
Differential susceptibility to chemically induced thymic lymphomas in SENCARB and SSIN inbred mice

Localization of QTLs for seed color usingrecombinant inbred lines of Brassica napus indifferent environments
Fu-You Fu, Lie-Zhao Liu, You-Rong Chai, Li Chen, Tao Yang, Meng-Yang Jin,Ai-Fen Ma, Xing-Ying Yan, Zheng-Sheng Zhang, and Jia-Na Li
Abstract: Yellow seed is one of the most important traits of Brassica napus L. Efficient selection of the yellow-seed traitis one of the most important objectives in oilseed rape breeding. Two recombinant inbred line (RIL) populations (RIL-1and RIL-2) were analyzed for 2 years at 2 locations. Four hundred and twenty SSR, RAPD, and SRAP marker loci cover-ing 1744 cM were mapped in 26 linkage groups of RIL-1, while 265 loci covering 1135 cM were mapped in 20 linkagegroups of RIL-2. A total of 19 QTLs were detected in the 2 populations. A major QTL was detected adjacent to the samemarker (EM11ME20/200) in both maps in both years. This major QTL could explain 53.71%, 39.34%, 42.42%, 30.18%,24.86%, and 15.08% of phenotypic variation in 6 combinations (location � year � population). BLASTn analysis of thesequences of the markers flanking the major QTL revealed that the homologous region corresponding to this major QTLwas anchored between genes At5g44440 and At5g49640 of Arabidopsis thaliana chromosome 5 (At C5). Based on compa-rative genomic analysis, the bifunctional gene TT10 is nearest to the homologue of EM11ME20/200 on At C5 and can beconsidered an important candidate gene for the major QTL identified here. Besides providing an effective strategy formarker-assisted selection of the yellow-seed trait in B. napus, our results also provide important clues for cloning of thecandidate gene corresponding to this major QTL.
Key words: Brassica napus L., quantitative trait locus (QTL), recombinant inbred line (RIL), seed color, sequence-relatedamplified polymorphism (SRAP), simple sequence repeat (SSR).
Resume : La graine jaune est un des caracteres les plus importants chez le Brassica napus L. Une selection efficace pourdes graines jaunes constitue un des objectifs les plus importants chez le colza oleagineux. Deux populations de lignees re-combinantes fixees (RIL-1 et RIL-2) ont ete analysees pendant deux ans sur deux sites. Quatre cent vingt marqueurs SSR,RAPD et SRAP couvrant 1744 cM ont ete assignes a 26 groupes de liaison chez RIL-1, tandis que 265 locus couvrant1135 cM ont ete assignes a 20 groupes de liaison chez RIL-2. Au total, 19 QTL ont ete detectes chez les deux populations.Un QTL majeur a ete detecte au voisinage du meme marqueur (EM11ME20/200) sur les deux cartes pour les deux annees.Ce QTL majeur expliquait 53,71 %, 39,34 %, 42,42 %, 30,18 %, 24,86 % et 15,08 % de la variation phenotypique au seindes 6 combinaisons (site � annee � population). Une analyse BLASTn effectuee a l’aide des sequences des marqueursbordant le QTL majeur a revele que la region homologue etait situee entre les genes At5g44440 et At5g49640 sur le chro-mosome 5 chez Arabidopsis thaliana. Sur la base d’une analyse genomique comparee, le gene bifonctionnel TT10 est leplus proche de l’homologue du marqueur EM11ME20/200 sur le chromosome 5 d’Arabidopsis thaliana et on peut leconsiderer comme un gene candidat serieux pour le QTL majeur identifie ici. En plus de fournir une strategie efficacepour la selection assistee pour les graines jaunes chez le B. napus, ces resultats contribuent egalement des indices impor-tants en vue du clonage du gene correspondant a ce QTL majeur.
Mots-cles : Brassica napus L., locus d’un caractere quantitatif (QTL), lignees recombinantes fixees (RIL), couleur desgraines, polymorphisme de sequence (SRAP), microsatellite (SSR).
[Traduit par la Redaction]
IntroductionBrassica napus L. (2n = 38, AACC) is one of the most
important oilseed crops grown worldwide. A primary objective
in the breeding of B. napus is to increase the oil yield perunit area, which is determined by seed yield and seed oilcontent. In recent years great attention has been paid to en-
Received 26 November 2006. Accepted 17 July 2007. Published on the NRC Research Press Web site at genome.nrc.ca on 19 September2007.
Corresponding Editor: G. Scoles.
F.-Y. Fu,1 L.-Z. Liu, Y.-R. Chai, L. Chen, T. Yang, M.-Y. Jin, A.-F. Ma, X.-Y. Yan, Z.-S. Zhang, and J.-N. Li. ChongqingRapeseed Technology Research Center, Chongqing Key Laboratory of Crop Quality Improvement, Key Laboratory of Biotechnology andCrop Quality Improvement of Ministry of Agriculture, College of Agronomy and Biotechnology, Southwest University, 216 TianshengRoad, Beibei, Chongqing, 400716, People’s Republic of China.
1Corresponding author (e-mail: [email protected]).
840
Genome 50: 840–854 (2007) doi:10.1139/G07-068 # 2007 NRC Canada

