
Topical isotretinoin vs. topical retinoic acid in the treatment of acne vulgaris
19
Cameo Nevocellular nevus associated with alopecia presenting as aplasia cutis congenita D. Neal Mastruserio, MD, Marc A. Cobb, MD, and Victor E. Ross, MD From Ohio State University, Division An 11-year-old white boy presented complaining of a patch of alopecia present on the of Dermatology, the Department of vertex of his scalp. According to the patient’s mother it had been present since birth. His Dermatology, National Naval Medical mother had noted no changes in the appearance of the patch over time and referred her Center, Bethesda, Maryland, and the son for diagnosis and possible cosmetic repair. The patient had no history of any physical Department of Dermatology, Beaufort or developmental problems; specifically, no extremity or ophthalmologic defects, Naval Hospital, Beaufort, North Carolina psychomotor retardation, or seizure disorder. The patient had received no prior medical or surgical therapy for the lesion. Correspondence Clinical examination revealed a 2 by 3 cm, flesh-colored, irregularly shaped patch of D. Neal Mastruserio, MD alopecia at the vertex of the scalp. Centrally, hair was rare to absent, blending to normal 456 West 10th Avenue hair density at the edges of the lesion (Fig. 1). Follicular orifices were absent centrally, Suite 4731 Columbus indicative of a scarring alopecia. The margin of the lesion was indistinct from normal OH 43210 scalp; no abnormalities of pigmentation were noted. Individual hairs centrally and at the margins appeared clinically normal. No sensory changes were detected. A 4 mm punch biopsy was taken from the center of the patch. Histopathology. Histological examination revealed an intradermal melanocytic nevus associated with an expanded fibrotic papillary dermis and the replacement of follicular structures with fibrotic tracts (Fig. 2). Theques of nevus cells were noted, coursing along the fibrotic tracts deep into the dermis (Fig. 3). One partially degenerated hair shaft, devoid of follicular epithelia and surrounded by theques of nevus cells, was observed at the periphery of the biopsy specimen. Discussion Given the location, appearance, and congenital nature of this patch of alopecia, the clinical impression was that of a case of aplasia cutis congenita (ACC). This condition can vary widely in both its physical appearance and in its histologic features. 1–4 According to Freiden’s system of classification for ACC, this case appeared clinically to fall under group 1: scalp ACC without multiple anomalies. 4 The microscopic features of this lesion were clearly consistent with an intradermal nevocellular nevus, excluding it from this group. No evidence of hyperplasia of the epidermis, sebaceous glands, or any other constituent of the skin was noted, clearly distinguishing this case from an organoid nevus. 2,3 Because of the relative homogeneity of the patch and the fact that the biopsy specimen was felt to be representative of the entire lesion, we feel justified in concluding that this is a case of alopecia associated with a nevocellular nevus. The presence of nevus cells deep in the reticular dermis, associated with fibrotic remnants of hair follicles, is consistent with the nevus being congenital. Whether the nevus cells played an etiologic role in the scarring alopecia or were simply accompanying this change is uncertain. © 1998 Blackwell Science Ltd International Journal of Dermatology 1998, 37, 37–51 37 Figure 1 A patch of alopecia measuring 0.8 3 2.0 cm, on the vertex of the scalp in an 11-year-old boy
Transcript of Topical isotretinoin vs. topical retinoic acid in the treatment of acne vulgaris

Cameo
Nevocellular nevus associated with alopecia presenting asaplasia cutis congenita
D. Neal Mastruserio, MD, Marc A. Cobb, MD, and Victor E. Ross, MD
From Ohio State University, Division An 11-year-old white boy presented complaining of a patch of alopecia present on theof Dermatology, the Department of vertex of his scalp. According to the patient’s mother it had been present since birth. HisDermatology, National Naval Medical mother had noted no changes in the appearance of the patch over time and referred herCenter, Bethesda, Maryland, and the
son for diagnosis and possible cosmetic repair. The patient had no history of any physicalDepartment of Dermatology, Beaufortor developmental problems; specifically, no extremity or ophthalmologic defects,Naval Hospital, Beaufort, North
Carolina psychomotor retardation, or seizure disorder. The patient had received no prior medical or
surgical therapy for the lesion.Correspondence Clinical examination revealed a 2 by 3 cm, flesh-colored, irregularly shaped patch ofD. Neal Mastruserio, MD
alopecia at the vertex of the scalp. Centrally, hair was rare to absent, blending to normal456 West 10th Avenuehair density at the edges of the lesion (Fig. 1). Follicular orifices were absent centrally,Suite 4731
Columbus indicative of a scarring alopecia. The margin of the lesion was indistinct from normalOH 43210 scalp; no abnormalities of pigmentation were noted. Individual hairs centrally and at the
margins appeared clinically normal. No sensory changes were detected. A 4 mm punch
biopsy was taken from the center of the patch.
Histopathology . Histological examination revealed an intradermal melanocytic nevus
associated with an expanded fibrotic papillary dermis and the replacement of follicular
structures with fibrotic tracts (Fig. 2). Theques of nevus cells were noted, coursing along
the fibrotic tracts deep into the dermis (Fig. 3). One partially degenerated hair shaft,
devoid of follicular epithelia and surrounded by theques of nevus cells, was observed at
the periphery of the biopsy specimen.
Discussion
Given the location, appearance, and congenital nature ofthis patch of alopecia, the clinical impression was that ofa case of aplasia cutis congenita (ACC). This condition canvary widely in both its physical appearance and in itshistologic features.1–4 According to Freiden’s system ofclassification for ACC, this case appeared clinically to fallunder group 1: scalp ACC without multiple anomalies.4 Themicroscopic features of this lesion were clearly consistentwith an intradermal nevocellular nevus, excluding it fromthis group. No evidence of hyperplasia of the epidermis,sebaceous glands, or any other constituent of the skin wasnoted, clearly distinguishing this case from an organoidnevus.2,3 Because of the relative homogeneity of the patchand the fact that the biopsy specimen was felt to berepresentative of the entire lesion, we feel justified inconcluding that this is a case of alopecia associated with anevocellular nevus. The presence of nevus cells deep in thereticular dermis, associated with fibrotic remnants of hairfollicles, is consistent with the nevus being congenital.Whether the nevus cells played an etiologic role in thescarring alopecia or were simply accompanying this changeis uncertain.
© 1998 Blackwell Science Ltd International Journal of Dermatology 1998, 37, 37–51
37
Figure 1 A patch of alopecia measuring 0.8 3 2.0 cm, on thevertex of the scalp in an 11-year-old boy

38 Cameo Aplasia cutis congenita Mastruserio, Cobb, and Ross
Figure 2 Nevocellular nevus associated with an expandedfibrotic papillary dermis and fibrotic tracts replacingfollicular units. (Hematoxylin and eosin; originalmagnification 340)
Figure 3 Nests of nevus cells andadjacent fibrotic changes.(Hematoxylin and eosin; originalmagnification 3100)
International Journal of Dermatology 1998, 37, 37–51 © 1998 Blackwell Science Ltd
Perinevoid alopecia has been described previously byYesudian and Thambiah in three case reports of alopeciaareata in association with central pigmented nevi.1 Ourcase differs from other cases previously described in anumber of key features. First, the cases described byYesudian and Thambiah were all associated with acquirednevocellular nevi and with findings consistent with alopeciaareata, i.e. ‘exclamation mark’ hairs and a tendency toappear spontaneously and to regress both with and withoutexcision of the nevi.1 The congenital nature of the patchof alopecia we have described and the lack of evidence insupport of alopecia areata make it different from casespreviously described in the literature.1,5–7
We have reported a case of alopecia associated with anonpigmented nevocellular nevus. Clinically, the diagnosisof ACC was suspected. The microscopic findings confirmedthe diagnosis of an intradermal nevocellular nevus associ-ated with dermal fibrosis and fibrotic replacement ofhair follicles. The congenital nature of the lesion clearlydistinguishes it from previously reported cases of perinevoidalopecia.1,5–7
Conclusions
Although it appears exceedingly rare, alopecia associatedwith nevocellular nevi should probably be considered inthe differential diagnosis of congenital, cicatricial alopecia.
References
1 Yesudian P, Thambiah AS. Perinevoid alopecia: an unusualvariety of alopecia areata. Arch Dermatol 1976; 112:1432–1434.

