
Deletion of retinoic acid receptor β (RARβ) impairs pancreatic endocrine differentiation
9
journal homepage: www.elsevier.com/locate/yexcr Available online at www.sciencedirect.com Research Article Deletion of retinoic acid receptor β (RARβ) impairs pancreatic endocrine differentiation Ronald J. Pérez a,1 , Yannick D. Benoit a,1 , Lorraine J. Gudas a,b,n a Pharmacology Department, Weill Medical College of Cornell University, New York, NY 10065, USA b Department of Medicine, Weill Medical College of Cornell University, New York, NY 10065, USA articleinformation Article Chronology: Received 12 March 2013 Received in revised form 30 May 2013 Accepted 31 May 2013 Available online 10 June 2013 Keywords: Retinoic acid RARβ Stem cell Endocrine Pancreas Islet cells abstract All-trans retinoic acid (RA) signals via binding to retinoic acid receptors (RARs α, β, and γ). RA directly influences expression of Pdx1, a transcription factor essential for pancreatic development and beta-cell (β-cell) maturation. In this study we follow the differentiation of cultured wild-type (WT) vs. RARβ knockout (KO) embryonic stem (ES) cells into pancreatic islet cells. We found that RARβ KO ES cells show greatly reduced expression of some important endocrine markers of differentiated islet cells, such as glucagon, islet amyloid polypeptide (Iapp), and insulin 1 (Ins1) relative to WT. We conclude that RARβ activity is essential for proper differentiation of ES cells to pancreatic endocrine cells. & 2013 Elsevier Inc. All rights reserved. Introduction In 2011 there were an estimated 366 million cases of diabetes worldwide, according to the International Diabetes Federation, and these cases are estimated to increase to 522 million by 2030 [1,2]. In the United States there were 23.7 million diagnosed cases, with an estimated healthcare cost of $113 billion [2,3]. Diabetes results when insulin production by pancreatic β-cells does not meet the metabolic demand of peripheral tissues such as the liver, fat, and muscle [4]. A reduction in β-cell number and function leads to hyperglycemia in both type 1 and type 2 diabetes [4]. In type 1 diabetes, insulin-producing pancreatic β-cells lose self-tolerance and this gives rise to hyperglycemia [5]. Each year in the United States there are over 30,000 new cases of type I diabetes diagnosed [6]. Patients with type I diabetes can control their blood glucose level with insulin supplements [7]. However, the differentiation of stem cells into pancreatic β-cells could be a long term, better solution [8,9]. Mouse embryonic stem (ES) cells are pluripotent cells derived from the inner cell mass of blastocyst-stage (day 3.5) embryos 0014-4827/$ - see front matter & 2013 Elsevier Inc. All rights reserved. http://dx.doi.org/10.1016/j.yexcr.2013.05.032 Abbreviations: ES, embryonic stem; Gcg, glucagon; Iapp, islet amyloid polypeptide; Ins1, insulin 1; KO, knockout; Ngn3, Neurogenin3; RA, all-trans retinoic acid; RAR, retinoic acid receptor; RARE, retinoic acid response element; Sst, somatostatin; WT, wild-type n Corresponding author at: Department of Pharmacology, Weill Cornell Medical College, New York, NY 10065, USA. Fax: +1 212 746 8835. E-mail address: [email protected] (L.J. Gudas). 1 These authors contributed equally to this research. EXPERIMENTAL CELL RESEARCH 319 (2013) 2196 – 2204
Transcript of Deletion of retinoic acid receptor β (RARβ) impairs pancreatic endocrine differentiation

Available online at www.sciencedirect.com
journal homepage: www.elsevier.com/locate/yexcr
E X P E R I M E N T A L C E L L R E S E A R C H 3 1 9 ( 2 0 1 3 ) 2 1 9 6 – 2 2 0 4
0014-4827/$ - see frohttp://dx.doi.org/10.1
Abbreviations: ES,RA, all-trans retinoi
nCorresponding auE-mail address: l1 These authors c
Research Article
Deletion of retinoic acid receptor β (RARβ) impairs pancreaticendocrine differentiation
Ronald J. Péreza,1, Yannick D. Benoita,1, Lorraine J. Gudasa,b,n
aPharmacology Department, Weill Medical College of Cornell University, New York, NY 10065, USAbDepartment of Medicine, Weill Medical College of Cornell University, New York, NY 10065, USA
a r t i c l e i n f o r m a t i o n
Article Chronology:
Received 12 March 2013Received in revised form30 May 2013Accepted 31 May 2013Available online 10 June 2013
Keywords:
Retinoic acidRARβStem cellEndocrinePancreasIslet cells
nt matter & 2013 Elsevier016/j.yexcr.2013.05.032
embryonic stem; Gcg, glc acid; RAR, retinoic acidthor at: Department of [email protected] (Lontributed equally to this
a b s t r a c t
All-trans retinoic acid (RA) signals via binding to retinoic acid receptors (RARs α, β, and γ).RA directly influences expression of Pdx1, a transcription factor essential for pancreaticdevelopment and beta-cell (β-cell) maturation. In this study we follow the differentiation ofcultured wild-type (WT) vs. RARβ knockout (KO) embryonic stem (ES) cells into pancreatic isletcells. We found that RARβ KO ES cells show greatly reduced expression of some importantendocrine markers of differentiated islet cells, such as glucagon, islet amyloid polypeptide (Iapp),and insulin 1 (Ins1) relative to WT. We conclude that RARβ activity is essential for properdifferentiation of ES cells to pancreatic endocrine cells.
& 2013 Elsevier Inc. All rights reserved.
Introduction
In 2011 there were an estimated 366 million cases of diabetesworldwide, according to the International Diabetes Federation,and these cases are estimated to increase to 522 million by 2030[1,2]. In the United States there were 23.7 million diagnosedcases, with an estimated healthcare cost of $113 billion [2,3].Diabetes results when insulin production by pancreatic β-cellsdoes not meet the metabolic demand of peripheral tissues such asthe liver, fat, and muscle [4]. A reduction in β-cell number
Inc. All rights reserved.
ucagon; Iapp, islet amyloireceptor; RARE, retinoic
armacology, Weill Cornell M.J. Gudas).research.
and function leads to hyperglycemia in both type 1 and type 2diabetes [4]. In type 1 diabetes, insulin-producing pancreaticβ-cells lose self-tolerance and this gives rise to hyperglycemia[5]. Each year in the United States there are over 30,000 new casesof type I diabetes diagnosed [6]. Patients with type I diabetes cancontrol their blood glucose level with insulin supplements [7].However, the differentiation of stem cells into pancreatic β-cellscould be a long term, better solution [8,9].
