
6Hydroxydopamine (6-OHDA) induces Drp1-dependent mitochondrial fragmentation in SH-SY5Y cells
10
Original Contribution 6-Hydroxydopamine (6-OHDA) induces Drp1-dependent mitochondrial fragmentation in SH-SY5Y cells Maria Gomez-Lazaro a,b , Nina A. Bonekamp b , Maria F. Galindo a,c , Joaquin Jordán a, ⁎, Michael Schrader b a Grupo de Neurofarmacología, Department Ciencias Médicas, Facultad de Medicina, Universidad de Castilla-La Mancha-Centro Regional de Investigaciones Biomédicas, Albacete, Spain b Centre for Cell Biology and Department of Biology, University of Aveiro, Aveiro, Portugal c Unidad Pfizer-Castilla-La Mancha de Neuropsicofarmacología Translacional, Complejo Hospitalario Universitario de Albacete, Spain article info abstract Article history: Received 12 October 2007 Revised 21 February 2008 Accepted 11 March 2008 Available online 20 March 2008 Mitochondrial alterations have been associated with the cytotoxic effect of 6-hydroxydopamine (6-OHDA), a widely used neurotoxin to study Parkinson's disease. Herein we studied the potential effects of 6-OHDA on mitochondrial morphology in SH-SY5Y neuroblastoma cells. By immunofluorescence and time-lapse fluorescence microscopy we demonstrated that 6-OHDA induced profound mitochondrial fragmentation in SH-SY5Y cells, an event that was similar to mitochondrial fission induced by overexpression of Fis1p, a membrane adaptor for the dynamin-related protein 1 (DLP1/Drp1). 6-OHDA failed to induce any changes in peroxisome morphology. Biochemical experiments revealed that 6-OHDA-induced mitochondrial fragmentation is an early event preceding the collapse of the mitochondrial membrane potential and cytochrome c release in SH-SY5Y cells. Silencing of DLP1/Drp1, which is involved in mitochondrial and peroxisomal fission, prevented 6-OHDA-induced fragmentation of mitochondria. Furthermore, in cells silenced for Drp1, 6-OHDA-induced cell death was reduced, indicating that a block in mitochondrial fission protects SH-SY5Y cells against 6-OHDA toxicity. Experiments in mouse embryonic fibroblasts deficient in Bax or p53 revealed that both proteins are not essential for 6-OHDA-induced mitochondrial fragmentation. Our data demonstrate for the first time an involvement of mitochondrial fragmentation and Drp1 function in 6-OHDA-induced apoptosis. Published by Elsevier Inc. Keywords: Dynamin Mitochondria Peroxisomes Organelle dynamics Apoptosis 6-Hydroxydopamine Parkinson Bax p53 Introduction Accumulating evidence suggests that neuronal apoptosis contri- butes to the pathogenesis of acute and chronic neurodegenerative diseases in the adult brain following metabolic or neurotoxic insults [1,2]. 6-Hydroxydopamine (6-OHDA), an oxidative metabolite of dopamine, is a neurotoxin which has been broadly used to generate experimental models of Parkinson's disease [3,4]. Although there is a consensus in the ability of 6-OHDA to induce cytotoxicity in different cell types [5,6], the mechanism involved is still controversial [4]. Among the mechanisms discussed, the generation of reactive oxygen species (ROS) is the most accepted [6]. Thus, 6-OHDA can either undergo extracellular autooxidation or intracellular enzymatic oxida- tion through the monoamine oxidase type B, yielding ROS and quinolinic products [7]. The mechanisms downstream of these toxic products are complex and might involve an increase in mitochondrial outer membrane permeability (MOMP), leading to the release of cytochrome c and other proapoptotic mitochondrial proteins that activate downstream effector proteins such as the effector caspase 3 [8–10]. Mitochondria form a highly dynamic semitubular network, the morphology of which is regulated by frequent fission and fusion events as well as by movements along the cytoskeleton [11]. Although the physiological significance of mitochondrial dynamics is not fully un- derstood, growing evidence indicates that maintaining correct mito- chondrial morphology by fission and fusion is critical for cell function [11–15]. This notion is supported by recent findings that link mutations in genes encoding fission/fusion proteins to human diseases [16,17,14]. Mitochondrial fission has also been implicated in synaptic and spine plasticity in neurons [18]. A remarkable episode occurring during neuronal apoptosis is the disintegration of the semireticular mito- chondrial network into small punctiform organelles. Evidence has been presented suggesting that mitochondrial fragmentation might play an active role in apoptotic cell death [19–21], although in some models mitochondrial fission is not a prerequisite for triggering the Free Radical Biology & Medicine 44 (2008) 1960–1969 ⁎ Corresponding author. Grupo de Neurofarmacología, Departamento de Ciencias Medicas, Universidad Castilla-La Mancha-Centro Regional de Investigaciones Biomédi- cas, Avda Almansa 14, Albacete 02006, Spain. Fax: +34 967599327. E-mail address: [email protected] (J. Jordán). Abbreviations: 6-OHDA, 6-hydroxydopamine; ABC, ATP-binding cassette; DLP1/ Drp1, dynamin-related protein 1; GDAP1, ganglioside-induced differentiation- associated protein 1; MARCH5, membrane-associated ring finger (C3HC4) 5; MEF, mouse embryonic fibroblast; Mfn2, mitofusin 2; MnSOD, manganese superoxide dismutase; MOMP, mitochondrial outer membrane permeability; PMP70, 70-kDa peroxisomal membrane protein; R123, Rhodamine 123; ROS, reactive oxygen species; TEMPOL, 4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl. 0891-5849/$ – see front matter. Published by Elsevier Inc. doi:10.1016/j.freeradbiomed.2008.03.009 Contents lists available at ScienceDirect Free Radical Biology & Medicine journal homepage: www.elsevier.com/locate/freeradbiomed
Transcript of 6Hydroxydopamine (6-OHDA) induces Drp1-dependent mitochondrial fragmentation in SH-SY5Y cells

Free Radical Biology & Medicine 44 (2008) 1960–1969
Contents lists available at ScienceDirect
Free Radical Biology & Medicine
j ourna l homepage: www.e lsev ie r.com/ locate / f reeradb iomed
Original Contribution
6-Hydroxydopamine (6-OHDA) induces Drp1-dependent mitochondrialfragmentation in SH-SY5Y cells
Maria Gomez-Lazaro a,b, Nina A. Bonekamp b, Maria F. Galindo a,c, Joaquin Jordán a,⁎, Michael Schrader b
a Grupo de Neurofarmacología, Department Ciencias Médicas, Facultad de Medicina, Universidad de Castilla-La Mancha-Centro Regional de Investigaciones Biomédicas, Albacete, Spainb Centre for Cell Biology and Department of Biology, University of Aveiro, Aveiro, Portugalc Unidad Pfizer-Castilla-La Mancha de Neuropsicofarmacología Translacional, Complejo Hospitalario Universitario de Albacete, Spain
a r t i c l e i n f o
⁎ Corresponding author. Grupo de NeurofarmacologMedicas, Universidad Castilla-La Mancha-Centro Regioncas, Avda Almansa 14, Albacete 02006, Spain. Fax: +34 9
E-mail address: [email protected] (J. Jordán).Abbreviations: 6-OHDA, 6-hydroxydopamine; ABC
Drp1, dynamin-related protein 1; GDAP1, gangliosassociated protein 1; MARCH5, membrane-associatedmouse embryonic fibroblast; Mfn2, mitofusin 2; Mndismutase; MOMP, mitochondrial outer membrane pperoxisomal membrane protein; R123, Rhodamine 123TEMPOL, 4-hydroxy-2,2,6,6-tetramethylpiperidine-N-o
0891-5849/$ – see front matter. Published by Elsevier Idoi:10.1016/j.freeradbiomed.2008.03.009
a b s t r a c t
Article history:Received 12 October 2007Revised 21 February 2008Accepted 11 March 2008Available online 20 March 2008
Mitochondrial alterations have been associated with the cytotoxic effect of 6-hydroxydopamine (6-OHDA), awidely used neurotoxin to study Parkinson's disease. Herein we studied the potential effects of 6-OHDA onmitochondrial morphology in SH-SY5Y neuroblastoma cells. By immunofluorescence and time-lapsefluorescence microscopy we demonstrated that 6-OHDA induced profound mitochondrial fragmentation inSH-SY5Y cells, an event that was similar to mitochondrial fission induced by overexpression of Fis1p, amembrane adaptor for the dynamin-related protein 1 (DLP1/Drp1). 6-OHDA failed to induce any changes inperoxisome morphology. Biochemical experiments revealed that 6-OHDA-induced mitochondrialfragmentation is an early event preceding the collapse of the mitochondrial membrane potential andcytochrome c release in SH-SY5Y cells. Silencing of DLP1/Drp1, which is involved in mitochondrial andperoxisomal fission, prevented 6-OHDA-induced fragmentation of mitochondria. Furthermore, in cellssilenced for Drp1, 6-OHDA-induced cell death was reduced, indicating that a block in mitochondrial fissionprotects SH-SY5Y cells against 6-OHDA toxicity. Experiments in mouse embryonic fibroblasts deficient in Baxor p53 revealed that both proteins are not essential for 6-OHDA-induced mitochondrial fragmentation.Our data demonstrate for the first time an involvement of mitochondrial fragmentation and Drp1 function in6-OHDA-induced apoptosis.
