
Formalin-Fixed Paraffin-Embedded (FFPE) Proteome Analysis Using Gel-Free and Gel-Based Proteomics
11
Formalin-Fixed Paraffin-Embedded (FFPE) Proteome Analysis Using Gel-Free and Gel-Based Proteomics Omid Azimzadeh,* ,† Zarko Barjaktarovic, † Michaela Aubele, ‡ Julia Calzada-Wack, ‡ Hakan Sarioglu, § Michael J. Atkinson, | and Soile Tapio* ,† Helmholtz Zentrum Mu ¨ nchen, German Research Center for Environmental Health, Institute of Radiation Biology, 85764 Neuherberg, Germany, Helmholtz Zentrum Mu ¨ nchen, German Research Center for Environmental Health, Institute of Pathology, 85764 Neuherberg, Germany, Helmholtz Zentrum Mu ¨ nchen, German Research Center for Environmental Health, Department of Protein Science, Proteomics Core Facility, Neuherberg, Germany, and Department of Radiation Oncology, Klinikum Rechts der Isar, Technische Universita ¨t Mu ¨ nchen, Munich, Germany Received May 7, 2010 Formalin-fixed paraffin-embedded (FFPE) tissue has recently gained interest as an alternative to fresh/ frozen tissue for retrospective protein biomarker discovery. However, during the fixation process, proteins undergo degradation and cross-linking, making conventional protein analysis technologies problematic. In this study, we have compared several extraction and separation methods for the analysis of proteins in FFPE tissues. Incubation of tissue sections at high temperature with a novel extraction buffer (20 mM Tris-HCl, pH 8.8, 2% SDS, 1% -octylglucoside, 200 mM DTT, 200 mM glycine, and a mixture of protease inhibitors) resulted in improved protein recovery. Protein separation by 1-DE followed by LC-ESI MS/MS analysis was the most effective approach to identify proteins, based on the number of peptides reliably identified. Interestingly, a number of peptides were identified in regions of the 1DE not corresponding to their native molecular weights. This is an indication of the formation of protein-protein complexes by cross-linking, and of protein fragmentation due to prolonged sample storage. This study will facilitate the development of future proteomic analysis of FFPE tissue and provide a tool for the validation in archival samples of biomarkers of exposure, prognosis and disease. Keywords: formalin-fixed paraffin-embedded (FFPE) • gel-based • gel-free • proteomics • protein extraction • peptide modification • cross-linking 1. Introduction Formalin-fixed paraffin-embedded (FFPE) tissues are rou- tinely prepared from biopsies and at autopsy, and are held in vast quantities for many years. These tissues are proving a valuable source for the retrospective analysis of DNA and RNA, where technologies exist to prepare usable samples from histologically defined tissues. Recently, FFPE tissues have also been considered as an alternative to fresh/frozen tissue for protein biomarker discovery, 1 especially when in many cases the large clinical archives represent the only source of bioma- terial available. However, the harsh and irreversible fixation conditions and the prolonged storage times have made retro- spective biological studies on a proteome level a difficult task. Simultaneously, the analysis of the FFPE proteome has been hampered by the chemical content of the extraction buffers needed to effectively extract proteins (e.g., detergents, reduc- tants, denaturants, and salts). These conditions render the protein extracts incompatible with the subsequent steps re- quired for a proteomics approach. Although several alternatives have been developed to optimize protein extraction and separation using FFPE tissue, 2-5,8 the qualitative and quantita- tive proteome analysis remains a challenge. In early studies, the heat-induced antigen retrieval procedure was used to extract proteins from FFPE tissue sections before separation by SDS-PAGE. 3-5 This method was used effectively for the detection of full-length proteins on Western blot, 6 reverse phase protein microarray analysis, 7 and post-transla- tional modification studies. 8 Unfortunately, this procedure fails to provide sufficient material that is compatible with conven- tional protein analysis methodologies, such as classical 2D gel electrophoresis. 5,9,10 Several studies have been conducted to overcome the adverse effects of cross-linking on the quality and quantity of the protein extracted from FFPE. To improve extraction pro- tocols Fowler et al. developed a tissue surrogate as a model system for FFPE samples to analyze the effect of the degree of cross-linking on the quality of the extraction The authors showed that exposure of the FFPE surrogate to the high temperatures (90 °C) in acidic (pH 5) buffers results in efficient protein extraction. 11 Using extraction buffers with various pH * To whom correspondence should be addressed. E-mail: omid.azimzadeh@ helmholtz-muenchen.de; [email protected]. † Institute of Radiation Biology. ‡ Institute of Pathology. § Department of Protein Science, Proteomics Core Facility. | Technische Universita ¨t Mu ¨ nchen. 4710 Journal of Proteome Research 2010, 9, 4710–4720 10.1021/pr1004168 2010 American Chemical Society Published on Web 07/06/2010
-
Upload
helmholtz-muenchen -
Category
Documents
-
view
0 -
download
0
Transcript of Formalin-Fixed Paraffin-Embedded (FFPE) Proteome Analysis Using Gel-Free and Gel-Based Proteomics

Formalin-Fixed Paraffin-Embedded (FFPE) Proteome Analysis Using
Gel-Free and Gel-Based Proteomics
Omid Azimzadeh,*,† Zarko Barjaktarovic,† Michaela Aubele,‡ Julia Calzada-Wack,‡
Hakan Sarioglu,§ Michael J. Atkinson,| and Soile Tapio*,†
Helmholtz Zentrum Munchen, German Research Center for Environmental Health, Institute of RadiationBiology, 85764 Neuherberg, Germany, Helmholtz Zentrum Munchen, German Research Center for
Environmental Health, Institute of Pathology, 85764 Neuherberg, Germany, Helmholtz Zentrum Munchen,German Research Center for Environmental Health, Department of Protein Science, Proteomics Core Facility,
Neuherberg, Germany, and Department of Radiation Oncology, Klinikum Rechts der Isar,Technische Universitat Munchen, Munich, Germany
Received May 7, 2010
Formalin-fixed paraffin-embedded (FFPE) tissue has recently gained interest as an alternative to fresh/frozen tissue for retrospective protein biomarker discovery. However, during the fixation process,proteins undergo degradation and cross-linking, making conventional protein analysis technologiesproblematic. In this study, we have compared several extraction and separation methods for the analysisof proteins in FFPE tissues. Incubation of tissue sections at high temperature with a novel extractionbuffer (20 mM Tris-HCl, pH 8.8, 2% SDS, 1% �-octylglucoside, 200 mM DTT, 200 mM glycine, and amixture of protease inhibitors) resulted in improved protein recovery. Protein separation by 1-DEfollowed by LC-ESI MS/MS analysis was the most effective approach to identify proteins, based on thenumber of peptides reliably identified. Interestingly, a number of peptides were identified in regionsof the 1DE not corresponding to their native molecular weights. This is an indication of the formationof protein-protein complexes by cross-linking, and of protein fragmentation due to prolonged samplestorage. This study will facilitate the development of future proteomic analysis of FFPE tissue and providea tool for the validation in archival samples of biomarkers of exposure, prognosis and disease.
Keywords: formalin-fixed paraffin-embedded (FFPE) • gel-based • gel-free • proteomics • proteinextraction • peptide modification • cross-linking
1. Introduction
Formalin-fixed paraffin-embedded (FFPE) tissues are rou-tinely prepared from biopsies and at autopsy, and are held invast quantities for many years. These tissues are proving avaluable source for the retrospective analysis of DNA and RNA,where technologies exist to prepare usable samples fromhistologically defined tissues. Recently, FFPE tissues have alsobeen considered as an alternative to fresh/frozen tissue forprotein biomarker discovery,1 especially when in many casesthe large clinical archives represent the only source of bioma-terial available. However, the harsh and irreversible fixationconditions and the prolonged storage times have made retro-spective biological studies on a proteome level a difficult task.Simultaneously, the analysis of the FFPE proteome has beenhampered by the chemical content of the extraction buffersneeded to effectively extract proteins (e.g., detergents, reduc-tants, denaturants, and salts). These conditions render the
protein extracts incompatible with the subsequent steps re-quired for a proteomics approach. Although several alternativeshave been developed to optimize protein extraction andseparation using FFPE tissue,2-5,8 the qualitative and quantita-tive proteome analysis remains a challenge.
In early studies, the heat-induced antigen retrieval procedurewas used to extract proteins from FFPE tissue sections beforeseparation by SDS-PAGE.3-5 This method was used effectivelyfor the detection of full-length proteins on Western blot,6
reverse phase protein microarray analysis,7 and post-transla-tional modification studies.8 Unfortunately, this procedure failsto provide sufficient material that is compatible with conven-tional protein analysis methodologies, such as classical 2D gelelectrophoresis.5,9,10
Several studies have been conducted to overcome theadverse effects of cross-linking on the quality and quantity ofthe protein extracted from FFPE. To improve extraction pro-tocols Fowler et al. developed a tissue surrogate as a modelsystem for FFPE samples to analyze the effect of the degree ofcross-linking on the quality of the extraction The authorsshowed that exposure of the FFPE surrogate to the hightemperatures (90 °C) in acidic (pH 5) buffers results in efficientprotein extraction.11 Using extraction buffers with various pH
* To whom correspondence should be addressed. E-mail: [email protected]; [email protected].
