
TPCs: Endolysosomal channels for Ca2+ mobilization from acidic organelles triggered by NAADP

NAADP as a second messenger: Neither CD38 nor the base-
exchange reaction are necessary for the in vivo generation of
NAADP in myometrial cells.
Sandra Soares*, Michael Thompson*, Thomas White*, Amir Isbell*, Michiko Yamasaki+, Yodeta Prakash*, Frances E Lund#, Antony Galione+, Eduardo N. Chini .*
*From the Signal Transduction Laboratory Departments of Anesthesiology and Physiology
Mayo Clinic and Foundation, Rochester, Minnesota 55905,
+Department of Pharmacology, University of Oxford, Mansfield Road, Oxford OX1 3QT, and
#Trudeau Institute, 154 Algonquin Avenue, Saranac Lake, New York 12983, USA.
Correspondence and reprint requests to: Eduardo Nunes Chini Department of Anesthesiology Mayo Clinic and Foundation Rochester, MN 55905 Phone: (507) 284-8130 Fax: (507) 284-8566 Email: [email protected] Mayo Foundation supported this research. Both Sandra Soares and Michael Thompson provided equal contributions to the study. KEY WORDS: cADPR, IP3, endoplasmic reticulum, ryanodine channel, NAADP, CD38, Base-exchange reaction
ABBREVIATIONS: cyclic-ADP-ribose (cADPR); Nicotinic acid adenine dinucleotide
phosphate (NAADP)
Page 1 of 44 Articles in PresS. Am J Physiol Cell Physiol (June 21, 2006). doi:10.1152/ajpcell.00638.2005
Copyright © 2006 by the American Physiological Society.

ABSTRACT
Nicotinic acid adenine dinucleotide phosphate (NAADP) has recently been shown to act as a
second messenger controlling intracellular Ca2+ responses in mammalian cells. Many questions
remain regarding this signaling pathway, including the role of the ryanodine receptor (RyR) in
NAADP-induced Ca2+ transients. Furthermore, the exact metabolic pathway responsible for the
synthesis of NAADP in vivo has not been determined. Here we demonstrate that the NAADP
mediated Ca2+ release system is present in human myometrial cells. We also demonstrate that
human myometrial cells use the NAADP- second messenger system to generate intracellular
Ca2+ transients in response to histamine. It has been proposed in the past that the NAADP-system
in mammalian cells is dependent on the presence of functional RyRs. Here we observed that the
histamine-induced Ca2+ transients are dependent on both the NAADP and IP3 signaling pathways
but are independent of RyRs. The enzyme CD38 has been shown to catalyze the synthesis of
NAADP in vitro by the base-exchange reaction. Furthermore, it has been proposed that this
enzyme is responsible for the intracellular generation of NAADP in vivo. Using CD38 knockout
mice, we observed that both the basal and histamine stimulated levels of NAADP are
independent of CD38 and the base-exchange reaction. Our group is the first to demonstrate that
NAADP is a second messenger for histamine-elicited Ca2+ transients in human myometrial cells.
Furthermore, the NAADP mediated mechanism in mammalian cells can be independent of RyRs
and CD38. Our data provides novel insights into the understanding of the mechanism of action
and metabolism of this new second messenger system.
Page 2 of 44

INTRODUCTION
Release of Ca2+ from intracellular stores is a widespread component of several signaling
pathways (Berridge and Lipp, 2000; Chini and deToledo, 2001). Nicotinic acid–adenine
dinucleotide phosphate (NAADP) is a recently discovered nucleotide with intracellular Ca2+-
releasing properties in several mammalian cells and tissues (Chini and deToledo, 2001; Chini et
al, 1995; Dousa et al, 1996; Galione et al, 2000). In fact, NAADP has been implicated as an
intracellular second messenger (Cancela et al, 1999; Chini, 2004; Churchill et al, 2003; Masgrau
et al, 2003; Yamasaki et al, 2004; Yamasaki et al, 2005; Kinnear et al, 2004).
In view of the potential role of NAADP as a second messenger, it is pivotal to determine
the mechanisms of NAADP induced Ca2+ release and its metabolism in mammalian tissues. In
fact, many aspects of the NAADP signaling system in mammalian cells have yet to be defined.
In regards to the mechanism of NAADP-induced Ca2+ transients, it has been proposed that
NAADP induces Ca2+ release from lysosomal like Ca2+ stores (Churchill et al, 2002; Yamasaki et
al, 2004; Kinnear et al, 2004). In addition, it has been proposed that the ryanodine receptor
(RyR) is necessary for NAADP-induced Ca2+ release in mammalian cells (Dammermann et al,
2005; Gerasimenko et al, 2003; Hohenegger et al, 2002). In fact, some authors have proposed
that the RyR is the site of action of NAADP in mammalian cells (Dammermann et al, 2005;
Galione and Petersen, 2005; Gerasimenko et al, 2003; Hohenegger et al, 2002). Here we
demonstrate that the agonist histamine induces Ca2+ transients in myometrial cells that are
dependent on NAADP but independent of the RyR. This new finding indicates that, similar to
Page 3 of 44

what is observed in invertebrates (Chini et al, 1995; Chini and Dousa, 1996; Lee and Aarhus,
1995), the RyR is not necessary for the effect of NAADP in mammalian cells.
Synthesis of this second messenger by the so-called base-exchange reaction has been described
in several mammalian tissues, including brain, heart, liver, spleen and kidney (Cheng et al, 2001;
Chini et al, 2002; Chini and Dousa, 1995). In the base-exchange reaction, the nicotinamide
residue from NADP is replaced by a nicotinic acid resulting in the formation of NAADP (Chini
and deToledo, 2002). This reaction is catalyzed by NADases in the presence of the substrates
NADP and nicotinic acid (Chini and deToledo, 2002). Furthermore, it has also been reported that
ADP-ribosyl cyclase is capable of catalyzing the base-exchange reaction leading to synthesis of
NAADP (Aarhus et al, 1995). This enzyme was first described as being responsible for the
synthesis of another second messenger, cADPR, and a link between these two signaling
pathways has been proposed (Aarhus et al, 1995; Chini et al, 2002). In mammalian cells ADP-
ribosyl cyclase, named CD38, is the major enzyme involved in the synthesis of cADPR in many
mammalian tissues (Takahashi et al, 2004). Furthermore, CD38 has also been reported to
catalyze the synthesis of NAADP (Aarhus et al, 1995; Chini et al, 2002). In addition, we have
recently described that CD38 is the major enzyme responsible for the in vitro synthesis of
NAADP catalyzed by the base-exchange reaction in mammalian tissues (Chini et al, 2002). In
fact, it has been proposed by many authors that the CD38 catalyzed base-exchange reaction is the
physiological pathway for the synthesis of NAADP (Aarhus et al, 1995; Chini et al, 2002).
However, whether CD38 can indeed generate NAADP via the base-exchange reaction in vivo has
not been described to date. Under the present experimental conditions, the concentrations of
substrate needed for the base-exchange reaction, namely nicotinic acid, are several times higher
than would be expected to be present in intact cells ((Chini and deToledo, 2002). Furthermore,
Page 4 of 44

the optimal pH for this reaction is out of the physiological range (Aarhus et al, 1995; Cheng et al,
2001). However, compartmentalization of nicotinic acid and NADP into an acidic environment
could theoretically provide a possible milieu for the synthesis of NAADP in vivo. With the recent
development of a sensitive and specific bioassay for the measurement of intracellular levels of
NAADP (Churchill et al, 2003), the question of whether CD38 and the base-exchange reaction
are responsible for the in vivo synthesis of NAADP can be now approached.
In the present study, we provide experimental evidence to suggest that CD38 and the base-
exchange reaction are not involved in the in vivo generation of NAADP. These data provide the
framework for the discovery of the true physiological metabolic pathway for the in vivo synthesis
of NAADP.
Page 5 of 44