Published by Elsevier Inc.
Keywords:DynaminMitochondriaPeroxisomesOrganelle dynamicsApoptosis6-HydroxydopamineParkinsonBaxp53
Introduction
Accumulating evidence suggests that neuronal apoptosis contri-butes to the pathogenesis of acute and chronic neurodegenerativediseases in the adult brain following metabolic or neurotoxic insults[1,2]. 6-Hydroxydopamine (6-OHDA), an oxidative metabolite ofdopamine, is a neurotoxin which has been broadly used to generateexperimental models of Parkinson's disease [3,4]. Although there is aconsensus in the ability of 6-OHDA to induce cytotoxicity in differentcell types [5,6], the mechanism involved is still controversial [4].Among the mechanisms discussed, the generation of reactive oxygenspecies (ROS) is the most accepted [6]. Thus, 6-OHDA can either
ía, Departamento de Cienciasal de Investigaciones Biomédi-67599327.
, ATP-binding cassette; DLP1/ide-induced differentiation-ring finger (C3HC4) 5; MEF,SOD, manganese superoxideermeability; PMP70, 70-kDa; ROS, reactive oxygen species;xyl.
nc.
undergo extracellular autooxidation or intracellular enzymatic oxida-tion through the monoamine oxidase type B, yielding ROS andquinolinic products [7]. The mechanisms downstream of these toxicproducts are complex and might involve an increase in mitochondrialouter membrane permeability (MOMP), leading to the release ofcytochrome c and other proapoptotic mitochondrial proteins thatactivate downstream effector proteins such as the effector caspase 3[8–10].
Mitochondria form a highly dynamic semitubular network, themorphologyofwhich is regulated by frequentfission and fusion eventsas well as by movements along the cytoskeleton [11]. Although thephysiological significance of mitochondrial dynamics is not fully un-derstood, growing evidence indicates that maintaining correct mito-chondrial morphology by fission and fusion is critical for cell function[11–15]. This notion is supported by recentfindings that linkmutationsin genes encoding fission/fusion proteins to human diseases [16,17,14].Mitochondrial fission has also been implicated in synaptic and spineplasticity in neurons [18]. A remarkable episode occurring duringneuronal apoptosis is the disintegration of the semireticular mito-chondrial network into small punctiform organelles. Evidence hasbeen presented suggesting that mitochondrial fragmentation mightplay an active role in apoptotic cell death [19–21], although in somemodels mitochondrial fission is not a prerequisite for triggering the

1961M. Gomez-Lazaro et al. / Free Radical Biology & Medicine 44 (2008) 1960–1969
apoptotic process [22]. Apoptosis can bemediated byan increase in thefission of tubular mitochondria and an inhibition of fusion, suggestingan involvement of the components of the “classical” mitochondrialfission machinery [19,23–25]. A reduction of the mitochondrialvolume had already been observed in pathological tissue [26–28],which was retrospectively found to undergo apoptosis [29–32].Furthermore, ultracondensation or fragmentation of mitochondriawas observed in a range of cell types undergoing apoptosis [33–35].Several components mediating mitochondrial fission in mammaliancells have already been identified, for example, dynamin-relatedprotein 1 (Drp1/DLP1), hFis1, MTP18 [36,37], GDAP1 [38,39], andMARCH5 [40]. Drp1 is a large cytosolic GTPase that translocates to themitochondria where it couples GTP hydrolysis with scission of themitochondrial tubule [41–43]. Its receptor at the mitochondrialmembrane is thought to be hFis1, a tail-anchored outer membraneprotein facing the cytoplasm [44–47]. Drp1 and hFis1 are also involvedin peroxisomal division and are shared by both mitochondria andperoxisomes [48].
Bax is a proapoptotic member of the Bcl-2 family that migrates tomitochondria after apoptotic insults, and it is accepted that Bax andBak are essential for MOMP [49,50]. In most cases studied, dis-integration of themitochondrial network occurs within the same timeframe as Bax activation and MOMP [51,52]. Indeed, Bax might play acritical role in the control of mitochondrial fission and morphology,since Bax colocalizes with Drp1 and Mfn2 in distinct foci, possiblyfuture scission sites, on the mitochondria after the induction of apop-tosis [53–55]. Furthermore, p53, a transcription factor that is up-regulated after DNA damage, and that translocates to the mitochon-dria during cell death [25], has been shown to increase after 6-OHDAtreatment [56].
Here we show that 6-OHDA induces profound mitochondrialfission in SH-SY5Y cell cultures, an event that precedes collapse of themitochondrial membrane potential and cytochrome c release. Weobserved that the lack of expression of Drp1, but not Bax or p53,prevented 6-OHDA-induced morphological mitochondrial changes.Furthermore, blocking mitochondrial fission by preventing Drp1expression alleviates cellular death induced by 6-OHDA.
Materials and Methods
cDNAs and antibodies
The construct pmitoGFP for the labeling of the mitochondrialmatrix was kindly provided by R. Lill (University of Marburg, Ger-many). hFis1 fused to the Myc epitope tag (Myc-hFis1) was describedpreviously [45]. pGFP-C1 and mito-DsRed2 were obtained fromClontech Laboratories, Inc. (Mountain View, CA).
Antibodies used were as follows: rabbit polyclonal antibodiesagainst PMP70, a peroxisomal membrane protein (kindly providedby A. Völkl, University of Heidelberg, Germany), anti-Cyt c (SantaCruz Biotechnology Inc., Santa Cruz), and DLP1/Drp1 (kindly providedby M.A. McNiven, Mayo Clinic, Rochester, MN). The followingmonoclonal antibodies were used: anti-tubulin DM1α (Sigma-Aldrich,St. Louis, MO), anti-MnSOD (Axxora GmbH, Loerrach, Germany), andanti-myc (Santa Cruz Biotechnology Inc.). Species-specific anti-IgGantibodies conjugated to HRP or to the fluorophores TRITC and Alexa488 were obtained from Bio-Rad (Hercules, CA) or Molecular ProbesEurope (Leiden, The Netherlands).
Cell culture and drug treatment procedures
SH-SY5Y cell cultures and mouse embryonic fibroblasts (MEFs)(WT, Bax-/-, and p53-/-) (kindly provided by Dr. M. Serrano, CNIO,Spain; see [57]) were grown in Dulbecco's modified Eagle's medium(DMEM) and Ham's F-12 supplemented with 2 mM L-glutamine,penicillin (20 units/ml), streptomycin (5 μg/ml), and 15% (v/v) fetal
bovine serum (Invitrogen, Carlsbad, CA) as previously described [57].Cells were grown in a humidified cell incubator at 37 °C under a 5%CO2 atmosphere.
Immediately before 6-OHDA (Sigma) addition, stocks of 6-OHDA(50 mM, 1000X) were made in ascorbic acid (0.01%) and added tothe culture medium to achieve the required final concentration(50 μM). In order to address an involvement of ROS in 6-OHDA-mediated mitochondrial fragmentation, we performed experimentswith “aged” (1 month) 6-OHDA stock solutions, from which ROShave been liberated by autooxidation [5]. Furthermore, dilutions of6-OHDA in culture medium were pretreated for 1 h with the anti-oxidants catalase (Sigma) (10 U/ml), ascorbic acid (Sigma) (0.01%), orTEMPOL (4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl) (Calbio-chem) (0.2 μM) before addition to the cells.
RNA interference and transfection experiments
To knock down the expression of Drp1 by RNA interference,21-nucleotide small interfering RNA (siRNA) was obtained fromDharmacon (Lafayette, CO) and annealed according to the ma-nufacturer's protocol. The siRNA sequence efficiently targetinghuman Drp1 (Accession No. NM_012062) corresponded to thecoding region 783–803 relative to the first nucleotide of thestart codon (siRNA, sense strand: 5′-UCCGUGAUGAGUAUGCUU-UdTdT-3′). Transfection of siRNA was carried out by electropora-tion. Cells grown to 90% confluence on an area of 75 cm2 (1×107
cells) were harvested by trypsination, washed in 10 ml of HBSsolution (21 mM Hepes, 137 mM NaCl, 5 mM KCl, 0.7 mM Na2HPO4,6 mM dextrose), resuspended in 0.5 ml HBS, and transferred to a0.4-cm-gap, sterile electroporation cuvette containing 10 µg ofDNA. Electroporation was performed in an Easyject Plus electro-porator (Peqlab, Erlangen, Germany) at 230 V, 150 0 µF, 25–30 msduration. After electroporation, cells were immediately resus-pended in complete medium and plated on coverslips. As a control,cells were transfected with siRNA duplexes targeting luciferase(Dharmacon). Cells were transfected with siRNA duplexes 24 hafter seeding and treated with 6-OHDA (50 μM) 72 h after elec-troporation, when tubular peroxisomes reached their maximum[58]. Successful Drp1 silencing was assayed by immunofluores-cence and immunoblotting. For transfection with plasmids, cellswere plated at a density of 5.3×104 cells/cm2 on poly-L-lysine-coated glass coverslips. After 24 h cells were transfected withLipofectamine reagent (Invitrogen) according to the manufacturer'sprotocol. After 4 h, the transfection mixture was replaced by freshcomplete medium, and the cells were treated with 6-OHDA.