hancing seed oil content. The yellow-seeded type ofB. napus has a significantly thinner seed coat, lower huskproportion, and higher oil content than the traditionalblack-seeded type (Stringam et al. 1974). It also has someother advantages, including a much lower content of pig-ments in the crude oil as well as a higher protein contentand a lower fiber content in the meal, resulting in betterfeeding value for livestock (Meng et al. 1998; Tang et al.1997). Consequently, selection of a stable yellow-seed traitis one of the most important breeding objectives. However,this process is very difficult because the yellow-seed traitis strongly influenced by the environment. Although theyellow-seed trait of B. napus was first reported in 1960(Olsson 1960), it is still not stable enough in phenotypesto be applied as a breeding germplasm for efficient selec-tion of commercial cultivars.
The hereditary pattern of the seed color trait of B. napusis still quite unclear. It was reported that black seed colorwas dominant over yellow seed color, that the seed colortrait was controlled by 3 independent genes, and that yellowseed color could result only from the homozygous recessivecondition of all 3 loci (Shrizadegan 1986; van Deynze andPauls 1994). Tang et al. (1997) and Li et al. (1998) reportedthat different plant materials contain different yellow seedcolor genes. Both dominant and recessive types of theyellow-seed trait have been found in B. napus (Shrizadegan1986; van Deynze and Pauls 1994; Wu et al. 1999). Rahman(2001) reported that 3 or 4 recessive genes were involved inthe determination of yellow seed color in B. napus. Somerset al. (2001) identified a single major gene in a cross be-tween a yellow-seeded and a black-seeded B. napus, andthis result was supported by Liu et al. (2005), who foundthat yellow seed color showed partial dominance over blackseed color and was controlled by a single gene. Liu et al.(2006) located 2 seed-color QTLs that explained 30.9% and46% of the phenotypic variation, respectively. Recently, Ba-dani et al. (2006) observed a clear correlation between seedcolor and acid detergent fibre (ADF) content, and they lo-cated a major QTL with a large effect on both seed colorand ADF content in multiple environments on chromosomeN18 by using 3 mapping populations developed from 2crosses between 2 distinct sources of true-breeding yellow-seeded B. napus and 2 different black-seeded genotypes. Inaddition, seed color shows tight correlations to other traits(Wang et al. 2003), for example, positive correlations toseed protein content and seed oil content and a negative cor-relation to seed husk percentage. During seed development,the expression of seed color gene(s) is strongly affected byenvironmental factors such as temperature (van Deynze etal. 1993) and red/blue light (Liang et al. 2003).
Nowadays, highly developed molecular marker technolo-gies have the potential to revolutionize genetic selection inplant breeding (Tanksley et al. 1989). Using common mo-lecular marker methods such as restriction fragment lengthpolymorphism (RFLP), random amplified polymorphicDNA (RAPD), simple sequence repeats (SSRs), and amplifiedfragment length polymorphism (AFLP), high-density geneticmaps have been constructed for many crops including rice,maize, and soybean. In recent years, certain achievementshave been made in map construction and molecular markeridentification in B. napus. For example, Ferreira et al.
(1994), Parkin et al. (1995), and Sharpe et al. (1995) con-structed RFLP maps; Lombard and Delourme (2001) con-structed a consensus map using isozymes, RAPD, AFLP,and RFLP; and Lowe et al. (2004) and Piquemal et al.(2005) constructed SSR maps of B. napus. Some QTLshave been identified in different populations, and marker-assisted selection (MAS) has been undertaken in breedingof rapeseed for a few traits (Delourme et al. 1994; Wanget al. 2000; Axelsson et al. 2001; Burns et al. 2003; Janejaet al. 2003; Zhao and Meng 2003; Liu et al. 2006; Zhao etal. 2006). However, no dense genetic map is available forB. napus to carry out fine-mapping and efficient MAS ofmany important traits of rapeseed. QTL mapping of theseed color trait has not been deeply studied, and MAS israrely applicable in the breeding of yellow-seeded rapeseedcultivars.
Sequence-related amplified polymorphism (SRAP) is anew codominant molecular marker technology (Li andQuiros 2001). SRAP primer pairs are designed according tocommon structural features of open reading frames (ORFs)of various known genes. SRAP technology has some advan-tages over other molecular marker methods, such as ease ofprimer development and the high possibility of direct ampli-fication in regions of functional genes. Since its emergence,it has been successfully used to construct genetic linkagemaps and to analyze genetic polymorphisms in many crops(McCouch et al. 1997; Ferriol et al. 2003; Lin et al. 2003;Budak et al. 2004; Yu et al. 2005).
The main objective of this research was to detect stableQTLs affecting seed color of B. napus in different genera-tions and different environments. Based on microsyntenyanalysis of marker sequences linked to the detected majorQTL, a candidate gene corresponding to this locus was alsoproposed.
Materials and methods
Mapping populationsGenetic mapping and segregation analysis were performed
in 2 segregating populations of B. napus derived from thesame female parent, GH 06, a yellow-seeded inbred linewith a completely dominant yellow-seed major gene andalso one parent of the famous yellow-seeded cultivar Yuhuang1 (Li et al. 2001). Two black-seeded inbred lines derived fromcultivars Zhongyou 821 and Youyan 2, respectively, wereused as male parents in population construction.
Inbred lines derived from the 2 crosses GH 06 � Zhon-gyou 821 and GH 06 � Youyan 2 were named RIL-1 andRIL-2, respectively. In each cross, the F1 plants were selfedto produce the F2 progenies in 1998, and F2 progenies werecontinuously selfed by single seed descent for 7 years. TheRIL-1 and RIL-2 populations comprised 185 and 183 lines,respectively.
In 2004 to 2005, all lines of the 2 RIL populations weregrown with 2 replicates in Beibei, Chongqing, China. In2005 to 2006, they were simultaneously grown with 2 repli-cates in both Beibei and Wanzhou, Chongqing, China.Although Beibei and Wanzhou are at almost the same lati-tude, the altitude of Wanzhou is about 700 m higher thanthat of Beibei. Each plot contained 3 rows with 15 plants
Fu et al. 841
# 2007 NRC Canada

