
iv) Management of benign bone tumours
10
(iv) Management of benign bone tumours Rob Pollock Abstract Benign primary bone tumours are rare, occurring most commonly in skel- etally immature patients, arising from cartilage or bone. The commonest locations are the distal femur, proximal tibia and proximal humerus. They present with pain, swelling or pathological fracture. Diagnosis is by plain x-rays, MRI scans and a core needle biopsy if indicated. More aggressive tumours may appear radiologically to be similar to malignant tumours. Treatment depends on the anatomical location, symptoms, the natural history of the tumour and the morbidity of treatment and in most cases involves either simple excision or curettage although occasionally it is necessary to perform a complete excision using the same principles as for malignant tumours. Keywords benign bone tumour; non-neoplastic tumour-like conditions of bone Introduction Primary bone tumours are extremely rare, accounting for only 0.2% of human tumours. 1 The majority are benign and affect the skeletally immature patient but some are difficult to distinguish from their malignant counterparts, have a significant incidence of local recurrence and may undergo malignant transformation. Diagnosis and treatment of bone tumours is complex and management of this group of patients is best undertaken at specialist centres in a multidisciplinary setting. Classification Two classification systems are commonly used, see Tables 1 and 2. The first is histological, based on the cell of origin. The second is more clinically orientated and based on the pattern of behaviour of the tumour. 2 While tumours may arise from chondrocytes, osteoblasts, osteoclasts, periosteum or soft tissue within bone such as fat or smooth muscle, some fall into a ‘‘miscellaneous’’ group and their pathogenesis is unclear. Their behaviour dictates their clinical presentation, varying from being an incidental finding on an x-ray in the case of a latent lesion to a rapidly growing, painful lesion associated with functional loss in the case of an aggressive lesion. This is reflected in Enneking’s classification. Aetiology The aetiology of the vast majority of benign bone tumours is unclear. Numerous theories have been proposed but none has ever been substantiated except multiple hereditary osteochon- dromatosis (also known as diaphyseal aclasis) which is inherited as an autosomal dominant. Two genes are responsible, namely the EXT 1 and EXT 2 genes found at the 8q24 and 11p11-12 loci respectively. Approximately 15% of patients with osteochon- dromas have the inherited form of the condition. Clinical features The clinical features are non-specific and variable. Some long standing lesions present as an incidental finding. For example, an adolescent who attends the accident and emergency department after a knee injury and a fibrous cortical defect is found on X-ray (Figure 1). Some tumours such as osteochondroma present with a long history of a painless swelling (Figure 2), but more aggressive lesions, such as giant cell tumour, usually present with a short history of a pain, swelling and loss of function. Investigation As some benign bone tumours can be difficult to distinguish from their malignant counterparts, investigation should be carried out in a multidisciplinary setting, ideally in a specialist musculo- skeletal tumour centre. 3 The bare minimum is adequate imaging by plain x-ray and a magnetic resonance imaging (MRI) scan. The radiological features may be so clear that, if compatible with the clinical picture, tissue diagnosis is not necessary before definitive treatment. However, if there is any doubt about the diagnosis, a tissue diagnosis should be obtained, which is best done by core needle biopsy using a Jamshedi needle. Management General principles Treatment is dependant on many factors, particularly: patient’s symptoms natural history of the tumour morbidity of treatment. Treatment varies from simple observation with repeat imaging through to wide excision using the same surgical principles as for malignant tumours. When treating bone tumours the surgeon has to balance excision margin against function. With wider margins, there may be greater functional loss but a lesser chance of local recurrence. Conversely, intra- lesional surgery has less morbidity but a greater risk of local recurrence. Non-operative Asymptomatic lesions that fall into the Enneking latent group of tumours can simply be observed, for example the natural history of lesions such as non-ossifying fibromas and is well documented and predictable and it is safe to leave them alone. If the diagnosis has been made on imaging alone and non-operative treatment has been chosen, obviously histological confirmation of the diagnosis is not going to be available. Thus it is advisable to repeat the plain x-ray after three to six months to ensure that the tumour is showing no signs of progression. If radiological or clinical progression does occur, then there must be a low threshold for biopsy. Rob Pollock BSc (Hons) FRCS (Tr & Orth) Consultant Orthopaedic Surgeon, Royal National Orthopaedic Hospital, Stanmore, UK. MINI-SYMPOSIUM: ORTHOPAEDIC ONCOLOGY ORTHOPAEDICS AND TRAUMA 23:4 248 Ó 2009 Elsevier Ltd. All rights reserved.
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of iv) Management of benign bone tumours

MINI-SYMPOSIUM: ORTHOPAEDIC ONCOLOGY
(iv) Management of benignbone tumoursRob Pollock
AbstractBenign primary bone tumours are rare, occurring most commonly in skel-
etally immature patients, arising from cartilage or bone. The commonest
locations are the distal femur, proximal tibia and proximal humerus. They
present with pain, swelling or pathological fracture. Diagnosis is by plain
x-rays, MRI scans and a core needle biopsy if indicated. More aggressive
tumours may appear radiologically to be similar to malignant tumours.
Treatment depends on the anatomical location, symptoms, the natural
history of the tumour and the morbidity of treatment and in most cases
involves either simple excision or curettage although occasionally it is
necessary to perform a complete excision using the same principles as
for malignant tumours.
