
Differential Role of PKC Isoforms in GnRH and Phorbol 12Myristate 13Acetate Activation of...
14
Differential Role of PKC Isoforms in GnRH and Phorbol 12-Myristate 13-Acetate Activation of Extracellular Signal-Regulated Kinase and Jun N-Terminal Kinase Masha Dobkin-Bekman, Liat Rahamim Ben-Navi, Boris Shterntal, Ludmila Sviridonov, Fiorenza Przedecki, Michal Naidich-Exler, Chaya Brodie, Rony Seger, and Zvi Naor Department of Biochemistry and Molecular Biology (M.D.-B., L.R.B.-N., B.S., L.S., F.P., M.N.-E., Z.N.), The George S. Wise Faculty of Life Sciences, Tel Aviv University, Ramat Aviv 69978, Israel; Gonda (Goldschmied) Medical Diagnosis Research Center (C.B.), The Mina and Everard Goodman Faculty of Life Sciences, Bar-Ilan University, Ramat-Gan 52900, Israel; and Department of Biological Regulation (R.S.), The Weizmann Institute of Science, Rehovot 76100, Israel GnRH is the first key hormone of reproduction. The role of protein kinase C (PKC) isoforms in GnRH- stimulated MAPK [ERK and Jun N-terminal kinase (JNK)] was examined in the T3-1 and LT2 gona- dotrope cells. Incubation of the cells with GnRH resulted in a protracted activation of ERK1/2 and a slower and more transient activation of JNK1/2. Gonadotropes express conventional PKC and con- ventional PKCII, novel PKC, novel PKC, and novel PKC, and atypical PKC-/. The use of green fluorescent protein-PKC constructs revealed that GnRH induced rapid translocation of PKC and PKCII to the plasma membrane, followed by their redistribution to the cytosol. PKC and PKC localized to the cytoplasm and Golgi, followed by the rapid redistribution by GnRH of PKC to the perinuclear zone and of PKC to the plasma membrane. Interestingly, PKC, PKCII, and PKC translocation to the plasma membrane was more pronounced and more prolonged in phorbol-12-myristate-13-acetate (PMA) than in GnRH-treated cells. The use of selective inhibitors and dominant-negative plasmids for the various PKCs has revealed that PKCII, PKC, and PKC mediate ERK2 activation by GnRH, whereas PKC, PKCII, PKC, and PKC mediate ERK2 activation by PMA. Also, PKC, PKCII, PKC, and PKC are involved in GnRH and PMA stimulation of JNK1 in a cell-context-dependent manner. We present preliminary evidence that persistent vs. transient redistribution of selected PKCs or redistribution of a given PKC to the perinuclear zone vs. the plasma membrane may dictate its selective role in ERK or JNK activation. Thus, we have described the contribution of selective PKCs to ERK and JNK activation by GnRH. (Endocrinology 151: 4894 – 4907, 2010) H ypothalamic GnRH is secreted in a pulsatile manner to stimulate the synthesis and release of LH and FSH (for review, see Refs. 1 and 2). Signaling of GnRH receptor (GnRHR) involves activation of phospholipase C (PLC)-, phospholipase D, phospholipase A 2 , enhanced phosphoinositide turnover, Ca 2 mobilization and influx, activation of protein kinase C (PKC), and the MAPK cas- cades and formation of prostaglandins and leukotrienes, culminating in gonadotropin synthesis and release (2–5). PKC is a family of 10 related isoforms (6, 7), which plays a significant role in signaling (6, 7). The PKCs are classified into conventional (cPKC; cPKC-, cPKCI, cPKCII, and cPKC), novel (nPKC; nPKC, nPKC, nPKC, and nPKC/), and atypical (aPKC; aPKC and ISSN Print 0013-7227 ISSN Online 1945-7170 Printed in U.S.A. Copyright © 2010 by The Endocrine Society doi: 10.1210/en.2010-0114 Received January 28, 2010. Accepted July 27, 2010. First Published Online September 1, 2010 Abbreviations: aPKC, Atypical PKCs; cPKC, conventional PKCs; DAG, diacylglycerol; DN, dominant negative; DP, diphospho; DUSP, dual-specificity phosphatase; FCS, fetal calf serum; FSH, FSH -subunit; G protein, guanine nucleotide binding protein; GFP, green fluorescent protein; GnRHR, GnRH receptor; GRASP65, Golgi reassembly stacking protein of 65 kDa; HA, hemagglutinin epitope; JNK, Jun N-terminal kinase; LH, LH -subunit; nPKC, novel PKCs; PKC, protein kinase C; PKCs, PKC isoforms; PMA, phorbol 12-myristate 13-acetate; PS, phosphatidylserine; siRNA, small interfering RNA. NEUROENDOCRINOLOGY 4894 endo.endojournals.org Endocrinology, October 2010, 151(10):4894 – 4907 The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 29 July 2015. at 01:57 For personal use only. No other uses without permission. . All rights reserved.
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Differential Role of PKC Isoforms in GnRH and Phorbol 12Myristate 13Acetate Activation of...

Differential Role of PKC Isoforms in GnRH andPhorbol 12-Myristate 13-Acetate Activation ofExtracellular Signal-Regulated Kinase and JunN-Terminal Kinase
Masha Dobkin-Bekman, Liat Rahamim Ben-Navi, Boris Shterntal,Ludmila Sviridonov, Fiorenza Przedecki, Michal Naidich-Exler, Chaya Brodie,Rony Seger, and Zvi Naor
Department of Biochemistry and Molecular Biology (M.D.-B., L.R.B.-N., B.S., L.S., F.P., M.N.-E., Z.N.),The George S. Wise Faculty of Life Sciences, Tel Aviv University, Ramat Aviv 69978, Israel; Gonda(Goldschmied) Medical Diagnosis Research Center (C.B.), The Mina and Everard Goodman Faculty of LifeSciences, Bar-Ilan University, Ramat-Gan 52900, Israel; and Department of Biological Regulation (R.S.),The Weizmann Institute of Science, Rehovot 76100, Israel
GnRH is the first key hormone of reproduction. The role of protein kinase C (PKC) isoforms in GnRH-stimulated MAPK [ERK and Jun N-terminal kinase (JNK)] was examined in the �T3-1 and L�T2 gona-dotrope cells. Incubation of the cells with GnRH resulted in a protracted activation of ERK1/2 and aslower and more transient activation of JNK1/2. Gonadotropes express conventional PKC� and con-ventional PKC�II, novel PKC�, novel PKC�, and novel PKC�, and atypical PKC-�/�. The use of greenfluorescentprotein-PKCconstructs revealedthatGnRHinducedrapidtranslocationofPKC�andPKC�IIto the plasma membrane, followed by their redistribution to the cytosol. PKC� and PKC� localized tothe cytoplasm and Golgi, followed by the rapid redistribution by GnRH of PKC� to the perinuclear zoneand of PKC� to the plasma membrane. Interestingly, PKC�, PKC�II, and PKC� translocation to theplasma membrane was more pronounced and more prolonged in phorbol-12-myristate-13-acetate(PMA) than in GnRH-treated cells. The use of selective inhibitors and dominant-negative plasmids forthe various PKCs has revealed that PKC�II, PKC�, and PKC� mediate ERK2 activation by GnRH, whereasPKC�, PKC�II, PKC�, and PKC� mediate ERK2 activation by PMA. Also, PKC�, PKC�II, PKC�, and PKC� areinvolved in GnRH and PMA stimulation of JNK1 in a cell-context-dependent manner. We presentpreliminary evidence that persistent vs. transient redistribution of selected PKCs or redistribution of agiven PKC to the perinuclear zone vs. the plasma membrane may dictate its selective role in ERK or JNKactivation. Thus, we have described the contribution of selective PKCs to ERK and JNK activation byGnRH. (Endocrinology 151: 4894–4907, 2010)
Hypothalamic GnRH is secreted in a pulsatile mannerto stimulate the synthesis and release of LH and FSH
(for review, see Refs. 1 and 2). Signaling of GnRH receptor(GnRHR) involves activation of phospholipase C(PLC)-�, phospholipase D, phospholipase A2, enhancedphosphoinositide turnover, Ca2� mobilization and influx,activation of protein kinase C (PKC), and the MAPK cas-
cades and formation of prostaglandins and leukotrienes,culminating in gonadotropin synthesis and release (2–5).
PKC is a family of 10 related isoforms (6, 7), whichplays a significant role in signaling (6, 7). The PKCs areclassified into conventional (cPKC; cPKC-�, cPKC�I,cPKC�II, and cPKC�), novel (nPKC; nPKC�, nPKC�,nPKC�, and nPKC/�), and atypical (aPKC; aPKC and
ISSN Print 0013-7227 ISSN Online 1945-7170Printed in U.S.A.Copyright © 2010 by The Endocrine Societydoi: 10.1210/en.2010-0114 Received January 28, 2010. Accepted July 27, 2010.First Published Online September 1, 2010
Abbreviations: aPKC, Atypical PKCs; cPKC, conventional PKCs; DAG, diacylglycerol; DN,dominant negative; DP, diphospho; DUSP, dual-specificity phosphatase; FCS, fetal calfserum; FSH�, FSH �-subunit; G protein, guanine nucleotide binding protein; GFP, greenfluorescent protein; GnRHR, GnRH receptor; GRASP65, Golgi reassembly stacking proteinof 65 kDa; HA, hemagglutinin epitope; JNK, Jun N-terminal kinase; LH�, LH �-subunit;nPKC, novel PKCs; PKC, protein kinase C; PKCs, PKC isoforms; PMA, phorbol 12-myristate13-acetate; PS, phosphatidylserine; siRNA, small interfering RNA.
