
dspace cover page - UNSWorks
273
Taking the lead from natural products: developing synthetic protocols to probe biological systems Author: Toop, Hamish Publication Date: 2014 DOI: https://doi.org/10.26190/unsworks/2627 License: https://creativecommons.org/licenses/by-nc-nd/3.0/au/ Link to license to see what you are allowed to do with this resource. Downloaded from http://hdl.handle.net/1959.4/53861 in https:// unsworks.unsw.edu.au on 2022-05-28
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of dspace cover page - UNSWorks

Taking the lead from natural products: developing syntheticprotocols to probe biological systems
Author:Toop, Hamish
Publication Date:2014
DOI:https://doi.org/10.26190/unsworks/2627
License:https://creativecommons.org/licenses/by-nc-nd/3.0/au/Link to license to see what you are allowed to do with this resource.
Downloaded from http://hdl.handle.net/1959.4/53861 in https://unsworks.unsw.edu.au on 2022-05-28

Taking the Lead from Natural Products :
Developing Synthetic Protocols to Probe
Biological Systems
Hamish D. Toop
A Thesis Submitted in Fulfilment of the Requirements for the Degree of
Doctor of Philosophy in Chemistry
The University of New South Wales
School of Chemistry
Faculty of Sciences
July 2014

i

ii
THE UNIVERSITY OF NEW SOUTH WALES
Thesis/Dissertation Sheet
Surname or Family name: TOOP
First name: HAMISH Other name/s: DAVID
Abbreviation for degree as given in the University calendar: PhD
School: CHEMISTRY Faculty: SCIENCES
Title: Taking the Lead from Natural Products : Developing Synthetic Protocols to Probe Biological Systems
Abstract 350 words maximum:
This thesis describes the development of synthetic protocols to gain access to and develop natural products as biomedical agents. Chapter 1
provides an introduction to natural products and the processes that are used to further them into the clinic. Three case studies are described which
highlight the importance of organic synthesis as a tool to gain access to, make analogs of, and determine the biological mechanism of these
compounds.
Chapter 2 describes the total synthesis of three isoquinoline alkaloids – phylline, dioncophylline E and ancistrotectorine C. The total synthesis of
phylline was completed in 7 steps and helped identify conditions to stereoselectively generate both the cis- and trans-1,3-dimethyl-1,2,3,4-
tetrahydroisoquinoline moieties present in this class of natural products. The total syntheses of dioncophylline E and ancistrotectorine C used the
Pinhey-Barton reaction as the key step to construct their biaryl bond. The synthesis of dioncophylline E involved construction of the biaryl bond then
formation of the trans-1,3-dimethyl-1,2,3,4-tetrahydroisoquinoline moiety using the protocol developed for phylline. For the synthesis of
ancistrotectorine C, an unprecedented intact cis-1,3-dimethyl-1,2,3,4-tetrahydroisoquinolinyllead triacetate species was synthesised and this was
used in the Pinhey-Barton reaction to generate the ancistrotectorine C scaffold in one step.
Chapter 3 discusses the development of AAL(S) as a new lead for the treatment of leukaemia. To access gram scale quantities of AAL(S) for
biological testing, a modified version of Hinterding’s protocol, which uses Schöllkopf’s reagent, was examined. Modification and extension of this
protocol was used to synthesise 20 analogs. These analogs had changes to the hydrophobic tail or the amino alcohol head group of AAL(S) and they
were tested for their biological activity against leukaemia cells and against specific ceramide synthases. Pleasingly, some of these analogs were
found to be more potent and selective than AAL(S). To determine AAL(S)’s mode of action three affinity chromatography probes were synthesised.
These probes were used to identify a novel target for AAL(S) and preliminary biological work has indicated that this could have implications for the
treatment of leukaemia.
A summary of the work described and the future directions for the projects are given in Chapter 4. Full experimental procedures have been provided
in Chapter 5.
Declaration relating to disposition of project thesis/dissertation
I hereby grant to the University of New South Wales or its agents the right to archive and to make available my thesis or dissertation in whole or in part in the University libraries in all forms of media, now or here after known, subject to the provisions of the Copyright Act 1968. I retain all property rights, such as patent rights. I also retain the right to use in future works (such as articles or books) all or part of this thesis or dissertation.
I also authorise University Microfilms to use the 350 word abstract of my thesis in Dissertation Abstracts International (this is applicable to doctoral theses only).
………………………………….
Signature
……………………………………..………………
Witness
……….……………………...…….…
Date
The University recognises that there may be exceptional circumstances requiring restrictions on copying or conditions on use. Requests for restriction for a period of up to 2 years must be made in writing. Requests for a longer period of restriction may be considered in exceptional circumstances and require the approval of the Dean of Graduate Research.
FOR OFFICE USE ONLY
Date of completion of requirements for Award:

iii
ORIGINALITY STATEMENT
‘I hereby declare that this submission is my own work and to the best of my knowledge it contains
no materials previously published or written by another person, or substantial proportions of
material which have been accepted for the award of any other degree or diploma at The
University of New South Wales or any other educational institution, except where due
acknowledgement is made in the thesis. Any contribution made to the research by others, with
whom I have worked with at The University of New South Wales or elsewhere, is explicitly
acknowledged in the thesis. I also declare that the intellectual content of this thesis is the product
of my own work, except to the extent that assistance from others in the project's design and
conception or in style, presentation and linguistic expression is acknowledged.’
Signed …………………………………………………………
Date ……………………………………………………………
AUTHENTICITY STATEMENT
‘I certify that the library deposit digital copy is a direct equivalent of the final officially approved
version of my thesis. No amendation of content has occurred and if there are any minor variation
in formatting, they are the result of the conversion to digital format.’
Signed …………………………………………………………
Date ……………………………………………………………

iv
COPYRIGHT STATEMENT
‘I hereby grant the University of New South Wales or its agents the right to archive and to make
available my thesis of dissertation in whole or part in the University libraries in all forms of media,
now or here after known, subject to the revisions of the Copyright Act 1968. I retain all propriety
rights, such as patent rights. I also retain the right to use in future works (such as articles or
books) all or part of this thesis or dissertation.
I also authorise University microfilms to use the 350 word abstract of my thesis in Dissertation
Abstract International.
I have either used no substantial portions of copyright material in my thesis or I have obtained
permission to use copyright material; where permission has not been granted I have applied/will
apply for a partial restriction of the digital copy of my thesis or dissertation.’
Signed …………………………………………………………
Date ……………………………………………………………

v

vi
Acknowledgements
Foremost, I would like to thank Associate Professor Jonathan Morris for his supervision and
guidance over the course of this PhD. I have appreciated the time that you have spent with me
talking about my projects and the effort that you have put into editing this thesis. I am very
grateful for your help making the transition to Sydney a smooth one, for helping get the CTx top-
up scholarship and for helping me keep the Tim-Tams off in the gym.
Thanks must go to all past and present members of the Morris group. In particular, I would like to
thank Milena and Belinda for allowing me to stay on your floor in the beginning and for numerous
discussions about my research. Thanks to Jason for showing an ongoing interest in the project.
Thank you to our collaborators on the AAL(S) project, Matt Dun, Nicole Verrills and Anthony Don.
A special thanks to Matt Dun for helping to get my head around the biology. Thanks to Jackie and
Steve who also contributed to this work and good luck to Elysha who is carrying on this research.
Thank you to all the technical staff at UNSW. Especially, Dr. Douglas Lawes, Dr. Donald Thomas
and Dr. Adelle Amoore at the UNSW NMR Facility and Lewis Adler and Leanne Stephensen at
the UNSW bioanalytical and mass spectrometry facility. Special thanks to Doug for keeping my
bike on the road over the course of this PhD.
I would like to thank UNSW for the APA and Cancer Therapeutics CTx for the top-up scholarship.
To my Sydney family, Sam and Tom, thanks for keeping me in trouble. Some weekends the only
motivation for going to the lab was the slab of beer (or three) that was promised at the end of the
day. The frantic ‘out-of-body’ work that started at 3 when I realised you had started without me
got me through – what parrots.. “We have a whole three story house, yet we are all in the same
square metre!”
A special thank you to Julz. I am so happy that you moved to Sydney. Thank you for
understanding, keeping me focussed and pushing me towards the end. Most of all thanks for
being as silly as me.
Thanks to Carla and Megan for the snapchats………….
Finally, I would like to thank my mum and dad. The last four years could not have been possible
without your continual love and support. I have missed being away from you and have loved
coming home to see you. For everything else I cannot list I cannot thank you enough.

vii

viii
This thesis is dedicated to my parents,
With whom I could have not done this without

ix

x
Abstract
This thesis describes the development of synthetic protocols to gain access to and develop
natural products as biomedical agents. Chapter 1 provides an introduction to natural products and
the processes that are used to further them into the clinic. Three case studies are described
which highlight the importance of organic synthesis as a tool to gain access to, make analogs of,
and determine the biological mechanism of these compounds.
Chapter 2 describes the total synthesis of three isoquinoline alkaloids – phylline, dioncophylline E
and ancistrotectorine C. The total synthesis of phylline was completed in 7 steps and helped
identify conditions to stereoselectively generate both the cis- and trans-1,3-dimethyl-1,2,3,4-
tetrahydroisoquinoline moieties present in this class of natural products. The total syntheses of
dioncophylline E and ancistrotectorine C used the Pinhey-Barton reaction as the key step to
construct their biaryl bond. The synthesis of dioncophylline E involved construction of the biaryl
bond then formation of the trans-1,3-dimethyl-1,2,3,4-tetrahydroisoquinoline moiety using the
protocol developed for phylline. For the synthesis of ancistrotectorine C, an unprecedented intact
cis-1,3-dimethyl-1,2,3,4-tetrahydroisoquinolinyllead triacetate species was synthesised and this
was used in the Pinhey-Barton reaction to generate the ancistrotectorine C scaffold in one step.
Chapter 3 discusses the development of AAL(S) as a new lead for the treatment of leukaemia. To
access gram scale quantities of AAL(S) for biological testing, a modified version of Hinterding’s
protocol, which uses Schöllkopf’s reagent, was examined. Modification and extension of this
protocol was used to synthesise 20 analogs. These analogs had changes to the hydrophobic tail
or the amino alcohol head group of AAL(S) and they were tested for their biological activity
against leukaemia cells and against specific ceramide synthases. Pleasingly, some of these
analogs were found to be more potent and selective than AAL(S). To determine AAL(S)’s mode
of action three affinity chromatography probes were synthesised. These probes were used to
identify a novel target for AAL(S) and preliminary biological work has indicated that this could
have implications for the treatment of leukaemia.
A summary of the work described and the future directions for the projects are given in Chapter 4.
Full experimental procedures have been provided in Chapter 5.
xi
Abbreviations
[ α ] Specific rotation EDCI 1-Ethyl-3-(3-dimethylamino- Ǻ Angstrom(s) propyl)carbodiimide Ac Acetyl EDTA Ethylenediaminetetraacetic aq Aqueous acid Ar Aryl ee Enantiomeric excess BINAP 1,1-binaphthalene EI Electron impact Bn Benzyl eq Equivalent Boc t-butoxycarbonyl er Enantiomer ratio bp Boiling point ESI Electrospray ionisation br Broad Et Ethyl n-Bu Primary butyl FAB Fast atom bombardment s-Bu sec-Butyl FDA US Food and Drug t-Bu tert-Butyl Administration Bz Benzoyl Fmoc 9-Fluorenylmethoxycarbonyl °C Degrees Celsius g Grams calcd Calculated GC Gas chromatography cAMP Cyclic adenosine monophosphate h Hours cat Catalytic HMBC Heteronuclear multiple bond Cbz Benzyloxycarbonyl correlation cm centimetres HMPA Hexamethylphosphoric cm-1 Wavenumbers triamide concd Concentrated HPLC High pressure liquid COSY Correlation spectroscopy chromatography Cp Cyclopentadienyl HRMS High resolution mass m-CPBA meta-chloroperbenzoic acid spectroscopy Cy Cyclohexyl HSQC Heteronuclear single quantum σ Chemical shift in parts per million correlation d Doublet Hz Hertz DBU 1,8-Diazabicyclo[5.4.0]undec-7-ene IC50 Half maximal inhibitory DCE 1,2-Dichloroethane concentration de Diastereomeric excess imid Imidazole DEAD Diethyl azodicarboxylate IR Infrared DDQ 2,3-dichloro-5,6-dicyano-1,4- J Coupling constant benzoquinone L litre DIAD Diisopropyl azodicarboxylate LDA Lithium N,N-diisopropylamide DIBAL Diisobutylaluminium hydride LHMDS Lithium hexamethyldisilylazide DIPEA N,N-Diisopropyl-N-ethylamine lit Literature DMAc N,N-Dimethylacetamide μ Micro DMAP 4-(N,N-Dimethylamino)pyridine m Multiplet DME 1,2-Dimethoxyethane M Molar DMF N,N-Dimethylformamide DMPU 1,3-Dimethyl-3,4,5,6-tetrahydro M+ Parent ion -2(1H)-pyrimidinone MALDI Matrix-assisted laser DMSO Dimethylsulfoxide Desorption ionisation DNA Deoxyribonucleic acid max Maximum dr Diastereomer ratio m meta ED50 Dose effective in 50 % of test Me Methyl subjects

xii
MES 2-(N-morpholino)ethanesulfonic TBDPS tert-Butyldiphenylsilyl acid TBS tert-Butyldimethylsilyl MHz Megahertz Troc 2,2,2-Triethoxycarbonyl min Minimum TES Triethylsilyl mM Millimolar Tf Trifluoromethanesulfonyl mol Moles TFA Trifluoroacetic acid MOM Methoxymethyl THF Tetrahydrofuran mp Melting point TIPS Triisopropylsilyl mRNA Messenger ribonucleic acid TLC Thin layer chromatography Ms Methylsulfonyl TMEDA N,N,N’,N’-tetramethyl-1,2- MS Mass spectroscopy ethylene diamine MTBE Methyl tert-butyl ether TMS Trimethylsilyl MW Molecular weight Teoc 2-(Trimethylsilyl)ethoxy m/z Mass-to-charge ratio carbonyl NBS N-Bromosuccinimide TOF Time of flight nm Nanometers p-tol para-Toluene NMR Nuclear magnetic resonance Ts para-Toluenesulfonic acid NOE Nuclear Overhauser effect TS Transition state NOESY Nuclear Overhauser effect UV Ultraviolet spectroscopy v/v Volume per unit volume Nu Nucleophile wt Weight o ortho w/w Weight per unit weight p para 2D Two-dimensional nuclear PDB Protein data bank NMR magnetic resonance PEG Polyethylene glycol spectroscopy Ph Phenyl piv Pivaloyl PMB para-Methoxybenzyl ppm Parts per million PPTS Pyridinium para-toluenesulfonate Pr Propyl i-Pr iso-Propyl PTC Phase transfer catalyst py Pyridine quant Quantitative q Quartet Rf Retention factor RP Reverse phase ROESY Rotating frame Overhauser effect spectroscopy rRNA Ribosomal ribonucleic acid rt Room temperature s Singlet SAR Structure-activity relationship sat Saturated SDS-PAGE
Sodium dodecyl sulfate Polyacrylamide gel electrophoresis
t Triplet TBAF Tetrabutylammonium fluoride

xiii
Table of Contents
Chapter 1 : Introduction 1
1.1. Natural Products in the Drug Discovery and Development Process 2
1.2. Natural Products and the Role of Organic Synthesis 4
1.2.1. Trabectedin (ET-743) 4
1.2.2. Halichondrin B 9
1.2.3. FK506 12
1.3. The Work Described in This Thesis 18
1.4. References for Chapter 1 20
Chapter 2 : Total Syntheses of Phylline, Dioncophylline E and Ancistrotectorine C 23
2.1. Introduction 24
2.1.1. Malaria 24
2.1.2. The Naphthylisoquinoline Alkaloids 24
2.1.3. Anti-Malarial Activity of the Naphthylisoquinoline Alkaloids 27
2.2. Total Syntheses of the Naphthylisoquinoline Alkaloids 29
2.2.1. The ‘Lactone Method’ 29
2.2.2. Meyers Biaryl Coupling 31
2.2.3. Suzuki-Miyara Cross Coupling Reaction 32
2.3. Previous Work in the Morris Group – Total Synthesis of 7,3’- and 5,3’-linked
Naphthylisoquinoline Alkaloids
35
2.4. The Work Described in Chapter 2 41
2.5. Initial Investigations into a Total Synthesis of Dioncophylline E 42
2.5.1. Retrosynthetic Analysis of Dioncophylline E 43
2.5.2. Synthesis of the Biaryl Bond 44
2.5.3. Examination of the Sulfinimine Cyclisation Protocol 53
2.6. Total Synthesis of Phylline 57
2.7. Completion of the Total Synthesis of Dioncophylline E 66
2.8. Investigations into the Total Synthesis of 5,3’-linked Naphthylisoquinoline
Alkaloids
69
2.9. Chapter Summary 76
2.10. References for Chapter 2 77

xiv
Chapter 3 : Development of AAL(S) as a New Lead for the Treatment of Leukaemia 81
3.1. Introduction 82
3.1.1. Discovery and Development of Myriocin as a Novel Immunosuppressant 82
3.1.2. Targeting Protein Phosphatase 2A for Cancer Therapy 86
3.1.3. AAL(S) as a Selective Cancer Therapeutic 88
3.2. Investigations into the Structure Activity Relationships of AAL(S) 91
3.2.1. Designing a Synthesis of AAL(S) 91
3.2.1.1. Investigations into an Alternative Synthesis of AAL(S) 93
3.2.1.2. Hinterding’s Synthesis of AAL(S) Using Schöllkopf’s Reagent 102
3.2.2. First Generation Synthesis of Hydrophobic Tail Analogs of AAL(S) 104
3.2.3. Structural Variations of the Amino Alcohol Head Group of AAL(S) 107
3.3. Biological Data 114
3.4. Inhibition of Ceramide Synthases by AAL(S) Analogs 118
3.4.1. Synthesis and Biological Data of Selective Ceramide Synthase Inhibitors 121
3.5. Determining the Protein Target of AAL(S) 125
3.5.1. Synthesis and Evaluation of an AAL(S) Affinity Chromatography Probe 125
3.5.2. Synthesis and Evaluation of an O-FTY720 Affinity Chromatography 133
Probe
3.6. Chapter Summary 137
3.7. References for Chapter 3 138
Chapter 4 : Summary and Future Work 143
4.1. Summary 144
4.2. Chapter 2 Summary and Future Work 144
4.3. Chapter 3 Summary and Future Work 150
4.4. References for Chapter 4 155
Chapter 5 : Experimental 157
5.1. General Experimental 158
5.2. Experiments Described in Chapter 2 161
5.3. Experiments Described in Chapter 3 198
5.4. References for Chapter 5 255

xv

1

Chapter 1 – Introduction
2
1. Introduction
An enormous cache of human biology remains to be explored following completion of the human
genome project. The expectation is that selective modulators for new targets will be discovered,
and that in some cases, these will be translated into therapeutics.[1] Small molecules are widely
considered to be the most valuable agents to understand and treat human disease. While small
molecules can be accessed from a variety of sources, the ones isolated from plants, fungi,
microbes or marine organisms have proved to be extremely useful in the discovery and
development of therapeutic agents. The utility of these secondary metabolites for the treatment of
diseases has been demonstrated for thousands of years through their use as traditional
medicines.[2] Natural products also make excellent starting points for medicinal chemistry
programs or to become drugs in their own right.[2] Their success can be attributed to their unique,
highly diverse molecular structures which have been designed through evolution to exert a
specialised function for the organism.[3]
1.1. Natural Products in the Drug Discovery and Development Process
The effectiveness of natural products as drugs has been highlighted in a series of reviews by
Newmann and Cragg.[4] Examples of complex natural products that have made it to the market
are the anti-cancer compounds Taxol™ (1.1), vinblastine (1.2), and anti-bacterial agent
erythromycin (1.3) (Figure 1.1). Of these, Taxol™ (1.1) alone has raised over $1 billion revenue
each year since its release onto the market in 1994.[2]
Figure 1.1: Blockbuster natural product drugs: Taxol™ (1.1), which was isolated from the Pacific yew tree (image
credit[5]), vinblastine (1.2), which was isolated from the Madagascan periwinkle (image credit[6]) and erythromycin
(1.3), which was isolated from the bacteria Saccharopolyspora erythraea (image credit[7]). Images were obtained
under free licence from Wikimedia commons.

Chapter 1 – Introduction
3
Unfortunately, discovery programs aimed at the isolation and identification of novel natural
products in large pharmaceutical companies have declined in recent years. This is largely due to
the amount of time and money required to get the natural product to the clinic which is often
longer than other approaches.[8] A general outline of the process and the quantity of material
needed at each step is outlined in Figure 1.2.
Figure 1.2: Chemical process for natural product drug discovery.[9]
Natural products are generally discovered through bioassay-guided purification. This can, in
some instances, be pre-meditated through prior knowledge about the sources involvement in
traditional medicine. However, most of the time, chemically impure natural product extracts are
screened in a high throughput fashion against a broad range of biological targets and the extracts
which have biological activity are identified. Only when the activity has been verified is effort
expended trying to purify and identify the active component of this extract. This process involves
a series of purification steps, generally using HPLC, which delivers a series of fractions which are
re-tested in the assay. Again, the active fraction(s) are identified, further purified and this process
repeated until a single active compound is obtained. After the active compound is identified, the
next issue is gaining access to significant quantities of it to allow for further testing. As can be
seen at the bottom of Figure 1.2, the amount of material required can escalate quite quickly and
is the stage that most natural product drug development programs are delayed. While some
compounds can be accessed readily by scaling up the extraction process, natural products are
often produced in such small quantities by the organism that this is an unrealistic undertaking.[9]

Chapter 1 – Introduction
4
In these cases, production of the natural product using a chemical synthesis (either using a total
synthesis or via a semi-synthetic process using a readily available material) is the only strategy
available. When developing such a synthesis the end goal is to devise a sequence which
proceeds in as few steps as possible, is high yielding, stereoselective (if that is required) and has
the potential to be scaled up.
The natural products in Figure 1.1 are examples of molecules that have made it to the clinic
without modification, but this is not always the case. Often the parent natural product does not
have the optimal properties and it is desirable to improve the pharmacological properties.
Chemical synthesis can aid in this aspect as, if designed appropriately, small modifications can
be made to the scaffold to improve properties such as selectivity for the target, solubility and
toxicity. Furthermore, a synthesis can allow for chemical biology techniques to be used to
determine the mode of action by which the compound exerts its biological activity. Both of these
are valuable in the drug discovery process.
1.2. Natural Products and the Role of Organic Synthesis
The importance of chemical synthesis in the development of natural products as therapeutics can
be illustrated by examining the case studies of trabectedin (ET-743) (1.4), halichondrin B (1.5)
and FK506 (1.6). Each of these natural products was isolated from a natural source in small
quantities, which meant their development as therapeutics was delayed. In each case, chemical
synthesis allowed the supply issue to be addressed and/or allowed the identification of analogs
with improved properties. The process for how each of these compounds was developed will be
discussed in the sections below.
1.2.1. Trabectedin (ET-743)
In 1970 the extracts from a marine ascidian Ecteinascidia turbinate, isolated from the Caribbean,
were tested for their anti-cancer activity on leukaemic mice. The results of the study found that 50
% of the mice were completely cured of their leukaemia. However, the structure of the active
component could not be solved using the analytical techniques that were available at the time
and so the identity of the molecule that gave rise to this potent anti-cancer activity remained a
mystery.[10] Some 20 years later, Professor Ken Rinehart re-examined the ascidian using more
advanced mass spectrometry and NMR techniques and was able to identify the structure which
gave rise to the biological activity. This compound is now known as trabectedin (ET-743) (1.4)
and its structure is presented in (Figure 1.3).[11]

Chapter 1 – Introduction
5
Figure 1.3: Structure of ET-743 (1.4) and the Caribbean ascidian, Ecteinascidia turbinata, which it is isolated from.[12]
Just 6 years later, Professor E. J. Corey and his group confirmed the structural assignment by
completing a 41 step total synthesis of ET-743 (1.4) (Figure 1.4).[13] With this key information,
coupled with the exciting biological data, the pharmaceutical company PharmaMar decided to
examine ET-743 (1.4) as a potential cancer therapeutic and set about carrying out a pre-clinical
evaluation.
Figure 1.4: Corey’s retrosynthetic analysis of ET743 (1.4).[13]
To take ET-743 (1.4) into pre-clinical trials PharmaMar required access to significant quantities of
the material for further testing. They initially employed an aquaculture method, growing the
ascidian and isolating ET-743 (1.4) from it. Although successful in delivering 1 gram of material
for testing, the process required 1 tonne of ascidian to be grown and as they would need much
more material for clinical trials and for subsequent use as a treatment, this made the isolation
route economically impractical.[14] At this point, PharmaMar decided that they would have to
pursue a chemical synthesis to access the material they required. Consequently, PharmaMar

Chapter 1 – Introduction
6
approached Professor Corey at Harvard University to carry out this task as he had already
established his synthesis.
Corey’s retrosynthetic analysis of ET-743 (1.4), shown in Figure 1.4, proposed construction of
ET-743 (1.4) through a biomimetic approach – potentially mimicking the way it was made in
nature. Here, the 10-membered lactone ring would be constructed through generation and
trapping of an ortho-quinonemethide intermediate 1.7 which would be accompanied by three
Pictet-Spengler reactions and one Mannich reaction to construct each of the three
tetrahydroisoquinoline units.[13] Corey completed the total synthesis of ET-743 (1.4) in 1996, and
it is outlined in Scheme 1.1. The synthesis started with a Mannich reaction between two readily
available fragments, 1.8 (synthesised by a Pictet-Spengler reaction) and 1.9, in the presence of
potassium cyanide and generated nitrile 1.10 in 61 % yield.
Scheme 1.1: Reagents and yields (a) KCN, AcOH, rt, 61 %; (b) Cs2CO3, CH2=CHCH2Br, DMF, rt, 87 %; (c) DIBAL-
H, PhMe, -78°C; (d) KF·2H2O, MeOH, rt; (e) MsOH, CH2Cl2, 3Ǻ Mol. Sieves, rt, 55 % (3 steps); (f) Tf2NPh, NEt3,
DMAP, CH2Cl2, rt, 72 %; (g) TBDPSCl, DMAP, CH2Cl2, rt, 89 %; (h) MOMBr, DIPEA, CH2Cl2, rt, 92 %; (i) Bu3SnH,
(Ph3P)2PdCl2, AcOH, CH2Cl2, rt, quant.; (j) CH2O, NaBH3CN, AcOH, MeCN, rt, 95 %; (k) SnMe4, (Ph3P)2PdCl2, LiCl,
DMF, 80°C, 83 %; (l) (PhSeO)2O, CH2Cl2, rt, 82 %; (m) TBAF, THF, rt, 91 %; (n) 1.13, EDCI·HCl, DMAP, CH2Cl2, rt,
91 %; (o) Tf2O, DMSO, CH2Cl2, -40°C; (p) DIPEA, 0°C; (q) t-BuOH, 0°C; (r) (Me2N)2C=N-t-Bu, rt; (s) Ac2O, rt, 79 %
(5 steps); (t) Bu3SnH, (Ph3P)2PdCl2, AcOH, CH2Cl2, rt, 84 %; (u) [N-methylpyridinium-4-carboxaldehyde]+ I-, DBU, 1:3
DMF/CH2Cl2, rt, 70 %; (v) 1.17, SiO2, EtOH, 82 %; (w) 4:1:1 TFA/THF/H2O, rt; (x) AgNO3, 3:2 MeCN/H2O, rt, 77 % (2
steps).[13]

Chapter 1 – Introduction
7
This was followed by allyl protection of the free phenol, reduction of the lactone to the lactol
(DIBAL-H, PhMe, -78°C) and global desilylation to generate the precursor for an intramolecular
Pictet-Spengler reaction. This reaction was achieved using MeSO3H in the presence of 3Ǻ
molecular sieves and generated the ET-743 pentacyclic core 1.11 in 48 % yield over the four
steps. The least hindered phenolic hydroxyl group was selectively reacted with Tf2NPh in the
presence of NEt3 and DMAP and a further three protecting group modifications were carried out:
(1) TBDPS protection of the primary hydroxyl group, (2) MOM protection of the free phenol and
(3) global allyl group deprotection. The N-Me substituent was introduced by reductive amination
(CH2O, NaBH3CN, AcOH, MeCN) and the triflate group replaced with a methyl group using
tetramethyltin under palladium catalysis to afford phenol 1.12 in 47 % yield over the five step
sequence.
The next stage of the synthesis was to construct the ten-membered lactone ring. This required
installation of the cysteine side chain followed by tandem o-quinonemethide formation,
deprotection and cyclisation. The first step in this sequence was oxidation and selective
hydroxylation of phenol 1.12 with (PhSeO)2O to afford o-hydroxydienone, which would serve as
the precursor to the o-quinonemethide, in 82 % yield. Deprotection of the primary TBDPS group
allowed for EDCI coupling of the alcohol with diprotected cysteine residue 1.13 in the presence of
DMAP to afford thiol 1.14 in 68 % yield over three steps. A five step-one pot procedure was
developed by Corey to complete the ten-membered lactone ring of 1.15. This involved addition of
a Swern-type reagent (Tf2O, DMSO) to generate an O-dimethylsulfonium intermediate which was
eliminated using N,N-diisopropyl-N-ethylamine to afford the o-quinonemethide moiety. After t-
BuOH was added to quench any excess Swern reagent, a selective deprotection of the thiol
Fmoc protecting group was achieved using a guanidine base. The free thiolate anion generated
from this deprotection underwent nucleophilic addition onto the o-quinonemethide generating
another free oxygen anion which was quenched with acetic anhydride. This sequence proceeded
in 79 % yield and retained the stereochemistry present in the starting material; that is, the thiolate
anion adds to the same face that the cysteine sidechain protrudes from in the starting material.
Allyl deprotection and oxidation of the resulting free amine using N-methylpyridinium-4-
carboxaldehyde, in the presence of DBU, afforded diketone 1.16 in 59 % yield over two steps.
Construction of the final tetrahydroisoquinoline unit was achieved using a stereoselective spiro-
Pictet-Spengler reaction with 1.17. The stereoselectivity of this reaction can be attributed to the
approach of the aromatic group to the least sterically hindered face of the imine. Finally, the spiro-
tetrahydroisoquinoline was transformed into ET-743 (1.4) by deprotection of the MOM protecting

Chapter 1 – Introduction
8
group and replacement of the primary nitrile with an alcohol using AgNO3 in 63 % yield over three
steps. This afforded ET-743 (1.4) in 0.72 % overall yield over 41 steps with a longest linear
sequence of 32 steps.[13] Four years later Corey published an improved protocol which, although
maintaining the same step count, improved the overall yield to 2.04 % by refining the opening
sequence to phenol 1.11.[15]
Corey’s synthetic protocol provided PharmaMar with an alternative route to gain access to more
ET-743 (1.4), thus potentially solving the problem of supply. However, if the synthesis was going
to prove useful for industrial production of ET-743 (1.4) it would need to be shorter and scalable.
To rectify this problem PharmaMar developed a semi-synthetic sequence, having cleverly
recognised the similarities between the pentacyclic core of ET-743 (1.4) and another natural
product of bacterial origin, safracin B (1.18). Optimisation of the fermentation process of the
Pseudomonas fluorescens bacteria which produces safracin B (1.18) enabled PharmaMar to
produce cyanosafracin B (1.19) on a kilogram scale. Cyanosafracin B (1.19) could be elaborated
in 15 steps into a similar o-quinonemethide precursor 1.20 as the one (1.14) used in Corey’s
synthesis (Scheme 1.2).
Scheme 1.2: Reagents and yields: (a) Boc2O, EtOH, rt, 81 %; (b) MOMBr, i-Pr2NEt, DMAP, MeCN, 40°C, 83 %; (c)
1M aq. NaOH, MeOH, rt, 68 %; (d) H2, 10% Pd/C, DMF, rt then ClCH2Br, Cs2CO3, 110°C; (e) CH2=CHCH2Br,
Cs2CO3, DMF, rt, 56 % (2 steps); (f) TFA, CH2Cl2, rt, 95 %; (g) phenyl isothiocyanate, CH2Cl2, rt, 87 %; (h) 4.3 M HCl
1,4-dioxane, rt, 82 %; (i) TrocCl, py, CH2Cl2, 0°C, 98 %; (j) MOMBr, i-Pr2Net, DMAP, MeCN, 40°C, 88 %; (k) Zn, 90
% aq. AcOH, rt, 83 %; (l) NaNO2, 90 % aq. AcOH, 1:1 THF/H2O, 0°C, 50 %; (m) 1.21, EDCI·HCl, DMAP, CH2Cl2, rt,
95 %; (n) Bu3SnH, (Ph3P)2PdCl2, AcOH, CH2Cl2, rt, 90 %; (o) (PhSeO)2O, CH2Cl2, rt, 91 %.[16]

Chapter 1 – Introduction
9
From this intermediate the principles of Corey’s synthesis were used to complete the semi-
synthesis of ET-743 (1.4) with some key changes made to the protecting groups used and the
sequence of reactions used to reach the final product. Overall, the semi-synthetic sequence
developed by PharmaMar proceeds in 21 overall steps and 0.96 % overall yield. This protocol is
currently carried out on an industrial scale to access large quantities of ET-743 (1.4) and has also
been used to develop analogs of ET-743 (1.4).[16]
Using the Corey total synthesis as a starting point has allowed PharmaMar to progress ET-743
(1.4) into a position that has allowed for the clinical trials and its approval as a cancer therapeutic.
In 2007 ET-743 (1.4) was approved by the European Union for the treatment of soft tissue
sarcoma under the trade name Yondelis®. Two years later, in 2009 it was approved for the
treatment of relapsing ovarian cancer and is currently the subject of a number of clinical trials for
breast, prostate, liposarcoma and paediatric sarcomas.[14]
Since Corey and PharmaMar’s efforts towards ET-743 (1.4) several other groups have reported
total syntheses (Fukuyama[17], Zhu[18] and Fukuyama[19]) and formal syntheses (Danishefsky,[20]
Williams[21], Takemoto[22] and Danishefsky[23]), further developing technology to efficiently access
this compound. More recently, it has been discovered that Candidatus endoecteinascidia
frumentensis, a bacteria which lives in symbiosis with the marine ascidian, found solely in the
Caribbean Sea is responsible for the biosynthesis of ET-743 (1.4). Furthermore, the gene cluster
responsible for ET-743’s biosynthesis in this bacteria has been determined, allowing for the
potential to produce ET-743 (1.4) through metabolic engineering.[24]
This case study serves as an excellent example of how a total synthesis provided a pathway to
access significant materials, which allowed the biomedical potential of this natural product to be
fully explored and ultimately allow this compound to be used as a drug.
1.2.2. Halichondrin B
The halichondrins are a small family of polyether macrolides that were discovered when the crude
extracts from a marine sponge Halichondria okadai were tested and found to exhibit excellent in
vivo anti-cancer activity (Figure 1.5). Halichondrin B (1.5) is the most potent member of the family
(IC50 of 0.0093 ng/mL against B-16 melanoma cells) and was first isolated by Uemura and co-
workers in 1986. Uemura isolated just 12.5 mg of halichondrin B (1.5) from 600 kg of wet sponge,
which allowed for initial, but not thorough, biological investigations to take place.[25] Like the
example of ET-743 (1.4), to be able to progress halichondrin B (1.5) into the clinic, access to

Chapter 1 – Introduction
10
more material was essential. Indeed, progress on this molecule was significantly delayed as a
result of the lack of material. Several options were pursued to generate significant quantities of
material and the total synthesis community certainly joined the quest to solve this issue.[26]
Figure 1.5: Structure of halichondrin B (1.5) and the marine sponge, Halichondria okadai, which it is isolated from.[26]
Professor Yoshito Kishi took on the challenge, reporting several communications on the synthesis
of fragments 1.22[27], 1.23[28] and 1.24[29] before publishing the first total synthesis of halichondrin
B (1.5) in 1992, outlined in Scheme 1.3.[30]
Scheme 1.3: Reagents and yields: (a) NiCl2, 0.5 mol % CrCl2, 1:5 DMF/THF, rt; (b) KH, DME, 80°C, 50–60 % (2
steps); (c) LiAlH4, Et2O, 0°C; (d) DMP, CH2Cl2, rt, 77 % (2 steps); (e) NiCl2, 0.1 mol % CrCl2, DMF, rt; (f) DMP,
CH2Cl2, rt; (g) DDQ, 10:1:100 pH = 7.0 phosphate buffer/t-BuOH/CH2Cl2, rt; (h) LiOH, 1:3 H2O/THF, rt; (i) NEt3, THF
then, DMAP, benzene, 63 % (5 steps); (j) TBAF, THF, rt; (k) PPTS, CH2Cl2, rt, 64 % (2 steps), (l) 4-NO2PhCOCl, py,
CH2Cl2, rt; (m) TBSOTf, NEt3, CH2Cl2, rt; (n) K2CO3, MeOH, rt; (o) DMP, CH2Cl2, rt; (p) NiCl2, 0.1 mol % CrCl2, DMF,
rt, 60 % (2 steps); (q) DMP, CH2Cl2, rt; (r) TBAF, DMF, rt; (s) DDQ, 10:1:100 pH = 7.0 phosphate buffer/t-
BuOH/CH2Cl2, rt; (t) CSA, CH2Cl2, rt, 50–60 % (4 steps).[30]

Chapter 1 – Introduction
11
Kishi’s total synthesis was very convergent, combining four individual fragments with limited
functional/protecting group manipulations after coupling. These fragments could be readily
accessed from carbohydrate-based precursors allowing for the chemistry of each individual
fragment to be improved and optimised irrespective of the whole synthesis. It also allowed Kishi
to access several members of the halichondrin family by varying the specific coupling partners.
Additionally, Kishi designed the synthesis to showcase the Nozaki-Hiyama-Kishi reaction which
had recently been developed. It was this reaction that was used to couple together each of the
fragments in high yield.
Once the halichondrin B (1.5) synthesis was complete Kishi sent the material and several of the
intermediates from the synthesis to Eisai Pharmaceuticals for evaluation of their anti-cancer
activity. Intriguingly, the results found that the macrolactone domain of halichondrin B 1.25,
generated from 1.26, had in vitro activity within an order of magnitude to that of halichondrin B
(1.5), as shown in Figure 1.6.[31] This data allowed for simplification of the true pharmacophore of
halichondrin B (1.5) which in turn opened up the opportunity to efficiently synthesise simpler
analogs of halichondrin B (1.5) with more potent activity.
Figure 1.6: Evolution of the right-hand fragment of halichondrin B (1.5) to Eribulin (1.27). Eribulin mesylate was
approved by the FDA under the trade name Halaven® in 2010.[26]
Early on in the development of macrolactone 1.25, Eisai Pharmaceuticals found that the lactone
unit was susceptible to hydrolysis, leading to an inactive compound. To rectify this problem the
lactone was replaced with a ketone which maintained the biological activity of the parent. Given

Chapter 1 – Introduction
12
that structural variation in the C1 – C14 portion, as evidenced by the investigation of other natural
halichondrins, resulted in weaker activity, little effort was expended on developing this part of the
molecule. Although not outlined in detail here, hundreds of analogs varying the C29 – C36
pyranopyran fragment of halichondrin B were synthesised. i This fragment was found to be
particularly important for the maintenance of the potency of the compound 10 hours after initial
treatment.[26] The investigation ultimately delivered E7389 (Eribulin) (1.27). Eribulin (1.27) could
be synthesised in 37 steps longest linear sequence (compared to halichondrin B’s 48 step longest
linear sequence) with improved binding properties and a better toxicity profile than halichondrin B
(1.5). Eribulin (1.27) was approved by the FDA for the treatment of metastatic breast cancer in
2010 under the trade name Halaven®.[32]
While Eisai Pharmaceuticals still use the foundations of Kishi’s original synthesis to access
Eribulin, there have been several formal and total syntheses of halichondrin B (1.5) and reports of
improved syntheses of fragments in the sequence. Perhaps the most exciting advancement has
been made by Phillips and co-workers who used an Achmatowicz oxidation/ionic hydrogenation
and tandem metathesis methodology to synthesise the pyran and pyranopyran fragments in their
total synthesis of norhalichondrin B.[33] This protocol proceeded in 37 steps longest linear
sequence and should allow for the synthesis of halichondrin B (1.5), Eribulin (1.27) and analogs
to be shortened even further.
1.2.3. FK506
Cyclosporin A (CsA) (1.28) was isolated in 1972 from a soil sample which contained the fungus
Tolypocladium inflatum.[34] It was originally pursued as an anti-fungal antibiotic but later found to
be a far more potent immunosuppressant. Cyclosporin A (1.28) inhibits a protein called
cyclophillin, a 14 KDa immunophillin. Cyclophillin has peptidyl-prolyl cis-trans isomerase (PPI)
activity, which catalyses the folding of ribonuclease and is also involved in the production of T-cell
derived interleukin 2 (IL-2), interleukin 3 (IL-3) and gamma-interferon (IFN-γ) induced by
antigens.[35] Inhibition of the production of these signalling molecules results in an inhibited
proliferative response of lymphocytes to antigen stimulation and inhibits the expression of the IL-2
receptor which results in immunosuppressant activity.[36] Subsequently, CsA (1.28) was approved
by the FDA to prevent graft rejection from transplantation in 1983.
i If the reader is interested in a more detailed review of the development of Eribulin (1.27) they are directed to reference 24.

Chapter 1 – Introduction
13
Figure 1.7: Structures of Cyclosporin A (1.28) and FK506 (1.6).
As part of a proposal founded on identifying compounds with similar immunosuppressant activity
as CsA (1.28), Goto and co-workers screened fermented broths from soil samples on their level
of IL-2 inhibition.[37] The outcome of this study resulted in the identification of FK506 (1.6) from a
Streptomyces tsukubaensis obtained from a Japanese soil sample in 1987.[38] Goto found FK506
(1.6) to inhibit this immune response in vitro in both mouse and human cells at concentrations
one hundred times lower than that of CsA (1.28).[39] Given the tremendous biological activity,
FK506 (1.6) was approved by the FDA in 1994 for use in organ transplantation under the trade
name Tacrolimus®. While it was an effective immunosuppressant drug, it was clear that
understanding the basis of how FK506 (1.6) and CsA (1.28) worked would be valuable. The
Schreiber research group embarked on this challenge and started their program by synthesising
an affinity chromatography probe of FK506 (1.6). Affinity chromatography would allow for the
intracellular binding partners of FK506 (1.6) to be identified. This technique requires covalent
attachment of a molecule or drug of interest to a solid matrix. This is incubated with cell lysate
after which any unbound extract is removed by a series of washing steps. The bound
components are eluted off the matrix with increasing concentrations of unbound drug and the
components identified by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-
PAGE) and sequencing, as shown in Figure 1.8.
Figure 1.8: Simplified representation of an affinity chromatography protocol.

Chapter 1 – Introduction
14
To apply this protocol to FK506 (1.6) Schreiber required access to sufficient quantities of FK506
(1.6) to attach to the solid matrix. Fortunately, large amounts of FK506 (1.6) could be isolated
through fermentation of the Streptomyces strain described by Goto which allowed Schreiber to
conduct a semi-synthesis of this probe, as shown in Scheme 1.4. This began first with a
stereoselective reduction of the C22 ketone using L-selectride. The C32 hydroxyl group was
selectively acylated with 3-azidopropanoyl chloride followed by chemoselective reduction of the
newly installed azide using 1,2-ethanedithiol. Finally, this FK506 derivative was attached to
Affigel-10 resin ready to be used in affinity chromatography experiments.[40]
Scheme 1.4: Reagents and yields: (a) L-selectride; (b) N3(CH2)2COCl; (c) HS(CH2)3SH; (e) Affigel-10.[40]
Investigations using affinity probe 1.29 revealed that a protein of ~14 KDa bound and could be
eluted off the matrix with FK506 (1.6). Given what was known about the CsA-cyclophillin
interaction this probe was originally assumed to be cyclophillin, but the protein could not be eluted
off the matrix with CsA (1.28). This suggested that a different protein had been isolated despite
the similar immunosuppressant activities. Sequencing of the isolated protein confirmed this
suspicion and identified it as a new class of protein called FK506 binding protein (FKBP).[40]
At this stage of the project, although FK506 (1.6) could be obtained in significant quantities from
fermentation and the direct target for FK506 (1.6) was known, to further develop FK506 (1.6) a
total synthesis was required that would allow for access to a range of analogs that could be
tested for their immunosuppressant activity and allow for further elucidation of the mechanism of
action. Schreiber’s retrosynthetic analysis for FK506 (1.6), shown in Figure 1.9, involved the
synthesis of five fragments which would be stitched together and cyclised to afford FK506
(1.6).[41] Emphasis was put on having a convergent coupling of precursors to allow for variations
to be readily made and allow efficient analog generation.

Chapter 1 – Introduction
15
Figure 1.9: Schreiber’s retrosynthetic analysis of FK506 (1.6).[41]
Although not outlined here, ultimately Schreiber was able to use this strategy to complete the total
synthesis of FK506 (1.6) in a total of 56 steps with the longest linear sequence being 32 steps.[41]
The intermediates synthesised and the protocol developed opened up investigations into FK506’s
structure-activity relationships. An early observation from Schreiber was that FKBP had the same
PPI activity as cyclophillin.[40] Instinctively, it was thought that this function of both FKBP and
cyclophillin constituted a signalling pathway of the immune system which when inhibited gave rise
to the immunosuppressant activity. Initial analog design, aided by an X-ray structure of the
FK506-FKBP complex, focussed on improving binding to the PPI domain of FKBP and resulted in
the synthesis of 506BD (1.30), as shown in Figure 1.10. Interestingly, as predicted, 506BD (1.30)
showed excellent inhibition of the PPI activity of FKBP. However, this compound lost all of the
immunosuppressant activity of FK506 (1.6) and was in fact, an antagonist for the
immunosuppressant activity.[42]
Figure 1.10: Design and synthesis of 506BD (1.30).[42]
This observation led Schreiber to believe that FK506 (1.6) had two protein binding surfaces. One,
that bound to FKBP and another that presented itself only when bound as the FK506-FKBP
complex. It was this second protein interaction which was suspected to be responsible for the
immunosuppressant activity. Subsequent investigations looking at the protein interactions of pre-

Chapter 1 – Introduction
16
complexed FK506-FKBP found this hypothesis to be correct and that the FK506-FKBP complex
bound a protein called calcineurin.[43] This interaction was also proven by using simplified analogs
of FK506 (1.6) such as 1.31 and 1.32. These acyclic analogs bound FKBP through the previously
described di-keto moiety but lacked the other part of FK506 (1.6) which led to no (in the case of
1.31) or very little binding to calcineurin. These analogs, as suspected, had no
immunosuppressant activity. As part of the same study Schreiber also showed that the CsA-
cyclophillin complex also bound calcineurin in a similar manner.[44]
Figure 1.11: Analogs synthesised as part of the evolution of FK506 (1.6) and their inhibition of FKBP and calcineurin
when bound to FKBP.[44]
The data that has been discussed for these compounds allowed Schreiber to propose a
mechanism for their immunosuppressant activity. Calcineurin is a calcium-dependent serine-
threonine phosphatase. Its role in the immune system is to dephosphorylate a transcription factor
called nuclear factor of activated T-cells (NF-AT) which is responsible for the transcription of the
IL-2 gene. Schreiber proposed that the FK506-FKBP and CsA-cyclophillin complexes
sequestered calcineurin, preventing it from dephosphorylating NF-AT and this resulted in no IL-2
transcription, as shown in Figure 1.12.[36]
Figure 1.12: Simplified representation of how FK506 (1.6) and CsA (1.28) inhibit calcineurin to exhibit their
immunosuppressant activity.[36]
Further work by Schreiber, taking into account the dual binding properties of these compounds,
resulted in the synthesis of SBL506 (1.33) (Figure 1.13). This was designed using data obtained

Chapter 1 – Introduction
17
from (1) site-directed mutagenesis experiments of FKBP and the effect it had on
immunosuppressant activity and (2) X-ray crystallographic data of the FK506-FKBP-calcineurin
complex. This acyclic analog was a poor inhibitor of FKBP’s PPI activity but formed an SBL506-
FKBP complex that could bind to calcineurin at nanomolar concentrations, 13 fold better than that
of FK506 (1.6).[44]
Figure 1.13: top: SBL506 (1.33);[44] bottom: Hausch group FKBP51 analog evolution.[45]
The Hausch group have also been working actively in the area, trying to develop analogs of
FK506 (1.6) that are selective for specific isoforms of FKBP (Figure 1.13). Their work has
resulted in three publications, two which focus heavily on X-ray crystallographic data of the
FK506-FKBP51 interaction to design analogs, such as compounds 1.34 and 1.35,[45a, 45c] and
another more recent report which improves the ligand efficiency of these molecules by putting in
conformational restraints, such as 1.36.[45b] It must be mentioned that all of the analogs discussed
above are still synthesised according to the principles originally developed by Schreiber for the
synthesis of FK506 (1.6). It is suspected that these reports will result in a selective and more
potent analog for FKBP.

Chapter 1 – Introduction
18
1.3. The Work Described in this Thesis
The three examples discussed here in the Introduction illustrate that natural products are an
important source of drugs to treat human disease. Moreover, these examples help to showcase
how development of organic syntheses can open up access to these materials, allow for analogs
of these materials to be synthesised and can help identify the mode of action of these materials
which can help direct future analog design.
Both of the projects described in this thesis are based on natural products that have potent
biological activity and require efficient syntheses so as to allow further work to proceed. As
illustrated in Figure 1.14, these targets are at different stages in the development pipeline.
Figure 1.14: Adaptation of Figure 1.2 to show where the two projects (the naphthylisoquinoline alkaloids and
AAL(S)) described in this thesis are positioned on the drug development timeline.
The first area of research that will be described is the total synthesis of the naphthylisoquinoline
alkaloids. This unique family of molecules have been found to have a broad range of biological
activities, with the most significant being their activity against malaria. Although a variety of total
syntheses have been reported for many members of the family, alkaloids containing a 7,3’- or
5,3’-linked biaryl bond, such as those shown in Figure 1.15, remain a challenge to synthesise.
This project was focussed on developing a short modular synthesis to gain access to these
natural products, which would allow for further biological investigations to be initiated.
Figure 1.15: Sterically hindered 7,3’- and 5,3’-linked naphthylisoquinoline alkaloids

Chapter 1 – Introduction
19
The second area of research is one that is far more advanced from a medicinal chemistry stand
point. AAL(S) (1.37) is a product of a medicinal chemistry program which started from isolation of
a natural product called myriocin (1.38) from a fungus. Myriocin (1.38) was found to be a potent
immunosuppressant and a medicinal chemistry program was initiated with the aim at developing
novel immunosuppressant compounds. Modifications of the synthesis ultimately allowed for
analogs to be synthesised with improved immunosuppressant activity with a compound, FTY720
(1.39), being clinically approved by the FDA for the treatment of relapsing multiple sclerosis in
2010 (Figure 1.16). While much work has been done to try and develop the immunosuppressant
activity of these compounds, they have also been found to have potent activity against several
cancer cell lines.
Figure 1.16: Evolution of AAL(S) (1.37) from myriocin (1.38).
Interestingly, AAL(S) (1.37) is found to retain this anti-cancer activity without any
immunosuppressant activity. The goals of this project were therefore to understand the
mechanism by which these compounds exert their anti-cancer activity and to develop analogs
based on the AAL(S) scaffold to improve the anti-cancer activity without the added complication
of the immunosuppressant activity.

Chapter 1 – Introduction
20
1.4. References for Chapter 1
[1] G. Vistoli, A. Pedretti, B. Testa, Drug Discovery Today 2008, 13, 285-294. [2] G. M. Cragg, D. J. Newman, Biochimica et Biophysica Acta (BBA) - General Subjects
2013, 1830, 3670-3695. [3] a) D. H. Williams, M. J. Stone, P. R. Hauck, S. K. Rahman, J. Nat. Prod. 1989, 52, 1189-
1208; b) V. Dančík, K. P. Seiler, D. W. Young, S. L. Schreiber, P. A. Clemons, J. Am. Chem. Soc. 2010, 132, 9259-9261.
[4] a) G. M. Cragg, D. J. Newman, K. M. Snader, J. Nat. Prod. 1997, 60, 52-60; b) D. J. Newman, G. M. Cragg, J. Nat. Prod. 2012, 75, 311-335; c) D. J. Newman, G. M. Cragg, J. Nat. Prod. 2007, 70, 461-477; d) D. J. Newman, G. M. Cragg, K. M. Snader, J. Nat. Prod. 2003, 66, 1022-1037.
[5] CatherineMunro at en.wikipedia [Public domain], 2006. [6] Venkatx5 (Own work) [CC-BY-SA-3.0 (http://creativecommons.org/licenses/by-sa/3.0)],
2012.
[7] 乌拉跨氪 (Own work) [CC-BY-SA-3.0 (http://creativecommons.org/licenses/by-sa/3.0)],
2012. [8] W. R. Strohl, Drug Discovery Today 2000, 5, 39-41. [9] F. E. Koehn, G. T. Carter, Nat Rev Drug Discov 2005, 4, 206-220. [10] M. M. Sigel, L. L. Welham, W. Lichter, L. E. Dudeck, L. E. Gargus, Food-Drugs From the
Sea, Marine Technological Society, Washington D. C., 1970. [11] K. L. Rinehart, T. G. Holt, N. L. Fregeau, J. G. Stroh, P. A. Keifer, F. Sun, L. H. Li, D. G.
Martin, J. Org. Chem. 1990, 55, 4512-4515. [12] PharmaMar, Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 32, Cover Image. [13] E. J. Corey, D. Y. Gin, R. S. Kania, J. Am. Chem. Soc. 1996, 118, 9202-9203. [14] C. Cuevas, A. Francesch, Nat. Prod. Rep. 2009, 26, 322-337. [15] E. J. Martinez, E. J. Corey, Org. Lett. 2000, 2, 993-996. [16] C. Cuevas, M. Pérez, M. J. Martín, J. L. Chicharro, C. Fernández-Rivas, M. Flores, A.
Francesch, P. Gallego, M. Zarzuelo, F. de la Calle, J. García, C. Polanco, I. Rodríguez, I. Manzanares, Org. Lett. 2000, 2, 2545-2548.
[17] A. Endo, A. Yanagisawa, M. Abe, S. Tohma, T. Kan, T. Fukuyama, J. Am. Chem. Soc. 2002, 124, 6552-6554.
[18] J. Chen, X. Chen, M. Bois-Choussy, J. Zhu, J. Am. Chem. Soc. 2005, 128, 87-89. [19] F. Kawagishi, T. Toma, T. Inui, S. Yokoshima, T. Fukuyama, J. Am. Chem. Soc. 2013,
135, 13684-13687. [20] B. Zhou, J. Guo, S. J. Danishefsky, Org. Lett. 2001, 4, 43-46. [21] W. Jin, S. Metobo, R. M. Williams, Org. Lett. 2003, 5, 2095-2098. [22] T. Enomoto, Y. Yasui, Y. Takemoto, J. Org. Chem. 2010, 75, 4876-4879. [23] S. Zheng, C. Chan, T. Furuuchi, B. J. D. Wright, B. Zhou, J. Guo, S. J. Danishefsky,
Angew. Chem. 2006, 118, 1786-1791. [24] C. M. Rath, B. Janto, J. Earl, A. Ahmed, F. Z. Hu, L. Hiller, M. Dahlgren, R. Kreft, F. Yu,
J. J. Wolff, H. K. Kweon, M. A. Christiansen, K. Håkansson, R. M. Williams, G. D. Ehrlich, D. H. Sherman, ACS Chem. Biol. 2011, 6, 1244-1256.
[25] Y. Hirata, D. Uemura, Pure Appl. Chem. 1986, 58, 701-710. [26] K. L. Jackson, J. A. Henderson, A. J. Phillips, Chem. Rev. 2009, 109, 3044-3079. [27] T. D. Aicher, Y. Kishi, Tetrahedron Lett. 1987, 28, 3463-3466. [28] T. D. Aicher, K. R. Buszek, F. G. Fang, C. J. Forsyth, S. H. Jung, Y. Kishi, P. M. Scola,
Tetrahedron Lett. 1992, 33, 1549-1552.

Chapter 1 – Introduction
21
[29] a) F. G. Fang, Y. Kishi, M. C. Matclich, P. M. Scola, Tetrahedron Lett. 1992, 33, 1557-1560; b) K. R. Buszek, F. G. Fang, C. J. Forsyth, S. H. Jung, Y. Kishi, P. M. Scola, S. K. Yoon, Tetrahedron Lett. 1992, 33, 1553-1556.
[30] T. D. Aicher, K. R. Buszek, F. G. Fang, C. J. Forsyth, S. H. Jung, Y. Kishi, M. C. Matelich, P. M. Scola, D. M. Spero, S. K. Yoon, J. Am. Chem. Soc. 1992, 114, 3162-3164.
[31] W. Zheng, B. M. Seletsky, M. H. Palme, P. J. Lydon, L. A. Singer, C. E. Chase, C. A. Lemelin, Y. Shen, H. Davis, L. Tremblay, M. J. Towle, K. A. Salvato, B. F. Wels, K. K. Aalfs, Y. Kishi, B. A. Littlefield, M. J. Yu, Bioorg. Med. Chem. Lett. 2004, 14, 5551-5554.
[32] T. K. Huyck, W. Gradishar, F. Manuguid, P. Kirkpatrick, Nat Rev Drug Discov 2011, 10, 173-174.
[33] K. L. Jackson, J. A. Henderson, H. Motoyoshi, A. J. Phillips, Angew. Chem. Int. Ed. 2009, 48, 2346-2350.
[34] M. Dreyfuss, E. Härri, H. Hofmann, H. Kobel, W. Pache, H. Tscherter, European J. Appl Microbiol. 1976, 3, 125-133.
[35] R. Handschumacher, M. Harding, J. Rice, R. Drugge, D. Speicher, Science 1984, 226, 544-547.
[36] A. M. Szpilman, E. M. Carreira, Angew. Chem. Int. Ed. 2010, 49, 9592-9628. [37] T. Kino, H. Hatanaka, M. Hashimoto, M. Nishiyama, T. Goto, M. Okuhara, M. Kohsaka,
H. Aoki, H. Imanaka, J. Antibiot. 1987, 40, 1249-1255. [38] H. Tanaka, A. Kuroda, H. Marusawa, H. Hatanaka, T. Kino, T. Goto, M. Hashimoto, T.
Taga, J. Am. Chem. Soc. 1987, 109, 5031-5033. [39] T. Kino, H. Hatanaka, S. Miyata, N. Inamura, M. Nishiyama, T. Yajima, T. Goto, M.
Okuhara, M. Kohsaka, H. Aoki, T. Ochiai, J. Antibiot. 1987, 40, 1256-1265. [40] M. W. Harding, A. Galat, D. E. Uehling, S. L. Schreiber, Nature 1989, 341, 758-760. [41] M. Nakatsuka, J. A. Ragan, T. Sammakia, D. B. Smith, D. E. Uehling, S. L. Schreiber, J.
Am. Chem. Soc. 1990, 112, 5583-5601. [42] P. K. Somers, T. J. Wandless, S. L. Schreiber, J. Am. Chem. Soc. 1991, 113, 8045-8056. [43] J. Liu, J. D. Farmer Jr, W. S. Lane, J. Friedman, I. Weissman, S. L. Schreiber, Cell 1991,
66, 807-815. [44] M. B. Andrus, S. L. Schreiber, J. Am. Chem. Soc. 1993, 115, 10420-10421. [45] a) R. Gopalakrishnan, C. Kozany, Y. Wang, S. Schneider, B. Hoogeland, A. Bracher, F.
Hausch, J. Med. Chem. 2012, 55, 4123-4131; b) Y. Wang, A. Kirschner, A.-K. Fabian, R. Gopalakrishnan, C. Kress, B. Hoogeland, U. Koch, C. Kozany, A. Bracher, F. Hausch, J. Med. Chem. 2013, 56, 3922-3935; c) R. Gopalakrishnan, C. Kozany, S. Gaali, C. Kress, B. Hoogeland, A. Bracher, F. Hausch, J. Med. Chem. 2012, 55, 4114-4122.

22

23

Chapter 2 – Naphthylisoquinoline Alkaloids
24
2.1. Introduction
2.1.1. Malaria
The World Health Organisation (WHO) has estimated that in 2012 there were 207 million cases of
malaria worldwide which, resulted in 627 thousand deaths.[1] Significantly, they have also
revealed that 3.4 billion people are still at risk of this debilitating disease.[1] Malaria in humans is
caused by infection with one of five species of parasite from the genus Plasmodium. These
species are P. falciparum, P. vivax, P. ovale, P. malariae and P. knowlesi.[2] The disease is
transmitted amongst humans primarily through being bitten by an infected female Anopheles
mosquito, which passes the infection on through their saliva. Once in the bloodstream, the
premature parasite, or sporozoite, migrates to the liver where it replicates and develops into the
adult parasite (merozoite). Approximately one week after infection the malarial merozoites exits
the liver into the bloodstream where they invade, replicate and rupture red blood cells, resulting in
anaemia.[3]
The standard treatment for malaria is with chloroquine (2.1) but, while this is effective at
eliminating malaria parasites of the P. vivax, P. ovale, P. malariae and P. knowlesi families, P.
falciparum has developed resistance to this drug and thus, P. falciparum is now the species
responsible for the most malaria fatalities. Fortunately, treatment of P. falciparum is still possible,
using artemesinin (2.2). However, the WHO has found that some strains of P. falciparum are
developing resistance to this drug and therefore, it is imperative that new alternative treatments
are discovered and developed.[1]
Figure 2.1: Structures of current first line malaria therapeutics chloroquine (2.1) and artemisinin (2.2).
2.1.2. The Naphthylisoquinoline Alkaloids
The Ancistrocaldaceae and Dioncophyllaceae plant families,[4] found in Africa and South-east
Asia,[5] have been used for hundreds of years as traditional herbal medicines to treat malaria and
dysentery.[6] Govindachari and co-workers were the first to identify the naphthylisoquinoline
alkaloids from these plants in the early 1970s.[7] However, it wasn’t until years later, in 1994, that
Bringmann and co-workers tested both the extracts of these plants and the isolated, pure

Chapter 2 – Naphthylisoquinoline Alkaloids
25
naphthylisoquinoline alkaloids for their anti-malarial activity. In a ground breaking discovery, the
naphthylisoquinoline alkaloids were found to be the active component of these extracts and
importantly, were effective at eliminating the K1 strain of P. falciparum which is resistant to
chloroquine (2.1).[6] Based on these results there has been intense work studying the vines that
these compounds are isolated from. To date, more than 100 of these molecules have been
discovered which have been found to have a broad range of biological activities including
fungicidal, anti-leishmanial, insect growth retardant and for some specific molecules anti-HIV
activity. Some of these molecules are represented in Figure 2.2.
Figure 2.2: Representative examples of naphthylisoquinoline alkaloids.[8]

Chapter 2 – Naphthylisoquinoline Alkaloids
26
As well as being biologically interesting, the naphthylisoquinoline alkaloids are also structurally
and biosynthetically unique.[9] Structurally, these molecules consist of a naphthalene fragment,
linked to an isoquinoline fragment through a biaryl bond. However, despite this simple definition
there is a very broad variation amongst the members of the family. While the position of the biaryl
bond on both the naphthalene and isoquinoline moiety is the most obvious difference (Figure
2.2), subtle variations are present depending on whether the alkaloid is from the
Ancistrocladaceae or Dioncophyllaceae plant families. Here, alkaloids isolated from the
Ancistrocladaceae plant family are oxygenated at C6 and have the S stereochemistry at C3
which, is in contrast to alkaloids isolated from the Dioncophyllaceae plant family which are not
oxygenated at C6 and have the R stereochemistry at C3.[9] Other common differences between
the families include the oxidation of the isoquinoline moiety and the stereochemistry associated
with C1 which can be cis or trans relative to the methyl at C3. Furthermore, depending on the
functionality ortho to the biaryl axis, rotation about this bond can be hindered, which results in
most of these molecules possessing the phenomenon of atropisomerism. A thorough analysis of
the thermodynamic stability of the biaryl bond for each class of the naphthylisoquinoline alkaloids
has not been carried out. However, observations made by Bringmann on a few examples has
found that generally those alkaloids with a di-substituted biaryl bond undergo free rotation, those
that are tri-substituted can either be stable or undergo free rotation depending on the substituents
and those that are tetra-substituted, such as 5,1’-linked ancistrocladine (Figure 2.2), decompose,
at temperatures above 200°C, before undergoing racemisation.[10] Racemisation has been
achieved under basic conditions (0.5 M aq. KOH, MeOH, rt, 6 days).[11] Interestingly, pairs of
atropisomers have been found to have different biological activities and recently, some of these
molecules have been isolated as dimers (Figure 2.2).[9]
In contrast to other alkaloids, which are synthesised biosynthetically from aromatic amino acids,
the naphthylisoquinoline alkaloids are synthesised from acetate units. This was proven
unambiguously by Bringmann and co-workers who fed [13C2]-labelled sodium acetate (2.3) to
Ancistrocladus heyneanus[12] and Triphyophyllum peltatum[13] plant cultures, members of the
Ancistrocladaceae and Dioncophyllaceae plant families respectively. After incubation, isolation of
the 13C-labelled components found that both the naphthalene 2.4 and isoquinoline fragment 2.5
contained complete incorporation of 13C and, in the T. peltatum case 13C-labelled dioncophylline
A (2.6) was isolated. A biosynthetic pathway was proposed whereby acetate precursors are
stitched together to afford a common polyketide intermediate 2.7. Cyclisation and aromatisation
of this intermediate will generate the naphthalene moiety 2.4, but if a transamination occurs
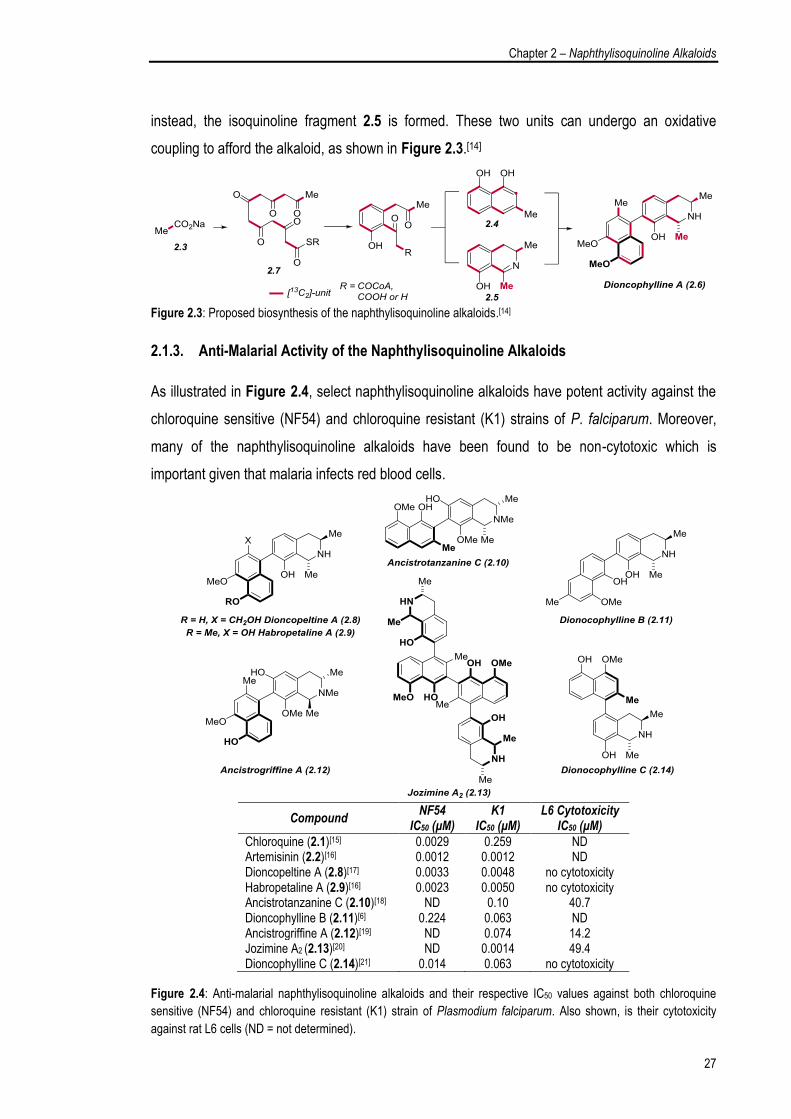
Chapter 2 – Naphthylisoquinoline Alkaloids
27
instead, the isoquinoline fragment 2.5 is formed. These two units can undergo an oxidative
coupling to afford the alkaloid, as shown in Figure 2.3.[14]
Figure 2.3: Proposed biosynthesis of the naphthylisoquinoline alkaloids.[14]
2.1.3. Anti-Malarial Activity of the Naphthylisoquinoline Alkaloids
As illustrated in Figure 2.4, select naphthylisoquinoline alkaloids have potent activity against the
chloroquine sensitive (NF54) and chloroquine resistant (K1) strains of P. falciparum. Moreover,
many of the naphthylisoquinoline alkaloids have been found to be non-cytotoxic which is
important given that malaria infects red blood cells.
Compound NF54
IC50 (μM) K1
IC50 (μM) L6 Cytotoxicity
IC50 (μM)
Chloroquine (2.1)[15] 0.0029 0.259 ND Artemisinin (2.2)[16] 0.0012 0.0012 ND Dioncopeltine A (2.8)[17] 0.0033 0.0048 no cytotoxicity Habropetaline A (2.9)[16] 0.0023 0.0050 no cytotoxicity Ancistrotanzanine C (2.10)[18] ND 0.10 40.7 Dioncophylline B (2.11)[6] 0.224 0.063 ND Ancistrogriffine A (2.12)[19] ND 0.074 14.2 Jozimine A2 (2.13)[20] ND 0.0014 49.4 Dioncophylline C (2.14)[21] 0.014 0.063 no cytotoxicity
Figure 2.4: Anti-malarial naphthylisoquinoline alkaloids and their respective IC50 values against both chloroquine
sensitive (NF54) and chloroquine resistant (K1) strain of Plasmodium falciparum. Also shown, is their cytotoxicity
against rat L6 cells (ND = not determined).

