Failure of Neuronal Maturation in Alzheimer Disease Dentate Gyrus
Upload
independentCategory
view
1download
0
Neuroscience Letters 384 (2005) 288–293
Palmitic and stearic fatty acids induce Alzheimer-likehyperphosphorylation of tau in primary rat cortical neurons
Sachin Patil, Christina Chan∗
Cellular and Biomolecular Laboratory, Department of Chemical Engineering and Material Science,Michigan State University, East Lansing, MI 48824, USA
Received 14 February 2005; received in revised form 26 April 2005; accepted 5 May 2005
Abstract
Epidemiological studies suggest that high fat diets significantly increase the risk of Alzheimer’s disease (AD). In addition, the AD brain ischaracterized by high fatty acid content compared to that of healthy subjects. Nevertheless, the basic mechanism relating elevated fatty acidsand the pathogenesis of AD remains unclear. The present study examines the role of fatty acids in causing hyperphosphorylation of the tauprotein, one of the characteristic signatures of AD pathology. Hyperphosphorylation of tau disrupts the cell cytoskeleton and leads to neuronaldegeneration. Here, primary rat cortical neurons and astrocytes were treated with saturated free fatty acids (FFAs), palmitic and stearic acids.T media fromF o-treatmento h a centralr©
K
Adbrfihdd[
i
aA(o5k
ighges
aysma
a-sma
orols
edthe
rainitic,b-
en
0d
here was no change in the levels of phosphorylated tau in rat cortical neurons treated directly with these FFAs. The conditionedFA-treated astrocytes, however, caused hyperphosphorylation of tau in the cortical neurons at AD-specific phospho-epitopes. Cf neurons withN-acetyl cysteine, an antioxidant, reduced FFA-induced hyperphosphorylation of tau. The present results establisole of FFAs in causing hyperphosphorylation of tau through astroglia-mediated oxidative stress.
2005 Elsevier Ireland Ltd. All rights reserved.
eywords:Alzheimer’s disease; Saturated fatty acids; Oxidative stress; Tau hyperphosphorylation; Primary cortical neurons
lzheimer’s disease (AD) is a progressive neurodegenerativeisorder, characterized by extracellular deposits of amyloideta (A�) protein and intracellular accumulation of neurofib-illary tangles (NFTs)[20]. NFTs consist of paired helicallaments of microtubule-associated tau protein, which isyperphosphorylated[16]. The hyperphosphorylation of tauisrupts the cytoskeleton of neurons, which leads to theiregeneration thus playing an important role in AD pathology
20].Epidemiological studies suggest that high fat diets signif-
cantly increase the risk of AD and the degree of saturation
Abbreviations: AD, Alzheimer’s disease; FFAs, free fatty acids; A�,myloid beta; NFTs, neurofibillary tangles; BBB, blood–brain barrier;poE4, apolipoprotein E4; NAC,N-acetyl cysteine; CM-H2DCFDA, 5-
6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate; ROS, reactivexygen species; H2O2, hydrogen peroxide; cdk5, cyclin-dependent kinase; GSK-3, glycogen synthase kinase-3; MAPK, mitogen-activated proteininase∗ Corresponding author. Tel.: +1 517 432 4530; fax: +1 517 432 1105.E-mail address:[email protected] (C. Chan).
of fatty acids is critical in determining the risk for AD[12].Animal studies support this notion; mice fed high-fat–hcholesterol diet develop AD-like pathophysiological chanin their brain [17,32,33]. Diets rich in saturated fats mincrease brain uptake of intact free fatty acids from the plathrough the blood brain barrier (BBB)[35]; the BBB is not abarrier for fatty acids[7]. It may be noteworthy that dibetes mellitus, which is characterized by elevated plalevels of saturated FFAs[22], is also a significant risk factfor AD [1]. Due to the interaction between the FFA poin the plasma and the brain[7,34,35], diabetes-associatincreases in plasma FFAs may affect the level of FFAs inbrain and increase the risk for AD. Likewise, traumatic binjury, which is associated with elevated levels of palmstearic and arachidonic acids in the brain[14], has been estalished as an independent risk factor for AD[13]. Followingtraumatic brain injury, palmitic acid increases from∼60 to180�M and stearic acid from∼50 to 350�M [18]. In addi-tion, the fatty acid profile of NFTs in AD brain has beshown to be rich in palmitic and stearic fatty acids[11].
304-3940/$ – see front matter © 2005 Elsevier Ireland Ltd. All rights reserved.oi:10.1016/j.neulet.2005.05.003

S. Patil, C. Chan / Neuroscience Letters 384 (2005) 288–293 289
Similarly, the white matter in AD brain is characterizedby high fatty acid content[29]. Finally, the genetic factor,apolipoprotein E4 (ApoE4), known to increase the risk ofAD, requires a hyperlipidemic lifestyle to manifest into AD[26]. Despite the accumulating data, the basic mechanismbehind the causal relationship between fatty acids and thepathogenesis of AD has not been established.