N E U R O E N D O C R I N O L O G Y
4894 endo.endojournals.org Endocrinology, October 2010, 151(10):4894–4907
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 29 July 2015. at 01:57 For personal use only. No other uses without permission. . All rights reserved.

aPKC�/�) PKCs. cPKCs are activated mainly by Ca2�, di-acylglycerol (DAG), or phorbol-12-myristate-13-acetate(PMA) and phosphatidylserine (PS) and are therefore re-garded as Ca2�-dependent PKCs. nPKCs are Ca2� inde-pendent and are activated by PS and DAG, or PMA. aPKCsare Ca2� and DAG, or PMA independent and are activatedby PS and some other lipid mediators (6, 7).
We and others have shown that GnRH activates gona-dotrope PKC and that PKC is involved in GnRH-stimulatedgonadotropin synthesis and release (2). In regard to specificPKCs, we have demonstrated that GnRH activates PKC�,PKC� and PKC� gene expression (8, 9) and that PKC�
and PKC� reconstituted PMA-induced LH secretion fromcultured rat pituitary cells using a depletion-insertionmethod (10). Both PMA and GnRH were reported to inducedown-regulation of PKCs in the gonadotrope cell lines (11,12). PKC� and PKC� were also reported to mediate GnRHactivation of cAMP in L�T2 cells (13).
Although PKC is implicated in MAPK activation bysome G protein-coupled receptors in general and byGnRH in particular (2), the nature of the PKCs mediatingGnRH activation of the MAPKs is still unknown. Thegonadotrope cell lines �T3-1 and L�T2 were reported toexpress PKC�, PKC�II, PKC�, PKC�, PKC�, PKC,PKC�/�, and PKC, which represent members of all groupsof the PKC superfamily (12, 14–16).
MAPKcascadesconsistofuptosixtiersofproteinkinases(17–19). At least four distinct MAPK cascades are known inmammals: ERK1/2 (p42 and p44), Jun N-terminal kinase(JNK; 1/2/3), p38 (�, �, �2, �, �), and ERK5 (17–19). Thehallmarkof theMAPKfamily is their ability to translocate tothenucleusandactivateavarietyof transcription factors,butcytosolic substrates are also activated during the retention ofMAPKs in the cytosol (19). MAPKs are thought to partici-pate in the transcriptional control of the gonadotropin sub-unit and GnRHR genes by GnRH (2, 20).
Here we demonstrate for the first time the relative con-tribution of selective PKCs to ERK and JNK activation byGnRH and PMA in �T3-1 and L�T2 cells. We show thatthe contribution of selective PKCs to MAPK activation isligand- and cell-context dependent. We also present someevidence that persistent vs. transient redistribution of se-lected PKCs or redistribution of a given PKC to the pe-rinuclear zone vs. the plasma membrane may dictate theiraccessibility to MAPK activation.
Materials and Methods
MaterialsGnRH, PMA, and rottlerin were obtained from Sigma (St.
Louis, MO). PD098059, Go6976, and GF109203X were ob-tained from A. G. Scientific, Inc. (San Diego, CA). Mouse mono-
clonal antiactive [anti-doubly phosphorylated (DP)]-ERK anti-bodies and rabbit polyclonal antibodies to general ERK, JNK,and PKC isoforms (�, �II, �, �, �, , �/�, and ) were obtainedfrom Sigma (Rehovot, Israel). Rabbit polyclonal anti-DP-JNKwas obtained from Cell Signaling Technology, Inc. (Danvers,MA). Mouse monoclonal antihemagglutinin epitope (HA) andanti-green fluorescent protein (GFP) antibodies were fromRoche Applied Science (Indianapolis, IN). Secondary horserad-ish peroxidase-conjugated goat antimouse antibodies or goatantirabbit antibodies were purchased from Jackson ImmunoRe-search Laboratories (West Grove, PA). All medium and serumwere obtained from Biological Industries (Beit-Hemeek, Israel).Vectashield mounting medium was from Vector Laboratories,Inc. (Burlingame, CA). ExGen 500 transfection reagent was ob-tained from Fermentas International, Inc. (Burlington, Canada),FuGENE6 was obtained from Roche Applied Science, and Tran-sIT-LT1 transfection reagent was obtained from Mirus BIOCorp. (Madison, WI). Protein A/G agarose beads were obtainedfrom Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
PlasmidsGFP-ERK2 and JNK1-HA constructs were previously de-
scribed (21, 22). GFP-PKCs and dominant-negative (DN)pHACE-PKCs constructs were described elsewhere (23–25).GRASP65-RFP construct was kindly provided by Dr. K. Hir-schberg (Tel-Aviv University).
Cell culture�T3-1 and L�T2 cells were grown in monolayer cultured in
DMEM supplemented with 10% fetal calf serum (FCS) and an-tibiotics in humidified 5% CO2 at 37 C. Cells were then starvedovernight in 0.1% serum DMEM before being pretreated withvarious inhibitors for 20 min. GnRH and PMA were then addedfor the length of time as indicated.
Activation of MAPK cascadesCells were harvested and proteins were separated. Western
blotting with anti-DP-MAPKs (pERK and pJNK) antibodies andtotal MAPKs were detected with polyclonal antibodies, followedby horseradish peroxidase-conjugated antimouse, or antirabbitFab antibodies, enhanced chemiluminescence (26) and densi-tometry (27). Results [percent of maximal phosphorylation(pERK and pJNK)] were first normalized to the levels obtainedwith total ERK and JNK, respectively, and the activation valueswere normalized for each treatment vs. its control (e.g.GnRH�drug vs. drug alone). Then the values obtained wereexpressed as percent of maximal activation that was observed ina particular experiment.
Cell transfectionIn general, �T3-1 cells were transiently transfected by ExGen
500 (Fermentas International) and L�T2 cells by FuGENE6(Roche Applied Science) or its analog, TransIT-LT1 transfectionreagent (Mirus BIO). For experiments with DN PKCs, �T3-1cells (in 60 mm plates) were transfected with 1.5 �g of GFP-ERK2 or JNK1-HA and 3 �g of control vector, pCDNA3, orwith 3 �g of the DN-PKCs constructs. L�T2 cell transfectionswere performed in 100-mm plates with 4 �g of GFP-ERK2 orJNK-HA along with 9 �g of control vector, pCDNA3, or 9 �g ofthe DN-PKCs constructs. Approximately 30 h after transfection,the cells were serum starved (0.1% FCS) for 16 h and then stim-
Endocrinology, October 2010, 151(10):4894–4907 endo.endojournals.org 4895
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 29 July 2015. at 01:57 For personal use only. No other uses without permission. . All rights reserved.

ulated with GnRH or PMA. They were then washed twice withice-cold PBS and treated with the lysis buffer, followed by onefreeze-thaw cycle. Cells were then harvested; after centrifugation(15,000 � g, 15 min, 4 C), supernatants were taken for immu-noprecipitation experiments.
Small interfering RNA (siRNA) and cell transfectionsiRNA for PKC� and HIV-1 trans-activator of transcription
(HIV-1 Tat) (as a control) were obtained from Dharmacon(Lafayette, CO). Transfection of the siRNA into cells was madeby using DharmaFECT I (Dharmacon) according to the manu-facturer’s instructions.
ImmunoprecipitationFor immunoprecipitation, protein A/G agarose beads were
mixed with anti-HA or anti-GFP antibodies at room temperaturefor 1 h, after which the beads were washed twice with PBS. Cell
lysates (300–500 �g) were added to thebeads and rotated at 4 C overnight. The im-munocomplexes were washed twice with ly-sis buffer and heated to 100 C in sodiumdodecyl sulfate-gel loading buffer.
Confocal microscopySubconfluent cells were plated on
18-mm coverslips and transfected with 2 �gGFP-PKCs constructs. Twenty-four hoursafter transfection, cells were serum starved(16 h, 0.1% FCS) and treated with GnRHand PMA for various time points. Subse-quently the cells were washed with PBS,fixed in 4% paraformaldehyde in PBS, andcoverslips placed on glass slides withVectashield mounting medium (VectorLab-oratories). Confocal fluorescent imageswere collected with a �40 magnificationlens on a Zeiss LSM510 confocal micro-scope (Zeiss, Jena, Germany).
Statistical analysisResults from three experiments were ex-
pressed as mean � SEM. Where appropriate,data were subjected to statistical analysis byStudent’s t test. Values of P � 0.05 wereconsidered statistically significant.
Results
ERK and JNK are activated byGnRH and PMA
Incubation of the gonadotrope�T3-1 cells (Fig. 1A) or the more ma-ture L�T2 cells (Fig. 1B) with GnRH(10 nM) resulted in a rapid and robustprotracted activation of ERK1/2, with aslower and more transient activation ofJNK1/2 (Fig. 1, Aa and Ab, and Ba andBb). Peak ERK1/2 activation was ob-
served 5 min after GnRH treatment, declining thereafter,but still detectable after 90 min (Fig. 1, A and B). JNK1/2activation by GnRH was more transient with maximalresponse occurring after 30 min, declining to basal levelsafter 90 min (Fig. 1, Ab and Bb). Because in many exper-iments the basal level was undetectable (e.g. Fig. 1Bb), itwas difficult to calculate the magnitude of MAPK activa-tion, which ranged between 10- and 40-fold. We thereforeexpressed the data as percent of maximal activation. Tostudy the role of PKC in MAPK activation, we stimulatedthe cells with the PKC activator, PMA, which mimics thebinding of DAG, the natural activator of PKC, to the C1region of the PKCs (6, 7, 28–33) (Fig. 1, Ac and Ad, andBc and Bd). As with GnRH, ERK1/2 activation was robust
FIG. 1. Time course of MAPK activation by GnRH and PMA in �T3-1 cells and L�T2 cells.�T3-1 (A) and L�T2 (B) cells were serum starved for 16 h before treatment with the GnRH (10nM) or PMA (50 nM) for 5–90 min. After the treatment, cell lysates were analyzed for ERK1/2(a and c) or JNK1/2 (b and d) activity by Western blotting using an antibody for phospho-ERK(pERK) or phospho-JNK (pJNK) accordingly. Total ERK (gERK) and JNK (gJNK) were detectedwith polyclonal antibodies as a control for sample loading. A representative blot is shown andsimilar results were observed in two other experiments. In this and subsequent figures, resultsfrom three experiments are shown as mean � SEM of maximal phosphorylation.
