
Pim3 negatively regulates glucose-stimulated insulin secretion
Upload
independentCategory
view
5download
0
DOI 10.1007/s00702-003-0015-9
J Neural Transm (2003) 110: 1029–1039
Inhibition of energy metabolism down-regulates
the Alzheimer related presenilin 2 gene
R. Ghidoni1, L. Gasparini1, A. Alberici1, L. Benussi1, L. Barbiero1,F. Mazzoli1, F. Nicosia1, D. Finazzi2, L. Benerini Gatta2,
A. Albertini2, O. Zanetti1, and G. Binetti1
1 Scientific Institute IRCCS ‘‘Centro San Giovanni di Dio-Fatebenefratelli’’, and2 Institute of Chemistry, Faculty of Medicine, University of Brescia, Brescia, Italy
Received August 25, 2002; accepted April 7, 2003Published online June 30, 2003; # Springer-Verlag 2003
Summary. Defects in energy metabolism and oxidative stress play an impor-tant role in the pathogenesis of Alzheimer’s Disease (AD). In sporadic ADcases, presenilin 2 (PS2) mRNA levels are decreased in brain areas affectedby the disease. The aim of the present study was to investigate whether mito-chondrial dysfunction might influence PS2 gene expression. We demonstratedthat the inhibition of energy metabolism by sodium azide down-regulates PS2gene expression through modification of promoter activity. No one of the ana-lyzed transcription factors, sensitive to redox status of the cell, could explainthis effect. Azide effect on PS2 expression was completely inhibited by theaddition of an antioxidant suggesting that the imbalance of the cellular redoxhomeostasis modulates the expression of this gene.
Keywords: PS2 expression, oxidative stress, nonradioactive RPA.
Introduction
As for most neurodegenerative diseases, the cause of the sporadic Alzheimer’sDisease (AD), is unknown. Only a minority of AD cases are clearly familial(FAD) and one half of them involves mutations in presenilin genes (PS1 andPS2) (Rogaev et al., 1995; Finckh et al., 2000). Even though ubiquitouslyexpressed, presenilins show tissue-specific transcriptional differences. The high-est levels of PS2 expression in brain were detected in hippocampus and cerebel-lum (Benkovic et al., 1997). Although this mRNA is expressed most prominentlyin neurons (Kovacs et al., 1996), lower but significant levels of transcript werealso detected in white matter glial cells. Significant decrease in PS2 mRNA wasobserved in pathologically affected areas of late-onset sporadic AD brains(McMillan et al., 1996; Takami et al., 1997). A recent study reported that thedecrease of PS2 expression is as severe in subjects with mild AD as it is in

subjects in late stage of the disease (McMillan et al., 2000): Thus the specificdown-regulation of PS2 gene expression is an early event in sporadic late-onsetAD and a possible involvement of presenilins in the etiology of sporadic AD hasbeen proposed.
Presenilin genes expression is modulated in AD pathology as well as in thebroader processes of brain development and response to injury (Lee et al.,1996). It has been demonstrated that the PS2 promoter contains several poten-tial binding sites for nuclear transcription factors sensitive to the redox status ofthe cell, i.e. Sp1, AP1, AP2 and NFkB (Pennypacker et al., 1998). Neverthelesslittle is known about factors and conditions that influence PS2 gene transcrip-tion. Aim of the present study was to investigate the role of mitochondrialdysfunction on PS2 gene expression. Reduction of activities of important mito-chondrial enzymes like cytochrome c oxidase (Parker et al., 1990; Mutisyaet al., 1994; Chandrasekaran et al., 1997) as well as increased levels of molec-ular species linked to oxidative stress, such as isoprostanes, protein carbonylsand homocysteine (Markesbery et al., 1999; Seshadri et al., 2002) have beendocumented in AD. In our experimental model we treated cells with sodiumazide (NaN3), an inhibitor of cytochrome c oxidase, to mimic the selectivereduction of activity of this enzyme observed in Alzheimer’s disease. Ourresults demonstrated that inhibition of energy metabolism down-regulatesPS2 gene expression.
Material and methods
Cell cultures, experimental treatments and cell viability
H4 human neuroglioma cells were cultured in Dulbecco’s Modified Essential Medium (DMEM-Gibco Brl) supplemented with 10% Fetal Calf Serum (Gibco Brl), Penicillin (100 U=ml), Strep-tomycin (100 mg=ml), at 37�C in 5% CO2=95% air. U373MG human glioblastoma cells werecultured in Minimum Essential Medium (MEM Gibco Brl); SH-SY5Y neuroblastoma cells werecultured in Minimum Essential Medium (MEM Gibco Brl) supplemented with Nutrient MixtureF12 (HAM) 1:1. Human neuroblastoma cells SH-SY5Y were differentiated by treatment withretinoic acid 10mM for 6 days. Experiments were performed by incubation of cell cultures with orwithout 10mM hydrogen peroxide (H2O2) (Fluka); with 500mM sodium azide (NaN3) (Sigma)and=or 100mM pyrrolidineditiocarbamate (PDTC) (Sigma) in serum free medium for the indi-cated time periods (7–24 h) at 37�C.
