
3D finite-difference dynamic-rupture modeling along nonplanar faults

N
Da
b
c
a
ARRA
KNRDIB
1
ttiTma
stctth
2
(
(
1d
Spectrochimica Acta Part A 79 (2011) 993– 1003
Contents lists available at ScienceDirect
Spectrochimica Acta Part A: Molecular andBiomolecular Spectroscopy
j ourna l ho me page: www.elsev ier .com/ locate /saa
onplanar property study of antifungal agent tolnaftate-spectroscopic approach
. Arul Dhasa, I. Hubert Joeb,∗, S.D.D. Roya, S. Balachandranc
Department of Physics, Nesamony Memorial Christian College, Marthandam 629 165, Tamil Nadu, IndiaCentre for Molecular and Biophysics Research, Department of Physics, Mar Ivanios College, Thiruvananthapuram 695015, Kerala, IndiaDepartment of Chemistry, M.G College, Thiruvananthapuram 695004, Kerala, India
r t i c l e i n f o
rticle history:eceived 21 December 2010eceived in revised form 7 April 2011ccepted 8 April 2011
eywords:BOaman
a b s t r a c t
Vibrational analysis of the thionocarbamate fungicide tolnaftate which is antidermatophytic, antitri-chophytic and antimycotic agent, primarily inhibits the ergosterol biosynthesis in the fungus, was carriedout using NIR FT-Raman and FTIR spectroscopic techniques. The equilibrium geometry, various bondingfeatures, harmonic vibrational wavenumbers and torsional potential energy surface (PES) scan studieshave been computed using density functional theory method. The detailed interpretation of the vibra-tional spectra has been carried out with the aid of VEDA.4 program. Vibrational spectra, natural bondingorbital (NBO) analysis and optimized molecular structure show the clear evidence for electronic interac-
FTCTioactivity
tion of thionocarbamate group with aromatic ring. Predicted electronic absorption spectrum from TD-DFTcalculation has been compared with the UV–vis spectrum. The Mulliken population analysis on atomiccharges and the HOMO–LUMO energy were also calculated. Vibrational analysis reveals that the simul-taneous IR and Raman activation of the C–C stretching mode in the phenyl and naphthalene ring provideevidence for the charge transfer interaction between the donor and acceptor groups and is responsiblefor its bioactivity as a fungicide.
. Introduction
Vibrational spectroscopic investigation with the help of quan-um chemical computational method has been used as an effectiveool for the structural analysis of pesticide molecules [1–4]. The tox-cologic and pharmacologic study of a new antitrichophyton agentolnaftate (TNF) [(2-naphthyl-N-methyl-N-(3-tolyl) thionocarba-ate] has been reported [5–8]. TNF has been widely used as topical
ntifungal drug in the treatment of cutaneous diseases [9–11].The present investigation aims to understand the molecular
tructure regarding intramolecular charge transfer (ICT) and elec-ron delocalization like �-conjugation and hyperconjugation of theompound using FT-Raman and IR along with the density functionalheory (DFT) calculation. The time-dependent density functionalheory (TD-DFT) calculation and natural bond orbital (NBO) analysisave been used to study the electronic effects.
. Experimental
The compound Tolnaftate was purchased from Sigma–AldrichU.S.A.). The NIR-FT Raman spectrum in the region 3500–50 cm−1
∗ Corresponding author. Tel.: +91 4712351053; fax: +91 4712530023.E-mail addresses: [email protected], [email protected]
I. Hubert Joe).
386-1425/$ – see front matter © 2011 Elsevier B.V. All rights reserved.oi:10.1016/j.saa.2011.04.011
© 2011 Elsevier B.V. All rights reserved.
was taken on a Bruker RFS 66v NIR FT-Raman Spectrometer. TheFTIR spectrum in the region 4000–400 cm−1 was recorded using aPerkin–Elmer Spectrum One FT-IR Spectrometer with samples inthe KBr. The UV–visible absorption spectrum was recorded usingJastrow V-550 UV–Visible Spectrophotometer.
3. Computational details
The DFT computation has been performed using Gaussian03W program package [12] using Becke’s three-parameter hybridmethod with the Lee, Yang and Parr’s correlation functionalmethods with the standard 6-31G (d) basis set. The simu-lated IR and Raman spectra were plotted using pure Lorentizianband shapes with a bandwidth of full width half-maximum(FWHM) of 10 cm−1. An empirical scaling factor of 0.9614 [13]was used to offset the systematic error caused by neglectingan harmonicity and electron correlation. The distributions ofassignment of the calculated wavenumbers were aided by VEDAprogram [14].
The Raman activities calculated by the Gaussian’03 programhave been converted to relative Raman intensities using the basictheory of Raman scattering [15,16]. The natural bonding orbital
(NBO) calculation was performed using NBO 3.1 program [17] andwas implemented in the Gaussian’03 package at the DFT/B3LYPlevel. The hyperconjugative interaction energy was deduced fromthe second order perturbation approach [18–20].
994 D. Arul Dhas et al. / Spectrochimica Acta Part A 79 (2011) 993– 1003
4
4
GTaitggtv2pCCtCsst
atNdcftiagp
4
sflTaactrcga(i
◦
Fig. 1. The optimized structure of TNF calculated at B3LYP/6-31G (d).
. Results and discussion
.1. Geometry optimization
The TNF structure was optimized using DFT method with 6–31 (d) basis set. The optimized structural parameters are given inable 1. The optimized molecular structure of the compound withtom numbering scheme adopted in the computation is shownn Fig. 1. The molecule consists of two separate ring systems viz,he naphthalene ring and tolyl ring connected by thionocarbamateroup and two methyl groups me1 (attached to thionocarbamateroup) and me2 (attached to tolyl group). The calculated struc-ural parameters are in good agreement with the experimentalalues reported in closely related compounds such as methyl-(7-benzyloxy-1-naphthyl)-2-oxacetate [21] and o-methyl n-henylthiocarbamate [22]. In o-methyl n-phenylthiocarbamate–O bond length is found out to be 1.443 A and that for TNF (i.e.12–O18) is 1.397 A, which indicates an extension of conjugation ofhe aromatic ring of naphthalene to oxygen. The C–N bond lengths19–N21 (1.357 A), C22–N21 (1.472 A) and C26–N21 (1.441 A) arehorter than the normal C–N single-bond length 1.480 A [23]. Thehortening of these C–N bonds reveal the effect of resonance inhionocarbamate part of the molecule.
The van-der Walls repulsion of H32 and H39 with H23 causes steric hindrance in achieving coplanarity for the tolyl ring withhe carbamate group and thus a twisting of tolyl group about21–C26 bond was noticed. Hence the tolyl group assumes the dihe-ral angles C22N21C26C31 (−95.4◦) and C22N21C26C27 (80.6◦). In theompound the hetero atoms O18 and N21 are slightly distortedrom the plane and the two rings are flipped in such a way thathe whole structure appears in the form of a boat with the S atomnside the boat. The methyl group attached to N21 and the sulphurtom attached to C19 is above and below the plane. Computationaleometry optimization reveals that the nonplanar conformation isreferred by a minimum energy −3314526 kJ mol−1.
.2. PES scan studies
The TNF molecule can adopt different conformations, mainly bypatially orienting the tolyl and methyl groups attached to N21 andipping the naphthyl group with respect to the ether oxygen (O18).he thionyl carbon is Sp2 hybridized and thus N–(C S)–O assume
planar configuration with 120◦ bond angle (Table 1). Rotationt C19–N21 bond of dihedral angle C26–N21–C19–S20 leads to twoonformers, the CH3 group attached to N21 is cis and trans to thehionocarbonyl group and are depicted in Fig. 2(A) and (C). Theotation at C18–O19 of dihedral angle C12–O18–C19–S20 results inis and trans naphthyl conformers with respect to thionocarbonyl
roup and are shown in Fig. 2(B) and (D). The four conformers can bessigned as cis–cis (A), cis–trans (B), trans–cis (C) and trans–transD). The rotational conformers in this kind of compounds contain-ng two aromatic rings, attached to two different hetero atoms,Fig. 2. Possible conformers of TNF.
via a thionocarbonyl group and two different groups attached tothe heteroatoms are influenced by different structural factors, suchas steric, dipolar, mesomeric and hyperconjugative effects includ-ing hydrogen bonding interactions. The optimized TNF structureappears in the trans–cis conformer (C) where the tolyl and naph-thyl rings are slightly out of plane (Table 1) to form of a boat withthe hetero S20 atom inside the boat.