† Institute of Radiation Biology.‡ Institute of Pathology.§ Department of Protein Science, Proteomics Core Facility.| Technische Universitat Munchen.
4710 Journal of Proteome Research 2010, 9, 4710–4720 10.1021/pr1004168 2010 American Chemical SocietyPublished on Web 07/06/2010

values, Shi et al. analyzed the FFPE proteomes of human breasttissue by mass spectrometry, combined with capillary isoelec-tric focusing (CIEF) and reversed-phase liquid chromatography(RPLC).12 Recently, Nirmalan et al. identified comparable butnot identical protein profiles of FFPE and fresh/frozen tissueusing surface-enhanced laser desorption ionization-time offlight mass spectroscopy (SELDI-TOF) analysis after boilingsamples in Laemmli buffer.13
Only a few studies so far have reported the application of aclassical 2D electrophoresis approach for the analysis of theFFPE tissue; these reports mainly confirming the unsuitabilityof this material for such an analysis.5,9,10 Ono et al. performeda quantitative study of FFPE versus fresh/frozen tissue usingtwo-dimensional difference in gel electrophoresis (2D-DIGE).The low quality of the stained 2D gel enabled the identificationof only a few deregulated protein spots.10 Recently, Addis etal. used sheep skeletal muscle and liver as standard tissuemodels to exclude the effect of the variability between FFPEsamples, arising mainly from different preparation proceduresbut also from other factors such as storage age. Using thisstandard FFPE model, the authors were able to improve the2D electrophoretic pattern of FFPE extracted proteins.14
Gel-free proteomic studies using label-free approach oralternative labeling strategies such as iTRAQ have been moresuccessful than the gel-based methods in the number ofidentified proteins in FFPE material.15,16 Several studies haveused shotgun proteomics strategy and liquid chromatographyafter a direct in-tissue digestion. In these studies, the compat-ibility of different extraction buffers with trypsin digestion wasexamined.17-21 Sprung et al. compared the protein profiles ofthe FFPE and fresh/frozen colon adenoma tissue by multidi-mensional LC-MS/MS after isoelectric focusing of the pep-tides.21 Using commercial protein preparation buffer (Liq-uidTissue) and stable isotopic labeling, Hood et al. performeda quantitative proteomic analysis on FFPE prostate tissue afternanoreverse-phase LC (nano-RPLC) tandem mass spectrom-etry.22 Scicchitano et al. applied the same buffer to comparethe protein profile of the FFPE and fresh/frozen rat liver. Theidentification of a greater number of proteins in FFPE samplescompared to fresh/frozen samples led the authors to concludethat the number of identified unique proteins in the FFPEsamples was not dramatically affected by the formalin-fixationand paraffin-embedding procedure.20 These data were consis-tent with a shotgun proteomic study done on formalin-fixedmouse liver tissue, the results indicating that fixation does notnecessarily compromise the proteome coverage.18
Sprung et al. used a detergent-free in-tissue digestionapproach for extraction of peptides form colon adenoma FFPEand fresh/frozen samples and separated the generated peptidesby isoelectric focusing before identification by LC MS-MS. Theresults showed a significant overlap in proteins identifiedbetween the two samples.21 Using multidimensional separationplatform including capillary isoelectric focusing (CIEF)/nano-RPLC, Guo et al. separated the tryptic peptides from micro-dissected FFPE glioblastoma.23 Negishi et al. described a label-free quantitative proteomics method using the 2-dimensionalimage-converted analysis of liquid chromatography and massspectrometry software (2DICAL) for the identification of de-regulated proteins obtained from FFPE oral squamous cellcarcinoma compared to a noncancerous oral tissue.15
In the present study, we comprehensively evaluated condi-tions for the optimal protein extraction and separation fromFFPE tissue using gel-free and gel-based proteomics. Moreover,
for the first time, the protein identifications obtained frommultidimensional approaches were compared to demonstratethe most reliable and reproducible method for FFPE proteomeanalysis. As a model tissue, we used the heart of the C57BL/6mouse that is easily available as both fresh/frozen and archivaltissue.
2. Experimental Section
2.1. Materials. �-Octylglucoside, Triton X-100, SDS, am-monium bicarbonate, and RIPA extraction buffer were obtainedfrom Sigma (St. Luis, MO); RapiGest from Waters; acetone,acetonitrile, formic acid, and trifluoroacetic acid (TFA) fromRoth (Karlsuhe, Germany); trifluoroethanol (TFE) from Fluka(Buches, Germany); dithiothreitol (DTT), tributylphosphine(TBP), iodoacetamide, urea, thiourea, tris-(hydroxymethyl)aminomethane (Tris), CHAPS, and IPG buffer 3-10 were fromGE Healthcare (Freiburg, Germany). QProteome extractionbuffer was purchased from Qiagen (Germany); sequencinggrade trypsin was obtained from Promega (Madison, WI);cyano-4-hydroxycinnamic acid was from Bruker Daltonik (Bre-men, Germany). All solutions were prepared using HPLC gradewater from Roth (Karlsuhe, Germany).
2.2. Tissue Samples. For all experiments, we used hearttissue obtained from male C57BL/6 mice. Tissues were fixedin 4% buffered formalin for 24 h, dehydrated with a gradedseries of ethanol before being embedded in paraffin. Blockswere stored in the dark at room temperature for between 1and 3 years. Sections (20 µm) of the archival tissue were cutfrom tissue blocks after initial trimming to remove exposedsurfaces. Fresh/frozen hearts used for comparison were re-moved and rinsed immediately with ice-cold buffer containing20 mM KCl, 2 mM K2HPO4, 1 mM EGTA, pH 6.8, to removeexcess blood. All experiments were done with three technicalreplicates.
2.3. Protein Extraction. FFPE mouse heart tissue sections(20 µm thick, 80 mm2 wide) were placed on microscope slidesand deparaffinized by incubating twice with xylene for 10 minat room temperature before rehydration in a graded series ofethanol (100%, 95% and 70%) for 10 min each. The tissuesections were scraped from the slides, washed with 0.5%�-octylglucoside and resuspended in a range of differentdetergent-containing buffers including (i) Laemmli buffercontaining 2% SDS,13 (ii) rehydration buffer containing 2%CHAPS,5 (iii) extraction buffer containing 0.2% Tween 20,2 (iv)RIPA buffer containing 2% SDS and 1% NP40,4 and (v)extraction buffer developed in this study containing 20 mMTris-HCl, pH 8.8, 2% SDS, 1% �-octylglucoside, 200 mM DTT,and 200 mM glycine. All buffers contained the mixture ofprotease inhibitor cocktail (Complete, Roche Diagnostics).
All samples were incubated in the buffers at 100 °C for 20min, and at 80 °C for 2 h with shaking. The extracts werecentrifuged for 30 min at 14 000g at 4 °C. The protein extractwas precipitated with the 2D clean up kit (GE Healthcare)following the manufacturer’s instructions and protein quan-tification was done from the pellet resuspended in Tris bufferin triplicate using both Bradford method and 2D Quant Kit (GEHealthcare) following the manufacturer’s instructions.