Keywords benign bone tumour; non-neoplastic tumour-like conditions
of bone
Introduction
Primary bone tumours are extremely rare, accounting for only
0.2% of human tumours.1 The majority are benign and affect the
skeletally immature patient but some are difficult to distinguish
from their malignant counterparts, have a significant incidence of
local recurrence and may undergo malignant transformation.
Diagnosis and treatment of bone tumours is complex and
management of this group of patients is best undertaken at
specialist centres in a multidisciplinary setting.
Classification
Two classification systems are commonly used, see Tables 1 and 2.
The first is histological, based on the cell of origin. The second is
more clinically orientated and based on the pattern of behaviour of
the tumour.2
While tumours may arise from chondrocytes, osteoblasts,
osteoclasts, periosteum or soft tissue within bone such as
fat or smooth muscle, some fall into a ‘‘miscellaneous’’ group
and their pathogenesis is unclear. Their behaviour dictates their
clinical presentation, varying from being an incidental finding on
an x-ray in the case of a latent lesion to a rapidly growing, painful
lesion associated with functional loss in the case of an aggressive
lesion. This is reflected in Enneking’s classification.
Aetiology
The aetiology of the vast majority of benign bone tumours is
unclear. Numerous theories have been proposed but none has
Rob Pollock BSc (Hons) FRCS (Tr & Orth) Consultant Orthopaedic Surgeon,
Royal National Orthopaedic Hospital, Stanmore, UK.
ORTHOPAEDICS AND TRAUMA 23:4 24
ever been substantiated except multiple hereditary osteochon-
dromatosis (also known as diaphyseal aclasis) which is inherited
as an autosomal dominant. Two genes are responsible, namely
the EXT 1 and EXT 2 genes found at the 8q24 and 11p11-12 loci
respectively. Approximately 15% of patients with osteochon-
dromas have the inherited form of the condition.
Clinical features
The clinical features are non-specific and variable. Some long
standing lesions present as an incidental finding. For example, an
adolescent who attends the accident and emergency department
after a knee injury and a fibrous cortical defect is found on X-ray
(Figure 1). Some tumours such as osteochondroma present with
a long history of a painless swelling (Figure 2), but more
aggressive lesions, such as giant cell tumour, usually present
with a short history of a pain, swelling and loss of function.
Investigation
As some benign bone tumours can be difficult to distinguish from
their malignant counterparts, investigation should be carried out
in a multidisciplinary setting, ideally in a specialist musculo-
skeletal tumour centre.3 The bare minimum is adequate imaging
by plain x-ray and a magnetic resonance imaging (MRI) scan.
The radiological features may be so clear that, if compatible with
the clinical picture, tissue diagnosis is not necessary before
definitive treatment. However, if there is any doubt about the
diagnosis, a tissue diagnosis should be obtained, which is best
done by core needle biopsy using a Jamshedi needle.
Management
General principles
Treatment is dependant on many factors, particularly:
� patient’s symptoms
� natural history of the tumour
� morbidity of treatment.
Treatment varies from simple observation with repeat
imaging through to wide excision using the same surgical
principles as for malignant tumours. When treating bone
tumours the surgeon has to balance excision margin against
function. With wider margins, there may be greater functional
loss but a lesser chance of local recurrence. Conversely, intra-
lesional surgery has less morbidity but a greater risk of local
recurrence.
Non-operative
Asymptomatic lesions that fall into the Enneking latent group of
tumours can simply be observed, for example the natural history
of lesions such as non-ossifying fibromas and is well documented
and predictable and it is safe to leave them alone. If the diagnosis
has been made on imaging alone and non-operative treatment
has been chosen, obviously histological confirmation of the
diagnosis is not going to be available. Thus it is advisable to
repeat the plain x-ray after three to six months to ensure that the
tumour is showing no signs of progression. If radiological or
clinical progression does occur, then there must be a low
threshold for biopsy.
8 � 2009 Elsevier Ltd. All rights reserved.

MINI-SYMPOSIUM: ORTHOPAEDIC ONCOLOGY
Curettage
Curettage is the treatment of choice for the majority of benign
bone tumours requiring surgical intervention. It is, by definition,
intralesional surgery. The intention is to achieve macroscopically
clear margins, accepting that microscopic disease is likely to be
left behind.
The technique involves exposing the affected bone and
creating a bone window with osteotomes. The window needs to
be big enough to obtain an adequate view of the tumour but
small enough to ensure that the grafting material can be con-
tained within the bone at the end of the procedure. It is helpful to
use an image intensifier per-operatively to ensure that the
curettage has cleared the entire lesion.
After curettage, dependant on the histology of the tumour, its
anatomical location of the tumour, the age of the patient, the
Classification of tumour by histological subtype
Cell type Tumour subtype
Chondrocyte Enchondroma
Osteochondroma
Chondroblastoma
Chondromyxoid fibroma
Osteoblast Osteoid osteoma
Osteoblastoma
Osteoclast Giant cell tumour
Periosteum Periosteal chondroma
Bizarre periosteal
osteochondromatous proliferation
Vascular Haemangioma
Fat Lipoma
Smooth muscle Leiomyoma
Miscellaneous Simple bone cyst
Aneurysmal bone cyst
Langerhans cell histiocytosis
Fibrous dysplasia
Osteofibrous dysplasia
Fibrous cortical defect
Non ossifying fibroma
Table 1
ORTHOPAEDICS AND TRAUMA 23:4 249
likelihood of local recurrence and the risk of pathological frac-
ture, the resulting cavity may be left unfilled or filled with iliac
crest autograft, morcellised allograft, synthetic bone substitute or
polymethylmethacrylate cement (PMMA). Additionally, the
curetted bone can be augmented with internal fixation to prevent
a pathological fracture (Figure 4).