The potential energy scan has been performed on the opti-mized geometry by rotating different spatially important groupswith respect to single bond present in TNF between 0 and360◦ with the increment of 10◦. The possible maxima and min-ima relative energies and the corresponding dihedral angles arelisted in Table S1 (Supporting Information). The rotation withrespect to other dihedral angles (C12O18C19S20, C13C12O18C19and O18C19N21C22) shows a restriction due to much interactionbetween the various groups.
The PES scan plot and the corresponding conformers for therotation of S20C19N21C22 with respect to the bond C19–N21 aregiven in Fig. S1 (Supporting Information). The staggered condi-tion (160◦) corresponds to the trans–cis conformer (C) and theeclipsed condition (0◦) to cis–cis conformer (A). Comparing to theeclipsed structure the trans–cis staggered conformer, which corre-sponds to the optimized structure, is stabilized by 296,189 kJ mol−1
(Table S1; Supporting Information). At the maximal position themethyl group attached to N21 and the ether oxygen O18 groups arecis to each other and H24. . .O18 and H25. . .O18 distances are 2.492and 2.615 A (Table S2a; Supporting Information), respectively. Theelectrostatic interaction and the double bond character of C19–N21bond due to the mesomeric shift of lone pair of electrons from N21along with the shift of double bonded electrons of C S group to Sattribute to the energy reduction of conformer (C) in comparisonto that of (A). The ether oxygen O18 can also interact via elec-tron donating mesomeric effect and as a result the thionocarbamtegroup will have an extended conjugation with both the aromaticrings via the tolyl and naphthyl. The steric strain however does notallow a planar structure which reduces the resonance stabilizationvia the extended conjugation and can be observed by the variousdihedral angles given in the optimized structure (Table 1).
The energy curve for the dihedral angle C12O18C19N21 (O18–C19bond) of TNF is shown in Fig. S2 (Supporting Information). It is
observed that the conformer at 0 corresponds to trans–cis con-former (C) and the rotation did not cause any hindrance till itreaches 280◦. The steric interactions due to van der Waals’ repulsionis comparatively weak since the distances between the interacting

D. Arul Dhas et al. / Spectrochimica Acta Part A 79 (2011) 993– 1003 995
Table 1Selected optimized geometric parameters of fungicide TNF on B3LYP/6-31G (d) basis set.
Bond lengths (Å) Bond angles (◦) Dihedral angles (◦)
B3LYP/6-31G(d) Experi mental B3LYP/6-31G(d) Experi mental B3LYP/6-31G(d) Experimental
C11–C12 1.371 1.375a H15–C11–C6 120.3 120.0a H16–C13–C12–C11 −179.8 −175.9a
C12–C13 1.413 1.418a H16–C13–C12 119.4 120.0a H12–C13–C14–C17 179.8 142.7a
C13–C14 1.374 1.363a O18–C12–C13 119.5 1125.2a C22–N21–C19–O18 0.9 −30.5b
C11–H15 1.086 0.930a C22–N21–C19 121.3 116.9b C22–N21–C26–C27 80.6 176.6b
C19–N21 1.357 1.331b H23–C22–N21 107.8 109.5b C22–N21–C26–C31 −95.4 −19.7b
C22–N21 1.472 1.337b H24–C22–N21 110.9 109.5b H23–C22–N21–C19 175.4 70.0b
C22–H23 1.090 0.970b H25–C22–N21 111.2 109.5b H24–C22–N21–C19 55.7 46.5b
C22–H24 1.093 0.970b C26–N21–C19 121.0 126.1b H25–C22–N21–C19 −64.9 180.0b
C22–H25 1.095 0.970b C27–C26–N21 119.6 117.0b H39–C27–C26–N21 4.2 0.0b
C26–N21 1.441 1.420b C26–N21–C22 117.6 1115.6b H27–C26–N21–C19 −101.9 −151.8b
C12–O18 1.397 1.361a N21–C19–O18 108.5 1112.5b C19–O18–C12–C11 −111.1 −153.5b
N21–C19–S20 126.5 1123.4b C26–N21–C19–S20 3.2 −177.6b
S20–C19–O18 124.9 1124.0b C26–N21–C19–O18 −176.4 3.0b
C31–C26–N21 119.8 1117.0b C22–N21–C19–S20 −179.5 −177.0b
C27–C26–N21 119.6 1123.9b C12–O18–C19–S20 4.9 5.7b
C12–O18–C19–N21 −175.5 −174.9b
O18–C19–N21–C22 0.9 −177.0b
gtbmrs
fiPtTpdHia(p
mCa(tHaiaHTbn(
pfibietaat
a Taken from [21].b Taken from [22].
roups are larger as shown in Table S2b (Supporting Informa-ion). The extended conjugation is the reason which causes a largerarrier for rotation 103,804 kJ mol−1 (Table S1; Supporting Infor-ation). The conformers B and D are not observed during the
otation since the groups interact very strongly and re-orient them-elves to attain stable trans–cis conformation.
When the tolyl group is rotated by keeping the remaining partxed with respect to N21–C26 bond, the possible conformers and theES curve are given in Fig. S3 (Supporting Information). Throughouthe rotation of tolyl group, the structure remains in conformer (C).wo energy barriers of 54 kJ mol−1 and 60 kJ mol−1 (Table S1; Sup-orting Information) are observed at 20◦ and 200◦ exactly at 180◦
ifference, which is due to van der Waals’ repulsion between39. . .H23(1.689 A) and H32. . .H23(1.689 A) (Table S2c; Support-
ng Information). At 80◦ and 130◦ and at 270◦ and 320◦ therere two additional lowering of energy of only less than 5 kJ mol−1
Table S1; Supporting Information) which can be ascribed to theossible van der Waals’ attraction of S20 with H39 and S20 with H32.
A half-chair conformation is observed (Fig. S4; Supporting Infor-ation), when the naphthyl group is rotated with respect to
12–O18 bond (dihedral angle C11C12O18C19) keeping the tolylnd thionocarbamate groups stationary. A barrier of 2917 kJ mol−1
Table S1; Supporting Information) is observed at eclipsed condi-ion and is due to the steric and electrostatic interactions between15 and H24 and S20 and H16 atoms. The extended conjugation maylso contribute to the increased energy barrier (Table 2d; Support-ng Information). There are two minimum energy areas which canscribe as before as due to the electrostatic attraction of S20 with, at 80◦ S20–H16 interaction and at 220◦ it is S20–H15 interaction.he conformer at 220◦ has the calculated energy nearest (largery 677 kJ mol−1) to the optimized structure even though there areumber of other conformers whose values are close to this valueTable S1; Supporting Information).
The possible conformations during the rotation of the com-ound by keeping me1 (methyl attached to nitrogen) groupxed and rotating the remaining part with respect to N21–C22ond is shown in Fig. S5(a) (Supporting Information). In fact,
t displays maxima for those conformations where the stericffect arising due to weak van der Waals repulsive interac-ion arises between the positively charged methyl hydrogen
nd phenyl ring hydrogen atoms (H23. . .H39 = 2.827 A at 50◦nd H24. . .H39 = 2.879 A at 170◦) (Table S2e; Supporting Informa-ion). The maximal interaction is for the rotation of 120◦ and
Fig. 3. Combined IR spectrum of TNF – (a) computed and (b) experimental.
is the bond angle of H23C22H24, for the remaining 240◦ there isno repulsive interaction. The similar effect can be observed inme2 (methyl attached to benzene ring) with respect to C30–C33bond (dihedral angle C29C30C33H36), where the maxima’s areat 90◦ (H34. . .H32 = 2.487 A and H35. . .H37 = 2.511 A) and 330◦
(H36. . .H32 = 2.496 A and H34. . .H37 = 2.505 A) (Table S2f; Support-ing Information). The barrier for me1 is 16,489 and 15,049 kJ mol−1
and me2 is having the barrier 21,558 and 30,556 kJ mol−1
(Table S1; Supporting Information). The energy barrier value showsthat the rotation about me1 group is easy while comparing to me2group, since there are more number of repulsive interactions inme2.
