
Treatment of Alzheimer’s Disease with Anti-Homocysteic acid Antibody
Upload
independentCategory
view
4download
0
Biochemical and Biophysical Research Communications 319 (2004) 993–1000
BBRCwww.elsevier.com/locate/ybbrc
The retinoic acid and brain-derived neurotrophic factordifferentiated SH-SY5Y cell line as a model for Alzheimer’s
disease-like tau phosphorylation
Anne J€ams€a,a,b Kristina Hasslund,b Richard F. Cowburn,a
Anders B€ackstr€om,b and Mervi Vas€angeb,*
a Karolinska Institutet, Neurotec Department, Division of Experimental Geriatrics, Novum, S-141 86 Huddinge, Swedenb AstraZeneca R&D, Forskargatan 20, Building 212, S-151 85 S€odert€alje, Sweden
Received 6 May 2004
Available online
Abstract
The paired helical filaments of highly phosphorylated tau protein are the main components of neurofibrillary tangles (NFT) in
Alzheimer’s disease (AD). Protein kinases including glycogen synthase kinase 3 beta (GSK3b), cyclin-dependent kinase 5 (Cdk5),
and c-Jun N-terminal kinase (JNK) have been implicated in NFT formation making the use of selective kinase inhibitors an at-
tractive treatment possibility in AD. When sequentially treated with retinoic acid (RA) and brain-derived neurotrophic factor
(BDNF), the human neuroblastoma SH-SY5Y differentiates to neuron-like cells. We found that coincident with morphologically
evident neurite outgrowth, both the content and phosphorylation state of tau increased in RA-BDNF differentiated SH-SY5Y cells.
Tau phosphorylation increased at all the examined sites ser-199, ser-202, thr-205, ser-396, and ser-404, all of which are hyper-
phosphorylated in AD brain. We also investigated whether GSK3b, Cdk5 or JNK was involved in tau phosphorylation in the
differentiated SH-SY5Y cells. We found that GSK3b contributed most and that Cdk5 made a minor contribution. JNK was not
involved in tau phosphorylation in this system. The GSK3b-inhibitor, lithium, inhibited tau phosphorylation in a concentration-
dependent manner and with good reproducibility, which enables ranking of substances in this cell model. RA-BDNF differentiated
SH-SY5Y cells could serve as a suitable model for studying the mechanisms of tau phosphorylation and for screening potential
GSK3b inhibitors.
� 2004 Elsevier Inc. All rights reserved.
Keywords: SH-SY5Y cells; GSK3b; Lithium; Cdk5; JNK
Neurofibrillary tangles (NFT) and amyloid plaques
are the pivotal lesions in Alzheimer’s disease (AD). The
major components of NFTs are paired helical filaments
(PHF) of highly phosphorylated microtubule-associated
protein tau [1,2]. Unlike normal tau that maintains the
structure of microtubules, hyperphosphorylated tau se-
questers normal tau and other microtubule-associated
proteins leading to microtubule disassembly and desta-bilization of neuronal cytoskeleton [3].
Many kinases including glycogen synthase kinase 3
beta (GSK3b), cyclin-dependent kinase 5 (Cdk5), and
* Corresponding author. Fax: +46-8-553-23-840.
E-mail address: [email protected] (M. Vas€ange).
0006-291X/$ - see front matter � 2004 Elsevier Inc. All rights reserved.
doi:10.1016/j.bbrc.2004.05.075
c-Jun N-terminal kinase (JNK) are implicated in AD
NFT formation. Furthermore, there is evidence of either
increased or deregulated activity of these three kinases in
AD brain. Increased levels of GSK3b have been found
in AD brain as compared to non-diseased controls and
immunohistochemical studies have co-localized GSK3bwith NFT in AD brains [4,5]. Additionally, active
GSK3b was found to accumulate in pretangle neurons[6]. Elevated Cdk5 enzyme activity in post mortem AD
brain has been reported in two independent studies [7,8]
and there is also evidence for accumulation of p25, a
cleavage product of the Cdk5 activator p35, in AD
brains [7,9]. P25 has been reported to cause prolonged
and mislocalized activation of Cdk5 [7]. Increased Cdk5

994 A. J€ams€a et al. / Biochemical and Biophysical Research Communications 319 (2004) 993–1000
immunoreactivity was also found colocalized to NFTsin AD brain [4,10]. The activated phospho-JNK
(p-JNK) in turn was detected in NFTs in hippocampal
and cortical regions in the brains from individuals with
severe AD [11].
Specific tau phosphorylation sites have been shown
to correlate with the severity of AD pathology [12]
with the AT8 and PHF-1 epitopes showing intense
staining in advanced AD cases. The AT8 epitopecomprises phosphorylated ser-199, ser-202, and thr-
205, whereas PHF-1 recognizes tau phosphorylated at
ser-396 and ser-404 sites. GSK3b and Cdk5 are able to
phosphorylate tau at all these sites in vitro [13] whereas
stress activated protein kinase (SAPK)/JNK has
been reported to phosphorylate ser-202, thr-205, and
ser-396 [14].