The number of viable cells was determined by tripan blue exclusion. Apoptosis was assayedby TUNEL system according to manufacturer’s instructions (Promega).
Protein extracts and Western blotting
Protein Extracts were performed as described previously (Alberici et al., 1999). Loading of thesamples (soluble cytosolic fraction and the particulate or membrane fraction) was normalized forthe total content of cellular proteins determined by using BCA assay (Pierce). Samples wereseparated on 15% SDS-PAGE gel, blotted onto polyvinylidene difluoride (PVDF) membrane(NEN) and probed with an anti-cytochrome c monoclonal antibody (PharMingen).
Ribonuclease Protection Assay (RPA)
The isolation of total RNA from cells was performed using TRIZOL Reagent (Life Technologies)according to the manufacturer’s protocol.
1030 R. Ghidoni et al.

PS2 exon 11 (285 bp including flanking introns) was amplified from genomic DNA by PCRand subcloned in TA cloning vector (Invitrogen). The plasmid with antisense DNA template waslinearized downstream of the insert using Xho I. The transcription reaction was performed byusing T7 RNA Polymerase. Hybridization of this transcript to human total RNA protects a 120 ntfragment of PS2 mRNA.
For b-actin probe transcription, pTRI-b-actin-Human plasmid (Ambion) was used. Transcrip-tion of linearized plasmid with T7 RNA Polymerase produced the antisense transcript 304 baseslength. Hybridization of the transcript to human total RNA protects a 245 nt fragment of humanb-actin mRNA.
For transcription reactions MAXIscript Kit (Ambion) was used. The probe was gel-purifiedon 5% polyacrylamide 8 M urea gel, eluted in Probe Elution Buffer (Ambion) and then labeled byusing the Psoralen-Biotin reagent (Ambion). The hybridization was carried out by overnightincubation of 300 pg of labeled probe and 30mg RNA samples at 56�C (RPA III, Ambion).The hybridization mixture was then treated with ribonuclease (Rnase A=T1 mix) for 30 min at37�C and hybridized RNA was separated on a 5% polyacrylamide-8 M urea gel followed byblotting onto Positively Charged Nylon Membranes (BrightStar-Plus, Ambion). After transfer, themembranes were baked at 80�C for 15 min and the bands were visualized by using secondarychemiluminescent detection (BrightStar BioDetect, Ambion).
Gene reporter assay
A 2966 bp fragment of the promoter region of PS2 (from � 2934 to þ 32) was amplified by PCRfrom a PAC Library (Genome Systems; PAC 292C15) and sub-cloned in p-Blue-TOPO expres-sion vector (Invitrogen) containing b-galactosidase as reporter gene, to obtain the construct PS2-p-Blue-Topo.
H4 cells were seeded in 12 well plates at a concentration of 12� 104=well. For each well,cells were transfected with 1mg of p-Blue-TOPO, PS2-p-Blue-TOPO or pcDNA3.1=HISB=lacZplasmids (Invitrogen) by using FuGENE 6 Transfection Reagent. Cells were exposed to theDNA-FuGENE complex until gene reporter assay. The treatment with NaN3 500mM was per-formed 14 and 24 h before harvesting cells. Forty-eight hours after transfection cells were har-vested with 100ml of lysis buffer (100 mM Potassium Phosphate, pH 7.8, DTT 1 mM). Proteinextracts were prepared by performing three freezing=thawing cycles and recovering the super-natant after centrifugation at 14.000� g (5 min at 4�C). The protein extracts were tested for b-galactosidase activity with a chemiluminescent assay (Chemiluminescent b-galactosidase assay-Clontech). Background levels of b-Gal activity were determined on H4 cells transfected by p-Blue-TOPO ‘‘basic vector’’ and then subtracted. Transfection efficiency was estimated in parallelby trasfections with pcDNA3.1=HISB=lacZ. All experiments were performed three times intriplicate. The specific activity of PS2 or CMV promoters in treated versus untreated cells isreported as mean of the three experiments.