4.3. Vibrational spectral analysis
The vibrational spectral analysis has been performed on thebasis of the characteristic group vibrations of naphthalene ring,phenyl ring, thionocarbamate group and methyl group. The exper-imental and scaled wavenumbers along with their respectivedominant normal modes and the corresponding (Potential Energy
Distribution) PED’s are presented in Table 2. The observed andsimulated FT-IR and Raman spectra are given in Figs. 3 and 4,respectively.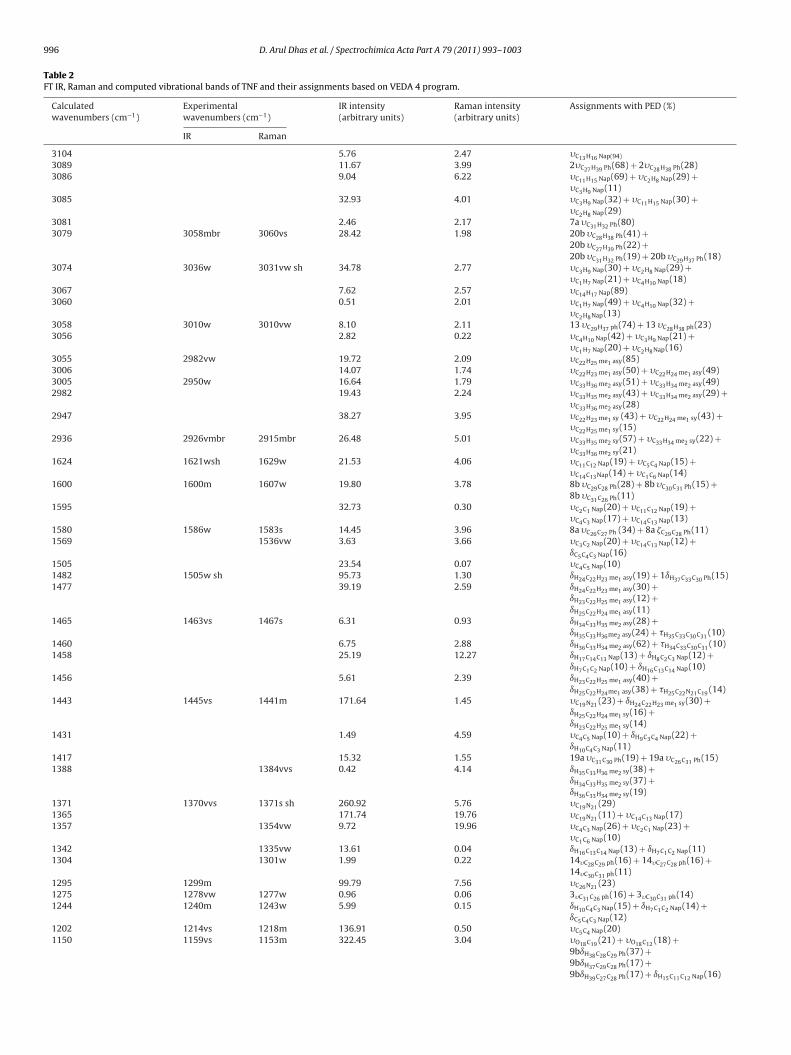
996 D. Arul Dhas et al. / Spectrochimica Acta Part A 79 (2011) 993– 1003
Table 2FT IR, Raman and computed vibrational bands of TNF and their assignments based on VEDA 4 program.
Calculatedwavenumbers (cm−1)
Experimentalwavenumbers (cm−1)
IR intensity(arbitrary units)
Raman intensity(arbitrary units)
Assignments with PED (%)
IR Raman
3104 5.76 2.47 �C13H16 Nap(94)
3089 11.67 3.99 2�C27H39 Ph(68) + 2�C28H38 Ph(28)3086 9.04 6.22 �C11H15 Nap(69) + �C2H8 Nap(29) +
�C3H9 Nap(11)3085 32.93 4.01 �C3H9 Nap(32) + �C11H15 Nap(30) +
�C2H8 Nap(29)3081 2.46 2.17 7a �C31H32 Ph(80)3079 3058mbr 3060vs 28.42 1.98 20b �C28H38 Ph(41) +
20b �C27H39 Ph(22) +20b �C31H32 Ph(19) + 20b �C29H37 Ph(18)
3074 3036w 3031vw sh 34.78 2.77 �C3H9 Nap(30) + �C2H8 Nap(29) +�C1H7 Nap(21) + �C4H10 Nap(18)
3067 7.62 2.57 �C14H17 Nap(89)3060 0.51 2.01 �C1H7 Nap(49) + �C4H10 Nap(32) +
�C2H8Nap(13)3058 3010w 3010vw 8.10 2.11 13 �C29H37 ph(74) + 13 �C28H38 ph(23)3056 2.82 0.22 �C4H10 Nap(42) + �C3H9 Nap(21) +
�C1H7 Nap(20) + �C2H8Nap(16)3055 2982vw 19.72 2.09 �C22H25 me1 asy(85)3006 14.07 1.74 �C22H23 me1 asy(50) + �C22H24 me1 asy(49)3005 2950w 16.64 1.79 �C33H36 me2 asy(51) + �C33H34 me2 asy(49)2982 19.43 2.24 �C33H35 me2 asy(43) + �C33H34 me2 asy(29) +
�C33H36 me2 asy(28)2947 38.27 3.95 �C22H23 me1 sy (43) + �C22H24 me1 sy(43) +
�C22H25 me1 sy(15)2936 2926vmbr 2915mbr 26.48 5.01 �C33H35 me2 sy(57) + �C33H34 me2 sy(22) +
�C33H36 me2 sy(21)1624 1621wsh 1629w 21.53 4.06 �C11C12 Nap(19) + �C5C4 Nap(15) +
�C14C13Nap(14) + �C1C6 Nap(14)1600 1600m 1607w 19.80 3.78 8b �C29C28 Ph(28) + 8b �C30C31 Ph(15) +
8b �C31C26 Ph(11)1595 32.73 0.30 �C2C1 Nap(20) + �C11C12 Nap(19) +
�C4C3 Nap(17) + �C14C13 Nap(13)1580 1586w 1583s 14.45 3.96 8a �C26C27 Ph (34) + 8a �C29C28 Ph(11)1569 1536vw 3.63 3.66 �C3C2 Nap(20) + �C14C13 Nap(12) +
ıC5C4C3 Nap(16)1505 23.54 0.07 �C4C5 Nap(10)1482 1505w sh 95.73 1.30 ıH24C22H23 me1 asy(19) + 1ıH37C33C30 Ph(15)1477 39.19 2.59 ıH24C22H23 me1 asy(30) +
ıH23C22H25 me1 asy(12) +ıH25C22H24 me1 asy(11)
1465 1463vs 1467s 6.31 0.93 ıH34C33H35 me2 asy(28) +ıH35C33H36me2 asy(24) + �H35C33C30C31 (10)
1460 6.75 2.88 ıH36C33H34 me2 asy(62) + �H34C33C30C31 (10)1458 25.19 12.27 ıH17C14C13 Nap(13) + ıH8C2C3 Nap(12) +
ıH7C1C2 Nap(10) + ıH16C13C14 Nap(10)1456 5.61 2.39 ıH23C22H25 me1 asy(40) +
ıH25C22H24me1 asy(38) + �H25C22N21C19 (14)1443 1445vs 1441m 171.64 1.45 �C19N21 (23) + ıH24C22H23 me1 sy(30) +
ıH25C22H24 me1 sy(16) +ıH23C22H25 me1 sy(14)
1431 1.49 4.59 �C4C5 Nap(10) + ıH9C3C4 Nap(22) +ıH10C4C3 Nap(11)
1417 15.32 1.55 19a �C31C30 Ph(19) + 19a �C26C31 Ph(15)1388 1384vvs 0.42 4.14 ıH35C33H36 me2 sy(38) +
ıH34C33H35 me2 sy(37) +ıH36C33H34 me2 sy(19)
1371 1370vvs 1371s sh 260.92 5.76 �C19N21 (29)1365 171.74 19.76 �C19N21 (11) + �C14C13 Nap(17)1357 1354vw 9.72 19.96 �C4C3 Nap(26) + �C2C1 Nap(23) +
�C1C6 Nap(10)1342 1335vw 13.61 0.04 ıH16C13C14 Nap(13) + ıH7C1C2 Nap(11)1304 1301w 1.99 0.22 14�C28C29 ph(16) + 14�C27C28 ph(16) +
14�C30C31 ph(11)1295 1299m 99.79 7.56 �C26N21 (23)1275 1278vw 1277w 0.96 0.06 3�C31C26 ph(16) + 3�C30C31 ph(14)1244 1240m 1243w 5.99 0.15 ıH10C4C3 Nap(15) + ıH7C1C2 Nap(14) +