MATERIAL AND METHODS
CD38 wild type and knockout mice. CD38 knockout mice (C57BL/6J.129 CD38-/- , N12
backcross) were produced as described previously (Partida-Sanchez et al, 2001) and were
maintained in the Mayo Clinic and Trudeau Institute Animal Breeding facility in accordance
with all Institutional Animal Care and Use Committee guidelines.
Cell preparation. In accordance with procedures reviewed and approved by the Mayo
Foundation Institutional Review Board, human myometrium was obtained from patients
undergoing elective hysterectomy. Human myometrial cells were isolated using techniques
previously described (Barata et al, 2004; Thompson et al, 2004). Briefly, the tissue was minced
in Hanks' balanced salt solution (HBSS) containing 1g/L glucose and 10 mM HEPES (pH 7.4).
The tissue was then suspended in fresh HBSS, aerated with 95% O2/5% CO2, and incubated in a
37o C water bath with gentle shaking for 2 h in the presence of 20 U/ml papain and 2,000 U/ml
DNase. Subsequently, the tissue was incubated for an additional 1 hour at 37o C, with the
addition of 1 mg/ml type IV collagenase. Human myometrial cells were released by triturating,
centrifuged, and resuspended in Dulbecco's Modified Eagle Medium (DMEM) containing 10%
fetal bovine serum (FBS), 100 U/liter penicillin, 100mg/liter streptomycin and 2.5 mg/ml
amphotericin B. Cultures were grown and maintained in 75 cm2 plastic flasks in a humidified
incubator supplied with 5% CO2/95% air at 37o C. Subcultures were obtained as needed by
detaching the cells with a Ca2+/Mg2+-free HBSS solution containing 0.25% trypsin and 5 mM
EDTA. Only cultures between passages 2 and 10 were used. Cells isolated by this procedure
stain positive for α-smooth muscle actin and negative for keratin. For experiments, cells were
made quiescent by replacing the growth medium with DMEM without serum. Cell medium was
Page 6 of 44

again replaced with DMEM containing test reagents solubilized in 0.1% DMSO or water added
to the final concentrations. HL60 cells were prepared as described previously (Munshi et al,
2002).
"Global" [Ca2+]i imaging in human myometrial cells. Cultured human myometrial cells were
plated on 9- x 22-mm coverslips for spectrofluorometer microscopy at a density of 25,000
cells/coverslip and grown until approximately 80% confluent in DMEM with 10% FBS. Cells
were made quiescent by changing the medium to serum free DMEM for 24 hours and then
incubated with 5 µM fura 2-AM for 2 h at 37 C. Cells were then washed in HBSS medium with
or without Ca2+, depending on the experiment and imaged alternately with 340/380 nm light
(emission 510 nm) using an F-2000 spectrofluorometer (Hitachi, Tokyo, Japan).
Isolation of microsomes. Microsomal fractions were isolated (at 0-4o C) from human
myometrial tissue. Myometrium, minced with a razor blade, was suspended in a buffer
containing 300 mM sucrose, 10 mM HEPES, 0.1 mM EDTA, and 0.5 mM
phemylmethylsulfonyl fluoride (PMSF) (pH 7.4), and homogenized using a Polytron
homogenizer. The homogenate was centrifuged for 10 min at 2,000 x g, and the pellet was
discarded. The supernatant was further centrifuged at 20,000x g for 20 min, and the supernatant
thus obtained was centrifuged at 100,000 x g for 1 h. The pellet was resuspended in a small
volume of homogenizing buffer by Dounce homogenizer. The microsomes were either used
fresh for measurement of Ca2+ release or were divided into aliquots, quickly frozen, and stored at
-70o C.
Ca2+ release in Human myometrium microsomes. Ca2+ uptake and release were measured in a
medium containing 250 mM N-methyl glucamine, 250 mM potassium gluconate, 20 mM HEPES
Page 7 of 44

buffer (pH 7.2), 1 mM MgCl2, 2 U/ml creatine kinase, 4 mM phosphocreatine, 1 mM ATP, 25
µg/ml leupeptin, 20 µg/ml aprotinin, 100 µg/ml soybean trypsin inhibitor, and 3 µM fluo-3.
Using a Hitachi F-2000 spectrofluorometer, fluo-3 fluorescence was monitored at 490 nm
excitation and 535 nm emission in a 250-µl cuvette contained in a 37oC temperature regulated
cuvette holder and mixed continuously with a magnetic stirring bar. The addition of stock
solutions of various reagents did not exceed 2% of the volume in the cuvette.
Confocal [Ca2+]i imaging and microinjection of NAADP. Dissociated human myometrial cells
attached to coverslips were incubated in 5 μM fluo-3/AM (Molecular Probes, Eugene OR) at
37oC for 30 min, and then placed on an open slide chamber (Warner Instruments, Hamden CT)
mounted on a Nikon Diaphot inverted microscope. The chamber was perfused with HBSS
containing 1 mM Ca2+ at 2-3 ml/min at room temperature.
Fluo-3 loaded cells were visualized using an Odyssey XL real-time confocal system
(Noran Instruments, Middleton WI) attached to the Nikon microscope, and equipped with an Ar-
Kr laser. In previous studies (Prakash et al, 2000), we have used real-time confocal imaging to
examine both spatial and temporal aspects of [Ca2+]i dynamics. However, in the present study,
the system was used predominantly to examine temporal aspects, within small regions of uterine
smooth muscle cells. Our previous experiments have determined that 30 frames/s was sufficient
to determine the dynamic [Ca2+]i response of myometrial cells without introducing frequency
aliasing. An Olympus 40X/1.3 oil-immersion objective lens was used for imaging with image
size set to 640x480 pixels (0.06 μm2/pixel). Optical section thickness was set to 1 μm. with
regions of interest (ROIs) of 5x5 pixels (1.5 μm2).
Myometrial cells displaying stable basal [Ca2+]i levels (i.e. no spontaneous fluctuations in
resting [Ca2+]i levels) were located, brought into the imaging field, and were impaled with single-
Page 8 of 44

lumen glass micropipettes (1.5 mm OD; World Precision Inc) that were pulled to a fine tip using
a Brown-Flaming electrode puller. The electrode tip resistance was found to be <100 KΩ when
filled with NAADP concentrations in 150mM KCl. The average tip diameter was estimated to be
<5 μm. The NAADP solution was injected into the myometrial cells using pressure injection
(General Valve Corp. PicoSpritzer) during simultaneous monitoring of [Ca2+]i responses. Since
the final NAADP concentration in the cell following injection was dependent on cell volume, the
volumes of ~10 myometrial cells was estimated a priori from online length and breadth
measurements. Based on this cell volume, the total time and amplitude of the pressure pulse was
then set such that the volume injected resulted in a final intracellular concentration of one of two
values (1 μM, and 1 mM).
Lysotracker red DND-99 and Bodipy-FL thapsigargin labeling. Simultaneous fluorescent
labelling of sarcoplasmic reticulum and lysosomes were performed using BODIPY-FL-
thapsigargin (Molecular Probes, B-7487) and Lysotracker red (Molecular Probes, L 7528). Cells
were grown to confluence in 8 well chambers slides and the medium was changed to fresh
growth medium prior to adding the fluorescent probes. The concentration of BODIPY-FL-
thapsigargin and Lysotracker red were respectively 1μΜ and 75nM. The cells were incubated
simultaneously with both labels for a period of 30 minutes to 1 hour under growth conditions and
imaging was done with an Olympus Fluoview confocal imaging station. (BODIPY-FL-
thapsigargin excitation was 488nm and emission 540nm and Lysotracker red excitation was 568
nm, and emission 590 nm). Cells were also labelled with each probe separately and the effect of
unlabelled excess TG (10 μM) and 50 μM GPN were determined.
Cyclase activity. ADP-ribosyl cyclase activity was measured using the NGD technique as
previously described (deToledo et al, 2000). Microsomal preparations were incubated in a
Page 9 of 44

