
Keywords: Micronuclei Mobile phone Plutonium Chromosome aberrations

Identi®cation of two distinct deleted regions on chromosome 13 in prostatecancer
Chunde Li1, Catharina Larsson2, Andrew Futreal3, Jonathan Lancaster3, Catherine Phelan2,Ulla Aspenblad4, Birgitta Sundelin4, Yie Liu5, Peter Ekman1, Gert Auer4 and Ulf SR Bergerheim1
Departments of 1Urology, 2Molecular Medicine, 4Pathology and 5Oncology, Karolinska Hospital, 171 76 Stockholm, Sweden;3Division of Gynecologic Oncology, Department of Surgery, Duke University Medical Center, Durham, North Carolina 27710,USA
Aberrations of 13q occur frequently in prostate cancerand this chromosome contains two known tumorsuppressor genes, BRCA2 and Rb1. This study analysed13q LOH, DNA ploidy, BRCA2 mutation and pRbexpression in prostate cancers. In total, 13q deletionswere found in 18 of 36 tumors but did not correlate withhistological grade, stage or DNA ploidy. Two smallestregions of overlapping deletions were de®ned: one ¯ankedby D13S218 and D13S153; the other ¯anked by D13S31and D13S137. BRCA2 was less frequently deletedwhereas Rb1 did have a high frequency of deletion.None of the two genes was located in any of these tworegions. Furthermore, BRCA2 mutation was not found inthe ®ve tumors where deletions had involved the BRCA2locus. Neither did the Rb1 deletion correlate with absentpRb expression. In addition, tetraploidy was found in 14out of 25 tumors analysed and correlated with aberrantpRb expression. Our results indicate that 13q deletion isan early non-random event. Tumor suppressor genesother than BRCA2 or Rb1 may be the target of 13qdeletions. Aberrant pRb expression may not re¯ect thetwo-hit Rb1 inactivation but may be involved in thetetraploidization of prostate cancer cells.
Keywords: prostate cancer; chromosome 13; BRCA2;Rb1; pRb; DBM
Introduction
Aberrations of the long arm of chromosome 13 occurfrequently in many types of tumors (Cletonjansen etal., 1995; Cooney et al., 1996; Li et al., 1994; Maestroet al., 1996; Pearce et al., 1996; Radford et al., 1995;Yang et al., 1992; Zhang et al., 1994). So far twotumor suppressor genes (TSG) have been identi®ed onthis chromosome: the retinoblastoma gene (Rb1) on13q14.2 and the breast cancer susceptibility locus 2(BRCA2) on 13q12.1 (Lee et al., 1987; MacGee et al.,1989; Wooster et al., 1995). Rb1 is a cell cycle regulatorwhich is involved in both hereditary and sporadicretinoblastoma, while BRCA2 mutations have beendescribed in a subset of families with site speci®c breastcancer only (Oddoux et al., 1996; Phelan et al., 1996;Thorlacius et al., 1996; Wooster et al., 1995; Yandell etal., 1989).
In prostate cancer, somatic deletions of chromosome13q have been frequently observed both by compara-tive genomic hybridization (CGH) and loss ofheterozygosity (LOH) analysis (Cher et al., 1996;Cooney et al., 1996; Visakorpi et al., 1995). The Rb1locus is frequently deleted, and in one prostate cancercell line a 103 bp deletion of the promoter region wasobserved with concomitant loss of the wild type allele,suggesting inactivation of this TSG by a two-hitmechanism (Bookstein et al., 1990; Riley et al., 1994).Furthermore, immunostaining studies have revealedthat the Rb1 expression is frequently decreased orabsent in a signi®cant proportion of sporadic prostatecancers (Phillips et al., 1994). However, screening forsomatic mutations in Rb1 have given negative resultsalso in the studies where larger series of tumors havebeen examined (Kubota et al., 1995; Sarkar et al.,1992; Tricoli et al., 1996). On the other hand, in onestudy on families with hereditary breast cancerattributable to BRCA2, the male carriers have a 3.3-fold increased risk of developing prostate cancer ascompared to non carriers (Wooster et al., 1995). Thus,the importance of the BRCA2 gene in sporadicprostate cancer can also be anticipated.Two approaches can be used to identify a candidate
gene to be a TSG in sporadic cancers. One is toidentify the somatic mutation coupled with loss of thenormal chromosome homologue and the other is toobserve the gene deletion in combination with absentor aberrant gene expression. These two approacheswere used in the present study to investigate theinvolvement of BRCA2 and Rb1 in sporadic prostatecancer. The results indicate that deletion of chromo-some 13q is an early non-random event, and thatneither BRCA2 nor Rb1 was the targets but insteadother putative TSGs might be located nearby.
Results
Overview of 13q losses in prostate cancer
The losses observed (Figure 1) either resulted incomplete absence of one of the constitutional allelesin the tumor (pure LOH) or a signi®cantly decreasedintensity, but not complete loss of one constitutionalallele (allelic imbalance). The decrease of the intensitywas not always related directly to the tumor cellcontent of the tissue sample from which the tumorDNA was derived. In both types of LOH, the retainedallele had usually stronger signal intensity as comparedto the corresponding allele in the normal DNA (Figure1).