4896 Dobkin-Bekman et al. Differential Role of PKC Isoforms in MAPK Activation by GnRH and PMAEndocrinology, October 2010, 151(10):4894–4907
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 29 July 2015. at 01:57 For personal use only. No other uses without permission. . All rights reserved.
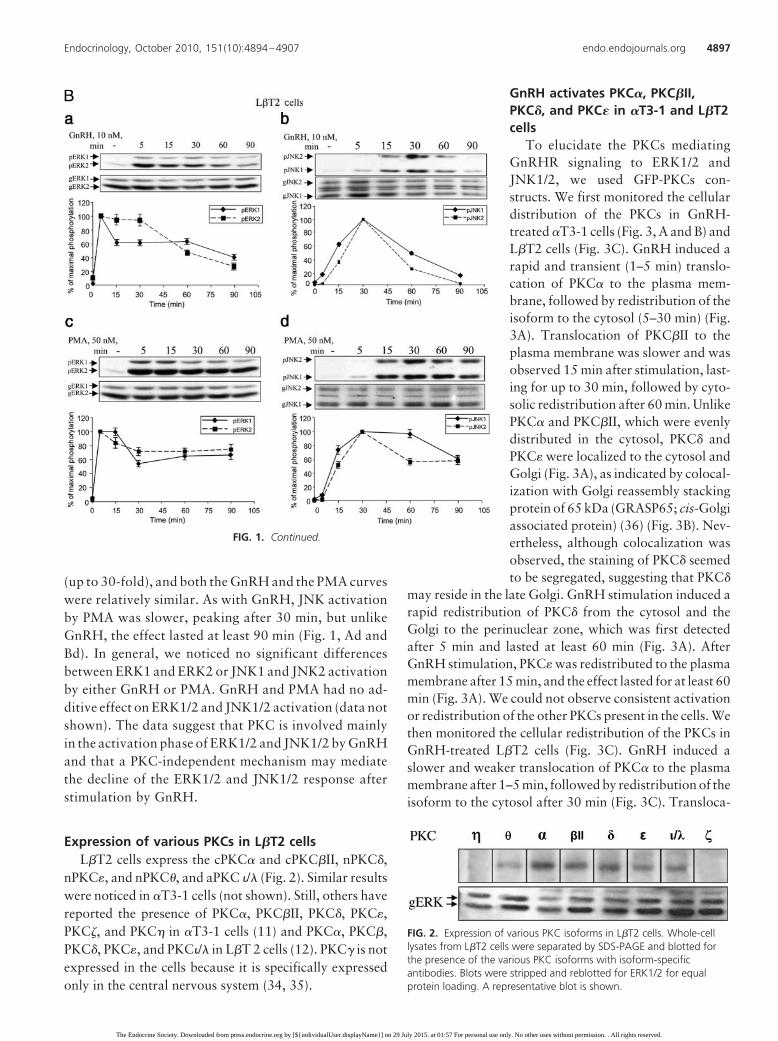
(up to 30-fold), and both the GnRH and the PMA curveswere relatively similar. As with GnRH, JNK activationby PMA was slower, peaking after 30 min, but unlikeGnRH, the effect lasted at least 90 min (Fig. 1, Ad andBd). In general, we noticed no significant differencesbetween ERK1 and ERK2 or JNK1 and JNK2 activationby either GnRH or PMA. GnRH and PMA had no ad-ditive effect on ERK1/2 and JNK1/2 activation (data notshown). The data suggest that PKC is involved mainlyin the activation phase of ERK1/2 and JNK1/2 by GnRHand that a PKC-independent mechanism may mediatethe decline of the ERK1/2 and JNK1/2 response afterstimulation by GnRH.
Expression of various PKCs in L�T2 cellsL�T2 cells express the cPKC� and cPKC�II, nPKC�,
nPKC�, and nPKC�, and aPKC �/� (Fig. 2). Similar resultswere noticed in �T3-1 cells (not shown). Still, others havereported the presence of PKC�, PKC�II, PKC�, PKC�,PKC, and PKC in �T3-1 cells (11) and PKC�, PKC�,PKC�, PKC�, and PKC�/� in L�T 2 cells (12). PKC� is notexpressed in the cells because it is specifically expressedonly in the central nervous system (34, 35).
GnRH activates PKC�, PKC�II,PKC�, and PKC� in �T3-1 and L�T2cells
To elucidate the PKCs mediatingGnRHR signaling to ERK1/2 andJNK1/2, we used GFP-PKCs con-structs. We first monitored the cellulardistribution of the PKCs in GnRH-treated �T3-1 cells (Fig. 3, A and B) andL�T2 cells (Fig. 3C). GnRH induced arapid and transient (1–5 min) translo-cation of PKC� to the plasma mem-brane, followed by redistribution of theisoform to the cytosol (5–30 min) (Fig.3A). Translocation of PKC�II to theplasma membrane was slower and wasobserved 15 min after stimulation, last-ing for up to 30 min, followed by cyto-solic redistribution after 60 min. UnlikePKC� and PKC�II, which were evenlydistributed in the cytosol, PKC� andPKC� were localized to the cytosol andGolgi (Fig. 3A), as indicated by colocal-ization with Golgi reassembly stackingprotein of 65 kDa (GRASP65; cis-Golgiassociated protein) (36) (Fig. 3B). Nev-ertheless, although colocalization wasobserved, the staining of PKC� seemedto be segregated, suggesting that PKC�
may reside in the late Golgi. GnRH stimulation induced arapid redistribution of PKC� from the cytosol and theGolgi to the perinuclear zone, which was first detectedafter 5 min and lasted at least 60 min (Fig. 3A). AfterGnRH stimulation, PKC� was redistributed to the plasmamembrane after 15 min, and the effect lasted for at least 60min (Fig. 3A). We could not observe consistent activationor redistribution of the other PKCs present in the cells. Wethen monitored the cellular redistribution of the PKCs inGnRH-treated L�T2 cells (Fig. 3C). GnRH induced aslower and weaker translocation of PKC� to the plasmamembrane after 1–5 min, followed by redistribution of theisoform to the cytosol after 30 min (Fig. 3C). Transloca-
FIG. 1. Continued.
FIG. 2. Expression of various PKC isoforms in L�T2 cells. Whole-celllysates from L�T2 cells were separated by SDS-PAGE and blotted forthe presence of the various PKC isoforms with isoform-specificantibodies. Blots were stripped and reblotted for ERK1/2 for equalprotein loading. A representative blot is shown.
Endocrinology, October 2010, 151(10):4894–4907 endo.endojournals.org 4897
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 29 July 2015. at 01:57 For personal use only. No other uses without permission. . All rights reserved.
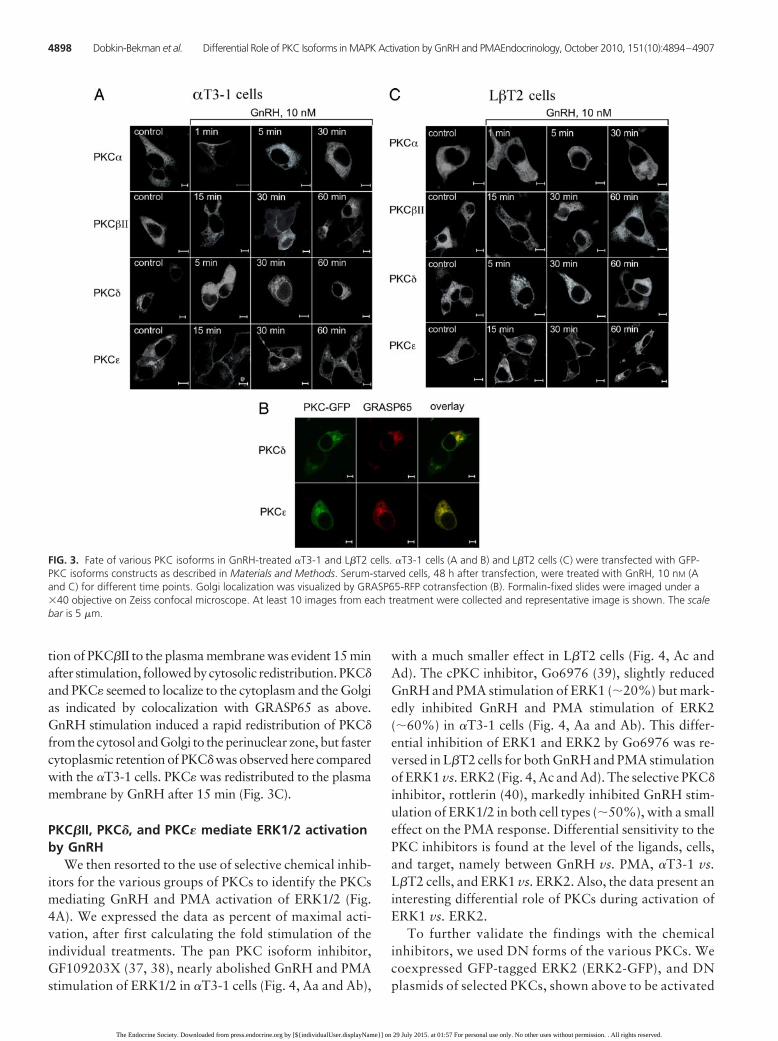
tion of PKC�II to the plasma membrane was evident 15 minafter stimulation, followedbycytosolic redistribution.PKC�
and PKC� seemed to localize to the cytoplasm and the Golgias indicated by colocalization with GRASP65 as above.GnRH stimulation induced a rapid redistribution of PKC�
from the cytosol and Golgi to the perinuclear zone, but fastercytoplasmic retention of PKC� was observed here comparedwith the �T3-1 cells. PKC� was redistributed to the plasmamembrane by GnRH after 15 min (Fig. 3C).