Previously, FFAs have been shown to stimulate the poly-merization of purified tau filaments in vitro[37]. The presentstudy focused on the role of FFAs in causing hyperphosphory-lation of tau in primary rat cortical neurons. Primary corticalneurons were isolated from one-day-old Sprague–Dawley ratpups and cultured according to the published methods asdescribed in Chandler et al.[3]. The cells were plated onpoly-d-lysine-coated, six-well plates at the concentration of2× 106 cells per well in fresh cortical medium [Dulbecco’sModified Eagle’s Medium (DMEM and all other media arefrom Invitrogen, CA, USA) supplemented with 10% horseserum (Sigma, MO, USA), 25 mM glucose, 10 mM HEPES(Sigma), 2 mM glutamine (BioSource International, CA,USA), 100 IU/ml penicillin, and 0.1 mg/ml streptomycine].Three days after incubation (37◦C, 5% CO2), the mediumwas subsequently replaced with 2 ml of cortical medium sup-plemented with 5�M cytosine-�-arabinofuranoside (Aracfrom Calbiochem, CA, USA). After 2 days, the neuronal cul-ture was switched back to cortical medium without Arac. Thee . Too ellsf ed inD um( mlscpa -fourh wasc
imesw nd5 ldr ri-t ris,pE mS ifu-g nc fromP einf GEg tin.T losem tw omD rceB rew ated
with appropriate HRP-linked secondary antibodies (PierceBiotechnology, IL, USA) diluted in PBS-T for 1 h. After anadditional three washes in PBS-T, blots were developed withthe Pierce SuperSignal West Femto Maximum SensitivitySubstrate (Pierce Biotechnology) and imaged with the Bio-Rad ChemiDoc.
To perform confocal immunofluorescence microscopicstudy, neurons and astrocytes cultures were fixed for 20 minin 4% paraformaldehyde and permeabilized with 0.1% Tri-ton X-100 and 5% goat serum (Invitrogen) in PBS. Cellswere then labeled overnight at 4◦C with appropriate pri-mary antibodies [1:50 MAP-2 (Santa Cruz Biotechnology,CA, USA) for neurons, 1:1000 GFAP (Dako, CA, USA) forastrocytes and 1:200 AT8 for hyperphosphorylated tau) in5% goat serum in PBS. After three PBS washes, primary anti-bodies were detected with rhodamine conjugated (Chemicon,CA, USA) or Alexa Flour 594 conjugated (Molecular Probes,OR, USA) secondary antibodies. The cells were visualizedwith confocal microscope Zeiss LSM 5 Pascal (Carl Zeiss,Jena, Germany) using a 40× (for MAP-2 and GFAP) or 63×(for AT8) oil-immersion objective lens.
Intracellular reactive oxygen species (ROS) weredetected by staining with the oxidant-sensitive dye 5-(6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate(CM-H2DCFDA, from Molecular Probes). H2DCFDA iscleaved of the ester groups by intracellular esterases and con-v riva-t nhcH nolr withP M 5P ford
oft atedw h.A lysisw per-p ls ofp ctlywF ctedb ain-i ityo cids[ igherc -c n toi opto-s igh( after2 wet ors edia
xperiments were performed on 6- to 7-day-old culturebtain primary cultures of astroglial cells, the cortical c
rom one-day-old Sprague–Dawley rat pups were culturMEM/Ham’s F12 medium (1:1), 10% fetal bovine ser
Biomeda, CA, USA), 100 IU/ml penicillin, and 0.1 mg/treptomycine[28]. The cells were plated on poly-d-lysineoated, 6-well plates at the concentration of 2× 106 cellser well. Cells were grown for 8–10 days (37◦C, 5% CO2)nd culture medium was changed every 2 days. Twentyours prior to treatment with fatty acids, the mediumhanged to neuronal cell culture medium.
For Western blot analysis, cells were washed three tith ice-cold TBS (25 mM Tris, pH 8.0, 140 mM NaCl, amM KCl) and lysed for 20 min by scraping into ice-co
adioimmunoprecipitation assay (RIPA) buffer [1% (v/v) Ton, 0.1% (w/v) SDS, 0.5% (w/v) deoxycholate, 20 mM TH 7.4, 150 mM NaCl, 100 mM NaF, 1 mM Na3VO4, 1 mMDTA, 1 mM EGTA, and 1 mM PMSF, all chemicals froigma][36]. The total cell lysate was obtained by centration at 12,000 rpm for 15 min at 4◦C. The total proteioncentration was measured by BCA protein assay kitierce (Rockford, IL, USA). Equal amounts of total prot
rom each condition were run at 200 V on 10% SDS–PAels (BioRad, CA, USA) for phosphorylated tau and ache separated proteins were transferred to nitrocelluembranes for 1 h at 100 V and incubated at 4◦C overnighith the appropriate primary antibodies [1:200 PHF-1 (frr. P. Davies, Albert Einstein, NY, USA), 1:200 AT8 (Pieiotechnology, IL, USA), 1:2000 actin (Sigma)]. Blots weashed three times in PBS–Tween (PBS-T) and incub
erted into membrane impermeable, nonfluorescent deive H2DCF. Oxidation of H2DCF by ROS results iighly fluorescent 2,7-dichlorofluorescein (DCF)[30]. Theells were incubated for 30 min at 37◦C with 2�M CM-2DCFDA in Hanks’ Balanced Salt Solution without phe
ed (Invitrogen). The cells were then washed three timesBS and analyzed with confocal microscopy (Zeiss LSascal). A 63× oil-immersion objective lens was usedata acquisition.