ıC5C4C3 Nap(12)1202 1214vs 1218m 136.91 0.50 �C5C4 Nap(20)1150 1159vs 1153m 322.45 3.04 �O18C19 (21) + �O18C12 (18) +
9bıH38C28C29 Ph(37) +9bıH37C29C28 Ph(17) +9bıH39C27C28 Ph(17) + ıH15C11C12 Nap(16)
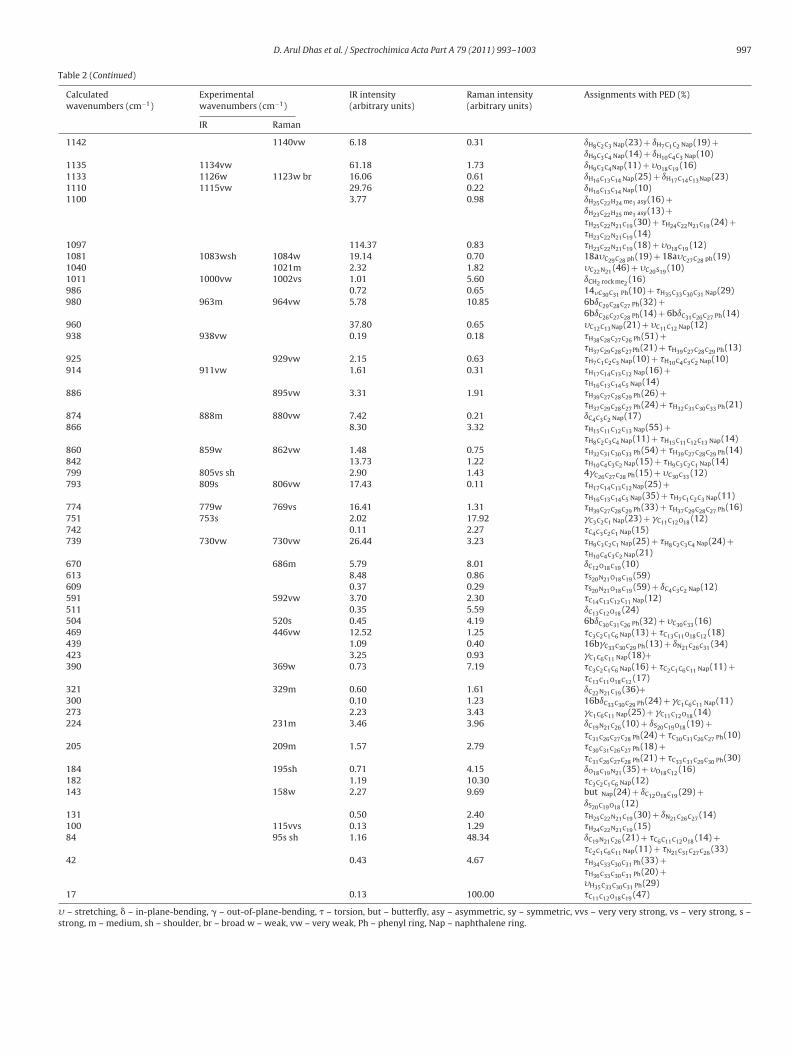
D. Arul Dhas et al. / Spectrochimica Acta Part A 79 (2011) 993– 1003 997
Table 2 (Continued)
Calculatedwavenumbers (cm−1)
Experimentalwavenumbers (cm−1)
IR intensity(arbitrary units)
Raman intensity(arbitrary units)
Assignments with PED (%)
IR Raman
1142 1140vw 6.18 0.31 ıH8C2C3 Nap(23) + ıH7C1C2 Nap(19) +ıH9C3C4 Nap(14) + ıH10C4C3 Nap(10)
1135 1134vw 61.18 1.73 ıH9C3C4Nap(11) + �O18C19 (16)1133 1126w 1123w br 16.06 0.61 ıH16C13C14 Nap(25) + ıH17C14C13Nap(23)1110 1115vw 29.76 0.22 ıH16C13C14 Nap(10)1100 3.77 0.98 ıH25C22H24 me1 asy(16) +
ıH23C22H25 me1 asy(13) +�H25C22N21C19 (30) + �H24C22N21C19 (24) +�H23C22N21C19 (14)
1097 114.37 0.83 �H23C22N21C19 (18) + �O18C19 (12)1081 1083wsh 1084w 19.14 0.70 18a�C29C28 ph(19) + 18a�C27C28 ph(19)1040 1021m 2.32 1.82 �C22N21 (46) + �C20S19 (10)1011 1000vw 1002vs 1.01 5.60 ıCH2 rock me2
(16)986 0.72 0.65 14�C30C31 Ph(10) + �H35C33C30C31 Nap(29)980 963m 964vw 5.78 10.85 6bıC29C28C27 Ph(32) +
6bıC26C27C28 Ph(14) + 6bıC31C26C27 Ph(14)960 37.80 0.65 �C12C13Nap(21) + �C11C12 Nap(12)938 938vw 0.19 0.18 �H38C28C27C26 Ph(51) +
�H37C29C28C27Ph(21) + �H39C27C28C29 Ph(13)925 929vw 2.15 0.63 �H7C1C2C3 Nap(10) + �H10C4C3C2 Nap(10)914 911vw 1.61 0.31 �H17C14C13C12 Nap(16) +
�H16C13C14C5 Nap(14)886 895vw 3.31 1.91 �H39C27C28C29 Ph(26) +
�H37C29C28C27 Ph(24) + �H32C31C30C33 Ph(21)874 888m 880vw 7.42 0.21 ıC4C3C2 Nap(17)866 8.30 3.32 �H15C11C12C13 Nap(55) +
�H8C2C3C4 Nap(11) + �H15C11C12C13 Nap(14)860 859w 862vw 1.48 0.75 �H32C31C30C33 Ph(54) + �H39C27C28C29 Ph(14)842 13.73 1.22 �H10C4C3C2 Nap(15) + �H9C3C2C1 Nap(14)799 805vs sh 2.90 1.43 4�C26C27C28 Ph(15) + �C30C33 (12)793 809s 806vw 17.43 0.11 �H17C14C13C12Nap(25) +
�H16C13C14C5 Nap(35) + �H7C1C2C3 Nap(11)774 779w 769vs 16.41 1.31 �H39C27C28C29 Ph(33) + �H37C29C28C27 Ph(16)751 753s 2.02 17.92 �C3C2C1 Nap(23) + �C11C12O18 (12)742 0.11 2.27 �C4C3C2C1 Nap(15)739 730vw 730vw 26.44 3.23 �H9C3C2C1 Nap(25) + �H8C2C3C4 Nap(24) +
�H10C4C3C2 Nap(21)670 686m 5.79 8.01 ıC12O18C19 (10)613 8.48 0.86 �S20N21O18C19 (59)609 0.37 0.29 �S20N21O18C19 (59) + ıC4C3C2 Nap(12)591 592vw 3.70 2.30 �C14C13C12C11 Nap(12)511 0.35 5.59 ıC13C12O18 (24)504 520s 0.45 4.19 6bıC30C31C26 Ph(32) + �C30C33 (16)469 446vw 12.52 1.25 �C3C2C1C6 Nap(13) + �C13C11O18C12 (18)439 1.09 0.40 16b�C33C30C29 Ph(13) + ıN21C26C31 (34)423 3.25 0.93 �C1C6C11 Nap(18)+390 369w 0.73 7.19 �C3C2C1C6 Nap(16) + �C2C1C6C11 Nap(11) +
�C13C11O18C12 (17)321 329m 0.60 1.61 ıC22N21C19 (36)+300 0.10 1.23 16bıC33C30C29 Ph(24) + �C1C6C11 Nap(11)273 2.23 3.43 �C1C6C11 Nap(25) + �C11C12O18 (14)224 231m 3.46 3.96 ıC19N21C26 (10) + ıS20C19O18 (19) +
�C31C26C27C28 Ph(24) + �C30C31C26C27 Ph(10)205 209m 1.57 2.79 �C30C31C26C27 Ph(18) +
�C31C26C27C28 Ph(21) + �C33C31C29C30 Ph(30)184 195sh 0.71 4.15 ıO18C19N21 (35) + �O18C12 (16)182 1.19 10.30 �C3C2C1C6 Nap(12)143 158w 2.27 9.69 but Nap(24) + ıC12O18C19 (29) +
ıS20C19O18 (12)131 0.50 2.40 �H25C22N21C19 (30) + ıN21C26C27 (14)100 115vvs 0.13 1.29 �H24C22N21C19 (15)84 95s sh 1.16 48.34 ıC19N21C26 (21) + �C6C11C12O18 (14) +
�C2C1C6C11 Nap(11) + �N21C31C27C26 (33)42 0.43 4.67 �H34C33C30C31 Ph(33) +
�H36C33C30C31 Ph(20) +�H35C33C30C31 Ph(29)
17 0.13 100.00 �C11C12O18C19 (47)
� – stretching, � – in-plane-bending, � – out-of-plane-bending, � – torsion, but – butterfly, asy – asymmetric, sy – symmetric, vvs – very very strong, vs – very strong, s –strong, m – medium, sh – shoulder, br – broad w – weak, vw – very weak, Ph – phenyl ring, Nap – naphthalene ring.