PKC�II, PKC�, and PKC� mediate ERK1/2 activationby GnRH
We then resorted to the use of selective chemical inhib-itors for the various groups of PKCs to identify the PKCsmediating GnRH and PMA activation of ERK1/2 (Fig.4A). We expressed the data as percent of maximal acti-vation, after first calculating the fold stimulation of theindividual treatments. The pan PKC isoform inhibitor,GF109203X (37, 38), nearly abolished GnRH and PMAstimulation of ERK1/2 in �T3-1 cells (Fig. 4, Aa and Ab),
with a much smaller effect in L�T2 cells (Fig. 4, Ac andAd). The cPKC inhibitor, Go6976 (39), slightly reducedGnRH and PMA stimulation of ERK1 (�20%) but mark-edly inhibited GnRH and PMA stimulation of ERK2(�60%) in �T3-1 cells (Fig. 4, Aa and Ab). This differ-ential inhibition of ERK1 and ERK2 by Go6976 was re-versed in L�T2 cells for both GnRH and PMA stimulationof ERK1 vs. ERK2 (Fig. 4, Ac and Ad). The selective PKC�
inhibitor, rottlerin (40), markedly inhibited GnRH stim-ulation of ERK1/2 in both cell types (�50%), with a smalleffect on the PMA response. Differential sensitivity to thePKC inhibitors is found at the level of the ligands, cells,and target, namely between GnRH vs. PMA, �T3-1 vs.L�T2 cells, and ERK1 vs. ERK2. Also, the data present aninteresting differential role of PKCs during activation ofERK1 vs. ERK2.
To further validate the findings with the chemicalinhibitors, we used DN forms of the various PKCs. Wecoexpressed GFP-tagged ERK2 (ERK2-GFP), and DNplasmids of selected PKCs, shown above to be activated
FIG. 3. Fate of various PKC isoforms in GnRH-treated �T3-1 and L�T2 cells. �T3-1 cells (A and B) and L�T2 cells (C) were transfected with GFP-PKC isoforms constructs as described in Materials and Methods. Serum-starved cells, 48 h after transfection, were treated with GnRH, 10 nM (Aand C) for different time points. Golgi localization was visualized by GRASP65-RFP cotransfection (B). Formalin-fixed slides were imaged under a�40 objective on Zeiss confocal microscope. At least 10 images from each treatment were collected and representative image is shown. The scalebar is 5 �m.
4898 Dobkin-Bekman et al. Differential Role of PKC Isoforms in MAPK Activation by GnRH and PMAEndocrinology, October 2010, 151(10):4894–4907
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 29 July 2015. at 01:57 For personal use only. No other uses without permission. . All rights reserved.

by GnRH. We then stimulated the cells with GnRH,lysed the cells, immunoprecipitated the tagged MAPKswith anti-GFP, and immunoblotted with anti-DP-ERKantibodies. As seen in Fig. 4B, the DN-PKC� had nosignificant effect on ERK2 activation by GnRH in�T3-1 and L�T2 cells (Fig. 4, Ba and Bb). On the otherhand, the DN-PKC�II markedly reduced ERK2 activa-tion by GnRH in both cell types (�50 – 60%). We there-fore concluded that the cPKC involved in GnRHR sig-
naling to ERK and identified throughthe use of Go6976 (Fig. 4A) wasPKC�II. The DN-PKC� and the DN-PKC� reduced ERK2 activation byGnRH in both cell types (�40 – 60%;Fig. 4B). Thus, PKC�II, PKC�, andPKC� are the major players in medi-ating ERK1/2 activation by GnRH.
As a proof of principle for the use ofthe DN-PKC approach, we also moni-tored the role of PKC� in GnRH-inducedERK activation using siRNA for PKC�.Incubation of �T3-1 cells with siRNA forPKC� for 96 h reduced PKC� expressionby 60%, whereas a nonrelated siRNAcontrol had no effect (Fig. 4C, upperlane). The reduction in PKC� expressionby the selective siRNA resulted also in47% inhibition of GnRH stimulation ofERK2 activation, whereas the controlsiRNAhadnoeffect (Fig.4C).Theresultsare in excellent agreement with the ef-fect obtained by the DN-PKC� (50%)(Fig. 4B).
PKC�, PKC�II, PKC�, and PKC� areinvolved in GnRH stimulation ofJNK1/2 in a cell-context-dependent manner
We then investigated the role of thePKCsinJNK1/2activationbyGnRHandPMA in �T3-1 and L�T2 cells (Fig. 5A).Unlike ERK1/2, in which the pan-PKCinhibitor, GF109203X, abolished stimu-lation by GnRH in �T3-1 cells and had alesser effect in L�T2 cells (Fig. 4A), theopposite was observed for JNK1/2 acti-vation. Here GF109203X produced alarger inhibition in the mature L�T2 cells(�70%), with a smaller effect in the im-mature �T3-1 cells (Fig. 5, Aa and Ac).Unlike ERK1/2 (Fig. 4A), the cPKC in-hibitor, Go6976, abolished GnRH stim-ulation of JNK1/2 in �T3-1 cells and dif-
ferentially reduced the activation of JNK1 (40%) and JNK2(80%) by GnRH in L�T2 cells (Fig. 5, Aa and Ac). Again,unlike ERK1/2 (Fig. 4A), the selective PKC� inhibitor, rot-tlerin, had little effect on JNK1/2 activation by GnRH in�T3-1 cells and differentially reduced GnRH-stimulatedJNK1 (20%) and JNK2 (70%) in L�T2 cells (Fig. 5, Aa andAc). Bypassing the GnRHR through the direct activation ofthe PKCs by PMA revealed that, similar to ERK1/2,
FIG. 4. Effect of selective PKC isoforms inhibitors and DN plasmids of various PKC isoformson ERK1/2 activation in �T3-1 and L�T2 cells. A, �T3-1 (a and b) and L�T2 cells (c and d)were serum starved for 16 h before pretreatment with the pan PKC inhibitor GF 109203X(GF; 3 �M) or the cPKC inhibitor Go6976 (Go; 1 �M) or the selective PKC� inhibitor rottlerin(Rot; 3 �M) for 30 min. Thereafter GnRH (10 nM, a and c) or PMA (50 nM, b and d) wasadded for 5 min. After the treatment, cell lysates were analyzed for ERK1/2 activity byWestern blotting using an antibody for phospho-ERK1/2 (pERK). Total ERK1/2 (gERK) wasdetected with polyclonal antibodies as a control for sample loading. B, �T3-1 (a) and L�T2cells (b) were transiently cotransfected with GFP-ERK2 and control vector or with GFP-ERK2and the DN plasmids of various PKCs as indicated. GnRH (10 nM) was added for 10 min toserum-starved cells approximately 48 h after transfection. After treatment, cell lysates wereanalyzed for ERK activity as described in Materials and Methods. A representative blot isshown and similar results were observed in two other experiments. Here and thereafter,results were normalized for each treatment vs. its control, as described in Materials andMethods. Results from three experiments are shown as mean � SEM of maximal activation.*, Statistical significance (P � 0.05); **, statistical significance (P � 0.01); ***, statisticalsignificance (P � 0.001) vs. maximal phosphorylation. C, Effect of knockdown of PKC� onGnRH-induced ERK activation. siRNA for PKC� and siRNA for HIV-1 trans-activator oftranscription (HIV-1 Tat) (as a control) were transfected into �T3-1 cells by using DharmaFECTI according to the manufacturer’s instructions (Dharmacon). Cells were incubated for 80 hand serum starved for an additional 16 h. GnRH (10 nM) was added for 10 min to the serum-starved cells. After treatment, cell lysates were analyzed for ERK activity as described inMaterials and Methods. A representative blot is shown and similar results were observed intwo other experiments.
Endocrinology, October 2010, 151(10):4894–4907 endo.endojournals.org 4899
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 29 July 2015. at 01:57 For personal use only. No other uses without permission. . All rights reserved.

GF109203XabolishedthePMA-inducedJNK1/2activationin both �T3-1 and L�T2 cells (Fig. 5, Ab and Ad). The cPKCinhibitor, Go6976, produced a much larger inhibition ofJNK1/2 activation by PMA (Fig. 5, Ab and Ad), comparedwith its inhibition of the ERK1/2 response as seen in Fig. 4.Likewise to the case of ERK1/2 (Fig. 4A), the selective PKC�
inhibitor, rottlerin, was the weakest inhibitor of PMA sig-naling to JNK1/2. Despite contributing to a 60% reductionto PMA stimulation of JNK1/2 in �T3-1 cells (Fig. 5Ab),rottlerin had no significant effect on PMA-induced JNK1/2activation in L�T2 cells (Fig. 5Ad).