Correspondence: C LiReceived 23 June 1997; revised 8 September 1997; accepted 9September 1997
Oncogene (1998) 16, 481 ± 487 1998 Stockton Press All rights reserved 0950 ± 9232/98 $12.00
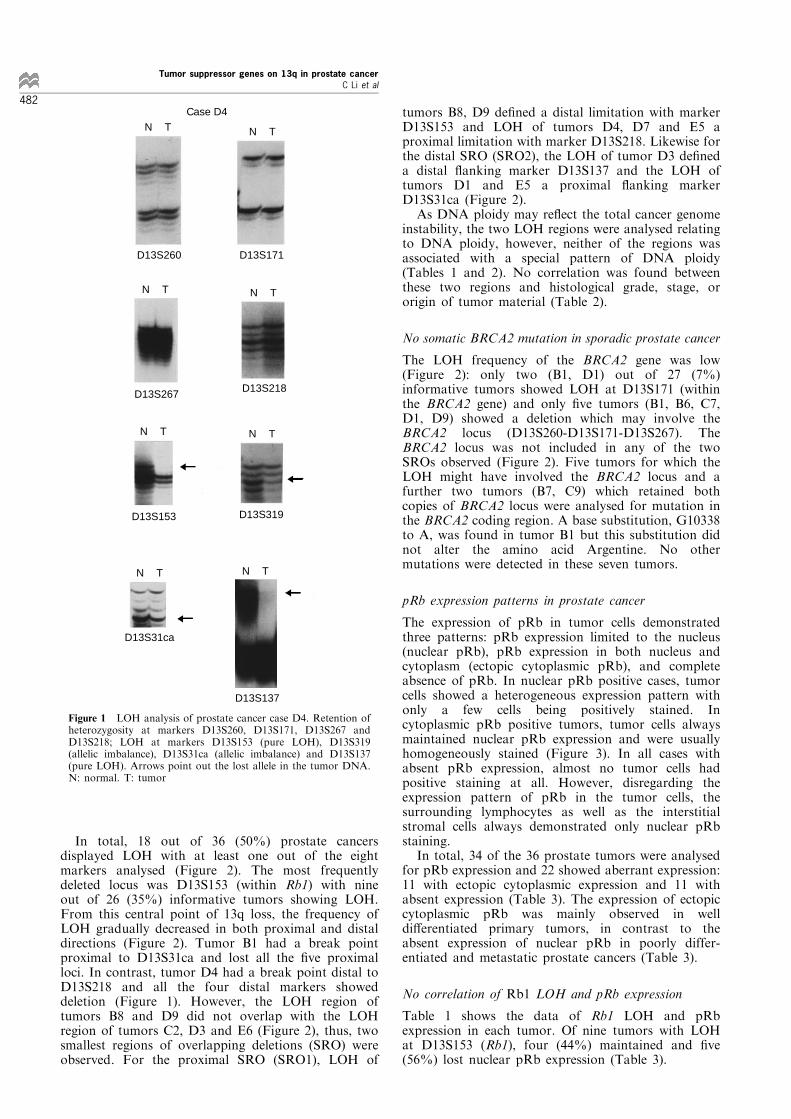
In total, 18 out of 36 (50%) prostate cancersdisplayed LOH with at least one out of the eightmarkers analysed (Figure 2). The most frequentlydeleted locus was D13S153 (within Rb1) with nineout of 26 (35%) informative tumors showing LOH.From this central point of 13q loss, the frequency ofLOH gradually decreased in both proximal and distaldirections (Figure 2). Tumor B1 had a break pointproximal to D13S31ca and lost all the ®ve proximalloci. In contrast, tumor D4 had a break point distal toD13S218 and all the four distal markers showeddeletion (Figure 1). However, the LOH region oftumors B8 and D9 did not overlap with the LOHregion of tumors C2, D3 and E6 (Figure 2), thus, twosmallest regions of overlapping deletions (SRO) wereobserved. For the proximal SRO (SRO1), LOH of
tumors B8, D9 de®ned a distal limitation with markerD13S153 and LOH of tumors D4, D7 and E5 aproximal limitation with marker D13S218. Likewise forthe distal SRO (SRO2), the LOH of tumor D3 de®neda distal ¯anking marker D13S137 and the LOH oftumors D1 and E5 a proximal ¯anking markerD13S31ca (Figure 2).As DNA ploidy may re¯ect the total cancer genome
instability, the two LOH regions were analysed relatingto DNA ploidy, however, neither of the regions wasassociated with a special pattern of DNA ploidy(Tables 1 and 2). No correlation was found betweenthese two regions and histological grade, stage, ororigin of tumor material (Table 2).
