
Low and high dietary folic acid levels perturb postnatal ...
Upload
independentCategory
view
0download
0
t
t
S
IM
r
EAC
4p
Neuroscience 190 (2011) 346–353
NEUROTOXICITY INDUCED BY DEXAMETHASONE IN THE HUMANNEUROBLASTOMA SH-SY5Y CELL LINE CAN BE PREVENTED BY
FOLIC ACIDttpflncnpaFie(
ssg
J. BUDNI,a A. ROMERO,b,c,d,e S. MOLZ,a
M. D. MARTÍN-DE-SAAVEDRA,b,c,d J. EGEA,b,c,d
L. DEL BARRIO,b,c,d C. I. TASCA,a
A. L. S. RODRIGUESa* AND M. G. LÓPEZb,c,d
aDepartamento de Bioquímica, Universidade Federal de Santa Ca-arina, 88040-900, Florianópolis, Santa Catarina, Brazil
bDepartamento de Farmacología y Terapéutica, Universidad Au-ónoma de Madrid, 28050, Madrid, Spain
cInstituto Teófilo Hernando, Universidad Autonoma de Madrid, Madrid,pain
dHIV Unit, Hospital La Paz/Autónoma University School of Medicine,diPAZ, Departamento de Farmacologia, Universidad Autonoma deadrid, Madrid, Spain
eDepartamento de Toxicología y Farmacología, Facultad de Veterina-ia, Universidad Complutense de Madrid, 28040, Madrid, Spain
Abstract—Folic acid (folate) is a vitamin of the B-complexgroup that is essential for cell replication. Folate is a majordeterminant of one-carbon metabolism, in which S-adenosyl-methionine donates methyl groups that are crucial for neu-rological function. Many roles for folic acid have been re-ported, including neuroprotective and antidepressant prop-erties. On the other hand, increased concentrations ofcorticoids have proven neurotoxic effects and hypersecre-tion of glucocorticoids has been linked to different mooddisorders. The purpose of this study was to investigate thepotential protective effect of folic acid on dexamethasone-induced cellular death in SH-SY5Y neuroblastoma cell lineand the possible intracellular signaling pathway involved insuch effect. Exposure to 1 mM dexamethasone for 48 hcaused a significant reduction of cell viability measured as3-[4,5 dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide(MTT) reduction. Exposure of SH-SY5Y cells for 72 h to in-creasing concentrations of folate (1–300 �M) was not cyto-toxic. However, pretreatment with folate (10–300 �M) re-duced dexamethasone-induced toxicity in a significant man-ner. To explore the putative intracellular signaling pathwaysimplicated in the protective effect of folate we used differentprotein kinase inhibitors. The protective effect of folic acid on
*Corresponding author. Tel: �55-48-3721-5043; fax: �55-48-3721-9672.-mail address: [email protected] (A. L. S. Rodrigues).bbreviations: BDNF, brain-derived neurotrophic factor; CaMKII,a2�/Calmodulin-dependent protein kinase II; CREB, cAMP re-
sponse element-binding; CRH, corticotropin-releasing hormone;EMEM, Eagle’s minimum essential medium; HPA, hypothalamic-pituitary-adrenal; H-89, N-[2-[[3-(4-bromophenyl)-2-propenyl]amino]ethyl]-5-isoquinolinesulfonamide hydrochloride; KN-93, N-[2-[[[3-(4-chlorophenyl)-2-propenyl]methylamino]methyl]phenyl]-N-(2-hydroxy-ethyl)-4-methoxybenzenesulphonamide; LY294002, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one hydrochloride; MEK1/2, mitogen-acti-vated protein/extracellular signal-regulated kinase kinase; MTT, 3-[4,5dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide; NMDA, N-methyl-D-aspartate; PD98059, 2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-
c-one; PI3K, phosphatidylinositol-3 kinase; PKA, protein kinase A; PKC,rotein kinase C; THF, tetrahydrofolic acid.
0306-4522/11 $ - see front matter © 2011 Published by Elsevier Ltd on behalf of IBdoi:10.1016/j.neuroscience.2011.05.053
346
dexamethasone-induced neurotoxicity was reversed by thephosphatidylinositol-3 kinase/Akt (PI3K/Akt, LY294002), Ca2�/Calmodulin-dependent protein kinase II (CaMKII, KN-93), andprotein kinase A (PKA, H-89) inhibitors, but not the mitogen-activated protein/extracellular signal-regulated kinase (MEK1/2,PD98059) and protein kinase C (PKC, chelerythrine) inhibitors.In conclusion, the results of this study show that folic acid canprotect against dexamethasone-induced neurotoxicity and itsprotective mechanism is related to a signaling pathway thatinvolves PI3K/Akt, CaMKII, and PKA. © 2011 Published byElsevier Ltd on behalf of IBRO.
Key words: folic acid, dexamethasone, neuroprotection,PI3K, CaMKII, PKA.
Folic acid (folate) is a water-soluble vitamin whose biolog-ically active form is tetrahydrofolic acid (THF), which par-ticipates in the transfer of one-carbon units (such asmethyl, methylene, and formyl groups) to the essentialsubstrates involved in the synthesis of DNA, RNA, andproteins, crucial for neurological function (Kronenberg etal., 2009; Fenech, 2010). The metabolism of folic acid inthe cell is initiated by dihydrofolate reductase in a two-stepreaction; the first step, conversion to dihydrofolate (DHF) is aslow and rate-limiting step. In the second step dihydrofolate isfurther reduced to THF. THF can then be converted intoadditional physiological folates including 5-methyl-THF, theform that is normally found in the circulation and in tissues.5-methyl-THF is also replenished by the conversion of folinicacid (5-formyltetrahydrofolate), an active metabolite of folicacid. Because de novo folate synthesis is not present inhe CNS, it depends on adequate folate transport acrosshe blood–brain barrier. Within neurons, part of the folateool will be catabolized by oxidation to dihydrofolates andolic acid, which can be reconverted to THF by dihydrofo-ate reductase (Ramaekers and Blau, 2004). A growingumber of epidemiological studies have linked folate defi-iency with an increased risk of neurodegenerative andeuropsychiatric diseases, including Alzheimer’s, de-ression, and schizophrenia (Reynolds, 2002; Mattsonnd Shea, 2003; Coppen and Gouaille-Bolander, 2005;arah, 2009). Recent preclinical studies by our group
ndicate that folic acid has antidepressant-like (Brocardot al., 2008a,b, 2009) and antimanic-like propertiesBrocardo et al., 2010).
Glucocorticoids are adrenal steroids secreted duringtress and their numerous actions are essential for thetress response (Lee et al., 2002). However, excess oflucocorticoids, through the activation of glucocorticoid re-
eptors, has been implicated in the reduction of the hip-RO.