Chapter 2 – Naphthylisoquinoline Alkaloids
28
The utility of the naphthylisoquinoline alkaloids as a treatment for malaria has been shown in an
in vivo mouse study by Bringmann and co-workers.[21] The study showed that if mice infected with
P. Berghei were treated with 20 mg/kg/day of dioncophylline C (2.14) they completely cleared
their infection within 4 days, in contrast to the control mice who all died within 18 days of infection,
as shown in Table 2.1. The investigation found that the oral ED50 of dioncophylline C (2.14) was
10.71 mg/kg which could be improved to 1.90 mg/kg if the compound was administered
intravenously. Moreover, a single 50 mg/kg oral dose of dioncophylline C (2.14) was found to be
sufficient to kill 99.6 % of the parasite, whereas 50 mg/kg/day for 4 days completely eradicated
the parasite without any detectable toxic effects.
Table 2.1: Inhibitory effects of 20 mg/kg/day of dioncophylline C (2.14) on the course of parasitemia of Plasmodium
berghei in mice.
Day
% Parasitemiaa
Control Dioncophylline C (2.14)
Mouse 1 Mouse 2 Mouse 3 Mouse 1 Mouse 2 Mouse 3
4 1 5 5 0 0 0 5 2 10 9 0 0 0 6 13 26 14 0 0 0 7 6 27 33 0 0 0 8 12 18 † 0 0 0
14 25 † † 0 b 0b 0 b 18 50 † † 0 0 0 21 † † † 0 0 0 28 † † † 0 0 0
K173-infected C57/Black/6J mice in comparison to control mice, which were not treated. Mice were infected
intraperitoneally with 106 parasites at day 0. Dioncophylline C (2.14) was delivered by osmotic pump containing 16.6
mg/mL, released at 1μL/h which was implanted subcutaneously at day -1. a Individual values for each mouse, † =
deceased. b No parasite present after isodiagnosis with samples from Swiss mice.[21]
Bringmann and co-workers have also reported on the mode of action of the naphthylisoquinoline
alkaloids. Ferriprotoporphyrin IX (FPIX) (2.15) (also known as heme) is released from
haemoglobin when it is digested by the parasite in red blood cells. However, the free FPIX
monomer 2.15 is toxic to the parasite, and as a consequence the parasite counters the toxicity by
converting 2.15 into a polymeric, crystalline structure called β-hematin or hemozoin. Through
NMR paramagnetic relaxation and structural modelling, Bringmann examined the relationship
between dioncophylline C (2.14) and FPIX (2.15) and showed that dioncophylline C (2.14) binds
to FPIX (2.15) to form a 1:1 complex. Bringmann speculated that the dioncophylline C:FPIX
complex stops the formation of non-toxic β-hematin polymers resulting in increased free FPIX
(2.15) which ultimately kills the parasite (Figure 2.5).[22] Furthermore, this mechanism of action is
similar to what other quinoline based anti-malarial drugs, such as chloroquine (2.1), have been
found to do.[23]

Chapter 2 – Naphthylisoquinoline Alkaloids
29
Figure 2.5: Simplified representation of how dioncophylline C (2.14) is proposed to exert its anti-malarial activity.[22]
While some naphthylisoquinoline alkaloids, such as dioncophylline A, can be readily isolated from
their natural source through plant culture[14] access to other molecules in the family has been
stymied due to the rare nature of the liana they derive from. In order to understand the key
structure-activity relationships of these compounds, access to them and their analogs is required.
Consequently, several groups, most notably Professor Bringmann’s at the Universität Würzburg,
have embarked on total syntheses of these molecules to remedy this.
2.2. Total Syntheses of Naphthylisoquinoline Alkaloids
In addition to their potent biological activity, the fascinating molecular architecture has made the
naphthylisoquinoline alkaloids a key source of inspiration for total synthesis. Of particular interest
has been the stereoselective construction of the biaryl bond of these molecules as this poses a
significant challenge.[8, 24] There have been a number of total syntheses reported which
successfully gain access to a variety of different classes of the naphthylisoquinoline alkaloids
which can be best drawn together by the strategy used to form the biaryl bond. Previous
successful strategies by other groups include the ‘lactone method’ developed by Bringmann, the
Meyer’s biaryl coupling and the Suzuki cross-coupling reaction.[8-9, 25]
2.2.1. The ‘Lactone Method’
The ‘lactone method’ is a two-step procedure which has been used by Bringmann to synthesise
over 20 naphthylisoquinoline alkaloids from the 5,1’-, 5,8’- and 7,1’-linked families.[26] The method
exploits a biaryl ester tether to efficiently construct the biaryl bond in an atropselective manner. In
the case of the naphthylisoquinoline alkaloids the naphthalene and the isoquinoline fragments

Chapter 2 – Naphthylisoquinoline Alkaloids
30
can be coupled as whole complete units[27] or constructed following assembly of the biaryl bond.
Scheme 2.1 shows application of the lactone method to the synthesis of the 5,8’-linked
naphthylisoquinoline alkaloids korupensamine A (2.16) and B (2.17). The ester tether serves two
purposes. Firstly, it is used to bring together the masked naphthalene fragment and the
isoquinoline fragment to allow for efficient construction of the C-C biaryl bond through an
intramolecular palladium-catalysed cyclisation. Under normal circumstances this biaryl bond
would exist as two non-interconverting atropisomers. However, the ester tether of 2.18 lowers the
rotational isomerisation barrier so that these interconvert freely at room temperature. The second
step installs the axial chirality by stereoselectively ring-opening the biaryl lactone 2.18 via a
dynamic kinetic resolution mechanism. The power of the methodology is that either enantiomer of
(R/S)-2.19 can be used to gain access to each atropisomer ((M/P)-2.20). Furthermore, the
atropisomers can be separated and the undesired atropisomer recycled by re-cyclisation to the
biaryl lactone 2.18. This allowed Bringmann and co-workers to synthesise both korupensamine A
(2.16) and B (2.17) from a common biaryl lactone intermediate 2.18 (Scheme 2.1).[28] While the
lactone method is a powerful strategy, it could not be successfully applied to the construction of
the biaryl bond of 7,3’-linked naphthylisoquinoline alkaloids.[29]
Scheme 2.1: Reagents and yields (a) Pd(OAc)2, P(p-tol)3, NaOAc, DMAc, 140°C, 74 %; (b) BH3, (R)-2.19, THF, -
30°C, 58 % (M:P = 6:94); (c) BH3, (S)-2.19, THF, 0°C, 57 % (M:P = 96:4).[28]

Chapter 2 – Naphthylisoquinoline Alkaloids
31
2.2.2. Meyers Biaryl Coupling
The Meyers biaryl coupling was reported by Meyers and co-workers in 1982 and involves the
nucleophilic aromatic substitution reaction of a methoxy group ortho- to a oxazoline with a
Grignard reagent.[30] The method has been refined so that it can be used to generate
atropisomers stereoselectively by using a chrial oxazoline moiety. It was first applied to the
synthesis of the naphthylisoquinoline alkaloids by Rizzacasa and Sargent in 1991.[31] Rizzacasa
expanded on this work, completing the total synthesis of (-)-O-methylancistrocladine (2.21).[32]
Here, the masked isoquinoline fragment 2.22 was coupled to naphthalene 2.23 to afford biaryl
2.24 in 76 % yield and great atropselectivity (M:P = 92:8), as shown in Scheme 2.2. After
synthesis of the biaryl bond, the isoquinoline moiety was constructed using a Bischler-Napieralski
reaction to afford (-)-O-methylancistrocladine (2.21). The protocol has since been optimised
further by Rizzacasa so that it is completely convergent by adding the complete isoquinoline
fragment 2.25 of (+)-O-methylancistrocline (2.26) into the naphthalene 2.23, as shown in Scheme
2.2. Using this synthesis to illustrate the mechanism, the Grignard reagent 2.25 adds to the least
sterically hindered face of the naphthalene 2.23, which is determined by the stereochemistry of
the oxazoline moiety. The atropselectivity of this reaction is a result of the steric and electronic
effects of the substituents ortho- to the Grignard reagent. In this case, the C6-methoxy group
coordinates to the magnesium of the azaenolate 2.27, and methoxy magnesium bromide is
eliminated to afford the product 2.28.[33] The strategy has been successfully applied to
naphthylisoquinoline alkaloids belonging to the 5,1’- and 7,1’-linked families. However, attempts
to extend the Meyers biaryl coupling reaction to the 7,3’-linked naphthylisoquinoline alkaloids
failed, with only starting materials being re-isolated.[34]

Chapter 2 – Naphthylisoquinoline Alkaloids
32
Scheme 2.2: Reagents and yields (a) THF, Δ, 70 % (M:P = 92:8); (b) 2.23, THF, Δ, (M:P = 82:18) (c) TFA, H2O,
THF; (d) Ac2O, py, 27 % (3 steps).[33]
2.2.3. Suzuki-Miyara Cross Coupling Reaction
Palladium catalysed cross-coupling reactions have become a valuable tool for the synthesis of
carbon-carbon bonds.[35] It is therefore the most broadly examined route to gain access to the
naphthylisoquinoline alkaloids with several variations of it developed.[36] Non-atropselective
reactions have been employed by Hoye,[37] Bringmann,[38] and Dawson[39] to synthesise a number
of 5,8’-linked naphthylisoquinoline alkaloids, where the atropisomers could be separated
chromatographically. Variations that have been investigated include using different chiral ligands
in the reaction and using the substrate to control the atropselectivity. Four representative
examples of these are shown in Scheme 2.3. Bringmann and co-workers have employed a chiral
(P)-BINAP ligand to synthesise the biaryl bond of ancistrotanzanine B (2.29) with reasonable
atropselectivity (M:P = 75:25). Interestingly, using the opposite enantiomer of the ligand did not
result in a preference for the formation of the opposite atropisomer and instead a diastereomeric

Chapter 2 – Naphthylisoquinoline Alkaloids
33
mixture was observed (Scheme 2.3, A). Uemera and co-workers have utilised a planar-chiral
(arene)Cr(CO)3 complex to control the stereochemical outcome of the Suzuki cross-coupling of
5,8’-linked naphthylisoquinoline alkaloids. Here, the tricarbonylchromium complex blocks one
face of the aromatic bromide 2.30 coupling partner so that naphthalene 2.31 joins from the least
hindered side, affording the biaryl compound 2.32 with complete atropselectivity (Scheme 2.3,
B). Lipshutz and co-workers have specifically designed isoquinoline substrates to obtain high
atropselectivities in the cross-coupling reaction. Their first report of this was in 1999 where a
phosphine tether was attached to C3 of the tetrahydroisoquinoline. The phosphine was able to
coordinate to the palladium in the cross-coupling reaction transition state which, when used with a
bulky dppf ligand system, forced the naphthalene moiety to be delivered from the least hindered
face.[40] More recently, they have employed the same principle with a 1-naphthyl tether at C3 of
the isoquinoline 2.33. Instead of coordinating to the palladium the naphthyl group loops around
and π-stacks with the isoquinoline in the transitions state 2.34, again forcing the naphthalene
moiety 2.35 to be delivered from the least hindered face, as shown in Scheme 2.3, C.[41] Lastly,
the most recent endeavour by Tang and co-workers has utilised a chiral monophosphorous ligand
in conjunction with an ortho-polar BOP group to achieve high atropselectivity. Scheme 2.3, D
shows Tang’s proposed transition state for the reaction (2.36). While the atropselectivity is
ultimately governed by the chiral ligand, which blocks one face of the palladium intermediate, it
relies on a strong polar-π interaction between the highly polarised ortho-BOP group of the
aromatic bromide coupling partner 2.37 and the extended π-system of the arylboronic acid 2.38
to hold the substituents in the specific conformation.
The 5,8’-linked naphthylisoquinoline alkaloids are the only members of the family that have been
synthesised via the Suzuki cross-coupling method at this stage due to a lower degree of steric
hindrance about their biaryl bond.[8] In addition, a palladium catalysed Stille reaction has been
used to synthesise the biaryl bond for the 7,6’-linked alkaloid dioncophylline B (2.11).[42] However,
the biaryl linkage of this natural product is semi-stable at room temperature and thus, also has a
lower degree of steric hindrance about its biaryl bond. Unfortunately, this methodology has never
been extended to naphthylisoquinoline alkaloids with a more hindered biaryl bond.

Chapter 2 – Naphthylisoquinoline Alkaloids
34
Sch
eme
2.3:
Rea
gent
s an
d yi
eld
s (a
) P
d 2(d
ba) 3
, (P
)-B
INA
P,
1:2
1 M
aq.
NaH
CO
3/P
hMe,
50
% (
M:P
= 7
5:25
); (
b) P
d(P
Ph 3
) 4,
1:10
1 M
aq.
NaH
CO
3/M
eOH
, 75
°C,
38 %
(M:P
= <
3:9
7); (
c) P
dI2,
SP
hos,
K3P
O4,
n-B
uOH
, rt,
72 %
(M
:P =
11:
1); (
d) P
d(O
Ac)
2, c
hira
l mon
opho
spho
rous
liga
nd, K
3PO
4, 5
:1 P
hMe/
H2O
, rt,
96 %
(M
:P =
3:9
7).

Chapter 2 – Naphthylisoquinoline Alkaloids
35
2.3. Previous Work in the Morris Group – Total Synthesis of 7,3’- and 5,3’-Linked
Naphthylisoquinoline Alkaloids
While the strategies outlined in Section 2.2 allow for efficient and atropselective syntheses of
most of the naphthylisoquinoline alkaloids, they do not allow for access to all of them. Most
notably, the 7,3’- and the 5,3’-linked members, which remain challenges due to increased steric
hindrance about the biaryl bond. The first alkaloid isolated from these families was 7,3’-linked
ancistrocladidine (2.39) by Govindachari and co-workers in 1973.[43] More recently Hua and co-
workers in 2013 reported the isolation of 5,3’-linked ancistrotectorines C (2.40) and
ancistrotectorine D (2.41).[44] Molecules from this class have been found to be some of the most
pharmacologically potent naphthylisoquinoline alkaloids (Figure 2.6). In particular, dioncophylline
E (2.42) has extremely potent activity against chloroquine resistant strains of malaria.[45]
Ancistrotanzanine A (2.43) and ancistrotectorines C (2.40) and D (2.41) have also been found to
have some cytotoxicity against several cancer cell lines.[44] However, thorough investigations into
their biological activity have not been reported and this is most likely due to the lack of access to
sufficient quantities of material. Consequently, to fully explore the biological potential of these
molecules efficient access to them is essential.[4] Therefore, a major focus of the Morris group
has been to develop a strategy to access these molecules.
Compound NF54
IC50 (μM) K1
IC50 (μM) L6 Cytotoxicity
IC50 (μM)
Ancistrocladidine (2.39)[18] 0.3 a 1.9 28.3 Ancistrotectorine C (2.40)[44] ND ND 10.24 b
Ancistrotectorine D (2.41)[44] ND ND 13.13 b
Dioncophylline E (2.42)[45] 0.0022 0.0021 ND Ancistrotanzanine A (2.43)[46] ND 0.3 6.4 (7.45 b) Ancistrotectorine (2.44)[18] 0.7 a 9.1 19.9 Ancistrotanzanine C (2.45)[18] 4.2 a 0.1 40.7
Figure 2.6: 7,3’- and 5,3’-linked naphthylisoquinoline alkalds and their respective IC50 values against both
chloroquine sensitive (NF54) and chloroquine resistant (K1) strain of Plasmodium falciparum. Also shown is their
cytotoxicity against rat L6 cells. a Against 3D7 strain of Plasmodium falciparum, a clone of NF54 b Against K562
leukaemia cells (ND: not determined).

Chapter 2 – Naphthylisoquinoline Alkaloids
36
Given the preceding discussion a new strategy was sought to construct the biaryl bond of the
7,3’- and 5,3’-linked naphthylisoquinoline alkaloids in an efficient manner. This ultimately led the
Morris group to examine other possibilities which culminated in examination the Pinhey-Barton
reaction. Pinhey and co-workers first reported the use of aryllead triacetates for the ortho-
arylation of phenols in 1976.[47] Pinhey established that relatively simple biaryl molecules could be
produced in high yields, with a regioselective preference for the ortho position, relative to the
phenol group using equimolar quantities of an aryllead triacetate, phenol and pyridine, which was
found to increase the rate of the reaction.[48] Barton and co-workers followed up these reports by
demonstrating that the reaction was extremely efficient at synthesising tetra-substituted biaryl
bonds, as shown in Scheme 2.4.[49] Moreover, Yamamoto and co-workers have shown that by
utilising a chiral base, such as brucine (2.46), the reaction can produce optically enriched
products (Scheme 2.5).[50]
Scheme 2.4: Reagents and yields (a) pyridine, CHCl3, rt, 87 %.[49]
Scheme 2.5: Reagents and yields (a) 2.47, 2.46, 4Ǻ Mol. Sieves, PhMe, -20°C; (b) 2.47 or 2.48, 2.46, 4Ǻ Mol.
Sieves, PhMe, -40°C.[50]
With this information Morris and Bungard decided to apply this methodology to the synthesis of
ancistrocladidine (2.39). For the synthesis of ancistrocladidine (2.39) the requisite aryllead
triacetate 2.49 was synthesised in 3 steps from iodide 2.50. Protection of the aldehyde as the
acetal using 1,2-ethandiol and catalytic TsOH under Dean-Stark conditions gave a substrate that
could undergo halogen-lithium exchange using t-BuLi in THF at -95°C. Quenching this
intermediate with Bu3SnCl afforded stannane 2.51 and transmetalation using Pb(OAc)4 and 10
mol % Hg(OAc)2 in CH2Cl2 generated the aryllead triacetate 2.49 in 78 % over the three step
sequence (Scheme 2.6). The aryllead triacetate was subjected to the standard Pinhey-Barton

Chapter 2 – Naphthylisoquinoline Alkaloids
37
coupling conditions with naphthol 2.52 and 3 equivalents of pyridine in CH2Cl2. Barton and co-
workers determined that this reaction happens through a ligand coupling mechanism, where the
lone pair of electrons from the phenol coordinates to the lead of the aryllead species. i[49, 51] The
pyridine employed in the reaction helps stabilise the aryloxylead intermediate 2.53 which allows
the aryllead species to come in close proximity to the ortho-position, relative to the phenol to form
the biaryl linkage, as shown in Scheme 2.6.[52] In this case, the crude acetal was immediately
hydrolysed using 3 % v/v aqueous H2SO4 in THF to afford the biaryl aldehyde 2.54 in 67 % yield
over two steps.[53]
Scheme 2.6: Reagents and yields (a) 1,2-ethanediol, TsOH, C6H6, Dean-Stark, 99%; (b) t-BuLi, Bu3SnCl, THF, -
95°C → rt, 85 %; (c) Pb(OAc)4, 10 mol % Hg(OAc)2, CH2Cl2, rt, 93 %; (d) 2.52, py, CH2Cl2, rt, 67 %.[53]
With the biaryl aldehyde 2.54 in hand Bungard and Morris required a stereoselective construction
of amphetamine 2.55 to complete the synthesis. This was achieved in 8 steps using a Katsuki-
Sharpless asymmetric epoxidation reaction as the key step to generate the C3 methyl group in an
asymmetric fashion. The synthesis was completed in a further 3 steps using a Bischler-
Napieralski reaction to afford ancistrocladidine as a 50:50 mix of atropisomers. Fortunately, the
atropisomers could be separated by recrystallization to afford ancistrocladidine (2.39) as a single
atropisomer (Scheme 2.6).[53]
Given the success of this synthesis, attention could now be focussed on the preparation of the
other naphthylisoquinoline alkaloids in this family. However, the 10 step end-game to complete
the dihydroisoquinoline fragement of ancistrocladidine (2.39) was felt to be too long, especially if
this protocol was going to be useful to synthesise analogs of this scaffold. A more convergent
synthesis, where the stereochemistry at C3 of the isoquinoline was already constructed was
i A radical mechanism has been independently discounted by both Pinhey[51] and Barton.[49]

Chapter 2 – Naphthylisoquinoline Alkaloids
38
therefore desired. Preliminary efforts to couple an intact 3,4-dihydro- or protected 1,2,3,4-
tetrahydroisoquinoline moiety failed due to the inability to form the required aryllead triacetate.
These reactions resulted in demetallation of the substrate which was proposed to be due to
coordination of the nitrogen to the lead species similarly to an observation of Barton and co-
workers[54] (Scheme 2.7).[53a]
Scheme 2.7: Reagents and yields (a) t-BuLi, Bu3SnCl, THF, -95°C → rt, 83 %, (b) Pb(OAc)4, 10 mol % Hg(OAc)2,
CH2Cl2, rt; (c) MeI, Me2CO, rt; (d) DIBAL-H, CH2Cl2, 78°C → rt, 85 % (2 steps); (e) t-BuLi, Bu3SnCl, THF, -95°C →
rt, 83 %; (f) NaBH4, MeOH, rt, 96 %; (g) ClCO2Me, NEt3, CH2Cl2, rt, 77 %.[53a]
Given that the synthesis could not be made convergent, it was decided to tackle the problem in a
different fashion and develop a new end-game strategy to access the isoquinoline moiety. As a
result, Jason Brusnahan, a PhD student in the Morris group, initiated a synthesis of
ancistrotanzanine A (2.43), a 5,3’-linked isomer of ancistrocladidine (2.39) which had recently
been isolated.[46] Brusnahan chose to examine a report by Davis and co-workers who had
developed a two-step synthesis of 3,4-dihydroisoquinoline 2.56 from o-tolylnitrile 2.57, using an
enantiopure sulfinimine 2.58 to stereoselectively construct the amphetamine sidechain, as shown
in Scheme 2.8.[55]
Scheme 2.8: Reagents and yields (a) LDA, diglyme, -78°C, then 2.58, diglyme -78°C, 68 %; (b) MeLi, Et2O, -20°C,
then 2 M aq HCl → rt, 65 %.[55]
Therefore, to apply this chemistry to ancistrotanzanine A (2.43), Brusnahan proposed generation
of an aryllead triacetate 2.59 which would be used in the Pinhey-Barton ortho-arylation reaction to
synthesise tolylnitrile 2.60. This substrate would be subject to Davis’ protocol to construct the 1,3-
dimethyl-3,4-dihydroisoquinoline moiety of ancistrotanzanine A in two steps (Figure 2.7).

Chapter 2 – Naphthylisoquinoline Alkaloids
39
Figure 2.7: Brusnahan’s retrosynthesis of ancistrotanzanine A (2.43).
Brusnahan synthesised the requisite aryllead triacetate 2.59 in a three step protocol similar to that
described for ancistrocladidine (2.39). Again, coupling of the aryllead triacetate 2.59 with naphthol
2.52 in the presence of pyridine constructed the sterically hindered 5,3’-linked biaryl bond in 77 %
yield. The resulting naphthol was protected as the MOM ether to afford tolylnitrile 2.60 in 91 %
yield using NaH and MOMCl in DMF. Initial efforts to deprotonate the methyl group ortho to the
nitrile using LDA were ineffective, but application of a less bulky base, LiNEt2, allowed the
reaction to proceed and generated amine 2.61 in 75 % yield. Preliminary work by Brusnahan
identified that this chemistry worked quite well but, it was more reliable using the t-butyl
sulfinimine (R)-2.62 which was a reflection of the quality of the reagents purchased from Aldrich.
However, disappointingly the stereoselectivity of this reaction was just 85:15. Despite this,
Brusnahan was able to complete the synthesis of ancistrotanzanine A (2.43) by treatment with
MeLi then working up with acid. This achieves three operations in one pot (1) addition of MeLi
into the nitrile to install the C1 methyl group, (2) cleavage of the t-butylsulfinyl group and (3)
intermolecular cyclisation of the amine onto the resulting ketimine/ketone. This afforded
ancistrotanzanine A (2.43) in 77 %, albeit as an inseparable mixture of atropodiastereomers, as
shown in Scheme 2.9.[56]
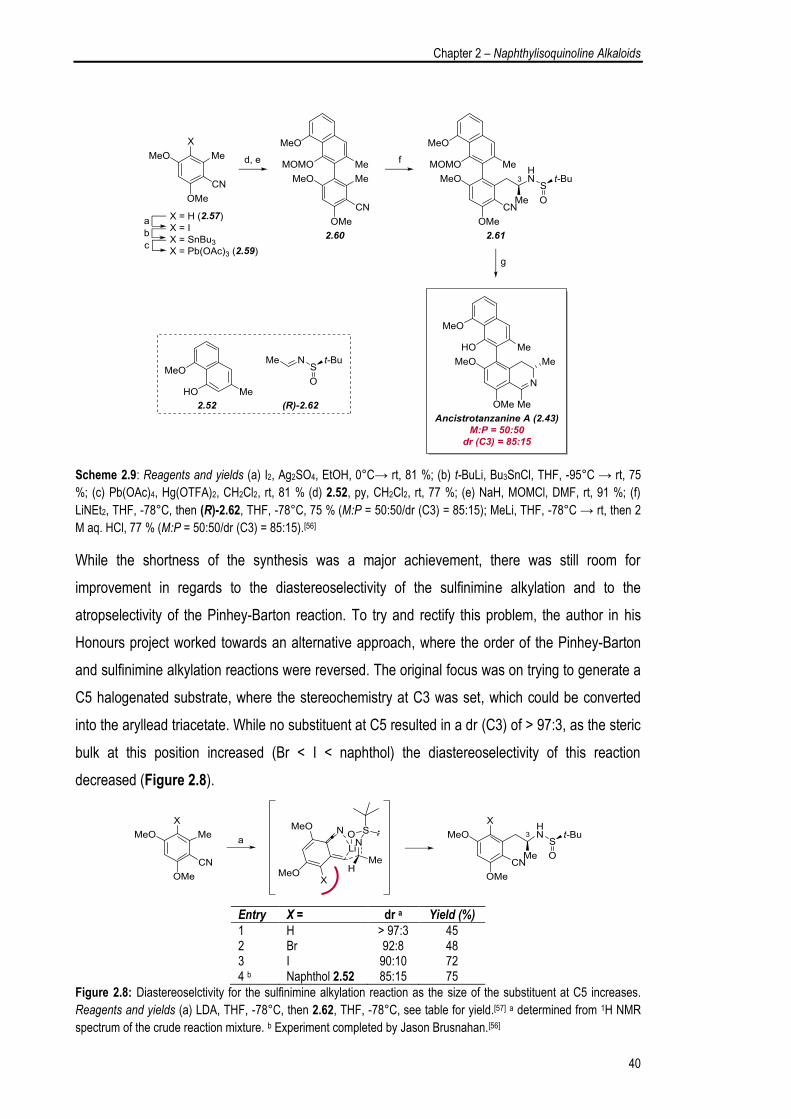
Chapter 2 – Naphthylisoquinoline Alkaloids
40
Scheme 2.9: Reagents and yields (a) I2, Ag2SO4, EtOH, 0°C→ rt, 81 %; (b) t-BuLi, Bu3SnCl, THF, -95°C → rt, 75
%; (c) Pb(OAc)4, Hg(OTFA)2, CH2Cl2, rt, 81 % (d) 2.52, py, CH2Cl2, rt, 77 %; (e) NaH, MOMCl, DMF, rt, 91 %; (f)
LiNEt2, THF, -78°C, then (R)-2.62, THF, -78°C, 75 % (M:P = 50:50/dr (C3) = 85:15); MeLi, THF, -78°C → rt, then 2
M aq. HCl, 77 % (M:P = 50:50/dr (C3) = 85:15).[56]
While the shortness of the synthesis was a major achievement, there was still room for
improvement in regards to the diastereoselectivity of the sulfinimine alkylation and to the
atropselectivity of the Pinhey-Barton reaction. To try and rectify this problem, the author in his
Honours project worked towards an alternative approach, where the order of the Pinhey-Barton
and sulfinimine alkylation reactions were reversed. The original focus was on trying to generate a
C5 halogenated substrate, where the stereochemistry at C3 was set, which could be converted
into the aryllead triacetate. While no substituent at C5 resulted in a dr (C3) of > 97:3, as the steric
bulk at this position increased (Br < I < naphthol) the diastereoselectivity of this reaction
decreased (Figure 2.8).
Entry X = dr a Yield (%)
1 H > 97:3 45 2 Br 92:8 48 3 I 90:10 72 4 b Naphthol 2.52 85:15 75
Figure 2.8: Diastereoselctivity for the sulfinimine alkylation reaction as the size of the substituent at C5 increases.
Reagents and yields (a) LDA, THF, -78°C, then 2.62, THF, -78°C, see table for yield.[57] a determined from 1H NMR
spectrum of the crude reaction mixture. b Experiment completed by Jason Brusnahan.[56]

Chapter 2 – Naphthylisoquinoline Alkaloids
41
Because the hydrogen substituted o-tolylnitrile 2.57 resulted in perfect diastereoselectivity in
sulfinamide 2.63 (Scheme 2.10, Entry 1) our second effort was to halogenate this material.
However, using either NBS or I2 and Ag2SO4 resulted in no halogenation and t-butyl sulfinyl
deprotected product 2.64, supposedly due to the generation of acid during the course of the
reaction. Although oxidation of the t-butyl sulfinyl group to a t-butyl sulfonamide 2.65 allowed for
bromination and subsequent generation of the stannane 2.66 again, the aryllead triacetate 2.67
could not be generated resulting in demetalated material 2.65, as shown in Scheme 2.10.[57]
While doubly protecting the nitrogen atom, or protecting it with a different group may have
resulted in formation of the aryllead triacetate, this was not examined as it would defeat the
purpose of trying to develop a shorter synthesis.
Scheme 2.10: Reagents and yields (a) NBS, CHCl3, rt; (b) I2, Ag2SO4, EtOH, 0°C → rt; (c) m-CPBA, CH2Cl2, 0°C,
98 %; (d) NBS, DMF, rt, 80 %; (e) t-BuLi, Bu3SnCl, THF, -95°C → rt, 29 % [57]
From the efforts described, it is clear that the Davis sulfinimine cyclisation strategy coupled with
the Pinhey-Barton ortho-arylation reaction might allow for ready access to the highly hindered
7,3- and 5,3’-linked members of the naphthylisoquinoline alkaloids. However, to do so, it is critical
that the sulfinimine alkylation can be carried out in a highly stereoselective fashion.
2.4. The Work Described in Chapter 2
Of the 7,3’- and 5,3’-linked naphthylisoquinoline alkaloids, dioncophylline E (2.42) is the most
potent and represents an attractive target. As it lacks the 6-OMe group that ancistrocladidine
(2.39) and ancistrotanzanine A (2.43) have, it can be thought to be structurally simpler and as
such, it was felt that developing a total synthesis could allow the synthetic strategy to be properly
evaluated. Once the validity of the stereoselectivity of the sulfinimine alkylation chemistry has
been established, it was felt that the challenges of ancistrotanzanine A (2.43) and the
ancistrotectorines could then be re-examined (Figure 2.9).

Chapter 2 – Naphthylisoquinoline Alkaloids
42
Figure 2.9: Naphthylisoquinoline alkaloids target proposed for total synthesis as part of this PhD project.
2.5. Initial Investigations into a Total Synthesis of Dioncophylline E
Dioncophylline E (2.42) was isolated from the roots of the rare West African liana Dioncophyllum
thollonii, a member of the Dioncophyllaceae plant family, in 2002. Purification of the extracts by
normal phase column chromatography identified several known naphthylisoquinoline alkaloids.
However, one of the more polar fractions, which appeared as one spot by TLC, consisted of two
new naphthylisoquinoline alkaloids, which were quite similar in the 1H NMR spectra. By using
reverse phase high pressure liquid chromatography (RP-HPLC) the two alkaloids could be
isolated. Upon examination of the ‘pure’ extracts by HPLC and 1H NMR spectroscopy, it was
found that each fraction gave rise to identical HPLC and 1H NMR spectra as the ‘impure’ extract
from the original purification procedure. Re-separation of the peaks by RP-HPLC and immediate
and subsequent monitoring of the fractions by RP-HPLC revealed that the compounds were in
fact one compound that was gradually equilibrating to the mixture at room temperature. This data,
in combination with the near identical NMR and the identical mass spectrometry data led
Bringmann and co-workers to conclude that this compound was a slowly interconverting mixture
of atropisomers. Bearing this in mind the 1H and 13C NMR data showed doubling up of some of
the peaks. In combination with the HRMS data, which indicated a molecular formula of
C23H25NO3, the compound was suggested to have one methoxy group (1H NMR = 4.00/4.04 ppm)
and two hydroxy substituents. The position of the biaryl bond was identified through thorough
analysis and comparison between the 1H-1H COSY and 1H-13C HMBC spectrums which showed
key interactions between H5 → H6, H6 → C3’ and also H5 → C7. These correlations
unambiguously assigned the biaryl linkage as 7,3’-, the first identified for a dioncophyllaceae-type
alkaloid. Furthermore, the semi-stable biaryl axis could be rationalised due to the lack of an
oxygen function at C6, in contrast to other 7,3’-linked naphthylisoquinoline alkaloids which exist
as single atropisomers. The relative stereochemistry of the tetrahydroisoquinoline moiety was
determined to be trans by a key ROESY correlation between H3 and Me1. The absolute
stereochemistry at these positions was determined by oxidative degradation and analysis of the
products by GC which resulted in the assignment of the 1-R and 3-R which was in agreement

Chapter 2 – Naphthylisoquinoline Alkaloids
43
with the relative trans stereochemistry assigned from the ROESY. The structure of dioncophylline
E (2.42), along with the original RP-HPLC traces reported by Bringmann and co-workers are
shown in Figure 2.10.[45]
Figure 2.10: Structure of dioncophylline E (2.42) and the original RP-HPLC traces collected by Bringmann which
indicated it as a mixture of slowly interconverting atropisomers.[45]
2.5.1. Retrosynthetic Analysis of Dioncophylline E
The retrosynthetic analysis of dioncophylline E (2.42) is shown in Figure 2.11. Unlike
ancistrocladidine (2.39) and ancistrotanzanine A (2.43), which are 1,3-dimethyl-3,4-
dihydroisoquinolines, dioncophylline E (2.42) has a trans-1,3-dimethyl-1,2,3,4-
tetrahydroisoquinoline moiety. To access this oxidation state and particularly, the trans geometry
of the 1,3-dimethyl groups of the isoquinoline a diastereoselective reduction of
dihydroisoquinoline 2.68 was proposed. This was based on some precedent in the literature by
Bringmann, who has developed conditions to access both the cis and the trans diastereomers
from a similarly substituted dihydroisoquinoline. Furthermore, having access to all three of these
substrates would also be beneficial for determining important SARs. This dihydroisoquinoline
fragment 2.68 could be obtained using the sulfinimine cyclisation protocol outlined for the
synthesis of ancistrotanzanine A (2.43) which would require access to biarylnitrile 2.69. This
could be synthesised using the Pinhey-Barton ortho-arylation reaction, using the naphthol
fragment 2.52 utilised in both the ancistrocladidine (2.39) and ancistrotanzanine A (2.43)
syntheses, with an appropriately protected aryllead triacetate reagent 2.70, to construct the 7,3’-
biaryl linkage. Consequently, the first goal for the synthesis was to gain efficient access to this
aryllead triacetate reagent 2.70.

Chapter 2 – Naphthylisoquinoline Alkaloids
44
Figure 2.11: Retrosynthetic analysis of dioncophylline E (2.42).
2.5.2. Synthesis of the Biaryl Bond
As described previously for the total syntheses of ancistrocladidine (2.39) and ancistrotanzanine
A (2.43), aryllead triacetates can be readily synthesised by transmetalation from the requisite
organostannane compounds using a mercury(II) salt as a catalyst. The stannane 2.71 can be
prepared by either halogen-lithium exchange and quenching with tri-n-butyltin chloride or by
palladium catalysed stannylation, both protocols of which require access to an appropriately
protected bromide 2.72. This could be synthesised from phenol 2.73 via a regioselective ortho-
bromination and protection protocol which, at the time this project was initiated, could be obtained
in two steps from commercially available aniline 2.74 through a Sandmeyer reaction and
methoxy-deprotection reactions. The methoxymethyl (MOM) protecting group was chosen to be
investigated as this had been utilised successfully for the protection of the naphthol group in both
of the before mentioned total syntheses, as shown in Figure 2.12.
Figure 2.10: Retrosynthetic analysis for the synthesis of aryllead triacetate 2.75.
Commercially available aniline 2.74 was subject to Sandmeyer-type conditions, using the
conditions reported by Giumanini and co-workers (t-BuONO, CuCN, DMSO, 60°C), to afford the
known nitrile 2.76 in 45 % yield.[58] While this methodology allowed access to the desired starting
material, it was a difficult reaction to scale up and required significant efforts to purify it. As such,
it was decided to examine alternate methods for accessing this material.
Sun and co-workers recently reported that it was possible to use a nitrile functional group as a
directing group for a palladium-catalysed C-H activation reaction and that an alkoxide could be

Chapter 2 – Naphthylisoquinoline Alkaloids
45
used as the nucleophile. Indeed, this allowed facile generation of ortho-alkoxybenzonitriles
including the desired nitrile 2.76.[59] Thus, using 2-methylbenzonitrile (2.77) with 10 mol %
Pd(OAc)2 and Na2S2O8 in MeOH at 70°C afforded the nitrile 2.76 in 77 % yield and based on
recovered starting material, this process actually generated the product in 98 % yield.
Subsequent deprotection of the methoxy substituent was achieved in excellent yield using either
BBr3 in dichloromethane at reflux or AlCl3 in dichlorobenzene at reflux. The latter protocol was
preferred as the material obtained after a base/acid work-up was sufficiently pure to use in the
next step, as shown in Scheme 2.11.
Scheme 2.11: Reagents and yields (a) CuCN, t-BuONO, DMSO, 60°C, 45 % (b) Pd(OAc)2, Na2S2O8, MeOH, 70°C,
77 %; (c) BBr3, CH2Cl2, Δ, 89 %; (d) AlCl3, C6H5Cl, Δ, 99 %; (e) NBS, HNi-Pr2, CH2Cl2, rt, 95 %.
Regioselective ortho-bromination of phenol 2.74 was achieved using a pre-formed NBS-HNi-Pr2
complex in dichloromethane, generating the bromide 2.78 in 95 % yield and on a multi-gram
scale. The regioselectivity was proven by a diagnostic ROESY correlation between the 3-Me and
H4, as shown in Figure 2.13.
Figure 2.13: 1H-1H ROESY spectrum of bromide 2.78. Highlighted is the key correlation between the 3-Me and H4
indicating that the position of the bromine atom is next to the hydroxy group.

Chapter 2 – Naphthylisoquinoline Alkaloids
46
Protection of the phenol as the MOM ether (MOMCl, K2CO3, DMF, rt) afforded bromide 2.72 in 90
% yield. Halogen-lithium exchange using t-BuLi in THF at -95°C, was carried out and subsequent
quenching with Bu3SnCl afforded stannane 2.71 in 63 % yield. Interestingly, if the reaction
mixture was allowed to warm to room temperature, rather than 0°C, addition of t-BuLi into the
nitrile group was observed. The stannane 2.71 could also be generated from a palladium-
catalysed stannylation reaction but this gave lower yields of the desired stannane 2.71 (50 %)
with considerable demetalated material (31%) (Scheme 2.12)
Scheme 2.12: Reagents and yields (a) MOMCl, K2CO3, DMF, rt, 90 %; (b) PdCl2(PPh3)2, KOAc, (Bu3Sn)2, DMF,
80°C, 50 % (c) t-BuLi, THF, -95°C, then ClSnBu3, -95°C → 0°C, 63 %.
With the requisite stannane 2.71 in hand attention was turned to generating the aryllead triacetate
2.75 from this material. The standard conditions were employed, stirring stannane 2.71 with 1
equivalent of freshly dried Pb(OAc)4 and 10 mol % Hg(OTFA)2 in dichloromethane at room
temperature for 24 h. (Scheme 2.13), and this resulted in material that from the 1H NMR
spectrum contained a new compound, but it was contaminated with unreacted starting material.
The crude material was purified by trituration with n-hexane to remove any stannane 2.71 and tri-
n-butyltin byproducts. The 1H NMR spectrum of this purified compound showed large upfield
shifts for all of the aromatic peaks which from the integration indicated there were still only two
aromatic protons and so there was still a substituent at C3. However, there was no peak at ~2.1
ppm representative of any acetate protons. On obtaining all of the characterisation data for this
compound it was unambiguously confirmed as the bisarylmercury compound 2.79 by HRMS
which showed the correct exact mass and importantly the diagnostic isotope profile for mercury
(Figure 2.14).
Scheme 2.13: Reagents and yields (a) Hg(OTFA)2, Pb(OAc)4, CH2Cl2, rt, 90 % (2.79).

Chapter 2 – Naphthylisoquinoline Alkaloids
47
HDTD023 #21 RT: 0.56 AV: 1 NL: 3.78E6T: FTMS + p NSI Full ms [150.00-2000.00]
567 568 569 570 571 572 573 574 575 576 577 578 579 580 581 582 583 584 585
m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100R
ela
tive
Ab
un
da
nce
577.10260
575.10083
574.10065576.10260
573.09906
579.10498
578.10559
580.10791571.09900 583.54291
572.14880
581.11102569.14618 584.54388567.29059
Figure 2.14: HRMS of bisarylmercury compound 2.79.
Pinhey has proposed that the transmetalation reaction of organotin compounds to aryllead
triacetate compounds that are catalysed by a mercury(II) salt proceed through a bisarylmercury
intermediate as shown in Figure 2.15.[60] Clearly, isolating and characterisation of the
bisarylmercury compound 2.79 provides strong evidence for this proposal. As it was surprising
that the second transmetalation reaction had not taken place, it was decided to determine what
the reason for this was. To do so, it was decided to examine the reaction of the methoxy-
protected stannane with lead tetraacetate and mercury trifluoroacetate as the Morris group had
successfully generated ortho-methoxy-protected aryllead species in the past when synthesising
ancistrocladidine (2.39) and ancistrotanzanine A (2.43).
Figure 2.15: Catalytic cycle proposed by Pinhey for the transmetalation of organotin compounds to aryllead
triacetates with mercury(II) acetate.[60]
The methoxy-protected stannane 2.80 was synthesised in 65 % yield over two steps using the
protocol developed for the MOM-protecting group (Scheme 2.14). Pleasingly, stirring this
compound with freshly dried Pb(OAc)4 and 10 mol % Hg(OTFA)2 in dichloromethane at room
temperature resulted in formation of a new product where the methoxy group singlet at 4.03 ppm
had moved downfield to 4.25 ppm. This was reminiscent of the shift observed for the ortho-

Chapter 2 – Naphthylisoquinoline Alkaloids
48
methoxy group of the aryllead triacetate used in the synthesis of ancistrocladidine (3.75 → 3.90
ppm).[53] However, even after 24 h the 1H NMR spectrum of the crude reaction mixture indicated
there was still a considerable amount of stannane starting material, as well as demetalated
material and what was assumed to be both the monoarylmercuryacetate and bisarylmercury
compounds.
Scheme 2.14: Reagents and yields (a) MeI, K2CO3, DMF, rt, 95 %; (b) t-BuLi, THF, -95°C, then Bu3SnCl, -95°C →
0°C, 68 %; (c) 10 mol % Hg(OTFA)2, Pb(OAc)4, CH2Cl2, rt.
To confirm that the aryllead triacetate had been formed, the crude material was subjected to the
Pinhey-Barton coupling conditions, utilising 1 equivalent of naphthol 2.52 i [61] as the coupling
partner and 3 equivalents of pyridine in dichloromethane at room temperature. This resulted in
the generation of a new compound in the 1H NMR spectrum of the crude reaction mixture.
Purification of the material by flash chromatography afforded arylated naphthol 2.81 in 16 % yield
over the two steps. Examination of the 1H NMR spectrum of the purified material revealed a new
methoxy peak at 3.63 ppm, significantly further upfield than that observed previously for the
trialkyltin compound and that the naphthol H3’ singlet at 7.10 ppm was absent, confirming that a
substituent had been added at this position. The position of the biaryl bond was unambiguously
confirmed by 2D NMR spectroscopy and comparison of this to the correlation observed for
natural dioncophylline E. Key 2D NMR correlations are shown in Figure 2.16.
Figure 2.16: Synthesis of arylated naphthol 2.81 with key 1H-1H ROESY and 1H-13C HMBC correlations shown which
helped prove the position of the biaryl bond. Reagents and yields (a) py, CH2Cl2, rt, 16 % (2 steps).
Given that the methoxy protected aryllead triacetate could be formed and used to synthesise the
biaryl bond, whereas the MOM-protected reagent 2.75 could not, lead us to speculate that the
i The naphthol 2.52 required for this reaction was synthesised in 21 % yield over 3 steps from commercially available 3,3-dimethylacrylic acid using the method described by Watanabe and Bringmann.[61] Naphthol 2.52 can also be synthesised using the protocol developed by Bungard and Morris which allows for differential substitution of the naphthalene fragment. An improvement of this synthesis was established using a Crabbe homolagation (see experimental section). However, the fact that Watanabe’s synthesis can be carried out on a large scale meant that it was preferred.

Chapter 2 – Naphthylisoquinoline Alkaloids
49
well-known chelating ability of the MOM group meant that a stabilised arylmercury complex was
being formed. This complex must not be suitable for further transmetalation reactions. A similar
observation has been made by Akkerman and co-workers who found that a bisarylmercury
compound with an ethylene glycol group ortho to the mercury was inert to transmetalation with
magnesium.[62] They obtained a crystal structure of their compound, which confirmed that the
mercury atom was chelating to the neighbouring oxygen atoms. However, the methoxy protected
substrate validated that this protocol could construct the biaryl linkage for dioncophylline E (2.42).
Unfortunately, using biarylnitrile 2.81 to complete the synthesis was problematic as it was not
possible to regioselectively deprotect the methoxy group adjacent to the nitrile. i A non-
coordinating protecting group that could be deprotected orthogonally, without affecting the
methoxy substituent of the naphthalene, was required. Therefore the isopropyl, benzyl and the
TBS protecting groups were examined. The isopropyl 2.82 and benzyl 2.83 protected stannanes
could be synthesised from bromide 2.78 similarly to the MOM-protected substrate, as shown in
Scheme 2.15.ii
Scheme 2.15: Reagents and yields (a) 2-bromopropane, K2CO3, DMF, 60°C, 99 %; (b) t-BuLi, THF, -95°C, then
Bu3SnCl, -95°C → 0°C, 75 %; (c) BnBr, K2CO3, DMF, rt, 97 %; (b) t-BuLi, THF, -95°C, then Bu3SnCl, -95°C → 0°C,
70 %.
With ready access to significant quantities of the protected stannanes (2.82 and 2.83),
examination of each of their behaviour in the transmetalation reaction could be carried out. The
transmetalation reactions were initially carried out in NMR tubes and progress followed by 1H
NMR spectroscopy. Each of the protected stannanes was examined using different loadings of
mercury(II) trifluoroacetate catalyst (10, 20, 30 and 50 mol %) over a 24 hour period with data
points taken every 6 hours. As can be seen in Table 2.2, the relative conversion of these
i Preliminary attempts to regioselectively deprotect the methoxy group adjacent to the nitrile over the methoxy group on the naphthalene moiety failed and resulted in sole regioselective deprotection of the methoxy group on the naphthalene. ii The TBS-protected bromide underwent a retro-[1,3]-Brook rearrangement when attempting to complete halogen-lithium-tin exchange. Here, the TBS protecting group migrated from the hydroxy group, to the position ortho to it – previously occupied by the bromine atom. In an effort to synthesise the TBS-stannane a palladium catalysed stannylation was examined. However, this led to no product and almost quantitative recovery of the starting material. Details of this can be found in the experimental section, Chapter 5.

Chapter 2 – Naphthylisoquinoline Alkaloids
50
stannanes to the corresponding aryllead triacetate after 24 h was quite similar for each of the
mercury(II) trifluoroacetate catalyst loadings. Because of this, for the sake of this discussion, only
the data collected for the benzyl-protected stannane 2.83 will be discussed. However, a more
detailed breakdown of this data can be found in the experimental section, Chapter 5.
Table 2.2: Percent relative abundance of aryllead triacetate formed after 24 h using both the isopropyl and benzyl
protected stannanes 2.82 and 2.83 with different loading of mercury(II) trifluoroacetate catalyst.
Stannane Substrate
Hg(OTFA)2 Isopropyl 2.82 Benzyl 2.83
10 mol % 13 14
20 mol % 32 32
30 mol % 35 45
50 mol % 42 38
Reactions were carried out in NMR tubes and progress followed by 1H NMR spectroscopy. Full experimental details
can be found in Chapter 5.
The benzylic methylene protons for stannane 2.83, monoarylmercury acetate 2.84,
bisarylmercury 2.85, aryllead triacetate 2.86 and the demetalated 2.87 material, were sufficiently
resolved in the 1H NMR spectrum of the crude reaction mixture so that their relative abundances
could be followed by integration of these peaks.i Similar to the result obtained for the methoxy-
protected stannane 2.80, using 10 and 20 mol % of Hg(OTFA)2 resulted in incomplete
consumption of the starting material and a considerable amount of demetalated material after 24
h. Using 30 mol % of Hg(OTFA)2 resulted in complete consumption of the starting material after
18 h with 35 % conversion to the aryllead triacetate reagent. This could not be improved upon,
even if using 50 mol % which, although resulting in complete consumption of the starting material
within 6 h, the outcome of conversion to the aryllead triacetate was similar to that observed for 30
mol %, as shown in Figure 2.17. Thus, 30 mol % Hg(OTFA)2 with one equivalent of freshly dried
Pb(OAc)4 in dichloromethane at room temperature for 18 h was determined to be the best
conditions to access the aryllead reagent 2.86.ii There were an additional two key facts that could
be concluded from the study: (1) although the reactions were left for 48 h (data not shown), the
reactions were generally complete after 18 h – even when stannane starting material remained in
i To differentiate between each of the intermediates in this reaction authentic compounds were synthesised. The experimental details and yields for these reactions can be found in the experimental section, Chapter 5. ii The reason for the increased demetalation in the NMR study is likely to be due to residual water present in the reaction mixture. Despite care being taken to carry out the reactions as true to larger scale reaction conditions as possible smaller scale thorough drying of glassware and unconventional transferring of solutions to the NMR tube could have all contribute to this. Although demetalated material was also observed upon scale up, it was fair less significant.

Chapter 2 – Naphthylisoquinoline Alkaloids
51
the reaction mixture, and (2) the amount of aryllead triacetate that was produced in the reaction
was proportional to the amount of mercury catalyst that was added.
Scheme 2.16: Reagents and yields (a) Hg(OTFA)2, Pb(OAc)4, CDCl3, rt
Figure 2.17: Reaction profiles for benzyl protected stannane 2.83 with different loadings of Hg(OTFA)2 catalyst, as
observed by 1H NMR (400 MHz, CDCl3)
0
10
20
30
40
50
60
70
80
90
100
0 6 12 18 24
Rel
ativ
e A
bu
nd
ance
(%
)
Time (h)
10 mol % Hg(OTFA)2
0
10
20
30
40
50
60
70
80
90
100
0 6 12 18 24
Rel
atiiv
e A
bu
nd
ance
(%
)
Time (h)
20 mol % Hg(OTFA)2
0
10
20
30
40
50
60
70
80
90
100
0 6 12 18 24
Rel
ativ
e A
bu
nd
ance
(%
)
Time (h)
30 mol % Hg(OTFA)2
0
10
20
30
40
50
60
70
80
90
100
0 6 12 18 24
Rel
ativ
e A
bu
nd
ance
(%
)
Time (h)
50 mol % Hg(OTFA)2

Chapter 2 – Naphthylisoquinoline Alkaloids
52
Considering that the data collected from this study was comparable for both the isopropyl and
benzyl protecting groups, the benzyl-protected stannane 2.83 was selected for scale up as the
benzyl group was felt to be more easily deprotected later in the sequence. The reaction was
carried out on a 1.7 g scale, affording the aryllead triacetate 2.86 which was coupled to naphthol
2.52 using the Pinhey-Barton conditions. Gratifyingly, the arylated naphthol 2.88 could be isolated
from this mixture in 40 % over the two steps, as shown in Scheme 2.17.
Scheme 2.17: Reagents and yields (a) 30 mol % Hg(OTFA)2, Pb(OAc)4, CH2Cl2, rt; (b) 2.52, py, CH2Cl2, 40 %.
While this result meant that the total synthesis could proceed, it was felt that an improvement in
the yield would be highly desirable. A general trend that was observed throughout this study was
that the stannane reagents investigated were not reactive enough to undergo a second
transmetalation reaction with the monoarylmercury acetate to form the bisarylmercury compound.
This is in contrast to the successful generation of the nitrile-containing aryllead triacetate 2.59 by
Brusnahan in his total synthesis of ancistrotanzanine A (2.43). The contrasting result can be
rationalised by examining the electronic nature of the substituents on the aromatic ring. The
electron withdrawing nitrile group has been documented to reduce the reactivity of the
stannane.[63] For the ancistrotanzanine A aryllead reagent 2.59 which was successfully prepared
by Brusnahan, the electron withdrawing nature of the nitrile is countered by having two electron
donating methoxy substituents, whereas on the substrate being investigated in this thesis, there
is only one alkoxy substituent. Clearly, this is not enough to activate the substrate for the
transmetalation process. As it is not possible to change the substrate, it was decided to examine
an alternative species to transmetalate to the aryllead triacetate 2.86.
While the tin-lead exchange route is the preferred method to synthesise aryllead triacetates due
to their ease of purification[60] the synthesis of aryllead triacetates can also be achieved by either
direct plumbation,[64] on substrates with electron donating substituents, or transmetalation from
either boron[63] or silicon species.[64] Pinhey has reported that boronic acids, in particular, are
superior to trialkyltin reagents in the mercury-catalysed transmetalation reactions on compounds
with electron withdrawing substituents.[63] Furthermore, the boronic acid could be synthesised in a
similar manner to the stannane 2.83.

Chapter 2 – Naphthylisoquinoline Alkaloids
53
As shown in Scheme 2.18, the boronic acid 2.89 was synthesised using a halogen-lithium
exchange from bromide 2.90, as before, and the resulting lithio species was quenched with B(Oi-
Pr)3. After an aqueous workup and purification the boronic acid 2.89 was isolated in 84 % yield.
Subjecting this material to the transmetalation conditions, using 10 mol % Hg(OTFA)2 with 1
equivalent of freshly dried Pb(OAc)4 in 1,2-DCE for 20 h, resulted in complete consumption of the
starting material and only the aryllead triacetate 2.86 was observed in the 1H NMR spectrum of
the crude reaction mixture. The crude material collected from this reaction was immediately
subjected to the Pinhey-Barton coupling conditions and the desired arylated naphthol 2.88 was
isolated in an improved 78 % overall yield.i
Scheme 2.18: Reagents and yields (a) t-BuLi, THF, -95°C, then ClSnBu3, -95°C → 0°C, %; (b) 10 mol %
Hg(OTFA)2, Pb(OAc)4, 1,2-DCE, rt; (c) 2.52, py, 1,2-DCE, 78 % (2 steps).
With efficient access to the arylated naphthol 2.88 now established, attention could be focussed
on the sulfinimine cyclisation protocol.
2.5.3. Examination of the Sulfinimine Cyclisation Protocol
Using Davis’ model for diastereoselection, the (S)-sulfinimine (S)-2.62 was used to generate the
R stereochemistry in the alkylated product.[55] The requisite (S)-sulfinimine (S)-2.62 was
synthesised from (S)-sulfinamide (S)-2.91 in one step using the protocol reported by
Brusnahan.[56] To examine the diastereoselectivity of the sulfinimine alkylation reaction the
arylated naphthol 2.88 had to first be protected. To allow for a late stage global deprotection, the
benzyl protecting group was again employed for this purpose. Deprotonation of naphthol 2.88
with sodium hydride and quenching the resulting anion with benzyl bromide in DMF at room
temperature afforded biarylnitrile in 87 % yield. Subsequently, the biarylnitrile was treated with 3
equivalents of LDA in THF at -78°C and the tolylnitrile anion was reacted with (S)-sulfinimine (S)-
2.62. The 1H NMR spectrum of the crude product confirmed the disappearance of the methyl
group adjacent to the nitrile (2.64 ppm) and the appearance of diagnostic multiplets representing
the new CH and CH2 protons at 3.67 – 3.8 and 2.98 – 3.18 ppm respectively. Interestingly,
instead of seeing one new singlet integrating for 9H for the t-butyl group of the sulfinamide at
~1.05 ppm and a doublet representing the C3-Me group at ~1.4 ppm in the 1H NMR spectrum of
i The success of the boronic acid prompted us to examine the potassium trifluoroborate and boronic acid ethyleneglycol ester as well. However, both of these performed inferiorly to the boronic acid.

Chapter 2 – Naphthylisoquinoline Alkaloids
54
the purified material, these were split into two in a ratio of 50:50. Because of the two benzyl-
protecting groups ortho to the biaryl axis these were identified as result of two atropisomers being
present. Importantly, examination of the 1H NMR spectrum between 1.1 and 2.0 ppm indicated
that only one diastereomer had been synthesised.
Having set the stereochemistry at C3, the dihydroisoquinoline 2.92 was generated by treating
sulfinamide 2.93 with 5 equivalents of MeLi in THF at -78°C, warming to 0°C and working up with
2 M aqueous hydrochloric acid. Again, a 50:50 mixture of atropisomers was observed in the 1H
NMR spectrum of the crude reaction mixture and formation of the dihydroisoquinoline was
confirmed by the appearance of two new atropisomeric peaks, representing the C1 methyl group,
at 2.35 and 2.46, which each integrated for 3H. Purification afforded the dihydroisoquinoline 2.92
in 45 % yield from arylated naphthol 2.88, as shown in Scheme 2.19.
Scheme 2.19: Reagents and yields (a) MeCHO, 4 Ǻ Mol. Sieves, CH2Cl2, rt, 88 % (b) NaH, BnBr, DMF, rt, 87 %; (c)
LDA, THF, -78°C, then (S)-2.62, THF, -78°C, 72 %; (d) MeLi, THF, -78°C → rt, then 2 M aq. HCl, rt, 71 %.
To complete the synthesis of dioncophylline E (2.42) a diastereoselective reduction of 1,3-
dimethyl-3,4-dihydroisoquinoline 2.92, to afford the trans-1,3-dimethyl-1,2,3,4-
tetrahydroisoquinoline, followed by a global deprotection of the two benzyl protecting groups, was
required. To achieve this, Bringmann has reported protocols, which exploit the stereoelectronic
preference for axial attack, to access both the cis- and the trans-1,3-dimethyl-1,2,3,4-
tetrahydroisoquinolines from 1,3-dimethyl-3,4-dihydroisoquinoline 2.94, as shown in Figure 2.18.
Reduction with NaBH4 in methanol gives rise to the cis-stereoisomer 2.95. The rationale behind
the stereoselectivity is that the nitrogen atom of the 1,3-dimethyl-3,4-dihydroisoquinoline
coordinates the NaBH4, this delivers the hydride source intramolecularly to the bottom face of the
1,3-dimethyl-3,4-dihydroisoquinoline. To access the trans-isomer 2.96, the 1,3-dimethyl-3,4-
dihydroisoquinoline 2.94 is complexed with the Lewis acid AlMe3, then LiAlH4 is added. As the
AlMe3 coordinates to the nitrogen, this blocks hydride attack from the bottom face and the only
option is attack by LiAlH4 through an open transition state to the top face of the 1,3-dimethyl-3,4-
dihydroisoquinoline, resulting in the trans-stereoisomer 2.96, as shown in Figure 2.18.
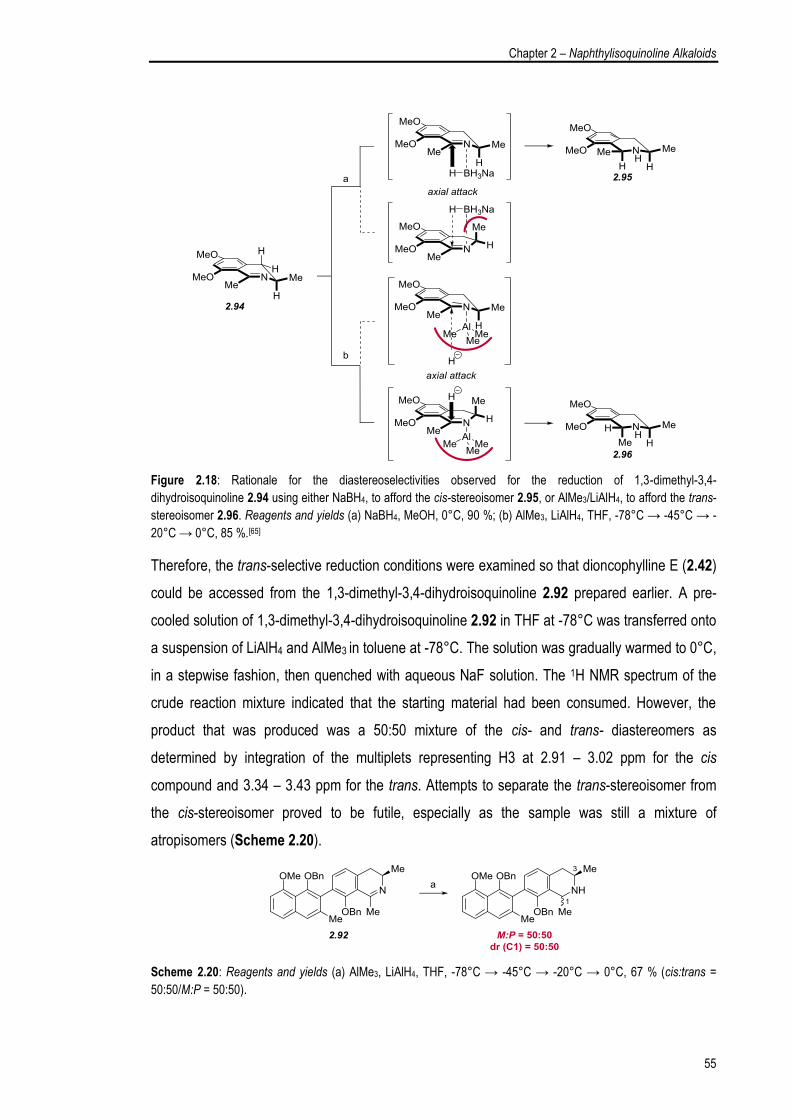
Chapter 2 – Naphthylisoquinoline Alkaloids
55
Figure 2.18: Rationale for the diastereoselectivities observed for the reduction of 1,3-dimethyl-3,4-
dihydroisoquinoline 2.94 using either NaBH4, to afford the cis-stereoisomer 2.95, or AlMe3/LiAlH4, to afford the trans-
stereoisomer 2.96. Reagents and yields (a) NaBH4, MeOH, 0°C, 90 %; (b) AlMe3, LiAlH4, THF, -78°C → -45°C → -
20°C → 0°C, 85 %.[65]
Therefore, the trans-selective reduction conditions were examined so that dioncophylline E (2.42)
could be accessed from the 1,3-dimethyl-3,4-dihydroisoquinoline 2.92 prepared earlier. A pre-
cooled solution of 1,3-dimethyl-3,4-dihydroisoquinoline 2.92 in THF at -78°C was transferred onto
a suspension of LiAlH4 and AlMe3 in toluene at -78°C. The solution was gradually warmed to 0°C,
in a stepwise fashion, then quenched with aqueous NaF solution. The 1H NMR spectrum of the
crude reaction mixture indicated that the starting material had been consumed. However, the
product that was produced was a 50:50 mixture of the cis- and trans- diastereomers as
determined by integration of the multiplets representing H3 at 2.91 – 3.02 ppm for the cis
compound and 3.34 – 3.43 ppm for the trans. Attempts to separate the trans-stereoisomer from
the cis-stereoisomer proved to be futile, especially as the sample was still a mixture of
atropisomers (Scheme 2.20).
Scheme 2.20: Reagents and yields (a) AlMe3, LiAlH4, THF, -78°C → -45°C → -20°C → 0°C, 67 % (cis:trans =
50:50/M:P = 50:50).

Chapter 2 – Naphthylisoquinoline Alkaloids
56
While separation of the mixture could have been attempted using HPLC, a 50:50 mixture for this
reduction was not considered good enough for a key step in the total synthesis of these
compounds. To achieve our goals, an alternate strategy that was highly diastereoselective was
essential. To allow us to achieve this, it was decided to focus on the total synthesis of phylline
(2.97), which is one of the biosynthetic precursors of the naphthylisoquinoline alkaloids. As it
does not have a biaryl linkage it will be easier to interpret the results that are obtained.