medium containing 0.2 mM NGD, 0.25 M sucrose, and 40 mM Tris-HCl (pH 7.2) at 37o C.
Activity was determined using a fluorescent assay at 300 nm excitation and 410 nm emission. In
key experiments, results were also confirmed with the use of NAD, which is the natural substrate
of the enzyme.
NAADP content. Tissues were removed from mice under anesthesia and immediately frozen in
liquid nitrogen. Tissues were then resuspended in 10% TCA and homogenized on ice using a
Potter-Elvehjem tissue grinder and the resultant homogenate was centrifuged for 2 minutes,
12,000-x g at 40C. The supernatant was removed and TCA was extracted using water saturated
ether. The pH of the supernatant was then adjusted to 8.0, injected onto an HPLC and the
NAADP fraction collected as stated previously. The sample was then dried under vacuum,
washed three times with 1ml methanol and resuspended in the same volume as injected onto the
column using GluIM (250mM N-methyl-glucamine, 250mM potassium gluconate, 1mM MgCl2,
20mM HEPES (pH 7.2)). NAADP content was then determined using a modification of the
binding protocol stated previously using GluIM. Briefly, 25μl of 0.5% (final concentration) sea
urchin egg homogenate was aliquoted into 12 x 75mm tubes followed by 25μl of cell extract.
The reaction is allowed to incubate on ice for 30 minutes. [32P]- NAADP (50,000-cpm/200μl) is
then added and the binding terminated after 20 additional minutes as described previously using
ice-cold GluIM. Concentration of NAADP in the cellular extract is then calculated by comparing
to a standard curve with known quantities of NAADP.
NAADP synthesis by the base-exchange reaction
Membrane fractions (1mg/ml) were incubated with 1mM NADP and 40mM nicotinic acid at
37°C in a buffer containing 40mM triethanolamine-acetic acid buffer (pH 7.2). Aliquots (3-7µl)
Page 10 of 44

were removed after different incubation times and NAADP content was determined using a
combination of the sea urchin egg homogenate bioassay and HPLC analysis of nucleotides
(Chini et al, 1995).
Sea urchin egg homogenate bioassay
Homogenates from Lytechinus pictus eggs were prepared as described previously, with minor
modification (Chini et al, 1995). Briefly, eggs were obtained by injection of 0.5M KCl into the
coelomic cavity, and collected in artificial sea water. The jelly coats were washed from the eggs
by several passages through 80-mm-mesh silk. The eggs were then washed once in artificial sea
water, twice in Ca2+-free sea water containing 1mM EGTA, twice in Ca2+-free water without
EGTA and once in GluIM. A suspension (25%, w/v) was prepared by homogenization with 4-5
strokes in a Dounce homogenizer with a type A pestle. The homogenate was then centrifuged for
10-12s at 13000 x g at 4°C and the supernatant was collected and stored in 1ml aliquots at -70°C.
Frozen homogenates were thawed in a 17°C water bath and diluted to 1.25% (v/v) with GluIM
containing 2units/ml creatine kinase, 4mM phosphocreatine, 1mM ATP, 3µg/ml oligomycin and
3µg/ml antimycin.
HPLC analysis of nucleotides
The synthesis of NAADP and cADPR by mammalian tissue extracts was verified by HPLC
analysis, performed by anion-exchange chromatography using an AG MP-1 column (Bio-Rad)
eluted with a non-linear gradient of trifluoroacetic acid, as described previously (Chini et al,
1995). The nucleotides were detected by UV absorption at 254nm. The authenticity of the
NAADP and cADPR produced were confirmed by co-elution with NAADP and cADPR
Page 11 of 44

standards and by the sea urchin egg homogenate bioassay. The NAADP and cADPR used on our
study were at least 99% pure as determined by HPLC analysis.
Western Blot. Human myometrial cell and HL60 extracts were incubated in lysis buffer
containing 0.05% IGEPAL-CA 630, 20 mM EDTA, 20 mM NaCl, 20mM Tris pH 7.0. After
10% SDS-PAGE, protein was electroblotted onto PVDF membrane, blocked with 5% non-fat
milk for 1 hour and probed with 1/100 dilution of mouse monoclonal antibody against human
CD38 (Santa Cruz, Cat No SC 7325) for 4 h. The immunoreactive bands were detected using a
1/20,000 dilution of horseradish peroxidase-conjugated anti-mouse IgG (Santa Cruz, SC 2020)
as secondary antibody and an enhanced chemiluminescence detection system. Western-blot in
mice tissues was performed with the antibody sc-7049 Ab from Santa Cruz. This Ab reacts with
mouse CD38.
Detection of cADPR levels in cells and tissues by cycling assay. Mouse tissue or human cells
were frozen in liquid N2, pulverized into a powder, and extracted with 10 % trichloroacetic acid
(TCA) at 4°C. TCA was removed with water-saturated ether. The aqueous layer containing the
cADPR was removed and adjusted to pH 8 with 1M Tris. To remove nucleotides except cADPR,
a mixture containing hydrolytic enzymes was added to the samples with the following final
concentrations: 0.44unit/ml pyrophosphatase, 12.5units/ml alkaline phosphatase, 0.0625unit/ml
NADase, 2.5mM MgCl2 and 20mM sodium phosphate, pH 8.0. The detection of cADPR was
performed by some modification of the cycling method described recently (Graeff and Lee,
2002).
Page 12 of 44

Materials. All other reagents, of the highest purity grade available, were supplied from Sigma
Chemical (St. Louis, MO), except when stated otherwise. The lysosomal inhibitors
glycylphenylalanine-2-naphthylamide (GPN), bafilomycin A (Baf A1), monesis, and the Ca2+
ATPase inhibitors thapsigargin (TG) and cyclopiazonic acid (CPA) were of the highest purity
available.
The reported experiments were repeated at least three times, data are expressed as means ± SE or
SD. Student’s t-test was used to evaluate statistical significance; P values <0.05 were considered
significant.
Page 13 of 44

RESULTS AND DISCUSSION
The NAADP-induced Ca2+releasing system in myometrial cells. We have previously described
the presence of the NAADP Ca2+ releasing system in smooth muscle cells (Kinnear et al 2004;
Yusufi et al, 2002). Here we found that human myometrial cells also contain the NAADP
system. Microinjection of μM concentrations of NAADP into myometrial cells leads to the
development of a cytoplasmic rise in intracellular free Ca2+ as described before for pulmonary
smooth muscle cells (Fig. 1A)(Boittin et al, 2002; Kinnear et et 2004.). However as described
before, the microinjection of high concentrations of NAADP (1 mM, HNAADP) had no effect
on intracellular Ca2+ transients in myometrial cells (Fig. 1A). In fact, it has been proposed that
the NAADP receptor from mammalian cells can be inactivated by high concentrations of
NAADP itself (Cancela et al, 1999).
To further characterize the NAADP system we used human myometrial cell microsomes. We
observed that NAADP can trigger Ca2+ release from microsomes. The NAADP-induced Ca2+
release in microsomes was inhibited by the lysosomal inhibitor glycylphenylalanine-2-
naphthylamide (GPN), which causes lysis of lysosomes (Fig. 1B and C), and by another
lysosomal inhibitor bafilomycin A (data not shown). In contrast, GPN had no effect on the Ca2+
transients induced by cADPR or IP3 (Fig 1C and data not shown). The NAADP-induced Ca2+
release in microsomes was not affected by the sarcoplasmic reticulum Ca2+ ATPase inhibitor
thapsigargin (TG) (Fig. 1B). In contrast, pre-treatment of microsomes with TG lead to a
complete inhibition of the IP3 and cADPR induced Ca2+ release (data not shown). NAADP-
induced Ca2+ release in microsomes was not affected by the IP3 -inhibitor (xestospongin C), or
by the cADPR inhibitor (8-Br-cADPR) (Fig. 1D). It has been previously shown that NAADP-
induced Ca2+ release can be inhibited by L-type Ca2+ channel blockers and by high (mM) or low
Page 14 of 44