2.4. Protein Separation. 2.4.1. Gel-Based Approaches.2.4.1.1. 1D SDS-PAGE. Samples (60 µg) of the protein extractfrom FFPE tissue with the different buffers were solubilized inSDS-PAGE sample buffer, and separated by 12% SDS-PAGEunder reducing conditions24 before staining with Brilliant blueG-250 or silver.25
FFPE Proteome Analysis Using Gel-Free and Gel-Based Proteomics research articles
Journal of Proteome Research • Vol. 9, No. 9, 2010 4711

2.4.1.2. IEF 2D SDS-PAGE. The protein extracts from FFPEand fresh/frozen material were precipitated with the 2D cleanup kit (GE Healthcare) following the manufacturer’s instruc-tions. A total of 200 µg of protein was dissolved in rehydrationbuffer containing 9 M urea, 4% CHAPS, 1% immobilized pHgradient buffer pH 3-10NL (GE Healthcare), and 50 mM DTTand incubated at room temperature for 1 h with shaking. Theprotein extracts were incubated on 13 cm 3-10NL strips O/Nunder low viscosity paraffin oil. Samples were separated by2-DE IEF that was performed using an IPGphor III IEF system(GE Healthcare) at 20 °C using voltages and running times asfollows: 2 h at 150 V (step), 3 h at 300 V (step), 4 h to 1,000 V(step), 2 h to 3500 (step), 2 h 10 000 V (step), and gradient to10 000 V, hold at 10 000 V until a total of at least 45 000 V/hwas reached. After focusing, the strips were equilibrated in 50mM Tris-HCl, pH 6.8, 2% SDS, 7 M urea, 10% glycerol, reducedwith 2% DTT for 15 min, and then alkylated with 2.5%iodoacetamide for 15 min. The equilibrated strips were trans-ferred to 12% slab gels, separated in the second dimensionunder reducing conditions at 10 mA for 1 h and then 20 mAuntil bromophenol blue dye had reached the end of the gel.The gel was stained with silver.25
2.4.1.3. LIEF 2D SDS PAGE. 200 µg of FFPE protein extractwas resuspended in 2.5 mL rehydration buffer containing 9 Murea, 4% CHAPS, 1% immobilized pH gradient buffer pH3-10NL (GE Healthcare), and 50 mM DTT and incubated atroom temperature for 1 h with gentle shaking and loaded intoa Microrotofor cell (Bio-Rad). LIEF was performed at 1 Wconstant power for 3 h under denaturing conditions. Tenfractions of 200 µL each were collected, and pH and proteinconcentration was determined from each fraction. The fractionswere precipitated O/N using cold acetone and then separatedby 12% SDS gel before staining with silver.25
2.4.1.4. In-Gel Digestion for MALDI Analysis. After stainingthe gels, the visible protein bands or spots were excised frompolyacrylamide gels and subjected to in-gel trypsin digestionbefore MS analysis as described26 with a slight modificationas follows. The gel spots were transferred to protein low bindtubes, and destained with 15 mM K3Fe(CN)6 and 50 mMNa2S2O3. The gel pieces were washed once with 500 µL of waterand afterward twice for 15 min with 200 mM NH4HCO3. Theliquid was removed and the gel pieces were shrunk with 25 µLof acetonitrile (ACN) for 5 min. Subsequently, the gel pieceswere dried and the samples were rehydrated with 10 µL oftrypsin (Promega, 10 ng/µL in 50 mM NH4HCO3) and digestedO/N at 37 °C. The resulting peptide mixture was extracted twicewith 50 µL of 50% ACN, 2.5% TFA by sonication for 10 min. Athird extraction was done by adding 10 µL of ACN andsonicating for 5 min. The supernatants were collected in a freshprotein low bind tube, frozen in liquid nitrogen, and reducedto a volume of 10-20 µL in a SpeedVac. The peptides werebound to C18ZipTips (Millipore) according to the manufac-turer’s instructions and eluted with 50% ACN, 0.1% TFA. A totalof 0.5 µL of sample was spotted onto a stainless steel MALDItarget plate by the dried droplet method. The matrix used was2.5 mg/mL R-cyano-4-hydroxycinnamic acid in 50% ACN, 0.1%TFA.
2.4.1.5. MALDI Analysis. Mass spectra were acquired usinga 4700 Proteomics Analyzer (MALDI-TOF/TOF) (Applied Bio-systems). Measurements were performed with a 355 nm Nb:YAG laser in positive reflector mode with a 20 kV accelerationvoltage. The mass range (m/z 900-4000) was externally cali-brated using the peptide calibration standard III (Applied
Biosystems). For each MS and MS/MS spectrum, 3000 lasershots were accumulated. Tandem mass spectrometry wasperformed by CID with air as collision gas. Precursor masseswere selected in a data-dependent manner using the 8 mostabundant ions excluding trypsin autolytic and common keratinpeptide masses. Two missed tryptic cleavages were allowed anda mass accuracy of 65 ppm was used for the searches andwithin 0.3 u for MS/MS. Spectra acquisition and processingwas done in an automatic fashion with the 4000 Series Explorersoftware (version 3.6, Applied Biosystems).
The GPS Explorer Software (version 3.6, Applied Biosystems)was used for spectra analyses. Database search was performedwith MASCOT (Version: 2.2.06) using the mouse UniRef100version from 20090718 (selected for Mus musculus, 78 401entries) and Swiss-Prot databases (Swiss-Prot version from20090212). We set carbamidomethylation of cysteine as thefixed modification and oxidized methionine and deamidationof asparagine and glutamine as variable modifications. Thevariable 30 Da modification methylol was set for the lysine,histidine, and arginine when searching the database. Precursortolerance was set to 75 ppm and MS/MS fragment toleranceto 0.3 Da.
2.4.2. Gel-Free Approach.2.4.2.1. In-Solution Digestion. The extracted proteins were
precipitated with cold acetone O/N, resuspended in 50 mMammonium bicarbonate (100 mM, pH 8.0) and 0.2% RapiGest(Waters). The protein mixture was reduced with TBP (10 mM)and DTT (25 mM) at 60 °C for 30 min and alkylated withiodoacetamide (50 mM) in the dark for 30 min at roomtemperature before digestion with trypsin at 1:20 (w/w) O/Nat 37 °C.
2.4.2.2. In-Tissue Digestion. Tryptic digestion was per-formed using detergent-free extraction buffer21 with a slightmodification as follows. FFPE tissue was deparaffinized andrehydrated as described before and the sections were incubatedin 100 µL of ammonium bicarbonate (100 mM, pH 8.0) at 80°C for 2 h, A total of 100 µL of trifluoroethanol (TFE) was addedand the sample was sonicated twice for 20 s on ice, heated for1 h at 60 °C, and sonicated again twice for 20 s on ice. Theprotein mixture was reduced with TBP (10 mM) and DTT (25mM) at 60 °C for 30 min, followed by alkylation with iodoac-etamide (50 mM) for 20 min at room temperature in the dark.The reduced and alkylated sample was digested with trypsinas in section 2.4.2.1.
The digested peptides from both in-solution and in-tissuedigestion were lyophilized before LC MS/MS analysis.
2.4.2.3. LC-MS/MS Analysis. For the identification of pro-teins, SDS-PAGE bands were cut out and in-gel digestion wasperformed as described before. The digested peptides wereseparated by reversed phase chromatography (PepMap, 15 cm×75 µm i.d., 3 µm/100 Å pore size, LC Packings) operated ona nano-HPLC (Ultimate 3000, Dionex) with a nonlinear 170 mingradient using 2% acetonitrile in 0.1% formic acid in water (A)and 0.1% formic acid in 98% acetonitrile (B) as eluted with aflow rate of 250 nL/min. The gradient settings were subse-quently: 0-140 min, 2-30% B; 140-150 min, 31-99% B;151-160 min, Stay at 99% B and equilibrate for 10 min atstarting conditions. The nano-LC was connected to a linearquadrupole ion trap-Orbitrap (LTQ Orbitrap XL) mass spec-trometer (ThermoFisher, Bremen, Germany) equipped with anano-ESI source. The mass spectrometer was operated in thedata-dependent mode to automatically switch between Orbi-trap-MS and LTQ-MS/MS acquisition. Survey full scan MS
research articles Azimzadeh et al.
4712 Journal of Proteome Research • Vol. 9, No. 9, 2010

spectra (from m/z 300 to 1500) were acquired in the Orbitrapwith resolution R ) 60 000 at m/z 400 (after accumulation toa target of 1 000 000 charges in the LTQ). The method usedallowed sequential isolation of the most intense ions, up to 10,depending on signal intensity, for fragmentation on the linearion trap using collision-induced dissociation at a target valueof 100 000 ions. High resolution MS scans in the Orbitrap andMS/MS scans in the linear ion trap were performed in parallel.Target peptides already selected for MS/MS were dynamicallyexcluded for 30 s. General mass spectrometry conditions were:electrospray voltage, 1.25-1.4 kV; no sheath and auxiliary gasflow. Ion selection threshold was 500 counts for MS/MS, andan activation Q-value of 0.25 and activation time of 30 ms werealso applied for MS/MS.
All MS/MS data were analyzed using MASCOT (MatrixScience) Version: 2.2.06 and Sequest (ThermoFinnigan, SanJose, CA; version SRF v. 5). Sequest was set up to search themouse.fasta database UniRef100 20090718 (8 663 575 se-quences; 3 067 231 997 residues) and Swiss-Prot 20090708(495 880 sequences; 174 780 353 residues) assuming the diges-tion enzyme trypsin and with a fragment ion mass toleranceof 1 Da and a parent ion tolerance of 10 PPM. Iodoacetamidederivative of cysteine as stable and oxidation of methionine,deamidation of arginine and glutamine as well as hydroxylmodification of lysine, arginine, and histidines as variablemodifications were specified in Sequest and MASCOT asvariable modifications.
Scaffold (version Scaffold_2_02_03, Proteome Software, Inc.,Portland, OR) was used to validate MS/MS-based peptide andprotein identifications. Peptide identifications were acceptedif they could be established at greater than 80.0% probabilityas specified by the Peptide Prophet algorithm.27 Proteinidentifications were accepted if they could be established atgreater than 95.0% probability and contained at least 2 identi-fied peptides. Protein probabilities were assigned by the ProteinProphet algorithm.28 Proteins that contained similar peptidesand could not be differentiated based on MS/MS analysis alonewere grouped to satisfy the principles of parsimony.