Mouse embryonic stem (ES) cells are pluripotent cells derivedfrom the inner cell mass of blastocyst-stage (day 3.5) embryos
d polypeptide; Ins1, insulin 1; KO, knockout; Ngn3, Neurogenin3;acid response element; Sst, somatostatin; WT, wild-typeedical College, New York, NY 10065, USA. Fax: +1 212 746 8835.

E X P E R I M E N T A L C E L L R E S E A R C H 3 1 9 ( 2 0 1 3 ) 2 1 9 6 – 2 2 0 4 2197
[9,10]. Upon LIF removal, ES cells spontaneously differentiate intoall three primary embryonic germ layers: endoderm, mesoderm,and ectoderm [9]. Several research groups have shown that thedirected differentiation of ES cells along the endocrine pathwaycan be achieved by using a wide range of growth/differentiationfactors, including retinoic acid (RA) treatment [11–16].
The effects of RA on cells and tissues are known to occurthrough RA binding and activation of retinoic acid receptors(RARα, RARβ, and RARγ) and their isoforms [17,18]. Each RARhas some specific functions and activates specific subsets of genes[19–21]. RA signaling is crucial for endocrine pancreatic develop-ment in Xenopus [22]. In addition, transgenic mice that express adominant negative RARα403 mutant, used to ablate all retinoicacid-dependent processes in vivo, lack both dorsal and ventralpancreas, and die at the neonatal stage [23]. Impaired pancreaticislet function was also observed in vitamin A deficiency andrepletion rodent models [24,25]. Another study, focused on therole of CRABP1 and RBP4 in pancreatic differentiation, showed anincrease in RARβ expression in early differentiation [10]. Whileprevious studies showed that RARβ is expressed during pancreasdevelopment, little is known about the role of RARβ in normalislet maintenance and function in adult animals [26,27].
The RARβ gene is frequently hypermethylated at CpG islands inhuman pancreatic adenocarcinoma [28] and this phenomenoncould also be associated with other pathologies, such as diabetes.We hypothesized that RARβ plays a key role in ES cell differentia-tion to pancreatic endocrine cells, and that this function of RARβmay be altered in pancreatic physiopathology. In this report, wemeasured the expression profiles of various pancreatic differen-tiation and retinoid signaling markers in WT and RARβKO ES cells.We show that the lack of all three isoforms of RARβ impairs thedifferentiation of cultured ES cells to pancreatic β-like cells.
Materials and methods
Cell culture and isolation of RARβ homozygous ES cell lines
Murine J1 wild-type ES cells were cultured as described pre-viously [29]. 129; C57BL/6 RARβ homozygous null mice were
Table 1 – Primer sequences used for RT-PCR.
Primer Application Forward sequence (5′–3′)
mIns1 RT-PCR TAGTGACCAGCTATAATCAGAGmGcg RT-PCR CCGCCGTGCCCAAGATTTTmSstn RT-PCR GAGCCCAACCAGACAGAGAAmNgn3n RT-PCR CTGCGCATAGCGGACCACAGCTTCmRARβ RT-PCR GATCCTGGATTTCTACACCGmRARγ2 RT-PCR ATGTACGACTGCATGGAATCGTmNanog RT-PCR AAAGGATGAAGTGCAAGCGGTGGmRex1 RT-PCR GAAAGCAGGATCGCCTCACTGTGCmCyp26a1 RT-PCR GAAACATTGCAGATGGTGCTTCAGmPax6 RT-PCR GCAACCCCCAGTCCCCAGTCAGAmIsl1n RT-PCR CCCGGGGGCCACTATTTGmIappn RT-PCR TGGGCTGTAGTTCCTGAAGCmPdx1 RT-PCR CTTTCCCGTGGATGAAATCCmNkx6.1 RT-PCR AGAGAGCACGCTTGGCCTATTCHPRT1 RT-PCR TGCTCGAGATGTGATGAAGG
All primers for RT-PCR are designed around introns, except those marked wi
provided by Dr. Pierre Chambon (Strasbourg-Cedex, France) [27].Mice were housed and treated according to appropriate WCMCIACUC guidelines. Blastocysts were harvested on day E3.5 andindividually cultured in ES cell medium as previously described[29] to generate RARβ KO ES cells by homologous recombination.These RARβ KO ES cells were karyotyped and shown by Southernanalysis to possess two RARβ KO alleles (not shown).
Pancreatic endocrine differentiation protocol
A slightly modified version of the established protocols publishedby Borowiak [13] and D'Amour [14] was used to carry outdifferentiation of hormone expressing endocrine cells from mouseESCs. Prior to differentiation, ESCs were seeded at 5�105 on30 mm gelatin-coated plates. After overnight culture, cells wereexposed to 250 nM BIO-Acetoxime (EMD Bioscience, San Diego,CA) +50 ng/mL activin A (R&D Systems, Minneapolis, MN) inAdvanced RPMI (GIBCO, Grand Island, NY) supplemented with1� L-Glu and 0.2% FBS (GIBCO) for 1 day, and then to activin Aalone in the same media. Cells were then cultured for 4 days toinduce endoderm differentiation. For pancreatic progenitor induc-tion, the cells were transferred to 50 ng/mL FGF10 (R&D Systems),7.5 μM cyclopamine (Calbiochem, San Diego, CA) in DMEM supple-mented with 1� L-Glu, 1X Pen/Strep, and 1X B27 (Invitrogen,Grand Island, NY) for 2 days. At day 7, cells were transferred toFGF10, cyclopamine, and 2 μM all-trans RA (Sigma, St. Louis, MO) inDMEM supplemented with 1� L-Glu, 1� Pen/Strep, and 1� B27(Invitrogen) for 4 days. At day 11, cells were cultured in thepresence of DMEM supplemented with 1� L-Glu, 1� Pen/Strep,and 1� B27 for 3 days. At day 14, CMRL (Invitrogen) medium wasadded and supplemented with 1� L-Glu, 1� Pen/Strep, 1� B27,50 ng/mL IGF-1 (R&D Systems), 50 ng/mL HGF (R&D Systems), and,in some experiments, 10 mM nicotinamide (Sigma) for 3 moredays. All stock compounds were made in either PBS or ethanol.