Isoda et al. Angiocentric T-cell lymphoma Cameo 39
2 Mehregan AH, Pinkus H. Life history of organoid nevi:special reference to nevus sebaceous of Jadassohn. ArchDermatol 1965; 91: 574–588.
3 Pinkus H. Life history of naevus syringadenomatosuspapilliferus. AMA Arch Dermatol 1954; 00: 305–322.
4 Freiden IJ. Aplasia cutis congenita: a clinical review andproposal for classification. J Am Acad Dermatol 1986; 14:646–660.
Cameo
EB virus-related angiocentric T-cell lymphoma
Ken-ichi Isoda, MD, Hitoshi Mizutani, MD, PhD, Takeshi Nishiguchi, MD, andMasayuki Shimizu, MD, PhD
From the Department of Dermatology, A 61-year-old woman developed an asymptomatic tumor on her right cheek. She had aMie University, Faculty of Medicine, history of surgical removal of a similar tumor on her left arm 2 years earlier. The facialTsu, Japan lesions were flesh-colored, with 232 cm ulcers (Fig. 1). The left arm had ulcerative
nodular lesions around the operation scar. Laboratory examinations revealed markedCorrespondenceleukocytosis (19,510/µL) with lymphocytosis (54.6%). A few abnormal blastoid cells withHitoshi Mizutani, MD, PhD
Department of Dermatology large nuclei were detected by hemogram (Fig. 2). The red blood cell count, platelet count,Faculty of Medicine coagulation tests, liver, and renal functions, including serum lysozyme, lipase, α1anti-Mie University trypsin, and angiotensin-converting enzyme activities, were normal, but a moderate2-174 Edobashi, Tsu, Mie 514
elevation of thymidinekinase activity was observed. Anti-nuclear human T-cell leukemiaJapanvirus type 1 (HTLV-1), human immunodeficiency virus (HIV), hepatitis B virus (HBV), and
hepatitis C virus (HCV) antibodies were not detected. Mantoux reaction was negative and
no DNA of tuberculosis was detected by polymerase chain reaction. Epstein–Barr virus
(EBV) titers revealed EBV viral capsid antigen (VCA)-immunoglobulin G (IgG): 3160;
VCA-IgM: undetectable level; early antigen (EA)-IgG: 310; and EBNA: 380. The
lymphocytes showed normal proliferation responses to lectins.
The tumor biopsy specimen revealed marked lymphocyte infiltration around vessels and
focal necrosis in the deep dermis (Fig. 3a). The infiltrate was composed mainly of large
blastoid lymphocytes with mitotic figures. Around the destroyed dermal vessels, tumor
cells and denatured endothelial cells concentrated characteristically and formed
glomeruloid lesions (Fig. 3b). Many auto-phagocytes forming ‘‘bean bag cells’’ were
electron microscopically detected in the tumor.
The proliferated cells in the dermis were CD31, CD41, CD71, CD21, CD51, WT311,
and CD45RO1; however, CD1, CD8, TCRd1, CD16, CD56, CD57, CD19, CD20, CD10,
L26, and LMP-1 were negative. Bone marrow cells showed lymphocytosis (39%) mixed
with abnormal lymphoid cells. Rearrangement of T cell receptor (TCR)-β or TCR-γ chain
was not detected in the samples from dermal tumors and peripheral blood.
EBV-encoded small nuclear RNA (EBER) was detected in the tumor cells from the
forearm by in situ hybridization.
Thus, the patient was diagnosed as having EB virus-related angiocentric lymphoma
with leukemic change, and was treated with bi-weekly cyclophosphamide, doxorubicin,
vincristine, and prednisone (CHOP) combination chemotherapy. The facial tumor
responded to CHOP markedly, and the peripheral blood leukemic cell count declined after
two cycles of CHOP. After the sixth cycle of CHOP, however, the patient developed acute
interstitial pneumonitis, and died from an intracranial hemorrhage.
The autopsy specimens revealed thickening of the alveolar wall, septal fibrosis, and
alveolar epithelial necrosis with focal lymphocyte infiltration. The bone marrow was
hypocellular, but infiltrated with blastoid tumor cells.
© 1998 Blackwell Science Ltd International Journal of Dermatology 1998, 37, 37–51
5 Quiroga VM, Pecoraro V. Alopecia perinaevica. Hautarzt1958; 9: 377–378.
6 Champion RH. Perinaevoid alopecia. Br J Dermatol 1962;74: 195. [Case presentation]
7 Weedon D. Unusual features of nevocellular nevi.J Cutaneous Pathol 1982; 9: 284–292.
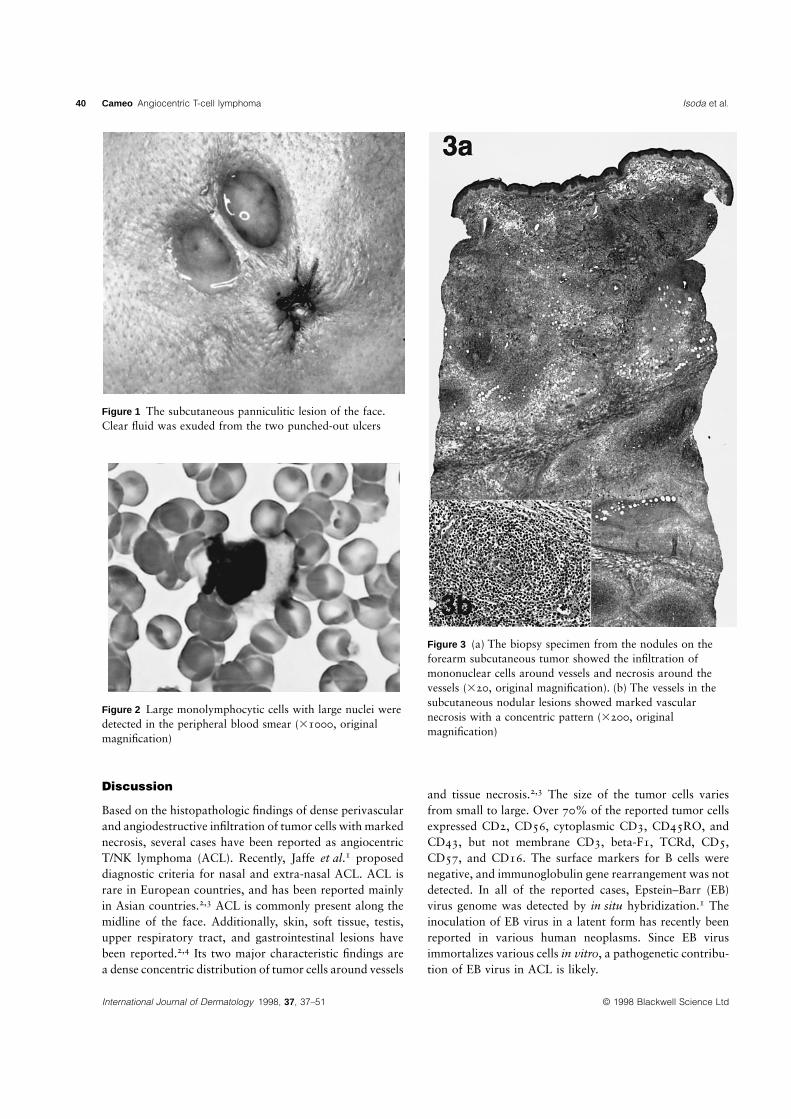
40 Cameo Angiocentric T-cell lymphoma Isoda et al.
Figure 1 The subcutaneous panniculitic lesion of the face.Clear fluid was exuded from the two punched-out ulcers
Figure 2 Large monolymphocytic cells with large nuclei weredetected in the peripheral blood smear (31000, originalmagnification)
Discussion
Based on the histopathologic findings of dense perivascularand angiodestructive infiltration of tumor cells with markednecrosis, several cases have been reported as angiocentricT/NK lymphoma (ACL). Recently, Jaffe et al.1 proposeddiagnostic criteria for nasal and extra-nasal ACL. ACL israre in European countries, and has been reported mainlyin Asian countries.2,3 ACL is commonly present along themidline of the face. Additionally, skin, soft tissue, testis,upper respiratory tract, and gastrointestinal lesions havebeen reported.2,4 Its two major characteristic findings area dense concentric distribution of tumor cells around vessels
International Journal of Dermatology 1998, 37, 37–51 © 1998 Blackwell Science Ltd
Figure 3 (a) The biopsy specimen from the nodules on theforearm subcutaneous tumor showed the infiltration ofmononuclear cells around vessels and necrosis around thevessels (320, original magnification). (b) The vessels in thesubcutaneous nodular lesions showed marked vascularnecrosis with a concentric pattern (3200, originalmagnification)
and tissue necrosis.2,3 The size of the tumor cells variesfrom small to large. Over 70% of the reported tumor cellsexpressed CD2, CD56, cytoplasmic CD3, CD45RO, andCD43, but not membrane CD3, beta-F1, TCRd, CD5,CD57, and CD16. The surface markers for B cells werenegative, and immunoglobulin gene rearrangement was notdetected. In all of the reported cases, Epstein–Barr (EB)virus genome was detected by in situ hybridization.1 Theinoculation of EB virus in a latent form has recently beenreported in various human neoplasms. Since EB virusimmortalizes various cells in vitro, a pathogenetic contribu-tion of EB virus in ACL is likely.

Lazaro et al. Cutaneous calcinosis Cameo 41
Our patient developed interstitial pneumonia. EB virushas been postulated as the causative agent for interstitialpneumonia.5 Since EB virus was inoculated in our patient’scutaneous tumor cells, it is hypothesized that it was reactiv-ated after the immunosuppressive therapy.
Subcutaneous panniculitic T-cell lymphoma6 has beenreported as a variant of lymphoma with panniculitis.The clinical and histopathologic findings of ACL andsubcutaneous panniculitic T-cell lymphoma are very similar,and therefore we cannot exclude the possibility of overlap ofthese two clinical entities. Lymphomatoid granulomatosis,which had been categorized in the angiocentric immuno-proliferative lesion group, was later determined aslymphoma of B-cell lineage.1 In our patient, CD4 and CD7were detected immunohistochemically, but CD56 was notdetected on the tumor cells, which were less characteristicof NK, but closer to T-cell lineage. Leukemic changes atthe late phase are atypical of ACL. Since the entity of ACLis still somewhat unclear, the determination of precisecriteria for ACL and possible subgroups of ACL is needed.
Cameo
Cutaneous calcinosis with transepithelial elimination in apatient with sarcoidosis
Teresa E. Lazaro, MD, Natalia Hernandez-Cano, MD, Francisco A. Rubio, MD,Goretti Robayna, MD, Felix Contreras, MD, Angel Pizarro, MD, and Mariano Casado, MD
From the Departments of A 44-year-old man who had worked as a plumber and was currently working as aDermatology and Pathology, La Paz bricklayer was referred to our hospital because of a 2-year history of progressiveUniversity Hospital, Autonomous dyspnea. Physical examination revealed cyanosis, clubbing of the fingers, and edema ofUniversity of Madrid, Madrid, Spain
the inferior extremities. Chest auscultation revealed bilateral inspiratory crackles
predominantly at the lung bases. Remarkably, there were several painful, firm papulesCorrespondenceMariano Casado, MD and nodular lesions over the fingertips of the hands, some with whitish crusts (Fig. 1).Department of Dermatology These lesions had appeared 2 years before and gradually increased in number and size.Hospital La Paz A full blood count was normal apart from moderate erythrocytosis and lymphopenia.Paseo de la Castellana, 261
The biochemical profile (including serum levels of calcium and phosphorus and daily28046 Madridurinary calcium excretion) was normal. Serum angiotensin converting enzyme activity wasSpainincreased at 60 U/mL (normal range, 6.1–21.1 U/mL). An antinuclear antibody test was
negative. Serum complement levels were normal. Chest X-ray film showed cardiomegaly,
bilateral hilar lymphadenopathy, and interstitial infiltrates (reticulo-nodular pattern) with a
predilection for the mid and lower lung zones. A computed tomography scan of the chest
confirmed mediastinal adenopathy and revealed bilateral and diffuse parenchymal
abnormalities consisting of nodular opacities along the bronchovascular bundles and
interlobular septa, irregular linear opacities oriented along the bronchovascular bundles,
and distortion of the lobules. A gallium-67 scan showed a ‘‘lambda’’ pattern created by
uniform uptake in bilateral intrathoracic and right paratracheal lymphadenopathy, which is
highly specific for the diagnosis of sarcoidosis.1 The recovery of bronchoalveolar lavage
© 1998 Blackwell Science Ltd International Journal of Dermatology 1998, 37, 37–51
References
1 Jaffe ES, Chan JK, Su IJ, et al. Report of the Workshopon Nasal and Related Extranodal AngiocentricT/Natural Killer Cell Lymphomas. Definitions,differential diagnosis, and epidemiology. Am J SurgPathol 1996; 20: 103–111.
2 Takeshita M, Kimura N, Suzumiya J, et al. Angiocentriclymphoma with granulomatous panniculitis in the skinexpressing natural killer cell and large granular T-cellphenotypes. Virchows Archiv 1994; 425: 499–504.
3 Tsai TF, Su IJ, Lu YC, et al. Cutaneous angiocentricT-cell lymphoma associated with Epstein–Barr virus.J Am Acad Dermatol 1992; 26: 31–38.
4 Su IJ, Tsai TF, Cheng AL, et al. Cutaneousmanifestations of Epstein–Barr virus-associated T-celllymphoma. J Am Acad Dermatol 1993; 29: 685–692.
5 Barbera JA, Hayashi S, Hegele RG, et al. Detection ofEpstein–Barr virus in lymphocytic interstitial pneumoniaby in situ hybridization. Am Rev Respir Dis 1992; 145:940–946.
6 Wang CE, Su WPD, Kurtin PJ. Subcutaneous panniculiticT-cell lymphoma. Int J Dermatol 1996; 35: 1–8.

