 lm dendrite, n = 7, p > 0.05; Fig. 5c).
In cultures depolarised in the presence of L-689,560, themajority of GluA1 puncta (� 80%) were also positive forGluA2 and this proportion remained the same in the LTP andKN-62 groups (LTP: 78.0 ± 5.3%, L-689,560: 81.3 ± 6.0%,KN-62: 76.7 ± 10.7%, p > 0.05, n = 7; Fig. 5d). A far lowerproportion of GluA2 puncta (� 40%) were also positive forGluA1 in cultures depolarised in the presence of L-689,560.However, there was a significant increase (� 46%) in thepercentage of GluA2 that co-localised with GluA1 in the LTPgroup (58.7 ± 6.7%) compared to the L-689,560-treatedgroup (40.0 ± 8.0%), an effect that was also prevented byKN-62 (40.0 ± 8.0%, p < 0.05, n = 7; Fig. 5e). These resultsindicate that endogenous GluA1-containing AMPARs areinserted into the plasma membrane mainly at sites thatalready contain GluA2. In addition, however, there is alsoinsertion into sites lacking GluA2 such that the proportion ofGluA1 positive, GluA2 negative sites remains similar.
To investigate if GluA1 is differentially recruited tosmaller or larger AMPAR clusters on dendrites, we per-formed cluster size analysis of GluA1 and GluA2 immuno-positive puncta. In cultures depolarised in the presence ofL-689,560, relatively more GluA1 (� 70%) than GluA2(� 50%) was associated with small puncta (� 1 lm3) with
Journal of Neurochemistry � 2011 International Society for Neurochemistry, J. Neurochem. (2011) 116, 530–543� 2011 The Authors
536 | V. J. Appleby et al.

GluA2GluA1
Pun
cta/
100
µm d
endr
ite
0
5
10
15
20
25
0
5
10
15
20
25
** **
5 min 30 min 5 min 30 min
GluA2GluA1 Merged
L-68
9,56
03 ×
1 s
K+
L-68
9,56
03 ×
1 s
K+
5 m
in30
min
(a)
(b)
(c)
LTP
LTP
Pun
cta/
100
µm d
endr
ite
GluA2GluA1
Ave
rage
pix
el in
tens
ity(a
rbitr
ary
units
)
0
0.1
0.2
0.3
0.4
0
0.1
0.2
0.3
** **
5 min 30 min
3 × 1 s K+
L-689,560+ + + +
+ +– –
3 × 1 s K+
L-689,560+ + + +
+ +– –3 × 1 s K+
L-689,560+ + + +
+ +– –
3 × 1 s K+
L-689,560+ + + +
+ +– –
5 min 30 min
Ave
rage
pix
el in
tens
ity(a
rbitr
ary
units
)
0.4
(e)
(d)
(f)
Fig. 4 Immunocytochemical analysis of
differential changes in the cell surface
expression of GluA1 and GluA2 AMPAR
subunits following transient depolarisation.
(a) Co-localisation of GluA1 (green) and
GluA2 (red) were carried out on the surface
of fixed, non-permeabilised hippocampal
neuronal cultures (21 DIV) using extracel-
lular N-terminal domain specific antibodies
5 min (a) and 30 min (b) following 3 · 1 s
pulses of 90 mM KCl in the absence of the
NMDAR antagonist L-689,560 (LTP) or in
the presence of 5 lM L-689,560 as indi-
cated on the left. GluA1 and GluA2 immu-
noreactivities were visualised by Alexa568
labelled anti-rabbit (green) and Alexa647
conjugated anti-mouse (red) secondary
antibodies, respectively. Scale bars: 10 lm.
(c,d) Quantitative analysis of pooled data
from six independent experiments indicate
significant increases in GluA1 puncta num-
bers (c; **p < 0.001, n = 6) at both 5 and
30 min following high K+ treatment but no
change in GluA2 immunoreactivity (d). (e,f)
Quantification of GluA1 and GluA2 average
pixel intensities on the surface of dendrites
indicate significant increases in GluA1 pixel
intensities (e; **p < 0.001, n = 6) at both 5
and 30 min following high K+ treatment but
no change in GluA2 immunoreactivity (f).
