
Broken Bracelets, Molien Series, Paraffin Wax, and an Elliptic Curve of Conductor 48
Upload
independentCategory
view
0download
0
ORIGINAL PAPERJournal of PathologyJ Pathol 2010; 221: 452–461Published online 7 June 2010 in Wiley InterScience(www.interscience.wiley.com) DOI: 10.1002/path.2728
Gene expression profiling of formalin-fixed, paraffin-embeddedfamilial breast tumours using the whole genome-DASL assayNic Waddell,1,2 Sibylle Cocciardi,1 Julie Johnson,1 Sue Healey,1 Anna Marsh,1 Joan Riley,3 Leonard da Silva,4 AnaCristina Vargas,4 Lynne Reid,4 kConFab Investigators,5 Peter T Simpson,4 Sunil R Lakhani4and Georgia Chenevix-Trench1*
1 The Queensland Institute of Medical Research, Brisbane, Australia2 Queensland Centre for Medical Genomics, Institute for Molecular Bioscience, Brisbane, Australia3 MRC Toxicology Unit, Leicester, UK4 Molecular and Cellular Pathology, University of Queensland Centre for Clinical Research, School of Medicine, Brisbane, Australia5 Peter MacCallum Cancer Centre, Melbourne, Australia
*Correspondence to: Georgia Chenevix-Trench, The Queensland Institute of Medical Research, Brisbane, Australiae-mail: [email protected]
AbstractTissue sample acquisition is a limiting step in many studies. There are many thousands of formalin-fixed,paraffin-embedded archival blocks collected around the world, but in contrast relatively few fresh frozen samplesin tumour banks. Once samples are fixed in formalin, the RNA is degraded and traditional methods for geneexpression profiling are not suitable. In this study, we have evaluated the ability of the whole genome DASL(cDNA-mediated Annealing, Selection, extension, and Ligation) assay from Illumina to perform transcriptomicanalysis of archived breast tumour tissue in formalin-fixed, paraffin-embedded (FFPE) blocks. We profiled 76familial breast tumours from cases carrying a BRCA1, BRCA2 or ATM mutation, or from non-BRCA1/2 families.We found that replicate samples correlated well with each other (r2 = 0.9–0.98). In 12/15 cases, the matchedformalin-fixed and frozen samples predicted the same tumour molecular subtypes with confidence. These resultsdemonstrate that the whole genome DASL assay is a valuable tool to profile degraded RNA from archival FFPEmaterial. This assay will enable transcriptomic analysis of a large number of archival samples that are stored inpathology archives around the globe and consequently will have the potential to improve our understanding andcharacterization of many diseases.Copyright 2010 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.
Keywords: BRCA1 and BRCA2; ATM; molecular subtypes; familial breast cancer; gene expression; formalin-fixed; paraffin-embeddedtissue; whole genome DASL
Received 8 February 2010; Revised 29 March 2010; Accepted 27 April 2010
No conflicts of interest were declared.
Introduction
Patient sample acquisition can be difficult and is alimiting step in many studies. Fresh frozen biospeci-mens yielding high-quality RNA for expression profil-ing are usually of a limited number and size, whereastissues archived in paraffin blocks are much more com-mon, but during the process of fixing tissues the RNAbecomes highly degraded. Protocols such as the cDNA-mediated Annealing, Selection, extension, and Ligation(DASL) assay from Illumina have been developedfor gene expression profiling of this degraded RNAfrom formalin-fixed, paraffin-embedded blocks (FFPE)[1–3]. The original DASL assay, targeting 502 cancer-related genes, was limited to 1536 probes that couldbe assayed at one time. Unique custom panels, lim-ited to a minimum of 512 and a maximum of 1536genes, can be designed, but the cost is high if only
a few samples are to be studied and the design pro-cess can be problematic. In an ambitious study, anapproximately 6000-gene custom panel consisting offour sets of 1536 probes (one probe per gene) wasused to profile breast tumour samples [4]. However,other customized studies of this scale have not beenperformed.
Huge numbers of FFPE blocks are potentiallyavailable around the world, representing an amazingresource for clinical research. However, these speci-mens are of varying age and have been subjected todifferent fixing protocols and processed by differentlaboratories, so the quality of the RNA may vary. TheRNA extracted from FFPE archival blocks is degraded,and tools such as the Agilent Bioanalyser that visual-ize the RNA and determine the RNA integrity score(RIN) may not predict performance in the DASL assay.The concentration and average size of the RNA prod-uct have been used to predict DASL performance [5].
Copyright 2010 Pathological Society of Great Britain and Ireland. J Pathol 2010; 221: 452–461Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com

Whole genome-DASL analysis of familial tumours 453
Alternatively, performance in the DASL assays can bepredicted with a real-time PCR assay to evaluate theexpression of RPL13A, because an inverse correlationbetween RPL13A cycle threshold (Ct) values from real-time PCR and DASL performance has been reported[1,5–10].
Recently, a whole genome-DASL assay was devel-oped which can assay approximately 24 000 probes persample. There are few published studies to date thathave used the whole genome-DASL assay. A studyby Illumina Inc demonstrated the assay reproducibil-ity [11]. A second analysed a total of 12 samples toidentify pathways altered in ovarian cancer; however,this study did not focus on the technical aspects of theDASL assay and included only two replicates, with nocomparison to fresh frozen samples or evaluation ofRNA quality assessment [12].
Molecular subtypes of ‘sporadic’ (unselected) humanbreast tumours and familial tumours have been iden-tified by gene expression profiling [13]. The fivemajor subtypes (luminal A, luminal B, HER2, basal-like, and normal breast-like) are associated with dif-ferent clinical outcomes [13–19]. Targeted thera-pies, such as tamoxifen, aromatase inhibitors, andtrastuzumab (Herceptin), are now routinely used inclinical practice for specific subtypes of breast cancer,and poly(ADP-ribose) polymerase (PARP) inhibitorsdirected at BRCA1- and BRCA2-deficient tumours arecurrently in clinical trials [20]. Therefore, increasinglycorrect classification of breast tumours will suggestoptimal therapeutic options, as well as improve ourunderstanding of the biology.