RT-PCR analysis
Various markers for endodermal (day 5), pancreatic progenitor(day 11), endocrine progenitor (day 14) and endocrine (day 17)
Reverse sequence (5′–3′) Product size (bp)
ACGCCAAGGTCTGAAGGTCC 289CCTGCGGCCGAGTTCCT 232GAAGTTCTTGCAGCCAGCTT 150CTTCACAAGAAGTCTGAGAACACCAG 233CACTGACGCCATAGTGGTA 248GATACAGTTTTTGTCACGGTGACAT 366CTGGCTTTGCCCTGACTTTAA 520CGATAAGACACCACAGTACACAC 641CGGCTGAAGGCCTGCATAATCAC 272AGTCCATTCCCGGGCTCCAGTTCA 399CGGGCACGCATCACGAA 397GCACTTCCGTTTGTCCATCT 199GTCAAGTTCAACATCACTGCC 205GTCGTCAGAGTTCGGGTCCAG 215TCCCCTGTTGACTGGTCATT 192
th n.

E X P E R I M E N T A L C E L L R E S E A R C H 3 1 9 ( 2 0 1 3 ) 2 1 9 6 – 2 2 0 42198
differentiation were analyzed by semi-quantitative RT-PCR in J1WT and RARβ KO ESCs. Specific primers used and amplificationconditions are listed in Table 1. Total RNA extraction, semi-quantitative, and quantitative PCR reactions were performed aspreviously described [30]. Amplified PCR products were resolvedon 1.5% agarose gels and visualized by staining with ethidiumbromide. PCR bands were sequenced for verification of the correctamplicon. Quantitation was performed using ImageJ software(National Institutes of Health) from three experimental, indepen-dent biological repeats.
Indirect immunofluorescence
Immunofluorescence assays on cells were performed as pre-viously described [31]. Briefly, differentiated samples were fixedusing 4% (w/v) paraformaldehyde and membrane permeabiliza-tion was done with 0.3% (w/v) Triton-X 100 (Sigma). Unspecificsites were blocked using 2% BSA for 30 min prior to incubationwith rabbit polyclonal anti–Pdx1 (Millipore, 06–1379, 1:1000),rabbit anti-C-Peptide (Cell Signaling, 4593, 1:500, Danvers, MA)and mouse monoclonal anti-Glucagon (Abcam, ab10988, 1:200)primary antibodies. Phalloidin-TRITC (Millipore, FAK100, 1:1000,Billerica, MA) was used to stain the actin stress fibers network (F-actin). Nuclei were stained using DAPI contained in Vectashields
mounting medium for fluorescence (Vector labs, Burlingame, CA).Quantitation of C-peptide positive stained cells and islet surfacearea was performed using NIS-Elements Advanced Research soft-ware (Nikon).
Microarray analysis
Total cellular RNA was isolated with Trizol reagent (Invitrogen),and concentration and integrity were assessed using Nanodrops
technology (Thermo Scientific). Preparation of cRNA, chip hybri-dization and scanning were carried out by the Microarray CoreFacility at Weill Cornell Medical College (WCMC). The microarrayanalyses were performed following the Affymetrix Genechipexpression analysis technical manual. The fragmented cRNA washybridized to the microarray chips (MG-430.2, Cat. #900496,Affymetrix, CA, USA), which include over 45,000 transcriptsrepresenting 34,000 substantiated mouse genes. Data analysiswas performed using the Genespring v7.0 software (AgilentTechnologies) as previously described [30].
Statistical analyses
All experiments were performed at least thrice using independentbiological triplicates. Results are presented as means7SEM. Allstatistical tests were performed using the GraphPad InStat softwareversion 3.10. A p-value of ≤0.05 indicates statistical significance.
Results
Pancreatic differentiation and assessment of pancreaticmarkers in WT murine ES cells
We selected the endocrine differentiation protocol of D'Amouret al. [14] because this protocol resulted in expression of laterstage endocrine markers using human ES cells [14]. We modified
the D'Amour et al. protocol by replacing Wnt3a with BIO-acetoxime. Wnt3a has been documented as being important formesendoderm specification and BIO-acetoxime acts in the samesignaling pathway [32,33]. Second, we included nicotinamideduring the last stage of differentiation; various published proto-cols included this reagent because of its reported effectiveness insupporting pancreatic differentiation [34,35] (Fig. 1A and B).
To characterize the differentiation of WT ES cells, we harvestedcellular extracts at various time points (Fig. 1B) and assessed themRNA levels of various differentiation markers by RT-PCR. LIFremoval combined with BIO and Activin A caused a majordecrease in transcripts of the stem cell markers Nanog [36] andRex1 (Zfp42) [37] (Fig. 1B, lane 6) compared to the levels in EScells cultured with LIF (Fig. 1B, lanes 1–4). Nanog and Rex1transcript levels remained low for the remainder of the differ-entiation process (Fig. 1B, lanes 7–12). While we observed robustexpression of transcripts for glucagon (Gcg), a functional markerof pancreatic α-cells, by day 14 (Fig. 1B, lane 8), somatostatin (Sst),a hormone secreted by δ-cells [38], was detectable as early as day5 (Fig. 1B, lane 6). Insulin-1 (Ins1), a β-cell marker [38], wasdetected by day 11 of the differentiation process and its expres-sion fluctuated depending on the presence of HGF, IGF1, or bothfactors together (Fig. 1B, lanes 9–11). The most consistentexpression of all three pancreatic endocrine differentiation mar-kers tested was observed in the presence of HGF, IGF1, andnicotinamide from days 14–17 (Fig. 1B, lane 12). As anothercontrol we cultured WT ES cells in absence of LIF for 17 daysbut with no other factors added; we observed a decrease in Nanogand Rex1 transcripts, but no induction of the differentiationmarkers tested (Fig. 1B, lane 5). For comparison, RNA from adultmouse pancreas is shown (Fig. 1B, right panel).
Thus, we demonstrated the differentiation of WT ES cells toendocrine cells capable of expressing pancreatic hormone-encoding genes. This differentiation protocol was then employedto investigate the role of RARβ at specific stages of pancreaticendocrine differentiation.
Lack of RARβ delays Pdx1 expression during pancreaticendocrine differentiation of ES cells
We next subjected WT and RARβKO ES cells to the endocrinedifferentiation protocol described above. We confirmed theabsence of RARβ transcripts in the RARβKO ES cells by RT-PCR(Fig. 2A). We detected an RA-associated increase in Cyp26a1mRNA level in both WT and RARβKO ES cells, indicating that theRARβKO ES cells can still respond to RA via RARγ [39] (Fig. 2A) toactivate Cyp26a1 transcriptionally.