(<1 nM) concentrations of NAADP itself (Bak et al, 1999; Patel et al, 2001). The channels
activated by NAADP show a bell-shaped activation curve, where sub-optimal or supra-optimal
concentrations of NAADP are inhibitory to channel activation and optimal NAADP
concentrations vary from one cell type to another. Here we found that pre-incubation of human
myometrial microsomes with nifedipine, a L-type channel blocker, or 1 mM NAADP
(HNAADP) leads to a complete inhibition of the subsequent Ca2+ release induced by 1 μM
NAADP (Fig 1D). In contrast high concentrations of NAADP had no effect upon the Ca2+
release induced by IP3 or cADPR (Fig. 1E). These data indicate that the NAADP-system is
present in myometrial cells and that it is independent of the RyR channel. Furthermore, it
indicates that the NAADP Ca2+ stores are derived from lysosomes.
Lysosomal Ca2+ stores in myometrial cells: role in histamine induced Ca2+ transients. As
previously described and demonstrated above, the NAADP Ca2+ stores are derived from
lysosomal-like structures (Churchill et al, 2002; Yamasaki et al, 2004; Kinnear et al, 2004). To
determine the role of lysosomes and NAADP on agonist stimulated Ca2+ transients in human
myometrial cells we first characterized the lysosomal Ca2+ stores in these cells. Using a double
stain with the lysosomal marker lysotraker red and with the SR marker bodipy-thapsigargin we
observed that myometrial cells contain both SR and lysosomal like structures (Fig. 2A). These
lysosomal like structures occur with a mostly perinuclear distribution in contrast with the more
diffuse distribution of the SR (Fig. 2A). Furthermore, we confirmed that the lysotracker red stain
was specific for lysosomes since the lysosomal inhibitor, GPN, can completely abolish the stain
obtained with lysotracker (Fig. 2C). In contrast, TG had no effect on the lysotracker stain (data
not shown). The reverse was also true, staining of SR was unaffected by GPN (Fig. 2C) while
being abolished by unlabeled TG (data not shown). To further assess the presence of lysosomal
Page 15 of 44

Ca2+ stores in myometrial cells, we determined the effects of GPN and TG on intracellular Ca2+
in intact human myometrial cells. We observed that both GPN and TG caused a transient
increase in intracellular Ca2+ (Fig. 2B). To determine the role of the SR Ca2+-ATPase on the
presence of Ca2+ in lysosomes and also to address any overlap between the SR and the lysosomal
Ca2+ stores, we determined the effect of TG on the lysosomal Ca2+ content. We found that
incubation of myometrial cells with TG leads to a transient increase in intracellular Ca2+
consistent with depletion of SR Ca2+ stores (Fig. 3A). However, after the intracellular free Ca2+
returns to its baseline further addition of TG fails to promote more Ca2+ release, indicating that
the SR were completely depleted by the first addition of TG (Fig. 3A). In contrast, pre-
incubation with GPN had no effect upon the TG induced Ca2+ transients indicating that the
lysosomal Ca2+ stores are independent from the SR stores (Fig. 3B). All these experiments were
carried out in the absence of extracellular Ca2+ so the observed increases in intracellular Ca2+ had
to be derived from intracellular stores. We also observed that the Ca2+ release induced by TG can
be prevented by pre-incubation with another SR Ca2+-ATPase inhibitor CPA but not with
bafilomycin A (BafA1), an inhibitor of the lysosomal H+-ATPase, or GPN (Fig. 3C). In contrast,
the Ca2+ release induced by GPN was abolished by pre-treatment with both GPN and bafilomicin
A but not with CPA, or TG (Fig. 3D)
To further determine the role of lysosomal Ca2+ stores in myometrial cells we studied the effect
of lysosomal Ca2+ inhibitors upon agonist induced Ca2+ transients. We first characterized the role
of different intracellular and extracellular signaling pathways on the Ca2+ release induced by
oxytocin, histamine, and acetyl choline (ACh)(Fig. 4). The Ca2+ release induced by histamine
was only slightly inhibited by removal of extracellular Ca2+ and almost abolished by depletion of
the SR stores with TG (Fig. 4B). These data indicate that intracellular Ca2+ mobilization and not
Page 16 of 44

extracellular Ca2+ influx play a major role in histamine induced Ca2+ transients. Furthermore,
histamine-induced Ca2+ release was inhibited by the IP3 antagonist xestospongin C (Fig. 4A, B),
but not by ryanodine. In contrast, we observed that the oxytocin-induced Ca2+ transients in
myometrial cells can be almost completely abolished by removal of extracellular Ca2+ (Fig. 4A).
These data indicates that both extracellular Ca2+ influx and intracellular Ca2+ release play a role
in oxytocin-induced Ca2+ transients. Ca2+ responses induced by ACh were not significantly
reduced by the omission of extracellular Ca2+ and were inhibited by inhibitors of both IP3R
(xestospongin C) and cADPR-RyRs pathway (8-Br-cADPR, and ryanodine) (Fig 4A), these data
indicates that the ACh induced Ca2+ release is dependent on intracellular Ca2+ stores regulated by
cADPR and IP3.
We further determined the role of the lysosomal Ca2+ store on oxytocin, and histamine-induced
Ca2+ transients. Using inhibitors of the lysosomal Ca2+ accumulation we observed that both the
histamine and oxytocin induced Ca2+ transients in myometrial cells were dependent on lysosomal
stores (Fig. 4 B and C). In Figure 4B we determined the effect of treatment with several
lysosomal inhibitors upon the histamine-induced Ca2+ release, including GPN, bafilomycin A1,
and monesin. This data clearly indicates that lysosomal Ca2+ contributes to Ca2+ transients
induced by certain agonists in myometrial cells. To confirm the specificity of the lysosomal
inhibitors we determined their effects on acetylcholine (ACh) induced Ca2+ transients in
myometrial cells, observing that they had no effect upon ACh induced Ca2+ transients (Fig. 4C).
Our data provides a chance to understand the role of lysosomal Ca2+ stores in RyR dependent
and RyR-independent Ca2+ transients. Furthermore, if one assumes that lysosomal Ca2+ release is
only mediated by NAADP it is possible to extrapolate our findings to the coupling between
NAADP and other channels. It has been previously described that NAADP-induced Ca2+
Page 17 of 44

transients may be mediated by direct stimulation of RyRs or by a mechanism coupled with the
NAADP dependent Ca2+ stores and the CICR systemmediated by RyRs (Dammermann et al,
2005; Galione and Petersen, 2005; Gerasimenko et al, 2003; Hohenegger et al, 2002). In fact, it
has been proposed that NAADP-induced Ca2+ transients are completely dependent on the
expression of RyR in cells (Dammermann et al, 2005; Galione and Petersen, 2005; Gerasimenko
et al, 2003; Hohenegger et al, 2002). Here we observed that NAADP can work in a RyR
dependent and independent manner. Oxytocin mediated Ca2+ transients appear to be dependent
on both IP3 and cADPR-RyR function (Fig. 4A). In contrast, inhibitors of the RyR-cADPR
system had no effect upon the histamine induced Ca2+ transients in myometrial cells (Fig.4A).
Furthermore, the histamine induced Ca2+ transients can be inhibited by the IP3 inhibitor
xestospongin C (Fig.4A and B). These data indicate that the lysosomal Ca2+ stores can work in a
RyR independent manner and that they may be coupled to the IP3 channel. It is possible that the
NAADP and the IP3 system can be couple by a CICR mechanism, since the IP3 system also
behaves as a CICR (Chini and Dousa, 1996).
NAADP as a second messenger. To further determine whether NAADP is a second messenger in
myometrial cells we determined NAADP levels in cells and examined the effect of histamine on
the intracellular accumulation of NAADP. We observed that NAADP is a nucleotide present in
myometrial cells and that stimulation of cells with histamine leads to a several fold increase in
intracellular levels of NAADP (Fig. 5). We further characterized the time course and the
coupling between histamine and NAADP accumulation (Fig, 5 inset). Stimulation of myometrial
cells with histamine led to a rapid increase in NAADP levels that peaked at 30 seconds and
declined back to basal levels in one minute (Fig. 5 inset). Furthermore, the histamine-induced
NAADP accumulation was inhibited by the H1 receptor antagonist diphenhydramine (Fig. 5).
Page 18 of 44