Wlfibc(Hec
pwlnte
pa
c2rphaSHdciaaffeamS
Mpm4
mn(
J. Budni et al. / Neuroscience 190 (2011) 346–353 347
pocampal volume observed in depression (Bremner et al.,2000; Sheline et al., 2003; Sterner and Kalynchuk, 2010).
hile the primary function of glucocorticoids is the mobi-ization of energy to respond to the stressor, they can alsoavor the cells to undergo apoptosis (Joëls, 2008). Specif-cally, dexamethasone, a synthetic glucocorticoid, haseen reported to induce cellular death in different kinds ofells, namely dexamethasone induces apoptosis in striatalMitchell et al., 1998), hippocampal (Hassan et al., 1996;aynes et al., 2001), cerebellar granule neurons (Jacobst al., 2006), glial cells (Tazik et al., 2009), and SH-SY5Yells (Tazik et al., 2009).
The human neuroblastoma cell line SH-SY5Y ex-resses several properties of neuronal cells and has beenidely used as a cellular model to investigate the intracel-
ular mechanisms mediating the actions of drugs on humaneurons (Xie et al., 2010). Therefore, this cell line is usefulo study mechanisms of neural death and protection (Kimt al., 2008; Romero et al., 2010).
Folic acid has previously been shown to exert neuro-rotective actions by preventing the damage caused bycute hyperhomocysteinemia in vivo (Tagliari et al., 2006).
It has also been reported to prevent neurotoxicity producedeither by glutamate and N-methyl-D-aspartate (NMDA) inultured mouse cerebellar granule neurons (Lin et al.,004) or by beta-amyloid peptide in cultured cortical neu-ons (Yu et al., 2009). Moreover, other studies have re-orted that deprivation of folic acid can compromise theealth of cultured dorsal root ganglion neurons (Tjiattas etl., 2004), embryonic cortical neurons, and differentiatedH-SY5Y human neuroblastoma cells (Ho et al., 2003).owever, more compelling evidence is needed for eluci-ating its neuroprotective role on dexamethasone-inducedell death in SH-SY5Y cells. Therefore, in this study, wenvestigated the effect of folic acid against neuronal dam-ge produced by dexamethasone in SH-SY5Y culturesnd the signaling transduction pathways regulating its ef-ect. Further elucidation of the neuroprotective effects ofolic acid on SH-SY5Y may lead to novel therapeutic strat-gies for some diseases, especially depression, taking intoccount that hypersecretion of glucocorticoids is linked toood disorders (Bremner et al., 2000; Kunugi et al., 2010;terner and Kalynchuk, 2010).
EXPERIMENTAL PROCEDURES
Drugs and chemicals
Folic acid was obtained from Sigma (Madrid, Spain). Cheleryth-rine, 2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one(PD98059), 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-onehydrochloride (LY294002), N-[2-[[[3-(4-chlorophenyl)-2-propenyl]methylamino]methyl]phenyl]-N-(2-hydroxyethyl)-4-methoxyben-zenesulphonamide (KN-93), and N-[2-[[3-(4-Bromophenyl)-2-pro-penyl]amino]ethyl]-5-isoquinolinesulfonamide hydrochloride (H-89)were purchased from Tocris (Biogen Científica, Madrid, Spain).Eagle’s minimum essential medium (EMEM), fetal bovine se-rum, and penicillin/streptomycin were purchased from GIBCO
(Madrid, Spain).Culture of SH-SY5Y cells
SH-SY5Y cells were maintained in a 1:1 mixture of F-12 NutrientMixture (Ham12) (Sigma Aldrich, Madrid, Spain), and EMEM sup-plemented with 15 nonessential amino acids, 1 mM sodium pyru-vate, 10% heat-inactivated fetal bovine serum (FBS), 100 U/mlpenicillin, and 100 �g/ml streptomycin (reagents from Invitrogen,
adrid, Spain). Cultures were seeded into flasks containing sup-lemented medium and maintained at 37 °C in a humidified at-osphere of 5% CO2 and 95% air. The cells were seeded into8-well culture plates at a seeding density 1�105 cells per well. All
treatments were performed when cells were grown to about 65%confluence and at the end of treatment, cells reached about80–90% confluence. All cells in this study were used at a lowpassage number (�13) and were maintained in 10% serum me-dium until the dexamethasone treatment.
SH-SY5Y cells treatment
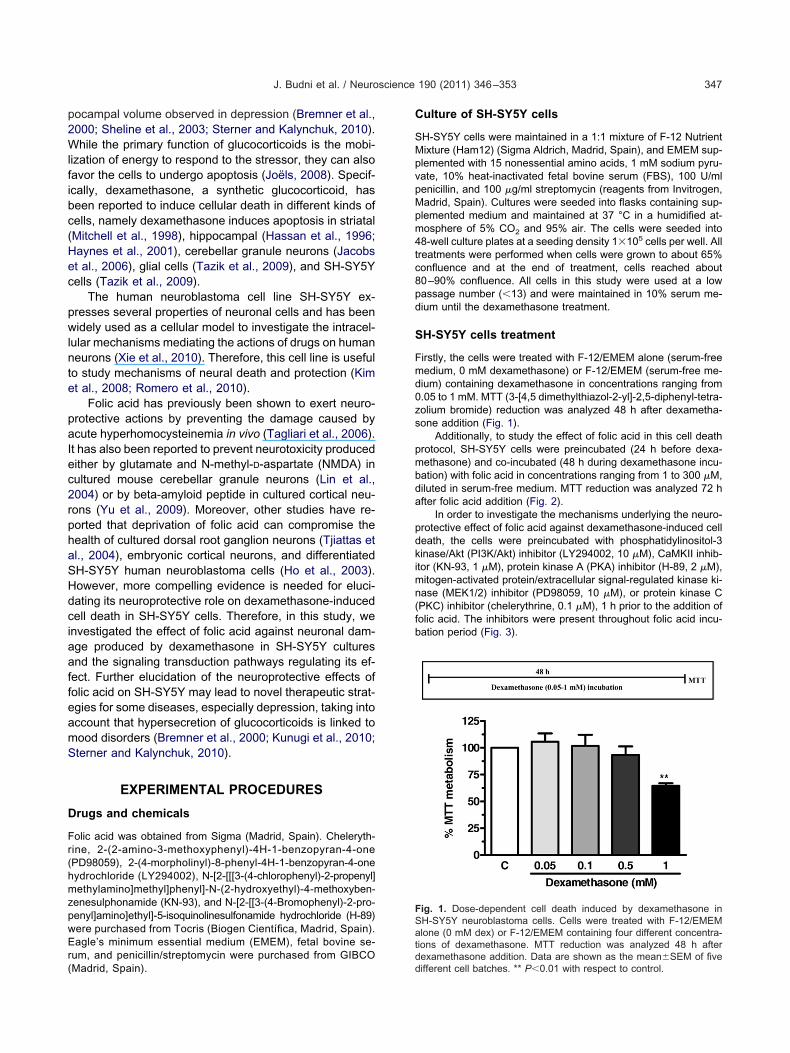
Firstly, the cells were treated with F-12/EMEM alone (serum-freemedium, 0 mM dexamethasone) or F-12/EMEM (serum-free me-dium) containing dexamethasone in concentrations ranging from0.05 to 1 mM. MTT (3-[4,5 dimethylthiazol-2-yl]-2,5-diphenyl-tetra-zolium bromide) reduction was analyzed 48 h after dexametha-sone addition (Fig. 1).