We evaluated the expression profile of Pdx1, a master regulatorof pancreatic development [40], in WT and RARβKO ES cells, in thepresence or absence of RA treatment (Fig. 2B). RT-PCR experi-ments revealed lower levels of Pdx1 in RARβKO (55.2%, p¼0.047)cells compared to WT in untreated conditions (Fig. 2B and C).Interestingly, RA treatment caused a remarkable increase in Pdx1expression in both WT (�3.3-fold, p¼0.0012) and RARβKO (�4.1-fold, p≤0.0001) cells compared to untreated conditions (Fig. 2Band C). Microarray-based transcriptome analyses performed onWTand RARβKO ES cells showed an increase in RARγ gene expressionin RARβ-null cells following RA treatment (Supplementary Table S1).Although we observed no statistically significant changes in RARαexpression in RARβKO ES cells compared to WT, RT-PCR analysis

Fig. 1 – Differentiation of WT ES cells to pancreatic endocrine cells. (A) Schematic representation of the endocrine differentiationprotocol [14]. WT murine ES cells are treated with different growth factors to differentiate the cells into definitive endoderm (DE),pancreatic progenitor (PP), endocrine progenitor (EP), and endocrine cells (EC). (B) WT ES cells were subjected to the 17-daydifferentiation protocol. Each lane represents a different condition at a specific time point. RT-PCR analyses were performed tomonitor the expression of pancreatic differentiation markers such as insulin-1 (Ins1), glucagon (Gcg), somatostatin (Sst),neurogenin-3 (Ngn3), Pdx1, and Sox17, as well as the ES cell markers Nanog and Rex1. HPRT1 was used as a loading control.Pancreas extracts from C57BL/6 WT mice were used as a positive control (far right lane). Sample lanes are labeled 1–12 at thebottom. This experiment was performed seven times, starting with fresh cells, with similar results.
E X P E R I M E N T A L C E L L R E S E A R C H 3 1 9 ( 2 0 1 3 ) 2 1 9 6 – 2 2 0 4 2199
confirmed the induction of RARγ2 expression in RA-treated RARβKOES cells (Fig. 2B). The absence of RARβ, combined with low RARγ2expression, lead to reduced Pdx1 levels (Fig. 2C). Moreover, the RA-dependent RARγ2 induction observed in RARβ-null cells (�5-fold,p¼0.0162) correlates with a restoration of Pdx1 expression (Fig. 2C).
WT and RARβKO ES cells were differentiated into panc-reatic endocrine cells, as described in Fig. 1, and we performedindirect immunofluorescence staining at different time points todetermine the Pdx1 protein expression profile (Fig. 2D). Wedetected Pdx1 protein in differentiating WT cells by day 5, andPdx1 was detectable at all of the other differentiation stagestested (Fig. 2D). In contrast, Pdx1 protein was absent from nucleiof differentiating RARβKO cells at days 5 and 11, was only detectedat day 14, and was only detected at a low level (Fig. 2D). Thesedata indicate that the lack of RARβ results in a delay in the
induction of Pdx1, which could potentially affect subsequent stepsin endocrine specialization.
Absence of RARβ expression impairs the pancreaticendocrine differentiation process
Decreased expression of pluripotency factors, including Nanog, inES cells is required for proper ES cell differentiation [41].By comparing Nanog transcript levels in WT and RARβKO ES cells,we observed the sustained expression of Nanog mRNA in RARβKOcells, while in WT Nanog mRNA levels declined over time(Fig. 3A). A similar phenomenon was also observed for otherpluripotency factors, such as Pou5f1 and Sox2, in microarrayexperiments comparing WT and RARβKO ES cell transcriptomes

Fig. 2 – Impact of RARβ deletion on Pdx1 protein expression. (A) RT-PCR analysis confirming the lack of RARβ transcripts inRARβKO ES cells. Analysis of Cyp26a1, a RA-responsive gene, demonstrates the presence of RA signaling activity via other receptorsin RARβKO cells. HPRT1 was used as a loading control. (B) Representative RT-PCR analysis of Pdx1 and RARγ2 expression in WT andRARβ KO ES cells, control untreated (NT) and RA treated (1 lM, 48 h). (C) Relative amounts of Pdx1 and RARγ2, normalized toHPRT1 levels, are shown in the histogram (n¼3; n: p≤0.047; nn p¼0.001; nnn: p≤0.0001). (D) Indirect immunofluorescence stainingfor Pdx1 protein (green) in WT and RARβKO cells at 5, 11, 14, and 17 days in the absence (untreated) or in the presence (treated) ofgrowth factors used in the differentiation protocol. Cells were counterstained using rhodamine-conjugated phalloidin, whichbinds to F-actin (red), and nuclei were stained with DAPI (blue) (Bars¼50 lm).
E X P E R I M E N T A L C E L L R E S E A R C H 3 1 9 ( 2 0 1 3 ) 2 1 9 6 – 2 2 0 42200
(Supplementary Table S1 and Fig. S2). Furthermore, Neurogenin-3(Ngn3), a master transcriptional regulator during the onset ofpancreatic islet differentiation [40,42,43], displayed a transientinduction pattern in WT but was not induced in RARβKO cells(Fig. 3A). The RARβ2 isoform mRNA was expressed during this ESdifferentiation protocol in WT cells, but not in RARβ KO cells(Fig. 3A and C). Hypoxanthine phosphoribosyltransferase 1(HPRT1) mRNA levels did not change during the ES differentia-tion, and thus this transcript was used as a loading control in RNAexpression assays (Fig. 3A and C).
Like Ngn3, Paired-box 6 (Pax6) and Islet1 (Isl1) are twotranscription factors which are expressed from the intermediate(“mid”) to the terminally differentiated (“late”) stages [43–46].While we noted no differences in Pax6 expression between WTand RARβKO, we detected the delayed expression of Isl1 inRARβKO as compared to WT cells (day 14 vs. day 11) (Fig. 3B).
