
Impaired sulphated glycosaminoglycan metabolism in a patient with GM2 gangliosidosis (Tay-Sachs...
11
J. 1nher. Metab. Dis. 13 (1990) 721 731 © SSIEM and Kluwer AcademicPublishers. Printed in the Netherlands Impaired Sulphated Glyeosaminoglycan Metabolism in a Patient with GM-2 Gangliosidosis (Tay-Sachs Disease) L. TOMA 1, W. PINTO 2, V. C. RODRIGUES 3, C. P. DIETRICH and H. B. NADER 1. 1Departamento de Bioquimica, Escola PauIista de Medicina, C.P. 20372, CEP 04023 SSo Paulo, S.P.; ZDepartamento de Gendtica, Universidade de Campinas, Campinas, S.P.; 3Departamento de Gendtica, Universidade Julio de Mesquita Filho, Araraquara, S.P., Brazil Summary: An abnormal urinary excretion of sulphated glycosaminoglycans in a patient with GM-2 gangliosidosis (Tay-Sachs disease) is described. Besides the accumulation of GM-2 ganglioside in liver and lack of hexosaminidase A, the patient shows an abnormal urinary excretion of an iduronic acid-rich low molecular weight heparan sulphate. Also, no dermatan sulphate could be detected in the urine, whereas this compound was the main sulphated glycosami- noglycan in the liver of the patient. Heparan sulphate was the main glycosamino- gtycan of normal liver. The total amount of sulphated glycosaminoglycans in the urine and liver of the patient did not differ significantly from the amounts found in the liver and urine of normal subjects. Several plasma glycosidases have been assayed and the activities did not differ significantly from the values obtained for the plasma of normal subjects. The mucopolysaccharidoses are a group of syndromes characterized by the intracellu- lar accumulation of specific sulphated glycosaminoglycans (SGAG) and the excretion of excessive quantities of largely undegraded SGAG in the urine (McKusick, 1969). The metabolic errors are due to the lack of activity of specific lysosomal enzymes which are required for glycosaminoglycan catabolism (Van Hoof, 1973; Neufeld, 1974). We have previously described a new mucopolysaccharidosis which seemed to be related to a defect in the synthesis of chondroitin 6-sulphate resulting in the excretion of this compound with a low degree of sulphation (Dietrich et al., 1972; Mourfio et al., 1973; Toledo et al., 1978). A decreased activity of chondro 6-sulphotransferase was later observed in fibroblasts in culture prepared from the skin of these patients (Mourfio et al., 1981). Contrary to the classical mucopolysaccharidoses, the total amounts of sulphated glycosaminoglycans excreted by the patients were within the normal range (Dietrich et al., 1972). MS received 7,11.89 Accepted 5.3.90 721
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Impaired sulphated glycosaminoglycan metabolism in a patient with GM2 gangliosidosis (Tay-Sachs...

J. 1nher. Metab. Dis. 13 (1990) 721 731 © SSIEM and Kluwer Academic Publishers. Printed in the Netherlands
Impaired Sulphated Glyeosaminoglycan Metabolism in a Patient with GM-2 Gangliosidosis (Tay-Sachs Disease) L. TOMA 1, W. PINTO 2, V. C. RODRIGUES 3, C. P. DIETRICH and H. B. NADER 1. 1Departamento de Bioquimica, Escola PauIista de Medicina, C.P. 20372, CEP 04023 SSo Paulo, S.P.; ZDepartamento de Gendtica, Universidade de Campinas, Campinas, S.P.; 3Departamento de Gendtica, Universidade Julio de Mesquita Filho, Araraquara, S.P., Brazil
Summary: An abnormal urinary excretion of sulphated glycosaminoglycans in a patient with GM-2 gangliosidosis (Tay-Sachs disease) is described. Besides the accumulation of GM-2 ganglioside in liver and lack of hexosaminidase A, the patient shows an abnormal urinary excretion of an iduronic acid-rich low molecular weight heparan sulphate. Also, no dermatan sulphate could be detected in the urine, whereas this compound was the main sulphated glycosami- noglycan in the liver of the patient. Heparan sulphate was the main glycosamino- gtycan of normal liver. The total amount of sulphated glycosaminoglycans in the urine and liver of the patient did not differ significantly from the amounts found in the liver and urine of normal subjects. Several plasma glycosidases have been assayed and the activities did not differ significantly from the values obtained for the plasma of normal subjects.
