
Neuroprotective Effect of Dexmedetomidine on Hyperoxia-Induced Toxicity in the Neonatal Rat Brain
Upload
independentCategory
view
1download
0
Biomedical and Life Sciences, Faculty of Health and Medicine, Lancaster University, Lancaster, UK
AbstractGlucagon-like peptide 1 (GLP-1) is a growth factor that hasdemonstrated neuroprotective properties in a range of studies.In an APPswe/PS1ΔE9 mouse model of Alzheimer’s disease(AD), we previously found protective effects on memoryformation, synaptic plasticity, synapse survival and a reductionof amyloid synthesis and plaque load in the brain. Here, weanalyse the neuroprotective properties of the GLP-1 analogueliraglutide in human neuroblastoma cell line SH-SY5Y duringmethyl glyoxal stress. We show for the first time that cellviability was enhanced by liraglutide (XTT assay) in a dose-dependent way, while cytotoxicity (LDH assay) and apoptosiswere reduced. Expression of the pro-survival Mcl1 signaling
protein was increased, as was activation of cell survivalkinases Akt, MEK1/2 and the transcription factor p90RSK.Liraglutide also decreased pro-apoptotic Bax and Bik expres-sion. In addition, the membrane potential and the influx ofcalcium into the cell were enhanced by liraglutide. GLP-1receptor expression was also increased by the drug. Theresults demonstrate a range of growth factor-related cytopro-tective processes induced by liraglutide, which is currently onthe market as a treatment for type 2 diabetes (Victoza�). It isalso tested in clinical trials in patients with Alzheimer disease.Keywords: Alzheimer’s disease, apoptosis, incretins, insulin,neurodegeneration, neuroprotection.J. Neurochem. (2014) 128, 459–471.
Glucagon-like peptide 1 (GLP-1) is a signaling peptide thathas a range of important physiological properties. Due to thefact that GLP-1 has beneficial effects on insulin release fromthe pancreas during episodes of hyperglycaemia (Druckerand Nauck 2006), a range of long-lasting GLP-1 mimeticshas been developed as treatments for type 2 diabetes.Currently, three such mimetics are on the market and areprescribed to diabetics, exendin-4 (Byeatta�), liraglutide(Victoza�), and lixisenatide (Lyxumia�) (Madsbad et al.2008; Vilsboll 2009; Christensen et al. 2011). The beneficialeffects of these drugs are not limited to the treatment ofdiabetes. GLP-1 has growth-factor properties (Greig et al.2004; Holscher 2011) and has been shown to improvecardiovascular conditions (Noyan-Ashraf et al. 2009; Kimet al. 2013). We found in a range of preclinical studies thatthe GLP-1 analogue liraglutide crosses the blood–brainbarrier into the brain and shows a range of neuroprotectiveeffects, eg. protection of memory formation in a mousemodel of Alzheimer’s disease (AD), protection of synapticplasticity in the hippocampus, prevention of synapse loss andreduction of amyloid plaque levels and soluble amyloidoligomer levels (McClean et al. 2010, 2011; Hunter andHolscher 2012; Han et al. 2013), normalisation of neuro-
genesis and progenitor proliferation in the hippocampus(Hamilton et al. 2011; Parthsarathy and Holscher 2013a),and a reduction of the chronic inflammation response in thebrain (McClean et al. 2011; Parthsarathy and Holscher2013b).In the present study, we are investigating the cellular
mechanisms and cell signaling pathways that underlie thecytoprotective and anti-inflammatory effect. While thesignaling mechanisms activated by GLP-1 in beta-cells inthe pancreas have been investigated in detail (Doyle andEgan 2007), very little is known about the signaling
Received July 13, 2013; revised manuscript received September 10,2013; accepted September 19, 2013.Address correspondence and reprint requests to Christian H€olscher,
Division of Biomedical and Life Sciences, Faculty of Health andMedicine, Lancaster University, Lancaster LA1 4YQ, UK.E-mail: [email protected] used: AD, Alzheimer’s disease; BBB, blood–brain
barrier; BDNF, brain derived neurotrophic factor; BrdU, 5-bromo-2-2deoxyuridine; ERK, extracellular signal-regulated kinase; LDH,lactate dehydrogenase; MG, methyl glyoxal; NGF, nerve growth factor;PBS, phosphate buffered saline; PI3k, phosphoinositide 3 kinase; PI,propidium iodide; PKA, protein kinase A; PKB, protein kinase B; SFM,serum free medium.
© 2013 International Society for Neurochemistry, J. Neurochem. (2014) 128, 459--471 459
JOURNAL OF NEUROCHEMISTRY | 2014 | 128 | 459–471 doi: 10.1111/jnc.12469

pathways in neurons. Few studies have investigated whatsecond messenger pathways are activated or modulated byGLP-1. Importantly, GLP-1 signaling affects the release ofother growth factors. In one in vivo study, peripheralinjection of GLP-1 releasing cells increased the levels ofmRNA expression of nerve growth factor-inducible proteinA in the brain (Fan et al. 2010). GLP-1 also enhanced therelease of brain derived neurotrophic factor (BDNF) at thesynapse (Mattson 2012). Importantly, insulin signaling isnormalised by GLP-1, and peripheral injection of exendin-4reversed the insulin desensitisation in the brain of APP/PS1mice (Bomfim et al. 2012). In a cell culture study of PC12cells, GLP-1 and exendin-4 were found to enhance nervegrowth factor (NGF)-induced cell differentiation into neu-rons (Perry et al. 2002). Activating the GLP-1 receptor inneurons activates an adenylyl cyclase and increases cAMPlevels, which in turn activates protein kinase A (PKA) andcyclic AMP response element binding protein (CREB) (Perryand Greig 2004; Hunter and Holscher 2012). Furthermore,neuroprotective effects of GLP-1 in PC12 cells were initiatedvia the phosphoinositide 3 kinase (PI3k) and Akt/PKBpathway and also involved mammalian target of rapamycinactivation (Perry and Greig 2005; Kimura et al. 2009), agrowth factor downstream pathway that is also activated byinsulin (Nelson and Alkon 2005). The PKA and PI3kpathway was activated and apoptosis via Caspase-3 wasreduced in this cell culture study (Li et al. 2010). Otherstudies confirmed the anti-apoptotic property of GLP-1mimetics in cultured neurons (During et al. 2003; Perryet al. 2003). In cultured neurons, exendin-4 was neuropro-tective in a PKA or PI3k activity dependent way. Inperipheral neurons, GLP-1 receptor activation increasedextracellular signal-regulated kinase (ERK) signaling andenhanced the total ERK to pERK ratio, comparable to NGFinduced ERK signaling (Jolivalt et al. 2011).Furthermore, cytokine release is reduced by the GLP-1
analogue liraglutide injected ip. in an x-ray inflammationresponse in mice, (Parthsarathy and Holscher 2013b), and inan in vivo study of APP/PS1 mice, exendin-4 reduced JNKcytokine cell signaling (Bomfim et al. 2012). Ca2+ is animportant second messenger in neurons, and abnormal Ca2+
levels are neurotoxic. Beta-amyloid disturbs the Ca2+
homeostasis, which is considered one of the molecularmechanisms of how beta-amyloid kills neurons in AD(Holscher 1998). GLP-1 analogues normalised Ca2+ levels inbeta cells stressed with methyl glyoxal (MG) (Selway et al.2012). We chose this stressor as it compromises energymetabolism in the cell, which is a common observation inneurons in AD (Hoyer 2004; Mattson 2012). In culturedneurons, GLP-1 normalised glutamate-induced voltage gatedcalcium channel dependent Ca2+ influx into neurons whichprotected the cells from glutamate induced toxicity (Gilmanet al. 2003). We therefore investigated if liraglutide haseffects on the Ca2+ levels of human neuroblastoma cells.
To further investigate the neuroprotective effects of theGLP-1 analogue liraglutide, we tested the drug in the humanneuroblastoma cell line SH-SY5Y, using MG as a stressor.We evaluated the effects of the drug on cell survival,proliferation, and apoptosis. We investigated the signalingpathways involved in the cytoprotective actions and mea-sured levels of growth factor kinases phosphorylated Akt,MEK1/2 and the transcription factor p90RSK, as well asapoptotic markers caspase-3, Bax and Bik, and survivalmarker Mcl1. In addition, changes in membrane potentialand total intracellular Ca2+ induced by MG stress weremeasured.