Chapter 2 – Naphthylisoquinoline Alkaloids
57
2.6. Total Synthesis of Phylline
The trans-1,3-dimethyl-1,2,3,4-tetrahydroisoquinoline moiety of dioncophylline E (2.42) is a
natural product in its own right. Phylline (2.97) was originally isolated from the stem of a member
of the Dioncophyllaceae plant family, Habropetalum dawei, by Bringmann and co-workers in
1999.[66] Its N-methyl 2.98 and N,O-dimethyl 2.99 structural variants have also been isolated from
a West African species of Ancistrocladaceae by Bringmann and co-workers in 2003.[67] The
structure and the stereochemistry of the 1,3-dimethyl substituents of these compounds was
unambiguously proven by a 1H NMR NOE correlation between H1 and Me3 and also by an X-ray
crystal structure of the hydrochloride salt of N,O-dimethylphylline 2.99 (Figure 2.19).[67] The most
efficient methods to access trans-1,2,3,4-tetrahydroisoquinolines are either the Pictet-Spengler
reaction or the combination of a Bischler-Naperialski reaction followed by a trans-selective
reduction (as was outlined briefly in the previous section).[68] As both reactions involve an
electrophilic aromatic substitution the cyclisation reaction will preferentially cyclise para to any
electron donating substituent present. Because phylline has just the C8 hydroxy group, this would
mean cyclisation will occur para to that functional group and generate an isomer which is
undesired, as shown in Figure 2.19.
Figure 2.19: Structure of phylline (2.97) and its methylated structural variants.
To circumvent this problem Bringmann and co-workers completed the first total synthesis of
phylline (2.97) starting from 3,5-dimethoxyphenylacetone (2.100).[65] They added extra
oxygenation to ensure an appropriate Pictet-Spengler reaction would take place, and once that
was achieved the extraneous group was deleted. The stereochemistry of C3 was constructed
using a two-step reductive amination protocol between α-methylbenzylamine and ketone 2.100 in
85 % yield over the two steps. The α-methylbenzyl auxiliary was replaced with a benzyl protecting

Chapter 2 – Naphthylisoquinoline Alkaloids
58
group and one of the two aromatic methoxy groups was deprotected using NaSEt in DMF to
afford the Pictet-Spengler substrate 2.101 in 92 % yield over four steps. By varying the pH of the
reaction mixture it proved possible to control the selectivity for the Pictet-Spengler reaction. While
good to excellent diastereoselectivity for the trans-stereoisomer was observed across the pH
range analysed, when basic (pH = 11.5) conditions were used, the major product was the result
of the cyclisation occurring para to the 6-OH group, which was the one desired to synthesise
phylline (2.97).
The optimised conditions afforded a 9:91 mixture of the cis:trans and an 84:16 mixture of the 6-
OH:8-OH compounds and this meant that the desired 1,3-dimethyl-1,2,3,4-tetrahydroisoquinoline
product 2.102 could be isolated in 78 % yield. The 6-OH group was removed in a two-step
procedure and in 89 % yield, utilising a reductive deoxygenation protocol originally developed by
Musliner and Gates. Conversion of the tetrahydroisoquinoline 2.103 to phylline (2.97) was
achieved in 58 % yield using a four step sequence. In summary, phylline (2.97) was synthesised
in 13 steps and in 32 % overall yield (Scheme 2.21).
Scheme 2.21: Reagents and yields (a) (R)-1-phenethylamine, PhMe, Δ; (b) H2 (5 bar), Raney Ni, EtOH, rt, 85 % (2
steps); (c) NH4HCO2, Pd/C, MeOH, Δ, 97 %; (d) PhCOCl, NEt3, CH2Cl2, rt; (e) LiAlH4, THF, Δ; (f) NaSEt, DMF, Δ, 95
% (3 steps); (g) MeCHO, pH = 11.5, 5:1 i-PrOH/H2O, 78 %; (h) 5-chloro-1-phenyltetrazole, PTC, 2 M aq. NaOH,
CH2Cl2; (i) H2 (5 bar), Pd/C, EtOH, 89 % (2 steps); (j) PhCOCl, NEt3, CH2Cl2, rt; (k) LiAlH4, THF, Δ (l) NaSEt, DMF, Δ;
(m) H2, Pd/C, MeOH, 58 % (4 steps).[65]
While this sequence achieved its goal, it is clear that such a sequence would not be appropriate
for our synthesis of dioncophylline E (2.42). Accordingly, it was decided to examine the synthetic
strategy detailed in Figure 2.20. If the Davis sulfinimine cyclisation protocol that we had used
could be applied here, it would immediately circumvent both the regioselectivity issue and the 6-
OH deletion steps as the nitrile group would ensure the correct product would be formed.
However, the immediate goal was to develop alternative conditions that would allow the reduction
of the 1,3-dimethyl-3,4-dihydroisoquinoline 2.104 to the trans-stereoisomer stereoselectively.

Chapter 2 – Naphthylisoquinoline Alkaloids
59
Achieving this would allow us to examine its application to a stereoselective synthesis of
dioncophylline E (2.42).
Figure 2.20: Retrosynthetic analysis of Phylline (2.97).
As detailed in Figure 2.21, the synthesis began with the benzyl ether 2.105. This was readily
obtained by reacting phenol 2.74, which was previously synthesised (Section 2.5.2.), with benzyl
bromide and potassium carbonate in DMF (97 % yield). Benzyl ether 2.105 was deprotonated
using LDA at -78°C and the resulting tolylnitrile anion was reacted with sulfinimine (S)-2.62 to
afford the sulfinamide 2.106, in high diastereoselectivity (> 97:3). Conversion to the
dihydroisoquinoline 2.104 was achieved in 84 % yield by first adding MeLi, followed by treatment
with 2 M aqueous HCl solution to implement cyclisation.
As noted earlier, reduction to the cis-stereoisomer was readily achieved by treating 1,3-dimethyl-
3,4-dihydroisoquinoline 2.104 with NaBH4 in methanol at -78°C and allowing the reaction solution
to slowly warm to room temperature. This resulted in complete consumption of the starting
material and a single new product formed. Analysis of the 1H NMR spectrum revealed a multiplet
at 2.84 – 2.95 ppm, which was assigned as H3. There was a strong correlation between H3 and
H1 in the 1H-1H NOE spectrum (Figure 2.21, entry 1), providing convincing evidence of the cis-
relationship of tetrahydroisoquinoline 2.107.
As discussed earlier, reaction of 1,3-dimethyl-3,4-dihydroisoquinoline 2.94 with AlMe3 and LiAlH4
is reported to favour the trans-isomer. While this was not the case in our first generation synthesis
of dioncophylline E (2.42) (Section 2.5.3.), it was felt prudent that these conditions be attempted
on the phylline system. Thus, a solution of 1,3-dimethyl-3,4-dihydroisoquinoline 2.104 in THF at -
78°C was added to a suspension of AlMe3 and LiAlH4 in toluene at -78°C. The solution was
warmed gradually to 0°C and quenched with saturated aqueous NaF. The 1H NMR spectrum of
the crude reaction mixture indicated complete consumption of the starting material but instead of
observing only the trans-configured 1,3-dimethyl-1,2,3,4-tetrahydroisoquinoline 2.108 as the
single new compound there was clearly cis-configured 1,3-dimethyl-1,2,3,4-tetrahydroisoquinoline
2.107 present in the reaction mixture. Integration of the multiplets at 2.84 – 2.95 ppm and that for
the trans compound at 3.25 – 3.37 ppm showed that these were in a ratio of 25:75 in favour of the
trans-configured 1,3-dimethyl-1,2,3,4-tetrahydroisoquinoline. Unfortunately, these two

Chapter 2 – Naphthylisoquinoline Alkaloids
60
diastereomers could not be separated by column chromatography and so the yield is reported for
both diastereomers (Figure 2.21, Entry 2). While this ratio is better than that observed using the
same conditions for dioncophylline E (2.42), it is still not as diastereoselective as the results
reported by Bringmann. In an attempt to improve the diastereoselectivity of this reduction a
bulkier Lewis acid was examined (Ali-Bu3) but this resulted in poorer diastereoselectivity (Figure
2.21, Entry 3). Ruthenium catalysed asymmetric transfer hydrogenation conditions were also
examined using both enantiomers of the ruthenium catalyst. However, in both cases, excellent
diastereoselectivity for the cis-configured 1,3-dimethyl-1,2,3,4-tetrahydroisoquinoline 2.107 was
achieved. Interestingly, the (S,S)-ligand system, which based on the known stereochemical
induction models should preferentially generate the trans-1,3-dimethyl-1,2,3,4-
tetrahydroisoquinoline, gave the cis- product but the reaction rate was appreciably slower. This
poor reactivity would suggest that the C3 methyl group disrupts the transition state for this
reaction, preventing the formation of the trans-stereoisomer (Figure 2.21, Entry 5).[69]
Entry Reagents (d) Conversion (%)a dr (cis:trans) Yield (%)
1 NaBH4, MeOH, -78°C → rt 100 > 97:3 92 2 AlMe3, LiAlH4, THF, -78°C → 0°C 100 25:75 68 3 Ali-Bu3, LiAlH4, THF, -78°C → 0°C 100 60:40 53
4 RuCl[(R,R)-TsDPEN](p-cymene), HCO2H:NEt3, MeCN, rt
100 > 97:3 87
5 RuCl[(S,S)-TsDPEN](p-cymene), HCO2H:NEt3, MeCN, rt
70 > 97:3 26
Figure 2.21: Synthesis of 1,3-dimethyl-3,4-dihydroisoquinoline 2.104 and a table of conditions examined to generate
the cis- and trans-1,3-dimethyl-1,2,3,4-tetrahydroisoquinolines. Also shown is a key nOe interaction observed in the
cis-stereoisomer to prove its structure. Reagents and yields (a) BnBr, K2CO3, DMF, rt, 97 % (b) LDA, THF, -78°C,
then (S)-2.62, THF, -78°C, 53 %; (c) MeLi, THF, -78°C → rt, then 2 M aq. HCl, rt, 84 %; (d) see table for reagents
and yields. a Determined by integration of the H3 signal in the 1H NMR spectrum of the crude reaction mixture.
Given the lack of success employing conventional Lewis acid or asymmetric transfer
hydrogenation approaches it was decided to trial a ‘covalent Lewis acid’ protocol. Polniaszek and
co-workers have pioneered the use of differentially substituted α-methylbenzyl derivatives as
auxiliaries for chiral reductions of 1-substituted-3,4-dihydroisoquinolinium ions,[70] and this has

Chapter 2 – Naphthylisoquinoline Alkaloids
61
been successfully applied by Hoye and coworkers to synthesise trans-1,3-dimethyl-1,2,3,4-
tetrahydroisoquinolines.[37] Unfortunately, an α-methylbenzyl derivative could not be directly
employed in this case as it was unlikely that synthesising it by alkylating 1,3-dimethyl-3,4-
dihydroisoquinoline 2.104 with a α-methylbenzyl bromide would be stereoselective. In Hoye’s
case, his synthesis required the use of α-methylbenzyl amine, which meant that this was not an
issue for him. However, other groups have shown that even benzyl groups that are not
substituted on the alpha-carbon can be used on 1,3-disubstituted-3,4-dihydroisoquinoline
systems and allow stereocontrol.[71] It was therefore hoped that in our work the methyl group at
C3 would force the benzyl group on the iminium ion into a position of least steric interaction,
similar to a conventional Lewis acid, so that reduction could occur from the opposite face.
Subsequently, dihydroisoquinoline 2.104 was reacted with BnBr in MeCN at reflux to form the
iminium ion 2.109, which was reduced with NaBH4 in MeOH at -78°C. After warming to room
temperature, the crude reaction mixture was isolated and examined by 1H NMR spectroscopy.
The 1H NMR spectrum of the crude reaction mixture gave a result similar to that observed for the
AlMe3/LiAlH4 case with a 25:75 mixture of the cis- and trans-1,3-dimethyl-1,2,3,4-
tetrahydroisoquinolines observed (Figure 2.22, Entry 1). Attempts to improve this ratio by using a
bulkier reducing agent (DIBAL-H) to help discriminate between the two conformations did not
have any effect (Figure 2.22, Entry 2) and using chloro substituted benzyl derivatives, which
were found to be superior by Polniaszek in related systems, resulted in poorer diastereoselectivity
(Figure 2.22, Entry 3) or a complex mixture of products (Figure 2.22, Entry 4).[70b]
Entry Reagents (a) R = Reagents (b) dr (cis:trans) Yield (%)
1 BnBr, MeCN, Δ Bn NaBH4, MeOH, -78°C → rt 25:75 92 2 BnBr, MeCN, Δ Bn DIBAL-H, CH2Cl2, -78°C → rt 25:75 68 3 2-ClBnBr, MeCN, Δ 2-ClBn NaBH4, MeOH, -78°C → rt 60:40 53 4 2,6-Cl2BnBr, MeCN, Δ 2,6-Cl2Bn NaBH4, MeOH, -78°C → rt ND ND
Figure 2.22: Table of conditions examined for the diastereoselective reduction of 1,3-dimethyl-3,4-
dihydroisoquinoline 2.104. See table for reagents and yields. ND: not determined.

Chapter 2 – Naphthylisoquinoline Alkaloids
62
By this stage it was clear that this strategy was not going to be as selective as we required for our
synthesis and prompted us to re-think our strategy. A recent report by Lipshutz and co-workers,
who had similar problems gaining diastereoselective access to trans-configured 1,3-disubstituted-
1,2,3,4-tetrahydroisoquinolines via the AlMe3/LiAlH4 approach reported by Bringmann, employed
a diastereoselective addition of methyl magnesium chloride into 3,4-dihydroisoquinoline 2.110 en
route to their total synthesis of korupensamine B (2.17), as shown in Scheme 2.22.[41] It was
therefore decided to examine this approach on our system.
Scheme 2.27: Reagents and yields (a) 8.4 eq. MeMgCl, -78°C → rt, Et2O, 85 % (< 1:20 cis:trans).[41]
Reduction of the nitrile group of sulfinamide 2.106 with DIBAL-H followed by an acidic workup
afforded 3-methyl-3,4-dihydroisoquinoline 2.111 in 67 % yield.[72] The 1H NMR spectrum of 3-
methyl-3,4-dihydroisoquinoline 2.111 confirmed the disappearance of the t-butyl group of the
sulfinamide and the appearance of a diagnostic singlet at 8.78 ppm representing the new H1
imine proton. Furthermore, there was a signal at 1622 cm-1 in the infrared spectrum confirming
that the molecule had an imine. Employing Lipshutz’s conditions and treating this compound with
MeMgBr in diethyl ether at room temperature for 18 h generated a new spot as observed by TLC,
but there was also a considerable amount of starting material remaining. Workup of the reaction
resulted in a crude reaction mixture and from examination of the 1H NMR spectrum revealed that
the trans-configured 1,3-methyl-1,2,3,4-tetrahydroisoquinoline 2.108 was the only product, albeit
in low yield. Nevertheless this could be isolated from the crude material in 13 % yield (Figure
2.23, Entry 1).
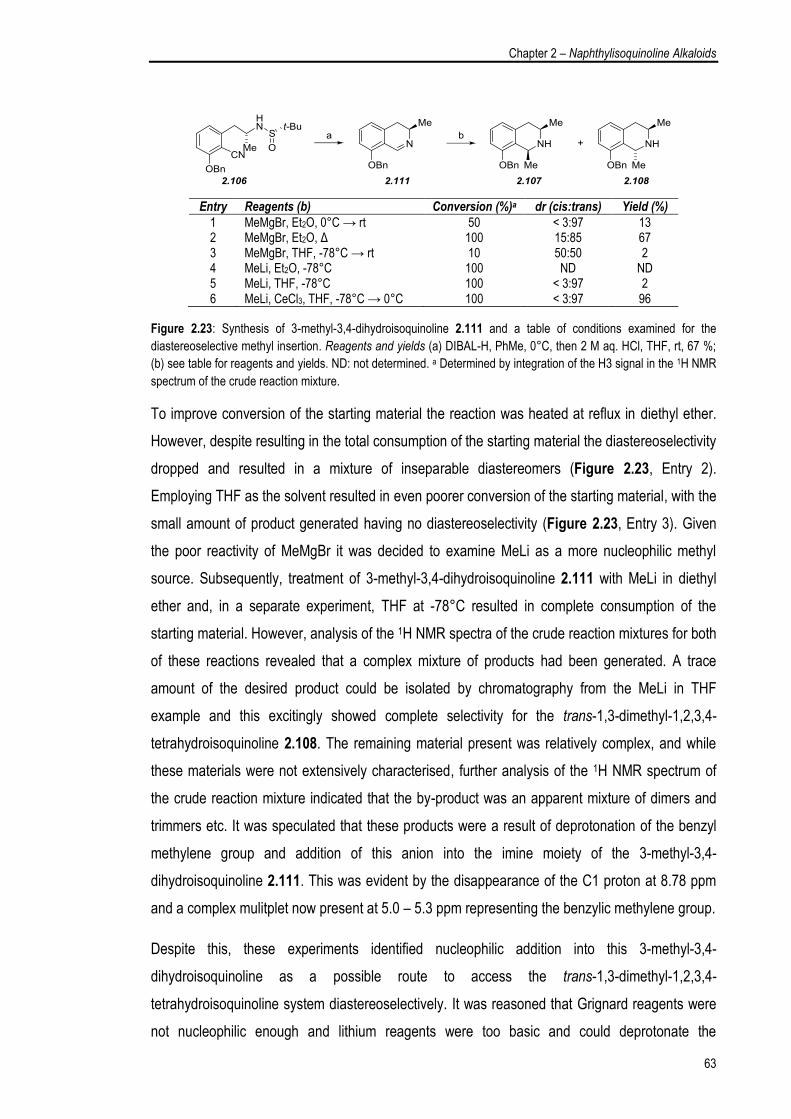
Chapter 2 – Naphthylisoquinoline Alkaloids
63
Entry Reagents (b) Conversion (%)a dr (cis:trans) Yield (%)
1 MeMgBr, Et2O, 0°C → rt 50 < 3:97 13 2 MeMgBr, Et2O, Δ 100 15:85 67 3 MeMgBr, THF, -78°C → rt 10 50:50 2 4 MeLi, Et2O, -78°C 100 ND ND 5 MeLi, THF, -78°C 100 < 3:97 2 6 MeLi, CeCl3, THF, -78°C → 0°C 100 < 3:97 96
Figure 2.23: Synthesis of 3-methyl-3,4-dihydroisoquinoline 2.111 and a table of conditions examined for the
diastereoselective methyl insertion. Reagents and yields (a) DIBAL-H, PhMe, 0°C, then 2 M aq. HCl, THF, rt, 67 %;
(b) see table for reagents and yields. ND: not determined. a Determined by integration of the H3 signal in the 1H NMR
spectrum of the crude reaction mixture.
To improve conversion of the starting material the reaction was heated at reflux in diethyl ether.
However, despite resulting in the total consumption of the starting material the diastereoselectivity
dropped and resulted in a mixture of inseparable diastereomers (Figure 2.23, Entry 2).
Employing THF as the solvent resulted in even poorer conversion of the starting material, with the
small amount of product generated having no diastereoselectivity (Figure 2.23, Entry 3). Given
the poor reactivity of MeMgBr it was decided to examine MeLi as a more nucleophilic methyl
source. Subsequently, treatment of 3-methyl-3,4-dihydroisoquinoline 2.111 with MeLi in diethyl
ether and, in a separate experiment, THF at -78°C resulted in complete consumption of the
starting material. However, analysis of the 1H NMR spectra of the crude reaction mixtures for both
of these reactions revealed that a complex mixture of products had been generated. A trace
amount of the desired product could be isolated by chromatography from the MeLi in THF
example and this excitingly showed complete selectivity for the trans-1,3-dimethyl-1,2,3,4-
tetrahydroisoquinoline 2.108. The remaining material present was relatively complex, and while
these materials were not extensively characterised, further analysis of the 1H NMR spectrum of
the crude reaction mixture indicated that the by-product was an apparent mixture of dimers and
trimmers etc. It was speculated that these products were a result of deprotonation of the benzyl
methylene group and addition of this anion into the imine moiety of the 3-methyl-3,4-
dihydroisoquinoline 2.111. This was evident by the disappearance of the C1 proton at 8.78 ppm
and a complex mulitplet now present at 5.0 – 5.3 ppm representing the benzylic methylene group.
Despite this, these experiments identified nucleophilic addition into this 3-methyl-3,4-
dihydroisoquinoline as a possible route to access the trans-1,3-dimethyl-1,2,3,4-
tetrahydroisoquinoline system diastereoselectively. It was reasoned that Grignard reagents were
not nucleophilic enough and lithium reagents were too basic and could deprotonate the

Chapter 2 – Naphthylisoquinoline Alkaloids
64
methylene group of the benzyl moiety. Accordingly, a more nucleophilic, but less basic, reagent
was required. Analysis of the literature revealed that organocerium reagents are known to be as
nucleophilic as lithium reagents yet much less basic.[73] Indeed, treating 3-methyl-3,4-
dihydroisoquinoline 2.111 with a freshly prepared methyl cerium reagent at -78°C (formed by
adding MeLi to a suspension of cerium(III) chloride in THF) resulted in complete consumption of
the starting material. Gratifyingly, analysis of the 1H NMR spectrum of the crude reaction mixture
indicated that the trans-configured 1,3-dimethyl-1,2,3,4-tetrahydroisoquinoline 2.108 was the only
product formed and after chromatography could be isolated in 96 % yield (Figure 2.23, Entry 6).
Analysis of the 1H-1H NOE spectrum of this material revealed a strong correlation between H3
and Me1 and Me3 (Figure 2.24), providing convincing evidence of the trans-relationship of 1,3-
dimethyl-1,2,3,4-tetrahydroisoquinoline 2.108. The rationale for the stereoselectivity for this
reaction was concluded to be similar to the NaBH4 reduction of the 1,3-dimethyl-3,4-
dihydroisoquinoline 2.111. Here, the organocerium reagent coordinates the nitrogen of the 3-
methyl-3,4-dihydroisoquinoline 2.111 which delivers the methyl group to the less hindered side,
as shown in Figure 2.24.
Figure 2.24: Proposed mechanism for the nucleophilic addition of organocerium reagent into 3-methyl-3,4-
dihydroisoquinoline 2.111 and part of the 1H-1H NOESY spectrum of 2.108 showing key correlation confirming trans
relationship of 1,3-dimethyl-1,2,3,4-tetrahydroisoquinoline.
Having gained access to the trans-1,3-dimethyl-1,2,3,4-tetrahydroisoquinoline 2.108 the total
synthesis of phylline (2.97) was completed by hydrogenolysis using H2 and Pd/C in MeOH.
Phylline (2.97) was formed in 7 steps and 21 % overall yield (Scheme 2.23).
ppm
1.52.02.53.03.54.04.55.0 ppm
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0

Chapter 2 – Naphthylisoquinoline Alkaloids
65
Scheme 2.23: Reagents and yields (a) H2, Pd/C, MeOH, 88 %.
Despite being noted in the literature, complete characterisation data for phylline (2.97) is
unavailable and as such, complete characterisation of synthetic phylline (2.97) was completed.
However, there has been much more work on the [1,1’-13C]-labelled analog of phylline which has
been completely characterised.[14] This data, along with the data obtained for the synthetic
phylline (2.97), is displayed in Table 2.3. The data obtained for synthetic phylline (2.97) overlays
quite well with the data reported for [1,1’-13C]-phylline, apart from the fact that it is not labelled
with 13C.
Table 2.3: Spectroscopic properties of phylline (2.97).
Phylline (2.97) Spectroscopic Data [1,1’-13C]-Phylline[14] Synthetic 1H NMR δ (400 MHz, MeOD) δ (400 MHz, MeOD) 1.46 (d, J = 6.4 Hz, 3H, 3-Me) 1.48 (d, J = 6.4 Hz, 3H) 1.79 (ddd, JHC = 130.0 and 4.4 Hz, JHH = 6.7 Hz,
3H, 1-Me) 1.79 (d, J = 6.8 Hz, 3H)
2.80 (dd, J = 17.6, 11.7 Hz, 1H, 4ax-H) 2.82 (dd, J = 17.6, 11.7 Hz, 1H) 3.11 (dd, J = 17.6, 4.6 Hz, 1H, 4eq-H) 3.10 (dd, J = 17.6, 4.7 Hz, 1H) 3.77 – 3.82 (m, 1H, 3-H) 3.76 – 3.84 (m, 1H) 4.89-4.95 and 4.53-4.59 (m, JHC = 146.4 Hz, 1H, 1-
H) 4.76 (q, J = 6.7 Hz, 1H)
6.68 (d, J = 7.8 Hz, 1H, 5-H) 6.68 (d, J = 7.9 Hz, 1H) 6.69 (d, J = 8.0 Hz, 1H, 6-H) 6.70 (d, J = 7.9 Hz, 1H) 7.10 (dd, J = 8.0 and 7.8 Hz, 1H, 7-H) 7.10 (dd, J = 7.9 and 7.9 Hz, 1H) 13C NMR δ (62.9 MHz, MeOD) δ (100 MHz, MeOD) 17.9 (d, JCC = 36.3 Hz, 1-Me) 17.9 19.2 (3-Me) 19.2 34.4 (4-C) 34.4 45.3 (3-C) 45.2 49.5 (d, JCC = 36.3 Hz, 1-C) 49.4 107.8 (6-C) 114.1 114.1 (4a-C) 120.7 120.7 (8-C) 121.3 130.0 (5-C) 129.9 132.9 (8a-C) 133.0 155.2 (7-C) 155.1
α [𝜶]𝑫𝟐𝟎 = -3.0 (0.08, MeOH) [𝜶]𝑫
𝟐𝟒 = -12.5 (0.08, MeOH)
IR 1139, 1205, 1677, 2853, 2924 1675, 3273
MS m/z calcd for [13C2]-C11H16NO [M+H]+ = 180.1299 m/z calcd for C11H16NO [M+H]+ = 178.1232 [M+H]+ = 180.1297 (HRMS-ESI) [M+H]+ = 178.1222 (HRMS-ESI)

Chapter 2 – Naphthylisoquinoline Alkaloids
66
2.7. Completion of the Total Synthesis of Dioncophylline E
The total synthesis of phylline (2.97) had provided us with an alternative set of conditions to
access the trans-1,3-dimethyl-1,2,3,4-tetrahydroisoquinoline system of dioncophylline E (2.42). In
contrast to before, employing this strategy would require access to 3-methyl-3,4-
dihydroisoquinoline 2.112 but this could be accessed from the same sulfinamide 2.93 that was
synthesised in Section 2.5.3, as shown in Figure 2.25.
Figure 2.25: Retrosynthetic analysis of dioncophylline E (2.42).
Subsequently, sulfinamide 2.93 was treated with DIBAL-H in toluene at 0°C which was followed
by an acidic workup to generate 3-methyl-3,4-dihydroisoquinoline 2.112 in 65 % yield. The methyl
cerium reagent was generated by adding MeLi to CeCl3 in THF at -78°C, then the 3-methyl-3,4-
dihydroisoquinoline 2.112 in THF at -78°C was added. Delightfully, analysis of the 1H NMR
spectrum of the crude reaction mixture revealed that the trans-configured 1,3-dimethyl-1,2,3,4-
tetrahydroisoquinoline 2.113 had been formed selectively, as was evident by the presence of the
multiplet for H3 at 3.34 – 3.43 ppm and the absence of the cis-1,3-dimethyl-1,2,3,4-
tetrahydroisoquinoline multiplet for H3 at 2.91 – 3.02 ppm. The synthesis was completed by a
global deprotection of the benzyl protecting, using H2 and Pd/C in 85 % yield (Scheme 2.24).
Scheme 2.24: Reagents and yields (a) DIBAL-H, PhMe, 0°C, then 2 M aq. HCl, THF, rt, 65 %; (b) MeLi, CeCl3, THF,
-78°C → 0°C, the 2.112, THF, -78°C → 0°C, 61 %; (c) H2, Pd/C, MeOH, rt, 85 %.
The total synthesis of dioncophylline E (2.42) was completed in a longest linear sequence of 12
steps from commercially available nitrile 2.77 and in 10 % overall yield. The synthetic
dioncophylline E (2.42) synthesised had analytical data that was in complete agreement with that

Chapter 2 – Naphthylisoquinoline Alkaloids
67
reported in the literature.[45] There was clearly a mixture of atropisomers as reported by
Bringmann.
Table 2.2: Spectroscopic properties of dioncophylline E (2.42).
Dioncophylline E (double values due to partial differences for the two atropisomeric forms)
Natural (TFA Salt) Synthetic (TFA Salt) 1H NMR δ (600 MHz, CDCl3) δ (400 MHz, CDCl3) 1.58 (d, J = 6.0 Hz, 3H, Me3) 1.59 (d, J = 6.0 Hz, 3H) 1.59 (d, J = 6.1 Hz, 3H, Me3) 1.60 (d, J = 5.9 Hz, 3H) 1.71 (d, J = 6.5 Hz, 3H, Me1) 1.70 (d, J = 6.6 Hz, 3H) 1.74 (d, J = 6.7 Hz, 3H, Me1) 1.74 (d, J = 6.7 Hz, 3H) 2.14 (s, 3H, Me2’) 2.14 (s, 3H) 2.15 (s, 3H, Me2’) 2.15 (s, 3H) 2.98 – 3.13 (m, 4H, H4ax/eq) 2.99 – 3.15 (m, 4H) 3.69 – 3.77 (m, 2H, H3) 3.71 – 3.77 (m, 2H) 4.00 (s, 3H, OMe5’) 3.99 (s, 3H) 4.04 (s, 3H, OMe5’) 4.04 (s, 3H) 4.89 – 4.96 (m, 2H, H1) 4.93 – 4.96 (m, 2H) 6.74 (m, 1H, H6’) 5.25 (1H, br s) 6.77 (m, 1H, H6’) 5.49 (1H, br s) 6.81 (d, J = 7.9 Hz, 1H, H5) 6.71 – 6.73 (m, 1H) 7.05 (d, J = 7.8 Hz, 1H, H6) 6.76 – 6.79 (m, 1H) 7.06 (d, J = 7.8 Hz, 1H, H6) 6.81 (d, J = 7.8 Hz, 2H) 7.24 (s, 1H, H1’) 7.04 – 7.06 (m, 2H) 7.27 (s, 1H, H1’) 7.23 (s, 1H) 7.29 – 7.37 (m, 4H, H7’ and H8’) 7.27 (s, 1H) 9.71 (s, 2H, OH4’) 7.30 (d, J = 4.3 Hz, 2H) 7.34 – 7.37 (m, 2H) 9.32 (br s, 1H) 9.47 (br s, 1H) 9.71 (2H, s) 9.90 (br s, 1H) 10.03 (br s, 1H) 13C NMR δ (150 MHz, CDCl3) δ (100 MHz, CDCl3) 17.8 (Me1) 18.0 18.1 (Me1) 18.3 18.4 (Me3) 18.7 18.8 (Me3) 19.0 20.6 (Me2’) 20.9 20.8 (Me2’) 21.0 33.8 33.8 (C4) 33.9 44.1 (C3) 44.4 44.4 (C3) 44.7 48.1 48.1 (C1) 48.5 56.2 56.2 (OMe5’) 56.3 104.0 104.0 (C6’) 104.0 113.3 113.3 (C4’a) 113.4 116.9 (C3’) 116.9 117.2 (C3’) 117.2 120.0 120.0 (C1’) 120.2 120.6 (C8a) 120.5 120.7 120.7 (C5) 120.8 121.1 (C8’) 121.1 121.2 (C8a) 121.2 121.3 (C8’) 121.3 122.0 (C7) 122.1 122.4 (C7) 122.6

Chapter 2 – Naphthylisoquinoline Alkaloids
68
126.6 126.6 (C7’) 126.8 130.2 (C6) 130.2 130.5 (C6) 130.4 131.3 (C4a) 130.7 131.4 (C4a) 131.3 136.3 136.4 136.5 136.5 137.4 137.5 138.1 138.1 149.5 (C8) 149.6 149.7 (C8) 149.8 151.8 151.8 (C4’) 151.8 156.0 156.0 (C5’) 151.9 156.1
IR 625, 805, 1088, 1358, 1464, 1578, 2851, 2919, 2961, 3376
1673, 3370
MS m/z calcd for C23H26NO3 [M+H]+ 364.1913 [M]+ = 363 (EI) [M+H]+ = 364.1900 [M – Me]+ = 348.1604 (HREIMS)

Chapter 2 – Naphthylisoquinoline Alkaloids
69
2.8. Investigation into the Total Synthesis of 5,3’-Linked Naphthylisoquinoline
Alkaloids
Completion of the total synthesis of dioncophylline E (2.42) signified the effectiveness of this
reaction sequence for the synthesis of the naphthylisoquinoline alkaloids. It was now possible to
revert our attention back to the 5,3’-linked members. While previously, ancistrotanzanine A (2.43)
was the only member of this family[46] a recent publication by Hui-Ming and co-workers has
identified two new members of the family – ancistrotectorine D (2.41), which is the atropisomer of
ancistrotanzanine A (2.43), and ancistrotectorine C (2.40), which is an N-methyl-cis-configured
1,3-dimethyl-1,2,3,4-tetrahydorisoquinoline variant, as shown in Figure 2.26.[44]
Figure 2.26: Structures of 5,3’-linked naphthylisoquinoline alkaloids ancistrotanzanine A (2.43),[46] ancistrotectorine D
(2.41)[44] and ancistrotectorine C (2.40).[44]
Previous work in the Morris group towards the total synthesis of the 5,3’-linked
naphthylisoquinoline alkaloids was discussed in Section 2.3. To reiterate this, Brusnahan found
that the diastereoselectivity of the sulfinimine alkylation reaction decreased as the size of the
substituent at C5 of the o-tolylnitrile increased. While Brusnahan was able to use the
methodology to synthesise ancistrotanzanine A (2.43), alkylation of arylated naphthol 2.60 with
the sulfinimine (R)-2.62 afforded sulfinamide 2.61 as a 85:15 mixture of stereoisomers at C3.[56]
Attempts to improve the diastereoselectivity of this reaction by the author were based around
synthesising an aryllead triacetate 2.67 where the stereochemistry at C3 had already been set.
While the stannane 2.66 was synthesised with complete stereochemical control at C3, the
aryllead triacetate 2.67 could not be generated from this material (Figure 2.27).[57]

Chapter 2 – Naphthylisoquinoline Alkaloids
70
Figure 2.27: Previous attempts to synthesise 5,3’-linked naphthylisoquinoline alkaloids as a single stereoisomer at
C3 of the isoquinoline.[56-57]
With this in mind, it was decided to re-examine generation of a completely intact isoquinolinyl lead
triacetate reagent, where the stereochemistry at C3 was already established. As discussed earlier
(Section 2.3) preliminary efforts towards this in the Morris group, resulted in the synthesis of
three differentially substituted isoquinoline stannanes 2.114, 2.115 and 2.116, as shown in Figure
2.28. However, attempts to transmetalate these to the aryllead triacetate resulted only in isolation
of demetalated material.[53a]
Figure 2.28: Stannanes generated by Bungard and Morris in an attempt to synthesise the 7,3’-linked biaryl bond of
ancistrocladidine in one-step.[53a]
It was concluded that the nitrogen atom on these systems was not sufficiently deactivated to stop
coordination of it to the lead reagents, resulting in demetalation. This is supported by results
reported by Konopelski and co-workers who have shown that aryllead triacetates can be
generated from amine containing molecules when the amine is protected with a Boc-protecting
group (Figure 2.29).[74]

Chapter 2 – Naphthylisoquinoline Alkaloids
71
Figure 2.29: Nitrogen containing aryllead triacetates synthesised in the group of Konopelski.[74]
Furthermore, our observation that boronic acids are superior to trialkyltin reagents in the
transmetalation reaction of these species to aryllead triacetates, prompted us to propose an
alternative synthesis to access these systems. Because the nitrogen atom needed to be
protected with the Boc-protecting group, a tetrahydroisoquinoline moiety was required. It was
envisaged that the cis-configured 1,3-dimethyl-1,2,3,4-tetrahydroisoquinoline could be utilised to
synthesise the arylated naphthol 2.117. This compound could be used to synthesise either
ancistrotectorine C, by using a deprotection/N-methylation strategy or, based on observations by
Bringmann, it may be possible to synthesise ancistrotanzanine A by oxidation of the
tetrahydroisoquinoline moiety (Figure 2.30). Therefore, the initial focus of this project was to
generate ancistrotectorine C and, if time permitted examine conditions to oxidise arylated
naphthol 2.117 to synthesise ancistrotanzanine A (2.43).
Figure 2.30: Retrosynthetic analysis for 5,3’-linked naphthylisoquinoline alkaloids.

Chapter 2 – Naphthylisoquinoline Alkaloids
72
To examine whether the isoquinolinyl lead triacetate 2.118 could be synthesised from this
substrate, boronic acid 2.119 was required. Starting from o-tolylnitrile 2.57 this was transformed
into 1,3-dimethyl-3,4-dihydroisoquinoline 2.120 using the sulfinimine cyclisation protocol with (R)-
sulfinimine (R)-2.62 in 39 % yield over the two steps. The 1,3-dimethyl-3,4-dihydroisoquinoline
2.120 was reduced using NaBH4 in MeOH, to generate the cis-configured 1,3-dimethyl-1,2,3,4-
tetrahydroisoquinoline (dr = >97:3) after which, the amine was immediately protected with the
Boc-protecting group (Boc2O, NEt3, CH2Cl2) and the amine 2.121 was obtained in 98 % yield over
the two steps. Regioselective iodination was achieved using the procedure reported by
Bringmann and co-workers.[75] Halogen-lithium exchange using t-BuLi in THF at -95°C in the
presence of B(Oi-Pr)3 formed the boronic ester, which was hydrolysed in situ to generate the
boronic acid 2.119 in 90 % yield (Scheme 2.25).
Scheme 2.25: Reagents and yields (a) LDA, THF, -78°C, then (R)-2.62, THF, -78°C, 63 %; (b) MeLi, THF, -78°C →
rt, then 2 M aq. HCl, 62 %; (c) NaBH4, MeOH, -10°C → rt; (d) Boc2O, NEt3, CH2Cl2, 98 % (2 steps); (e) I2, Ag2SO4,
EtOH, 0°C→ rt, 70%; (f) t-BuLi, B(Oi-Pr)3, THF, -95°C → 0°C, 90 %.
Gratifyingly, stirring boronic acid 2.119 with 10 mol % Hg(OTFA)2 and 1 equivalent of freshly
dried Pb(OAc)4 in 1,2-DCE at room temperature protected from light for 18 h resulted in a crude
reaction mixture, which from examination of the 1H NMR spectrum indicated that complete
consumption of the boronic acid 2.119 starting material had occurred. Although there was a
considerable amount of demetalated material, there was another new aromatic singlet at 6.59
ppm which had diagnostic 207Pb-satellites at 92.0 Hz either side of the parent peak. Without
purification of the mixture, the residue was dissolved in 1,2-DCE and added to a solution of 1
equivalent of naphthol 2.52 (relative to starting boronic acid) and 3 equivalents of pyridine in 1,2-
DCE. The reaction was again stirred at room temperature protected from light for 18 h. The 1H
NMR spectrum obtained from the crude reaction mixture from this reaction indicated that there
were two new peaks, in a ratio of 50:50, representing OH-4’ (9.38 and 9.39 ppm) which had
shifted downfield to the normal naphthol peak (9.24 ppm). The doubling of signals was suspected
to be due to two atropisomers that were formed as a result of the coupling reaction. Examination
of the 1H NMR spectrum of the purified material clearly revealed that two compounds were
present. Again, the naphthol H3’ singlet at 7.10 ppm was absent, confirming that a substituent
had been added at this position. The position of the biaryl bond was confirmed by 2D NMR

Chapter 2 – Naphthylisoquinoline Alkaloids
73
spectroscopy particularly, the 1H-13C HMBC correlations between H7 → C5 and C8 proving that
there was no substitution at this position. Other key 2D NMR correlations used to ascertain the
position of the biaryl bond are shown in Figure 2.31. The arylated naphthol 2.117 was collected
as a mixture of atropisomers and in 50 % yield over the two steps.
Figure 2.31: Synthesis of arylated naphthol 2.117 with key 1H-13C HMBC and 1H-1H ROESY correlations shown
which helped prove the position of the biaryl bond. Reagents and yields: (a) Hg(OTFA)2, Pb(OAc)4, CH2Cl2, rt; (b)
2.52, py, 1,2-DCE, rt, 50 % (2 steps);
With access to arylated naphthol 2.117 the total synthesis of ancistrotectorine C (2.40) was
completed by removal of the Boc-protecting group using TFA in CH2Cl2 at 0°C, followed by
reductive amination with formaldehyde and NaBH4 in methanol to add the N-methyl substituent,
as shown in Scheme 2.26. The 1H NMR spectrum of the purified material indicated that the Boc
group had been removed, ascertained by the absence of the t-butyl peaks at 1.49 and 1.50 ppm,
and the appearance of two new singlets at 2.31 and 2.32 ppm integrating for 3 protons each
representing the N-Me substituent of the two atropisomers. Furthermore, the spectra could be
compared directly to the spectra for ancistrotectorine C (2.40) found in the literature, which
confirmed that ancistrotectorine C (2.40) had been synthesised. Unfortunately, all attempts at
separating the two atropisomers by flash chromatography and recrystallisation were
unsuccessful.
Scheme 2.26: Reagents and yields (a) TFA, CH2Cl2, -10°C; (b) CH2O, NaBH4, MeOH, 83 % (2 steps).
The total synthesis of ancistrotectorine C (2.40) and its atropisomer was completed in 10 steps
from commercially available o-tolylnitrile 2.57 and in 10 % overall yield. The synthesis solved two
of our initial ‘big picture’ goals associated with the project. These were (1) generation of an intact

Chapter 2 – Naphthylisoquinoline Alkaloids
74
isoquinolinyllead triacetate which could be used directly in the Pinhey-Barton reaction and (2)
synthesis of the 5,3’-linked biaryl bond with complete stereocontrol at C3 of the isoquinoline
moiety. However, while the ancistrotectorine C synthesis has established that this strategy is a
worthwhile endeavour it raises some intriguing questions – can we construct the biaryl bond of
these molecules atropselectively and can the 1,2,3,4-tetrahydroisoquinoline moiety be oxidised to
the 3,4-dihydroisoquinoline. In the short time remaining in this PhD project some insight into both
of these questions was learnt.
As ancistrotectorine C and its atropisomer were inseparable it would be advantageous to be able
to synthesise each atropisomer atropselectively. It was initially hoped that the stereochemistry
present at C3 of the cis-1,3-dimethyl-1,2,3,4-tetrahydroisoquinoline moiety might induce some
‘natural’ atropselectivity. However, this proved not to be the case, similar to an observation
recorded by Rizzacasa and co-workers for their total synthesis of (+)-(O)-methylancistrocline
(2.26).[33] Interestingly, attempts to utilise brucine (2.46), which Yamamoto has pioneered as a
suitable promoter (Section 2.3), in place of pyridine as the chiral ligand in the Pinhey-Barton
coupling reaction also resulted in no atropselectivity. It was speculated that this was due to either
an increased steric interaction in the transition state or a mismatched relationship between
brucine and the C3 stereocentre of the cis-1,3-dimethyl-1,2,3,4-tetrahydroisoquinoline moiety. It is
suggested that an alternative chiral base may solve this problem and this is an ongoing area of
research in our laboratories.
To see if the 1,2,3,4-tetrahydroisoquinoline moiety of ancistrotectorine C could be oxidised to gain
access into the ancistrotanzanine A/ancistrotectorine D scaffold the conditions reported by
Bringmann were examined.[65] With the small amount of material remaining amine 2.122 was
treated with KMnO4 in H2O and THF (Figure 2.32). Examination of the 1H NMR of the crude
reaction mixture indicated that the 1,2,3,4-tetrahydroisoquinoline moiety had indeed been
oxidised to the 3,4-dihydroisoquinoline. However, the spectrum did not match that reported for
ancistrotanzanine A (2.43) and/or ancistrotectorine D (2.41). While the material collected was not
sufficient for complete characterisation, the 1H NMR spectra indicated that there was no longer a
resonance representing H1’ (7.21 ppm) and the C2’-Me was shifted significantly upfield. Thus, it
was speculated that under the reaction conditions the naphthol was also oxidised to the p-
quinone 2.123.

Chapter 2 – Naphthylisoquinoline Alkaloids
75
Figure 2.32: Attempt to oxidise 1,3-dimethyl-1,2,3,4-tetrahydroisoquinoline 2.122 to ancistrotanzanine A
(2.43)/ancistrotectorine D (2.41). Reagents and yields (a) KMnO4, 1:2 H2O/THF, rt.
Clearly, to circumvent this problem either alternative oxidation conditions are required or the
naphthol could be protected before oxidation. This is currently the focus of this work.

Chapter 2 – Naphthylisoquinoline Alkaloids
76
2.9. Chapter Summary
In summary, three viable synthetic routes to phylline (2.97), dioncophylline E (2.42) and
ancistrotectorine C (2.40) have been established. The total synthesis of phylline (2.97)
investigated the diastereoselective synthesis of the trans-1,3-dimethyl-1,2,3,4-
tetrahydroisoquinoline moiety. This allowed us to utilise this chemistry, in combination with the
Pinhey-Barton reaction, to synthesise dioncophylline E (2.42). Using the knowledge gained from
both of these syntheses it was realised that aryllead triacetates are more readily prepared by
transmetalation from the boronic acid rather than the trialkyltin compounds. This allowed for the
generation of an isoquinolinyllead triacetate species, a substrate that previously could not be
generated, and this could be used in the Pinhey-Barton reaction to synthesise ancistrotectorine C
(2.40).
The strategy that has been developed now allows us to synthesise these complex structures in a
‘one-step’ process. Importantly, the method allows for variations to be made to each of the
coupling partners to quickly and efficiently investigate the SAR of these molecules. Figure 3.33
demonstrates a study that could be carried out to investigate the SAR for dioncophylline E (2.42)
using the designed protocol. Variations that could be made include different phenol coupling
partners in the Pinhey-Barton reaction, the stereochemistry and substituent at C1 and C3 of the
1,2,3,4-tetrahydroisoquinoline subunit and differentially substituted tolylnitriles.
Figure 2.33: Proposed synthesis of analogs of dioncophylline E using the designed synthetic protocol indicating
positions in the synthesis that could be varied to probe its SAR.

Chapter 2 – Naphthylisoquinoline Alkaloids
77
2.10. References for Chapter 2
[1] World Malaria Report: 2013, World Health Organisation, 2013. [2] P. D. Crompton, J. Moebius, S. Portugal, M. Waisberg, G. Hart, L. S. Garver, L. H. Miller,
C. Barillas, S. K. Pierce, Annu. Rev. Immunol. 2014, 32, 157-187. [3] I. S. Tantular, Indonesian Journal of Tropical and Infectious Disease 2010, 1, 122-127. [4] G. Bringmann, C. Rummey, J. Chem. Inf. Comput. Sci 2003, 43, 304-316. [5] G. Bringmann, M. Dreyer, J. H. Faber, P. W. Dalsgaard, D. Staerk, J. W. Jaroszewski, H.
Ndangalaski, F. Mbago, R. Brun, M. Reichert, K. Maksimenka, S. B. Christensen, J. Nat. Prod. 2003, 66, 1159-1165.
[6] G. François, G. Bringmann, J. D. Phillipson, L. A. Assi, C. Dochez, M. Rübenacker, C. Schneider, M. Wéry, D. C. Warhurst, G. C. Kirby, Phytochemistry 1994, 35, 1461-1464.
[7] T. R. Govindachari, P. C. Parthasarathy, Tetrahedron 1971, 27, 1013-1026. [8] G. Bringmann, T. Gulder, T. A. M. Gulder, M. Breuning, Chemical Reviews (Washington,
DC, United States) 2010, 111, 563-639. [9] G. Bringmann, F. Pokorny, in The Alkaloids: Chemistry and Pharmacology, Vol. Volume
46 (Ed.: A. C. Geoffrey), Academic Press, 1995, pp. 127-271. [10] J. Fleischhauer, A. Koslowski, B. Kramer, E. Zobel, G. Bringmann, K. P. Gulden, T.
Ortmann, B. Peter, Zeitschrift fur Naturforschung B 1993, 48, 140-148. [11] M. R. Boyd, Y. F. Hallock, J. H. Cardellina, K. P. Manfredi, J. W. Blunt, J. B. McMahon,
R. W. Buckheit, G. Bringmann, M. Schaeffer, J. Med. Chem. 1994, 37, 1740-1745. [12] G. Bringmann, M. Wohlfarth, H. Rischer, M. Rückert, J. Schlauer, Tetrahedron Lett.
1998, 39, 8445-8448. [13] G. Bringmann, M. Wohlfarth, H. Rischer, M. Grüne, J. Schlauer, Angew. Chem. Int. Ed.
2000, 39, 1464-1466. [14] G. Bringmann, J. Mutanyatta-Comar, M. Greb, S. Rüdenauer, T. F. Noll, A. Irmer,
Tetrahedron 2007, 63, 1755-1761. [15] M. Xu , T. Bruhn, B. Hertlein, R. Brun, A. Stich, J. Wu, G. Bringmann, Chem. Eur. J.
2010, 16, 4206-4216. [16] G. Bringmann, K. Messer, B. Schwöbel, R. Brun, L. Aké Assi, Phytochemistry 2003, 62,
345-349. [17] G. Bringmann, W. Saeb, M. Wohlfarth, K. Messer, R. Brun, Tetrahedron 2000, 56, 5871-
5875. [18] G. Bringmann, M. Dreyer, J. H. Faber, P. W. Dalsgaard, J. W. Jaroszewski, H.
Ndangalasi, F. Mbago, R. Brun, S. B. Christensen, J. Nat. Prod. 2004, 67, 743-748. [19] G. Bringmann, M. Wohlfarth, H. Rischer, J. Schlauer, R. Brun, Phytochemistry 2002, 61,
195-204. [20] G. Bringmann, G. Zhang, T. Büttner, G. Bauckmann, T. Kupfer, H. Braunschweig, R.
Brun, V. Mudogo, Chem. Eur. J. 2013, 19, 916-923. [21] G. François, G. Timperman, W. Eling, L. A. Assi, J. Holenz, G. Bringmann, Antimicrob.
Agents Chemother. 1997, 41, 2533-2539. [22] K. F. Schwedhelm, M. Horstmann, J. H. Faber, Y. Reichert, G. Bringmann, C. Faber,
ChemMedChem 2007, 2, 541-548. [23] I. Weissbuch, L. Leiserowitz, Chem. Rev. 2008, 108, 4899-4914. [24] G. Bringmann, A. J. Price Mortimer, P. A. Keller, M. J. Gresser, J. Garner, M. Breuning,
Angew. Chem. Int. Ed. 2005, 44, 5384-5427. [25] M. A. Rizzacasa, in Studies in Natural Products Chemistry, Vol. Volume 20, Part F (Ed.:
R. Atta ur), Elsevier, 1997, pp. 407-455. [26] G. Bringmann, D. Menche, Acc. Chem. Res. 2001, 34, 615-624. [27] G. Bringmann, W. Saeb, M. Rübenacker, Tetrahedron 1999, 55, 423-432.

Chapter 2 – Naphthylisoquinoline Alkaloids
78
[28] G. Bringmann, M. Ochse, R. Götz, J. Org. Chem. 2000, 65, 2069-2077. [29] a) G. Bringmann, H. Reuscher, Tetrahedron Lett. 1989, 30, 5249-5252; b) G. Bringmann,
J. R. Jansen, H.-P. Rink, Angewandte Chemie International Edition in English 1986, 25, 913-915.
[30] T. G. Gant, A. I. Meyers, Tetrahedron 1994, 50, 2297-2360. [31] a) M. A. Rizzacasa, M. V. Sargent, J. Chem. Soc., Perkin Trans. 1 1991, 2773-2781; b)
M. A. Rizzacasa, M. V. Sargent, J. Chem. Soc., Perkin Trans. 1 1991, 845-854; c) M. A. Rizzacasa, M. V. Sargent, J. Chem. Soc., Perkin Trans. 1 1991, 841-844.
[32] B. N. Leighton, M. A. Rizzacasa, J. Org. Chem. 1995, 60, 5702-5705. [33] P. Chau, I. R. Czuba, M. A. Rizzacasa, G. Bringmann, K.-P. Gulden, M. Schäffer, J. Org.
Chem. 1996, 61, 7101-7105. [34] M. F. Comber, J. C. Morris, M. V. Sargent, Aust. J. Chem. 1998, 51, 19-22. [35] N. Miyaura, A. Suzuki, Chemical Reviews (Washington, DC, United States) 1995, 95,
2457-2483. [36] O. Baudoin, Eur. J. Org. Chem. 2005, 2005, 4223-4229. [37] T. R. Hoye, M. Chen, B. Hoang, L. Mi, O. P. Priest, J. Org. Chem. 1999, 64, 7184-7201. [38] G. Bringmann, R. Götz, S. Harmsen, J. Holenz, R. Walter, Liebigs Annalen 1996, 1996,
2045-2058. [39] P. D. Hobbs, V. Upender, M. I. Dawson, Synlett 1997, 1997, 965-967. [40] B. H. Lipshutz, J. M. Keith, Angew. Chem. Int. Ed. 1999, 38, 3530-3533. [41] S. Huang, T. B. Petersen, B. H. Lipshutz, J. Am. Chem. Soc. 2010, 132, 14021-14023. [42] G. Bringmann, C. Günther, E.-M. Peters, K. Peters, Tetrahedron 2001, 57, 1253-1259. [43] T. R. Govindachari, P. C. Parthsarathy, H. K. Desai, Indian Journal of Chemistry 1973,
11, 1190. [44] C. Jiang, Z.-L. Li, P. Gong, S.-L. Kang, M.-S. Liu, Y.-H. Pei, Y.-K. Jing, H.-M. Hua,
Fitoterapia 2013, 91, 305-312. [45] G. Bringmann, K. Messer, K. Wolf, J. Mühlbacher, M. Grüne, R. Brun, A. M. Louis,
Phytochemistry 2002, 60, 389-397. [46] G. Bringmann, M. Dreyer, J. H. Faber, P. W. Dalsgaard, J. W. Jaroszewski, H.
Ndangalasi, F. Mbago, R. Brun, M. Reichert, K. Maksimenka, S. B. Christensen, J. Nat. Prod. 2003, 66, 1159-1165.
[47] a) H. C. Bell, G. L. May, J. T. Pinhey, S. Sternhell, Tetrahedron Lett. 1976, 17, 4303-4306; b) H. Bell, J. Pinhey, S. Sternhell, Aust. J. Chem. 1979, 32, 1551-1560.
[48] H. C. Bell, J. T. Pinhey, S. Sternhell, Aust. J. Chem. 1979, 32, 1551-1560. [49] D. H. R. Barton, D. M. X. Donnelly, P. J. Guiry, J.-P. Finet, J. Chem. Soc., Perkin Trans.
1 1994, 0, 2921-2926. [50] a) S. Saito, T. Kano, H. Muto, M. Nakadai, H. Yamamoto, J. Am. Chem. Soc. 1999, 121,
8943-8944; b) T. Kano, Y. Ohyabu, S. Saito, H. Yamamoto, J. Am. Chem. Soc. 2002, 124, 5365-5373.
[51] a) J. Morgan, J. T. Pinhey, J. Chem. Soc., Perkin Trans. 1 1993, 1673-1676; b) J. Morgan, T. W. Hambley, J. T. Pinhey, J. Chem. Soc., Perkin Trans. 1 1996, 2173-2177.
[52] D. H. R. Barton, D. M. X. Donnelly, P. J. Guiry, J. H. Reibenspies, J. Chem. Soc., Chem. Commun. 1990, 1110-1111.
[53] a) C. J. Bungard, J. C. Morris, J. Org. Chem. 2006, 71, 7354-7363; b) C. J. Bungard, J. C. Morris, Org. Lett. 2002, 4, 631-633.
[54] D. H. R. Barton, D. M. X. Donnelly, J.-P. Finet, P. J. Guiry, J. Chem. Soc., Perkin Trans. 1 1991, 2095-2102.
[55] F. A. Davis, P. K. Mohanty, D. M. Burns, Y. W. Andemichael, Org. Lett. 2000, 2, 3901-3903.
[56] J. S. Brusnahan, The University of Adelaide (Adelaide, South Australia), 2009.

Chapter 2 – Naphthylisoquinoline Alkaloids
79
[57] H. D. Toop, The University of Adelaide (Adelaide, South Australia), 2009. [58] A. G. Giumanini, G. Verardo, P. Geatti, P. Strazzolini, Tetrahedron 1996, 52, 7137-7148. [59] W. Li, P. Sun, J. Org. Chem. 2012, 77, 8362-8366. [60] R. Kozyrod, J. Morgan, J. Pinhey, Aust. J. Chem. 1985, 38, 1147-1153. [61] a) M. Watanabe, S. Hisamatsu, H. Hotokezaka, S. Furukawa, Chem. Pharm. Bull. 1986,
34, 2810-2820; b) G. Bringmann, G. Zhang, A. Hager, M. Moos, A. Irmer, R. Bargou, M. Chatterjee, Eur. J. Med. Chem. 2011, 46, 5778-5789.
[62] P. R. Markies, A. Villena, O. S. Akkerman, F. Bickelhaupt, W. J. J. Smeets, A. L. Spek, J. Organomet. Chem. 1993, 463, 7-21.
[63] J. Morgan, J. T. Pinhey, J. Chem. Soc., Perkin Trans. 1 1990, 715-720. [64] H. Bell, J. Kalman, J. Pinhey, S. Sternhell, Aust. J. Chem. 1979, 32, 1521-1530. [65] G. Bringmann, R. Weirich, H. Reuscher, J. R. Jansen, L. Kinzinger, T. Ortmann, Liebigs
Ann. Chem. 1993, 1993, 877-888. [66] G. Bringmann, K. Messer, M. Wohlfarth, J. Kraus, K. Dumbuya, M. Rückert, Anal. Chem.
1999, 71, 2678-2686. [67] G. Bringmann, C. Schneider, U. Mohler, R.-M. Pfeifer, R. Gotz, L. Ake Assi, E.-M. Peters,
K. Peters, Zeitschrift fur Naturforschung B 2003, 58B, 577-584. [68] M. Chrzanowska, M. D. Rozwadowska, Chemical Reviews (Washington, DC, United
States) 2004, 104, 3341-3370. [69] N. Uematsu, A. Fujii, S. Hashiguchi, T. Ikariya, R. Noyori, J. Am. Chem. Soc. 1996, 118,
4916-4917. [70] a) R. P. Polniaszek, J. A. McKee, Tetrahedron Lett. 1987, 28, 4511-4514; b) R. P.
Polniaszek, C. R. Kaufman, J. Am. Chem. Soc. 1989, 111, 4859-4863. [71] J. Van der Eycken, J. P. Bosmans, D. Van Haver, M. Vandewalle, A. Hulkenberg, W.
Veerman, R. Nieuwenhuizen, Tetrahedron Lett. 1989, 30, 3873-3876. [72] F. A. Davis, P. K. Mohanty, J. Org. Chem. 2002, 67, 1290-1296. [73] a) R. Bloch, Chemical Reviews (Washington, DC, United States) 1998, 98, 1407-1438; b)
H.-J. Liu, K.-S. Shia, X. Shang, B.-Y. Zhu, Tetrahedron 1999, 55, 3803-3830. [74] a) H. Deng, J. P. Konopelski, Org. Lett. 2001, 3, 3001-3004; b) J. P. Konopelski, J. M.
Hottenroth, H. M. Oltra, E. A. Véliz, Z.-C. Yang, Synlett 1996, 1996, 609-611; c) J. Lin, B. S. Gerstenberger, N. Y. T. Stessman, J. P. Konopelski, Org. Lett. 2008, 10, 3969-3972.
[75] G. Bringmann, S. Rüdenauer, T. Bruhn, L. Benson, R. Brun, Tetrahedron 2008, 64, 5563-5568.

80

81

Chapter 3 – AAL(S)
82
3.1. Introduction
3.1.1. Discovery and Development of Myriocin as a Novel Immunosuppressant
In 2010, FTY720 (Fingolimod, Gilenya) (1.39) was approved by the US FDA for the treatment of
relapsing multiple sclerosis.[1] It was the culmination of 16 years of research that started with the
isolation of a natural product myriocin (1.38) by Fujita and co-workers.[2] As discussed in Section
1.2, the discovery of cyclosporin A (CsA) (1.28) and FK506 (1.6) validated the search for
immunosuppressive compounds from fungi and microorganisms.[3] Inspired by this, Fujita and co-
workers examined the entomopathogenic fungus Isaria sinclarii which has been used for
thousands of years in Chinese traditional medicine. Using a bioassay developed in his group,
aimed at evaluating the proliferation of T-cells in vitro and in vivo, Fujita was able to isolate
myriocin (1.38) in 1994.[2] This compound was found to be 10 to 100 times more effective at
inhibiting the growth of T cells than CsA (1.28) but, in contrast to CsA (1.28), myriocin (1.38) was
toxic and had other properties that were undesirable.
Prompted by the exciting immunosuppressant activity that myriocin (1.38) had, Fujita sought to
optimise the structure to improve its pharmacological profile. Although not outlined in detail here,
Figure 3.1 shows the structural modifications that were made to improve myriocin (1.38). Initial
key changes were made based on the immunosuppressant activity of myriocin homologues, the
mycestericins which were also isolated by Fujita and co-workers from a similar fungus to the one
that yielded myriocin (1.38).[4] These structural changes involved replacement of the head group
carboxylic acid with a hydroxymethyl group, removal of the C14-ketone, 6-double bond and the 4-
hydroxy group as well as shortening the carbon backbone to C14 (compared to myriocin’s C20).
These changes afforded ISP-I-55 (3.1) which was more effective than myriocin (1.38) and 30-fold
less toxic. Finally, a phenyl group was inserted into the carbon chain backbone in a bid to prevent
the compound from being metabolised and to increase rigidity. The positioning of this phenyl ring
was found to be integral as when moving it one carbon atom towards, or away from the aminodiol
head group led to a 10-fold loss in immunosuppressant activity. The outcome of this investigation
was the development of FTY720 (1.39). Not only was FTY720 (1.39) more potent than myriocin
(1.38) but, it was also non-toxic and was more water soluble.[3, 5] Furthermore, FTY720 (1.39)
could be administered at a dose ten times lower than that of the other analogs while still
maintaining potent immunosuppressant activity.