� 2011 The AuthorsJournal of Neurochemistry � 2011 International Society for Neurochemistry, J. Neurochem. (2011) 116, 530–543
CaMKII-dependent recruitment of GluA1 in LTP | 537
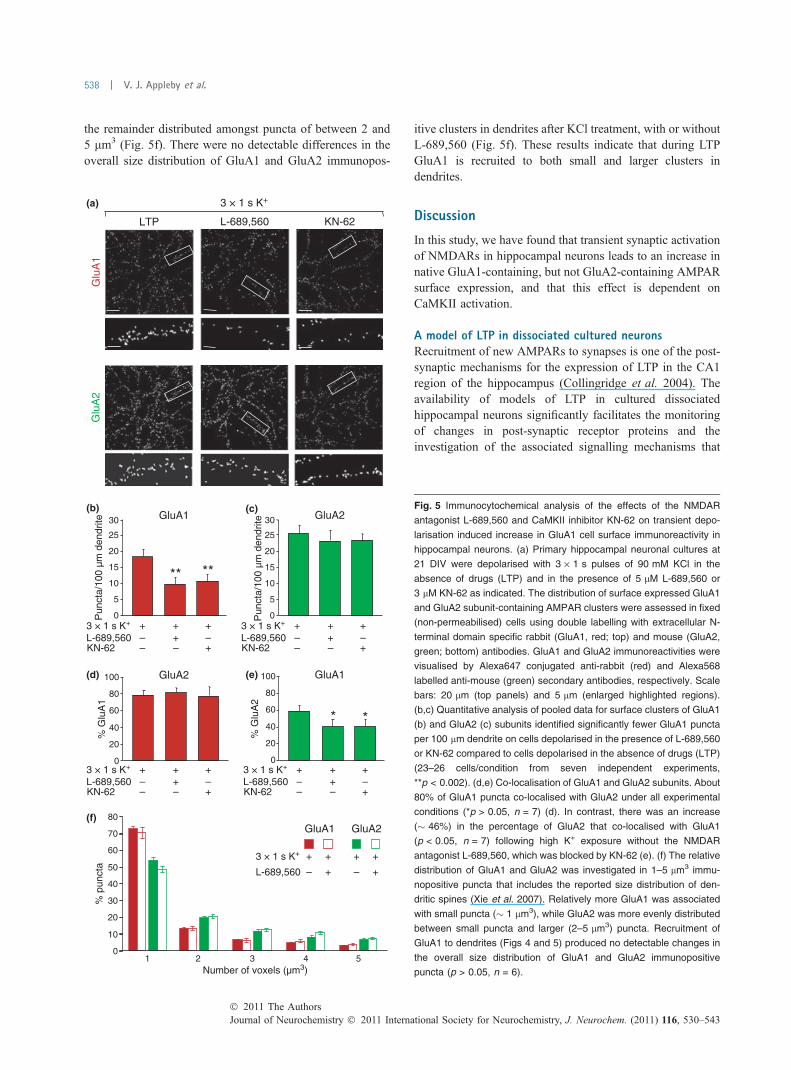
the remainder distributed amongst puncta of between 2 and5 lm3 (Fig. 5f). There were no detectable differences in theoverall size distribution of GluA1 and GluA2 immunopos-
itive clusters in dendrites after KCl treatment, with or withoutL-689,560 (Fig. 5f). These results indicate that during LTPGluA1 is recruited to both small and larger clusters indendrites.
Discussion
In this study, we have found that transient synaptic activationof NMDARs in hippocampal neurons leads to an increase innative GluA1-containing, but not GluA2-containing AMPARsurface expression, and that this effect is dependent onCaMKII activation.
A model of LTP in dissociated cultured neuronsRecruitment of new AMPARs to synapses is one of the post-synaptic mechanisms for the expression of LTP in the CA1region of the hippocampus (Collingridge et al. 2004). Theavailability of models of LTP in cultured dissociatedhippocampal neurons significantly facilitates the monitoringof changes in post-synaptic receptor proteins and theinvestigation of the associated signalling mechanisms that
Pun
cta/
100
µm d
endr
ite
0
5
10
15
20
25
GluA130 GluA2
GluA2
0
20
40
60
80
100
% G
luA
1
GluA1
0
20
40
60
80
100
% G
luA
2
0
5
10
15
20
25
30
** **
(b)
* *
Pun
cta/
100
µm d
endr
ite
3 × 1 s K+
L-689,560KN-62
+ ++–
– –
+
+–
3 × 1 s K+
L-689,560KN-62
+ ++–
– –
+
+–
3 × 1 s K+
L-689,560KN-62
+ ++–
– –
+
+–
3 × 1 s K+
L-689,560KN-62
+ ++–
– –
+
+–
Glu
A1
Glu
A2
KN-62
3 × 1 s K+(a)
L-689,560LTP
0
10
20
30
40
50
60
70
80
1 2 3 4 5
3 × 1 s K+
L-689,560 +– +–
+ + + +
GluA1 GluA2
% p
unct
a
Number of voxels (µm3)
(f)
(c)
(d) (e)
Fig. 5 Immunocytochemical analysis of the effects of the NMDAR
antagonist L-689,560 and CaMKII inhibitor KN-62 on transient depo-
larisation induced increase in GluA1 cell surface immunoreactivity in
hippocampal neurons. (a) Primary hippocampal neuronal cultures at
21 DIV were depolarised with 3 · 1 s pulses of 90 mM KCl in the
absence of drugs (LTP) and in the presence of 5 lM L-689,560 or
3 lM KN-62 as indicated. The distribution of surface expressed GluA1
and GluA2 subunit-containing AMPAR clusters were assessed in fixed
(non-permeabilised) cells using double labelling with extracellular N-
terminal domain specific rabbit (GluA1, red; top) and mouse (GluA2,
green; bottom) antibodies. GluA1 and GluA2 immunoreactivities were
visualised by Alexa647 conjugated anti-rabbit (red) and Alexa568
labelled anti-mouse (green) secondary antibodies, respectively. Scale
bars: 20 lm (top panels) and 5 lm (enlarged highlighted regions).