We also observed that the NAADP accumulation induced by histamine appears to be Ca2+ and -
calmodulin dependent, as the accumulation of NAADP was inhibited by the Ca2+ chelator
BAPTA and the calmidazolium, a calmodulin antagonist (Fig. 5). These data were surprising
since the only enzyme known to generate NAADP in mammalian cells, CD38, appears not to be
Ca2+ or calmodulin-dependent (data not shown). These data led us to explore the role of the
enzyme CD38 on the in vivo generation of NAADP.
CD38 is not necessary for histamine induced NAADP accumulation in myometrial cells. It has
been previously shown that the enzyme CD38 is capable of generating NAADP in vitro (Aarhus
et al, 1995; Chini et al, 2002). CD38 catalyses the so-called base-exchange reaction where the
nicotinamide from NADP is substituted by nicotinic acid generating NAADP. To date, CD38
and the base-exchange reaction are the only enzyme and reaction known to be able to generate
NAADP (Aarhus et al, 1995; Chini et al, 2002). CD38 is also the enzyme responsible for the
synthesis of the second messenger cADPR (Aarhus et al, 1995; Chini et al, 2002). We observed
that extracts prepared from the tissues of CD38 knock out mice are not capable of synthesis of
NAADP (by the base-exchange reaction) or cGDPR in vitro (Fig. 6). cGDPR is a marker of the
activity of the ADPribosyl cyclase from CD38 (Chini et al, 2002), in previous studies we have
shown that synthesis of cADPR (Askoy , et al, 2006, Chini et al, 2002) from NAD is nearly
abolished in several tissues from CD38 knock out mice (Askoy , et al, 2006, Chini et al, 2002).
Furthermore, we determined the levels of NAADP and cADPR present in situ in tissues from
both wild type and knock out mice. Not surprisingly, the presence of intracellular levels of
cADPR is essentially eliminated in CD38 knockout tissues (Fig. 7), the only tissue where
cADPR levels were detected in equal amounts in both wild type and CD38 knock out mice was
Page 19 of 44

in the brain (data not shown). However, the in vivo cellular content of NAADP was not reduced
by the absence of CD38 (Fig. 7). The data from figures 8 and 9 indicate that neither CD38 nor
the base-exchange reaction is necessary for the in vivo generation of NAADP.
We further determined the role of the CD38 enzyme upon the agonist (histamine)-induced
intracellular in vivo accumulation of NAADP. Using myometrial cells cultured from CD38 wild
type and CD38 knockout mice we determine the role of this enzyme on the histamine-induced
accumulation of NAADP in vivo. As shown in figure 8 (inset) CD38 is normally expressed in
myometrial cells from wild type mice but is absent in myometrial cells from CD38 knockout
mice. Furthermore, the absence of CD38 does not prevent the in vivo generation and
accumulation of NAADP in response to histamine (Fig. 8), as expected histamine-induced Ca2+
transients are not altered by the absence of CD38 (Fig. 8B). In fact, myometrial cells from the
CD38 knockouts appear to have a higher content of NAADP than wild type cells in both the
basal and agonist stimulated states (Fig 8). At present, the mechanism behind this is not known
but one possibility is compensation for the absence of the second messenger cADPR. These data
indicate that CD38 is not necessary for basal and agonist-induced generation of NAADP in vivo.
We also determined the effect of over-expression of the enzyme CD38 upon intracellular levels
of NAADP in cells. Treatment of HL60 cells with retinoic acid (RA) has been shown to increase
the expression of CD38, and subsequently the synthesis and intracellular levels of cADPR (Fig.
9A, B, C)(Munshi et al, 2002). Furthermore, over-expression of CD38 with RA leads to an
increase of the in vitro generation of NAADP catalyzed by the base-exchange reaction (Fig. 9D).
In contrast, the intracellular levels of NAADP were not increased by over-expression of CD38
with RA (Fig. 9E). Our data presents a new aspect of the NAADP second messenger system,
indicating that CD38 is not the enzyme responsible for the in vivo generation of this nucleotide.
Page 20 of 44

Our experiments open a new avenue for the discovery of the in vivo pathway responsible for the
synthesis of NAADP.
The present study demonstrates that the NAADP-system is present and functional in human
myometrial cells, and may play a role as a second messenger transducing the generation of
[Ca2+]i transients in response to histamine and oxytocin. Furthermore, we clearly indicated that
the acidcalciossomes (the NAADP-sensitive Ca2+ stores) are present in myometrial cells and play
a role in intracellular Ca2+ homeostasis. In contrast with other authors (Dammermann et al, 2005;
Galione and Petersen, 2005; Gerasimenko et al, 2003; Hohenegger et al, 2002), that proposed
that the NAADP-system is dependent on the RyR, we provide evidence that the NAADP system
can also work in a RyR-independent way, however more direct work needs to be done to further
determine the role of the RyR on the NAADP functions. Recently extracellular effects of
NAADP have also been described (Heidemann et al, 2005), however in our studies extracellular
application of NAADP has not been explored and the possible role of lysosomal and IP3 channels
upon extracellular effect of NAADP in myometrial cells can not be assumed without
experimental evidence.
The majority of the experiments in the first part of our study were obtained with the use of
pharmacologic inhibitors. In these regard our conclusions have the limitations of any studies that
depend heavily on the use pharmacological inhibitors. However, the inhibitors used here have
been extensively used by us and others and have been shown to have an acceptable specificity
for such studies (Churchill et al, 2002; Yamasaki et al, 2004; Kinnear et al, 2004).
In another aspect of our study we determined the role of the enzyme CD38 on the in vivo
accumulation of NAADP. We (Cheng et al, 2001; Chini et al, 2002; Chini and Dousa, 1995)
previously described the synthesis of NAADP in several tissues including brain, liver, spleen,
Page 21 of 44

heart, and kidney glomeruli. Synthesis of NAADP can be catalyzed in vitro by a NAD(P)ase,
analog to the lymphocyte antigen CD38 (Cheng et al, 2001; Chini et al, 2002; Chini and Dousa,
1995), in a reaction called the base-exchange reaction (Fig.12; Chini and deToledo, 2002). The
enzyme catalyzes the exchange of nicotinamide for nicotinic acid on the molecule of NADP+,
generating NAADP (Fig.10; see also;Chini and deToledo, 2002).
For these reasons, it is important to consider other theoretical pathways for the synthesis of
NAADP in vivo (Fig. 10). Conceivably, NAADP might be generated by deamination of NADP+
(Fig. 10) or phosphorylation of NAAD+. The latter is a particularly attractive hypothetical route
because NAAD+ is a compound present in cells and NAADP might be then catalyzed by NAD+
kinase with ATP as a 2'-phosphate donor (Fig.10). These alternative synthetic pathways ought to
be explored in future studies. Notably, Lerner et al. (Lerner et al, 2001) characterized a human
NAD+ kinase in vitro and found no evidence that it could synthesize NAADP by phosphorylation
of NAAD+. Nevertheless, these data do not completely exclude the possibility that NAAD+
phosphorylation might perhaps occur in vivo, for example, if putative intracellular cofactors
and/or other physiological conditions exist in vivo that are required to enable the reaction. They
also do not exclude the possibility that other isoforms might catalyze the reaction. Therefore, the
postulated NAAD+ phosphorylation pathway seems unlikely at this point but cannot be ruled out.
Despite the limitations discussed, the base-exchange reaction is the only pathway currently
described for the synthesis of NAADP in biological systems (Chini and deToledo, 2002). In this
regard, an important observation is that enzymes with ADP-ribosyl cyclase activity (capacity for
synthesis of cyclic ADP-ribose) are also able to catalyze the synthesis of NAADP through the
base-exchange reaction (Chini and deToledo, 2002). In fact, the mammalian version of ADP-
ribosyl cyclase, CD38, is capable of generating both NAADP and cADPR (Chini and deToledo,
Page 22 of 44