42 Cameo Cutaneous calcinosis Lazaro et al.
showed 61% macrophages (normal range, 80%–90%) and 35% lymphocytes (normal
value, ,10%). The CD4/CD8 lymphocyte ratio was increased at 3.6. Smears and cultures
of the bronchial washings failed to demonstrate any infectious organisms. Cytologic
examination of three sputum samples gave negative results for malignant cells, as well as
for hyphae and acid-fast bacilli.
A biopsy specimen of a papular lesion of the fingertips revealed acanthosis and
orthokeratotic hyperkeratosis in the epidermis. Remarkably, there were deposits of a
basophilic and von Kossa-positive material in the upper and mid dermis (Fig. 2). In some
areas, there were calcific deposits perforating into the epidermis, in the manner of
transepithelial elimination.2
The patient was diagnosed as having cutaneous calcinosis with transepithelial
elimination associated with pulmonary sarcoidosis. Progressive pulmonary fibrosis
developed despite treatment with high-dose oral glucocorticoids. The patient died 10
months after the initiation of treatment due to severe pulmonary hypertension and right
heart failure. During this period, no new cutaneous lesions appeared on the fingertips and
there was a reduction in size with symptomatic improvement in some of them.
Discussion
Sarcoidosis is a systemic granulomatous disease of un-known etiology. Skin lesions occur in 20%–40% ofpatients in most series.3–6 Two types of cutaneous manifesta-tions of sarcoidosis are distinguished: nonspecific (such aserythema nodosum, which is observed in acute sarcoidosis)and specific, peculiar to a chronic form of the disease.Interestingly, hypercalcemia occurs in around 20% of cases,but cutaneous calcinosis has rarely been described.7–9 Thepresent case is unique because of the location of the lesions(fingertips) and the presence of transepidermal eliminationof calcific deposits. To the best of our knowledge, thisis the first case of calcinosis cutis of the fingers withtransepithelial elimination associated with sarcoidosis
Figure 1 Papular and nodularlesions with whitish crusts overthe fingertips of the hands
International Journal of Dermatology 1998, 37, 37–51 © 1998 Blackwell Science Ltd
reported so far; however, tissue calcification of the fingersis a common problem as a part of certain multisystemicdiseases, such as scleroderma and the CREST syndrome.7,10
The concurrent presentation of pulmonary sarcoidosisand cutaneous calcinosis in this patient suggests a possiblepathogenic relationship between them. Hypercalcemia isfrequently found in patients with sarcoidosis. Abnormalcalcium metabolism may lead to metastatic calcifications.7,8
As our patient had normal calcium and phosphorus levelsat the time of diagnosis, the metastatic nature of thecalcifying disorder may be questioned. Thus, we cannotcompletely exclude, at least in part, other possibilities, suchas dystrophic calcifications related to previous local tissueinjuries. Indeed, previous jobs of this patient (plumber and

Lazaro et al. Cutaneous calcinosis Cameo 43
Figure 2 Skin biopsy specimenshowing deposits of a basophilicmaterial in the dermis. There arecalcific deposits perforating intothe epidermis (arrow)(hematoxylin and eosin stain,380)
bricklayer) could facilitate repeated microtraumatisms tothe fingertips. Interestingly, cases of both cutaneous cal-cinosis9 and nephrocalcinosis with glomerular microcalci-fications11 have been reported previously in normocalcemicsarcoidosis patients, which suggests that abnormal calcify-ing deposits in sarcoidosis may be related to factors otherthan serum calcium levels; however, the presence of thelesions in normocalcemic patients does not provide conclus-ive evidence against a direct role of serum calcium levelsin tissue calcinosis. It is noteworthy that calcium levels areknown to fluctuate because of several factors, such asseasonal exposure to ultraviolet radiation from sunlight.11
In fact, this phenomenon has been observed in patientswith sarcoidosis,12 and could have occurred in our caseover the months preceding the laboratory study.
References
1 Sulavik SB, Spencer RP, Palestro CJ, et al. Specificity andsensitivity of distinctive chest radiographic and/or 67Gaimages in the noninvasive diagnosis of sarcoidosis. Chest1993; 103: 403–409.
2 Patterson JW. The perforating disorders. J Am AcadDermatol 1984; 10: 561–585.
© 1998 Blackwell Science Ltd International Journal of Dermatology 1998, 37, 37–51
3 Elgart ML. Cutaneous lesions of sarcoidosis. PrimaryCare 1978; 5: 249–262.
4 Kerdel FA, Moschella SL. Sarcoidosis. An updatedreview. J Am Acad Dermatol 1984; 11: 1–19.
5 Sharma OP. Cutaneous sarcoidosis: clinical features andmanagement. Chest 1972; 61: 320–325.
6 Samtsov AV. Cutaneous sarcoidosis. Int J Dermatol1992; 31: 385–391.
7 Walsh JS, Fairley JA. Calcifying disorders of the skin.J Am Acad Dermatol 1995; 33: 693–706.
8 Cribier B, Grosshans E. Calcinosees cutanees. AnnDermatol Venereol 1992; 119: 151–168.
9 Kroll JJ, Shapiro L, Koplon B, et al. Subcutaneoussarcoidosis with calcification. Arch Dermatol 1972; 106:894–895.
10 Bottomley WW, Goodfield MJD, Sheehan-Dare RA.Digital calcification in systemic sclerosis: effectivetreatment with good tissue preservation using the carbondioxide laser. Br J Dermatol 1996; 135: 302–304.
11 Trillo A, Orozco R, Jindal K. Glomerular calcinosis insarcoidosis. Arch Pathol Lab Med 1992; 116:1221–1225.
12 Taylor RL, Lynch HJ, Wysor WG. Seasonal influence ofsunlight on the hypercalcemia of sarcoidosis. Am J Med1963; 34: 221–227.

44 Cameo Regional ganglionar metastasis Vignale et al.
Cameo
Regional ganglionar metastasis 24 years after surgicalresection of a malignant melanoma
Raul Vignale, MD, Silvia Brugnini, MD, Miguel Martinez, MD, Omar Alonso, MD, andFernando Fleurquin, MD
From the Clinica Dermatologica, A 40-year-old woman, with type I skin, came to the Dermatology Clinic after noticing aHospital Maldonado, MSP, tumor in the armpit. The physical examination revealed a hard, stretched-shaped tumoralMontevideo, Uruguay node with a diameter of 30 mm, located in the left armpit. It moved on the superficial and
deeper layers. In addition, the scar of a large graft was found in the homolateralCorrespondencesupraclavicular region, where a malignant melanoma had been excised 24 years earlier.Raul Vignale, MD
Palmar 2542 At that time, the primary tumoral lesion had the characteristics of an extensive superficialCP 11600 Montevideo melanoma with an infiltrating core with vertical growth. The pathologic study of the tumorUruguay confirmed the diagnosis of a malignant melanoma with a Clark level II and a 1.17 mm
Breslow thickness. Surgery had a margin of 3 cm. At present, the patient is in excellent
condition and laboratory tests are normal. Immunologic tests performed showed a slight
increase in CD41, CD45A1, DR1 lymphocytes, CD161, CD561, NK cells, and a
marked increase in tumor necrosis factor-α (TNF-α) and γ-interferon (IFN-γ) levels (Table
1). Imaging studies (chest X-rays, abdominal ultrasound, bone scintigraphy) did not show
any abnormal signs. Computerized tomography confirmed the ganglionic mass, without
any other abnormalities. Tc-99m methoxy-isobutyl-isonitrile (MIBI) scintigraphy was
performed, showing a focally increased concentration of radiomarker in the left armpit,
indicating a thick adenopathic conglomerate (Fig. 1). No other areas of abnormal uptake
were observed, not even in the original area of the primary tumor. The surgical
exploration of the axillar contents confirmed the presence of adenopathies. Pathology
studies showed metastatic invasion of intensely melanin-pigmented and pleomorphic cells,
with atypical mitosis.
Discussion
Malignant melanoma has special characteristics with regardto its frequency and relapse rates, which are related to theacquisition of an invading cell phenotype, the possibilityof lymphatic or hematic migration, and the multiplicity ofpotential target organs.1,2 There is a wide range of variation:the survival rate at 5 years is, according to different authors,between 14% and 79%.3–5 Reports of metastases with avery late onset (10–20 years) are scarce and isolated.6 Onlyin very rare cases have recurrences been reported after 20,30, or more years free from symptoms.1,2,7–9 How can sucha long symptom-free interval be explained?
In our patient, we believe that primary tumoral cellsinvaded the lymph node at the very beginning, and remainedinactive in G0–G1. We do not believe that there are anytumoral cells remaining in the location of the primarytumor, based on the absence of Tc-99m methoxy-isobutyl-isonitrile (MIBI) uptake and on the fact that no malignantcells were observed on pathologic observation of a seriesof biopsy samples from the same area. Perhaps the laterecurrence in this case is due to the fact that the patient
International Journal of Dermatology 1998, 37, 37–51 © 1998 Blackwell Science Ltd
did not show any signs of systemic immunodepression. Incontrast, some T lymphocytes, NK cells, tumor necrosisfactor-α (TNF-α) and γ-interferon (INF-γ), which are
Figure 1 Tc-99m MIBI scintigraphy, showing a focallyincreased concentration of radiomarker in the left armpit