This involvement of protein kinases in tau hyper-phosphorylation makes the use of selective kinase in-
hibitors an attractive treatment possibility in AD
(reviewed in [15]). To enable screening for potential ki-
nase inhibitors, a well-characterized cell model is nee-
ded. SH-SY5Y is a human neuroblastoma cell line that
upon sequential treatment with retinoic acid (RA) and
brain-derived neurotrophic factor (BDNF) differentiates
to neuron-like cells [16]. The RA-treatment inducesexpression of functional TrkB-receptors making cells
responsive to BDNF [17]. BDNF, in turn, activates
phosphatidylinositol 3-kinase (PI 3-K) and extracellular
regulated kinase (ERK) pathways that mediate survival
and neuritogenesis, respectively [18]. RA-BDNF differ-
entiated SH-SY5Y cells express neuronal markers
including neuron-specific enolase (NSE) and growth-
associated protein-43 (GAP-43), and neuronal polaritymarkers such as microtubule-associated protein 2
(MAP2) and tau [16]. Tau immunoreactivity was pre-
viously reported to increase dramatically in RA-predif-
ferentiated SH-SY5Y cells that were exposed to 7-day
treatment with BDNF as compared to undifferentiated
control cells [16].
We wanted to investigate whether differentiated SH-
SY5Y cells could serve as a model for testing selectiveinhibitors targeting kinases proposed to be involved in
aberrant tau phosphorylation in AD. First, we tested
whether a shorter differentiation protocol would induce
appearance of tau in SH-SY5Y cells. We found that
48 h BDNF treatment of RA-predifferentiated SH-
SY5Y cells was enough to increase tau content in the
cells. In addition, tau phosphorylation increased at all
the examined sites ser-199, ser-202, thr-205, ser-396,and ser-404. We also investigated whether GSK3b,Cdk5 or JNK was involved in tau phosphorylation
and found that GSK3b contributed most to the tau
phosphorylation in the differentiated SH-SY5Y cells
whereas the contribution by Cdk5 was minor. JNK
was not involved in tau phosphorylation in this
system.
Materials and methods
Cell cultures and treatments. The human neuroblastoma SH-SY5Y
cell line from European collection of cell cultures (ECACC) was used
for these experiments. SH-SY5Y cells were cultured in medium with
equal amount of minimum essential medium (MEM, Gibco) and
Nutrient Mixture Ham’s F-12 (Gibco), supplemented with 1% non-
essential amino acids (Gibco) and 0–10% heat-inactivated fetal calf
serum (FCS) (HyClone). No antibiotics were added to the medium.
Cells were plated at a density of 5.0� 103 cells/cm2 in 60mm-diameter
culture dishes (Corning) using cell medium with 10% FCS. From day 1
after plating, cells were differentiated in the presence of 10 lM all-
trans-retinoic acid (RA, Sigma–Aldrich) for 6 days in the cell medium
containing 1% FCS. Thereafter, cells were switched to serum-free
medium supplemented with 2 nM brain-derived neurotrophic factor
(BDNF, Sigma–Aldrich) in which they were grown for 48 h. Cells were
maintained at 37 �C in a humidified atmosphere containing 95% air
and 5% CO2.
Cell cultures were treated with 0.5, 1, 2, 10 or 20mM lithium
(Sigma–Aldrich), 1, 10 or 20lM Roscovitine (Biomol) or 1, 10 or
50 lM SP600125 (synthesized in-house under the name AR-
CO82659YY, AstraZeneca) for 2 h before the cells were lysed. Lithium
was diluted in distilled water as a 1M stock solution, whereas Ros-
covitine and SP600125 were prepared as 10mM and 25mM stock
solutions, respectively, in dimethyl sulfoxide (DMSO, Sigma–Aldrich).
All cultures treated with Roscovitine or SP600125 received equal
volumes of DMSO including the control cells, the final concentration
being 0.2%. UV-treatment was carried out for 20 s in UV Crosslinker
(Hoefer, UVC 500) 30min before lysis of the cells. UV light intensity
was 254 nm and the energy was 8mJ/cm2.
Western blot. Cells were lysed in buffer containing 50mM Tris–
HCl, pH 8.0, 150mM NaCl, 1% Triton X-100, 1mM EDTA, 10mM
NaF, 1mM Na3VO4, and one complete protease inhibitor cocktail
tablet (Roche Diagnostics)/10ml buffer. Cell lysates were centrifuged
at 14,000 rpm (Eppendorf 5417R) for 15min. The protein content in
the supernatants was measured using BCA Protein Assay kit (Pierce).
Samples containing 35 lg protein were resolved in 10% NuPage Bis–
Tris gels (Invitrogen). To detect p-JNK 70 lg protein was loaded in
each well. The proteins were transfered to Hybond nitrocellulose
membranes (Amersham Biosciences) using a TRANS-BLOT Semi-dry
Transfer Cell (Bio-Rad). Membranes were blocked in PBS with 0.05%
Tween 20 containing 5% non-fat dried milk for 1 h at room tempera-
ture (RT). Primary antibodies were diluted in either 5% BSA or 5%
milk and incubations were carried out at 4 �C overnight. Primary an-
tibodies were used at the following dilutions: GSK3a=b [pY279=216]
(Biosource International) 1:1500, GSK3b (Santa Cruz, H-76) 1:1500,
Cdk5 (Santa Cruz, C-8) 1:2000, p35 (Sigma) 1:1000, p-SAPK/JNK
(Cell Signaling) 1:1000, SAPK/JNK (Cell Signaling) 1:1000, Tau-5
(RDI) 1:200, Tau[pS199] 1:3000, Tau[pS202] 1:1000, Tau[pT205] 1:500,
Tau[pS396]1:4000, and Tau[pS404] 1:1000. All anti-pTau antibodies
were from Biosource International. Horseradish peroxidase (HRP)
conjugated secondary antibodies (Amersham Biosciences) were incu-
bated for 1 h at RT in 5% milk at the dilution of 1:5000 for anti-rabbit
and 1:10000 for anti-mouse antibody. Blots were developed using the
enhanced chemiluminescence (ECL) Western blotting detection system
(Amersham Biosciences). When needed, membranes were stripped with
Restore Western blot stripping buffer (Pierce) for 30min at 50 �C.Average density of the bands was measured in Fluor-S MultiIm-
ager (Bio-Rad) by using Quantity One software. Density of pTau at
the different residues was normalized to total tau levels detected with
the phosphorylation-independent Tau-5 antibody. When using phos-
phorylation-dependent and site-specific tau antibodies Tau[pS199],
Tau[pS202], and Tau[pT205] two tau species were detected, the lower one
of which was interpreted as non-phosphorylated tau since it over-
lapped with the tau band from the Tau-5 antibody. Our interpretation
is that although the phospho-antibodies are reported to be specific,

A. J€ams€a et al. / Biochemical and Biophysical Research Communications 319 (2004) 993–1000 995
they might not be specific enough for a total discrimination of p-tau
and unphosphorylated tau. Tau[pS396] and Tau[pS404] antibodies de-
tected only one tau band. The degree of phosphorylation was ex-
pressed as a percent of untreated control. All the results were from
three to four separate experiments and the data presented as mean-
s� SEM. The program Graph Pad Prism was used to calculate IC50
values and for statistical analyses. Statistical significance was deter-
mined using one way ANOVA with Dunnett’s test.