After curettage some tumours have a higher incidence of local
recurrence than others, such as giant cell tumours, osteo-
blastomas, chondroblastomas and aneurysmal bone cysts. When
treating these, it has been shown that using some form of
Figure 1 Fibrous cortical defect in the medial aspect of the proximal tibia.
Classification of tumour by biological behaviour according to Enneking2
Classification Behaviour Example of tumour
Latent Slow growth with spontaneous healing. Often incidental
finding on x-ray. No treatment required
Fibrous cortical defect, non ossifying fibroma,
Active Progressive growth over time and usually symptomatic.
Treatment of choice; curettage. Low incidence of local recurrence
Chondromyxoid fibroma, enchondroma, LCH,
simple bone cyst
Aggressive Rapid growth of tumour often extending beyond periosteum into
the soft tissues. Treatment of choice curettage or excision.
10e15% chance of local recurrence
Chondroblastoma, osteoblastoma, giant
cell tumour, aneurysmal bone cyst
Table 2
� 2009 Elsevier Ltd. All rights reserved.
MINI-SYMPOSIUM: ORTHOPAEDIC ONCOLOGY
adjuvant treatment reduces the risk of local recurrence.4 These
include treating the cavity wall with a high speed burr, phenol,
hydrogen peroxide or liquid nitrogen and filling the defect with
PMMA. Which of these adjuvants is used depends on the sur-
geon’s preference, but the incidence of local recurrence after
curettage of these more aggressive tumours is 10e15% even with
adjuvant treatment.
Radiofrequency ablation (RFA)
RFA is the technique of choice for small, symptomatic lesions
less than 1.5 cm in diameter e.g. osteoid osteomas. It causes
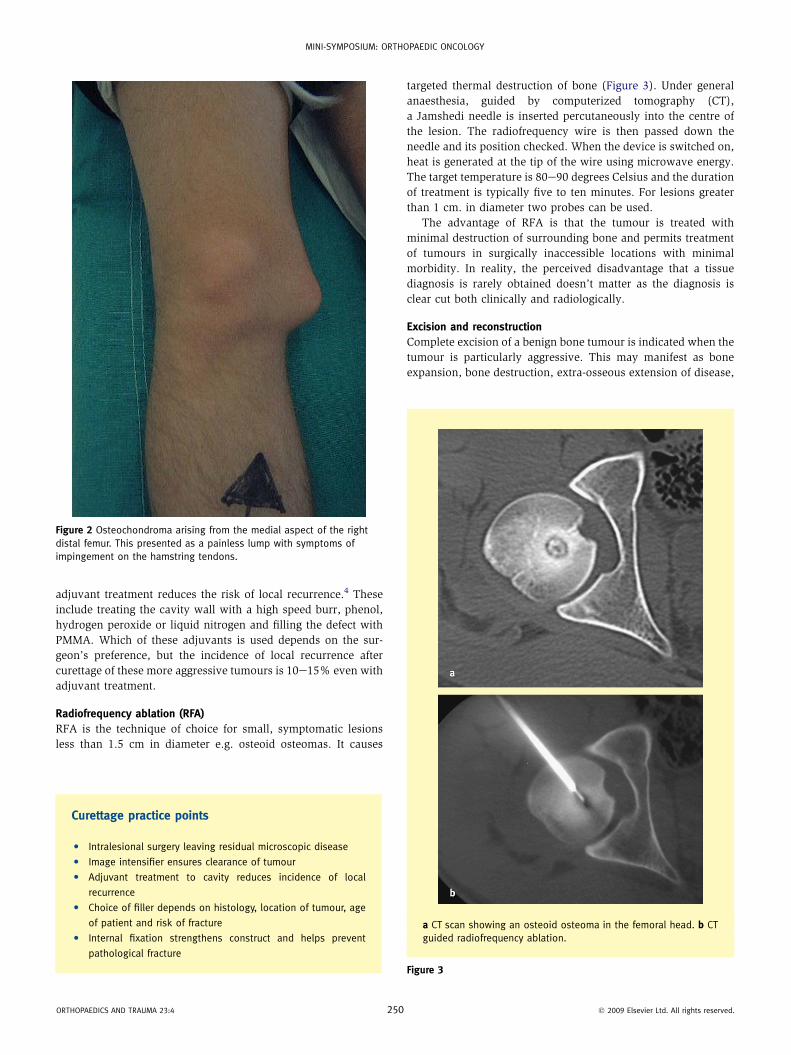
Figure 2 Osteochondroma arising from the medial aspect of the right
distal femur. This presented as a painless lump with symptoms of
impingement on the hamstring tendons.