We have recently shown from the analysis of freshfrozen tumours that non-BRCA1/2 familial tumourswithout mutations in the breast cancer-associated genesBRCA1 and BRCA2 are heterogeneous, but predom-inantly of the luminal A subtype. In contrast, thosewith mutations in BRCA1 or BRCA2 are predomi-nantly basal-like, or luminal A and B, respectively[21]. These findings suggest that the gene that har-bours a mutation may predispose to specific types ofbreast cancer. If this were the case, it would haveimportant implications for the identification of addi-tional breast cancer susceptibility genes, suggestingthat linkage analysis and other approaches should strat-ify families by molecular subtype. In this study, wehave used the whole genome-DASL assay to anal-yse a large cohort of FFPE familial breast tumours,to determine whether we can identify the molecu-lar subgroups within FFPE tumours and to evaluatethe intra-family distribution of subtypes. Successfulapplication of whole genome-DASL opens up manyopportunities for research including rare tumour types,stratification of families for genetic studies, and retro-spective analysis of clinical trial specimens for study-ing cohorts with long-term clinical follow-up.
Materials and methods
Patient material and RNA extractionWe extracted RNA from 86 individual FFPE blocksand analysed 85 familial FFPE primary breast tumours(15 in duplicate) and a single metastasis which orig-inated from one of the primaries, to give a totalof 101 arrays. Fifteen of the FFPE tumours havematching fresh frozen material which has been anal-ysed previously [21]. Successful whole genome-DASLprofiling was performed on the metastasis and 76primary tumours (BRCA1, n = 18; BRCA2, n = 19;non-BRCA1/2, n = 29; ATM V2424G, n = 6; andunknown mutation type, n = 4). Eight of these RNAsamples were arrayed successfully in duplicate. Allclinical data collected and information regarding theFFPE blocks and RNA hybridizations may be found inthe Supporting information (Supplementary Table 1).Ethics approval was obtained from the Peter MacCal-lum Cancer Centre and QIMR Ethics Committees.
RNA was extracted from FFPE material on the sameday that sections (10 µm) were cut from the paraffinblocks. The mean number of sections extracted wassix, with a range of 2–20 sections; the section num-ber was determined by a pathologist based on the sizeof the tumour. All sections were microdissected witha needle to enrich for tumour cells and only blockswith more than 50% tumour cells after microdissec-tion were extracted. Cells were lysed in 260 µl of lysisbuffer for 1 h at 70 ◦C, proteinase K was added, andcells were incubated for a further 1 h at 55 ◦C. 500 µlof Tri reagent and 200 µl of chloroform were addedto each sample; the samples were mixed vigorouslyand incubated for 3 min at room temperature. Sam-ples were centrifuged, the upper phase was transferredto a fresh tube, and an equal volume of isopropanolwas added to precipitate the RNA overnight at −80 ◦C.RNA was pelleted by centrifugation, washed twice with70% ethanol, dried, redissolved in water, and stored at−80 ◦C. DNA was removed from the RNA by digest-ing samples with DNA-free (Ambion Inc., Austin, TX,USA). Illumina (San Diego, CA, USA) suggest theuse of the High Pure RNA Paraffin kit (Roche, Cas-tle Hill, Australia) for RNA extraction. However, ourlaboratory routinely uses the above Tri method for theisolation of total RNA and miRNA from fresh frozentissue and FFPE; consequently, we opted to use thismethod for this study. During all steps, standard pre-cautions were followed when working with RNA.
Quality control of RNA using real-time PCRand the bioanalyserRNA samples were evaluated using a bioanalyser (Agi-lent, Forest Hill, Australia) with the RNA Nano kitfollowing the manufacturer’s protocols. We used amodified real-time PCR protocol from Illumina on77 FFPE RNA samples and two RNA samples fromfresh frozen tumours in an attempt to quality con-trol RNA and determine whether we could predict
Copyright 2010 Pathological Society of Great Britain and Ireland. J Pathol 2010; 221: 452–461Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com

454 N Waddell et al
which samples would perform well on the DASLassay. Real-time PCR using primers for a 90 bp prod-uct of RPL13A (GTTGGTGTTCATCCGCTTG andGTACGCTGTGAAGGCATCAA) was performed [9].First-strand synthesis from 250 µg of total RNA usingSuperscript III RT (Invitrogen, Mount Waverly, Victo-ria, Australia) was followed by real-time PCR usingthe SYBR qPCR SuperMix-UDG kit (Invitrogen) with2 µl of undiluted cDNA and 2.5 µM forward andreverse primers. Triplicate reactions were performedon a Lightcycler480 (Roche, Basel, Switzerland), with40 cycles of 15 s extension at 72 ◦C. The RNA qual-ity of ten samples was also assessed using a TaqManassay for RPL13A (Assay Id: Hs03043885_g1; AppliedBiosystems, Foster City, CA, USA), using triplicate10 µl reactions containing 2 µl of undiluted cDNA andstandard TaqMan cycling conditions on a StepOnePlusReal-Time PCR Machine (Applied Biosystems).
Whole genome-DASL and data analysisThe Illumina Whole Genome-DASL (cDNA-mediatedAnnealing, Selection, Extension, and Ligation) assay,which quantifies approximately 24 000 transcripts,was performed using 250 ng of RNA following themanufacturer’s instructions (WGDASL Assay Guide11 322 443_RevA.pdf). Degraded RNA from FFPE tis-sue can be used in DASL because it does not dependon an intact poly-A tail for cDNA synthesis. All dataare available on GEO (Accession number GSE21921).
The data were quality controlled using Genomes-tudio (Illumina) and the lumi bioconductor softwarepackages [22]. Failed samples (n = 16/101) wereremoved from analysis, due to a low average signalintensity or a low p95 (<250 and <800, respectively).Data was quantile normalized and probes were fil-tered based on the Illumina detection score (>0.99in at least two of 77 individual samples), whichyielded 22 752/24 526 probes. Probes with discordantexpression patterns between the eight replicate samples(>two-fold) were determined. The probes were initiallyfiltered based on the Illumina detection score (>0.99in at least four of the 16 samples), which yielded18 227/24 526 probes. There were nine probes with ahigh detection score which were discordant between alleight replicate samples and 3556/18 227 probes werediscordant in at least four out of eight replicate sam-ples, which left 14 671 probes.
Genespring 7.2 was used to perform unsupervisedhierarchical clustering, whereby transcripts which hadmore than a two-fold change versus the mean wereclustered. Distance weighted discrimination (DWD)[23] coupled with Single Sample Predictor (SSP) [14]was used to predict molecular breast tumour subtype,whereby 296/306 of the previously described Intrinsicgene list [14] were mapped to the Illumina version3 human-8 bead arrays used in the whole genome(WG)-DASL assay: 283/306 were mapped via RefSeqnumbers; 13/306 were matched by gene name; and10/306 could not be mapped to the Illumina platform.