per row. Seeds were harvested from self-pollinated plantsfor seed color analysis.
DNA extractionGenomic DNA was extracted from 1 g of leaves of each
RIL plant using the protocol of Santoni and Berville (1992).The concentration and purity of each DNA sample weremeasured using a GeneSpec I spectrophotometer at wave-lengths of 260 nm and 280 nm.
Assay of DNA markersSequences of public SSR primer pairs were downloaded
from the Brassica database (http://brassica.bbsrc.ac.uk/cgi-bin/ace/searches/browser/BrassicaDB; accessed 1 May2005), whereas sequences of other SSR primer pairs wereobtained from Piquemal et al. (2005). PCR was carried outon a PTC-200 Thermal Cycler in a total volume of 10 mL.The composition of the mixture and the PCR procedurewere the same as those reported by Piquemal et al. (2005).The SRAP procedure was performed according to a previousdescription (Ferriol et al. 2003). In total, 64 forward primersand 64 reverse primers were used, resulting in 4096 primercombinations. The SSR and SRAP primers were synthesizedby Shanghai Sangon (Shanghai, China). RAPD primers weresynthesized by Invitrogen Company (Shanghai, China). The10 mL RAPD reaction mixture was initially denatured at94 8C for 1 min and then amplified by 35 cycles at 94 8Cfor 30 s, 38 8C for 1 min, and 72 8C for 1 min and a finalextension at 72 8C for 10 min. All PCR products were de-tected using non-denaturing polyacrylamide gel electropho-resis (8% polyacrylamide) and silver staining (Zhang et al.2002).
Marker codes and designationsIf a primer or primer pair detected multiple loci, numbers
were assigned to these loci according to descending frag-ment size. SRAP and SSR loci were named after the primernames followed by a reverse slant line and a number that in-dicated the fragment length (bp).
Map construction and QTL mappingChi square analysis was used to test goodness of fit be-
tween segregation ratios of all marker loci and the ratio ex-pected for RILs (1:1, � ‡ 0.05).
JoinMap 3.0 (van Ooijen and Voorrips 2001) was used tobuild the genetic linkage maps. A minimum logarithm ofodds (LOD) score of 3.0 with a maximum genetic distanceof 45 cM was first used to integrate SSR and SRAP lociinto initial linkage groups. Genetic distances were calculatedaccording to the Kosambi (1944) formula. QTLs of seedcolor were resolved by composite interval mapping usingWindows QTL Cartographer version 2.5 (Wang et al. 2006)with a LOD threshold of 2.0 (likelihood ratio ‡ 9.2). Confir-mation of linkage group designations was achieved by com-parison with the map positions of public SSR markers(Bancroft 2006; Lowe et al. 2004; Piquemal et al. 2005).The linkage group order and QTLs in the map were proc-essed by Mapchart 2.1 (Voorrips 2002). QTL nomenclaturewas in accord with that for rice (McCouch et al. 1997).
Measurement of seed colorThe same amount of seed from each plant of the 2 RILs
in 3 environments (i.e., 3 location � year combinations) wasscanned with a Uniscan A688 color scanner (Thunis, Peking,China) in 24 real color mode at 300 dpi. The RGB values ofthe seed picture were obtained using the software packageAdobe Photoshop and transformed to HSB values. The yel-low-seeded degree (YSD) of the seeds was calculated by thefollowing formula:
½1� YSD ¼ ½ðSi þ BiÞ � ðSb þ BbÞ�=½ðSy þ ByÞ�ðSb þ BbÞ� � 100
where Si and Bi are saturation (S) and brightness (B) valuesof the seed from plant i, Sb and Bb are S and B values ofseed from the black-seeded parent, and Sy and By are S andB values of yellow seed from B. rapa. The YSD value ofB. rapa was defined as 100% and the YSD value of theblack-seeded parent was set as zero.
Statistical analysis of the phenotype data was performedusing the SPSS1 13.0 software package (SPSS Inc. 2004).
Comparative genomic analysis of the major QTL withArabidopsis chromosome sequences
The marker bands flanking the major QTL of RIL-1 werere-separated on agarose gel, recovered, and cloned in thepMD 18-T vector (TaKaRa, Dalian, China). Positive cloneswere sequenced by Invitrogen China (Shanghai, China). Thesequences were used to carry out BLASTn searches withA. thaliana chromosomes (Altschul et al. 1997).
Results
Traits analysisSegregation data for seed color in the 2 RILs followed a
normal distribution and could be used for QTL analysis(Figs. 1a, 1b). Histograms that illustrate the segregation ofseed color in the 2 populations in 3 environments (3 location �year combinations) (Figs. 1a, 1b) show 2 peaks, 1 forgenotypes with black seed and 1 for genotypes with yellowseed.
Construction of the linkage mapsFour hundred and forty-one SSR primer pairs, 500 RAPD
primers, and 4096 SRAP primer combinations were used toscreen for polymorphisms between black-seeded and yellow-seeded parents. Of the 4096 SRAP primer combinations,2536 did not yield distinct amplification bands.
In RIL-1, 92 SSR primer pairs (20.86%), 70 RAPD pri-mers (14%), and 260 SRAP primer combinations (16.67%)yielded 106, 98, and 416 unambiguous polymorphic bands,respectively. Four hundred and twenty loci (65 for SSR, 65for RAPD, and 290 for SRAP) were mapped on 26 linkagegroups ranging from 32 cM to 142 cM with an averagelength of 67.08 cM and containing 3 to 51 markers each.The map covered a total of 1744 cM, about 69.76% of thegenome (Lombard and Delourme 2001), and the averagedistance between 2 adjacent markers was 4.15 cM (Fig. 2).
In RIL-2, 114 SSR primer pairs (25.85%) and 121 SRAPprimer combinations (7.76%) yielded 145 and 198 unambig-uous polymorphic bands, respectively. Two hundred and
842 Genome Vol. 50, 2007
# 2007 NRC Canada

sixty-five loci (105 for SSR and 160 for SRAP) weremapped on 20 linkage groups ranging from 17 cM to94 cM with an average length of 56.75 cM and containing3 to 32 markers each. The map covered a total of 1135 cM,about 45.40% of the genome, and the average distance be-tween 2 adjacent markers was 4.28 cM (Fig. 3).
The linkage groups were designated using the standardN1–N19 nomenclature system for B. napus based on con-sensus SSR markers (Lowe et al. 2004; Piquemal et al.2005; Bancroft 2006). Because of a lack of consensusmarkers, linkage groups LG03, LG06, LG07, LG09, LG15,LG17, LG18, LG19, LG22, LG24, and LG25 from RIL-1
and LG12, LG14, and LG19 from RIL-2 were not assignedwithin the N1–N19 nomenclature system. Therefore, moreconsensus SSR markers and some RLFP markers must beapplied to designate all linkage groups using the standard.In our results, the major markers constituting the 2 linkagemaps were SRAP markers (61.31%), and SSR markers weredistributed among the SRAP markers.
QTL analysis
RIL-1A list of the QTLs identified in RIL-1 is presented in
Fig. 1. Phenotypic distributions of 2 populations of B. napus recombinant inbred lines (a, RIL-1; b, RIL-2) in 3 different environments forthe degree of yellow seed color. SCW 06, Wanzhou 2006; SC 06, Beibei 2006; SC 05, Beibei 2005.
Fu et al. 843
# 2007 NRC Canada