Curettage practice points
C Intralesional surgery leaving residual microscopic disease
C Image intensifier ensures clearance of tumour
C Adjuvant treatment to cavity reduces incidence of local
recurrence
C Choice of filler depends on histology, location of tumour, age
of patient and risk of fracture
C Internal fixation strengthens construct and helps prevent
pathological fracture
ORTHOPAEDICS AND TRAUMA 23:4 250
targeted thermal destruction of bone (Figure 3). Under general
anaesthesia, guided by computerized tomography (CT),
a Jamshedi needle is inserted percutaneously into the centre of
the lesion. The radiofrequency wire is then passed down the
needle and its position checked. When the device is switched on,
heat is generated at the tip of the wire using microwave energy.
The target temperature is 80e90 degrees Celsius and the duration
of treatment is typically five to ten minutes. For lesions greater
than 1 cm. in diameter two probes can be used.
The advantage of RFA is that the tumour is treated with
minimal destruction of surrounding bone and permits treatment
of tumours in surgically inaccessible locations with minimal
morbidity. In reality, the perceived disadvantage that a tissue
diagnosis is rarely obtained doesn’t matter as the diagnosis is
clear cut both clinically and radiologically.
Excision and reconstruction
Complete excision of a benign bone tumour is indicated when the
tumour is particularly aggressive. This may manifest as bone
expansion, bone destruction, extra-osseous extension of disease,
a CT scan showing an osteoid osteoma in the femoral head. b CT
guided radiofrequency ablation.
Figure 3
� 2009 Elsevier Ltd. All rights reserved.

MINI-SYMPOSIUM: ORTHOPAEDIC ONCOLOGY
pathological fracture or combinations of the above. If the tumour
is subarticular, there may even be no alternative but to sacrifice
the joint.
aPlain x-ray showingahaemangiomaof theproximal femur. Thepatient
is at risk of pathological fracture. b Post-operative x-ray following
curettage and internal fixationwith a dynamic condylar screwandplate.
Figure 4
ORTHOPAEDICS AND TRAUMA 23:4 25
The surgical principles are similar to those used when treating
a malignant bone tumour. The intention is to perform a marginal
but complete excision and remove both macroscopic and
microscopic disease. After a section of bone has been excised,
some form of reconstruction will be necessary, which may be
a biological reconstruction such as a vascularised fibula graft or
a massive endoprosthesis (Figure 5). The reconstruction used is
dependant on many factors, particularly anatomical location and
the individual surgeon’s preferences.
Management of specific tumours
Cartilage tumours
Osteochondroma (synonym: exostosis) Osteochondromas are
cartilage capped bony projections with a marrow cavity contin-
uous with the underlying bone. They account for approximately
35% of all benign bone tumours and present within the first three
decades of life.5 Most occur close to the physis of long bones but
they can also arise from flat bones such as the scapula and pelvis.
The commonest sites are the distal femur and proximal tibia,
followed by the proximal humerus and proximal fibula. Symp-
toms are most commonly mechanical and caused by impinge-
ment of the osteochondroma on nearby tendons, especially
around the knee.
In about 15% of cases they are multiple, a condition known as
diaphyseal aclasis (Figure 6), which in most of cases are familial
and inherited as an autosomal dominant, but sporadic cases are
well recognised. The genetic defect again appears to be in the
tumour suppressor genes EXT1 and EXT2.
Malignant transformation of a solitary osteochondroma into
chondrosarcoma occurs in approximately 1% of cases and up to
3% patients with diaphyseal aclasis.5 The warning signs include
rapid increase in size of an osteochondroma associated with
increasing pain. Of particular concern are pelvic osteochon-
dromas that may be large and silent until they transform. If there
is any suspicion of malignant transformation the osteochon-
droma should be imaged with an MRI scan and the cartilage cap
assessed. Cartilage caps greater than 1 cm suggest a high prob-
ability of malignant transformation.
Surgical excision of osteochondromas is usually curative but it
is important to excise the cartilage cap in its entirety or local
recurrence is likely. In patients with diaphyseal aclasis it is
impractical to excise every osteochondroma and surgery is
reserved for those which are symptomatic, unsightly or which
are suspicious for malignant transformation.
Enchondroma Enchondromas are benign, intramedullary
cartilage neoplasms which account for approximately 10e25% of
all benign bone tumours and present at any age.5 They mainly
affect the long bones and are most commonly solitary. The hands
and feet are most commonly affected followed by the proximal
humerus and proximal and distal femur.
While enchondromas in the larger long bones such as the
humerus or femur may be asymptomatic and discovered inci-
dentally, the most common presentation is with a palpable
swelling on the hands or feet. Pain may or may not be present
and some are associated with a pathological fracture.
The radiological features of enchondromas of the hands and
feet are those of a well defined, radiolucent lesion exhibiting
punctate mineralization, and may be associated with bone
1 � 2009 Elsevier Ltd. All rights reserved.

MINI-SYMPOSIUM: ORTHOPAEDIC ONCOLOGY
expansion. They are usually ‘‘hot’’ on bone scan. In larger bones
chondral tissue is seen in the metaphyseal region. Lesions greater
than 5 cm in length and causing endosteal scalloping on MRI
should be considered as possible low-grade chondrosarcoma.
The majority of enchondromas are successfully treated with
curettage and the local recurrence rate is extremely low.