(b,c) Quantitative analysis of pooled data for surface clusters of GluA1
(b) and GluA2 (c) subunits identified significantly fewer GluA1 puncta
per 100 lm dendrite on cells depolarised in the presence of L-689,560
or KN-62 compared to cells depolarised in the absence of drugs (LTP)
(23–26 cells/condition from seven independent experiments,
**p < 0.002). (d,e) Co-localisation of GluA1 and GluA2 subunits. About
80% of GluA1 puncta co-localised with GluA2 under all experimental
conditions (*p > 0.05, n = 7) (d). In contrast, there was an increase
(� 46%) in the percentage of GluA2 that co-localised with GluA1
(p < 0.05, n = 7) following high K+ exposure without the NMDAR
antagonist L-689,560, which was blocked by KN-62 (e). (f) The relative
distribution of GluA1 and GluA2 was investigated in 1–5 lm3 immu-
nopositive puncta that includes the reported size distribution of den-
dritic spines (Xie et al. 2007). Relatively more GluA1 was associated
with small puncta (� 1 lm3), while GluA2 was more evenly distributed
between small puncta and larger (2–5 lm3) puncta. Recruitment of
GluA1 to dendrites (Figs 4 and 5) produced no detectable changes in
the overall size distribution of GluA1 and GluA2 immunopositive
puncta (p > 0.05, n = 6).
Journal of Neurochemistry � 2011 International Society for Neurochemistry, J. Neurochem. (2011) 116, 530–543� 2011 The Authors
538 | V. J. Appleby et al.

underlie LTP. While most of these models require prolongedpharmacological stimulation of neurons (Lu et al. 2001;Otmakhov et al. 2004a,b; Oh et al. 2006), we use briefdepolarisation to induce an increase in synaptic activity inhippocampal neurons (Fitzjohn et al. 2001; Pickard et al.2001; Nash et al. 2010). The transient depolarisation alsobriefly removes the Mg2+ block of NMDARs, which is likelyto facilitate the induction of LTP. Of course, high K+ mayrelease other factors in addition to L-glutamate that arepresent in dissociated cell cultures and can be released bybrief depolarisation in a TTX-independent manner. In termsof an in vitro model of LTP, the high K+ treatment releasesendogenous glutamate with temporal characteristics thatresemble that which would occur during the induction of LTPin more intact preparations, such as in hippocampal slicesand in vivo. By using an antibody that recognised allAMPAR subtypes (GluA1–4) we have shown previously thatthis form of LTP is associated with the insertion of AMPARsat sites that previously lacked AMPARs (Pickard et al.2001). These results are consistent with the reported increasein GluA1 on the cell surface identified by immunolabelling(Liao et al. 2001; Lu et al. 2001), direct visualisation ofexocytosed pH-sensitive green fluorescent protein tagged-GluA1 (Yudowski et al. 2007) and single-particle tracking ofsurface GluA1 subunits (Petrini et al. 2009) in hippocampalneurons following NMDAR activation. In the present study,we have studied two subunits, GluA1 and GluA2, to obtaininformation on their potential differential trafficking.
CaMKII activation is associated with LTP in hippocampalneuronal culturesThe present study provides direct visualisation of endoge-nous P-CaMKII in hippocampal neurons and demonstratesthat transient depolarisation-induced LTP is associated withthe activation of CaMKII. Transient depolarisation failed toactivate CaMKII in the presence of the NMDAR glycine-binding site antagonist L-689,560. This is consistent with thepreviously established role of NMDARs in the activation ofCaMKII (Fukunaga et al. 1993; Barria et al. 1997; Lee et al.2009). Our results are also consistent with the idea thatactivated CaMKII enzymes are translocated to synapses(Strack et al. 1997; Inagaki et al. 2000; Gardoni et al. 2001;Liao et al. 2001; Otmakhov et al. 2004b; Merrill et al. 2005;Skelding and Rostas 2009; Bingol et al. 2010). CaMKIIphosphorylation was particularly noticeable in NMDAR-containing synapses following LTP induction. This isconsistent with the hypothesis that CaMKII binds to GluN2BNMDAR subunits (Bayer et al. 2001, 2006; Barria andMalinow 2005; Zhou et al. 2007).
While the transient depolarisation-induced increase inactivated CaMKII was readily detectable in hippocampalneurons maintained for 2–3 weeks in vitro, it was notapparent in younger (< 10 DIV) cultures. Interestingly, LTPis not observed in these younger cultures either.
CaMKII-mediated regulation of AMPARs in NMDAR-LTPIt has been widely postulated that in hippocampal neuronsCaMKII provides a link between NMDAR activation andchanges in AMPARs in NMDAR-LTP (Lisman and Zhab-otinsky 2001; Lisman et al. 2002; Collingridge et al. 2004;Derkach et al. 2007). There are a number of possible, non-exclusive mechanisms by which this could occur.
Activated CaMKII can directly phosphorylate GluA1 atSer831 and this can lead to an increase in the mean singlechannel conductance of AMPARs and thereby potentiationof synaptic transmission (McGlade-McCulloh et al. 1993;Barria et al. 1997; Derkach et al. 1999, 2007). An increasein single channel conductance has been observed duringLTP (Benke et al. 1998) and CaMKII has been shown toincrease synaptic AMPAR single channel conductance(Poncer et al. 2002). However, inhibition of CaMKII doesnot affect LTP at a stage in development when the increasein single channel conductance is prominent (Wikstromet al. 2003). Furthermore, the functional regulation ofAMPARs by GluA1-Ser831 phosphorylation is lost in thepresence of the GluA2 subunit (Oh and Derkach 2005).Therefore, the extent to which a CaMKII-mediated increasein AMPAR single channel conductance is involved in LTPis unknown.