Table 1. The positions of the QTLs are shown in Fig. 2. Al-together, 12 significant QTLs for seed color were detected.
In 2006 in Wanzhou, 2 QTLs were detected. They weredesignated qSCWZ06A-2-1 and qSCWZ06A-20-2, weremapped on LG02 (N5) and LG20 (N9), and explained7.16% and 53.71% of the phenotypic variation, respectively.The major QTL, qSCWZ06A-20-2, was located in the inter-val between EM30ME10/120 and EM30ME01/100 within a5.91 cM genetic region. The allele originating from GH 06increased the seed color phenotype value by 9.84.
In 2005 in Beibei, 6 QTLs were detected. They were des-ignated qSCBB05A-2-1, qSCBB05A-7-2, qSCBB05A-8-3,qSCBB05A-8-4, qSCBB05A-15-5, and qSCBB05A-20-6,were mapped on LG02 (N5), LG07, LG08 (N6), LG15, andLG20 (N9), and explained 7.4%, 4.43%, 3.02%, 3.13%,3.02%, and 39.34% of the phenotypic variation, respec-tively. The major QTL, qSCBB05A-20-6, was located in theinterval between EM30ME10/120 and EM57ME40/580within a 13.03 cM genetic region. The allele originatingfrom GH 06 increased the seed color phenotype value by7.10.
In 2006 in Beibei, 4 QTLs were detected. They were des-ignated qSCBB06A-9-1, qSCBB06A-15-2, qSCBB06A-18-3,and qSCBB06A-20-4, were mapped on LG09, LG15, LG18,and LG20 (N9), and explained 2.98%, 3.12%, 3.42%, and42.42% of the phenotypic variation, respectively. The majorQTL, qSCBB06A-20-4, was located in the interval betweenEM30ME10/120 and EM31ME03/150 within a 14.05 cMgenetic region. The allele originating from GH 06 increasedthe seed color phenotype value by 9.23.
The minor QTLs (e.g., qSCBB05A-7-2, explaining only4.43% of the phenotypic variation) were strongly affectedby environmental conditions and were not reproducible inthese environments.
RIL-2In this population, 7 significant QTLs were identified
(Table 2 and Fig. 3): 3 for Wanzhou in 2006, 2 for Beibeiin 2005, and 2 for Beibei in 2006. They were designatedqSCWZ06B-2-1, qSCWZ06B-5-2, qSCWZ06B-10-3,qSCBB05B-10-1, qSCBB05B-13-2, qSCBB06B-8-1, andqSCBB06B-10-2 and explained 8.06%, 6.64%, 30.18%,7.39%, 24.86%, 6.28%, and 15.08% of the phenotypic varia-tion, respectively. Three of these QTLs were located in thesame marker interval between EM11ME20/200 andCB10092/550 and explained a high proportion of phenotypicvariation in the environment. They were adjacent to thesame SRAP marker, EM11ME20/200, and were consideredmajor QTLs.
Comparison of QTLs across different populations anddifferent environments
In this study, a total of 19 QTLs were identified in 2
populations across 2 locations and 2 years. More QTLswere detected from RIL-1 than from RIL-2, and QTLs fromRIL-1 also showed higher hereditability. Nevertheless, onesignificant major QTL was detected in both populations inall environments. It could explain a very high proportion ofthe phenotypic variation and was adjacent to the samemarker, EM11ME20/200, in both populations and all envi-ronments. Therefore, our results show the convincing andstable reproducibility of this QTL in different populationsand environments.
Comparative genomic analysis of the major yellow-seedQTL with Arabidopsis chromosome sequences
In this study, the fragments of markers flanking the majorQTL of RIL-1 (EM30ME10/120, EM11ME20/200,EM30ME01/100, and EM43ME16/220) were sequenced.BLASTn analysis with A. thaliana chromosomes revealedthat the 4 marker sequences showed homology to functionalgenes located within the region of 17.9–24.8 Mb on A. thali-ana chromosome 5 (At C5). The exact homology relation-ships are as follows. EM30ME10/120: At5g44440 (electroncarrier, 17.9 Mb); EM11ME20/200: At5g49640 (unknownprotein, 20.2 Mb); EM30ME01/100: At5g61590 (a memberof the ERF (ethylene response factor) subfamily B-3 of theERF/AP2 transcription factor family); and EM43ME16/220:At5g56500 (ATP binding / protein binding / unfolded pro-tein binding; similar to chaperonin). The 4 markers cover6.2 cM (53.9–60.1 cM) on LG20 (N9), while the corre-sponding homologous genes cover 6.9 Mb on At C5. Withinthis region of At C5, there is an important TRANSPARENTTESTA (TT) gene, TT10, which encodes laccase 15(AtLAC15), an enzyme involved in accumulation of bothpigment and lignin in the seed coat (Liang et al. 2006). Themajor yellow-seed QTL identified here is mapped betweenthe markers EM30ME01/100 and EM11ME20/200, andTT10 is located within the corresponding homologous regionbetween At5g61590 and At5g49640 (Fig. 4). MarkerEM11ME20/200 was also found to be linked with the majoryellow-seed QTL in the second population, RIL-2. MarkerEM11ME20/200 was repeatedly detected in both popula-tions, and QTL detection with Windows QTL Cartographeralso showed that this marker was the one most tightly linkedto the major QTL.
DiscussionDoubled haploid, F2, and backcross populations have been
reported as materials to construct genetic linkage maps ofB. napus (Ferreira et al. 1994; Parkin et al. 1995; Sharpe etal. 1995; Mikkelsen et al. 1996; Lombard and Delourme2001; Lowe et al. 2004; Piquemal et al. 2005). Here we suc-cessfully constructed a genetic linkage map of B. napus byusing 2 high-generation RIL populations. This is the first re-port of the application of B. napus RIL populations to map
Fig. 2. A genetic linkage map of RIL-1 based on 26 SRAP and SSR linkage groups, which span 1744 cM and a total of 420 loci (65 forSSR, 65 for RAPD, and 290 for SRAP). Linkage groups (e.g., LG01) were constructed using JoinMap 3.0; numbers in parentheses (e.g.,N11) are chromosome numbers according to Piquemal et al. (2005), Lowe et al. (2004), and Bancroft (2006). The letters ‘‘P’’, ‘‘L’’, and‘‘B’’ in parentheses following some marker names indicate that these markers are synonymous with the markers of Piquemal et al. (2005),Lowe et al. (2004), and Bancroft (2006), respectively. The QTLs for seed color are indicated by boxes with the name of the QTL on theright-hand side of the linkage group.
844 Genome Vol. 50, 2007
# 2007 NRC Canada

Fu et al. 845
# 2007 NRC Canada

Fig. 2 (continued).
846 Genome Vol. 50, 2007
# 2007 NRC Canada

Fig. 2 (concluded).
Fu et al. 847
# 2007 NRC Canada

Fig. 3. A genetic linkage map of RIL-2 based on 20 SRAP and SSR linkage groups, which span 1135 cM and a total of 265 loci (105 forSSR, 160 for SRAP). Other annotations are the same as those in Fig. 2.
848 Genome Vol. 50, 2007
# 2007 NRC Canada