Fluorescence microscopy
Cells grown on glass coverslips were washed twice with PBS,pH 7.35, fixed with 4% paraformaldehyde in PBS, pH 7.35, for 20 minat RT and then washed three times with PBS. Next, cellular mem-branes were permeabilized by incubation in 0.2% Triton-X100 for10min at RT. Cells werewashed three times in PBS and then incubatedfor 10 min with 1% BSA. After washing once with PBS, cells wereincubated for 1 h with primary antibodies diluted in PBS. After ex-tensive washing in PBS, cells were incubated with fluorescentlylabeled secondary antibodies diluted in PBS (Alexa-488 or TRITC),washed an additional 3 times, and mounted in Mowiol 4-88 contain-ing n-propylgalate as described [59]. Samples were examined using anOlympus BX-61 fluorescence microscope (Olympus Optical Co. GmbH,Hamburg, Germany) equipped with the appropriate filter combina-tions and a 100× objective (Olympus Plan-Neofluar; numerical aper-ture, 1.35). Fluorescence images were acquired with a F-view II CCDcamera (Soft Imaging System GmbH, Münster, Germany) driven bySoft imaging software. Digital images were optimized for contrast andbrightness using Adobe Photoshop software.

Fig. 1. Effects of hFis1 overexpression and 6-OHDA on the mitochondrial fission pattern. SH-SY5Y cell cultures were fixed with 4% paraformaldehyde and mitochondrial morphologywas assessed by immunofluorescence microscopy using antibodies against the mitochondrial matrix protein MnSOD. (A) Control cultures exhibit mainly mitochondria with afilamentous morphology. (B,C) SH-SY5Y cells overexpressing hFis1-myc for 24 h (B) or treated with 6-OHDA (6 h, 50 μM) presented dramatic fragmentation of mitochondria (C). Theimages shown are representative of results obtained in four separate experiments, each performed in triplicate.
1962 M. Gomez-Lazaro et al. / Free Radical Biology & Medicine 44 (2008) 1960–1969
Mitochondrial morphology
To examine mitochondrial morphology we used Mitotracker Red-CMX-ROS staining (Invitrogen). Cells grown on 24-mm-diameterpoly-L-lysine-coated glass coverslips were stained with 300 nMMitoTracker Red CMX-ROS for 30 min in Hepes buffer with thefollowing ionic composition (in mM): NaCl 140, KCl 5.9, MgCl2 1.2,Hepes 15, glucose 10, CaCl2 2.5, pH 7.4. Cells were thenwashed in freshHepes buffer and mounted in a chamber for confocal microscopy. Theexcitation/emission wavelengths for MitoTracker Red CMX-ROS were578/599 nm. Images were captured with a Leica DMIRE2 confocalmicroscope, using a 63× 1.4 NA objective. Digital images wereoptimized for contrast and brightness using Adobe Photoshop soft-ware. To quantify the occurrence of mitochondrial fission, we countedthe number of cells with fragmented mitochondria of each coversliptotalling ∼150 cells/coverslip. The percentage of cells with fragmentedmitochondria was determined on three coverslips for each conditionand normalized to parallel controls. Each independent coverslip wastreated as a single observation. The average relative percentage of cellswith fragmented mitochondria from at least three separate cultureswas determined. Results are expressed as mean±SE values, and
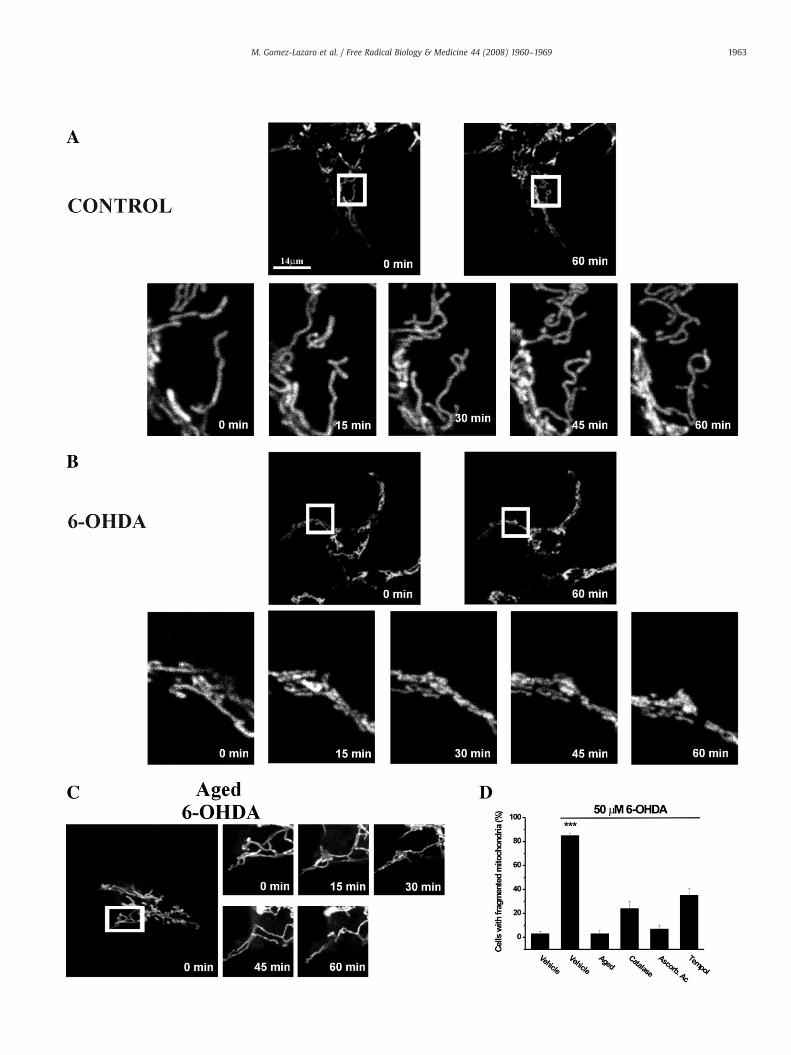
Fig. 2. Time course of 6-OHDA-induced mitochondrial fission. SH-SY5Y cells were transfmonitored by confocal laser scanning microscopy. Zoomed confocal time-lapse images wereand fresh (B) or “aged” (C) 50 μM 6-OHDA. Only fresh 6-OHDA (B,D) was able to induce changfragmentation. 6-OHDA (50 μM) was exposed to the antioxidants catalase (10 U/ml), ascorbicCells exhibiting fragmented mitochondria were counted 3 h after the addition of 6-OHDA. Reat least five experiments, each performed in triplicate.
significance was determined by Student's t test. Statistical significancewas considered at the Pb0.05 level.
Assessment of apoptotic cell death
To assess apoptotic cell death, SH-SY5Y cells were plated on poly-L-lysine-coated glass coverslips, and were treated with 6-OHDA(50 μM) for 1, 3, 6, and 24 h. Nuclei were stained with 0.5 µg/ml ofHoechst 33342 in PBS for 5 min. Cells were examined with an UVillumination microscope (Leica DMRXA). Uniformly stained nucleiwere scored as healthy, viable cells. Condensed or fragmented nucleiwere scored as apoptotic. Viable and dead cells were counted onadjacent fields of each coverslip totaling ∼350–450 cells. Thepercentage of dying cells was determined on four coverslips foreach condition and normalized to parallel controls. Each independentcoverslip was treated as a single observation, and at least fourcoverslips were used in each experiment. The average relativepercentage of apoptotic cells from at least three separate cultureswas determined. Results are expressed as mean±SE values, andsignificance was determined by Student's t test. Statistical signifi-cance was considered at the Pb0.05 level.
ected with DsRed2-mito and after 24 h changes in mitochondrial morphology wereacquired every 15 min for 1 h from cells cultured under different conditions: Control (A),es in mitochondrial morphology. (D) ROS participate in 6-OHDA-induced mitochondrialacid (0.01%), and TEMPOL (0.2 μM) for 1 h before the addition to cultured SH-SY5Y cells.sults are given as percentage of total cells (mean±SE). Experiments are representative of
1963M. Gomez-Lazaro et al. / Free Radical Biology & Medicine 44 (2008) 1960–1969

Fig. 3. 6-OHDA failed to induce changes in peroxisome morphology. SH-SY5Y cells were treated with 50 μM 6-OHDA for 3 h and fixed with 4% paraformaldehyde.Immunofluorescence microscopy using antibodies to PMP70, a peroxisomal membrane protein, revealed no apparent morphological changes of peroxisomes (1 and 3) while MnSODimmunofluorescence revealed changes in mitochondrial morphology (4). Expression of Pex11pβ-myc induced peroxisomal elongation (5), constriction, and division into smallspherical peroxisomes (6), indicating that SH-SY5Y cells are capable of multiplying their peroxisomes by growth and division.