Materials and methods
Materials
Liraglutide was purchased from GL Biochem (Shanghai) Ltd. Thepurity of the peptide was analysed by reversed-phase HPLC andcharacterized using matrix-assisted laser desorption/ionization timeof flight (MALDI-TOF) mass spectrometry. Cell proliferation kit II(XTT) and cell proliferation ELISA 5-bromo-2-2deoxyuridine(BrdU) (colorimetric) kit were purchased from Roche and theCytoTox 96 Non-Radioactive Cytotoxicity Assay assay kit fromPromega (Madison, WI, USA). Image-iT LIVE Green poly caspasedetection kit, anti-rabbit goat Alexa555 secondary antibody andphosphate buffered saline (PBS tablets) were purchased fromInvitrogen (Paisley, UK). FLIPR Membrane Potential Assay kit andFLIPR calcium assay kit were purchased from Molecular devices(Berkshire, UK). Cleaved caspase-3 (Asp175), Mcl1, Bax, Bikpolyclonal and phospho-AKT (Ser 473), phospho-MEK1/2(Ser217/221), phospho-p90RSK (Ser380) monoclonal, primaryantibodies raised in rabbit and horseradish peroxidase labeledsecondary antibody, were purchased from Cell signaling technology(Hertfordshire, UK). Millicell EZ 8-well sterile glass slides werepurchased from Merck Millipore (Watford, UK). Cell lysis bufferwas obtained from Cell signaling technology, Quick start proteinassay reagent from Biorad (Dublin, Ireland), X-Ray film, polyv-inylidene difluoride membranes and Amersham ECL Primewestern blotting detection reagent from GE, Healthcare Lifescienc-es (Buckinghamshire, UK). Other materials for western blotting andcell culture were obtained from Invitrogen.
Cell culture
The human neuroblastoma cell line SH-SY5Y was obtained fromLGC standards (ATCC No. CRL-2266), and maintained inDulbecco’s minimum essential medium, DMEM+F12 (1 : 1)GlutaMax supplemented with 10% heat-inactivated foetal bovineserum (FBS) and 100 Units per ml of Penicillin and 100 lg per mLof Streptomycin at 37°C in a humidified incubator with 95% air and5% CO2. The cells were sub-cultured when 80–90% confluent andseeded at 1 : 5 ratio. Media was changed every 4–5 days.
Measurement of cell viability, cytotoxicity and proliferation
Cell viability, cytotoxicity, and proliferation were assessed using 2,3-Bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide(XTT, Cell proliferation kit II), Lactate dehydrogenase (LDH,
© 2013 International Society for Neurochemistry, J. Neurochem. (2014) 128, 459--471
460 M. K. Sharma et al.

CytoTox 96 assay kit) and 5-bromo-2-deoxyuridine (BrdU, Cellproliferation ELISA colorimetric kit) incorporation, respectively.
The assay was formatted by seeding SH-SY5Y cells in laminin-coated 96-well plates at a density of 5 9 104 cells/well for 24 h.The cells were serum starved for 12 h in serum free medium (SFM)and were pre-treated for 4 h with different concentrations ofLiraglutide. Medium was removed and the cells were exposed tovarious concentrations of MG for 12 h. Supernatant was removed(to be used for LDH assay) and 49 lL of XTT and 1 lL of electron-coupling reagent diluted 1 : 1 with SFM was added to each well andthe plate incubated at 37°C for 4 h. XTT measures cell viabilitybased on the activity of mitochondrial enzymes in a live cell thatreduces XTT to a highly water-soluble product, kmax 492 nm, whichis directly proportional to the number of living cells in the sample.The plate was gently shaken for 5 min on Microtitre plate shaker(Stuart, Staffordshire, UK) and absorbance measured at 492 nm and690 nm in a FLUOstar Omega microplate reader (BMG LAB-TECH, Aylesbury, Bucks, UK).
Cell proliferation was assessed in actively proliferating cells bymeasuring the amount of BrdU incorporated in newly synthesizedDNA strands using a BrdU cell proliferation kit. BrdU incorporationwas measured as per the manufacturer’s instructions. Briefly, after12 h of MG stress, BrdU labeling reagent was added for 4 h and theplate incubated at 37°C. BrdU incorporation was measured immu-nochemically by peroxidase labeled anti-BrdU antibody followed bycolorimetric substrate detection. The plate was read at 370/492 nm kusing microplate reader. The amount of BrdU incorporated in theproliferating cells was directly proportional to the increasing opticaldensity.
The CytoTox 96 assay kit was used to quantitatively measure theLDH, a stable cytosolic enzyme, which is released upon cell lysis,according to the manufacturer’s instructions. LDH in supernatant ismeasured in an enzymatic assay that results in the formation of a redformazan product and the amount of colour formed is proportionalto the number of lysed cells.
Immunocytochemistry
SH-SY5Y cells were grown on laminin coated eight-well chamberslides and pre-treated for 4 h with 100 nM Liraglutide. Mediumwas removed and the cells were exposed to 600 lM MG for 8 h.Cells were washed with 1X PBS (pH 7.4) and fixed in 4%paraformaldehyde for 10 min followed by permeabilization in 0.3%Triton-X-100 for 5 min. Blocking was done in 1% bovine serumalbumin and 5% goat serum for 1 h at 25�C. Cleaved caspase-3(Asp175) rabbit polyclonal antibody was added at dilution 1 : 400and incubated at 25�C for 1 h. Cells were then washed three times(5 min each) in 1X PBS followed by incubation at 25�C for 1 h ingoat anti-rabbit Alexa555 secondary antibody at dilution 1 : 800,counterstaining with Hoechst 33342 (nuclear stain) for 10 min.Mounting was done in Vectashield mounting medium and micros-copy was performed using Axio Scope 1 (Zeiss, Cambridge, UK).Imaging of GLP-1R was performed using a confocal microscope(Leica Microsystems, Milton Keynes, UK; SP5 LAS IF Software).
Apoptotic insult
Apoptotic cells were assessed by differential Hoechst 33342 andpropidium iodide (PI) staining. SH-SY5Y cells were grown onlaminin coated eight-well chamber slides and pre-treated for 4 h
with 100 nM Liraglutide. Medium was removed and the cells wereexposed to 600 lM MG for 8 h. Cells were washed with 1X PBS(pH 7.4) and fixed in 4% paraformaldehyde for 10 min. Fixed cellswere washed twice with 1X PBS (pH 7.4) followed by addition of1 lg/ml Hoechst 33342 and PI and incubated at 25�C for 15 min.Mounting was done in Vectashield mounting medium and observedunder Fluorescence microscope. Hoechst 33342 stains nuclei blueand shows typical apoptotic features such as chromatin condensa-tion and fragmentation. The intact membrane of the healthy cellsexcludes PI but necrotic cells are stained brightly as their membraneintegrity is compromised. So, the apoptotic cells were quantified bycounting the magenta coloured nuclei.
Measurement of total caspase
Image-iT LIVE Green poly caspase detection kit was used to detectactive caspases based on a fluorescent inhibitor of caspases, FAM-VAD-FMK poly caspases reagent (FLICA). Cells were grown onlaminin-coated eight-well chamber slides and pre-treated for 4 hwith 100 nM Liraglutide. Medium was removed and the cells wereexposed to 600 lM MG for 8 h and thereafter, 1X FLICA reagentwas added in sufficient amount to cover the cells followed byincubation at 37°C for 1 h. After gently removing the solution andgently rinsing the cells with SFM, Hoechst 33342 was used tocounterstain the nucleus and mounted in apoptosis fixative solutionand observed under a Fluorescence microscope. FLICA reagent hasapproximate excitation/emission maxima of 488/530 nm and asso-ciates a fluoromethyl ketone (FMK) moiety, which can react with acaspase-specific amino acid sequence (valine-alanine-aspartic acid,VAD) and a carboxyfluorescein group (FAM) is attached as areporter. Thus, green fluorescent signal observed is a direct measureof the total active caspases present.
Measurement of total intracellular calcium and membrane
potential
Intracellular calcium and membrane potential were measured usingFLIPR calcium assay and Membrane Potential Assay kit. 5 9 104
cells/well were seeded in Laminin pre-coated clear bottom 96 wellblack plates for 24 h and pre-treated for 4 h with differentconcentrations of Liraglutide. Cell culture assay medium wasremoved and the cells were exposed to 300 lM and 600 lM MGfor 8 h. Intracellular Ca2+ responses and membrane potential weremonitored using FlexStation scanning fluorimeter with integratedfluid transfer workstation (Ojo et al. 2011).
Western blotting
2 9 106 cells were grown in Laminin pre-coated 100 mm platesand total protein was extracted. Cells were washed with cold 1XPBS buffer followed by addition of cell lysis buffer containingprotease inhibitors. After two freeze thaw cycles, and incubation at4°C on a shaking platform, the lysate was collected and totalproteins isolated after centrifugation at 20 000 g for 15 min. Quickstart protein assay reagent was used to estimate the proteinconcentration based on the principle of Bradford method thatinvolves binding of Commasie Brilliant Blue G-250 dye to proteins.A 10 mg/mL stock solution of bovine serum albumin was preparedand further diluted to 1.5, 1.0, 0.75, 0.5, 0.25, 0.125, and0.0625 mg/mL using double distilled water. 5 lL of the dilutedsample was added into a 96 well clear bottom plate in quadruplicate
© 2013 International Society for Neurochemistry, J. Neurochem. (2014) 128, 459--471
Neuroprotective effects of liraglutide in cell culture 461

along with 20 lL of double distilled water and 150 lL of the Quickstart protein assay reagent and the plate read after 5 min at 595 nm.