Hashimoto et al. Oral–facial–digital syndrome Cameo 45
Table 1 Immunologic tests
Value observed Normal value
T LymphocytesCD31 2215/mm3 (1200–2100)CD41 1480/mm3 (700–1250)CD81 1010/mm3 (300–950)CD251 25/mm3 (5–35)CD45A1 805/mm3 (300–750)CD711 20/mm3 (10–95)DR1 165/mm3 (5–125)
B LymphocytesCD191 155/mm3 (60–560)CD561 30/mm3 (15–75)
NK cellsCD161 890/mm3 (215–590)CD561 150/mm3 (5–65)
CitokinesC-reactive protein 1 mg/ml (,6)IL-4 1 µg/mL (0–1)IL-8 1 µg/mL (0–2)TNF-α 109 µg/mL (0–2)IFN-γ 257 UV/mL (0–1)
IL, interleukin; TNF, tumor necrosis factor; IFN, interferon.
related to possible antitumoral effects, were found in highlevels. We wish to emphasize the usefulness of Tc-99mMIBI scintigraphy for initial staging and follow-up ofpatients with melanoma.10–13 Clinical surveillance and theTc-99m MIBI method will be maintained in order toidentify early signs of dissemination.
References
1 Balch ChM, Milton GM. Diagnosis of metastaticmelanoma at distant sites. In: Balch ChM, Milton GW,eds. Cutaneous Melanoma. Clinical Management and
Cameo
Oral–facial–digital syndrome (OFDS) type I in a patient withWerdnig–Hoffman diseaseYoshio Hashimoto, MD, Takayuki Kashiwagi, MD, Hidetoshi Takahashi, MD, andHajime Iizuka, MD
From the Department of Dermatology, An infant with various craniofacial anomalies was referred to our department at the age ofAsahikawa Medical College, 10 days. She was born as a floppy infant with a symmetrical muscle weakness that wasAsahikawa, Hokkaido, Japan more extensive in the proximal part of the limbs. Her brother had Werdnig–
© 1998 Blackwell Science Ltd International Journal of Dermatology 1998, 37, 37–51
Treatment. Results Worldwide. Philadelphia: JBLippincott, 1985: 221–250.
2 Balch ChM, Soong S. Characteristics of melanoma thatpredict the risk of metastasis. In: Costanzi JJ, ed.Malignant Melanoma 1. The Hague: Martinus Nijhoff,1983: 117–149.
3 Shaw HM, Beattie CW, McCarthy WH, Milton GM. Laterelapse from cutaneous stage I malignant melanoma. ArchSurg 1985; 120: 1155–1159.
4 Gutman M, Klausner JM, Inbar M, Rozin RR. Laterecurrence Stage I malignant melanoma. J Surg Oncol1989; 42: 96–98.
5 Saiag P, Avril MF, Bonerandi JJ, Drena B. Le melanomaapres la conference de consensus. Nouv Dermatol 1996;15: 156–158.
6 Avril MF, Nguyen T, Duvillard P, et al. Late recurrence ofmelanoma beyond 10 years. Ann Dermatol Venereol1994; 121: 454–458.
7 Rosenkranz L, Schroeder C. Recurrent malignantmelanoma following a 46 years-disease-free interval. N YState J Med 1985; 85: 95.
8 Tahery DP, Moy RL. Recurrent malignant melanomafollowing a 35-years disease free interval. J Dermatol SurgOncol 1993; 19: 161–163.
9 Bouffard D, Barnhill RL, Mihm MC, Sober AJ. Very latemetastasis (27 years) of cutaneous malignant melanomaarising in a halo giant congenital nevus. Dermatology1994; 189: 162–166.
10 Alonso O, Martinez M, Delgado L, Lago G. Tc-99msestamibi uptake by malignant melanoma lesions. NuclMed Comm 1996; 17: 542.
11 Alonso O, Martinez M, Lago G, et al. Scintigraphicdetection of invasive malignant melanoma with Tc-99mMIBI. Clin Nucl Med 1996; 21: 557–559.
12 Perrot J-L, Soler C, Tiffet O, et al. Detection of malignantmelanoma metastasis with 99m Tc-sestamibi SPECT(single photon emission tomography) during follow-up ofmalignant melanoma in 17 patients. Eur J Dermatol 1996;6: 505–508.
13 Schadendorf D, Herford R, Czarenetzky BM.P-glycoprotein expression in primary and metastaticmalignant melanoma. Br J Dermatol 1995; 132: 551–555.

46 Cameo Oral–facial–digital syndrome Hashimoto et al.
Correspondence Hoffman disease and died of respiratory failure at the age of 4 months. No family historyYoshio Hashimoto, MD of malformations was detected.Department of Dermatology
Physical examination revealed the following abnormalities: a high arched palate,Asahikawa Medical Collegemicrognathia, a medial cleft of the upper lip, cleft palate, and low set ears (Fig. 1). She3–11 Nishikagura, 078
Japan also had clinodactyly and partial syndactyly of toes II–III, III–IV on the right foot (Fig. 2).
Radiographs of the right foot revealed polydactyly of the first toe. Nail hypoplasia was
observed on the bilateral second toes (Fig. 2). No abnormalities were seen on both
hands. The face, trunk, and limbs were symmetrical. There was no evidence of
abnormalities of the genitalia.
Wrinkled skin was conspicuous on the neck and four extremities (Fig. 1). The hair was
sparse and brittle. Fine scale and dry skin were conspicuous on the scalp and the upper
part of the trunk. Numerous milia were seen on the face. Skin biopsy could not be
performed. Hypohidrosis was not found clinically. Because of the accompanying skin
manifestations, she was given a diagnosis of oral–facial–digital syndrome (OFDS) type I.
Chromosome analysis revealed a normal female karyotype. Chest roentgenogram and
a computerized tomographic (CT) scan of the brain detected no abnormal findings. No
malformations of the cardiovascular, gastro-intestinal, or urinary systems were detected.
Because of the craniofacial anomalies and general muscle weakness, she had feeding
problems and had stopped gaining weight at the age of 3 months. She had considerable
difficulty in breathing because of chest muscle weakness.
Electromyogram (EMG) showed a typical neurogenic pattern. Because of the
progressive muscle weakness, typical EMG findings, and a family history, she was
diagnosed as having Werdnig–Hoffman disease. Her clinical course was complicated by
recurrent pneumonia, apnea, and difficulty with feeding. At the age of 7 months, she died
of respiratory failure.
Discussion
Since the first report by Papillon-Leage in 1954,1 nineclinical types of oral–facial–digital syndrome (OFDS) havebeen described.2 The delineation of OFDS is usually basedon the presence of particular oral, facial, and digitalmalformations and on the types of inheritance. Because
Figure 1 Cleft lip, cleft palate, andlow set ears are shown. Wrinkledskin and dry skin are conspicuouson the neck and scalp. The hair issparse and brittle. Several milia-like papules are visible on the leftcheek
International Journal of Dermatology 1998, 37, 37–51 © 1998 Blackwell Science Ltd
there are no precise biochemical or genetic markers andthere is significant overlap of the clinical features, thedifferentiation between OFDS types I–IX is provisional atpresent. OFDS type I is the most common, with X-linked dominant heredity, and is fatal in male children.Malformations of OFDS include midline cleft lip, highlyarched palate with midline cleft of the soft palate, bifid or

Hashimoto et al. Oral–facial–digital syndrome Cameo 47
Figure 2 Clinodactyly and partialsyndactyly of the toes are shown.Nail hypoplasia is visible on thebilateral second toes
multilobed tongue, micrognathia, hypertelorism, hypo-plasia of the nasal alae, low set ears, syndactyly, polydactyly,polycystic kidney, cardiac anomalies, agenesis of corpuscallosum, and microcephaly. OFDS type I is the only typewith cutaneous manifestations, such as scaly skin, facialmilia, and brittle hairs (all detected in our case). Becausethe cutaneous manifestations of OFDS type I might notnecessarily be conspicuous, without definite informationabout heredity, the diagnosis of OFDS type I is occasionallyhampered. Our case was clinically diagosed as OFDS typeI because of cutaneous manifestations. Our case alsoshowed a mild wrinkled skin, which is an atypical cutaneoussymptom of OFDS type I. Otherwise, it was consistentwith OFDS type I.
Differential diagnoses of OFDS include ectodermaldysplasias associated with cleft lip or palate: EEC syndrome(ectrodactyly, ectodermal dysplasia, and cleft lip and/orpalate), Rapp–Hodgkin syndrome (ectodermal dysplasiaassociated with distinctive craniofacial features, cleft lipand/or palate, and hypospadias), and AEC syndrome(ankyloblepharon associated with ectodermal dysplasia andcleft lip and/or palate). These are autosomal-dominantdisorders and share many clinical features associated withcleft lip and/or palate, syndactyly or clinodactyly, scalpdermatitis, ear anomalies, palmoplantar keratoderma,polythelia, reduced sweating, and abnormalities of thegenitalia.3
In our patient, the absence of ectrodactyly, hypo-hidrosis, ankyloblepharon, scalp dermatitis, genitourinaryanomalies, and palmoplantar keratoderma excluded EECsyndrome, Rapp–Hodgkin syndrome, and AEC syndrome.OFDS type I is known to be assocaited with variouscerebro-spinal abnormalities, such as agenesis of the corpus
© 1998 Blackwell Science Ltd International Journal of Dermatology 1998, 37, 37–51
callosum, microcephaly, macrocephaly, cerebellar defects,and interhemispheric cysts, that may be accompanied byhypotonia;4,5 however, in our case, no cerebro-spinalabnormalities were detected in the computerized tomo-graphic (CT) scan, suggesting that the muscle weaknesshad its origin in the spinal muscle atrophy (SMA).
The patient also had Werdnig–Hoffman disease (acuteform of hereditary childhood SMA: type I)6 with progressivemuscle weakness and typical electromyogram (EMG) find-ings. Werdnig–Hoffman disease is a degenerative diseaseof anterior horn cells of the spinal cord, that develops atbirth or within 3 months. This results in systemic muscleweakness and hypotonia and patients usually die of respirat-ory failure within 2 years. Recently, a deletion in the 5q13 region has been detected in Werdnig–Hoffman disease.7
To the best of our knowledge, our patient is the firstreported case of OFDS accompanied by Werdnig–Hoffmandisease. Leao and Ribeiro-Silva5 suggested that the OFDStype I gene is located on Xp22. A defect in Xp22-relatedgenes has been related to Aicardi syndrome, Goltz syn-drome, and microphthalmus with linear skin defect (MLS).8
The phenotypic differences in the three disorders probablyreflect variations in X-inactivation. These disorders mayshow agenesis of the corpus callosum,8–10 which was foundin some patients with OFDS type I.4,5 In our case, wrinkledskin and brittle hair were observed, both of which are thecutaneous symptoms in Goltz syndrome. Although thepresent case suggets an incidental association of OFDStype I and Werdnig–Hoffman disease, the sporadic natureof our case hampers the definite diagnosis of OFDS ‘‘typeI’’. This is especially because OFDS-like anomalies11 havebeen associated with the deletion of 5q, which is also