Fig. 1. Effect of differentiation on morphological appearance of SH-
SY5Y cells. (A) Undifferentiated SH-SY5Y cells exhibited roundish
features with few short processes. (B) RA alone induced modest degree
of neuronal differentiation resulting in cells that acquired a more
neuron-like phenotype with loss of the round morphology and signs of
neurite outgrowth. (C) Sequential treatment with RA and BDNF gave
rise to cells with mature neuronal morphology with neurite extensions
that occasionally connected the cells.
Results
BDNF enhances the differentiating effects of RA
Undifferentiated SH-SY5Y cells exhibited roundish
features (Fig. 1A) with few short processes. Treatment
with 10 lM RA for 6 days induced a modest degree of
neuronal differentiation resulting in cells that acquired a
more neuron-like phenotype with loss of the round
morphology and signs of neurite outgrowth (Fig. 1B).When treated sequentially with 10 lM RA for 6 days
and 2 nM BDNF for 2 days the cells displayed a mature
neuronal morphology with neurite extensions that
occasionally connected the cells (Fig. 1C).
The content and phosphorylation state of tau are
increased in RA-BDNF differentiated SH-SY5Y cells
The tau isoform detected in undifferentiated SH-
SY5Y cells was of approximately 48 kDa, which
corresponds to the molecular weight of fetal tau [19].
Treatment with either RA or RA-BDNF did not gen-
erate any additional tau isoforms (data not shown).Differentiation with RA or RA-BDNF increased tau
content in the cells although the increase was significant
only in the RA-BDNF treated cells (p < 0:05) (Fig. 2A,
n ¼ 4). Tau phosphorylation occurred only at low levels
in undifferentiated cells. A slight increase was seen in cells
treated with RA, whereas RA differentiation followed by
48 h BDNF treatment robustly increased tau phosphor-
ylation (Figs. 2B, and C). Twenty-four hour BDNFtreatment after RA differentiation was also tested but
showed similar results to RA alone (data not shown). RA
increased tau phosphorylation at all examined sites by
20–30% as compared to undifferentiated control (Fig. 2B,
n ¼ 4). A more prominent increase was seen in the
RA-BDNF differentiated cells, the increase in tau phos-
phorylation being statistically significant (p < 0:05) forser-199, ser-202, ser-396, and ser-404 (Fig. 2B,n ¼ 4).Thr-205 did not quite reach statistical significance (p ¼ 0:08).
Levels of GSK3b and p35/Cdk5 but not of JNK are
increased in RA and RA-BDNF differentiated SH-SY5Y
cells
Levels of total GSK3b and GSK3b Y216 phosphory-
lation that denotes active enzyme increased when the
cells were differentiated with RA but no additional in-
crease was seen after treatment with RA+BDNF
(Fig. 3A, n ¼ 3). Levels of p35, a neuron-specific acti-vator of Cdk5, were increased in RA differentiated cells
and an additional slight increase was seen after sub-
sequent treatment with BDNF (Fig. 3B, n ¼ 3). Levels
of Cdk5 were slightly higher in differentiated cells as
compared to undifferentiated controls (Fig. 3B, n ¼ 3).
There was, however, no difference in Cdk5 levels

Fig. 2. Differentiation of SH-SY5Y cells with RA or RA-BDNF in-
creased the content and the phosphorylation state of tau. (A) Differ-
entiation with RA increased tau with 32% whereas sequential
treatment with RA and BDNF increased tau content in the cells with
69% (p < 0:05) as compared to undifferentiated control cells. (B) RA
increased tau phosphorylation at all the examined sites by 15–25%.
More prominent increase in tau phosphorylation was seen when RA
predifferentiation was followed by 48 h BDNF treatment. Increase in
tau phosphorylation was statistically significant (p < 0:05) at ser-199,
ser-202, ser-396, and ser-404 whereas thr-205 did not quite reach sta-
tistical significance (p ¼ 0:08). (C) Representative Western blots
showing phosphorylation at ser-199, ser-202, thr-205, ser-396, and ser-
404 and total tau as assessed with Tau-5 antibody in undifferentiated,
RA and RA-BDNF differentiated SH-SY5Y cells. Lane 1, undiffer-
entiated; lane 2, RA; and lane 3 RA+BDNF. *p < 0:05.