Electrophoretic Mobility Shift Assay (EMSA)
H4 human neuroglioma cells were exposed for 3, 7, 14 and 21 h to NaN3 (500mM). Nuclearextracts were prepared as previously described (Andrews et al., 1991) in the presence or not of0.25 mM DTT. Gel mobility shift assays were performed by using DIG Gel Shift Kit (Roche)according to the manufacturer’s instruction. Briefly, Sp1, AP1, AP2 and NFkB double strandednucleotides (Promega) were labeled with digoxigenin-11-ddUTP. DNA binding reactions werecarried out by combining 2–4mg of nuclear extracts with 60 fmol of Sp1, AP1, AP2, NFkBlabeled oligonucleotides in binding buffer (50 mM NaCl, 10 mM Tris-HCl, 1 mM MgCl2, 0.5 mMEDTA, 4% glycerol, with or without 0.5 mM DTT) containing 0.5mg Poly(dI-dC) and 2mg ofbovine serum albumin (BSA) in a total volume of 20ml for 15 min at room temperature (RT). Thespecificity of each DNA–protein interaction was tested by competition with 50 and 200-foldmolar excess of unlabeled nucleotides (data not shown). Protein–DNA complexes were resolvedon 6% DNA Retardation Gels (Novex Pre-Cast Gels). After transfer, the positively charged nylon(Roche) membranes were baked at 120�C for 15–30 min. Bands were visualized by using achemiluminescent detection procedure (Wash and Block Buffer Set-Roche).
Energy metabolism and presenilin 2 expression 1031

Results
Inhibition of energy metabolism down-regulated PS2 mRNAexpression in different cell lines
We treated H4 human neuroglioma cells with sodium azide, an inhibitor ofcomplex IV of the mitochondrial respiratory chain. In order to determinewhether azide induces cell death, cell viability was measured after 7, 14, 24and 48 h by tripan blue exclusion. Sodium azide did not alter significantly cellviability after 7 and 14 h of treatment (p>0,05) as shown in Fig. 1A. Forty-eight hours of exposure to azide resulted in a massive cell death. The release ofcytochrome c from mitochondria was evaluated after 14 and 24 h of incubationwith sodium azide: a strong release was observed solely after 24 h of treatment(Fig. 1B). This phenomenon induced, later on, apoptosis in roughly 50% of H4cells as demonstrated by TUNEL assay (data not shown).
Fig. 1. A Effect of sodium azide on cell viability. Cell viability was evaluated by tripan blueexclusion after incubation for 7, 14, 24, 48 h in serum free medium (control condition) and inthe presence of 500 mM sodium azide (NaN3). The ordinate represents the percentage of deadcells. Data are mean� SD values of at least three independent measurements. Sodium azide didnot alter significantly cell viability after 7 and 14 h of treatment (p>0,05); Statistical signifi-cance was reached at 24 h (�p<0,05) and 48 h (�p<0,05). Statistical analysis was performedwith one-way ANOVA. B Release of cytochrome c from mitochondria to cytosol after azidetreatment. Cytochrome c is released in the soluble fraction after 24 h of incubation with500 mM sodium azide. Lane 1: control condition (serum free medium); lane 2: sodium azide
(NaN3)
1032 R. Ghidoni et al.
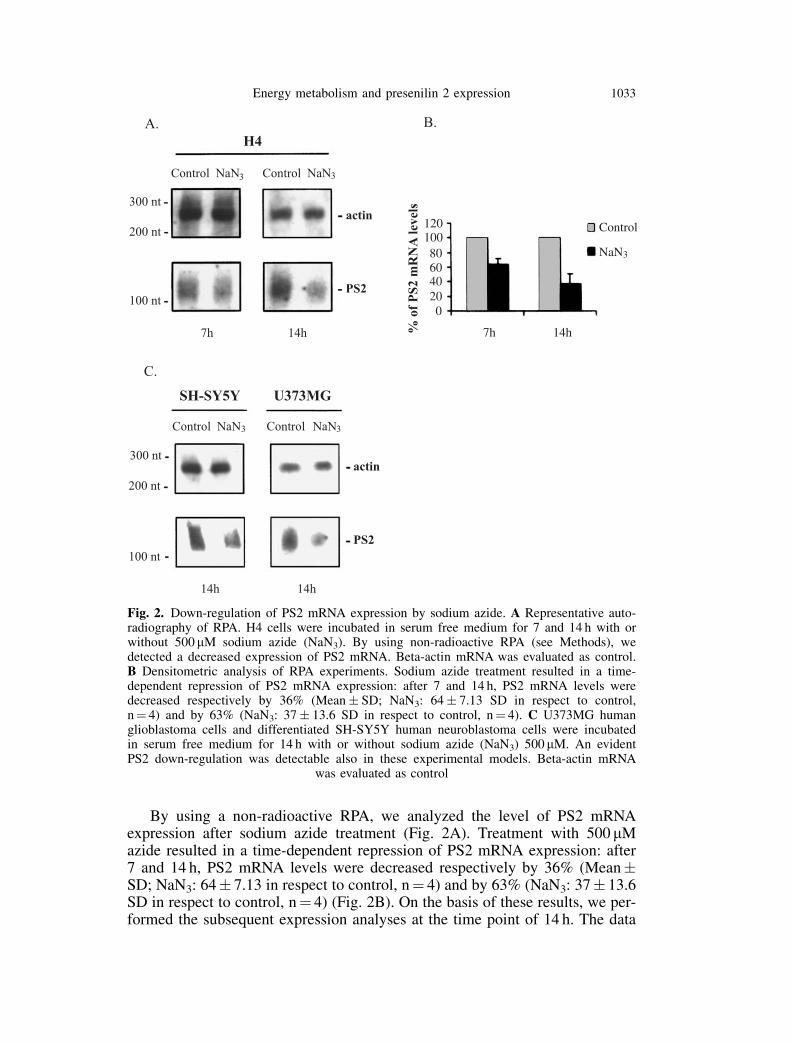