The mucopolysaccharidoses are a group of syndromes characterized by the intracellu- lar accumulation of specific sulphated glycosaminoglycans (SGAG) and the excretion of excessive quantities of largely undegraded SGAG in the urine (McKusick, 1969). The metabolic errors are due to the lack of activity of specific lysosomal enzymes which are required for glycosaminoglycan catabolism (Van Hoof, 1973; Neufeld, 1974).
We have previously described a new mucopolysaccharidosis which seemed to be related to a defect in the synthesis of chondroitin 6-sulphate resulting in the excretion of this compound with a low degree of sulphation (Dietrich et al., 1972; Mourfio et al., 1973; Toledo et al., 1978). A decreased activity of chondro 6-sulphotransferase was later observed in fibroblasts in culture prepared from the skin of these patients (Mourfio et al., 1981). Contrary to the classical mucopolysaccharidoses, the total amounts of sulphated glycosaminoglycans excreted by the patients were within the normal range (Dietrich et al., 1972).
MS received 7,11.89 Accepted 5.3.90
721

722 Toma et al.
This paper reports another syndrome characterized as GM-2 gangliosidosis (Tay Sachs disease, McKusick 27280) showing an abnormal urinary excretion of an iduronic acid-rich low molecular weight heparan sulphate without increase in the total amount of SGAG. In addition, no dermatan sulphate could be detected in the urine of the patient.
MATERIALS AND METHODS
Case report: P.H.V., a male, was born at term with a birth weight of 3.5 kg and a length of 42 cm. The parents were non-consanguineous. The first sign 3 months after birth was the appearance of a distended abdomen due to hepatosplenomegaly. The patient showed early psychomotor regression with rapidly progressing neurological symptoms including motor weakness and hypotonia besides hyperactive deep tendon reflexes. Appetite was poor and weight gain abnormal. The cornea was clear, The child had coarse facial features and low-set ears and the gums were hypertrophied. Dorsolumbar kyphosis and inguinal hernia were also present. Recurrent infections, especially bronchopneumonia, were constant problems. Clonic-tonic convulsions started at the age of 1.5 years and were difficult to control. The musculature became deficient and atrophic; marked atrophy of the interosseous muscles of the hands was observed. The amount of sulphated glycosaminoglycans excreted by the patient was within the normal range. The child died at the age of 2 years. Histological analysis including electromicroscopy of the liver has shown a large number of vacuoles in the hepatocytes (Figures 1 and 2). In some of the cells the nucleus was displaced to the periphery, probably due to the large number of vacuoles. Staining with PAS and toluidine blue was negative. Since the specimens were prepared in paraffin no specific staining for lipids was performed. After pertinent radiological examination and other tests it was concluded that the child's symptoms were indistinguishable from those of GM-2 gangliosidosis (Tay-Sachs disease). GM-2 gangliosidosis was confirmed by the findings that the hexosaminidase A levels were well below the normal values and that liver extracts have shown the accumulation of GM-2 ganglioside.