Fig. 3. Levels of GSK3b and p35/Cdk5 but not of JNK were in-
creased in RA and RA-BDNF differentiated SH-SY5Y cells. (A)
Levels of total GSK3b and activation-associated GSK3b Y216 phos-
phorylation increased when the cells were differentiated with RA but
no additional increase was seen after subsequent treatment with
BDNF. (B) Levels of p35, a neuron-specific activator of Cdk5, were
increased in RA differentiated cells and an additional slight increase
was seen after subsequent treatment with BDNF. Similar levels of
Cdk5 were detected in RA and RA-BDNF differentiated cells and
were only slightly higher than in undifferentiated control cells. (C)
Levels of JNK were unaffected by differentiation. p-JNK was unde-
tectable in undifferentiated cells and was barely above the detection
level in RA and RA-BDNF differentiated cells as compared to UV
treated cells that showed a strong p-JNK signal. Lane 1, undiffer-
entiated; lane 2, RA; lane 3, RA+BDNF; and lane 4, RA+
BDNF+UV.
996 A. J€ams€a et al. / Biochemical and Biophysical Research Communications 319 (2004) 993–1000
between RA and RA-BDNF differentiated cells. JNK
levels were unaffected by differentiation (Fig. 3C, n ¼ 3).
P-JNK was undetectable in undifferentiated cells and
was barely above the detection level in RA andRA-BDNF differentiated cells. Cells treated with UV as
a positive control showed a strong p-JNK signal
(Fig. 3C).
GSK3b contributes most to the tau phosphorylation in
RA-BDNF differentiated SH-SY5Y cells
To pharmacologically assess the role of GSK3b,Cdk5, and JNK in tau phosphorylation in the RA-
BDNF differentiated SH-SY5Y cells, the cell cultures
were treated with kinase inhibitors: lithium for GSK3b,Roscovitine for Cdk5, and SP600125 for JNK. Initially
the cell cultures were treated for 2 and 4 h. However, 4 htreatment was discontinued since it resulted either in
unchanged (lithium) or decreased (Roscovitine and
SP600125) inhibition of tau phosphorylation as com-
pared to 2 h treatment.
Lithium at 10 and 20mM induced prominent inhi-
bition of tau phosphorylation whereas inhibition was
modest at the lowest lithium concentrations 0.5, 1, and
2mM. Inhibition of tau phosphorylation by lithium wasstatistically significant for ser-199 at the concentration
of 20mM (p < 0:001), for ser-202 at 10 and 20mM
(p < 0:001), for thr-205 at 10 and 20mM (p < 0:001),and for ser-396 at 10mM (p < 0:05) and 20mM
(p < 0:001) (Fig. 4, n ¼ 3). Lithium did not inhibit
phosphorylation significantly at ser-404, the maximal
inhibition being about 20%. The IC50 values for lithium
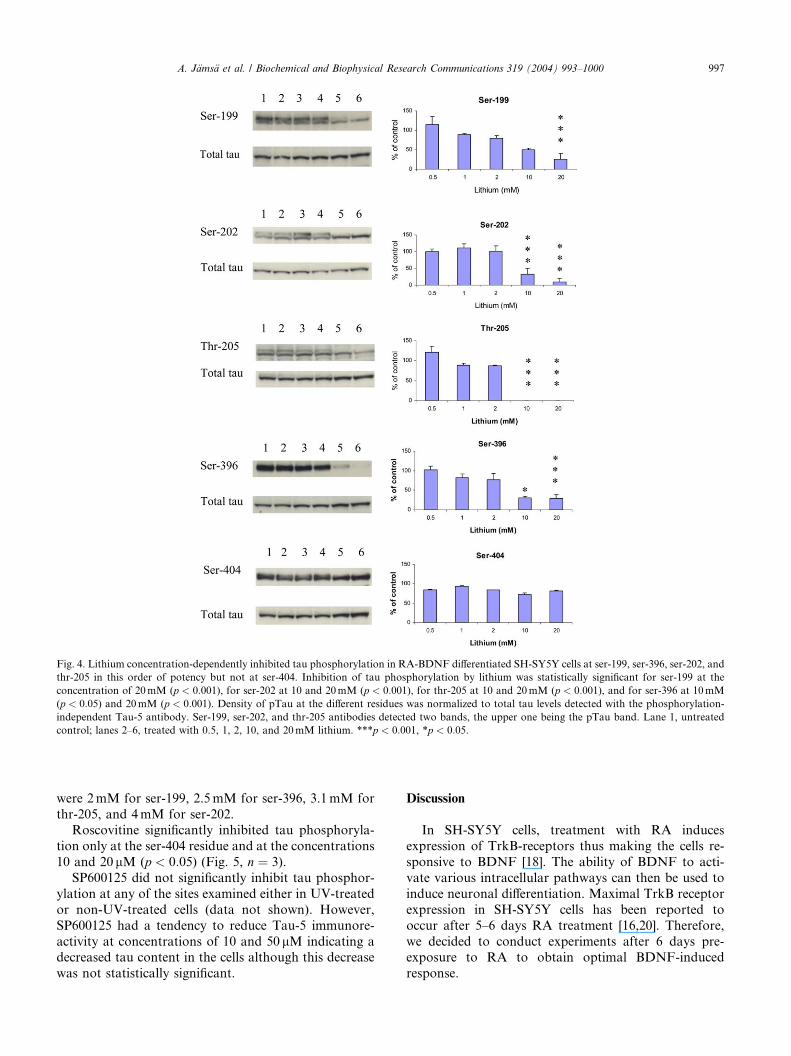
Fig. 4. Lithium concentration-dependently inhibited tau phosphorylation in RA-BDNF differentiated SH-SY5Y cells at ser-199, ser-396, ser-202, and
thr-205 in this order of potency but not at ser-404. Inhibition of tau phosphorylation by lithium was statistically significant for ser-199 at the
concentration of 20mM (p < 0:001), for ser-202 at 10 and 20mM (p < 0:001), for thr-205 at 10 and 20mM (p < 0:001), and for ser-396 at 10mM
(p < 0:05) and 20mM (p < 0:001). Density of pTau at the different residues was normalized to total tau levels detected with the phosphorylation-
independent Tau-5 antibody. Ser-199, ser-202, and thr-205 antibodies detected two bands, the upper one being the pTau band. Lane 1, untreated
control; lanes 2–6, treated with 0.5, 1, 2, 10, and 20mM lithium. ***p < 0:001, *p < 0:05.