Additionally, to study the effect of folic acid in this cell deathprotocol, SH-SY5Y cells were preincubated (24 h before dexa-methasone) and co-incubated (48 h during dexamethasone incu-bation) with folic acid in concentrations ranging from 1 to 300 �M,diluted in serum-free medium. MTT reduction was analyzed 72 hafter folic acid addition (Fig. 2).
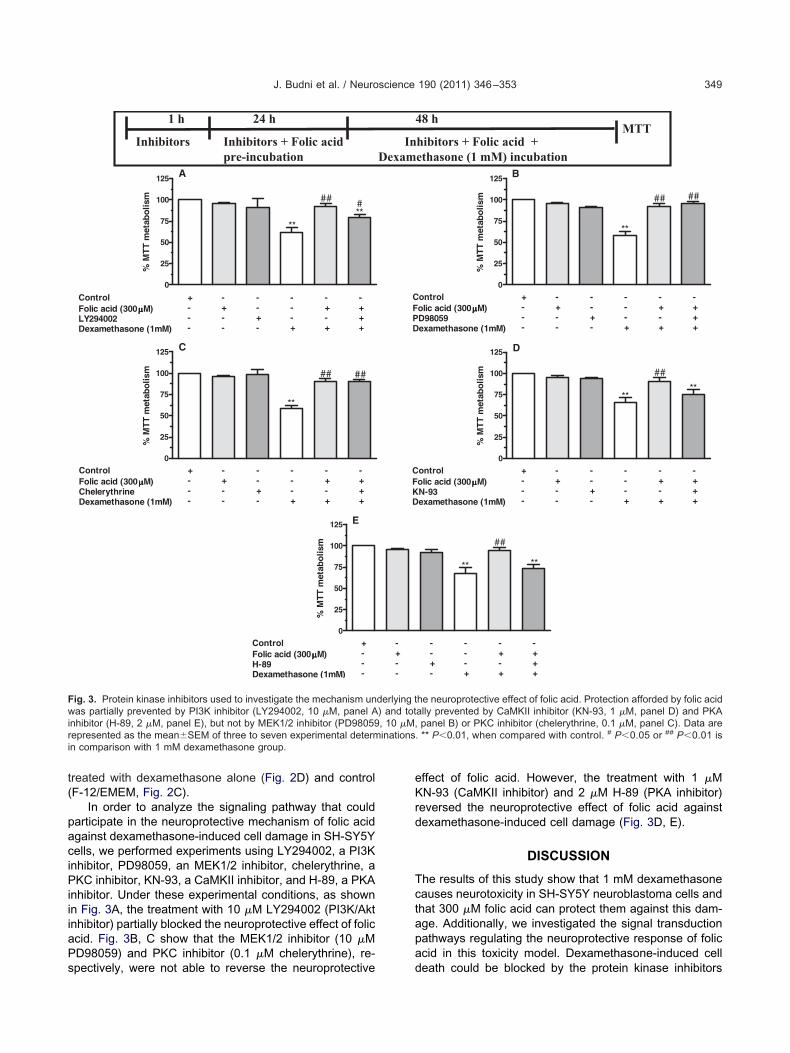
In order to investigate the mechanisms underlying the neuro-protective effect of folic acid against dexamethasone-induced celldeath, the cells were preincubated with phosphatidylinositol-3kinase/Akt (PI3K/Akt) inhibitor (LY294002, 10 �M), CaMKII inhib-itor (KN-93, 1 �M), protein kinase A (PKA) inhibitor (H-89, 2 �M),
itogen-activated protein/extracellular signal-regulated kinase ki-ase (MEK1/2) inhibitor (PD98059, 10 �M), or protein kinase CPKC) inhibitor (chelerythrine, 0.1 �M), 1 h prior to the addition of
folic acid. The inhibitors were present throughout folic acid incu-bation period (Fig. 3).
Fig. 1. Dose-dependent cell death induced by dexamethasone inSH-SY5Y neuroblastoma cells. Cells were treated with F-12/EMEMalone (0 mM dex) or F-12/EMEM containing four different concentra-tions of dexamethasone. MTT reduction was analyzed 48 h afterdexamethasone addition. Data are shown as the mean�SEM of five
different cell batches. ** P�0.01 with respect to control.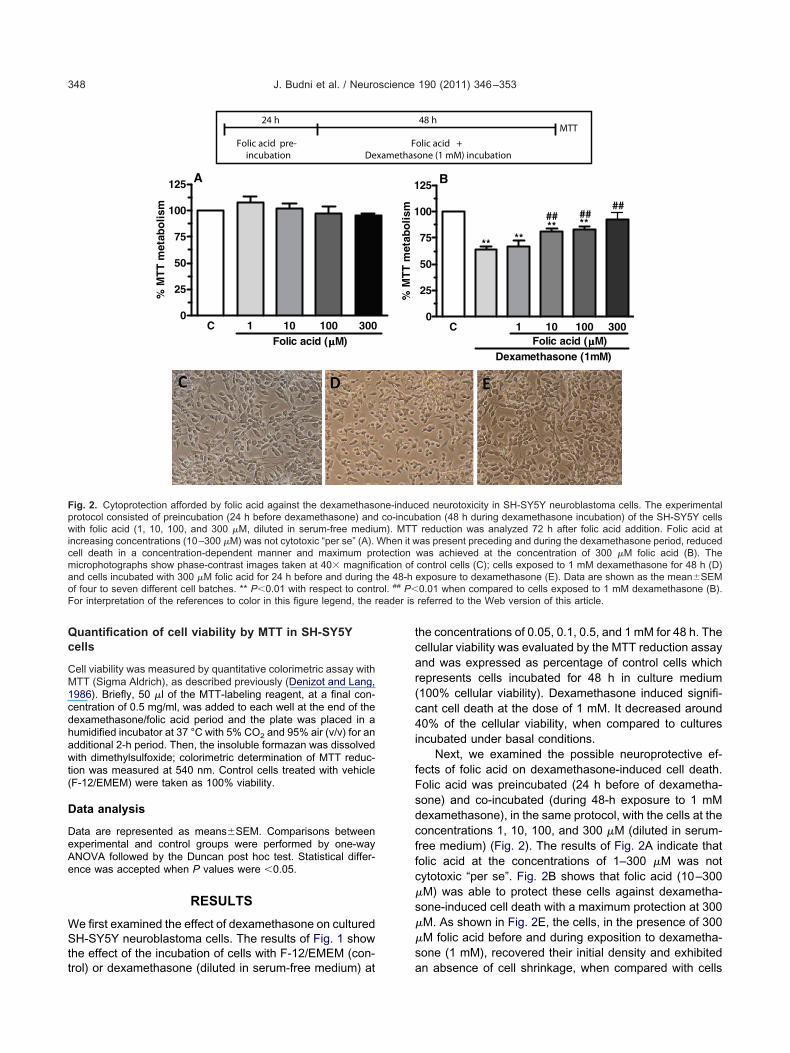
tt
ffc
s
s
ma
rol. P�F eader is
J. Budni et al. / Neuroscience 190 (2011) 346–353348
Quantification of cell viability by MTT in SH-SY5Ycells
Cell viability was measured by quantitative colorimetric assay withMTT (Sigma Aldrich), as described previously (Denizot and Lang,1986). Briefly, 50 �l of the MTT-labeling reagent, at a final con-centration of 0.5 mg/ml, was added to each well at the end of thedexamethasone/folic acid period and the plate was placed in ahumidified incubator at 37 °C with 5% CO2 and 95% air (v/v) for anadditional 2-h period. Then, the insoluble formazan was dissolvedwith dimethylsulfoxide; colorimetric determination of MTT reduc-tion was measured at 540 nm. Control cells treated with vehicle(F-12/EMEM) were taken as 100% viability.