48 Cameo LEOPARD syndrome Jozwiak et al.
consistent with the genetic defect of Werdnig–Hoffmandisease.
References
1 Papillon-Leage, Psaume J. Une malformation hereditairede la muqueuse biccale, brides et fret anormaux. RevStomatol (Paris) 1954; 55: 209–227.
2 Toriello HV. Review: oral–facial–digital syndrome. ClinDysmorph 1993; 2: 95–105.
3 Cambiaghi S, Tadini G, Barbareschi M, et al. Rapp–Hodgkin syndrome and AEC syndrome: are they the sameentity? Br J Dermatol 1994; 130: 97–101.
4 Towfighi J, Berlin CM, Ladda RL, et al. Neuropathologyof oral–facial–digital syndromes. Arch Pathol Lab Med1985; 109: 642–646.
5 Leao MJ, Ribeiro-Silva ML. Orofaciodigital syndrometype I in a patient with severe CNS defects. Pediatr Neurol1995; 13: 247–251.
Cameo
Familial occurrence of the LEOPARD syndromeSergiusz Jozwiak, MD, PhD, Robert A. Schwartz, MD, MPH, Camila KrysickaJanniger, MD, and Jacek Zaremba, MD, PhD
From Neurology, Monument Case 1 A 10-year-old boy was admitted to the Neurology Department at the Child’sChildren’s Hospital, Warsaw, Poland; Health Center in Warsaw, Poland, for a diagnosis of mental retardation. Since infancy,Dermatology and Pediatrics, UMD— small, dark brown, irregularly shaped macules, varying in size from pinpoint to 5 mm inNew Jersey Medical School, Newark,
diameter, had been observed on the face, the sclerae, and the neck (Fig. 1).New Jersey; and Genetics, Institute ofHe was born full-term by spontaneous delivery after uncomplicated pregnancy. TheNeurology and Psychiatry, Warsaw,
Poland birth weight was 3000 g. The Apgar score was 5 at the first minute. In the first hours after
delivery, the infant demonstrated peripheral cyanosis. A heart murmur, graded 3/6Correspondence according to the Levine scale, was heard. Chest roentgenogram revealed theRobert A. Schwartz, MD, MPH
enlargement of both heart ventricles. Electrocardiography and echocardiography carriedDermatologyout several times in the first 3 years of life were within normal limits. His psychomotorNew Jersey Medical School
185 South Orange Avenue development was delayed—he sat at 1 year and walked at 2 years of age. There was noNewark, NJ 07103-2714 history of epileptic seizures.
At admission, multiple lentigines on the face and neck were seen. There was a slight
disproportion between his height (50–75 percentile) and weight (10–25 percentile). The boy
demonstrated poor skeletal muscle development with scoliosis, pectus carinatum, and
winged scapulae. Neurologic status was normal, except for slight mental retardation (IQ 5
63, according to Wechsler’s Intelligence Scale). No cardiac murmur was appreciated at that
time. Ophthalmologic examination, audiogram, and electroencephalography were within
normal limits. Computerized tomography revealed no brain abnormalities. There was
abnormal repolarization on electrocardiography, but echocardiography remained normal.
The family was composed of two natural parents, two brothers, and three sisters. Both
parents were alive and well. The father did not reside with the family and was unavailable
for examination. No lentigines were observed by the mother on his skin. Examination of
the mother did not reveal significant skin findings. She had a history of progressive loss of
International Journal of Dermatology 1998, 37, 37–51 © 1998 Blackwell Science Ltd
6 Pearn JH, Carter CO, Wilson J. The genetic identity ofacute infantile spinal muscular atrophy. Brain 1973; 96:463–470.
7 Melki J, Lefebvre S, Burglen L, et al. De novo andinherited deletion of the 5q 13 region in spinal muscularatrophies. Science 1994; 264: 1474–1477.
8 Lindsay EA, Grillo A, Ferrero GB, et al. Microphthalmiawith linear skin defects (MLS) syndrome: clinical,cytogenetic, and molecular characterization. Am J MedGenet 1994; 49: 229–234.
9 Naritomi K, Izumikawa Y, Nagataki S, et al. CombinedGoltz and Aicardi syndromes in a terminal Xp deletion:are they a contiguous gene syndrome? Am J Med Genet1992; 43: 839–843.
10 Friedman PA, RaoKW, Teplin SW, et al. Provisional deletionmapping of the focal dermal hypoplasia (FDH) gene to XP22.31 (abstract). Am J Hum Genet 1988; 43: A50.
11 Kleczkowska A, Fryns JP, Van Den Berghe H. A distinctmultiple congenital anomalies syndrome associated withdistal 5q deletion (q 35.1 qter). Ann Genet 1993; 36: 126–128.