998 D. Arul Dhas et al. / Spectrochimica A
F
4
uaa2wi2sc[tcititwt
o1gi1iRpw
4
buis[1iitfth
ig. 4. Combined Raman spectrum of TNF – (a) computed and (b) experimental.
.3.1. Methyl group vibrationThe position of the CH3 vibration is almost entirely dependent
p on the nature of the element to which the methyl groups arettached. The asymmetric C–H stretching mode of me2 is expectedround 2980 cm−1 and the symmetric stretching is expected at870 cm−1 [24,25]. The me2 asymmetric stretching is observed as aeak band in IR at 2950 cm−1 and the symmetric stretching mode
s observed as a medium broad band at 2926 cm−1 in IR and at915 cm−1in Raman. The blue shifting (56 cm−1) of me2 symmetrictretching is due to the electron donating inductive effect and hyperonjugative effect of methyl group attached to the aromatic ring26,27]. These effects imply electron delocalization, which may beaken in to account by a molecular orbital approach, this can point tohanging polarizability and dipole moment due to electron delocal-zation [28]. The asymmetric C–H stretching of me1 is expected inhe range 2820–2760 cm−1 [29]. The asymmetric stretching modes shifted towards higher wavenumber 2982 cm−1 (PED 85%) inhe IR spectrum. This blue shifting (162 cm−1) of methyl stretchingavenumber is due to the hyperconjugation interaction between
he � (C22–H24) → �* (N21–C26) bond.The asymmetric deformation mode of the hydrogen atoms
f me2 group results very intense band at 1463 cm−1 in IR and467 cm−1 in Raman which are extremely stable, since the methylroup is attached to another carbon atom. The symmetric bend-ng vibrations of me2 group are expected to appear in the region390–1370 cm−1 [24,25]. The intense band at 1384 cm−1 in Raman
s correlated to symmetric bending mode. The relatively largeaman intensity of the symmetric bending mode suggests a largeositive charge localized on the hydrogen atom of me2 group,hich further supports the presence of hyperconjugation.
.3.2. Naphthalene ring vibrationsNaphthalene ring vibrations are found to make a major contri-
ution in IR and Raman [30]. Naphthalene ring stretching vibrationssually occur in the region 1640–1400 cm−1. C C stretching is an
mportant marker band and its wavenumber is a genuine mea-ure of the degree of conjugation through the �-electron chain31]. In this compound the C C stretching mode is identified at621 and 1214 cm−1 in IR and at 1629, 1536, 1354 and 1218 cm−1
n Raman, respectively. Simultaneous activation of C C stretch-ng mode of the naphthalene ring (1621:1629, 1214:1218 cm−1) in
he IR and Raman spectra provides evidence for the charge trans-er interaction between the donor and acceptor groups throughhe � system [32]. The characteristic C–H stretching vibrations ofeteromatic structure are expected to appear in the wavenum-cta Part A 79 (2011) 993– 1003
ber range 3000–3100 cm−1 [29]. The observed band at 3031 cm−1
in Raman and 3036 cm−1 in IR are assigned to C–H stretchingvibration.
4.3.3. Thionocarbamate group vibrationsIn thionocarbamate related compound [33], C S stretching
mode is expected at 1050 cm−1. The medium band observed at1021 cm−1 in Raman is assigned to C S stretching mode. The C–Ostretching vibration is expected at 1149 cm−1 [34]. In TNF, the C–Ostretching vibration is observed as very strong band at 1159 cm−1
in IR and medium band at 1153 cm−1 in Raman.The vibrational band corresponds to C–N stretching in tertiary
amine occur in the range 1250–1020 cm−1 [24]. In TNF compoundC19–N21 stretching vibration is observed as very strong band at1445 cm−1 in IR and medium band at 1441 cm−1 in Raman. Thisblue shift of about 195 cm−1 is due to the charge transfer fromelectron donor me1 group to acceptor aromatic rings through anextended hyperconjugation involving the thionocarbonyl group.Also a very strong band of C19–N21 stretching vibration is observedat 1370 cm−1 in IR and 1371 cm−1 in Raman. The stretching vibra-tion N21–C26 is observed as a medium band in IR at 1299 cm−1,which is blue shifted (49 cm−1) due to hyperconjugation interactionbetween the � (C22–H24) → �* (N21–C26) bond. These hypercon-jugative charge transfer interactions contribute to the fungicidalactivity of TNF.
4.3.4. Phenyl ring vibrationVarious normal modes of vibration of the substituted phenyl
ring have been comprehensively studied according to Wilson’snumbering convention [29,35]. TNF consists of meta disubstitutedphenyl ring whose normal modes of C–H in-plane bending areclassified as 3, 9b, 18a and 18b. The observed weak bands at1277 cm−1 (IR) and 1278 cm−1 (Raman) are correlated to the C–Hin-plane bending mode3. The benzene mode 9b is expected in theregion 1149–1166 cm−1 [29], and is observed as medium bandat 1153 cm−1 in Raman and as very intense band at 1159 cm−1
in IR. The corresponding calculated value lies at 1150 cm−1 (veryintense). The intense IR band is due to strong electron–donormethyl group attached to benzene ring [29]. The ring mode 18aappears at 1083 and 1084 cm−1 as a weak band in IR and Ramanrespectively. The in-plane bending mode 3, 9b and 18a are foundto be simultaneously active in both IR and Raman because ofsymmetry lowering of the molecule; in addition such deforma-tion changes the bond length between the carbon atoms, whichbuild the charge transfer axis and the polarizability of such �conjugated system.
The selection rule allows five normal modes 8a, 8b, 19a, 19b and14 for the tangential C–C stretching mode in asymmetrically dis-ubstituted benzene derivatives. In meta disubstituted benzene thewavenumber of 8a is smaller than that of 8b [29]. DFT computa-tion shows vibrational modes 8a and 8b at 1580 and 1600 cm−1;such splitting can be observed experimentally and is allowed forthe selection rule for asymmetrically meta disubstituted ring. Thephenyl ring mode 8a manifests as very strong band in Raman at1583 cm−1 and weak band in IR at 1586 cm−1 and its companion8b appears as weak and medium band at 1607 and 1600 cm−1 inRaman and IR, respectively. The calculated values also agree wellwith the experimental data. The mode 20b is observed as veryintense band in Raman at 3060 cm−1 and medium broad band inIR at 3058 cm−1.
5. Electronic absorption spectra
Electronic transitions have been investigated by UV–visiblespectroscopy. Absorption maximum (�max) was calculated by TD-

D. Arul Dhas et al. / Spectrochimica A
Fe
Dss
a
TC
L
ig. 5. Combined UV–visible absorption spectrum of TNF – (a) computed and (b)xperimental.
FT method. The combined UV–visible absorption spectrum of the
ample is shown in Fig. 5. The electronic transitions and the corre-ponding excitation energies are listed in Table 3.The UV–visible spectrum was measured in methanol solutionnd it is found that the absorption bands are observed at 258 and
able 3alculated electronic absorption spectrum of TNF molecule.