1964 M. Gomez-Lazaro et al. / Free Radical Biology & Medicine 44 (2008) 1960–1969
Cytochrome c determination
Cytosolic extracts from nontreated and 6-OHDA-treated cultureswere obtained as previously described [60]. Cells were washed oncewith PBS and collected by centrifugation (2000 ×g; 5 min). The cellpellet was resuspended in 200 μl of extraction buffer containing(mM): sucrose 250, Tris–HCl 50, EGTA 1, EDTA 1, DTT 1, PMSF 0.1, pH7.4. Cells were homogenized in a Teflon-glass homogenizer (fivestrokes) and, after 15 min on ice, the suspension was centrifuged(15,000 ×g; 15 min). The cytoplasmic supernatant was removed andstored at - 80 °C until analysis by gel electrophoresis. The amount of15 μg of protein from control and treated samples was run on 15%SDS–polyacrylamide gels and transferred to a PVDF membrane, thatwas incubated with anti-cytochrome c (1:1000 dilution of rabbitpolyclonal IgG, Santa Cruz Biotechnology Inc.). Mitochondrial con-tamination was addressed with an anti-cytochrome c oxidase subunitIV antibody (COX-IV; BD Biosciences). The signal was detected usingan enhanced chemiluminescence detection kit (Amersham ECL RPN2106 Kit, GE Healthcare, Waukesha, WI).
Mitochondrial potential
For the determination of mitochondrial membrane potential,SH-SY5Y cells were gently resuspended in PBS, collected by centrifuga-tion at 300 ×g. Control and 6-OHDA-treated cells were resuspended inPBS (0.5×106cells/ml) and incubated for 30min at 37°CwithRhodamine123 (0.004 μg/ml) (Molecular Probes, Inc.). After twowashes in PBS, thefluorescence was measured in an EPICS flow cytometer (BDLSR,, BectonDickinson), and results were analyzed in real time using the BD
CellQuest Pro programand displayed as two-parameter histograms: cellnumber versus fluorescence.
Gel electrophoresis and immunoblotting
Protein samples were separated by SDS-PAGE, transferred tonitrocellulose or PVDF membranes using a semidry apparatus andanalyzed by immunoblotting. Immunoblots were processed usingHRP-conjugated secondary antibodies and enhanced chemilumines-cence reagents (Amersham Corp., Arlington Heights, IL).
Results
6-OHDA induces mitochondrial fragmentation in SH-SY5Y cells
In order to study the potential effects of 6-hydroxydopamine onmitochondria in SH-SY5Y cells, we first characterized the normalmitochondrial morphology in these cells. SH-SY5Y cells were pre-pared for indirect immunofluorescence and stained with specificantibodies to MnSOD, a mitochondrial matrix protein. As shown inFig. 1A, mitochondria in control cells exhibit an elongated-tubular andfilamentous morphology and show an even distribution within thecytoplasm and along cellular processes. In these cultures less than 5%of the cells present mitochondrial fragmentation hallmarks. To de-monstrate that mitochondrial fission can be induced in SH-SY5Y cells,and to analyze the typical fission pattern in these cells, we over-expressed Myc-tagged hFis1, a known inducer of mitochondrial di-vision [45]. At 24 h after hFis1 transfection SH-SY5Y cells presentedmitochondria with striking fission hallmarks including small and

1965M. Gomez-Lazaro et al. / Free Radical Biology & Medicine 44 (2008) 1960–1969
spherical structures (Fig. 1B). Next we exposed cells to 50 μM 6-OHDAfor 6 h. Remarkably, exposure to 6-OHDA triggered a dramatic frag-mentation of mitochondria (Fig. 1C).
To track the effects of 6-OHDA on single mitochondria in real time,SH-SY5Y cells were transfected with a vector encodingmito-DsRed2, ared fluorescent protein variant specifically labeling the mitochondrialmatrix (Fig. 2). Fluorescence time-lapse microscopy revealed thatmitochondria of nontreated cells exhibit a typical elongated morphol-ogy during the course of the experiment (Fig. 2A). By 1 h after theaddition of 50 μM 6-OHDA, we observed a pronounced fragmentationof single elongated mitochondria into small and spherical organelles(Fig. 2B). Once initiated, mitochondrial fragmentation is fast andsynchronous in the 6-OHDA-treated cultures. At 3 h after 6-OHDAaddition, around 85% (±6%) of the cells showed fragmented mito-chondria with altered morphology.
To provide evidence for an involvement of ROS in 6-OHDA-mediated mitochondrial fragmentation, we performed experimentswith “aged” 6-OHDA, from which ROS have been liberated by auto-oxidation [5], and with 6-OHDA pretreated with several antioxidants(Fig. 2D). “Aged” 6-OHDA was unable to induce mitochondrial frag-mentation (Figs. 2C and D). Interestingly, addition of the antioxidantscatalase (10 U/ml), ascorbic acid (0.01%), or TEMPOL (0.2 μM) reducedthe promoting effect of 6-OHDA on mitochondrial fragmentation(Fig. 2D). These findings support the involvement of ROS in 6-OHDA-mediated mitochondrial fragmentation in SH-SY5Y cells.
Recent studies have revealed that mitochondria and peroxisomesshare components of their division machinery, for example, Drp1 andhFis1 [48].We therefore examined the effect of 6-OHDAonperoxisomemorphology in SH-SY5Y cells. Peroxisomes in controls and 6-OHDA-treated cells were stained with specific antibodies to PMP70, an ABCtransporter of the peroxisomal membrane. Peroxisomes in SH-SY5Y
Fig. 4.Mitochondrial fission is not related tomitochondrial membrane potential collapse and3 h and stained with Rhodamine 123 (R123). (A) Mitochondrial morphology changes wereR123. SH-SY5Y cells were treated with 6-OHDA (50 μM) for 3 h. Statistical analysis of the Rcultures. (C) 6-OHDA-induced cytochrome c release is not evident by the time mitochondrialdifferent time points were subjected to gel electrophoresis and immunoblotting with an antcontamination. The figure shows a representative experiment that was repeated three time
cells were mainly of spherical and/or rod-shaped morphology (Fig. 3).Treatment with 50 μM 6-OHDA failed to induce any changes inperoxisome morphology, while mitochondria showed evident frag-mentation (Fig. 3, lower panels). However, typical hallmarks ofperoxisomal growth and division, such as elongation, constriction,and division into small, spherical peroxisomes were observed afterexpression of Pex11pβ (Fig. 3D). This peroxin is involved inperoxisomeproliferation and in the regulation of peroxisome abundance [59]. Inaddition, small rod shaped and “beads on a string”-like peroxisomeswere observed in untreated SH-SYS5 cells, which are indicative forgrowth anddivision (not shown) [58]. These observations demonstratethat SH-SYS5 cells are capable of multiplying their peroxisomes bygrowth and division (see also Fig. 5B).
Mitochondrial fragmentation precedes the collapse of mitochondrialmembrane potential and cytochrome c release
To establish the temporal relationship between 6-OHDA-inducedmitochondrial fragmentation and events linked to cell death, we as-sayed the collapse of the mitochondrial trans-membrane potential aswell as the release of cytochrome c.
A collapse of the mitochondrial trans-membrane potential hasbeen linked to several models for apoptosis [61]. To examine if thisevent accompanies mitochondrial fragmentation, SH-SY5Y cells wereincubated with Rhodamine 123 (R123), a fluorescent dye that accu-mulates rapidly and selectivelywithinmitochondria depending on themembrane potential. Its uptake is directly proportional to the latterparameter, and was assayed by flow cytometry. Statistical analysis ofthe R123 fluorescence intensity of 10,000 cells/condition showed nodifferences between control and 50 μM 6-OHDA-treated samples after3 and 6 h of treatment (102±2.3% and 90±1.14%, respectively, of R123
cytochrome c release. SH-SY5Y cell cultures were treated or not with 50 μM6-OHDA foranalyzed by confocal microscopy. (B). Flow cytometry of SH-SY5Y cells incubated with123 fluorescence intensity showed no difference between control and 6-OHDA-treatedfission was observed. Cytoplasmic extracts from SH-SY5Y cells treated with 6-OHDA foribody to cytochrome c. COX-IV protein levels were addressed to monitor mitochondrials with similar results.