240 of the 296 intrinsic probes present on the arrayhad a high detection score (>0.99). The five centroidtraining set of 249 breast tumour samples [14] was thenused in SSP to classify the tumours.
Results
Overall success of the WG-DASL assay
Of the FFPE samples selected for RNA extraction,86/92 (93%) yielded sufficient RNA (250 ng) for theDASL assay. Fifteen of these samples were arrayed induplicate (n = 101 in total). The majority of archivalsamples performed well in the WG-DASL assay, withan 85% (85/101) success rate, including eight ofthe duplicate pairs of samples. The criteria that weused to determine whether a sample passed qualitycontrol were an average signal intensity and p95 of>250 and >800, respectively (see the Materials andmethods section). The remaining samples failed dataquality control. For seven of these samples, theirduplicate sample passed quality control, suggesting thatthe RNA quality was acceptable. A summary of thetumours that passed the DASL assay is presented inthe Supporting information (Supplementary Figure 1and Supplementary Table 1).
Evaluation of block age, RNA yield, and blocksource on WG-DASL success
There are several factors that have the potential toalter the RNA integrity from FFPE blocks. It hasbeen suggested that older paraffin blocks have moredegraded RNA and may fail the DASL assay morefrequently [1]. We found that blocks prepared during orafter 2002 (n = 33) passed the DASL quality control,whereas paraffin blocks prepared prior to 2002 weremore likely to fail (Figure 1A). The concentration ofall available RNA (ng/µl) was measured in parallel andalthough not a significant predictor of pass rate in theDASL assay, it also showed a trend (Figure 1B). Themean concentrations for samples that failed and passedthe DASL assay were 95 and 152 ng/µl, respectively,and all samples with a concentration greater than189 ng/µl (n = 21) were successful in the DASL assay.The fixing protocol may also have an effect on theRNA quality. The tumours analysed in this study camefrom 45 different pathology centres from Australia(range 1–13 tumours per pathology laboratory), with18 pathology centres supplying two or more blocks(Supporting information, Supplementary Table 1). Thenine samples that failed the DASL quality controlwere prepared by seven different pathology centres.Three of these tumours came from a single pathologylaboratory that had an overall DASL failure rateof 3/7 (43%). These data suggest that local tissueprocessing methods may affect the success of theDASL assay.
Copyright 2010 Pathological Society of Great Britain and Ireland. J Pathol 2010; 221: 452–461Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com
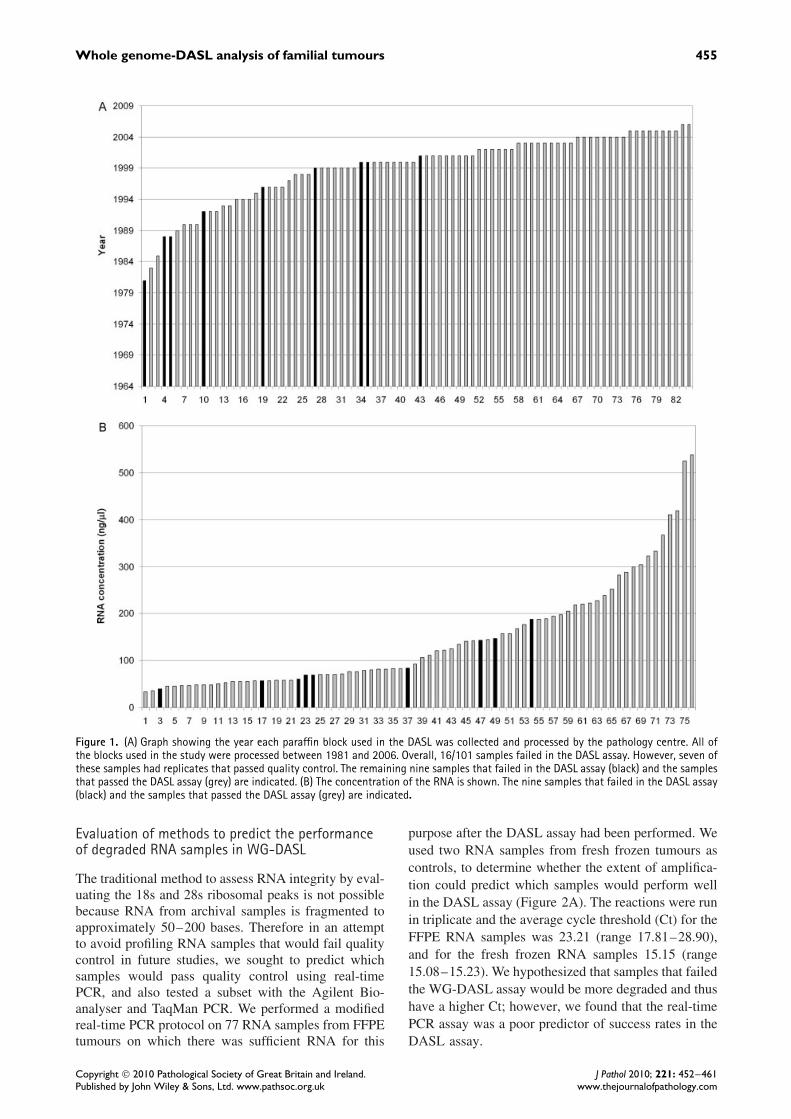
Whole genome-DASL analysis of familial tumours 455
Figure 1. (A) Graph showing the year each paraffin block used in the DASL was collected and processed by the pathology centre. All ofthe blocks used in the study were processed between 1981 and 2006. Overall, 16/101 samples failed in the DASL assay. However, seven ofthese samples had replicates that passed quality control. The remaining nine samples that failed in the DASL assay (black) and the samplesthat passed the DASL assay (grey) are indicated. (B) The concentration of the RNA is shown. The nine samples that failed in the DASL assay(black) and the samples that passed the DASL assay (grey) are indicated.