By using a non-radioactive RPA, we analyzed the level of PS2 mRNAexpression after sodium azide treatment (Fig. 2A). Treatment with 500 mMazide resulted in a time-dependent repression of PS2 mRNA expression: after7 and 14 h, PS2 mRNA levels were decreased respectively by 36% (Mean�SD; NaN3: 64� 7.13 in respect to control, n¼ 4) and by 63% (NaN3: 37� 13.6SD in respect to control, n¼ 4) (Fig. 2B). On the basis of these results, we per-formed the subsequent expression analyses at the time point of 14 h. The data
Fig. 2. Down-regulation of PS2 mRNA expression by sodium azide. A Representative auto-radiography of RPA. H4 cells were incubated in serum free medium for 7 and 14 h with orwithout 500 mM sodium azide (NaN3). By using non-radioactive RPA (see Methods), wedetected a decreased expression of PS2 mRNA. Beta-actin mRNA was evaluated as control.B Densitometric analysis of RPA experiments. Sodium azide treatment resulted in a time-dependent repression of PS2 mRNA expression: after 7 and 14 h, PS2 mRNA levels weredecreased respectively by 36% (Mean� SD; NaN3: 64� 7.13 SD in respect to control,n¼ 4) and by 63% (NaN3: 37� 13.6 SD in respect to control, n¼ 4). C U373MG humanglioblastoma cells and differentiated SH-SY5Y human neuroblastoma cells were incubatedin serum free medium for 14 h with or without sodium azide (NaN3) 500 mM. An evidentPS2 down-regulation was detectable also in these experimental models. Beta-actin mRNA
was evaluated as control
Energy metabolism and presenilin 2 expression 1033

obtained in H4 cells were confirmed in other cell lines, such as U373MGhuman glioblastoma cells and differentiated SH-SY5Y human neuroblastomacells (Fig. 2C). Beta-actin mRNA, evaluated as control, didn’t decrease in theseexperimental conditions.
Repression of PS2 gene transcription is mediated by oxidative stress
We co-incubated H4 cells with sodium azide and 100 mM pyrrolidinedithiocar-bamate (PDTC) for 14 h in serum free medium. The antioxidant PDTC rescuedthe effects of sodium azide on PS2 mRNA expression in H4 cells (Fig. 3). Wealso investigated whether exposure to an oxygen radical generator, such ashydrogen peroxide (H2O2), also resulted in a down-regulation of PS2 geneexpression. H4 cells were therefore treated for 14 h with 10 mM H2O2 in serumfree medium. We observed a decrease of PS2 mRNA levels comparable to theones detected following azide treatment (data not shown).
Fig. 3. Antioxidant PDTC rescues the effect of sodium azide on PS2 mRNA expression. In thisexperiment we evaluated the effects of antioxidant pyrrolidinedithiocarbamate (PDTC). ARepresentative autoradiography of RPA. Experiments were performed by incubation of cellcultures with 500 mM sodium azide (NaN3) (lane 2) and=or 100 mM PDTC (lane 4=lane 3respectively) for 14 h in serum free medium. In the lane 1 (control) the cells were incubatedfor 14 h in serum free medium. We observed that PDTC rescued the effect of sodium azide(500 mM) on PS2 expression. B Densitometric analysis of RPA experiments. Data are expressedas percentage in respect to controls (mean� SD). NaN3 (column 2): 37� 13,6 (n¼ 4); PDTC
(column 3): 100,51� 20,27 (n¼ 3); NaN3þ PDTC (column 4): 87,91� 2,52 (n¼ 3)