Ollier’s disease and Mafucci syndrome Ollier’s disease is
a developmental disorder characterized by multiple
Osteochondroma practice points
C Commonest benign bone tumour
C Most commonly seen in the metaphyseal region of long bones
C Distal femur and proximal tibia are the commonest sites
C 15% cases are multiple
C Autosomal dominant inheritance
C 1% risk of malignant transformation in solitary cases (3% if
multiple)
C Surgical excision if symptomatic, unsightly or suspicious for
malignant transformation
ORTHOPAEDICS AND TRAUMA 23:4 25
enchondromas in the long bones of the hands, feet and limbs.
When associated with soft tissue or visceral haemangiomas the
condition is known as Mafucci syndrome (Figure 7). The aeti-
ology is unclear; most cases are sporadic rather than inherited.
The condition usually presents in early childhood with lumps in
the hands and feet, limb deformity and/or multiple pathological
fractures.
Diagnosis is based on the clinical picture and x-ray appear-
ances. Treatment is aimed at maintaining function, preventing
deformity and careful surveillance in order to pick up malignant
transformation early. The incidence of malignant transformation
into chondrosarcoma is approximately 15e30% in patients with
Ollier’s and probably even greater in those with Mafucii’s.6
Chondroblastoma Chondroblastoma is a cartilage producing
tumour typically arising in the epiphysis of skeletally immature
patients. There is a slight male preponderance. 75% occur in
long bones and the commonest sites are the proximal and distal
femur, the proximal tibia and the proximal humerus.7
Symptoms vary from mild pain of many years duration to
recent onset of severe pain. Clinically, patients may develop an
effusion in the hip or knee associated with stiffness. Plain x-rays
show a well-defined, lytic lesion within the epiphysis and MRI
a Lateral view of the wrist showing a GCT of the distal radius. b Axial T1 weighted MRI scan showing expansion of the distal radius with cortical
thinning. The extensor tendons are closely applied to the pseudo-capsule of the lesion. c A free, vascularised fibula graft has been inserted with
a paddle of soft tissue to aid wound closure. d One year post-operative x-ray showing complete bone union and successful wrist arthrodesis.
Figure 5
2 � 2009 Elsevier Ltd. All rights reserved.

MINI-SYMPOSIUM: ORTHOPAEDIC ONCOLOGY
shows a sharply demarcated chondral lesion with surrounding
oedema (Figure 8). Core needle biopsy will confirm the diagnosis.
Curettage is the treatment of choice and is successful in
80e90%.
Chondromyxoid fibroma This is one of the least common bone
tumours. It most commonly affects the long bones, particularly
around the knee, of patients in their second and third decades.
The clinical presentation is very similar to a chondroblastoma
and the treatment and prognosis is also the same.
Bone tumours
Osteoid osteoma This is a small, bone forming tumour which
can occur in any bone but is most commonly seen in the prox-
imal femur (See Figure 3). It usually affects skeletally immature
patients but it is also seen in young adults. Histologically the
features are the same as an osteoblastoma.8
The clinical presentation is classic with a history of constant,
unremitting pain often worse at night, which usually responds to
aspirin and non-steroidal anti-inflammatory drugs (NSAIDs).
The site of the lesion often determines the clinical findings; in
the spine it may cause scoliosis, in fingers it may cause signifi-
cant swelling and loss of function and if the lesion is sub-
articular, it may cause a joint effusion. The differential diagnosis
includes Brodie’s abscess, stress fracture or subtle area of
osteomyelitis.
Figure 6 Classic appearances of diaphyseal aclasis.
ORTHOPAEDICS AND TRAUMA 23:4 25
They are too small to biopsy and the diagnosis is made on the
basis of clinical presentation and imaging. The findings of scle-
rosis on plain x-ray, high uptake on bone scan or single photon
emission computed tomography (SPECT), a nidus on CT and the
presence of oedema on MRI are enough to make the diagnosis.
The treatment of choice is percutaneous, CT guided radio-
frequency ablation described above.4 The success rate in most
series is 90e95% after a single treatment and 100% after two
treatments. The complication rate is extremely low.9
Osteoblastoma This is a rare, bone forming tumour that typically
affects young adults aged 10e30 years. It is twice as common in
men as women. Over 50% of cases affect the posterior elements
of the spine, pelvis or the sacrum (Figure 9). In the rest of the
skeleton the proximal and distal femur and the proximal tibia are
the commonest sites.10
The clinical features are similar to those of an osteoid osteoma
i.e. chronic pain. Imaging reveals a well defined, lytic lesion
somewhere between 3 and 10 cm in diameter. Some osteo-
blastomas are associated with aneurysmal bone cyst (ABC)
change. Osteoblastomas can be hard to differentiate from oste-
oblastic osteosarcoma and thus core needle biopsy is recom-
mended before definitive treatment is carried out.
The treatment of choice is curettage and cementation. The
prognosis is good and the incidence of local recurrence after
curettage is low.
Figure 7 AP x-ray of the foot showing multiple enchondromas and a hae-
mangioma in the 4th web space. These are the features of Mafuccci
syndrome.
3 � 2009 Elsevier Ltd. All rights reserved.