No somatic BRCA2 mutation in sporadic prostate cancer
The LOH frequency of the BRCA2 gene was low(Figure 2): only two (B1, D1) out of 27 (7%)informative tumors showed LOH at D13S171 (withinthe BRCA2 gene) and only ®ve tumors (B1, B6, C7,D1, D9) showed a deletion which may involve theBRCA2 locus (D13S260-D13S171-D13S267). TheBRCA2 locus was not included in any of the twoSROs observed (Figure 2). Five tumors for which theLOH might have involved the BRCA2 locus and afurther two tumors (B7, C9) which retained bothcopies of BRCA2 locus were analysed for mutation inthe BRCA2 coding region. A base substitution, G10338to A, was found in tumor B1 but this substitution didnot alter the amino acid Argentine. No othermutations were detected in these seven tumors.
pRb expression patterns in prostate cancer
The expression of pRb in tumor cells demonstratedthree patterns: pRb expression limited to the nucleus(nuclear pRb), pRb expression in both nucleus andcytoplasm (ectopic cytoplasmic pRb), and completeabsence of pRb. In nuclear pRb positive cases, tumorcells showed a heterogeneous expression pattern withonly a few cells being positively stained. Incytoplasmic pRb positive tumors, tumor cells alwaysmaintained nuclear pRb expression and were usuallyhomogeneously stained (Figure 3). In all cases withabsent pRb expression, almost no tumor cells hadpositive staining at all. However, disregarding theexpression pattern of pRb in the tumor cells, thesurrounding lymphocytes as well as the interstitialstromal cells always demonstrated only nuclear pRbstaining.In total, 34 of the 36 prostate tumors were analysed
for pRb expression and 22 showed aberrant expression:11 with ectopic cytoplasmic expression and 11 withabsent expression (Table 3). The expression of ectopiccytoplasmic pRb was mainly observed in welldi�erentiated primary tumors, in contrast to theabsent expression of nuclear pRb in poorly differ-entiated and metastatic prostate cancers (Table 3).
No correlation of Rb1 LOH and pRb expression
Table 1 shows the data of Rb1 LOH and pRbexpression in each tumor. Of nine tumors with LOHat D13S153 (Rb1), four (44%) maintained and ®ve(56%) lost nuclear pRb expression (Table 3).
N T N T
N T N T
D13S260 D13S171
D13S267 D13S218
N TN T
D13S153 D13S319
N T N T
D13S31ca
D13S137
Case D4
Figure 1 LOH analysis of prostate cancer case D4. Retention ofheterozygosity at markers D13S260, D13S171, D13S267 andD13S218; LOH at markers D13S153 (pure LOH), D13S319(allelic imbalance), D13S31ca (allelic imbalance) and D13S137(pure LOH). Arrows point out the lost allele in the tumor DNA.N: normal. T: tumor
Tumor suppressor genes on 13q in prostate cancerC Li et al
482
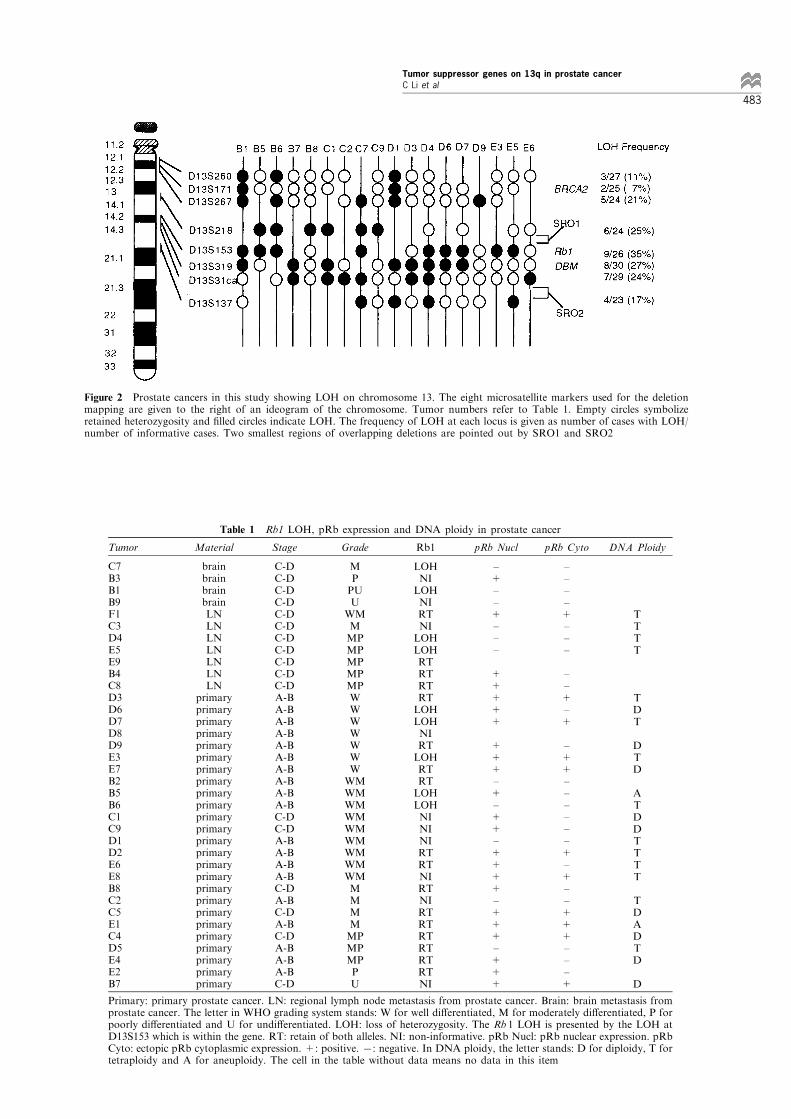
Table 1 Rb1 LOH, pRb expression and DNA ploidy in prostate cancer
Tumor Material Stage Grade Rb1 pRb Nucl pRb Cyto DNA Ploidy
C7B3B1B9F1C3D4E5E9B4C8D3D6D7D8D9E3E7B2B5B6C1C9D1D2E6E8B8C2C5E1C4D5E4E2B7
brainbrainbrainbrainLNLNLNLNLNLNLN
primaryprimaryprimaryprimaryprimaryprimaryprimaryprimaryprimaryprimaryprimaryprimaryprimaryprimaryprimaryprimaryprimaryprimaryprimaryprimaryprimaryprimaryprimaryprimaryprimary
C-DC-DC-DC-DC-DC-DC-DC-DC-DC-DC-DA-BA-BA-BA-BA-BA-BA-BA-BA-BA-BC-DC-DA-BA-BA-BA-BC-DA-BC-DA-BC-DA-BA-BA-BC-D
MPPUU
WMMMPMPMPMPMPWWWWWWWWMWMWMWMWMWMWMWMWMMMMMMPMPMPPU