In addition to the modification of existing AMPARs viaphosphorylation, CaMKII may also be involved in therecruitment of new AMPARs at synapses during LTP(Hayashi et al. 2000). The mechanism by which CaMKIIdrives AMPARs into synapses is not fully understood.Mutation of Ser831, the CaMKII phosphorylation site on theC-terminal domain of GluA1, does not affect the delivery ofAMPARs, whereas deletion of the postsynaptic density95/disc-large/zona occludens (PDZ) binding domain on theC-terminus of GluA1 prevents synaptic insertion (Hayashiet al. 2000). One protein that binds this region of GluA1 isSAP97, a membrane associated guanylate kinase that isimportant for the trafficking of AMPARs to synapses(Rumbaugh et al. 2003; Nakagawa et al. 2004; Regaladoet al. 2006). CaMKII binds directly to SAP97 and over-expression of aCaMKII-Thr286Asp dramatically increasedSAP97 levels at post-synaptic terminals (Mauceri et al.2004). These observations indicate that CaMKII may beinvolved in the SAP97-mediated synaptic delivery of GluA1.
Another function of CaMKII may be to provide anchorageof AMPARs and tether them close to NMDARs (Lisman andZhabotinsky 2001). Indeed, recent evidence suggests a rolefor CaMKII in the trapping of laterally diffusing AMPARs atsynapses (Opazo et al. 2010).
Our cell culture model provides direct evidence thatCaMKII activation is necessary for the cell surface deliveryof endogenous AMPARs in NMDAR-LTP in hippocampalneurons, but does not preclude additional roles for CaMKIIin synaptic plasticity, including the involvement of CaMKIIin metaplasticity (Bortolotto and Collingridge 1998), hete-
� 2011 The AuthorsJournal of Neurochemistry � 2011 International Society for Neurochemistry, J. Neurochem. (2011) 116, 530–543
CaMKII-dependent recruitment of GluA1 in LTP | 539

rosynaptic plasticity (Rose et al. 2009) and synaptic tagging(Redondo et al. 2010).
GluA1-containing AMPARs are preferentially recruited tothe cell surface during NMDAR-LTPPrevious work has suggested that AMPAR trafficking isregulated by subunit-specific mechanisms (Nishimune et al.1998). For example, it has been suggested that GluA1/GluA2-containing AMPARs are inserted into the synapseduring LTP, whereas GluA3/GluA2-containing AMPARs areconstitutively inserted into the synapse (Shi et al. 2001;Molnar and Isaac 2002). In this model, GluA2 remains anintegral component of all AMPAR heteromers. Typically, theQ/R edited GluA2 subunit blocks Ca2+ permeability of mostAMPARs in glutamatergic principal neuron synapses underphysiological conditions (Cull-Candy et al. 2006; Gregerand Esteban 2007). However, previous electrophysiologicalstudies of CA1 hippocampal pyramidal neurons raised theintriguing possibility that AMPARs newly inserted duringLTP can be Ca2+-permeable (Plant et al. 2006). The transientexpression of Ca2+-permeable AMPARs would provide apossible mechanism to ‘tag’ newly potentiated synapses in anactivity-dependent manner and to drive the subsequent Ca2+-dependent changes in protein synthesis and gene expressionrequired for a stable increase in synaptic strength (Plant et al.2006; Isaac et al. 2007; Liu and Zukin 2007). However,other electrophysiological studies concluded that Ca2+-per-meable, GluA2-lacking AMPARs are not inserted duringLTP and these AMPARs are not required to maintain theincrease in synaptic strength (Adesnik and Nicoll 2007). Tofurther investigate this contentious issue, we studied relativechanges in the cell surface expression of GluA1 and GluA2in our LTP model. Our results lend support to the idea thatLTP can be associated with an increase in Ca2+-permeableAMPARs.
The proposed change in the GluA2 content of AMPARsfollowing LTP induction (Plant et al. 2006; Isaac et al. 2007;Liu and Zukin 2007) could be because of either a replace-ment of GluA2-containing with GluA2-lacking receptors, orthe addition of GluA2-lacking AMPARs to the existingcomplement. Our results indicate that the GluA1-containingAMPARs that are recruited to the plasma membrane duringLTP lack GluA2 subunits. It is likely, therefore, that therelative increase in the number of GluA1 increases thenumber of Ca2+-permeable AMPARs. Indeed, previous co-immunoprecipitation studies identified a substantial popula-tion of homomeric GluA1-containing AMPARs in total brainhomogenates (Wenthold et al. 1996). Our GluA1 and GluA2co-localisation experiments indicate that GluA1-containingAMPARs are mainly, but not exclusively, inserted at sitesthat contain GluA2. The selective increase in GluA1 content,at both GluA2 containing and GluA2 lacking sites, is likelyto correspond to an increase in the proportion of Ca2+-permeable AMPARs at synapses. Our studies showed that
this insertion of GluA1 happens rapidly (within 5 min) andthat the increase in the GluA1 to GluA2 ratio can persist forat least 30 min. In electrophysiological studies, the increasein Ca2+-permeable AMPARs was transient (Plant et al.2006). Under our experimental conditions internalisation ofsurface AMPARs was inhibited (Pickard et al. 2001; Glad-ding et al. 2009), therefore the replacement of GluA2 lackingwith GluA2 containing AMPARs has not been recapitulatedin this cell culture model.