2.5. Immunoblot Analysis. Proteins separated by SDS-PAGEwere transferred to nitrocellulose membranes (GE Healthcare)using a TE 77 semidry blotting system (GE Healthcare) at 1mA/cm for 1 h .29 The membranes were blocked using 3% BSAin PBS, pH 7.4, for 1 h at room temperature, washed three timesin 10 mM Tris-HCl, pH 7.4, 150 mM NaCl for 5 min, andincubated O/N at 4 °C with antibodies against actin (Santa CruzBiotechnology), cardiac heavy chain myosin (Affinity Bioreagent),14-3-3 protein (Abcam), or tubulin (Sigma Aldrich) usingdilutions recommended by the manufacturer. After washingthree times as described above, the blots were incubated withhorseradish peroxidise-conjugated anti-mouse, anti-rabbit, oranti-goat secondary antibody (Santa Cruz Biotechnology) for1 h at room temperature and developed using the ECL system(GE Healthcare) following standard procedures.
Data Availability. Access to raw MS data is provided inPRIDE (http://www.ebi.ac.uk/pride). Here the PRIDE XML filesconverted with the PRIDE converter tool from the Mascot.datfiles for each MALDI spot of all relevant experiments for thispublication can be found. Accession numbers: 12227-12237.Reviewer account: Username, review72549; Password, gt9wNc_2.
3. Results and Discussion
3.1. Extraction of Proteins from FFPE Heart Tissue. Toestablish an optimized extraction method for FFPE samples,
we used C57BL/6 murine heart tissue as a model system. Forthis purpose, we used both archival samples and fresh/frozentissue for comparison.
To test the reproducibility and effectiveness of proteinextraction, different combinations of detergents, denaturants,reductants, pH values, and temperatures were examined onthe same batch of tissue material. The protein yield and the1D pattern using different extraction buffers are shown inFigure 1. The extraction buffer developed for this study (Figure1, lane l) resulted in maximal protein yield and the clearestband pattern when compared to other buffers publishedpreviously (Figure 1, lanes a-k).
To scavenge cross-linking, we used glycine in our extractionbuffer in a concentration of 200 mM. Treatment of archivaltissue with buffers containing glycine often resulted in extrabands and a smear in low molecular weight region (Figure 1,lanes e-i). However, this was not the case when glycine wasused as a component of our buffer (lane l).
It has been reported that protein recovery after extractionis affected by pH and it therefore needs to be adjusted basedon the tissue type.11 In this study, maximal yield was obtainedby using a basic Tris buffer (pH 8.8). Incubation of the FFPEsection with acidic buffer (pH 4) generated a smeared proteinpattern after separation (Figure 1, lanes e and h).
Most of the proteomics studies on FFPE have used extractionbuffers containing SDS4,11,13,30 due to its functions as adetergent and a protein denaturant. We showed that a mixtureof nonionic detergent (1% �-octylglucoside) together with 2%SDS resulted in optimal protein release from FFPE sections;increasing the amount of SDS (>4%) did not enhance theprotein yield further, on the contrary, it generated a diffusedprotein pattern on 1DE (not shown).
Treatment of FFPE tissue with disulfide reducing agents suchas DTT, TBP, and �-mercaptoethanol during extraction resultedin an enhanced protein recovery. However, we discovered thatthe majority of the detected proteins remained as cross-linkedprotein complexes (see below), indicating that extraction inreducing conditions did not efficiently reverse the intra- andintermolecular cross-linking resulting from formalin fixation.
The optimal extraction conditions were incubation underconstant shaking at 100 °C for 20 min followed by incubationat 80 °C for 2 h. The benefit of using high temperatures for theproteinextractionfromFFPEtissuehasbeenshownpreviously.6,7,12
Heating may renature the cross-linked proteins by a partialthermal hydrolysis of methylene bridges.23,31
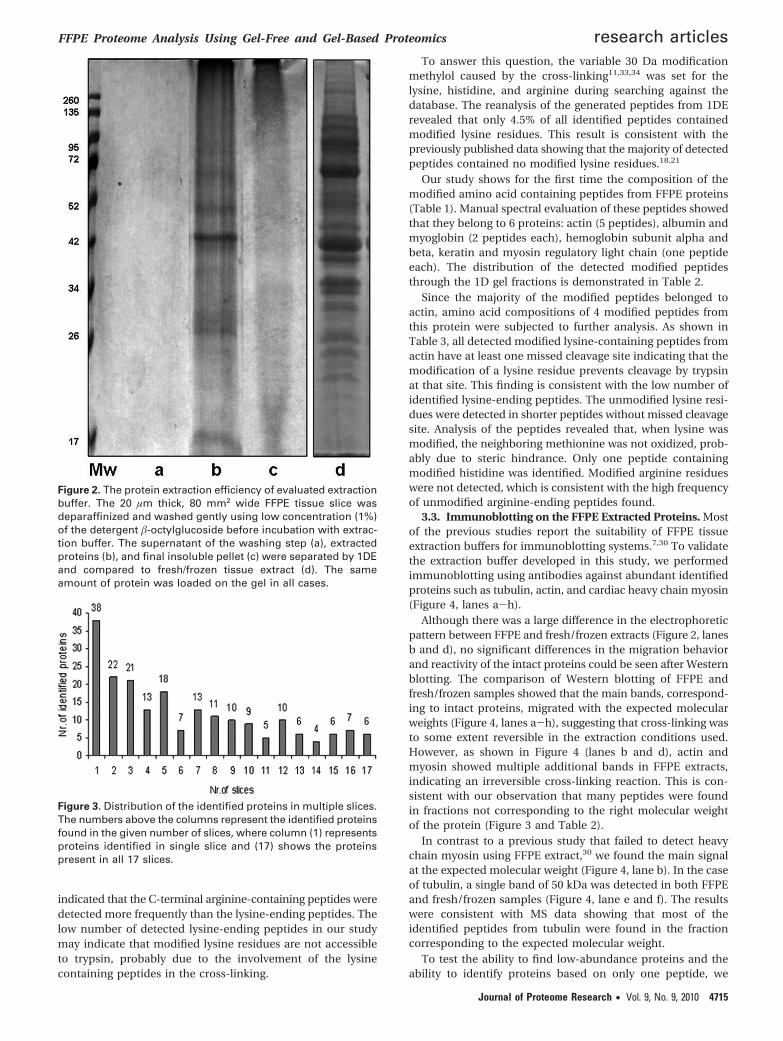
The average protein yield from a 20 µm thick, 80 mm2 widetissue slice was 48 µg/slice, a yield significantly greater thanwhen using the commercial buffer (25 µg/slice). The extractionefficiency is demonstrated in Figure 2 where, after the firstprotein extraction using our buffer (lane b), the insolublematerial was resuspended in the same buffer and separatedwith 1D-PAGE (lane c). Lane c had no significant proteincontent, indicating an efficient extraction was completed in thefirst step.
An additional washing step using a low concentration (1%)of �-octylglucoside improved the resolution of the 1D pattern.The analysis of the supernatant of this washing step by 1D gelshowed no protein bands, suggesting that proteins were notreleased during the washing (Figure 2, lane a).
3.2. 1DE Protein Separation and Protein Identification. Toanalyze the quality of the electrophoretic separation using 1DSDS-PAGE, the patterns of FFPE and fresh/frozen tissue werecompared (Figure 2, lanes b and d). Although we have shown
FFPE Proteome Analysis Using Gel-Free and Gel-Based Proteomics research articles
Journal of Proteome Research • Vol. 9, No. 9, 2010 4713

(Figures 1 and 2) that using the buffer developed in this studyresults in a relatively clear 1DE pattern and efficient extractionof archival tissue proteins, the quality and the quantity of theproteins are clearly inferior when compared to those of fresh/frozen tissue. Clearly visible bands migrating in the molecularweight range 10-150 kDa were present in both fresh/frozenand FFPE sampes on silver-stained 1D SDS-PAGE, but most ofthe protein bands from FFPE tissue appeared less sharp thanthose from fresh/frozen tissue (Figure 2, lanes b and d). Anintense signal in the higher molecular weight region suggestedthe accumulation of cross-linked proteins. The low quality ofthe FFPE protein pattern separated on SDS-PAGE has beenreported previously.13,23,30 However, in contrast to previousstudies where no intact molecular mass bands were observed,we were able to show distinct bands even in the high molecularweight region.
To determine the integrity and compatibility of the FFPEextracted proteins with mass spectrometry approach, the 1 DEgel, consisting of protein bands in the range of 10-200 kDa,was cut into 17 slices (Figure 1) and digested with trypsin. Thefractions were analyzed by LC-MS/MS as described in section2.4.2.3. A total of 1312 unique peptides were isolated; basedon them, 192 proteins were identified by two or more peptides(Supplementary Table 1). The number of identified proteinscould be increased significantly by manually validating the dataof the proteins identified by a single peptide. An example ofthis is the 14-3-3-like protein. Although this protein wasidentified by only one peptide, the MS/MS analysis showedgood sequence coverage of the y and b ion series (see Sup-porting Information).
The theoretical molecular weight of the identified proteinsranged from 8 kDa (mitochondrial ATP synthase subunit e) upto 312 kDa (fibrillin 1). The isoelectric point (pI) values ranged
from 4.09 (calsequestrin) to 12.02 (ribosomal protein L28).Approximately 75% of the identified proteins had a pI between4 and 7.