Chapter 3 – AAL(S)
83
Figure 3.1: Evolution of myriocin (1.38) to FTY720 (1.39). Data shown for rat skin allograft survival. a MST: mean
survival time; b MST in vehicle was 8.5 ± 0.3 days.[3, 5]

Chapter 3 – AAL(S)
84
The Novartis Institute for BioMedical Research decided to investigate FTY720 (1.39) further and
quickly realised that its immunosuppressant activity was bought about by a mechanism that was
different to both CsA (1.28) and FK506 (1.6) which was an exciting discovery.[6] Furthermore,
researchers recognised the structural similarity between myriocin (1.38) and sphingosine (3.2), an
endogenous lipid that is part of a large family of molecules known as the sphingolipids (Figure
3.2). They therefore postulated that FTY720 (1.39) may mimic sphingosine (3.2).
Figure 3.2: The sphingosine metabolism pathway; Alk = alkyl chain.[7]
The sphingolipids are an important class of signalling and cell recognition molecules.
Sphingosine-1-phosphate (S1P) (3.3), in particular, is the result of phosphorylation of sphingosine
(3.2) by sphingosine kinase 2 (SphK2). S1P (3.3) activates a group of five G-protein coupled
receptors called S1P1-5 which are involved in lymphocyte trafficking from the thymus and
secondary lymphoid tissue. Activation of these receptors by S1P (3.3) results in retention of
lymphocytes, suppressing the immune response. Brinkmann and co-workers at Novartis
proposed that FTY720 (1.39) may mimic S1P (3.3) and thus, may be able to activate S1P
receptors resulting in an immunosuppressant response.[7] Subsequent investigation into this
hypothesis revealed that FTY720 (1.39) was in fact phosphorylated by SphK2, as shown in
Figure 3.3. This was achieved by incubating FTY720 (1.39) with SphK2 and radiolabelled [γ-
32P]ATP. The products were resolved using TLC, with the phosphorus labelled compounds
detected using autoradiography and confirmed as FTY720-P (3.4) using mass spectrometry.[8]

Chapter 3 – AAL(S)
85
Figure 3.3: Different concentrations of FTY720 (1.39) were incubated with recombinant mouse SphK2 after which,
TLC was used to resolve the products (shown on the right). The alcohol products were detected by staining the TLC
plate with Fluram®. The phosphorylated analog was radiolabelled with 32P and detected using autoradiography. The
TLC plate shown in the Figure is the result of two overlapping images.[8]
Brinkmann and co-workers also tested two other compounds in their phosphorylation assay,
AAL(R) (3.5) and AAL(S) (1.37) – which were synthesised as part of the original structure-activity
relationship study that identified FTY720 (1.39). In comparison to FTY720 (1.39), AAL(R) (3.5)
and AAL(S) (1.37) are deoxy derivatives of FTY720 (1.39), each having one of the hydroxymethyl
groups on the FTY720 (1.39) head group replaced with a methyl group. A consequence of this is
that a stereogenic centre is generated to afford either the R or S stereochemistry. Interestingly,
the original publication reported AAL(R) (3.5) to have improved immunosuppressant activity
compared to FTY720 (1.39). However, AAL(S) (1.37) was found to have no immunosuppressant
activity at all.[9] The results of Brinkmann and co-workers showed that, like FTY720 (1.39),
AAL(R) (3.5) was phosphorylated by SphK2 (AAL(R)-P (3.6)) and that AAL(S) (1.37) was not
(Figure 3.4). Additionally, Don and co-workers have since found that AAL(R) (3.5), in comparison
to FTY720 (1.39), is much more rapidly phosphorylated by SphK2. They also showed that
FTY720 (1.39), when compared to AAL(R) (3.5), is dephosphorylated much more readily by
endothelial lipid phosphate phosphatases in vitro.[10]
Figure 3.4: Different concentrations of AAL(R) (3.5) and AAL(S) (1.37) were incubated with recombinant mouse
SphK2 after which, TLC was used to resolve the products (shown on the right). The alcohol products were detected
by staining the TLC plate with Fluram®. The phosphorylated analogs were radiolabelled with 32P and detected using
autoradiography. The TLC plate shown in the Figure is the result of two overlapping images.[8]

Chapter 3 – AAL(S)
86
At the same time that Novartis were performing these experiments, Rosen and co-workers at
Merck Research Laboratories showed that FTY720 (1.39) did not activate any of the S1P
receptors but, in fact it was the phosphorylated analog (FTY720-P (3.4)) that had low nanomolar
binding to four of the five S1P receptors. Furthermore, by synthesising each enantiomer of
FTY720-P (3.4) they showed that the (S)-FTY720-P ((S)-3.4) had all of the biological activity, as
shown in Table 3.1. These results have been reiterated by Kiuchi and co-workers at Yoshitomi
Pharmaceutical Industries[11] and Brinkmann and co-workers at Novartis who, in conjunction with
their work regarding AAL(R) (3.5) and AAL(S) (1.37), showed that this phosphorylation process
by SphK2 is stereoselective.[12]
Table 3.1: IC50 values for the ligand competition of S1P (3.3), FTY720 (1.39), (R)-FTY720-P ((R)-3.4) and (S)-
FTY720-P ((S)-3.4) to S1P1-5 in the presence of [33P]-S1P (nm).[13]
Compound S1P1 S1P2 S1P3 S1P4 S1P5
S1P (3.3) 0.67 0.35 0.26 34 0.55 FTY720 (1.39) 840 > 10000 > 10000 > 10000 > 10000 (R)-FTY720-P ((R)-3.4) 25 > 10000 120 380 49 (S)-FTY720-P ((S)-3.4) 0.28 1100 6.3 17 0.77
These studies by Rosen and Brinkmann confirmed that (S)-FTY720-P ((S)-3.4) was the
immunosuppressant active metabolite and that this molecule activates S1P1 resulting in the
sequestering of lymphocytes. A comprehensive review regarding the biology of FTY720-P (3.4)
activation of S1P1 and its role in the immune system was published in 2004 and the reader is
directed to that if they wish to learn more on this aspect.[14]
3.1.2. Targeting Protein Phosphatase 2A for Cancer Therapy
Protein phosphatase 2A (PP2A) is a Ser/Thr protein phosphatase which dephosphorylates a
range of proteins in the cell. PP2A is a trimeric enzyme composed of three subunits – a
conserved catalytic (PP2A-C) and structural (PP2A-A) subunit and one of several different
regulatory (PP2A-B) subunits which differentiate between substrates (Figure 3.5). PP2A, along
with protein phosphatase 1 (PP1), constitutes 90 % of all Ser/Thr phosphatase activity in the
cell.[15] In contrast to kinases, which mostly generate signals for cell growth and proliferation,
phosphatases can inhibit and regulate these activities. Consequently, inhibition of PP2A has been
found to have implications in cancer and thus, makes PP2A an important tumour suppressor. [16]
For example, inhibition of PP2A is crucial for the oncogenic effects of leukaemia associated with
tyrosine kinases such as BCR/ABL, c-KIT and FLT3-ITD, as shown in Figure 3.5.[17] Furthermore,
down regulation of PP2A has been found to be a common factor in acute myeloid leukaemia
(AML) patients.[18]

Chapter 3 – AAL(S)
87
Figure 3.5: The structure of Protein Phosphatase 2A (PDB accession number : 2NYM) and a simplified model for
PP2A inhibition in myeloid leukaemias.
While the focus of the development of many cancer therapies has been on trying to selectively
inhibit kinases, it is now recognised that activation of phosphatases may be a valid alternative. As
such, it is recognised that activation of PP2A could be a potential treatment for cancer. Current
examples of strategies that are available to activate PP2A are outlined in Figure 3.6.
Figure 3.6: Pharmacological activation of PP2A. (A) SET is a protein which, when activated by BCR/ABL, interacts
with PP2A to inactivate it. Subsequently, when SET is knocked down using a short hairpin RNA (shRNA) the activity
of PP2A is restored;[19] (B) forskolin/3-isobutyl-1-methylxanthine (IBMX) stimulate production of cAMP which
activates PP2A;[20] (C) PP2A is activated by overexpression of the PP2A-C catalytic subunit;[19] (D) FTY720 (1.39) is
a small molecule activator of PP2A.[17a, 21]
The first report that FTY720 could activate PP2A was in 2003 by Shinomiya and co-workers who
demonstrated that FTY720 (1.39) initiated apoptosis in several leukaemia cell lines.[21] Their work
revealed that this activity was due to the dephosphorylation of Akt (also known as protein kinase
B), a Ser/Thr protein kinase which is involved in promoting cell growth and survival. The authors
showed that this activity was not due to direct inhibition of Akt, nor as a result of inhibition of

Chapter 3 – AAL(S)
88
upstream protein kinases, such as phosphatidylinositol 3’-kinase (PI 3-kinase) and
phosphatidylinosityl-dependent kinase (PDK1). Moreover, they were able to demonstrate that
FTY720 (1.39) actually activated a Ser/Thr protein phosphatase. An initial screen of protein
phosphatase 1 (PP1) and protein phosphatase 2A (PP2A) found that FTY720 (1.39) increased
the activity of PP2A by 175 % at a concentration of 10 μM. In contrast, PP1 was not affected.
However, activation of PP2A was found to be indirectly associated with FTY720 (1.39) binding to
PP2A and they concluded that the mechanism for this remains to be elucidated.[21]
This publication led to a series of reports documenting FTY720’s ability to induce apoptosis in
several other cancer cell lines, but with no further explanation for how the molecule exerted its
activity. Finally, Perotti and co-workers made a breakthrough discovery when investigating
FTY720’s effects on chronic myeloid leukaemia cells.[17a] They found that as well as
dephosphorylating or inactivating Akt, FTY720 (1.39) also inactivated other important oncogenes
including BCR/ABL, STAT5 and ERK. Perhaps more interestingly however, was that they found
that inactivation of BCR/ABL was independent of FTY720 phosphorylation and in fact, treatment
of these cells with FTY720-P (3.4) resulted in no BCR/ABL inactivation. This observation has
since been confirmed in ovarian cancer,[22] acute lymphoblastic leukaemia cells,[23] and acute
myeloid leukaemia where, although FTY720 (1.39) induced cell death, FTY702-P (3.4) had no
effect on these cells.
Importantly, FTY720 (1.39) can eliminate cancer cells which are resistant to current therapeutics
without disruption to normal blood and bone marrow cells.[24] While there has been a lot of work
on discovering what FTY720 (1.39) does, there has been little explanation of why or how it does
this. It is clear from almost all of the data that has been generated on this issue that activation of
PP2A is critical. This is what attracted the research groups of Dr. Anthony Don (The Lowy Cancer
Research Centre), Dr. Nicole Verrills (The University of Newcastle) and A/Prof Jonathan Morris to
start a collaboration to tackle this problem.
3.1.3. AAL(S) as a Selective Cancer Therapeutic
Our collaborators, Dr. Matt Dun and Dr. Nicole Verrills at the University of Newcastle, have also
investigated whether phosphorylation of FTY720 (1.39) is necessary for activation of PP2A.
Instead of using inactivation of the BCR/ABL oncogene as a marker, they have previously shown
that FDC.P1 myeloid cells (normal bone marrow cells) expressing a mutation in an important
oncogene called c-KIT, are more sensitive to PP2A activation than wild type c-KIT+ FDC.P1
myeloid or empty vector (EV) cells. The mutation in this enzyme is where an aspartic acid residue
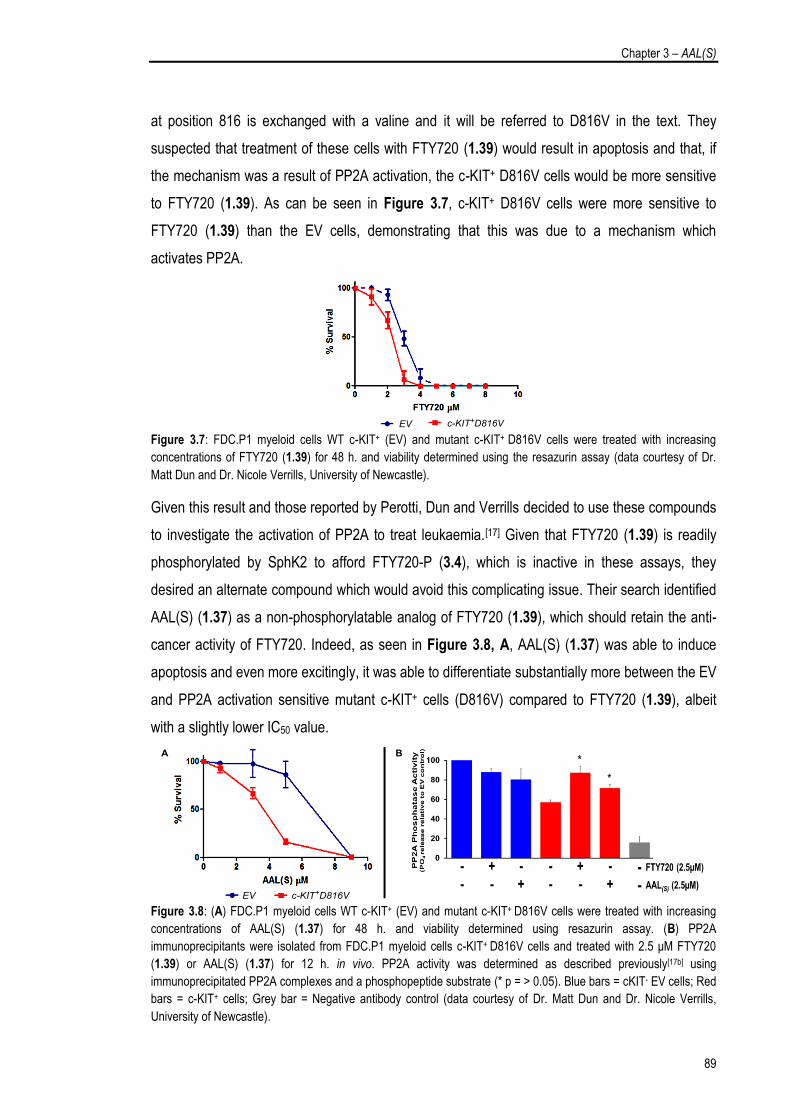
Chapter 3 – AAL(S)
89
at position 816 is exchanged with a valine and it will be referred to D816V in the text. They
suspected that treatment of these cells with FTY720 (1.39) would result in apoptosis and that, if
the mechanism was a result of PP2A activation, the c-KIT+ D816V cells would be more sensitive
to FTY720 (1.39). As can be seen in Figure 3.7, c-KIT+ D816V cells were more sensitive to
FTY720 (1.39) than the EV cells, demonstrating that this was due to a mechanism which
activates PP2A.
Figure 3.7: FDC.P1 myeloid cells WT c-KIT+ (EV) and mutant c-KIT+ D816V cells were treated with increasing
concentrations of FTY720 (1.39) for 48 h. and viability determined using the resazurin assay (data courtesy of Dr.
Matt Dun and Dr. Nicole Verrills, University of Newcastle).
Given this result and those reported by Perotti, Dun and Verrills decided to use these compounds
to investigate the activation of PP2A to treat leukaemia.[17] Given that FTY720 (1.39) is readily
phosphorylated by SphK2 to afford FTY720-P (3.4), which is inactive in these assays, they
desired an alternate compound which would avoid this complicating issue. Their search identified
AAL(S) (1.37) as a non-phosphorylatable analog of FTY720 (1.39), which should retain the anti-
cancer activity of FTY720. Indeed, as seen in Figure 3.8, A, AAL(S) (1.37) was able to induce
apoptosis and even more excitingly, it was able to differentiate substantially more between the EV
and PP2A activation sensitive mutant c-KIT+ cells (D816V) compared to FTY720 (1.39), albeit
with a slightly lower IC50 value.
Figure 3.8: (A) FDC.P1 myeloid cells WT c-KIT+ (EV) and mutant c-KIT+ D816V cells were treated with increasing
concentrations of AAL(S) (1.37) for 48 h. and viability determined using resazurin assay. (B) PP2A
immunoprecipitants were isolated from FDC.P1 myeloid cells c-KIT+ D816V cells and treated with 2.5 μM FTY720
(1.39) or AAL(S) (1.37) for 12 h. in vivo. PP2A activity was determined as described previously[17b] using
immunoprecipitated PP2A complexes and a phosphopeptide substrate (* p = > 0.05). Blue bars = cKIT- EV cells; Red
bars = c-KIT+ cells; Grey bar = Negative antibody control (data courtesy of Dr. Matt Dun and Dr. Nicole Verrills,
University of Newcastle).
*
*

Chapter 3 – AAL(S)
90
Confirmation that these compounds were inducing apoptosis by activation of PP2A was obtained
when purified PP2A complexes from mutant c-KIT+ D816V cells were treated with a
phosphopeptide and 2.5 μM of either FTY720 (1.39) and AAL(S) (1.37) (Figure 3.8, B). The level
of phosphate released from the phosphopeptide, due to dephosphorylation by PP2A, was
determined and showed that both compounds increased the level of phosphate release relative to
the control.[25]
From the results discussed above, it is hypothesised that AAL(S) (1.37) will enable us to
selectively target apoptosis in leukaemia cells without the complication of immunosuppressant
activity (in contrast to FTY720 (1.39) and AAL(R) (3.5)). However, it should be noted that AAL(S)
(1.37) is not as active as FTY720 (1.39). Given its relationship to FTY720 (1.39), it is surprising
that there have not been more structural modifications made to AAL(S) (1.37) to improve its anti-
cancer properties.
It was at this point that the author became involved in this project and as part of his PhD program
decided that two key questions needed to be answered.
- What are the key structural-activity requirements of AAL(S)’s for biological activity?
- What are the intermolecular binding partners of AAL(S) (1.37) and how do these activate
PP2A?
The following sections will outline our efforts towards addressing these questions.

Chapter 3 – AAL(S)
91
3.2. Investigation into the Structure-Activity Relationships of AAL(S)
3.2.1. Designing a Synthesis of AAL(S)
Our first goal was to synthesise analogs of AAL(S) (1.37) that could be tested for their anti-cancer
activity. As can be seen in Figure 3.9, there are three regions of AAL(S) that could be modified –
the hydrophobic tail, aromatic linker and the amino alcohol head group. The areas which we
chose to target as part of our initial structure-activity relationship study were the hydrophobic tail
and the amino alcohol head group. Given the success with FTY720’s tail optimisation (Figure
3.1) it was surprising that no one has done the same thing for AAL(S) (1.37). While it had been
established that the C8 chain length in FTY720 was optimal for immunosuppressant activity, what
was the optimal length required to activate PP2A?
Figure 3.9: Regions of AAL(S) (1.37) that could be targeted as part of a structure-activity relationship study.
Also, for the amino alcohol head group, the S stereochemistry is important but can the
substituents be varied to improve activity? Before we embarked on this study, a synthesis was
required which allows for significant quantities of AAL(S) (1.37) to be produced and allow for
manipulations to be made for analog production.
Examination of the literature revealed that there was one reported stereoselective synthesis of
AAL(R) (3.5) by Hinterding and co-workers and it utilised Schöllkopf’s protocol[26] for the synthesis
of chiral aminoesters (Figure 3.10).[27] Schöllkopf’s protocol relies on the stereoselective
alkylation of Schöllkopf’s reagent, which is a masked aminoester fragment. In the case of AAL(R)
(3.5), deprotonation of Schöllkopf’s reagent (R)-3.7 using n-butyllithium, generates the bis-lactim
anion 3.8. As shown in Figure 3.10, the isopropyl group of Schöllkopf’s reagent (R)-3.7 blocks
one face of the reagent so that the subsequent alkylation can only occur at the opposite face.
This anion was quenched with iodide 3.9 to afford mono-alkylated bis-lactim ether 3.10 in 71 %
yield and as a single diastereomer. Deprotonation of bis-lactim ether 3.10 and quenching with
methyl iodide adds the methyl group, again to the side opposite of the isopropyl group. With the R
stereochemistry established the Schöllkopf moiety was hydrolysed using 0.5 M HCl in 1,4-
dioxane to afford the chiral aminoester 3.11 in 58 % yield. Subsequently, reduction with LiAlH4 in
THF at 65°C provided AAL(R) (3.5) in 95 % yield.

Chapter 3 – AAL(S)
92
Figure 3.10: Hinterding’s synthesis of AAL(R) using Schöllkopf’s reagent. Reagents and yields (a) n-BuLi, THF, -
78°C → 0°C; (b) 3.9, THF, -78°C → 0°C, 71 %; (c) n-BuLi, THF, -78°C → 0°C; (d) MeI, -78°C → 0°C, 65 %; (e)
0.5 M HCl in 1,4-dioxane, rt, 58 %; (f) LiAlH4, THF, 65°C, 95 %.[28]
Hinterding’s synthesis of AAL(R) (3.5) proceeds in seven steps and 17 % overall yield. While this
route would obviously allow for ready access to AAL(S) (1.37) and for structural analogs to be
made, it was thought prudent to explore alternative methods which would gain rapid access to
AAL(S) (1.37) in shorter steps and improved yields.
The stereoselective synthesis of quaternary α-aminoesters and α-aminoalcohols has been an
intense area of interest.[29] However, for us to consider a switch from the Schöllkopf protocol to
any of the other methods that are available they had to abide to some criteria:
The synthesis must be reliable,
Highly stereoselective,
Less than five steps, and
Modular so as to allow for efficient synthesis of analogs
Given that all chiral reagents require several steps to generate the quaternary centre, it was felt
that seeking an alternative to Schöllkopf’s reagent would not be useful. Therefore, a multi-
component or catalytic method, particularly where the amino alcohol head group was constructed
in one step would be more attractive. From the various protocols available, the Petasis reaction
and the Sharpless asymmetric aminohydroxylation reaction were chosen as worthy of further
investigation as alternative routes to AAL(S) (1.37) (Figure 3.11). In principle, both protocols
allow for additional manipulations to be made for subsequent SAR investigations.

Chapter 3 – AAL(S)
93
Figure 3.11: Proposed alternative retrosyntheses of AAL(S) (1.37).
3.2.1.1. Investigations into an Alternative Synthesis of AAL(S)
The first procedure we examined was the Petasis reaction. The Petasis reaction is a boron
variation of the Mannich reaction. The process involves the one-pot combination of an
organoboronic acid, an amine and a α-hydroxyaldehyde or ketone in one-pot for the synthesis of
1,2-amino acids/alcohols (Figure 3.12).[30] Moreover, the process can be carried out
stereoselectively using a chiral amine or ligand.[30b]
Figure 3.12: The Petasis reaction.
Our motivation to examine the Petasis reaction originated from two recent reports in the literature.
The first, by Ishii and co-workers, utilised the Petasis reaction to synthesise FTY720 (1.39), as
shown in Scheme 3.1. Here, they employed dihydroxyacetone (3.12), benzylamine (3.13) and
vinylboronic acid 3.14 to construct the tertiary aminodiol head group of FTY720 in one step.
Removal of the benzyl protecting group and hydrogenation of the internal alkene was achieved in
one step and afforded FTY720 (1.39) in excellent yield. However, Ishii and co-workers also
investigated hydroxyacetone (3.15) as a substrate under these conditions, in an attempt to
synthesise AAL(S/R), but this resulted in no product and only isolation of the starting materials.[31]
Ketone (a) Yield (%) (b) Yield (%)
3.12 40 90 3.15 NR -
Scheme 3.1: Reagents and yields (a) EtOH, rt, see table for yield; (b) 10 % Pd/C, H2, 1:100 10 % aq. HCl/EtOH, rt,
see table for yield (NR: no reaction).[31]

Chapter 3 – AAL(S)
94
The second report was by Hutton and co-workers who utilised vinylboronic acid 3.16, glyoxylic
acid (3.17) and Ellman’s t-butyl sulfinamide 2.62 as the amine source in their synthesis of β,γ-
dihydroxytyrosine derivatives. Hutton found that utilising enantiopure sulfinamides in the reaction
resulted in isolation of the secondary amino acids in high yield and with excellent
diastereoselectivity (Scheme 3.2).[32]
Scheme 3.2: Reagents and yields (a) 0.33 M in CH2Cl2, rt, 12 h, 99 % (dr = >20:1).[32]
Given these reports and our success with the t-butyl sulfinamide reagent ourselves in Chapter 2
we decided to apply this reaction to an asymmetric synthesis of AAL(S) (1.37). Our retrosynthetic
analysis is outlined in Figure 3.13. This would require access to styrylboronic acid 3.18 and,
based on Hutton’s model for diastereoselectivity, the (S)-enantiomer of the sulfinamide to afford
the (S)-stereochemistry in the product. From the onset, it was recognised that the challenge in
this synthesis would be either formation or the reactivity of the required ketimine intermediate and
so hydroxyacetone (3.15), dihydroxyacetone (3.12) and pyruvic acid (3.19) were all examined as
viable coupling partners. Before committing resources to the synthesis of the required boronic
acid, A/Prof Hutton was kind enough to supply us with 4-acetoxystyryl boronic acid (3.20) to
examine the reaction conditions.
Figure 3.13: Retrosynthetic analysis of AAL(S) (1.37) using the Petasis reaction.
Consequently, it was decided to pursue the hydroxyacetone derivatives first as they would allow
for direct access to AAL(S)’s amino alcohol head group. Thus, using Hutton’s optimised
conditions of equivalent amounts of (S)-t-butyl sulfinamide (S)-2.62, hydroxyacetone (3.15) and
boronic acid 3.20 in dichloromethane at room temperature for 16 hours resulted in no product
formed nor any ketimine intermediate observed. This result is in agreement with Ishii’s data. This
is further supported by an observation, made by us and by Ellman, that a Lewis acid is required
for the synthesis of inactivated ketimines with t-butyl sulfinamides. While the ketimine of TBS-
protected hydroxyacetone 3.21 could be synthesised, using the method reported by Ellman and

Chapter 3 – AAL(S)
95
co-workers ((S)-t-butyl sulfinamide (S)-2.62 and TBS-protected hydroxyacetone in the presence
of titanium isopropoxide in THF at reflux),[33] this was still unreactive in the Petasis reaction
(Scheme 3.3).
Scheme 3.3: Reagents and yields (a) TBSCl, imid., DMF, rt, 60 %; (b) Ti(Oi-Pr)4, (S)-2.62, THF, Δ, 35 %; (c) 3.20,
CH2Cl2, rt.
Similarly, using dihydroxyacetone (3.12) resulted in no Petasis product either. However, in this
case the ketimine intermediate was observed in the 1H NMR spectrum of the crude reaction
mixture. This was determined by the disappearance of the CH2 singlet of dihydroxyacetone (3.12)
and inclusion of a new AB system for the diastereotopic CH2 group now next to the sulfinamide
stereocentre.
Given these poor results using the hydroxyacetone derivatives, our attention turned to using
pyruvic acid (3.19), using the exact conditions used for the hydroxyacetone example. While the
1H NMR spectrum of the crude reaction mixture indicated that the product had formed, albeit with
poor diastereoselectivity, there was also poor consumption of the boronic acid 3.20 starting
material. Attempts to purify this material from the boronic acid 3.20 to determine the exact
diastereoselectivity were problematic. As a result efforts were focussed on trying to increase the
consumption of the boronic acid to allow for easier purification. It has been shown that
1,1,1,3,3,3-hexafluoroisopropanol (HFIP) is able to increase the reaction rate and improve the
yields of the Petasis reaction by assisting formation of ionic intermediates and stabilising polar
transition states.[34] This was also shown in Hutton’s work where he found using 10 % v/v
HFIP:CH2Cl2 could improve the yield whilst maintaining high diastereoselectivity. Indeed, using
this solvent system in the presence of pyruvic acid (3.19) resulted in complete consumption of the
starting material. However, there were multiple products in the 1H NMR spectrum of the crude
reaction mixture. It was suspected that this was due to partial acidic cleavage of the t-butyl
sulfinyl group in the product. Given the suspected product(s) were carboxylic acids it was difficult
to separate and identify the components by TLC and so, in an attempt to resolve the mixture, the
crude mixture was treated with TMS-diazomethane. Examination of the 1H NMR spectrum of the
crude reaction mixture between 3 and 4 ppm indicated that there were at least five different

Chapter 3 – AAL(S)
96
methyl ester singlets, indicating a complex mixture which was difficult to purify.i Based on our
criteria for our AAL(S) synthesis it was felt that this was not a worthwhile pursuit. This preliminary
investigation suggested that the Petasis approach was not going to be conducive to completing a
short synthesis and therefore, it was decided to discontinue our investigation.
Given the unsuccessful foray with the Petasis reaction, our attention turned to utilising the
Sharpless asymmetric aminohydroxylation reaction. The Sharpless asymmetric
aminohydroxylation reaction allows for the catalytic and asymmetric preparation of α-
aminoalcohols from alkenes. Similar to the Sharpless asymmetric dihydroxylation, the reaction
typically uses catalytic potassium osmate dihydrate as the oxidant and a chiral ligand, such as
those shown in Figure 3.14. To introduce the amine functionality, a protected ammonia
equivalent is also added.[35]
Figure 3.14: The Sharpless asymmetric aminohydroxylation reaction and the structures of common ligands and
amine sources. Reagents and yields (a) Amine source, Ligand, K2OsO4.2H2O, 1,3-dichloro-5,5-dimethylhydantoin,
NaOH, 1:1 n-PrOH/H2O.[35a]
Sharpless’ original report optimised the reaction conditions on terminal mono-substituted alkenes
and found that the amine generally attached to the least hindered position of the alkene.[36] Due to
the potential in generating such products as single stereo and regioisomers much work has gone
into developing ligand-amine source combinations for specific substrates. It was felt that applying
this process to AAL(S) (1.37) might identify a system that could allow for ready access to it.
i Employing 10 % v/v HFIP/CH2Cl2 as the solvent for both hydroxyacetone and dihydroxyacetone did not change the results observed using neat CH2Cl2.

Chapter 3 – AAL(S)
97
Furthermore, accessing the regioisomer of AAL(S) 3.22 would be interesting from a structure-
activity relationship standpoint (Figure 3.15).
Figure 3.15: Retrosynthetic analysis of AAL(S) (1.37) and its regioisomer 3.22 using the Sharpless asymmetric
aminohydroxylation.
An excellent example of the stereo- and regiocontrol that the Sharpless asymmetric
aminohydroxylation can achieve has been reported by McLeod and co-workers. Their focus was
on the preparation of chiral amino alcohols which are found in the GABA neurotransmitter class of
natural products. As seen in Scheme 3.4, McLeod showed that the stereochemistry of the
dihydroquinidine (DHQ or DHQD) fragment of the chiral ligand could control the stereochemistry
of the product and that variation of the aromatic linker (anthraquinone (AQN) or phthalazine
(PHAL)) between these two units could control which regioisomer (such as 3.23 or 3.24) was
produced when the reaction was carried out on terminal and 1,2-disubstituted alkenes.[37]
Entry Ligand Ratio (3.23:3.24) Yield Major (%) ee Major (%)
1 (DHQD)2AQN (3.27) 2.6:1 48 84 2 (DHQ)2AQN 2.3:1 43 81a 3 (DHQD)2PHAL (3.25) 1:8 56 89 4 (DHQ)2PHAL 1:10 38 91a
Scheme 3.4: Reagents and yields (a) TMSEOCONH2 (3.26), Ligand (see table), K2OsO4.2H2O, 1,3-dichloro-5,5-
dimethylhydantoin, NaOH, 1:1 n-PrOH/H2O, rt, see table for yield. a (R,R)-configuration.[37]
McLeod’s reasoning for the regioselectivity with these aromatic linkers was based on whether the
dihydroquinidine or the aromatic linker had a stronger π-bonding interaction with the substrate
aromatic portion, as shown in Figure 3.16.
Figure 3.16: McLeod model for regioselectivity (R = Teoc).[37]

Chapter 3 – AAL(S)
98
Theoretical calculations indicated that the dihydroquinidine unit had a stronger π-bonding
interaction in the PHAL aromatic linker case, which results in the terminal hydroxyl compound
being synthesised. In contrast to this, the AQN system has a stronger π-bonding interaction with
the substrate resulting in the terminal amine regioisomer.[38]
To utilise this reaction for the synthesis of AAL(S) (1.37) would require conducting the Sharpless
asymmetric aminohydroxylation on a 1,1-disubstituted alkene. Currently, there are no examples
of this reported in the literature. However, McLeod and co-workers’ results open up the intriguing
possibility that by varying the ligand in the reaction the natural preference for regioselectivity
could be overcome. It was felt that this would be an excellent starting point for our study.
To examine the scope of this procedure an initial study was carried out on a simplified substrate
to determine the best conditions to access both AAL(S) regioisomers. Using McLeod’s system as
a model, we hypothesised that the (DHQD)2PHAL ligand (3.25) with 2-(trimethylsilyl)ethyl
cabamate (TMSEOCONH2) (3.26) as the amine source would afford the (S)-stereochemistry and
hydroxy-terminal regioisomer as the major product. In contrast, the (DHQD)2AQN (3.27) ligand
should generate the amine-terminal regioisomer. The commercially available (DHQD)2Pyr (3.28)
ligand was also included as part of this study (Scheme 3.5).
Entry Amine Source (P) Ligand Ratio (3.31:3.32) Yield Major (%)
1 TMSEOCONH2 3.26 (Teoc) (DHQD)2PHAL 3.25 >20:1 35 2 TMSEOCONH2 3.26 (Teoc) (DHQD)2AQN 3.27 >20:1 54 3 TMSEOCONH2 3.26 (Teoc) (DHQD)2Pyr 3.28 >20:1 50 4 BocNH2 3.33 (Boc) (DHQD)2PHAL 3.25 >20:1 32 5 BocNH2 3.33 (Boc) (DHQD)2AQN 3.27 >20:1 57 6 BocNH2 3.33 (Boc) (DHQD)2Pyr 3.28 >20:1 52 7 CbzNH2 3.34 (Cbz) (DHQD)2PHAL 3.25 >20:1 35 8 CbzNH2 3.34 (Cbz) (DHQD)2AQN 3.27 >20:1 48 9 CbzNH2 3.34 (Cbz) (DHQD)2Pyr 3.28 >20:1 50
Scheme 3.5: Reagents and yields (a) n-BuLi, MePPh3Br, THF, rt, 84 %; (b) Amine source, Ligand, K2OsO4.2H2O,
1,3-dichloro-5,5-dimethylhydantoin, NaOH, 1:1 n-PrOH/H2O, rt, see table for yield.
The model substrate 3.29 was readily synthesised in 84 % yield from the commercially available
ketone 3.30 via a Wittig methylenation reaction with methyltriphenylphosphonium ylide in THF. To
our surprise, employing this substrate, the (DHQD)2PHAL (3.25) ligand system and carbamate
3.26 as the amine source afforded a single regioisomer which was proven to be the amine-

Chapter 3 – AAL(S)
99
terminal regioisomer 3.31 by comparison of the 1H NMR spectrum to McLeod’s data and by using
2D NMR techniques (Scheme 3.5, Entry 1). Particularly, the 1H-1H COSY spectrum, showed a
diagnostic correlation between the NH and the 1-CH2 group, as shown in Figure 3.17.
NH 1’-CH2 OMe 1-CH2 4-CH2 OH 3-CH2 2-Me 2’-CH2
Figure 3.17: 1H-1H COSY NMR spectrum of compound (Teoc)-3.31 between the region 0.5 – 5.5 ppm. The key
correlation between the NH and 1-CH2 group, indicating that the compound was the amine-terminal regioisomer, is
highlighted.
In an attempt to try and generate the hydroxy-terminal regioisomer 3.32 the two other ligand
systems were trialled (Scheme 3.5, Entries 2 and 3) but without success. In McLeod’s study it
was found that the amine source can also influence the regioselectivity and so, in a final attempt,
the Boc 3.33 and Cbz 3.34 protected amine equivalents were examined, using the three ligands,
but these also failed to achieve the desired result (Scheme 3.5 Entries 4 – 9).
In a bid to determine if the substrate was the reason for the high degree of natural preference for
the N-terminal regioisomer, it was decided to examine the Sharpless aminohydroxylation reaction
on two other substrates. These included 1,1-disubstituted alkene 3.35, which is a system similar
to McLeod’s, to see if the alkene-aromatic spacer length was important and another where the
methyl group was removed from the AAL(S) model substrate 3.36, to determine if the steric
interaction of this group was preventing formation of the hydroxyl-terminal regioisomer.
ppm
1.01.52.02.53.03.54.04.55.05.5 ppm
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5

Chapter 3 – AAL(S)
100
Substrates 3.35 and 3.36 were synthesised using a Mitsonobu reaction and a Grignard cross-
coupling reaction respectively, as detailed in Scheme 3.6.
As can be seen from the results in Scheme 3.6, no regioselectivity for the hydroxy-terminal
regioisomer 3.38 was seen by extending the alkene-aromatic distance, no matter which ligand
was used. However, by removing the methyl group on the AAL(S) simplified substrate some of
the hydroxy-terminal regioisomer 3.40 was observed when using the (DHQD)2PHAL (3.25)
ligand. Interestingly, this reaction also resulted in incomplete consumption of the starting material
which was even more evident when the reaction was scaled up. Unfortunately, these
regioisomers could not be separated by column chromatography but could again be
characterised using 2D NMR techniques (as was done for 3.31).
Entry Ligand Ratio (3.37:3.38) Yield Major (%)
1 (DHQD)2PHAL 3.25 >20:1 47 2 (DHQD)2AQN 3.27 >20:1 44 3 (DHQD)2Pyr 3.28 >20:1 75
Entry Ligand Ratio (3.39:3.40) Yield Major (%)
1 (DHQD)2PHAL 3.25 5:1 37a,b 2 (DHQD)2AQN 3.27 >20:1 93 3 (DHQD)2Pyr 3.28 >20:1 65
Scheme 3.6: Reagents and yields (a) 3.5 M DIAD in PhMe, 3-methyl-3-buten-1-ol, PPh3, THF, Δ, 99 %; (b)
TMSEOCONH2, Ligand, K2OsO4.2H2O, 1,3-dichloro-5,5-dimethylhydantoin, NaOH, 1:1 n-PrOH/H2O, rt, see table for
yield; (c) AllylMgBr, THF, 0°C → rt, 45 %. a reaction did not go to completion; b mixture of regioisomers.
In this section of work the goal was to develop a new efficient, stereoselective synthesis of
AAL(S) (1.37). The Petasis reaction was examined first, using conditions developed by Hutton
and co-workers. While there is precedent in the literature for using aldehyde substrates it was
found that the analogous ketones, which are required for the AAL(S) synthesis, were much less
reactive. Clearly, in this case Ellman’s t-butyl sulfinamide auxiliary is not the ideal reagent for

Chapter 3 – AAL(S)
101
these types of substrates and future work should be focussed on identifying a chiral amine that
generates a more reactive ketimine.
The Sharpless asymmetric aminohydroxylation methodology was also examined as a catalytic
strategy to gain access to AAL(S) (1.37). In contrast to McLeod’s work, examination of different
ligand and amine source combinations did not allow for access to the hydroxy-terminal
regioisomer to be achieved. By investigation with two model substrates the reason for the poor
regioselectivity was concluded to be attributed to factors concerning both the steric nature of the
1,1-disubstituted alkene and the alkene-aromatic spacer, as shown in Figure 3.18.
Figure 3.18: Proposed model for regioselectivity on AAL(S) 1,1-disubstituted alkene system using (DHQD)2PHAL (R
= Teoc).
While it was not possible to generate the hydroxy-terminal regioisomer of AAL(S) (1.37) using this
strategy the investigation was able to achieve impressive regioselectivity for the amine-terminal
regioisomer in a short efficient manner. Aspects of this work will be returned to later in Section
3.2.3. when addressing structure-activity relationships of the AAL(S)’s head group.

Chapter 3 – AAL(S)
102
3.2.1.2. Hinterding’s Synthesis of AAL(S) Using Schöllkopf’s Reagent
With this brief interlude into alternative syntheses of AAL(S) (1.37) concluded, our attention
turned back to using the Hinterding/Schöllkopf strategy to synthesise AAL(S) (1.37), (Section
3.2.1.) To apply this chemistry to AAL(S) (1.37) it was recognised that the (S)-enantiomer of
Schöllkopf’s reagent (S)-3.7 and iodide 3.9 would be required. While (S)-3.7 was available
commercially,i iodide 3.9 was synthesised from tyrosol 3.41 in 84 % yield over three steps using
the procedure reported by Fujita, as shown in Figure 3.19.
Figure 3.19: Examination of alkylation of Schöllkopf’s reagent (S)-3.7 with iodide 3.9. Shown are key nOe
correlations observed which proved the stereochemistry of the alkylation products. Reagents and yields (a) H15C7Br,
NaOEt, EtOH, Δ, 96 %; (b) MsCl, NEt3, CH2Cl2, 0°C → rt; (c) NaI, Me2CO, rt, 87 % (2 steps); (d) n-BuLi, THF, -78°C
then 3.9, THF, -78°C 15 min, then 0°C 2.5 h, 3.42 = 71 %, 3.43 = 21 % (dr = 3:1); (e) n-BuLi, THF, -78°C then 3.9,
THF, -78°C → 0°C, 3.42 = 84 %.
Subsequently, treatment of (S)-3.7 with n-butyllithium at -78°C followed by addition of iodide 3.9
at -78°C for 15 minutes then replacing the cold bath with a 0°C cold bath for two and a half hours
resulted in complete consumption of the starting material by TLC. Following work-up of the
reaction, analysis of the 1H NMR spectrum of the crude reaction mixture revealed that two
compounds were present in a ratio of 3:1 (Figure 3.20, top). These could be attributed to the two
diastereomers possible from the reaction. Indeed, purification of the mixture by flash
chromatography on silica gel afforded two similar compounds (Figure 3.20 middle and bottom).
By means of 2D NMR spectroscopy, the compounds were assigned as the two possible
diastereomers and importantly, the 1H-1H NOESY spectrum revealed that the major diastereomer
isolated was the result of alkylation at the least sterically hindered face of (S)-3.7 (important 1H-1H
NOESY correlations of both diastereomers are shown in Figure 3.19).
i Over the course of this PhD 5 g of (S)-3.7 could be obtained through Acros Organics for $1291.30 (AUD).

Chapter 3 – AAL(S)
103
Figure 3.20: top: Part of the 1H NMR spectrum of the crude reaction between (S)-3.7 and 3.9 (Figure 3.19) when the
-78°C cold bath was immediately replaced with a 0°C cold bath. The region shows the ratio of diastereomers;
middle: Part of the 1H NMR spectrum for the major diastereomer 3.42 from this reaction; bottom: Part of the 1H NMR
spectrum for the minor diastereomer 3.43 from this reaction.
Compared to the results reported by Hinterding, the diastereoselectivity of this reaction was much
lower than expected. It was presumed that this result was due to the drastic temperature
adjustment made after addition of the iodide 3.9 (-78°C then 0°C). To improve this, a more
moderate temperature increase was employed. Pleasingly, retaining the conditions as before but
2.402.452.502.552.602.652.702.752.802.852.90 ppm
1.0
0
0.3
1
1.01.52.02.53.03.54.0 ppm
1.01.52.02.53.03.54.0 ppm

Chapter 3 – AAL(S)
104
allowing the mixture to slowly warm to 0°C in the -78°C cold bath over 5 hours resulted in a
single diastereomer in the 1H NMR spectrum of the crude reaction mixture affording the product
3.42 in 84 % yield after purification.
With these optimised conditions for the alkylation of (S)-3.7 in hand, attention turned to the
second alkylation step and completion of the synthesis of AAL(S) (1.37). Again, deprotonation of
3.42 with n-butyllithium at -78°C, addition of methyl iodide and slowly warming the reaction
mixture to 0°C in the -78°C cold bath afforded a single diastereomer as was observed in the 1H
NMR spectrum of the crude reaction mixture. The stereochemistry of this compound was proven
to be the proposed diastereomer 3.44 by a key 1H-1H NOESY correlation between a proton on
the isopropyl group of the Schöllkopf reagent and a proton on the phenethyl side chain of AAL(S),
as shown in Figure 3.21.
Figure 3.21: Reagents and yields (a) n-BuLi, THF, -78°C, then MeI, -78°C → 0°C, 72 %; (b) TFA, H2O, MeCN, rt,
87 %; (c) LiAlH4, THF, 0°C → rt, 86 %.
Having served its purpose the Schöllkopf core was hydrolysed using TFA and water in MeCN to
afford aminoester 3.45 which was reduced to the amino alcohol 1.37 using LiAlH4 in 75 % yield
over the two steps. The AAL(S) (1.37) generated using this strategy had spectroscopic data
identical to that reported in the literature. In particular, the optical rotation (- 4 (0.5, CHCl3))
matched that reported by Fujita demonstrating that the S stereoisomer had been successfully
made.[9] Starting from commercially available tyrosol 3.41, AAL(S) (1.37) could be generated in
38 % yield over seven steps. Significantly, the synthesis could be carried out on a multi-gram
scale allowing access to substantial quantities of AAL(S) (1.37) to be synthesised for biological
testing.
3.2.2. First Generation Synthesis of Hydrophobic Tail Analogs of AAL(S)
With an efficient synthesis of AAL(S) (1.37) in hand, our attention turned to synthesising analogs.
While the direct target for AAL(S) (1.37) was not known at this point, to explore the role and the

Chapter 3 – AAL(S)
105
space that the hydrophobic tail of AAL(S) (1.37) occupies, a broad range of analogs were
proposed. These included varying the length of the alkyl chain, to determine an optimum chain
length, a benzyl and cyclohexyl analog, to determine if the pocket that AAL(S) binds can
accommodate larger substituents as well as introducing some conformational rigidity and a C7-
length ethylene glycol tail to try and improve the solubility of AAL(S) (1.37) (Figure 3.22).
Figure 3.22: The questions that were proposed to be answered as part of our first generation AAL(S) hydrophobic
tail analog synthesis.
While installation of the hydrophobic tail was efficient early in the synthesis for AAL(S) (1.37),
using this strategy for the purpose of analog syntheses would require a multistep synthesis for
each analog. It was therefore desirable to have a more convergent synthesis, where the
hydrophobic tail could be installed as late as possible in the synthesis. As shown in Figure 3.23,
there were two strategies considered to gain access to these analogs. The first was alkylation of
phenol 3.46, where the Schöllkopf reagent is still part of the molecule, and the second was
alkylation of a complete amino alcohol substrate 3.47. While strategy 2 is much more convergent,
that is the hydrophobic tail is installed later in the synthesis, there were concerns that the amino
alcohol head group may also be alkylated under the conditions required to alkylate the phenol.
Strategy 1 was appealing as the amino alcohol head group is protected by the Schöllkopf reagent
which could be deprotected as part of the overall synthesis after alkylation. With this insight it was
decided to examine Strategy 1 first.
Figure 3.23: Retrosynthesis of possible routes to access AAL(S) hydrophobic tail analogs.
To employ Strategy 1 for the synthesis of AAL(S) hydrophobic tail analogs would require a
suitably protected iodotyrosol (Figure 3.23). The TBS protecting group was chosen for this

Chapter 3 – AAL(S)
106
purpose as this could be deprotected orthogonally to the Schöllkopf reagent. Consequently, the
iodotyrosol 3.48 was synthesised in four steps, similarly to those used in the synthesis of
iodotyrosol 3.9 (Scheme 3.7). Synthesis of the bis-lactim ether 3.49 was achieved using the two-
step alkylation protocol optimised for the synthesis of AAL(S) in 85 % yield over two steps.
With the bis-lactim ether 3.49 in hand, development of a deprotection/alkylation protocol could be
carried out. Oriyama and co-workers showed that arylsilyl ethers could be deprotected using
cesium fluoride and the resulting phenoxide anion alkylated with various alkyl and benzyl
bromides in one-pot.[39] Reaction of 3.49 with 1-bromoheptane, as a direct comparison to the
AAL(S) synthesis, generated 3.44 in 88 % yield after purification (Scheme 3.7). Accordingly, this
procedure was applied to several other alkyl-halides, each of which resulted in the generation of
alkylated products in good to excellent yields (71 – 98 %, Scheme 3.7).
Final Analog R = (c) Yield (%) (d) Yield (%) (e) Yield (%)
3.51 Me 98 96 88 3.52 C4H9 71 95 68 AAL(S) (1.37) C7H15 88 - - 3.53 C8H17 81 64 63 3.54 C9H19 89 66 45 3.55 C10H21 77 91 69 3.56 C12H25 98 54 56 3.57 Bn 83 85 69 3.58 C4H8Cy 88 55 33 3.59 (CH2CH2O)2Me see scheme 71 78
Scheme 3.7: Reagents and yields (a) TBSCl, imidazole, DMF, rt; (b) I2, MeOH, rt, 96 % (two steps); (c) MsCl, NEt3,
CH2Cl2, 0°C → rt; (d) NaI, (Me)2CO, rt, 78 % (two steps); (e) n-BuLi, THF, -78°C, then 3.48, -78°C → 0°C, 89 %; (f)
n-BuLi, THF, -78°C, then MeI, -78°C → 0°C, 96 %; (g) CsF, RX, DMF, rt, see table for yield; (h) CsF, DMF, rt, 77 %;
(i) Br(CH2CH2O)2Me, K2CO3, DMF, rt, 44 %; (j) TFA, H2O, MeCN, rt, see table for yield; (k) LiAlH4, THF, 0°C → rt,
see table for yield.

Chapter 3 – AAL(S)
107
However, reaction with a C7-length ethylene glycol tail as the alkyl halide resulted in only isolation
of the TBS-deprotected material 3.50. It was speculated that this could possibly be due to
coordination of the ethylene glycol chain to the cesium cation preventing reaction with the
phenoxide. To overcome this problem, the synthesis of this analog was carried out in two steps.
Deprotection of the TBS group was achieved using CsF in DMF, and subsequent alkylation using
1-bromo-2-(2-methoxyethoxy)ethane and K2CO3 in DMF afforded the bis-lactam ether in 34 %
yield over the two steps. Completion of the synthesis of AAL(S) analogs was achieved using the
procedure outlined for AAL(S) – hydrolysis (TFA, H2O, MeCN) of the lactim and reduction (LiAlH4,
THF) to afford the amino alcohols 3.51 – 3.59, as shown in Scheme 3.7.
This methodology was successful in generating nine hydrophobic tail analogs of AAL(S) for
biological testing, which will be discussed in Section 3.3.
3.2.3. Structural Variations of the Amino Alcohol Head Group of AAL(S)
Similar to the analog design process that was laid out for the synthesis of hydrophobic tail
analogs of AAL(S), the analogs proposed for synthesis of the amino alcohol head group were
aimed at learning key characteristics about each of the three groups on the head group. While it
is obvious that the stereochemistry of this moiety is important, the size of the methyl group and
whether the amine and the alcohol group are proton donors or acceptors was initially examined
(Figure 3.24).
Figure 3.24: The questions that were proposed to be answered as part of our first generation AAL(S) amino alcohol
head group analog synthesis.
Outlined in Figure 3.25 are the strategies that will allow us to gain access to these analogs. It
was envisaged that the most straightforward analogs to synthesise would be those resulting from
direct alkylation of the 1-alcohol and 2-amino functional groups of the AAL(S) scaffold. To
synthesise analogs of the 1-alcohol group a nucleophilic ring opening of a chiral aziridine 3.60
was planned. Using the previously mentioned asymmetric Sharpless chemistry (starting from
alkene 3.61) would allow generation of regioisomers, which would provide insight into the role of
the amine and alcohol groups. Lastly, replacement of the 2-methyl group could be achieved by
diastereoselective alkylation of the mono-alkylated bis-lactim ether 3.42 that had been prepared
earlier as part the AAL(S) synthesis.

Chapter 3 – AAL(S)
108
Figure 3.25: Strategies proposed to make AAL(S) amino alcohol head group analogs.
Our initial focus was to functionalise the amine of the AAL(S) head group. Using AAL(S) (1.37) as
the starting material, a dimethylamino analog 3.62 and N-acetyl analog 3.63 were synthesised.
This would help to determine if the 2-amino group was acting as a hydrogen bond donor or
acceptor. The dimethylamino analog 3.62 was synthesised using a reductive amination protocol
(NaCNBH3, (CH2O)n, AcOH, MeCN). This process afforded a single product in the 1H NMR
spectrum of the crude material, that after purification, was confirmed to be the dimethylamino
analog by the observation of a singlet at 2.28 ppm integrating for 6H in the 1H NMR spectrum.
Treating AAL(S) (1.37) with AcCl and NEt3 in CH2Cl2 at 0°C afforded the N-acetyl analog 3.63 in
35 % yield (Scheme 3.8). The structure was confirmed by 1H NMR spectroscopy, with the
presence of a new singlet integrating for 3H at 1.91 ppm and a broad singlet at 5.50 ppm. The
singlet at 5.50 ppm was assigned to the 1-hydroxy group and this was confirmed when a 1H—1H
COSY correlation to the terminal CH2 group was observed.
Scheme 3.8: Reagents and yields (a) NaCNBH3, (CH2O)n, AcOH, MeCN, 0°C → rt, 54 %; (b) AcCl, NEt3, CH2Cl2,
0°C → rt, 35 %.
With those analogs prepared, attention turned to synthesising the required aziridine 3.60 so the
desired 1-alcohol analogs could be accessed. To determine whether the 1-alcohol group of

Chapter 3 – AAL(S)
109
AAL(S) was a proton donor or acceptor a fluorinated analog 3.64 and methyl ether analog 3.65
were proposed. As terminal aziridines have been shown to undergo nucleophilic ring opening at
the least sterically hindered end, retaining the stereochemistry in the starting material,[40] these
analogs should be able to be generated from aziridine 3.60. To synthesise aziridine 3.60, the
amine group of AAL(S) had to first be protected. This was achieved using the protocol reported
by Nishi and co-workers for the synthesis of Boc-AAL(R) in 83 % yield (Scheme 3.9).[41]
However, heating Boc-AAL(S) 3.66 with TsCl and KOH in Et2O at reflux did not generate the
desired aziridine 3.60. From the 1H NMR spectrum of the isolated material it was notable that the
new compound did not contain a t-butyl group and the CH2 group adjacent to the 1-alcohol group
had moved upfield from 3.6 to 4.1 ppm. The 13C NMR spectrum of the purified material provided
evidence that the carbonyl group was still present with a peak at 159.7 ppm but the definitive
evidence was provided by the HRMS which indicated a molecular ion where the product had lost
the t-butyl group but retained the CO2 of the Boc group. The product was thus concluded to be
oxazolidinone 3.67 which results from reaction of the 1-alcohol group with the carbonyl group of
the Boc group. This reaction has been reported in the literature on similar quaternary carbamate
protected α-aminoalcohols.[41-42]
Scheme 3.9: Reagents and yields (a) Boc2O, sat. aq. NaHCO3, EtOAc, Δ, 83 %; (b) KOH, TsCl, Et2O, Δ, 90 %
(3.67); (c) NaH, DMF, rt, then MeI, rt, 88 %; (d) 6 M aq. HCl, Δ.
Although this reaction did not gain access to the aziridine 3.60, oxazolidinones have also been
known to undergo nucleophilic ring opening at the least sterically hindered carbon, similarly to
aziridines, releasing CO2 in the process.[43] However, heating oxazolidinone 3.67 with either
TBAF in MeCN, NaOMe in MeOH or 6 M aqueous HCl, in a bid to regenerate AAL(S) (1.37), did
not afford any ring opened products with only starting material being recovered. Despite this, the
compound generated provided us with another interesting analog with which to test in our assays.
The N-methyl oxazolidinone 3.68 was also prepared in 88 % yield, as shown in Scheme 3.9.

Chapter 3 – AAL(S)
110
While this route provided us with two new analogs a strategy was still needed to gain access to
analogs of the 1-alcohol group. While an alternative N-protecting group, such as a tosylate
protecting group, would allow for the formation of the aziridine the Boc compound served as a
central part of some of the other syntheses and thus, it was chosen to examine a report by
Hinterding and co-workers who showed that cyclic sulfamidates can also undergo ring opening
reactions with nucleophiles on the AAL(R) substrate.[44] To investigate this strategy Boc-AAL(S)
3.66 was treated with thionyl chloride and pyridine in MeCN which afforded a 50:50 mixture of
diastereomers. These diastereomers were oxidised with RuCl3 and NaIO4 in MeCN:H2O, which
abolished the stereochemistry about the sulfur. i With the cyclic sulfamidate 3.69 in hand, the
nucleophilic ring opening could be examined. Gratifyingly, treating this material with TBAF in
MeCN at reflux resulted in a new compound. TLC analysis of the reaction mixture revealed a new
spot that was lower in Rf than that of the starting material. Without isolation of this compound the
crude residue was treated with 6 M aqueous HCl solution to cleave off the sulfate and the Boc
protecting group. Purification of this material by flash chromatography on silica gel afforded the
fluoro analog 3.64 (Scheme 3.10). Confirmation that this had been synthesised was evident by
the coupling of the terminal-CH2-F (JH-F = 17.7 Hz) in the 1H NMR spectrum and the terminal-C-F
(J = 17.5 Hz), in the 13C NMR spectrum.
Scheme 3.10: Reagents and yields (a) SOCl2, py, MeCN, -40°C → -10°C, diastereomer one 48 %: diastereomer
two 46 %; (b) RuCl3, NaIO4, 3:1 MeCN/H2O, 0°C, 85 %; (c) n-Bu4NF, MeCN, Δ, then 6 M aq HCl, Δ, 88 %; (d) MeI, n-
Bu4NSO4, 50 % w/v aq. NaOH/THF, rt; (e) 2 M aq. HCl, MeCN, Δ, 82 % (2 steps).
To synthesise the methyl ether analog the cyclic sulfamidate 3.69 was treated with an excess of
freshly prepared NaOMe in methanol at reflux. However, this provided none of the desired ring
opened material and only starting material was isolated. Fortunately, the methyl ether analog
could be synthesised by direct alkylation of the hydroxyl group of Boc-AAL(S) 3.66 with MeI in 50
% w/v aqueous NaOH and THF using n-Bu4NSO4 as a phase transfer catalyst. Again, TLC
i A two-step protocol, without purification of the cyclic sulfamidite, was also employed and afforded the product cyclic sulfamidate 3.69 in 99 % yieldover two steps.

Chapter 3 – AAL(S)
111
analysis of the crude reaction mixture indicated a new spot which had an Rf lower to that of the
starting material and, after removal of the Boc-protecting group with 6 M aqueous HCl solution,
methyl ether 3.65 was isolated in 82 % yield (Scheme 3.10). The 1H and 13C NMR spectra
confirmed that the methyl ether, and not the N-methylated compound, had been synthesised as a
new signal was observed (3.36 ppm in the 1H NMR spectrum and 59.4 ppm in the 13C NMR
spectrum) which is indicative of the new methyl ether group. Furthermore, this is in contrast to the
dimethylamino analog 3.62 discussed earlier which has an N-methyl resonance at 2.28 ppm in
the 1H NMR spectrum, showing the difference between N- and O-alkylated products.
Having already established that the Sharpless asymmetric aminohydroxylation reaction could be
used to synthesise regioisomers of the amino alcohol moiety, it was decided to use this
methodology to prepare a regioisomer of AAL(S) (Scheme 3.11). The alkene precursor 3.61 was
therefore required. This was synthesised in two steps from commercially available phenol 3.70.
The phenol was alkylated using Cs2CO3 and 1-bromoheptane in THF (91 % yield) and the
resulting ketone was converted to the alkene 3.61 in 83 % yield via a Wittig reaction using
methyltriphenylphosphonium ylide in THF. With the alkene in hand, the chemistry developed in
Section 3.2.1.2. could now be utilised. Here, the best results to access the amine-terminal
regioisomer were found to be when (DHQD)2AQN 3.27 was used as the chiral ligand and
TMSEOCONH2 3.26 was the amine source. As suspected, utilising these conditions afforded the
amine-terminal regioisomer (confirmed by 2D 1H-1H COSY NMR) which was subsequently
deprotected using 2 M etheral HCl in methanol to afford the amino alcohol 3.21 in 35 % yield over
the two steps. Similarly, a dihydroxy analog 3.71 was synthesised, to determine if the amine was
important, using the Sharpless asymmetric dihydroxylation. Employing Sharpless’ standard
conditions and again utilising the DHQD ligand to afford the S stereochemistry in the product[45]
afforded the dihydroxy analog 3.71 in 83 % yield. At this stage there was no attempt to optimise
the optical purity of both regio-AAL(S) 3.21 and dihydroxy-AAL(S) 3.71. However, the compounds
isolated were optically enriched as indicated by their optical rotations. Finally, using the same
alkene substrate an analog where the alcohol was deleted was synthesised by exploiting the
Ritter reaction.[46] Using Ritter’s standard conditions (KCN, H2SO4, AcOH, under a static
atmosphere) generated a 50:50 mixture of amide rotamers which after deprotection, by heating
the material neat in neat 6 M aqueous HCl solution at reflux, afforded amine 3.72 in 93 % yield
over the two steps. The analog 3.72 was established to be the alcohol-deleted analog by the
absence of AAL(S)’s terminal CH2OH group and a singlet at 1.16 ppm integrating for 6H in the 1H
NMR spectrum.

Chapter 3 – AAL(S)
112
Scheme 3.11: Reagents and yields (a) Cs2CO4, H15C7Br, DMF, rt, 91 %; (b) n-BuLi, MePPh3Br, THF, rt, 83 %; (c)
TMSEOCONH2, (DHQD)2AQN, K2OsO4.2H2O, 1,3-dichloro-5,5-dimethylhydantoin, NaOH, 1:1 n-PrOH/H2O, 77 %; (d)
2 M HCl in Et2O, MeOH, rt, 45 %; (e) K2OsO4.2H2O, K2CO3, K3Fe(CN)6, (DHQD)2PHAL, 1:1 t-BuOH/H2O, 0°C → rt,
83 %; (f) KCN, H2SO4, AcOH, 0°C → rt, 97 %; (g) 6 M aq. HCl, Δ, 96 %.
Lastly, variation of the 2-methyl substituent of the AAL(S) head group was examined. The
analogs proposed for this position were a des-methyl analog 3.73, where the methyl group was
replaced with a hydrogen atom (consequently, changing this carbon from a quaternary to a
tertiary centre) and an ethyl group analog 3.74, to see if the pocket that the methyl group projects
into was open to larger substituents. Because, at this point, a considerable amount of the TBS-
protected mono-alkylated bis-lactim ether 3.75 had been stockpiled for the synthesis of
hydrophobic tail analogs this was employed in this study. However, while the ethyl analog could
be readily synthesised by lithiation and quenching of the common intermediate with ethyl iodide to
afford bis-lactim ether 3.76 in 84 % yield (Scheme 3.12), the des-methyl bis-lactim ether could
not be synthesised in this way. While the opposite (R)-enantiomer of Schöllkopf’s reagent (R)-3.7
could be employed to construct the stereochemistry of this group, an alternative strategy where
the current bis-lactim ether’s stereochemistry was interconverted was investigated. This would
require quenching of the common lithiated bis-lactim ether intermediate with a proton source that
would react at a low temperature. While water was considered for this purpose it would freeze at
the temperature required for the lithiation and would only react when warmed up, at which stage
the lithiated intermediate would have decomposed. Subsequently, lithiation and quenching of the
common intermediate 3.75 with methanol was investigated and this resulted in a 3:7 mixture of
diastereomers (in favour of the starting material) where the desired diastereomer 3.77 could be
isolated in 27 % yield. In an attempt to try and improve this ratio it was thought that increasing the
steric bulk of the proton source (so that it would interact more readily with the isopropyl group of

Chapter 3 – AAL(S)
113
the bis-lactim ether and deliver the proton to the least sterically hindered face) would afford the
interconverted material as the major product. However, utilising t-butanol on this system resulted
in no improvement in diastereoselectivity. Regardless of this the required diastereomer could be
separated via flash chromatography and carried through the three step sequence described
earlier to afford the des-methyl analog 3.76 in 31 % yield over two steps. While the ethyl analog
bis-lactim ether 3.73 was easier to synthesise, hydrolysis of the Schöllkopf reagent proved
troublesome due to increased steric bulk.i Extension of the reaction time to 72 hours resulted in
isolation of the aminoester product in 25 % yield with the remainder of the material consisting of
of partially hydrolysed products.[47] The desired ethyl analog 3.74 was synthesised by reduction of
the aminoester in 60 % yield.
Final Analog R = (b) Yield (%) (c) Yield (%) (d) Yield (%)
3.73 H 72 99 44 3.74 Et 84 25 60
Scheme 3.12: Reagents and yields (a) n-BuLi, THF, -78°C, then MeOH, -78°C, then 0°C, 27 % (59 % recovered
starting material); (b) CsF, H15C7Br, DMF, rt, see table for yield; (c) TFA, H2O, MeCN, rt, see table for yield; (d)
LiAlH4, THF, 0°C → rt, see table for yield; (e) n-BuLi, THF, -78°C, then EtI, -78°C → 0°C, 87 %.
i This problem has also been reported by Hinterding and co-workers for the synthesis of chiral FTY720 analogs where the substituent at this position is any bigger than a methyl – see reference 47 for further details.

Chapter 3 – AAL(S)
114
3.3. Biological Data
The compounds synthesised in Section 3.2. were tested in a number of assays by our
collaborators. These were a PP2A activation assay and cell viability assays against various
cancer cell lines. The collaborator that completed the assays is acknowledged in the text and
under the respective data in the Figure legend.
The PP2A activation assay was completed by Dr. Matt Dun and Dr. Nicole Verrills at the
University of Newcastle. The data for a sample selection of representative compounds is shown
in Figure 3.26. This shows that all of the analogs that were synthesised activate PP2A at least
one and a half times that observed for the control D816V cells in vitro. The cytotoxicity of the
compounds were tested on FDC.P1 myeloid cells (EV) and FDC.P1 myeloid cells expressing
mutant cKIT+ (D816V) by Dr. Matt Dun and Dr. Nicole Verrills at the University of Newcastle and
on the Bcr/Abl+ K562 chronic myeloid leukaemia cell line by Dr. Anthony Don at the Lowy Cancer
Research Centre. For clarity, presentation of this data has been split up between the hydrophobic
tail analogs and the amino alcohol head group analogs.
Figure 3.26: PP2A immunoprecipitatants were isolated from D816V-cKIT+ myeloid cells and treated with 2.5 μM of
either FTY720 (1.39), AAL(S) (1.37) or analog for 30 min. in vitro. PP2A activity was determined by detecting the
amount of free inorganic phosphate (PO4-) released from a PP2A specific phosphopeptide and presented relative to
the control (D816V).
In Section 3.2.2., nine hydrophobic tail analogs were synthesised (Figure 3.27). The data
obtained for the cell viability for these compounds is shown in Figures 3.28 and 3.29. The data
for all of the cell types tested clearly shows that the eight atom length hydrophobic tail analog
3.53 is more potent than AAL(S) (1.37) but, it has less efficacy for the EV myeloid cells.
Interestingly, although all of the analogs synthesised were able to activate isolated PP2A
complexes in vitro, only those with a tail between 7 and 9 atoms long have cytotoxic activity. It is
postulate that this could be due to the solubility of the compounds and/or their ability to cross the
0
50
100
150
200
250
D81
6V
FT
Y72
0
AA
L(S
)
3.51
3.55
3.56
3.71
3.72
PP
2A A
ctiv
ity
(% r
elat
ive
to D
816V
)
Compound

Chapter 3 – AAL(S)
115
cell membrane into the cell. Furthermore, the tail moiety has to be hydrophobic as shown by the
complete loss of activity observed for the ethylene glycol analog 3.59, which has the same
number of atoms in the tail as AAL(S) (1.37).
R = R =
3.51 Me 3.56 C12H25
3.52 C4H9 3.57 Bn
3.53 C8H17 3.58 (CH2)4Cy
3.54 C9H19 3.59 (CH2CH2O)2Me
3.55 C10H21
Figure 3.27: Summary of the hydrophobic analogs that were made in Section 3.2.2.
Figure 3.28: FDC.P1 myeloid cells WT c-KIT+ (EV) and mutant c-KIT+ D816V cells were treated with increasing
concentrations of either AAL(S) (1.37) or hydrophobic tail analogs for 48 h. and viability determined using the
resazurin assay. From this data the IC50 values were determined and are presented in the graph (data courtesy of Dr.
Matt Dun and Dr. Nicole Verrills, University of Newcastle).
Figure 3.29: K562 cells were treated with 3 μM or 10 μM of either AAL(S) (1.37) or hydrophobic tail analogs for 24 h.
and viability determined using Annexin V/PI assay, n = 3 (data courtesy of Dr. Anthony Don, Lowy Cancer Research
Centre).
0
1
2
3
4
5
6
7
8
9
3.51
3.52
AA
L(S
)
3.53
3.54
3.55
3.56
3.57
3.58
3.59
Cyt
oto
xici
ty (
IC50
μM
)
Compound
EV D816V
0
20
40
60
80
100
120
140
160
180
200
3.51
3.52
AA
L(S
)
3.53
3.54
3.55
3.56
3.57
3.58
3.59
Cel
l Via
bili
ty (
%)
Compound
3 μM 10 μM

Chapter 3 – AAL(S)
116
In Section 3.2.3. eleven amino alcohol head group analogs were synthesised (Figure 3.30).
Similar to the hydrophobic tail analogs, these show a similar trend between the D816V cells and
the K562 leukaemia cells, as shown in Figures 3.31 and 3.32. Compounds 3.73 and 3.74 were
synthesised to determine the role of the methyl group on the AAL(S) head group. These analogs
have either the methyl group removed and replaced with a hydrogen (ie. 3.73) or extended to an
ethyl group (ie. 3.74). Both of these analogs have decreased activity relative to AAL(S)
suggesting that the methyl group is the optimal size.
3.62 3.63 3.67 3.68 3.64 3.65 3.21 3.71 3.72 3.73 3.74
X Me Me Me Me Me Me Me Me Me H Et
Y OH OH -OC(O)
N(H)-
-OC(O)
N(Me)-
F OMe NH2 OH H OH OH
Z NMe2 NHAc NH2 NH2 OH OH NH2 NH2 NH2
Figure 3.30: Summary of the hydrophobic analogs that were made in Section 3.2.3.
Figure 3.31: FDC.P1 myeloid cells WT c-KIT+ (EV) and mutant c-KIT+ D816V cells were treated with increasing
concentrations of either FTY720 (1.39), AAL(S) (1.37) or head group analogs for 48 h. and viability determined using
the resazurin assay. From this data the IC50 values were determined and are presented in the graph. At the time this
thesis was written there was currently no data available for compounds 3.63 and 3.73 (data courtesy of Dr. Matt Dun
and Dr. Nicole Verrills, University of Newcastle).
0123456789
10
FT
Y72
0
AA
L(S
)
3.62
3.63
3.67
3.68
3.64
3.65
3.21
3.71
3.72
3.73
3.74
Cyt
oto
xici
ty (
IC50
μM
)
Compound
EV D816V

Chapter 3 – AAL(S)
117
Figure 3.32: K562 cells were treated with 3 μM or 10 μM of either FTY720 (1.39), AAL(S) (1.37) or head group
analogs for 24 h. and viability determined using Annexin V/PI assay, n = 3 (data courtesy of Dr. Anthony Don, Lowy
Cancer Research Centre).
To determine whether the alcohol moiety is a hydrogen bond donor or acceptor a fluoro analog
3.64 and methyl ether analog 3.65 were synthesised. Both of these analogs have similar activity
to AAL(S) (1.37) demonstrating that this group is a hydrogen bond acceptor and not a hydrogen
bond donor. The methyl ether analog 3.65 has particularly good activity against the K562 cell line
being equipotent to FTY720 (1.39). Interestingly, analog 3.72 where the hydroxy group has been
removed completely shows improved cytotoxic activity to AAL(S) (1.37). However, its cytotoxic
activity against the EV cell line is also considerable. The data for compounds 3.63, 3.67, and 3.68
where the amine has been converted to an amide and dihydroxy analog 3.71 were found to have
completely lost their cytotoxic activity. This indicates that the amine of AAL(S) is a hydrogen bond
donor. The analog where the hydroxy and amine functionalities are swapped (3.21) was slightly
less cytotoxic to AAL(S) (1.37).
The data presented above is summarised in Figure 3.33. While analogs with improved activity
have been synthesised, a concern that has been highlighted with these molecules is their
cytotoxicity against the EV cell line. It is suspected that some analogs, such as 3.72, are not
performing via the same mechanism as AAL(S) (1.37) and instead are acting more like
detergents to kill cells. This is also highlighted with the hydrophobic tail analogs in that although
analogs from C7 – C9 all exhibit excellent cytotoxicity against the D816V cells their selectivity
against the EV cells decreases as the chain length increases, or becomes more hydrophobic.
0
20
40
60
80
100
120
140
160
180
200
FT
Y72
0
AA
L(S
)
3.62
3.63
3.67
3.68
3.64
3.65
3.21
3.71
3.72
3.73
3.74
Cel
l Via
bili
ty (
%)
Compound
3 μM 10 μM

Chapter 3 – AAL(S)
118
Figure 3.33: Summary of structure-activity data obtained for AAL(S).
In conclusion, the best analogs synthesised as part of this project were determined to be those
with the highest selectivity between the D816V myeloid leukaemia cells and the EV cell types.
These are shown in Figure 3.34 with their selectivity index between the EV and D816V cells.
While some analogs, such as the methyl ether analog 3.65, have much greater cytotoxicity than
AAL(S) against K562 cells, this molecule is unfortunately quite effective at killing normal EV cells.
This observation has raised some concerns with the hydrophobicity of these molecules which will
have ramifications for how future analog design is tackled.
Figure 3.34: Selectivity index for AAL(S) analogs between EV and D816V cells. NB: only those analogs with
selectivity greater than 10 % are shown.
3.4. Inhibition of Ceramide Synthases by AAL(S) Analogs
Ceramides are an important class of bioactive signalling lipids which are involved in the
sphingolipid metabolism pathway (see Figure 3.2). Their formation is catalysed by an enzyme
called ceramide synthase (CerS). This enzyme exists as six isoforms and each of these isoforms
catalyses the addition of different length fatty acid groups onto the amine group of sphingosine,
as shown in Figure 3.35.[48]
05
101520253035404550
FT
Y72
0
AA
L(S
)
3.53
3.58
3.62
3.65
3.74
Sel
ecti
vity
Ind
ex (
%)
Compound

Chapter 3 – AAL(S)
119
Ceramide Synthase Isoform
CerS1 CerS2 CerS3 CerS4 CerS5 CerS6
Alk C18 C22 – C26 C26 – C28 C18 – C20 C14 – C18 C16
Figure 3.35: Part of the sphingolipid metabolism pathway demonstrating the role of individual ceramide synthase
isoforms in the production of ceramides.
Variation in the expression of specific CerS isoforms has been found to be unique amongst
different human tissues. For example, CerS3 is found primarily in the skin and male reproductive
organs and CerS6 is found in the intestine, spleen and lymph nodes.[48] Deregulation of these can
have implication in several diseases including cancer, multiple sclerosis, diabetes, and
neurological disorders.[49] Perhaps surprisingly, very few selective CerS inhibitors have been
reported in the literature.[50] The groups of Berdyshev[51] and Futerman[52] have independently
shown that FTY720 (1.39) inhibits CerS, particularly CerS1. However, as has been previously
discussed, FTY720 (1.39) is readily phosphorylated by SphK2 and this phosphorylated analog
does not have any activity against CerS isoforms. Our collaborator, Dr. Anthony Don has an
interest in compounds that inhibit CerS. Unpublished data from his group has shown that AAL(S)
(1.37) also inhibits CerS1. It was this observation that prompted us to examine our analogs
activity against specific CerS isoforms. The data presented in Figure 3.36 is an ongoing
collaboration with the Don group and so, some of the analogs have not been tested against all of
the CerS isoforms available (CerS1, CerS2 and Cer5/6). While the data will not be discussed in
detail the analogs show varying degrees of activity against CerS1. Interestingly, an inverse
relationship is observed between the activity of analogs against CerS1 and CerS2. All of the
analogs tested are poor inhibitors of CerS5/6.