Jozwiak et al. LEOPARD syndrome Cameo 49
vision in the right eye from very early childhood. She had this eyeball removed at 18
years of age. She had not reported epileptic seizures. Her mental status was normal. The
younger brother had a congenital cataract of the left eye and amblyopia. Ultrasonography
of the eyeball revealed the presence of arteria hyaloidea persistens. He did not
demonstrate lentigines on the skin. His psychomotor development so far is normal. He is
now 6 years old. The youngest sister is 10 years old. She has no skin features of the
LEOPARD syndrome. Her development is normal. Features of the LEOPARD syndrome
have been found in the other siblings, as delineated below.
Case 2 Multiple lentigines on the face and neck were found in the middle sister. She had
been born after uneventful pregnancy with a weight of 2800 g. Since early childhood, her
physical and mental development was delayed. At 18 months, her weight was 8650 g. At
7 years, during routine pediatric examination, a heart murmur was evident. Except for this
observation and pectus excavatum, a careful cardiologic examination, electrocardiography
and echocardiography were normal; the murmur was regarded as innocent. At 10 years of
age, her height and weight remained between the 10th and 25th percentile. Her
neurologic status was normal, but psychologic examination found slight mental retardation
(IQ 5 57). There was no history of epilepsy. Electroencephalography was normal. At 17
years of age she had several syncopal attacks. Admitted to the regional hospital, she
died, with death attributed to probable acute cardiopulmonary insufficiency.
Case 3 The oldest sister was born at term after uneventful pregnancy. Her birth weight
was 2550 g. There were no lentigines seen on her skin. Since early childhood, slight
mental retardation has been observed. At 9 years of age, she was admitted to the
Cardiology Department because of signs of cardiac insufficiency. At admission, her weight
and height were very low (between the 3rd and 10th percentile). Pectus excavatum was
seen. She had a tachycardia of 160/min and hepatosplenomegaly. There was a heart
murmur, graded 3/6 by the Levine scale, and enlargement of the whole heart on chest
roentgenogram. A large tumor in the left cardiac atrium was diagnosed and subsequently
resected. Histologically, a cardiac myxoma was diagnosed. The postoperative period was
uneventful. At 13 years of age, she underwent a second operation because of tumor
regrowth. She demonstrated slight mental retardation with an IQ of 67.
Electroencephalography was abnormal with slow activity in the frontal lobes. There was
no history of epilepsy. Presently, she is without cardiac symptoms. Her mental status has
not changed.
Discussion
The LEOPARD syndrome, also known as cardiocutaneouslentiginosis syndrome or multiple lentigines syndrome, isa complex dysmorphogenetic disorder transmitted as anautosomal-dominant trait with variable penetrance andexpressively.1–10 The name ‘‘LEOPARD syndrome’’ wasintroduced in 1969 by Gorlin et al.2 in order to recallthe various features of this syndrome: L, lentigines; E,electrocardiographic conduction abnormalities; O, ocularhypertelorism; P, pulmonary stenosis; A, abnormalitiesof genitalia; R, retardation of growth; and D, deafness(sensorineural). The syndrome is regarded as a relativelyrare entity, but reports on oligosymptomatic cases,4–32 andour description of clinical signs of the LEOPARD syndromein members of the same family, suggest that its trueprevalence may be underestimated.
In spite of the rather long list of features included in the
© 1998 Blackwell Science Ltd International Journal of Dermatology 1998, 37, 37–51
LEOPARD syndrome, the diagnosis is difficult, as mostpatients meet only three to five of these criteria.3 Lentiginesare the most typical and one of the most frequent featuresof the syndrome. Voron et al.4 evaluated 80 patients fromthe literature, with 74 having lentiginosis. According tothese authors, if lentigines are absent, a diagnosis of thesyndrome may be made if the patient has an immediaterelative with multiple lentigines syndrome and has featuresin at least three other categories.
In the reported family, two individuals have the LEO-PARD syndrome with typical lentiginosis. The diagnosiscan also be made in the oldest sister without lentigines, butwith cardiac myxoma, delayed growth, mental retardation,and skeletal anomalies. Cardiac abnormalities, structuralor electrocardiographic, are among the most frequentfeatures of the LEOPARD syndrome. In the reported family,all three patients had a heart murmur, although a structuralabnormality, a cardiac myxoma, was demonstrated only
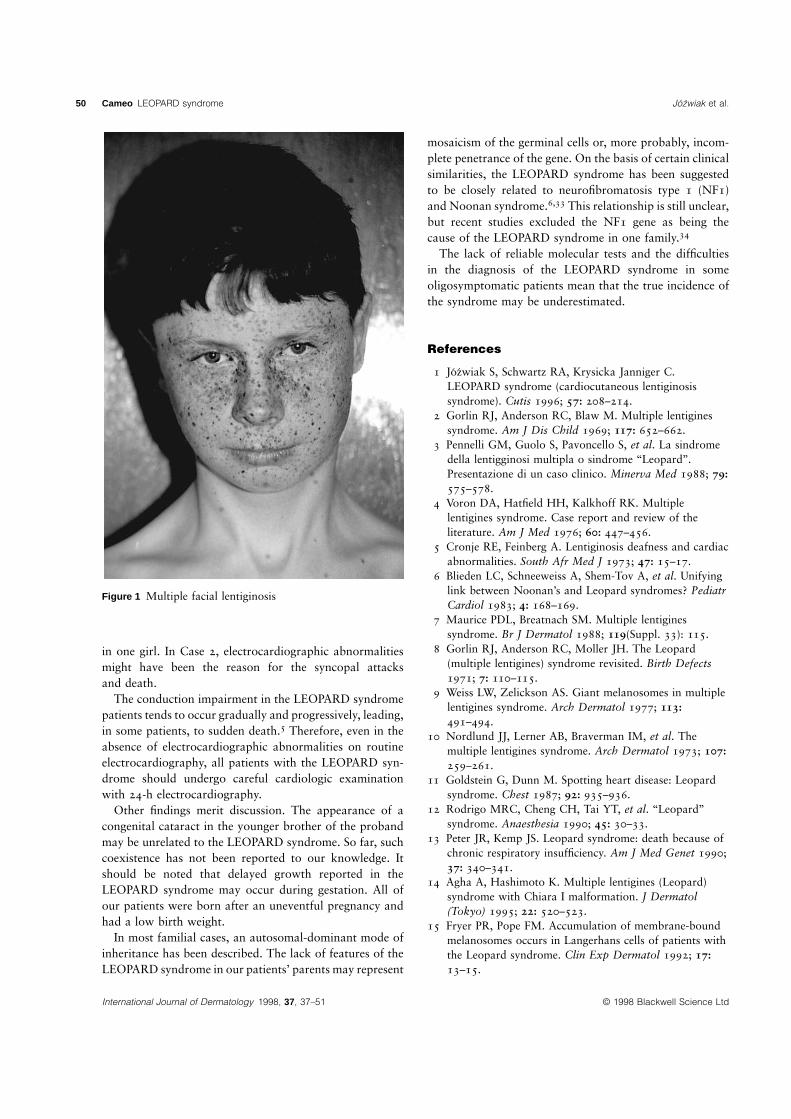
50 Cameo LEOPARD syndrome Jozwiak et al.
Figure 1 Multiple facial lentiginosis
in one girl. In Case 2, electrocardiographic abnormalitiesmight have been the reason for the syncopal attacksand death.
The conduction impairment in the LEOPARD syndromepatients tends to occur gradually and progressively, leading,in some patients, to sudden death.5 Therefore, even in theabsence of electrocardiographic abnormalities on routineelectrocardiography, all patients with the LEOPARD syn-drome should undergo careful cardiologic examinationwith 24-h electrocardiography.
Other findings merit discussion. The appearance of acongenital cataract in the younger brother of the probandmay be unrelated to the LEOPARD syndrome. So far, suchcoexistence has not been reported to our knowledge. Itshould be noted that delayed growth reported in theLEOPARD syndrome may occur during gestation. All ofour patients were born after an uneventful pregnancy andhad a low birth weight.
In most familial cases, an autosomal-dominant mode ofinheritance has been described. The lack of features of theLEOPARD syndrome in our patients’ parents may represent
International Journal of Dermatology 1998, 37, 37–51 © 1998 Blackwell Science Ltd
mosaicism of the germinal cells or, more probably, incom-plete penetrance of the gene. On the basis of certain clinicalsimilarities, the LEOPARD syndrome has been suggestedto be closely related to neurofibromatosis type 1 (NF1)and Noonan syndrome.6,33 This relationship is still unclear,but recent studies excluded the NF1 gene as being thecause of the LEOPARD syndrome in one family.34
The lack of reliable molecular tests and the difficultiesin the diagnosis of the LEOPARD syndrome in someoligosymptomatic patients mean that the true incidence ofthe syndrome may be underestimated.
References
1 Jozwiak S, Schwartz RA, Krysicka Janniger C.LEOPARD syndrome (cardiocutaneous lentiginosissyndrome). Cutis 1996; 57: 208–214.
2 Gorlin RJ, Anderson RC, Blaw M. Multiple lentiginessyndrome. Am J Dis Child 1969; 117: 652–662.
3 Pennelli GM, Guolo S, Pavoncello S, et al. La sindromedella lentigginosi multipla o sindrome ‘‘Leopard’’.Presentazione di un caso clinico. Minerva Med 1988; 79:575–578.
4 Voron DA, Hatfield HH, Kalkhoff RK. Multiplelentigines syndrome. Case report and review of theliterature. Am J Med 1976; 60: 447–456.
5 Cronje RE, Feinberg A. Lentiginosis deafness and cardiacabnormalities. South Afr Med J 1973; 47: 15–17.
6 Blieden LC, Schneeweiss A, Shem-Tov A, et al. Unifyinglink between Noonan’s and Leopard syndromes? PediatrCardiol 1983; 4: 168–169.
7 Maurice PDL, Breatnach SM. Multiple lentiginessyndrome. Br J Dermatol 1988; 119(Suppl. 33): 115.
8 Gorlin RJ, Anderson RC, Moller JH. The Leopard(multiple lentigines) syndrome revisited. Birth Defects1971; 7: 110–115.
9 Weiss LW, Zelickson AS. Giant melanosomes in multiplelentigines syndrome. Arch Dermatol 1977; 113:491–494.
10 Nordlund JJ, Lerner AB, Braverman IM, et al. Themultiple lentigines syndrome. Arch Dermatol 1973; 107:259–261.
11 Goldstein G, Dunn M. Spotting heart disease: Leopardsyndrome. Chest 1987; 92: 935–936.
12 Rodrigo MRC, Cheng CH, Tai YT, et al. ‘‘Leopard’’syndrome. Anaesthesia 1990; 45: 30–33.
13 Peter JR, Kemp JS. Leopard syndrome: death because ofchronic respiratory insufficiency. Am J Med Genet 1990;37: 340–341.
14 Agha A, Hashimoto K. Multiple lentigines (Leopard)syndrome with Chiara I malformation. J Dermatol(Tokyo) 1995; 22: 520–523.
15 Fryer PR, Pope FM. Accumulation of membrane-boundmelanosomes occurs in Langerhans cells of patients withthe Leopard syndrome. Clin Exp Dermatol 1992; 17:13–15.

Dutz and Lui Calcipotriol and anthralin Clinical trial 51
16 Shelley WB. Factitial dermatitis as the presenting sign ofmultiple lentiginosis syndrome. Therapeutic effect ofautodermabrasion. Arch Dermatol 1982; 118: 260–262.
17 Kolvekar S, Williams BT, Venn GE. Hypertrophicobstructive cardiomyopathy with LEOPARD(Moynahan’s) syndrome: surgical treatment. J Roy SocMed 1993; 86: 115–116.
18 Kubeyinje EP, Onunu AN, Obasohan AD. Multiplelentigines syndrome in a Nigerian family. TropGeograph Med 1993; 45: 135–137.
19 Munoz J, Ibanez M, Bodi V. Cardiomyopathiclentiginosis: an echo-Doppler report. Int J Cardiol 1993;48: 317–319.
20 Torok L, Szentendrei L, Szili M, et al. Progressivekariomyopatische lentiginosis (LEOPARD-syndrom) bei3 Patienten, kombiniert mit Marfan-Syndrom. ZeitschrHautkrankh 1989; 65: 197–201.
21 Torok L, Szentendrei L, Szili M, et al. Progressivecardiomyopathias lentiginosis (LEOPARD-syndroma,Moynahen-syndromea). Orvosi Hetilap 1989; 130:1531–1536.
22 Galaction-Nitelea O, Dociu I. Lentiginoses et anomaliesde developpement. Observation familiale de lentiginoseavec dysraphisme et debilite mentale. Dermatologica(Basel) 1974; 148: 108–115.
23 Andre P, Janin-Mercier A, Souteyrand P, et al.Lentiginose cutaneo-muqueuse, ephelides et myxomecardiaque. Ann Dermatol Venereol (Paris) 1988; 115:151–157.
24 Uhle P, Norvell S Jr. Generalized lentiginosis. J Am AcadDermatol 1988; 18: 444–447.
25 Schievink WI, Michels V, Mokri B, et al. Brief report: a
Clinical trial
A comparative study of calcipotriol and anthralin for chronicplaque psoriasis in a day care treatment center
Jan P. Dutz, MD, FRCPC, and Harvey Lui, MD, FRCPC
From the Division of Dermatology, Eighteen patients with symmetric plaque-type psoriasis were recruited for an open,Psoriasis and Phototherapy Clinic, controlled, bilateral half-body comparison study to evaluate the efficacy of calcipotriol/tar/Vancouver Hospital and Health UVB vs. anthralin/tar/UVB in a day care treatment setting. No patient had been onSciences Centre, University of British
systemic antipsoriatic agents for at least 3 months prior to enrolment. One half-body wasColumbia, Vancouver, Britisharbitrarily assigned to treatment with gradually increasing concentrations of anthralin asColumbia, Canadatolerated. The other half-body received calcipotriol ointment twice daily. Both sides
Presented at the Photomedicine received UVB and additional coal tar distillate in accordance with our standard day careSociety Annual Meeting, February regimen.1996, Washington DC
© 1998 Blackwell Science Ltd International Journal of Dermatology 1998, 37, 51–55
familial syndrome of arterial dissections withlentiginosis. N Engl J Med 1995; 332: 576–579.
26 Holder JE, Graham-Brown RA, Camp RD. Partialunilateral lentiginosis associated with blue naevi. Br JDermatol 1994; 130: 390–393.
27 Pique E, Aquilar A, Farina MC, et al. Partial unilaterallentiginosis: report of seven cases and review of theliterature. Clin Exp Dermatol 1995; 20: 319–322.
28 Fressinaud C, Preux PM, Milor AM, et al. Complexefamilial myxome-lentiginose revele par un probableaccident ischemique transitoire. Rev Neurol (Paris)1993; 149: 219–221.
29 Zahorcsek Z, Schneider I. Generalized lentiginosismanifesting through three generations. Int J Dermatol1996; 35: 357–359.
30 Torok L. Hautveranderungen als Hinweis auf kardialeStorungen. Zeitschr Hautkrankh 1989; 65: 1001–1006.
31 Van Voolen GA, Selmanowitz VJ. Lentiginosis profusa.III. Aesthetic and sanguineous aspects. Cutis 1976; 17:325–329.
32 Arnsmeier SL, Paller AS. Pigmentary anomalies in themultiple lentigines syndrome: is it distinct fromLEOPARD syndrome? Ped Dermatol 1996; 13:100–104.
33 Selmanowitz VJ. Lentigines profusa syndrome (multiplelentigines syndrome) II. Histological findings, modifiedCrowe’s sign and possible relationship to vonRecklinghausen’s disease. Acta Derm Venereol (Stockh)1971; 51: 387–393.
34 Ahlbom EB, Dahl N, Zetterqvist P, Anneren G. Noonansyndrome with cafe-au-lait spots and multiple lentiginessyndrome are not linked to the neurofibromatosis type 1locus. Clin Genet 1995; 48: 85–89.