To examine the FFA-induced hyperphosphorylationau, primary rat cortical neurons were left untreated or treith 0.2 mM of either palmitic or stearic acids for 24fter 24 h, the cells were lysed and Western blot anaas performed to determine the cellular levels of hyhosphorylated tau. There was no change in the levehosphorylated tau in rat cortical neurons treated direith palmitic or stearic acid, as compared to controls (Fig. 1).urthermore, the morphology of the neurons was not affey the FFA treatment, as shown by MAP-2 immunost
ng (Fig. 2A). This may be attributed to the low capacf primary neurons to take up and metabolize fatty a
2]. The astrocytes, in contrast, possess a relatively hapacity to utilize fatty acids[2]. Primary rat cortical astroytes treated with 0.2 mM palmitic acid have been showncrease de novo synthesis of ceramide and undergo apis in a time-dependent manner; the cell viability was habout 85–90%) until 12 h and decreased to 65–70%4 h of treatment[2]. Therefore, in the present study,
reated the rat cortical astrocytes with 0.2 mM palmitictearic acid for 12 h and transferred the conditioned m
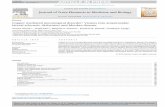
290 S. Patil, C. Chan / Neuroscience Letters 384 (2005) 288–293
Fig. 1. Direct treatment of neurons with saturated FFAs. Primary rat corti-cal neurons were treated for 24 h with 0.2 mM of either palmitic acid (PA)or stearic acid (SA) or with 5% bovine serum albumin (BSA), vehicle forFFAs (control). Detergent cell lysates from fatty acid-treated and controlcells were immunoblotted with PHF-1 and AT8 antibodies, which recognizephosphorylated tau.�-actin is shown as a marker for protein loading.
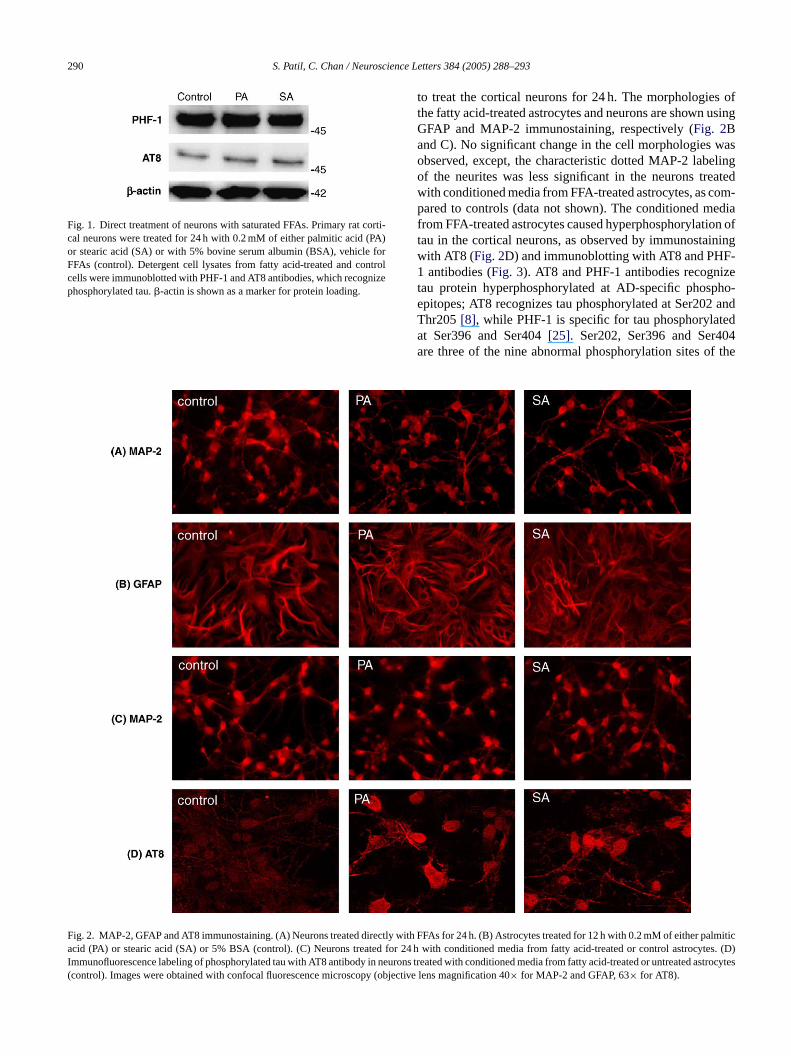
to treat the cortical neurons for 24 h. The morphologies ofthe fatty acid-treated astrocytes and neurons are shown usingGFAP and MAP-2 immunostaining, respectively (Fig. 2Band C). No significant change in the cell morphologies wasobserved, except, the characteristic dotted MAP-2 labelingof the neurites was less significant in the neurons treatedwith conditioned media from FFA-treated astrocytes, as com-pared