A. J€ams€a et al. / Biochemical and Biophysical Research Communications 319 (2004) 993–1000 997
were 2mM for ser-199, 2.5mM for ser-396, 3.1mM for
thr-205, and 4mM for ser-202.Roscovitine significantly inhibited tau phosphoryla-
tion only at the ser-404 residue and at the concentrations
10 and 20 lM (p < 0:05) (Fig. 5, n ¼ 3).
SP600125 did not significantly inhibit tau phosphor-
ylation at any of the sites examined either in UV-treated
or non-UV-treated cells (data not shown). However,
SP600125 had a tendency to reduce Tau-5 immunore-
activity at concentrations of 10 and 50 lM indicating adecreased tau content in the cells although this decrease
was not statistically significant.
Discussion
In SH-SY5Y cells, treatment with RA induces
expression of TrkB-receptors thus making the cells re-
sponsive to BDNF [18]. The ability of BDNF to acti-
vate various intracellular pathways can then be used to
induce neuronal differentiation. Maximal TrkB receptor
expression in SH-SY5Y cells has been reported to
occur after 5–6 days RA treatment [16,20]. Therefore,
we decided to conduct experiments after 6 days pre-exposure to RA to obtain optimal BDNF-induced
response.
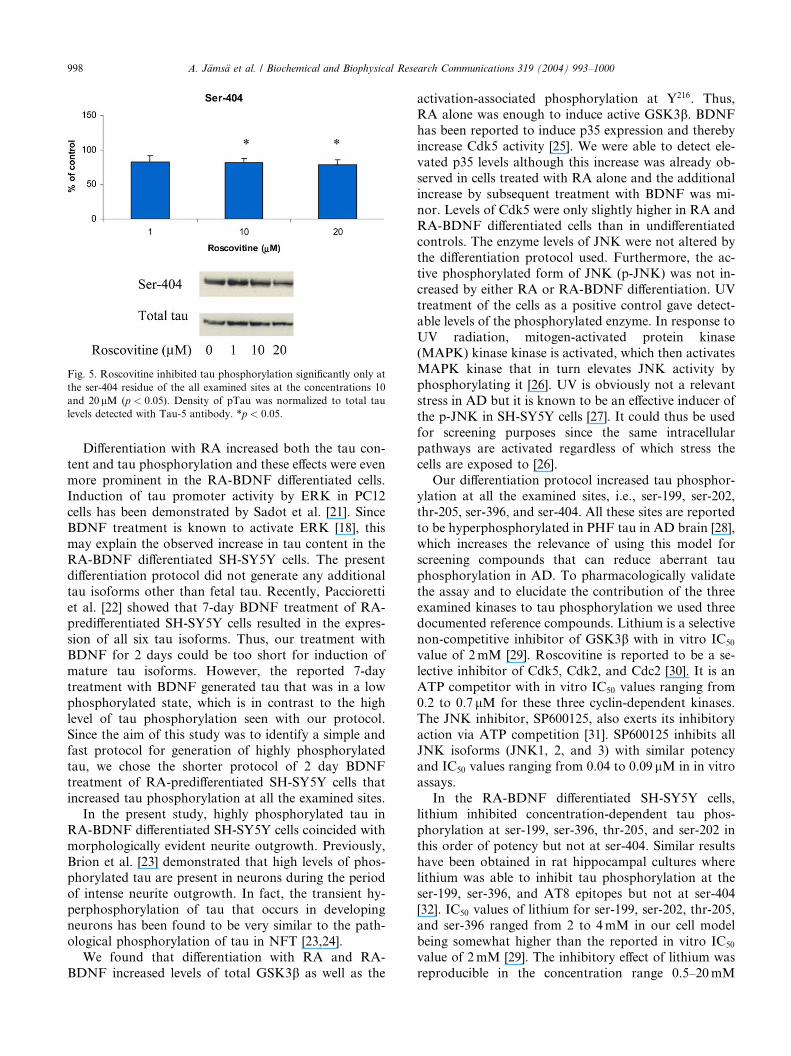
Fig. 5. Roscovitine inhibited tau phosphorylation significantly only at
the ser-404 residue of the all examined sites at the concentrations 10
and 20 lM (p < 0:05). Density of pTau was normalized to total tau
levels detected with Tau-5 antibody. *p < 0:05.