Excitation CI expansion coefficient Excitation energies (kJ m
Excited state 1HOMO − 2 → LUMO 0.40514HOMO − 1 → LUMO 0.35848HOMO − 1 → LUMO + 1 0.27321HOMO − 1 → LUMO + 2 −0.10113 359
HOMO → LUMO 0.37352HOMO → LUMO + 1 −0.19625Excited state 2HOMO − 2 → LUMO + 1 −0.24525HOMO − 1 → LUMO 0.50836HOMO − 1 → LUMO + 1 −0.16720 374
HOMO → LUMO −0.35227HOMO → LUMO + 1 −0.24615Excited state 3HOMO − 4 → LUMO 0.13291HOMO − 4 → LUMO + 1 0.18287 435
HOMO − 4 → LUMO + 2 0.59163HOMO − 4 → LUMO + 4 0.21633Excited state 4HOMO − 2 → LUMO + 1 0.33508HOMO − 1 → LUMO 0.23511HOMO → LUMO −0.11335 480
HOMO → LUMO + 1 0.36662HOMO → LUMO + 2 −0.11859Excited state 5HOMO − 5 → LUMO + 4 −0.12652HOMO − 3 → LUMO + 2 −0.11597HOMO − 3 → LUMO + 3 −0.23275HOMO − 2 → LUMO + 2 −0.33378HOMO − 2 → LUMO + 4 0.10735 488
HOMO − 1 → LUMO + 2 0.18375HOMO → LUMO + 1 0.21031HOMO → LUMO + 2 0.36655Excited state 6HOMO − 6 → LUMO 0.31953HOMO − 4 → LUMO −0.11200HOMO − 2 → LUMO + 3 0.11891HOMO − 1 → LUMO + 1 0.43584 512
HOMO − 1 → LUMO + 5 0.10474HOMO → LUMO + 1 −0.11449
UMO is No.82 Orbital. HOMO is No.81 Orbital.
cta Part A 79 (2011) 993– 1003 999
222 nm. The very strong band at 222 nm is a characteristic peakof aromatic system due to �–�* transition (E band) [36]. TD-DFTcalculation shows that the corresponding band lies at 234 nm. Theweak band at 258 nm is due to n–�* transition (R-band), whichis attributed to sulphur atom [37]. The corresponding calculatedvalue lies at 245 nm.
5.1. HOMO–LUMO
Spatial distribution of molecular orbitals, especially those ofhighest occupied molecular orbital (HOMO) and lowest unoccupiedmolecular orbital (LUMO) are excellent indicators of electron trans-port in molecular systems. In the molecule HOMO–LUMO energygap is 222 kJ mol−1. The lowering of the HOMO–LUMO band gap isessentially a consequence of the large stabilization of LUMO dueto strong electron-accepting ability of the electron-acceptor group.The atomic orbital components of the frontier molecular orbitalsare shown in Fig. 6.
The absorption band in the long wavelength region (258 nm)is primarily due to HOMO → LUMO + 2 transition, in which theelectronic charge density is withdrawn from the thionocarbamate
group (i.e. 3p-orbit of S atom in C S group) to the tolyl ring, i.e.the absorption band near 258 nm of UV–visible spectra resultedfrom the electron transitions from the initial state is mainly con-tributed by HOMO and the final state is contributed by LUMO. Theol−1) Wavelength (nm) Oscillator strength (f) Assignment
Calc. Expt.
333 – 0.0941 –
320 – 0.0492 –
275 – 0.0012 –
249 – 0.1171 –
245 258 0.3337 n→�*
234 222 0.4398 � → �*

1000 D. Arul Dhas et al. / Spectrochimica A
ciHttoTrw
6
oui(
�jdT(e
bt((st4
ai(l
Fig. 6. HOMO–LUMO plot of TNF.
harge transfer from thionocarbamate group (i.e. 3p-orbit of S atomn C S group) to naphthalene ring is also very pronounced in theOMO − 1 → LUMO + 1 transition, which predominantly occurs in
he band observed at 222 nm. The HOMO and LUMO plot showshat both the HOMO and LUMO orbitals predominantly localizen the naphthalene ring, thionocarbamate group and phenyl ring.he small HOMO–LUMO gap (222 kJ mol−1) reflects the chemicallyeactive [38] nature with a low electron density at the sulphur atomhich accounts its fungicidal activity.
. NBO analysis
NBO results showing the formation of Lewis and non-Lewisrbital by the valence hybrids corresponding to the intramolec-lar bonds are given in Table 4. In addition the most important
nteractions between ‘filled’ (donors) Lewis-type NBOs and ‘empty’acceptors) non-Lewis NBOs are reported in Table 5.
There is strong intramolecular hyperconjugative interaction of electrons in the aromatic ring, i.e. C3–C4 and C12–C13 bonds con-
ugate to the �* (C5–C14) bond of the naphthalene ring. The electronensity (ED) 0.480 e leads to a stabilization energy 79.5 kJ mol−1.his enhancement of �* (C5–C14) NBO further conjugates with �*C3–C4) and �* (C6–C11) resulting to an enormous stabilizationnergy of 260.8 and 432.4 kJ mol−1, respectively.
The weakening of C22–H24 bond (1.093 A) is due to rehy-ridization, which is revealed by low value of ED (0.012 e) inhe �* (C22–H24) orbitals. The hyperconjugative interaction energy12.6 kJ mol−1), due to interaction between � (C22–H24) and �*N21–C26) leads to blue shifting (162 cm−1) of methyl asymmetrictretching wavenumber, which is confirmed by vibrational spec-ral analysis. This is further supported by the blue shifting of about9 cm−1 in the N21–C26 stretching (Table 2).
The hyperconjugative interaction between the oxygen lone pair
nd C–C antibonding orbital of the naphthalene ring is maximum,.e. n1 (O18) → �* (C12–C13) increases ED in C–C antibonding orbital0.506 e) that weakens the respective bond (C12–C13 = 1.413 A)eading to stabilization energy of 16.4 kJ mol−1. The existence ofcta Part A 79 (2011) 993– 1003
weak intramolecular C–H. . .S hydrogen bonds due to the interac-tion between the lone pair of sulphur n1 (S20) with the antibondingorbitals �* (C13–H16)(6.3 kJ mol−1) and (* (C27–H39)(9.6 kJ mol−1)have been confirmed by the results of NBO analysis. This wassubstantiated by PES analysis during the rotation with respect toN21–C26 bond. The most important interaction energy related tothe resonance of the molecule is electron donation from n1 (N21)(1.625 e) to the antibonding acceptor �* (C26–C27) of the tolylring (124.9 kJ mol−1) and �* (C19–S20) of thionocarbamate group(245.8 kJ mol−1). These interactions lead to stability and in turn tothe bioactivity of fungicide.
6.1. NHO directional analysis
The bending angles of different bonds expressed as the angle ofdeviation from the direction of the line joining the two nuclei cen-ters are given in Table 6. For maximum resonance interaction tooccur O18 should be in the same plane with naphthalene ring, butdue to the weak C22H24. . .O18 interaction (2.6 kJ mol−1) oxygen of�CO is bent away from the line of C12–O18 centers by 17.8◦ as aresult of lying in the strong charge transfer path towards naphtha-lene ring, whereas the carbon NHO is approximately aligned withthe C–O axis. A little lower bending effect is also noticed at theC12–C13 (14.8◦) and C5–C14 (13.0◦) bonds of the naphthalene ring.The bending of the bonds within the naphthalene ring, phenyl ringand thionocarbamate group attributes to the increase of strains atthe active center of the compound. Also in � (O18–C19), the bent of�OC bond orbital from the line of O–C centers decreases (1.8◦) dueto the presence of sulphur atom close to it.
7. Atomic net charges
A comparative study of Mulliken’s net charges and the atomicnatural charges for TNF have been carried out by B3LYP/6-31 G (d)basis set and the results are shown in Table S3 (Supporting Informa-tion). The Mulliken population analysis shows the presence of threelarge electronegative atoms such as O18, S20 and N21, which imposemore positive charge on C19 (0.373 e). The C22–N21 bond length con-necting the donor-subunit with � conjugating path is computed tobe 1.472 A, while for the acceptor moiety C26–N21 bond distanceis about 1.441 A. The shortening of C26–N21 is mainly due to thehyperconjugative interaction between phenyl ring and the strongelectron donating group (N-methyl). Since the H24 has compara-tively high positive charge, this leads to increase the distance of H24and O18 atoms by about 2.497 A. The increase of electron densityon O18 (1.949 e) seems to indicate a strong interaction of electrondonating methyl group towards O18. It is observed that the inter-action between H24 and O18 atoms contribute extra stability to theinclined structure.