To further confirm the findings, we coexpressedJNK1-HA with the DN-PKCs (Fig. 5B), similar to the ap-proach described in Fig. 4B. However, unlike ERK2, inwhich the DN-PKC� had no significant effect on the GnRH
response (Fig. 4B), a stronger inhibitoryeffect on JNK1 activation by GnRH wasobserved for both �T3-1 and L�T2 cells(Fig. 5, Ba and Bb). Again, unlike ERK2,in which the DN-PKC�II was the stron-gest inhibitor of the GnRH response (Fig.4B), in the case of JNK1 activation, theDN-PKC�II had no effect in �T3-1 cells,and a 40% inhibitory effect was noticedin L�T 2 cells. Only the inhibition of thenPKC family by the DNs of PKC� andPKC� gave a similar magnitude of inhi-bition for both GnRH-stimulated ERK2(Fig. 4B) and JNK1 (�50%) (Fig. 5B).The combined contribution of membersof the cPKC (� and �II) and nPKC (� and�) seems therefore to be required for fullactivation of JNK1 by GnRH in L�T2cells, whereas PKC�, PKC�, and PKC�
are sufficient to mediate JNK1 activationin �T3-1 cells (Fig. 5B). Hence, common(PKC�II, PKC�, and PKC�) and distinct(PKC�) PKCs participate in ERK andJNK activation by GnRH.
PKC�, PKC�, PKC�II, and PKC�
mediate ERK2 activation by PMAin a cell-context-dependentmanner
We then resorted to the use of the DNplasmids of the PKCs to identify the rel-evant PKCs participating in PMA stimu-lation of ERK2 (Fig. 6A). The DN-PKC�
and the DN-PKC�II reduced ERK2 acti-vation by PMA (60 and 40%, respec-tively) in �T3-1 cells. The DN-PKC� andthe DN-PKC� reduced ERK2 activationby PMA (50 and 65%, respectively). We
then repeated the experiment in L�T2 cells (Fig. 6Ab). TheDN-PKC� reduced ERK2 activation by PMA (25%),whereas the DN-PKC�II had no significant effect. The DN-PKC� and the DN-PKC� reduced ERK2 activation by PMA(25 and 50%, respectively). Thus, whereas PKC�II, PKC�,PKC�, and PKC� are the major players in mediating ERK2activation by PMA in �T3-1 cells, PKC�, PKC�, and PKC�
are sufficient in mediating ERK2 activation by PMA inL�T2cells.
PKC�, PKC�, PKC�II, and PKC� mediate JNK1activation by PMA in a cell-context-dependentmanner
We then used a similar approach to identify the relevantPKCs participating in PMA stimulation of JNK1 (Fig. 6B).
FIG. 4. Continued.
4900 Dobkin-Bekman et al. Differential Role of PKC Isoforms in MAPK Activation by GnRH and PMAEndocrinology, October 2010, 151(10):4894–4907
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 29 July 2015. at 01:57 For personal use only. No other uses without permission. . All rights reserved.

The DN-PKC� and the DN-PKC�II re-duced JNK1 activation by PMA (50 and30%, respectively) in �T3-1 cells. TheDN-PKC� and the DN-PKC� reducedJNK1 activation by PMA (50%). Quitedifferent results were found in L�T2cells (Fig. 6Bb). Whereas the DN-PKC�
had no significant effect, that of PKC�IIreduced JNK1 activation by PMA by20%. Surprisingly, the DN-PKC� ele-vated JNK1 activation by PMA, in goodagreement with the lack of inhibitionobtained by the PKC� inhibitor, rot-tlerin, in Fig. 5A. The DN-PKC� gavethe maximal inhibition and reducedJNK1 activation by PMA by 50%.Hence, whereas PKC�II, PKC�, PKC�,and PKC� participate in JNK1 activa-tion by PMA in �T3-1 cells, onlyPKC�II and PKC� play a role in JNK1activation by PMA in L�T2cells.
Differential translocation of PKCsby PMA
It was interesting to find out whetherPKC activation was sufficient to deter-mine its participation in MAPK activa-tion or whether the participation wasdependent on the nature of the activat-ing ligand. This is important becausecommon (PKC�II, PKC�, and PKC�)and separate (PKC�) PKCs were foundto mediate GnRH vs. PMA signaling toERK. Also, whereas PKC�, PKC�II,PKC�, and PKC� participate in JNK1activation by GnRH in L�T2 cells, onlyPKC�II and PKC� play a role in JNK1activation by PMA in the same cells. Wereasoned that differential localizationof the relevant PKCs by the activatingligand may dictate whether a givenPKCs will participate in ERK or JNKactivation in a ligand- and cell-context-dependent manner. To this end we an-alyzed the translocation of variousPKCs by PMA in �T3-1 and L�T2 cells(Fig. 7). PKC� translocation to theplasma membrane induced by PMAwas rapid and more pronounced thanthat obtained with GnRH in Fig. 3 andwas not followed by redistribution ofPKC� to the cytosol (Fig. 7A). Similar
FIG. 5. Effect of selective PKC isoforms inhibitors and DN plasmids of various PKC isoformson JNK1/2 activation in �T3-1 and L�T2 cells. A, �T3-1 (a and b) and L�T2 cells (c and d)were serum starved for 16 h before pretreatment with the pan PKC inhibitor GF 109203X(GF; 3 �M), or the cPKC inhibitor Go6976 (Go; 1 �M) or the selective PKC� inhibitor rottlerin(Rot; 3 �M) for 30 min. Thereafter GnRH (10 nM, a and c) or PMA (50 nM, b and d) wasadded for 30 min. After the treatment, cell lysates were analyzed for JNK1/2 activity byWestern blotting using an antibody for phospho-JNK (pJNK). Total JNK (gJNK) were detectedwith polyclonal antibodies as a control for sample loading. B, �T3-1 (a) and L�T2 cells (b)were transiently cotransfected with HA-JNK1 and control vector or with HA-JNK1 and the DNplasmids of various PKCs as indicated. GnRH (10 nM) was added for 30 min to serum-starvedcells approximately 48 h after transfection. After treatment, cell lysates were analyzed forJNK1 activity as described in Materials and Methods. A representative blot is shown andsimilar results were observed in two other experiments. Bars shown are mean � SEM from thethree experiments. *, Statistical significance (P � 0.05) vs. maximal phosphorylation.
Endocrinology, October 2010, 151(10):4894–4907 endo.endojournals.org 4901
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 29 July 2015. at 01:57 For personal use only. No other uses without permission. . All rights reserved.

differences were also seen in L�T2 cells, but here the trans-location was slower (15 min), albeit persistent (Fig. 7B).PKC�II translocation to the plasma membrane by PMAwas more persistent and evenly distributed at 60 min (Fig.7A), compared with that obtained with GnRH in Fig. 3.Weaker translocation of PKC�II to the plasma membranewas observed in L�T2 cells but also included the nuclearenvelope at 30–60 min (Fig. 7B). PKC� was redistributed
by PMA from the cytosol and Golgi to the perinuclear zoneand the plasma membrane after 15 min (Fig. 7A). In L�T2cells, PKC� was mainly distributed to the plasma mem-brane by PMA (5–60 min) (Fig. 7B). PKC� was redistrib-uted from the cytosol and Golgi to the plasma membraneafter 15–30 min (Fig. 7).
Hence, PKC�, PKC�II, and PKC� translocation to theplasma membrane was more pronounced and more pro-
FIG. 6. Effect of DN plasmids of various PKC isoforms on PMA-induced ERK2 and JNK1 activation in �T3-1 and L�T2 cells. A, �T3-1 (a) and L�T2cells (b) were transiently cotransfected with GFP-ERK2 and control vector or with GFP-ERK2 and the DN plasmids of various PKCs as indicated.PMA (50 nM) was added for 10 min to serum-starved cells approximately 48 h after transfection. After treatment, cell lysates were analyzed forERK2 activity as described in Materials and Methods. B, �T3-1 (a) and L�T2 cells (b) were transiently cotransfected with HA-JNK1 and controlvector or with HA-JNK1 and the DN plasmids of various PKCs as indicated. PMA (50 nM) was added for 30 min to serum-starved cellsapproximately 48 h after transfection. After treatment, cell lysates were analyzed for JNK1 activity as described in Materials and Methods. Arepresentative blot is shown and similar results were observed in two other experiments. Bars shown are mean � SEM from the three experiments.*, Statistical significance (P � 0.05); **, statistical significance (P � 0.01); ***, statistical significance (P � 0.001) vs. maximal phosphorylation.
4902 Dobkin-Bekman et al. Differential Role of PKC Isoforms in MAPK Activation by GnRH and PMAEndocrinology, October 2010, 151(10):4894–4907
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 29 July 2015. at 01:57 For personal use only. No other uses without permission. . All rights reserved.

longed in PMA- than GnRH-treated cells. Thus, the fate ofthe various PKCs seems to be ligand dependent. Also, thetranslocation of PKC� by PMA differs between �T3-1 andL�T2 cells, suggesting that the fate of selective PKCs maybe also cell-context dependent.