Data analysis
Data are represented as means�SEM. Comparisons betweenexperimental and control groups were performed by one-wayANOVA followed by the Duncan post hoc test. Statistical differ-ence was accepted when P values were �0.05.
RESULTS
We first examined the effect of dexamethasone on culturedSH-SY5Y neuroblastoma cells. The results of Fig. 1 showhe effect of the incubation of cells with F-12/EMEM (con-
De
h 42
Folic acid pre-incubation
C 1 10 100 3000
25
50
75
100
125
**
Folic acid (µµµµM)
A%
MT
T m
etab
olis
m
Fig. 2. Cytoprotection afforded by folic acid against the dexamethasoprotocol consisted of preincubation (24 h before dexamethasone) andwith folic acid (1, 10, 100, and 300 �M, diluted in serum-free mediuincreasing concentrations (10–300 �M) was not cytotoxic “per se” (A).cell death in a concentration-dependent manner and maximum pr
icrophotographs show phase-contrast images taken at 40� magnifind cells incubated with 300 �M folic acid for 24 h before and during
of four to seven different cell batches. ** P�0.01 with respect to contor interpretation of the references to color in this figure legend, the r
rol) or dexamethasone (diluted in serum-free medium) at a
the concentrations of 0.05, 0.1, 0.5, and 1 mM for 48 h. Thecellular viability was evaluated by the MTT reduction assayand was expressed as percentage of control cells whichrepresents cells incubated for 48 h in culture medium(100% cellular viability). Dexamethasone induced signifi-cant cell death at the dose of 1 mM. It decreased around40% of the cellular viability, when compared to culturesincubated under basal conditions.
Next, we examined the possible neuroprotective ef-fects of folic acid on dexamethasone-induced cell death.Folic acid was preincubated (24 h before of dexametha-sone) and co-incubated (during 48-h exposure to 1 mMdexamethasone), in the same protocol, with the cells at theconcentrations 1, 10, 100, and 300 �M (diluted in serum-ree medium) (Fig. 2). The results of Fig. 2A indicate thatolic acid at the concentrations of 1–300 �M was notytotoxic “per se”. Fig. 2B shows that folic acid (10–300
�M) was able to protect these cells against dexametha-one-induced cell death with a maximum protection at 300
�M. As shown in Fig. 2E, the cells, in the presence of 300�M folic acid before and during exposition to dexametha-one (1 mM), recovered their initial density and exhibited
olic acid +one (1 mM) incubation
MTTh 84
C 1 10 100 3000
25
50
75
00
25
**
Folic acid (µµµµM)
B
**
** **
#### ##
Dexamethasone (1mM)
ed neurotoxicity in SH-SY5Y neuroblastoma cells. The experimentalation (48 h during dexamethasone incubation) of the SH-SY5Y cellsreduction was analyzed 72 h after folic acid addition. Folic acid atas present preceding and during the dexamethasone period, reduced
was achieved at the concentration of 300 �M folic acid (B). Thecontrol cells (C); cells exposed to 1 mM dexamethasone for 48 h (D)exposure to dexamethasone (E). Data are shown as the mean�SEM0.01 when compared to cells exposed to 1 mM dexamethasone (B).referred to the Web version of this article.
Fxamethas
1
1
% M
TT
met
abo
lism
ne-inducco-incub
m). MTTWhen it wotectioncation ofthe 48-h
##
n absence of cell shrinkage, when compared with cells
paciPii
K
r inations
J. Budni et al. / Neuroscience 190 (2011) 346–353 349
treated with dexamethasone alone (Fig. 2D) and control(F-12/EMEM, Fig. 2C).
In order to analyze the signaling pathway that couldarticipate in the neuroprotective mechanism of folic acidgainst dexamethasone-induced cell damage in SH-SY5Yells, we performed experiments using LY294002, a PI3Knhibitor, PD98059, an MEK1/2 inhibitor, chelerythrine, aKC inhibitor, KN-93, a CaMKII inhibitor, and H-89, a PKA
nhibitor. Under these experimental conditions, as shownn Fig. 3A, the treatment with 10 �M LY294002 (PI3K/Aktinhibitor) partially blocked the neuroprotective effect of folicacid. Fig. 3B, C show that the MEK1/2 inhibitor (10 �MPD98059) and PKC inhibitor (0.1 �M chelerythrine), re-
Inhibitors + Folic acid pre-incubation
h 421 h
Inhibitors
0
25
50
75
100
125
**
##**#
A
ControlFolic acid (300µµµµM)LY294002Dexamethasone (1mM)
+---
-+--
--+-
-+-+
---+
-+++
% M
TT
met
abo
lism
0
25
50
75
100
125
**
## ##
C
ControlFolic acid (300µµµµM)ChelerythrineDexamethasone (1mM)
+---
-+--
--+-
-+-+
---+
-+++
% M
TT
met
abo
lism
0
25
50
75
100
125 E
ControlFolic acid (300µµµµM)H-89Dexamethasone (1mM)
+---
% M
TT
met
abo
lism
Fig. 3. Protein kinase inhibitors used to investigate the mechanism unwas partially prevented by PI3K inhibitor (LY294002, 10 �M, panel Ainhibitor (H-89, 2 �M, panel E), but not by MEK1/2 inhibitor (PD98059epresented as the mean�SEM of three to seven experimental determ
in comparison with 1 mM dexamethasone group.
spectively, were not able to reverse the neuroprotective
effect of folic acid. However, the treatment with 1 �MN-93 (CaMKII inhibitor) and 2 �M H-89 (PKA inhibitor)
reversed the neuroprotective effect of folic acid againstdexamethasone-induced cell damage (Fig. 3D, E).