to controls (data not shown). The conditioned mediafrom FFA-treated astrocytes caused hyperphosphorylation oftau in the cortical neurons, as observed by immunostainingwith AT8 (Fig. 2D) and immunoblotting with AT8 and PHF-1 antibodies (Fig. 3). AT8 and PHF-1 antibodies recognizetau protein hyperphosphorylated at AD-specific phospho-epitopes; AT8 recognizes tau phosphorylated at Ser202 andThr205[8], while PHF-1 is specific for tau phosphorylatedat Ser396 and Ser404[25]. Ser202, Ser396 and Ser404are three of the nine abnormal phosphorylation sites of the
FaI(
ig. 2. MAP-2, GFAP and AT8 immunostaining. (A) Neurons treated directly wcid (PA) or stearic acid (SA) or 5% BSA (control). (C) Neurons treated for
mmunofluorescence labeling of phosphorylated tau with AT8 antibody in neucontrol). Images were obtained with confocal fluorescence microscopy (obj
ith FFAs for 24 h. (B) Astrocytes treated for 12 h with 0.2 mM of either palmitic24 h with conditioned media from fatty acid-treated or control astrocytes. (D)
rons treated with conditioned media from fatty acid-treated or untreatedastrocytesective lens magnification 40× for MAP-2 and GFAP, 63× for AT8).
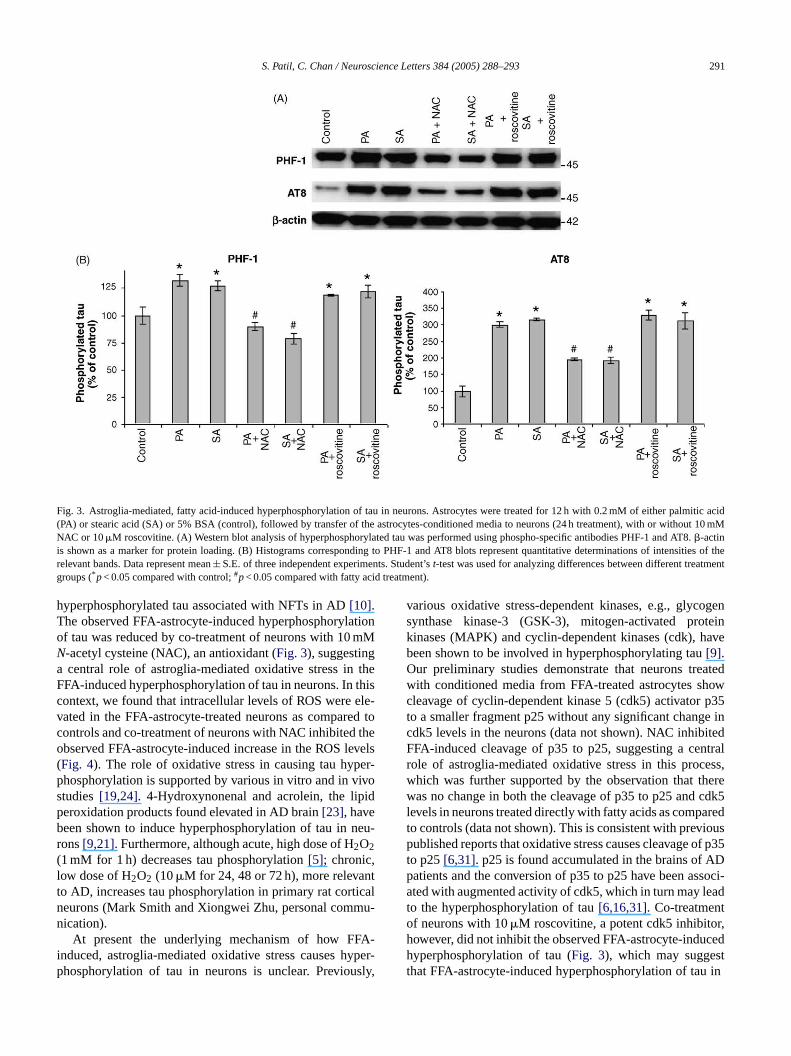
S. Patil, C. Chan / Neuroscience Letters 384 (2005) 288–293 291
Fig. 3. Astroglia-mediated, fatty acid-induced hyperphosphorylation of tau in neurons. Astrocytes were treated for 12 h with 0.2 mM of either palmitic acid(PA) or stearic acid (SA) or 5% BSA (control), followed by transfer of the astrocytes-conditioned media to neurons (24 h treatment), with or without 10mMNAC or 10�M roscovitine. (A) Western blot analysis of hyperphosphorylated tau was performed using phospho-specific antibodies PHF-1 and AT8.�-actinis shown as a marker for protein loading. (B) Histograms corresponding to PHF-1 and AT8 blots represent quantitative determinations of intensities of therelevant bands. Data represent mean± S.E. of three independent experiments. Student’st-test was used for analyzing differences between different treatmentgroups (*p< 0.05 compared with control;#p< 0.05 compared with fatty acid treatment).