Discussion
The role of PKC in MAPK activation by GnRH has beenstudied extensively (2, 41). Here we further examined thespecific PKC isoforms activated by GnRH and participatingin MAPK activation. MAPK (ERK and JNK) activation byGnRH is robust (20- to 50-fold). GnRH induced a rapid andprotracted activation of ERK1/2 but a slower and more tran-sient activation of JNK1/2. Interestingly, after 90 min ofGnRH stimulation, only ERK1/2 but not JNK1/2 was stillactivated, which may be significant in terms of transcrip-tional control and signal specificity (18). To elucidate thesignaling from PKC to MAPK, we used PMA, which mimicsthe action of the naturally occurring DAG by binding to theC1 region of PKC, thus activating the enzyme (7, 29). PMAmimicked the activation of ERK and JNK by GnRH but notthe subsequentdeclineof the response.Wethereforeproposethat the activation, but not the termination of ERK and JNKsignaling from the GnRH receptor, is mediated by a PKC-dependent mechanism.
It was tempting to suggest that the other arm ofGnRHR signaling involving Ca2�, which plays a criticalrole in GnRH-induced gonadotropin secretion (3, 42),was involved in signal termination. We therefore treated
the cells individually with the Ca2� ionophores A23187and ionomycin or in various combinations with GnRH, orPMA, to pharmacologically reconstitute the GnRH acti-vation profile but failed to mimic the MAPK signal ter-mination (data not shown). Inactivation of MAPK can beexplained by activation of dual-specificity phosphatases(DUSPs), particularly the MAPK phosphatases and atyp-ical DUSPs that can both dephosphorylate ERK1/2. InHeLa cells, 12 of 16 DUSPs influenced ERK2 responses, asshown by a short inhibitory RNA screen (43). However,there is no evidence for termination of GnRHR signalingby the nuclear-inducible MAPK phosphatases, DUSP1, -2,or -4. The siRNA screen revealed that GnRH effects can beinfluenced by members of each of the major DUSP sub-types, although these proteins do not have a direct role indephosphorylation of ERK in GnRH responses (44).MAPK activation kinetics is thought to play a role in signalspecificity (18). Our data suggest that the profile of MAPKactivation by GnRH observed here cannot be responsiblefor gonadotrope maturation because we could not observesignificant differences when comparing the �T3-1 cells,which were derived from mouse embryonic day 11.5 (45)vs. the more mature L�T2 cells, which were derived frommouse embryonic day 16.5 (46).
We then searched for the PKC isoforms activated byGnRH and participating in ERK and JNK activation. Us-ing GFP-PKC constructs (23, 25), we followed the fate ofthe various PKCs in GnRH- and PMA-treated cells. Wecould thus show that GnRH and PMA activate PKC�,PKC�II, PKC�, and PKC�. Interestingly, PKC� and PKC
FIG. 7. Fate of various PKC isoforms in PMA-treated �T3-1 and L�T2 cells. �T3-1 cells (A) and L�T2 cells (B) were transfected with GFP-PKCisoforms constructs as described in Materials and Methods. Serum-starved cells, 48 h after transfection, were treated with PMA, 50 nM fordifferent time points. Formalin-fixed slides were imaged under a �40 objective on Zeiss confocal microscope. At least 10 images from eachtreatment were collected and representative image is shown. The scale bar is 5 �m.
Endocrinology, October 2010, 151(10):4894–4907 endo.endojournals.org 4903
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 29 July 2015. at 01:57 For personal use only. No other uses without permission. . All rights reserved.

are highly homologous and both have been reported tocolocalize to the Golgi in which they induce protein kinaseD activation (47, 48). Once activated, protein kinase D isthought to be involved in the process of fission in transportof vesicles from the trans-Golgi network to the plasmamembrane (49). Hence, it would be interesting to inves-tigate whether GnRH-activated PKC� and PKC� in theGolgi participate in pituitary gonadotrope secretory gran-ules formation and transport during GnRH activation ofgonadotropin secretion.
We then resorted to the use of isoform-specific chemicalinhibitors and DN plasmids of the relevant PKCs to iden-tify the PKCs mediating GnRH and PMA signaling to ERKand JNK. Interestingly, gonadotrope maturation has pro-duced a switch in the relative sensitivity of ERK1 andERK2 to the cPKC inhibitor, Go6976, which was inde-pendent of activation of the GnRHR. Also the role of PKCin GnRH to ERK1/2 signaling appears to diminish withthe maturation of pituitary gonadotropes, as evidenced bya decrease in the response to the pan PKC isoforms inhib-itor, GF109203X, in L�T2 cells as compared with theimmature �T3-1 cells.
Interestingly, common and separate PKCs seem to par-ticipate in GnRH and PMA signaling to ERK2 and JNK1.In the case of GnRH, PKC�II, PKC�, and PKC� contributeto ERK2 activation in �T3-1 and L�T2 cells. However,bypassing the GnRHR by direct activation of PKC byPMA implicated PKC�, PKC�II, PKC�, and PKC� inERK2 activation in �T3-1 cells, whereas PKC�, PKC�,and PKC� participate in ERK2 activation by PMA inL�T2 cells (Fig. 8). Similarly, whereas PKC�, PKC�, andPKC� participate in JNK1 activation by GnRH in �T3-1cells, PKC�, PKC�II, PKC�, and PKC� participate in
JNK1 activation by GnRH in L�T2cells. A somewhat different profile wasobtained for PMA. Whereas PKC�,PKC�II, PKC�, and PKC� participate inJNK1 activation by PMA in �T3-1cells, only PKC�II and PKC� partici-pate in JNK1 activation by PMA inL�T2 cells. Hence, common and sepa-rate PKCs mediate ERK and JNK acti-vation in a ligand- and cell-context-de-pendent manner. We are thereforepresented with a paradox: both GnRHand PMA activate PKC�; however, theactivated PKC� is used by PMA but notGnRH for ERK2 activation in L�T2cells. The most likely explanation to re-solve the paradox is that both ligandshave differentially directed PKC� interms of spatio/temporal distributionaffecting its accessibility to the ERK
cascade. Indeed, when comparing the effect of GnRH andPMA, we found that persistent redistribution of PKC� tothe plasma membrane may be needed to implicate the iso-form to ERK activation and that this requirement is met byPMA but not by GnRH.
It is important to notice that the spatio/temporal dis-tribution of the various PKCs affects their intracellularsignaling. Alternatively, or in addition, selective PKCsmay be targeted to different signaling scaffolds in a ligand-and cell-context-dependent manner, and the scaffoldsmay differ in their affinity to ERK and JNK. The impor-tance of the scaffolds has been demonstrated in varioussystems (50–52), including that of the GnRHR. Studiesare therefore in progress to compare the GnRH- and thePMA-activated so-called signalosomes in pituitary gona-dotropes, in the hope of further resolving the above-men-tioned fate of the various PKCs.
Surprisingly, the DN-PKC� elevated JNK1 activation byPMA in L�T2 cells. These data suggest that endogenousPKC� is inhibitory to JNK1 activation by PMA but not byGnRH and in L�T2 but not in �T3-1 cells. Therefore, it wasinteresting to find out whether PMA-induced translocationof PKC� in L�T2 cells is unique. Both GnRH (in �T3-1 andL�T2 cells) and PMA (only in �T3-1 cells) stimulation re-sulted in a rapid redistribution of PKC� from the cytosol andthe Golgi to the perinuclear zone. In contrast, PKC� wasmainly distributed to the plasma membrane by PMA inL�T2 cells. It is therefore possible that this differential dis-tribution may dictate the fate of PKC�.
Various PKCs have been implicated in MAPK activation(53–55). How do the PKCs participate in ERK activation?PKC� can phosphorylate and activate Raf-1 (56). Indeed,
FIG. 8. A proposed model for the relative role of the various PKC isoforms in GnRH- andPMA-stimulated ERK1/2 and JNK1/2 activation in �T3-1 and L�T2 cells.
4904 Dobkin-Bekman et al. Differential Role of PKC Isoforms in MAPK Activation by GnRH and PMAEndocrinology, October 2010, 151(10):4894–4907
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 29 July 2015. at 01:57 For personal use only. No other uses without permission. . All rights reserved.

we have previously shown that GnRH activates Raf-1 in aPKC-dependent manner (57). Also, Ras guanyl nucleotide-releasing proteins can be activated by direct binding of DAGand/or by PKC phosphorylation, leading to Ras/Raf/MAPKkinase/ERK activation (58, 59). Alternatively, or in addi-tion, c-Src may participate in the transmission of signalsfrom PKCs to Raf-1 (60), and this pathway is supported byour findings that 4-amino-5-(4-chlorophenyl)-7-(t-bu-tyl)pyrazolo[3,4-d] pyrimidine blocked GnRH and PMAactivation of ERK and JNK (data not shown).
The use of chemical inhibitors and coexpression of HA-JNK1 and DNs of the various PKCs revealed that membersof both cPKC (PKC� and PKC�II) and nPKC (PKC� andPKC�) are implicated in JNK1 activation in a ligand(GnRH vs. PMA) and cell-context (�T3-1 vs. L�T2 cells)-dependent manner. As with ERK activation, differentialspatio/temporal distribution or binding to different scaf-folds may be responsible for the selective role of a givenPKC in JNK activation. Nevertheless, our implication ofthe major role of PKCs in GnRHR to JNK signaling differsfrom previous reports that found that GnRH activation ofJNK in �T3-1 cells does not involve mediation by PKC (3,61). The differences in growth conditions, passage num-bers, and the use of PKC isoform-specific reagents mayexplain the conflicting results.
How do the PKCs participate in JNK activation? Byserving as a docking site for both PKC and JNK (via dif-ferent tryptophan-aspartic acid domains) the receptor foractivated C kinase 1 promotes PKC phosphorylation ofJNK on S219, which potentiates JNK phosphorylation onresidues 183 and 185 by MAPK kinase 4 and MAPK ki-nase 7, culminating in maximal JNK activation (62). Ourresults have identified the participating PKCs and addedc-Src downstream to the PKCs on the way to JNK.