LOHNILOHNIRTNILOHLOHRTRTRTRTLOHLOHNIRTLOHRTRTLOHLOHNININIRTRTNIRTNIRTRTRTRTRTRTNI
±+±±+±±±
+++++
+++±+±++±++++±+++±+++
±±±±+±±±
±±+±+
±++±±±±±±+±+±±+++±±±+
TTTT
TDT
DTD
ATDDTTTT
TDADTD
D
Primary: primary prostate cancer. LN: regional lymph node metastasis from prostate cancer. Brain: brain metastasis fromprostate cancer. The letter in WHO grading system stands: W for well di�erentiated, M for moderately di�erentiated, P forpoorly di�erentiated and U for undi�erentiated. LOH: loss of heterozygosity. The Rb 1 LOH is presented by the LOH atD13S153 which is within the gene. RT: retain of both alleles. NI: non-informative. pRb Nucl: pRb nuclear expression. pRbCyto: ectopic pRb cytoplasmic expression. +: positive. 7: negative. In DNA ploidy, the letter stands: D for diploidy, T fortetraploidy and A for aneuploidy. The cell in the table without data means no data in this item
Figure 2 Prostate cancers in this study showing LOH on chromosome 13. The eight microsatellite markers used for the deletionmapping are given to the right of an ideogram of the chromosome. Tumor numbers refer to Table 1. Empty circles symbolizeretained heterozygosity and ®lled circles indicate LOH. The frequency of LOH at each locus is given as number of cases with LOH/number of informative cases. Two smallest regions of overlapping deletions are pointed out by SRO1 and SRO2
Tumor suppressor genes on 13q in prostate cancerC Li et al
483
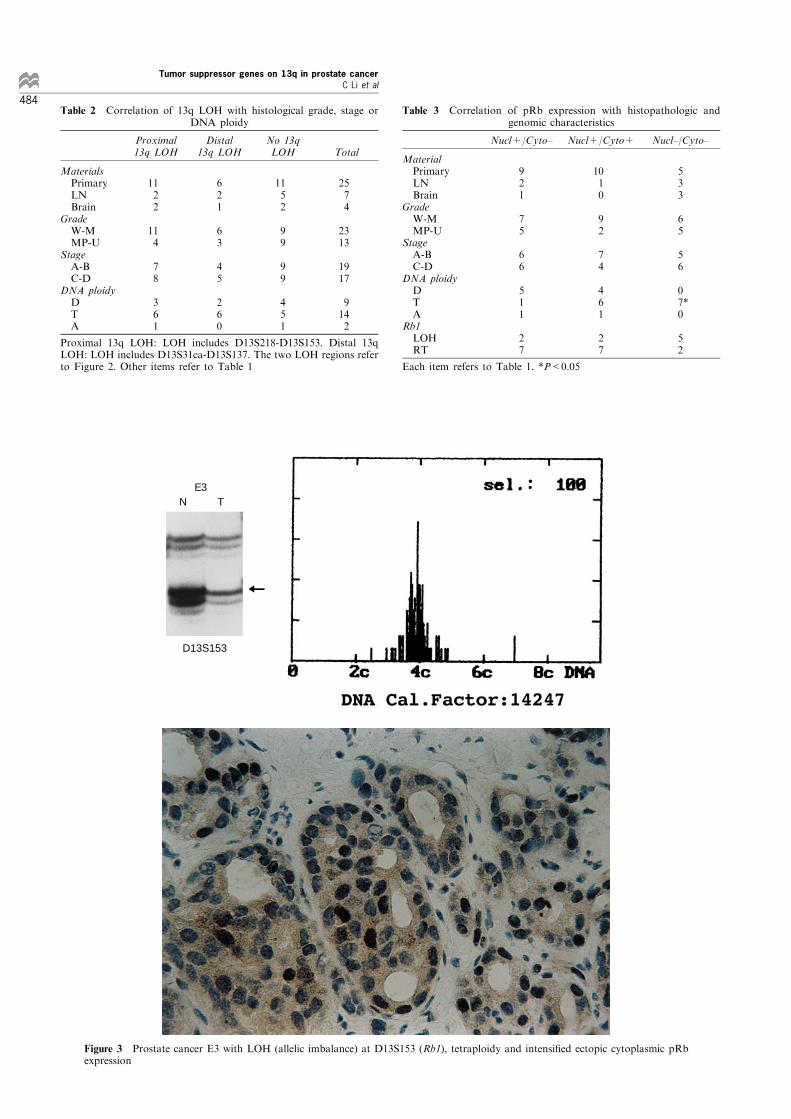
Table 2 Correlation of 13q LOH with histological grade, stage orDNA ploidy
Proximal13q LOH
Distal13q LOH
No 13qLOH Total
MaterialsPrimaryLNBrain
1122
621
1152
2574
GradeW-MMP-U
114
63
99
2313
StageA-BC-D
78
45
99
1917
DNA ploidyDTA
361
260
451
9142
Proximal 13q LOH: LOH includes D13S218-D13S153. Distal 13qLOH: LOH includes D13S31ca-D13S137. The two LOH regions referto Figure 2. Other items refer to Table 1
Table 3 Correlation of pRb expression with histopathologic andgenomic characteristics
Nucl+/Cyto± Nucl+/Cyto+ Nucl±/Cyto±
MaterialPrimaryLNBrain
921
1010
533
GradeW-MMP-U
75
92
65
StageA-BC-D
66
74
56
DNA ploidyDTA
511
461
07*0
Rb1LOHRT
27
27
52
Each item refers to Table 1. *P<0.05
N TE3
D13S153
Figure 3 Prostate cancer E3 with LOH (allelic imbalance) at D13S153 (Rb1), tetraploidy and intensi®ed ectopic cytoplasmic pRbexpression
Tumor suppressor genes on 13q in prostate cancerC Li et al
484

pRb expression in tetraploid tumors
DNA ploidy was analysed in 25 of the 36 tumors.Fourteen (56%) were tetraploid. Most of the tetraploidcases were primary tumors with well to moderatedi�erentiation (Table 4). Interestingly, tetraploidy wascorrelated to pRb expression. Tetraploidy was seen in14% of tumors with normal nuclear pRb expression,55% of tumors with ectopic cytoplasmic pRbexpression, and 100% of tumors with absent pRbexpression (Table 3, P50.05 in Pearson's test).
Discussion
LOH presents the imbalance ratio between thenumbers of the homologous chromosomes. In spite ofindicating the secondary event to speci®c TSGmutation, LOH may also re¯ect the general instabilityof the cancer cell's genome. This type of LOH occursrandomly and is related to parameters which roughlyre¯ect the whole genomic alteration, such as DNAploidy. We have found 50% of prostate tumorsshowing LOH at one or more 13q markers and nocorrelation between 13q LOH and DNA ploidy,indicating 13q deletion to be an early, non-random,and independent chromosomal alteration.The frequently deleted region on 13q in these 36