Concluding remarks
We have demonstrated, using a dissociated cell culture modelto enable the immunolabelling of endogenous AMPARs, thatNMDAR-LTP is associated with a CaMKII-dependentinsertion of GluA1-containing AMPARs into the plasmamembrane. Our experiments indicate, therefore, that CaMKIIactivation plays an important role in the preferential recruit-ment of GluA1-containing AMPARs to the cell surface inNMDAR-LTP.
Acknowledgements
This work was supported by the Medical Research Council (MRC)
UK (grants 80049 and 57294) and the Wellcome Trust, UK (grant
056059). GLC is a World Class University International Scholar.
Supporting information
Additional Supporting information may be found in the online
version of this article:
Figure S1. Co-localisation of autophosphorylated CaMKII,
NMDAR subunit GluN1 and synaptotagmin in hippocampal
neurons.
Figure S2. Immunocytochemical analysis of GluA1 AMPA
receptor subunit clusters on the surface of hippocampal neurons
with and without transient depolarisation.
As a service to our authors and readers, this journal provides
supporting information supplied by the authors. Such materials are
peer-reviewed and may be re-organized for online delivery, but are
not copy-edited or typeset. Technical support issues arising from
supporting information (other than missing files) should be
addressed to the authors.
References
Adesnik H. and Nicoll R. A. (2007) Conservation of glutamate receptor2-containing AMPA receptors during long-term potentiation.J. Neurosci. 27, 4598–4602.
Anderson W. W. and Collingridge G. L. (2001) The LTP program: a dataacquisition program for on-line analysis of long-term potentiationand other synaptic events. J. Neurosci. Meth. 108, 71–83.
Barria A. and Malinow R. (2005) NMDA receptor subunit compositioncontrols synaptic plasticity by regulating binding to CaMKII.Neuron 48, 289–301.
Barria A., Muller D., Derkach V., Griffith L. C. and Soderling T. R.(1997) Regulatory phosphorylation of AMPA-type glutamate
Journal of Neurochemistry � 2011 International Society for Neurochemistry, J. Neurochem. (2011) 116, 530–543� 2011 The Authors
540 | V. J. Appleby et al.

receptors by CaM-KII during long-term potentiation. Science 276,2042–2045.
Bayer K. U., De Koninck P., Leonard A. S., Hell J. W. and Schulman H.(2001) Interaction with the NMDA receptor locks CaMKII in anactive conformation. Nature 411, 801–805.
Bayer K. U., LeBel E., McDonald G. L., O’Leary H., Schulman H. andDe Koninck P. (2006) Transition from reversible to persistentbinding of CaMKII to postsynaptic sites and NR2B. J. Neurosci.26, 1164–1174.
Benke T. A., Luthi A., Isaac J. T. and Collingridge G. L. (1998) Mod-ulation of AMPA receptor unitary conductance by synaptic activity.Nature 393, 793–797.
Bennett M. K., Erondu N. E. and Kennedy M. B. (1983) Purification andcharacterization of a calmodulin-dependent protein kinase that ishighly concentrated in brain. J. Biol. Chem. 258, 12735–12744.
Bingol B., Wang C., Amott D., Cheng D., Peng J. and Sheng M. (2010)Autophosphorylated CaMKIIa acts as a scaffold to recruit pro-teasomes to dendritic spine. Cell 140, 567–578.
Bortolotto Z. A. and Collingridge G. L. (1998) Involvement of calcium/calmodulin-dependent protein kinases in the setting of a molecularswitch involved in hippocampal LTP. Neuropharmacology 37,535–544.
Collingridge G. L., Isaac J. T. R. and Wang Y. T. (2004) Receptortrafficking and synaptic plasticity. Nat. Rev. Neurosci. 5, 952–962.
Cull-Candy S., Kelly L. and Farrant M. (2006) Regulation of Ca2+-permeable AMPA receptors: synaptic plasticity and beyond. Curr.Opin. Neurobiol. 16, 288–297.
Derkach V., Barria A. and Soderling T. R. (1999) Ca2+/calmodulin-kinase II enhances channel conductance of alpha-amino-3-hydro-xy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc.Natl Acad. Sci. USA 96, 3269–3274.
Derkach V. A., Oh M. C., Guire E. S. and Soderling T. R. (2007)Regulatory mechanisms of AMPA receptors in synaptic plasticity.Nat. Rev. Neurosci. 8, 101–113.
Fitzjohn S. M., Pickard L., Duckworth J. K., Molnar E., Henley J. M.,Collingridge G. L. and Noel J. (2001) An electrophysiologicalcharacterization of long-term potentiation in cultured dissociatedhippocampal neurons. Neuropharmacology 41, 693–699.
Fukunaga K., Stoppini L., Miyamoto E. and Muller D. (1993) Long-termpotentiation is associated with an increased activity of Ca2+/cal-modulin-dependent protein kinase II. J. Biol. Chem. 268, 7863–7867.
Fukunaga K., Muller D. and Miyamoto E. (1995) Increased phosphor-ylation of Ca2+/calmodulin-dependent protein kinase II and itsendogenous substrates in the induction of long-term potentiation.J. Biol. Chem. 270, 6119–6124.
Gardoni F., Schrama L. H., Kamal A., Gispen W. H., Cattabeni F. andDi Luca M. (2001) Hippocampal synaptic plasticity involvescompetition between Ca2+/calmodulin-dependent protein kinase IIand postsynaptic density 95 for binding to the NR2A subunit of theNMDA receptor. J. Neurosci. 21, 1501–1509.