Fig. 3 (concluded).
Fu et al. 849
# 2007 NRC Canada
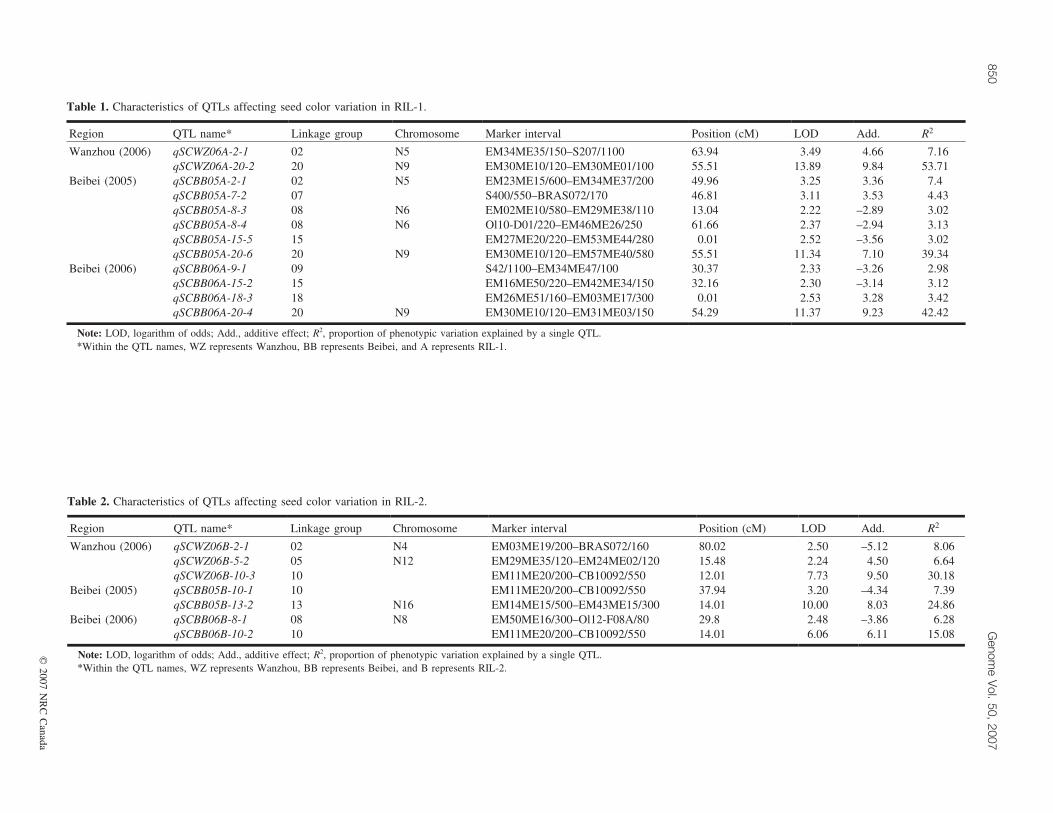
Table 1. Characteristics of QTLs affecting seed color variation in RIL-1.
Region QTL name* Linkage group Chromosome Marker interval Position (cM) LOD Add. R2
Wanzhou (2006) qSCWZ06A-2-1 02 N5 EM34ME35/150–S207/1100 63.94 3.49 4.66 7.16qSCWZ06A-20-2 20 N9 EM30ME10/120–EM30ME01/100 55.51 13.89 9.84 53.71
Beibei (2005) qSCBB05A-2-1 02 N5 EM23ME15/600–EM34ME37/200 49.96 3.25 3.36 7.4qSCBB05A-7-2 07 S400/550–BRAS072/170 46.81 3.11 3.53 4.43qSCBB05A-8-3 08 N6 EM02ME10/580–EM29ME38/110 13.04 2.22 –2.89 3.02qSCBB05A-8-4 08 N6 Ol10-D01/220–EM46ME26/250 61.66 2.37 –2.94 3.13qSCBB05A-15-5 15 EM27ME20/220–EM53ME44/280 0.01 2.52 –3.56 3.02qSCBB05A-20-6 20 N9 EM30ME10/120–EM57ME40/580 55.51 11.34 7.10 39.34
Beibei (2006) qSCBB06A-9-1 09 S42/1100–EM34ME47/100 30.37 2.33 –3.26 2.98qSCBB06A-15-2 15 EM16ME50/220–EM42ME34/150 32.16 2.30 –3.14 3.12qSCBB06A-18-3 18 EM26ME51/160–EM03ME17/300 0.01 2.53 3.28 3.42qSCBB06A-20-4 20 N9 EM30ME10/120–EM31ME03/150 54.29 11.37 9.23 42.42
Note: LOD, logarithm of odds; Add., additive effect; R2, proportion of phenotypic variation explained by a single QTL.*Within the QTL names, WZ represents Wanzhou, BB represents Beibei, and A represents RIL-1.
Table 2. Characteristics of QTLs affecting seed color variation in RIL-2.
Region QTL name* Linkage group Chromosome Marker interval Position (cM) LOD Add. R2
Wanzhou (2006) qSCWZ06B-2-1 02 N4 EM03ME19/200–BRAS072/160 80.02 2.50 –5.12 8.06qSCWZ06B-5-2 05 N12 EM29ME35/120–EM24ME02/120 15.48 2.24 4.50 6.64qSCWZ06B-10-3 10 EM11ME20/200–CB10092/550 12.01 7.73 9.50 30.18
Beibei (2005) qSCBB05B-10-1 10 EM11ME20/200–CB10092/550 37.94 3.20 –4.34 7.39qSCBB05B-13-2 13 N16 EM14ME15/500–EM43ME15/300 14.01 10.00 8.03 24.86
Beibei (2006) qSCBB06B-8-1 08 N8 EM50ME16/300–Ol12-F08A/80 29.8 2.48 –3.86 6.28qSCBB06B-10-2 10 EM11ME20/200–CB10092/550 14.01 6.06 6.11 15.08
Note: LOD, logarithm of odds; Add., additive effect; R2, proportion of phenotypic variation explained by a single QTL.*Within the QTL names, WZ represents Wanzhou, BB represents Beibei, and B represents RIL-2.
850G
enome
Vol.
50,2007
#2007
NR
CC
anada