52 Clinical trial Calcipotriol and anthralin Dutz and Lui
Correspondence Patients who were admitted to the day care program attended the clinic for UVB,Harvey Lui, MD anthralin, and calcipotriol on weekdays for two consecutive weeks. Anthralin was appliedDivision of Dermatology
to psoriatic plaques on one side in the following fashion: anthralin 0.1% with salicylic acidUniversity of British Columbia3% in zinc oxide paste on days 1 and 2; anthralin 0.2% with salicylic acid 3% in zinc835 West 10th Avenue
Vancouver BC, V5Z 4E8 oxide paste on days 3–5; anthralin 1% with salicylic acid 3% in hydrophilic petrolatum forCanada 60 min on days 8–10 to thicker lesions; and anthralin 2% with salicylic acid 3% in
hydrophilic petrolatum for 60 min on day 11 to thicker lesions. On the contralateral side,
calcipotriol ointment 0.05 µg/mL (Leo Pharmaceuticals, Ajax, Ontario) was applied to
lesions twice daily. No anthralin or calcipotriol was applied on weekends. All patients
applied coal tar oil 50% (Doak Oil Forte, Trans CanaDerm, St-Laurent, Quebec,
equivalent to 5% coal tar distillate) with salicylic acid 5% in hydrophilic petrolatum to their
lesions at home in the evenings and on weekends. UVB (FSX72T12 lamps, National
Biologic Corporation, Twinsburg, Ohio) was administered twice daily on weekdays in
increasing doses as tolerated (to erythema) prior to the application of the topical
medications. No trial medications were applied to the face, scalp, or genital regions.
For clinical evaluation, the standard Psoriasis Activity and Severity Index (PASI) score1
was modified by splitting the score for area under 10%; the modified score (mPASI) for an
area of coverage of 1%–4% was 0.5 and for an area of 5%–9% was 1. The head and
neck area was excluded from the analysis since neither anthralin nor calcipotriol was
used at these sites. Each half-body was considered to represent 100% in the area score
determination. The maximum modified score for each side was 64.8 (vs. 72 in the
standard PASI scoring system1). Clinical evaluations were completed at days 0 (baseline),
3, 7, 10, and 42. The primary end-point was day 10. On day 10, patients were asked to
compare the calcipotriol ointment to the anthralin on a five-point scale in terms of efficacy
and irritancy and to state their future preferred treatment modality. Following discharge
from day care, patients were continued on outpatient UVB and tar treatments three times
weekly and asked to return for a repeat clinical assessment after 4 weeks (day 42). Blood
samples taken prior to treatment and at day 10 were analyzed for serum calcium.
Comparisons of treatment efficacy were based on changes in the mPASI scores from
onset of treatment to day 10, as well as on the corresponding percentage changes.
Analyses were carried out using the Wilcoxon test. Subjective patient comparisons of
effectiveness and irritancy, as well as patient preference, were tested for equiprobability
using the chi-square goodness-of-fit test with an examination of the adjusted residuals.
Results
Eighteen patients were recruited from a total of 46 admittedto the day care program during the 4-month period of thisstudy. The mean age of the patients was 40 years (range,16–72 years), and the average disease duration was 13years (range, 2–30 years). The average body surface areainvolvement at baseline was 15% (range, 4%–69%). Calci-potriol ointment use was recorded in eight patients andaveraged 120 g over 2 weeks for each half-body (range,82–245 g). No hypercalcemia was noted during the study.One patient developed intolerable erythema, irritation, andincreasing scale within 7 days due to the calcipotriol andhad to be withdrawn from the study.
Morphologic differences were noted that could not bequantitated by the mPASI scoring system. There appearedto be slightly more flattening and erythema on the calcipo-
International Journal of Dermatology 1998, 37, 51–55 © 1998 Blackwell Science Ltd
triol side, while the anthralin side demonstrated character-istic staining by day 5. At day 10, these differences werediminished. Differences in mean mPASI scores between thetwo treatments were slight, with a trend to more rapidimprovement early with calcipotriol. The average mPASIscore on initiation was 14 and improved to 8 in each arm(Table 1). The range of mPASI scores between patients waswide, but the differences between scores at baseline andday 10 were highly significant for both anthralin andcalcipotriol (p , 0.0001). The mean percentage improve-ment was approximately 50% on each side (Table 1) withno significant difference between the two treatments(p 5 0.21). Although the primary end-point of the studywas day 10, patients were continued on daily tar and twice-weekly UVB for the next 4 weeks and reassessed. Elevenpatients presented for this later follow-up evaluation.Again, no difference was detected between the two treat-

Dutz and Lui Calcipotriol and anthralin Clinical trial 53
Table 1 Modified PASI scores at baseline (day 0) and day10 (D10) and percentage improvement for calcipotriol andanthralin treatment
Statistic Day 0* Calcipotriol Anthralin(D10) (D10)
Mean (6 S.D.) 14.5 (8.1) 7.8 (6.0) 7.6 (4.9)Range 4.6–35.4 1.15–24.4 1.7–17.8Percentage improvement N/A 49 (17) 48 (15)
(6 S.D.)
*Baseline mPASI scores on the right and left half-bodieswere equal.
ments (data not shown). Fifteen patients completed thepatient preference questionnaire. Patients subjectively feltthat calcipotriol was more effective than anthralin(p50.01), while anthralin use resulted in more irritation(p50.001). The majority (11/15) preferred calcipotriol overanthralin (p50.001).
Discussion
Recent studies addressing the combined use of UVB andtopical calcipotriol for the treatment of chronic plaquepsoriasis have been contradictory. In an open right/leftcomparison study of 20 patients, Kragballe2 found nostatistical difference in outcome between calcipotriol aloneand calcipotriol with broad-band UVB. Kerscher et al.3
subsequently showed that calcipotriol and narrow-bandUVB were more efficacious than calcipotriol alone. Morerecently, Kokelj et al.4 and Hecker and Lebwohl5 haveshown that UVB and calcipotriol are significantly moreeffective than UVB therapy alone. When used alone, calci-potriol has been shown to be more effective than eithershort contact anthralin6 or coal tar solution.7
We sought to compare the performance of calcipotrioland UVB/tar with anthralin and UVB/tar in a population ofpatients referred for intensive outpatient day care treatment.Both combined treatment modalities led to a similar signi-ficant improvement of approximately 50% over 10 days.The liberal use of calcipotriol combined with UVB did notresult in a high rate of adverse effects. McKenna and Stern8
reported the occurrence of photosensitivity associated withcombined UVB and calcipotriol therapy in 27% of theirpatients in whom calcipotriol was added to ongoing high-
© 1998 Blackwell Science Ltd International Journal of Dermatology 1998, 37, 51–55
dose UVB therapy. The one patient in our study whodeveloped significant irritation had not been receiving highdoses of UVB prior to study enrolment. Further, we didnot see evidence of hyperpigmentation due to calcipotriolas reported by Kokelj et al.9
Based on our experience, we believe calcipotriol to be asafe and effective adjunct to intensive UVB light treatmentin a day care treatment setting. Overall, patients preferredcalcipotriol to anthralin when used in combination withUVB and tars, although the objective efficacy was similar.
Acknowledgments
The Clinical Resource Nurses of the Psoriasis and Photo-therapy Clinic provided assistance, and Dr Michael Schulzerperformed the statistical analysis. The Vancouver Hospitaland Health Sciences Center and the University of BritishColumbia provided financial support.
References
1 Fredriksson T, Petterson U. Severe psoriasis—oraltherapy with a new retinoid. Dermatologica 1978; 157:238–244.
2 Kragballe K. Combination of topical calcipotriol (MC903) and UVB radiation for psoriasis vulgaris.Dermatologica 1990; 181: 211–214.
3 Kerscher M, Volkenandt M, Plewig G, Lehmann P.Combination phototherapy of psoriasis with calcipotrioland narrow-band UVB [letter]. Lancet 1993; 342: 923.
4 Kokelj F, Lavaroni G, Guadagnini A. UVB versus UVBplus calcipotriol (MC 903) therapy for psoriasis vulgaris.Acta Derm Venereol (Stockh) 1995; 75: 386–387.
5 Hecker DI, Lebwohl M. Topical calcipotriene incombination with UVB phototherapy for psoriasis. Int JDermatol 1997; 36: 302–303.
6 Berth-Jones J, Chu AC, Dodd WA, et al. A multicentre,parallel-group comparison of calcipotriol ointment andshort-contact dithranol therapy in chronic plaquepsoriasis. Br J Dermatol 1992; 127: 266–271.
7 Tham SN, Lun KC, Cheong WK. A comparative studyof calcipotriol ointment and tar in chronic plaquepsoriasis. Br J Dermatol 1994; 131: 673–677.
8 McKenna KE, Stern RS. Photosensitivity associated withcombined UV-B and calcipotriene therapy. ArchDermatol 1995; 131: 1305–1307.
9 Kokelj F, Lavaroni G, Perkan V, Plozzer C.Hyperpigmentation due to calcipotriol (MC 903) plusheliotherapy in psoriatic patients. Acta Derm Venereol(Stockh) 1995; 75: 307–309.