hyperphosphorylated tau associated with NFTs in AD[10].The observed FFA-astrocyte-induced hyperphosphorylationof tau was reduced by co-treatment of neurons with 10 mMN-acetyl cysteine (NAC), an antioxidant (Fig. 3), suggestinga central role of astroglia-mediated oxidative stress in theFFA-induced hyperphosphorylation of tau in neurons. In thiscontext, we found that intracellular levels of ROS were ele-vated in the FFA-astrocyte-treated neurons as compared tocontrols and co-treatment of neurons with NAC inhibited theobserved FFA-astrocyte-induced increase in the ROS levels(Fig. 4). The role of oxidative stress in causing tau hyper-phosphorylation is supported by various in vitro and in vivostudies[19,24]. 4-Hydroxynonenal and acrolein, the lipidperoxidation products found elevated in AD brain[23], havebeen shown to induce hyperphosphorylation of tau in neu-rons[9,21]. Furthermore, although acute, high dose of H2O2(1 mM for 1 h) decreases tau phosphorylation[5]; chronic,low dose of H2O2 (10�M for 24, 48 or 72 h), more relevantto AD, increases tau phosphorylation in primary rat corticalneurons (Mark Smith and Xiongwei Zhu, personal commu-nication).
At present the underlying mechanism of how FFA-induced, astroglia-mediated oxidative stress causes hyper-phosphorylation of tau in neurons is unclear. Previously,
various oxidative stress-dependent kinases, e.g., glycogensynthase kinase-3 (GSK-3), mitogen-activated proteinkinases (MAPK) and cyclin-dependent kinases (cdk), havebeen shown to be involved in hyperphosphorylating tau[9].Our preliminary studies demonstrate that neurons treatedwith conditioned media from FFA-treated astrocytes showcleavage of cyclin-dependent kinase 5 (cdk5) activator p35to a smaller fragment p25 without any significant change incdk5 levels in the neurons (data not shown). NAC inhibitedFFA-induced cleavage of p35 to p25, suggesting a centralrole of astroglia-mediated oxidative stress in this process,which was further supported by the observation that therewas no change in both the cleavage of p35 to p25 and cdk5levels in neurons treated directly with fatty acids as comparedto controls (data not shown). This is consistent with previouspublished reports that oxidative stress causes cleavage of p35to p25[6,31]. p25 is found accumulated in the brains of ADpatients and the conversion of p35 to p25 have been associ-ated with augmented activity of cdk5, which in turn may leadto the hyperphosphorylation of tau[6,16,31]. Co-treatmentof neurons with 10�M roscovitine, a potent cdk5 inhibitor,however, did not inhibit the observed FFA-astrocyte-inducedhyperphosphorylation of tau (Fig. 3), which may suggestthat FFA-astrocyte-induced hyperphosphorylation of tau in
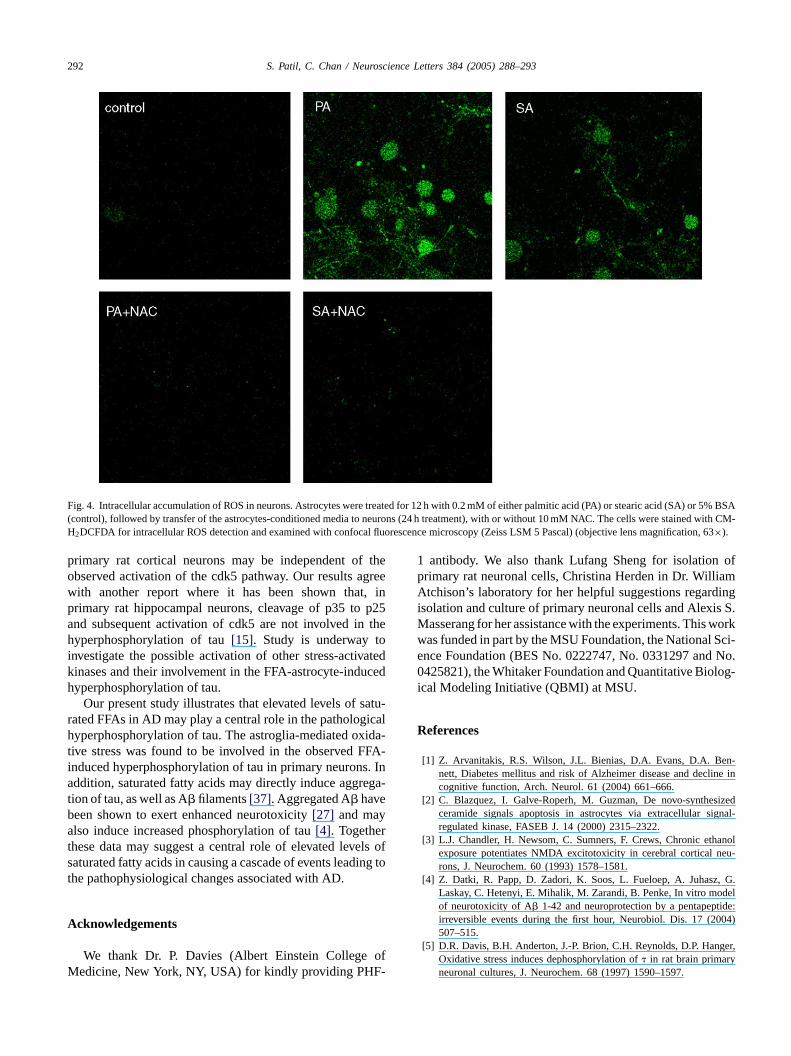
292 S. Patil, C. Chan / Neuroscience Letters 384 (2005) 288–293