Only 18% of the identified proteins migrated in a singlefraction corresponding to the predicted molecular weight(Figure 3, lane 1), suggesting that they were in an intactmonomeric form. The remaining (82%) were present either ashigher molecular weight moieties, suggesting formaldehydecross-linking, or were smaller than predicted, indicating proteinfragmentation due to heating, aging, or possible contact withproteases during the fixation and embedding (Figure 3, lanes2-17). The MS/MS analysis showed that the high molecularweight region of the 1D gel was enriched for identified peptidesoriginating from proteins with unexpected molecular weight.However, the “top hit” identified proteins in each fractionmigrated with the correct molecular weight. Peptides originat-ing from the most abundant proteins such as actin, ATPsynthase subunits, and albumin, or contaminating proteinssuch as hemoglobin and keratin, were found in all 17 fractions(Figure 3, lane 17). Tandem mass spectrometry showed thatin most of the cases the number of identifications correspond-ing to the number of identified spectra was considerably lowerthan 30%, indicating the low intensity of the detected peaks.This is a general problem originating from the low quality ofthe FFPE sample and may not be totally overcome by anymeasures. 1DE fractions 4-9 corresponding to the 50-90 kDamolecular regions resulted in quantitatively and qualitativelybest proteins identifications by MS/MS analysis.
Krause et al. observed a general preferential detection oftryptic peptides with the C-terminal arginine over lysine inMALDI MS.32 In FFPE tissue, the percentage of lysine residuesis even lower (about 10%) than that found in fresh/frozentissue.18 In good agreement with previously published datausing FFPE,18,21 a comparison of the identified peptides
Figure 1. Comparing the electrophoretic pattern of extracted protein using different detergent containing buffers. Proteins from FFPEmouse heart were extracted using different extraction buffers and 20 µg of FFPE extracted proteins was separated on 12% 1D-PAGEunder reducing condition before staining by coomassie (lines a-l); Tris buffer containing 1% �-octylglucoside (a), rehydration buffercontaining 2% CHAPS (b), Laemmli buffer containing 2% SDS (c), RIPA buffer containing 2% SDS and 1% NP40 (d), acidic Tris buffercontaining Glycine, 2% SDS (e) or 0.2% Tween 20 (h), neutral Tris buffer containing Glycine, 2% SDS (f) or 0.2% Tween 20 (g), basicTris buffer containing 2% SDS (or 0.2% Tween 20 and Glycine with DTT (i) and without DTT (j), commercial extraction buffer (k) andextraction buffer used in this study containing 20 mM Tris-HCl, pH 8.8, 2% SDS, 1% �-octylglucoside, 200 mM DTT, 200 mM glycineand a mixture of protease inhibitor cocktail (l). The numbers indicate the gel slices were cut for ESI-LC-MS/MS analysis.
research articles Azimzadeh et al.
4714 Journal of Proteome Research • Vol. 9, No. 9, 2010
indicated that the C-terminal arginine-containing peptides weredetected more frequently than the lysine-ending peptides. Thelow number of detected lysine-ending peptides in our studymay indicate that modified lysine residues are not accessibleto trypsin, probably due to the involvement of the lysinecontaining peptides in the cross-linking.
To answer this question, the variable 30 Da modificationmethylol caused by the cross-linking11,33,34 was set for thelysine, histidine, and arginine during searching against thedatabase. The reanalysis of the generated peptides from 1DErevealed that only 4.5% of all identified peptides containedmodified lysine residues. This result is consistent with thepreviously published data showing that the majority of detectedpeptides contained no modified lysine residues.18,21
Our study shows for the first time the composition of themodified amino acid containing peptides from FFPE proteins(Table 1). Manual spectral evaluation of these peptides showedthat they belong to 6 proteins: actin (5 peptides), albumin andmyoglobin (2 peptides each), hemoglobin subunit alpha andbeta, keratin and myosin regulatory light chain (one peptideeach). The distribution of the detected modified peptidesthrough the 1D gel fractions is demonstrated in Table 2.
Since the majority of the modified peptides belonged toactin, amino acid compositions of 4 modified peptides fromthis protein were subjected to further analysis. As shown inTable 3, all detected modified lysine-containing peptides fromactin have at least one missed cleavage site indicating that themodification of a lysine residue prevents cleavage by trypsinat that site. This finding is consistent with the low number ofidentified lysine-ending peptides. The unmodified lysine resi-dues were detected in shorter peptides without missed cleavagesite. Analysis of the peptides revealed that, when lysine wasmodified, the neighboring methionine was not oxidized, prob-ably due to steric hindrance. Only one peptide containingmodified histidine was identified. Modified arginine residueswere not detected, which is consistent with the high frequencyof unmodified arginine-ending peptides found.
3.3. Immunoblotting on the FFPE Extracted Proteins. Mostof the previous studies report the suitability of FFPE tissueextraction buffers for immunoblotting systems.7,30 To validatethe extraction buffer developed in this study, we performedimmunoblotting using antibodies against abundant identifiedproteins such as tubulin, actin, and cardiac heavy chain myosin(Figure 4, lanes a-h).
Although there was a large difference in the electrophoreticpattern between FFPE and fresh/frozen extracts (Figure 2, lanesb and d), no significant differences in the migration behaviorand reactivity of the intact proteins could be seen after Westernblotting. The comparison of Western blotting of FFPE andfresh/frozen samples showed that the main bands, correspond-ing to intact proteins, migrated with the expected molecularweights (Figure 4, lanes a-h), suggesting that cross-linking wasto some extent reversible in the extraction conditions used.However, as shown in Figure 4 (lanes b and d), actin andmyosin showed multiple additional bands in FFPE extracts,indicating an irreversible cross-linking reaction. This is con-sistent with our observation that many peptides were foundin fractions not corresponding to the right molecular weightof the protein (Figure 3 and Table 2).
In contrast to a previous study that failed to detect heavychain myosin using FFPE extract,30 we found the main signalat the expected molecular weight (Figure 4, lane b). In the caseof tubulin, a single band of 50 kDa was detected in both FFPEand fresh/frozen samples (Figure 4, lane e and f). The resultswere consistent with MS data showing that most of theidentified peptides from tubulin were found in the fractioncorresponding to the expected molecular weight.
To test the ability to find low-abundance proteins and theability to identify proteins based on only one peptide, we
Figure 2. The protein extraction efficiency of evaluated extractionbuffer. The 20 µm thick, 80 mm2 wide FFPE tissue slice wasdeparaffinized and washed gently using low concentration (1%)of the detergent �-octylglucoside before incubation with extrac-tion buffer. The supernatant of the washing step (a), extractedproteins (b), and final insoluble pellet (c) were separated by 1DEand compared to fresh/frozen tissue extract (d). The sameamount of protein was loaded on the gel in all cases.
Figure 3. Distribution of the identified proteins in multiple slices.The numbers above the columns represent the identified proteinsfound in the given number of slices, where column (1) representsproteins identified in single slice and (17) shows the proteinspresent in all 17 slices.
FFPE Proteome Analysis Using Gel-Free and Gel-Based Proteomics research articles
Journal of Proteome Research • Vol. 9, No. 9, 2010 4715

validated the MS/MS data of 14-3-3-like protein by Westernblotting. The immunoblotting detected a single signal of 27 kDacorresponding to the expected molecular weight in both FFPE(Figure 4, lane g) and fresh/frozen (lane h) protein extracts.This confirmed the compatibility of the extraction method andthe subsequent mass spectrometry strategy for identificationof very low abundance proteins.
3.4. Separation and Identification of Proteins by IEF-2DE. Although previously not proven to be a successful methodto analyze FFPE material, conventional 2D electrophoresis forprotein separation was used as a comparison with othermethods. Figure 5a shows that only a few spots ranging from20 to 80 kDa were detected on the 2D-PAGE gel after silverstaining. Most of the detected spots were migrating in the acidicarea, while the basic proteins were missing in comparison tothe fresh/frozen tissue (Figure 5b). Thirty-four spots wereexcised and subjected to MALDI MS/MS identification after in-gel digestion. Mass spectrometry analysis identified 9 proteins(Table 4). Intense protein spots were identified at the correct
molecular weight and pI. Five identified proteins were presentin multiple spots (a total of 15 spots) showing the potentialprotein modifications.
The comparison with previously published results using 2DE-PAGE to analyze FFPE material shows that the extraction bufferused in this study improves the separation; it has been shownthat proteins extracted by rehydration buffer containing urea andCHAPS were not resolved and no clear spot pattern wasdetected.5,9,10 Hood et al. concluded that the classical 2DEmethodology for proteins extracted from FFPE tissue yieldedessentially no results.22 Our study showed a comparable patternto that published recently by Addis et al.14 However, as we showclear benefits from other strategies, we suggest that classical 2D-PAGE is not the optimal method for a proteomic analysis of FFPE.