DISCUSSION
The results of this study show that 1 mM dexamethasonecauses neurotoxicity in SH-SY5Y neuroblastoma cells andthat 300 �M folic acid can protect them against this dam-age. Additionally, we investigated the signal transductionpathways regulating the neuroprotective response of folicacid in this toxicity model. Dexamethasone-induced cell
hibitors + Folic acid +ethasone (1 mM) incubation
MTTh 84
0
25
50
75
100
125
**
## ##
B
ontrololic acid (300µµµµM)D98059examethasone (1mM)
+---
-+--
--+-
-+-+
---+
-+++
% M
TT
met
abo
lism
0
25
50
75
100
125
**
##
**
D
ontrololic acid (300µµµµM)N-93examethasone (1mM)
+---
-+--
--+-
-+-+
---+
-+++
% M
TT
met
abo
lism
**
##
**
--+-
-+-+
---+
-+++
he neuroprotective effect of folic acid. Protection afforded by folic acidally prevented by CaMKII inhibitor (KN-93, 1 �M, panel D) and PKApanel B) or PKC inhibitor (chelerythrine, 0.1 �M, panel C). Data are
. ** P�0.01, when compared with control. # P�0.05 or ## P�0.01 is
InDexam
CFPD
CFKD
-+--
derlying t) and tot, 10 �M,
death could be blocked by the protein kinase inhibitors

Cs(ddisnr
mStaamhh
pct
(2rc2sc
J. Budni et al. / Neuroscience 190 (2011) 346–353350
LY294002, KN-93, and H-89, but not by PD98059 or chel-erythrine. Therefore, we showed that the protection af-forded by folic acid was likely mediated through the acti-vation of PI3K/Akt, CaMKII, and PKA signaling pathwayrather than MEK1/2 and PKC pathways.
Physical and psychological stressors stimuli activatethe hypothalamic-pituitary-adrenal (HPA) axis by increas-ing the production and release of corticotropin-releasinghormone (CRH) and arginine vasopressin from the para-ventricular nucleus of the hypothalamus. CRH stimulatesthe pituitary to produce adrenocorticotropic hormone(ACTH), which enters the bloodstream and activates theadrenal glands to release glucocorticoids, including corti-sol in humans and corticosterone in rodents. Glucocortico-ids, in turn, exert inhibitory feedback effects mainly at thehypothalamus and pituitary glands to inhibit the synthesisand secretion of CRH and ACTH, respectively. The hip-pocampus also confers an inhibitory effect on the HPA axis(Kunugi et al., 2010). The sites in the brain where gluco-corticoids act are determined by the distribution of gluco-corticoid receptors. The hippocampus is a region with highlevels of these receptors (Conrad, 2008; Joëls, 2011).Depending on the intensity or duration of the stress, as wellas individual qualities (genetics, psychological state, etc.),the endocrine response to stress (which is supposed to beadaptive) becomes pathological because it involves hyper-secretion of glucocorticoids by HPA axis overstimulation,triggering disorders such as Cushing’s disease, posttrau-matic stress disorder, bipolar disorder, and depression(Bremner et al., 2000; Conrad, 2008; Kunugi et al., 2010).
orroborating the relationship between the HPA axis over-timulation and depression, a study from Haynes et al.2004) showed that chronic treatment with different anti-epressants, such as tranylcypromine (a monoamine oxi-ase inhibitor), fluoxetine (a selective serotonin reuptake
nhibitor), and desipramine (a tricyclic antidepressant) re-ulted in marked protection from dexamethasone-inducedeuronal damage in the striatum and the hippocampus ofats.
In this study we used a model of neurotoxicity thatimics the hypersecretion of glucocorticoids by incubatingH-SY5Y cells with dexamethasone, a synthetic glucocor-
icoid (Mitchell et al., 1998; Haynes et al., 2001; Jacobs etl., 2006; Zhu et al., 2006; Tazik et al., 2009), taking intoccount that a glucocorticoid hypersecretion occurs inost of depressive patients (Wolkowitz et al., 2009). Theippocampal neuronal damage caused by glucocorticoidsas been well documented in vivo and in vitro (Woolley et
al., 1990; Virgin et al., 1991; Hassan et al., 1996; Mitchellet al., 1998; Ahlbom et al., 2000; Joëls, 2001; Lu et al.,2003; Crochemore et al., 2005; Zhu et al., 2006). As pre-viously reported in the literature, our results show thatdexamethasone induces neuronal damage at the concen-tration of 1 mM. Literature data indicate that several puta-tive mechanisms may be implicated in this damage. One isthat the inhibition of the PI3K/Akt signaling pathway en-hances dexamethasone-induced cell death (Nuutinen etal., 2006). Another possibility is that glucocorticoid induces
the increase of extracellular glutamate in the hippocampus(Sapolsky, 2000) which can be prevented by blockingNMDA receptors (Armanini et al., 1990). Moreover, exces-sive glucocorticoid reduces the expression and impairsbrain-derived neurotrophic factor (BDNF) function, whichdamages the hippocampus and other brain areas (Ku-mamaru et al., 2008; Kunugi et al., 2010). It is interesting tomention that glucocorticoids exert antiproliferative effectsin many cell types (Crochemore et al., 2002; Hong et al.,2011) by inhibition of cell cycle progression or induction ofcell death (Crochemore et al., 2002). Our results indicatedthat dexamethasone induces morphological alteration inSH-SY5Y cells which is indicative of cell death rather thanreduced proliferation, but additional studies are necessaryfor confirm these results.
Under the experimental conditions of this study, 24-hpreincubation with folic acid, at increasing concentrations(10–300 �M) preceding the dexamethasone period (seerotocol on top of Fig. 2), reduced neurotoxicity in a con-entration-dependent manner (Fig. 2B). Maximum protec-ion was achieved at the concentration of 300 �M of folicacid (29% protection; Fig. 2B, E). Fig. 2D shows thattreatment with 1 mM dexamethasone for 48 h changedhealthy birefringence cells (Fig. 2C) into cells without bire-fringence and with granular cytoplasm. The literature re-ports that folic acid deficiency induces neurotoxicity bymultiple routes. It was reported that folic acid deprivationincreased cytosolic Ca2� and reactive oxygen speciesROS) and impaired mitochondrial function (Ho et al.,003; Tjiattas et al., 2004). Folic acid plays a protectiveole against glutamate and NMDA-induced cytotoxicity inultured mouse cerebellar granule neurons (Lin et al.,004). Moreover, a previous study of Yu et al. (2009)howed that folic acid protects neurons from the damageaused by A�25-35 peptide by maintaining mitochondrial
function and DNA integrity and regulating apoptosis-re-lated genes.
The neuroprotective actions of folic acid described inthis study are relevant in the context of previous results: (a)the dexamethasone-induced cell death protocol mimicsthe hypersecretion of glucocorticoids in vitro (Joëls, 2001,2008; Jacobs et al., 2006; Tazik et al., 2009); (b) chronictreatment with antidepressants resulted in protection fromdexamethasone-induced neuronal damage in hippocam-pal and striatal neuronal populations of rats (Haynes et al.,2004); (c) preclinical studies of our group indicate that folicacid has antidepressant-like properties (Brocardo et al.,2008a,b, 2009); (d) folic acid was reported to have neuro-protective properties (Ho et al., 2003; Lin et al., 2004;Tjiattas et al., 2004; Yu et al., 2009).