prostate cancers was in agreement with the lost regiondetected by previous CGH analyses (Cher et al., 1994;Visakorpi et al., 1995). However, in the LOH mapping(Figure 2), two independent smallest regions ofoverlapping deletions were further de®ned. The distalregion was ¯anked by marker D13S31ca and D13S137(Figure 2) which overlaps the region described in aprevious LOH analysis (Cooney et al., 1996). Theproximal region ¯anked by markers D13S218 andD13S153 (Figure 2) has not been described before(Cooney et al., 1996). These two regions may suggestthe existence of more than one TSG.The BRCA2 gene has been suggested to be a
candidate but it was not located within these minimalregions (Figure 2). Lack of mutation in the codingregion in tumors with LOH at the BRCA2 locusfurther exclude the BRCA2 gene as being involved insporadic prostate cancer.The Rb1 gene spans approximately 200 kb DNA of
13q14 (Lee et al., 1987; McGee et al., 1989). MarkerD13S153 which is located within the gene had thehighest frequency of LOH (Figure 2). However, thismarker seems not within the two SROs observed
(Figure 1). We also observed a lack of concordancebetween LOH with D13S153 (Rb1) and aberrant pRbexpression (Tables 1 and 3) as reported in a previousstudy (Cooney et al., 1996). In particular, tumors withRb1 LOH presented also normal nuclear pRbexpression (case B5, D6, D7 and E3), indicating theexistence of an intact copy of the Rb1 gene (Table 1)and contradicting the two-hit inactivation mechanism(Tables 1 and 3). This may further disqualify the Rb1gene to be the target of 13q deletions.Other candidate genes in the proximal critical region
include the serotonin receptor subtype 2 (HTR2) gene andthe gene for an orphan G-protein coupled receptor (U16)which may need to be investigated. Both genes encode G-protein coupled receptors which have wide biologicalactivities (Hsieh et al., 1990; Herzog et al., 1996). HTR2is mapped centromeric to Rb1 on 13q14.2, while U16 islocated in the intron 17 or Rb1 (Hsieh et al., 1990;Herzog et al., 1996; Schuler et al., 1996). In the distalcritical region, one TSG candidate may be theDBM genewhich has been found to be homozygously deleted in Bcell malignancy (Liu et al., 1995). However, the exactphysical distance of the marker D13S319 from the DBMgene is unknown (Figure 2).A previous study has shown that mouse embryonic
®broblast (MEF) cell line with null pRb function whenexposed to mitotic spindle inhibitors has a strongtendency of polyploidization via whole genome duplica-tion (Leonardo et al., 1997). Our results further showedan association between pRb aberration and tetraploidyin a subset of tumor materials from prostate cancerpatients (Table 3). The clinical importance of thisobservation needs further investigation.In summary, this study has shown the importance of
13q deletion in prostate cancer. The alteration ofBRCA2 gene is rare and does not play an importantrole in this tumor. Aberrant expression of pRb maynot re¯ect a two-hit inactivation of the Rb1 gene butmay cause the tetraploidization of a subset of prostatecancers. The result suggests the existence andimportance of other yet unidenti®ed genes onchromosome 13q in prostate cancer.