3.5. Separation and Identification of Proteins by LIEF-2DE. The lack of basic proteins on 2D gel samples may becaused by insolubility of the protein extract, a high percentageof detergent in the sample, and/or incomplete isoelectricfocusing in the strip. To test the latter, the extracted proteins
Table 1. The Sequences of the Modified Peptides after 1DE ESI LC-MS/MSa
protein name sequence
MASCOTMOWSEion score
MASCOTidentity
scoreobserved
mass actual mass charge
Actin (K)YPIEHGIITNWDDMEK(I) 28.0 33.0 664.3128 1989.9151 3(K)EITALAPSTMKIK(I) 31.0 29.0 716.9082 1431.8008 2(R)MQKEITALAPSTMK(I) 59.0 33.0 797.9133 1593.811 2(R)HQGVMVGMGQKDSYVGDEAQSK(R) 32.0 32.0 799.6987 2396.0728 3(R)DLTDYLMKILTER(G) 46.0 33.0 820.9328 1639.85 2
Albumin (K)ATAEQLKTVMDDFAQFLDTCCK(A) 29.0 24.0 745.3006 1488.5856 2(R)ENYGELADCCTK(Q) 42.0 33.0 874.7348 2621.1811 3
Hemoglobin subunit beta-1 (R)YFDSFGDLSSASAIMGNAK(V) 98.0 33.0 1,005.96 2009.9054 2Hemoglobin subunit alpha (R)MFASFPTTKTYFPHFDVSHGSAQVK(G) 35.0 35.0 954.131 2859.3697 3Myoglobin (R)HSGDFGADAQGAMSK(A) 86.0 28.0 754.826 1507.6364 2
(K)HGCTVLTALGTILK(K) 34.0 30.0 757.4252 1512.8348 2Myosin regulatory light chain 2 (K)EAPGPINFTVFLTMFGEK(L) 40.0 33.0 1,014.51 2027.0094 2Keratin, type cytoskeletal 2 (R)TAAENEFVGLK(K) 44.0 33.0 604.81 1207.6078 2
a The modified amino acids are shown in bold.
Table 2. Distribution of the Modified Amino Acids Containing Peptides in 17 1DE Slicesa
slice
no. proteins description accession number Mw (kDa) 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17
1 Actin Q3TG92 42 0 0 1 4 5 0 1 6 2 0 0 0 1 0 0 1 02 Hemoglobin subunit alpha Q91VB8 15 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 03 Hemoglobin subunit beta-1 A8DV59 16 1 0 1 1 1 0 1 0 0 0 0 0 1 1 0 0 04 Keratin, type II cytoskeletal 2 P04104 63 0 0 0 1 1 0 0 0 0 0 0 1 0 0 0 1 15 Myoglobin P04247 17 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 16 Myosin regulatory light chain 2 P51667 19 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 07 Serum albumin P07724 69 0 0 0 0 2 0 0 0 0 0 0 0 0 0 0 0 0
a The number of unique peptides containing the modified amino acids is shown.
Table 3. The Identified FFPE Heart Proteins after IEF-2DE-MALDI-MS/MS Analysisa
no. protein description accession number Mw (kDa) protein score pIsequencecoverage
uinque peptidesmatched
1 Actin alpha Q3TG92 42 198 5.23 19% 72 Alpha-Crystallin B chain P23927 20 110 6.76 29% 83 ATP synthase subunit alpha Q03265 55 131 9.22 16% 74 ATP synthase subunit beta P56480 56 244 5.19 24% 135 ATP synthase subunit O Q9DB20 23 790 10.00 29% 106 Hemoglobin subunit beta-1 A8DV59 15 59 7.9 21% 57 Myosin light chain 3 P09542 225 283 5.03 43% 118 Myosin regulatory light chain 2 P51667 18 102 4.86 27% 139 Troponin Isoform A1 P50752 34 118 5.16 22% 7
a Theoretical isoelectric points and molecular weights are derived from the amino acid sequences in Swiss-Prot.
research articles Azimzadeh et al.
4716 Journal of Proteome Research • Vol. 9, No. 9, 2010

were focused in liquid phase. The proteins were separated in10 fractions by LIEF and stained with silver (Figure 5c). Theoverall pattern of focused proteins was comparable to that ofthe 2D PAGE, the proteins mainly being detected in the acidic
fractions. The accumulation of the proteins on the top of theeach lane suggested precipitation of protein complexes. Thevisible bands from LIEF-1D PAGE were analyzed by MALDIMS/MS. Twenty-four bands were cut for the tryptic cleavage
Figure 4. Immunoblotting analysis of FFPE extracted proteins. The extracted proteins from fresh/frozen (lanes a, c, e, and g) and FFPE(lines b, d, f, and h) tissue samples were separated by 12% SDS-PAGE and analyzed by immunoblotting using anti-myosin (a and b),anti-actin (c and d), anti-tubulin (e and f), and anti-14-3-3(g and h). The same amount of protein was used in all cases.
Figure 5. Comparing of the 2D electrophoretic pattern of FFPE extracts using different focusing methods. A total of 200 µg of totalextracted proteins from FFPE samples was focused on pH gradient 3-10 NL using either IEF (a) or LIEF (c) and separated on 2D PAGE.Protein detection was performed by staining with silver. The FFPE electrophoretic patterns were then compared with 200 µg of thefresh/frozen tissue extracts (b) focused on 3-10 NlL strip and separated under the identical condition.
Table 4. The Identified FFPE Heart Proteins after LIEF-1DE-MALDI-MS/MS Analysisa
no. protein description accession number Mw (kDa) protein score pIsequencecoverage
unique peptidesmatched
1 Actin alpha Q3TG92 42 423 5.23 63% 322 ATP synthase subunit alpha Q03265 59 138 9.22 44% 263 ATP synthase subunit beta P56480 56 523 5.19 45% 304 Hemoglobin subunit beta-1 A8DV59 15 59 7.9 21% 55 Keratin, type II cytoskeletal P04104 60 520 8.51 32% 146 Myosin light chain 3 P09542 225 140 5.03 67% 287 Serum albumin P07724 70 900 5.75 9% 6
a Theoretical isoelectric points and molecular weights are derived from the amino acid sequences in Swiss-Prot.
FFPE Proteome Analysis Using Gel-Free and Gel-Based Proteomics research articles
Journal of Proteome Research • Vol. 9, No. 9, 2010 4717

and 7 proteins were identified with significant scores (Table5). Mass spectrometry analysis showed that a significantnumber of peptides generated from abundant proteins suchas actin subunits and ATP synthase subunit alpha wereidentified several times in unexpected positions. Proteins suchas albumin, myosin light chain, and keratin were found inexpected molecular weight and pI but with low sequencecoverage. Most of the protein identifications could be verifiedonly by single MS/MS, indicating the poor quality and quantityof the FFPE samples. The absence of the basic proteins on both2D PAGE and LIEF strongly suggests the involvement of basicamino acid containing proteins in the cross-linking reaction.
We reanalyzed the MS data of peptides originating from actinand appearing at unexpected molecular weight regions usingLIEF-1DE. The methylol modification was used as described
above. The analysis confirmed that the same modified lysineresidues were detected as in 1DE analysis (not shown).
The separation of FFPE proteins using LIEF is presentedfor the first time in this study. However, the low resolutionof the protein bands makes LIEF-2DE a suboptimal separa-tion method for this material. Furthermore, MALDI/TOF-MS analysis in both strip and liquid focusing approachesshowed that the intensity of low-abundance proteins wassuppressed by the presence of dominant peptides of abun-dant proteins making the use of MALDI-TOF/TOF for FFPEprotein identification problematic.