1034 R. Ghidoni et al.
Sodium azide treatment reduced PS2 promoter transcriptionalactivity in H4 cells
To investigate the mechanism of reduced PS2 gene expression, we transientlytransfected H4 cells with a plasmid containing the PS2 promoter upstream of
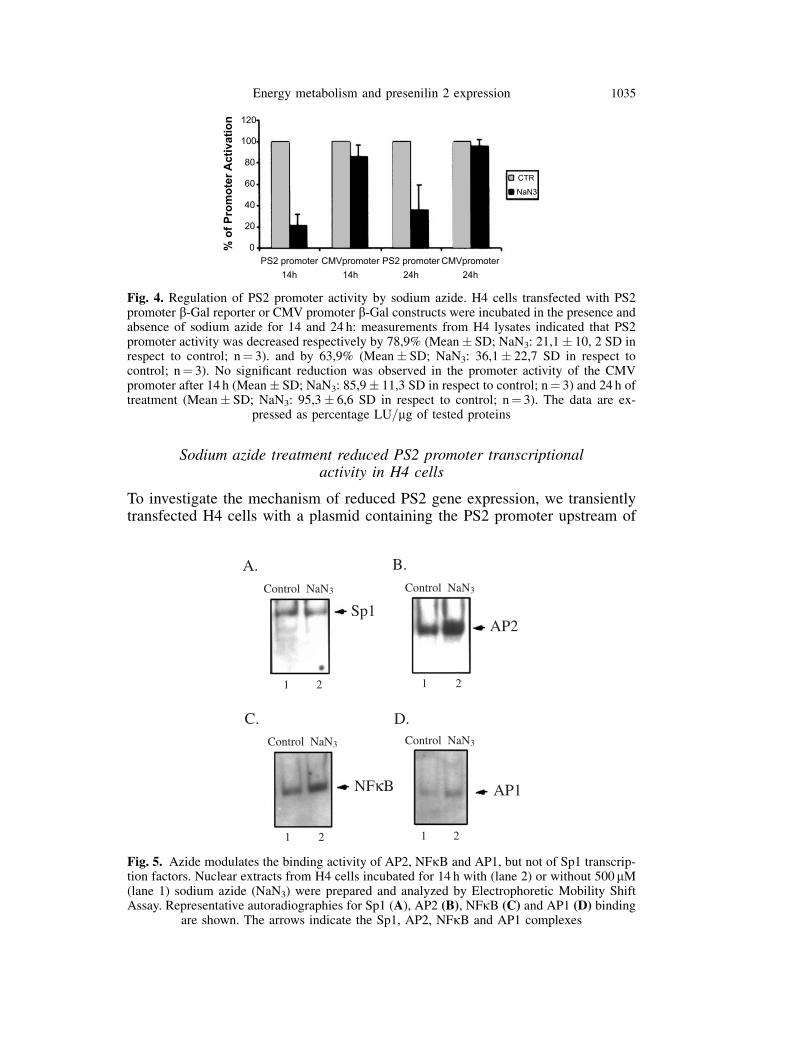
Fig. 4. Regulation of PS2 promoter activity by sodium azide. H4 cells transfected with PS2promoter b-Gal reporter or CMV promoter b-Gal constructs were incubated in the presence andabsence of sodium azide for 14 and 24 h: measurements from H4 lysates indicated that PS2promoter activity was decreased respectively by 78,9% (Mean� SD; NaN3: 21,1� 10, 2 SD inrespect to control; n¼ 3). and by 63,9% (Mean� SD; NaN3: 36,1� 22,7 SD in respect tocontrol; n¼ 3). No significant reduction was observed in the promoter activity of the CMVpromoter after 14 h (Mean� SD; NaN3: 85,9� 11,3 SD in respect to control; n¼ 3) and 24 h oftreatment (Mean� SD; NaN3: 95,3� 6,6 SD in respect to control; n¼ 3). The data are ex-
pressed as percentage LU=mg of tested proteins
Fig. 5. Azide modulates the binding activity of AP2, NFkB and AP1, but not of Sp1 transcrip-tion factors. Nuclear extracts from H4 cells incubated for 14 h with (lane 2) or without 500 mM(lane 1) sodium azide (NaN3) were prepared and analyzed by Electrophoretic Mobility ShiftAssay. Representative autoradiographies for Sp1 (A), AP2 (B), NFkB (C) and AP1 (D) binding
are shown. The arrows indicate the Sp1, AP2, NFkB and AP1 complexes
Energy metabolism and presenilin 2 expression 1035

b-Gal reporter gene. Incubation for 14 and 24 h with 500 mM sodium azidedramatically decreased the activity of PS2 promoter respectively by 78,9%and 63,9%. No significant reduction was observed in the promoter activityof the CMV promoter (pcDNA3.1=HISB=lacZ) following azide treatment(Fig. 4).