We also analyzed the expression of different, functional, endo-crine differentiation markers, such as glucagon (Gcg; α-cells),insulin-1 (Ins1; β-cells) and islet amyloid polypeptide (IAPP;β-cells) [14,47,48] in WT and RARβ KO cells (Fig. 3C). RARβ KO
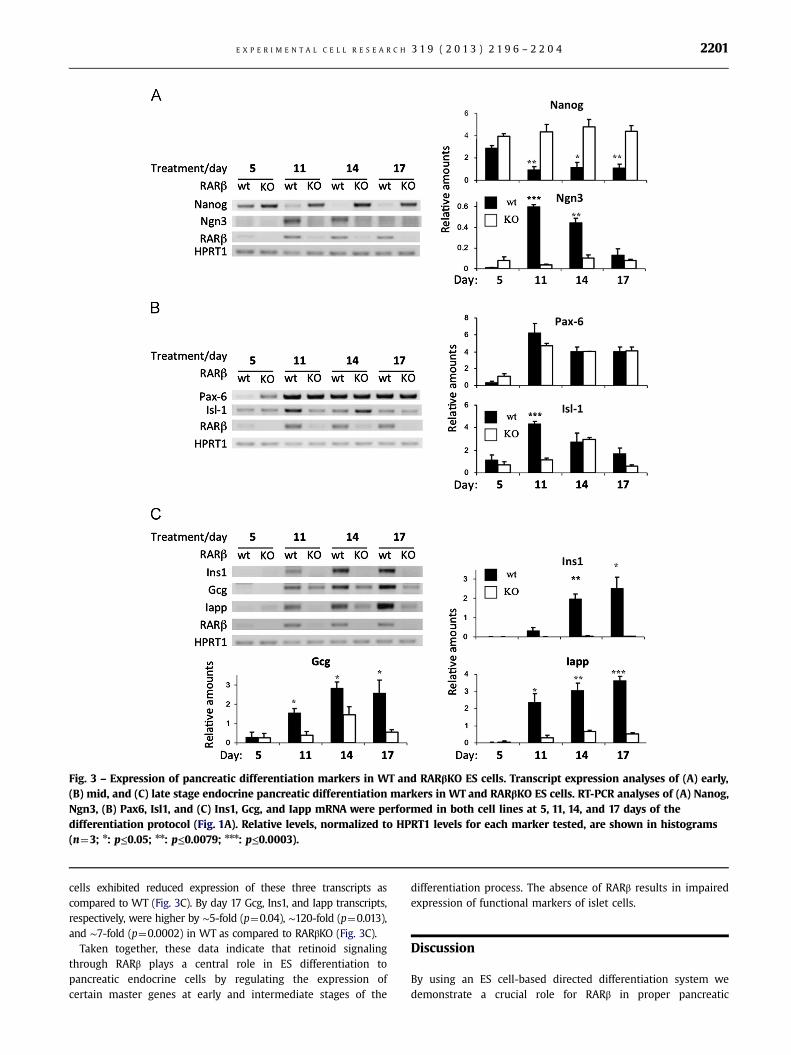
Fig. 3 – Expression of pancreatic differentiation markers in WT and RARβKO ES cells. Transcript expression analyses of (A) early,(B) mid, and (C) late stage endocrine pancreatic differentiation markers in WT and RARβKO ES cells. RT-PCR analyses of (A) Nanog,Ngn3, (B) Pax6, Isl1, and (C) Ins1, Gcg, and Iapp mRNA were performed in both cell lines at 5, 11, 14, and 17 days of thedifferentiation protocol (Fig. 1A). Relative levels, normalized to HPRT1 levels for each marker tested, are shown in histograms(n¼3; n: p≤0.05; nn: p≤0.0079; nnn: p≤0.0003).
E X P E R I M E N T A L C E L L R E S E A R C H 3 1 9 ( 2 0 1 3 ) 2 1 9 6 – 2 2 0 4 2201
cells exhibited reduced expression of these three transcripts ascompared to WT (Fig. 3C). By day 17 Gcg, Ins1, and Iapp transcripts,respectively, were higher by ∼5-fold (p¼0.04), ∼120-fold (p¼0.013),and ∼7-fold (p¼0.0002) in WT as compared to RARβKO (Fig. 3C).
Taken together, these data indicate that retinoid signalingthrough RARβ plays a central role in ES differentiation topancreatic endocrine cells by regulating the expression ofcertain master genes at early and intermediate stages of the
differentiation process. The absence of RARβ results in impairedexpression of functional markers of islet cells.
Discussion
By using an ES cell-based directed differentiation system wedemonstrate a crucial role for RARβ in proper pancreatic

E X P E R I M E N T A L C E L L R E S E A R C H 3 1 9 ( 2 0 1 3 ) 2 1 9 6 – 2 2 0 42202
endocrine cell differentiation. The absence of RARβ leads to adecrease in terminal pancreatic differentiation and to a greatreduction in expression of functional markers, such as insulin1and glucagon. We also conclude that Pdx1 expression during thepancreatic differentiation process is delayed in the absence ofRARβ (Fig. 2). Pdx1 is a key transcription factor in the earlydetermination of pancreatic progenitors and bud expansion[43,44,49,50]. RA directly induces Pdx1 expression in WT ES cells[49]. Moreover, a putative retinoic acid response element (RARE)is located at �3 kb upstream of the transcription start site of Pdx1in F9 teratocarcinoma cells by ChIP-chip analyses (unpublisheddata). We speculate that RARα and RARβ together participate inthe Pdx1 biphasic expression pattern during endocrine pancreaticdifferentiation, as reviewed by Soria [43]. However, the lack ofRARβ would need to be compensated by RARγ, which is inducedby RA treatment from day 8 through day 11 (Figs. 1A and 2B andC). This would explain the delayed Pdx1 expression we observe inthe RARβKO cells (Fig. 2D).Pdx1 mis-expression was previously associated with severe
β-cell dysfunction and increased cell death [51]. Accordingly, weshow that the lack of RARβ causes a reduction in pancreatic β-cellterminal differentiation in this cell culture system, as assessed bymarkers such as Ins1 and Iapp (Fig. 3C). Recent findings by Dalginet al. [52] in zebrafish also link RA signaling and endocrine cellfate. Our data suggest that the lack of RARβ also results in adecrease in α-cell differentiation, characterized by reducedexpression of glucagon in our cell culture system (Fig. 3C).Like Pdx1, the bHLH transcription factor Neurogenin3 (Ngn3) is
a key protein in the commitment of endoderm cells to becomepancreatic precursors [42,44,48]. Among the transcription factorsinvolved in pancreas development, Ngn3 is the earliest to beexpressed in the endocrine differentiation pathway [44,53].We found that the RARβKO ES cells displayed decreased levelsof Ngn3 transcripts during pancreatic differentiation (Fig. 3A).Pax6 and Isl1 are two transcription factors that play roles in
endocrine lineage specification after bud formation [46,54]. SincePax6 and Nkx6.1 transcript levels are not altered in the RARβKOcells (Figs. 3B and S1B) and Isl1 expression is only delayed (Fig. 3),we speculate that the absence of RA signaling through RARβ is notsufficient to abrogate completely endocrine differentiation, butmay lead to significant defects in islet cell function. Moreover, thelow, residual expression of Ngn3 mRNA detected in the RARβKOES cells could account for the low levels of Gcg and Iapp mRNAobserved in these cells (Fig. 3C).According to our observations, the directed-differentiation
protocol used in our experiments gives rise to a mixed populationof α, β, and δ-cells, considering the simultaneous detection of theirrespective markers Gcg, Ins1, and Sst (Figs. 1 and 3). Interestingly,the absence of RARβ in our model seems to specifically impair αand β-cell terminal differentiation, while Sst expression (δ-cellmarker) is unchanged in RARβKO cells (Fig. S1A).Another aspect of cell differentiation that is altered by the
absence of RARβ is the pluripotency marker Nanog; Nanogexpression persists in the mutant RARβ cells throughout theendocrine pancreatic differentiation protocol (Fig. 3A). Microarrayanalyses also highlighted the diminished capacity of RARβKO cellsto repress other pluripotency genes, such as Pou5f1 and Sox2,upon RA treatment (Fig. S2). This was accompanied by thereduced induction of early pancreatic endoderm markers, suchas Gata4, Gata6, and FoxA1 (Fig. S2) [44,55]. Thus, our data
suggest that RARβ, in addition to its role in Pdx1 transcriptionalregulation, affects other aspects of ES cell differentiation in thiscell culture system.