Gladding C. M., Collett V. J., Jia Z., Bashir Z. I., Collingridge G. L.and Molnar E. (2009) Tyrosine dephosphorylation regulatesAMPAR internalisation in mGluR-LTD. Mol. Cell. Neurosci. 40,267–279.
Greger I. H. and Esteban J. A. (2007) AMPA receptor biogenesis andtrafficking. Curr. Opin. Neurobiol. 17, 289–297.
Hayashi Y., Shi S. H., Esteban J. A., Piccini A., Poncer J. C. andMalinow R. (2000) Driving AMPA receptors into synapses by LTPand CaMKII: requirement for GluR1 and PDZ domain interaction.Science 287, 2262–2267.
Hinds H. L., Tonegawa S. and Malinow R. (1998) CA1 long-term po-tentiation is diminished but present in hippocampal slices fromalpha-CaMKII mutant mice. Learn. Mem. 5, 344–354.
Inagaki N., Nishizawa M., Arimura N., Yamamoto H., Takeuchi Y.,Miyamoto E., Kaibuchi K. and Inagaki M. (2000) Activation ofCa2+/calmodulin-dependent protein kinase II within post-synapticdendritic spines of cultured hippocampal neurons. J. Biol. Chem.275, 27165–27171.
Isaac J. T. R., Ashby M. and McBain C. J. (2007) The role of the GluR2subunit in AMPA receptor function and synaptic plasticity. Neuron54, 859–871.
Ito I., Hidaka H. and Sugiyama H. (1991) Effects of KN-62, a specificinhibitor of calcium:calmodulin-dependent protein kinase II, onlong-term potentiation in the rat hippocampus. Neurosci. Lett. 121,119–121.
Kelly P. T., McGuinness T. L. and Greengard P. (1984) Evidence that themajor postsynaptic density protein is a component of a Ca2+/cal-modulin-dependent protein kinase. Proc. Natl Acad. Sci. USA 81,945–949.
Kennedy M. B., McGuinness T. and Greengard P. (1983) A calcium/calmodulin-dependent protein kinase from mammalian brain thatphosphorylates Synapsin I: partial purification and characteriza-tion. J. Neurosci. 3, 818–831.
Lee S. R., Escobedo-Lozoya E., Szatmari E. M. and Yasuda R. (2009)Activation of CaMKII in single dendritic spines during long-termpotentiation. Nature 458, 299–304.
Lengyel I., Voss K., Cammarota M., Bradshaw K., Brent V., MurphyK. P., Giese K. P., Rostas J. A. and Bliss T. V. (2004) Autonomousactivity of CaMKII is only transiently increased following theinduction of long-term potentiation in the rat hippocampus. Eur. J.Neurosci. 20, 3063–3072.
Liao D., Scannevin R. H. and Huganir R. (2001) Activation of silentsynapses by rapid activity-dependent synaptic recruitment ofAMPA receptors. J. Neurosci. 21, 6008–6017.
Lisman J. E. andZhabotinskyA.M. (2001)Amodel of synapticmemory: aCaMKII/PP1 switch that potentiates transmission by organizing anAMPA receptor anchoring assembly. Neuron 31, 191–201.
Lisman J., Schulman H. and Cline H. (2002) The molecular basis ofCaMKII function in synaptic and behavioural memory. Nat. Rev.Neurosci. 4, 175–190.
Liu S. J. and Zukin R. S. (2007) Ca2+-permeable AMPA receptors insynaptic plasticity and neuronal death. Trends Neurosci. 30, 126–134.
Lledo P. M., Hjelmstad G. O., Mukherji S., Soderling T. R., Malenka R.C. and Nicoll R. A. (1995) Calcium/calmodulin-dependentkinase II and long-term potentiation enhance synaptic transmissionby the same mechanism. Proc. Natl Acad. Sci. USA 92, 11175–11179.
Lou L. L., Lloyd S. J. and Schulman H. (1986) Activation of the mul-tifunctional Ca2+/calmodulin-dependent protein kinase by autop-hosphorylation: ATP modulates production of an autonomousenzyme. Proc. Natl Acad. Sci. USA 83, 9497–9501.
Lu W., Man H., Ju W., Trimble W. S., McDonald J. F. and Wang Y. T.(2001) Activation of synaptic NMDA receptors induces membraneinsertion of new AMPA receptors and LTP in cultured hippocampalneurons. Neuron 29, 243–254.
Malinow R., Schulman H. and Tsien R. W. (1989) Inhibition of post-synaptic PKC or CaMKII blocks induction but not expression ofLTP. Science 245, 862–866.
Mauceri D., Cattabeni F., Di Luca M. and Gardoni F. (2004) Calcium/calmodulin-dependent protein kinase II phosphorylation drivessynapse-associated protein 97 into spines. J. Biol. Chem. 279,23813–23821.
McGlade-McCulloh E., Yamamoto H., Tan S. E., Brickey D. A. andSoderling T. R. (1993) Phosphorylation and regulation of gluta-mate receptors by calcium/calmodulin-dependent protein kinase II.Nature 362, 640–642.
� 2011 The AuthorsJournal of Neurochemistry � 2011 International Society for Neurochemistry, J. Neurochem. (2011) 116, 530–543
CaMKII-dependent recruitment of GluA1 in LTP | 541

McGuinness T. L., Lai Y. and Greengard P. (1985) Ca2+/calmodulin-dependent protein kinase II. Isozymic forms from rat forebrain andcerebellum. J. Biol. Chem. 260, 1696–1704.