2002). This observation led to the proposal of cross talk between these two signaling pathways.
However, as discussed above, whether the base-exchange reaction occurs under physiological
conditions is still an unanswered question. Using CD38 knockout mice, we determined that
CD38 is the major enzyme responsible for the base-exchange reaction in mouse tissues in vitro
(Chini et al, 2002). However, in that study, the capacity for synthesis of NAADP by the base-
exchange reaction in cells did not correlate with the presence of NAADP-induced Ca2+ release in
the same cells. As a result, this discrepancy raised doubts about the role of the base-exchange
reaction as the physiological route for the synthesis of NAADP. Our present data clearly
indicates that there is no correlation between the expression of CD38 and the in vivo intracellular
content of NAADP in basal and agonist induced NAADP accumulation. It is important for future
work to explore and determine the true pathway responsible for the in vivo generation of this new
intracellular second messenger.
Page 23 of 44

FIGURE LEGENDS
Figure 1. The NAADP-system in human myometrial cells and microsomes. In (A) freshly
isolated intact human myometrial cells were microinjected with 1 μM or 1 mM NAADP and
Ca2+ transients were determined using Fluo-3 as described in the material and methods section.
In (B, C, D, and E) human myometrial microsomes were used and Ca2+ release was determined
using Fluo-3. In (B) Ca2+ release-induced 1 μM NAADP was determined before and after the
depletion of SR stores with 1μM thapsigargin (TG) or depletion of lysosomal Ca2+ stores with 50
μM GPN. In (C) a characteristic fluorescence tracing from experiment used to derive that for the
graph in B, units are change from base-line fluo-3 fluorescence (arbitrary units). In (C) we also
show that the Ca2+ release induced by 1μM cADPR is not affected by depletion of lysosomal
stores with 50 μM GPN. In (D) Ca2+ release-induced 1 μM NAADP was determined before and
after the 1 minute pre-incubation with the Ca2+ releasing channel antagonist for NAADP, high
concentration (1 mM) NAADP (HNAADP) or 10 μM nifedipine, or with 100 μM 8-Br-cADPR
(a cADPR antagonist), or with the IP3 antagonist xestospongin C (100 μM). In (E) the effect of
pre-incubation with 1 mM NAADP upon the Ca2+ release induced by 10 μM cADPR or IP3 data
is average of 3-4 independent experiments.
Figure 2. Lysosomal Ca2+ stores in human myometrial cells. In (A) myometrial cells were co-
stained with lysotracker (a lysosomal specific fluorescent probe that appears orange in the
perinuclear region) and with bodipy-TG that appears as a green diffuse labeling in the SR. In (B)
intact cells were labeled with the intracellular Ca2+ indicator Fura-2 AM, the Ca2+ release
induced by depletion of the intracellular stores was determined. Ca2+ release was induced by 10
μM TG (a SR Ca2+ ATPase inhibitor) or 50 μM GPN (a lysosomal inhibitor). All Ca2+ release
experiments were performed in the absence of extracellular Ca2+, so the intracellular Ca2+
Page 24 of 44

transients observed were derived from depletion of intracellular stores and not influx of
extracellular Ca2+. In (C) we show the effect of incubation of lysotracker (upper panel) or
bodipy-TG (lower panel) labeled myometrial cells with 50 μM GPN. All data shown in this
figure are the typical results from at least 4 different experiments.
Figure 3. Characterization of the lysosomal Ca2+ stores in human myometrial cells. In (A and B)
we show a representative tracing of a typical experiment, intact human myometrial cells were
loaded with the Ca2+ fluorescent probe Fura-2 AM and the Ca2+ release induced by 10μM
thapsigargin (TG) and 30 μM GPN was determined. In (C and D) we show the average and
standard error (SE) of 4 independent experiments. In (C) we show the Ca2+ release induced by
10μM thapsigargin (TG) before (CTL) or after 2 minute pre-incubation with 10μM CPA (a SR
Ca2+ ATPase inhibitor), 50 μM GPN, or 5 μM bafilomicin A1 (BafA1, a lysosomal H+-ATPase
inhibitor). In (B) Ca2+ release induced by 50 μM GPN was determined before (CTL) and after
the same pre-incubation as described in B, except for the pre-incubation with 10μM thapsigargin
(TG). All Ca2+ release experiments were performed in the absence of extracellular Ca2+, so the
intracellular Ca2+ transients observed a derived from depletion of intracellular stores and not
influx of extracellular Ca2+.
Figure 4. Role of different signaling pathways in agonist-induced Ca2+ transients in human
myometrial cells.
In (A) intact human myometrial cells were loaded with the Ca2+ fluorescent probe Fura-2 AM
and the Ca2+ release induced by 100μM histamine, oxytocin or acetylcholine was determined
before (CTL) or after the 3 min pre-incubation in the absence of extracellular Ca2+ (“0” Ca), or in
the presence of 100 μM 8-Br-cADPR (8-Br), 100 μM ryanodine (Ry), 100 μM xestospongin C
Page 25 of 44

(XesC), 1 μM thapsigargin (TG), or 50 μM GPN. We show the average and standard error (SE)
of 4 independent experiments.
In (B) we tested the role of lysosomal Ca2+ stores in histamine induced Ca2+ transients in human
myometrial cells. All data shown figure 4B are the typical results from at least 8 different
experiments. Intact human myometrial cells were loaded with the Ca2+ fluorescent probe Fura-2
AM and the Ca2+ release induced by 100μM histamine was determined in the presence of 2.2
mM extracellular CaCl2, in the absence of extracellular Ca2+, after 5 minutes pre-incubation with
100 μM xestospongin C, 10 μM thapsigargin, 50 μM GPN, 5 μM bafilomicin A1, or 50 μM
monesin (a lysosomal Ca2+ accumulation inhibitor). With exception of the experiment in A in the
presence of 2.2 mM extracellular Ca2+, all other experiments were performed in the absence of
extracellular Ca2+.
In (C) we tested the role of lysosomal Ca2+ stores in agonist induced Ca2+ transients in human
myometrial cells. Intact human myometrial cells were loaded with the Ca2+ fluorescent probe
Fura-2 AM and the Ca2+ release induced by 100μM histamine, oxytocin or acetylcholine were
determined before (CTL) or after the 3 min pre-incubation with 50 μM GPN or 5 μM
bafilomycin A1. We show the average and standard error (SE) of 4 independent experiments.
Figure 5. Agonist-induced NAADP accumulation in human myometrial cells. Human
myometrial cells received treatment for 30 seconds with vehicle or histamine (100 μM) alone or
histamine in the presence of, 100 μM diphenhydramine (H1 blocker), 1 μM calmidazolium
(CMDZ), or 5 μM BAPTA-AM. We show the average and standard error (SE) of 4 independent
experiments. Inset shows the time course of the NAADP accumulation induced by 100 μM
histamine.
Page 26 of 44

Figure 6. Base-Exchange reaction and cyclase activity in wild type and CD38 knockout mice. In
(A) Cyclase activity was determined using 0.4 mM NGD (nicotinamide guanine dinucleotide) as
a substrate. In (B) tissues (1 mg/ml) were incubated with NADP and nicotinic acid for the base
exchage reaction as described in material and methods. The abbreviations correspond to uterus
(Ut), heart (H), liver (L), kidney (K), spleen (S). KO refers to CD38 knockout mice. We show
the average and standard error (SE) of 4 independent experiments. The inset shows the western
blot analysis of the presence of CD38 in wild type (cd38+/+) and CD38 knockout mice (cd38-/-).
ND refers to not detected.
Figure. 7 Levels of NAADP and cADPR in wild type and CD38 knockout mice. NAADP levels
and cAPDR levels were detected in several tissues from wild type and CD38 knockout mice as
described in material and methods. Abbreviations are the same as figure 6. We show the average
and standard error (SE) of 4 independent experiments.
Figure 8. Histamine stimulated NAADP accumulation in wild type and CD38 knockout
myometrial cells. In (A) myometrial cells were isolated from wild type and CD38 knockout
mice and treated with for 30 seconds with vehicle (control) or 100 μM histamine. The inset
shows a western blot analysis for CD38 in wild type and CD38 knockout (KO) myometrial cells.
In (B) Ca2+ transients induced by histamine in wild type and CD38 knock out myometrial cells
were determine using Fura-2 as a probe as described in material and methods. We show the
typical tracing of 4 independent experiments.
Figure 9. Over-expression of CD38 in HL60 cells. HL-60 cells were treated with 1 μM retinoic
acid (RA) and cellular extracts were analyzed for CD38 expression (A, shows western blot for
CD38), activity of the ADP-ribosyl cyclase (B, shows the NGD cyclase activity), intracellular
levels of cADPR (C), synthesis of NAADP by the base-exchange reaction (D), and levels of
Page 27 of 44