RARβ expression is known to depend on epigenetic regulation[56,57]. Aberrant hypermethylation of the RARβ2 promoter wasreported in different pancreatic disorders, such as cancer, dia-betes, and chronic pancreatitis [58–60]. Therefore, we suggestthat the epigenetic silencing of RARβ, combined with partialvitamin A deficiency, could play a causal role in various diseasesinvolving the pancreas, including diabetes and pancreaticadenocarcinoma.
Conclusions
The production of insulin secreting endocrine cells from ES cellsusing RA-based protocols is a promising tool for diabetic therapy.Our findings provide new insights into the role of RARβ inpancreatic endocrine differentiation.
Acknowledgments
We thank Dr. Lavoisier Ramos for providing INS-1E cells, andDr. Pierre Chambon for the RARβKO mice. We also thank WeixunWang, Drs. Kristian B. Laursen, Steve Trasino, and Naira Rezendefor their scientific input, and Tamara Weissman for editorialassistance. This research was supported by funds from WeillCornell Medical College, and in part by NIH T32 DK07313, NIHT32 CA062948, NCI R01CA043796 (to L.J.G.) and by the Fonds deRecherche en Santé du Québec (PF1-Benoit-25389) (to Y.D.B.).
Appendix A. Supporting information
Supplementary data associated with this article can be found inthe online version at http://dx.doi.org/10.1016/j.yexcr.2013.05.032.
r e f e r e n c e s
[1] L. Guariguata, D. Whiting, C. Weil, N. Unwin, et al., The Interna-tional Diabetes Federation diabetes atlas methodology for esti-mating global and national prevalence of diabetes in adults,Diabetes Res. Clin. Pract. 94 (2011) 322–332.
[2] D.R. Whiting, L. Guariguata, C. Weil, J. Shaw, et al., IDF diabetesatlas: global estimates of the prevalence of diabetes for 2011 and2030, Diabetes Res. Clin. Pract. 94 (2011) 311–321.
[3] E.S. Huang, A. Basu, M. O'Grady, J.C. Capretta, et al., Projecting thefuture diabetes population size and related costs for the U.S.,Diabetes Care 32 (2009) 2225–2229.
[4] J.M. Oliver-Krasinski, D.A. Stoffers, et al., On the origin of the betacell, Genes Dev. 22 (2008) 1998–2021.
[5] F. Waldron-Lynch, K.C. Herold, et al., Immunomodulatory therapyto preserve pancreatic beta-cell function in type 1 diabetes, Nat.Rev. Drug Discov. 10 (2011) 439–452.
[6] F. Waldron-Lynch, M. von Herrath, K.C. Herold, et al., Towards acurative therapy in type 1 diabetes: remission of autoimmunity,maintenance and augmentation of beta cell mass, NovartisFound. Symp. 292 (2008) 146–155 (discussion 155-148,202-143).

E X P E R I M E N T A L C E L L R E S E A R C H 3 1 9 ( 2 0 1 3 ) 2 1 9 6 – 2 2 0 4 2203
[7] B. Charbonnel, A. Penfornis, M. Varroud-Vial, O. Kusnik-Joinville,B. Detournay, et al., Insulin therapy for diabetes mellitus:treatment regimens and associated costs, Diabetes Metab. (2011).
[8] K.S. Zaret, M. Grompe, et al., Generation and regeneration of cellsof the liver and pancreas, Science 322 (2008) 1490–1494.
[9] G.C. Weir, C. Cavelti-Weder, S. Bonner-Weir, et al., Stem cellapproaches for diabetes: towards beta cell replacement, GenomeMed. 3 (2011) 61.
[10] J. Sui, M. Mehta, B. Shi, G. Morahan, F.X. Jiang, et al., Directeddifferentiation of embryonic stem cells allows exploration ofnovel transcription factor genes for pancreas development, StemCell Rev. 1 (2012) 1–10.
[11] A. Ben-Yehudah, C. White, C.S. Navara, C.A. Castro, D. Ize-Ludlow,B. Shaffer, M. Sukhwani, C.E. Mathews, J.R. Chaillet, S.F. Witchel,et al., Evaluating protocols for embryonic stem cell differentiationinto insulin-secreting beta-cells using insulin II-GFP as a specificand noninvasive reporter, Cloning Stem Cells 11 (2009) 245–257.
[12] P. Blyszczuk, J. Czyz, G. Kania, M. Wagner, U. Roll, L. St-Onge,A.M. Wobus, et al., Expression of Pax4 in embryonic stem cellspromotes differentiation of nestin-positive progenitor andinsulin-producing cells, Proc. Natl. Acad. Sci. U.S.A. 100 (2003)998–1003.
[13] M. Borowiak, R. Maehr, S. Chen, A.E. Chen, W. Tang, J.L. Fox,S.L. Schreiber, D.A. Melton, et al., Small molecules efficientlydirect endodermal differentiation of mouse and humanembryonic stem cells, Cell Stem Cell 4 (2009) 348–358.
[14] K.A. D'Amour, A.G. Bang, S. Eliazer, O.G. Kelly, A.D. Agulnick,N.G. Smart, M.A. Moorman, E. Kroon, M.K. Carpenter, E.E. Baetge,et al., Production of pancreatic hormone-expressing endocrinecells from human embryonic stem cells, Nat. Biotechnol. 24(2006) 1392–1401.