construction. Because B. napus is a polyploid species andidentical alleles often occur at 2 (or more) homoeologousloci, it is very difficult to construct a genetic linkage mapof B. napus using the main current molecular marker meth-ods, especially dominant marker systems such as SRAP andAFLP (and even many dominantly inherited Brassica spp.SSRs). This can lead to large errors in B. napus maps unlessmarkers showing unexpected segregation are deleted beforethe linkage groups are built. In this study, the w2 test wasused to delete some markers that showed unexpected segre-gation, which reduced the above-mentioned problem tosome extent.
Phenotype is determined by the interaction of genotypeand environment, since environment influences the level ofgene expression. Therefore, different QTLs can be detectedin different environments (Huang et al. 1997), and it is diffi-cult to identify the genetic effect of a QTL with high repro-ducibility across different environments. Since each QTLhas a certain degree of genetic stability in a certain environ-ment, a trait with high genetic stability is more likely to bedetected across different environments. Tanksley et al.(1991) found that some major QTLs can be detected in dif-ferent environments, whereas a single-environment test can-not reveal how environment affects the variation of a QTL.Therefore, a QTL detected in a single environment shouldbe of low value in breeding selection. In our study, 2 RIL
populations, which resulted from crossing the same femaleparent (donor of yellow-seed genes) with different male pa-rents, were grown in 2 different locations (Wanzhou andBeibei) for 2 years (2005 and 2006). Nineteen QTLs weredetected by a composite interval mapping model with aLOD threshold of 2.5, and a major QTL with the sametightly linked marker (EM11ME20/200) within the samelinkage group was detected in both populations in both loca-tions, strongly implying that it is a major QTL of yellowseed color (Figs. 2 and 3).
As mentioned in the Introduction, seed color has differentinheritance models in different B. napus materials. In ourstudy, the major QTL for seed color was rather stably de-tected across locations, years, and populations, and someQTLs with minor effects on seed color variation were alsofound. The major QTL might be responsible for a structuralor regulatory gene involved in catabolism or regulation ofseed coat pigment biosynthesis and deposition, so it deservesto be cloned and functionally identified in the future. Cli-mate and harvest time generally affect the seed color ofB. napus to some degree, which results in instability of thistrait. The minor-effect QTLs with low reproducibility de-tected in our study might be responsible for other genescausing the above-mentioned environmental variations ofseed color.
Interestingly, A. thaliana chromosome 5 includes several
Fig. 4. Microsynteny between a 6.2 cM (53.9–60.1 cM) region on LG20 (N9) of B. napus and the 6.9 Mb (17.9–24.8 Mb) homologousregion on Arabidopsis thaliana chromosome 5 (At C5). The major yellow-seed QTL identified here is mapped between EM30ME01/100and EM11ME20/200, closest to EM11ME20/200.
Fu et al. 851
# 2007 NRC Canada

key functional genes related to seed color. Among the 17functionally characterized TT genes determining seed coatpigmentation of A. thaliana, AT5G07990 (TT7/F3’H),AT5G13930 (TT4/CHS), AT5G17220 (TT19/GST26),AT5G23260 (TT16/MADS BSISTER), AT5G24520 (TTG1/WD40), AT5G35550 (TT2/MYB123), AT5G42800 (TT3/DFR), AT5G43650 (TT8/bHLH), and AT5G48100 (TT10/LAC15) are located on chromosome 5 (Fig. 4). In this study,one major yellow-seed QTL was located within a 6.2 cM re-gion on LG20 (N9), and this region showed good microsyn-teny to a 6.9 Mb region on At C5. In a previous study of thesegmental structure of the B. napus genome based on com-parative analysis with A. thaliana, Parkin et al. (2005) iden-tified a homologous relationship between segments withpositions similar to those of the 2 above-mentioned regions,supporting the microsynteny revealed here. In both RILpopulations, marker EM11ME20/200 had the tightest link-age to the major QTL. Based on comparative genomic anal-ysis, the bifunctional gene TT10, with the nearest distance tothe homologue of EM11ME20/200, can be considered animportant candidate gene for the major QTL identified here.This gene is involved in both the seed coat pigment trait andthe seed coat lignin trait, conforming to the phenomenon ofsimultaneous reduction of both pigment and lignin in theseed coat of GH 06. Whether this assumption is correct andhow GH 06 differs from black-seeded lines at the TT10 locus(if at all) need to be determined through comparative clon-ing and functional characterization of the TT10 gene se-quences. Because this At C5 region is also near to someother TT genes such as TT8, TT3, and TT2, the possibilitythat the major QTL might encode another TT gene or anon-TT gene cannot be excluded. Similarly, the majorQTL on chromosome N18 detected by Badani et al.(2006) also has a large effect on both seed color and aciddetergent fibre content in multiple environments, but itschromosome location is different from that of the majorQTL located in this study. In future study, we will investi-gate whether these 2 QTLs encode proteins with the sameor similar functions (possibly TT10).
Seed color is an important trait in the breeding of B. napus,but it is very difficult to measure exactly. Some researchersidentify seed color by visual observation (Rahman 2001),which tends to be highly subjective. A colorimeter hasalso been used to classify the grades of yellow-seeded degree(Liu et al. 2005), but the cost of such an instrument shouldbe considered. Yellow-seeded degree can also be evaluatedusing the exposure time of seed under a dissecting micro-scope (Liu et al. 2006), but the result is only an indirectprofiling of the seed color that deviates somewhat fromthe actual color. By using a scanner to obtain a seed pic-ture with a stable beam and transforming the RGB valuesto HSB values, we used direct color information to evalu-ate the yellow-seeded degree of a seed sample, which effi-ciently reduced experimental error.
In this study, using 2 RIL populations, we successfullyidentified a stable major QTL that explained a large propor-tion of the phenotypic variation in 2 locations, and thecandidate gene was identified based on comparativegenomic analysis. Besides providing an effective strategyfor marker-assisted selection of the yellow-seed trait inB. napus, our results also provide important clues for
cloning of the candidate gene corresponding to this majorQTL.
AcknowledgementsThis research was supported by the Major Program of
National Natural Science Foundation (30330400), theNational Basic Research Program of China (973 Program2006CB101604), the National High Technology Researchand Development Program of China (863 Program2006AA100106), and the Major Program of ChongqingMunicipal Natural Science Foundation (8446).
ReferencesAltschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z.,
Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs.Nucleic Acids Res. 25: 3389–3402. doi:10.1093/nar/25.17.3389.PMID:9254694.
Axelsson, T., Shavorskaya, O., and Lagercrantz, U. 2001. Multipleflowering time QTLs within several Brassica species could bethe result of duplicated copies of one ancestral gene. Genome,44: 856–864. doi:10.1139/gen-44-5-856. PMID:11681610.
Badani, A.G., Snowdon, R.J., Wittkop, B., Lipsa, F.D., Baetzel, R.,Horn, R., et al. 2006. Colocalization of a partially dominantgene for yellow seed colour with a major QTL influencing aciddetergent fibre (ADF) content in different crosses of oilseed rape(Brassica napus). Genome, 49: 1499–1509. doi:10.1139/G06-091.
Bancroft, I. 2006. Bancroft home page. Linkage maps of oilseedrape (Brassica napus) [online]. Available from http://www.jic.bbsrc.ac.uk/staff/ian-bancroft/research_page3.htm [accessed 4April 2006].
Budak, H., Shearman, R.C., Parmaksiz, I., and Dweikat, I. 2004.Comparative analysis of seeded and vegetative biotype buffalo-grasses based on phylogenetic relationship using ISSRs, SSRs,RAPDs, and SRAPs. Theor. Appl. Genet. 109: 280–288. doi:10.1007/s00122-004-1630-z. PMID:15024466.
Burns, M.J., Barnes, S.R., Bowman, J.G., Clarke, M.H., Werner,C.P., and Kearsey, M.J. 2003. QTL analysis of an intervarietalset of substitution lines in Brassica napus: seed oil content andfatty acid composition. Heredity, 90: 39–48. doi:10.1038/sj.hdy.6800176. PMID:12522424.
Delourme, R., Bouchereau, A., Hubert, N., Renard, M., andLandry, B.S. 1994. Identification of RAPD markers linked to afertility restorer gene for the Ogura radish cytoplasmic malesterility of rapeseed (Brassica napus L.). Theor. Appl. Genet.88: 741–748. doi:10.1007/BF01253979.
Ferreira, M.E., Williams, P.H., and Osborn, T.C. 1994. RFLPmapping of Brassica napus using doubled haploid lines. Theor.Appl. Genet. 89: 615–621. doi:10.1007/BF00222456.
Ferriol, M., Pico, B., and Nuez, F. 2003. Genetic diversity of agermplasm collection of Cucurbita pepo using SRAP and AFLPmarkers. Theor. Appl. Genet. 107: 271–282. doi:10.1007/s00122-003-1242-z. PMID:12845442.
Huang, N., Angeles, E.R., Doming, J., Magpantay, G., Singh, S.,Zhang, G., et al. 1997. Pyramiding of bacterial blight resistancegenes in rice: marker-assisted selection using RFLP and PCR.Theor. Appl. Genet. 95: 313–320. doi:10.1007/s001220050565.
Janeja, H.S., Banga, S.S., and Lakshmikumaran, M. 2003. Identifi-cation of AFLP markers linked to fertility restorer genes fortournefortii cytoplasmic male-sterility system in Brassica napus.Theor. Appl. Genet. 107: 148–154. PMID:12835940.
Kosambi, D.D. 1944. The estimation of map distances from recom-bination values. Ann. Eugen. 12: 172–175.
852 Genome Vol. 50, 2007
# 2007 NRC Canada