MINI-SYMPOSIUM: ORTHOPAEDIC ONCOLOGY
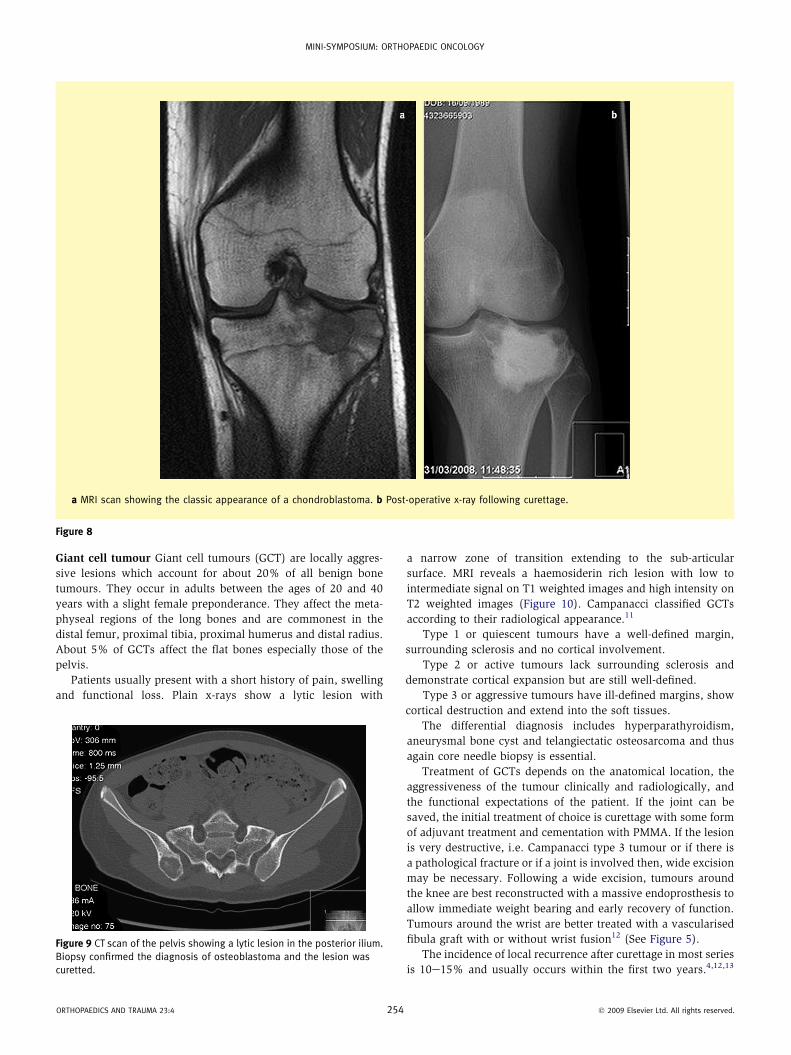
a MRI scan showing the classic appearance of a chondroblastoma. b Post-operative x-ray following curettage.
Figure 8
Giant cell tumour Giant cell tumours (GCT) are locally aggres-
sive lesions which account for about 20% of all benign bone
tumours. They occur in adults between the ages of 20 and 40
years with a slight female preponderance. They affect the meta-
physeal regions of the long bones and are commonest in the
distal femur, proximal tibia, proximal humerus and distal radius.
About 5% of GCTs affect the flat bones especially those of the
pelvis.
Patients usually present with a short history of pain, swelling
and functional loss. Plain x-rays show a lytic lesion with
Figure 9 CT scan of the pelvis showing a lytic lesion in the posterior ilium.
Biopsy confirmed the diagnosis of osteoblastoma and the lesion was
curetted.
ORTHOPAEDICS AND TRAUMA 23:4 25
a narrow zone of transition extending to the sub-articular
surface. MRI reveals a haemosiderin rich lesion with low to
intermediate signal on T1 weighted images and high intensity on
T2 weighted images (Figure 10). Campanacci classified GCTs
according to their radiological appearance.11
Type 1 or quiescent tumours have a well-defined margin,
surrounding sclerosis and no cortical involvement.
Type 2 or active tumours lack surrounding sclerosis and
demonstrate cortical expansion but are still well-defined.
Type 3 or aggressive tumours have ill-defined margins, show
cortical destruction and extend into the soft tissues.
The differential diagnosis includes hyperparathyroidism,
aneurysmal bone cyst and telangiectatic osteosarcoma and thus
again core needle biopsy is essential.
Treatment of GCTs depends on the anatomical location, the
aggressiveness of the tumour clinically and radiologically, and
the functional expectations of the patient. If the joint can be
saved, the initial treatment of choice is curettage with some form
of adjuvant treatment and cementation with PMMA. If the lesion
is very destructive, i.e. Campanacci type 3 tumour or if there is
a pathological fracture or if a joint is involved then, wide excision
may be necessary. Following a wide excision, tumours around
the knee are best reconstructed with a massive endoprosthesis to
allow immediate weight bearing and early recovery of function.
Tumours around the wrist are better treated with a vascularised
fibula graft with or without wrist fusion12 (See Figure 5).
The incidence of local recurrence after curettage in most series
is 10e15% and usually occurs within the first two years.4,12,13
4 � 2009 Elsevier Ltd. All rights reserved.

MINI-SYMPOSIUM: ORTHOPAEDIC ONCOLOGY
a Plain x-ray showing a giant cell tumour of the proximal tibia. Note the narrow zone of transition and extension of tumour up to the sub-chondral
region. b MRI scan showing classical appearances of a GCT.