Materials and methods
Prostate cancer patients
Diagnoses of prostate cancer in 36 patients were con®rmedby histopathological examination of the surgical specimen.The mean age at diagnosis was 63 years (ranges 52 ± 72).The disease extent (Table 1) was determined according toWhitemore and Jewitt staging system (Jewitt, 1975;Whitemore, 1956). None of the 36 patients was subjectedto any hormonal or radiation therapies prior to surgery.
Prostate cancer tissue
Immediately after the specimen was removed, a horizontalsection across the tumor nodular lesion was made. Tumortissue was sampled inside the nodule to avoid contamina-tion of surrounding normal tissue. One piece of the samplewas para�n embedded and used for pRb immunostaining,and DNA ploidy measurement. The other piece was snapfrozen in 7708C and used for DNA extraction. Therepresentativity of the tissue was estimated by histopatho-logic examination and only samples which contained 50%or more tumor cells were included in the study.
Table 4 Correlation between DNA ploidy pattern and pathologicalfeatures
Diploidy Tetraploidy Aneuploidy
MaterialPrimaryLNBrain
900
1040
200
GradeW-MMP-U
63
113
20
StageA-BC-D
45
104
20
Each item refers to Table 1
Tumor suppressor genes on 13q in prostate cancerC Li et al
485

Of 36 tumor samples, 25 were from primary tumors,seven were from lymph node metastases, and four were frombrain metastates. The di�erentiation of prostate cancer(Table 1) was estimated according to the World HealthOrganization's prostate cancer grading system (Mosto® etal., 1980).
DNA isolation
High molecular weight DNA was isolated from matchedpairs of peripheral leukocytes and tumor samples usingstandard methods.
LOH analysis
Eight markers on 13q were used: D13S260-D13S171 (with-in BRCA2)-D13S267-D13S218-D13S153 (within Rb1)-D13S319-D13S31ca-D13S137 (Figure 1). The order of themarkers is according to the Human Genome Data Base(GDB). D13S319 is located within the homozygouslydeleted DBM (disrupted in B cell malignancy) region.D13S31ca was synthesized by the Nordic Human GenomeMapping Program at the Uppsala University Hospital.Other markers were supplied by Research Genetic Inc.(Huntsville, AL). The sequences and location of all themarkers used can be found in the GDB.
The technical aspects of the PCR and microsatelliteanalysis were described previously (Liu et al., 1995). LOHwas de®ned as a complete absence of or signi®cantlydecreased signal intensity of one of the constitutional allelesin the tumor DNA and can be unambiguously detected bythe naked eye (Figure 2). The result was reviewed by threeinvestigators (C. Li, CL and USRB).
The BRCA2 gene mutation analysis
The details for the single strand conformational analysis(SSCA) and direct DNA sequencing of the BRCA2 genehave been published before (Phelan et al., 1996). In short,26 coding exons of the gene were ampli®ed using 55di�erent fragments of 200 ± 300 bp each, and the PCRproducts were run on MDE gels. When a SSCA shift wasseen, the shifted band was excised from the gel, puri®edand sequenced using a 377 Automated FluorescentSequencer (Applied BioSystems, CA).
pRb immunostaining
The immunostaining was performed according to astandard avidin-biotin complex (ABC) procedure on3 mm thick slide of para�n embedded tissue selection.The pRb monoclonal antibody, PMG3-245 (PharMin-gen), speci®cally recognizes the pRb epitope of aminoacid residue 300 ± 380 (DeCaprio et al., 1988). Thedilution of the primer was 1 : 200. One normal braintissue and 10 tissues of histologically con®rmed benignprostatic hyperplasia were used as controls in eachexperiment. The corresponding slide of hematoxylin and
eosin staining was used to distinguish the tumor cellsfrom surrounding stroma as well as in®ltratinglymphocytes.
If the cell nucleus alone or together with cytoplasm waspositively stained it was regarded as a pRb positive cell. Atumor with pRb negative staining was de®ned as in a ®eldunder a 10610 times magni®cation less than 1% of over alltumor cells was pRb positive. Three investigators (C Li, CLand USRB) independently evaluated all the immunostainingslides and the ®nal judgement was reviewed by thepathologist (GA).