[15] E. Kroon, L.A. Martinson, K. Kadoya, A.G. Bang, O.G. Kelly, S.Eliazer, H. Young, M. Richardson, N.G. Smart, J. Cunningham, A.D.Agulnick, K.A. D'Amour, M.K. Carpenter, E.E. Baetge, et al.,Pancreatic endoderm derived from human embryonic stem cellsgenerates glucose-responsive insulin-secreting cells in vivo, Nat.Biotechnol. 26 (2008) 443–452.
[16] S.J. Micallef, M.E. Janes, K. Knezevic, R.P. Davis, A.G. Elefanty, E.G.Stanley, et al., Retinoic acid induces Pdx1-positive endoderm indifferentiating mouse embryonic stem cells, Diabetes 54 (2005)301–305.
[17] N.P. Mongan, L.J. Gudas, et al., Diverse actions of retinoidreceptors in cancer prevention and treatment, Differentiation 75(2007) 853–870.
[18] K. Niederreither, P. Dollé, et al., Retinoic acid in development:towards an integrated view, Nat. Rev. Genet. 9 (2008) 541–553.
[19] J.F. Boylan, D. Lohnes, R. Taneja, P. Chambon, L.J. Gudas, et al., Lossof retinoic acid receptor gamma function in F9 cells by genedisruption results in aberrant Hoxa-1 expression and differen-tiation upon retinoic acid treatment, Proc. Natl. Acad. Sci. U.S.A.90 (1993) 9601–9605.
[20] A.L. Means, L.J. Gudas, et al., The roles of retinoids in vertebratedevelopment, Annu. Rev. Biochem. 64 (1995) 201–233.
[21] V. Kashyap, L.J. Gudas, F. Brenet, P. Funk, A. Viale, J.M. Scandura,et al., Epigenomic reorganization of the clustered Hox genes inembryonic stem cells induced by retinoic acid, J. Biol. Chem. 286(2011) 3250–3260.
[22] Y. Chen, F.C. Pan, N. Brandes, S. Afelik, M. Solter, T. Pieler, et al.,Retinoic acid signaling is essential for pancreas development andpromotes endocrine at the expense of exocrine cell differentia-tion in Xenopus, Dev. Biol. 271 (2004) 144–160.
[23] M. Ostrom, K.A. Loffler, S. Edfalk, L. Selander, U. Dahl, C. Ricordi,J. Jeon, M. Correa-Medina, J. Diez, H. Edlund, et al., Retinoic acidpromotes the generation of pancreatic endocrine progenitor cellsand their further differentiation into beta-cells, PLoS One 3(2008) e2841.
[24] K.A. Matthews, W.B. Rhoten, H.K. Driscoll, B.S. Chertow, et al.,Vitamin A deficiency impairs fetal islet development and causes
subsequent glucose intolerance in adult rats, J. Nutr. 134 (2004)1958–1963.
[25] B.S. Chertow, W.S. Blaner, N.G. Baranetsky, W.I. Sivitz,M.B. Cordle, D. Thompson, P. Meda, et al., Effects of vitamin Adeficiency and repletion on rat insulin secretion in vivo andin vitro from isolated islets, J. Clin. Invest. 79 (1987) 163–169.
[26] P. Dolle, E. Ruberte, P. Leroy, G. Morriss-Kay, P. Chambon, et al.,Retinoic acid receptors and cellular retinoid binding proteins. I. Asystematic study of their differential pattern of transcriptionduring mouse organogenesis, Development 110 (1990) 1133–1151.
[27] N.B. Ghyselinck, V. Dupé, A. Dierich, N. Messaddeq, J.M. Garnier,C. Rochette-Egly, P. Chambon, M. Mark, et al., Role of the retinoicacid receptor beta (RARbeta) during mouse development, Int. J.Dev. Biol. 41 (1997) 425–447.
[28] T. Ueki, M. Toyota, T. Sohn, C.J. Yeo, J.P. Issa, R.H. Hruban,M. Goggins, et al., Hypermethylation of multiple genes inpancreatic adenocarcinoma, Cancer Res. 60 (2000) 1835–1839.
[29] E. Martinez-Ceballos, P. Chambon, L.J. Gudas, et al., Differences ingene expression between wild type and Hoxa1 knockoutembryonic stem cells after retinoic acid treatment or leukemiainhibitory factor (LIF) removal, J. Biol. Chem. 280 (2005) 16484–16498.
[30] K.B. Laursen, P.M. Wong, L.J. Gudas, et al., Epigenetic regulationby RARalpha maintains ligand-independent transcriptionalactivity, Nucleic Acids Res. 40 (2012) 102–115.
[31] Y.D. Benoit, C. Lussier, P.A. Ducharme, S. Sivret, L.M. Schnapp,N. Basora, J.F. Beaulieu, et al., Integrin alpha8beta1 regulatesadhesion, migration and proliferation of human intestinal cryptcells via a predominant RhoA/ROCK-dependent mechanism, Biol.Cell 101 (2009) 695–708.
[32] R. Spokoini, S. Kfir-Erenfeld, E. Yefenof, R.V. Sionov, et al.,Glycogen synthase kinase-3 plays a central role in mediatingglucocorticoid-induced apoptosis, Mol. Endocrinol. 24 (2010)1136–1150.
[33] T.P. Yamaguchi, S. Takada, Y. Yoshikawa, N. Wu, A.P. McMahon, et al.,T (Brachyury) is a direct target of Wnt3a during paraxial mesodermspecification, Genes Dev. 13 (1999) 3185–3190.
[34] T. Otonkoski, G.M. Beattie, M.I. Mally, C. Ricordi, A. Hayek, et al.,Nicotinamide is a potent inducer of endocrine differentiation incultured human fetal pancreatic cells, J. Clin. Invest. 92 (1993)1459–1466.
[35] N. Lumelsky, O. Blondel, P. Laeng, I. Velasco, R. Ravin, R. McKay, et al.,Differentiation of embryonic stem cells to insulin-secreting structuressimilar to pancreatic islets, Science 292 (2001) 1389–1394.
[36] I. Chambers, D. Colby, M. Robertson, J. Nichols, S. Lee, S. Tweedie,A. Smith, et al., Functional expression cloning of Nanog, apluripotency sustaining factor in embryonic stem cells, Cell 113(2003) 643–655.
[37] B.A. Hosler, G.J. LaRosa, J.F. Grippo, L.J. Gudas, et al., Expression ofREX-1, a gene containing zinc finger motifs, is rapidly reduced byretinoic acid in F9 teratocarcinoma cells, Mol. Cell. Biol. 9 (1989)5623–5629.