Figure 10
Some studies have suggested that bisphosphonates post-opera-
tively may reduce the incidence of local recurrence.14
Despite them being histologically benign, pulmonary metas-
tases are seen in up to 2% of patients with giant cell tumours, on
average three to four years after diagnosis. Most grow slowly,
some regress and some progress leading to the death of the
patient. True malignant transformation of a giant cell tumour is
extremely rare.
Miscellaneous lesions
Non-ossifying fibroma/fibrous cortical defect By convention,
a fibrous cortical defect measures less than 1 cm in diameter and
a non-ossifying fibroma is larger than 1 cm but histologically
these two conditions are identical (Figure 5). Their aetiology is
GCT practice points
C Locally aggressive
C Distal femur, proximal tibia, proximal humerus, distal radius
C Differential diagnosis ABC, telangiectatic osteosarcoma,
hyperparathyroidism
C Campanacci types 1, 2 & 3
C Treat with curettage and some form of adjuvant
C 10e15% local recurrence rate
C Bisphosphonates may reduce incidence of local recurrence
ORTHOPAEDICS AND TRAUMA 23:4 255
unclear. They fall into the latent category by Enneking’s classi-
fication and are most commonly seen as an incidental finding in
skeletally immature patients. They are rarely symptomatic and
can be safely observed. Occasionally a large non-ossifying
fibroma can cause a stress fracture in the adjacent bone in which
case curettage and internal fixation are indicated.
Simple bone cyst (synonym: unicameral bone cyst) Simple
bone cysts (SBC) are fluid filled, metaphyseal cysts of unclear
aetiology. They are most commonly seen in the fist two decades
of life with a male to female ratio of 3:1. The proximal femur,
proximal tibia and proximal humerus account for 90% of cases.15
Most patients present with a pathological fracture after
minimal trauma (Figure 11). Plain X-rays are diagnostic and
show a metaphyseal lucency often extending into the diaphysis
but never involving the epiphysis. The cortex is usually thin and
may be mildly expanded but never beyond the width of the
physis. If a fracture is present a ‘‘fallen fragment’’ sign may be
seen.
Numerous methods of treating SBCs have been described
including; aspiration and injection with steroid, injection with
bone marrow, inserting a cannulated screw to decompress the
cyst, fixation with Nancy nails and curettage combined with
internal fixation.16 Even after curettage recurrence rates of
10e20% are reported. By the time the patient is skeletally mature
the cyst will have healed. The treatment modality chosen
depends partly on the anatomical location of the cyst. Cysts of
weight bearing bones such as those in the femoral neck are more
� 2009 Elsevier Ltd. All rights reserved.

MINI-SYMPOSIUM: ORTHOPAEDIC ONCOLOGY
frequently curetted, grafted and internally fixed than those in the
non- weight bearing bones.
Aneurysmal bone cyst Aneurysmal bone cysts (ABC) are mul-
tilocular, expansile, aggressive, destructive lesions that may
expand into the soft tissues. They are most common in the first
two decades of life and typically affect the metaphyseal regions of
the long bones (Figure 12). They can arise de novo or they can
complicate other benign tumours i.e. they show ‘‘ABC change’’.
Most patients present with a short history of pain and
swelling. The characteristic radiological findings are those of an
eccentric, expansile, well-defined, lytic lesion on plain x-ray with
the presence of multiple fluid-fluid levels on MRI. The differential
diagnosis includes giant cell tumour and telangiectatic osteosar-
coma. Biopsy should be attempted although, frequently, only
blood clot is obtained.
The majority of ABCs are suitable for curettage and grafting.
They can be extremely vascular and it is advisable to perform
arteriography and embolisation pre-operatively in lesions where
a tourniquet cannot be used. Local recurrence occurs in
approximately 25% and is usually apparent within a year of
surgery.17 ABCs at the aggressive end of the spectrum may
require marginal but complete excision.
Langerhans cell histiocytosis. (Synonyms: Histiocytosis X,
Eosinophilic granuloma) Langerhans cell histiocytosis (LCH) is
a rare disorder that can affect any bone and be monostotic or
polyostotic. Visceral structures can also be involved. There is
a wide age range from the first to the eighth decade but 80% of
patients are under thirty.
Figure 11 Typical x-ray appearance of a simple bone cyst. Note the mild
expansion but not wider than the physis.
ORTHOPAEDICS AND TRAUMA 23:4 256
Patients present with pain and swelling and the imaging
features may be aggressive. The differential diagnosis includes
osteomyelitis and Ewing’s sarcoma and thus biopsy is essential.
Treatment varies from surveillance through to curettage and for
those with multiple sites of disease, chemotherapy.
Fibrous dysplasia Fibrous dysplasia (FD) is a developmental
abnormality rather than a tumour and may affect any bone.
Clinical presentation with pain, deformity or pathological frac-
ture can occur at any age. It may be monostotic or polyostotic.
McCune-Albright syndrome is polyostotic FD, pigmented skin
lesions resembling ‘‘the coast of Maine’’ and endocrinopathy.
X-ray appearances are diagnostic showing a ground glass
appearance of the affected bone. ABC change can occur.
Treatment is aimed at symptom control. Bisphosphonates
have been shown to be effective in reducing pain.18 Surgery is
reserved for pathological fractures or severe deformity.