Li, G., and Quiros, C.F. 2001. Sequence-related amplified poly-morphism (SRAP), a new marker system based on a simplePCR reaction: its application to mapping and gene tagging inBrassica. Theor. Appl. Genet. 103: 455–461. doi:10.1007/s001220100570.
Li, J.N., Zhang, X.K., Chen, L., Wang, R., and Chui, C. 1998. In-itial study on the genetics of seed color of yellow-seeded lines(Brassica napus L.) from different genetic sources. Chin. J. OilCrop Sci. 20: 16–19.
Li, J.N., Chen, L., Tang, Z.L., Zhang, X.K., and Yan, S.J. 2001.Genetic study and commercial application of the yellow-seededrapeseed (Brassica napus L.). In Proceedings of the InternationalSymposium on Rapeseed Science, Science Press, New York,19–23 April 2001. pp. 28–34.
Liang, Y., Li, J.N., and Chen, L. 2003. Influence of red and bluelight on seedcoat color of yellow and black-seed in B. napus.Chin. J. Oil Crop Sci. 25: 21–24.
Liang, M., Davis, E., Gardner, D., Cai, X., and Wu, Y. 2006. In-volvement of AtLAC15 in lignin synthesis in seeds and in rootelongation of Arabidopsis. Planta, 224: 1185–1196. doi:10.1007/s00425-006-0300-6. PMID:16779554.
Lin, Z.X., Zhang, X.L., Nie, Y.C., He, D.H., and Wu, M.Q. 2003.Construction of a genetic linkage map for cotton based onSRAP. Chin. Sci. Bull. 48: 2063–2067. doi:10.1360/03wc0193.
Liu, L.Z., Meng, J.L., Lin, N., Chen, L., Tang, Z.L., Zhang, X.K.,and Li, J.N. 2006. QTL mapping of seed coat color for yellow-seeded Brassica napus. Acta Genet. Sin. 33: 181–187. doi:10.1016/S0379-4172(06)60037-1. PMID:16529302.
Liu, Z.W., Fu, T.D., Tu, J.X., and Chen, Y. 2005. Inheritance ofseed color and identification of RAPD and AFLP markers linkedto the seed color gene in rapeseed (Brassica napus L.). Theor.Appl. Genet. 110: 303–310. doi:10.1007/s00122-004-1835-1.PMID:15565377.
Lombard, V., and Delourme, R. 2001. A consensus linkage map forrapeseed (Brassica napus L.): construction and integration ofthree individual maps from DH populations. Theor. Appl. Genet.103: 491–507. doi:10.1007/s001220100560.
Lowe, A.J., Moule, C., Trick, M., and Edwards, K.J. 2004. Effi-cient large-scale development of microsatellites for marker andmapping applications in Brassica crop species. Theor. Appl.Genet. 108: 1103–1112. doi:10.1007/s00122-003-1522-7. PMID:15067397.
McCouch, S.R., Cho, Y.G., Yano, M., Paul, E., Blinstrub, M.,Mori-shima, H., and Kinoshita, T. 1997. Report on QTL nomen-clature. Rice Genet. Newsl. 14: 11–13.
Meng, J.L., Shi, S.W., Gan, L., Li, Z.Y., and Qu, X.S. 1998. The pro-duction of yellow-seeded Brassica napus (AACC) through cross-ing interspecific hybrids of B. campestris (AA) and B. carinata(BBCC) with B. napus. Euphytica, 103: 329–333. doi:10.1023/A:1018646223643.
Mikkelsen, T.R., Jensen, J., and Jørgensen, R.B. 1996. Inheritanceof oilseed rape (Brassica napus) RAPD markers in a backcrossprogeny with Brassica campestris. Theor. Appl. Genet. 92:492–497. doi:10.1007/s001220050154.
Olsson, G. 1960. Species crosses within the genus Brassica. II. Ar-tificial Brassica napus L. Hereditas, 46: 351–396.
Parkin, I.A.P., Sharpe, A.G., Keith, D.J., and Lydiate, D.J. 1995.Identification of the A and C genomes of amphidiploid Brassicanapus (oilseed rape). Genome, 38: 1122–1131.
Parkin, I.A.P., Gulden, S.M., Sharpe, A.G., Lukens, L., Trick, M.,Osborn, T.C., and Lydiate, D.J. 2005. Segmental structure of theBrassica napus genome based on comparative analysis withArabidopsis thaliana. Genetics, 171: 765–781. doi:10.1534/genetics.105.042093. PMID:16020789.
Piquemal, J., Cinquin, E., Couton, F., Rondeau, C., Seignoret, E.,Doucet, I., et al. 2005. Construction of an oilseed rape (Brassicanapus L.) genetic map with SSR markers. Theor. Appl. Genet.111: 1514–1523. doi:10.1007/s00122-005-0080-6. PMID:16187118.
Rahman, M.H. 2001. Production of yellow-seeded Brassica napusthrough interspecific crosses. Plant Breed. 120: 463–472.doi:10.1046/j.1439-0523.2001.00640.x.
Santoni, S., and Berville, A. 1992. Characterization of the nuclearribosomal DNA units and phylogeny of Beta L. wild forms andcultivated beets. Theor. Appl. Genet. 83: 533–542. doi:10.1007/BF00226896.
Sharpe, A.G., Parkin, I.A.P., Keith, D.J., and Lydiate, D.J. 1995.Frequent non-reciprocal translocations in the amphidiploid gen-ome of oilseed rape (Brassica napus). Genome, 38: 1112–1121.
Shrizadegan, M. 1986. Inheritance of seed color in Brassica napusL. Z. Pflanzenzuecht. 96: 140–146.
Somers, D.J., Rakow, G., Prabhu, V.K., and Friesen, K.R. 2001.Identification of a major gene and RAPD markers for yellow-seeded coat colour in Brassica napus. Genome, 44: 1077–1082.doi:10.1139/gen-44-6-1077. PMID:11768211.
SPSS Inc. 2004. SPSS1. Version 13.0 [computer program]. SPSSInc., Chicago.
Stringam, G.R., Mcgregor, D.I., and Pawlowski, S.H. 1974. Chemi-cal and morphological characteristics associated with seed coatcolour in rapeseed. In Proceedings of the 4th International Rape-seed Conference, Giessen, Germany, 4–8 June 1974. pp. 99–108.
Tang, Z.L., Li, J.N., Zhang, X.K., Chen, L., and Wang, R. 1997.Genetic variation of yellow-seeded rapeseed lines (Brassicanapus L.) from different genetic sources. Plant Breed. 116:471–474. doi:10.1111/j.1439-0523.1997.tb01033.x.
Tanksley, S.D., Young, N.D., Paterson, A.H., and Bonierbale,M.W. 1989. RFLP mapping in plant breeding: new tools for anold science. Biol. Technology, 7: 257–264.
Tanksley, S.D., Ahn, N., Causse, M., Coffman, R., Fulton, T.,McCouch, S.R., et al. 1991. RFLP mapping of the rice genome.In Rice genetics. International Rice Research Institute, LosBanos, Laguna, Phillipines. pp. 435–442.
van Deynze, A., and Pauls, K.P. 1994. The inheritance of seedcolor and vernalization requirement in Brassica napus and usingdoubling haploid populations. Euphytica, 74: 77–83. doi:10.1007/BF00033770.
van Deynze, A.E., Beversdorf, W.D., and Pauls, K.P. 1993. Tem-perature effects on seed color in black-and yellow-seeded rape-seed. Can. J. Plant Sci. 73: 383–387.
van Ooijen, J.W., and Voorrips, R.E. 2001. JoinMap 3.0: softwarefor the calculation of genetic linkage maps. Plant Research In-ternational BV, Wageningen, the Netherlands. Available fromhttp://www.kyazma.nl.
Voorrips, R.E. 2002. MapChart: software for the graphical presen-tation of linkage maps and QTLs. J. Hered. 93: 77–78.
Wang, J.X., Yang, G.S., Fu, T.D., and Meng, J.L. 2000. Develop-ment of PCR-based markers linked to the fertility restorer genefor the Polima cytoplasmic male sterility in rapeseed (Brassicanapus L.). Acta Genet. Sin. 27: 1012–1016. PMID:11209683.
Wang, R., Li, J.N., Chen, L., Tang, Z.L., and Zhang, X.K. 2003.Genetic correlation analysis for main characters in yellow-seeded rapeseed lines (Brassica napus L.). Chin. J. Oil CropSci. 25: 8–11.
Wang, S., Basten, C.J., and Zeng, Z.B. 2006. Windows QTL Carto-grapher. Version 2.5 [computer program]. Department of Statis-tics, North Carolina State University, Raleigh, N.C. Availablefrom http://statgen.ncsu.edu/qtlcart/WQTLCart.htm.
Fu et al. 853
# 2007 NRC Canada