NAADP (E). All experiments were determined as described in material and methods. The gel is
the representative data from 3 experiments all other graphs are the average and standard error
(SE) of 4 independent experiments.
Figure 10. The base-exchange reaction and alternative pathways for the synthesis of NAADP.
(A) The base-exchange reaction is catalyzed by the enzyme CD38 in the presence of NAADP
and excess nicotinic acid. (B) Alternatively NAADP synthesis could be catalyzed by
phosphorylation of NAAD by ATP catalyzed by a NAAD kinase or by deamination of NADP.
Page 28 of 44

REFERENCES
1. Aarhus, R., Graeff, R.M., Dickey, D.M., Walseth, T.F., and Lee, H.C. ADP-ribosyl cyclase and
CD38 catalyze the synthesis of a calcium-mobilizing metabolite from NADP. J Biol Chem 270: 30327-30333, 1995.
2. Askoy, P., White, T., Thompson, M., and Chini, E.N. Regulation of intracellular levels of NAD: a novel role for CD38. Biochem. Biophys. Res. Commun.(In press),2006.
3. Bak, J., White, P., Timar, G., Missiaen, L., Genazzani, A.A., and Galione, A. Nicotinic acid adenine dinucleotide phosphate triggers Ca2+ release from brain microsomes. Curr Biol 9:751-754, 1999.
4. Barata, H., Thompson, M., Zielinska, W., Han, Y.S., Mantilla, C.B., Prakash, Y.S., Feitoza, S., Sieck, G., Chini, E.N. The role of cyclic-ADP-ribose-signaling pathway in oxytocin-induced Ca2+ transients in human myometrium cells. Endocrinology. 145:881-9, 2004.
5. Berridge, M.J., Lipp, P., and Bootman, M.D. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 1: 11-21, 2000.
6. Boittin, F.X., Galione, A., Evans, A.M. Nicotinic acid adenine dinucleotide phosphate mediates Ca2+ signals and contraction in arterial smooth muscle via a two-pool mechanism. Circ Res. 91:1168-75, 2002.
7. Cancela, J.M., Churchill, G.C., and Galione, A. “Coordination of agonist-induced Ca2+-signalling patterns by NAADP in pancreatic acinar cells” Nature. 398:74-76, 1999.
8. Cheng J, Yusufi AN, Thompson MA, Chini EN, Grande JP.Nicotinic acid adenine dinucleotide phosphate: a new Ca2+ releasing agent in kidney. J Am Soc Nephrol.;12: 54-60, 2001.
9. Chini, E.N. To be or not to be ... nicotinic acid adenine dinucleotide phosphate (NAADP): a new intracellular second messenger?. Biol Res;37:559-563, 2004.
10. Chini, E.N., Beers, K.W., and Dousa, T.P. Nicotinate adenine dinucleotide phosphate (NAADP) triggers a specific calcium release system in sea urchin eggs. J Biol Chem 270: 3216-3223, 1995.
11. Chini, E.N., Chini, C.C., Kato, I., Takasawa, S., Okamoto, H. CD38 is the major enzyme responsible for synthesis of nicotinic acid-adenine dinucleotide phosphate in mammalian tissues. Biochem J 362: 125-130, 2002.
12. Chini, E.N., Chini, C.C., Barata da Silva, H., Zielinska, W. cyclic-ADP-ribose signaling pathway in human myometrium. Arch Biochem Biophys. 407:152-9, 2002.
13. Chini, E.N., and De Toledo, F. G. S. The new calcium-mobilizing nucleotides cyclic ADP-ribose and NAADP. In Recent Res Devel Biophys Biochem. Trivandrum, India: Research Signpost, pp. 43-57, 2001.
14. Chini, E.N., De Toledo, F.G. Nicotinic acid adenine dinucleotide phosphate: a new intracellular second messenger? Am. J. Physiol. 282:C1191-8, 2002
15. Chini, E.N., and Dousa, T.P. Enzymatic synthesis and degradation of nicotinate adenine dinucleotide phosphate (NAADP), a Ca2+-releasing agonist, in rat tissues. Biochem Biophys Res Commun 205: 167-174, 1995.
16. Chini, E.N., and Dousa, T.P. Nicotinate-adenine dinucleotide phosphate-induced Ca2+-release does not behave as a Ca2+-induced Ca2+-release system. Biochem J 316: 709-711, 1996.
Page 29 of 44

17. Churchill, G.C., and Galione, A. NAADP induces Ca2+ oscillation via a two-pool mechanism by priming IP3 and cADPR-senstive Ca2+ stores. EMBO J. 20: 2666-2671, 2001
18. Churchill, G.C., Okada, Y., Thomas, J.M., Genazzani, A.A., Patel, S., Galione, A. NAADP mobilizes Ca(2+) from reserve granules, lysosome-related organelles, in sea urchin eggs. Cell.111(5):703-8, 2002.
19. Churchill, G.C., O'Neill, J.S., Masgrau, R., Patel, S., Thomas, J.M., Genazzani, A.A., Galione, A.. Sperm deliver a new second messenger: NAADP. Current Biology 13:125-8, 2003.
20. Dammermann, W., Guse, A.H. Functional ryanodine receptor expression is required for NAADP-mediated local Ca2+ signaling in T-lymphocytes. J Biol Chem.;280:21394-21399, 2005.
21. De Toledo, F.G., Cheng, J., Liang, M., Chini, E.N., Dousa, T.P.. ADP-Ribosyl cyclase in rat vascular smooth muscle cells: properties and regulation. Circ Res 86: 1153-1159, 2000.
22. Dousa, T.P., Chini, E.N., and Beers, K.W. Adenine nucleotide diphosphates: emerging second messengers acting via intracellular Ca2+ release. Am J Physiol 271: C1007-1024, 1996.
23. Galione, A., Patel, S., and Churchill, G.C. NAADP-induced calcium release in sea urchin eggs. Biol Cell 92: 197-204, 2000.
24. Galione A, Petersen OH. The NAADP Receptor: New Receptors or New Regulation? Mol Interv;5:73-79, 2005.
25. Gerasimenko JV, Maruyama Y, Yano K, Dolman NJ, Tepikin AV, Petersen OH, Gerasimenko OV. NAADP mobilizes Ca2+ from a thapsigargin-sensitive store in the nuclear envelope by activating ryanodine receptors. J Cell Biol.;163:271-282, 2003.
26. Graeff R, Lee HC. A novel cycling assay for cellular cADP-ribose with nanomolar sensitivity. Biochem J. 361:379-384, 2002..
27. Heidemann, A.C., Schipke, C.G., and Kettenmann, H. Extracellular application of nicotinic acid adenine dinucleotide phosphate induces Ca2+ signaling in astrocytes in situ. J. Biol. Chem. 280:35630-35640, 2005.
28. Hohenegger M, Suko J, Gscheidlinger R, Drobny H, Zidar A. Nicotinic acid-adenine dinucleotide phosphate activates the skeletal muscle ryanodine receptor. Biochem J. 367:423-431, 2002.
29. Kinnear NP, Boittin FX, Thomas JM, Galione A, Evans AM. Lysosome-sarcoplasmic reticulum junctions. A trigger zone for calcium signaling by nicotinic acid adenine dinucleotide phosphate and endothelin-1. J Biol Chem.;279:54319-54326, 2004
30. Lee, H.C., and Aarhus, R. A derivative of NADP mobilizes calcium stores insensitive to inositol trisphosphate and cyclic ADP-ribose. J Biol Chem 270: 2152-2157, 1995
31. Lee, H.C., Walseth, T.F., Bratt, G.T., Hayes, R.N., and Clapper, D.L. Structural determination of a cyclic metabolite of NAD+ with intracellular Ca2+-mobilizing activity. J Biol Chem 264: 1608-1615, 1989.
32. Lerner F, Niere M, Ludwig A, Ziegler M Structural and functional characterization of human NAD kinase. Biochem Biophys Res Commun;288: 69-74, 2001.
33. Masgrau, R., Churchill, G.C., Morgan, A.J., Ashcroft, S.J., Galione, A. NAADP: a new second messenger for glucose-induced Ca2+ responses in clonal pancreatic beta cells. Current Biology 13: 247-51, 2003.
34. Munshi, C.B., Graeff, R., Lee, H.C. Evidence for a causal role of CD38 expression in granulocytic differentiation of human HL-60 cells. J Biol Chem. 277: 49453-8, 2002
Page 30 of 44