Cell lysate containing 5 lg of protein was separated on 4–12%gradient Bis-Tris gel with Novex pre-stained marker and electro-phoresed in running buffer at 200 mV for 35 min followed bytransfer to polyvinylidene difluoride membrane. Following proteintransfer, the membrane was washed in 1X TBST (tris-bufferedsaline with 0.05% Tween-20, pH 8) and blocked in 5% skimmedmilk for 1 h at 25�C. The membrane was then incubated with anti-Mcl1 (1 : 200), anti-Bax (1 : 200), anti-Bik (1 : 200), anti-pAkt(Ser473) (1 : 400), anti-phospho-MEK1/2 (Ser217/221) (1 : 1000),anti-phospho-p90RSK (Ser380) (1 : 1000) antibodies at 25�C for1 h and after three washes (10 min each) in TBST furtherincubated with 1 : 400 horseradish peroxidase-conjugated anti-rabbit IgG. All the primary antibodies used were generated inrabbit. The protein bands were visualized by Amersham ECLPrime western blotting detection reagent according to the manu-facturer’s recommendation. Image J was used to perform thedensitometric analysis of each band density. Selecting each bandon the scan created the profile plot and the peaks corresponded tothe dark bands in the original image. GAPDH was used as loadingcontrol and relative density of the peaks were calculated in Excelspreadsheet after normalizing with GAPDH. To reprobe, themembranes were incubated in a 0.5 M Tris HCl buffer pH 6.8containing 10% SDS with 0.8% ß-mercaptoethanol with someagitation at 50°C for 45 min followed by thorough rinse in waterfor 1 h and extensive washing in TBST for 5 min. The membraneswere checked with chemiluminescent detection preceding incuba-tion in another primary antibody of interest.
Statistical analysis
Statistical analysis was done using Prism 5.0 (GraphPad softwareInc., La Jolla, CA, USA) with the 95% level of probability and theresults were presented as mean � SEM. Data were analysed byunpaired Student’s t-test and one-way ANOVA, followed by Bonfer-roni’s multiple comparison post-hoc.
Results
Liraglutide pre-treatment increases cell survival and cell
proliferation and reduces cell cytotoxicity
To determine the neuroprotective effect of Liraglutide in theSH-SY5Y human neuroblastoma cells from oxidative stressby Methylglyoxal (MG), we performed the XTT and LDHassays to study cell viability and cytotoxicity, respectively.To examine whether GLP-1 analogue, Liraglutide canprotect SH-SY5Y cells from MG toxicity, cells were pre-treated with 10, 100, and 200 nM Liraglutide and afterstressed with 300, 600 and 1200 lM MG for 12 h and cellviability determined by XTT assay (Fig. 1a). Liraglutidepre-treatment significantly ameliorated 600 lM MG inducedstress at 100 (p < 0.001) and 200 nM (p < 0.01) whencompared to 0 nM Liraglutide. Although 1200 lM MGconcentration has the highest impact on reducing the cellviability, 200 nM Liraglutide was still effective significantly(p < 0.05). A 50% decrease in cell viability at 600 lM MG
was restored to 80% upon pre-treatment with 100 nMLiraglutide. For additional assessment of cell viability, LDHlevels were quantified in the supernatant collected after 12 h
(a)
100
100
200
300
ns nsns
ns
ns
ns
**** **
* **
* *
** **
***
*** ***
*
∂
∂∂
∂
∂
∂
50
0
0
200
600
400
Cel
l pro
lifer
atio
n(%
of c
ontro
l)
0
0 300 600 1200
0 nM Lira10 nM Lira100 nM Lira200 nM Lira
0 nM Lira10 nM Lira100 nM Lira200 nM Lira
0 nM Liraglutide10 nM Liraglutide100 nM Liraglutide200 nM Liraglutide
Concentration of methylglyoxal (μM)
0 300 600 1200Concentration of methylglyoxal (μM)
0 300 600Concentration of methylglyoxal (μM)
Sur
viva
l (%
of c
ontro
l)LD
H le
vels
(% o
f con
trol)
(b)
(c)
Fig. 1 Neuroprotective effect of glucagon-like peptide 1 analogue,
Liraglutide against methyl glyoxal (MG) induced cell death. Liraglutidepre-treatment increase cell survival and cell proliferation and reducescell cytotoxicity. (a) Liraglutide protected SH-SY5Y cells from 300, 600,
and 1200 lM concentration of MG induced stress. Cells were pre-treated with 10, 100, and 200 nM Liraglutide for 4 h followed byexposure to different concentration of MG for 12 h. (b) Liraglutide (100
and 200 nM) pre-treatment prevented MG (300, 600, and 1200 lM)induced increase in levels of lactate dehydrogenase (LDH). (c) Cellproliferation [5-bromo-2-2deoxyuridine (BrdU)]: Increasing concentra-
tion of Liraglutide (10, 100, 200 nM) pre-treatmemt for 4 h elevatedBrdU concentration after 12 h versus untreated controls. 100 nMLiraglutide dose induced maximum BrdU incorporation followed by 0and 300 lM MG induced stress. Data are presented as mean � SEM
and as a percentage of control. Statistical analysis was done byone-way ANOVA followed by Bonferroni’s Multiple Comparison test(*p < 0.05, **p < 0.01, ***p < 0.001), n = 5. (a–b) Statistical analyses
of 0 lM MG versus 300, 600 and 1200 lM MG without Liraglutide(op < 0.001).
© 2013 International Society for Neurochemistry, J. Neurochem. (2014) 128, 459--471
462 M. K. Sharma et al.

of MG stress. As shown in Fig. 1b, pre-treatment of SH-SY5Y cells with 100 and 200 nM Liraglutide conferredsimilar protective effects, almost reversing the increasedLDH levels, especially at 600 lM MG stress (p < 0.001).Together, these results demonstrate that 100 nM Liraglutideis protective against 600 lM MG induced stress in SH-SY5Y cells. We considered 600 lM concentration of MGstress for further studies as 100 nM Liraglutide exhibitedhighly significant effect at this stress, both shown by XTTand LDH assays.After showing successful neuroprotective effects by
Liraglutide pre-treatment, we next thought to examine theproliferative effects by monitoring BrdU incorporation. Adose dependent increase in cell proliferation was observedafter Liraglutide treatment. Figure 1c shows a threefoldincrease in cell proliferation at 100 nM Liraglutide(p < 0.01) concentration when stressed with 300 lM MG.Interestingly, there was no significant difference in prolifer-ation in cells stressed with 600 lM MG, demonstrating cellprotection by Liraglutide.
Liraglutide pre-treatment increases GLP-1R expressionTo confirm the presence of GLP-1 receptor on SH-SY5Ycells, we performed an immunostaining with the GLP-1Rantibody in cells exposed to 0 nM (Fig. 2a) and 100 nM(Fig. 2b) Liraglutide for 4 h. GLP-1R fluorescence wasquantified to compare total cell fluorescence in Liraglutidetreated (68881.27 � SEM) v/s un-treated cells(39034.9 � SEM) (Fig. 2c). A 1.76 fold increase in theGLP-1R expression was observed (p < 0.01).
Liraglutide pre-treatment protects against MG induced
apoptosis
To elucidate whether the neuroprotection conferred byLiraglutide pre-treatment against MG induced stressinvolves protection from apoptosis, we performed Hoechst33342 and PI staining on the cells pre-treated with 100 nMLiraglutide, after stressed for 12 h with 600 lM MG stress.Figure 3d shows a decrease in apoptotic cells in theLiraglutide treated (19.65 � SEM) as compared toun-treated ones (31.67 � SEM) (Fig. 3c). Number of
(a)
(b)
(c) 100 000
Cor
rect
ed to
tal c
ell
fluor
esce
nce
(CT
CF
)
80 000 **
60 000
40 000
20 000
0Liraglutide (nM) 0 100
Hoechst 33342 0 μm 100 GLP-1R 0 μm 100 Overlay 0 μm 100
Hoechst 33342 0 μm 100 GLP-1R 0 μm 100 Overlay 0 μm 100
Fig. 2 Representative photomicrographs showing GLP-1R immuno-staining in SH-SY5Y cells, 40X. (a) 0 nM Liraglutide (Control), (b)100 nM Liraglutide pre-treatment for 4 h. Nuclei were stained withHoechst 33342 and imaging performed using a confocal microscope.