Gtycosaminogtyeans: The urinary glycosaminoglycans were extracted from the urine with cetyltrimethylammonium bromide according to the method of Meyer et al. (1958). Liver tissue samples from the patient and normal subjects were obtained at autopsy and kept frozen until extraction as follows: the tissue was ground with 5 volumes of acetone and the mixture maintained at room temperature for 24 h. The precipitate formed was collected by centrifugation, washed once with acetone and dried. The acetone powder (1 g) was suspended in 10ml of a solution containing 0.15mol/L sodium chloride in 0.05mol/L Tris-HC1 buffer, pH 8.0 and 10rag of Superase (a proteolytic enzyme from Chas Pfizer Co., New York). The mixture was incubated for 18 h at 60°C. After incubation trichloroacetic acid was added to the mixture to a final concentration of 10% (w/v) and kept at 5°C for 15 rain. The mixture was centrifuged at 5000g for 15min. To the supernatant 2 volumes of ethanol were then added and the mixture was kept for 8 h at - 20°C. The precipitate formed was collected by centrifugation, washed twice with 80% (v/v) ethanol-water and dried (Cassaro and Dietrich, 1977).
J. lnher, Metab. Dis. 13 (1990)

Gtycosaminoglycan Metabolism in GM-2 Gangtiosidosis 723
A
!N
N
I N
B C
100 tJm Figure ! Histological analysis of the patient's liver. Liver autopsy sample, fixed in tbrmalin, included in paraffin and sections stained with haematoxylin-eosin (A), PAS (B) and toluidine blue (C). Arrows: (1) hepatocyte containing cytoplasmic vacuoles and the nucleus displaced towards the periphery; (2) Kupffer cell; (3) cytoplasmic vacuoles.
Figure 2 Electron microscopy of the patient's liver. Arrows: (1) hepatocyte nucleus; (2) Kupffer cell nucleus; (3) cytoplasmic vacuoles.
Analytical: The sutphated gtycosaminoglycans were analysed and quantitated by a combination of agarose gel electrophoresis in three different buffer systems and enzymatic degradation with chondroitinases and heparitinases, essentially as previ- ously described (Bianchini et aI., 1980). The chondroitinase AC acts upon N- acetylhexosamine/%l,4-glucuronic acid linkages whereas the chondroitinase ABC is
J. lnher. Metab. Dis. 13 (1990)

724 Toma et at.
less specific, acting also upon N-acetylhexosamine/%l,4-iduronic acid linkages. The heparitinases I and II act upon N-sulphated or N-acetytated glucosamine c~-I,4- glucuronic acid linkages whereas the heparinase acts upon N-sulphated hexosamine ~-l,4-iduronic acid linkages. For further details see Nader et al. (1987). The products formed from these reactions were visualized by silver nitrate and toluidine blue staining after chromatography of the incubates in Whatman No. 1 paper using as descending solvent isobutyric acid: 1 mol/L ammonia, 5:3, (v/v) as described previously (Cassaro and Dietrich, 1977). Gangtiosides from the patient's liver and from normal subjects who died of accidental causes were extracted and characterized as described by Ikeno et al. (1982)
Enzyme assays: Enzyme determinations were performed as described by Hall and Neufeld (1973) with minor modifications as follows: 0.5 #tool of synthetic substrates were incubated with 50-100#1 of plasma and/or serum of the patient and normal controls in 0.05 mol/L sodium acetate buffer, pH 5.0 at 37°C. After 1 h the reaction was stopped by addition of 1 ml of 0.05 mol/L sodium hydroxide, any precipitate was removed by centrifugation and the absorbance was measured at 400nm. For each substrate a control incubation was carried out in which plasma or serum was added to the reaction mixture after the addition of sodium hydroxide. The following substrates were used: p-nitrophenyl-/~-D-glucuronide, p-nitrophenyl-/?-D-mannoside, p-nitrophenyl-/%D-N-acetylglucosaminide and p-nitrophenyl-fi-D-N-acetylgalacto- side, all purchased from the Sigma Chemical Co., Saint Louis, USA. e-L-iduronidase activity was determined in leukocytes using phenyl-c~-L-iduronide (Kyoto Medical Scientific Laboratory Co. Ltd., Kyoto, Japan) in 0.05 mol/L sodium formate, pH 3.5, containing 0.15mol/L sodium chloride and 0.1% sodium azide. After 18h at 25°C Folin-Ciocalteau reagent (Merck Co., Darmstadt, FRG) was added and the assay performed according to Hall and Neufeld (1973). Hexosaminidases A and B were assayed in liver extracts essentially as described by O'Brien et al. (1970) using 4- methylumbelliferyl-N-acetyl-/%D-glucosaminide as substrate.