998 A. J€ams€a et al. / Biochemical and Biophysical Research Communications 319 (2004) 993–1000
Differentiation with RA increased both the tau con-
tent and tau phosphorylation and these effects were even
more prominent in the RA-BDNF differentiated cells.Induction of tau promoter activity by ERK in PC12
cells has been demonstrated by Sadot et al. [21]. Since
BDNF treatment is known to activate ERK [18], this
may explain the observed increase in tau content in the
RA-BDNF differentiated SH-SY5Y cells. The present
differentiation protocol did not generate any additional
tau isoforms other than fetal tau. Recently, Paccioretti
et al. [22] showed that 7-day BDNF treatment of RA-predifferentiated SH-SY5Y cells resulted in the expres-
sion of all six tau isoforms. Thus, our treatment with
BDNF for 2 days could be too short for induction of
mature tau isoforms. However, the reported 7-day
treatment with BDNF generated tau that was in a low
phosphorylated state, which is in contrast to the high
level of tau phosphorylation seen with our protocol.
Since the aim of this study was to identify a simple andfast protocol for generation of highly phosphorylated
tau, we chose the shorter protocol of 2 day BDNF
treatment of RA-predifferentiated SH-SY5Y cells that
increased tau phosphorylation at all the examined sites.
In the present study, highly phosphorylated tau in
RA-BDNF differentiated SH-SY5Y cells coincided with
morphologically evident neurite outgrowth. Previously,
Brion et al. [23] demonstrated that high levels of phos-phorylated tau are present in neurons during the period
of intense neurite outgrowth. In fact, the transient hy-
perphosphorylation of tau that occurs in developing
neurons has been found to be very similar to the path-
ological phosphorylation of tau in NFT [23,24].
We found that differentiation with RA and RA-
BDNF increased levels of total GSK3b as well as the
activation-associated phosphorylation at Y216. Thus,RA alone was enough to induce active GSK3b. BDNF
has been reported to induce p35 expression and thereby
increase Cdk5 activity [25]. We were able to detect ele-
vated p35 levels although this increase was already ob-
served in cells treated with RA alone and the additional
increase by subsequent treatment with BDNF was mi-
nor. Levels of Cdk5 were only slightly higher in RA and
RA-BDNF differentiated cells than in undifferentiatedcontrols. The enzyme levels of JNK were not altered by
the differentiation protocol used. Furthermore, the ac-
tive phosphorylated form of JNK (p-JNK) was not in-
creased by either RA or RA-BDNF differentiation. UV
treatment of the cells as a positive control gave detect-
able levels of the phosphorylated enzyme. In response to
UV radiation, mitogen-activated protein kinase
(MAPK) kinase kinase is activated, which then activatesMAPK kinase that in turn elevates JNK activity by
phosphorylating it [26]. UV is obviously not a relevant
stress in AD but it is known to be an effective inducer of
the p-JNK in SH-SY5Y cells [27]. It could thus be used
for screening purposes since the same intracellular
pathways are activated regardless of which stress the
cells are exposed to [26].
Our differentiation protocol increased tau phosphor-ylation at all the examined sites, i.e., ser-199, ser-202,
thr-205, ser-396, and ser-404. All these sites are reported
to be hyperphosphorylated in PHF tau in AD brain [28],
which increases the relevance of using this model for
screening compounds that can reduce aberrant tau
phosphorylation in AD. To pharmacologically validate
the assay and to elucidate the contribution of the three
examined kinases to tau phosphorylation we used threedocumented reference compounds. Lithium is a selective
non-competitive inhibitor of GSK3b with in vitro IC50
value of 2mM [29]. Roscovitine is reported to be a se-
lective inhibitor of Cdk5, Cdk2, and Cdc2 [30]. It is an
ATP competitor with in vitro IC50 values ranging from
0.2 to 0.7 lM for these three cyclin-dependent kinases.
The JNK inhibitor, SP600125, also exerts its inhibitory
action via ATP competition [31]. SP600125 inhibits allJNK isoforms (JNK1, 2, and 3) with similar potency
and IC50 values ranging from 0.04 to 0.09 lM in in vitro
assays.
In the RA-BDNF differentiated SH-SY5Y cells,
lithium inhibited concentration-dependent tau phos-
phorylation at ser-199, ser-396, thr-205, and ser-202 in
this order of potency but not at ser-404. Similar results
have been obtained in rat hippocampal cultures wherelithium was able to inhibit tau phosphorylation at the
ser-199, ser-396, and AT8 epitopes but not at ser-404
[32]. IC50 values of lithium for ser-199, ser-202, thr-205,
and ser-396 ranged from 2 to 4mM in our cell model
being somewhat higher than the reported in vitro IC50
value of 2mM [29]. The inhibitory effect of lithium was
reproducible in the concentration range 0.5–20mM

A. J€ams€a et al. / Biochemical and Biophysical Research Communications 319 (2004) 993–1000 999
lithium, which indicates that this model can be used torank substances.
Roscovitine was able to significantly reduce tau
phosphorylation only at ser-404, the maximal inhibition
being about 20%. This indicates that Cdk5 plays a minor
role in tau phosphorylation in the described system, at
least at the sites examined here. Despite the elevated
p-JNK levels in UV-treated cells, the JNK inhibitor
SP600125 did not have any significant effect on tauphosphorylation. Neither could we see any effect on tau
phosphorylation in non-UV-treated cells. As a conclu-
sion, JNK is either not involved in tau phosphorylation
in our cell system or does not phosphorylate the sites
examined here.