It can be hypothesized that folic acid-induced cell sur-vival was dependent on different signaling pathways,which are key elements of signal transduction involved oncell proliferation, differentiation, and stress response suchas PI3K/Akt, PKA, MAPK/ERK, CaMKII, or PKC pathways(Cantley, 2002; Vivanco and Sawyers, 2002; Einat et al.,2003; Cañas et al., 2007; Gokce et al., 2009; Romero etal., 2010).
Our results indicate that exposure of SH-SY5Y cells
with LY294002, a PI3K inhibitor, partially suppressed the
aeti2
aa
tSat
J. Budni et al. / Neuroscience 190 (2011) 346–353 351
neuroprotective effects of folic acid in dexamethasone-exposed cells. A large body of genetic and pharmacolog-ical evidence indicates that the PI3K/Akt signaling pathwayis critical for cell growth and survival (Hennessy et al.,2005). The PI3K/Akt pathway is particularly important formediating neuronal survival under a wide variety of circum-stances (Brunet et al., 2001; Shao et al., 2011) and hasbeen found to be sufficient and, in some cases, necessaryfor the protection mediated by trophic factors such asinsulin-like growth factor-1 (IGF-1) on neuronal cell deathinduced by corticosterone in hippocampal neurons (Nitta etl., 2004). Moreover, dexamethasone can decrease prolif-ration and increase apoptosis in chondrocyte cell cultureshrough inhibition of Akt phosphorylation and thereforenhibition of the PI3K/Akt signaling pathway (Chrysis et al.,005). To reinforce our results, a previous study of Seto et
al. (2010) showed that in an animal model of diabetesmellitus-associated hypertension, oral folic acid supple-mentation restored the blunted acetylcholine-induced aor-tic relaxation observed in mice, probably via enhancementof the activity of PI3K/Akt cascade. In line with this finding,our results indicate that LY294002 partially blocked theneuroprotective effect of folic acid against dexametha-sone-induced cell injury in SH-SY5Y cells. This result re-inforces that the PI3K/Akt signaling pathway is implicatedin the neuroprotective effect of folic acid.
Additionally, our results show that PD98059 and chel-erythrine were not able to block the protective action offolic acid against dexamethasone-induced damage. Theseresults suggest that this signaling pathway does not par-ticipate in the neuroprotective effect of folic acid in thisprotocol.
Moreover, CaMKII is the most abundant protein kinasein the brain involved in neuronal plasticity (Popoli et al.,2000; Cammarota et al., 2002). Downstream calcium-de-pendent responses to BDNF involve the activation of cal-cium/calmodulin-dependent kinases (CaMKs) (Spencer etal., 2008). It involves downstream calcium-dependentmechanisms and the CaMK-dependent phosphorylationinduces activation of cAMP response element-binding(CREB) transcription factor. Our results show that KN-93suppressed the neuroprotective effect of folic acid, there-fore this signaling pathway seems to be involved in theneuroprotective mechanism related to folic acid in this celldeath protocol.
We also found that H-89 suppressed the neuroprotec-tive effect of folic acid against dexamethasone-inducedneurotoxicity. H-89 is an isoquinolinesulfonamide that actsas a competitive inhibitor against ATP binding to the cat-alytic subunit of PKA (Chijiwa et al., 1990). This enzyme isinvolved in several physiologic functions in the brain, in-cluding neurotransmitter synthesis and release, gene ex-pression, synaptic plasticity, memory, cell growth and dif-ferentiation, and cell survival. The major mechanism ofPKA-mediated function is through the phosphorylation ofspecific substrates, which include CREB (D’Sa and Du-man, 2002; Gould and Manji, 2002; Blendy, 2006). Thectivation of CREB causes the expression of proteins such
s BDNF, which has been implicated in the maintenance ofneurons, cell survival, and neuronal plasticity (D’Sa andDuman, 2002; Blendy, 2006). In our study H-89 was ableto reverse the dexamethasone-induced SH-SY5Y neuro-toxicity, suggesting that the activation of this signalingpathway by folic acid is also likely implicated in its neuro-protective effect.
As depicted in Fig. 4 this study presents clear evidencehat folic acid can protect the human neuroblastoma SH-Y5Y cell line against dexamethasone-induced cell dam-ge and that PI3K/Akt, CaMKII, and PKA are molecularargets implicated in the neuroprotective effect of folic acid.
Acknowledgments—This study was supported by the FINEP re-search grant CAPES-DGU (Project 173/08) and also by the Span-ish Ministry of Education Ref. PHB2007-0004-PC (ProgramaHispanobrasileño de Cooperación Interuniversitaria), the Span-ish Ministry of Science and Innovation Ref. SAF2009-12150and the Spanish Ministry of Health (Instituto de Salud Carlos III)RETICS-RD06/0026.Contributors—Josiane Budni projected the study, performed theexperiments, analyzed the data, and wrote the paper; AlejandroRomero performed the human neuroblastoma SH-SY5Y cells cul-ture and reviewed the manuscript; Simone Molz performed theexperiments and reviewed the manuscript; Maria Dolores Martín-de-Saavedra projected the study and reviewed the manuscript;Javier Egea, projected the study; Laura Del Barrio reviewed themanuscript; Carla Inês Tasca analyzed the data and reviewed themanuscript; Ana Lúcia Severo Rodrigues analyzed the data andreviewed the manuscript; Manuela Garcia Lopéz provided finan-cial support for the conduct of the research, analyzed the data andreviewed the manuscript.
REFERENCES
Ahlbom E, Gogvadze V, Chen M, Celsi G, Ceccatelli S (2000) Prenatal
Folic acid
PI3K
Akt
Neuroprotection against dexamethasone-induced neurotoxicity in the SH-SY5Y cells
LY 294002
+ + +CAMKII PKA
H-89KN-93
?
++
+
Fig. 4. Schematic diagram illustrating the putative intracellular sur-vival pathways activated by folic acid to prevent dexamethasone-induced neurotoxicity in the SH-SY5Y cell line. Neuroprotection af-forded by folic acid in this neurotoxicity protocol involves moleculartargets such as PI3K/Akt, CaMKII, and PKA activation. For interpre-tation of the references to color in this figure legend, the reader isreferred to the Web version of this article.
exposure to high levels of glucocorticoids increases the suscepti-

J
J
J
K
K
K
K
L
J. Budni et al. / Neuroscience 190 (2011) 346–353352
bility of cerebellar granule cells to oxidative stress-induced celldeath. Proc Natl Acad Sci U S A 97(26):14726–14730.
Armanini MP, Hutchins C, Stein BA, Sapolsky RM (1990) Glucocorti-coid endangerment of hippocampal neurons is NMDA-receptordependent. Brain Res 532(1–2):7–12.
Blendy JA (2006) The role of CREB in depression and antidepressanttreatment. Biol Psychiatry 59:1144–1150.
Bremner JD, Narayan M, Anderson ER, Staib LH, Miller HL, CharneyDS (2000) Hippocampal volume reduction in major depression.Am J Psychiatry 157(1):115–118.
Brocardo PS, Budni J, Kaster MP, Santos ARS, Rodrigues ALS(2008a) Folic acid administration produces an antidepressant-likeeffect in mice: evidence for the involvement of the serotonergic andnoradrenergic systems. Neuropharmacology 54(2):464–473.