Evaluation of methods to predict the performanceof degraded RNA samples in WG-DASL
The traditional method to assess RNA integrity by eval-uating the 18s and 28s ribosomal peaks is not possiblebecause RNA from archival samples is fragmented toapproximately 50–200 bases. Therefore in an attemptto avoid profiling RNA samples that would fail qualitycontrol in future studies, we sought to predict whichsamples would pass quality control using real-timePCR, and also tested a subset with the Agilent Bio-analyser and TaqMan PCR. We performed a modifiedreal-time PCR protocol on 77 RNA samples from FFPEtumours on which there was sufficient RNA for this
purpose after the DASL assay had been performed. Weused two RNA samples from fresh frozen tumours ascontrols, to determine whether the extent of amplifica-tion could predict which samples would perform wellin the DASL assay (Figure 2A). The reactions were runin triplicate and the average cycle threshold (Ct) for theFFPE RNA samples was 23.21 (range 17.81–28.90),and for the fresh frozen RNA samples 15.15 (range15.08–15.23). We hypothesized that samples that failedthe WG-DASL assay would be more degraded and thushave a higher Ct; however, we found that the real-timePCR assay was a poor predictor of success rates in theDASL assay.
Copyright 2010 Pathological Society of Great Britain and Ireland. J Pathol 2010; 221: 452–461Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com
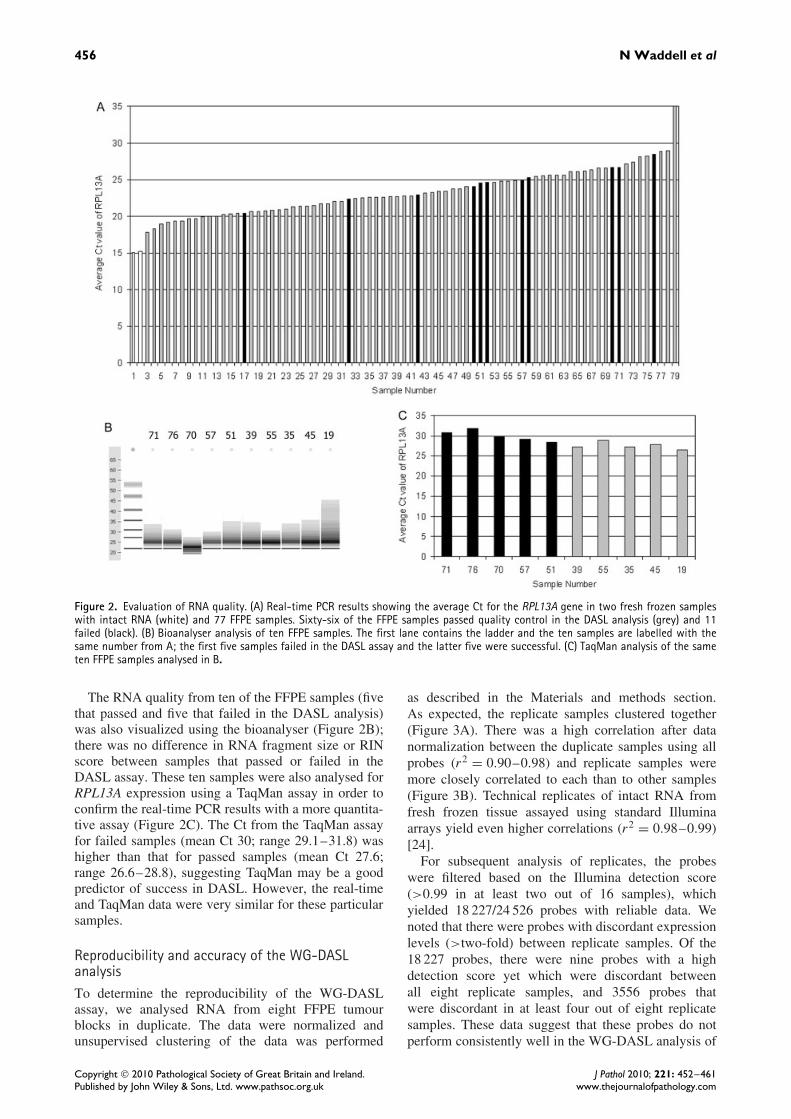
456 N Waddell et al
Figure 2. Evaluation of RNA quality. (A) Real-time PCR results showing the average Ct for the RPL13A gene in two fresh frozen sampleswith intact RNA (white) and 77 FFPE samples. Sixty-six of the FFPE samples passed quality control in the DASL analysis (grey) and 11failed (black). (B) Bioanalyser analysis of ten FFPE samples. The first lane contains the ladder and the ten samples are labelled with thesame number from A; the first five samples failed in the DASL assay and the latter five were successful. (C) TaqMan analysis of the sameten FFPE samples analysed in B.
The RNA quality from ten of the FFPE samples (fivethat passed and five that failed in the DASL analysis)was also visualized using the bioanalyser (Figure 2B);there was no difference in RNA fragment size or RINscore between samples that passed or failed in theDASL assay. These ten samples were also analysed forRPL13A expression using a TaqMan assay in order toconfirm the real-time PCR results with a more quantita-tive assay (Figure 2C). The Ct from the TaqMan assayfor failed samples (mean Ct 30; range 29.1–31.8) washigher than that for passed samples (mean Ct 27.6;range 26.6–28.8), suggesting TaqMan may be a goodpredictor of success in DASL. However, the real-timeand TaqMan data were very similar for these particularsamples.
Reproducibility and accuracy of the WG-DASLanalysisTo determine the reproducibility of the WG-DASLassay, we analysed RNA from eight FFPE tumourblocks in duplicate. The data were normalized andunsupervised clustering of the data was performed
as described in the Materials and methods section.As expected, the replicate samples clustered together(Figure 3A). There was a high correlation after datanormalization between the duplicate samples using allprobes (r2 = 0.90–0.98) and replicate samples weremore closely correlated to each than to other samples(Figure 3B). Technical replicates of intact RNA fromfresh frozen tissue assayed using standard Illuminaarrays yield even higher correlations (r2 = 0.98–0.99)[24].