8. Bioactivity
The thionocarbamate antimycotic agent, tolnaftate has potentantidermatophytic activity [39], which is primarily inhibiting theergosterol biosynthesis in the fungus. The exact mode of actionhas not been reported, but similar to that of allylamines naftifineand terbinafine, TNF is known to inhibit selectively the fungalsqualene epoxidase reaction, i.e., the conversion of squalene into2,3-oxidosqualene catalyzed by squalene epoxidase, ultimatelyresulting in the lack of ergosterol, which is an indispensable lipidcomponent of fungal cell membrane. The thionocarbamate moi-
ety is the active center as in the carbamate pesticides which arecholinesterase inhibitor [40–44]. The electron density variation ofC O group is well studied experimentally in carbamate and thereduction in electron density at the O atom of C O enhances its
D. Arul Dhas et al. / Spectrochimica Acta Part A 79 (2011) 993– 1003 1001
Table 4NBO results showing the formation of Lewis and non-Lewis orbitals by the valence hybrids corresponding to the intramolecular bonds of TNF.
Bond (A–B) ED/Energy (a.u.) EDA (%) EDB (%) NBO s (%) P(%)
�C3–C4 1.741 48.12 51.88 0.694 (sp 99.99)C 0.18 99.77−0.259 +0.720 (sp 99.99)C 0.50 99.45
�C12–C13 1.581 62.05 37.95 0.788 (sp 94.26)C 5.69 94.26−0.246 +0.616 (sp 99.86)C 0.08 99.86
�*C5–C14 0.480 57.51 42.49 0.758 (sp99.99)C 0.05 99.91
−0.002 −0.652 (sp18.88)C 5.03 94.89�*
C12–C13 0.506 37.95 62.05 0.616 (sp16.58)C 5.69 94.26−0.012 −0.788 (sp99.99)C 0.08 99.86
�*C26–C27 0.365 48.95 51.05 0.670 (sp 99.99)C 0.55 99.42
0.023 −0.715 (sp 99.99)C 0.88 99.06�*
C13–H16 0.020 38.16 61.84 0.622 (sp 1.99)C 22.79 77.120.713 −0.783 (s)H 100.00 –
�*C27–H39 0.031 36.52 63.48 0.604 (sp 2.16)C 31.62 68.33
0.467 −0.797 (s)H 100.00 –n1(S20) 1.977 – – sp99.99 0.39 99.56
−0.706n1(O18) 1.949 – – sp1.44 40.90 59.05
−0.575n2(O18) 1.825 – – sp17.38 5.44 94.49
−0.352
Table 5Second order perturbation theory analysis of Fock matrix in NBO basis.
Donor NBO (i) ED(i) (e) Acceptor NBO (j) ED(j) (e) E(2)(kJ mol−1) E(j)–E(i) (kJ mol−1)a F(i,j) (kJ mol−1)b
� (C1–C2) 1.733 � * (C3–C4) 0.263 82.6 761 176� * (C6–C11) 0.346 70.3 709 160
� (C3–C4) 1.741 � * (C1–C2) 0.257 67.8 788 163� * (C5–C14) 0.480 79.5 683 173
� (C5–C14) 1.556 � * (C6–C11) 0.346 32.8 709 110� * (C12–C13) 0.506 153.5 630 223
� (C12–C13) 1.581 � * (C5–C14) 0.480 50.8 630 129� * (C6–C11) 0.346 75.9 683 165
� * (C5–C14) 0.480 � * (C3–C4) 0.263 260.8 105 181� * (C6–C11) 0.346 432.4 53 163
� * (C12–C13) 0.506 � * (C6–C11) 0.346 252.9 79 147� * (C26–C27) 0.365 � * (C28–C29) 0.317 464.6 53 173
� * (C30–C31) 0.353 499.2 53 192� (C13–C14) 1.976 �*(C12–O18) 0.050 8.6 2652 108� (C30–C31) 1.972 �*(N21–C26) 0.037 16.9 2862 155� (C22–H24) 1.994 �*(N21–C26) 0.037 12.6 2416 123n1(S20) 1.977 �*(C13–H16) 0.020 6.3 2625 92
�*(C27–H39) 0.031 9.6 2863 116n1(O18) 1.949 �*(C19–S20) 0.414 23.1 2363 165
�* (C6–C11) 0.022 13.3 2967 142� * (C6–C11) 0.346 10.9 1549 100�* (C12–C13) 0.040 10.8 2862 123� * (C12–C13) 0.506 16.4 1470 126
n1(N21) 1.625 � * (C26–C27) 0.018 124.9 761 218�* (C26–C31) 0.020 3.4 2127 66�* (C19–S20) 0.028 9.8 1549 95� * (C19–S20) 0.414 245.8 551 260
ED – electron density. E(2) means energy of hyperconjugative interactions (stabilization energy).a Energy difference between donor and acceptor i and j NBO orbitals.b F (i, j) is the Fock matrix element between i and j NBO orbitals.
Table 6NHO directionality and “bond bending” (deviations from line of nuclear centers) of TNF.
NBO Bond A–B Line of centers Hybrid 1 Hybrid 2
� (◦) ϕ (◦) � (◦) � (◦) Deviation at A (◦) � (◦) ϕ (◦) Devia tion at B (◦)
Around thionocarbamate group�C12–O18 C12–O18 124.4 161.2 108.6 170.6 17.8 49.0 340.4 6.6� O18–C19 O18–C19 72.8 200.2 72.4 198.3 1.8 107.0 18.9 1.3Around phenyl ring�C26–C31 C26–C31 73.6 133.2 78.4 128.2 6.8 109.2 317.3 4.8�C28–C29 C28–C29 73.9 132.9 80.5 134.7 6.8 110.7 309.9 5.4Around naphthalene ring�C5–C14 C5–C14 99.6 227.7 106.5 232.6 8.4 91.1 55.3 13.0�C12–C13 C12–C13 102.2 298.7 88.7 292.3 14.8 70.6 116.2 7.6

1 ica A
ptadH(i
tStdcogostapwcbttdtpoiistafpgjac
9
hsaemnmocttitlfattst
[[
[
[[
[
[
[
[[
[[
[[
[
[
[[
[[
[[[
002 D. Arul Dhas et al. / Spectrochim
esticide activity. Analogues to carbamates the variation of elec-ron density is observed in C S group when different groups arettached to the thionocarbamate group. The calculated Chargeensity, Dipole Moment, Mulliken Charge of S, SCF Energy andomo–Lumo Energy gap for various thionocarbamate derivatives
Fig. S6; Supporting Information) are listed in Table S4 (Support-ng Information).
From Table S4 (Supporting Information), it is clearly observedhat the parent compound (TNF) has the lowest charge density at
atom. The maximum charge density is observed for dimethylhionocarbamate, where both the methyl groups are electrononating and enhances the charge at sulphur through the hyper-onjugated structure. In carbamates due to higher electronegativityf oxygen (3.5) the �-electrons of C O is shifted more towards oxy-en but in thionocarbamates the nearly equal electronegativitesf carbon and sulphur (2.5) give equal probability for �-electronhift. The phenyl and naphthyl group on the other hand is elec-ron withdrawing and thus decreases the electron density at C19nd which in turn abstract the �-electrons of C S. The near co-lanarity of aromatic rings with hetero atoms O18 and N21 andith C19 (Table 1) allows an extended conjugation throughout the
ompound. The shifted electrons from sulphur towards the C19 car-on moves towards naphthyl group via oxygen atom while towardsolyl group via nitrogen atom, the lone pair of electron present inhese hetero atoms help in these electron flow. The lesser electronensity at S20 enhances its biological activity as shown experimen-ally [45]. The substitution of electron donating methyl group inhenyl ring increases the electron density in S when they are atrtho and para positions. When it is at meta position (TNF) theres comparatively smaller electron density at S (Table S4; Support-ng Information). Thionocarbamates with electron-withdrawingubstituent groups are highly fungicidal active [45]. In tolnaf-ate there are two electron-withdrawing substituent groups suchs benzene and naphthalene ring, hence tolnaftate is highlyungicidal active compound. Also the large energy barriers inotential energy scan, the lowering of HOMO–LUMO energyap, influence of electronic effect resulting from the hypercon-ugation and induction of methyl group in the aromatic ringre the main factors which decide the bioactive nature of theompound.