Merrill M. A., Chen Y., Strack S. and Hell J. W. (2005) Activity-drivenpostsynaptic translocation of CaMKII. Trends Pharmacol. Sci. 26,645–653.
Migues P. V., Lehmann I. T., Fluechter L., CammarotaM., Gurd J.W., SimA. T. R., Dickson P.W. and Rostas J. A. P. (2006) Phosphorylation ofCaKII at Thr253 occurs in vivo and enhances binding to isolatedpostsynaptic densities. J. Neurochem. 98, 289–299.
Miller S. G. and Kennedy M. B. (1986) Regulation of brain type II Ca2+/calmodulin-dependent protein kinase by autophosphorylation: aCa2+-triggered molecular switch. Cell 44, 861–870.
Molnar E. and Isaac J. T. R. (2002) Developmental and activity de-pendent regulation of ionotropic glutamate receptors at synapses.TheScientificWorldJOURNAL 2, 27–47.
Molnar E., Baude A., Richmond S. A., Patel P. B., Somogyi P. andMcIlhinney R. A. J. (1993) Biochemical and immunocytochemicalcharacterization of anti-peptide antibodies to a cloned GluR1 glu-tamate receptor subunit: cellular and subcellular distribution in therat forebrain. Neuroscience 53, 307–326.
Molnar E., Varadi A., McIlhinney R. A. J. and Ashcroft S. J. H. (1995)Identification of functional ionotropic glutamate receptor proteinsin pancreatic b-cells and in islets of Langerhans. FEBS Lett. 371,253–257.
Nakagawa T., Futai K., Lashuel H. A., Lo I., Okamoto K., Walz T.,Hayashi Y. and Sheng M. (2004) Quaternary structure, proteindynamics, and synaptic function of SAP97 controlled by L27 do-main interactions. Neuron 44, 453–467.
Nash J. E., Appleby V. J., Correa S. A. L., Wu H., Fitzjohn S. M., GarnerC. C., Collingridge G. L. and Molnar E. (2010) Disruption of theinteraction between myosin VI and SAP97 is associated with areduction in the number of AMPARs at hippocampal synapses.J. Neurochem. 112, 677–690.
Nishimune A., Isaac J. T., Molnar E., Noel J., Nash S. R., Tagaya M.,Collingridge G. L., Nakanishi S. and Henley J. M. (1998) NSFbinding to GluR2 regulates synaptic transmission. Neuron 21, 87–97.
Noel J., Ralph G. S., Pickard L., Williams J., Molnar E., Uney J. B.,Collingridge G. L. and Henley J. M. (1999) Surface expression ofAMPA receptors in hippocampal neurons is regulated by an NSF-dependent mechanism. Neuron 23, 365–376.
Oh M. C. and Derkach V. A. (2005) Dominant role of the GluR2 subunitin regulation of AMPA receptors by CaMKII. Nat. Neurosci. 8,853–854.
Oh M. C., Derkach V. A., Guire E. S. and Soderling T. R. (2006)Extrasynaptic membrane trafficking regulated by GluR1 serine 845phosphorylation primes AMPA receptors for long-term potentia-tion. J. Biol. Chem. 281, 752–758.
Opazo P., Labrecque S., Tigaret C. M., Frouin A., Wiseman P. W.,De Koninck P. and Choquet D. (2010) CaMKII triggers the dif-fusional trapping of surface AMPARs through phosphorylation ofstargazing. Neuron 67, 239–252.
Otmakhov N., Griffith L. C. and Lisman J. E. (1997) Postsynaptic in-hibitors of calcium/calmodulin-dependent protein kinase type IIblock induction but not maintenance of pairing-induced long-termpotentiation. J. Neurosci. 17, 5357–5365.
Otmakhov N., Khibnik L., Otmakhova N., Carpenter S., Riahi S., As-rican B. and Lisman J. (2004a) Forskolin-induced LTP in the CA1hippocampal region is NMDA receptor dependent. J. Neurophy-siol. 91, 1955–1962.
Otmakhov N., Tao-Cheng J., Carpenter S., Asrican B., Dosemeci A.,Reese T. S. and Lisman J. (2004b) Persistent accumulation ofcalcium/calmodulin-dependent protein kinase II in dendritic spines
after induction of NMDA receptor-dependent chemical long-termpotentiation. J. Neurosci. 24, 9324–9331.
Petrini E. M., Lu J., Cognet L., Lounis B., Ehlers M. D. and Choquet D.(2009) Endocytotic trafficking and recycling maintain a pool ofmobile surface AMPA receptors required for synaptic potentiation.Neuron 63, 92–105.
Pettit D. L., Perlman S. and Malinow R. (1994) Potentiated transmissionand prevention of further LTP by increased CaMKII activityin postsynaptic hippocampal slice neurons. Science 266, 1881–1885.
Pickard L., Noel J., Henley J. M., Collingridge G. L. and Molnar E.(2000) Developmental changes in synaptic AMPA and NMDAreceptor distribution and AMPA receptor subunit composition inliving hippocampal neurons. J. Neurosci. 20, 7922–7931.