Electrophoretic mobility-shift analysis
Several potential Sp1, AP2, AP1 and NFkB regulatory sites are located in thePS2 promoter. EMSA for SP1, AP2, AP1 and NFkB binding activity wasperformed after incubation of H4 cells with 500mM sodium azide.
In our experimental model, treatment with azide for 3, 7, 14 and 21 h did notmodulate the Sp1 DNA binding activity (Fig. 5A). Incubation with sodiumazide for 3, 7 and 14 h resulted in an evident increase in AP2 (Fig. 5B), NFkB(Fig. 5C) and AP1 binding (Fig. 5D).
Discussion
Our data provide evidence that the inhibition of energy metabolism down-reg-ulates PS2 gene expression. We analyzed PS2 mRNA levels in different celllines: in H4 human neuroglioma cells, U373MG human glioblastoma cells andin differentiated SH-SY5Y cells. The metabolic impairment mediated bysodium azide produced a specific down-regulation of PS2 gene expression.Lee et al. (1996) reported that, in human cortex, PS1 and PS2 mRNA areexpressed at comparable levels and change as a function of age. While PS1mRNA is expressed at significantly higher levels in developing brain, the levelof PS2 mRNA increases during postnatal development, suggesting a differentialtranscriptional regulation of these genes during processes of brain maturation.Presenilins are functionally regulated not only during development but also insporadic AD and brain injury. Two studies (McMillan et al., 1996; Takami et al.,1997) reported that, in subjects with late onset sporadic AD, PS2 mRNA levelsare decreased in brain areas severely affected by the disease such as hippocam-pus. Recently McMillan et al. (2000) demonstrated that mild AD cases showprominent down-regulation of PS2 mRNA levels in hippocampal regions evenin the absence of severe neuropathology suggesting that this decrease is an earlystep in the progression of the disease. There are presently few data about thefactors and conditions that influence PS2 expression: Understanding the regula-tion of the PS2 gene might thus be essential for the elucidation of its patho-physiological role in AD.
In the last years several study pointed out that there is a selective deficit inthe azide-sensitive mitochondrial complex IV (complex IV or cytochrome oxi-dase) in the cerebral cortex of patients with AD (Parker et al., 1990; Mutisyaet al., 1994; Chandrasekaran et al., 1997). We found a causal relation betweenimpairment of oxidative energy metabolism and down-regulation of PS2expression providing a conceptual framework that relates presenilin expressionand sporadic AD. In H4 cells, we observed that PS2 mRNA levels were reducedrespectively by 36% and by 63% after 7 and 14 hours of treatment. This expres-sion pattern might closely reflect what observed in human AD brains in which
1036 R. Ghidoni et al.

PS2 mRNA levels are reduced but not completely removed (McMillan et al.,2000). The decrease is an early event which might be part of an acute cellresponse mechanism following azide treatment and, as a matter of fact, it pre-ceded the release of cytochrome c and the consequent reduction of cell via-bility. The cytochrome oxidase inhibitor sodium azide generates damagingreactive oxygen species (ROS) in vivo and in vitro (Smith et al., 1997; Partridgeet al., 1994). In our experimental setting, the possible involvement of oxidativestress in repression of PS2 gene transcription is supported by two lines ofevidence. First, the antioxidant PDTC rescued the effect of sodium azide onPS2 mRNA expression. Second, the exposure to an oxygen radical generator,such as hydrogen peroxide (H2O2), also resulted in a down-regulation of PS2gene transcription. Thus the imbalance of redox homeostasis within the cellseems to modulate PS2 gene expression.
A large number of transcription factors are sensitive to modulation of thecellular redox status and a growing number of studies have shown that chronicoxidative stress specifically down-regulates the expression of various genes(reviewed in Morel et al., 1999). We observed a strong decrease of PS2 pro-moter activity following azide treatment. PS2 basal promoter activity residesbetween � 403 and þ 13, a TATA less, GC-rich region containing numerousputative AP2 and Sp1 sites (Pennypacker et al., 1998). Sp1 transcription factoris a sensitive target of oxidative stress (Ammendola et al., 1994). Gel shiftassays revealed that Sp1 DNA-binding efficiency was not modulated by sodiumazide. Conversely, inhibition of energy metabolism resulted in AP2, NFkB andAP1 activation. Given these results, we can only speculate about other hypothet-ical mechanisms involved in PS2 gene repression. The chronic oxidative stresscan repress gene expression not only by influencing the activity of a transcrip-tion factor, but also altering the promoter sequence. A study (Ghosh andMitchell, 1999) showed that the oxidative damage in Sp1 (and AP1) consensusbinding sequence of a promoter element inhibits transcription factors binding.Thus a mechanism involving a damage in PS2 promoter sequence might beresponsible for the observed gene repression, modifying the PS2 basal promoteractivity; otherwise there may be noncanonical regulatory sites that mediateazide effect.