Wu, J.S., Shi, S.W., and Wu, D.W. Liu, and H.L. 1999. Studies onthe inheritance of yellow seedcoat in rapeseed (Brassica napusL). In Proceedings of the 10th International Rapeseed Congress,September 1999, Canberra, Australia. Edited by N. Wratten andP.A. Salisbury. The Regional Institute Ltd., Gosford, NSW, Aus-tralia. Available from http://www.regional.org.au/au/gcirc/4/146.htm [accessed 17 May 2006].
Yu, C.Y., Hu, S.W., Zhao, H.X., Guo, A.G., and Sun, G.L. 2005.Genetic distances revealed by morphological characters, iso-zymes, proteins and RAPD markers and their relationships withhybrid performance in oilseed rape (Brassica napus L.). Theor.Appl. Genet. 110: 511–518. doi:10.1007/s00122-004-1858-7.PMID:15578151.
Zhang, J., Guo, W., and Zhang, T.Z. 2002. Molecular linkage mapof allotetraploid (Gossypium hirsutum L. � Gossypium barba-dense L.) with a haploid population. Theor. Appl. Genet. 105:1166–1174. doi:10.1007/s00122-002-1100-4. PMID:12582895.
Zhao, J., Becker, H.C., Zhang, D., Zhang, Y., and Ecke, W. 2006.Conditional QTL mapping of oil content in rapeseed with re-spect to protein content and traits related to plant developmentand grain yield. Theor. Appl. Genet. 113: 33–38. doi:10.1007/s00122-006-0267-5. PMID:16614833.
Zhao, J.W., and Meng, J.L. 2003. Genetic analysis of loci asso-ciated with partial resistance to Sclerotinia sclerotiorum in rape-seed (Brassica napus L.). Theor. Appl. Genet. 106: 759–764.PMID:12596007.
854 Genome Vol. 50, 2007
# 2007 NRC Canada
Copyright © 2022 FDOKUMEN















![Increased [CO 2] does not compensate for negative effects on yield caused by higher temperature and [O 3] in Brassica napus L](https://static.fdokumen.com/doc/165x107/631794e7831644824d038e8a/increased-co-2-does-not-compensate-for-negative-effects-on-yield-caused-by-higher.jpg)