1966 M. Gomez-Lazaro et al. / Free Radical Biology & Medicine 44 (2008) 1960–1969
fluorescence level of controls, n=5). However, at these time pointsmitochondrial fragmentation was evident (Figs. 4A and B). After 12 h6-OHDA (50 μM) was observed to induce significant changes inmitochondrial membrane potential in SH-SY5Y cells (67±2.3% of R123fluorescence level of controls, Pb0.001, n=5).
We then examined the release of cytochrome c frommitochondria.We have recently shown that 6-OHDA induces cytochrome c releasefrom mitochondria in SH-SY5Y cells [62]. Immunoblotting of cyto-
Fig. 5. Mitochondrial fragmentation on 6-OHDA treatment is abolished by silencing of Drp1.processed for immunofluorescence after 72 h. (A) Immunoblots of cell lysates prepared fromEqual amounts of protein (40 µg/lane) were loaded onto the gels. (B,C) Immunofluorescencemembrane protein PMP70 (B) and the mitochondrial matrix protein MnSOD (C). (D) MitochoCells were processed for immunofluorescence using antibodies toMnSOD. (E) Percentage of cand cells silenced for Drp1. Data are presented as mean±SE of percentage of control SH-SY5after silencing of Drp1. Percentage of apoptotic nuclei after Hoechst 33342 staining of 6-Opresented as mean±SE of at least five different experiments, each performed in triplicate (⁎
plasmic fractions revealed that after 3 h of 6-OHDA treatment mito-chondrial fragmentationwas not accompanied by a prominent releaseof cytochrome c from mitochondria (Fig. 4B). Cytochrome c wasdetected in the cytoplasmic fraction only after 9 h of 6-OHDA expo-sure. In summary, our data indicate that 6-OHDA-induced mitochon-drial fragmentation is an early event preceding the collapse of thetrans-membrane potential and mitochondrial cytochrome c release inSH-SY5Y cells.
SH-SY5Y cells were transfected with Drp1 siRNA duplexes by electroporation and werecontrols and transfected cells (siRNA) using anti-Drp1 and anti-tubulin (Tub) antibodies.microscopy of control and Drp1-silenced cells using antibodies against the peroxisomalndrial morphology in control and 6-OHDA-treated cells without or with Drp1 silencing.ells with fragmentedmitochondria at 3 h after the addition of 50 μM6-OHDA in controlsY cells (n=3) (⁎⁎⁎Pb0.001). (F) 6-OHDA-induced cell death is reduced in SH-SY5Y cellsHDA (50 μM)-treated and nontreated SH-SY5Y cells (3 and 24 h treatment). Data arePb0.05; ⁎⁎⁎Pb0.001).

Fig. 6. Loss of Bax or p53 does not inhibit 6-OHDA-induced mitochondrial fragmenta-tion. Embryonic fibroblast cultures from Bax-/- or p53-/- mice were treated for 3 h with6-OHDA (50 μM). MEF were incubated with MitoTracker Red and mitochondrialmorphology was analyzed by confocal microscopy. Confocal images were capturedusing a 63× oil immersion objective. MitoTracker Red staining revealed mainly typicalelongated filamentous morphology of mitochondria in controls (left panels), whereasafter 3 h of 50 μM6-OHDA treatment a small and round pattern is evident (right panels)in wild-type (wt) (A), Bax-/- (B), and p53-/- (C) MEF. The images shown arerepresentative of results obtained in four separate experiments, each performed intriplicate.
1967M. Gomez-Lazaro et al. / Free Radical Biology & Medicine 44 (2008) 1960–1969
Dynamin-like protein 1 is required for 6-OHDA-induced mitochondrialfragmentation
To ascertain if the 6-OHDA-induced mitochondrial fragmentation ismediated by components of the mitochondrial fission machinery, weperformed RNA interference (RNAi) experiments to reduce Drp1 mRNAand protein levels in SH-SY5Y cells [41–43,63,64]. DLP1/Drp1 siRNAduplexes specific for human Drp1 were transfected into SH-SY5Y cellsusing a recently described electroporation protocol [63,65]. Cells wereprocessed for immunofluorescence microscopy and immunoblotting72 h after transfection. Immunoblots of cell homogenates demonstratedthat the Drp1 protein level was reduced to 20–30% of the control level(Fig. 5A). A significant reduction of Drp1 protein level was not observedin controls transfected with a luciferase siRNA duplex (Fig. 5A). Efficientsilencing of Drp1 can easily be monitored and quantified by a mor-phological analysis of the peroxisomal (and mitochondrial) compart-ment. Silencing of Drp1, which has recently been shown to be involvedin the fission process of peroxisomes (and mitochondria) [63,65], leadsto a pronounced elongation of peroxisomes (and mitochondria). Afterimmunofluorescence microscopy using an antibody to PMP70, elon-gated peroxisomes were observed in 90% of the SH-SY5Y cells silencedfor Drp1 compared to none in control cell cultures, indicating that thetransfection and knock down efficiencies were high (Fig. 5B, see also[63]). Consistent with a role for Drp1 in mitochondrial fission,immunostaining for mitochondrial MnSOD also revealed elongatedand entangled mitochondrial tubules in cells silenced for Drp1 (Fig. 5B).Silenced SH-SY5Y cells were now treated with 50 μM 6-OHDA.Interestingly, cells lacking Drp1 maintained elongated mitochon-dria after 6-OHDA treatment for 3 h (Figs. 5D and E). An inhibition of6-OHDA-inducedmitochondrial fragmentationwas also evident at latertime points after 6-OHDA addition (6, 12, and 24 h; data not shown).
Next, we investigated the effect of Drp1 silencing on 6-OHDA-induced cell death. We used the DNA-binding dye Hoechst 33342 toanalyze the effects of 6-OHDA on cell viability. Three days aftertransfection with the Drp1 siRNA the cells and appropriate controlswere exposed to 50 μM 6-OHDA or vehicle, and cell viability wasassayed (after 1, 3, 6, and 24 h). At 24 h after the addition of 6-OHDA,cell death in SH-SY5Y cell cultures was evidenced by condensed orfragmented chromatin. However, in cells silenced for Drp1 cell deathwas reduced (Fig. 5F), indicating that a reduced expression andprotein level of Drp1, and thus, reducedmitochondrial fission, protectsSH-SY5Y cells against 6-OHDA toxicity.
We would like to point out that 6-OHDA treatment per se did notinduce changes in the cellular protein levels of Drp1 at any of thetimes tested (3, 6, and 12 h) (not shown). It has been previouslyreported that an increase in the expression of Drp1, which is abundantin the cytosol, does not induce mitochondrial (or peroxisomal) fission,indicating that mitochondrial (or peroxisomal) fragmentation ismainly mediated by the recruitment of Drp1 to the organelle mem-brane as well as by Drp1 assembly/oligomerization and regulation ofits activity [45–48,65].
Bax and p53 are not involved in 6-OHDA-induced mitochondrial fission
Bax protein is related to mitochondrial fission events and mightparticipate in the activation of Drp1 [66–68]. Thus, on induction ofapoptotic cell death, Bax translocates to mitochondria in foci where itcolocalizes with mitochondrial fission and fusion proteins such as Drp1andMfn2 [53]. In the next set of experimentwewanted to ascertain therole of Bax protein in 6-OHDA-induced mitochondrial fission. To do so,we used embryonic fibroblasts from Bax-/- mice. As indicated in Fig. 6B,the lack of Baxprotein expression did not preventmitochondrialfission/fragmentation after the addition of 50 μM 6-OHDA.
The transcription factor p53 belongs to the cytoplasmic proteinsthat migrate to the mitochondria after apoptotic insults [69], and hasbeen reported to be increased after 6-OHDA stimulation [56]. Mito-
chondrial staining revealed that in MEF p53-/- cells the mitochondrialmorphology was similar to that observed in native MEF cells treatedwith 50 μM 6-OHDA (Fig. 6C).
Discussion
In this study, we demonstrate that 6-OHDA induces profoundmitochondrial fission in neuronal SH-SY5Y cells. Furthermore, weprovide evidence that mitochondrial fission is an early and requiredprocess in 6-OHDA-induced SH-SY5Y cell death.
In the first set of experiments we analyzed the morphologicalchanges of mitochondria in SH-SY5Y cells which were induced by theneurotoxin 6-OHDA. Immunofluorescence studieswith antibodies to themitochondrial matrix protein MnSOD revealed that in untreated SH-SY5Y cells mitochondria exhibited an overwhelmingly elongated andfilamentous morphology. Strikingly, after addition of 50 μM 6-OHDAmitochondria formed short and spherical structures, due to the frag-mentation of single filamentous mitochondria into multiple isolatedorganelles. This new morphology was similar to that observed afteroverexpression of Myc-hFis1, a tail-anchored adaptor protein of themitochondrialfissionmachinery [45],whichwas used to corroborate theability of SH-SY5Y cells to undergomitochondrialfission and to visualizethe typical fission morphology of these mitochondria. Furthermore,time-lapse fluorescence microscopy revealed that 6-OHDA-inducedmitochondrial fragmentation occurred rapidly and synchronous within15 min after 6-OHDA addition, and was visible in approximately 80% ofthe treated cells after 3 h.