The etiology of AD is complex, with both genetic and environmental factorsinfluencing the phenotype of the disease (Rogaev et al., 1995; Finckh et al.,2000; Hoyer, 2000). Expression of both presenilins is highly regulated (Theunset al., 2000) and might represent the point of convergence of several signalingpathways contributing to the pathogenesis of AD. Our data allow us to arguethat two major pathogenetic pathways, which are likely to contribute to theneuropathology associated with AD (mitochondrial dysfunction and oxidativestress), share a common step represented by the down-regulation of PS2 expres-sion. The significance of this process is controversial. Mutations as well asvariations leading to altered expression of this AD gene could have majoreffects on protein functioning, affecting, for example, amyloid precursor pro-tein (APP) processing and amyloid b-peptide (Abeta) deposition. In this regard,Refolo et al. (1999) reported that the antisense-induced reduction of presenilin1 expression affects the APP processing leading to an increased production of
Energy metabolism and presenilin 2 expression 1037

Abeta 42 in transfected cells. Another possibility is that PS2 directly partici-pates in the biochemical pathway regulating apoptosis (Deng et al., 1996;Janicki et al., 1997). A recent paper reported that wild-type and mutatedpresenilins 2 trigger p53-dependent apoptosis (Alves da Costa et al., 2002).Wolozin et al. (1996) demonstrated that transfection of antisense PS2 conferredprotection against apoptotic cell death. Thus we can hypothesize that, in ourexperimental model, cells are attempting to reverse the process of incipient celldeath by down-regulating PS2 expression. Since PS2 expression is regulated bystress factors, we can argue that PS2 is one of the genes coordinately modulatedin brain in response to situations that require a defensive reaction. At presentnone of these different hypotheses can be excluded. Further studies about thefactors which affect presenilins expression and the consequent analysis of itstranscriptional regulation will lead to more insight into AD etiology.
References
Alberici A, Moratto D, Benussi L, Gasparini L, Ghidoni R, Gatta LB, Finazzi D, Frisoni GB,Trabucchi M, Growdon JH, Nitsch RM, Binetti G (1999) Presenilin 1 protein directlyinteracts with Bcl-2. J Biol Chem 274(43): 30764–30769
Alves da Costa C, Paitel E, Mattson MP, Amson R, Telerman A, Ancolio K, Checler F, MattsonMP (2002) Wild-type and mutated presenilins 2 trigger p53-dependent apoptosis and down-regulate presenilin 1 expression in HEK293 human cells and in murine neurons. PNAS 99(6):4043–4048
Ammendola R, Mesuraca M, Russo T, Cimino F (1994) The DNA-binding efficiency of Sp1 isaffected by redox changes. Eur J Biochem 225: 483–489
Andrews NC, Faller DV (1991) A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucl Acids Res 19: 2499
Benkovic SA, McGowan EM, Rothwell NJ, Hutton M, Morgan DG, Gordon MN (1997)Regional and cellular localization of presenilin-2 RNA in rat and human brain. Exp Neurol145: 555–564
Chandrasekaran K, Hatanpaa K, Rapoport SI, Brady DR (1997) Decreased expression of nuclearand mitochondrial DNA-encoded genes of oxidative phosphorylation in association neo-cortex in Alzheimer disease. Brain Res Mol Brain Res 44: 99–104
Deng G, Pike CJ, Cotman CW (1996) Alzheimer-associated presenilin-2 confers increasedsensitivity to apoptosis in PC12 cells. FEBS Lett 397(1): 50–54
Finckh U, Alberici A, Antoniazzi M, Benussi L, Fedi V, Giannini C, Gal A, Nitsch RM, Binetti G(2000) Variable expression of familial Alzheimer disease associated with presenilin 2mutation M239I. Neurology 54: 2006–2008
Ghosh R, Mitchell DL (1999) Effect of oxidative DNA damage in promoter elements ontranscription factor binding. Nucl Acids Res 27(15): 3213–3218
Hoyer S (2000) Brain glucose and energy metabolism abnormalities in sporadic Alzheimerdisease. Causes and consequences: an update. Exp Gerontol 35(9–10): 1363–1372
Janicki S, Monteiro MJ (1997) Increased apoptosis arising from increased expression of theAlzheimer’s disease-associated presenilin-2 mutation (N141I). J Cell Biol 139(2): 485–495
Kovacs DM, Fausett HJ, Page KJ, Kim TW, Moir RD, Merriam DE, Hollister RD, Hallmark OG,Mancini R, Felsenstein KM, Hyman BT, Tanzi RE, Wasco W (1996) Alzheimer-associatedpresenilins 1 and 2: neuronal expression in brain and localization to intracellular membranesin mammalian cells. Nat Med 2(2): 224–229
Lee M, Slunt H, Martin L, Thinkaran G, Kim G, Gandy S, Seeger M, Koo E, Price D, Sisodia S(1996) Expression of presenilin 1 and 2 (PS1 and PS2) in human and murine tissues.J Neurosci 16(23): 7513–7525
Markesbery WR, Carney JM (1999) Oxidative alterations in Alzheimer’s disease. Brain Pathol9(1): 133–146