For subsequent analysis of replicates, the probeswere filtered based on the Illumina detection score(>0.99 in at least two out of 16 samples), whichyielded 18 227/24 526 probes with reliable data. Wenoted that there were probes with discordant expressionlevels (>two-fold) between replicate samples. Of the18 227 probes, there were nine probes with a highdetection score yet which were discordant betweenall eight replicate samples, and 3556 probes thatwere discordant in at least four out of eight replicatesamples. These data suggest that these probes do notperform consistently well in the WG-DASL analysis of
Copyright 2010 Pathological Society of Great Britain and Ireland. J Pathol 2010; 221: 452–461Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com
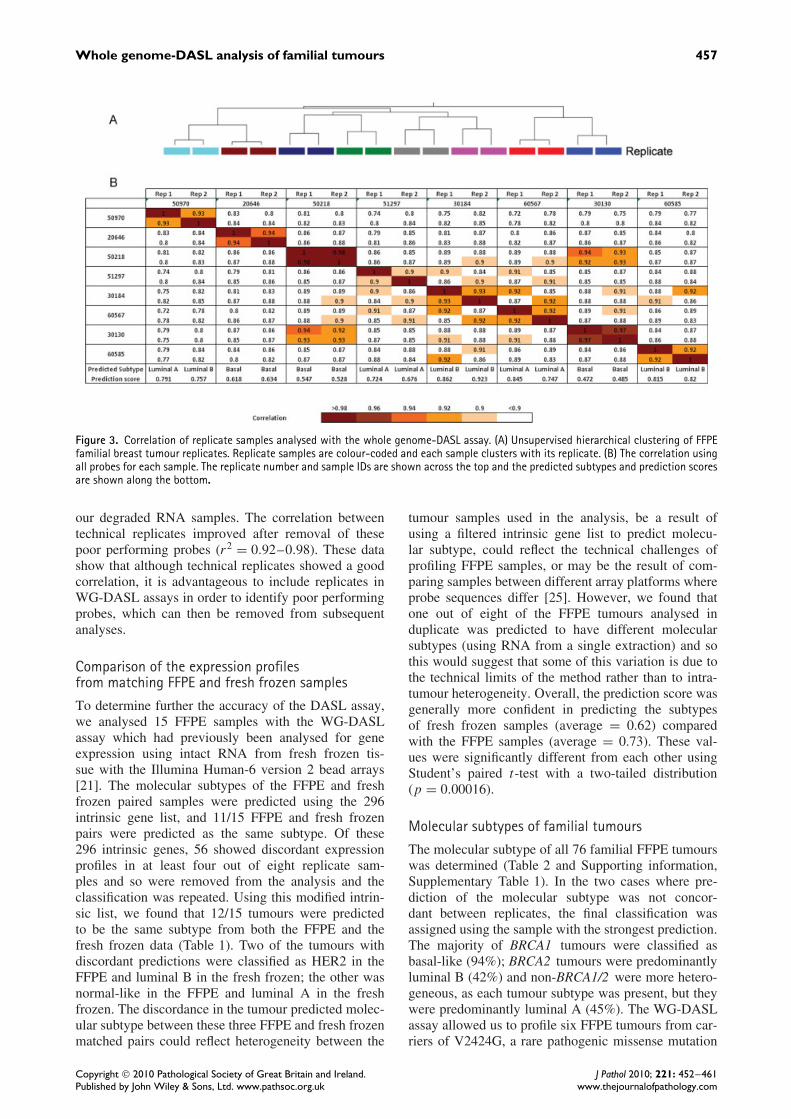
Whole genome-DASL analysis of familial tumours 457
Figure 3. Correlation of replicate samples analysed with the whole genome-DASL assay. (A) Unsupervised hierarchical clustering of FFPEfamilial breast tumour replicates. Replicate samples are colour-coded and each sample clusters with its replicate. (B) The correlation usingall probes for each sample. The replicate number and sample IDs are shown across the top and the predicted subtypes and prediction scoresare shown along the bottom.
our degraded RNA samples. The correlation betweentechnical replicates improved after removal of thesepoor performing probes (r2 = 0.92–0.98). These datashow that although technical replicates showed a goodcorrelation, it is advantageous to include replicates inWG-DASL assays in order to identify poor performingprobes, which can then be removed from subsequentanalyses.
Comparison of the expression profilesfrom matching FFPE and fresh frozen samples
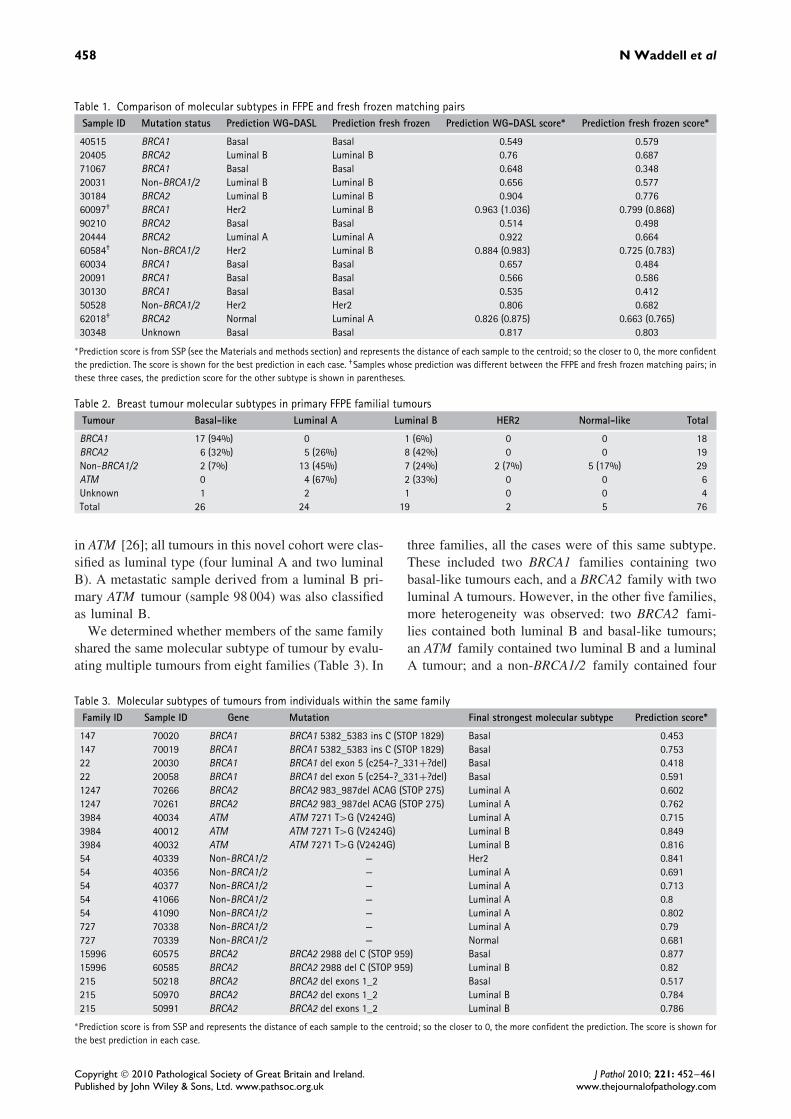
To determine further the accuracy of the DASL assay,we analysed 15 FFPE samples with the WG-DASLassay which had previously been analysed for geneexpression using intact RNA from fresh frozen tis-sue with the Illumina Human-6 version 2 bead arrays[21]. The molecular subtypes of the FFPE and freshfrozen paired samples were predicted using the 296intrinsic gene list, and 11/15 FFPE and fresh frozenpairs were predicted as the same subtype. Of these296 intrinsic genes, 56 showed discordant expressionprofiles in at least four out of eight replicate sam-ples and so were removed from the analysis and theclassification was repeated. Using this modified intrin-sic list, we found that 12/15 tumours were predictedto be the same subtype from both the FFPE and thefresh frozen data (Table 1). Two of the tumours withdiscordant predictions were classified as HER2 in theFFPE and luminal B in the fresh frozen; the other wasnormal-like in the FFPE and luminal A in the freshfrozen. The discordance in the tumour predicted molec-ular subtype between these three FFPE and fresh frozenmatched pairs could reflect heterogeneity between the
tumour samples used in the analysis, be a result ofusing a filtered intrinsic gene list to predict molecu-lar subtype, could reflect the technical challenges ofprofiling FFPE samples, or may be the result of com-paring samples between different array platforms whereprobe sequences differ [25]. However, we found thatone out of eight of the FFPE tumours analysed induplicate was predicted to have different molecularsubtypes (using RNA from a single extraction) and sothis would suggest that some of this variation is due tothe technical limits of the method rather than to intra-tumour heterogeneity. Overall, the prediction score wasgenerally more confident in predicting the subtypesof fresh frozen samples (average = 0.62) comparedwith the FFPE samples (average = 0.73). These val-ues were significantly different from each other usingStudent’s paired t-test with a two-tailed distribution(p = 0.00016).