Chapter 3 – AAL(S)
120
Figure 3.36: Untransfected HEK293 cell lysates were treated with 10 μM AAL(S) (1.37) or AAL(S) analogs, 10 μM
NBD-sphingosine and either 50 μM C18:0-CoA (CerS1 data), C24:1 (CerS2 data) or C16:0-CoA (CerS5 or 6 data).
Products formed were quantified by TLC with a fluorescent imager and normalised relative to the control as
described.[53] (Data courtesy of Dr. Anthony Don, Lowy Cancer Research Centre).
CerS1 is primarily expressed in the brain and skeletal muscle.[48] Up regulation of CerS1
increases levels of C18-ceramide which has been shown to be associated with neurological
disorders such as Alzheimer’s disease and stroke.[54] While some analogs were quite effective at
inhibiting CerS1 if they are going to be of therapeutic benefit, particularly in the case of these
neurological disorders, they would have to be non-cytotoxic. Consequently, the data for the
analogs against CerS1 was plotted against the cytotoxicity data obtained for the K562 leukaemia
cells (Figure 3.37).
Figure 3.37: Comparison of the cytotoxicity of AAL(S) (1.37) and AAL(S) analogs against K562 cells (adapted from
Figures 3.29 and 3.32) and CerS1 activity (adapted from Figure 3.36) at 10 μM. The benzyl analog 3.57 is
highlighted in the yellow box as this analog has no cytotoxicity but is quite potent against CerS1.
0
20
40
60
80
100
120
140
160
AA
L(S
)
3.51
3.52
3.53
3.54
3.55
3.56
3.57
3.58
3.59
3.62
3.63
3.67
3.68
3.64
3.65
3.21
3.71
3.72
3.73
3.74
Cer
S a
ctiv
ity
(% r
elat
iv t
o c
on
tro
l)
Compound
CerS1 CerS2 CerS5/6
0
20
40
60
80
100
120
140
AA
L(S
)
3.51
3.52
3.53
3.54
3.55
3.56
3.57
3.58
3.59
3.62
3.63
3.67
3.68
3.64
3.65
3.21
3.71
3.72
3.73
3.74
Via
bili
ty/A
ctiv
ity
(% r
elat
ive
to c
on
tro
l)
Compound
K562 CerS1

Chapter 3 – AAL(S)
121
This data identified that benzyl hydrophobic tail analog 3.57 (data highlighted in the yellow box in
Figure 3.37) did not have any cytotoxic activity but it was one of the most potent inhibitors
against CerS1. While selective, if this compound is going to be useful as an effective inhibitor of
CerS1 its potency must be improved.
3.4.1. Synthesis and Biological Data of Selective Ceramide Synthase Inhibitors
The data shown in Section 3.4. indicates that the benzyl hydrophobic tail analog 3.57 was a
selective inhibitor of CerS1. A second generation of analogs, examining the effect of substitution
on the benzyl aromatic ring, were proposed using the Topliss decision tree as a guide (Figure
3.38).
Figure 3.38: Topliss decision tree for aromatic substituents. (L: less active; E: equiactive; M: more active).[55]
The Topliss Tree was developed by Professor J. G. Topliss in 1972 to aid medicinal chemists in
systematic analog design.[56] It was based on an original concept developed by Hantsch, which
takes into account the lipophilicity, electronic and steric properties of substituents on aromatic
rings to guide substitution based on biological results. Traditionally, the Topliss protocol is a linear
process in that the 4-chloro analog is made and its activity compared to the parent aromatic.
Comparison of the activity of the new analog (ie less, equal or more active) to the parent dictates
which part of the tree is examined. Thus, if the activity was higher, the 3,4-dichloro analog is
made and the process repeated. However, further work by Topliss indicated that preparing the 4-
chloro analog and the first tier of analogs can be more efficient at learning what to synthesise
next.[55] Therefore, for the purpose of this study it was decided to synthesise the 4-chloro analog
3.78 and the first tier (4-methoxy 3.79, 4-methyl 3.80 and 3,4-dichlorobenzyl 3.81 analogs) of the
Topliss tree.

Chapter 3 – AAL(S)
122
While the chemistry discussed in Section 3.2.2 could be used to synthesise these four analogs it
would require a total of 12 steps from advanced TBS-protected intermediate 3.49 (Figure 3.39). It
was therefore decided to examine the more convergent pathway (Strategy 2) that was briefly
discussed in Section 3.2.2.
Figure 3.39: Number of steps required to synthesise 4 benzyl hydrophobic tail analogs using the previous strategy.
Hinterding and co-workers had shown that protection of the amino group of AAL(R) (3.4) with the
Boc protecting group allowed alkylation of the phenol moiety to be achieved.[47] Thus, efforts were
focussed on synthesising amine-protected phenol 3.82 (Figure 3.40).
Figure 3.40: Retrosynthesis of benzyl-hydrophobic tail analogs of AAL(S).
As a considerable amount of TBS-bislactim ether 3.49 had been stockpiled this was investigated
as a starting material to access phenol 3.82, but this proved not to be successful. i A new
protecting group was needed for the phenol that could withstand the conditions required to
remove the Schöllkopf reagent. It was considered that the benzyl group could fulfil this task as the
benzyl analog 3.57 had already been synthesised via this route. To streamline access to the
benzyl analog 3.57 the TBS-iodotyrosol 3.48, used in the first generation synthesis, was
substituted with a Bn-iodotyrosol 3.83 in the opening sequence which resulted in Bn-amino
alcohol 3.57 being synthesised in 24 % yield over 7 steps, as shown in Scheme 3.13. The 2-
amino group was subsequently protected with the Boc protecting group and the benzyl group
removed using H2 and 10 % Pd/C in MeOH to afford phenol 3.82 in 57 % yield over the two
steps. Phenol 3.82 could be selectively alkylated with the four benzyl bromides chosen as part of
this study in the presence of K2CO3 in DMF. Deprotection of the Boc protecting group using
methanolic HCl was efficient at generating the 4-chloro 3.78, 4-methyl 3.80 and 3,4-dichloro
benzyl 3.81 analogs. However, the 4-methoxybenzyl analog 3.79 did not survive the acid
i Jaqueline Liu, a third year summer project student working in the group, examined the chemistry of this, trying a deprotection of the TBS ether on the TBS bis-lactim ether 3.49, followed by hydrolysis and the alternate strategy of hydrolysis of the lactim, then deprotection. While deprotection of the TBS group could be carried out, hydrolysis of the lactim, in the presence of the free phenol, was problematic, and a complex mixture was obtained. Using the hydrolysis conditions on this substrate also resulted in a complex mixture of products.

Chapter 3 – AAL(S)
123
deprotection step.[57] This was perhaps not that surprising as the 4-methoxybenzyl group is also
used as a protecting group which can be cleaved under acidic conditions. While this analog could
be synthesised by modification of the Boc group to an alternative protecting group earlier in the
synthesis, the subsequent biological data of the three analogs obtained indicated that synthesis
of this analog was unnecessary.
Final Analog R = (g and h) Yield (%)
3.78 4-Cl 51a
3.79 4-OMe - 3.80 4-Me 49a
3.81 3,4-Cl2 57
Scheme 3.13: Reagents and yields (a) BnBr, K2CO3, MeOH, rt, 97 %; (b) MsCl, NEt3, CH2Cl2, 0°C → rt; (c) NaI,
Me2CO, rt, 68 %; (d) n-BuLi, THF, -78°C, then 3.83, -78°C → 0°C, 92 %; (e) n-BuLi, THF, -78°C, then MeI, -78°C
→ 0°C, 67 %; (f) see Scheme 3.7; (g) see Scheme 3.7; (h) Boc2O, sat. aq. NaHCO3, EtOAc, Δ, 91 %; (b) H2, 10 %
Pd/C, MeOH, rt, 63 %; (c) R-C6H4CH2-X, K2CO3, DMF, rt; (d) 2M HCl in MeOH, Δ, see table for yield over two steps. a Synthesised by Stephen Butler, a second year CHEM3998 student working in the group.
This procedure is currently used to synthesise hydrophobic tail analogs of AAL(S). The phenol
3.82 can be synthesised on a gram scale and in just two more steps, which can be completed in
one-pot, the hydrophobic tail analogs can be generated.
These compounds were tested for their activity against CerS1 at 1 μM, as shown in Figure 3.41.
Following the Topliss scheme logic, the 4-chlorobenzyl analog 3.78 has better activity than 3.57
which would indicate that the 3,4-dichlorobenzyl analog 3.81 should be the next synthesised in
the series. It was therefore not surprising that the 4-methylbenzyl analog 3.80 had slightly poorer
activity than 3.57. Pleasingly, the 3,4-dichlorobenzyl analog 3.81 had improved activity compared
to 3.57 and was considerably more active than AAL(S) (1.37).

Chapter 3 – AAL(S)
124
Figure 3.41: Untransfected HEK293 cell lysates treated with 1 μM AAL(S) (1.37) or AAL(S) benzyl analogs, 10 μM
NBD-sphingosine and 50 μM C18:0-CoA. Products formed were quantified by TLC with a fluorescent imager and
normalised relative to the control as described.[53] (Data courtesy of Dr. Anthony Don, Lowy Cancer Research
Centre).
The CerS1 activity data collected for these analogs indicates that the benzyl aromatic group of
AAL(S) follows either a π, 2π-π2, σ or π+σ-dependency according to Topliss’s scheme (Table
3.2).[55] It is not possible to be more precise due to the lack of data for the 4-methoxybenzyl
analog 3.79. However, in the 1977 Topliss paper, a series of new analogs are suggested. This
work is currently the focus of another PhD student in the Morris group, Elysha Taylor.
Table 3.2: Potency order for various aromatic parameter dependencies. Highlighted in yellow are those series that
the benzyl hydrophobic tail group of AAL(S) seems to follow.[55]
Parameters
Substituents π 2π-π2 σ -σ π+σ 2π-σ π-σ π-2σ π-3σ E4α
3,4-Cl2 1 1-2 1 5 1 1 1-2 3-4 5 2-5
4-Cl 2 1-2 2 4 2 2-3 3 3-4 3-4 2-5
4-Me 3 3 4 2 3 2-3 1-2 1 1 2-5
4-OMe 4-5 4-5 5 1 5 4 4 2 2 2-5
H 4-5 4-5 3 3 4 5 5 5 3-4 1
0
10
20
30
40
50
60
70
80
AA
L(S
)
3.57
3.78
3.8O
3.81
Cer
S1
acti
vity
(%
rel
ativ
e to
co
ntr
ol)
Compound

Chapter 3 – AAL(S)
125
3.5. Determining the Protein Target of AAL(S)
The biological data presented in Section 3.4. has given some insight into the structural
requirements essential for AAL(S)’s cytotoxic activity. The mechanism of how it exerts this activity
is still unknown. As elucidation of this is essential for the development of new improved analogs,
it was proposed to use affinity chromatography.
3.5.1. Synthesis and Evaluation of an AAL(S) Affinity Chromatography Probe
Affinity chromatography is a technique which can be used to determine all of the intermolecular
binding partners of a small molecule from a cell extract. This technique was discussed in Section
1.2.3 (Figure 1.8) for its application in determining the intermolecular binding partners of FK506
(1.6). To apply this technique to AAL(S) (1.37) a derivatised AAL(S) molecule would have to be
synthesised which would be attached via a linker moiety to either a biotin molecule, to be used
with streptavidin to ‘pull-down’ the protein of interest, or covalently to a solid support.[58]
Recent methodology developed by Finn and co-workers has shown that the solid phase approach
can be performed using the click reaction to couple an agarose bead to a molecule of interest for
affinity chromatography.[59] Moreover, the methodology they have developed allows for the
amount of compound attached to the solid support to be quantified. This is achieved by utilising
either a p-nitrophenoxide leaving group in a reagent (releasing 3.84) or Fmoc protecting group
which, when removed, yields fluorene (3.85). These groups can be detected using a UV visible
spectrometer whereby, after completion of reaction, the reaction solution can be removed from
the column and the absorbance measured. The absorbance detected can be directly correlated,
using the Beer-Lambert law, to the amount of released chromophore which is proportional to the
amount of compound loaded onto the solid support. This is demonstrated using the example
reported by Finn and co-workers who attached the C-terminal peptide of a HIV protease substrate
to a solid support for affinity chromatography using this methodology, as shown in Scheme 3.14.

Chapter 3 – AAL(S)
126
Scheme 3.14: Reagents and yields (a) 1:20 pH 4.7 0.1 M aq. MES Buffer/DMF, rt; 20 – 30 % (b) 2,6-lutidine, 2,2-
bipyridyl, CuBr, sodium ascorbate, DMF, rt; (c) 1:5 piperidine/DMF, rt, 39 % (2 steps).[59]
To apply this methodology to our system an appropriately derivatised azide/alkyne AAL(S) and
complementarily functionalised solid support were required. Key considerations when designing
these coupling partners were the position to derivatise/attach the solid support to AAL(S) (1.37)
and the hydrophobicity and length of the linker moiety. As the head group of AAL(S) was
considered important for biological activity, attachment from either the aromatic ring or the
hydrophobic tail was considered. At the time that this work was initiated in this PhD project the
first generation hydrophobic tail synthesis had just been completed. Consequently, it was chosen
to synthesise a terminal-alkyne hydrophobic tail analog as this would be compatible with this
synthesis. With this decided, a complementary terminal azide-solid support was required. The
linker moiety has been found to drastically influence the success of an affinity chromatography
project. As shown in Figure 3.42, if the linker is too short the drug-target interaction might be
susceptible to undesired steric hindrance.[58, 60] Conversely, if the linker is too hydrophobic,
particularly under aqueous incubation conditions, the linker may aggregate, preventing binding
and therefore detection of the binding partners.

Chapter 3 – AAL(S)
127
Figure 3.42: Factors that are important to consider when choosing a solid support-drug linker for affinity
chromatography.[60]
To circumvent potential problems regarding the linker, two terminal azide linker-solid supports
were proposed. These were (C5)-3.86 and (PEG4)-3.86. The PEG4-linker was chosen as a
hydrophilic linker which would prevent aggregation. However, as the biological data for the
hydrophobic tail analogs of AAL(S) found that this group must be hydrophobic a C5-linker was
also chosen as this may be better suited to the AAL(S) binding pocket. Additionally, synthesis of a
negative control bead 3.87, where the head group of AAL(S) was completely removed, was also
proposed. Our retrosynthesis for these affinity chromatography probes is shown in Figure 3.43.
Figure 3.43: Retrosynthesis for AAL(S) affinity chromatography probes.
The requisite terminal azide-linker agarose beads 3.86 were synthesised using the protocol
described by Finn and co-workers, as shown in Figure 3.44.[59] While the yields obtained for the
coupling of agarose bead 3.88 with both p-nitrophenylester linkers 3.89 were comparable to those
obtained by Finn, Bach and co-workers have reported that the optimal loading of a drug onto a
solid support for affinity chromatography is 3 μmol of drug/mL of solid support.[60] Assuming that
the subsequent click reaction would also proceed in similar yield to the example of Finn’s in

Chapter 3 – AAL(S)
128
Scheme 3.14 (39 %), this would mean a loading of ~ 1 - 2 μmol drug/mL onto the solid support. It
was therefore decided to repeat the procedure on each column to increase the loading of azide.
This second procedure proceeded almost identically to the first and resulted in a total yield of 48
% (9.69 μmol/mL) for (C5)-3.86 and 65 % (12.85 μmol/mL) for (PEG4)-3.86.
Linker (a) Yield (%) 1(b) Yield (%) 2(b) Yield (%) Total N3 (%) a
-(CH2)4- (C5) 75 25 23 48 (9.69 μmol/mL) -(CH2CH2O)4- (PEG4) 65 36 29 65 (12.85 μmol/mL)
Figure 3.44: Synthesis of azide terminal agarose beads. Images of the Carboxylink columns before and after
reaction with p-nitrophenylester linkers 3.89 showing release of the p-nitrophenoxide fluorophore 3.84. Reagents and
yields (a) 4-nitrophenol, EDCI·HCl, CH2Cl2, rt, see table for yield; (b) (C5)-3.89 or (PEG4)-3.89, 1:20 pH 4.7 0.1 M
aq. MES buffer/DMF, rt, see table for yield. a Total azide loaded onto the agarose beads after two iterations of (b).
To synthesise the requisite alkyne-AAL(S) precursor 3.90, access to the terminal-alkyne
hydrophobic tail was required. This was synthesised in three steps from commercially available
alcohol 3.91 (Scheme 3.15). The first step in the sequence was a zipper reaction.[61] This reaction
typically uses a strong base, such as sodium 2-aminoethylamide in 1,2-ethylenediamine, to
isomerise the position of an internal triple bond. The isomerisation reaction proceeds through an
allene intermediate which is generated by simultaneous deprotonation of a CH2 group adjacent
the triple bond and protonation of the opposite end of the triple bond. The reaction exists in an
equilibrium which terminates when the anion reaches the terminal position of the alkyl chain
which is thermodynamically stable. The resulting alcohol 3.92 was converted to the mesylate
which was substituted with sodium iodide to afford iodide 3.93 in 62 % yield over the three steps.

Chapter 3 – AAL(S)
129
Scheme 3.15: Reagents and yields (a) NaH, H2N(CH2)2NH2, 80°C, 88 %; (b) MsCl, NEt3, CH2Cl2, 0°C → rt; (c) NaI,
Me2CO, rt, 70 %; (d) 3.93, CsF, DMF, rt, 87 %; (e) TFA, H2O, MeCN, rt, 82 %; (f) LiAlH4, THF, 0°C → rt, 77 %; (g)
FmocCl, sat. aq. NaHCO3, EtOAc, Δ, 63 %; (h) 3.96, CuI, TBTA, DIPEA, DMF, rt, 53 %.
With this in hand the protocol developed in Section 3.2.2. was employed to synthesise the
requisite terminal-alkyne AAL(S) analog 3.94. Proof that this had been synthesised with the
terminal-alkyne group intact was evident by the diagnostic triplet at 1.94 ppm (J = 2.6 Hz)
representing the terminal alkyne proton in the 1H NMR spectrum of the purified material.
Additionally, the infrared spectrum had an absorbance at 2115 cm-1 representing the triple bond.
The amine group was protected with the Fmoc group using FmocCl in saturated aqueous
NaHCO3 and ethyl acetate at reflux to afford the click precursor 3.90 in 35 % yield in five steps
from TBS-bis lactim ether 3.49.
Because of the expense invested in the terminal-azide solid support beads it was thought that a
model substrate, where the click reaction had been carried out, should be synthesised to see if
this retained biological activity. Synthesis of the model click substrate 3.95 using equimolar
amounts of both the alkyne AAL(S) analog 3.94 and PEG2-azide 3.96, 10 mol % CuI and DIPEA
in DMF at rt resulted in only 30 % conversion of the starting materials, with 15 % of the desired
triazole 3.95 could be isolated. It was thought that this could be due to the amino alcohol moiety
coordinating to the copper(I) species. To rectify this tris(benzyltriazoloylmethyl)amine (TBTA) was
employed as a ligand as this has been reported to help stabilise the copper(I) species in the
reaction.[62] Using the conditions stated above, but with TBTA present resulted in complete
consumption of the starting material and the desired triazole 3.95 could be isolated in 54 % yield
(Scheme 3.15). Triazole 3.95 was tested in the cytotoxicity assays and PP2A activation assay
that were discussed in Section 3.3. The data is shown in Figure 3.45 and shows that although
triazole 3.95 did not exhibit any cytotoxicity against both the EV and D816V myeloid cells (up to 9
μM) it was still able to activate PP2A in vitro.
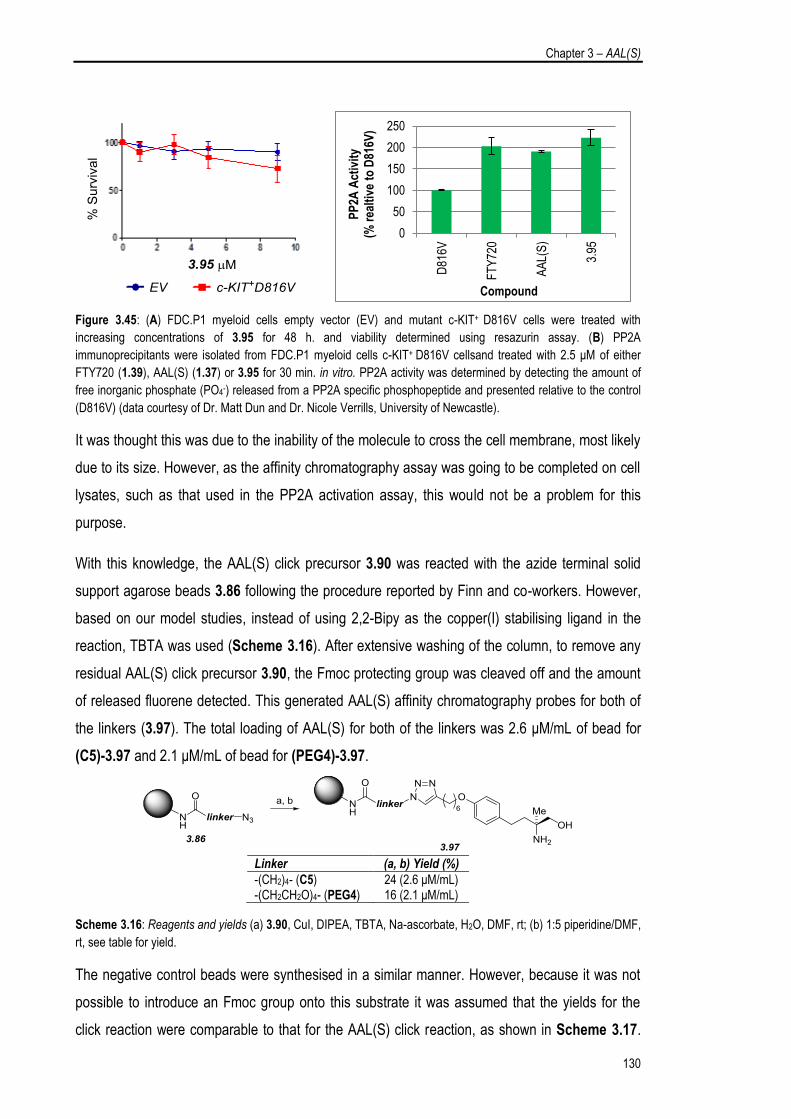
Chapter 3 – AAL(S)
130
Figure 3.45: (A) FDC.P1 myeloid cells empty vector (EV) and mutant c-KIT+ D816V cells were treated with
increasing concentrations of 3.95 for 48 h. and viability determined using resazurin assay. (B) PP2A
immunoprecipitants were isolated from FDC.P1 myeloid cells c-KIT+ D816V cellsand treated with 2.5 μM of either
FTY720 (1.39), AAL(S) (1.37) or 3.95 for 30 min. in vitro. PP2A activity was determined by detecting the amount of
free inorganic phosphate (PO4-) released from a PP2A specific phosphopeptide and presented relative to the control
(D816V) (data courtesy of Dr. Matt Dun and Dr. Nicole Verrills, University of Newcastle).
It was thought this was due to the inability of the molecule to cross the cell membrane, most likely
due to its size. However, as the affinity chromatography assay was going to be completed on cell
lysates, such as that used in the PP2A activation assay, this would not be a problem for this
purpose.
With this knowledge, the AAL(S) click precursor 3.90 was reacted with the azide terminal solid
support agarose beads 3.86 following the procedure reported by Finn and co-workers. However,
based on our model studies, instead of using 2,2-Bipy as the copper(I) stabilising ligand in the
reaction, TBTA was used (Scheme 3.16). After extensive washing of the column, to remove any
residual AAL(S) click precursor 3.90, the Fmoc protecting group was cleaved off and the amount
of released fluorene detected. This generated AAL(S) affinity chromatography probes for both of
the linkers (3.97). The total loading of AAL(S) for both of the linkers was 2.6 μM/mL of bead for
(C5)-3.97 and 2.1 μM/mL of bead for (PEG4)-3.97.
Linker (a, b) Yield (%)
-(CH2)4- (C5) 24 (2.6 μM/mL) -(CH2CH2O)4- (PEG4) 16 (2.1 μM/mL)
Scheme 3.16: Reagents and yields (a) 3.90, CuI, DIPEA, TBTA, Na-ascorbate, H2O, DMF, rt; (b) 1:5 piperidine/DMF,
rt, see table for yield.
The negative control beads were synthesised in a similar manner. However, because it was not
possible to introduce an Fmoc group onto this substrate it was assumed that the yields for the
click reaction were comparable to that for the AAL(S) click reaction, as shown in Scheme 3.17.
0
50
100
150
200
250
D81
6V
FT
Y72
0
AA
L(S
)
3.95
PP
2A A
ctiv
ity
(% r
ealt
ive
to D
816V
)
Compound

Chapter 3 – AAL(S)
131
The biological experiments using the affinity chromatography beads synthesised were carried out
by Dr. Matt Dun and Dr. Nicole Verrills at the University of Newcastle. Because the author didn’t
do this work this will not be discussed in great detail in this thesis. However, there are some key
findings and implications of the affinity chromatography results that will be discussed.
Scheme 3.17: Reagents and yields (a) 3.93, K2CO3, DMF, rt, 66 %; (b) 3.98, CuI, DIPEA, TBTA, Na-ascorbate, H2O,
DMF, rt, yield not determined.
A simplified example of a typical experimental procedure with the affinity chromatography beads
involves incubation of both the negative control 3.99 and AAL(S) affinity chromatography beads
3.97 with the cell lysates from both EV and D816V cells. The cellular protein extracts from both of
these cell types were used simultaneously so that direct comparisons could be made to identify
how PP2A is reactivated by AAL(S) in the D816V acute myeloid leukaemia (AML) model cell line
compared to the normal cells (EV). The negative control beads were used to identify proteins that
bound non-specifically to the solid support matrix. After incubation, the beads were washed
thoroughly, to remove any components that were not specifically bound to the beads. Bound
components were eluted off the beads using increasing concentrations of AAL(S) (1.37). The
filtrates from these washings were collected and subjected to sodium dodecyl sulphate-
polyacrylamide gel electrophoresis (SDS-PAGE) to separate the components by their molecular
weight.
A representative SDS-PAGE gel obtained from one of these experiments using the PEG4 affinity
chromatography beads (PEG4)-3.97 and (PEG4)-3.99 is shown in Figure 3.46. It should be
noted that the C5 affinity chromatography linker (C5)-3.97 and (C5)-3.99 gave similar results to
the PEG4 linker. Lanes 1 (negative control (PEG4)-3.99) and 2 (AAL(S) (PEG4)-3.97) in Figure
3.46A represent beads incubated with the EV cell lysates and lanes 3 (negative control (PEG4)-
3.99) and 4 (AAL(S) (PEG4)-3.97) represent proteins isolated from D816V AML cell lysates.
Comparison of the lanes using the AAL(S) beads with the EV cells (lane 2) and the D816V cells
(lane 4) revealed that one band (at 28 kDa) was observed only in the D816V cell lysates.
Subsequent isolation, reduction, alkylation, digestion and microsequencing of this protein by
liquid chromatography mass spectrometry (LCMS/MS) identified it as a protein whose function is

Chapter 3 – AAL(S)
132
relatively understudied. However, its aberrant expression has previously suggested it as a cancer
predisposing disorder. Unfortunately, at the time that this thesis was printed patenting
implications prevented disclosure of the real name of this protein and so, it will be identified in the
text as AAL(S)-binding protein (AAL(S)-BP). As the protein is known, an antibody (which we will
refer to as anti-AAL(S)-BP) had already been generated and this could be used in a Western blot
to further confirm the identity of the AAL(S)-binding protein (Figure 3.46B). The sensitivity of the
Western blot revealed that low levels of AAL(S)-BP are also present in the EV cells. However,
there is substantial enrichment of the protein in D816V cells AAL(S) bead pull down.
Figure 3.46: (A) Affinity chromatography results using PEG4-linker AAL(S) beads (PEG4)-3.96 and PEG4-linker
negative control beads (PEG4)-3.98. Lanes 1 and 2: EV cell lysate incubated with affinity chromatography beads.
Lanes 3 and 4: D816V cell lysate incubated affinity chromatography beads. All targets eluted with 2.5 μM AAL(S) in
activity buffer. (B) Western blot confirming the LCMS result (anti-AAL(S)-BP).
Having identified that AAL(S)-BP was an intermolecular target of AAL(S) (1.37), further
experiments were undertaken to determine whether it was this interaction that was responsible
for AAL(S)’s cytotoxic activity. Firstly, Dun and Verrills wanted to determine if AAL(S)-BP
interacted with PP2A. This was achieved by conducting an AAL(S)-BP co-immunoprecipitation
experiment using a non-reduced D816V cell lysate. The results identified several proteins that
interact with AAL(S)-BP, as shown in Figure 3.47. Again, these were isolated and identified using
LC-MS/MS to determine their identity. Excitingly, of the proteins identified these included the
A
B

Chapter 3 – AAL(S)
133
catalytic subunit of PP2A (PP2A-C), the structural subunit (PP2A-A) and a regulatory subunit of
PP2A (PP2A-B55α), clearly indicating that AAL(S)-BP binds to PP2A. Identification of the
individual subunits was confirmed by using each of their respective antibodies in a Western blot
(Figure 3.47).
Figure 3.47: (A) Co-immunoprecipitation (coIP) of AAL(S)-BP intramolecular binding partners in D816V cells using
anti-AAL(S)-BP Gt pAB Lane 1: D816V cell lysate. Lane 2: negative control. Lane 3: results obtained from AAL(S)-
BP coIP, includes proteins identified from LCMS and sequencing. (B) Western blot confirming the LCMS results.
PP2A-B56α subunit was included as a negative control.
To further validate the AAL(S)-AAL(S)-BP interaction it was decided to apply the affinity
chromatography methodology developed to FTY720 (1.39), to examine whether AAL(S)-BP is a
common target for these molecules. Clearly, if this was the case then AAL(S)-BP may be a novel
target which, when overexpressed has implications in cancer.
3.5.2. Synthesis and Evaluation of an FTY720 Affinity Chromatography Probe
Given the success of the AAL(S) affinity chromatography beads 3.97 it was decided to apply this
same protocol to FTY720 (1.39) to determine if it also bound AAL(S)-BP. To apply our protocol to
FTY720 would require access to FTY720 click precursor 3.100. It was initially envisaged that this
could be synthesised from alkyne 3.101 using a zipper reaction which could be obtained from a
Sonagashira reaction with iodide 3.102 and 1-octyne (Figure 3.48).

Chapter 3 – AAL(S)
134
Figure 3.48: Proposed retrosynthesis of FTY720 affinity chromatography click precursor 3.100.
Initial investigation into this sequence, attempting to synthesise the terminal alkyne required for
the complementary negative control affinity chromatography beads were unsuccessful. While the
Sonagashira reaction between iodobenzene (3.103) and 1-octyne successfully afforded alkyne
3.104, all attempts at the zipper reaction resulted in re-isolation of the starting material along with
several other unidentifiable products (Scheme 3.18). This was perhaps not surprising as there
are no reports of zipper reactions of alkynes next to aromatic groups reported in the literature.
This is possibly due to the triple bond being strongly conjugated with the aromatic group.
Scheme 3.18: Reagents and yields (a) 1-octyne, CuI, Pd(PPh3)4, NEt3, rt, 88 %; (b) NaH, H2N(CH2)2NH2, 80°C.
This result suggested that the synthesis of an FTY720 affinity chromatography probe was not
going to be a trivial process. While it was not unreasonable to design another route to access this
material it was decided to synthesise an FTY720/AAL(S) hybrid probe 3.105 which could be
synthesised similarly to AAL(S) (Figure 3.49).
Figure 3.49: Proposed retrosynthesis of O-FTY720 affinity chromatography click precursor 3.105.
Again, before synthesising the affinity chromatography probe, the O-FTY720 analog 3.106 was
synthesised to make sure it retained the biological activity of FTY720 (1.39). This compound has
been previously prepared in the literature and so, this general protocol was followed.[50] The
synthesis was modified to incorporate the TBS one-pot deprotection/alkylation strategy that was
developed in Section 3.2.2. so that other hydrophobic tail analogs, including the required alkyne
hydrophobic tail could be installed later in the synthetic sequence. Thus, TBS-iodide 3.48 was
alkylated with diethyl 2-acetamidomalonate 3.107 in the presence of NaH in DMF at room
temperature to afford malonate 3.108 in 70 % yield. One-pot deprotection of the TBS group and
alkylation with 1-bromoheptane successfully installed the hydrophobic tail before the malonate
head group was converted into the dihydroxy amino head group of FTY720 using the literature

Chapter 3 – AAL(S)
135
reaction sequence.[50] This procedure afforded O-FTY720 (3.106) in 41 % overall yield over 4
steps (Scheme 3.19).
Scheme 3.19: Reagents and yields (a) NaH, DMF, rt, then 3.48, rt, 70 %; (b) CsF, H15C7Br, DMF, rt, 94 %; (c) LiAlH4,
THF, 0°C → rt; (d) conc. HCl, EtOH, Δ, 62 % ( 2 steps).
O-FTY720 (3.106) was tested on K562 leukaemia cells by Dr. Anthony Don, as shown in Figure
3.50. While this analog was not as effective as FTY720 (1.39) at killing these cells it was
comparable to AAL(S) (1.37). The difference between FTY720 (1.39) and O-FTY720 (3.106) is
quite intriguing but nevertheless the O-FTY720 substrate was deemed satisfactory to be used in
our affinity chromatography assay to see if this compound also associates with AAL(S)-BP.
Figure 3.50: K562 cells were treated with 3 μM or 10 μM of either FTY720 (1.39), AAL(S) (1.37) or O-FTY720
(3.106) for 24 h. and viability determined using Annexin V/PI assay, n = 3 (data courtesy of Dr. Anthony Don, Lowy
Cancer Research Centre).
The O-FTY720 affinity chromatography probe 3.108 was synthesised in six steps using the
methodology discussed previously (Figure 3.51). This chemistry proceeded uneventfully.
Utilisation of the probe in the affinity chromatography experiments revealed that this compound
also associates with AAL(S)-BP which was confirmed by Western blot using anti-AAL(S)-BP.
Interestingly, a biotin-FTY720 probe has recently been reported by Ogretmen and co-workers to
pull down I2PP2A/SET, a complex which our AAL(S) affinity chromatography beads also pulled
down. However, the experiment was not completed on whole cell lysates and only with purified
I2PP2A/SET complexes and so the interaction with AAL(S)-BP was not detected.[63]
0
10
20
30
40
50
60
70
80
90
FTY720 AAL(S) O-FTY720
Ce
ll V
iab
ility
(%
)
Compound
3 μM 10 μM

Chapter 3 – AAL(S)
136
Figure 3.51: Synthesis of O-FTY720 affinity chromatography bead and western blot with anti-AAL(S)-BP confirming
that this bead also binds to AAL(S)-BP Reagents and yields (a) 3.93, CsF, H15C7Br, DMF, rt, 82 %; (b) LiAlH4, THF,
0°C → rt; (c) conc. HCl, EtOH, Δ, 62 % ( 2 steps); (d) FmocCl, sat. aq. NaHCO3, EtOAc, Δ, 44 %; (e) (PEG4)-3.86,
CuI, DIPEA, TBTA, Na-ascorbate, H2O, DMF, rt; (f) 1:5 piperidine/DMF, rt, 45 % (2 steps).
The data collected by Dun and Verrills using our affinity chromatography beads shows strong
evidence that AAL(S)-BP may be a novel target for cancer therapeutics. Preliminary work has
identified that the interaction between AAL(S)-BP and PP2A renders PP2A catalytically inactive
(data not shown). At this stage, it is speculated that AAL(S) (1.37) exerts its cytotoxic biological
activity by binding AAL(S)-BP in a manner that disrupts its interaction with PP2A. The current
focus of this work is aiming to biologically up-regulate AAL(S)-BP in EV cells (by retroviral
transduction of cells with an AAL(S)-BP expression vector) to see if this leads to the inactivation
of PP2A and induces cancer commensurate. This is being done in conjunction with the
downregulation of AAL(S)-BP in D816V cells (using short hairpin RNA’s) to see if this restores
PP2A activity resulting in PP2A mediated programmed cell death.

Chapter 3 – AAL(S)
137
3.6. Chapter Summary
In conclusion, chemical synthesis has been used to investigate AAL(S) as a new lead for the
treatment of leukaemia. This was achieved in two ways. Firstly, synthetic protocols were
designed and implemented to access analogs of AAL(S). The analogs that have been prepared
have been evaluated for their PP2A activation activity, anti-leukaemia activity as well as their
activity against specific ceramide synthases. Some analogs have been found to have improved
activity to AAL(S) and the data obtained has helped add to our structure activity relationship study
of AAL(S). Secondly, to determine AAL(S)’s mode of action, an AAL(S)-alkyne analog was
prepared and ‘clicked’ to an agarose bead for affinity chromatography. This probe was used to
identify a protein, called AAL(S)-BP, which shows affinity for AAL(S) in leukaemia cells compared
to healthy bone marrow cells. An O-FTY720 affinity chromatography probe was synthesised and
further confirmed that AAL(S)-BP is a common target for these molecules. Furthermore, Dun and
Verrills have examined patients with acute myeloid leukaemia to see if AAL(S)-BP is
overexpressed in these cells. The results, found in Figure 3.52, identified that AAL(S)-BP was
significantly upregulated in 9 out of the 12 patient samples that were examined. Importantly, a
further investigation has found that AAL(S)-BP interacts directly with PP2A in vitro. This data
indicates that AAL(S)-BP may be a novel target which, when overexpressed, has implications in
cancer.
Figure 3.52: Western blot of acute myeloid leukaemia patients sample’s whole cell lysates probed with anti-AAL(S)-
BP.

Chapter 3 – AAL(S)
138
3.7. References for Chapter 3
[1] J. A. Cohen, F. Barkhof, G. Comi, H.-P. Hartung, B. O. Khatri, X. Montalban, J. Pelletier, R. Capra, P. Gallo, G. Izquierdo, K. Tiel-Wilck, A. de Vera, J. Jin, T. Stites, S. Wu, S. Aradhye, L. Kappos, N. Engl. J. Med. 2010, 362, 402-415.
[2] T. Fujita, K. Inoue, S. Yamamoto, T. Ikumoto, S. Sasaki, R. Toyama, M. Yoneta, Y. Hoshino, T. Okumoto, Journal of Antibiotics 1994, 47, 208-215.
[3] K. Adachi, K. Chiba, Perspectives in Medicinal Chemistry 2007, 1, 11-23. [4] T. Fujita, N. Hamamichi, M. Kiuchi, T. Matsuzaki, Y. Kitao, K. Inoue, R. Hirose, M.
Yoneta, S. Sasaki, K. Chba, Journal of Antibiotics 1996, 49, 846-853. [5] C. R. Strader, C. J. Pearce, N. H. Oberlies, J. Nat. Prod. 2011, 74, 900-907. [6] T. Fujita, R. Hirose, M. Yoneta, S. Sasaki, K. Inoue, M. Kiuchi, S. Hirase, K. Chiba, H.
Sakamoto, M. Arita, J. Med. Chem. 1996, 39, 4451-4459. [7] V. Brinkmann, A. Billich, T. Baumruker, P. Heining, R. Schmouder, G. Francis, S.
Aradhye, P. Burtin, Nat. Rev. Drug Discov. 2010, 9, 883-897. [8] V. Brinkmann, M. D. Davis, C. E. Heise, R. Albert, S. Cottens, R. Hof, C. Bruns, E.
Prieschl, T. Baumruker, P. Hiestand, C. A. Foster, M. Zollinger, K. R. Lynch, J. Biol. Chem. 2002, 277, 21453-21457.
[9] M. Kiuchi, K. Adachi, T. Kohara, K. Teshima, Y. Masubuchi, T. Mishina, T. Fujita, Bioorg. Med. Chem. Lett. 1998, 8, 101-106.
[10] E. Jary, T. Bee, S. R. Walker, S. Chung, K. Seo, J. C. Morris, A. S. Don, Mol. Pharm. 2010, 78, 685 - 692.
[11] M. Kiuchi, K. Adachi, A. Tomatsu, M. Chino, S. Takeda, Y. Tanaka, Y. Maeda, N. Sato, N. Mitsutomi, K. Sugahara, K. Chiba, Bioorg. Med. Chem. 2005, 13, 425-432.
[12] R. Albert, K. Hinterding, V. Brinkmann, D. Guerini, C. Müller-Hartwieg, H. Knecht, C. Simeon, M. Streiff, T. Wagner, K. Welzenbach, F. Zécri, M. Zollinger, N. Cooke, E. Francotte, J. Med. Chem. 2005, 48, 5373-5377.
[13] a) S. Mandala, R. Hajdu, J. Bergstrom, E. Quackenbush, J. Xie, J. Milligan, R. Thornton, G.-J. Shei, D. Card, C. Keohane, M. Rosenbach, J. Hale, C. L. Lynch, K. Rupprecht, W. Parsons, H. Rosen, Science 2002, 296, 346-349; b) J. J. Hale, L. Yan, W. E. Neway, R. Hajdu, J. D. Bergstrom, J. A. Milligan, G.-J. Shei, G. L. Chrebet, R. A. Thornton, D. Card, M. Rosenbach, H. Rosen, S. Mandala, Bioorg. Med. Chem. 2004, 12, 4803-4807.
[14] V. Brinkmann, J. G. Cyster, T. Hla, American Journal of Transplantation 2004, 4, 1019-1025.
[15] P. J. A. Eichhorn, M. P. Creyghton, R. Bernards, Biochimica et Biophysica Acta 2009, 1795, 1-15.
[16] C. Bialojan, A. Takai, Biochem. J. 1988, 256, 283-290. [17] a) P. Neviani, R. Santhanam, J. J. Oaks, A. M. Eiring, M. Notari, B. W. Blaser, S. Liu, R.
Trotta, N. Muthusamy, C. Gambacorti-Passerini, B. J. Druker, J. Cortes, G. Marcucci, C.-S. Chen, N. M. Verrills, D. C. Roy, M. A. Caligiuri, C. D. Bloomfield, J. C. Byrd, D. Perotti, J. Clin. Invest. 2007, 117, 2408-2421; b) K. G. Roberts, A. M. Smith, F. McDougall, H. Carpenter, M. Horan, P. Neviani, J. A. Powell, D. Thomas, M. A. Guthridge, D. Perrotti, A. T. R. Sim, L. K. Ashman, N. M. Verrills, Cancer Res. 2010, 70, 5438-5447.
[18] I. Cristobal, L. Garcia-Orti, C. Cirauqui, M. M. Alonso, M. J. Calasanz, M. D. Odero, Leukemia 2011, 25, 606-614.
[19] P. Neviani, R. Santhanam, R. Trotta, M. Notari, B. W. Blaser, S. Liu, H. Mao, J. S. Chang, A. Galietta, A. Uttam, D. C. Roy, M. Valtieri, R. Bruner-Klisovic, M. A. Caligiuri, C. D. Bloomfield, G. Marcucci, D. Perrotti, Cancer Cell 2005, 8, 355-368.
[20] M. S. Feschenko, E. Stevenson, A. C. Nairn, K. J. Sweadner, J. Pharmacol. Exp. Ther. 2002, 302, 111-118.

Chapter 3 – AAL(S)
139
[21] Y. Matsuoka, Y. Nagahara, M. Ikekita, T. Shinomiya, Br. J. Pharmacol. 2003, 138, 1303-1312.
[22] N. Zhang, Y. Qi, C. Wadham, L. Wang, A. Warren, W. Di, P. Xia, Autophagy 2010, 6, 1157-1167.
[23] a) C. T. Wallington-Beddoe, J. Hewson, K. F. Bradstock, L. J. Bendall, Autophagy 2011, 7, 707-715; b) Y. Yang, Q. Huang, Y. Lu, X. Li, S. Huang, J. Cell. Biochem. 2012, 113, 1314-1322.
[24] Q. Liu, X. Zhao, F. Frissora, Y. Ma, R. Santhanam, D. Jarjoura, A. Lehman, D. Perrotti, C.-S. Chen, J. T. Dalton, N. Muthusamy, J. C. Byrd, Blood 2008, 111, 275-284.
[25] A. Collison, L. Hatchwell, N. Verrills, P. A. B. Wark, A. P. de Siqueira, M. Tooze, H. Carpenter, A. S. Don, J. C. Morris, N. Zimmermann, N. W. Bartlett, M. E. Rothenberg, S. L. Johnston, P. S. Foster, J. Mattes, Nat Med 2013, 19, 232-237.
[26] a) U. Schöllkopf, U. Groth, C. Deng, Angew. Chem. Int. Ed. 1981, 20, 798-799; b) U. Schöllkopf, Tetrahedron 1983, 39, 2085-2091.
[27] K. Hinterding, R. Albert, S. Cottens, Tetrahedron Lett. 2002, 43, 8095 - 8098. [28] K. Hinterding, R. Albert, S. Cottens, Tetrahedron Lett. 2002, 43, 8095-8097. [29] a) C. Cativiela, M. D. Díaz-de-Villegas, Tetrahedron: Asymmetry 2007, 18, 569-623; b) C.
Cativiela, M. Ordóñez, Tetrahedron: Asymmetry 2009, 20, 1-63; c) C. Gaul, B. W. Schweizer, P. Seiler, D. Seebach, Helv. Chim. Acta 2002, 85, 1546-1566; d) C. L. Hugelshofer, K. T. Mellem, A. G. Myers, Org. Lett. 2013, 15, 3134-3137; e) C. Spino, Chem. Commun. 2011, 47, 4872-4883.
[30] a) N. A. Petasis, I. A. Zavialov, J. Am. Chem. Soc. 1998, 120, 11798-11799; b) N. R. Candeias, F. Montalbano, P. M. S. D. Cal, P. M. P. Gois, Chemical Reviews (Washington, DC, United States) 2010, 110, 6169-6193.
[31] S. Sugiyama, S. Arai, M. Kiriyama, K. Ishii, Chem. Pharm. Bull. 2005, 53, 100-102. [32] Q. I. Churches, J. M. White, C. A. Hutton, Org. Lett. 2011, 13, 2900-2903. [33] T. P. Tang, S. K. Volkman, J. A. Ellman, J. Org. Chem. 2001, 66, 8772-8778. [34] K. K. Nanda, B. Wesley Trotter, Tetrahedron Lett. 2005, 46, 2025-2028. [35] a) J. A. Bodkin, M. D. McLeod, J. Chem. Soc., Perkin Trans. 1 2002, 2733-2746; b) K. B.
Sharpless, D. W. Patrick, L. K. Truesdale, S. A. Biller, J. Am. Chem. Soc. 1975, 97, 2305-2307.
[36] G. Li, H.-T. Chang, K. B. Sharpless, Angew. Chem. Int. Ed. 1996, 35, 451-454. [37] M. Harding, J. A. Bodkin, F. Issa, C. A. Hutton, A. C. Willis, M. D. McLeod, Tetrahedron
2009, 65, 831-843. [38] J. A. Bodkin, G. B. Bacskay, M. D. McLeod, Org. Biomol. Chem. 2008, 6, 2544-2553. [39] T. Oriyama, K. Noda, K. Yatabe, Synlett 1997, 1997, 701-703. [40] B. G. M. Burgaud, D. C. Horwell, A. Padova, M. C. Pritchard, Tetrahedron 1996, 52,
13035-13050. [41] T. Nakamura, T. Tsuji, Y. Iio, S. Miyazaki, T. Takemoto, T. Nishi, Tetrahedron:
Asymmetry 2006, 17, 2781-2792. [42] T. Tsuji, Y. Iio, T. Takemoto, T. Nishi, Tetrahedron: Asymmetry 2005, 16, 3139-3142. [43] G. S. Poindexter, D. A. Owens, P. L. Dolan, E. Woo, J. Org. Chem. 1992, 57, 6257-6265. [44] K. Högenauer, K. Hinterding, P. Nussbaumer, Bioorg. Med. Chem. Lett. 2010, 20, 1485-
1487. [45] E. N. Jacobsen, I. Marko, W. S. Mungall, G. Schroeder, K. B. Sharpless, J. Am. Chem.
Soc. 1988, 110, 1968-1970. [46] J. J. Ritter, P. P. Minieri, J. Am. Chem. Soc. 1948, 70, 4045-4048. [47] K. Hinterding, S. Cottens, R. Albert, F. Zecri, P. Buehlmayer, C. Spanka, V. Brinkmann,
P. Nussbaumer, P. Ettmayer, K. Hoegenauer, N. Gray, S. Pan, Synthesis 2003, 2003, 1667-1670.

Chapter 3 – AAL(S)
140
[48] M. Levy, A. H. Futerman, IUBMB Life 2010, 62, 347-356. [49] a) A. H. Futerman, Y. A. Hannun, EMBO Rep. 2004, 5, 777-782; b) J.-W. Park, W.-J.
Park, A. H. Futerman, Biochimica et Biophysica Acta - Molecular and Cell Biology of Lipids 2014, 1841, 671-681.
[50] A. Zivkovic, H. Stark, Tetrahedron Lett. 2010, 51, 3769-3771. [51] E. V. Berdyshev, I. Gorshkova, A. Skobeleva, R. Bittman, X. Lu, S. M. Dudek, T.
Mirzapoiazova, J. G. N. Garcia, V. Natarajan, J. Biol. Chem. 2009, 284, 5467-5477. [52] S. Lahiri, H. Park, E. L. Laviad, X. Lu, R. Bittman, A. H. Futerman, J. Biol. Chem. 2009,
284, 16090-16098. [53] H. J. Kim, Q. Qiao, H. D. Toop, J. C. Morris, A. S. Don, J. Lipid Res. 2012, 53, 1701-
1707. [54] N. Haughey, NeuroMol. Med. 2010, 12, 301-305. [55] J. G. Topliss, J. Med. Chem. 1977, 20, 463-469. [56] J. G. Topliss, J. Med. Chem. 1972, 15, 1006-1011. [57] P. G. M. Wuts, T. W. Greene, Greene's Protective Groups in Organic Synthesis, 4 ed.,
John Wiley & Sons, Inc., 2007. [58] S. Ziegler, V. Pries, C. Hedberg, H. Waldmann, Angew. Chem. Int. Ed. 2013, 52, 2744-
2792. [59] S. Punna, E. Kaltgrad, M. G. Finn, Bioconjugate Chem. 2005, 16, 1536-1541. [60] D. Guiffant, D. Tribouillard, F. Gug, H. Galons, L. Meijer, M. Blondel, S. Bach, Biotechnol.
J. 2007, 2, 68-75. [61] C. A. Brown, A. Yamashita, J. Am. Chem. Soc. 1975, 97, 891-892. [62] T. R. Chan, R. Hilgraf, K. B. Sharpless, V. V. Fokin, Org. Lett. 2004, 6, 2853-2855. [63] S. A. Saddoughi, S. Gencer, Y. K. Peterson, K. E. Ward, A. Mukhopadhyay, J. Oaks, J.
Bielawski, Z. M. Szulc, R. J. Thomas, S. P. Selvam, C. E. Senkal, E. Garrett‐Mayer, R. M. De Palma, D. Fedarovich, A. Liu, A. A. Habib, R. V. Stahelin, D. Perrotti, B. Ogretmen, EMBO Mol. Med. 2012, 5, 105-121.

141

142

143

Chapter 4 – Summary and Future Work
144
4.1. Summary
This thesis has described efforts to utilise organic synthesis to help gain access to and develop
natural products as biomedical agents. The discussion in Chapter 1 illustrated the importance of
natural products as a significant source of novel leads in the drug discovery and development
process. The molecules discussed were chosen to demonstrate that total synthesis can be used
to overcome problems with supply of these molecules. Moreover, development of efficient
syntheses can allow for analogs to be synthesised and for elucidation of their biological
mechanism to be achieved.
4.2. Chapter 2 Summary and Future Work
The work discussed in Chapter 2 resulted in the completion of the total syntheses of three
isoquinoline alkaloids – phylline (2.97), dioncophylline E (2.42) and ancistrotectorine C (2.40).
Initially, the goal of this study was to complete a total synthesis of the 7,3’-linked
naphthylisoquinoline alkaloid, dioncophylline E (2.42), with a particular emphasis on completing
this stereoselectively. A preliminary investigation towards the synthesis of the required aryllead
triacetate coupling partner, for the required Pinhey-Barton ortho-arylation reaction, involved a
screen of both the protecting group ortho- to the transmetalation site and also of the functionality
used to transmetalate to the lead triacetate. The results of this study identified benzyl-protected
boronic acid 2.89 to be the most efficient substrate to gain access to arylated naphthol 2.88.
Construction of 1,3-dimethyl-3,4-dihydroisoquinoline 2.92 was achieved using a two-step
sulfinimine cyclisation protocol, with complete stereocontrol at C3. This result would suggest that
the poor diastereoselectivity observed with Brusnahan’s synthesis of ancistrotanzanine A (2.43)
(discussed in Section 2.3.) was a result of substrate effects, rather than the methodology not
being able to generate the material with high stereoselectivity. It is speculated that the
naphthalene moiety is disrupting the transition state of the sulfinimine alkylation reaction of
Brusnahan’s substrate. However, in the case of dioncophylline E, the methodology proved to be
robust and highly stereoselective, so attention could then be focussed on the completion of the
total synthesis.
Reduction of 1,3-dimethyl-3,4-dihydroisoquinoline 2.92 using the conditions reported in the
literature resulted in 50:50 mixture of cis:trans diastereomers, which could not be separated
(Figure 4.1). Efforts to improve the selectivity failed.

Chapter 4 – Summary and Future Work
145
Figure 4.1: Synthesis of the biaryl bond of dioncophylline E (2.42) and investigation into reduction of 1,3-dimethyl-
3,4-dihydroisoquinoline 2.92.
To rectify this problem it was decided that phylline (2.97), a simpler version of the system under
investigation, would be an ideal target to use to develop a better solution to this challenge. The
specific goals of this synthesis were to develop conditions that could be applied to dioncophylline
E (2.42) and allow the construction of the the trans-1,3-dimethyl-1,2,3,4-tetrahydroisoquinoline
moiety stereoselectively. While the cis-1,3-dimethyl-1,2,3,4-tetrahydroisoquinoline could be
successfully synthesised from 1,3-dimethyl-3,4-dihydroisoquinoline 2.104, using the literature
reduction conditions, all attempts to reduce 2.104 to the trans-1,3-dimethyl-1,2,3,4-
tetrahydroisoquinoline 2.108 resulted in, at best, a 75:25 mixture (trans:cis). A new approach,
taking inspiration from a recent report from Lipshutz and co-workers,[1] resulted in an investigation
into the addition of methyl nucleophiles into 3-methyl-3,4-dihydroisoquinoline 2.111. This study
identified that a methyl cerium reagent could be used to generate the trans-1,3-dimethyl-1,2,3,4-
tetrahydroisoquinoline 2.108 diastereoselectively. Deprotection of this material yielded phylline
(2.97) in 7 steps and 21 % overall yield (Figure 4.2).
Figure 4.2: Preparation of cis- and trans-1,3-dimethyl-1,2,3,4-tetrahydroisoquinolines and total synthesis of phylline.

Chapter 4 – Summary and Future Work
146
Consequently, the conditions developed as part of the phylline (2.97) synthesis were applied to
the total synthesis of dioncophylline E (2.42). Subsequently, addition of the methyl cerium reagent
into 3-methyl-3,4-dihydroisoquinoline 2.112 led to the trans-1,3-dimethyl-1,2,3,4-
tetrahydroisoquinoline moiety of dioncophylline E in high diastereoselectivity. This substrate was
deprotected to afford dioncophylline E in 12 overall steps and 12 % overall yield (Figure 4.3).
Figure 4.3: Completion of the total synthesis of dioncophylline E.
At this point, our attention turned back to the 5,3’-linked naphthylisoquinoline alkaloids. The total
synthesis of dioncophylline E had taught us that aryllead triacetates are much more readily
generated from boronic acids than tri-alkyltin compounds. This, in combination with results by
Konopelski and co-workers (discussed in Section 2.8),[2] led us to propose a new synthesis to
these molecules. This required access to Boc-protected cis-1,3-dimethyl-1,2,3,4-
tetrahydroisoquinolinylboronic acid 2.119 which could undergo transmetalation, to cis-1,3-
dimethyl-1,2,3,4-tetrahydroisoquinolinyllead triacetate 2.118, and coupling to naphthol 2.52 to
afford arylated naphthol 2.117. A deprotection/N-methylation protocol of 2.117 would lead to
ancistrotectorine C (2.40). Alternatively, based on a literature precedent by Bringmann and co-
workers,[3] amine 2.117 could be oxidised to afford ancistrotanzanine A (2.43) and
ancistrotectorine D (2.41) (Figure 4.4).
Figure 4.4: Retrosynthetic analysis for 5,3’-linked naphthylisoquinoline alkaloids.

Chapter 4 – Summary and Future Work
147
The cis-1,3-dimethyl-1,2,3,4-tetrahydroisoquinolinyllead triacetate 2.118 was synthesised in
seven steps using the sulfinimine cyclisation protocol, a cis-selective reduction and
transmetalation from the boronic acid 2.119. This substrate 2.118 was successfully coupled to
naphthol 2.52 which afforded a 1:1 mixture of atropisomers. The arylated naphthol 2.117 was
elaborated to ancistrotectorine C (2.40) and its atropisomer in 10 steps and 10 % overall yield,
albeit as an inseparable mixture of atropisomers (Figure 4.5). Unfortunately, using the literature
oxidation conditions did not result in formation of ancistrotanzanine A (2.43) and/or
ancistrotectorine D (2.41) and instead over oxidation to p-quinone 2.123 was observed.
Figure 4.5: Total synthesis of ancistrotectorine C and its atropisomer.
The work described in Chapter 2 opens up several avenues to be explored for future work.
Clearly, the ancistrotectorine C (2.40) synthesis opens up the possibility that this strategy could
become completely convergent and as a result, should be examined on the 7,3’-linked
naphthylisoquinoline alkaloids. In particular, application of this strategy to dioncophylline E (2.42)
would require synthesis of trans-1,3-dimethyl-1,2,3,4-tetrahydroisoquinolinyllead triacetate 4.1
which could be coupled to naphthol 2.52, as shown in Figure 4.6.

Chapter 4 – Summary and Future Work
148
Figure 4.6: Proposed second generation synthesis of dioncophylline E (2.42) and examples of other ‘naphthalene’
fragments which could be used to generate a library of analogs.
Furthermore, it is speculated that the 1,3-dimethyl-1,2,3,4-tetrahydroisoquinoline moiety of
dioncophylline E is important for its biological activity and so, this strategy would allow for rapid
access into several naphthalene analogs (Figure 4.6) and allow the medicinal chemistry program
to be initiated.
To complete the total synthesis of ancistrotanzanine A (2.43) and ancistrotanzanine D (2.41) a
selective oxidation strategy needs to be investigated (Figure 4.7). While protection of the
naphthol would allow for the oxidation to take place without formation of the p-quinone 2.123, it is
envisaged that another, more mild method could allow for this to take place. This is currently
being examined in our laboratories.
Figure 4.7: Proposed synthesis of ancistrotanzanine A (2.43) and ancistrotectorine D (2.41).
Finally, for this strategy to join other leading methods, especially for its application to the
synthesis of other naphthylisoquinoline alkaloids, the atropselectivity of the Pinhey-Barton
reaction needs to be controlled. To solve this problem our group is examining chiral pyridine
derivatives such as 4.2, 4.3 and 4.4 as alternative ligands to be used in the Pinhey-Barton
reaction. These have the advantage that either enantiomer of the ligand can be synthesised to
generate either atropisomer.

Chapter 4 – Summary and Future Work
149
Figure 4.8: Representative chiral pyridine derivatives being synthesised for their use in the Pinhey-Barton reaction.
If these problems can be solved it is suspected that an efficient, general protocol to access these
families of molecules will be achieved.

Chapter 4 – Summary and Future Work
150
4.3. Chapter 3 Summary and Future Work
In Chapter 3, AAL(S) (1.37) was examined as a new lead for the treatment of leukaemia. The
goals of this project were:
- What are the key structural-activity requirements of AAL(S)’s for biological activity?
- What are the intermolecular binding partners of AAL(S) (1.37) and how do these activate
PP2A?
To achieve these goals, a gram scale synthesis of AAL(S) (1.37) was required. Initial
investigations into the Petasis reaction and the Sharpless asymmetric aminohydroxylation
reaction as alternative strategies to access AAL(S) (1.37) were unsuccessful. However, synthesis
of AAL(S) (1.37) was achieved using the protocol reported by Hinterding, which uses Schöllkopf’s
protocol for the synthesis of chiral amino esters, as shown in Figure 4.9. Significantly, the
synthesis could be carried out on a multi-gram scale to gain sufficient quantities of AAL(S) (1.37)
for biological testing.
Figure 4.9: Synthesis of AAL(S).
By making variations to Hinterding’s synthesis, a first generation of hydrophobic tail and amino
alcohol head group analogs of AAL(S) (1.37) was completed. This led to the synthesis of 9
hydrophobic tail analogs and 11 amino alcohol head group analogs being synthesised. The
analogs were tested by our collaborators which identified some key structure-activity relationships
for the anti-cancer activity of AAL(S) (1.37), as shown in Figure 4.10.

Chapter 4 – Summary and Future Work
151
Figure 4.10: Summary of anti-leukaemia structure-activity data obtained for AAL(S) (1.37) and some of the best
analogs that were synthesised as part of the study.
The results from these biological assays identified the benzyl hydrophobic tail analog 3.57 as
being non-cytotoxic while having activity against CerS1. A series of benzyl analogs were
synthesised based on the Topliss decision tree. This investigation identified 3,4-dichlorobenzyl
analog 3.81 as being far more potent than both benzyl analog 3.57 and AAL(S) (1.37) at inhibiting
CerS1 (Figure 4.11).
Figure 4.11: Evolution of ‘AAL(S)’ as a CerS1 inhibitor.
To identify the intermolecular binding partners of AAL(S) (1.37) and, as such, determine how it
exerts it’s anti-cancer activity, an affinity chromatography probe was made. This was synthesised
using the methodology developed as part of our SAR study of AAL(S) (1.37) (the affinity
chromatography probes that were synthesised can be found in Figure 4.12). The assays
conducted with this probe identified a novel target, referred to in this thesis as AAL(S)-BP.

Chapter 4 – Summary and Future Work
152
Figure 4.12: AAL(S) and O-FTY720 affinity chromatography probes synthesised to determine their intermolecular
binding partners.
Synthesis of an O-FTY720 affinity chromatography probe confirmed that AAL(S)-BP is a novel
target of these molecules. Further work by our collaborators has found that this protein is up-
regulated significantly in leukaemia patient cells and has also identified that this protein binds
directly to PP2A which, at this stage, is speculated to be the reason for AAL(S)’s anti-cancer
activity.
Identifying the key biological target for AAL(S) (1.37) has set the stage to allow for a more rational
drug design program to be initiated. Fortunately, there is a crystal structure for AAL(S)-BP
available which has led to some initial computational docking investigations being carried out. As
shown in Figure 4.13, a putative docking site has been identified and this is currently being used
to rationalise the biological data that we have collected.
Figure 4.13: Preliminary computer model of AAL(S) bound to AAL(S)-BP.
To validate that we have identified the correct binding site for AAL(S) in our docking analysis a
photoaffinity probe such as 4.5 or 4.6 would be very valuable (Figure 4.14). In combination with
this, our collaborators are also working towards getting a co-crystal structure of AAL(S)-AAL(S)-
BP.

Chapter 4 – Summary and Future Work
153
Figure 4.14: Proposed AAL(S) photoaffinity probes.
Our work on selective CerS inhibitors identified the 3,4-dichlorobenzyl hydrophobic tail analog
3.81 as a selective inhibitor of CerS1. While the Topliss series of these compounds is currently
being extended, an investigation into the head group has also been initiated. A recent report from
Stark and co-workers has shown that O-FTY720 analogs with variation on the head group,
particularly alkylation or acylation of the amine, can increase the selectivity for specific CerS’s.[4]
The analogs they have synthesised are shown in Figure 4.15. While their analogs were not
tested on CerS1, their data suggests that N-alkyl analogs (4.7 and 4.8) inhibit CerS2 and 4, 4-
phenylbutanamide 4.9 inhibits CerS4 and 6 and heptadecanamide 4.10 inhibits CerS2. Taking
this into account, a thorough examination of the head group with the benzyl hydrophobic tail
analogs that are being synthesised is expected to further tune their selectivity and potency for
specific CerSs. Some of the analogs that are proposed for synthesis are shown in Figure 4.15.
Figure 4.15: Selective CerS O-FTY720 analogs synthesised by Stark and co-workers and highlighted in the box are
analogs that are proposed based on their biological data.[4]
Parallel to this work, the ability of AAL(S) to up-regulate PP2A has attracted attention from the
Mattes group at the University of Newcastle. Recent work by the Mattes group has shown that a
reduction of PP2A activity is observed in allergic airway inflammation as a result from a rhinovirus
or house dust mite stimuli.[5] By treating mice exposed to either rhinovirus or house dust mite with
AAL(S) they were able to restore PP2A activity which lead to a reduction in airway hyperreactivity
and inflammation. While their results found that this occurs by up-regulation of E3 ubiquitin ligase
midline 1 (MID1) and tumor necrosis factor-related apoptosis inducing ligand (TRAIL) by the

Chapter 4 – Summary and Future Work
154
rhinovirus and house dust mite stimuli, the current focus of this collaboration is trying to determine
whether this response occurs dependently or independently of AAL(S)-BP and if the analogs that
were synthesised as part of this project have the same effect.
Completion of the above suggestions should further our understanding of how AAL(S) exerts its
biological activity. While these views are from an organic synthesis stand point much of the
biological mechanism of AAL(S)-BP is still not understood. Particularly, how it interacts with PP2A
and the biological reason for its up-regulation in cancer cells is required to further this project.
These are goals that are currently being investigated within the collaboration.

Chapter 4 – Summary and Future Work
155
4.4. References Chapter 4
[1] S. Huang, T. B. Petersen, B. H. Lipshutz, J. Am. Chem. Soc. 2010, 132, 14021-14023. [2] a) H. Deng, J. P. Konopelski, Org. Lett. 2001, 3, 3001-3004; b) J. Lin, B. S.
Gerstenberger, N. Y. T. Stessman, J. P. Konopelski, Org. Lett. 2008, 10, 3969-3972; c) J. P. Konopelski, J. M. Hottenroth, H. M. Oltra, E. A. Véliz, Z.-C. Yang, Synlett 1996, 1996, 609-611.
[3] G. Bringmann, R. Weirich, H. Reuscher, J. R. Jansen, L. Kinzinger, T. Ortmann, Liebigs Ann. Chem. 1993, 1993, 877-888.
[4] S. Schiffmann, D. Hartmann, S. Fuchs, K. Birod, N. Ferreiròs, Y. Schreiber, A. Zivkovic, G. Geisslinger, S. Grösch, H. Stark, Biochimie 2012, 94, 558-565.
[5] a) A. Collison, L. Hatchwell, N. Verrills, P. A. B. Wark, A. P. de Siqueira, M. Tooze, H. Carpenter, A. S. Don, J. C. Morris, N. Zimmermann, N. W. Bartlett, M. E. Rothenberg, S. L. Johnston, P. S. Foster, J. Mattes, Nat Med 2013, 19, 232-237; b) A. Collison, J. Li, A. Pereira de Siqueira, J. Zhang, H. D. Toop, J. C. Morris, P. S. Foster, J. Mattes, Am. J. Respir. Cell Mol. Biol. 2014, 51, 86-93; c) L. Hatchwell, J. Girkin, M. D. Dun, M. Morten, N. Verrills, H. D. Toop, J. C. Morris, S. L. Johnston, P. S. Foster, A. Collison, J. Mattes, J. Allergy Clin. Immunol. 2014, 133, 1720-1727.