(c) Quantification of GLP-1R fluorescence by Image J. 100 nM
Liraglutide treated SH-SY5Y cells (68881.27 � SEM) when comparedwith non-treated cells (39034.9 � SEM) shows a significant 1.76 foldincrease in the corrected total cell fluorescence (CTCF) of GLP-1Rexpression. Data are presented as mean � SEM. Statistical evalua-
tion done by Unpaired t-test, **p < 0.01.
© 2013 International Society for Neurochemistry, J. Neurochem. (2014) 128, 459--471
Neuroprotective effects of liraglutide in cell culture 463

apoptotic cells was quantified and a significant decrease(p < 0.001) of 37.9% in the % apoptotic cells wasobserved (Fig. 3f).
Liraglutide pre-treatment decreases caspase expression
To confirm the effect of Liraglutide in reducing theapoptotic effect induced by MG, we performed caspase-3
(a)
(b)
(c)
(d)
(e)
(f)
Hoechst 33342 PI Overlay
Overlay
Overlay
Overlay
Overlay
PI
PI
PI
PI
Hoechst 33342
Hoechst 33342
Hoechst 33342
Hoechst 33342
10 μm
10 μm
10 μm
10 μm
2 μm
Concentration of methyly glyoxal (µM)
0 600
ns
0 nM Lira100 nM Lira
40
30
20
% A
popt
otic
cel
ls
10
0
***
Fig. 3 Liraglutide pre-treatment protects against MG induced apop-tosis. Representative images of three independent experimentsshowing staining of SHSY5Y cells with Hoechst 33342 and propidiumiodide (PI), 20X. (a) 0 nM Liraglutide followed by 0 lM MG (Control),
(b) 100 nM Liraglutide followed by 0 lM MG, (c) 0 nM Liraglutidefollowed by 600 lM MG, (d) Pre-treated with 100 nM Liraglutide for4 h followed by 600 lM MG for 12 h. (e) Representative image
showing apoptotic (thick arrow) and normal (thin arrow) nuclei, 100X,(f) Quantification of apoptotic cells. 100 nM Liraglutide pre-treatedSHSY5Y cells (19.65 � SEM) when compared with non-treated600 lM MG stressed cells (31.67 � SEM) shows a significant
decrease of 37.9% in the%age of apoptotic cells. Data analysed byOne-way ANOVA followed by Bonferroni’s Multiple Comparison test,***p < 0.001.
© 2013 International Society for Neurochemistry, J. Neurochem. (2014) 128, 459--471
464 M. K. Sharma et al.

immunostaining in 100 nM Liraglutide pre-treated SH-SY5Y cells stressed with 600 lM MG for 8 h. Figure 4dand e shows a decrease in the expression of cleaved form ofcaspase-3 in the Liraglutide treated (32966.47 � SEM) as
compared to the un-treated cells (44258.13 � SEM) (Fig. 4cand e). Corrected total cell fluorescence of caspase-3expression was measured (Fig. 4e) and a significant decreaseof 25.5% was observed.
(a) (e)
(j)
(b)
(c)
(d)
(f) (g)
(h) (i)
Hoechst 33342 Caspase-3 Overlay 10 μm
Hoechst 33342 Caspase-3 Overlay 10 μm
Hoechst 33342 Caspase-3 Overlay 10 μm
Hoechst 33342 Caspase-3 Overlay 10 μm
10 μm10 μm
10 μm10 μm
Concentration of methyly glyoxal (µM)0 0 600 600
ns
0 nM Lira100 nM Lira
50 000
40 000
30 000
20 000
10 000Cor
rect
ed to
tal c
ell
fluor
esce
nce
(CT
CF
)
0
**
Concentration of methylglyoxal (µM)0 0 600 600
ns
0 nM Lira100 nM Lira
200
150
100
50Inte
grat
ed d
ensi
ty
0
**
Fig. 4 Representative photomicrographs showing caspase-3 immu-nostaining and caspase Live Imaging in SH-SY5Y cells stressed with
600 lM Methylglyoxal for 8 h, 20X. Caspase-3 immunostaining (a)0 nM Liraglutide followed by 0 lMMG (Control), (b) 100 nM Liraglutidefollowed by 0 lM MG, (c) 0 nM Liraglutide followed by 600 lM MG, (d)
Pre-treated with 100 nM Liraglutide for 4 h followed by 600 lMMG, (e)Quantification of caspase-3 fluorescence by Image J. 100 nM Liraglu-tide pre-treated SHSY5Y cells (32966.47 � SEM) when compared
with non-treated 600 lM MG stressed cells (44258.13 � SEM) showsa significant decrease of 25.5% in the corrected total cell fluorescence(CTCF) of caspase expression. Caspase Live Imaging (using Image-iT
Live Green Poly Caspases Detection kit) (f) 0 nM Liraglutide followedby 0 lMMG (Control), (g) 100 nM Liraglutide followed by 0 lMMG, (h)
0 nM Liraglutide followed by 600 lM MG, (i) Pre-treated with 100 nMLiraglutide for 4 h followed by 600 lM MG, (j) Quantification ofcaspase fluorescence by Image J. 100 nM Liraglutide pre-treated
SHSY5Y cells (93.2 � SEM) when compared with non-treated 600 lMMG stressed cells (136.3 � SEM) shows a significant decrease of32% in the measured Integrated density of caspase expression. Data
analysed by One-way ANOVA followed by Bonferroni’s multiple compar-ison test, **p < 0.01.
© 2013 International Society for Neurochemistry, J. Neurochem. (2014) 128, 459--471
Neuroprotective effects of liraglutide in cell culture 465

To further corroborate a change in the caspase-3 levels, weperformed a polycaspase Live Imaging in 100 nM Liraglu-tide pre-treated SH-SY5Y cells stressed with 600 lM MGfor 8 h. Figure 4i shows a decrease in the expression ofcaspases in the treated as compared to the un-treated cells(Fig. 4h). Quantification of caspase fluorescence showed asignificant decrease of 32% (p < 0.01) in the measuredintegrated density of caspase expression (Fig. 4j).
Liraglutide pre-treatment decreases pro-apoptotic (Bax andBik) and increases pro-survival Mcl1 expression
Two other representative markers for apoptotic cell death,Bax and Bik were assessed by western blot analysis.Figure 5a shows altered Bax and Bik expression when cellspre-treated with 100 nM Liraglutide followed by 600 lMMG were compared to untreated cells. Densitometric analy-sis of Bax (Fig. 5b) and Bik (Fig. 5c) showed a significantdecrease (p < 0.001) in the 100 nM Liraglutide treated cells.
MG stress resulted in almost a twofold increase in the proteinlevels of Bax and Bik that was markedly ameliorated byLiraglutide pre-treatment.In addition to the pro-apoptotic markers, we also
performed western blot analysis of Mcl1, a member ofpro-survival Bcl-2 family and found a significant decrease(~ 1.8-fold) in its levels in MG stressed cells, which waspartially restored to control levels by Liraglutide pre-treatment (Fig. 5d). Densitometric analysis of Mcl1 (Fig. 5e)showed a significant increase (p < 0.001) in the 100 nMLiraglutide treated cells, thus, further confirming an overalldecrease in the cell apoptosis as a mechanism involved inneuroprotection conferred by Liraglutide.
Liraglutide pre-treatment restores membrane potential and
normalises intracellular calcium levels
The FLIPR membrane potential assay kit was used to detection channel modulation by varying the fluorescent signal in
(a)Bax (20 kDa)
Bik (20 kDa)
GAPDH (37 kDa)
Liraglutide 100 nM ––
+–
–+
++
––
+–
–+
++
MG 600 nM
Mcl1 (40 kDa)GAPDH (37 kDa)Liraglutide 100 nMMG 600 nM
0 nM Lira100 nM Lira
ns
250
200
150
100
50
Bax
den
sity
(% o
f con
trol
)
00
600Concentration of MG (μM)
***0 nM Lira100 nM Lira
ns
200
150
100
50
Bik
den
sity
(% c
ontr
ol)
00
600Concentration of MG (μM)
***
0 nM Lira100 nM Lira
ns
150
100
50
Mcl
1 de
nsity
(% o
f con
trol
)
00
600Concentration of MG (μM)
***
(b) (c)
(d) (e)
Fig. 5 Western blot analysis of pro-apoptotic and pro-survival mark-ers. Analysis of pro-apoptotic markers, Bax and Bik (a). Western blotshowing altered Bax and Bik expression when cells were pre-treated
with 100 nM Liraglutide followed by 600 lM MG as compared tountreated cells, (b) Densitometric analysis of Bax and (c). Bik. Both thepro-apoptotic markers show a significant decrease. Analysis of
pro-survival marker, Mcl1 (d). Western blot showing altered Mcl1expression when cells were pre-treated with 100 nM Liraglutidefollowed by 600 lM MG as compared to untreated cells, (e). Densi-tometric analysis of Mcl1. A significant increase in the Mcl1 expressionwas observed. Analysis by One-way ANOVA followed by Bonferroni’smultiple comparison test, ***p < 0.001.