. Conclusions
Predicted electronic absorption spectra from TD-DFT calculationave been analysed comparing with the experimental UV–visiblepectrum and they are mainly derived from the contribution of n-�*nd �–�* band. The lowering of HOMO–LUMO energy gap clearlyxplains the charge transfer interactions taking place within theolecule and which leads to its enhanced bioactivity. The simulta-
eous IR and Raman activation of the phenyl ring C–C stretchingode, in-plane bending mode and symmetric stretching mode
f methyl groups also provide evidences for the intramolecularharge transfer interaction between the donor and acceptor grouphrough the � system. NBO analysis reveals that the most impor-ant interaction energy related to the resonance of the molecules electron donation from n1 (N21) to the antibonding accep-or �* (C26–C27) of the tolyl ring (124.9 kJ mol−1), and whicheads to the stability and in turn to the bioactive nature of TNFungicide. The near co-planarity of aromatic rings with heterotoms O18 and N21 and with C19 allows an extended conjuga-
ion throughout the compound, the PES scan result also supporthis observation. The two electron-withdrawing substituent groupsuch as benzene and naphthalene increase the fungicidal activity ofolnaftate.[[[
cta Part A 79 (2011) 993– 1003
Acknowledgements
The author D. Arul Dhas thanks the University Grants Commis-sion (UGC), India, for the award of a Teacher Fellowship under FIPscheme leading to the Ph.D. degree. Authors also thank Prof. M.H.Jamroz, Institute of Industrial Chemistry Research, Warsaw, Poland,for providing VEDA 4 Program.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.saa.2011.04.011.
References
[1] S. De-Qing, Z. Xiao-Fei, S. Yuan-Zhi, Spectrochim. Acta Mol. Biomol. Spectrosc.A 71 (2008) 1011–1020.
[2] V.L. Furer, I.I. Vandyukova, A.E. Vandyukov, S. Fuchs, J.P. Majoral, A.M. Cami-nade, V.I. Kovalenko, J. Mol. Struct. 932 (2009) 97–104.
[3] M. Karabacak, D. Karagöz, M. Kurt, J. Mol. Struct. 892 (2008) 25–31.[4] D. Arul Dhas, I. Hubert Joe, S.D.D. Roy, T.H. Freeda, Spectrochim. Acta Part A 77
(2010) 36–44.[5] Y. Hashimoto, T. Noguchi, H. Kitagawa, G. Ohta, Toxicol. Appl. Pharmacol. 8
(1966) 380–385.[6] T. Noguchi, Y. Hashimoto, T. Makita, T. Tanimura, Toxicol. Appl. Pharmacol. 8
(1966) 386–397.[7] A.E. Czeizel, Z. Kazy, E. Puho, Reprod. Toxicol. 18 (2004) 443–444.[8] R. Munguia, S. Daniel, J. Interferon J. Ped. Otorhinolaryngol 72 (2008) 453–459.[9] F. Chapeland, R. Fritz, C. Lanen, M. Gredt, P. Leroux, Pest. Biochem. Phys. 64
(1999) 85–100.10] B. Tang, X. Wang, G. Wang, C. yu, Z. Chen, Talanta 69 (2006) 113–120.11] T. Bo, W. Xu, W. Jing, Y. Chengguang, C. Zhenzhen, D. Yi, J. Phys. Chem. B110
(2006) 8877–8884.12] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman,
V.G. Zakrzewski, J.A. Montgomery Jr., R. E/Stratmann, J.C. Burant, S. Dapprich,J.M. Millam, A.D. Daniels, K.N. Kudin, M.C. Strain, O. Farkas, J. Tomasi, V. Barone,M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski,G.A. Petersson, P.Y. Ayala, Q. Cui, K. Morokuma, D.K. Malick, A.D. Rabuck, K.Raghavachari, B. Foresman, J. Cioslowski, J.V. Ortiz, A.G. Baboul, B.B. Stefanov,G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R.L. Martin, D.J. Fox, T.Keith, M.A. Al-Laham, C.Y. Peng, A. Nanayakkara, M. Challacombe, P.M.W. Gill,B. Johnson, W. Chen, M.W. Wong, J.L. Andres, C. Gonzalez, M. Head-Gordon, E.S.Replogle, J.A. Pople, Gaussian 03, Revision C. 02, Gaussian, Inc, Wallingford CT,2004.
13] A.P. Scott, L. Radom, J. Phys. Chem. 100 (1996) 16502–16513.14] M.H. Jamroz, Vibrational Energy Distribution Analysis: VEDA 4 Program, Drug
Institute, Warsaw, Poland, 2004.15] G. Keresztury, S. Holly, J. Varga, G.A. Besenyei, Y. Wang, J.R. Durig, Spectrochim.
Acta A 49 (1993), 2007–2017, 2019–2026.16] G. Keresztury, J.M. Chalmers, P.R. Griffith, Raman Spectroscopy, Theory in Hand-
book of Vibrational Spectroscopy, vol. 1, John Wiley & Sons Ltd, New York, 2002,71–87.
17] E.D. Glendening, A.E. Reed, J.E. Carpenter, F. Weinhold, NBO Version 3.1,TCI,University of Wisconsin, Madison, 1998.
18] X.-H. Li, R.-Z. Zhang, X.-Z. Zhang, Struct. Chem. 20 (2009) 1049–1054.19] J. Chocholousova, V. Vladimin Spirko, P. Hobza, Phys. Chem. Chem. Phys. 6
(2004) 37–41.20] A.E. Reed, L.A. Curtiss, F. Weinhold, Chem. Rev. 88 (1988) 899–926.21] F. Hoong-Kun, S. Chantrapromma, L. Shu-Xian, L. Hua-Min, Acta Crystallogr.
E64 (2008) 1409.22] S.Y. Ho, C.S. Lai, E.R.T. Tiekink, Acta Crystallogr. E59 (2003) 1155–1156.23] H. Arslan, U. Florke, N. Kulcu, G. Binzet, Spectrochim. Acta A 68 (2007)
1347–1355.24] B. Smith, Infrared Spectral Interpretation. A Systematic Approach, CRC Press,
New York, DC, 1999.25] N.B. Colthup, L.H. Daly, S.E. Wiberley, Introduction to Infrared and Raman Spec-
troscopy, 3rd ed., Academic Press, New York, 1990.26] Y. Huang, D.F.R. Gilson, I.S. Butler, J. Chem. Phys. 97 (1993) 1998–2001.27] M. Gussoni, C. Castiglioni, M.N. Ramos, M.C. Rui, G. Zerbi, J. Mol. Struct. 224
(1990) 445–470.28] V. Hernandz, C. Castiglioni, G. Zerbi, J. Mol. Struct. 324 (1994) 189–198.29] G. Varsanyi, Vibrational Spectra of Benzene Derivatives, Academic Press, New
York, 1969.30] A.J. Barnes, Spectrochim. Acta A 41 (1985) 629–635.31] L. Rimai, M.E. Heyde, D. Gill, J. Am. Chem. Soc. 95 (1973) 4493–4501.32] M. Snehalatha, C. Ravikumar, N. Sekhar, V.S. Jayakumar, I. Hubert Joe, J. Raman
Spectrosc. 39 (2008) 928–936.33] G. Fairthorne, D. Fornasiero, J. Ralston, Int. J. Miner. Process. 46 (1996) 137–153.34] H. Arslan, Ulrich, N. Kulcu, Spectrochim. Acta A 67 (2007) 936–943.35] G. Socrates, Infrared Characteristic Group frequencies, Wiley-Interscience Pub-
lication, New York, 1980.

ica A
[[[[
[
[
[
[43] M.J. Weinstein, E.M. Oden, E. Moss, Antimicrob. Agents Chemother. 10 (1964)595–601.
D. Arul Dhas et al. / Spectrochim
36] L. Gomati Devi, G. Krishnamurthy, J. Hazard. Mater. 162 (2009) 899–905.37] H.M. Pinheiro, E. Touraud, O. Thomas, Dyes Pigments 61 (2004) 121–139.38] N.S. Mills, A. Levy, B.F. Plummer, J. Org. Chem. 69 (2004) 6623–6633.39] K. Iwata, T. Yamashita, H. Uehara, Antimicrob. Agents Chemother. 33 (1989)
2118–2125.40] W. Iwatani, T. Arika, H. Yamaguchi, Antimicrob. Agents Chemother. 37 (1993)
785–788.41] M.H. Weiden, J. Bull. Org. Mond. Sante, Bull. World Health Org. 44 (1971)
203–213.
[[
cta Part A 79 (2011) 993– 1003 1003
42] N.S. Ryder, I.F. Marie, C.D. Pont, Antimicrob. Agents Chemother. 29 (1986)858–860.
44] K.J. Barretti-Bee, A.C. Lane, R.W. Turner, J. Med. Vet. Mycol. 24 (1987) 155–160.45] M. Albores-Velsco, J. Thorne, R.L. Wain, J. Agric. Food Chem. 43 (1995)
2260–2261.
Copyright © 2022 FDOKUMEN




