Molecular subtypes of familial tumours
The molecular subtype of all 76 familial FFPE tumourswas determined (Table 2 and Supporting information,Supplementary Table 1). In the two cases where pre-diction of the molecular subtype was not concor-dant between replicates, the final classification wasassigned using the sample with the strongest prediction.The majority of BRCA1 tumours were classified asbasal-like (94%); BRCA2 tumours were predominantlyluminal B (42%) and non-BRCA1/2 were more hetero-geneous, as each tumour subtype was present, but theywere predominantly luminal A (45%). The WG-DASLassay allowed us to profile six FFPE tumours from car-riers of V2424G, a rare pathogenic missense mutation
Copyright 2010 Pathological Society of Great Britain and Ireland. J Pathol 2010; 221: 452–461Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com
458 N Waddell et al
Table 1. Comparison of molecular subtypes in FFPE and fresh frozen matching pairsSample ID Mutation status Prediction WG-DASL Prediction fresh frozen Prediction WG-DASL score∗ Prediction fresh frozen score∗
40515 BRCA1 Basal Basal 0.549 0.57920405 BRCA2 Luminal B Luminal B 0.76 0.68771067 BRCA1 Basal Basal 0.648 0.34820031 Non-BRCA1/2 Luminal B Luminal B 0.656 0.57730184 BRCA2 Luminal B Luminal B 0.904 0.77660097† BRCA1 Her2 Luminal B 0.963 (1.036) 0.799 (0.868)90210 BRCA2 Basal Basal 0.514 0.49820444 BRCA2 Luminal A Luminal A 0.922 0.66460584† Non-BRCA1/2 Her2 Luminal B 0.884 (0.983) 0.725 (0.783)60034 BRCA1 Basal Basal 0.657 0.48420091 BRCA1 Basal Basal 0.566 0.58630130 BRCA1 Basal Basal 0.535 0.41250528 Non-BRCA1/2 Her2 Her2 0.806 0.68262018† BRCA2 Normal Luminal A 0.826 (0.875) 0.663 (0.765)30348 Unknown Basal Basal 0.817 0.803
∗Prediction score is from SSP (see the Materials and methods section) and represents the distance of each sample to the centroid; so the closer to 0, the more confidentthe prediction. The score is shown for the best prediction in each case. †Samples whose prediction was different between the FFPE and fresh frozen matching pairs; inthese three cases, the prediction score for the other subtype is shown in parentheses.
Table 2. Breast tumour molecular subtypes in primary FFPE familial tumoursTumour Basal-like Luminal A Luminal B HER2 Normal-like Total
BRCA1 17 (94%) 0 1 (6%) 0 0 18BRCA2 6 (32%) 5 (26%) 8 (42%) 0 0 19Non-BRCA1/2 2 (7%) 13 (45%) 7 (24%) 2 (7%) 5 (17%) 29ATM 0 4 (67%) 2 (33%) 0 0 6Unknown 1 2 1 0 0 4Total 26 24 19 2 5 76
in ATM [26]; all tumours in this novel cohort were clas-sified as luminal type (four luminal A and two luminalB). A metastatic sample derived from a luminal B pri-mary ATM tumour (sample 98 004) was also classifiedas luminal B.
We determined whether members of the same familyshared the same molecular subtype of tumour by evalu-ating multiple tumours from eight families (Table 3). In
three families, all the cases were of this same subtype.These included two BRCA1 families containing twobasal-like tumours each, and a BRCA2 family with twoluminal A tumours. However, in the other five families,more heterogeneity was observed: two BRCA2 fami-lies contained both luminal B and basal-like tumours;an ATM family contained two luminal B and a luminalA tumour; and a non-BRCA1/2 family contained four
Table 3. Molecular subtypes of tumours from individuals within the same familyFamily ID Sample ID Gene Mutation Final strongest molecular subtype Prediction score∗
147 70020 BRCA1 BRCA1 5382_5383 ins C (STOP 1829) Basal 0.453147 70019 BRCA1 BRCA1 5382_5383 ins C (STOP 1829) Basal 0.75322 20030 BRCA1 BRCA1 del exon 5 (c254-?_331+?del) Basal 0.41822 20058 BRCA1 BRCA1 del exon 5 (c254-?_331+?del) Basal 0.5911247 70266 BRCA2 BRCA2 983_987del ACAG (STOP 275) Luminal A 0.6021247 70261 BRCA2 BRCA2 983_987del ACAG (STOP 275) Luminal A 0.7623984 40034 ATM ATM 7271 T>G (V2424G) Luminal A 0.7153984 40012 ATM ATM 7271 T>G (V2424G) Luminal B 0.8493984 40032 ATM ATM 7271 T>G (V2424G) Luminal B 0.81654 40339 Non-BRCA1/2 — Her2 0.84154 40356 Non-BRCA1/2 — Luminal A 0.69154 40377 Non-BRCA1/2 — Luminal A 0.71354 41066 Non-BRCA1/2 — Luminal A 0.854 41090 Non-BRCA1/2 — Luminal A 0.802727 70338 Non-BRCA1/2 — Luminal A 0.79727 70339 Non-BRCA1/2 — Normal 0.68115996 60575 BRCA2 BRCA2 2988 del C (STOP 959) Basal 0.87715996 60585 BRCA2 BRCA2 2988 del C (STOP 959) Luminal B 0.82215 50218 BRCA2 BRCA2 del exons 1_2 Basal 0.517215 50970 BRCA2 BRCA2 del exons 1_2 Luminal B 0.784215 50991 BRCA2 BRCA2 del exons 1_2 Luminal B 0.786
∗Prediction score is from SSP and represents the distance of each sample to the centroid; so the closer to 0, the more confident the prediction. The score is shown forthe best prediction in each case.