Although RA alone was able to increase the levels of
active GSK3b, we chose a protocol with RA and BDNF
because this treatment gave highly phosphorylated tauand thus a better signal to noise ratio for screening
purposes than RA alone. However, the prominent in-
crease in tau phosphorylation after BDNF treatment
cannot solely be attributed to GSK3b, since BDNF per
se did not increase the levels of GSK3b. In addition to
the three kinases we studied, other proline directed Ser/
Thr kinases could contribute to tau phosphorylation in
our system. For instance, ERK1/2 that is activated byBDNF [16] has been shown to be capable of phos-
phorylating tau in human neuroblastoma SK-N-SH
cells [33]. However, this enzyme was not within the
scope of the present investigation.
In conclusion, the present differentiation protocol
promotes neuron-like cell morphology and increases tau
phosphorylation at relevant epitopes for AD. The model
was thus investigated for involvement of GSK3b, Cdk5,and JNK, kinases implicated in AD tau phosphoryla-
tion. GSK3b was shown to be the only one of these
kinases with a clear involvement in tau phosphorylation.
Lithium, a GSK3b-inhibitor, was able to inhibit tau
phosphorylation in a concentration-dependent manner
with good reproducibility, which would enable ranking
of substances in this cell model. RA-BDNF differenti-
ated SH-SY5Y cells could thus serve as a suitable modelfor studying the mechanisms of tau phosphorylation
and for screening potential GSK3b inhibitors.
References
[1] I. Grundke-Iqbal, K. Iqbal, Y.C. Tung, M. Quinlan, H.M.
Wisniewski, L.I. Binder, Abnormal phosphorylation of the
microtubule-associated protein tau (tau) in Alzheimer cytoskeletal
pathology, Proc. Natl. Acad. Sci. 83 (1986) 4913–4917.
[2] K.S. Kosik, C.L. Joachim, D.J. Selkoe, Microtubule-associated
protein tau (tau) is a major antigenic component of paired helical
filaments in Alzheimer disease, Proc. Natl. Acad. Sci. 83 (1986)
4044–4048.
[3] A.C. Alonso, I. Grundke-Iqbal, H.S. Barra, K. Iqbal, Abnormal
phosphorylation of tau and the mechanism of Alzheimer neuro-
fibrillary degeneration: sequestration of microtubule-associated
proteins 1 and 2 and the disassembly of microtubules by the
abnormal tau, Proc. Natl. Acad. Sci. 94 (1997) 298–303.
[4] H. Yamaguchi, K. Ishiguro, T. Uchida, A. Takashima, C.A.
Lemere, K. Imahori, Preferential labeling of Alzheimer neurofi-
brillary tangles with antisera for tau protein kinase (TPK) I/
glycogen synthase kinase-3 beta and cyclin-dependent kinase 5, a
component of TPK II, Acta Neuropathol. (Berl) 92 (1996) 232–
241.
[5] J.J. Pei, T. Tanaka, Y.C. Tung, E. Braak, K. Iqbal, I. Grundke-
Iqbal, Distribution, levels, and activity of glycogen synthase
kinase-3 in the Alzheimer disease brain, J. Neuropathol. Exp.
Neurol. 56 (1997) 70–78.
[6] J.J. Pei, E. Braak, H. Braak, I. Grundke-Iqbal, K. Iqbal, B.
Winblad, R.F. Cowburn, Distribution of active glycogen synthase
kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease
neurofibrillary changes, J. Neuropathol. Exp. Neurol. 58 (1999)
1010–1019.
[7] G.N. Patrick, L. Zukerberg, M. Nikolic, S. de la Monte, P.
Dikkes, L.H. Tsai, Conversion of p35 to p25 deregulates Cdk5
activity and promotes neurodegeneration, Nature 402 (1999) 615–
622.
[8] K.Y. Lee, A.W. Clark, J.L. Rosales, K. Chapman, T. Fung, R.N.
Johnston, Elevated neuronal Cdc2-like kinase activity in the
Alzheimer disease brain, Neurosci. Res. 34 (1999) 21–29.
[9] H.C. Tseng, Y. Zhou, Y. Shen, L.H. Tsai, A survey of Cdk5
activator p35 and p25 levels in Alzheimer’s disease brains, FEBS
Lett. 523 (2002) 58–62.
[10] J.J. Pei, I. Grundke-Iqbal, K. Iqbal, N. Bogdanovic, B. Winblad,
R.F. Cowburn, Accumulation of cyclin-dependent kinase 5 (cdk5)
in neurons with early stages of Alzheimer’s disease neurofibrillary
degeneration, Brain Res. 797 (1998) 267–277.
[11] X. Zhu, A.K. Raina, C.A. Rottkamp, G. Aliev, G. Perry, H.
Boux, M.A. Smith, Activation and redistribution of c-jun N-
terminal kinase/stress activated protein kinase in degenerating
neurons in Alzheimer’s disease, J. Neurochem. 76 (2001) 435–441.
[12] J.C. Augustinack, A. Schneider, E.M. Mandelkow, B.T. Hyman,
Specific tau phosphorylation sites correlate with severity of
neuronal cytopathology in Alzheimer’s disease, Acta Neuropa-
thol. (Berl) 103 (2002) 26–35.
[13] F. Liu, K. Iqbal, I. Grundke-Iqbal, C.X. Gong, Involvement of
aberrant glycosylation in phosphorylation of tau by cdk5 and
GSK-3beta, FEBS Lett. 530 (2002) 209–214.
[14] C.H. Reynolds, M.A. Utton, G.M. Gibb, A. Yates, B.H.
Anderton, Stress-activated protein kinase/c-jun N-terminal kinase
phosphorylates tau protein, J. Neurochem. 68 (1997) 1736–
1744.