Osteofibrous dysplasia Osteofibrous dysplasia (OFD) is a slow
growing, fibro-osseous lesion classically affecting the cortical
bone of the tibial diaphysis. It is most commonly seen in skele-
tally immature males. Presentation varies from an incidental
finding to a painful deformity. It is usually self limiting and
spontaneous healing often occurs. The differential diagnosis
includes adamantinoma; a low-grade malignant lesion with
similar radiological appearances. Indeed there is controversy as
to whether OFD has the potential to transform into ada-
mantinoma, but others think they are two separate entities. A
Figure 12 Plain x-ray showing an aneurysmal bone cyst. Note that the
bone is expanded beyond the width of the physis.
� 2009 Elsevier Ltd. All rights reserved.

MINI-SYMPOSIUM: ORTHOPAEDIC ONCOLOGY
REFERENCES
1 Dorfman H, Vanel D, Czerniak B, Park Y, Kotz R, Unni K. WHO classifi-
cation of bone tumours. In: Fletcher C, Unni K, Mertens F, eds. World
Health Organization classification of tumours. Pathology and genetics
of tumours of soft tissue and bone. Lyon: IARC Press; 2002. p. 226e32.
2 Enneking W. Musculoskeletal tumor surgery, vol. 1. New York:
Churchill Livingstone; 1983. 1e60.
3 Mankin H, Lange T, Spanier S. The hazards of biopsy in patients with
malignant primary bone and soft tissue tumors. J Bone Joint Surg
1982; 64A: 1121e7.
4 Balke M, Schremper L, Gebert C, et al. Giant cell tumor of bone:
treatment and outcome of 214 cases. J Cancer Res Clin Oncol 2008;
134(9): 969e78.
5 Unni K. Dahlin’s bone tumors: general aspects and data on 11,087
cases. Philadelphia: Lippincott-Raven; 1996.
6 Schwartz H, Zimmerman N, Simon M, Wroble R, Miller E, Bonfiglio M.
The malignant potential of enchondromatosis. J Bone Joint Surg Am
1987; 69: 269e74.
7 Springfield D, Capanna R, Gherlinzoni F, Picci P, Campanacci M.
Chondroblastoma. A review of seventy cases. J Bone Joint Surg Am
1985; 67: 748e55.
8 Klein M, Parisien M, Schneider-Stock R. Osteoid osteoma. In:
Fletcher C, Unni K, Mertens F, eds. World Health Organization
classification of tumours. Pathology and genetics of tumours of soft
tissue and bone. Lyon: IARC Press; 2002. p. 260e1.
9 Lindner N, Ozaki, Roedl R, Gosheger G, Winkelman W, Worler K.
Percutaneous radiofrequency ablation in osteoid osteoma. J Bone
Joint Surg 2001; 83(3): 391e6.
10 Malcolm A, Schiller A, Schneider-Stock R. Osteoblastoma. In:
Fletcher C, Unni K, Mertens F, eds. World Health Organization clas-
sification of tumours. Pathology and genetics of tumours of soft
tissue and bone. Lyon: IARC Press; 2002. p. 262e3.
11 Campanacci M, Baldini N, Boriani S, Sudanese A. Giant-cell tumor of
bone. J Bone Joint Surg Am 1987; 69: 106e14.
12 Pollock R, Stalley P, Lee K, Pennington D. Free vascularized fibula
grafts in limb salvage surgery. J Reconstr Microsurg 2005; 21(2):
79e84.
ORTHOPAEDICS AND TRAUMA 23:4 25
13 Meldenhall W, Zlotecki R, Scarborough M, Gibbs C,
Meldenhall N. Giant cell tumor of bone. Am J Clin Oncol 2006;
29(1): 96e9.
14 Tse L, Wong K, Kumta S, Huang L, Chow T, Griffith J. Bisphosphonates
reduce local recurrence in extremity giant cell tumour of bone: a case
control study. Bone 2008; 42(1): 68e73.
15 Kalil R, Araujo E. Simple bone cyst. In: Fletcher C, Unni K, Mertens F,
eds. World Health Organization classification of tumours. Pathology
and genetics of tumours of soft tissue and bone. Lyon: IARC Press;
2002. p. 340.
16 Wilkins R. Unicameral bone cysts. J Am Acad Orthop Surg 2000; 8(4):
217e24.
17 Kransdorf M, Sweet D. Aneurysmal bone cyst: concept, controversy,
clinical presentation and imaging. AJR Am J Roentgenol 1995; 164(3):
573e80.
18 DiMeglio L. Bisphosphonate therapy for fibrous dysplasia. Pediatr
Endocrinol Rev 2007; 4(Suppl 4): 440e5.
FURTHER READING
Fletcher C, Unni K, Mertens F, eds. World Health Organization classification
of tumours. Pathology and genetics of tumours of soft tissue and
bone. Lyon: IARC Press; 2002.
Research directions
C Phase 2 clinical trial studying the use of Denosumab in the
management of GCTs. This agent is a monoclonal antibody
that targets the RANK ligand, part of the signaling pathway
that stimulates osteoblasts
C Use of novel bone substitutes to promote bone healing after
curettage
C Molecular genetic studies to identify genes and chromosomal
aberrations responsible for these tumours
7 � 2009 Elsevier Ltd. All rights reserved.




