Other methods: Protein was determined in the serum and plasma by the method of Lowry et al. (1951). Uronic acid was determined according to the method of DiFerrante et al. (1971). Molecular weight determinations were performed as previously described (Dietrich and Nader, 1974). The sulphated glycosaminoglycans were fractionated in large scale by agarose gel electrophoresis as described elsewhere (Dietrich and Nader, 1974).
RESULTS Urinary sulphated gtycosaminoglyeans: Figure 3 shows the electrophoretic migration of the SGAG excreted by the patient with GM-2 gangliosidosis compared with the pattern of SGAG excreted by normal subjects. The compound migrating as chondro- itin sulphate standard from both normals and the patient was susceptible to the action of chondroitinase ABC. For the SGAG from normal urine, a compound migrating as dermatan sulphate became apparent after degradation with chondroitin- ase AC. This compound was totally degraded by chondroitinase ABC, characterizing
J. Inher. Metab. Dis. 13 (1990)

GlycosaminogIycan Metabolism in GM-2 GangIiosidosis 725
E C
t l ) v
40-20... PATI 2
0 10- o- J'~., 10- 0
40
2
0 2 4
No 2 _..,--/X
HS DS CHS
I
; 0 2 4 6
CONTROL
CHONDROITINASE AC
CHONDROITINASE ABC
STANDARDS
ELECTROPHORETIC MIGRATION (cm) Figure 3 Gel electrophoresis of urinary sulphated glycosaminoglycans after enzymatic degradation with mucopolysaccharidases. About 50 #g of urinary glycosaminoglycans excreted by the patient and normal subjects were incubated with 0.01 U of chondroitinases AC and ABC (Sigma Chemical Co., St. Louis, MO) at 37°C for t8h in a final volume of 20~& 5/~1 aliquots were submitted to agarose gel electrophoresis in 0.05 mol/L diaminopropane-acetate buffer, pH 9.0 for 60rain at 120V. After drying and staining with toluidine blue the glycosaminoglycans were subjected to densitometry at 525 nm. HS: heparan sulphate; DS: dermatan sulphate; CHS: chondroitin sulphate.
it as dermatan sulphate by this differential susceptibility to the action of the chondroitinases. On the other hand, it was absent in the patient's urine. A third band migrating as heparan sulphate is present in both urines. This band was identified as heparan sulphate due to its susceptibility to heparitinases (see below).
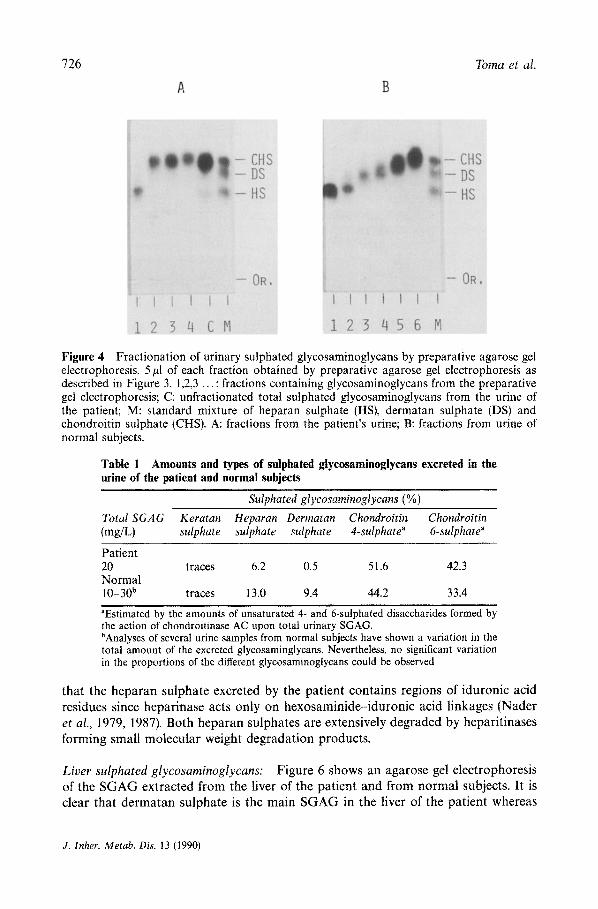
The absence of dermatan sulphate in the patient's urine was further confirmed by the fractionation of the SGAG as shown in Figure 4. No compound migrating as dermatan sulphate could be observed after the fractionation of the SGAG from the patient, contrasting with the profile shown by normal individuals where at least two fractions with the same electrophoretic mobility as dermatan sulphate could be observed. Table 1 shows the relative amounts of the urinary SGAG of the patient and normal subjects.