Fig. 4. Intracellular accumulation of ROS in neurons. Astrocytes were treated for 12 h with 0.2 mM of either palmitic acid (PA) or stearic acid (SA) or 5%BSA(control), followed by transfer of the astrocytes-conditioned media to neurons (24 h treatment), with or without 10 mM NAC. The cells were stained with CM-H2DCFDA for intracellular ROS detection and examined with confocal fluorescence microscopy (Zeiss LSM 5 Pascal) (objective lens magnification, 63×).
primary rat cortical neurons may be independent of theobserved activation of the cdk5 pathway. Our results agreewith another report where it has been shown that, inprimary rat hippocampal neurons, cleavage of p35 to p25and subsequent activation of cdk5 are not involved in thehyperphosphorylation of tau[15]. Study is underway toinvestigate the possible activation of other stress-activatedkinases and their involvement in the FFA-astrocyte-inducedhyperphosphorylation of tau.
Our present study illustrates that elevated levels of satu-rated FFAs in AD may play a central role in the pathologicalhyperphosphorylation of tau. The astroglia-mediated oxida-tive stress was found to be involved in the observed FFA-induced hyperphosphorylation of tau in primary neurons. Inaddition, saturated fatty acids may directly induce aggrega-tion of tau, as well as A� filaments[37]. Aggregated A� havebeen shown to exert enhanced neurotoxicity[27] and mayalso induce increased phosphorylation of tau[4]. Togetherthese data may suggest a central role of elevated levels ofsaturated fatty acids in causing a cascade of events leading tothe pathophysiological changes associated with AD.
Acknowledgements
We thank Dr. P. Davies (Albert Einstein College ofM -
1 antibody. We also thank Lufang Sheng for isolation ofprimary rat neuronal cells, Christina Herden in Dr. WilliamAtchison’s laboratory for her helpful suggestions regardingisolation and culture of primary neuronal cells and Alexis S.Masserang for her assistance with the experiments. This workwas funded in part by the MSU Foundation, the National Sci-ence Foundation (BES No. 0222747, No. 0331297 and No.0425821), the Whitaker Foundation and Quantitative Biolog-ical Modeling Initiative (QBMI) at MSU.
References
[1] Z. Arvanitakis, R.S. Wilson, J.L. Bienias, D.A. Evans, D.A. Ben-nett, Diabetes mellitus and risk of Alzheimer disease and decline incognitive function, Arch. Neurol. 61 (2004) 661–666.
[2] C. Blazquez, I. Galve-Roperh, M. Guzman, De novo-synthesizedceramide signals apoptosis in astrocytes via extracellular signal-regulated kinase, FASEB J. 14 (2000) 2315–2322.
[3] L.J. Chandler, H. Newsom, C. Sumners, F. Crews, Chronic ethanolexposure potentiates NMDA excitotoxicity in cerebral cortical neu-rons, J. Neurochem. 60 (1993) 1578–1581.