Pickard L., Noel J., Duckworth J. K., Fitzjohn S. M., Henley J. M.,Collingridge G. L. and Molnar E. (2001) Transient synaptic acti-vation of NMDA receptors leads to the insertion of native AMPAreceptors at hippocampal neuronal plasma membranes. Neuro-pharmacology 41, 700–713.
Plant K., Pelkey K. A., Bortolotto Z. A., Morita D., Terashima A.,McBain C., Collingridge G. L. and Isaac J. T. (2006) Transientincorporation of native GluR2-lacking AMPA receptors duringhippocampal long-term potentiation. Nat. Neurosci. 9, 602–604.
Poncer J. C., Esteban J. A. and Malinow R. (2002) Multiple mechanismsfor the potentiation of AMPA receptor-mediated transcription byalpha-Ca2+/calmodulin-dependent protein kinase II. J. Neurosci.22, 4406–4411.
Redondo R. L., Okuno H., Spooner P. A., Frenguelli B. G. and MorrisR. G. (2010) Synaptic tagging and capture: differential role ofdistinct calcium/calmodulin kinases in protein synthesis-dependentlong-term potentiation. J. Neurosci. 30, 4981–4989.
Regalado M. P., Terry-Lorenzo R. T., Waites C. L., Garner C. C. andMalenka R. C. (2006) Transsynaptic signaling by postsynapticsynapse-associated protein 97. J. Neurosci. 26, 2343–2357.
Richmond S. A., Irving A. J., Molnar E., McIlhinney R. A. J., Miche-langeli F., Henley J. M. and Collingridge G. L. (1996) Localizationof the glutamate receptor subunit GluR1 on the surface of livingand within cultured hippocampal neurons. Neuroscience 75, 69–82.
Rose J., Jin S. X. and Craig A. M. (2009) Heterosynaptic moleculardynamics: locally induced propagating synaptic accumulation ofCaM kinase II. Neuron 61, 351–358.
Rumbaugh G., Sia G., Garner C. C. and Huganir R. L. (2003) Synapse-associated protein-97 isoform-specific regulation of surface AMPAreceptors and synaptic function in cultured neurons. J. Neurosci.23, 4567–4576.
Shi S., Hayashi Y., Esteban J. A. and Malinow R. (2001) Subunit-specific rules governing AMPA receptor trafficking to synapses inhippocampal pyramidal neurons. Cell 105, 331–343.
Silva A. J., Stevens C. F., Tonegawa S. and Wang Y. (1992) Deficienthippocampal long-term potentiation in alpha-calcium-calmodulinkinase II mutant mice. Science 257, 201–206.
Skelding K. A. and Rostas J. A. (2009) Regulation of CaMKII in vivo:The importance of targeting and the intracellular microenviron-ment. Neurochem. Res. 34, 1792–1804.
Strack S., Choi S., Lovinger D. M. and Colbran R. J. (1997) Translo-cation of autophosphorylated calcium/calmodulin-dependent pro-tein kinase II to the postsynaptic density. J. Biol. Chem. 272,13467–13470.
Tokumitsu H., Chijiwa T., Hagiwara M., Mizutani A., Terasawa M. andHidaka H. (1990) KN-62, 1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-L-tyrosyl]-4-phenylpiperazi ne, a specific inhibitor of Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 265, 4315–4320.
Journal of Neurochemistry � 2011 International Society for Neurochemistry, J. Neurochem. (2011) 116, 530–543� 2011 The Authors
542 | V. J. Appleby et al.

Vissavajjhala P., Janssen W. G. M., Hu Y., Gazzaley A. H., Moran T.,Hof P. R. and Morrison J. H. (1996) Synaptic distribution of theAMPA-GluR2 subunit and its colocalization with calcium-bindingproteins in rat cerebral cortex: an immunohistochemical studyusing a GluR2-specific monoclonal antibody. Exp. Neurol. 142,296–312.
Wenthold R. J., Petralia R. S., Blahos II J. and Niedzielski A. S. (1996)Evidence for multiple AMPA receptor complexes in hippocampalCA1/CA2 neurons. J. Neurosci. 16, 1982–1989.
Wikstrom M. A., Matthews P., Roberts D., Collingridge G. L. andBortolotto Z. A. (2003) Parallel kinase cascades are involved in theinduction of LTP at hippocampal CA1 synapses. Neuropharma-cology 45, 828–836.
Xie Z., Srivastava D. P., Photowala H., Kai L., Cahill M. E., Woolfrey K.M., Shum C. Y., Surmeier D. J. and Penzes P. (2007) Kalirin-7controls activity-dependent structural and functional plasticity ofdendritic spines. Neuron 56, 640–656.
Yudowski G. A., Puthenveedu M. A., Leonoudakis D., Panicker S.,Thorn K. S., Beattie E. C. and von Zastrow M. (2007) Real-timeimaging of discrete exocytic events mediating surface delivery ofAMPA receptors. J. Neurosci. 27, 11112–11121.
Zhou Y., Takahashi E., Li W., Halt A., Wiltgen B., Ehninger D., LiG., Hell J. W., Kennedy M. B. and Silva A. J. (2007)Interactions between the NR2B receptor and CaMKII modulatesynaptic plasticity and spatial learning. J. Neurosci. 27, 13843–13853.
� 2011 The AuthorsJournal of Neurochemistry � 2011 International Society for Neurochemistry, J. Neurochem. (2011) 116, 530–543
CaMKII-dependent recruitment of GluA1 in LTP | 543




