© 2013 International Society for Neurochemistry, J. Neurochem. (2014) 128, 459--471
466 M. K. Sharma et al.
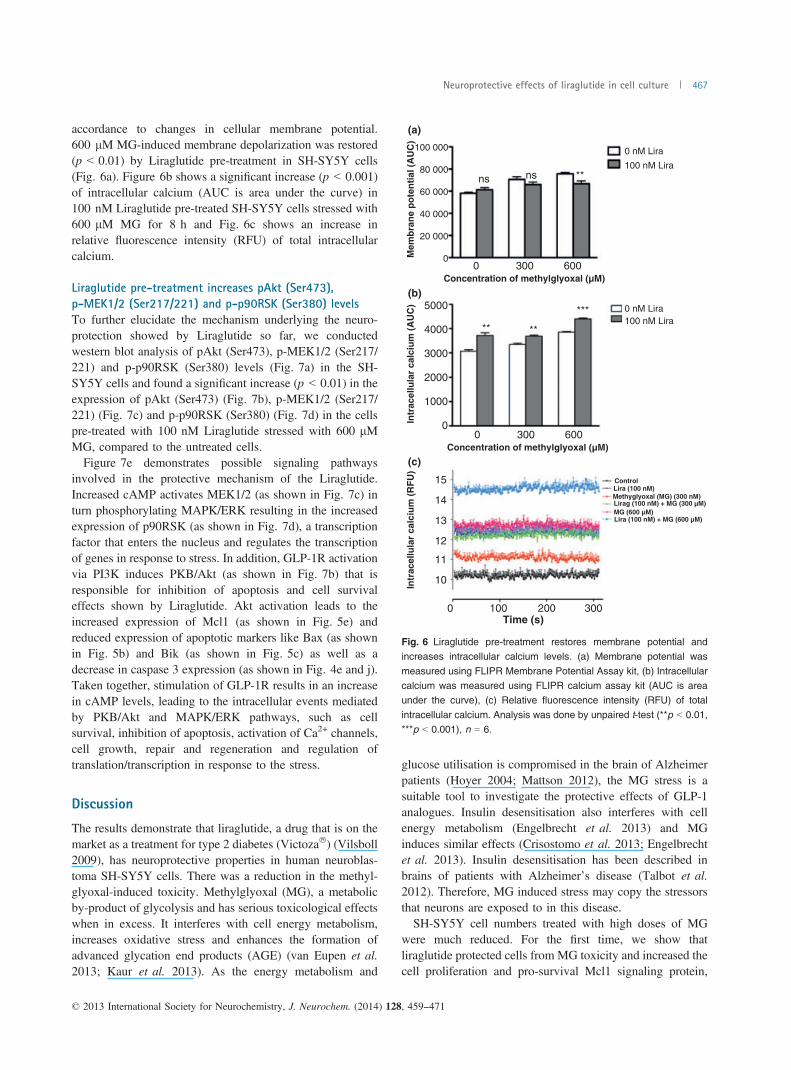
accordance to changes in cellular membrane potential.600 lM MG-induced membrane depolarization was restored(p < 0.01) by Liraglutide pre-treatment in SH-SY5Y cells(Fig. 6a). Figure 6b shows a significant increase (p < 0.001)of intracellular calcium (AUC is area under the curve) in100 nM Liraglutide pre-treated SH-SY5Y cells stressed with600 lM MG for 8 h and Fig. 6c shows an increase inrelative fluorescence intensity (RFU) of total intracellularcalcium.
Liraglutide pre-treatment increases pAkt (Ser473),
p-MEK1/2 (Ser217/221) and p-p90RSK (Ser380) levels
To further elucidate the mechanism underlying the neuro-protection showed by Liraglutide so far, we conductedwestern blot analysis of pAkt (Ser473), p-MEK1/2 (Ser217/221) and p-p90RSK (Ser380) levels (Fig. 7a) in the SH-SY5Y cells and found a significant increase (p < 0.01) in theexpression of pAkt (Ser473) (Fig. 7b), p-MEK1/2 (Ser217/221) (Fig. 7c) and p-p90RSK (Ser380) (Fig. 7d) in the cellspre-treated with 100 nM Liraglutide stressed with 600 lMMG, compared to the untreated cells.Figure 7e demonstrates possible signaling pathways
involved in the protective mechanism of the Liraglutide.Increased cAMP activates MEK1/2 (as shown in Fig. 7c) inturn phosphorylating MAPK/ERK resulting in the increasedexpression of p90RSK (as shown in Fig. 7d), a transcriptionfactor that enters the nucleus and regulates the transcriptionof genes in response to stress. In addition, GLP-1R activationvia PI3K induces PKB/Akt (as shown in Fig. 7b) that isresponsible for inhibition of apoptosis and cell survivaleffects shown by Liraglutide. Akt activation leads to theincreased expression of Mcl1 (as shown in Fig. 5e) andreduced expression of apoptotic markers like Bax (as shownin Fig. 5b) and Bik (as shown in Fig. 5c) as well as adecrease in caspase 3 expression (as shown in Fig. 4e and j).Taken together, stimulation of GLP-1R results in an increasein cAMP levels, leading to the intracellular events mediatedby PKB/Akt and MAPK/ERK pathways, such as cellsurvival, inhibition of apoptosis, activation of Ca2+ channels,cell growth, repair and regeneration and regulation oftranslation/transcription in response to the stress.
Discussion
The results demonstrate that liraglutide, a drug that is on themarket as a treatment for type 2 diabetes (Victoza�) (Vilsboll2009), has neuroprotective properties in human neuroblas-toma SH-SY5Y cells. There was a reduction in the methyl-glyoxal-induced toxicity. Methylglyoxal (MG), a metabolicby-product of glycolysis and has serious toxicological effectswhen in excess. It interferes with cell energy metabolism,increases oxidative stress and enhances the formation ofadvanced glycation end products (AGE) (van Eupen et al.2013; Kaur et al. 2013). As the energy metabolism and
glucose utilisation is compromised in the brain of Alzheimerpatients (Hoyer 2004; Mattson 2012), the MG stress is asuitable tool to investigate the protective effects of GLP-1analogues. Insulin desensitisation also interferes with cellenergy metabolism (Engelbrecht et al. 2013) and MGinduces similar effects (Crisostomo et al. 2013; Engelbrechtet al. 2013). Insulin desensitisation has been described inbrains of patients with Alzheimer’s disease (Talbot et al.2012). Therefore, MG induced stress may copy the stressorsthat neurons are exposed to in this disease.SH-SY5Y cell numbers treated with high doses of MG
were much reduced. For the first time, we show thatliraglutide protected cells from MG toxicity and increased thecell proliferation and pro-survival Mcl1 signaling protein,
(a)
(b)
(c)
100 000
Mem
bra
ne
po
ten
tial
(A
UC
)
80 000**nsns
0 nM Lira
100 nM Lira
60 000
40 000
20 000
00 300 600
Concentration of methylglyoxal (µM)
5000
Intr
acel
lula
r ca
lciu
m (
AU
C)
4000 ** **
*** 0 nM Lira100 nM Lira
3000
2000
1000
00 300 600
Concentration of methylglyoxal (µM)
0
15
14
13
12
11
10Intr
acel
lula
r ca
lciu
m (
RF
U)
100 200
ControlLira (100 nM)
Lirag (100 nM) + MG (300 µM)
Lira (100 nM) + MG (600 µM)MG (600 µM)
Methyglyoxal (MG) (300 nM)
300Time (s)
Fig. 6 Liraglutide pre-treatment restores membrane potential andincreases intracellular calcium levels. (a) Membrane potential was
measured using FLIPR Membrane Potential Assay kit, (b) Intracellularcalcium was measured using FLIPR calcium assay kit (AUC is areaunder the curve), (c) Relative fluorescence intensity (RFU) of total
intracellular calcium. Analysis was done by unpaired t-test (**p < 0.01,***p < 0.001), n = 6.