54 Clinical trial Topical isotretinoin vs. topical retinoic acid Domınguez et al.
Clinical trial
Topical isotretinoin vs. topical retinoic acid in the treatmentof acne vulgaris
Judith Domınguez, MD, Marıa Teresa Hojyo, MD, Jose Luıs Celayo, MD,Luciano Domınguez-Soto, MD, and Fernanda Teixeira, MD
From the Department of Dermatology, This is a clinical, prospective, and longitudinal study comparing the efficacy and incidence ofHospital General ‘‘Dr Manuel Gea averse effects of topical isotretinoin against those of topical retinoic acid in the treatment of acneGonzalez’’, Mexico City, Mexico vulgaris. The 30 participants were recruited from the patients attending the outpatient clinic of the
Department of Dermatology of ‘‘Dr Manuel Gea Gonzalez’’ General Hospital in Mexico City. TheyCorrespondencebelonged to either sex and any race, their ages ranged between 13 and 30 years, and theyLuciano Domınguez-Soto, MD
Department of Dermatology presented with 15 to 100 facial inflammatory lesions (papulo-pustules) and/or 15 to 100Hospital General ‘‘Dr Manuel Gea noninflammatory lesions (comedones) and no more than three nodulo-cystic lesions. The criteriaGonzalez’’ of exclusion were as follows: pregnancy or lactation, systemic treatment with steroids,Calzada de Tlalpan 4800
antibiotics, antiandrogens, or oral retinoids in the preceding 24 months, treatment with ultravioletMexico 14,000, D.F.radiation, hypersensitivity to retinoids, or a severe systemic illness. From 44 interviewed patients,Mexico14 were excluded. A detailed clinical history was obtained from the remaining individuals, the
Drug names degree of seborrhea was recorded, and acne lesions were counted. Each patient received eitherisotretinoin: Isotrex (topical) isotretinoin gel 0.05% or retinoic acid cream 0.05%. The patients were instructed to wash theirretinoic acid: Retin-A
faces in the mornings and evenings with a neutral soap, and to apply the product after the
evening cleansing.
The patients were examined again after 2, 4, 8, and 12 weeks of treatment and, at each
appointment, the number of lesions was recorded and the severity of acne was graded
according to the classification of Plewig and Kligman.1 The seriousness of the adverse effects,
such as stinging, pruritus, erythema, xerosis, and desquamation, was evaluated blindly by an
investigator who did not know what group the patient belonged to, and graded as 15mild, 25
moderate, and 35severe.
The efficacy of each drug was determined by the reduction in the number of lesions between
weeks 0 and 12 of treatment. An excellent response corresponded to a 76%–100% reduction of
the lesions, a good response to a 51%–75% reduction, a fair response to a 26%–50% reduction,
and a poor response to a 0%–25% reduction. The results were analyzed statistically using the
chi-square test, the exact test of Fisher and the test of Wilcoxon–Mann–Whitney. The changes in
the numbers of lesions between weeks 0 and 12 were analyzed separately for each group of
treatment, and the level of statistical significance was fixed at 0.05. The analysis was performed
with the aid of a Stat program, version 4.0.
Results The patients were assigned randomly to either Group I (isotretinoin) or Group II (retinoic
acid). Each group was composed of 15 individuals and, as a coincidence, in each group there
were nine women and six men. The clinical differences between the groups at the first visit were
not statistically significant.
In both groups, there was, in general, a good response to treatment (Fig. 1). Both drugs had a
similar degree of efficacy on inflammatory lesions. At the first visit, grades III and IV
predominated, whereas, after 12 weeks of treatment, most patients were classified in grades I or
II (Fig. 2). Similar results were observed regarding noninflammatory lesions (Fig. 3).
Ten of the patients of Group II complained of stinging associated with the treatment, especially
at weeks 8 and 12, as well as erythema and desquamation at the 12th week. Erythema and
stinging lasted for minutes or hours, whereas desquamation persisted for several days. Seven
individuals receiving isotretinoin mentioned irritation, which was of a mild degree.
International Journal of Dermatology 1998, 37, 51–55 © 1998 Blackwell Science Ltd
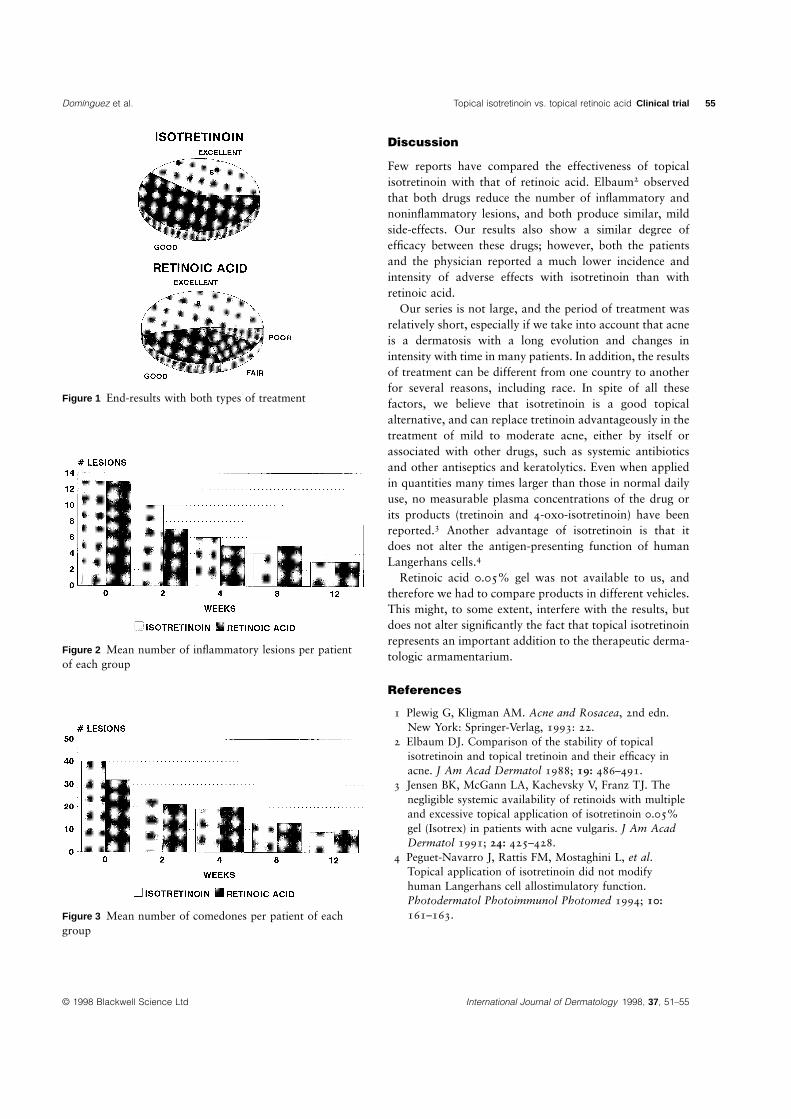
Domınguez et al. Topical isotretinoin vs. topical retinoic acid Clinical trial 55
Figure 1 End-results with both types of treatment
Figure 2 Mean number of inflammatory lesions per patientof each group
Figure 3 Mean number of comedones per patient of eachgroup
© 1998 Blackwell Science Ltd International Journal of Dermatology 1998, 37, 51–55
Discussion
Few reports have compared the effectiveness of topicalisotretinoin with that of retinoic acid. Elbaum2 observedthat both drugs reduce the number of inflammatory andnoninflammatory lesions, and both produce similar, mildside-effects. Our results also show a similar degree ofefficacy between these drugs; however, both the patientsand the physician reported a much lower incidence andintensity of adverse effects with isotretinoin than withretinoic acid.
Our series is not large, and the period of treatment wasrelatively short, especially if we take into account that acneis a dermatosis with a long evolution and changes inintensity with time in many patients. In addition, the resultsof treatment can be different from one country to anotherfor several reasons, including race. In spite of all thesefactors, we believe that isotretinoin is a good topicalalternative, and can replace tretinoin advantageously in thetreatment of mild to moderate acne, either by itself orassociated with other drugs, such as systemic antibioticsand other antiseptics and keratolytics. Even when appliedin quantities many times larger than those in normal dailyuse, no measurable plasma concentrations of the drug orits products (tretinoin and 4-oxo-isotretinoin) have beenreported.3 Another advantage of isotretinoin is that itdoes not alter the antigen-presenting function of humanLangerhans cells.4
Retinoic acid 0.05% gel was not available to us, andtherefore we had to compare products in different vehicles.This might, to some extent, interfere with the results, butdoes not alter significantly the fact that topical isotretinoinrepresents an important addition to the therapeutic derma-tologic armamentarium.
References
1 Plewig G, Kligman AM. Acne and Rosacea, 2nd edn.New York: Springer-Verlag, 1993: 22.
2 Elbaum DJ. Comparison of the stability of topicalisotretinoin and topical tretinoin and their efficacy inacne. J Am Acad Dermatol 1988; 19: 486–491.
3 Jensen BK, McGann LA, Kachevsky V, Franz TJ. Thenegligible systemic availability of retinoids with multipleand excessive topical application of isotretinoin 0.05%gel (Isotrex) in patients with acne vulgaris. J Am AcadDermatol 1991; 24: 425–428.
4 Peguet-Navarro J, Rattis FM, Mostaghini L, et al.Topical application of isotretinoin did not modifyhuman Langerhans cell allostimulatory function.Photodermatol Photoimmunol Photomed 1994; 10:161–163.




