1968 M. Gomez-Lazaro et al. / Free Radical Biology & Medicine 44 (2008) 1960–1969
Ourdata point out thatmitochondrial fragmentation appears to be anearly event in 6-OHDA-induced cell death. After 3 h treatment (whenmitochondrial fragmentation was prominent), we neither observed acollapse of the mitochondrial membrane potential nor a prominentrelease of cytochrome c. As in controls, mitochondria in treated cells stillmaintained Rhodamine 123 fluorescence after 3 h. Furthermore, sig-nificant changes in the chromatin structure were not detected early on.6-OHDA (50 μM) had to be present more than 9 h to initiate significantchanges in mitochondrial membrane potential in SH-SY5Y cells.
We cannot exclude the possibility that mitochondria had started torelease amounts of cytochrome c too low to be detected by im-munoblotting. However, in line with our observations, it has beenobserved that cytochrome c filled fragmented mitochondria during theapoptotic process, thus indicating that mitochondrial fission occursbefore MOMP [70]. Furthermore, Gao et al. [71] reported that thedetection of fragmented mitochondria that were depleted of cyto-chrome c under their apoptotic conditions was a rare event. Very recentfindings indicate that although fission appears to be required for celldeath, fission in itself is not sufficient for cell death induction [72–74]. Inneurons, it is also suggested that mitochondrial fission is required, butdoes not per se lead to neuronal death [21,75]. Furthermore, in the lineofour previous work, we can exclude the participation of mitochondrialswelling, since we did not observe mitochondria with typical hallmarksof swelling in 6-OHDA-treated cells [62].
Our data are consistent with recent findings that multiple neuronalstressors including nitric oxide, rotenone, N-methyl-D-aspartate, andβ-amyloid peptide induce profound mitochondrial fission prior toneurodegeneration [21,76].
The nature of the mechanisms involved in 6-OHDA-inducedmitochondrial fragmentation remains largely unknown. One inter-pretation is that 6-OHDA oxidation induces ROS increase, which mayact on mitochondrial dynamics. In support of this hypothesis we haveshown that “aged” 6-OHDA, from which ROS have been liberated byautooxidation, failed to induce mitochondrial fragmentation. In ad-dition, incubation with the antioxidants catalase, ascorbic acid, orTEMPOL reduced the promoting effect of 6-OHDA on mitochondrialfragmentation in SH-SY5Y cells.
Further experiments revealed the requirement of the dynamin-likeGTPase Drp1 for 6-OHDA to induce mitochondrial division. Whenwe transfected SH-SY5Y cells with Drp1 siRNA duplexes to silenceDrp1, 6-OHDA-induced mitochondrial fragmentation was inhibited.Furthermore, 6-OHDA-induced cell death was reduced after silencing ofDrp1. These data indicate that Drp1 is required for 6-OHDA-inducedmitochondrialfissionandcell death. In linewith thesefindings, inhibitionof Drp1 function in other experimental models has also been shown topreventmitochondrialfission and cell death [19,23,77]. In a recent report,a block in mitochondrial fission by the expression of dominant-negativeDrp1 or wild-type Mfn1 prevented mitochondrial fragmentation andrescued neurons from NO-induced degeneration and cell death [21].
Herein we also studied a putative role for Bax and p53 in 6-OHDA-induced mitochondrial fragmentation. These proteins are known tomigrate to the mitochondria after apoptotic insults and interestingly,6-OHDA has been shown to increase the expression levels of bothproteins [56,78]. The participation of Bax in mitochondrial fission iscontroversially discussed [50]. Bax was found to colocalize with Drp1and Mfn2 at mitochondrial fission sites in apoptotic cells [53], and hasbeen suggested to participate directly in apoptotic mitochondrialfission. In support of this, Drp1 or Fis1 knockdown resulted in areduced accumulation of Bax on mitochondria [20,54]. In other stud-ies, however, Drp1 knockdown or Drp1K38A exhibited no effects onBax dynamics [19,20]. Our findings obtained with MEF cell culturesfrom mice lacking Bax or p53 indicate that these proteins are notrequired for 6-OHDA-induced mitochondrial fragmentation. It isnoteworthy that Bax expression appears to be essential for 6-OHDAto induced cell death in these MEF [79], raising the possibility that Baxmight act in a latter step downstream of mitochondrial fission.
Acknowledgments
We are grateful to Remedios Sanchis and Juana Rozalen for tech-nical assistance. This work was supported by SAF2002-04721 andSAF2005-07919-C02-01 from CICYT and 04005-00 and PI2007/55Consejería de Sanidad from Junta de Comunidades de Castilla LaMancha (to J.J.) and by the German Research Foundation (DFG) (SCHR518/6-1,2) and the Portuguese Foundation for Science and Technology(FCT) (PTDC/BIA-BCM/71932/2006) (to M.S.). M.G.-L. has been a fellowfrom JCCM.
References
[1] Bredesen, D. E. Keeping neurons alive, the molecular control of apoptosis. Neur-oscientist 2:181–190; 1996.
[2] Mandemakers, W.; Morais, V. A.; De Strooper, B. A cell biological perspective onmitochondrial dysfunction in Parkinson disease and other neurodegenerativediseases. J. Cell Sci. 120:1707–1716; 2007.
[3] Bove, J.; Prou, D.; Perier, C.; Przedborski, S. Toxin-induced models of Parkinson'sdisease. NeuroRx 2:484–494; 2005.
[4] Blum, D.; Torch, S.; Lambeng, N.; Nissou, M.; Benabid, A. L.; Sadoul, R.; Verna, J. M.Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine andMPTP, contribution to the apoptotic theory in Parkinson's disease. Prog. Neurobiol.65:135–172; 2001.
[5] Fernandez-Gomez, F. J.; Pastor, M. D.; Garcia-Martinez, E. M.; Melero-Fernandez deMera, R.; Gou-Fabregas,M.; Gomez-Lazaro, M.; Calvo, S.; Soler, R.M.; Galindo, M. F.;Jordan, J. Pyruvate protects cerebellar granular cells from 6-hydroxydopamine-induced cytotoxicity by activating the Akt signaling pathway and increasingglutathione peroxidase expression. Neurobiol. Dis. 24:296–307; 2006.
[6] Galindo, M. F.; Jordan, J.; Gonzalez-Garcia, C.; Cena, V. Chromaffin cell deathinduced by 6-hydroxydopamine is independent of mitochondrial swelling andcaspase activation. J. Neurochem. 84:1066–1073; 2003.
[7] Woodgate, A.; MacGibbon, G.; Walton, M.; Dragunow, M. The toxicity of6-hydroxydopamine onPC12andP19 cells.BrainRes.Mol. BrainRes.69:84–92; 1999.
[8] Zhang, D.; Zhang, J. J.; Liu, G. T. The novel squamosamide derivative FLZ protectsagainst 6-hydroxydopamine-induced apoptosis through inhibition of relatedsignal transduction in SH-SY5Y cells. Eur. J. Pharmacol. 561:1–6; 2007.
[9] Ochu, E. E.; Rothwell, N. J.; Waters, C. M. Caspases mediate 6-hydroxydopamine-induced apoptosis but not necrosis in PC12 cells. J. Neurochem. 70:2637–2640;1998.
[10] Hanrott, K.; Gudmunsen, L.;O'Neill,M. J.;Wonnacott, S. 6-Hydroxydopamine-inducedapoptosis is mediated via extracellular auto-oxidation and caspase 3-dependentactivation of protein kinase Cdelta. J. Biol. Chem. 281:5373–5382; 2006.
[11] Hoppins, S.; Lackner, L.; Nunnari, J. The machines that divide and fusemitochondria. Annu. Rev. Biochem. 76:751–780; 2007.
[12] Rouiller, C. Physiological and pathological changes in mitochondrial morphology.Int. Rev. Cytol. 9:227–292; 1960.
[13] Chen, H.; Chan, D. C. Emerging functions of mammalian mitochondrial fusion andfission. Hum. Mol. Genet. 14:R283–R289; 2005.
[14] Kiefel, B. R.; Gilson, P. R.; Beech, P. L. Cell biology of mitochondrial dynamics. Int.Rev. Cytol. 254:151–213; 2006.
[15] Chan, D. C. Mitochondria, dynamic organelles in disease, aging, and development.Cell 125:1241–1252; 2006.