3.6. Protein Identification with Gel-Free Approach. Weused gel-free proteomics performing either in-tissue or in-solution tryptic digestion. The tryptic peptides thus generatedwere analyzed by LC-MS/MS. The numbers of proteins identi-
Table 5. The Shared Identified FFPE Heart Proteins after In-Solution and In-Tissue Digestiona
identified unique peptides
no. protein description accession number Mw (kDa) pIin-tissuedigstion
in-solutiondigestion
1 2-oxoglutarate dehydrogenase E1 component, mitochondrial Q60597 116 6.36 3 12 Acetyl-Coenzyme A acyltransferase 2 Q3TIT9 42 8.33 4 33 Aconitate hydratase, mitochondrial Q99KI0 85 8.08 4 24 Actin alpha Q3TG92 42 5.23 7 65 Actinin alpha 2 Q8K3Q4 104 5.34 8 36 ADP/ATP translocase 1 P48962 33 9.73 3 47 ATP synthase subunit alpha, mitochondrial Q03265 60 9.22 6 118 ATP synthase subunit beta, mitochondrial P56480 56 5.19 7 99 Citrate synthase, mitochondrial Q9CZU6 52 8.72 3 1
10 Cytochrome b-c1 complex subunit 2, mitochondrial Q9DB77 48 9.26 1 211 Cytochrome c oxidase subunit 6B1 P56391 10 8.96 2 012 Desmin n ) 2 P31001 53 5.21 3 313 Dihydrolipoyllysine-residue acetyltransferase component of
pyruvate dehydrogenase complex, mitochondrialQ8BMF4 68 8.81 2 0
14 Electron transfer flavoprotein subunit alpha, mitochondrial Q99LC5 35 8.62 2 315 Enoyl-CoA hydratase/isomerase Q8BH95 50 9.61 2 016 Fatty acid-binding protein, heart P11404 15 6.11 2 117 Hemoglobin subunit alpha Q91VB8 15 7.97 1 218 Hemoglobin subunit beta-1 A8DV59 16 7.85 4 519 Histone H2A type 1-F Q64426 14 11.05 1 220 Histone H2B type 1-F P10853 14 10.31 0 221 Isocitrate dehydrogenase [NADP], mitochondrial P54071 51 8.88 4 522 Long-chain specific acyl-CoA dehydrogenase, mitochondrial P51174 48 8.53 3 323 Malate dehydrogenase, mitochondrial P08249 36 8.93 3 724 M-protein O55124 165 5.51 2 025 Myoglobin n ) 2 P04247 17 7.06 3 526 Myosin binding protein C, cardiac A2AGQ1 141 6.18 3 027 Myosin light chain 3 P09542 22 5.03 4 428 Myosin regulatory light chain 2, ventricular/cardiac muscle
isoformP51667 19 4.86 2 1
29 Myosin-6 Q02566 224 5.57 32 930 Myozenin-2 Q9JJW5 30 8.53 2 031 NADH dehydrogenase (Ubiquinone) 1 alpha subcomplex Q6GTD3 43 9.75 2 132 NADH dehydrogenase [ubiquinone] iron-sulfur protein 3,
mitochondrialQ9DCT2 30 6.67 2 0
33 NADH-ubiquinone oxidoreductase 75 kDa subunit Q3TIU7 80 5.51 2 034 Putative uncharacterized protein Q9D6U7 43 6.58 5 035 Sarcoplasmic/endoplasmic reticulum calcium ATPase 2 O55143 115 5.23 2 036 Serum albumin P07724 69 5.75 4 337 Succinate dehydrogenase [ubiquinone] flavoprotein subunit,
mitochondrialQ8K2B3 73 7.06 2 2
38 Triosephosphate isomerase P17751 27 6.9 2 039 Tropomyosin1 alpha Q8BP43 33 4.69 2 240 Troponin T, cardiac muscle P50752 36 4.98 0 341 Tubulin Q3TIZ0 50 4.96 2 0
a Theoretical isoelectric points and molecular weights are derived from the amino acid sequences in Swiss-Prot.
research articles Azimzadeh et al.
4718 Journal of Proteome Research • Vol. 9, No. 9, 2010

fied were 111 and 93 for the in-tissue and in-solution digestedsamples, respectively. Almost one-third of the proteins wereidentified by at least two peptides; 45% of identified proteinsoverlapped between the two approaches (Table 5). The slightlyhigher number of proteins identified after in-tissue digestionmay result from disturbance by detergents in the case of thein-solution digestion. Several factors such as denaturation,solubility of the proteins, and accessibility of tryptic sites mayinfluence the number of identified peptides when using eitherin-tissue or in-solution digestion. The question which methodis superior may also be tissue-specific.
Again, less peptides with a C-terminal lysine residue weredetected in comparison to arginine-ending peptides. Analysisof the MS/MS data showed that 45% of all detected spectraresulted in a protein identification. The theoretical molecu-lar weight of the identified proteins varied between 10 kDa(cytochrome C oxidase) and 224 kDa (myosin). The pI valuesranged from 4.7 (tropomyosin 1 alpha) to 11.05 (histone H2Atype 1-F).
The number of proteins identified by gel-free approach wassmaller than by 1DE combined with LC-MS/MS, the gel-basedseparation step presumably reducing the complexity of thesample in a beneficial manner. The gel-free approach couldequally benefit from the introduction of an additional frac-tionation step such as Reverse phase or Strong Cation Exchangechromatography preceding the mass spectrometry analysis.
Recently, several studies have been published using in-tissuedigestion with a commercial extraction buffer; most of thesestudies applied sophisticated downstream mass spectrometrymethods to identify FFPE peptides.15,20,22,23 Although these gel-free proteomic studies provided a large number of identifiedproteins, most identifications were based on a single peptideassignment22,23 or used nonarchival formalin-fixed sampleswithout paraffin embedding,18,22 all these parameters probablyinfluencing the number of identified proteins in a positivemanner.
3.7. Overlapping of Protein Profiles Using Different Sepa-ration Methods. The Venn diagram shown in Figure 6 dem-onstrates the overlap among of the proteins identified by thedifferent approaches. Only proteins identified by at least twopeptides are shown in the figure. The largest number ofproteins was identified by LC-MS/MS after 1DE separation ofproteins. Almost all of the identified proteins using othermethods were also found by the 1DE/LC-MS/MS approach.Five abundant proteins, namely, actin, ATP synthase alpha andbeta subunits, hemoglobin subunit beta, and myosin light chainwere identified by all methods. All proteins identified by IEF-2DE were also detected by other methods.
4. Conclusions
This study provides extraction and separation methods forFFPE material that are compatible with downstream proteom-ics analyses. The protocol validated in this study using differentgel-free and gel-based approaches enables highly efficientextraction of proteins and peptides from FFPE tissues, gener-ates reproducible protein patterns, and indicates potentialmodification sites on the peptides.
We confirm previous results showing that LC/MS basedmethods are in general superior to 2D/MS based methods. Thestudy presents evidence of cross-linked complexes, consistingespecially of basic proteins, and protein fragmentation that forthe first time gives an explanation to problems seen with 2Dgel-based methods. We have developed an optimized extraction
procedure using a new buffer in combination with 1D and LC/MS; this resulted in the greatest number of peptides identified.Differences in the FFPE tissue, experimental procedure, anddownstream mass spectrometry approaches make direct com-parisons between published studies difficult. Nevertheless, theeasy-to-use, reproducible, rapid and relatively inexpensiveextraction and separation method presented here is, consider-ing the number of reliably identified proteins in the FFPEmaterial, comparable with the best data published so far.
Abbreviations: ESI-MS/MS, electrospray ionization tandemmass spectrometry; HPLC, high performance liquid chroma-tography; IEF, isoelectric focusing; LIEF, liquid isoelectricfocusing; MS; mass spectrometry; MALDI TOF/TOF, matrixassisted laser desorption/ionisation time of flight; Mw, Molec-ular weight LC-MS, liquid chromatography-mass spectrometry;SDS, sodium dodecyl sulphate; O/N, overnight; pI, isoelectricpoint.
Acknowledgment. The research leading to these resultsis supported by a grant from the European Community’s Sev-enth Framework Programme (EURATOM) contract no. 232628(STORE).
Supporting Information Available: The identifiedFFPE heart proteins after 1DE-ESI-LCMS/ MS analysis; MS/MS analysis of peptide 1532.7 from 14-3-3 protein. This materialis available free of charge via the Internet at http://pubs.acs.org.
References(1) Tapio, S.; Atkinson, M. J. Molecular information obtained from
radiobiological tissue archives: achievements of the past andvisions of the future. Radiat Environ Biophys 2008, 47 (2), 183–7.
(2) Aerni, H. R.; Cornett, D. S.; Caprioli, R. M. High-throughputprofiling of formalin-fixed paraffin-embedded tissue using parallelelectrophoresis and matrix-assisted laser desorption ionizationmass spectrometry. Anal. Chem. 2009, 81 (17), 7490–5.
(3) Conti, C. J.; Larcher, F.; Chesner, J.; Aldaz, C. M. Polyacrylamidegel electrophoresis and immunoblotting of proteins extracted fromparaffin-embedded tissue sections. J Histochem Cytochem 1988,36 (5), 547–50.
Figure 6. Venn diagram of proteins identified from different FFPEheart protein separations by different approaches. The proteinsused to provide the diagram were identified by two or morepeptides.
FFPE Proteome Analysis Using Gel-Free and Gel-Based Proteomics research articles
Journal of Proteome Research • Vol. 9, No. 9, 2010 4719

(4) Ikeda, K.; Monden, T.; Kanoh, T.; Tsujie, M.; Izawa, H.; Haba, A.;Ohnishi, T.; Sekimoto, M.; Tomita, N.; Shiozaki, H.; Monden, M.Extraction and analysis of diagnostically useful proteins fromformalin-fixed, paraffin-embedded tissue sections. J HistochemCytochem 1998, 46 (3), 397–403.
(5) Ahram, M.; Flaig, M. J.; Gillespie, J. W.; Duray, P. H.; Linehan,W. M.; Ornstein, D. K.; Niu, S.; Zhao, Y.; Petricoin, E. F., 3rd;Emmert-Buck, M. R. Evaluation of ethanol-fixed, paraffin-embed-ded tissues for proteomic applications. Proteomics 2003, 3 (4), 413–21.
(6) Yamashita, S.; Okada, Y. Mechanisms of heat-induced antigenretrieval: analyses in vitro employing SDS-PAGE and immunohis-tochemistry. J Histochem Cytochem 2005, 53 (1), 13–21.