© 2013 International Society for Neurochemistry, J. Neurochem. (2014) 128, 459--471
Neuroprotective effects of liraglutide in cell culture 467

(a)
(b)
(e)
(c) (d)0 nM Lira100 nM Lira
0 nM Lira100 nM Lira
ns
ns 200
250
150
100
50
p-M
EK
1/2
(Ser
217/
221)
dens
ity (%
of c
ontro
l)
p-MEK1/2 (Ser217/221) (45 kDa)pA
kt (S
er47
3)de
nsity
(% o
f con
trol)
pAkt (Ser473) (60 kDa)
00
150
100
50
0600
Concentration of MG (μM)0 600
Concentration of MG (μM)
L Liraglutide
GLP-1R
PI3KBax
Bik
Casp-9
Casp-3
P
P
P
P
P
P
P
P
PAktPKB/
MAPK/ERK
AC
ADP
EPAC
CREB
c-Raf
CytoplasmNucleus
MEK1/2
p90RSK
p90RSK
cAMPATP
ATP
Mcl1
PKAInhibition ofApoptosis
Regulation of translation andtranscription in response to
stress
Cell growth, synapsegrowth,
repair and regeneration
Cell survival
Extracellular membrane
Activation of Ca2+ channels
Ca2+
Ca2+
Ca2+
Ca2+
****0 nM Lira100 nM Lira
ns
200
150
100
50
Pho
spho
-p90
RS
K (S
er38
0)de
nsity
(% o
f con
trol)
p-p90RSK (Ser380) (90 kDa)
GAPDH (37 kDa)
MG 600 μM
Liraglutide 100 nM
00
600Concentration of MG (μM)
**
–
–
+
–
–
+
+
+
Fig. 7 Western blot analysis of pAkt (Ser473), p-MEK1/2, p-p90RSK.
(a) Western blot showing altered pAkt (Ser473), p-MEK1/2 (Ser217/221) and p-p90RSK (Ser380) expression when cells were pre-treatedwith 100 nM Liraglutide followed by 600 lM MG as compared tountreated cells, (b–d) Densitometric analysis of pAkt (Ser473), p-
MEK1/2 (Ser217/221), and p-p90RSK (Ser380), respectively, shows asignificant increase in their expression levels as analysed by One-wayANOVA followed by Bonferroni’s multiple comparison test, **p < 0.01. (e)
Diagrammatic representation of the potential pathways that underliesthe neuroprotective effects of Liraglutide, mediated by PKB/Akt andMAPK/ERK pathways. Liraglutide stimulates GLP-1R resulting in an
increase in the cAMP further leading to intracellular events such as cell
survival, inhibition of apoptosis, activation of Ca2+ channels, cell
growth, repair and regeneration and regulation of translation/transcrip-tion in response to stress. GLP-1R, GLP-1 receptor; PKA, proteinkinase A; PI3K, phosphoinositide 3 kinase; PKB, protein kinase B; AC,adenylate cylase; EPAC, exchange proteins directly activated by
cAMP; MAPK, mitogen-activated protein kinase; ERK, extracellularsignal-regulated kinase; CREB, cyclic AMP response element bindingprotein; P90RSK, ribosomal S6 kinase; MEK1/2, MAPK or Erk kinases;
c-Raf, cellular Raf gene (Rapidly accelerated fibrosarcoma); Mcl1,myeloid cell leukaemia protein-1; Casp-9, caspase 9; Casp-3, caspase3; Bax, Bcl2 associated X protein; Bik, Bcl2-interacting killer; Ca2+,
calcium ions.
© 2013 International Society for Neurochemistry, J. Neurochem. (2014) 128, 459--471
468 M. K. Sharma et al.

which is a member of the Bcl-2 family (Vlacic-Zischke et al.2011). Activation of the growth factor signal and cellsurvival kinases Akt and MEK1/2 was also increased byliraglutide. These kinases activate the transcription factorp90RSK. In addition, liraglutide reduced the induction ofapoptosis and caspase and pro-apoptotic Bax and Bikexpression (see summary Fig. 7e). Also, the membranepotential and the influx of Ca2+ into the cell was enhanced byliraglutide treatment. In neurons, Ca2+ influx after theinitiation of action potentials is an important intracellularmessenger for the activation of key enzymes and themodulation of synaptic activity and signal transduction(Bliss and Collingridge 1993). Dysregulation of Ca2+
signaling and of the membrane potential can severelyinterfere with neuronal firing, synaptic plasticity and signalprocessing (Stanton 1996). The normalisation of Ca2+ levelsin neurons is a major factor in protecting neurons andpreserving their functionality. As beta-amyloid is known todestabilise Ca2+ levels in neurons (Holscher 1998), GLP-1mimetics may be protective as well. Previous research foundthat GLP-1 activates L-type voltage dependent calciumchannels and thereby activates ERK growth factor signaling(Kennedy 1989; Selway et al. 2012). It is of interest to notethat GLP-1 receptor expression was increased after treatmentwith liraglutide, thereby enhancing GLP-1 signaling andpotentiating the effects of liraglutide over time. Treatment ofthe cells with liraglutide furthermore increased the expres-sion of GLP-1 receptors, as had been reported in a previousstudy that showed an increase in GLP-1R expression in vivoafter drug treatment (Romani-Perez et al. 2013). Thisincrease of expression suggests a positive feedback loop toenhance GLP-1 signaling.The activation of growth factor signaling and the reduction
of apoptosis is most likely the underlying molecularmechanism for the neuroprotective effects of liraglutidefound in several studies. An additional property of growthfactors is that they reduce inflammation. We have shown thatliraglutide reduces a chronic inflammation response (Parth-sarathy and Holscher 2013b), which also contributes to theneuroprotective effects. Other growth factors have shown avery similar profile of activating cell growth genes, energyutilization, synapse protection, and synaptogenesis, and aprotection of memory formation (Holscher 2011). BDNF hasbeen shown to protect synapses in mouse models of AD.Injecting BDNF icv improved memory formation, reducedthe observed impairments of synaptic plasticity in this mousemodel, and increased synaptic numbers (Blurton-Jones et al.2009). Enhancing the BDNF levels in the brain by genedelivery vectors also has demonstrated neuroprotectiveeffects on synapses. The elevation of BDNF levels reversedsynapse loss, improves synaptic plasticity and restoreslearning abilities of a mouse model of AD (Nagahara et al.2009; Poon et al. 2009). BDNF also activates growth factorcell signaling such as the PI3K and PKB/Akt pathway
(Foulstone et al. 1999; Duan et al. 2012). This suggests thatgrowth factors activate similar pathways and can inducesimilar physiological changes. However, BDNF does notcross the blood–brain barrier (BBB), and therefore it wouldneed to be injected into the brain in order to be effective(Schulte-Herbruggen et al. 2007; Zuccato and Cattaneo2009). There is also an interaction between growth factors.Recent studies have shown that peripheral injection of GLP-1 mimetics enhance BDNF levels in the brain (Mattson2012). This is an additional pathway of how GLP-1 canenhance growth factor cell signaling in neurons. A differentgrowth factor that has very similar properties is NGF. NGFwas found to protect synapses, synaptic plasticity, andlearning abilities in AD mouse models or in nonprimatemonkeys (Clarris et al. 1994; Kordower et al. 1997;Covaceuszach et al. 2009). However, NGF does not crossthe BBB either, which severely limits the potential use ofthis growth factor to protect the brain (Bradbury 2005; Heeseet al. 2006; Schulte-Herbruggen et al. 2007; Covaceuszachet al. 2009). Therefore, enhancing the GLP-1 signal withmimetics that can cross the BBB may compensate for IGF-1and Insulin desensitisation, which has been observed in thebrains of AD patients (Hoyer 1998; de la Monte 2011;Talbot et al. 2012). Phosphorylation of the insulin receptor ßchain was reduced at positions IRb pY1150/1151 and IRbpY960, while the insulin receptor substrate 1 (IRS-1) washyperphosphorylated at positions IRS-1 pS616 and IRS-1pS636, which deactivates IRS-1 signaling, and IRS-1 bindingto PI3K p85a was also much reduced, reducing theactivation of PI3K (Talbot et al. 2012). A histological studyof brain tissue of AD patients found similar effects (Moloneyet al. 2010). Insulin signaling impairment has detrimentaleffects on cognition in AD patients (Carro and Torres-Aleman 2004; Craft 2005; Reger et al. 2008). Based onthese findings, a phase II clinical trial testing nasal insulinapplication in patients with AD had been conducted andshowed improvement in key biomarkers and cognition (Craftet al. 2012; Freiherr et al. 2013).The same profile of insulin desensitisation found in the
brains of AD patients was also found in the APP/PS1 mousemodel of AD (Bomfim et al. 2012), and treatment with theGLP-1 mimetic exendin-4 (Bomfim et al. 2012) or withliraglutide (Wang et al. 2012) reversed the insulin desensi-tisation profile of insulin cell signaling proteins. In addition,we have shown in previous studies that liraglutide enhancessynaptic plasticity in the hippocampus of the rat (McCleanet al. 2010; Han et al. 2013), progenitor cell proliferationand neurogenesis in the dentate gyrus in the mouse brain(Hamilton et al. 2011) and reduced amyloid plaque load,amyloid oligomer levels, activated microglia chronic inflam-mation in the brains of APP/PS1 mice, while preservingspatial learning, synapse numbers in the cortex andhippocampus, and enhancing synaptic plasticity and normal-ising neurogenesis in the hippocampus (McClean et al.