[16] Olichon, A.; Guillou, E.; Delettre, C.; Landes, T.; Arnaune-Pelloquin, L.; Emorine, L. J.;Mils, V.; Daloyau, M.; Hamel, C.; Amati-Bonneau, P.; Bonneau, D.; Reynier, P.;Lenaers, G.; Belenguer, P. Mitochondrial dynamics and disease, OPA1. Biochim.Biophys. Acta 1763:500–509; 2006.
[17] Santel, A. Get the balance right, mitofusins roles in health and disease. Biochim.Biophys. Acta 1763:490–499; 2006.
[18] Li, Z.; Okamoto, K.; Hayashi, Y.; Sheng, M. The importance of dendriticmitochondria in the morphogenesis and plasticity of spines and synapses. Cell119:873–887; 2004.
[19] Frank, S.; Gaume, B.; Bergmann-Leitner, E. S.; Leitner, W.W.; Robert, E. G.; Catez, F.;Smith, C. L.; Youle, R. J. The role of dynamin-related protein 1, a mediator ofmitochondrial fission, in apoptosis. Dev. Cell. 1:515–525; 2001.
[20] Lee, Y. J.; Jeong, S. Y.; Karbowski, M.; Smith, C. L.; Youle, R. J. Roles of the mam-malian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 inapoptosis. Mol. Biol. Cell 15:5001–5011; 2004.
[21] Barsoum, M. J.; Yuan, H.; Gerencser, A. A.; Liot, G.; Kushnareva, Y.; Graber, S.;Kovacs, I.; Lee, W. D.; Waggoner, J.; Cui, J.; White, A. D.; Bossy, B.; Martinou, J. C.;Youle, R. J.; Lipton, S. A.; Ellisman, M. H.; Perkins, G. A.; Bossy-Wetzel, E. Nitricoxide-induced mitochondrial fission is regulated by dynamin-related GTPases inneurons. EMBO J. 25:3900–3911; 2006.
[22] Arnoult, D. Mitochondrial fragmentation in apoptosis. Trends Cell Biol.17:6–12; 2007.[23] Breckenridge, D. G.; Stojanovic, M.; Marcellus, R. C.; Shore, G. C. Caspase cleavage
product of BAP31 induces mitochondrial fission through endoplasmic reticu-lum calcium signals, enhancing cytochrome c release to the cytosol. J. Cell Biol.160:1115–1127; 2003.
[24] Karbowski, M.; Arnoult, D.; Chen, H.; Chan, D. C.; Smith, C. L.; Youle, R. J.Quantitation of mitochondrial dynamics by photolabeling of individual organellesshows that mitochondrial fusion is blocked during the Bax activation phase ofapoptosis. J. Cell Biol. 164:493–499; 2004.

1969M. Gomez-Lazaro et al. / Free Radical Biology & Medicine 44 (2008) 1960–1969
[25] Karbowski, M.; Jeong, S. Y.; Youle, R. J. Endophilin B1 is required for themaintenance of mitochondrial morphology. J. Cell Biol. 166:1027–1039; 2004.
[26] Arbustini, E.; Diegoli, M.; Fasani, R.; Grasso, M.; Morbini, P.; Banchieri, N.; Bellini,O.; Dal Bello, B.; Pilotto, A.; Magrini, G.; Campana, C.; Fortina, P.; Gavazzi, A.;Narula, J.; Vigano, M. Mitochondrial DNA mutations and mitochondrial abnorm-alities in dilated cardiomyopathy. Am. J. Pathol. 153:1501–1510; 1998.
[27] Nishino, I.; Kobayashi, O.; Goto, Y.; Kurihara, M.; Kumagai, K.; Fujita, T.; Hashimoto,K.; Horai, S.; Nonaka, I. A new congenital muscular dystrophy with mitochondrialstructural abnormalities. Muscle Nerve 21:40–47; 1998.
[28] Tandler, B.; Hoppel, C. L. Studies on giant mitochondria. Ann. N. Y. Acad. Sci.488:65–81; 1986.
[29] Hackenbrock, C. R. Chemical and physical fixation of isolated mitochondria in low-energy and high-energy states. Proc. Natl. Acad. Sci. U. S. A. 61:598–605; 1968.
[30] Wachstein, M.; Besen, M. Electron microscopy of renal coagulative necrosis dueto Dl-serine, with special reference to mitochondrial pyknosis. Am. J. Pathol.44:383–400; 1964.
[31] Rouiller, C. Electron microscopy for the study of the normal and pathological liver.Ann. Anat. Pathol. (Paris) 2:548–562; 1957.
[32] Mannweiler, K.; Bernhard,W. Research on ultrastructures in an experimental renaltumor in the hamster. J. Ultrastruct. Res. 1:158–169; 1957.
[33] James, T. N.; Terasaki, F.; Pavlovich, E. R.; Vikhert, A. M. Apoptosis and pleomorphicmicromitochondriosis in the sinus nodes surgically excised from five patients withthe long QT syndrome. J. Lab. Clin. Med. 122:309–323; 1993.
[34] Mancini, M.; Anderson, B. O.; Caldwell, E.; Sedghinasab, M.; Paty, M. B.;Hockenbery, D. M. Mitochondrial proliferation and paradoxical membranedepolarization during terminal differentiation and apoptosis in a human coloncarcinoma cell line. J. Cell Biol. 138:449–469; 1997.
[35] Zhuang, J.; Dinsdale, D.; Cohen, G. M. Apoptosis, in human monocytic THP.1 cells,results in the release of cytochrome c from mitochondria prior to theirultracondensation, formation of outer membrane discontinuities and reductionin inner membrane potential. Cell Death Differ. 5:953–962; 1998.
[36] Tondera, D.; Santel, A.; Schwarzer, R.; Dames, S.; Giese, K.; Klippel, A.; Kaufmann, J.Knockdown of MTP18, a novel phosphatidylinositol 3-kinase-dependent protein,affects mitochondrial morphology and induces apoptosis. J. Biol. Chem.279:31544–31555; 2004.
[37] Tondera, D.; Czauderna, F.; Paulick, K.; Schwarzer, R.; Kaufmann, J.; Santel, A. Themitochondrial protein MTP18 contributes to mitochondrial fission in mammaliancells. J. Cell Sci. 118:3049–3059; 2005.
[38] Niemann, A.; Ruegg, M.; La Padula, V.; Schenone, A.; Suter, U. Ganglioside-induceddifferentiation associated protein 1 is a regulator of the mitochondrial network, newimplications for Charcot–Marie–Tooth disease. J. Cell Biol.170:1067–1078; 2005.
[39] Pedrola, L.; Espert, A.;Wu, X.; Claramunt, R.; Shy, M. E.; Palau, F. GDAP1, the proteincausing Charcot–Marie–Tooth disease type 4A, is expressed in neurons and isassociated with mitochondria. Hum. Mol. Genet. 14:1087–1094; 2005.
[40] Karbowski, M.; Neutzner, A.; Youle, R. J. The mitochondrial E3 ubiquitin ligaseMARCH5 is required for Drp1 dependent mitochondrial division. J. Cell Biol.178:71–84; 2007.
[41] Pitts, K. R.; Yoon, Y.; Krueger, E. W.; McNiven, M. A. The dynamin-like protein DLP1is essential for normal distribution and morphology of the endoplasmic reticulumand mitochondria in mammalian cells. Mol. Biol. Cell. 10:4403–4417; 1999.
[42] Smirnova, E.; Griparic, L.; Shurland, D. L.; van der Bliek, A. M. Dynamin-relatedprotein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol.Cell 12:2245–2256; 2001.
[43] Smirnova, E.; Shurland, D. L.; Ryazantsev, S. N.; van der Bliek, A. M. A humandynamin-related protein controls the distribution of mitochondria. J. Cell Biol.143:351–358; 1998.
[44] Yu, T.; Fox, R. J.; Burwell, L. S.; Yoon, Y. Regulation of mitochondrial fission andapoptosis by the mitochondrial outer membrane protein hFis1. J. Cell Sci.118:4141–4151; 2005.
[45] Yoon, Y.; Krueger, E. W.; Oswald, B. J.; McNiven, M. A. The mitochondrial proteinhFis1 regulates mitochondrial fission in mammalian cells through an interactionwith the dynamin-like protein DLP1. Mol. Cell. Biol. 23:5409–5420; 2003.
[46] Stojanovski, D.; Koutsopoulos, O. S.; Okamoto, K.; Ryan, M. T. Levels of human Fis1at the mitochondrial outer membrane regulate mitochondrial morphology. J. CellSci. 117:1201–1210; 2004.
[47] James, D. I.; Parone, P. A.; Mattenberger, Y.; Martinou, J. C. hFis1, a novel component ofthemammalianmitochondrialfissionmachinery. J. Biol. Chem.278:36373–36379; 2003.
[48] Schrader, M. Shared components of mitochondrial and peroxisomal division.Biochim. Biophys. Acta 1763:531–541; 2006.