[15] L.F. Lau, J.B. Schachter, P.A. Seymour, M.A. Sanner, Tau
protein phosphorylation as a therapeutic target in Alzheimer’s
disease, Curr. Top. Med. Chem. 2 (2002) 395–415.
[16] M. Encinas, M. Iglesias, Y. Liu, H. Wang, A. Muhaisen, V. Cena,
C. Gallego, J.X. Comella, Sequential treatment of SH-SY5Y
cells with retinoic acid and brain-derived neurotrophic factor
gives rise to fully differentiated, neurotrophic factor-dependent,
human neuron-like cells, J. Neurochem. 75 (2000) 991–
1003.
[17] D.R. Kaplan, K. Matsumoto, E. Luracelli, C.J. Thiele, Induction
of TrkB by retinoic acid mediates biologic responsiveness to
BDNF and differentiation of human neuroblastoma cells, Neuron
11 (1993) 321–331.
[18] M. Encinas, M. Iglesias, N. Llecha, J.X. Comella, Extracellular-
regulated kinases and phosphatidylinositol 3-kinase are involved
in brain-derived neurotrophic factor-mediated survival and neu-
ritogenesis of the neuroblastoma cell line SH-SY5Y, J. Neuro-
chem. 73 (1999) 1409–1421.
[19] M. Goedert, R. Jakes, Expression of separate isoforms of human
tau proteins: correlation with the tau pattern in brain and effects
on tubulin polymerisation, EMBO J. 9 (1990) 4225–4230.

1000 A. J€ams€a et al. / Biochemical and Biophysical Research Communications 319 (2004) 993–1000
[20] L. Mai, R.S. Jope, X. Li, BDNF-mediated signal transduction is
modulated by GSK3beta and mood stabilizing agents, J. Neuro-
chem. 82 (2002) 75–83.
[21] E. Sadot, H. Jaaro, R. Seger, I. Ginzburg, Ras-signaling
pathways: positive and negative regulation of tau expression in
PC12 cells, J. Neurochem. 70 (1998) 428–431.
[22] S. Paccioretti, D. Uberti, M. Memo, A novel in vitro model of
human neurons to study tau abnormal post-transcriptional
modification, Alzheimer’s and Parkinson’s Disease Sixth Interna-
tional Conference, Book of Abstracts (2003) p. 31.
[23] J.P. Brion, J.N. Octave, A.M. Couck, Distribution of the
phosphorylated microtubule-associated protein tau in developing
cortical neurons, Neuroscience 63 (1994) 895–909.
[24] J.P. Brion, C. Smith, A.M. Couck, J.M. Gallo, B.M. Anderton,
Developmental changes in tau phosphorylation: fetal tau is
transiently phosphorylated in a manner similar to paired helical
filament-tau characteristic of Alzheimer’s disease, J. Neurochem.
61 (1993) 2071–2080.
[25] H. Tokuoka, T. Saito, H. Yorifuji, F. Wei, T. Kishimoto, S.
Hisanaga, Brain-derived neurotrophic factor-induced phosphory-
lation of neurofilament-H subunit in primary cultures of embryo
rat cortical neurons, J. Cell Sci. 113 (2000) 1059–1068.
[26] Z.G. Liu, J. Lewis, T.H. Wang, A. Cook, Role of c-Jun N-
terminal kinase in apoptosis, Methods Cell Biol. 66 (2001) 187–
195.
[27] K. Mielke, A. Damm, D.D. Yang, T. Herdegren, Selective
expression of JNK isoforms and stress-specific JNK activity in
different neural cell lines, Brain Res. Mol. Brain Res. 75 (2000)
128–137.
[28] S. Lovestone, C.H. Reynolds, The phosphorylation of tau: a
critical stage in neurodevelopment and neurodegenerative pro-
cesses, Neuroscience 78 (1997) 309–324.
[29] P.S. Klein, D.A. Melton, A molecular mechanism for the effect of
lithium on development, Proc. Natl. Acad. Sci. 93 (1996) 8455–
8459.
[30] L. Meijer, A. Borgne, O. Mulner, J.P. Chong, J.J. Blow, N.
Inagaki, M. Inagaki, J.G. Delcros, J.P. Moulinoux, Biochemical
and cellular effects of roscovitine, a potent and selective inhibitor
of the cyclin-dependent kinases cdc2, cdk2 and cdk5, Eur. J.
Biochem. 243 (1997) 527–536.
[31] B.L. Bennett, D.T. Sasaki, B.W. Murray, E.C. O’Leary, S.T.
Sakata, W. Xu, J.C. Leisten, A. Motiwala, S. Pierce, Y. Satoh,
S.S. Bhagwat, A.M. Manning, D.W. Anderson, SP600125, an
anthrapyrazolone inhibitor of Jun N-terminal kinase, Proc. Natl.
Acad. Sci. 98 (2001) 13681–13686.
[32] M. Takahashi, K. Yasutake, K. Tomizawa, Lithium inhibits
neurite growth and tau protein kinase I/glycogen synthase
kinase-3beta-dependent phosphorylation of juvenile tau in
cultured hippocampal neurons, J. Neurochem. 73 (1999) 2073–
2083.
[33] S. Guise, D. Braguer, G. Carles, A. Delacourte, C. Briand,
Hyperphosphorylation of tau is mediated by ERK activation
during anticancer drug-induced apoptosis in neuroblastoma cells,
J. Neurosci. Res. 63 (2001) 257–267.
Copyright © 2022 FDOKUMEN




