Brocardo PS, Budni J, Lobato KR, Kaster MP, Santos ARS, RodriguesALS (2008b) Antidepressant-like effect of folic acid: involvement ofNMDA receptors and L-arginine-nitric oxide-cyclic guanosinemonophosphate pathway. Eur J Pharmacol 598(1–3):37–42.
Brocardo PS, Budni J, Lobato KR, Santos ARS, Rodrigues ALS (2009)Evidence for the involvement of the opioid system in the antide-pressant-like effect of folic acid in the mouse forced swimming test.Behav Brain Res 200(1):122–127.
Brocardo PS, Budni J, Pavesi E, Franco JL, Uliano-Silva M, TrevisanR, Terenzi MG, Dafre AL, Rodrigues ALS (2010) Folic acid admin-istration prevents ouabain-induced hyperlocomotion and altera-tions in oxidative stress markers in the rat brain. Bipolar Disord12(4):414–424.
Brunet A, Datta SR, Greenberg ME (2001) Transcription-dependentand -independent control of neuronal survival by the PI3K-Aktsignaling pathway. Curr Opin Neurobiol 11(3):297–305.
Cammarota M, Bevilaqua LR, Viola H, Kerr DS, Reichmann B, Teix-eira V, Bulla M, Izquierdo I, Medina JH (2002) Participation ofCaMKII in neuronal plasticity and memory formation. Cell MolNeurobiol 22(3):259–267.
Cañas N, Valero T, Villarroya M, Montell E, Vergés J, García AG,López MG (2007) Chondroitin sulfate protects SH-SY5Y cells fromoxidative stress by inducing heme oxygenase-1 via phosphatidyl-inositol 3-kinase/Akt. J Pharmacol Exp Ther 323(3):946–953.
Cantley LC (2002) The phosphoinositide 3-kinase pathway. Science296(5573):1655–1657.
Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, NaitoK, Toshioka T, Hidaka H (1990) Inhibition of forskolin-inducedneurite outgrowth and protein phosphorylation by a newly synthe-sized selective inhibitor of cyclic AMP-dependent protein kinase,N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J Biol Chem 265:5267–5272.
Chrysis D, Zaman F, Chagin AS, Takigawa M, Sävendahl L (2005)Dexamethasone induces apoptosis in proliferative chondrocytesthrough activation of caspases and suppression of the Akt-phos-phatidylinositol 3=-kinase signaling pathway. Endocrinology 146(3):1391–1397.
Conrad CD (2008) Chronic stress-induced hippocampal vulnerability:the glucocorticoid vulnerability hypothesis. Rev Neurosci 19(6):395–411.
Coppen A, Gouaille-Bolander C (2005) Treatment of depression: timeto consider folic acid and vitamin B12. J Psychopharmacol 19:59–65.
Crochemore C, Lu J, Wu Y, Liposits Z, Sousa N, Holsboer F, AlmeidaOF (2005) Direct targeting of hippocampal neurons for apoptosisby glucocorticoids is reversible by mineralocorticoid receptor acti-vation. Mol Psychiatry 10(8):790–798.
Crochemore C, Michaelidis TM, Fischer D, Loeffler JP, Almeida OF(2002) Enhancement of p53 activity and inhibition of neural cellproliferation by glucocorticoid receptor activation. FASEB J16(8):761–770.
D’sa C, Duman RS (2002) Antidepressants and neuroplasticity. Bipo-
lar Disord 4:183–194.Denizot F, Lang R (1986) Rapid colorimetric assay for cell growth andsurvival. Modifications to the tetrazolium dye procedure givingimproved sensitivity and reliability. J Immunol Methods 89:271–277.
Einat H, Yuan P, Gould TD, Li J, Du JH, Zhang L, Manji HK, Chen G(2003) The role of the extracellular signal-regulated kinase signal-ing pathway in mood modulation. J Neurosci 23:7311–7316.
Farah A (2009) The role of L-methylfolate in depressive disorders.CNS Spectr 14(2):2–7.
Fenech M (2010) Folate, DNA damage and the aging brain. MechAgeing Dev 131(4):236–241.
Gokce O, Runne H, Kuhn A, Luthi-Carter R (2009) Short-term striatalgene expression responses to brain-derived neurotrophic factorare dependent on MEK and ERK activation. PLoS One 4(4):e5292.
Gould TD, Manji HK (2002) Signaling networks in the pathophysiologyand treatment of mood disorders. J Psychosom Res 53:687–697.
Hassan AH, von Rosenstiel P, Patchev VK, Holsboer F, Almeida OF(1996) Exacerbation of apoptosis in the dentate gyrus of the agedrat by dexamethasone and the protective role of corticosterone.Exp Neurol 140(1):43–52.
Haynes LE, Barber D, Mitchell IJ (2004) Chronic antidepressant med-ication attenuates dexamethasone-induced neuronal death andsublethal neuronal damage in the hippocampus and striatum.Brain Res 1026(2):157–167.
Haynes LE, Griffiths MR, Hyde RE, Barber DJ, Mitchell IJ (2001)Dexamethasone induces limited apoptosis and extensive sublethaldamage to specific subregions of the striatum and hippocampus:implications for mood disorders. Neuroscience 104(1):57–69.
Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB (2005) Exploiting thePI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Dis-cov 4(12):988–1004.
Ho PI, Ashline D, Dhitavat S, Ortiz D, Collins SC, Shea TB, Rogers E(2003) Folate deprivation induces neurodegeneration: roles of ox-idative stress and increased homocysteine. Neurobiol Dis 14(1):32–42.
Hong D, Chen HX, Yu HQ, Wang C, Deng HT, Lian QQ, Ge RS (2011)Quantitative proteomic analysis of dexamethasone-induced effectson osteoblast differentiation, proliferation, and apoptosis inMC3T3-E1 cells using SILAC. Osteoporos Int 22(7):2175–2186.
Jacobs CM, Trinh MD, Rootwelt T, Lømo J, Paulsen RE (2006) Dexa-methasone induces cell death which may be blocked by NMDAreceptor antagonists but is insensitive to Mg2� in cerebellar gran-ule neurons. Brain Res 1070(1):116–123.
oëls M (2001) Corticosteroid actions in the hippocampus. J Neuroen-docrinol 13(8):657–669.
oëls M (2008) Functional actions of corticosteroids in the hippocam-pus. Eur J Pharmacol 583(2–3):312–321.
oëls M (2011) Impact of glucocorticoids on brain function: relevancefor mood disorders. Psychoneuroendocrinology 36(3):406–414.
im MS, Kim MK, Kim KS, Chung JH, Kim SJ, Kim JH, Kim JR, Lee J,Yu BP, Chung HY (2008) Cytotoxicity of 1,2-diacetylbenzene inhuman neuroblastoma SHSY5Y cells is mediated by oxidativestress. Toxicology 243(1–2):216–223.
ronenberg G, Colla M, Endres M (2009) Folic acid, neurodegenera-tive and neuropsychiatric disease. Curr Mol Med 9(3):315–323.
umamaru E, Numakawa T, Adachi N, Yagasaki Y, Izumi A, Niyaz M,Kudo M, Kunugi H (2008) Glucocorticoid prevents brain-derivedneurotrophic factor-mediated maturation of synaptic function indeveloping hippocampal neurons through reduction in the activityof mitogen-activated protein kinase. Mol Endocrinol 22(3):546–558.
unugi H, Hori H, Adachi N, Numakawa T (2010) Interface betweenhypothalamic-pituitary-adrenal axis and brain-derived neurotrophicfactor in depression. Psychiatry Clin Neurosci 64(5):447–459.
ee AL, Ogle WO, Sapolsky RM (2002) Stress and depression: pos-sible links to neuron death in the hippocampus. Bipolar Disord
4(2):117–128.