[38] M. Marchand, I.S. Schroeder, S. Markossian, A. Skoudy, D. Negre,F.L. Cosset, P. Real, C. Kaiser, A.M. Wobus, P. Savatier, et al., MouseES cells over-expressing the transcription factor NeuroD1 showincreased differentiation towards endocrine lineages and insulin-expressing cells, Int. J. Dev. Biol. 53 (2009) 569–578.
[39] S. Langton, L.J. Gudas, et al., CYP26A1 knockout embryonic stemcells exhibit reduced differentiation and growth arrest inresponse to retinoic acid, Dev. Biol. 315 (2008) 331–354.
[40] A.S. Bernardo, C.W. Hay, K. Docherty, et al., Pancreatic tran-scription factors and their role in the birth, life and survival ofthe pancreatic beta cell, Mol. Cell Endocrinol. 294 (2008) 1–9.
[41] V. Kashyap, N.C. Rezende, K.B. Scotland, S.M. Shaffer, J.L. Persson,L.J. Gudas, N.P. Mongan, et al., Regulation of stem cell pluripo-tency and differentiation involves a mutual regulatory circuit ofthe NANOG, OCT4, and SOX2 pluripotency transcription factors

E X P E R I M E N T A L C E L L R E S E A R C H 3 1 9 ( 2 0 1 3 ) 2 1 9 6 – 2 2 0 42204
with polycomb repressive complexes and stem cell microRNAs,Stem Cells Dev. 18 (2009) 1093–1108.
[42] J.M. Rukstalis, J.F. Habener, et al., Neurogenin3: a master reg-ulator of pancreatic islet differentiation and regeneration, Islets1 (2009) 177–184.
[43] B. Soria, et al., In-vitro differentiation of pancreatic beta-cells,Differentiation 68 (2001) 205–219.
[44] D. Van Hoof, K.A. D'Amour, M.S. German, et al., Derivation ofinsulin-producing cells from human embryonic stem cells, StemCell Res. 3 (2009) 73–87.
[45] Y. Gosmain, L.S. Katz, M.H. Masson, C. Cheyssac, C. Poisson,J. Philippe, et al., Pax6 is crucial for beta-cell function, insulinbiosynthesis, and glucose-induced insulin secretion, Mol. Endo-crinol. 26 (2012) 696–709.
[46] U. Ahlgren, S.L. Pfaff, T.M. Jessell, T. Edlund, H. Edlund, et al.,Independent requirement for ISL1 in formation of pancreaticmesenchyme and islet cells, Nature 385 (1997) 257–260.
[47] O. Naujok, F. Francini, S. Picton, C.J. Bailey, S. Lenzen, A. Jorns, et al.,Changes in gene expression and morphology of mouse embryonicstem cells on differentiation into insulin-producing cells in vitroand in vivo, Diabetes Metab. Res. Rev. 25 (2009) 464–476.
[48] R. Gasa, C. Mrejen, N. Leachman, M. Otten, M. Barnes, J. Wang,S. Chakrabarti, R. Mirmira, M. German, et al., Proendocrine genescoordinate the pancreatic islet differentiation program in vitro,Proc. Natl. Acad. Sci. U.S.A. 101 (2004) 13245–13250.
[49] J. Cai, C. Yu, Y. Liu, S. Chen, Y. Guo, J. Yong, W. Lu, M. Ding,H. Deng, et al., Generation of homogeneous PDX1(+) pancreaticprogenitors from human ES cell-derived endoderm cells, J. Mol.Cell. Biol. 2 (2010) 50–60.
[50] J. Jonsson, L. Carlsson, T. Edlund, H. Edlund, et al., Insulin-promoter-factor 1 is required for pancreas development in mice,Nature 371 (1994) 606–609.
[51] K. Fujimoto, K.S. Polonsky, et al., Pdx1 and other factors thatregulate pancreatic beta-cell survival, Diabetes Obes. Metab. 11(Suppl. 4) (2009) 30–37.
[52] G. Dalgin, A.B. Ward, T. Hao le, C.E. Beattie, A. Nechiporuk,V.E. Prince, et al., Zebrafish mnx1 controls cell fate choice in thedeveloping endocrine pancreas, Development 138 (2011) 4597–4608.
[53] A. Vetere, E. Marsich, M. Di Piazza, R. Koncan, F. Micali, S. Paoletti,et al., Neurogenin3 triggers beta-cell differentiation of retinoicacid-derived endoderm cells, Biochem. J. 371 (2003) 831–841.
[54] C. Dohrmann, P. Gruss, L. Lemaire, et al., Pax genes and thedifferentiation of hormone-producing endocrine cells in thepancreas, Mech. Dev. 92 (2000) 47–54.
[55] C. Chen, Y. Zhang, X. Sheng, C. Huang, Y.Q. Zang, et al.,Differentiation of embryonic stem cells towards pancreaticprogenitor cells and their transplantation into streptozotocin-induced diabetic mice, Cell. Biol. Int. 32 (2008) 456–461.
[56] S.M. Sirchia, M. Ren, R. Pili, E. Sironi, G. Somenzi, R. Ghidoni,S. Toma, G. Nicolo, N. Sacchi, et al., Endogenous reactivation ofthe RARbeta2 tumor suppressor gene epigenetically silenced inbreast cancer, Cancer Res. 62 (2002) 2455–2461.
[57] E.M. Youssef, M.R. Estecio, J.P. Issa, et al., Methylation and regulationof expression of different retinoic acid receptor beta isoforms inhuman colon cancer, Cancer Biol. Ther. 3 (2004) 82–86.
[58] M.G. House, J.G. Herman, M.Z. Guo, C.M. Hooker, R.D. Schulick,K.D. Lillemoe, J.L. Cameron, R.H. Hruban, A. Maitra, C.J. Yeo, et al.,Aberrant hypermethylation of tumor suppressor genes in pan-creatic endocrine neoplasms, Ann. Surg. 238 (2003) 423–431(discussion 431-422).
[59] N. Sato, N. Fukushima, R.H. Hruban, M. Goggins, et al., CpG islandmethylation profile of pancreatic intraepithelial neoplasia, Mod.Pathol. 21 (2008) 238–244.
[60] M. Volkmar, S. Dedeurwaerder, D.A. Cunha, M.N. Ndlovu,M. Defrance, R. Deplus, E. Calonne, U. Volkmar, M. Igoillo-Esteve,N. Naamane, S. Del Guerra, M. Masini, M. Bugliani, P. Marchetti,M. Cnop, D.L. Eizirik, F. Fuks, et al., DNA methylation profilingidentifies epigenetic dysregulation in pancreatic islets from type2 diabetic patients, EMBO J. 31 (2012) 1405–1426.




