Copyright 2010 Pathological Society of Great Britain and Ireland. J Pathol 2010; 221: 452–461Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com

Whole genome-DASL analysis of familial tumours 459
individuals with tumours classified as luminal A (atages 79, 43, 34, and 47 years) and an individual witha HER2 tumour at age 26. The remaining family was anon-BRCA1/2 family which contained individuals withtumours classified as normal-like or luminal A.
Discussion
We have shown that the whole genome-DASL assayis a robust tool for the analysis of degraded RNA fromarchival FFPE blocks. Eighty-five per cent of tumoursanalysed produced successful DASL gene expressiondata. The correlation between technical replicates isgood (0.90–0.98) and the predicted molecular sub-type is consistent with that predicted by analysis ofthe matched fresh frozen tumour sample in 12 out of15 cases.
The correlation between technical replicates is, how-ever, lower than that obtained with standard geneexpression profiling data of intact RNA on Illuminaarrays. This is likely due to a combination of factors:the use of degraded RNA; subsequent PCR amplifica-tion bias during the DASL assay; and the inclusion ofunreliable probes. Importantly, we identified more than3000 unreliable probes as having discordant expressionlevels between technical replicate RNA samples. Wefound that the correlation between replicates and theconcordance of the prediction of molecular subtypes ofmatching FFPE and fresh frozen pairs increased whenthe poor performing probes were excluded from theanalysis. Therefore developing a means of identifyingthese poor probes will improve WG-DASL analysisand highlights the need for several technical replicatesper assay run.
Characteristics of samples which fail DASL analysisThe failure of samples in the WG-DASL assay couldbe explained by a number of factors such as particularlypoor RNA integrity, different fixing methods, andsample handling during the DASL assay. We triedto use a real-time PCR assay to predict whether asample was likely to fail in the DASL assay. While therange of Ct values from the real-time PCR overlappedfor samples that passed and failed quality control inthe DASL assay, the mean Ct value was lower forthe samples that passed, suggesting that this couldbe a useful way of prioritizing samples for DASLanalysis in large studies even though it is not absolutelypredictive. We also analysed a subset of the sampleswith the bioanalyser and found little correlation withRNA size and DASL success, since all the RNAsamples were highly degraded. All FFPE blocks usedin this study were collected during 1981–2006 andcame from multiple different pathology laboratoriesin Australia. We found that the older blocks weremore likely to fail DASL and that all blocks processedduring or after 2002 passed DASL. Although notsignificant, we also found that RNA samples with a
higher concentration were more likely to be successfulin the DASL assay. Blocks from several pathologycentres had a poor pass rate in the DASL assay, whichcould suggest that sample fixing could have had animpact on RNA integrity. However, the numbers fromeach centre in this study were small and there may beother factors that predict success in the DASL assay.
Molecular subtypes of familial tumours
The proportion of molecular subtypes within the famil-ial tumours was similar to that which has previ-ously been reported by us and others [21,27]. BRCA1tumours were predominantly basal-like, and BRCA2and non-BRCA1/2 tumours were predominantly lumi-nal. We also assayed a small cohort of tumours derivedfrom carriers of a pathogenic mutation in the breastcancer-associated gene ATM [26] and found that allof these tumours were luminal. Breast tumours fromATM carriers are rare, and the DASL assay allowedus to obtain an expression profile and classify thesearchival tumours, which would have been impossibleif we had to depend on frozen tissue.
Although we have only analysed a small numberof families so far, our data suggest that carriers ofmutations within specific genes may show a tendencyto form specific tumour types, suggesting that loss offunction of certain genes could give rise to specifictumour types. Thus, stratification of tumours wherethe predisposition gene or genes is unknown maybetter enable the identification of other breast cancer-associated genes. The availability of the WG-DASLassay opens up the potential to evaluate this issue on amuch larger scale and to determine whether there arefamilies that appear to have inherited a predispositionto a particular molecular subtype of breast cancer.
In summary, WG-DASL technology appears to be arobust assay for gene expression profiling of degradedRNA samples such as those obtained from archivalclinical samples. There is now the potential to studycomplex molecular features, clinical scenarios such asrare disorders, and cohorts from clinical trials or withlong-term clinical follow-up.
Acknowledgment
We wish to thank Heather Thorne, Eveline Niedermayr,all the kConFab research nurses and staff, the headsand staff of the Family Cancer Clinics, and theClinical Follow Up Study (funded by NHMRC grants145684, 288704, and 454508) for their contributionsto this resource, and the many families who contributeto kConFab. kConFab is supported by grants fromthe National Breast Cancer Foundation, the NationalHealth and Medical Research Council (NHMRC); andby the Queensland Cancer Fund; the Cancer Councilsof New South Wales, Victoria, Tasmania, and SouthAustralia; and the Cancer Foundation of WesternAustralia. This work was supported by grants from theNational Breast Cancer Foundation and the National
Copyright 2010 Pathological Society of Great Britain and Ireland. J Pathol 2010; 221: 452–461Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com

460 N Waddell et al
Health and Medical Research Council. GCT is a SeniorPrincipal Research Fellow of the NHMRC.
Author Contributions
NW was involved in study design, data collection,data analysis, data interpretation, literature search, gen-eration of figures, and writing of the manuscript;SC—data collection, data analysis, and data interpre-tation; JJ and SH—data collection and data analysis;AM, JR, LdS, ACV, and LR—data collection; kCon-Fab Investigators—sample acquisition; PTS—studydesign, literature search, and writing of the manuscript;SRL—study design and writing of the manuscript;and GC-T—study design, data interpretation, literaturesearch, and writing of the manuscript.