J. Budni et al. / Neuroscience 190 (2011) 346–353 353
Lin Y, Desbois A, Jiang S, Hou ST (2004) Group B vitamins protectmurine cerebellar granule cells from glutamate/NMDA toxicity.Neuroreport 15:2241–2244.
Lu J, Goula D, Sousa N, Almeida OF (2003) Ionotropic and metabo-tropic glutamate receptor mediation of glucocorticoid-induced ap-optosis in hippocampal cells and the neuroprotective role of syn-aptic N-methyl-D-aspartate receptors. Neuroscience 121(1):123–131.
Mattson MP, Shea TB (2003) Folate and homocysteine metabolism inneural plasticity and neurodegenerative disorders. Neuroscience26:137–146.
Mitchell IJ, Cooper AJ, Griffiths MR, Barber DJ (1998) Phencyclidineand corticosteroids induce apoptosis of a subpopulation of striatalneurons: a neural substrate for psychosis? Neuroscience 84(2):489–501.
Nitta A, Zheng Wh, Quirion R (2004) Insulin-like growth factor 1prevents neuronal cell death induced by corticosterone throughactivation of the PI3k/Akt pathway. J Neurosci Res 76(1):98–103.
Nuutinen U, Postila V, Mättö M, Eeva J, Ropponen A, Eray M,Riikonen P, Pelkonen J (2006) Inhibition of PI3-kinase-Akt pathwayenhances dexamethasone-induced apoptosis in a human follicularlymphoma cell line. Exp Cell Res 312(3):322–330.
Popoli M, Brunello N, Perez J, Racagni G (2000) Second messenger-regulated protein kinases in the brain: their functional role and theaction of antidepressant drugs. J Neurochem 74:21–33.
Ramaekers VT, Blau N (2004) Cerebral folate deficiency. Dev MedChild Neurol 46(12):843–851.
Reynolds EH (2002) Folic acid, ageing, depression, and dementia.BMJ 324(7352):1512–1515.
Romero A, Egea J, García AG, López MG (2010) Synergistic neuro-protective effect of combined low concentrations of galantamineand melatonin against oxidative stress in SH-SY5Y neuroblastomacells. J Pineal Res 49(2):141–148.
Sapolsky RM (2000) The possibility of neurotoxicity in the hippocam-pus in major depression: a primer on neuron death. Biol Psychiatry48(8):755–765.
Seto SW, Lam TY, Or PM, Lee WY, Au AL, Poon CC, Li RW, ChanSW, Yeung JH, Leung GP, Lee SM, Ngai SM, Kwan YW (2010)Folic acid consumption reduces resistin level and restores bluntedacetylcholine-induced aortic relaxation in obese/diabetic mice. JNutr Biochem 21(9):872–880.
Shao JL, Wan XH, Chen Y, Bi C, Chen HM, Zhong Y, Heng XH, Qian JQ
(2011) H(2)S protects hippocampal neurons from anoxia-reoxygen-ation through cAMP-mediated PI3K/Akt/p70S6K cell-survivalsignaling pathways. J Mol Neurosci 43(3):453–460.
Sheline YI, Gado MH, Kraemer HC (2003) Untreated depression andhippocampal volume loss. Am J Psychiatry 160(8):1516–1518.
Spencer TK, Mellado W, Filbin MT (2008) BDNF activates CaMKIVand PKA in parallel to block MAG-mediated inhibition of neuriteoutgrowth. Mol Cell Neurosci 38:110–116.
Sterner EY, Kalynchuk LE (2010) Behavioral and neurobiological con-sequences of prolonged glucocorticoid exposure in rats: relevanceto depression. Prog Neuropsychopharmacol Biol Psychiatry 34(5):777–790.
Tagliari B, Zamin LL, Salbego CG, Netto CA, Wyse AT (2006) Hyper-homocysteinemia increases damage on brain slices exposed to invitro model of oxygen and glucose deprivation: prevention by folicacid. Int J Dev Neurosci 24(4):285–291.
Tazik S, Johnson S, Lu D, Johnson C, Youdim MB, Stockmeier CA, OuXM (2009) Comparative neuroprotective effects of rasagiline andaminoindan with selegiline on dexamethasone-induced brain cellapoptosis. Neurotox Res 15(3):284–290.
Tjiattas L, Ortiz DO, Dhivant S, Mitton K, Rogers E, Shea TB (2004)Folate deficiency and homocysteine induce toxicity in cultureddorsal root ganglion neurons via cytosolic calcium accumulation.Aging Cell 3:71–76.
Virgin CE Jr, Ha TP, Packan DR, Tombaugh GC, Yang SH, HornerHC, Sapolsky RM (1991) Glucocorticoids inhibit glucose transportand glutamate uptake in hippocampal astrocytes: implications forglucocorticoid neurotoxicity. J Neurochem 57(4):1422–1428.
Vivanco I, Sawyers CL (2002) The phosphatidylinositol 3-kinase AKTpathway in human cancer. Nat Rev Cancer 2:489–501.
Wolkowitz OM, Burke H, Epel ES, Reus VI (2009) Glucocorticoids.Mood, memory, and mechanisms. Ann N Y Acad Sci 1179:19–40.
Woolley CS, Gould E, Mcewen BS (1990) Exposure to excess gluco-corticoids alters dendritic morphology of adult hippocampal pyra-midal neurons. Brain Res 531(1–2):225–231.
Xie HR, Hu LS, Li GY (2010) SH-SY5Y human neuroblastoma cell line:in vitro cell model of dopaminergic neurons in Parkinson’s disease.Chin Med J (Engl) 123(8):1086–1092.
Yu HL, Li L, Zhang XH, Xiang L, Zhang J, Feng JF, Xiao R (2009)Neuroprotective effects of genistein and folic acid on apoptosis ofrat cultured cortical neurons induced by beta-amyloid 31-35. Br JNutr 102(5):655–662.
Zhu MY, Wang WP, Bissette G (2006) Neuroprotective effects ofagmatine against cell damage caused by glucocorticoids in cul-
tured rat hippocampal neurons. Neuroscience 141(4):2019–2027.(Accepted 22 May 2011)(Available online 31 May 2011)
Copyright © 2022 FDOKUMEN




