[4] Z. Datki, R. Papp, D. Zadori, K. Soos, L. Fueloep, A. Juhasz, G.Laskay, C. Hetenyi, E. Mihalik, M. Zarandi, B. Penke, In vitro modelof neurotoxicity of A� 1-42 and neuroprotection by a pentapeptide:irreversible events during the first hour, Neurobiol. Dis. 17 (2004)507–515.
[5] D.R. Davis, B.H. Anderton, J.-P. Brion, C.H. Reynolds, D.P. Hanger,Oxidative stress induces dephosphorylation of� in rat brain primary
edicine, New York, NY, USA) for kindly providing PHF
neuronal cultures, J. Neurochem. 68 (1997) 1590–1597.
S. Patil, C. Chan / Neuroscience Letters 384 (2005) 288–293 293
[6] S.M. de la Monte, N. Ganju, N. Feroz, T. Luong, K. Banerjee,J. Cannon, J.R. Wands, Oxygen free radical injury is sufficient tocause some Alzheimer-type molecular abnormalities in human CNSneuronal cells, J. Alzheimer’s Dis. 2 (2000) 261–281.
[7] G.A. Dhopeshwarkar, J.F. Mead, Uptake and transport of fatty acidsinto the brain and role of the blood–brain barrier system, Adv. LipidRes. 11 (1973) 109–142.
[8] M. Goedert, R. Jakes, E. Vanmechelen, Monoclonal antibody AT8recognizes tau protein phosphorylated at both serine 202 and threo-nine 205, Neurosci. Lett. 189 (1995) 167–170.
[9] A. Gomez-Ramos, J. Diaz-Nido, M.A. Smith, G. Perry, J. Avila,Effect of the lipid peroxidation product acrolein on tau phosphory-lation in neural cells, J. Neurosci. Res. 71 (2003) 863–870.
[10] C.X. Gong, T.J. Singh, I. Grundke-Iqbal, K. Iqbal, Alzheimer’s dis-ease abnormally phosphorylated� is dephosphorylated by proteinphosphatase-2B (calcineurin), J. Neurochem. 62 (1994) 803–806.
[11] W.J. Goux, S. Rodriguez, D.R. Sparkman, Analysis of the core com-ponents of Alzheimer paired helical filaments. A gas chromatogra-phy/mass spectrometry characterization of fatty acids, carbohydratesand long-chain bases, FEBS Lett. 366 (1995) 81–85.
[12] W.B. Grant, Dietary links to Alzheimer’s disease: 1999 update, J.Alzheimer’s Dis. 1 (1999) 197–201.
[13] Z. Guo, L.A. Cupples, A. Kurz, S.H. Auerbach, L. Volicer, H. Chui,R.C. Green, A.D. Sadovnick, R. Duara, C. DeCarli, K. Johnson, R.C.Go, J.H. Growdon, J.L. Haines, W.A. Kukull, L.A. Farrer, Headinjury and the risk of AD in the MIRAGE study, Neurology 54(2000) 1316–1323.
[14] P. Homayoun, E.B.R. De Turco, N.E. Parkins, D.C. Lane, J.Soblosky, M.E. Carey, N.G. Bazan, Delayed phospholipid degra-dation in rat brain after traumatic brain injury, J. Neurochem. 69(1997) 199–205.
[ tila,doesom-
[ ai,405
[ sedallyalth
[ . 79
[ uc-cul-ase-3,
[ ease,
[ nal,of
997)
[ .P.ialand
siol.
[23] T.J. Montine, M.D. Neely, J.F. Quinn, M.F. Beal, W.R. Markes-bery, L.J. Roberts, J.D. Morrow, Lipid peroxidation in aging brainand Alzheimer’s disease, Free Radic. Biol. Med. 33 (2002) 620–626.
[24] H. Nakashima, T. Ishihara, O. Yokota, S. Terada, J.Q. Trojanowski,V.M.Y. Lee, S. Kuroda, Effects of�-tocopherol on an animal modelof tauopathies, Free Radic. Biol. Med. 37 (2004) 176–186.
[25] L. Otvos Jr., L. Feiner, E. Lang, G.I. Szendrei, M. Goedert, V.M.Y.Lee, Monoclonal antibody PHF-1 recognizes tau protein phospho-rylated at serine residues 396 and 404, J. Neurosci. Res. 39 (1994)669–673.
[26] G.J. Petot, F. Traore, S.M. Debanne, A.J. Lerner, K.A. Smyth, R.P.Friedland, Interactions of apolipoprotein E genotype and dietary fatintake of healthy older persons during mid-adult life, Metabolism 52(2003) 279–281.
[27] C.J. Pike, A.J. Walencewicz, C.G. Glabe, C.W. Cotman,Aggregation-related toxicity of synthetic�-amyloid protein in hip-pocampal cultures, Eur. J. Pharmacol. 207 (1991) 367–368.