References
1. Bibikova M, Talantov D, Chudin E, et al . Quantitative geneexpression profiling in formalin-fixed, paraffin-embedded tissuesusing universal bead arrays. Am J Pathol 2004; 165: 1799–1807.
2. Bibikova M, Yeakley JM, Wang-Rodriguez J, et al . Quantita-tive expression profiling of RNA from formalin-fixed, paraffin-embedded tissues using randomly assembled bead arrays. Methods
Mol Biol 2008; 439: 159–177.3. Fan JB, Yeakley JM, Bibikova M, et al . A versatile assay for high-
throughput gene expression profiling on universal array matrices.Genome Res 2004; 14: 878–885.
4. Duenwald S, Zhou M, Wang Y, et al . Development of a microar-ray platform for FFPET profiling: application to the classificationof human tumors. J Transl Med 2009; 7: 65.
5. Conway C, Mitra A, Jewell R, et al . Gene expression profilingof paraffin-embedded primary melanoma using the DASL assayidentifies increased osteopontin expression as predictive of reducedrelapse-free survival. Clin Cancer Res 2009; 15: 6939–6946.
6. Abramovitz M, Ordanic-Kodani M, Wang Y, et al . Optimizationof RNA extraction from FFPE tissues for expression profiling inthe DASL assay. Biotechniques 2008; 44: 417–423.
7. Da Silva L, Parry S, Reid L, et al . Aberrant expression of E-cadherin in lobular carcinomas of the breast. Am J Surg Pathol
2008; 32: 773–783.8. Di Cesare S, Nantel A, Marshall JC, et al . Expression profiling of
formalin-fixed paraffin embedded primary human uveal melanomasusing DASL matrices. J Cancer Res Clin Oncol 2010; 136:577–586.
9. Haller AC, Kanakapalli D, Walter R, et al . Transcriptional profil-ing of degraded RNA in cryopreserved and fixed tissue samplesobtained at autopsy. BMC Clin Pathol 2006; 6: 9.
10. Ravo M, Mutarelli M, Ferraro L, et al . Quantitative expressionprofiling of highly degraded RNA from formalin-fixed, paraffin-embedded breast tumor biopsies by oligonucleotide microarrays.Lab Invest 2008; 88: 430–440.
11. April C, Klotzle B, Royce T, et al . Whole-genome gene expres-sion profiling of formalin-fixed, paraffin-embedded tissue samples.PLoS One 2009; 4: e8162.
12. Chien J, Fan JB, Bell DA, et al . Analysis of gene expression instage I serous tumors identifies critical pathways altered in ovariancancer. Gynecol Oncol 2009; 114: 3–11.
13. Perou CM, Jeffrey SS, van de Rijn M, et al . Distinctive geneexpression patterns in human mammary epithelial cells and breastcancers. Proc Natl Acad Sci U S A 1999; 96: 9212–9217.
14. Hu Z, Fan C, Oh DS, et al . The molecular portraits of breasttumors are conserved across microarray platforms. BMC Genomics2006; 7: 96.
15. Perou CM, Sorlie T, Eisen MB, et al . Molecular portraits ofhuman breast tumours. Nature 2000; 406: 747–752.
16. Sorlie T, Perou CM, Tibshirani R, et al . Gene expression patternsof breast carcinomas distinguish tumor subclasses with clinicalimplications. Proc Natl Acad Sci U S A 2001; 98: 10869–10874.
17. Sorlie T, Tibshirani R, Parker J, et al . Repeated observation ofbreast tumor subtypes in independent gene expression data sets.Proc Natl Acad Sci U S A 2003; 100: 8418–8423.
18. Sotiriou C, Neo SY, McShane LM, et al . Breast cancer classifi-cation and prognosis based on gene expression profiles from apopulation-based study. Proc Natl Acad Sci U S A 2003; 100:10393–10398.
19. van ‘t Veer LJ, Dai H, van de Vijver MJ, et al . Gene expressionprofiling predicts clinical outcome of breast cancer. Nature 2002;415: 530–536.
20. Fong PC, Boss DS, Yap TA, et al . Inhibition of poly(ADP-ribose)polymerase in tumors from BRCA mutation carriers. N Engl J Med
2009; 361: 123–134.21. Waddell N, Arnold J, Cocciardi S, et al . Subtypes of familial
breast tumours revealed by expression and copy number profiling.Breast Cancer Res Treat 2009; [Epub ahead of print].
22. Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illuminamicroarray. Bioinformatics 2008; 24: 1547–1548.
23. Benito M, Parker J, Du Q, et al . Adjustment of systematicmicroarray data biases. Bioinformatics 2004; 20: 105–114.
24. Kuhn K, Baker SC, Chudin E, et al . A novel, high-performancerandom array platform for quantitative gene expression profiling.Genome Res 2004; 14: 2347–2356.
25. Mecham BH, Klus GT, Strovel J, et al . Sequence-matched probesproduce increased cross-platform consistency and more repro-ducible biological results in microarray-based gene expressionmeasurements. Nucleic Acids Res 2004; 32: e74.
26. Tavtigian SV, Oefner PJ, Babikyan D, et al . Rare, evolutionarilyunlikely missense substitutions in ATM confer increased risk ofbreast cancer. Am J Hum Genet 2009; 85: 427–446.
27. Honrado E, Osorio A, Milne RL, et al . Immunohistochemicalclassification of non-BRCA1/2 tumors identifies different groupsthat demonstrate the heterogeneity of BRCAX families. Mod
Pathol 2007; 20: 1298–1306.
Copyright 2010 Pathological Society of Great Britain and Ireland. J Pathol 2010; 221: 452–461Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com

Whole genome-DASL analysis of familial tumours 461
SUPPORTING INFORMATION ON THE INTERNET
The following supporting information may be found in the online version of this article.
Table S1. All clinical data collected and information regarding the FFPE blocks and RNA hybridizations
Figure S1. A schematic diagram showing the number of samples that were processed in the WG-DASL assay, with a breakdown of thenumber of samples that failed. The number of samples used at each quality control step is also shown.
Copyright 2010 Pathological Society of Great Britain and Ireland. J Pathol 2010; 221: 452–461Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com
Copyright © 2022 FDOKUMEN




