(7) Becker, K. F.; Schott, C.; Hipp, S.; Metzger, V.; Porschewski, P.;Beck, R.; Nahrig, J.; Becker, I.; Hofler, H. Quantitative proteinanalysis from formalin-fixed tissues: implications for translationalclinical research and nanoscale molecular diagnosis. J Pathol 2007,211 (3), 370–8.
(8) Becker, K. F.; Kremmer, E.; Eulitz, M.; Becker, I.; Handschuh, G.;Schuhmacher, C.; Muller, W.; Gabbert, H. E.; Ochiai, A.; Hirohashi,S.; Hofler, H. Analysis of E-cadherin in diffuse-type gastric cancerusing a mutation-specific monoclonal antibody. Am. J. Pathol.1999, 155 (6), 1803–9.
(9) Bellet, V.; Boissiere, F.; Bibeau, F.; Desmetz, C.; Berthe, M.; Rochaix,P.; Maudelonde, T.; Mange, A.; Solassol, J., Proteomic analysis ofRCL2 paraffin-embedded tissues. J Cell Mol Med 2007.
(10) Ono, A.; Kumai, T.; Koizumi, H.; Nishikawa, H.; Kobayashi, S.;Tadokoro, M. Overexpression of heat shock protein 27 in squa-mous cell carcinoma of the uterine cervix: a proteomic analysisusing archival formalin-fixed, paraffin-embedded tissues. HumPathol 2009, 40 (1), 41–9.
(11) Fowler, C. B.; Cunningham, R. E.; O’Leary, T. J.; Mason, J. T. ‘Tissuesurrogates’ as a model for archival formalin-fixed paraffin-embed-ded tissues. Lab Invest 2007, 87 (8), 836–46.
(12) Shi, S. R.; Liu, C.; Balgley, B. M.; Lee, C.; Taylor, C. R. Proteinextraction from formalin-fixed, paraffin-embedded tissue sections:quality evaluation by mass spectrometry. J Histochem Cytochem2006, 54 (6), 739–43.
(13) Nirmalan, N. J.; Harnden, P.; Selby, P. J.; Banks, R. E. Developmentand validation of a novel protein extraction methodology forquantitation of protein expression in formalin-fixed paraffin-embedded tissues using western blotting. J Pathol 2009, 217 (4),497–506.
(14) Addis, M. F.; Tanca, A.; Pagnozzi, D.; Rocca, S.; Uzzau, S. 2-D PAGEand MS analysis of proteins from formalin-fixed, paraffin-embed-ded tissues. Proteomics 2009, 9 (18), 4329–39.
(15) Negishi, A.; Masuda, M.; Ono, M.; Honda, K.; Shitashige, M.; Satow,R.; Sakuma, T.; Kuwabara, H.; Nakanishi, Y.; Kanai, Y.; Omura, K.;Hirohashi, S.; Yamada, T. Quantitative proteomics using formalin-fixed paraffin-embedded tissues of oral squamous cell carcinoma.Cancer Sci 2009, 100 (9), 1605–11.
(16) Xiao, Z.; Li, G.; Chen, Y. ; Li, M.; Peng, F. ; Li, C. ; Li, F.; Yu, Y.;Ouyang, Y. ; Xiao, Z. ; Chen, Z. , Quantitative Proteomic Analysisof Formalin-fixed and Paraffin-embedded Nasopharyngeal Car-cinoma Using iTRAQ Labeling, Two-dimensional Liquid Chroma-tography, and Tandem Mass Spectrometry. J Histochem Cytochem.
(17) Hwang, S. I.; Thumar, J.; Lundgren, D. H.; Rezaul, K.; Mayya, V.;Wu, L.; Eng, J.; Wright, M. E.; Han, D. K. Direct cancer tissueproteomics: a method to identify candidate cancer biomarkersfrom formalin-fixed paraffin-embedded archival tissues. Oncogene2007, 26 (1), 65–76.
(18) Jiang, X.; Jiang, X.; Feng, S.; Tian, R.; Ye, M.; Zou, H. Developmentof efficient protein extraction methods for shotgun proteomeanalysis of formalin-fixed tissues. J Proteome Res 2007, 6 (3), 1038–47.
(19) Balgley, B. M.; Guo, T.; Zhao, K.; Fang, X.; Tavassoli, F. A.; Lee,C. S. Evaluation of archival time on shotgun proteomics offormalin-fixed and paraffin-embedded tissues. J Proteome Res2009, 8 (2), 917–25.
(20) Scicchitano, M. S.; Dalmas, D. A.; Boyce, R. W.; Thomas, H. C.;Frazier, K. S. Protein extraction of formalin-fixed, paraffin-embed-ded tissue enables robust proteomic profiles by mass spectrom-etry. J Histochem Cytochem 2009, 57 (9), 849–60.
(21) Sprung, R. W., Jr.; Brock, J. W.; Tanksley, J. P.; Li, M.; Washington,M. K.; Slebos, R. J.; Liebler, D. C. Equivalence of protein inventoriesobtained from formalin-fixed paraffin-embedded and frozen tissuein multidimensional liquid chromatography-tandem mass spec-trometry shotgun proteomic analysis. Mol Cell Proteomics 2009,8 (8), 1988–98.
(22) Hood, B. L.; Darfler, M. M.; Guiel, T. G.; Furusato, B.; Lucas, D. A.;Ringeisen, B. R.; Sesterhenn, I. A.; Conrads, T. P.; Veenstra, T. D.;Krizman, D. B. Proteomic analysis of formalin-fixed prostate cancertissue. Mol Cell Proteomics 2005, 4 (11), 1741–53.
(23) Guo, T.; Wang, W.; Rudnick, P. A.; Song, T.; Li, J.; Zhuang, Z.; Weil,R. J.; DeVoe, D. L.; Lee, C. S.; Balgley, B. M. Proteome analysis ofmicrodissected formalin-fixed and paraffin-embedded tissue speci-mens. J Histochem Cytochem 2007, 55 (7), 763–72.
(24) Laemmli, U. K. Cleavage of structural proteins during the assemblyof the head of bacteriophage T4. Nature 1970, 227 (5259), 680–5.
(25) Heukeshoven, J.; Dernick, R. Simplified method for silver stainingof proteins in polyacrylamide gels and the mechanism of silverstaining. ELECTROPHORESIS 1985, 6 (3), 103–12.
(26) Gharahdaghi, F.; Weinberg, C. R.; Meagher, D. A.; Imai, B. S.;Mische, S. M. Mass spectrometric identification of proteins fromsilver-stained polyacrylamide gel: a method for the removal ofsilver ions to enhance sensitivity. Electrophoresis 1999, 20 (3), 601–5.
(27) Keller, A.; Nesvizhskii, A. I.; Kolker, E.; Aebersold, R. Empiricalstatistical model to estimate the accuracy of peptide identificationsmade by MS/MS and database search. Anal. Chem. 2002, 74 (20),5383–92.
(28) Nesvizhskii, A. I.; Aebersold, R. Analysis, statistical validation anddissemination of large-scale proteomics datasets generated bytandem MS. Drug Discov Today 2004, 9 (4), 173–81.
(29) Towbin, H.; Staehelin, T.; Gordon, J. Electrophoretic transfer ofproteins from polyacrylamide gels to nitrocellulose sheets: pro-cedure and some applications. Proc Natl Acad Sci U S A 1979, 76(9), 4350–4.
(30) Addis, M. F.; Tanca, A.; Pagnozzi, D.; Crobu, S.; Fanciulli, G.; Cossu-Rocca, P.; Uzzau, S. Generation of high-quality protein extractsfrom formalin-fixed, paraffin-embedded tissues. Proteomics 2009,9 (15), 3815–23.
(31) Shi, S. R.; Cote, R. J.; Taylor, C. R. Antigen retrieval immunohis-tochemistry: past, present, and future. J Histochem Cytochem 1997,45 (3), 327–43.
(32) Krause, E.; Wenschuh, H.; Jungblut, P. R. The dominance ofarginine-containing peptides in MALDI-derived tryptic massfingerprints of proteins. Anal. Chem. 1999, 71 (19), 4160–5.
(33) Nirmalan, N. J.; Harnden, P.; Selby, P. J.; Banks, R. E. Mining thearchival formalin-fixed paraffin-embedded tissue proteome: op-portunities and challenges. Mol Biosyst 2008, 4 (7), 712–20.
(34) Metz, B.; Kersten, G. F.; Hoogerhout, P.; Brugghe, H. F.; Timmer-mans, H. A.; de Jong, A.; Meiring, H.; ten Hove, J.; Hennink, W. E.;Crommelin, D. J.; Jiskoot, W. Identification of formaldehyde-induced modifications in proteins: reactions with model peptides.J. Biol. Chem. 2004, 279 (8), 6235–43.
PR1004168
research articles Azimzadeh et al.
4720 Journal of Proteome Research • Vol. 9, No. 9, 2010




