Besides the absence of dermatan sulphate in the patient's urine, the amount of heparan sulphate seemed also somewhat decreased. In order to study this result some analyses of the heparan sulphate excreted by the patient were performed. The purified heparan sulphates from normal urine and the patient's urine (Figure 4) were subjected to degradation with heparinase and heparitinases as shown in Figure 5. The heparan sulphate from normal individuals has an average molecular weight of 68 000 whereas that from the patient is smaller, with an average molecular weight of 21 000 as judged by the migration in polyacrylamide gel electrophoresis (Figure 5). Furthermore, the patient's heparan sulphate is partially susceptible to heparinase forming products of smaller molecular weight, whereas the heparan sulphate present in the urine of normal individuals is resistant to the action of this enzyme (Figure 5). This indicates
J. Inher. Metab. Dis. 13 (t990)
726 Toma et al.
A B
..... C H $
• - - D ~
~i!!!! !ilii i ̧
Figure 4 Fractionation of urinary sulphated glycosaminoglycans by preparative agarose gel electrophoresis. 5/A of each fraction obtained by preparative agarose gel electrophoresis as described in Figure 3. 1,2,3 ... : fractions containing glycosaminoglycans from the preparative gel electrophoresis; C: unfractionated total sulphated glycosaminoglycans from the urine of the patient; M: standard mixture of heparan sulphate (HS), dermatan sulphate (DS) and chondroitin sulphate (CHS). A: fractions from the patient's urine; B: fractions from urine of normal subjects.
Table 1 Amounts and types of sulphated glycosaminoglycans excreted in the urine of the patient and normal subjects
Sulphated glycosaminoglycans (%)
Total SGAG Keratan Heparan Dermatan Chondroitin Chondroitin (rag/L) sulphate sulphate sulphate 4-sulphate" &sulphate"
Patient 20 traces 6.2 0.5 51.6 42.3 Normal 10-30 b traces ! 3.0 9.4 44.2 33.4
"Estimated by the amounts of unsaturated 4- and 6-sulphated disaccharides formed by the action of chondroitinase AC upon total urinary SGAG. bAnalyses of several urine samples from normal subjects have shown a variation in the total amount of the excreted glycosaminglycans, Nevertheless, no significant variation in the proportions of the different glycosaminoglycans could be observed
that the heparan sulphate excreted by the patient contains regions of iduronic acid residues since heparinase acts only on hexosaminide-iduronic acid linkages (Nader et aL, 1979, 1987). Both heparan sulphates are extensively degraded by heparitinases forming small molecular weight degradation products.
Liver sulphated glycosaminoglycans: Figure 6 shows an agarose gel electrophoresis of the SGAG extracted from the liver of the patient and from normal subjects. It is clear that dermatan sulphate is the main SGAG in the liver of the patient whereas
J. Inher. Metab. Dis. 13 (1990)

Glycosaminogtycan Metabolism in GM-2 Gangtiosidosis 727
20-
O.