1038 R. Ghidoni et al.

McMillan PJ, Leverenz JB, Poorkaj P, Schellenberg GD, Dorsa DM (1996) Neuronal expressionof STM2 mRNA in human brain is reduced in Alzheimer’s disease. J Histochem Cytochem44(11): 1215–1222
McMillan PJ, Leverenz JB, Dorsa DM (2000) Specific downregulation of presenilin 2 geneexpression is prominent during early stages of sporadic late-onset Alzheimer’s disease. BrainRes Mol Brain Res 78(1–2): 138–145
Morel Y, Barouki R (1999) Repression of gene expression by oxidative stress. Biochem J 342:481–496
Mutisya EM, Bowling AC, Beal MF (1994) Cortical cytochrome oxidase activity is reduced inAlzheimer’s disease. J Neurochem 63(6): 2179–2184
Parker WD Jr, Filley CM, Parks JK (1990) Cytochrome oxidase deficiency in Alzheimer’sdisease. Neurology 40(8): 1302–1303
Partridge RS, Monroe SM, Parks JK, Johnson K, Parker WD Jr, Eaton GR, Eaton SS (1994) Spintrapping of azidyl and hydroxyl radicals in azide-inhibited rat brain submitochondrialparticles. Arch Biochem Biophys 310(1): 210–217
Pennypacker KR, Fuldner R, Xu R, Hernandez H, Dawbarn D, Mehta N, Perez-Tur J, Bake M,Hutton M (1998) Cloning and characterization of the presenilin-2 gene promoter. Brain ResMol Brain Res 56(1–2): 57–65
Refolo LM, Eckman C, Prada CM, Yager D, Sambamurti K, Mehta N, Hardy J, Younkin SG(1999) Antisense-induced reduction of presenilin 1 expression selectively increases theproduction of amyloid beta42 in transfected cells. J Neurochem 73(6): 2383–2388
Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K,Tsuda T, Mar L, Sorbi S, Nacmias B, Piacentini S, Amaducci L, Chumakov I, Cohen D,Lannfelt L, Fraser PE, Rommens JM, George-Hyslop PH (1995) Familial Alzheimer’sdisease in kindreds with missense mutations in a gene on chromosome 1 related to theAlzheimer’s disease type 3 gene. Nature 376: 775–778
Seshadri S, Beiser A, Selhub J, Jacques PF, Rosenberg IH, D’Agostino RB, Wilson PW, Wolf PA(2002) Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N Engl JMed 346(7): 476–483
Smith TS, Bennett JP Jr (1997) Mitochondrial toxins in models of neurodegenerative diseases. I.In vivo brain hydroxyl radical production during systemic MPTP treatment or followingmicrodialysis infusion of methylpyridinium or azide ions. Brain Res 765(2): 183–188
Takami K, Terai K, Matsuo A, Walker DG, McGeer PL (1997) Expression of presenilin-1 and -2mRNAs in rat and Alzheimer’s disease brains. Brain Res 748: 122–130
Theuns J, Van Broeckhoven C (2000) Transcriptional regulation of Alzheimer’s disease genes:implications for susceptibility. Hum Mol Genet 9(16): 2383–2394
Wolozin B, Iwasaki K, Vito P, Ganjei JK, Lacan�aa E, Sunderland T, Zhao B, Kusiak JW, Wasco W,D’Adamio L (1996) Participation of presenilin 2 in apoptosis: enhanced basal activityconferred by an Alzheimer mutation. Science 274(5293): 1710–1713
Authors’ address: Dr. G. Binetti, Neurobiology Lab, Memory Clinic, Scientific InstituteIRCCS ‘‘Centro San Giovanni di Dio-Fatebenefratelli’’, Via Pilastroni, 4, I-25125 Brescia, Italy,e-mail: [email protected]
Energy metabolism and presenilin 2 expression 1039
Copyright © 2022 FDOKUMEN




