© 2013 International Society for Neurochemistry, J. Neurochem. (2014) 128, 459--471
Neuroprotective effects of liraglutide in cell culture 469

2011). Based on these encouraging preclinical findings, aclinical trial of liraglutide in patients with AD has started (seehttp://clinicaltrials.gov/ct2/show/NCT01843075?term=liraglu-tide+and+alzheimer&rank=1.The data presented in this study on the neuroprotective
effects in SH-SY5Y cells add important information on thecellular mechanisms that underlie the neuroprotective effectsobserved in the previous studies (Holscher 2012).
Acknowledgement/conflict of interest
The work has been supported by Vice Chancellor’s ResearchScholarship, University of Ulster, NI, UK and Alzheimer’s ResearchUK. The authors MKS and JJ do not declare a conflict of interest.CH is a named inventor on a patent application by Ulster Universitythat lists GLP-1 analogues as a potential treatment for neurodegen-erative diseases.
References
Bliss T. V. P. and Collingridge G. L. (1993) A synaptic model ofmemory: long-term potentiation in the hippocampus. Nature 361,31–39.
Blurton-Jones M., Kitazawa M., Martinez-Coria H., Castello N. A.,Muller F. J., Loring J. F., Yamasaki T. R., Poon W. W., Green K.N. and Laferla F. M. (2009) Neural stem cells improve cognitionvia BDNF in a transgenic model of Alzheimer disease. Proc. NatlAcad. Sci. USA 106, 13594–13599.
Bomfim T. R., Forny-Germano L., Sathler L. B. et al. (2012) An anti-diabetes agent protects the mouse brain from defective insulinsignaling caused by Alzheimer’s disease-associated Abetaoligomers. J. Clin. Invest. 122, 1339–1353.
Bradbury J. (2005) Hope for AD with NGF gene-therapy trial. LancetNeurol. 4, 335.
Carro E. and Torres-Aleman I. (2004) The role of insulin and insulin-likegrowth factor I in the molecular and cellular mechanismsunderlying the pathology of Alzheimer’s disease. Eur. J.Pharmacol. 490, 127–133.
Christensen M., Knop F. K., Vilsboll T. and Holst J. J. (2011)Lixisenatide for type 2 diabetis mellitus. Expert Opin. Investig.Drugs 20, 549–557.
Clarris H. J., Nurcombe V., Small D. H., Beyreuther K. and Masters C. L.(1994) Secretion of nerve growth factor from septum stimulatesneurite outgrowth and release of the amyloid protein precursor ofAlzheimer’s disease from hippocampal explants. J. Neurosci. Res.38, 248–258.
Covaceuszach S., Capsoni S., Ugolini G., Spirito F., Vignone D. andCattaneo A. (2009) Development of a non invasive NGF-basedtherapy for Alzheimer’s disease. Curr. Alzheimer Res. 6, 158–170.
Craft S. (2005) Insulin resistance syndrome and Alzheimer’s disease:age- and obesity-related effects on memory, amyloid, andinflammation. Neurobiol. Aging 26, 65–69.
Craft S., Baker L. D., Montine T. J. et al. (2012) Intranasal insulintherapy for Alzheimer disease and amnestic mild cognitiveimpairment: a pilot clinical trial. Arch. Neurol. 69, 29–38.
Crisostomo J., Matafome P., Santos-Silva D., Rodrigues L., Sena C. M.,Pereira P. and Seica R. (2013) Methylglyoxal chronicadministration promotes diabetes-like cardiac ischaemia diseasein Wistar normal rats. Nutr. Metab. Cardiovasc. Dis. in press. doi:10.1016/j.numecd.2013.01.005.
Doyle M. E. and Egan J. M. (2007) Mechanisms of action of glucagon-like peptide 1 in the pancreas. Pharmacol. Ther. 113, 546–593.
Drucker D. J. and Nauck M. A. (2006) The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4inhibitors in type 2 diabetes. Lancet 368, 1696–1705.
Duan B., Liu D. S., Huang Y., Zeng W. Z., Wang X., Yu H., Zhu M. X.,Chen Z.Y. and Xu T. L. (2012) PI3-kinase/Akt pathway-regulatedmembrane insertion of acid-sensing ion channel 1a underliesBDNF-induced pain hypersensitivity. J. Neurosci. 32, 6351–6363.
During M. J., Cao L., Zuzga D. S. et al. (2003) Glucagon-like peptide-1receptor is involved in learning and neuroprotection. Nat. Med. 9,1173–1179.
Engelbrecht B., Stratmann B., Hess C., Tschoepe D. and Gawlowski T.(2013) Impact of GLO1 knock down on GLUT4 trafficking andglucose uptake in L6 myoblasts. PLoS ONE 8, e65195.
van Eupen M. G., Schram M. T., Colhoun H. M., Hanssen N. M.,Niessen H. W., Tarnow L., Parving H. H., Rossing P., StehouwerC. D. and Schalkwijk C. G. (2013) The methylglyoxal-derivedAGE tetrahydropyrimidine is increased in plasma of individualswith type 1 diabetes mellitus and in atherosclerotic lesions and isassociated with sVCAM-1. Diabetologia 56, 1845–1855.
Fan H., Wang L., Guo F., Wei S. and Zhao R. (2010) Neonatalintramuscular injection of plasmid encoding glucagon-like peptide-1 affects anxiety behaviour and expression of the hippocampalglucocorticoid receptor in adolescent rats. J. Biosci. 35, 63–71.
Foulstone E. J., Tavare J. M. and Gunn-Moore F. J. (1999) Sustainedphosphorylation and activation of protein kinase B correlates withbrain-derived neurotrophic factor and insulin stimulated survival ofcerebellar granule cells. Neurosci. Lett. 264, 125–128.
Freiherr J., Hallschmid M., Frey W. H., 2nd, Brunner Y. F., Chapman C. D.,Holscher C., Craft S., De Felice F. G. and Benedict C. (2013)Intranasal insulin as a treatment for Alzheimer’s disease: a reviewof basic research and clinical evidence. CNS Drugs 27, 505–514.
Gilman C. P., Perry T., Furukawa K., Grieg N. H., Egan J. M. andMattson M. P. (2003) Glucagon-like peptide 1 modulates calciumresponses to glutamate and membrane depolarization inhippocampal neurons. J. Neurochem. 87, 1137–1144.
Greig N. H., Mattson M. P., Perry T., Chan S. L., Giordano T.,Sambamurti K., Rogers J. T., Ovadia H. and Lahiri D. K. (2004)New therapeutic strategies and drug candidates forneurodegenerative diseases: p53 and TNF-{alpha} inhibitors, andGLP-1 receptor agonists. Ann. N. Y. Acad. Sci. 1035, 290–315.
Hamilton A., Patterson S., Porter D., Gault V. A. and Holscher C. (2011)Novel GLP-1 mimetics developed to treat type 2 diabetes promoteprogenitor cell proliferation in the brain. J. Neurosci. Res. 89, 481–489.
Han W.-N., Holscher C., Yuan L., Yang W., Wang X.-H., Wu M.-N.and Qi J.-S. (2013) Liraglutide protects against ß-amyloid inducedimpairment of spatial learning and memory in rats. Neurobiol.Aging 32, 576–588.
Heese K., Low J. W. and Inoue N. (2006) Nerve growth factor, neuralstem cells and Alzheimer’s disease. Neurosignals 15, 1–12.
Holscher C. (1998) Possible causes of Alzheimer’s disease: amyloidfragments, free radicals, and calcium homeostasis. Neurobiol. Dis.5, 129–141.
Holscher C. (2011) Diabetes as a risk factor for Alzheimer’s disease:insulin signalling impairment in the brain as an alternative modelof Alzheimer’s disease. Biochem. Soc. Trans. 39, 891–897.
Holscher C. (2012) Potential role of glucagon-like peptide-1 (GLP-1) inneuroprotection. CNS Drugs 26, 871–882.
Hoyer S. (1998) Risk factors for Alzheimer’s disease during aging.Impacts of glucose/energy metabolism. J. Neural Transm. Suppl.54, 187–194.
© 2013 International Society for Neurochemistry, J. Neurochem. (2014) 128, 459--471
470 M. K. Sharma et al.

Hoyer S. (2004) Glucose metabolism and insulin receptor signaltransduction in Alzheimer disease. Eur. J. Pharmacol. 490, 115–125.
Hunter K. and Holscher C. (2012) Drugs developed to treat diabetes,liraglutide and lixisenatide, cross the blood brain barrier andenhance neurogenesis. BMC Neurosci. 13, 33–38.