2 0 -
0
E 2 0 -
68.0 21.0 9.3 4.0 150 J M.W. x 10 - 3
I cONTROL
HEPARINASE
HEPARITINASES t , [1
to ~ATIE 20 - CONTROL
t.u 0 0
20 - . ~ ~ HEPARINASE z O ....
0 20 - HEPARITINASES I ". I I
'= , - . ' r " - ' > < 0 I ~ ' 0 1 2 3 4
ELECTROPHORETIC MIGRATION (cm)
Figure 5 Polyacrylamide gel electrophoresis of urinary heparan sulphate obtained from the patient and normal subjects after enzymatic degradation, About 10#g of heparan sulphate (fraction 1, Figure 4) from the patient and normal subjects were incubated with 0.01 U of heparinase and heparitinases I + II at 30°C for I8 h in a final volume of 20#1. The incubation mixtures were then subjected to polyacrylamide gel electrophoresis as referred to in the Methods section. The compounds were visualized after toluidine blue staining and quantitated by densitometry at 525 nm,
40 pArlENT FORMAt̂
0 E
A to 2 0 '
to - O-
Lu 4 0 - HS D~ j~CHS HS D S t C H S
0-2/ CO _
< O- • i ~ i i ' 0 2 4 6 0 2 4 6
ELECTROPHORETIC MIGRATION (cm)
CONTROL
CHONDROITINASE ABC
STANDARDS
Figure 6 Degradation of liver glycosaminoglycans by chondroitinase ABC. The experiment was performed as described in Figure 3 except that 20#g of sulphated gtycosaminogtycans obtained from the liver of the patient and 50#g from the liver of normal subjects were used. Abbreviations as in Figure 3.
heparan sulphate and dermatan sulphate are the main SGAG in the liver of normal subjects. This is confirmed by the susceptibility of the compound migrating as dermatan sulphate to the action of chondroitinase ABC as shown in Figure 6. The
J. Inher. Metab. Dis. 13 (1990)

728 Toma et al.
band migrating as heparan sulphate is totally degraded by the combined action of the heparitinases I and II (results not shown). Thus no keratan sulphate could be detected in the liver of the patient. The total amounts of sulphated glycosaminoglycans in the liver of the patient were within the normal range (290#g/g of dry tissue) compared with 250-320/zg/g of dry tissue in liver of normal individuals. Analyses of several livers obtained from normal subjects have shown no significant variations in the proportions of the different glycosaminoglycans. These combined results lead to the conclusion that the patient's liver contains relatively more dermatan sulphate and relatively less heparan sulphate when compared to the livers of normal subjects.
Serum, plasma and liver gtycosidases: Table 2 shows the activity of different glycosidases in the serum and plasma of the patient compared to the activity of these enzymes in normal subjects. The activity obtained for the several glycosidases in the patient's plasma did not differ significantly from the values obtained for the plasma of normal subjects. Likewise, the activity of L-iduronidase in leukocytes did not differ significantly between the patient and normal controls. The two forms of hexosaminidase, A and B, are shown in Table 3. It is clear that the patient exhibits ahnost exclusively the B form whereas the normal control shows the activity of both hexosaminidase A and B.
Gangliosides: The thin layer chromatography of the gangliosides extracted from the liver of the patient and from a normal control is shown in Figure 7. GM-2 ganglioside is present in large amounts in the liver of the patient and is almost absent from the control liver.