156

157

Chapter 5: Experimental
158
5.1. General Experimental
Melting points were obtained on OptiMelt Automated Melting Point System with Digital Image
Processing Technology and are uncorrected. 1H NMR and 13C NMR were recorded at the
Nuclear Magnetic Resonance Facility within the Mark Wainwright Analytical Centre at The
University of New South Wales on a Bruker Avance III 300 (300 MHz), Bruker DPX 300 (300
MHz), Bruker Avance III 400 (400 MHz), Bruker Avance III 500 (500 MHz) or Bruker Avance III
600 (600 MHz), with data acquired and processed using TopSpin 3.0 software. Chemical shifts
are expressed in parts per million (PPM) on the δ scale. Chemical shifts in (a) CDCl3 were
referenced relative to CHCl3 (7.26 ppm) for 1HNMR and CHCl3 (77.16 ppm) for 13CNMR, (b)
MeOD were referenced relative to CH3OH (3.31 ppm) for 1HNMR and CD3OD (49.00 ppm) for
13CNMR, and (c) (CD3)2SO were referenced relative to (CH3)2SO (2.50 ppm) for 1HNMR and
(CD3)2SO (39.52 ppm) for 13CNMR.[1] Infrared spectra were obtained on a ThermoNicolet Avatar
370 FT–IR spectrometer and are reported in wavenumbers (cm–1). Spectra were recorded from
thin films using NaCl plates. HRMS were performed at the Bioanalytical Mass Spectrometry
Facility within the Mark Wainwright Analytical Centre at The University of New South Wales on an
Orbitrap LTQ XL (Thermo Fisher Scientific, San Jose, Ca, USA) ion trap mass spectrometer
using a nanospray (nano-electrospray) ionization source to generate ions from the analyte in
solution. The instrument was calibrated with a standard calibration solution (as outlined in the
instrument manual) on the day of analysis using direct infusion with the nanospray source. The
instrument conditions were optimized for sensitivity on each compound of interest using LC tune
software. The analysis was carried out in positive ion mode using the orbitrap FTMS analyser at a
resolution of 100000. Samples, 5 L, (1 g/mL in methanol or acetonitrile), were injected into a
glass needle and inserted into the nanospray source. Ions generated were measured over the
mass range 150 to 2000. Data was acquired in full scan mode over 60 seconds. Data was
analyzed using the Qual Browser feature in Xcaliber 2.1 (Thermo Fisher Scientific, San Jose, Ca,
USA). Optical rotations (𝛼) were recorded on Rudolph Research Analytical Autopol 1 Automatic
Polarimeter. Samples were prepared in 10 or 5 mL volumetric flasks at stated concentration
(g/100 mL) in chloroform. Measurements were taken at 589 nm (sodium D line), at the stated
temperature in a 1.0 or 0.5 dm path length optical cell. Values are reported as specific rotations
([𝛼]).
Unless otherwise stated all reactions were performed in flame dried glassware under an
atmosphere of dry argon. Reaction temperatures refer to the external bath temperature.

Chapter 5: Experimental
159
Concentration of solvents was performed under reduced pressure on a rotary evaporator after
which, residual solvent was removed under high vacuum (~0.1 mm/Hg).
Reagents and solvents were purchased from commercial sources and used without further
purification, unless stated below. Reagents and solvents used in reactions were purified
according to well established procedures.[2] In particular, tetrahydrofuran (THF), 1,4-dioxane and
toluene were freshly distilled from sodium and benzophenone under an inert atmosphere of
argon. N,N-Dimethylformamide (DMF) was dried sequentially over three batches of 4Ǻ molecular
sieves (3 × 24 h), before finally being stored over a fourth batch of 4Ǻ molecular sieves, under
argon. To remove residual N,N-dimethylamine from DMF, the solvent was evacuated (~0.1
mm/Hg) for at least 30 min prior to use. Methanol was distilled from magnesium and stored over
3Ǻ molecular sieves, under argon. N,N-Diisopropylamine, pyridine, dichlorormethane and 1,2-
dichloroethane were distilled from calcium hydride immediately prior to use. Triisopropylborate
and ethylenediamine were distilled from sodium immediately before use. (S)-Schöllkopf reagent
was distilled immediately before use (53 – 55°C at 0.1 mm/Hg). Copper(I) iodide was purified by
extraction with dichloromethane using a Soxhlet extractor then dried under high vacuum (~0.1
mm/Hg). Sodium hydride (60 % in mineral oil) was washed free of mineral oil by placing in a
reaction flask to which dry n-hexane was added. The suspension was stirred for 5 min then the
stirring was stopped, the suspension allowed to settle and the liquid removed (this procedure was
repeated two more times). Excess n-hexane was removed under high vacuum (~0.1 mm/Hg)
before use. Lead(IV) tetraacetate was recrystallised from acetic acid, then dried over potassium
hydroxide under high vacuum (~0.1 mmHg) for 2 h prior to use. n-Butyllithium in hexanes, t-
butyllithium in pentanes and methyl lithium in diethyl ether were purchased from Sigma Aldrich
and titrated using menthol and 2,2’-bipyridyl in THF as described by Eastham.[3] Similarly,
Methylmagnesium bromide in diethyl ether and allylmagnesium bromide in diethyl ether were
purchased from Sigma Aldrich and titrated using menthol and 1,10-phenanthroline in THF.
Diisobutylaluminium hydride in hexanes was purchased from Sigma Aldrich and titrated using 4-
anisaldehyde in THF as described by Hoye.[4] The following compounds and catalysts were
prepared following literature procedures: RuCl[(R,R)-TsDPEN](p-cymene) and RuCl[(S,S)-
TsDPEN](p-cymene),[5] Pd(PPh3)4,[6] chloromethyl methyl ether,[7] 2-trimethylsilyl)ethyl
carbamate,[8] benzyl carbamate,[9] TBTA.[10]
Analytical thin layer chromatography was conducted on Merck, aluminium-backed silica plates 60
F254 or silica gel 60 RP-C18 F254 plates and visualised using UV light and stained with a dip of
either a potassium permanganate, vanillin or phosphomolybdic acid. Flash chromatography was

Chapter 5: Experimental
160
routinely performed using Grace Davison Discovery Sciences, Davisil LC60A 40 – 63 micron
silica gel, following published guidelines.[11] Solvent was eluted using a Thomson SINGLE StEP
pump at the flow rate recommended by the manufacturer (Thomson Instrument Company,
Oceanside, Ca, USA). Deactivated silica gel was prepared by washing a column packed with
silica gel with neat triethylamine (5 column volumes). After drying, the column was washed with n-
hexane to remove any residual triethylamine. Reverse phase flash chromatography was
conducted using Thomson SINGLE StEP C18 Flash Column. Reverse phase C18 silica was
recycled by successively washing with DMSO, dichloromethane, methanol + 1 % TFA and
methanol.

Chapter 5: Experimental
161
5.2. Experiments Described in Chapter 2
2-Methoxy-6-methylbenzonitrile (2.76)[12]
Method 1: Sandmeyer Reaction[13]
A solution of aniline 2.74 (8.10 g, 59.08 mmol) in dry DMSO (100 mL) was added dropwise to a
solution of copper(I) cyanide (11.02 g, 123.04 mmol) and t-butyl nitrite (23 mL, 193.60 mmol) in
dry DMSO (600 mL) at 60°C. The solution was stirred at 60°C for 17 h then cooled to 0°C. The
pH of the solution was adjusted to pH 1using 1 M aqueous hydrochloric acid solution [CAUTION:
conduct in well ventilated fumehood to avoid contact with hydrogen cyanide]. The mixture was
filtered through a short plug of Celite, eluting with water and dichloromethane. The filtrate was
extracted with dichloromethane (× 3). The organic extracts were combined and washed with brine
(× 3), then dried (Na2SO4). The solvent was removed under reduced and the crude material was
purified 2.76 by flash chromatography on silica gel, eluting with 10 % ethyl acetate/n-hexane, to
afford the product as a white solid (3.93 g, 45 %) with all the analytical data matching that
reported in the literature.[12]
Method 2: Palladium catalysed alkoxylation[14]
Palladium(II) acetate (0.19 g, 0.86 mmol) and sodium persulfate (10.06 g, 42.22 mmol) were
placed in a Youngs tube, which was evacuated and purged with argon (× 3). Dry methanol (28
mL) and o-tolulnitrile (2.77) (1.0 mL, 8.44 mmol) were added successively and the tube was
sealed. The suspension was stirred [NB: for maximum conversion a large stirrer bar stirring at
1000 rpm to form a vortex was used] at 70°C for 17 h. The black solution was allowed to cool to
room temperature and the methanol was removed under reduced pressure. The residue was
diluted with 1M aqueous hydrochloric acid solution and extracted with ethyl acetate (× 3). The
organic extracts were combined and washed with water and brine, then dried (Na2SO4). The
solvent was removed under reduced pressure and the crude material was purified by flash
chromatography on silica gel, eluting with 10 % ethyl acetate/n-hexane, to afford first, the starting
material 2.77 (0.21 g, 21 % recovered) then the product 2.76 as a white solid (0.76 g, 77 %) with
all the analytical data matching that reported in the literature.[12] Mp 59-61°C (lit.[12] 61-63°C); 1H

Chapter 5: Experimental
162
NMR (400 MHz; CDCl3) δ 2.50 (s, 3H), 3.91 (s, 3H), 6.78 (d, J = 8.5 Hz, 1H), 6.87 (d, J = 7.7 Hz,
1H), 7.39 (dd, J = 8.5 and 7.7 Hz, 1H); 13C NMR (75 MHz; CDCl3) δ 20.6, 56.2, 102.6, 108.3,
115.8, 122.2, 133.6, 144.1, 161.8.
2-Hydroxy-6-methylbenzonitrile (2.73)[15]
Method 1: Using boron tribromide
Boron tribromide (7.5 mL, 77.84 mmol) was added dropwise to a solution of nitrile 2.76 (2.30 g,
15.60 mmol) in dry dichloromethane (30 mL) at 0°C. The solution was allowed to warm to room
temperature then heated at reflux for 12 h. The dark red solution was cooled to 0°C and
quenched dropwise with ice water. The pH of the reaction mixture was adjusted to pH 13 with 2M
aqueous sodium hydroxide solution and extracted with dichloromethane (× 3). The aqueous layer
was acidified to pH 1 with concentrated hydrochloric acid solution (32 %) and extracted with ethyl
acetate (× 3). The respective organic extracts were combined and washed with brine, then dried
(Na2SO4). The solvent was removed under reduced pressure and the crude material was
recrystallised from dichloromethane to afford the product 2.73 as a white solid (0.93 g, 89 %) with
all the analytical data matching that reported in the literature.[15]
Method 2: Using aluminium trichloride
Aluminium chloride (10.87 g, 81.55 mmol) was added as a solid in one portion to a solution of
nitrile 2.76 (2.99 g, 20.34 mmol) in chlorobenzene (200 mL) at room temperature. The solution
was heated at reflux for 4 h then allowed to cool to room temperature. The reaction mixture was
quenched dropwise with water then the pH of the solution was adjusted to pH 14 with 2 M
aqueous sodium hydroxide solution. The mixture was extracted with dichloromethane (× 3). The
aqueous layer was acidified to pH 1 with concentrated hydrochloric acid solution (32 %) and
extracted with ethyl acetate (× 3). The respective organic extracts were combined and washed
with brine, then dried (Na2SO4). The solvent was removed under reduced pressure to afford the
product 2.73 as a white solid (2.71 g, 99 %) which was of sufficient purity to use in the next step,
with all the analytical data matching that reported in the literature.[15] Mp 109-111°C (lit.[15] 111-
112°C); 1H NMR (400 MHz; CDCl3) δ 2.50 (s, 3H), 6.05 (br s, 1H), 6.79 (d, J = 8.4 Hz, 1H), 6.85
(d, J = 7.6 Hz, 1H), 7.33 (dd, J = 8.4 and 7.6 Hz, 1H); 13C NMR (75 MHz; CDCl3) δ 20.7, 100.7,
113.6, 122.3, 128.3, 134.0, 143.2, 158.6.

Chapter 5: Experimental
163
3-Bromo-2-hydroxy-6-methylbenzonitrile (2.78)
N,N-Diisopropylamine (3.3 mL, 23.55 mmol) was added in one portion to a solution of N-
bromosuccinimide (2.78 g, 15.60 mmol) in dry dichloromethane (160 mL) at room temperature.
The solution was allowed to stir at room temperature for 30 min. before being added dropwise to
a solution of nitrile 2.73 (2.01 g, 15.60 mmol) in dichloromethane (80 mL). After addition, the
reaction solution was stirred at room temperature for 10 h. 1M Aqueous hydrochloric acid solution
was added and the solution was extracted with dichloromethane (× 3). The organic extracts were
combined and washed with water and brine, then dried (Na2SO4). The solvent was removed
under reduced pressure and the crude product 2.78 was purified by flash chromatography on
silica gel, eluting with dichloromethane, to afford the title compound as a white solid (3.15 g, 95
%). Mp 114-115°C; 1H NMR (300 MHz; CDCl3) δ 2.46 (s, 3H, 6-Me), 5.71 (br s, 1H, 2-OH), 6.75
(d, J = 8.3 Hz, 1H, 5-ArH), 7.52 (d, J = 8.3 Hz, 1H, 4-ArH); 13C NMR (75 MHz; CDCl3) δ 20.5 (6-
Me), 101.7 (3-ArC), 107.4 (1-ArC), 114.6 (1-CN), 123.1 (5-ArC), 136.0 (4-ArC), 143.4 (6-ArC),
154.8 (2-ArC); IR (NaCl, neat) 3274, 2235 cm-1; HRMS (ESI-MS): m/z calcd for C8H679BrNONa
[M+Na]+ 233.9530, found 233.9525.
3-Bromo-2-(methoxymethoxy)-6-methylbenzonitrile (2.72)
Chloromethyl methyl ether (0.32 mL, 4.26 mmol) was added dropwise to a suspension of nitrile
2.78 (0.60 g, 2.84 mmol) and potassium carbonate (1.18 g, 8.53 mmol) in DMF (30 mL) at room
temperature. The suspension was stirred at room temperature for 17 h then diluted with water
and extracted with ethyl acetate (× 3). The organic extracts were combined and washed with
water and brine, then dried (Na2SO4). The solvent was removed under reduced pressure and the
crude material purified, as indicated, to afford the product 2.72 by flash chromatography on silica
gel, eluting with 5 % ethyl acetate/n-hexane, to afford the product as a clear colourless oil (0.66 g,
90 %). 1H NMR (300 MHz; CDCl3) δ 2.50 (s, 3H), 3.74 (s, 3H), 5.27 (s, 2H), 6.96 (d, J = 8.4 Hz,
1H), 7.64 (dd, J = 8.4 and 8.1 Hz, 1H); 13C NMR (75 MHz; CDCl3) δ 20.7, 58.8, 100.6, 111.0,
114.5, 115.3, 127.2, 137.6, 143.5, 156.9; IR (NaCl, neat) 2228 cm-1; HRMS (ESI-MS): m/z calcd
for C10H10NO279BrNa [M+Na]+ 277.9792, found 277.9787.

Chapter 5: Experimental
164
3-(Tri-n-butylstannyl)-2-methoxymethoxy-6-methylbenzonitrile (2.71)
Method 1: Palladium catalysed stannylation
Bis(triphenylphosphine)palladium(II) dichloride (13 mg, 18.5 μmol) and potassium acetate (54 mg,
0.55 mmol) were added sequentially to a solution of bromide 2.72 (46 mg, 0.18 mmol) in DMF
(0.5 mL). The reaction mixture was frozen (liquid N2) and evacuated for 10 min. The evacuation
was ceased and the mixture allowed to thaw. This was repeated (× 2) and once thawed the argon
atmosphere was restored. Bis(tri-n-butyltin) (0.18 mL, 0.36 mmol) was added before the mixture
was heated at 80°C for 15 h. The solution was cooled to room temperature, diluted with ethyl
acetate and filtered through a short pad of Celite, eluting with ethyl acetate. The filtrate was
collected and washed with water (× 2) and brine, then dried (Na2SO4). The solvent was removed
under reduced pressure and the crude material was purified by flash chromatography on silca
gel, eluting with 5 % ethyl acetate/n-hexane to first, afford the product (42 mg, 50 %) with all of
the analytical data matching that reported later for 2.71. Further elution afforded the demetallated
material 5.1 (10 mg, 31 %) with all of the analytical data matching that reported for 5.1.
Method 2: Halogen-lithium-tin exchange
A solution of t-butyllithium in pentane (2.7 mL, 1.2 M, 3.24 mmol) was added dropwise to a
solution of bromide 2.72 (0.49 g, 1.93 mmol) in THF (20 mL) at -95°C. The reaction solution was
stirred at -95°C for 15 min after which, tri-n-butyltin chloride (0.58 mL, 2.14 mmol) was added
dropwise. The solution was allowed to warm slowly to -10°C over 4 h where it was quenched with
saturated aqueous sodium bicarbonate solution. The mixture was allowed to warm to room
temperature then extracted with ethyl acetate (× 3). The organic extracts were combined and
washed with water and brine, then dried (Na2SO4). The solvent was removed under reduced
pressure and the crude material purified by flash chromatography on silica gel, eluting with 5 %
ethyl acetate/n-hexane, to afford the product 2.71 as a clear colourless oil (0.56 g, 63 %). 1H
NMR (400 MHz; CDCl3) δ 0.89 (t, J = 7.2 Hz, 9H), 1.05 – 1.22 (m, 6H), 1.31 – 1.41 (m, 6H), 1.51
– 1.59 (m, 6H), 2.52 (s, 3H), 3.63 (s, 3H), 5.19 (s, 2H), 7.02 (d, J = 7.4 Hz, 1H), 7.47 (td, J = 10.8
and 7.4 Hz, 1H); 13C NMR (100 MHz; CDCl3) δ 10.6 (t, JC-Sn = 172.3 Hz), 14.0, 20.9, 27.6 (t, JC-Sn
= 30.7 Hz), 29.3 (t, JC-Sn = 9.9 Hz), 58.4, 100.4, 107.1, 117.1, 125.7, 133.3, 142.0, 144.2, 164.7;
IR (NaCl, neat) 2221 cm-1; HRMS (ESI-MS): m/z calcd for C22H39NO2120Sn [M+H]+ 468.1925,
found 468.1916.

Chapter 5: Experimental
165
Bis(3-cyano-(2-methoxymethoxy)-4-methylphenyl)mercury (2.79)
Mercury(II) trifluoroacetate (29 mg, 68.0 μmol) was added as a solid in one portion to a solution to
a solution of stannane 2.71 (64 mg, 0.14 mmol) in dichloromethane (1.3 mL) at room
temperature. The solution was stirred at room temperature protected from light for 16 h. The
solvent was removed under reduced pressure and the residue was triturated with n-hexane (× 3)
to afford the product 2.79 as a white solid (27 mg, 69 %). 1H NMR (300 MHz; CDCl3) δ 2.54 (s,
3H), 3.57 (s, 3H), 5.25 (s, 2H), 7.14 (d, J = 7.6 Hz, 1H), 7.51 (d, J = 7.6 Hz, 1H); 13C NMR (75
MHz; CDCl3) δ 20.6, 57.4, 99.5, 100.2, 125.9, 126.7, 135.7, 142.2, 143.6, 155.9; IR (NaCl, neat)
2218 cm-1; HRMS (ESI-MS): m/z calcd for C20H20HgN2O4Na [M+Na]+ 577.1027, found 577.1026
2-(Methoxymethoxy)-6-methylbenzonitrile (5.1)
Chloromethyl methyl ether (8 μL, 0.11 mmol) was added dropwise to a suspension of nitrile 2.73
(10 mg, 75.7 μmol) and potassium carbonate (31 mg, 0.23 mmol) in DMF (1.5 mL) at room
temperature. The suspension was stirred at room temperature for 17 h then diluted with water
and extracted with ethyl acetate (× 3). The organic extracts were combined and washed with
water and brine, then dried (Na2SO4). The solvent was removed under reduced pressure and the
crude material purified by flash chromatography on silica gel, eluting with 5 % ethyl acetate/n-
hexane, to afford the product as a clear colourless oil (10 mg, 77 %). 1H NMR (400 MHz; CDCl3)
δ 2.49 (s, 3H), 3.50 (s, 3H), 5.26 (s, 2H), 6.91 (d, J = 7.6 Hz, 1H), 7.02 (d, J = 8.4 Hz, 1H), 7.34
(dd, J = 8.4 and 7.6 Hz, 1H); 13C NMR (100 MHz; CDCl3) δ 20.8, 56.8, 95.0, 103.8, 112.1, 115.8,
123.4, 133.7, 144.0, 159.6; IR (NaCl, neat) 2223 cm-1; HRMS (ESI-MS): m/z calcd for
C10H11NO2Na [M+Na]+ 200.0687, found 200.0682.
3-Bromo-2-methoxy-6-methylbenzonitrile (5.2)
Methyl iodide (0.20 mL, 2.15 mmol) was added dropwise to a suspension of nitrile 2.78 (0.23 g,
1.08 mmol) and potassium carbonate (0.30 g, 2.15 mmol) in DMF (8 mL) at room temperature.

Chapter 5: Experimental
166
The suspension was stirred at room temperature for 17 h then diluted with water and extracted
with ethyl acetate (× 3). The organic extracts were combined and washed with water and brine,
then dried (Na2SO4). The solvent was removed under reduced pressure and the crude material
purified by flash chromatography on silica gel, eluting with 1 % ethyl acetate/n-hexane, to afford
the product 5.2 as a clear colourless oil (0.23 g, 95 %). 1H NMR (400 MHz; CDCl3) δ 2.49 (s, 3H),
4.03 (s, 3H), 6.94 (d, J = 8.4 Hz, 1H), 7.63 (d, J = 8.4 Hz, 1H); 13C NMR (100 MHz; CDCl3) δ
20.7, 62.4, 109.6, 114.4, 115.1, 126.9, 137.7, 143.6, 159.8; IR (NaCl, neat) 2224 cm-1; HRMS
(ESI-MS): m/z calcd for C9H879BrNONa [M+Na]+ 247.9687, found 247.9680.
3-(Tri-n-butylstannyl)-2-methoxy-6-methylbenzonitrile (2.80)
A solution of t-butyllithium in pentane (0.64 mL, 1.2 M, 0.77 mmol) was added dropwise to a
solution of nitrile 5.2 (0.17 g, 0.76 mmol) in THF (12 mL) at -95°C. The reaction solution was
stirred at -95°C for 15 min after which, tri-n-butyltin chloride (0.25 mL, 0.92 mmol) was added
dropwise. The solution was allowed to warm slowly to -10°C over 5 h where it was quenched with
saturated aqueous sodium bicarbonate solution. The mixture was allowed to warm to room
temperature then extracted with ethyl acetate (× 3). The organic extracts were combined and
washed with water and brine, then dried (Na2SO4). The solvent was removed under reduced
pressure and the crude material purified by flash chromatography on silica gel, eluting with 1 %
ethyl acetate/n-hexane, to afford the product 2.80 as a clear colourless oil (0.22 g, 68 %). 1H
NMR (400 MHz; CDCl3) δ 0.88 (t, J = 7.3 Hz, 9H), 0.98 – 1.15 (m, 6H), 1.27 – 1.37 (m, 6H), 1.47
– 1.54 (m, 6H), 2.51 (s, 3H), 4.03 (s, 3H), 6.99 (d, J = 7.2 Hz, 1H), 7.63 (d, J = 8.4 Hz, 1H); 13C
NMR (100 MHz; CDCl3) δ 10.1 (JC-Sn = 176.9 Hz), 13.8, 20.7, 27.5 (JC-Sn = 29.6 Hz), 29.2 (JC-Sn =
9.8 Hz), 61.9, 104.3, 116.8, 124.9, 132.6, 141.4, 144.3, 167.5; IR (NaCl, neat) 2221 cm-1; HRMS
(ESI-MS): m/z calcd for C21H35120SnNONa [M+Na]+ 460.1638, found 460.1636.
7-Ethynyl-5-methoxybicyclo[4.2.0]octa-1,3,5-trien-7-ol (5.4)[16]
A solution of ethynylmagnesium bromide in THF (6.7 mL, 0.4 M, 2.95 mmol) was added dropwise
to a solution of benzocyclobutenone 5.3[17] (0.22 g, 1.48 mmol) in THF (10 mL) at -10°C
(ice/acetone). The solution was allowed to warm slowly, in the cold bath, to 10°C over 4 h. The
solution was cooled to -40°C and quenched with saturated aqueous ammonium chloride. The

Chapter 5: Experimental
167
solution was allowed to warm to room temperature and extracted with ethyl acetate. The
remaining aqueous layer was acidified to pH 1 with 1 M aqueous hydrochloric acid and extracted
further with ethyl acetate (× 2). The organic extracts were combined and washed with water and
brine, then dried (Na2SO4). The solvent was removed under reduced pressure and the crude
material was purified by flash chromatography on silica gel, eluting with 10 % ethyl acetate/n-
hexane, to afford the product 5.4 as a white solid (0.18 g, 70 %) with all the analytical data
matching that reported in the literature.[16] 1H NMR (300 MHz; CDCl3) δ 2.65 (s, 1H), 3.23 (br s,
1H), 3.40 (d, J = 14.0 Hz, 1H), 3.74 (d, J = 14.0 Hz, 1H), 4.05 (s, 3H), 6.71 – 6.76 (m, 2H), 7.25
(dd, J = 8.4, 7.2 Hz, 1H).
8-Methoxy-3-methylnaphthalen-1-ol (2.52)[18]
Copper(I) iodide (59 mg, 0.31 mmol) was added to a solution of alkyne 5.4 (54 mg, 0.31 mmol) in
freshly distilled 1,4-dioxane (4 mL) at room temperature. The solution was stirred at room
temperature for 15 min where a white precipitate had formed. Paraformaldehyde (10 mg, 0.33
mmol) and freshly distilled N,N-diisopropylamine (45 μL, 0.32 mmol) were added successively at
room temperature after which, the solution had turned homogeneous. The solution was slowly
warmed to reflux temperature where it was stirred for 18 h. The solution was cooled to room
temperature and diluted with water. The mixture was extracted with diethyl ether (× 3). The
organic extracts were combined and washed with water and brine, then dried (Na2SO4). The
solvent was removed under reduced pressure and the crude material was purified by flash
chromatography on silica gel, eluting with 4 % ethyl acetate/n-hexane, to afford the product 2.52
as a white solid (51 mg, 88 %) with all the analytical data matching that reported in the
literature.[18] 1H NMR (300 MHz; CDCl3) δ 2.43 (s, 3H), 4.03 (s, 3H), 6.70 (dd, J = 7.4, 1.0 Hz,
1H), 6.75 (d, J = 1.0 Hz, 1H), 7.10 (s, 1H), 7.24 – 7.35 (m, 2H), 9.24 (s, 1H).
N,N-Diethyl senecioamide (5.7)[18]
a) Oxalyl chloride (21.1 mL, 0.25 mol) was added dropwise to a solution of 3,3-
dimethylacrylic acid (5.5) (22.40 g, 0.22 mol) in dichloromethane (150 mL) at room temperature.
The reaction was stirred at room temperature for 12 h. The dichloromethane was removed by

Chapter 5: Experimental
168
distillation. Excess oxalyl chloride was removed by dissolution of the residue in chloroform and
distillation of both the oxalyl chloride and chloroform (× 3) to afford a yellow oil.
b) Freshly distilled N,N-diethylamine (30 mL, 0.57 mol) was added dropwise to a solution of
the crude residue 5.6 in freshly distilled THF (450 mL) at 0°C. The mixture was stirred at 0°C for
15 min. then at room temperature for 14 h. The resulting precipitate was removed by filtration
under reduced pressure. The THF was removed from the filtrate under reduced pressure before
the residue was distilled under reduced pressure (bp 102 -104°C, 14 mmHg) to afford the product
5.7 as a clear colourless oil (21.78 g, 63 % over two steps) with all of the analytical data matching
that reported in the literature.[18] 1H NMR (300 MHz; CDCl3) δ 1.13 (t, J = 7.1 Hz, 6H), 1.83 (d, J =
1.2 Hz, 3H), 1.92 (d, J = 1.1 Hz, 3H), 3.29 – 3.43 (m, 4H), 5.79 – 5.80 (m, 1H).
8-Methoxy-3-methylnaphthalen-1-ol (2.52)[18-19]
A solution of n-butyllithium in hexanes (67.6 mL, 0.142 mol) was added dropwise to a solution of
freshly distilled N-cyclohexyl-N-isopropylamine (13.8 mL, 0.142 mol) in freshly distilled THF (280
mL) at -78°C (dry ice/acetone). The cold bath was removed and immediately replaced with a 0°C
cold bath where it was stirred for 30 min. The 0°C cold bath was replaced with a -78°C cold bath
and a solution of N,N-diethyl senecioamide (5.7) (6.30 g, 40.6 mmol) in freshly distilled THF (160
mL) was added dropwise. The bright yellow solution was stirred at -78°C for 1 h then the cold
bath was replaced with a -10°C cold bath (ice/acetone) and stirred for an additional 10 min. A
solution of 3-bromoanisole (5.8) (10.2 mL, 81.2 mmol) in freshly distilled THF (81 mL) was added
dropwise. The dark red reaction mixture was allowed to slowly warm to room temperature over 20
h. The reaction was quenched with saturated aqueous ammonium chloride solution, till the colour
dissipated, then the solution was acidifed to pH 1 using 1 M aqueous hydrochloric acid solution.
The mixture was stirred vigorously for 30 min. The THF was removed under reduced pressure
and the residue was extracted with dichloromethane (× 3). The organic extracts were combined
and washed with brine the dried (Na2SO4). The solvent was removed under reduced pressure
and the crude material was purified by flash chromatography on silica gel, eluting with 5 % ethyl
acetate/n-hexane, to afford the product 2.52 as a white solid (2.52 g, 33 %) with all the analytical
data matching that reported before and in the literature.[18]

Chapter 5: Experimental
169
2-Methoxy-3-(1’-hydroxy-8’-methoxy-3’-methylnaphthalen-2’-yl)-6-methylbenzonitrile (2.81)
(a) A solution of stannane 2.80 (41 mg, 94.0 μmol) in dry dichloromethane (0.5 mL) was
added dropwise to a solution of freshly dried lead(IV) tetraacetate (44 mg, 99.2 μmol) and
mercury(II) trifluoroacetate (12 mg, 28.1 μmol) in dry dichloromethane (0.5 mL). The solution was
protected from light and stirred at room temperature for 17 h. The reaction mixture was filtered
through a short plug of Celite, eluting with dichloromethane. The solvent was removed under
reduced pressure and the residue was triturated with n-hexane to afford crude aryllead triacetate
5.9 as a light yellow gum. Due to the lability of this compound it was used immediately in the next
step.
(b) A solution of crude aryllead triacetate 5.9 in dry 1,2-dichloroethane (0.5 mL) was added
dropwise to a solution of naphthol 2.52 (18 mg, 95.6 μmol) and dry pyridine (30 μL, 0.37 mmol) in
dry 1,2-dichloroethane (0.5 mL) at room temperature. The solution was protected from light and
stirred at room temperature for 16 h. The reaction mixture was poured onto 1M aqueous
hydrochloric acid and extracted with dichloromethane (× 3). The organic extracts were combined
and washed with 1M aqueous hydrochloric acid solution, water and brine, then dried (Na2SO4).
The solvent was removed under reduced pressure and the crude material was purified by flash
chromatography on deactivated silica gel, eluting with 20 % ethyl acetate/n-hexane, to afford the
product 2.81 as a white solid (5 mg, 16 % over two steps). 1H NMR (300 MHz; CDCl3) δ 2.15 (s,
3H), 2.58 (s, 3H), 3.63 (s, 3H), 4.03 (s, 3H), 6.76 (d, J = 7.3 Hz, 1H), 7.11 (d, J = 8.0 Hz, 1H),
7.24 (s, 1H), 7.29 – 7.39 (m, 3H), 9.60 (s, 1H); 13C NMR (75 MHz; CDCl3) δ 20.6, 20.8, 56.2,
61.5, 103.8, 107.8, 113.4, 116.2, 119.1, 119.8, 121.4, 125.2, 126.3, 128.4, 136.4, 136.8, 137.2,
143.0, 151.3, 156.3, 160.8; IR (NaCl, neat) 2224, 3378 cm-1; HRMS (ESI-MS): m/z calcd for
C21H19NO3Na [M+Na]+ 356.1262, found 356.1258.

Chapter 5: Experimental
170
3-Bromo-2-(t-butyldimethylsilyloxy)-6-methylbenzonitrile (5.10)
Nitrile 2.78 (0.31 g, 1.46 mmol) was added as a solid in one portion to a solution of t-
butyldimethylsilyl chloride (0.33 g, 2.19 mmol) and imidazole (0.15 g, 2.19 mmol) in DMF (2 mL)
at room temperature. The solution was stirred at room temperature for 16 h. The solution was
diluted with water and the mixture was extracted with ethyl acetate (× 3). The organic extracts
were combined and washed with water and brine, then dried (Na2SO4). The solvent was removed
under reduced pressure and the crude material purified by flash chromatography on silica gel,
eluting with 3 % ethyl acetate/n-hexane, to afford the product 5.10 as a light yellow oil (0.41 g, 86
%). 1H NMR (300 MHz; CDCl3) δ 0.37 (s, 6H), 1.08 (s, 9H), 2.46 (s, 3H), 6.78 (d, J = 8.4 Hz, 1H),
7.57 (dd, J = 8.4 Hz, 1H); 13C NMR (75 MHz; CDCl3) δ -2.4, 18.9, 20.8, 26.2, 108.1, 112.9, 116.2,
124.2, 137.6, 143.4, 155.3; IR (NaCl, neat) 2225 cm-1; HRMS (ESI-MS): m/z calcd for
C14H2079BrNOSiNa [M+Na]+ 348.0395, found 348.0391.
3-(t-butyldimethylsilyl)-2-hydroxy-6-mthylbenzonitrile (5.11)
A solution of t-butyllithium in pentane (1.34 mL, 1.5 M, 2.01 mmol) was added dropwise to a
solution of nitrile 5.10 (0.41 g, 1.26 mmol) in freshly distilled THF (15 mL) at -95°C (liquid
nitrogen/ethanol). The solution was allowed to warm to -10°C, in the cold bath, over 5 h. The
reaction was quenched with saturated aqueous sodium bicarbonate solution and extracted with
ethyl acetate (× 3). The organic extracts were combined and washed with water and brine, then
dried (Na2SO4). The solvent was removed under reduced pressure and the crude material
purified by flash chromatography on silica gel, eluting with 3 % ethyl acetate/n-hexane, to afford
the product 5.11 as a clear colourless oil (0.30 g, 95 %). 1H NMR (300 MHz; CDCl3) δ 0.32 (s, 6H,
Si(CH3)2), 0.89 (s, 9H, SiC(CH3)3), 2.48 (s, 3H, 6-CH3), 6.02 (br s, 1H, 2-OH), 6.84 (d, J = 7.5 Hz,
1H, 5-H), 7.42 (d, J = 7.5 Hz, 1H, 6-H); 13C NMR (75 MHz; CDCl3) δ -4.7, 17.8, 20.8, 27.0, 100.2,
116.3, 121.7, 121.9, 141.2, 143.9, 163.3; IR (NaCl, neat) 2227 cm-1; HRMS (ESI-MS): m/z calcd
for C14H2079BrNOSiNa [M+Na]+ 348.0395, found 270.1283.

Chapter 5: Experimental
171
2-(t-Butyldimethylsilyloxy)-6-methylbenzonitrile (5.12)
Nitrile 2.73 (22 mg, 0.17 mmol) was added as a solid in one portion to a solution of t-
butyldimethylsilyl chloride (38 mg, 0.25 mmol) and (15 mg, 0.25 mmol) in DMF (0.3 mL) at room
temperature. The solution was stirred at room temperature for 16 h. The solution was diluted with
water and the mixture was extracted with ethyl acetate (× 3). The organic extracts were combined
and washed with water and brine, then dried (Na2SO4). The solvent was removed under reduced
pressure and the crude material purified by flash chromatography on silica gel, eluting with 5 %
ethyl acetate/n-hexane, to afford the product 5.12 as a light yellow oil (32 mg, 76 %). 1H NMR
(400 MHz; CDCl3) δ 0.27 (s, 6H), 1.05 (s, 9H), 2.49 (s, 3H), 6.71 (d, J = 8.4 Hz, 1H), 6.86 (d, J =
8.0 Hz, 1H), 7.29 (dd, J = 8.5 and 7.7 Hz, 1H); 13C NMR (100 MHz; CDCl3) δ -4.1, 18.4, 20.9,
25.8, 106.4, 116.4, 116.9, 122.7, 133.4, 143.9, 158.6; IR (NaCl, neat) 2224 cm-1; HRMS (ESI-
MS): m/z calcd for C14H21NOSiNa [M+Na]+ 270.1290, found 270.1284.
3-Bromo-2-isopropoxy-6-methylbenzonitrile (5.13)
2-bromopropane (0.58 mL, 6.18 mmol) was added dropwise to a suspension of nitrile 2.78 (0.87
g, 4.12 mmol) and potassium carbonate (1.71 g, 12.36 mmol) in DMF (25 mL) at room
temperature. The suspension was stirred at 60°C for 17 h then allowed to cool to room
temperature. then diluted with water and extracted with ethyl acetate (× 3). The organic extracts
were combined and washed with water and brine, then dried (Na2SO4). The solvent was removed
under reduced pressure and the crude material purified by flash chromatography on silica gel,
eluting with 2 % ethyl acetate/n-hexane, to afford the product 5.13 as a clear colourless oil (1.04
g, 99 %). 1H NMR (300 MHz; CDCl3) δ 1.41 (d, J = 6.3 Hz, 6H), 2.48 (s, 3H), 4.75 (sept, J = 6.3
Hz, 1H), 6.90 (d, J = 8.4 Hz, 1H), 7.61 (d, J = 8.4 Hz, 1H); 13C NMR (75 MHz; CDCl3) δ 20.5,
22.5, 78.9, 110.6, 115.0, 115.4, 126.2, 137.5, 143.1, 157.6; IR (NaCl, neat) 2226 cm-1; HRMS
(ESI-MS): m/z calcd for C11H12NO79BrNa [M+Na]+ 276.0000, found 275.9997.

Chapter 5: Experimental
172
3-(Tri-n-butylstannyl)-2-isopropoxy-6-methylbenzonitrile (2.82)
A solution of t-butyllithium in pentane (5.0 mL, 1.1 M, 5.50 mmol) was added dropwise to a
solution of nitrile 5.13 (0.87 g, 3.42 mmol) in THF (30 mL) at -95°C. The reaction solution was
stirred at -95°C for 15 min after which, tri-n-butyltin chloride (1.0 mL, 3.69 mmol) was added
dropwise. The solution was allowed to warm slowly to -10°C over 5 h where it was quenched with
saturated aqueous sodium bicarbonate solution. The mixture was allowed to warm to room
temperature then extracted with ethyl acetate (× 3). The organic extracts were combined and
washed with water and brine, then dried (Na2SO4). The solvent was removed under reduced
pressure and the crude material purified by flash chromatography on silica gel, eluting with 3 %
ethyl acetate/n-hexane, to afford the product 2.82 as a clear colourless oil (1.20 g, 75 %). 1H
NMR (300 MHz; CDCl3) δ 0.81 (t, J = 7.1 Hz, 9H), 0.96 – 1.18 (m, 6H), 1.26 – 1.40 (m, 6H), 1.33
(d, J = 6.1 Hz, 6H), 1.45 – 1.56 (m, 6H), 2.50 (s, 3H), 5.03 (sept, J = 6.1 Hz, 1H), 6.94 (d, J = 7.3
Hz, 1H), 7.41 (td, J = 19.9 and 7.3 Hz, 1H); 13C NMR (75 MHz; CDCl3) δ 10.7 (t, JC-Sn = 171.4
Hz), 14.0, 21.0, 22.8, 27.7 (t, JC-Sn = 30.5 Hz), 29.3 (t, JC-Sn = 10.0 Hz), 75.4, 104.5, 117.6, 124.2,
133.2, 141.8, 144.3, 165.0; IR (NaCl, neat) 2219 cm-1; HRMS (ESI-MS): m/z calcd for
C23H39NO120SnNa [M+Na]+ 488.1951, found 488.1948.
2-Isopropoxy-6-methylbenzonitrile (5.14)
2-bromopropane (56 μL, 0.60 mmol) was added dropwise to a suspension of nitrile 2.73 (39 mg,
0.30 mmol) and potassium carbonate (82 mg, 0.59 mmol) in DMF (2 mL) at room temperature.
The suspension was stirred at 60°C for 17 h then allowed to cool to room temperature. then
diluted with water and extracted with ethyl acetate (× 3). The organic extracts were combined and
washed with water and brine, then dried (Na2SO4). The solvent was removed under reduced
pressure and the crude material purified by flash chromatography on silica gel, eluting with 1 %
ethyl acetate/n-hexane, to afford the product 5.14 as a clear colourless oil (46 mg, 89 %). 1H
NMR (400 MHz; CDCl3) δ 1.39 (d, J = 6.0 Hz, 6H), 2.48 (s, 3H), 4.62 (sept, J = 6.0 Hz, 1H), 6.76
(d, J = 8.7 Hz, 1H), 6.82 (d, J = 7.8 Hz, 1H), 7.35 (dd, J = 8.7 and 7.8 Hz, 1H); 13C NMR (100

Chapter 5: Experimental
173
MHz; CDCl3) δ 20.9, 22.2, 72.0, 104.0, 110.9, 116.2, 122.0, 133.6, 144.3, 160.6; IR (NaCl, neat)
2222 cm-1; HRMS (ESI-MS): m/z calcd for C11H13NONa [M+Na]+198.0894, found 198.0891.
3-Bromo-2-benzyloxy-6-methylbenzonitrile (2.90)
Benzyl bromide (1.9 mL, 15.64 mmol) was added dropwise to a suspension of nitrile 2.78 (3.15 g,
5.88 mmol) and potassium carbonate (6.16 g, 44.56 mmol) in DMF (148 mL) at room
temperature. The suspension was stirred at room temperature for 17 h then diluted with water
and extracted with ethyl acetate (× 3). The organic extracts were combined and washed with
water and brine, then dried (Na2SO4). The solvent was removed under reduced pressure and the
crude material purified by flash chromatography on silica gel, eluting with 8 % ethyl acetate/n-
hexane, to afford the product 2.90 as a white solid (1.73 g, 97 %). Mp 77-78°C; 1H NMR (300
MHz; CDCl3) δ 2.50 (s, 3H), 5.17 (s, 2H), 6.96 (d, J = 8.3 Hz, 1H), 7.38-7.45 (m, 3H), 7.59-7.61
(m, 2H), 7.66 (d, J = 8.3 Hz, 1H); 13C NMR (75 MHz; CDCl3) δ 20.5, 76.7, 110.1, 114.7, 115.0,
127.0, 128.7, 128.9, 129.1, 135.5, 137.5, 143.4, 158.0; IR (NaCl, neat) 2226 cm-1; HRMS (ESI-
MS): m/z calcd for C15H1279BrNONa [M+Na]+ 324.0000, found 323.9992.
3-(Tri-n-butylstannyl)-2-benzyloxy-6-methylbenzonitrile (2.83)
A solution of t-butyllithium in pentane (1.8 mL, 1.1 M, 1.98 mmol) was added dropwise to a
solution of nitrile 2.90 (0.37 g, 1.22 mmol) in THF (15 mL) at -95°C. The reaction solution was
stirred at -95°C for 15 min after which, tri-n-butyltin chloride (0.36 mL, 1.33 mmol) was added
dropwise. The solution was allowed to warm slowly to -10°C over 5 h where it was quenched with
saturated aqueous sodium bicarbonate solution. The mixture was allowed to warm to room
temperature then extracted with ethyl acetate (× 3). The organic extracts were combined and
washed with water and brine, then dried (Na2SO4). The solvent was removed under reduced
pressure and the crude material purified by flash chromatography on silica gel, eluting with 3 %
ethyl acetate/n-hexane, to afford the product 2.83 as a clear colourless oil (0.44 g, 70 %). 1H
NMR (400 MHz; CDCl3) δ 0.84 (t, J = 7.1 Hz, 9H), 0.88 – 1.01 (m, 6H), 1.19 – 1.31 (m, 6H), 1.35
– 1.48 (m, 6H), 2.55 (s, 3H), 5.20 (s, 2H), 7.04 (d, J = 7.4 Hz, 1H), 7.35 – 7.43 (m, 3H), 7.48 (d, J
= 7.4 Hz, 1H), 7.50 – 7.53 (m, 2H); 13C NMR (100 MHz; CDCl3) δ 10.2 (t, JC-Sn = 172.2 Hz), 13.8,
20.7, 27.4 (t, JC-Sn = 32.1 Hz), 29.1 (t, JC-Sn = 9.7 Hz), 76.7, 105.1, 116.9, 125.1, 128.3, 128.4,

Chapter 5: Experimental
174
128.6, 132.8, 136.5, 141.6, 144.4, 166.1; IR (NaCl, neat) 2216 cm-1; HRMS (ESI-MS): m/z calcd
for C27H39NO120SnNa [M+H]+ 536.1951, found 536.1956.
2-Benzyloxy-6-methylbenzonitrile (2.87)
Benzyl bromide (0.73 mL, 6.14 mmol) was added dropwise to a suspension of nitrile 2.74 (0.81 g,
6.10 mmol) and potassium carbonate (2.53 g, 18.30 mmol) in DMF (60 mL) at room temperature.
The suspension was stirred at room temperature for 17 h then diluted with water and extracted
with ethyl acetate (× 3). The organic extracts were combined and washed with water and brine,
then dried (Na2SO4). The solvent was removed under reduced pressure and the crude material
purified by flash chromatography on silica gel, eluting with 10 % ethyl acetate/n-hexane, to afford
the product 2.87 as a clear colourless oil (3.53 g, 97 %). 1H NMR (300 MHz; CDCl3) δ 2.52 (s,
3H), 5.19 (s, 2H), 6.81 (d, J = 8.5 Hz, 1H), 6.87 (d, J = 7.7 Hz, 1H), 7.30 – 7.48 (m, 6H); 13C NMR
(75 MHz; CDCl3) δ 20.6, 70.6, 103.1, 109.9, 115.7, 122.4, 127.0, 128.2, 128.8, 133.5, 136.0,
144.1, 160.7; IR (NaCl, neat) 2222 cm-1; HRMS (ESI-MS): m/z calcd for C15H13NONa [M+Na]+
246.0894, found 246.0888.
3-Cyano-2-benzyloxy-4-methylphenylmercury acetate (2.84)
Mercury(II) acetate (0.19 g, 0.58 mmol) was added as a solid in one portion to a solution of
stannane 2.83 (0.30 g, 0.59 mmol) in chloroform (5 mL) at room temperature. The solution was
stirred at room temperature protected from light for 16 h. The solvent was removed under
reduced pressure and the crude material triturated with n-hexane to afford the product as a white
solid (0.28 g, 99 %). 1H NMR (300 MHz; CDCl3) δ 2.09 (s, 3H), 2.62 (s, 3H), 5.27 (s, 2H), 7.10 (d,
J = 7.7 Hz, 1H), 7.26 (d, J = 7.7 Hz, 1H), 7.30 – 7.41 (m, 5H); 13C NMR (75 MHz; CDCl3) δ 20.7,
23.0, 78.1, 108.1, 115.9, 126.4, 129.1, 129.37, 129.41, 129.5, 135.8, 140.5, 145.2, 163.4, 177.4;
IR (NaCl, neat) 1577, 2219 cm-1; HRMS (ESI-MS): m/z calcd for C17H16HgNO3 [M+H]+ 484.0836,
found 484.0231.
Bis(3-cyano-(2-benzyloxy)-4-methylphenyl)mercury (2.85)

Chapter 5: Experimental
175
Mercury(II) trifluoroacetate (38 mg, 89.1 μmol) was added as a solid in one portion to a solution of
stannane 2.83 (92 mg, 0.18 mmol) in chloroform (2 mL) at room temperature. The solution was
stirred at room temperature protected from light for 14 h. The solvent was removed under
reduced pressure and the crude material was recrystallised (EtOH/n-hexane) to afford the
product 2.85 as a white solid (50 mg, 85 %). 1H NMR (400 MHz; CDCl3) δ 2.57 (s, 6H), 5.15 (s,
4H), 7.01 (d, J = 7.6 Hz, 2H), 7.16 (d, J = 7.6 Hz, 2H), 7.25 – 7.26 (m, 6H), 7.31 – 7.33 (m, 4H);
13C NMR (100 MHz; CDCl3) δ 20.7, 77.6, 107.5, 116.9, 125.9, 128.8, 129.05, 129.08, 136.4,
141.5, 143.8, 158.6, 165.8; IR (NaCl, neat) 2219 cm-1; HRMS (ESI-MS): m/z calcd for
C30H24HgN2O2Na [M+Na]+ 669.1442, found 669.1437.
General Procedure for the Conversion of Stannanes to Aryllead Triacetates that were
Monitored by 1H NMR Spectroscopy.
Stannane 2.80, 2.82 or 2.83 (60 μmol) was dissolved in deuterated chloroform (0.6 mL) and
placed in a dry NMR tube. A 1H NMR spectrum (400 MHz: d1 = 3, ns = 2) was taken representing
the 0 h time point. Freshly dried lead tetraacetate (60 μmol) and mercury trifluoroacetate (either
10, 20, 30 or 50 mol %) were added as solids, sequentially to the tube. The tube was purged with
argon, sealed and shaken before being placed in a 30°C water bath protected from light. The
reactions were removed from the water bath momentarily to record 1H NMR spectra at 6, 12, 18
and 24 h time intervals.
The raw data for these experiments can be found in Table 5.1 (Me), 5.2 (i-Pr) and 5.3 (Bn) below.

Chapter 5: Experimental
176
Table 5.1: Relative abundances (%) of species present in the transmetalation reaction of stannane 2.80 to aryllead
triacetate using different loadings of mercury(II) trifluoroacetate catalyst recorded over time. Experiments were
conducted using the general procedure outlined above. The relative abundances were determined by integration of
the methyl ether signal in the 1H NMR spectrum. In this case the mercury compounds could not be distinguished and
thus, these are given as a combined total (R: OAc or Ar).
Relative Abundance (%)
Time (h) ArSnBu3 ArHgR ArPb(OAc)3 ArH
10 mol % 0 100 0 0 0
6 61 9 22 8
12 38 12 38 12
18 32 9 45 14
24 25 10 50 15
20 mol % 0 100 0 0 0
6 56 13 21 10
12 32 18 36 14
18 18 18 46 18
24 10 20 50 20
30 mol % 0 100 0 0 0
6 40 13 33 14
12 19 14 48 19
18 6 17 55 22
24 6 17 55 22
50 mol % 0 100 0 0 0
6 41 12 29 18
12 13 17 43 26
18 0 15 50 35
24 0 15 50 35

Chapter 5: Experimental
177
Table 5.2: Relative abundances (%) of species present in the transmetalation reaction of stannane 2.82 to aryllead
triacetate using different loadings of mercury(II) trifluoroacetate catalyst recorded over time. Experiments were
conducted using the general procedure outlined above. The relative abundances were determined by integration of
the isopropyl ether CH signal in the 1H NMR spectrum. In this case the mercury compounds could not be
distinguished and thus, these are given as a combined total (R: OAc or Ar).
Relative Abundance (%)
Time (h) ArSnBu3 ArHgR ArPb(OAc)3 ArH
10 mol % 0 100 0 0 0
6 64 10 8 18
12 60 9 9 22
18 55 10 11 25
24 50 12 13 25
20 mol % 0 100 0 0 0
6 48 17 11 24
12 28 20 20 32
18 14 22 28 36
24 6 23 32 39
30 mol % 0 100 0 0 0
6 11 34 23 32
12 0 33 33 33
18 0 29 36 36
24 0 27 35 35
50 mol % 0 100 0 0 0
6 10 43 20 27
12 0 37 33 30
18 0 31 38 31
24 0 25 42 33

Chapter 5: Experimental
178
Table 5.3: Relative abundances (%) of species present in the transmetalation reaction of stannane 2.83 to aryllead
triacetate using different loadings of mercury(II) trifluoroacetate catalyst recorded over time. Experiments were
conducted using the general procedure outlined above. The relative abundances were determined by integration of
the benzyl ether CH2 signal in the 1H NMR spectrum.
Relative Abundance (%)
Time (h) ArSnBu3 ArHgOAc ArHgAr ArPb(OAc)3 ArH
10 mol % 0 100 0 0 0 0
6 71 8 4 6 9
12 63 8 4 10 13
18 59 7 4 12 16
24 53 8 4 14 19
20 mol % 0 100 0 0 0 0
6 43 9 15 16 17
12 25 10 14 25 25
18 15 12 13 30 28
24 13 13 13 32 30
30 mol % 0 100 0 0 0 0
6 32 9 13 23 21
12 8 12 12 38 31
18 0 17 6 43 34
24 0 14 5 45 36
50 mol % 0 100 0 0 0 0
6 0 17 14 34 34
12 0 19 7 37 37
18 0 15 6 38 42
24 0 12 4 38 46

Chapter 5: Experimental
179
2-Benzyloxy-3-(4’-hydroxy-5’-methoxy-2’-methylnaphthalen-3’-yl)-6-methylbenzonitrile
(2.88)
(a) A solution of stannane 2.83 (1.72 g, 3.36 mmol) in dry chloroform (17 mL) was added
dropwise to a solution of freshly dried lead(IV) tetraacetate (1.49 g, 3.35 mmol) and mercury(II)
trifluoroacetate (0.43 mg, 1.01 mmol) in dry dichloromethane (17 mL). The solution was protected
from light and stirred at room temperature for 18 h. The reaction mixture was filtered through a
short plug of Celite, eluting with dichloromethane. The solvent was removed under reduced
pressure and the crude material was triturated with n-hexane to afford crude aryllead triacetate
2.86 as a light yellow gum (1.57 g). Due to the lability of this compound it was used immediately
in the next step. All of the analytical data matched that reported for later for 2.86.
(b) A solution of crude aryllead triacetate 2.86 (1.57 g) in dry chloroform (12 mL) was added
dropwise to a solution of naphthol 2.52 (0.32 g, 1.70 mmol) and dry pyridine (0.59 mL, 7.31
mmol) in dry chloroform (12 mL) at room temperature. The solution was protected from light and
stirred at room temperature for 16 h. The reaction mixture was poured onto 1M aqueous
hydrochloric acid and extracted with dichloromethane (× 3). The organic extracts were combined
and washed with 1M aqueous hydrochloric acid solution, water and brine, then dried (Na2SO4).
The solvent was removed under reduced pressure and the crude material was purified by flash
chromatography on deactivated silica gel, eluting with 10 % ethyl acetate/n-hexane, to afford the
product 2.88 as a white solid (0.26 g, 38 % over two steps). All the analytical data matched that
reported later for 2.88.
2-Benzyloxy-3-cyano-4-methylphenylboronic acid (2.89)
A solution of t-butyllithium in pentane (2.4 mL, 1.3 M, 3.12 mmol) was added dropwise to a
solution of bromide 2.90 (0.68 g, 2.23 mmol) in freshly distilled THF (22 mL) at -95°C (liquid
N2/ethanol). The dark red/purple reaction solution was stirred at -95°C for 15 min, then freshly

Chapter 5: Experimental
180
distilled triisopropylborate (1.0 mL, 4.33 mmol) was added dropwise. The solution was allowed to
slowly warm to 0°C over 6 h. then quenched dropwise with water. The mixture was stirred
vigorously at room temperature for 2 h. The mixture was extracted with ethyl acetate (× 3). The
organic extracts were combined and washed with water and brine, then dried (Na2SO4). The
solvent was removed under reduced pressure and the crude material was purified by flash
chromatography on silica gel, eluting with 30 % ethyl acetate/n-hexane, to afford the product 2.89
as a white solid (0.50 g, 84 %). Due to the lability of this compound it was used immediately in the
next step. Mp 131 - 132°C; 1H NMR (400 MHz; CDCl3) δ 2.60 (s, 3H), 5.23 (s, 2H), 5.89 (br s,
2H), 7.15 (d, J = 7.6 Hz, 1H), , 7.42-7.46 (m, 3H), 7.52-7.54 (m, 2H), 7.95 (d, J = 7.6 Hz, 1H); 13C
NMR (100 MHz; CDCl3) δ 21.0, 78.8, 106.8, 116.0, 126.1, 129.1, 129.4, 129.5, 134.9, 140.8,
147.8, 166.6; IR (NaCl, neat) 3333, 2246 cm-1; HRMS (ESI-MS): m/z calcd for C15H14BNO3Na
[M+Na]+ 290.0964, found 290.0960.
2-Benzyloxy-3-(4’-hydroxy-5’-methoxy-2’-methylnaphthalen-3’-yl)-6-methylbenzonitrile
(2.88)
(a) A solution of boronic acid 2.89 (0.35 g, 1.32 mmol) in dry dichloromethane (3.5 mL) was
added dropwise to a solution of freshly dried lead(IV) tetraacetate (0.62 g, 1.39 mmol) and
mercury(II) trifluoroacetate (56 mg, 0.13 mmol) in dry dichloromethane (3.5 mL). The solution was
protected from light and stirred at room temperature for 13 h. The reaction mixture was diluted
with dichloromethane and washed with water, then dried (Na2SO4). The solution was filtered
through a short plug of Celite, eluting with dichloromethane. The solvent was removed under
reduced pressure to afford crude aryllead triacetate 2.86 as a light yellow gum (0.72 g). Due to
the lability of this compound it was used immediately in the next step. 1H NMR (300 MHz; CDCl3)
δ 1.98 (s, 9H), 2.61 (t, JH-Pb = 8.4 Hz, 3H), 5.44 (s, 2H), 7.31 (d, J = 8.1 Hz, 1H), 7.38 – 7.45 (m,
3H), 7.65 – 7.68 (m, 2H), 7.96 (d, J = 8.1 Hz, 1H); 13C NMR (75 MHz; CDCl3) δ 19.9, 20.8, 77.9,
107.5, 114.9, 127.1, 128.5, 128.8, 129.3, 134.5, 136.1, 148.4, 152.9, 160.0, 180.1.
(b) A solution of crude aryllead triacetate 2.86 (0.72 g 1.18 mmol) in dry 1,2-dichloroethane
(3 mL) was added dropwise to a solution of naphthol 2.52 (0.22 g, 1.18 mmol) and dry pyridine

Chapter 5: Experimental
181
(0.38 mL, 4.7 mmol) in dry 1,2-dichloroethane (3 mL) at room temperature. The solution was
protected from light and stirred at room temperature for 16 h. The reaction mixture was poured
onto 1M aqueous hydrochloric acid and extracted with dichloromethane (× 3). The organic
extracts were combined and washed with 1M aqueous hydrochloric acid solution, water and
brine, then dried (Na2SO4). The solvent was removed under reduced pressure and the crude
material was purified by flash chromatography on deactivated silica gel, eluting with 15 % ethyl
acetate/n-hexane, to afford the product 2.88 as a white solid (0.31 g, 77 % over two steps). Mp
105 - 107°C; 1H NMR (500 MHz; CDCl3) δ 2.19 (s, 3H), 2.62 (s, 3H), 4.02 (s, 3H), 4.74 (d, J =
10.8 Hz, 1H), 4.88 (d, J = 10.8 Hz, 1H), 6.77 (d, J = 7.5 Hz, 1H), 7.03 – 7.04 (m, 2H), 7.13 – 7.18
(m, 4H), 7.28 (s, 1H), 7.34 – 7.38 (m, 1H), 7.40 – 4.43 (m, 2H), 9.62 (s, 1H); 13C NMR (75 MHz;
CDCl3) δ 20.6, 20.7, 56.1, 76.1, 103.7, 108.6, 113.3, 116.1, 119.1, 119.6, 121.3, 125.5, 126.3,
128.0, 128.2, 128.4, 129.2, 136.3, 136.4, 136.8, 137.3, 142.9, 151.3, 156.2, 159.5; IR (NaCl,
neat) 2224, 3374 cm-1; HRMS (ESI-MS): m/z calcd for C27H23NO3Na [M+Na]+ 432.1576, found
432.1577.
2-Benzyloxy-3-(4’-benzyloxy-5’-methoxy-2’-methylnaphthalen-3’-yl)-6-methylbenzonitrile
(5.15)
A solution of arylated naphthol 2.88 (0.46 g, 1.13 mmol) in dry DMF (16 mL) was added dropwise
to a suspension of freshly washed sodium hydride (0.30 g, 1.25 mmol) in dry DMF (5 mL). The
solution was stirred at room temperature for 1 h resulting in a dark green solution. Benzyl bromide
(0.15 mL, 1.26 mmol) was added dropwise at room temperature and the reaction solution
continued to stir at room temperature for a further 15 h. The reaction mixture was poured onto
saturated aqueous sodium bicarbonate solution and the mixture extracted with ethyl acetate (×
4). The organic extracts were combined and washed with water and brine, then dried (Na2SO4).
The solvent was removed under reduced pressure and the crude material was purified by flash
chromatography on silica gel, eluting with 15 % ethyl acetate/n-hexane, to afford the product 5.15
as a white solid (0.49 g, 87 %). Mp 121 - 123°C; 1H NMR (500 MHz; CDCl3) δ 2.20 (s, 3H), 2.64
(s, 3H), 3.87 (s, 3H), 4.69 (d, J = 10.6 Hz, 1H), 4.89 (d, J = 10.5 Hz, 2H), 4.96 (d, J = 10.6 Hz,
1H), 6.88 – 6.89 (m, 1H), 7.03 – 7.07 (m, 3H), 7.11 – 7.14 (m, 3H), 7.17 – 7.20 (m, 2H), 7.29 –
7.30 (m, 3H), 7.43 – 7.47 (m, 3H), 7.57 (s, 1H); 13C NMR (125 MHz; CDCl3) δ 20.6, 20.9, 56.0,
75.8, 76.3, 105.6, 108.0, 116.3, 118.9, 120.5, 124.8, 124.9, 126.7, 127.6, 128.0, 128.16, 128.20,

Chapter 5: Experimental
182
128.24, 129.1, 129.5, 136.2, 136.4, 137.3, 138.0, 143.1, 152.6, 156.2, 159.6; IR (NaCl, neat)
2224 cm-1; HRMS (ESI-MS): m/z calcd for C34H29NO3Na [M+Na]+ 522.2045, found 522.2025.
(S)-(+)-N-Ethene-t-butylsulfinimine ((S)-2.62)[20]
Acetaldehyde (0.23 mL, 4.11 mmol) was added to a suspension of (S)-(+)-2-methylpropane-2-
sulfinamide ((S)-2.91) (0.17 g, 1.39 mmol) and crushed 4Å molecular sieves (6 g) in dry
dichloromethane (14 mL). The solution was stirred at room temperature for 10 h where further
acetaldehyde (0.23 mL, 4.11 mmol) was added. After stirring for a further 15 h. the reaction
mixture was filtered through a short plug of Celite, which was washed thoroughly with
dichloromethane. The solvent was removed under reduced pressure and the material was
purified by flash chromatography on silica gel, eluting with dichloromethane, to afford the product
(S)-2.62 as a light yellow oil (0.18 g, 88 %) with all the analytical data matching that reported in
the literature.[20] 𝛼𝐷24 + 218 (0.5, CHCl3), 1H NMR (300 MHz; CDCl3) δ 1.19 (s, 9H), 2.24 (d, J =
5.0 Hz, 3H), 8.09 (q, J = 5.0 Hz, 1H); 13C NMR (100 MHz; CDCl3) δ 22.5, 22.6, 56.7, 166.1.
(S)-N-[(2R)-1-(3-Benzyloxy-2-cyano-4-(4’-benzyloxy-5’-methoxy-2’-methylnaphthalen-3’-
yl)phenyl)propan-2-yl]-2-methylpropane-2-sulfinamide (2.93)
A solution of n-butyllithium in hexanes (0.47 mL, 1.4 M, 0.66 mmol) was added dropwise to a
solution of freshly distilled N,N-diisopropylamine (93 μL, 0.66 mmol) in freshly distilled THF (1.3
mL) at -78°C (dry ice/acetone). After addition, the -78°C cold bath was replaced with a 0°C cold
bath and the solution was stirred at 0°C temperature for 15 min. After this time the solution was
cooled back down to -78°C and to this, a solution of nitrile 5.15 (0.11 g, 0.22 mmol) in freshly
distilled THF (0.5 mL) at -78°C was added dropwise. The solution turned dark red and was
continued to stir at -78°C for 30 min. After this time a solution of (S)-sulfinimine (S)-2.62 (49 mg,
0.33 mmol) in freshly distilled THF (0.5 mL) at -78°C was added dropwise and the solution was
stirred at -78°C for a further 2 h. The reaction was quenched at -78°C with saturated aqueous
ammonium chloride solution and allowed to warm to room temperature. The mixture was diluted
with water and extracted with dichloromethane (× 4). The organic extracts were combined and
washed with water and brine, then dried (Na2SO4). The solvent was removed under reduced
pressure and the crude material was purified by flash chromatography on deactivated silica gel,

Chapter 5: Experimental
183
eluting with 60 % ethyl acetate/n-hexane, to afford the product 2.93 as a mixture of atropisomers
as a yellow gum (0.10 g, 72 %). [𝛼]𝐷23.3 = - 34 (0.5, CHCl3); 1H NMR (500 MHz; CDCl3) δ 1.07 (s,
9H, one atropisomer), 1.08 (s, 9H, one atropisomer), 1.45 (d, J = 6.5 Hz, 3H, one atropisomer),
1.47 (d, J = 6.5 Hz, 3H, one atropisomer), 2.13 (s, 6H, both atropisomers), 2.98 – 3.18 (m, 6H,
both atropisomers), 3.67 – 3.73 (m, 1H, one atropisomers), 3.74 – 3.80 (m, 1H, one
atropisomers), 3.83 (s, 6H, both atropisomers), 4.66 (d, J = 10.3 Hz, 2H, both atropisomers), 4.80
– 4.92 (m, 6H, both atropisomers), 6.85 (d, J = 5.1 Hz, 1H, one atropisomers), 6.86 (d, J = 5.1 Hz,
1H, one atropisomer), 6.99 – 7.17 (m, 16H, both atropisomers), 7.24 – 7.29 (m, 6H, both
atropisomers), 7.42 – 7.43 (m, 4H, both atropisomers), 7.46 - 7.49 (m, 2H, both atropisomers),
7.54 (s, 2H, both atropisomers); 13C NMR (125 MHz; CDCl3) δ 20.8, 22.6, 23.65, 23.70, 42.9,
43.3, 54.8, 55.4, 55.93, 55.97, 55.99, 56.01, 75.9, 76.25, 76.27, 105.6, 105.7, 108.6, 109.0,
116.4, 116.6, 118.8, 118.9, 120.5, 124.8, 124.9, 125.4, 126.9, 127.4, 127.5, 127.6, 127.7, 128.02,
128.06, 128.13, 128.20, 128.21, 128.23, 128.27, 128.32, 128.6, 128.83, 128.86, 130.4, 130.5,
135.9, 136.0, 136.18, 136.22, 136.5, 136.6, 137.29, 137.31, 137.8, 137.9, 143.8, 144.0, 152.47,
152.53, 156.2, 159.5, 159.6; IR (NaCl, neat) 2224, 3261, 3405 cm-1; HRMS (ESI-MS): m/z calcd
for C40H42N2O4SNa [M+Na]+ 669.2763, found 669.2731.
(3R)-8-Benzyloxy-7-(4’-benzyloxy-5’-methoxy-2’-methylnaphthalen-3’-yl)-1,3-dimethyl-3,4-
dihydroisoquinoline (2.92)
A solution of methyl lithium in diethyl ether (0.81 mL, 0.6 M, 0.49 mmol) was added dropwise to a
solution of sulfinamide 2.93 (63 mg, 97.4 μmol) in THF (1 mL) at -78°C (liquid N2/ethyl acetate).
The solution was stirred at -78°C for 10 min. then, the cold bath was removed and replaced with
a 0°C cold bath. The solution was stirred at 0°C temperature for a further 30 min. then 2 M
aqueous hydrochloric acid solution (3 mL) was added dropwise. The cold bath was removed and
the solution was stirred at room temperature for 2.5 h. The mixture was neutralised with solid
sodium bicarbonate then diluted with water and extracted with dichloromethane (× 4). The
organic extracts were combined and washed with water and brine, then dried (Na2SO4). The
solvent was removed under reduced pressure and the crude material was purified by flash
chromatography on deactivated silica gel, eluting with 30 % ethyl acetate/n-hexane, to afford the
product 2.92 as a light yellow gum (37 mg, 71 %). [𝛼]𝐷25.6 = -16 (0.5, CHCl3); 1H NMR (400 MHz;
CDCl3) δ 1.44 (d, J = 6.7 Hz, 3H, one atropisomer), 1.49 (d, J = 6.7 Hz, 3H, one atropisomer),

Chapter 5: Experimental
184
2.23 (s, 3H, one atropisomer), 2.29 (s, 3H, one atropisomer), 2.35 (d, J = 1.8 Hz, 3H, one
atropisomer), 2.46 (d, J = 1.8 Hz, 3H, one atropisomer), 2.39 – 2.45 (m, 2H, both atropisomers),
2.70 – 2.77 (m, 2H, both atropisomers), 3.37 – 3.51 (m, 2H, both atropisomers), 3.82 (s, 3H, one
atropisomer), 3.88 (s, 3H, one atropisomer), 4.48 (d, J = 10.8 Hz, 1H, one atropisomer), 4.54 (d, J
= 10.8 Hz, 1H, one atropisomer), 4.60 (d, J = 10.8 Hz, 1H, one atropisomer), 4.63 (d, J = 10.8 Hz,
1H, one atropisomer), 4.73 (d, J = 10.3 Hz, 1H, one atropisomer), 4.74 (d, J = 10.3 Hz, 1H, one
atropisomer), 4.94 (d, J = 10.3 Hz, 1H, one atropisomer), 5.01 (d, J = 10.3 Hz, 1H, one
atropisomer), 6.77 – 6.86 (m, 6H, both atropisomers), 6.97 – 6.99 (m, 2H, both atropisomers),
7.01 – 7.13 (m, 10H, both atropisomers), 7.19 – 7.21 (m, 3H, both atropisomers), 7.24 – 7.25 (m,
3H, both atropisomers), 7.30 (d, J = 7.5 Hz, 1H, one atropisomer), 7.38 (d, J = 7.5 Hz, 1H, one
atropisomer), 7.37 – 7.44 (m, 4H, both atropisomers), 7.45 (s, 1H, one atropisomer), 7.57 (s, 1H,
one atropisomer); 13C NMR (100 MHz; CDCl3) δ 21.1, 21.3, 21.7, 22.3, 26.77, 26.79, 34.5, 34.6,
51.9, 52.3, 56.06, 56.08, 76.18, 76.23, 76.3, 105.5, 105.6, 120.4, 120.5, 122.6, 122.7, 124.6,
124.9, 126.41, 126.44, 127.5, 127.69, 127.74, 127.76, 127.82, 127.9, 128.11, 128.14, 128.17,
128.19, 128.24, 130.7, 130.9, 131.1, 131.3, 133.9, 136.0, 136.6, 136.8, 137.02, 137.06, 138.25,
138.33, 140.1, 140.6, 152.4, 152.8, 155.1, 155.5, 156.2, 156.3, 163.82, 163.84; IR (NaCl, neat)
1613 cm-1; HRMS (ESI-MS): m/z calcd for C37H36NO3 [M+H]+ 542.2695, found 542.2671.
(S)-N-((R)-1-(3-Benzyloxy-2-cyanophenyl)propan-2-yl)-2-methylpropane-2-sulfinamide
(2.106)
A solution of n-butyllithium in hexanes (0.81 mL, 2.2 M, 1.78 mmol) was added dropwise to a
solution of freshly distilled N,N-diisopropylamine (0.25 mL, 1.77 mmol) in freshly distilled THF (8
mL) at -78°C (dry ice/acetone). After addition, the -78°C cold bath was replaced with a 0°C cold
bath and the solution was stirred at 0°C temperature for 15 min. After this time the solution was
cooled back down to -78°C and to this, a solution of nitrile 2.87 (0.26 g, 1.17 mmol) in freshly
distilled THF (4 mL) at -78°C was added dropwise. The solution turned dark red and was
continued to stir at -78°C for 30 min. After this time a solution of (S)-sulfinimine (S)-2.62 (0.18 g,
1.19 mmol) in freshly distilled THF (4 mL) at -78°C was added dropwise and the solution was
stirred at -78°C for a further 2 h. The reaction was quenched at -78°C with saturated aqueous
ammonium chloride solution and allowed to warm to room temperature. The mixture was diluted
with water and extracted with dichloromethane (× 4). The organic extracts were combined and

Chapter 5: Experimental
185
washed with water and brine, then dried (Na2SO4). The solvent was removed under reduced
pressure and the crude material was purified by flash chromatography on silica gel, eluting with
70 % ethyl acetate/n-hexane, to afford the product 2.106 as colourless gum (0.23 g, 53 %).
[𝛼]𝐷25.5 + 16 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 1.05 (s, 9H), 1.38 (d, J = 6.5 Hz, 3H),
2.87 – 3.01 (m, 2 H), 3.15 (d, J = 8.1 Hz, 1H), 3.57 – 3.71 (m, 1H), 5.17 (s, 2H), 6.83 (d, J = 8.4
Hz, 1H), 6.89 (d, J = 7.7 Hz, 1H), 7.29 – 7.43 (m, 6H); 13C NMR (75 MHz; CDCl3) δ 22.6, 23.6,
43.1, 54.9, 56.0, 70.8, 103.9, 110.7, 116.0, 122.7, 127.0, 128.3, 128.9, 133.7, 135.9, 145.0,
160.8; IR (NaCl, neat) 2222, 3252, 3414 cm-1; HRMS (ESI-MS): m/z calcd for C21H26N2O2SNa
[M+Na]+ 393.1612, found 393.1594.
(R)-8-Benzyloxy-1,3-dimethyl-3,4-dihydroisoquinoline (2.104)
A solution of methyl lithium in diethyl ether (1.2 mL, 0.6 M, 0.72 mmol) was added dropwise to a
solution of sulfinamide 2.106 (69 mg, 0.19 mmol) in THF (4 mL) at -78°C (liquid N2/ethyl acetate).
The solution was stirred at -78°C for 10 min. then, the cold bath was removed and replaced with
a 0°C cold bath. The solution was stirred at 0°C temperature for a further 30 min. then 2 M
aqueous hydrochloric acid solution (12 mL) was added dropwise. The cold bath was removed
and the solution was stirred at room temperature for 2.5 h. The mixture was neutralised with solid
sodium bicarbonate then diluted with water and extracted with ethyl acetate (× 3). The organic
extracts were combined and washed with water and brine, then dried (Na2SO4). The solvent was
removed under reduced pressure and the crude material was purified by flash chromatography
on deactivated silica gel, eluting with 50 % ethyl acetate/n-hexane, to afford the product 2.104 as
a light yellow gum (41 mg, 84 %). [𝛼]𝐷27.4 - 28 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 1.37 (d,
J = 6.7 Hz, 3H), 2.35 (dd, J = 15.8, 12.8 Hz, 1H), 2.46 (d, J = 2.0 Hz, 3H), 2.63 (dd, J = 15.8, 4.4
Hz, 1H), 3.28 – 3.40 (m, 1H), 5.11 (s, 2H), 6.79 (d, J = 7.5 Hz, 1H), 6.90 (d, J = 8.4 Hz, 1H), 7.26
(dd, J = 8.4, 7.5 Hz, 1H), 7.31 – 7.45 (m, 5H); 13C NMR (75 MHz; CDCl3) δ; 22.0, 28.1, 34.7,
51.7, 70.8, 111.5, 119.8, 120.2, 127.7, 127.9, 128.2, 128.8, 131.2, 136.6, 141.1, 156.6, 163.4; IR
(NaCl, neat) 1613 cm-1; HRMS (ESI-MS): m/z calcd for C18H20NO [M+H]+ 266.1544, found
266.1532.