[49] Wei, M. C.; Zong, W. X.; Cheng, E. H.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A. J.;Roth, K. A.; MacGregor, G. R.; Thompson, C. B.; Korsmeyer, S. J. Proapoptotic BAXand BAK: a requisite gateway to mitochondrial dysfunction and death. Science292:727–730; 2001.
[50] Antignani, A.; Youle, R. J. How do Bax and Bak lead to permeabilization of theouter mitochondrial membrane? Curr. Opin. Cell Biol. 18:685–689; 2006.
[51] Lalier, L.; Cartron, P. F.; Juin, P.; Nedelkina, S.; Manon, S.; Bechinger, B.; Vallette, F. M.Bax activation and mitochondrial insertion during apoptosis. Apoptosis 12:887–896;2007.
[52] Youle, R. J.; Karbowski, M. Mitochondrial fission in apoptosis. Nat. Rev., Mol. Cell.Biol. 6:657–663; 2005.
[53] Karbowski, M.; Lee, Y. J.; Gaume, B.; Jeong, Y.; Frank, S.; Nechushtan, A.; Santel,A.; Fuller, M.; Smith, C. L.; Youle, R. J. Spatial and temporal association of Baxwith mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J. Cell Biol.159:931–938; 2002.
[54] Neuspiel, M.; Zunino, R.; Gangaraju, S.; Rippstein, P.; McBride, H. Activatedmitofusin2 signals mitochondrial fusion, interferes with Bax activation, and reducessusceptibility to radical induced depolarization. J. Biol. Chem. 280:25060–25070;2005.
[55] Karbowski, M.; Norris, K. L.; Cleland, M. M.; Jeong, S. Y.; Youle, R. J. Role of Bax andBak in mitochondrial morphogenesis. Nature 443:658–662; 2006.
[56] Biswas, S. C.; Ryu, E.; Park, C.; Malagelada, C.; Greene, L. A. Puma and p53 playrequired roles in death evoked in a cellular model of Parkinson disease. Neuro-chem. Res. 30:839–845; 2005.
[57] Gomez-Lazaro, M.; Galindo, M. F.; Melero-Fernandez de Mera, R. M.; Fernandez-Gomez, F. J.; Concannon, C. G.; Segura, M. F.; Comella, J. X.; Prehn, J. H.; Jordan, J.Reactive oxygen species and p38 mitogen-activated protein kinase activate Bax toinducemitochondrial cytochrome c release and apoptosis in response tomalonate.Mol. Pharmacol. 71:736–743; 2007.
[58] Schrader, M.; Burkhardt, J. K.; Baumgart, E.; Luers, G.; Spring, H.; Volkl, A.; Fahimi,H. D. Interaction of microtubules with peroxisomes. Tubular and sphericalperoxisomes in HepG2 cells and their alterations induced by microtubule-activedrugs. Eur. J. Cell Biol. 69:24–35; 1996.
[59] Schrader, M.; Reuber, B. E.; Morrell, J. C.; Jimenez-Sanchez, G.; Obie, C.; Stroh, T. A.;Valle, D.; Schroer, T. A.; Gould, S. J. Expression of PEX11beta mediates peroxisomeproliferation in the absence of extracellular stimuli. J. Biol. Chem. 273:29607–29614;1998.
[60] Maestre, I.; Jordan, J.; Calvo, S.; Reig, J. A.; Cena, V.; Soria, B.; Prentki, M.; Roche, E.Mitochondrial dysfunction is involved in apoptosis induced by serum withdrawaland fatty acids in the beta-cell line INS-1. Endocrinology 144:335–345; 2003.
[61] Prehn, J. H.; Bindokas, V. P.; Jordan, J.; Galindo, M. F.; Ghadge, G. D.; Roos, R. P.;Boise, L. H.; Thompson, C. B.; Krajewski, S.; Reed, J. C.; Miller, R. J. Protective effectof transforming growth factor-beta 1 on beta-amyloid neurotoxicity in rathippocampal neurons. Mol. Pharmacol. 49:319–328; 1996.
[62] Jordan, J.; Galindo, M. F.; Tornero, D.; Gonzalez-Garcia, C.; Cena, V. Bcl-x L blocksmitochondrial multiple conductance channel activation and inhibits 6-OHDA-induced death in SH-SY5Y cells. J. Neurochem. 89:124–133; 2004.
[63] Boll, A.; Schrader, M. Elongation of peroxisomes as an indicator for efficientdynamin-like protein 1 knock down in mammalian cells. J. Histochem. Cytochem.53:1037–1040; 2005.
[64] Yoon, Y.; Pitts, K. R.; McNiven, M. A. Mammalian dynamin-like protein DLP1tubulates membranes. Mol. Biol. Cell. 12:2894–2905; 2001.
[65] Koch, A.; Schneider, G.; Luers, G. H.; Schrader, M. Peroxisome elongation andconstriction but not fission can occur independently of dynamin-like protein 1.J. Cell Sci. 117:3995–4006; 2004.
[66] Wasiak, S.; Zunino, R.; McBride, H. M. Bax/Bak promote sumoylation of DRP1 andits stable association with mitochondria during apoptotic cell death. J. Cell Biol.177:439–450; 2007.
[67] Yuan, H.; Gerencser, A. A.; Liot, G.; Lipton, S. A.; Ellisman, M.; Perkins, G. A.; Bossy-Wetzel, E. Mitochondrial fission is an upstream and required event for bax fociformation in response to nitric oxide in cortical neurons. Cell Death Differ.14:462–471; 2007.
[68] Arnoult, D.; Rismanchi, N.; Grodet, A.; Roberts, R. G.; Seeburg, D. P.; Estaquier, J.;Sheng, M.; Blackstone, C. Bax/Bak-dependent release of DDP/TIMM8a promotesDrp1-mediated mitochondrial fission and mitoptosis during programmed celldeath. Curr. Biol. 15:2112–2118; 2005.
[69] Zhao, Y.; Chaiswing, L.; Velez, J. M.; Batinic-Haberle, I.; Colburn, N. H.; Oberley, T. D.;St Clair, D. K. p53 translocation to mitochondria precedes its nuclear translocationand targets mitochondrial oxidative defense protein-manganese superoxidedismutase. Cancer Res. 65:3745–3750; 2005.
[70] Martinou, J. C.; Youle, R. J. Which came first, the cytochrome c release or themitochondrial fission? Cell Death Differ. 13:1291–1295; 2006.
[71] Gao, W.; Pu, Y.; Luo, K. Q.; Chang, D. C. Temporal relationship between cytochromec release and mitochondrial swelling during UV-induced apoptosis in living HeLacells. J. Cell Sci. 114:2855–2862; 2001.
[72] Delivani, P.; Adrain, C.; Taylor, R. C.; Duriez, P. J.;Martin, S. J. Role for CED-9 and Egl-1as regulators of mitochondrial fission and fusion dynamics. Mol. Cell. 21:761–773;2006.
[73] Parone, P. A.; James, D. I.; Da Cruz, S.; Mattenberger, Y.; Donze, O.; Barja, F.;Martinou, J. C. Inhibiting the mitochondrial fission machinery does not preventBax/Bak-dependent apoptosis. Mol. Cell Biol. 26:7397–7408; 2006.
[74] Alirol, E.; James, D.; Huber, D.; Marchetto, A.; Vergani, L.; Martinou, J. C.; Scorrano,L. The mitochondrial fission protein hFis1 requires the endoplasmic reticulumgateway to induce apoptosis. Mol. Biol. Cell. 17:4593–4605; 2006.
[75] Cheung, E. C.; McBride, H. M.; Slack, R. S. Mitochondrial dynamics in the regulationof neuronal cell death. Apoptosis 12:979–992; 2007.
[76] Yan, S. D.; Xiong, W. C.; Stern, D. M. Mitochondrial amyloid-beta peptide,pathogenesis or late-phase development? J. Alzheimers Dis. 9:127–137; 2006.
[77] Germain, M.; Mathai, J. P.; McBride, H. M.; Shore, G. C. Endoplasmic reticulum BIKinitiates DRP1-regulated remodelling of mitochondrial cristae during apoptosis.EMBO J. 24:1546–1556; 2005.
[78] Mladenovic, A.; Perovic, M.; Raicevic, N.; Kanazir, S.; Rakic, L.; Ruzdijic, S. 6-Hydroxydopamine increases the level of TNFalpha and bax mRNA in the striatumand induces apoptosis of dopaminergic neurons in hemiparkinsonian rats. BrainRes. 996:237–245; 2008.
[79] Gomez-Lazaro, M.; Galindo, M. F.; Concannon, C. G.; Segura, M. F.; Fernandez-Gomez, F. J.; Llecha, N.; Comella, J. X.; Prehn, J. H. .; Jordan, J. 6-Hydroxydopamineactivates the mitochondrial apoptosis pathway through p38 MAPK-mediated,p53-independent activation of Bax and PUMA. J. Neurochem.104:1599–1612; 2008.




