DNA ploidy analysis
The detailed procedure was described before (Steinbecket al., 1994). Brie¯y, paralleled with pRb immunostain-ing, a 4 mm thick section was prepared for Feulgenstaining. The DNA content of individual tumor cell wasmeasured in a TV-based microscopic image analysissystem. In each slide, at least 100 tumor cell nuclei werecounted (Figure 3). The DNA content pro®le wasrecorded and compared with an internal diploidstandard obtained from the measurement of at least 20normal cells.
Statistical analysis
The 36 tumors were divided into three subgroups accordingto the origin of the tumor material analysed: primarytumors, lymph node metastases and brain metastases(Tables 1 ± 4). To facilitate the statistical analysis, theWHO tumor grading system was integrated into twogroups (Tables 2 ± 4): well to moderately di�erentiated(W-M) and moderately poor to undi�erentiated (MP-U).For the same reason, the prostate cancer stage wasintegrated into two groups (Tables 1 ± 4): stage A-B andstage C-D. Fisher's and Pearson's tests were used toanalyse these categories each in association with 13q LOH,DNA ploidy pattern and pRb expression pattern. The sametests were also used to analyse the association between pRbexpression pattern, the Rb1 LOH, and DNA ploidypattern.
AcknowledgementsDr Lena SpaÊ nberg at the Department of Clinic Genetics ofUppsala University Hospital is acknowledged for kind andfree supplement of microsatellite markers. Thanks shouldalso go to colleagues at the Department of Urology inKarolinska Hospital for their collaboration in collectingthe special prostate cancer materials. This study wassupported in part by research grants from the SwedishCancer Foundation, Swedish Cancer Society and NCI/Duke University SPORE in breast cancer, P50-CA68438.
References
Bookstein R, Rio P, Madreperla SA, Hong F, Allred C,Grizzle WE and Lee W-H. (1990). Proc. Natl. Acad. Sci.USA, 87, 7762 ± 7766.
Cher ML, Bova GS, Moore DH, Small EJ, Carroll PR, PinSS, Epstein JI, Isaacs WB and Jensen RH. (1996). CancerRes., 56, 3091 ± 3102.
Cher ML, MacGrogan D, Bookstein R, Brown JA, JenkinsRB and Jensen RH. (1994). Genes Chrom. Cancer, 11,153 ± 162.
Cletonjansen AM, Collins N, Lakhani SR, Weissenbach J,Devilee P, Cornelisse CJ and Stratton MR. (1995). Br. J.Cancer, 72, 1241 ± 1244.
Cooney KA, Wetzel JC, Merajver SD, Macoska JA,Singleton TP and Wojno KJ. (1996). Cancer Res., 56,1142 ± 1145.
DeCaprio JA, Ludlow JW, Figge J, Shew J-Y, Huang C-M,Lee W-H, Marsilio E, Paucha E and Livingston DM.(1988). Cell, 54, 275 ± 283.
Tumor suppressor genes on 13q in prostate cancerC Li et al
486

Herzog H, Darby K, Hort YJ and Shine J. (1996). GenomeRes., 6, 858 ± 861.
Hsieh CL, Bowcock AM, Farrer LA, Huang KN, Cavalli-Sforza LL, Julius D and Francke U. (1990). 16, 567 ±574.
Jewitt HJ. (1975). Urol. Clin. North. Am., 2, 105.Kubota Y, Fujinami K, Uemura H, Dobashi Y, MiyamotoH, Iwasaki Y, Kitamura H and Shuin T. (1995). Prostate,27, 314 ± 320.
Leonardo AD, Khan SH, Linke SP, Creco V, Seidita G andGMW. (1997). Cancer Res., 57, 1013 ± 1019.
Lee WH, Bookstein R, Hong F, Young LJ, Shew JY and LeeEY. (1987). Science, 235, 1394 ± 1399.
Li X, Lee NK, Ye YW, Waber PG, Schweitzer C, Cheng QCand Nisen PD. (1994). J. Natl. Cancer Inst., 86, 1524 ±1529.
Liu Y, Hermanson M, Grander D, Merup M, Wu XS,Heyman M, Rasool O, Juliusson G, Gahrton G,Detlofsson R, Nikiforova N, Buys C, Soderhall S,Yankovsky N, Zabarovsky E and Einhorn S. (1995).Blood, 86, 1911 ± 1915.
Maestro R, Piccinin S, Doglioni C, Gasparotto D,Vukosavljevic T, Sulfaro S, Barzan L and Boiocchi M.(1996). Cancer Res., 56, 1146 ± 1150.
McGee TL, Yandell DW and Dryja TP. (1989). Gene, 80,119 ± 128.
Mosto® FK, Sesterhenn I and Sobin LH. (1980). Interna-tional Histological Classi®cation Of Tumors. World HealthOrganization. No. 10.
Oddoux C, Struewing JP, Clayton CM, Neuhausen S, BrodyLC, Kaback M, Haas B, Norton L, Borgen P, Jhanwar S,Goldgar D, Ostrer H and O�t K. (1996). Nature Genet.,14, 188 ± 190.