Once activated by GnRH via the PKCs identified here,ERK1/2 and JNK1/2 participate in GnRH-stimulated go-nadotropin subunit (common �, LH�, and FSH�) geneexpression (63–66). This results in LH and FSH synthesisand release, leading to ovulation and spermatogenesis.
In addition to ameliorating disease through inhibiting go-nadotropin and sex hormone production, GnRH analogshave direct antiproliferative and apoptotic effects on repro-ductive tissues such as prostate cancer cells (67–70). ThePKC/MAPK pathway has been implicated in the antiprolif-erative effects of GnRH analogs in cancer cells. It was dem-onstrated that JNK1/2 and p38 (70) mediate the apoptoticeffects of GnRH in human prostate cancer cells and thatERK1/2activationviaaPKC-dependentpathway isessentialfor GnRH inhibition of proliferation in ovarian cancer cells(71). Therefore, it will be interesting to identify the PKC iso-forms involved in MAPK activation by GnRH analogs in sexhormone-dependent cancers, which mediate a death signal
vs. those identified here that mediate MAPK activation inpituitarycells leading toa life signal (gonadotropinsynthesis,ovulation, and spermatogenesis).
Acknowledgments
We thank P. Mellon for the gonadotrope cell lines and Stefan Limand Zhong Yao for the help and interest in our study.
Address all correspondence and requests for reprints to: Pro-fessor Zvi Naor, Department of Biochemistry, Tel Aviv Univer-sity, Tel-Aviv 69978, Israel. E-mail: [email protected].
This work was supported by The Israel Science FoundationGrant 221/05, the German-Israeli Foundation for Research andDevelopment Grant I-751-168.2/2002, U.S.-Israel BinationalScience Foundation Grant 2007057, and the Adams Super-Cen-ter for Brain Studies at Tel-Aviv University. R.S. is the incumbentof the Yale S. Lewine and Ella Miller Lewine Professorial chairfor cancer research. Z.N. is the incumbent of the Abraham E.Kazan Chair in Structural Biology.
Disclosure Summary: The authors have nothing to disclose.
References
1. Millar RP, Lu ZL, Pawson AJ, Flanagan CA, Morgan K, MaudsleySR 2004 Gonadotropin-releasing hormone receptors. Endocr Rev25:235–275
2. Naor Z 2009 Signaling by G-protein-coupled receptor (GPCR):studies on the GnRH receptor. Front Neuroendocrinol 30:10–29
3. Mulvaney JM, Roberson MS 2000 Divergent signaling pathwaysrequiring discrete calcium signals mediate concurrent activation oftwo mitogen-activated protein kinases by gonadotropin-releasinghormone. J Biol Chem 275:14182–14189
4. Ruf F, Fink MY, Sealfon SC 2003 Structure of the GnRH receptor-stimulated signaling network: insights from genomics. Front Neu-roendocrinol 24:181–199
5. Ferris HA, Shupnik MA 2006 Mechanisms for pulsatile regulationof the gonadotropin subunit genes by GNRH1. Biol Reprod 74:993–998
6. Nishizuka Y 1992 Membrane phospholipid degradation and pro-tein kinase C for cell signalling. Neurosci Res 15:3–5
7. Newton ACa 2003 Regulation of the ABC kinases by phosphory-lation: protein kinase C as a paradigm. Biochem J 370:361–371
8. Shraga-Levine Z, Ben-Menahem D, Naor Z 1994 Activation of pro-tein kinase C beta gene expression by gonadotropin-releasing hor-mone in �T3-1 cell line. Role of Ca2� and autoregulation by proteinkinase C. J Biol Chem 269:31028–31033
9. Harris D, Reiss N, Naor Z 1997 Differential activation of proteinkinase C� and epsilon gene expression by gonadotropin-releasinghormone in �T3-1 cells. Autoregulation by protein kinase C. J BiolChem 272:13534–13540
10. Naor Z, Dan-Cohen H, Hermon J, Limor R 1989 Induction ofexocytosis in permeabilized pituitary cells by �- and �-type proteinkinase C. Proc Natl Acad Sci USA 86:4501–4504
11. Poulin B, Rich N, Mas JL, Kordon C, Enjalbert A, Drouva SV 1998GnRH signalling pathways and GnRH-induced homologous desen-sitization in a gonadotrope cell line (�T3-1). Mol Cell Endocrinol142:99–117
12. Liu F, Austin DA, Webster NJ 2003 Gonadotropin-releasing hor-mone-desensitized L�T2 gonadotrope cells are refractory to acute
Endocrinology, October 2010, 151(10):4894–4907 endo.endojournals.org 4905
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 29 July 2015. at 01:57 For personal use only. No other uses without permission. . All rights reserved.

protein kinase C, cyclic AMP, and calcium-dependent signaling.Endocrinology 144:4354–4365
13. Lariviere S, Garrel G, Simon V, Soh JW, Laverriere JN, Counis R,Cohen-Tannoudji J 2007 Gonadotropin-releasing hormone couplesto 3�,5�-cyclic adenosine-5�-monophosphate pathway throughnovel protein kinase C� and -� in L�T2 gonadotrope cells. Endo-crinology 148:1099–1107
14. Kratzmeier M, Poch A, Mukhopadhyay AK, McArdle CA 1996Selective translocation of non-conventional protein kinase C isoen-zymes by gonadotropin-releasing hormone (GnRH) in the gonado-trope-derived �T3-1 cell line. Mol Cell Endocrinol 118:103–111
15. Naor Z, Harris D, Shacham S 1998 Mechanism of GnRH receptorsignaling: combinatorial cross-talk of Ca2� and protein kinase C.Front Neuroendocrinol 19:1–19
16. Maccario H, Junoy B, Poulin B, Boyer B, Enjalbert A, Drouva SV2004 Protein kinase C� as gonadotropin-releasing hormone targetisoenzyme in the �T3-1 gonadotrope cell line. Neuroendocrinology79:204–220
17. Seger R, Krebs EG 1995 The MAPK signaling cascade. FASEB J9:726–735
18. Murphy LO, Blenis J 2006 MAPK signal specificity: the right placeat the right time. Trends Biochem Sci 31:268–275
19. Yoon S, Seger R 2006 The extracellular signal-regulated kinase:multiple substrates regulate diverse cellular functions. Growth Fac-tors 24:21–44
20. Caunt CJ, Finch AR, Sedgley KR, McArdle CA 2006 Seven-trans-membrane receptor signalling and ERK compartmentalization.Trends Endocrinol Metab 17:276–283
21. Avraham A, Jung S, Samuels Y, Seger R, Ben-Neriah Y 1998 Co-stimulation-dependent activation of a JNK-kinase in T lymphocytes.Eur J Immunol 28:2320–2330
22. Rubinfeld H, Hanoch T, Seger R 1999 Identification of a cytoplas-mic-retention sequence in ERK2. J Biol Chem 274:30349–30352
23. Kronfeld I, Kazimirsky G, Lorenzo PS, Garfield SH, Blumberg PM,Brodie C 2000 Phosphorylation of protein kinase C� on distincttyrosine residues regulates specific cellular functions. J Biol Chem275:35491–35498
24. Blass M, Kronfeld I, Kazimirsky G, Blumberg PM, Brodie C 2002Tyrosine phosphorylation of protein kinase C� is essential for itsapoptotic effect in response to etoposide. Mol Cell Biol 22:182–195
25. Okhrimenko H, Lu W, Xiang C, Ju D, Blumberg PM, Gomel R,Kazimirsky G, Brodie C 2005 Roles of tyrosine phosphorylation andcleavage of protein kinase C� in its protective effect against tumornecrosis factor-related apoptosis inducing ligand-induced apopto-sis. J Biol Chem 280:23643–23652
26. Yang JY, Widmann C 2001 Antiapoptotic signaling generated bycaspase-induced cleavage of RasGAP. Mol Cell Biol 21:5346–5358
27. Kraus S, Benard O, Naor Z, Seger R 2003 c-Src is activated by theepidermal growth factor receptor in a pathway that mediates JNKand ERK activation by gonadotropin-releasing hormone in COS7cells. J Biol Chem 278:32618–32630
28. Kikkawa U, Ogita K, Shearman MS, Ase K, Sekiguchi K, Naor Z,Kishimoto A, Nishizuka Y, Saito N, Tanaka C 1988 The family ofprotein kinase C: its molecular heterogeneity and differential ex-pression. Cold Spring Harb Symp Quant Biol 53(Pt 1):97–102
29. Nishizuka Y 1992 Intracellular signaling by hydrolysis of phospho-lipids and activation of protein kinase C. Science 258:607–614
30. Dekker LV, Parker PJ 1994 Protein kinase C—a question of speci-ficity. Trends Biochem Sci 19:73–77
31. Newton AC 1995 Protein kinase C: structure, function, and regu-lation. J Biol Chem 270:28495–28498
32. Newton AC 1997 Regulation of protein kinase C. Curr Opin CellBiol 9:161–167
33. Newton AC 2003 The ins and outs of protein kinase C. MethodsMol Biol 233:3–7
34. Shearman MS, Naor Z, Kikkawa U, Nishizuka Y 1987 Differentialexpression of multiple protein kinase C subspecies in rat centralnervous tissue. Biochem Biophys Res Commun 147:911–919
35. Naor Z, Shearman MS, Kishimoto A, Nishizuka Y 1988 Calcium-independent activation of hypothalamic type I protein kinase C byunsaturated fatty acids. Mol Endocrinol 2:1043–1048