Table 2 Enzymatic activity of different glycosidases in serum, plasma and leukocytes of the patient and normal subjects
Enzyme (nmol p-nitrophenol/ mg protein/h)
Patient Normal
Serum Plasma Leukocyte Serum Plasma Leukocyte
fl-glucuronidase 0.99 1.17 0.77 fl-N-acetylglucosaminidase 1.57 1.43 1.87 3.46 fl-N-aeetylgalactosaminidase 0.67 2.82 0.95 2.82 fl-mannosidase 1.08 2.58 1.29 2.66 e-L-iduronidase" 3.1 2.0
"nmol of iduronic acid formed/mg protein/h
Table 3 Hexosaminidase forms A and B in liver extracts of the patient and from normal subjects
Hexosaminidase A a Hexosaminidase B a
Normal 12.13 14.03 Patient 2.54 20.37
ammol of umbelliferone formed/mg protein/h
J. lnher. Metab. Dis. 13 (1990)

Glycosaminoglycan Metabolism in GM-2 Gangliosidosis 729
GM I GM2 GM3
I I I I I
N P GMI + N P
GM3
Figure 7 Thin layer chromatography of gangliosides. N: gangliosides from normal liver; P: gangliosides from the patient's liver; GM1 + GM3: mixture of standard GM-1 and GM-3 ganglioside.
DISCUSSION
The patient's clinical, radiological and histological findings together with the virtual absence of hexosaminidase A activity and accumulation of GM-2 ganglioside in the liver are compatible with GM-2 gangliosidosis (Tay-Sachs disease). We now show that these patients also have an impaired metabolism of glycosaminoglycans showing a qualitative change in the urinary SGAG pattern with the presence of an iduronic acid-rich low molecular weight heparan sulphate and lack of dermatan sulphate in the urine together with its relative accumulation in liver. The finding that dermatan sulphate is the main SGAG in the liver together with its absence in the urine leads to the suggestion of a possible metabolic defect in the transport of this compound across the cell membranes in the present syndrome, besides the well established deficiency of hexosaminidase A and accumulation of GM-2 ganglioside. The impair- ment of the dermatan sulphate metabolism does not seem to be related to L- iduronidase activity, as it occurs in Hurler and Scheie syndromes (Neufeld, 1974), since this enzymatic activity is within the normal range at least in the patient's leukocytes. The metabolism of heparan sulphate seems also to be impaired by the finding that it is decreased in the liver and urine besides showing a chemically abnormal structure.
So far, the pathogenetic processes leading to the accumulation of GM-2 ganglioside and dermatan sulphate in the liver and the structural changes in heparan sulphate in this syndrome are obscure. It has been reported that in Hurler, Hunter and San Filippo syndromes, besides the well-known accumulation of glycosaminoglycans in tissues, there is also accumulation of GM-1, GM-2 and GM-3 gangliosides in brain and tissues, as reviewed by Ikeno et al. (1982). These results therefore suggest that hexosaminidase A belongs to a common pathway in the degradation of gangliosides and processing of the sulphated glycosaminoglycans.
The impairment in the metabolism of the sulphated glycosaminoglycans of the present case was not obvious, and only after specific methods for the identification
J. lnher. Metab. Dis. 13 (1990)

730 Toma et aI.
of the glycosaminoglycans and enzyme degradation was it possible to show a defect in the structure of the compounds. It is possible that other patients with Tay-Sachs disease might exhibit the metabolic errors described here.