Pearce SH, Trump D, Wooding C, Sheppard MN, ClaytonRN and Thakker RV. (1996). Clin. Endocrinol., 45, 195 ±200.
Phelan CM, Lancaster J, Tonin P, Gumbs C, Carter R,Ghadirian P, Peter C, Moslehi R, Dion F, Faucher M-C,Dole K, Karimi S, Lounis H, Warner E, Goss P, AndersonD, Larsson C, Narod SA and Futreal PA. (1996). NatureGenet., 13, 120 ± 122.
Phillips SM, Barton CM, Lee SJ, Morton DG, Wallace DM,Lemoine NR and Neoptolemos JP. (1994). Br. J. Cancer,70, 1252 ± 1257.
Radford DM, Fair KL, Phillips NJ, Ritter JH, SteinbrueckT, Holt MS and Doniskeller H. (1995). Cancer Res., 55,3399 ± 3405.
Riley DJ, Lee EY-HP and Lee W-H. (1994). Annu. Rev. CellBiol., 10, 1 ± 29.
Sarkar FH, Sakr W, Li YW, Macoska J, Ball DE andCrissman JD. (1992). Prostate, 21, 145 ± 152.
Schuler CD, Boguski MS, Stewart EA, Stein LD, Gyapay G,Rice K, White RE, Rodriguez-Tome P, Aggarwal A,Bajorek E, Bentolila S, Birren BB, Butler A, Castle AB,Chiannikulchai N, Chu A, Clee C, Cowles S, Day PJR,Dibling T, Drouot N, Dunham I, Duprat S, East C,Edwards C, Fan J-B, Fang N, Fizames C, Garrett C,Green L, Hadley D, Harris M, Harrison P, Brady S, HicksA, Holloway E, Hui L, Hussain S, Louis-Dit-Sully C, MaJ, MacGilvery A, Mader C, Maratukulam A, Matise TC,McKusick KB, Morissette J, Mugall A, Muselet D,Nusbaum HC, Page DC, Peck A, Perkins S, Piercy M,Qin F, Quackenbush J, Ranby S, Reif T, Rozen S, SandersC, She X, Silva J, Slonim DK, Soderlund C, Sun W-L,Tabar P, Thangarajah T, Vega-Czarny N, Vollrath D,Voyticky S, Wilmer T, Wu X, Adams MD, Ao�ray C,Walter NAR, Brandon R, Dhejia A, Goodfellow PN,Houlgatte R, Hudson Jr JR, Ide SE, Iorio KR, Lee WY,Seki N, Nagase T, Ishikawa K, Nomura N, Phillips C,Polymeropoulos MH, Sandusky M, Schmitt K, Berry R,Swanson K, Torres R, Venter JC, Sikela JM, BeckmannJS, Weissenbach J, Myers RM, Cox DR, James MR,Bentley D, Deloukas P, Lander ES and Hudson TJ.(1996). Science, 274, 540 ± 546.
Steinbeck RG, Heselmeyer KM and Auer GU. (1994). Anal.Quant. Cyto., 16, 196 ± 202.
Thorlacius S, Olafsdottir G, Tryggvadottir L, Neuhausen S,Jonasson JG, Tavtigian SV, Tulinius H, OgmundsdottirHM and Eyfjord JE. (1996). Nature Genet., 13, 117 ± 119.
Tricoli JV, Gumerlock PH, Yao JL, Chi SG, D'Souza SA,Nestok BR and de Vere WR. (1996). Genes Chrom.Cancer, 15, 108 ± 114.
Visakorpi T, Kallioniemi AH, Syvanen AC, Hyytinen ER,Karhu R, Tammela T, Isola JJ and Kallioniemi OP.(1995). Cancer Res., 55, 342 ± 347.
Whitemore WFJ. (1956). Am. J. Med., 21, 697.Wooster R, Bignell G, Lancaster J, Swift S, Seal SJM,Collins N, Gregory S, Gumbs C, Micklem G, Barfoot R,Hamoudi R, Patel SRC, Biggs P, Hashim Y, Smith A,Connor F, Arason AJG, Ficenec D, Kelsell D, Ford D,Tonin P, Bishop DT, Spurr NK, Ponder BAJ, Eels R, PetoJ, Devilee P, Cornelisse C, Lynch H, Narod S, Lenoir G,Egilsson V, Barkadottir RS, Easton DF, Bentley DR,Futreal PA, Ashworth A and Stratton MR. (1995).Nature, 378, 789 ± 792.
Yandell DW, Campbell TA, Dayton SH, Petersen R, WaltonD, Little JB, McConikie-Rosell A, Buckley EG and DryjaTP. (1989). N. Engl. J. Med., 321, 1689 ± 1695.
Yang FT, Li S, Han H and Schwartz PE. (1992). Int. J.Cancer, 52, 575 ± 580.
Zhang X, Xu HJ, Murakami Y, Sachse R, Yashima K,Hirohashi S, Hu SX, Benedict WF and Sekiya T. (1994).Cancer Res., 54, 4177 ± 4182.
Tumor suppressor genes on 13q in prostate cancerC Li et al
487
Copyright © 2022 FDOKUMEN




