36. Wang Y, Satoh A, Warren G 2005 Mapping the functional domainsof the Golgi stacking factor GRASP65. J Biol Chem 280:4921–4928
37. Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T,Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F 1991 Thebisindolylmaleimide GF 109203X is a potent and selective inhibitorof protein kinase C. J Biol Chem 266:15771–15781
38. Gekeler V, Boer R, Uberall F, Ise W, Schubert C, Utz I, Hofmann J,Sanders KH, Schachtele C, Klemm K, Grunicke H 1996 Effects of theselective bisindolylmaleimide protein kinase C inhibitor GF109203X on P-glycoprotein-mediated multidrug resistance. Br JCancer 74:897–905
39. Gschwendt M, Dieterich S, Rennecke J, Kittstein W, Mueller HJ,Johannes FJ 1996 Inhibition of protein kinase C mu by variousinhibitors. Differentiation from protein kinase C isoenzymes. FEBSLett 392:77–80
40. Gschwendt M, Muller HJ, Kielbassa K, Zang R, Kittstein W, RinckeG, Marks F 1994 Rottlerin, a novel protein kinase inhibitor. Bio-chem Biophys Res Commun 199:93–98
41. Ferris HA, Shupnik MA 2006 Mechanisms for pulsatile regulationof the gonadotropin subunit genes by GNRH1. Biol Reprod 74:993–998
42. Stojilkovic SS, Reinhart J, Catt KJ 1994 Gonadotropin-releasinghormone receptors: structure and signal transduction pathways. En-docr Rev 15:462–499
43. Caunt CJ, Rivers CA, Conway-Campbell BL, Norman MR,McArdle CA 2008 Epidermal growth factor receptor and proteinkinase C signaling to ERK2: spatiotemporal regulation of ERK2 bydual specificity phosphatases. J Biol Chem 283:6241–6252
44. Armstrong SP, Caunt CJ, McArdle CA 2009 Gonadotropin-releas-ing hormone and protein kinase C signaling to ERK: spatiotemporalregulation of ERK by docking domains and dual-specificity phos-phatases. Mol Endocrinol 23:510–519
45. Windle JJ, Weiner RI, Mellon PL 1990 Cell lines of the pituitarygonadotrope lineage derived by targeted oncogenesis in transgenicmice. Mol Endocrinol 4:597–603
46. Turgeon JL, Kimura Y, Waring DW, Mellon PL 1996 Steroid andpulsatile gonadotropin-releasing hormone (GnRH) regulation of lu-teinizing hormone and GnRH receptor in a novel gonadotrope cellline. Mol Endocrinol 10:439–450
47. Valette A, Gas N, Jozan S, Roubinet F, Dupont MA, Bayard F 1987Influence of 12-O-tetradecanoylphorbol-13-acetate on prolifera-tion and maturation of human breast carcinoma cells (MCF-7): re-lationship to cell cycle events. Cancer Res 47:1615–1620
48. Waldron RT, Rey O, Iglesias T, Tugal T, Cantrell D, Rozengurt E2001 Activation loop Ser744 and Ser748 in protein kinase D aretransphosphorylated in vivo. J Biol Chem 276:32606–32615
49. Ghanekar Y, Lowe M 2005 Protein kinase D: activation for Golgicarrier formation. Trends Cell Biol 15:511–514
50. Sim AT, Scott JD 1999 Targeting of PKA, PKC and protein phos-phatases to cellular microdomains. Cell Calcium 26:209–217
51. Chen D, Purohit A, Halilovic E, Doxsey SJ, Newton AC 2004 Cen-trosomal anchoring of protein kinase C �II by pericentrin controlsmicrotubule organization, spindle function, and cytokinesis. J BiolChem 279:4829–4839
52. Buensuceso CS, Obergfell A, Soriani A, Eto K, Kiosses WB, Arias-Salgado EG, Kawakami T, Shattil SJ 2005 Regulation of outside-insignaling in platelets by integrin-associated protein kinase C�. J BiolChem 280:644–653
53. Lang W, Wang H, Ding L, Xiao L 2004 Cooperation betweenPKC-� and PKC-� in the regulation of JNK activation in human lungcancer cells. Cell Signal 16:457–467
54. Andrassy M, Belov D, Harja E, Zou YS, Leitges M, Katus HA,Nawroth PP, Yan SD, Schmidt AM, Yan SF 2005 Central role ofPKC� in neointimal expansion triggered by acute arterial injury.Circ Res 96:476–483
4906 Dobkin-Bekman et al. Differential Role of PKC Isoforms in MAPK Activation by GnRH and PMAEndocrinology, October 2010, 151(10):4894–4907
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 29 July 2015. at 01:57 For personal use only. No other uses without permission. . All rights reserved.

55. Campbell M, Trimble ER 2005 Modification of PI3K- and MAPK-dependent chemotaxis in aortic vascular smooth muscle cells byprotein kinase C�II. Circ Res 96:197–206
56. Kolch W, Heidecker G, Kochs G, Hummel R, Vahidi H, Mischak H,Finkenzeller G, Marme D, Rapp UR 1993 Protein kinase C� acti-vates RAF-1 by direct phosphorylation. Nature 364:249–252
57. Benard O, Naor Z, Seger R 2001 Role of dynamin, Src, and Ras inthe protein kinase C-mediated activation of ERK by gonadotropin-releasing hormone. J Biol Chem 276:4554–4563
58. Ebinu JO, Bottorff DA, Chan EY, Stang SL, Dunn RJ, Stone JC 1998RasGRP, a Ras guanyl nucleotide-releasing protein with calcium-and diacylglycerol-binding motifs. Science 280:1082–1086
59. Teixeira C, Stang SL, Zheng Y, Beswick NS, Stone JC 2003 Inte-gration of DAG signaling systems mediated by PKC-dependentphosphorylation of RasGRP3. Blood 102:1414–1420
60. Naor Z, Benard O, Seger R 2000 Activation of MAPK cascades byG-protein-coupled receptors: the case of gonadotropin-releasinghormone receptor. Trends Endocrinol Metab 11:91–99
61. Yokoi T, Ohmichi M, Tasaka K, Kimura A, Kanda Y, Hayakawa J,Tahara M, Hisamoto K, Kurachi H, Murata Y 2000 Activation ofthe luteinizing hormone � promoter by gonadotropin-releasing hor-mone requires c-Jun NH2-terminal protein kinase. J Biol Chem 275:21639–21647
62. Lopez-Bergami P, Habelhah H, Bhoumik A, Zhang W, Wang LH,Ronai Z 2005 RACK1 mediates activation of JNK by protein kinaseC. Mol Cell 19:309–320
63. Kanasaki H, Bedecarrats GY, Kam KY, Xu S, Kaiser UB 2005Gonadotropin-releasing hormone pulse frequency-dependent acti-vation of extracellular signal-regulated kinase pathways in perifusedL�T2 cells. Endocrinology 146:5503–5513
64. Xie J, Bliss SP, Nett TM, Ebersole BJ, Sealfon SC, Roberson MS2005 Transcript profiling of immediate early genes reveals a uniquerole for activating transcription factor 3 in mediating activation of
the glycoprotein hormone �-subunit promoter by gonadotropin-releasing hormone. Mol Endocrinol 19:2624–2638
65. Dobkin-Bekman M, Naidich M, Pawson AJ, Millar RP, Seger R,Naor Z 2006 Activation of mitogen-activated protein kinase(MAPK) by GnRH is cell-context dependent. Mol Cell Endocrinol252:184–190
66. Maudsley S, Naor Z, Bonfil D, Davidson L, Karali D, Pawson AJ,Larder R, Pope C, Nelson N, Millar RP, Brown P 2007 Proline-richtyrosine kinase 2 mediates gonadotropin-releasing hormone signal-ing to a specific extracellularly regulated kinase-sensitive transcrip-tional locus in the luteinizing hormone �-subunit gene. Mol Endo-crinol 21:1216–1233
67. Emons G, Muller V, Ortmann O, Schulz KD 1998 Effects of LHRH-analogues on mitogenic signal transduction in cancer cells. J SteroidBiochem Mol Biol 65:199–206
68. Limonta P, Moretti RM, Marelli MM, Dondi D, Parenti M, MottaM 1999 The luteinizing hormone-releasing hormone receptor inhuman prostate cancer cells: messenger ribonucleic acid expression,molecular size, and signal transduction pathway. Endocrinology140:5250–5256
69. Kraus S, Levy G, Hanoch T, Naor Z, Seger R 2004 Gonadotropin-releasing hormone induces apoptosis of prostate cancer cells: role ofc-Jun NH2-terminal kinase, protein kinase B, and extracellular sig-nal-regulated kinase pathways. Cancer Res 64:5736–5744
70. Maudsley S, Davidson L, Pawson AJ, Chan R, Lopez de MaturanaR, Millar RP 2004 Gonadotropin-releasing hormone (GnRH) an-tagonists promote proapoptotic signaling in peripheral reproductivetumor cells by activating a G�i-coupling state of the type I GnRHreceptor. Cancer Res 64:7533–7544
71. Kim KY, Choi KC, Auersperg N, Leung PC 2006 Mechanism ofgonadotropin-releasing hormone (GnRH)-I and -II-induced cellgrowth inhibition in ovarian cancer cells: role of the GnRH-I recep-tor and protein kinase C pathway. Endocr Relat Cancer 13:211–220
Sign up for eTOC alerts today to get the latest articles as soon as they are online.
http://mend.endojournals.org/subscriptions/etoc.shtml
Endocrinology, October 2010, 151(10):4894–4907 endo.endojournals.org 4907
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 29 July 2015. at 01:57 For personal use only. No other uses without permission. . All rights reserved.




