Chapter 5: Experimental
186
(1S,3R)-8-Benzyloxy-1,3-dimethyl-1,2,3,4-tetrahydroisoquinoline (2.107)
Sodium borohydride (19 mg, 0.50mmol) was added in one portion to a solution of 1,3-dimethyl-
3,4-dihydroisoquinoline 2.104 (68 mg, 0.26 mmol) in methanol (12 mL) at -10°C. The solution
was allowed to slowly warm, in the cold bath, to room temperature over 15 h. The methanol was
removed under reduced pressure. The residue was dissolved in dichloromethane and filtered
through a short pad of Celite, eluting with dichloromethane. The solvent was removed under
reduced pressure to afford the product 2.107 as a clear colourless gum (47 mg, 68 %). [𝛼]𝐷25.8 =
-92 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 1.24 (d, J = 6.3 Hz, 3H, 3-CH3), 1.52 (d,J = 6.3 Hz,
3H, 1-CH3), 2.48 (dd, J = 15.6, 10.7 Hz, 1H, 4-Heq), 2.70 (dd, J = 15.6, 2.5 Hz, 1H, 4-Hax), 2.84 –
2.95 (m, 1H, 3-H), 4.42 (q, J = 6.3 Hz, 1H, 1-H), 5.06 (d,J = 12 Hz, 1H, 8-OCH(H)Ph), 5.11 (d, J =
12 Hz, 1H, 8-OCH(H)Ph), 6.71 (d, J = 7.6 Hz, 1H, 5-H), 6.77 (d, J = 8.2 Hz, 1H, 7-H), 7.09 (dd,J =
8.2, 7.6 Hz, 1H, 6-H), 7.29 – 7.45 (m, 5H, Bn-ArH); 13C NMR (75 MHz; CDCl3) δ; 22.6 (3-CH3),
23.0 (1-CH3), 39.6 (4-C), 48.4 (3-C), 49.8 (1-C), 70.0 (O-CH2Ph), 109.5 (7-C), 121.8 (5-C), 126.5
(6-C), 127.2 (Bn-ArC), 127.9 (Bn-ArC), 128.7 (Bn-ArC), 129.1 (8a-C), 137.4 (Bn-CH2-ArC), 137.9
(4a-C), 156.3 (8-C); IR (NaCl, neat) 3303 cm-1; HRMS (ESI-MS): m/z calcd for C18H22NO [M+H]+
268.1701, found 268.1685.
(R)-8-Benzyloxy-3-methyl-3,4-dihydroisoquinoline (2.111)
A solution of diisobutylaluminium hydride in hexanes (1.18 mL, 0.8 M, 0.94 mmol) was added
dropwise to a solution of sulfinamide 2.106 (0.11 g, 0.29 mmol) in dry toluene (14 mL) at -10°C.
The solution was stirred at -10°C for 1 h. After this time the solution was cooled back down to
0°C, diluted with diethyl ether (1 mL) and water (16 μL) was added dropwise. 1M Aqueous
sodium hydroxide solution (16 μL) was then added followed by more water (35 μL). The cold bath
was removed and the solution was stirred at room temperature for 15 min, then dried (Na2SO4).
The solution was filtered and the solvent was removed under reduced pressure. The residue was
dissolved in THF (2 mL) and 3 M aqueous hydrochloric acid solution (2 mL) was added dropwise.
The solution was stirred vigorously at room temperature for 13 h. The mixture was diluted with
water and neutralised with solid sodium bicarbonate. The mixture was extracted with
dichloromethane (× 5). The organic extracts were combined and washed with brine, then dried

Chapter 5: Experimental
187
(Na2SO4). The solvent was removed under reduced pressure and the crude material was purified
by flash chromatography on deactivated silica gel, eluting with 20 % ethyl acetate/n-hexane, to
afford the product 2.111 as a light yellow gum (49 mg, 67 %). [𝛼]𝐷25.2 = +32 (0.5, CHCl3); 1H
NMR (400 MHz; CDCl3) δ 1.39 (d, J = 6.8 Hz, 3H), 2.47 (dd, J = 16.4, 12.2 Hz, 1H), 2.63 (dd, J =
16.4, 5.6 Hz, 1H), 3.59 – 3.69 (m, 1H), 5.14 (s, 2H), 6.74 (d, J = 7.5 Hz, 1H), 6.84 (d, J = 8.4 Hz,
1H), 7.26 (dd, J = 8.4, 7.5 Hz, 1H), 7.31 – 7.45 (m, 5H), 8.78 (d, J = 2.6 Hz, 1H); 13C NMR (75
MHz; CDCl3) δ 21.8, 32.8, 51.9, 70.3, 110.9, 120.2, 127.3, 128.2, 128.8, 132.2, 136.7, 138.5,
154.8, 156.3; IR (NaCl, neat) 1622 cm-1; HRMS (ESI-MS): m/z calcd for C17H18NO [M+H]+
252.1388, found 252.1395.
(1R,3R)-8-Benzyloxy-1,3-dimethyl-1,2,3,4-tetrahydroisoquinoline (2.108)
Cerium chloride heptahydrate (0.24 g, 0.64 mmol) was heated at 140°C under reduced pressure
(0.1 mm/Hg) for 16 hours. The flask was allowed to cool to room temperature and freshly distilled
THF (3 mL) was added. The suspension was stirred vigorously at room temperature for 2 hours
then cooled to -78°C (dry ice/acetone). A solution of methyl lithium in diethyl ether (0.63 mL, 1.0
M, 0.63 mmol) was added dropwise. The light yellow suspension was stirred at -78°C for 30 min.,
then the -78°C cold bath was replaced with a 0°C cold bath and the suspension was stirred for 1
h. at that temperature. After this time the light yellow suspension was cooled back down to -78°C
and a solution of 3-methyl-3,4-dihydroisoquinoline 2.111 (16 mg, 63.7 μmol) in freshly distilled
THF (2.5 mL) at -78°C was added dropwise. The solution was allowed to warm slowly in the cold
bath to 0°C, over 5 h. The yellow reaction solution was quenched at 0°C temperature with water
and allowed to warm to room temperature. The THF was removed under reduced pressure and
the mixture was extracted with dichloromethane (× 4). The organic extracts were combined and
washed with water and brine, then dried (Na2SO4). The solvent was removed under reduced
pressure and the crude material was purified by flash chromatography on deactivated silica gel,
eluting with 60 % ethyl acetate/n-hexane, to afford the product 2.108 as a light yellow gum (16
mg, 92 %). [𝛼]𝐷25.2 = -22 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 1.22 (d, J = 6.3 Hz, 3H, 3-
CH3), 1.48 (d, J = 6.7 Hz, 3H, 1-CH3), 2.45 (dd, J = 16.6, 11.0 Hz, 1H, 4-Heq), 2.79 (dd, J = 16.6,
4.0 Hz, 1H, 4-Hax), 3.26 – 3.38 (m, 1H, 3-H), 4.47 (q, J = 6.7 Hz, 1H, 1-H), 5.06 (d, J = 12.1 Hz,
1H, 8-OCH(H)Ph), 5.12 (d, J = 12.1 Hz, 1H, 8-OCH(H)Ph), 6.71 (d, J = 7.6 Hz, 1H, 5-H), 6.73 (d,
J = 8.0 Hz, 1H, 7-H), 7.09 (dd, J 8.0, 7.6 Hz, 1H, 7-H), 7.30 – 7.45 (m, 5H, BnAr-H); 13C NMR (75

Chapter 5: Experimental
188
MHz; CDCl3) δ 21.6 (1-CH3), 23.0 (3-CH3), 37.6 (4-C), 42.0 (3-C), 47.6 (1-C), 69.8 (O-CH2Ph),
108.8 (7-C), 121.7 (5-C), 126.7 (6-C), 127.1 (Bn-ArC), 127.8 (Bn-ArC), 128.9 (Bn-ArC), 129.4
(8a-C), 136.1 (4a-C), 137.5 (Bn-CH2-ArC), 155.2 (8-C); IR (NaCl, neat) 3295 cm-1; HRMS (ESI-
MS): m/z calcd for C18H22NO [M+H]+ 268.1701, found 268.1694.
Phylline (2.97)
Palladium on carbon (50 mg, 10 wt. %) was added to a solution of 1,3-dimethyl-1,2,3,4-
tetrahydroisoquinoline 2.108 (51 mg, 0.19 mmol) in dry methanol (1.9 mL) at room temperature. A
hydrogen balloon was attached to the flask and the flask was evacuated and purged with
hydrogen (× 3). The solution was stirred at room temperature for 14 h. The reaction mixture was
diluted with dichloromethane and filtered through a short pad of Celite, eluting with
dichloromethane. The solvent was removed under reduced pressure to afford the free base of
phylline which was immediately converted to the trifluoroacetate salt by adding trifluoroacetic acid
(20 μL) to the residue in methanol (1.9 mL) at room temperature. The solution was stirred at this
temperature for 1 h then the solvent was removed under reduced pressure to afford the product
2.97 as a clear colourless gum (33 mg, 97 %). [𝛼]𝐷24.0 = - 12.5 (0.08, MeOH); 1H NMR (400 MHz;
MeOD) δ 1.48 (d, J = 6.4 Hz, 3H), 1.64 (d, J = 6.8 Hz, 3H), 2.82 (dd, J = 17.6, 11.7 Hz, 1H), 3.10
(dd, J = 17.6, 4.7 Hz, 3H), 3.76 – 3.84 (m, 1H), 4.76 (q, J = 6.7 Hz, 1H), 6.68 (d, J = 7.9 Hz, 1H),
6.70 (d, J = 7.9 Hz, 1H), 7.10 (dd, J = 7.9, 7.9 Hz, 1H); 13C NMR (100 MHz; MeOD) δ 17.9, 19.2,
34.4, 45.2, 49.4, 114.1, 120.7, 121.3, 129.9, 133.0, 155.1; IR (NaCl, neat) 1675, 3273 cm-1;
HRMS (ESI-MS): m/z calcd for C11H16NO [M+H]+ 178.1232, found 178.1222.
(3R)-8-Benzyloxy-7-(4’-benzyloxy-5’-methoxy-2’-methylnaphthalen-3’-yl)-3-methyl-3,4-
dihydroisoquinoline (2.112)
A solution of diisobutylaluminium hydride in hexanes (0.28 mL, 0.8 M, 0.22 mmol) was added
dropwise to a solution of sulfinamide 2.93 (60 mg, 92.8 μmol) in dry toluene (0.5 mL) at 0°C. The
solution was stirred at 0°C for 15 min then at room temperature for 2 h. After this time the solution
was cooled back down to 0°C, diluted with diethyl ether (1 mL) and water (16 μL) was added
dropwise. 1M Aqueous sodium hydroxide solution (16 μL) was then added followed by more

Chapter 5: Experimental
189
water (35 μL). The cold bath was removed and the solution was stirred at room temperature for
15 min, then dried (Na2SO4). The solution was filtered and the solvent was removed under
reduced pressure. The residue was dissolved in THF (2 mL) and 3 M aqueous hydrochloric acid
solution (2 mL) was added dropwise. The solution was stirred vigorously at room temperature for
13 h. The mixture was diluted with water and neutralised with solid sodium bicarbonate. The
mixture was extracted with dichloromethane (× 5). The organic extracts were combined and
washed with brine, then dried (Na2SO4). The solvent was removed under reduced pressure and
the crude material was purified by flash chromatography on deactivated silica gel, eluting with 35
% ethyl acetate/n-hexane, to afford the product 2.112 as a mixture of atropisomers as a yellow
gum (32 mg, 65 %) [𝛼]𝐷26.7 + 18 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 1.40 (d, J = 6.8 Hz,
3H, one atropisomer), 1.46 (d, J = 6.8 Hz, 3H, one atropisomer), 2.23 (s, 3H, one atropisomer),
2.27 (s, 3H, one atropisomer), 2.49 – 2.60 (m, 2H, both atropisomers), 2.77 – 2.88 (m, 2H, both
atropisomers), 3.60 – 3.69 (m, 1H, one atropisomer), 3.71 – 3.80 (m, 1H, one atropisomer), 3.84
(s, 3H, one atropisomer), 3.87 (s, 3H, one atropisomer), 4.59 – 4.71 (m, 6H, both atropisomers),
4.91 (d, J = 10.1 Hz, 1H, one atropisomer), 4.95 (d, J = 10.1 Hz, 1H, one atropisomer), 6.83 –
6.86 (m, 2H, both atropisomers), 6.90 – 6.93 (m, 4H, both atropisomers), 6.96 – 7.00 (m, 6H, both
atropisomers), 7.05 – 7.17 (m, 6H, both atropisomers), 7.20 – 7.23 (m, 6H, both atropisomers),
7.30 (d, J = 7.6 Hz, 1H, one atropisomer), 7.34 (d, J = 7.6 Hz, 1H, one atropisomer), 7.37 – 7.45
(m, 4H, both atropisomers), 7.55 (d, J = 9.2 Hz, 2H, both atropisomers), 8.61 (d, J = 2.8 Hz, 1H,
one atropisomer), 8.62 (d, J = 2.8 Hz, 1H, one atropisomer); 13C NMR (75 MHz; CDCl3) δ 21.11,
21.14, 21.5, 22.1, 32.6, 32.8, 52.0, 52.3, 56.08, 56.09, 76.4, 76.5, 76.6, 77.4, 105.6, 119.1,
120.50, 120.51, 121.8, 121.9, 122.8, 122.9, 124.71, 124.74, 126.5, 127.56, 127.59, 127.9, 128.0,
128.20, 128.22, 128.3, 128.4, 130.3, 130.4, 134.7, 134.8, 136.4, 136.5, 136.7, 136.8, 137.2,
137.4, 137.9, 138.1, 138.2, 152.6, 152.7, 155.36, 155.40, 155.6, 156.32, 156.34; IR (NaCl, neat)
1622 cm-1; HRMS (ESI-MS): m/z calcd for C36H34NO3 [M+H]+ 528.2539 found 528.2520.
(1R,3R)-8-Benzyloxy-7-(4’-benzyloxy-5’-methoxy-2’-methylnaphthalen-3’-yl)-1,3-dimethyl-
1,2,3,4-tetrahydroisoquinoline (2.113)
Cerium chloride heptahydrate (0.23 g, 0.61 mmol) was heated at 140°C under reduced pressure
(0.1 mm/Hg) for 15 hours. The flask was allowed to cool to room temperature and freshly distilled
THF (3 mL) was added. The suspension was stirred vigorously at room temperature for 2 hours

Chapter 5: Experimental
190
then cooled to -78°C (dry ice/acetone). A solution of methyl lithium in diethyl ether (1.0 mL, 0.6 M,
0.60 mmol) was added dropwise. The light yellow suspension was stirred at -78°C for 30 min.,
then the -78°C cold bath was replaced with a 0°C cold bath and the suspension was stirred for 1
h. at that temperature. After this time the light yellow suspension was cooled back down to -78°C
and a solution of 3-methyl-3,4-dihydroisoquinoline 2.112 (32 mg, 60.7 μmol) in freshly distilled
THF (2.4 mL) at -78°C was added dropwise. The solution was allowed to warm slowly in the cold
bath to 0°C, over 5 h. The yellow reaction solution was quenched at 0°C temperature with water
and allowed to warm to room temperature. The THF was removed under reduced pressure and
the mixture was extracted with dichloromethane (× 4). The organic extracts were combined and
washed with water and brine, then dried (Na2SO4). The solvent was removed under reduced
pressure to afford a light yellow gum which was purified by flash chromatography on deactivated
silica gel, eluting with 60 % ethyl acetate/n-hexane, to afford the product 2.113 as a mixture of
atropisomers as a light yellow gum (20 mg, 61 %). [𝛼]𝐷26.1 - 12 (0.5 , CHCl3); 1H NMR (500 MHz;
CDCl3) δ 1.24 – 1.27 (m, 6H, both atropisomers), 1.38 (d, J = 6.6 Hz, 3H, one atropisomer), 1.47
(d, J = 6.5 Hz, 3H, one atropisomer), 2.23 (s, 3H, one atropisomer), 2.31 (s, 3H, one
atropisomer), 2.51 – 2.60 (m, 2H, both atropisomers), 2.82 – 2.91 (m, 2H, both atropisomers),
3.34 – 3.43 (m, 2H, both atropisomers), 3.79 (s, 3H, one atropisomer), 3.86 (s, 3H, one
atropisomer), 4.35 – 4.62 (m, 7H, both atropisomers), 4.84 (d, J = 10.0 Hz, 1H, one atropisomer),
4.93 – 4.97 (m, 2H, both atropisomers), 6.79 – 6.83 (m, 2H, both atropisomers), 6.91 – 6.95 (m,
8H, both atropisomers), 7.06 – 7.07 (m, 9H, both atropisomers), 7.19 – 7.21 (m, 4H, both
atropisomers), 7.24 – 7.29 (m, 4H, both atropisomers), 7.36 – 7.38 (m, 4H, both atropisomers),
7.52 – 7.54 (m, 2H, both atropisomers); 13C NMR (75 MHz; CDCl3) δ 20.9, 21.4, 22.4, 22.5,
23.06, 23.11, 37.6, 37.7, 41.97, 42.02, 48.5, 56.0, 56.1, 74.2, 74.6, 76.06, 76.11, 77.4, 105.45,
105.49, 119.2, 119.4, 120.4, 120.5, 124.2, 124.4, 124.5, 124.8, 126.2, 126.3, 127.4, 127.45,
127.49, 127.51, 127.55, 127.7, 127.86, 127.93, 128.0, 128.14, 128.15, 128.20, 128.22, 128.5,
129.9, 130.0, 131.5, 131.7, 133.9, 135.48, 135.50, 136.2, 136.97, 137.01, 137.4, 137.9, 138.0,
138.48, 138.51, 152.1, 152.9, 153.8, 154.3, 156.3, 156.4; IR (NaCl, neat) 3307, 3370 cm-1;
HRMS (ESI-MS): m/z calcd for C37H38NO3 [M+H]+ 544.2852 found 544.2846.
Dioncophylline E (2.42)[21]

Chapter 5: Experimental
191
Palladium on carbon (20 mg, 10 wt. %) was added to a solution of 1,3-dimethyl-1,2,3,4-
tetrahydroisoquinoline 2.113 (20 mg, 36.8 μmol) in dry methanol (0.5 mL) at room temperature. A
hydrogen balloon was attached to the flask and the flask was evacuated and purged with
hydrogen (× 3). The solution was stirred at room temperature for 14 h. The reaction mixture was
diluted with dichloromethane and filtered through a short pad of Celite, eluting with
dichloromethane. The solvent was removed under reduced pressure to afford the free base of
dioncophylline E (2.42) which was immediately converted to the trifluoroacetate salt by adding
trifluoroacetic acid (15 μL) to the residue in methanol (1.0 mL) at room temperature. The solution
was stirred at this temperature for 1 h then the solvent was removed under reduced pressure to
afford the product as a clear colourless gum (12 mg, 85 %). 1H NMR (400 MHz; CDCl3) δ 1.59 (d,
J = 6.0 Hz, 3H, one atropisomer), 1.60 (d, J = 5.9 Hz, 3H, one atropisomer), 1.70 (d, J = 6.6 Hz,
3H, one atropisomer), 1.74 (d, J = 6.7 Hz, 3H, one atropisomer), 2.14 (s, 3H, one atropisomer),
2.15 (s, 3H, one atropisomer), 2.99 – 3.15 (m, 4H, both atropisomers), 3.71 – 3.77 (m, 2H, both
atropisomers), 3.99 (s, 3H, one atropisomer), 4.04 (s, 3H, one atropisomer), 4.93 - 4.96 (m, 2H,
both atropisomers), 5.25 (br s, 1H, one atropisomer), 5.49 (br s, 1H, one atropisomer), 6.71 –
6.73 (m, 1H, one atropisomer), 6.76 – 6.79 (m, 1H, one atropisomer), 6.81 (d, J = 7.8 Hz, 2H,
both atropisomers), 7.04 – 7.06 (m, 2H, both atropisomers), 7.23 (s, 1H, one atropisomer), 7.27
(s, 1H, one atropisomer), 7.30 (d, J = 4.3 Hz, 2H, both atropisomers), 7.34 – 7.37 (m, 2H, both
atropisomers), 9.32 (br s, 1H, one atropisomer), 9.47 (br s, 1H, one atropisomer), 9.71 (s, 1H,
one atropisomer), 9.74 (s, 1H, one atropisomer), 9.90 (br s, 1H, one atropisomer), 10.03 (br s,
1H, one atropisomer); 13C NMR (100 MHz; CDCl3) δ 18.0, 18.3, 18.7, 19.0, 20.9, 21.0, 33.9, 44.4,
44.7, 48.5, 56.3, 104.0, 113.4, 116.9, 117.2, 120.2, 120.5, 120.8, 121.1, 121.2, 121.3, 122.1,
122.6, 126.8, 130.2, 130.4, 130.7, 131.3, 136.4, 136.5, 137.5, 138.1, 149.6, 149.8, 151.8, 151.9,
156.1. IR (NaCl, neat) 3370, 1674 cm-1; HRMS (ESI-MS): m/z calcd for C23H26NO3 [M+H]+
364.1913, found 364.1900.
(R)-(-)-N-(ethene)-t-butylsulfinimine ((R)-2.62)[22]
Acetaldehyde (0.54 mL, 9.61 mmol) was added to a solution of (R)-(+)-2-methyl-2-
propanesulfinamide ((R)-2.91) (0.388 g, 3.20 mmol) and crushed 4A molecular sieves (15 g) in
dry dichloromethane (40 mL). The solution was stirred at room temperature for 24 h after which
further acetaldehyde (0.54 mL, 9.61 mmol) was added and the reaction solution stirred o.n. After
this time the reaction solution was filtered though a plug of Celite which was washed thoroughly

Chapter 5: Experimental
192
with ethyl acetate. The solvent was removed under reduced pressure to afford the product (R)-
2.62 as a clear yellow oil which was used without any further purification (0.457 g, 97 %). [𝛼]𝐷23 =
-326 (0.5, CHCl3) (lit.[23] -409 (1.0, CHCl3). All of the analytical data matched that reported
previously for the enantiomer (S)-2.62.
(Rs)-N-((S)-1-(2-cyano-3,5-dimethoxyphenyl)propan-2-yl)-2-methyl-2-propanesulfinamide
(2.63)[23]
A solution of n-butyllithium in hexanes (7.1 mL, 1.4 M, 9.94 mmol) was added dropwise to a
solution of freshly distilled N,N-diisopropylamine (1.4 mL, 9.94 mmol) in freshly distilled THF (50
mL) at -78°C (dry ice/acetone). After addition, the -78°C cold bath was replaced with a 0°C cold
bath and the solution was stirred at 0°C temperature for 15 min. After this time the solution was
cooled back down to -78°C and to this, a solution of nitrile 2.57[24] (0.88 g, 4.95 mmol) in freshly
distilled THF (10 mL) at -78°C was added dropwise. The solution turned dark red and was
continued to stir at -78°C for 30 min. After this time a solution of (R)-sulfinimine (R)-2.91 (0.77 g,
5.20 mmol) in freshly distilled THF (20 mL) at -78°C was added dropwise and the solution was
stirred at -78°C for a further 1 h. The reaction was quenched at -78°C with saturated aqueous
ammonium chloride solution and allowed to warm to room temperature. The mixture was diluted
with water and extracted with dichloromethane (× 4). The organic extracts were combined and
washed with water and brine, then dried (Na2SO4). The solvent was removed under reduced and
the crude material was purified by flash chromatography on silica gel, eluting with 75 % ethyl
acetate/n-hexane, to afford the product 2.63 as colourless gum (1.02 g, 63 %) with all analytical
data matching that reported in the literature.[25] [𝛼]𝐷23 = - 4 (0.5, CHCl3) (lit.[25] -3.7 (4.3, CHCl3);
1H NMR (300 MHz, CDCl3) δ 1.10 (s, 9H), 1.38 (d, J = 6.4 Hz, 3H), 2.84 – 3.02 (m, 2H), 3.11 (br
d, J = 7.6 Hz, 1H), 3.59 – 3.72 (m, 1H), 3.84 (s, 3H), 3.87 (s, 3H), 6.32 (d, J = 2.0 Hz, 1H), 6.44
(d, J = 2.0 Hz, 1H).
(S)-6,8-Dimethoxy-1,3-dimethyl-3,4-dihydroisoquinoline (2.120)
A solution of methyl lithium in diethyl ether (10.4 mL, 0.8 M, 8.32 mmol) was added dropwise to a
solution of sulfinamide 2.63 (0.67 g, 2.07 mmol) in THF (13 mL) at -78°C. The dark red solution

Chapter 5: Experimental
193
was stirred at -78°C for 15 min then the cold bath was removed and the solution was allowed to
stir at room temperature for 2 h. 2 M Aqueous hydrochloric acid solution (30 mL) was added
dropwise and the solution was stirred at room temperature for 3 h. The mixture was neutralised
using solid sodium bicarbonate, diluted with water and extracted with dichloromethane (× 4). The
organic extracts were combined and washed with water and brine, then dried (Na2SO4). The
solvent was removed under reduced pressure and the crude material was purified by flash
chromatography on deactivated silica gel, eluting with 60 % ethyl acetate/n-hexane, to afford the
product 2.120 as a clear gum (0.28 g, 62 %) with all analytical data matching that reported in the
literature.[24] [𝛼]𝐷25.1 = - 115 (0.5, CHCl3); 1H NMR (300 MHz, CDCl3) δ 1.37 (d, J = 6.7 Hz, 3H),
2.35 (dd, J = 15.8, 13.1 Hz, 1H), 2.44 (d, J = 1.6 Hz, 3H), 2.60 (dd, J = 15.8, 4.5 Hz, 1H), 3.29 –
3.39 (m, 1H), 3.83 (s, 3H), 3.84 (s, 3H), 6.31 (d, J = 2.3 Hz, 1H), 6.36 (d, J = 2.3 Hz, 1H).
t-Butyl (1R,3S)-6,8-dimethoxy-1,3-dimethyl-1,2,3,4-tetrahydroisoquinoline carboxylate
(2.121)[26]
a) Sodium borohydride (0.25 g, 6.69 mmol) was added as a solid in one portion to a solution
of 1,3-dimethyl-3,4-dihydroisoquinoline 2.120 (0.29 g, 1.11 mmol) in methanol (55 mL) at -10°C.
The solution was allowed to slowly warm to room temperature, in the cold bath, over 16 h. The
methanol was removed under reduced pressure. The residue was dissolved in dichloromethane
and filtered through a short plug of Celite, eluting with dichloromethane. The solvent was
removed under reduced pressure to afford the product 5.16 as a clear colourless gum which was
of sufficient purity to use in the next step without further purification. 1H NMR (400 MHz, CDCl3) δ
1.21 (d, J = 6.3 Hz, 3H), 1.43 (d, J = 6.3 Hz, 3H), 2.43 (dd, J = 15.8, 10.9 Hz, 1H), 2.62 (dd, J =
15.8, 2.5 Hz, 1H), 2.81 – 2.89 (m, 1H), 3.78 (s, 6H), 4.22 (q, J = 6.3 Hz, 1H), 6.20 (d, J = 2.2 Hz,
1H), 6.31 (d, J = 2.2 Hz, 1H).
b) The residue was dissolved in dichloromethane (11 mL) and triethylamine (0.43 mL, 3.31
mml) and di-t-butyl dicarbonate (0.27 g, 1.22 mmol) were added successively at room
temperature. The solution was stirred at room temperature for 18 h then poured onto water. The
mixture was extracted with dichloromethane (× 4). The organic extracts were combined and
washed with water and brine, then dried (Na2SO4). The solvent was removed under reduced
pressure and the crude material purified by flash chromatography on silica gel, eluting with 20 %
ethyl acetate/n-hexane, to afford the product 2.121 as a clear colourless gum (0.35 g, 98 %) with

Chapter 5: Experimental
194
all of the analytical data matching that reported in the literature. [𝛼]𝐷26.4 = - 10 (0.5, CHCl3); 1H
NMR (300 MHz, CDCl3) δ 1.33 (d, J = 6.5 Hz, 3H), 1.40 (d, J = 6.8 Hz, 3H), 1.48 (s, 9H), 2.68
(dd, J = 15.8, 6.5 Hz, 1H), 2.93 (dd, J = 15.8, 6.5 Hz, 1H), 3.78 (s, 3H), 3.79 (s, 3H), 4.34 (br s,
1H), 5.41 (br s, 1H), 6.26 (d, J = 2.3 Hz, 1H), 6.32 (d, J = 2.3 Hz, 1H).
t-Butyl (1R,3S)-5-iodo-6,8-dimethoxy-1,3-dimethyl-1,2,3,4-tetrahydroisoquinoline
carbamate (5.17)
A solution of iodine (0.29 g, 1.14 mmol) in dry ethanol (1.5 mL) was added dropwise to a solution
of 1,3-dimethyl-1,2,3,4-tetrahydroisoquinoline 2.121 (0.35 g, 1,09 mmol) and silver sulfate (0.68
g, 2.17 mmol) in dry ethanol (7.2 mL) at 0°C. The reaction mixture was allowed to slowly warm to
room temperature in the cold bath over 16 h. The solution was filtered through a plug of Celite
which was washed thoroughly with dichloromethane. The solvent was removed under reduced
pressure and the residue was dissolved in dichloromethane and washed with 10% aqueous
sodium thiosulfate solution, water and brine, then dried (Na2SO4). The solvent was removed
under reduced pressure and the crude material purified using flash chomatography on silica gel,
eluting with 15 % ethyl acetate/n-hexane, to afford the product 5.17 as a gum (0.34 g, 70 %).
[𝛼]𝐷25.7 = + 22 (0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 1.36 (d, J = 6.5 Hz, 3H), 1.37 (d, J =
6.8 Hz, 3H), 1.47 (s, 9H), 2.76 (dd, J = 16.5, 7.3 Hz, 1H), 3.19 (br s, 1H), 3.84 (s, 3H), 3.88 (s,
3H), 4.20 (br s, 1H), 5.52 (br s, 1H), 6.35 (s, 1H); 13C NMR (100 MHz, CDCl3, 50°C) δ 21.6, 23.5,
28.7, 40.7, 45.6, 46.8, 55.8, 56.8, 79.6, 82.0, 94.5, 122.8, 138.2, 154.9, 156.7, 157.8; IR (NaCl,
neat) 1686 cm-1; HRMS (ESI-MS): m/z calcd for C18H26NO4INa [M+Na]+ 470.0804, found
470.0785.
((1R,3S)-2-(t-Butoxycarbonyl)-6,8-dimethoxy-1,3-dimethyl-1,2,3,4-tetrahydroisoquinolin-5-
yl) boronic acid (2.119)
A solution of t-butyllithium in n-pentane (0.35 mL, 1.4 M, 0.49 mmol) was added dropwise to a
solution of iodide 5.17 (0.11 g, 0.25 mmol) [iodide 5.17 was dried azeotropically with benzene (×
3) and residual benzene was removed under reduced pressure immediately before use] and

Chapter 5: Experimental
195
freshly distilled B(Oi-Pr)3 (0.12 mL, 0.52 mmol) in dry THF (5 mL) at -95°C. The solution was
stirred for 15 min at this temperature and then the cold bath was replaced with a -10°C cold bath.
The solution was stirred for a further 30 min then quenched with saturated aqueous ammonium
chloride solution. The mixture was extracted with dichloromethane (× 4). The organic extracts
were combined and washed with water and brine, then dried (Na2SO4). The solvent was removed
under reduced pressure and the crude material purified by flash chromatography on silica gel,
eluting with 50 % ethyl acetate/n-hexane, to afford the product 2.119 as a clear colourless gum
(82 mg, 90 %). [𝛼]𝐷26.4 = + 20 (0.5, MeOH); 1H NMR (600 MHz, MeOD) δ 1.55 – 1.57 (m, 6H),
1.67 (s, 9H), 2.86 (dd, J = 15.9, 6.9 Hz, 1H), 2.96 (dd, J = 15.9, 6.9 Hz, 1H), 3.99 (s, 3H), 4.06 (s,
3H), 4.34 (br s, 1H), 5.70 (br s, 1H), 6.67 (s, 1H); 13C NMR (125 MHz, MeOD, 40°C) δ 21.9, 23.6,
28.84, 29.86, 36.2, 47.0, 48.3, 55.97, 55.99, 80.9, 93.9, 114.6, 121.0, 138.1, 156.7, 158.2, 162.7;
IR (NaCl, neat) 1666, 3373 cm-1; HRMS (ESI-MS): m/z calcd for C20H32BNO6Na [M-
(2×OH)+(2×MeO)+Na]+ 416.2220, found 416.2219.
t-Butyl (1R,3S)-5-(4’-hydroxy-5’-methoxy-2’-methylnaphthalen-3’-yl)-6,8-dimethoxy-1,3-
dimethyl-1,2,3,4-tetrahydroisoquinoline carboxylate (2.117)
(a) A solution of boronic acid 2.119 (0.13 g, 0.36 mmol) in dry 1,2-dichloroethane (1 mL) was
added dropwise to a solution of freshly dried lead(IV) tetraacetate (0.17 g, 0.38 mmol) and
mercury(II) trifluoroacetate (15 mg, 35.2 μmol) in dry 1,2-dichloroethane (2.5 mL). The solution
was protected from light and stirred at room temperature for 14 h. The reaction mixture was
diluted with dichloromethane and washed with water, then dried (Na2SO4). The solution was
filtered through a short plug of Celite, eluting with dichloromethane. The solvent was removed
under reduced pressure to afford crude aryllead triacetate 2.118 as a light yellow gum. Due to the
lability of this compound it was used immediately in the next step.
(b) A solution of crude aryllead triacetate 2.118 in dry 1,2-dichloroethane (1 mL) was added
dropwise to a solution of naphthol 2.52 (68 mg, 0.36 mmol) and dry pyridine (0.12 mL, 1.49
mmol) in dry 1,2-dichloroethane (2.5 mL) at room temperature. The solution was protected from
light and stirred at room temperature for 18 h. The reaction mixture was poured onto saturated
aqueous ammonium chloride solution and extracted with dichloromethane (× 3). The organic

Chapter 5: Experimental
196
extracts were combined and washed with water and brine, then dried (Na2SO4). The solvent was
removed under reduced pressure and the crude material was purified by flash chromatography
on silica gel, eluting with 15 % ethyl acetate/n-hexane, to afford the product 2.117 as a mixture of
atropisomers as a clear colourless gum (90 mg, 49 % over two steps). [𝛼]𝐷24.8 = + 42 (0.5,
CHCl3); 1H NMR (400 MHz, CDCl3) δ 1.23 (d, J = 6.5 Hz, 3H, one atropisomer), 1.28 (d, J = 6.4
Hz, 3H, one atropisomer), 1.37 (d, J = 6.5 Hz, 3H, one atropisomer), 1.41 (d, J = 6.9 Hz, 3H, one
atropisomer), 1.49 (s, 9H, one atropisomer), 1.50 (s, 9H, one atropisomer), 2.08 (s, 3H, one
atropisomer), 2.10 (s, 3H, one atropisomer), 2.45 (dd, J = 16.3, 6.6 Hz, 1H, one atropisomer),
2.70 (dd, J = 16.3, 6.6 Hz, 1H, one atropisomer), 2.84 (dd, J = 15.5, 7.2 Hz, 1H, one
atropisomer), 2.92 (dd, J = 15.5, 7.2 Hz, 1H, one atropisomer), 3.72 (s, 6H, both atropisomers),
3.90 (s, 3H, one atropisomer), 3.91 (s, 3H, one atropisomer), 4.00 (s, 3H, one atropisomer), 4.01
(s, 3H, one atropisomer), 4.23 (br s, 1H, one atropisomer),4.29 (br s, 1H, one atropisomer), 5.52
(br s, 1H, one atropisomer), 5.59 (br s, 1H, one atropisomer), 6.50 (s, 1H, one atropisomer), 6.52
(s, 1H, one atropisomer), 6.72 (d, J = 7.6 Hz, 1H, one atropisomer), 6.73 (d, J = 7.6 Hz, 1H, one
atropisomer), 7.24 (s, 1H, one atropisomer), 7.25 (s, 1H, one atropisomer), 7.27 (dd, J = 8.4, 7.6
Hz, 2H, both atropisomers), 7.37 (d, J = 8.4 Hz, 2H, both atropisomers), 9.38 (s, 1H, one
atropisomer), 9.39 (s, 3H, one atropisomer); 13C NMR (100 MHz, CDCl3, 50°C) δ 20.5, 20.6,
21.8, 22.1, 23.7, 28.8, 32.4, 32.6, 45.9, 47.0, 55.6, 55.7, 55.8, 56.2, 56.3, 79.1, 79.2, 93.4, 94.1,
94.4, 103.5, 113.7, 113.8, 118.7, 119.58, 119.62, 121.4, 121.5, 125.49, 125.51, 134.4, 134.9,
136.2, 136.3, 138.5, 138.6, 151.3, 151.4, 155.0, 155.2, 155.4, 155.7, 156.4, 156.61, 156.63,
157.7, 161.2; IR (NaCl, neat) 1681, 3392 cm-1; HRMS (ESI-MS): m/z calcd for C30H37NO6Na
[M+Na]+ 530.2518, found 530.2515.
3-((1R,3S)-6,8-Dimethoxy-1,2,3-trimethyl-1,2,3,4-tetrahydroisoquinolin-5-yl)-5’-methoxy-2’-
methylnaphthalen-1’-ol (2.41)
(a) Trifluoruacetic acid (0.14 mL, 1.83 mmol) was added dropwise to a solution of Boc-
protected tetrahydroisoquinoline 2.117 (73 mg, 0.14 mmol) in dichloromethane (2,9 mL) at 0°C.
The solution was stirred at this temperature for 2 h. then neutralised with saturated aqueous
sodium bicarbonate solution. The mixture was extracted with dichloromethane (× 4). The organic

Chapter 5: Experimental
197
extracts were combined and washed with water and brine, then dried (Na2SO4). The solvent was
removed under reduced pressure to afford crude tetrahydroisoquinoline 5.18 as a light yellow
gum. Due to the lability of this compound it was used immediately in the next step.
(b) Concentrated aqueous formaldehyde solution (0.11 mL, 38%, 1.44 mmol) was added to
a solution of crude tetrahydroisoquinoline 5.20 in methanol (2.9 mL) at room temperature. The
solution was stirred at this temperature for 1 h then sodium borohydride (33 mg, 0.87 mmol) was
added in one portion at room temperature. The solution was stirred at this temperature for 15 h.
then quenched with 1M aqueous hydrochloric acid solution. The mixture was neutralised with
solid sodium bicarbonate then extracted with dichloromethane (× 4). The organic extracts were
combined and washed with brine, then dried (Na2SO4). The solvent was removed under reduced
pressure and the crude material was purified by flash chromatography on deactivated silica gel,
eluting with 50 % ethyl acetate/n-hexane, to afford the product 2.41 as a mixture of atropisomers
as a light yellow gum (50 mg, 83 % over two steps). [𝛼]𝐷22.2 = + 15 (0.2, MeOH); 1H NMR (400
MHz, (CD3)2SO) δ 0.91 – 0.93 (m, 6H, both atropisomers), 1.27 (d, J = 6.3 Hz, 6H, both
atropisomers), 1.91 (s, 3H, one atropisomer), 1.95 (s, 3H, one atropisomer), 1.97 – 2.29 (m, 6H,
both atropisomers), 2.31 (s, 3H, one atropisomer), 2.32 (s, 3H, one atropisomer), 3.60 – 3.66 (m,
2H, both atropisomers), 3.62 (s, 3H, one atropisomer), 3.63 (s, 3H, one atropisomer), 3.86 (s, 3H,
both atropisomers), 3.98 (s, 3H, both atropisomers), 6.59 (s, 2H, both atropisomers), 6.85 – 6.88
(m, 2H, both atropisomers), 7.21 (s, 1H, one atropisomer), 7.22 (s, 1H, one atropisomer), 7.29 –
7.35 (m, 4H, both atropisomers), 9.29 (s, 1H, one atropisomer), 9.35 (s, 1H, one atropisomer);
13C NMR (100 MHz, (CD3)2SO) δ 20.2, 20.4, 21.3, 21.5, 23.1, 23.2, 35.6, 36.0, 40.9, 41.0, 54.2,
54.5, 55.26, 55.37, 55.39, 56.1, 56.3, 93.8, 94.0, 103.6, 103.7, 112.9, 113.0, 115.6, 116.1, 118.1,
118.2, 119.2, 119.3, 120.0, 120.2, 120.4, 125.86, 125.89, 135.32, 135.34, 135.4, 136.0, 137.3,
137.7, 150.5, 151.1, 155.25, 155.29, 155.50, 155.52, 155.7; IR (NaCl, neat) 3391 cm-1; HRMS
(ESI-MS): m/z calcd for C25H28NO4 [M-CH4]+ 406.2012, found 406.1997.

Chapter 5: Experimental
198
5.3. Experiments Described in Chapter 3
General Procedure A for Sharpless Asymmetric Aminohydroxylation
A solution of aqueous sodium hydroxide solution (0.4 M, 3 eq) was added dropwise to a solution
of carbamate (3 eq) in n-propanol (0.7 M) at room temperature. 1,3-Dichloro-5,5-
dimethylhydantoin (2 eq) was added as a solid in one portion, followed by a solution of chiral
ligand (5 mol %) in n-propanol (0.02 M) and a solution of alkene (1 eq) in n-propanol (1.3 M). The
solution was stirred vigorously at room temperature until it became homogeneous, then
potassium osmate dihydrate (4 mol %) was added as a solid in one portion. The solution
immediately turned dark green and was stirred at room temperature for 24 h after which, it had
turned light yellow. Sodium sulphite (10 eq) was added and the suspension was stirred for a
further 10 min. The mixture was diluted with water and extracted with ethyl acetate (× 3). The
organic extracts were combined and washed with brine, then dried (Na2SO4). The solvent was
removed under reduced pressure and the crude material was purified, as indicated, to afford the
product.
1-Methoxy-4-(3’-methylbut-3’-enyl)benzene (3.29)[27]
A solution of n-butyllithium in hexanes (11.30 mL, 1.4 M, 15.82 mmol) was added dropwise to a
solution of methyltriphenylphosphonium bromide (6.05 g, 16.94 mmol) in freshly distilled THF
(150 mL) at room temperature. The solution was stirred at room temperature for 15 min. where it
had turned dark orange. A solution of ketone 3.30 (2.01 g, 11.29 mmol) in freshly distilled THF
(50 mL) was added dropwise and the resulting reaction solution was stirred at room temperature
for an additional 4 h. The reaction was quenched with water and the mixture extracted with n-
pentane (× 3). The organic extracts were combined and washed with water and brine, then dried
(Na2SO4). The solvent was removed under reduced pressure and the crude material was purified
by flash chromatography on silica gel, eluting with 5 % ethyl acetate/n-hexane, to afford the
product 3.29 as a clear colourless oil (1.68 g, 84 %) with all the analytical data matching that
reported in the literature.[27] 1H NMR (300 MHz; CDCl3) δ 1.77 (s, 3H), 2.26 – 2.32 (m, 2H), 2.67 –
2.73 (m, 2H), 3.79 (s, 3H), 4.70 – 4.71 (m, 1H), 4.73 – 4.74 (m, 1H), 6.81 – 6.85 (m, 2H), 7.09 –
7.14 (m, 2H).

Chapter 5: Experimental
199
Examination of Regioselectivity of Sharpless Asymmetric Aminohydroxylation on 1-
Methoxy-4-(3’-methylbut-3’-enyl)benzene (3.29)
Using 2-(trimethylsilyl)ethyl carbamate as the carbamate source (P = Teoc).
All experiments were conducted using general procedure A on a 0.5 mmol scale of alkene 3.29.
The ratio of regioisomers was determined by analysis of the 1H NMR spectrum of the crude
reaction mixture. The crude material was purified by flash chromatography on silica gel, eluting
with dichloromethane, to remove any residual 2-(trimethylsilyl)ethyl carbamate (3.26), then 40 %
ethyl acetate/n-hexane, to afford the product as a clear colourless gum.
Ligand Ratio (3.31:3.32) Yield Major (%)
(DHQD)2PHAL (3.25) >20:1 35 (DHQD)2AQN (3.27) >20:1 54 (DHQD)2Pyr (3.28) >20:1 50
(2S)-2-(Trimethylsilyl)ethyl-(2-hydroxy-4-(4’-methoxyphenyl)-2-methylbutyl)carbamate
1H NMR (300 MHz; CDCl3) δ 0.03 (s, 9H, Si(CH3)3), 0.95 – 1.01 (m, 2H, CO2CH2CH2Si(CH3)3),
1.23 (s, 3H, CH3), 1.71 – 1.80 (m, 2H, ArCH2CH2), 2.34 (br s, 1H, OH), 2.62 – 2.67 (m, 2H,
ArCH2CH2), 3.15 – 3.28 (m, 2H, CH2NHTeoc), 3.78 (s, 3H, OCH3), 4.13 – 4.19 (m, 2H,
CO2CH2CH2Si(CH3)3), 5.06 (br t, J = 5.5 Hz, 1H, NHTeoc), 6.80 – 6.84 (m, 2H, ArH-2/6), 7.09 –
7.12 (m, 2H, ArH-3/5); 13C NMR (75 MHz; CDCl3) δ -1.36 (Si(CH3)3), 17.9 (CO2CH2CH2Si(CH3)3),
24.6 (CH3), 29.3 (ArCH2CH2), 42.1 (ArCH2CH2), 50.6 (CH2NHTeoc), 55.4 (OCH3), 63.5
(CO2CH2CH2Si(CH3)3), 72.9 (ArC4), 114.0 (ArC-2/6), 129.3 (ArC3/5), 134.3 (ArC1), 157.9 (ArC4),
157.9 (CO2CH2CH2Si(CH3)3); IR (NaCl, neat) 1697, 3407 cm-1; HRMS (ESI-MS): m/z calcd for
C18H31NO4SiNa [M+Na]+ 376.1920, found 376.1914.
Using t-Butylcarbamate as the carbamate source (P = Boc).
All experiments were conducted using general procedure A on a 0.5 mmol scale of alkene 3.29.
The ratio of regioisomers was determined by analysis of the 1H NMR spectrum of the crude
reaction mixture. The crude material was purified by flash chromatography on silica gel, eluting
with dichloromethane, to remove any residual t-butyl carbamate (3.33), then 30 % ethyl acetate/n-
hexane, to afford the product as a clear colourless gum.

Chapter 5: Experimental
200
Ligand Ratio (3.31:3.32) Yield Major (%)
(DHQD)2PHAL (3.25) >20:1 32 (DHQD)2AQN (3.27) >20:1 57 (DHQD)2Pyr (3.28) >20:1 52
(2S)-t-Butyl-(2-hydroxy-4-(4’-methoxyphenyl)-2-methylbutyl)carbamate
1H NMR (300 MHz; CDCl3) δ 1.22 (s, 3H, CH3), 1.45 (s, 9H, C(CH3)3), 1.71 – 1.76 (m, 2H,
ArCH2CH2), 2.41 (br s, 1H, OH), 2.61 – 2.67 (m, 2H, ArCH2CH2), 3.10 – 3.24 (m, 2H, CH2NH),
3.78 (s, 3H, OCH3), 4.95 (br s, 1H, NHBoc), 6.80 – 6.84 (m, 2H, ArH2/6), 7.09 – 7.12 (m, 2H,
ArH3/5); 13C NMR (75 MHz; CDCl3) δ 24.6 (CH3), 28.5 (C(CH3)3), 29.4 (ArCH2CH2), 42.2
(ArCH2CH2), 50.2 (CH2NHBoc), 55.4 (OCH3), 73.0 (ArC4), 79.8 (C(CH3)3), 114.0 (ArC2/6), 129.3
(ArC3/5), 134.4 (ArC1), 157.2 (NHCO2C(CH3)3), 157.9 (ArC4); IR (NaCl, neat) 1693, 3415 cm-1;
HRMS (ESI-MS): m/z calcd for C17H27NO4Na [M+Na]+ 332.1838, found 332.1825.
Using Benzyloxy carbamate as the carbamate source (P = Cbz).
All experiments were conducted using general procedure A on a 0.5 mmol scale of alkene 3.29.
The ratio of regioisomers was determined by analysis of the 1H NMR spectrum of the crude
reaction mixture. The crude material was purified by flash chromatography on silica gel, eluting
with dichloromethane, to remove any residual benzyloxy carbamate (3.34), then 20 % ethyl
acetate/n-hexane, to afford the product as a clear colourless gum.
Ligand Ratio (3.31:3.32) Yield Major (%)
(DHQD)2PHAL (3.25) >20:1 35 (DHQD)2AQN (3.27) >20:1 48 (DHQD)2Pyr (3.28) >20:1 50
(2S)-Benzyl-(2-hydroxy-4-(4’-methoxyphenyl)butyl)carbamate
1H NMR (300 MHz; CDCl3) δ 1.23 (s, 3H, CH3), 1.71 – 1.77 (m, 2H, ArCH2CH2), 2.28 (br s, 1H,
OH), 2.61 – 2.67 (m, 2H, ArCH2CH2), 3.18 – 3.31 (m, 2H, CH2NH), 3.78 (s, 3H, OCH3), 5.12 (s,
2H, Cbz-CH2), 5.26 (br t, J = 5.5 Hz, 1H, NHCbz), 6.80 – 6.85 (m, 2H, ArH2/6), 7.08 – 7.11 (m,
2H, ArC3/5), 7.31 – 7.36 (m, 5H, ArH-Cbz); 13C NMR (75 MHz; CDCl3) δ 24.6 (CH3), 29.3
(ArCH2CH2), 42.0 (ArCH2CH2), 50.6 (CH2NHCbz), 55.4 (OCH3), 67.0 (Cbz-CH2), 72.9 (ArC4),
114.0 (ArC2/6), 128.2 (Cbz ArC), 128.3 (Cbz ArC), 128.7 (Cbz ArC), 129.3 (ArC3/5), 134.2
(ArC1), 136.5 (Cbz 1-ArC), 157.4 (NHCO2CH2Ar), 157.9 (ArC4); IR (NaCl, neat) 1704, 3360,
3419 cm-1; HRMS (ESI-MS): m/z calcd for C20H25NO4Na [M+Na]+ 366.1681, found 366.1676.

Chapter 5: Experimental
201
1-(But-3-enyl)-4-methoxybenzene (3.36)[28]
A solution of allylmagnesium bromide in diethyl ether (7.00 mL, 1 M, 7.00 mmol) was added
dropwise to a solution of 4-methoxybenzyl chloride (5.19) (0.65 mL, 4.79 mmol) in freshly distilled
THF (10 mL) at 0°C. The solution was allowed to stir at 0°C for 2 h. then, at room temperature for
a further 21 h. The dark brown solution was quenched with saturated aqueous ammonium
chloride solution. The mixture was extracted with diethyl ether (× 3). The organic extracts were
combined and washed with water and brine, then dried (Na2SO4). The solvent was removed
under reduced pressure and the crude material was purified by flash chromatography on silica
gel, eluting with n-pentane, to afford the product 3.36 as a clear colourless oil (0.35 g, 45 %). with
all the analytical data matching that reported in the literature.[28] 1H NMR (300 MHz; CDCl3) δ
2.30- 2.38 (m, 2H), 2.63 – 2.68 (m, 2H), 3.79 (s, 3H), 4.97 (ddt, J = 10.3, 2.0, 1.2 Hz, 1H), 5.03
(ddt, J = 17.0, 1.9, 1.2 Hz, 1H), 5.85 (ddt, J = 17.0, 10.3, 6.5 Hz, 1H), 6.80 – 6.85 (m, 2H), 7.08 –
7.13 (m, 2H).
Examination of Regioselectivity of Sharpless Asymmetric Aminohydroxylation on 1-(But-3-
enyl)-4-methoxybenzene
All experiments were conducted using general procedure A on a 0.1 mmol scale of alkene 3.36
and using 2-(trimethylsilyl)ethyl carbamate as the carbamate source. The ratio of regioisomers
was determined by analysis of the 1H NMR spectrum of the crude reaction mixture. The crude
material was purified by flash chromatography on silica gel, eluting with dichloromethane, to
remove any residual 2-(trimethylsilyl)ethyl carbamate (3.26), then 30 % ethyl acetate/n-hexane, to
afford the product as a clear colourless gum.
Ligand Ratio (3.39:3.40) Yield Major (%)
(DHQD)2PHAL (3.25) 10:1 37 (DHQD)2AQN (3.27) >20:1 93 (DHQD)2Pyr (3.28) >20:1 65

Chapter 5: Experimental
202
(2S)-2-(Trimethylsilyl)ethyl-(2-hydroxy-4-(4’-methoxyphenyl)-2-methylbutyl)carbamate (10:1
mixture of regioisomers)
1H NMR (300 MHz; CDCl3) δ 0.03 (s, 9H, SiMe3, major regioisomer), 0.04 (s, 9H, SiMe3, minor
regioisomer), 0.94 – 1.00 (m, 2H, CH2SiMe3, both regioisomers), 1.69 – 1.80 (m, 2H, PhCH2CH2,
both regioisomers), 2.55 – 2.78 (m, 3H, PhCH2CH2 and OH, both regioisomers), 3.05 – 3.14 (m,
1H, CH(OH)C(H)HNH, both regioisomers), 3.34 (ddd, J = 14.0, 6.3, 2.9 Hz, 1H,
CH(OH)C(H)HNH, both regioisomers), 3.54 – 3.60 (m, 1H, CH2CH(OH), minor regioisomer), 3.66
– 3.74 (m, 1H, CH2CH(OH), major regioisomer), 3.78 (s, 3H, OMe, both regioisomers), 4.12 –
4.18 (m, 2H, OCOCH2, both regioisomers), 4.84 (br d, 1H, J = 7.8 Hz, NH, minor regioisomer),
5.07 (br t, J = 5.6 Hz, 1H, NH), 6.80 – 6.84 (m, 2H, ArH2/6, both regioisomers), 7.08 – 7.13 (m,
2H, ArH3/5, both regioisomers); 13C NMR (75 MHz; CDCl3) δ -1.36, 17.9, 31.0, 36.6, 47.1, 55.4,
63.5, 71.0, 114.0, 129.4, 133.8, 157.8, 158.0; IR (NaCl, neat) 1696, 3425 cm-1; HRMS (ESI-MS):
m/z calcd for C17H29NO4SiNa [M+Na]+ 362.1764, found 362.1756.
1-Methoxy-4-(3’-methylbut-3’-enyloxy)benzene (3.35)[29]
A solution of diisopropyl azodicarboxylate (0.67 mL, 4.27 mmol) in dry toluene (1.2 mL) was
added dropwise to a solution of 4-methoxyphenol (5.20) (0.82 g, 6.60 mmol), 3-methyl-3-buten-1-
ol (0.33 mL, 3.27 mmol) and triphenylphosphine (1.13 g, 4.29 mmol) in freshly distilled THF (10
mL) at room temperature. The solution was heated at reflux for 1.5 h. then cooled to room
temperature. The solvent was removed under reduced pressure and the crude material purified
by flash chromatography on silica gel, eluting with 5 % ethyl acetate/n-hexane, to afford the
product 3.35 as a clear colourless oil (0.62 g, 99 %) with all the analytical data matching that
reported in the literature.[29] 1H NMR (300 MHz; CDCl3) δ 1.80 (s, 3H), 2.48 (t, J = 6.9 Hz, 2H),
3.77 (s, 3H), 4.03 (t, J = 6.9 Hz, 2H), 4.79 – 4.80 (m, 1H), 4.83 – 4.84 (m, 1H), 6.80 – 6.88 (m,
4H).
Examination of Regioselectivity of Sharpless Asymmetric Aminohydroxylation on 1-
Methoxy-4-(3-methylbut-3-enyloxy)benzene

Chapter 5: Experimental
203
All experiments were conducted using general procedure A on a 0.5 mmol scale of alkene 3.35
and using 2-(trimethylsilyl)ethyl carbamate as the carbamate source. The ratio of regioisomers
was determined by analysis of the 1H NMR spectrum of the crude reaction mixture. The crude
material was purified by flash chromatography on silica gel, eluting with dichloromethane, to
remove any residual 2-(trimethylsilyl)ethyl carbamate, then 40 % ethyl acetate/n-hexane, to afford
the product as a clear colourless gum.
Ligand Ratio (3.37:3.38) Yield Major (%)
(DHQD)2PHAL >20:1 47 (DHQD)2AQN >20:1 44 (DHQD)2Pyr >20:1 75
(2S)-2-(Trimethylsilyl)ethyl-(2-hydroxy-4-(4’-methoxyphenoxy)-2-methylbutyl)carbamate
1H NMR (300 MHz; CDCl3) δ 0.02 (s, 9H, Si(CH3)3), 0.93 – 0.99 (m, 2H, CO2CH2CH2Si(CH3)3),
1.23 (s, 3H, CH3), 1.83 – 1.92 (m, 1H, ArOCH2C(H)H), 1.97 – 2.06 (m, 1H, PhOCH2C(H)H), 3.24
(br d, J = 6.2 Hz, 2H, CH2NHTeoc), 3.30 (br s, 1H, OH), 3.74 (s, 3H, OCH3), 4.04 – 4.17 (m, 4H,
CO2CH2CH2Si(CH3)3 and ArOCH2CH2), 5.30 (br t, J = 5.1 Hz, 1H, NHTeoc), 6.78 – 6.84 (m, 4H,
ArH); 13C NMR (75 MHz; CDCl3) δ -1.42 (Si(CH3)3), 17.8 (CO2CH2CH2Si(CH3)3), 25.0 (CH3), 31.0
(ArCH2CH2), 50.7 (CH2NHTeoc), 55.7 (OCH3), 63.3 (CO2CH2CH2Si(CH3)3 or ArOCH2CH2), 65.4
(CO2CH2CH2Si(CH3)3 or ArOCH2CH2), 72.4 (C4), 114.8 (ArC2/6 or 3/5), 115.5 (ArC2/6 or 3/5),
152.5 (ArC1), 154.2 (ArC4), 157.8 (CO2CH2CH2Si(CH3)3); IR (NaCl, neat) 1694, 3431 cm-1;
HRMS (ESI-MS): m/z calcd for C18H31NO5SiNa [M+Na]+ 392.1869, found 392.1862.
2-(4’-Heptyloxyphenyl)ethanol (5.21)[30]
Tyrosol 3.41 (4.60 g, 33.31 mmol) was added as a solid in one portion to a solution of sodium
ethoxide in ethanol, prepared in situ by dissolving sodium (0.85 g, 36.96 mmol) in ethanol (200
mL). The solution was stirred at room temperature for 30 min. after which, 1-bromoheptane (5.8
mL, 36.88 mmol) was added dropwise. The solution was heated at reflux for 14 h. The
suspension was cooled to room temperature and the ethanol was removed under reduced
pressure. 1M Aqueous sodium hydroxide was added to the residue which, was extracted with
ether (× 3). The organic extracts were combined and washed with brine, then dried (Na2SO4).
The solvent was removed under reduce pressure to afford the product 5.21 as a white solid (7.59
g, 96 %) with all the analytical data matching that reported in the literature. [30] This was used
without further purification. 1H NMR (300 MHz; CDCl3) δ 0.90 (t, J = 6.8 Hz, 3H), 1.31 – 1.48 (m,

Chapter 5: Experimental
204
9H), 1.73 -1.82 (m, 2H), 2.80 (t, J = 6.6 Hz, 2H), 3.81 (t, J = 6.6 Hz, 2H), 3.93 (t, J = 6.6 Hz, 2H),
6.83 – 6.87 (m, 2H), 7.10 – 7.15 (m, 2H).
2-(4’-Heptyloxyphenyl)-1-iodoethane (3.9)[30]
(a) Methanesulfonyl chloride (2.75 mL, 35.53 mmol) was added dropwise to a solution of
alcohol 5.21 (7.59 g, 30.06 mmol) and triethylamine (13.50 mL, 96.86 mmol) in dichloromethane
(400 mL) at 0°C. The solution was stirred at 0°C for 15 min. then the cold bath was removed and
the solution stirred at room temperature for 3 h. The reaction mixture was poured onto brine and
extracted with dichloromethane (× 2). The organic extracts were combined and dried (Na2SO4).
The solvent was removed under reduced pressure to afford a light orange residue (8.73 g) which
was used in the next step without further purification.
(b) The crude material (8.73 g) was dissolved in acetone (260 mL) and sodium iodide (45.06
g, 300.63 mmol) was added. The solution was stirred at room temperature, protected from light
for 16 h. The acetone was removed under reduced pressure. Water was added to the residue
which was extracted with dichloromethane (× 3). The organic extracts were combined and
washed with water and brine, then dried (Na2SO4). The solvent was removed under reduced
pressure and the crude material was purified by flash chromatography on silica gel, eluting with 2
% ethyl acetate/n-hexane, to afford the product 3.9 as a clear colourless oil (9.05 g, 87 %) with all
the analytical data matching that reported in the literature.[30] 1H NMR (300 MHz; CDCl3) δ 0.90 (t,
J = 6.8 Hz, 3H), 1.26 – 1.47 (m, 9H), 1.73 -1.82 (m, 2H), 3.11 (t, J = 7.8 Hz, 2H), 3.31 (t, J = 7.8
Hz, 2H), 3.94 (t, J = 6.6 Hz, 2H), 6.82 – 6.86 (m, 2H), 7.07 – 7.12 (m, 2H).
(2R,5S)-5-Isopropyl-3,6-dimethoxy-2-(4;-heptyloxyphenethyl)-2,5-dihydropyrazine (3.42)
Method 1: Warming to 0°C to give a mixture of diastereomers
A solution of n-butyllithium in hexanes (0.56 mL, 1.5 M, 0.82 mmol) was added dropwise to a
solution of freshly distilled (S)-Schollkopf’s reagent (S)-3.7 (0.15 g, 0.81 mmol) in freshly distilled
THF (1 mL) at -78°C (dry ice/acetone). The solution was stirred at -78°C for 15 min after which, it
had turned dark yellow. A solution of iodide 3.9 (0.25 g, 0.73 mmol) in freshly distilled THF (1 mL)

Chapter 5: Experimental
205
at -78°C was added dropwise. The solution was stirred at -78°C for 30 min then the -78°C cold
bath was replaced with a 0°C cold bath where it was stirred for a further 2.5 h. The reaction was
quenched with saturated aqueous sodium bicarbonate solution and allowed to warm to room
temperature. The THF was removed under reduced pressure and the residue extracted with
dichloromethane (× 4). The organic extracts were combined and dried (Na2SO4). The solvent was
removed under reduced pressure to afford a yellow oil. 1HNMR analysis showed the
diastereometric ratio to be 79:21 through integration of the benzylic multiplets at δ 2.47 – 2.63
and δ 2.66 – 2.81 respectively. The crude material was purified by flash chromatography on silica
gel, eluting with 3 % ethyl acetate/n-hexane, to afford the major diastereomer 3.42 as a clear
colourless oil (0.21 g, 71 %). [𝛼]𝐷25.0 = - 6 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.70 (d, J =
6.8 Hz, 3H), 0.89 (t, J = 6.8 Hz, 3H), 1.05 (d, J = 6.8 Hz, 3H), 1.25 – 1.49 (m, 8H), 1.71 – 1.81 (m,
2H), 1.89 – 2.01 (m, 1H), 2.07 – 2.18 (m, 1H), 2.27 (septd, J = 6.8, 3.3 Hz, 1H), 2.47 – 2.63 (m,
2H), 3.69 (s, 3H), 3.70 (s, 3H), 3.92 (t, J = 6.6 Hz, 2H), 3.95 – 3.98 (m, 1H), 4.02 – 4.07 (m, 1H),
6.78 – 6.83 (m, 2H), 7.07 – 7.11 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 14.2, 16.8, 19.2, 22.8,
26.2, 29.2, 29.5, 30.2, 31.9, 36.2, 52.50, 52.51, 55.1, 61.0, 68.2, 114.5, 129.5, 134.2, 157.4,
163.7, 163.9; IR (NaCl, neat) 1697 cm-1; HRMS (ESI-MS): m/z calcd for C24H39N2O3 [M+H]+
403.2961, found 403.2951.
Further elution afforded the minor diastereomer 3.43 as a clear colourless oil (62 mg, 21 %).
[𝛼]𝐷25.0 = + 44 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.74 (d, J = 6.8 Hz, 3H), 0.89 (t, J = 6.8
Hz, 3H), 1.07 (d, J = 6.8 Hz, 3H), 1.25 – 1.49 (m, 8H), 1.70 – 1.81 (m, 3H), 2.12 – 2.29 (m, 2H),
2.66 – 2.81 (m, 2H), 3.68 (s, 3H), 3.71 (s, 3H), 3.92 (t, J = 6.6 Hz, 2H), 3.94 – 4.02 (m, 2H), 6.79
– 6.84 (m, 2H), 7.11 – 7.15 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 14.2, 17.7, 19.7, 22.8, 26.2,
29.2, 29.5, 31.4, 31.5, 31.9, 37.7, 52.39, 52.42, 55.2, 61.0, 68.2, 114.5, 129.5, 134.2, 157.5,
163.2, 163.9; IR (NaCl, neat) 1693 cm-1; HRMS (ESI-MS): m/z calcd for C24H39N2O3 [M+H]+
403.2961, found 403.2959.
Method 2: Warming slowly to -15°C to give one diastereomer
A solution of n-butyllithium in hexanes (1.70 mL, 1.4 M, 2.38 mmol) was added dropwise to a
solution of freshly distilled (S)-Schollkopf’s reagent (S)-3.7 (0.44 g, 2.38 mmol) in freshly distilled
THF (3 mL) at -78°C (dry ice/acetone). The solution was stirred at -78°C for 15 min after which, it
had turned dark yellow. A solution of iodide 3.9 (0.75 g, 2.16 mmol) in freshly distilled THF (2 mL)
at -78°C was added dropwise. The solution was stirred at -78°C for 30 min then allowed to slowly
warm to -15°C over 4 h. The reaction was quenched with saturated aqueous sodium bicarbonate

Chapter 5: Experimental
206
solution and allowed to warm to room temperature. The THF was removed under reduced
pressure and the residue extracted with dichloromethane (× 4). The organic extracts were
combined and dried (Na2SO4). The solvent was removed under reduced pressure to afford a
yellow oil which was purified by flash chromatography on silica gel, eluting with 3 % ethyl
acetate/n-hexane, to afford the product 3.42 as a clear colourless oil (0.73 g, 84 %) with all
analytical data matching that reported previously for 3.42.
(2S,5S)-5-Isopropyl-3,6-dimethoxy-2-(4’-heptyloxyphenethyl)-2-methyl-2,5-dihydropyrazine
(3.44)
A solution of n-butyllithium in hexanes (2.3 mL, 2.4 M, 5.52 mmol) was added dropwise to a
solution of bis-lactim ether 3.42 (1.72 g, 4.27 mmol) in freshly distilled THF (15 mL) at -78°C (dry
ice/acetone). The solution was stirred at -78°C for 15 min after which it had turned a dark yellow.
Methyl iodide (0.35 mL, 5.62 mmol) was added dropwise. The solution was allowed to slowly
warm to -15°C over 4 h. The reaction was quenched with saturated aqueous sodium bicarbonate
solution and allowed to warm to room temperature. The THF was removed under reduced
pressure and the residue extracted with dichloromethane (× 4). The organic extracts were
combined and dried (Na2SO4). The solvent was removed under reduced pressure and the crude
material was purified by flash chromatography on silica gel, eluting with 2 % ethyl acetate/n-
hexane, to afford the product 2.44 as a clear colourless oil (1.28 g, 72 %). [𝛼]𝐷26.7 = +46 (0.5,
CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.70 (d, J = 6.8 Hz, 3H), 0.89 (t, J = 6.8 Hz, 3H), 1.12 (d, J
= 6.8 Hz, 3H), 1.25 – 1.47 (m, 11H), 1.71 – 1.81 (m, 2H), 1.85 (td, J = 12.9, 5.0 Hz, 1H), 2.08 (td,
J = 12.9, 4.3 Hz, 1H), 2.23 (td, J = 12.9, 4.3 Hz, 1H), 2.32 – 2.47 (m, 2H), 3.70 (s, 3H), 3.71 (s,
3H), 3.92 (t, J = 6.6 Hz, 2H) 3.94 (d, J = 3.3 Hz, 1H), 6.78 – 6.82 (m, 2H), 7.04 – 7.07 (m, 2H);
13C NMR (75 MHz; CDCl3) δ 14.2, 17.1, 19.7, 22.8, 26.2, 28.7, 29.2, 29.5, 30.7, 30.8, 31.9, 43.0,
52.4, 58.4, 60.5, 68.2, 114.5, 129.3, 134.7, 157.3, 162.1, 165.7; IR (NaCl, neat) 1691 cm-1;
HRMS (ESI-MS): m/z calcd for C25H41N2O3 [M+H]+ 417.3117, found 417.3094.