35. Partida-Sanchez S, Cockayne DA, Monard S, Jacobson EL, Oppenheimer N, Garvy B, Kusser K, Goodrich S, Howard M, Harmsen A, Randall TD, Lund FE.Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat Med;7:1209-1126, 2001.
36. Patel S., Churchill G.C., Galione, A. Coordination of Ca2+ signaling by NAADP Trends Biochem Sci. 26(8):482-489, 2001
37. Prakash, Y.S., Kannan, M.S., Walseth, T.F., and Sieck, G.C. cADP ribose and [Ca(2+)](i) regulation in rat cardiac myocytes. Am J Physiol Heart Circ Physiol 279: H1482-1489, 2000.
38. Takahashi, J., Kagaya, Y., Kato, I., Ohta, J., Isoyama S., Miura, M., Sugai, Y., Hirose, M., Wakayama, Y., Ninomiya, M., Watanabe, J., Takasawa, S., Okamoto, H., Shirato, K. Deficit of CD38/cyclic ADP-ribose is differentially compensated in hearts by gender. Biochem Biophys Res Commun. 312:434-40, 2004.
39. Thompson, M, Barata, H, White, T, Zielinska, W, Bailey, JP, Olund, FE, Sieck G, and Chini EN. Role of CD38 in myometrial Ca2+ transients: Modulation by progesterone. Am. J. Physiol. 287:E1142-1148, 2004.
40. Yamasaki. M, Masgrau. R, Morgan. A.J, Churchill, G.C, Patel S, Ashcroft, S.J, Galione, A. Organelle selection determines agonist-specific Ca2+ signals in pancreatic acinar and beta cells. J Biol Chem. 279:7234-40, 2004.
41. Yamasaki M, Thomas JM, Churchill GC, Garnham C, Lewis AM, Cancela JM, Patel S, Galione A. Role of NAADP and cADPR in the induction and maintenance of agonist-evoked Ca2+ spiking in mouse pancreatic acinar cells. Curr Biol.;15:874-878, 2005.
42. Yusufi, A.N., Cheng, J., Thompson, M.A., Burnett, J.C., and Grande, J.P. Differential mechanisms of Ca(2+) release from vascular smooth muscle cell microsomes. Exp Biol Med (Maywood) 227: 36-44, 2002.
Page 31 of 44

Figure 1.
Page 32 of 44

Y Data
1.6
1.8
2.0
2.2
2.4
TG GPN
100 sec
ALysotrack Lysotrack + GPN
Thaps. Thaps.+GPNB
C
D
Figure 2.
Page 33 of 44
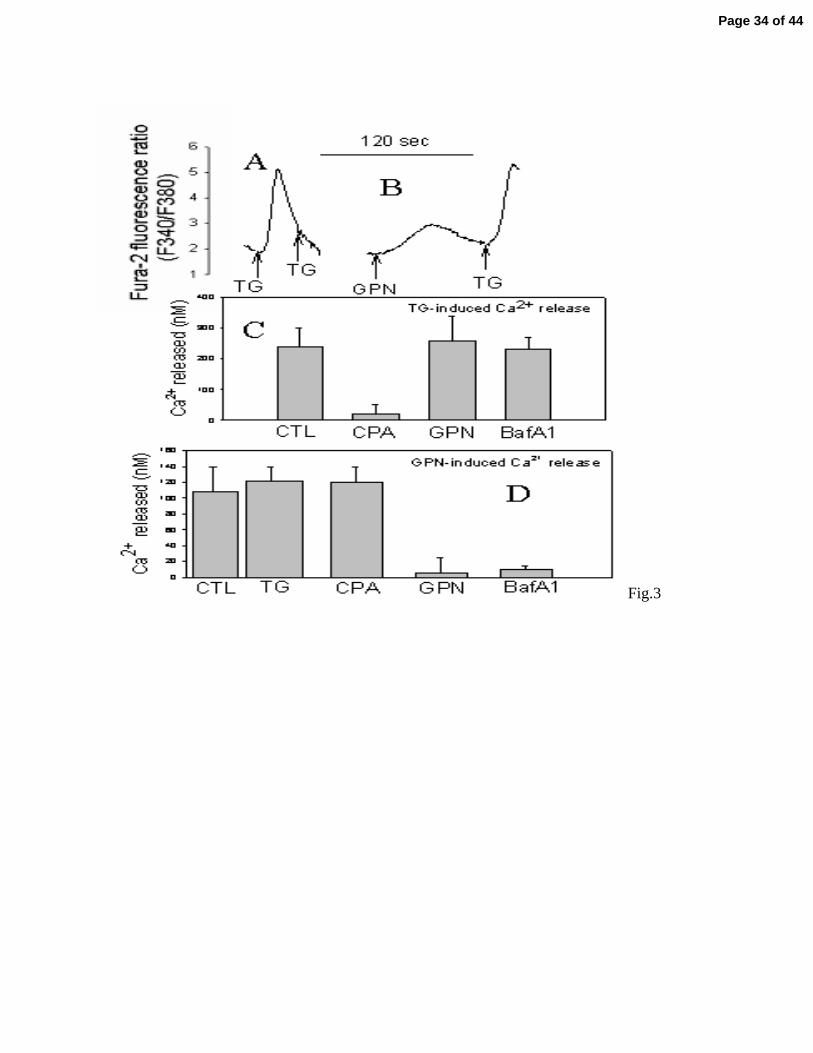
Fig.3
Page 34 of 44

Fig.4A
Ca2+
rele
ase
(%) o
f con
trol
0
20
40
60
80
100
120
0
20
40
60
80
100
120
Ca2+
rele
ase
(%) o
f con
trol
0
20
40
60
80
100
120
Ca2+
rele
ase
(%) o
f con
trol
Oxytocin
Histamine
ACh
"0"Ca
8-Br Ry XesCCTL CTL
CTL
"0"Ca
"0"Ca
8-Br
8-Br
Ry
Ry
XesC
XesC
Page 35 of 44

Fig.4B
Page 36 of 44

Figure 4C
Page 37 of 44

Fig.5
Page 38 of 44

Fig.6
Page 39 of 44

Figure 7
NA
AD
P le
vels
(fm
oles
/mg
of p
rote
in)
0
2
4
6
8
cA
DP
R le
vels
(pm
oles
/mg
of p
rote
in)
0
200
400
600
800
1000
Ut H
L
K S Ut H L K SWild type KO
Ut UtH
L
K S H L K SWild type KO
Page 40 of 44

Fig. 8
A
Page 41 of 44

Fig.9
Page 42 of 44

Fig.10
Page 43 of 44

Page 44 of 44
Copyright © 2022 FDOKUMEN




