A C K N O W L E D G E M E N T S
We wish to thank Dr Hello K. Takahashi for help with the determination of the gangliosides. This work was aided by grants from F I N E P (Financiadora de Estudos e Projetos), CNPq (Conselho Nacional de Desenvolvimento Cientifico e Tecnologico) and FAPESP (Fundag~o de Amparo a Pesquisa do Estado de S~o Paulo), Brazil
REFERENCES
Bianchini, P., Nader, H. B., Takahashi, H. K., Osima, B., Straus, A. H. and Dietrich, C. P. Fractionation and identification of heparin and other acidic mucopolysaccharides by a new discontinuous etectrophoretic method. J. Chromatog. 196 (1980) 455-462
Cassaro, C. M. F. and Dietrich, C. P. Distribution of sulfated mucopolysaccharides in invertebrates. J. BioL Chem. 252 (1977) 2254--2261
Dietrich, C. P., Mour~o, P. A. S. and Toledo, S. P. A. A new mucopolysaccharidosis characterized by excretion of chondroitin sulfate C of low sulfate content. In Piras, R. and Pontis, G. H. (eds.), Biochemistry of the Glycosidic Linkage, Academic Press, New York, 1972, pp. 751-758
Dietrich, C. P. and Nader, H. B. Fractionation and properties of four heparitin sulfates from beef lung tissue. Isolation and partial characterization of a homogenous species of heparitin sulfate. Biochim. Biophys. Acta 343 (1974) 3444
DiFerrante, N., Nichols, B. L., Donnelly, P. V., Neri, G., Argovic, R. and Berglund, R. K. Induced degradation of glycosaminoglycans in Hurlers and Hunters syndromes by plasma infusion. Proc. Natl. Acad. Sci. USA 68 (1971) 303-307
Hall, C. W. and Neufeld, E. F. L-Iduronidase activity in cultured skin fibroblasts and amniotic fluid cells. Arch. Biochem. Biophys. 158 (1973) 8t7-821
Ikeno, T., Minami, R., Tsugawa, S. K. and Nakao, T. Acidic glycosaminoglycans and gangliosides in the brain from four patients with genetic mucopolysaccharidosis. Tohoku J. Exp. Med. 137 (1982) 253-260
Lowry, O. H., Rosebrough, N. J., Farr, A. L. and Randall, R. J. Protein measurement with the folin phenol reagent. J. Biol. Chem. 193 (1951) 265-275
McKusick, V. A. The nosology of the mucopolysaccharidoses. Am. J. Med. (1969) 730-747 Meyer, K., Grumbak, M. M., Linker, A. and Hoffman, P. Excretion of sulfated mucopolysacch-
arides in Gargoylism (Hurlers syndrome). Proc. Soc. Exp. Biol. Med. 97 (1958) 275-279 Mourfio, P. A. S., Toledo, S. P. A., Nader, H. B. and Dietrich, C. P. Excretion of chondroitin
sulfate C with low sulfate content by patients with generalized platyspondily (brachyolmia). Biochem. Med. 7 (1973) 415-423
Mour~o, P. A. S., Kato, S. and Donelly, P. V. Spondyloepiphyseat dysplasia, chondroitin sulfate type: a possible defect of PAPS-chondroitin sulfate sulfotransferase in humans. Biochem. Biophys. Res. Commun. 98 (1981) 388-396
Nader, H. B., Cohen, D. M. and Dietrich, C. P. Chemistry of heparitin sulfate and heparin from normal tissues and from patients with Hunter syndrome Biochim. Biophys. Acta 582 (1979) 33-43
Nader, H. B., Dietrich, C. P., Buonassisi, V. and Colburn, P. Heparin sequences in the heparan sulfate chains of an endothelial cell proteoglycan. Proc. Natl. Acad. Sci. USA 84 (1987) 3565-3569
Neufeld, E. F. The biochemical basis for mucopolysaccharidoses and mucolipidoses. In Steinberg, A. G. and Bearn, A. C. (eds.), Progress in Medical Genetics 10 (t974) 81-t01
J. lnher. Metab. Dis. 13 (1990)

Giycosaminoglycan Metabolism in GM-2 GangIiosidosis 731
O'Brien, J. S., Odara, S., Chen, A. and Fillerup, B. S. Tay-Sachs disease. Detection of heterozygotes and homozygotes by serum hexosaminidase assay. N. Engl. J. Med. 283 (1970) 15-20
Toledo, S. P. A., Mourao, P. A. S., Lamego, C., Alves, C. A., Dietrich, C.P., Assis, L. M. and Mattar, E. Recessively inherited, late onset, spondylar dysplasia and peripheral corneal opacity with anomalies in the urinary mucopolysaccharides. A possible error of chondroitin sulfate synthesis. Am. J. Med. Genet. 2 (1978) 385-395
Van Hoof, F. Mucopolysaccharidoses. In Hers, H. G. and Van Hoof, F. (eds.), Lysosomes and Storage Diseases, Academic Press, New York, 1973, 217-259
J. lnher. Metab. Dis. 13 (1990)




