[28] S.P. Pshenichkin, B.C. Wise, Okadaic acid increases nerve growthfactor secretion, mRNA stability, and gene transcription in primarycultures of cortical astrocytes, J. Biol. Chem. 270 (1995) 5994–5999.
[29] A.E. Roher, N. Weiss, T.A. Kokjohn, Y.-M. Kuo, W. Kalback, J.Anthony, D. Watson, D.C. Luehrs, L. Sue, D. Walker, M. Emmer-ling, W. Goux, T. Beach, Increased A� peptides and reducedcholesterol and myelin proteins characterize white matter degen-eration in Alzheimer’s disease, Biochemistry 41 (2002) 11080–11090.
[30] H. Sauer, B. Klimm, J. Hescheler, M. Wartenberg, Activation ofp90RSK and growth stimulation of multicellular tumor spheroidsare dependent on reactive oxygen species generated after puriner-
39–
[ naseibits
ss, J.
[ iet-
[rt 14
[ n in001)
[ offatty
[ lds,ll,einsoid-Neu-
[ iza-on. 150
15] P. Kerokoski, T. Suuronen, A. Salminen, H. Soininen, T. PirtCleavage of the cyclin-dependent kinase 5 activator p35 to p25not induce tau hyperphosphorylation, Biochem. Biophys. Res. Cmun. 298 (2002) 693–698.
16] M.-S. Lee, Y.T. Kwon, M. Li, J. Peng, R.M. Friedlander, L.-H. TsNeurotoxicity induces cleavage of p35 to p25 by calpain, Nature(2000) 360–364.
17] J.A. Levin-Allerhand, C.E. Lominska, J.D. Smith, Increaamyloid-� levels in APPSWE transgenic mice treated chronicwith a physiological high-fat high-cholesterol diet, J. Nutr. HeAging 6 (2002) 315–319.
18] P. Lipton, Ischemic cell death in brain neurons, Physiol. Rev(1999) 1431–1568.
19] M.A. Lovell, S. Xiong, C. Xie, P. Davies, W.R. Markesbery, Indtion of hyperphosphorylated tau in primary rat cortical neurontures mediated by oxidative stress and glycogen synthase kinJ. Alzheimers Dis. 6 (2004) 659–671.
20] M.P. Mattson, Pathways towards and away from Alzheimer’s disNature 431 (2004) 631–639.
21] M.P. Mattson, W. Fu, G. Waeg, K. Uchida, 4-Hydroxynonea product of lipid peroxidation, inhibits dephosphorylationthe microtubule-associated protein tau, NeuroReport 8 (12275–2281.
22] L.D. Monti, C. Landoni, E. Setola, E. Galluccio, P. Lucotti, ESandoli, A. Origgi, G. Lucignani, P. Piatti, F. Fazio, Myocardinsulin resistance associated with chronic hypertriglyceridemiaincreased FFA levels in Type 2 diabetic patients, Am. J. Phy287 (2004) H1225–H1231.
gic receptor stimulation by ATP, FASEB J. 15 (2001) 252541.
31] T.B. Shea, Y.-L. Zheng, D. Ortiz, H.C. Pant, Cyclin-dependent ki5 increases perikaryal neurofilament phosphorylation and inhneurofilament axonal transport in response to oxidative streNeurosci. Res. 76 (2004) 795–800.
32] F.-S. Shie, L.-W. Jin, D.G. Cook, J.B. Leverenz, R.C. LeBoeuf, Dinduced hypercholesterolemia enhances brain A� accumulation intransgenic mice, NeuroReport 13 (2002) 455–459.
33] F.-S. Shie, R.C. LeBoeur, L.-W. Jin, Early intraneuronal A� depo-sition in the hippocampus of APP transgenic mice, NeuroRepo(2003) 123–129.
34] Q.R. Smith, H. Nagura, Fatty acid uptake and incorporatiobrain: studies with the perfusion model, J. Mol. Neurosci. 16 (2167–172.
35] S.-W. Wang, M. Wang, B.M. Grossman, R.J. Martin, Effectsdietary fat on food intake and brain uptake and oxidation ofacids, Physiol. Behav. 56 (1994) 517–522.
36] R. Williamson, T. Scales, B.R. Clark, G. Gibb, C.H. ReynoS. Kellie, L.N. Bird, L.M. Varndell, P.W. Sheppard, I. EveraI.B.H. Anderton, Rapid tyrosine phosphorylation of neuronal protincluding tau and focal adhesion kinase in response to amyl�
peptide exposure: involvement of src family protein kinases, J.rosci. 22 (2002) 10–20.
37] D.M. Wilson, L.I. Binder, Free fatty acids stimulate the polymertion of tau and amyloid� peptides. In vitro evidence for a commeffector of pathogenesis in Alzheimer’s disease, Am. J. Pathol(1997) 2181–2195.
Copyright © 2022 FDOKUMEN