Jolivalt C. G., Fineman M., Deacon C. F., Carr R. D. and Calcutt N. A.(2011) GLP-1 signals via ERK in peripheral nerve and preventsnerve dysfunction in diabetic mice. Diabetes Obes. Metab. 13,990–1000.
Kaur S., Zilmer K., Leping V. and Zilmer M. (2013) Serummethylglyoxal level and its association with oxidative stress anddisease severity in patients with psoriasis. Arch. Dermatol. Res.305, 489–494.
Kennedy M. B. (1989) Regulation of neuronal function by calcium.Trends Neurosci. 12, 417–420.
Kim M., Platt M. J., Shibasaki T., Quaggin S. E., Backx P. H., Seino S.,Simpson J. A. and Drucker D. J. (2013) GLP-1 receptor activationand Epac2 link atrial natriuretic peptide secretion to control ofblood pressure. Nat. Med. 19, 567–575.
Kimura R., Okouchi M., Fujioka H. et al. (2009) Glucagon-like peptide-1 (GLP-1) protects against methylglyoxal-induced PC12 cellapoptosis through the PI3K/Akt/mTOR/GCLc/redox signalingpathway. Neuroscience 162, 1212–1219.
Kordower J. H., Mufson E. J., Fox N., Martel L. and Emerich D. F.(1997) Cellular delivery of NGF does not alter the expression ofbeta-amyloid immunoreactivity in young or aged nonhumanprimates. Exp. Neurol. 145, 586–591.
Li Y., Tweedie D., Mattson M. P., Holloway H. W. and Greig N. H.(2010) Enhancing the GLP-1 receptor signaling pathway leads toproliferation and neuroprotection in human neuroblastoma cells. J.Neurochem. 113, 1621–1631.
Madsbad S., Krarup T., Deacon C. F. and Holst J. J. (2008) Glucagon-like peptide receptor agonists and dipeptidyl peptidase-4 inhibitorsin the treatment of diabetes: a review of clinical trials. Curr. Opin.Clin. Nutr. Metab. Care 11, 491–499.
Mattson M. P. (2012) Energy intake and exercise as determinants ofbrain health and vulnerability to injury and disease. Cell Metab. 16,706–722.
McClean P. L., Gault V. A., Harriott P. and Holscher C. (2010)Glucagon-like peptide-1 analogues enhance synaptic plasticity inthe brain: a link between diabetes and Alzheimer’s disease. Eur. J.Pharmacol. 630, 158–162.
McClean P., Parthsarathy V., Faivre E. and H€olscher C. (2011) Thediabetes drug Liraglutide prevents degenerative processes in amouse model of Alzheimer’s disease. J. Neurosci. 31, 6587–6594.
Moloney A. M., Griffin R. J., Timmons S., O’Connor R., Ravid R. andO’Neill C. (2010) Defects in IGF-1 receptor, insulin receptor andIRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol. Aging 31, 224–243.
de la Monte S. M. (2011) Contributions of brain insulin resistance anddeficiency in amyloid-related neurodegeneration in Alzheimer’sdisease. Drugs 72, 49–66.
Nagahara A. H., Merrill D. A., Coppola G. et al. (2009) Neuroprotectiveeffects of brain-derived neurotrophic factor in rodent and primatemodels of Alzheimer’s disease. Nat. Med. 15, 331–337.
Nelson T. J. and Alkon D. L. (2005) Insulin and cholesterol pathways inneuronal function, memory and neurodegeneration. Biochem. Soc.Trans. 33(Pt 5), 1033–1036.
Noyan-Ashraf M. H., Momen M. A., Ban K., Sadi A. M., Zhou Y. Q.,Riazi A. M., Baggio L. L., Henkelman R. M., Husain M. andDrucker D. J. (2009) GLP-1R agonist liraglutide activatescytoprotective pathways and improves outcomes afterexperimental myocardial infarction in mice. Diabetes 58, 975–983.
Ojo O. O., Abdel-Wahab Y. H., Flatt P. R., Mechkarska M. and ConlonJ. M. (2011) Tigerinin-1R: a potent, non-toxic insulin-releasingpeptide isolated from the skin of the Asian frog, Hoplobatrachusrugulosus. Diabetes Obes. Metab. 13, 1114–1122.
ParthsarathyV. andHolscher C. (2013a) Chronic treatment with the GLP1analogue liraglutide increases cell proliferation and differentiationinto neurons in an AD mouse model. PLoS ONE 8, e58784.
Parthsarathy V. and Holscher C. (2013b) The type 2 diabetes drugliraglutide reduces chronic inflammation induced by irradiation inthe mouse brain. Eur. J. Pharmacol. 700, 42–50.
Perry T. A. and Greig N. H. (2004) A new Alzheimer’s diseaseinterventive strategy: GLP-1. Curr. Drug Targets 5, 565–571.
Perry T. and Greig N. H. (2005) Enhancing central nervous systemendogenous GLP-1 receptor pathways for intervention inAlzheimer’s disease. Curr. Alzheimer Res. 2, 377–385.
Perry T., Lahiri D. K., Chen D., Zhou J., ShawK. T., Egan J. M. and GreigN. H. (2002) A novel neurotrophic property of glucagon-like peptide1: a promoter of nerve growth factor-mediated differentiation inPC12 cells. J. Pharmacol. Exp. Ther. 300, 958–966.
Perry T., Lahiri D. K., Sambamurti K., Chen D., Mattson M. P., Egan J.M. and Greig N. H. (2003) Glucagon-like peptide-1 decreasesendogenous amyloid-beta peptide (Abeta) levels and protectshippocampal neurons from death induced by Abeta and iron. J.Neurosci. Res. 72, 603–612.
Poon W. W., Blurton-Jones M., Tu C. H., Feinberg L. M., ChabrierM. A., Harris J. W., Jeon N. L. and Cotman C. W. (2009) beta-Amyloid impairs axonal BDNF retrograde trafficking. Neurobiol.Aging 39, 821–833.
Reger M. A., Watson G. S., Green P. S. et al. (2008) Intranasal insulinimproves cognition and modulates beta-amyloid in early AD.Neurology 70, 440–448.
Romani-Perez M., Outeirino-Iglesias V., Gil-Lozano M., Gonzalez-Matias L. C., Mallo F. and Vigo E. (2013) Pulmonary GLP-1receptor increases at birth and exogenous GLP-1 receptor agonistsaugmented surfactant-protein levels in litters from normal andnitrofen-treated pregnant rats. Endocrinology 154, 1144–1155.
Schulte-Herbruggen O., Braun A., Rochlitzer S., Jockers-Scherubl M. C.and Hellweg R. (2007) Neurotrophic factors–a tool for therapeuticstrategies in neurological, neuropsychiatric and neuroimmuno-logical diseases? Curr. Med. Chem. 14, 2318–2329.
Selway J., Rigatti R., Storey N., Lu J., Willars G. B. and Herbert T. P.(2012) Evidence that Ca2+ within the microdomain of the L-typevoltage gated Ca2 + channel activates ERK in MIN6 cells inresponse to glucagon-like peptide-1. PLoS ONE 7, e33004.
Stanton P. K. (1996) LTD., LTP, and the sliding threshold for long-termsynaptic plasticity. Hippocampus 6, 35–42.
Talbot K., Wang H. Y., Kazi H. et al. (2012) Demonstrated brain insulinresistance in Alzheimer’s disease patients is associated with IGF-1resistance, IRS-1 dysregulation, and cognitive decline. J. Clin.Invest. 122, 1316–1338.
Vilsboll T. (2009) Liraglutide: a new treatment for type 2 diabetes.Drugs Today (Barc) 45, 101–113.
Vlacic-Zischke J., Hamlet S. M., Friis T., Tonetti M. S. and Ivanovski S.(2011) The influence of surface microroughness and hydrophilicityof titanium on the up-regulation of TGFbeta/BMP signalling inosteoblasts. Biomaterials 32, 665–671.
Wang H.-Y. A., Stucky J., Kvasic S. M., Shah K. P., Bakshi P., McCleanC., Holscher C. and Talbot K. (2012) The Diabetes DrugLiraglutide Ameliorates Insulin Resistance in the HippocampalFormation (HF) of the APP/PS1 Mouse Model of Alzheimer’sDisease (AD). Society for Neuroscience Annual Meeting, NewOrleans, USA.
Zuccato C. and Cattaneo E. (2009) Brain-derived neurotrophic factor inneurodegenerative diseases. Nat. Rev. Neurol. 5, 311–322.
© 2013 International Society for Neurochemistry, J. Neurochem. (2014) 128, 459--471
Neuroprotective effects of liraglutide in cell culture 471
Copyright © 2022 FDOKUMEN




