Chapter 5: Experimental
207
Methyl (2S)-2-amino-4-(4’-heptyloxyphenyl)-2-methylbutanoate (3.45)
A solution of TFA (10 mL) in water (20 mL) was added dropwise to a solution of bis-lactim ether
3.44 (0.84 g, 2.02 mmol) in acetonitrile (55 mL). The solution was stirred at room temperature for
4 h after which the acetonitrile was removed under reduced pressure. The residue was diluted
with water and neutralised with portions of solid sodium bicarbonate, then extracted with
dichloromethane (× 4). The organic extracts were combined and dried (Na2SO4). The solvent was
removed under reduced pressure and the crude material was purified by flash chromatography
on silica gel, eluting with ethyl acetate, to afford the product 3.45 as a clear colourless oil (0.56 g,
87 %). [𝛼]𝐷23.3 = +4 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) 0.88 (t, J = 6.8 Hz, 3H), 1.25 – 1.48
(m, 8H), 1.36 (s, 3H), 1.71 – 1.80 (m, 4H), 1.85 (td, J = 12.6, 5.5 Hz, 1H), 2.00 (td, J = 12.6, 5.1
Hz, 1H), 2.23 (td, J = 12.6, 5.1 Hz, 1H), 2.58 (td, J = 12.6, 5.5 Hz, 1H), 3.70 (s, 3H), 3.91 (t, J =
6.6 Hz, 2H) 3.94 (d, J = 3.3 Hz, 1H), 6.78 – 6.82 (m, 2H), 7.04 – 7.09 (m, 2H); 13C NMR (75 MHz;
CDCl3) δ 14.2, 22.7, 26.1, 26.6, 29.2, 29.4, 29.8, 29.9, 31.9, 43.2, 52.27, 52.33, 57.9, 68.1, 114.6,
129.3, 133.5, 157.5, 178.1; IR (NaCl, neat) 1732, 3314, 3378 cm-1; HRMS (ESI-MS): m/z calcd
for C19H32NO3 [M+H]+ 322.2382, found 322.2382.
AAL(S) (1.37)[30]
Lithium aluminium hydride (0.10 g, 2.69 mmol) was added portion wise to a solution of
aminoester 3.45 (0.58 g, 1.80 mmol) in freshly distilled THF (18 mL) at 0°C. The solution was
stirred at 0°C for 20 min then the cold bath was removed and the solution stirred at room
temperature for 1 h. The reaction was quenched with saturated aqueous sodium sulfate solution
and the mixture was extracted with ethyl acetate (× 4). The organic extracts were combined and
washed with saturated aqueous sodium bicarbonate solution, water and brine, then dried
(Na2SO4). The solvent was removed under reduced pressure and the crude material was purified
by flash chromatography on silica gel, eluting with 2 % methanol/5 %
triethylamine/dichloromethane, to afford the product 1.37 as a white solid (0.45 g, 86 %) with all
the analytical data matching that reported in the literature.[30] [𝛼]𝐷24.9 = +4 (0.5, CHCl3); 1H NMR
(300 MHz; CDCl3) δ 0.89 (t, J = 6.8 Hz, 3H), 1.14 (s, 3H), 1.26 – 1.37 (m, 6H), 1.41 – 1.46 (m,

Chapter 5: Experimental
208
2H), 1.62 – 1.78 (m, 6H), 2.56 – 2.61 (m, 2H), 3.34 (d, J = 10.5 Hz, 1H), 3.39 (d, J -= 10.5 Hz,
1H), 3.92 (t, J = 6.6 Hz, 2H), 6.80 – 6.82 (m, 2H), 7.08 – 7.10 (m, 2H); HRMS (ESI-MS): m/z
calcd for C18H32NO2 [M+H]+ 294.2433, found 294.2429.
2-(4’-t-Butyldimethylsilyloxyphenyl)ethanol (3.48)[31]
(a) Tyrosol 3.41 (7.18 g, 51.94 mmol) was added as a solid in one portion to a solution of
t-butyldimethylsilyl chloride (19.57 g, 129.84 mmol) and imidazole (8.84 g, 129.85 mmol) in dry
DMF (50 mL) at room temperature. The solution was stirred for 12 h after which, water was
added and the mixture extracted with n-hexane (× 3). The organic extracts were combined and
washed with water, brine then dried (Na2SO4). The solvent was removed under reduced pressure
to afford the crude bis-TBS tyrosol compound 5.23 as a light yellow oil (17.62 g), which was used
in the next step without further purification.
(b) The crude material (17.62 g) was dissolved in methanol (140 mL) and iodine (1.76 g,
10 (wt/wt) %, 6.94 mmol) was added. The solution was stirred at room temperature for 4 h. 10 %
Aqueous sodium thiosulfate solution was added until the solution remained colourless. The
methanol was removed under reduced pressure. The residue was extracted with diethyl ether (×
3). The organic extracts were combined and washed with water, brine then dried (Na2SO4). The
solvent was removed under reduced pressure and the crude material was purified by flash
chromatography on silica gel, eluting with 15 % ethyl acetate/n-hexane, to afford the product 5.24
as a clear colourless oil (12.56 g, 96 %) with all the analytical data matching that reported in the
literature.[31] 1H NMR (300 MHz; CDCl3) δ 0.19 (s, 6H), 0.98 (s, 9H), 1.42 (br s, 1H), 2.80 (t, J =
6.5 Hz, 2H),3.82 (br t, J = 6.5 Hz, 2H), 6.76 – 6.81 (m, 2H), 7.06 – 7.10 (m, 2H).
2-(4’-t-Butyldimethylsilyloxyphenyl)-1-iodoethane (3.48)[32]
(a) Methanesulfonyl chloride (0.34 mL, 4.39 mmol) was added dropwise to a solution of
alcohol 5.24 (1.01 g, 4.00 mmol) and triethylamine (1.7 mL, 12.20 mmol) in dichloromethane (60
mL) at 0°C. The solution was stirred at 0°C for 15 min then the cold bath was removed and the
solution stirred at room temperature for 3 h. The reaction mixture was poured onto brine and the
organic layer was removed. The aqueous layer was extracted further with dichloromethane (× 2).
The organic extracts were combined washed with brine then dried (Na2SO4). The solvent was

Chapter 5: Experimental
209
removed under reduced pressure to afford an orange residue (1.36 g), which was used in the
next step without further purification.
(b) The crude material (1.36 g) was dissolved in acetone (30 mL) and sodium iodide
(6.00 g, 40.02 mmol) was added in one portion. The solution was stirred at room temperature
protected from light for 14 h. The acetone was removed under reduced pressure. Water was
added to the residue which was extracted with dichloromethane (× 3). The organic extracts were
combined and washed with water, brine then dried (Na2SO4). The solvent was removed under
reduced pressure and the crude material was purified by flash chromatography on silica gel,
eluting with 1 % ethyl acetate/n-hexane, to afford the product 3.48 as a clear colourless oil (1.13
g, 78 %) with all the analytical data matching that reported in the literature.[33] 1H NMR (400 MHz;
CDCl3) δ 0.19 (s, 6H), 0.98 (s, 9H), 3.18 (t, J = 7.7 Hz, 2H), 3.82 (t, J = 7.7 Hz, 2H), 6.76 – 6.79
(m, 2H), 7.02 – 7.06 (m, 2H).
(2R,5S)-5-isopropyl-3,6-dimethoxy-2-(4’-t-butyldimethylsilyloxyphenethyl)-2,5-
dihydropyrazine (3.75)
A solution of n-butyllithium in hexanes (3.4 mL, 2.4 M, 8.16 mmol) was added dropwise to a
solution of freshly distilled (S)-Schöllkopf’s reagent (S)-3.7 (1.50 g, 8.15 mmol) in freshly distilled
THF (8 mL) at -78°C (dry ice/acetone). The solution was stirred at -78°C for 15 min, where it had
turned dark yellow. A solution of iodide 3.48 (2.81 g, 7.75 mmol) in freshly distilled THF (6 mL) at
-78°C was added dropwise. The solution was stirred for a further 30 min at -78°C then allowed to
slowly warm to -15°C over 4 h. The reaction mixture was quenched with saturated aqueous
sodium bicarbonate solution and allowed to warm to room temperature. The THF was removed
under reduced pressure and the residue extracted with dichloromethane (× 4). The organic
extracts were combined and dried (Na2SO4). The solvent was removed under reduced pressure
and the crude material was purified by flash chromatography on silica gel, eluting with 2 % ethyl
acetate/n-hexane, to afford the product 3.75 as a clear colourless oil (2.89 g, 89 %). [𝛼]𝐷25.0 = - 6
(0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.18 (s, 6H), 0.70 (d, J = 6.8 Hz, 3H), 0.97 (s, 9H),
1.05 (d, J = 6.8 Hz, 3H), 1.90 – 2.02 (m, 1H), 2.07 – 2.18 (m, 1H), 2.27 (septd, J = 6.8, 3.3 Hz,
1H), 2.46 – 2.62 (m, 1H), 3.69 (s, 3H), 3.70 (s, 3H), 3.95 (t, J = 3.3 Hz, 1H), 4.02 – 4.07 (m, 1H),
6.71 – 6.76 (m, 2H), 7.01 – 7.06 (m, 2H); 13C NMR (75 MHz; CDCl3) δ -4.3, 16.8, 18.3, 19.2, 25.9,
30.3, 31.9, 36.0, 52.50, 52.51, 55.1, 61.0, 119.9, 129.4, 134.9, 153.7, 163.7, 163.9; IR (NaCl,

Chapter 5: Experimental
210
neat) 1697 cm-1; HRMS (ESI-MS): m/z calcd for C23H39N2O3Si [M+Na]+ 419.2729, found
419.2710.
(2S,5S)-5-Isopropyl-3,6-dimethoxy-2-(4’-t-butyldimethylsilyloxyphenethyl)-2-methyl-2,5-
dihydropyrazine (3.49)
A solution of n-butyllithium in hexanes (3.7 mL, 2.4 M, 8.88 mmol) was added dropwise to a
solution of bis-lactim ether 3.75 (2.89 g, 6.90 mmol) in freshly distilled THF (25 mL) at -78°C (dry
ice/acetone). The solution was stirred at -78°C for 15 min, where it had turned dark yellow.
Methyl iodide (0.55 mL, 8.83 mmol) was added dropwise. The solution was stirred for a further 30
min at -78°C then allowed to slowly warm to -15°C over 4 h. The reaction mixture was quenched
with saturated aqueous sodium bicarbonate solution and allowed to warm to room temperature.
The THF was removed under reduced pressure and the residue extracted with dichloromethane
(× 4). The organic extracts were combined and dried (Na2SO4). The solvent was removed under
reduced pressure and the crude material was purified by flash chromatography on silica gel,
eluting with 2 % ethyl acetate/n-hexane, to afford the product 3.49 as a clear colourless oil (2.85
g, 96 %). [𝛼]𝐷25.3 = + 54 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.17 (s, 6H), 0.70 (d, J = 6.8
Hz, 3H), 0.98 (s, 9H), 1.12 (d, J = 6.8 Hz, 3H), 1.31 (s, 3H), 1.85 (td, J = 12.8, 5.0 Hz, 1H), 2.08
(td, J = 12.9, 4.3 Hz, 1H), 2.23 (td, J = 12.9, 4.3 Hz, 1H), 2.32 – 2.46 (m, 2H), 3.70 (s, 3H), 3.71
(s, 3H), 3.94 (d, J = 3.3 Hz, 1H), 6.71 – 6.75 (m, 2H), 6.99 – 7.02 (m, 2H); 13C NMR (75 MHz;
CDCl3) δ -4.3, 17.1, 18.3, 19.7, 25.9, 28.7, 30.7, 30.9, 42.8, 52.4, 58.4, 60.5, 119.9, 129.3, 135.5,
153.6, 162.1, 165.7; IR (NaCl, neat) 1690 cm-1; HRMS (ESI-MS): m/z calcd for C24H41N2O3Si
[M+H]+ 433.2886, found 433.2887.
General procedure B for the one-pot TBS-deprotection/alkylation procedure
Cesium fluoride (2 eq) was added in one portion to a solution of bis-lactim ether 3.49 (1 eq) in dry
DMF (0.15 M) at room temperature. The solution was stirred for 15 min, where it had turned dark
orange, before alkyl-halide (1.05 eq) was added dropwise. The solution was stirred at room

Chapter 5: Experimental
211
temperature for 14 h. Water was added and the mixture was extracted with ethyl acetate (× 3).
The organic extracts were combined and washed with water and brine, then dried (Na2SO4). The
solvent was removed under reduced pressure and the crude material was purified, as indicated,
to afford the product 5.26 – 5.33.
(2S,5S)-5-Isopropyl-3,6-dimethoxy-2-(4’-methoxyphenethyl)-2-methyl-2,5-dihydropyrazine
(5.26)
Prepared using general procedure B using cesium fluoride (51 mg, 0.34 mmol), bis-lactim ether
3.49 (71 mg, 0.16 mmol), methyl iodide (22 μL, 0.35 mmol) and dry DMF (2 mL). The crude
material was purified by flash chromatography on silica gel, eluting with 3 % ethyl acetate/n-
hexane, to afford the product 5.26 as a clear colourless oil (54 mg, 98 %). [𝛼]𝐷23.0 = + 3 (0.5,
CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.71 (d, J = 6.8 Hz, 3H), 1.13 (d, J = 6.8 Hz, 3H), 1.32 (s,
3H), 1.86 (td, J = 12.8, 5.0 Hz, 1H), 2.09 (td, J = 12.8, 4.3 Hz, 1H), 2.26 (td, J = 12.8, 4.3 Hz, 1H),
2.33 – 2.49 (m, 2H), 3.71 (s, 3H), 3.72 (s, 3H), 3.78 (s, 3H), 3.95 (d, J = 3.3 Hz, 1H), 6.80 – 6.84
(m, 2H), 7.07 – 7.11 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 17.1, 19.7, 28.7, 30.7, 30.8, 43.0, 52.4,
55.4, 58.4, 60.5, 113.9, 129.3, 134.9, 157.8, 162.1, 165.7; IR (NaCl, neat) 1690 cm-1; HRMS
(ESI-MS): m/z calcd for C19H29N2O3 [M+H]+ 333.2178, found 333.2175.
(2S,5S)-5-Isopropyl-3,6-dimethoxy-2-(4’-butoxyphenethyl)-2-methyl-2,5-dihydropyrazine
(5.27)
Prepared using general procedure B using cesium fluoride (67 mg, 0.44 mmol), bis-lactim ether
3.49 (95 mg, 0.2 mmol), 1-bromobutane (48 μL, 0.44 mmol) and dry DMF (2 mL). The crude
material was purified by flash chromatography on silica gel, eluting with 2 % ethyl acetate/n-
hexane, to afford the product 5.27 as a clear colourless oil (58 mg, 71 %). [𝛼]𝐷21.6 = + 4 (0.5,
CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.70 (d, J = 6.8 Hz, 3H), 0.97 (t, J = 7.4 Hz, 3H), 1.12 (d, J
= 6.8 Hz, 3H), 1.30 (s, 3H), 1.42 – 1.54 (m, 2H), 1.70 – 1.90 (m, 3H), 2.08 (td, J = 12.8, 4.3 Hz,
1H), 2.24 (td, J = 12.8, 4.3 Hz, 1H), 2.32 – 2.47 (m, 2H), 3.70 (s, 3H), 3.71 (s, 3H), 3.91 – 3.95
(m, 3H), 6.78 – 6.83 (m, 2H), 7.04 – 7.07 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 14.0, 17.1, 19.4,
19.7, 28.7, 30.7, 30.8, 31.5, 43.0, 52.4, 58.4, 60.5, 67.9, 114.5, 129.3, 134.7, 157.3, 162.1, 165.7;
IR (NaCl, neat) 1691 cm-1; HRMS (ESI-MS): m/z calcd for C22H35N2O3 [M+H]+ 375.2648, found
375.2645.

Chapter 5: Experimental
212
(2S,5S)-5-Isopropyl-3,6-dimethoxy-2-(4’-heptoxyphenethyl)-2-methyl-2,5-dihydropyrazine
(3.44)
Prepared using general procedure B using cesium fluoride (0.48 g, 3.15 mmol), bis-lactim ether
3.49 (0.67 g, 1.56 mmol), 1-bromoheptane (0.30 mL, 1.91 mmol) and dry DMF (10 mL). The
crude material was purified by flash chromatography on silica gel, eluting with 5 % ethyl
acetate/n-hexane, to afford the product 3.44 as a clear colourless oil (0.54 g, 88 %) with all the
analytical data matching that reported previously for 3.44.
(2S,5S)-5-Isopropyl-3,6-dimethoxy-2-(4’-octoxyphenethyl)-2-methyl-2,5-dihydropyrazine
(5.28)
Prepared using general procedure B using cesium fluoride (0.15 g, 0.99 mmol), bis-lactim ether
3.49 (0.21 g, 0.49 mmol), 1-bromooctane (0.10 mL, 0.58 mmol) and dry DMF (3 mL). The crude
material was purified by flash chromatography on silica gel, eluting with 5 % ethyl acetate/n-
hexane, to afford the product 5.28 as a clear colourless oil (0.17 g, 81 %). [𝛼]𝐷25.1 = + 60 (0.5,
CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.70 (d, J = 6.8 Hz, 3H), 0.89 (t, J = 6.8 Hz, 3H), 1.12 (d, J
= 6.8 Hz, 3H), 1.23 – 1.47 (m, 13H), 1.71 – 1.81 (m, 2H), 1.85 (td, J = 12.8, 5.0 Hz, 1H), 2.08 (td,
J = 12.8, 4.3 Hz, 1H), 2.24 (td, J = 12.8, 4.3 Hz, 1H), 2.32 – 2.48 (m, 2H), 3.70 (s, 3H), 3.71 (s,
3H), 3.90 – 3.95 (m, 3H), 6.78 – 6.83 (m, 2H), 7.04 – 7.07 (m, 2H); 13C NMR (75 MHz; CDCl3) δ
14.2, 17.1, 19.7, 22.8, 26.2, 28.7, 29.4, 29.49, 29.52, 30.7, 30.8, 32.0, 43.0, 52.4, 58.4, 60.5,
68.2, 114.5, 129.3, 134.7, 157.3, 162.1, 165.7; IR (NaCl, neat) 1691 cm-1; HRMS (ESI-MS): m/z
calcd for C26H43N2O3 [M+H]+ 431.3274, found 431.3256.
(2S,5S)-5-Isopropyl-3,6-dimethoxy-2-(4’-nonoxyphenethyl)-2-methyl-2,5-dihydropyrazine
(5.29)
Prepared using general procedure B using cesium fluoride (81 mg, 0.53 mmol), bis-lactim ether
3.49 (0.12 g, 0.27 mmol), 1-bromononane (0.10 mL, 0.54 mmol) and dry DMF (3 mL). The crude
material was purified by flash chromatography on silica gel, eluting with 2 % ethyl acetate/n-
hexane, to afford the product 5.29 as a clear colourless oil (0.11 g, 89 %). [𝛼]𝐷26.5 = + 32 (0.5,
CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.70 (d, J = 6.8 Hz, 3H), 0.89 (t, J = 6.8 Hz, 3H), 1.12 (d, J
= 6.8 Hz, 3H), 1.23 – 1.49 (m, 12H), 1.31 (s, 3H), 1.71 – 1.81 (m, 2H), 1.85 (td, J = 12.8, 5.0 Hz,
1H), 2.08 (td, J = 12.8, 4.3 Hz, 1H), 2.24 (td, J = 12.8, 4.3 Hz, 1H), 2.32 – 2.48 (m, 2H), 3.70 (s,
3H), 3.71 (s, 3H), 3.90 – 3.95 (m, 3H), 6.78 – 6.83 (m, 2H), 7.04 – 7.07 (m, 2H); 13C NMR (75
MHz; CDCl3) δ 14.3, 17.1, 19.7, 22.8, 26.2, 28.7, 29.4, 29.5, 29.6, 29.7, 30.7, 30.8, 32.0, 43.0,
52.4, 58.4, 60.5, 68.2, 114.5, 129.3, 134.7, 157.3, 162.1, 165.7; IR (NaCl, neat) 1691 cm-1;
HRMS (ESI-MS): m/z calcd for C27H45N2O3 [M+H]+ 445.3430, found 445.3418.

Chapter 5: Experimental
213
(2S,5S)-5-Isopropyl-3,6-dimethoxy-2-(4’-decoxyphenethyl)-2-methyl-2,5-dihydropyrazine
(5.30)
Prepared using general procedure B using cesium fluoride (67 mg, 0.44 mmol), bis-lactim ether
3.49 (95 mg, 0.22 mmol), 1-bromodecane (92 μL, 0.44 mmol) and dry DMF (2 mL). The crude
material was purified by flash chromatography on silica gel, eluting with 2 % ethyl acetate/n-
hexane, to afford the product 5.30 as a clear colourless oil (78 mg, 77 %). [𝛼]𝐷21.9 = + 4 (0.5,
CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.70 (d, J = 6.8 Hz, 3H), 0.88 (t, J = 6.8 Hz, 3H), 1.12 (d, J
= 6.8 Hz, 3H), 1.23 – 1.48 (m, 14H), 1.30 (s, 3H), 1.71 – 1.81 (m, 2H), 1.85 (td, J = 12.8, 5.0 Hz,
1H), 2.08 (td, J = 12.8, 4.3 Hz, 1H), 2.24 (td, J = 12.8, 4.3 Hz, 1H), 2.32 – 2.47 (m, 2H), 3.70 (s,
3H), 3.71 (s, 3H), 3.90 – 3.94 (m, 3H), 6.79 – 6.82 (m, 2H), 7.04 – 7.07 (m, 2H); 13C NMR (75
MHz; CDCl3) δ 14.3, 17.1, 19.7, 22.8, 26.2, 28.7, 29.5, 29.6, 29.71, 29.73, 30.7, 30.8, 32.0, 43.0,
52.4, 58.4, 60.5, 68.2, 114.5, 129.3, 134.7, 157.3, 162.1, 165.7; IR (NaCl, neat) 1694 cm-1;
HRMS (ESI-MS): m/z calcd C28H47N2O3 [M+H]+ 459.3587, found 459.3587.
(2S,5S)-5-Isopropyl-3,6-dimethoxy-2-(4’-dodecoxyphenethyl)-2-methyl-2,5-dihydropyrazine
(5.31)
Prepared using general procedure B using cesium fluoride (81 mg, 0.53 mmol), bis-lactim ether
3.49 (0.12 g, 0.27 mmol), 1-bromododecane (0.13 mL, 0.53 mmol) and dry DMF (3 mL). The
crude material was purified by flash chromatography on silica gel, eluting with 2 % ethyl
acetate/n-hexane, to afford the product 5.31 as a clear colourless oil (0.13 g, 98 %). [𝛼]𝐷26.5 = +
32 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.70 (d, J = 6.8 Hz, 3H), 0.89 (t, J = 6.8 Hz, 3H),
1.12 (d, J = 6.8 Hz, 3H), 1.27 – 1.49 (m, 18H), 1.31 (s, 3H), 1.71 – 1.81 (m, 2H), 1.85 (td, J =
12.8, 5.0 Hz, 1H), 2.08 (td, J = 12.8, 4.3 Hz, 1H), 2.24 (td, J = 12.8, 4.3 Hz, 1H), 2.32 – 2.48 (m,
2H), 3.70 (s, 3H), 3.71 (s, 3H), 3.90 – 3.95 (m, 3H), 6.78 – 6.83 (m, 2H), 7.04 – 7.07 (m, 2H); 13C
NMR (75 MHz; CDCl3) δ 14.3, 17.1, 19.7, 22.8, 26.2, 28.7, 29.5, 29.6, 29.7, 29.75, 29.79, 29.81,
30.7, 30.8, 32.1, 43.0, 52.4, 58.4, 60.5, 68.2, 114.5, 129.3, 134.7, 157.4, 162.1, 165.7; IR (NaCl,
neat) 1691cm-1; HRMS (ESI-MS): m/z calcd for C28H47N2O3 [M+H]+ 487.3900, found 487.3883.
(2S,5S)-5-Isopropyl-3,6-dimethoxy-2-(4’-benzyloxyphenethyl)-2-methyl-2,5-dihydropyrazine
(5.32)
Prepared using general procedure B using cesium fluoride (0.14 g, 0.89 mmol), bis-lactim ether
3.49 (0.19 g, 0.44 mmol), benzylbromide (0.11 mL, 0.92 mmol) and dry DMF (4 mL). The crude
material was purified by flash chromatography on silica gel, eluting with 2 % ethyl acetate/n-
hexane, to afford the product 5.32 as a clear colourless oil (0.15 g, 83 %). [𝛼]𝐷22.3 = + 4 (0.5,
CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.71 (d, J = 6.8 Hz, 3H), 1.13 (d, J = 6.8 Hz, 3H), 1.31 (s,

Chapter 5: Experimental
214
3H), 1.86 (td, J = 12.8, 5.0 Hz, 1H), 2.09 (td, J = 12.8, 4.3 Hz, 1H), 2.25 (td, J = 12.8, 4.3 Hz, 1H),
2.33 – 2.49 (m, 2H), 3.71 (s, 3H), 3.72 (s, 3H), 3.95 (d, J = 3.3 Hz, 1H), 5.04 (s, 2H), 6.87 – 6.92
(m, 2H), 7.07 – 7.10 (m, 2H), 7.29 – 7.45 (m, 5H); 13C NMR (75 MHz; CDCl3) δ 17.1, 19.7, 28.7,
30.7, 30.8, 42.9, 52.4, 58.4, 60.5, 70.2, 114.8, 127.6, 128.0, 128.7, 129.4, 135.2, 137.4, 157.0,
162.1, 165.7; IR (NaCl, neat) 1694 cm-1; HRMS (ESI-MS): m/z calcd for C25H33N2O3 [M+H]+
409.2491, found 409.2490.
(2S,5S)-5-Isopropyl-3,6-dimethoxy-2-(4’-cyclohexylbutoxyphenyl)-2-methyl-2,5-
dihydropyrazine (5.33)
Prepared using general procedure B using cesium fluoride (0.15 g, 0.97 mmol), bis-lactim ether
3.49 (0.21 g, 0.48 mmol), (0.13 g, 0.59 mmol) and dry DMF (3 mL). The crude material was
purified by flash chromatography on silica gel, eluting with 5 % ethyl acetate/n-hexane, to afford
the product 5.33 as a clear colourless oil (0.19 g, 88 %). [𝛼]𝐷25.1 = + 50 (0.5, CHCl3); 1H NMR
(300 MHz; CDCl3) δ 0.70 (d, J = 6.8 Hz, 3H), 0.82 – 0.92 (m, 2H), 1.12 (d, J = 6.8 Hz, 3H), 1.15 –
1.28 (m, 6H), 1.31 (s, 3H), 1.40 – 1.50 (m, 2H), 1.58 – 1.79 (m, 8H), 1.85 (td, J = 12.8, 5.0 Hz,
1H), 2.08 (td, J = 12.8, 4.3 Hz, 1H), 2.24 (td, J = 12.8, 4.3 Hz, 1H), 2.32 – 2.48 (m, 2H), 3.70 (s,
3H), 3.71 (s, 3H), 3.90 – 3.95 (m, 3H), 6.78 – 6.83 (m, 2H), 7.04 – 7.07 (m, 2H); 13C NMR (75
MHz; CDCl3) δ 17.1, 19.7, 23.5, 26.6, 26.9, 28.7, 29.8, 30.7, 30.8, 33.5, 37.4, 37.8, 43.0, 52.4,
58.4, 60.5, 68.2, 114.5, 129.3, 134.7, 157.4, 162.1, 165.7; IR (NaCl, neat) 1692 cm-1; HRMS
(ESI-MS): m/z calcd for C28H45N2O3 [M+H]+ 457.3430, found 457.3419.
(2S,5S)-5-Isopropyl-3,6-dimethoxy-2-(4’-hydroxyphenethyl)-2-methyl-2,5-dihydropyrazine
(3.50)
Cesium fluoride (72 mg, 0.47 mmol) was added in one portion to a solution of bis-lactim ether
3.49 (0.10 g, 0.24 mmol) in dry DMF (3 mL) at room temperature. The solution was stirred for13
h. Water was added and the mixture was extracted with ethyl acetate (× 3). The organic extracts
were combined and washed with water and brine, then dried (Na2SO4). The solvent was removed
under reduced pressure and the crude material purified by flash chromatography on silica gel,
eluting with 10 % ethyl acetate/n-hexane, to afford the product 3.50 as a clear colourless oil (57
mg, 76 %). [𝛼]𝐷21.7 = + 4 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.72 (d, J = 6.8 Hz, 3H), 1.12
(d, J = 6.8 Hz, 3H), 1.34 (s, 3H), 1.86 (td, J = 12.9, 4.9 Hz, 1H), 2.09 (td, J = 12.9, 4.3 Hz, 1H),

Chapter 5: Experimental
215
2.23 (td, J = 12.9, 4.3 Hz, 1H), 2.33 – 2.47 (m, 2H), 3.72 (s, 6H), 3.97 (d, J = 3.3 Hz, 1H), 6.04 (s,
1H), 6.71 – 6.75 (m, 2H), 6.99 – 7.01 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 17.2, 19.7, 28.7, 30.8,
42.7, 52.5, 52.6, 58.6, 60.6, 115.4, 129.5, 134.5, 153.9, 162.6, 165.7; IR (NaCl, neat) 1691, 3349
cm-1; HRMS (ESI-MS): m/z calcd for C18H27N2O3 [M+H]+ 319.2022, found 319.2016.
(2S,5S)-5-Isopropyl-3,6-dimethoxy-2-(4’-(2’-(2’-methoxyethoxy)ethoxy)phenethyl)-2-methyl-
2,5-dihydropyrazine (5.34)
1-Bromo-2-(2-methoxyethoxy)ethane (74 μL, 0.55 mmol) was added dropwise to a suspension of
bis-lactim ether 3.50 (0.12 g, 0.36 mmol) and potassium carbonate (0.15 g, 1.09 mmol) in dry
DMF (3 mL). The suspension was stirred at room temperature for 15 h then 60°C for 6 h. The
solution was allowed to cool to room temperature and water was added. The solution was
extracted with ethyl acetate (× 3). The organic extracts were combined and washed with water
and brine, then dried (Na2SO4). The solvent was removed under reduced pressure and the crude
material was purified by flash chromatography on silica gel, eluting with 15 % ethyl acetate/n-
hexane, to afford the product 5.34 as a clear colourless oil (68 mg, 44 %). [𝛼]𝐷24.6 = + 54 (0.5,
CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.70 (d, J = 6.8 Hz, 3H), 1.11 (d, J = 6.8 Hz, 3H), 1.30 (s,
3H), 1.84 (td, J = 12.9, 5.0 Hz, 1H), 2.07 (td, J = 12.9, 4.4 Hz, 1H), 2.23 (td, J = 12.9, 4.4 Hz, 1H),
2.32 – 2.47 (m, 2H), 3.39 (s, 3H), 3.56 – 3.59 (m, 2H), 3.70 – 3.73 (m, 8H), 3.82 – 3.86 (m, 2H),
3.72 (s, 6H), 3.93 (d, J = 3.3 Hz, 1H), 4.09 – 4.13 (m, 2H), 6.79 – 6.84 (m, 2H), 7.03 – 7.08 (m,
2H); 13C NMR (75 MHz; CDCl3) δ 17.1, 19.7, 28.7, 30.7, 30.8, 42.9, 52.4, 58.4, 59.2, 60.5, 67.6,
70.0, 70.9, 72.1, 114.6, 129.3, 135.1, 157.0, 162.1, 165.7; IR (NaCl, neat) 1691 cm-1; HRMS
(ESI-MS): m/z calcd for C23H37N2O5 [M+H]+ 421.2703, found 421.2695.
General Procedure C for the preparation of aminoesters

Chapter 5: Experimental
216
A solution of TFA (50 eq) in water (200 % vol/vol of TFA) was added dropwise to a solution of bis-
lactim ether 5.35 – 5.44 (1 eq) in acetonitrile (0.03 M). The solution was stirred at room
temperature for 4 h after which the acetonitrile was removed under reduced pressure. The
residue was diluted with water and neutralised with portions of solid sodium bicarbonate, then
extracted with dichloromethane (× 4). The organic extracts were combined and dried (Na2SO4).
The solvent was removed under reduced pressure and the crude material was purified, as
indicated, to afford the product.
Methyl (2S)-2-amino-4-(4’-methoxyphenyl)-2-methylbutanoate (5.35)
Prepared using general procedure C using TFA (1 mL), water (2 mL) bis-lactim ether 5.26 (67
mg, 0.20 mmol) and acetonitrile (6 mL). The crude material was purified by flash chromatography
on silica gel, eluting with ethyl acetate, to afford the product 5.35 as a clear colourless oil (46 mg,
96 %). [𝛼]𝐷25.6 = + 12 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 1.36 (s, 3H), 1.74 (s, 2H), 1.79 –
1.90 (m, 1H), 1.95 – 2.05 (m, 1H), 2.40 – 2.50 (m, 1H), 2.54 – 2.65 (m, 1H), 3.70 (s, 3H), 3.77 (s,
3H), 6.79 – 6.84 (m, 2H), 7.06 – 7.10 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 26.6, 29.9, 43.2, 52.4,
55.4, 57.9, 114.0, 129.4, 133.7, 158.0, 178.1; IR (NaCl, neat) 1729, 3315, 3372 cm-1; HRMS
(ESI-MS): m/z calcd for C13H20NO3 [M+H]+ 238.1443, found 238.1438.
Methyl (2S)-2-amino-4-(4’-butoxyphenyl)-2-methylbutanoate (5.36)
Prepared using general procedure C using TFA (1 mL), water (2 mL) bis-lactim ether 5.27 (58
mg, 0.16 mmol) and acetonitrile (6 mL). The crude material was purified by flash chromatography
on silica gel, eluting with ethyl acetate, to afford the product 5.36 as a clear colourless oil (41 mg,
95 %). [𝛼]𝐷23.5 = + 12 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.96 (t, J = 7.3 Hz, 3H), 1.36 (s,
3H), 1.41 – 1.54 (m, 2H), 1.69 (br s, 2H), 1.72 – 1.79 (m, 2H), 1.81 – 1.90 (m, 2H), 1.95 – 2.05
(m, 1H), 2.40 – 2.50 (m, 1H), 2.54 – 2.64 (m, 1H), 3.70 (s, 3H), 3.92 (t, J = 6.5 Hz, 2H), 6.78 –
6.83 (m, 2H), 7.04 – 7.09 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 14.0, 19.4, 26.7, 29.9, 31.5, 43.2,
52.3, 57.9, 67.8, 114.6, 129.3, 133.5, 157.5, 178.1; IR (NaCl, neat) 1732, 3310, 3378 cm-1;
HRMS (ESI-MS): m/z calcd for C16H26NO3 [M+H]+ 280.1913, found 280.1913.
Methyl (2S)-2-amino-4-(4’-octoxyphenyl)-2-methylbutanoate (5.37)
Prepared using general procedure C using TFA (3 mL), water (6 mL) bis-lactim ether 5.28 (0.17
g, 0.40 mmol) and acetonitrile (8 mL). The crude material was purified by flash chromatography
on silica gel, eluting with ethyl acetate, to afford the product 5.37 as a clear colourless oil (95 mg,
64 %). [𝛼]𝐷27.9 = + 16 (0.5, CHCl3); 1H NMR (400 MHz; CDCl3) δ 0.88 (t, J = 6.8 Hz, 3H), 1.26 –
1.46 (m, 10H), 1.35 (s, 3H), 1.68 (br s, 2H), 1.71 – 1.78 (m, 2H), 1.80 – 1.88 (m, 1H), 1.96 – 2.03

Chapter 5: Experimental
217
(m, 1H), 2.40 – 2.48 (m, 1H), 2.54 – 2.62 (m, 1H), 3.69 (s, 3H), 3.90 (t, J = 6.6 Hz, 2H), 6.78 –
6.81 (m, 2H), 7.05 – 7.07 (m, 2H); 13C NMR (100 MHz; CDCl3) δ 14.2, 22.7, 26.1, 26.6, 29.3,
29.38, 29.43, 29.9, 31.9, 43.2, 52.3, 57.8, 68.1, 114.5, 129.2, 133.4, 157.5, 178.1; IR (NaCl, neat)
1732, 3316, 3376 cm-1; HRMS (ESI-MS): m/z calcd for C20H34NO3 [M+H]+ 336.2539, found
336.2518.
Methyl (2S)-2-amino-4-(4’-nonoxyphenyl)-2-methylbutanoate (5.38)
Prepared using general procedure C using TFA (2 mL), water (4 mL) bis-lactim ether 5.29 (0.11
g, 0.24 mmol) and acetonitrile (7 mL). The crude material was purified by flash chromatography
on silica gel, eluting with ethyl acetate, to afford the product 5.38 as a clear colourless oil (55 mg,
66 %). [𝛼]𝐷26.3 = + 12 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.88 (t, J = 6.8 Hz, 3H), 1.27 –
1.48 (m, 12H), 1.36 (s, 3H), 1.71 – 1.90 (m, 5H), 1.95 – 2.05 (m, 1H), 2.40 – 2.50 (m, 1H), 2.54 –
2.64 (m, 1H), 3.70 (s, 3H), 3.91 (t, J = 6.6 Hz, 2H), 6.78 – 6.83 (m, 2H), 7.04 – 7.09 (m, 2H); 13C
NMR (75 MHz; CDCl3) δ 14.2, 22.8, 26.2, 26.6, 29.37, 29.43, 29.5, 29.7, 29.9, 32.0, 43.2, 52.3,
57.9, 68.1, 114.6, 129.3, 133.5, 157.5, 178.1; IR (NaCl, neat) 1732, 3314, 3379 cm-1; HRMS
(ESI-MS): m/z calcd for C21H36NO3 [M+H]+ 350.2695, found 350.2685.
Methyl (2S)-2-amino-4-(4’-decoxyphenyl)-2-methylbutanoate (5.39)
Prepared using general procedure C using TFA (1 mL), water (2 mL) bis-lactim ether 5.30 (78
mg, 0.17 mmol) and acetonitrile (6 mL). The crude material was purified by flash chromatography
on silica gel, eluting with ethyl acetate, to afford the product 5.39 as a clear colourless oil (54 mg,
91 %). [𝛼]𝐷23.6 = + 12 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.88 (t, J = 6.8 Hz, 3H), 1.27 –
1.48 (m, 14H), 1.36 (s, 3H), 1.71 – 1.79 (m, 4H), 1.80 – 1.90 (m, 1H), 1.95 – 2.05 (m, 1H), 2.40 –
2.50 (m, 1H), 2.54 – 2.64 (m, 1H), 3.70 (s, 3H), 3.91 (t, J = 6.6 Hz, 2H), 6.78 – 6.83 (m, 2H), 7.04
– 7.09 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 14.2, 22.8, 26.2, 26.6, 29.4, 29.5, 29.66, 29.69,
29.91, 32.0, 43.2, 52.3, 57.9, 68.1, 114.6, 129.3, 133.5, 157.5, 178.1; IR (NaCl, neat)1733, 3317,
3379 cm-1; HRMS (ESI-MS): m/z calcd for C22H38NO3 [M+H]+ 364.2852, found 364.2849.
Methyl (2S)-2-amino-4-(4’-dodecoxyphenyl)-2-methylbutanoate (5.40)
Prepared using general procedure C using TFA (2 mL), water (4 mL) bis-lactim ether 5.31 (0.13
g, 0.27 mmol) and acetonitrile (7 mL). The crude material was purified by flash chromatography
on silica gel, eluting with ethyl acetate, to afford the product 5.40 as a clear colourless oil (56 mg,
54 %). [𝛼]𝐷26.3 = + 12 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.88 (t, J = 6.8 Hz, 3H), 1.26 –
1.48 (m, 18H), 1.36 (s, 3H), 1.71 – 1.90 (m, 5H), 1.95 – 2.05 (m, 1H), 2.40 – 2.50 (m, 1H), 2.54 –
2.64 (m, 1H), 3.70 (s, 3H), 3.91 (t, J = 6.6 Hz, 2H), 6.78 – 6.83 (m, 2H), 7.04 – 7.09 (m, 2H); 13C

Chapter 5: Experimental
218
NMR (75 MHz; CDCl3) δ 14.2, 22.8, 26.2, 26.6, 29.4, 29.46, 29.52, 29.70, 29.71, 29.75, 29.77,
29.9, 32.0, 43.2, 52.3, 57.9, 68.1, 114.6, 129.3, 133.5, 157.5, 178.1; IR (NaCl, neat) 1732, 3326,
3380 cm-1; HRMS (ESI-MS): m/z calcd for C24H42NO3 [M+H]+ 392.3165, found 392.3154.
Methyl (2S)-2-amino-4-(4’-benzyloxyphenyl)-2-methylbutanoate (5.41)
Prepared using general procedure C using TFA (1.5 mL), water (3 mL) bis-lactim ether 5.32 (0.15
g, 0.37 mmol) and acetonitrile (10 mL). The crude material was purified by flash chromatography
on silica gel, eluting with ethyl acetate, to afford the product 5.41 as a clear colourless oil (98 mg,
85 %). [𝛼]𝐷23.5 = + 12 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 1.38 (s, 3H), 1.70 (br s, 2H),
1.81 – 1.91 (m, 1H), 1.97 – 2.07 (m, 1H), 2.42 – 2.52 (m, 1H), 2.56 – 2.67 (m, 1H), 3.70 (s, 3H),
5.04 (s, 2H), 6.88 – 6.93 (m, 2H), 7.07 – 7.12 (m, 2H), 7.29 – 7.45 (m, 5H); 13C NMR (75 MHz;
CDCl3) δ 26.6, 29.9, 43.1, 52.3, 57.8, 70.1, 114.9, 127.5, 127.9, 128.6, 129.3, 134.0, 137.2,
157.1, 178.0; IR (NaCl, neat) 1733 cm-1; HRMS (ESI-MS): m/z calcd for C19H24NO3 [M+H]+
314.1756, found 314.1754.
Methyl (2S)-2-amino-4-(4’-cyclohexylbutoxyphenyl)-2-methylbutanoate (5.42)
Prepared using general procedure C using TFA (3 mL), water (6 mL) bis-lactim ether 5.33 (0.19
g, 0.43 mmol) and acetonitrile (9 mL). The crude material was purified by flash chromatography
on silica gel, eluting with ethyl acetate, to afford the product 5.42 as a clear colourless oil (85 mg,
55 %). [𝛼]𝐷27.9 = + 16 (0.5 , CHCl3); 1H NMR (400 MHz; CDCl3) δ 0.81 – 0.92 (m, 2H), 1.10 –
1.26 (m, 6H), 1.35 (s, 3H), 1.39 – 1.47 (m, 2H), 1.61 – 1.76 (m, 9H), 1.80 – 1.88 (m, 1H), 1.95 –
2.03 (m, 1H), 2.40 – 2.48 (m, 1H), 2.54 – 2.62 (m, 1H), 3.69 (s, 3H), 3.90 (t, J = 6.6 Hz, 2H), 6.78
– 6.80 (m, 2H), 7.04 – 7.07 (m, 2H); 13C NMR (100 MHz; CDCl3) δ 23.4, 26.5, 26.6, 26.8, 29.7,
29.9, 33.4, 27.3, 37.7, 43.2, 52.2, 57.8, 68.1, 114.5, 129.2, 133.4, 157.5, 178.1; IR (NaCl, neat)
1732, 3315, 3377 cm-1; HRMS (ESI-MS): m/z calcd for C22H36NO3 [M+H]+ 362.2695, found
362.2673.
Methyl (2S)-2-amino-4-(4’-(2’-(2’-methoxyethoxy)ethoxy)phenyl)-2-methylbutanoate (5.43)
Prepared using general procedure C using TFA (1 mL), water (2 mL) bis-lactim ether 5.34 (68
mg, 0.16 mmol) and acetonitrile (6 mL). The crude material was purified by flash chromatography
on silica gel, eluting with ethyl acetate, to afford the product 5.43 as a clear colourless oil (37 mg,
71 %). [𝛼]𝐷24.0 = + 16 (0.5, CHCl3); 1H NMR (500 MHz; CDCl3) δ 1.35 (s, 3H), 1.75 (br s, 2H),
1.80 – 1.86 (m, 1H), 1.95 – 2.01 (m, 1H), 2.40 – 2.46 (m, 1H), 2.54 – 2.60 (m, 1H), 3.37 (s, 3H),
3.54 – 3.56 (m, 2H), 3.68 – 3.70 (m, 5H), 3.81 – 3.83 (m, 2H), 4.08 – 4.10 (m 2H), 6.80 – 6.83 (m,
2H), 7.04 – 7.06 (m, 2H); 13C NMR (125 MHz; CDCl3) δ 26.6, 29.9, 43.1, 52.3, 57.8, 59.1, 67.5,

Chapter 5: Experimental
219
69.9, 70.8, 72.0, 114.7, 129.3, 133.9, 157.1, 178.1; IR (NaCl, neat) 1730, 3310, 3374 cm-1;
HRMS (ESI-MS): m/z calcd for C17H28NO5 [M+H]+ 326.1968, found 326.1967.
General Procedure D for the preparation of aminoalcohols
Lithium aluminium hydride (1.5 eq) was added as a solid in one portion to a solution of
aminoester (1 eq) in freshly distilled THF (0.05 M) at 0°C. The solution was stirred at 0°C for 20
min then the cold bath was removed and the solution stirred at room temperature for 1 h. The
reaction was quenched with saturated aqueous sodium sulfate solution and the mixture was
extracted with ethyl acetate (× 4). The organic extracts were combined and washed with
saturated aqueous sodium bicarbonate solution, water and brine, then dried (Na2SO4). The
solvent was removed under reduced pressure and the crude material was purified, as indicated,
to afford the product 3.51 – 3.59.
(2S)-2-Amino-4-(4’-methoxyphenyl)-2-methyl-1-butanol (3.51)
Prepared using general procedure D using lithium aluminium hydride (14 mg, 0.36 mmol),
aminoester 5.35 (46 mg, 0.19 mmol) and freshly distilled THF (2 mL). The crude material was
recrystallised (EtOH/n-hexane) to afford the product 3.51 as a clear colourless oil (36 mg, 88 %).
[𝛼]𝐷25.6 = + 2 (0.5, MeOH); 1H NMR (300 MHz; CDCl3) δ 1.14 (s, 3H), 1.62 – 1.74 (m, 2H), 2.33
(br s, 3H), 2.59 (t, J = 8.5 Hz, 2H), 3.34 (d, J = 10.6 Hz, 1H), 3.40 (d, J = 10.6 Hz, 1H), 3.78 (s,
3H), 6.81 – 6.84 (m, 2H), 7.09 – 7.12 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 24.3, 29.4, 42.1, 53.5,
55.4, 70.1, 114.0, 129.3, 134.3, 157.9; IR (NaCl, neat) 3445 cm-1; HRMS (ESI-MS): m/z calcd for
C12H20NO2 [M+H]+ 210.1494, found 210.1491.
(2S)-2-Amino-4-(4’-butoxyphenyl)-2-methyl-1-butanol (3.52)
Prepared using general procedure D using lithium aluminium hydride (8 mg, 0.21 mmol),
aminoester 5.36 (41 mg, 0.15 mmol) and freshly distilled THF (2 mL). The crude material was
purified by flash chromatography on silica gel, eluting with 3 % methanol/6 %
triethylamine/dichloromethane, to afford the product 3.52 as a clear colourless oil (25 mg, 68 %).
[𝛼]𝐷25.5 = - 2 (0.5 , CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.96 (t, J = 7.3 Hz, 3H), 1.14 (s, 3H),

Chapter 5: Experimental
220
1.41 – 1.54 (m, 2H), 1.63 – 1.79 (m, 4H), 2.55 – 2.61 (m, 6H), 3.35 (d, J = 10.7 Hz, 1H), 3.41 (d, J
= 10.7 Hz, 1H), 3.92 (t, J = 6.5 Hz, 2H), 6.78 (m, 2H), 7.06 – 7.11 (m, 2H); 13C NMR (75 MHz;
CDCl3) δ 14.0, 19.4, 24.2, 29.4, 31.5, 41.9, 53.7, 67.8, 69.8, 114.6, 129.2, 134.1, 157.5; IR (NaCl,
neat) 3386 cm-1; HRMS (ESI-MS): m/z calcd for C15H26NO2 [M+H]+ 252.1964, found 252.1963.
(2S)-2-Amino-4-(4’-octoxyphenyl)-2-methyl-1-butanol (3.53)
Prepared using general procedure D using lithium aluminium hydride (16 mg, 0.42 mmol),
aminoester 5.37 (95 mg, 0.28 mmol) and freshly distilled THF (2 mL). The crude material was
purified by flash chromatography on silica gel, eluting with 2 % methanol/5 %
triethylamine/dichloromethane, to afford the product 3.53 as a clear colourless oil (55 mg, 63 %).
[𝛼]𝐷26.9 = + 6 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.88 (t, J = 6.8 Hz, 3H), 1.11 (s, 3H),
1.28 – 1.46 (m, 10H), 1.60 – 1.80 (m, 4H), 2.24 (br s, 3H), 2.54 – 2.60 (m, 2H), 3.32 (d, J = 10.6
Hz, 1H), 3.38 (d, J =10.6 Hz, 1H), 3.91 (t, J = 6.6 Hz, 2H), 6.78 – 6.83 (m, 2H), 7.06 – 7.11 (m,
2H); 13C NMR (75 MHz; CDCl3) δ 14.2, 22.8, 24.5, 26.2, 29.3, 29.4, 29.5, 31.9, 42.2, 53.2, 68.1,
70.2, 114.6, 129.2, 134.2, 157.4; IR (NaCl, neat) 3184, 3264, 3333 cm-1; HRMS (ESI-MS): m/z
calcd for C19H34NO2 [M+H]+ 308.2589, found 308.2572.
(2S)-2-Amino-4-(4’-nonoxyphenyl)-2-methyl-1-butanol (3.54)
Prepared using general procedure D using lithium aluminium hydride (12 mg, 0.32 mmol),
aminoester 5.38 (55 mg, 0.16 mmol) and freshly distilled THF (3 mL). The crude material was
purified by flash chromatography on silica gel, eluting with 2 % methanol/5 %
triethylamine/dichloromethane, to afford the product 3.54 as a clear colourless oil (23 mg, 45 %).
[𝛼]𝐷26.6 = + 4 (0.5, CHCl3); 1H NMR (600 MHz; CDCl3) δ 0.88 (t, J = 6.8 Hz, 3H), 1.14 (s, 3H),
1.27 – 1.35 (m, 10H), 1.41 – 1.46 (m, 2H), 1.62 – 1.78 (m, 4H), 2.10 (br s, 1H), 2.13 (br,s, 3H),
2.57 – 2.59 (m, 2H), 3.34 (d, J = 10.6 Hz, 1H), 3.39 (d, J = 10.6 Hz, 1H), 3.91 (t, J = 6.6 Hz, 2H),
6.80 – 6.82 (m, 2H), 7.08 – 7.09 (m, 2H); 13C NMR (150 MHz; CDCl3) δ 14.3, 22.8, 24.4, 26.2,
29.4, 29.5, 29.6, 29.7, 32.0, 42.1, 53.4, 68.2, 70.1, 114.7, 129.2, 134.2, 157.5; IR (NaCl, neat)
3150, 3264, 3333 cm-1; HRMS (ESI-MS): m/z calcd for C20H36NO2 [M+H]+ 322.2746, found
322,2738.
(2S)-2-Amino-4-(4’-decoxyphenyl)-2-methyl-1-butanol (3.55)
Prepared using general procedure D using lithium aluminium hydride (9 mg, 0.24 mmol),
aminoester 5.39 (54 mg, 0.16 mmol) and freshly distilled THF (2 mL). The crude material was
purified by flash chromatography on silica gel, eluting with 2 % methanol/4 %
triethylamine/dichloromethane, to afford the product 3.55 as a clear colourless oil (36 mg, 69 %).

Chapter 5: Experimental
221
[𝛼]𝐷25.5 = - 2 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.88 (t, J = 6.8 Hz, 3H), 1.12 (s, 3H),
1.27 – 1.48 (m, 14H), 1.58 – 1.80 (m, 4H), 2.32 (br s, 3H), 2.55 – 2.60 (m, 2H), 3.33 (d, J = 10.6
Hz, 1H), 3.39 (d, J = 10.6 Hz, 1H), 3.91 (t, J = 6.6 Hz, 2H), 6.78 – 6.83 (m, 2H), 7.06 – 7.11 (m,
2H); 13C NMR (75 MHz; CDCl3) δ 14.2, 22.8, 24.4, 26.2, 29.45, 29.53, 29.68, 29.70, 32.0, 42.1,
53.4, 68.2, 70.1, 114. 6, 129.2, 134.2, 157.5; IR (NaCl, neat) 3177, 3264, 3333 cm-1; HRMS (ESI-
MS): m/z calcd for C21H38NO2 [M+H]+ 336.2903, found 336.2899.
(2S)-2-Amino-4-(4’-dodecoxyphenyl)-2-methyl-1-butanol (3.56)
Prepared using general procedure D using lithium aluminium hydride (11 mg, 0.29 mmol),
aminoester 5.40 (56 mg, 0.14 mmol) and freshly distilled THF (3 mL). The crude material was
purified by flash chromatography on silica gel, eluting with 1 % methanol/5 %
triethylamine/dichloromethane, to afford the product 3.56 as a clear colourless oil (29 mg, 56 %).
[𝛼]𝐷26.6 = + 4 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.88 (t, J = 6.8 Hz, 3H), 1.13 (s, 3H),
1.26 – 1.36 (m, 16H), 1.41 – 1.46 (m, 2H), 1.61 – 1.78 (m, 4H), 2.13 (br s, 3H), 2.56 – 2.59 (m,
2H), 3.34 (d, J = 10.6 Hz, 1H), 3.39 (d, J = 10.6 Hz, 1H), 3.91 (t, J = 6.6 Hz, 2H), 6.80 – 6.82 (m,
2H), 7.08 – 7.09 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 14.3, 22.8, 24.5, 26.2, 29.47, 29.48, 29.6,
29.72, 29.74, 29.77, 29.80, 32.1, 42.2, 53.4, 68.2, 70.2, 114.6, 129.2, 134.2, 157.5; IR (NaCl,
neat) 3176, 3264, 3333 cm-1; HRMS (ESI-MS): m/z calcd for C23H42NO2 [M+H]+ 364.3216, found
364.3208.
(2S)-2-Amino-4-(4’-benzyloxyphenyl)-2-methyl-1-butanol (3.57)
Prepared using general procedure D using lithium aluminium hydride (18 mg, 0.47 mmol),
aminoester 5.41 (98 mg, 0.31 mmol) and freshly distilled THF (3 mL). The crude material was
purified by flash chromatography on silica gel, eluting with 2 % methanol/4 %
triethylamine/dichloromethane, to afford the product 3.57 as a clear colourless oil (61 mg, 69 %).
[𝛼]𝐷25.5 = - 2 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 1.13 (s, 3H), 1.58 – 1.76 (m, 2H), 2.07
(br s, 3H), 2.55 – 2.64 (m, 2H), 3.33 (d, J = 10.6 Hz, 1H), 3.39 (d, J = 10.6 Hz, 1H), 5.04 (s, 2H),
6.88 – 6.93 (m, 2H), 7.09 – 7.14 (m, 2H), 7.29 – 7.45 (m, 5H); 13C NMR (75 MHz; CDCl3) δ 24.6,
29.5, 42.2, 53.1, 70.2, 70.3, 115.0, 127.6, 128.0, 128.7, 129.3, 134.8, 137.3, 157.1; IR (NaCl,
neat) 3425 cm-1; HRMS (ESI-MS): m/z calcd for C [M+H]+ 286.1807, found 286.1807.
(2S)-2-Amino-4-(4’-cyclohexylbutoxyphenyl)-2-methyl-1-butanol (3.58)
Prepared using general procedure D using lithium aluminium hydride (14 mg, 0.37 mmol),
aminoester 5.42 (85 mg, 0.24 mmol) and freshly distilled THF (2 mL). The crude material was
purified by flash chromatography on silica gel, eluting with 2 % methanol/5 %

Chapter 5: Experimental
222
triethylamine/dichloromethane, to afford the product 3.58 as a clear colourless oil (26 mg, 33 %).
[𝛼]𝐷26.9 = + 6 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.85 – 0.92 (m, 2H), 1.12 (s, 3H), 1.15 –
1.30 (m, 7H), 1.39 – 1.50 (m, 2H), 1.62 – 1.78 (m, 9H), 1.99 (br s, 3H), 2.55 – 2.61 (m, 2H), 3.35
(br s, 2H), 3.91 (t, J = 6.6 Hz, 2H), 6.79 – 6.83 (m, 2H), 7.07 – 7.10 (m, 2H); 13C NMR (75 MHz;
CDCl3) δ 23.5, 26.5, 26.9, 29.5, 29.8, 33.5, 37.3, 37.7, 68.2, 114.6, 129.2, 134.2, 157.5; IR (NaCl,
neat) 3169, 3268, 3332 cm-1; HRMS (ESI-MS): m/z calcd for C21H36NO2 [M+H]+ 334.2746, found
334.2729.
(2S)-2-Amino-4-(4’-(2’-(2’-methoxyethoxy)ethoxy)phenyl)-2-methyl-1-butanol (3.59)
Prepared using general procedure D using lithium aluminium hydride (6 mg, 0.16 mmol),
aminoester 5.43 (36 mg, .11 mmol) and freshly distilled THF (2 mL). The crude material was
purified by flash chromatography on silica gel, eluting with 5 % methanol/5 %
triethylamine/dichloromethane, to afford the product 3.59 as a clear colourless oil (25 mg, 78 %).
[𝛼]𝐷25.5 = + 2 (0.5 , CHCl3); 1H NMR (300 MHz; CDCl3) δ 1.15 (s, 3H), 1.63 – 1.77 (m, 2H), 2.54 –
2.60 (m, 2H), 2.72 – 2.83 (m, 4H), 3.38 (s, 3H), 3.55 – 3.58 (m, 2H), 3.69 – 3.73 (m, 2H), 3.82 –
3.85 (m, 2H), 4.08 – 4.11 (m, 2H), 6.80 – 6.83 (m, H), 7.07 – 7.10 (m, 2H); 13C NMR (75 MHz;
CDCl3) δ 23.9, 29.4, 41.6, 53.9, 59.2, 67.5, 69.6, 69.9, 70.8, 72.0, 114.7, 129.2, 134.4, 157.1; IR
(NaCl, neat) 3286, 3349 cm-1; HRMS (ESI-MS): m/z calcd for C16H28NO4 [M+H]+ 298.2018, found
298.2018.
(2S)-2-(Dimethylamino)-4-(4’-heptyloxyphenyl)-2-methylbutanol (3.62)
Sodium cyanoborohydride (42 mg, 0.67 mmol) was added as a solid in one portion to a solution
of AAL(S) 1.37 (49 mg, 0.17 mmol), paraformaldehyde (20 mg, 0.67 mmol) and acetic acid (0.2
mL) in acetonitrile (2 mL) at 0°C. The solution was stirred for 15 min. at 0°C then room
temperature for 3 h. After diluting the solution with saturated aqueous sodium bicarbonate
solution the acetonitrile was removed under reduced pressure. The resulting solution was
extracted with ethyl acetate (× 3). The organic extracts were combined and washed with water
and brine, then dried (Na2SO4). The solvent was removed under reduced pressure and the crude
material purified by flash chromatography in silica gel, eluting with 1 % methanol/1 %
triethylamine/dichloromethane, to afford the product 3.62 as a clear colourless oil (29 mg, 54 %).
[𝛼]𝐷25.5 = -2 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.89 (t, J = 6.8 Hz, 3H), 1.06 (s, 3H), 1.26
– 1.49 (m, 9H), 1.64 – 1.70 (m, 2H), 1.72 – 1.81 (m, 2H), 2.28 (s, 6H), 2.51 – 2.57 (m, 2H), 2.74

Chapter 5: Experimental
223
(br s, 1H), 3.41 (d, J = 10.6 Hz, 1H), 3.49 (d, J = 10.6 Hz, 1H), 3.92 (t, J = 6.6 Hz, 2H), 6.79 –
6.84 (m, 2H), 7.06 – 7.11 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 14.2, 17.6, 22.7, 26.2, 29.2, 29.5,
30.1, 31.9, 36.7, 38.1, 59.3, 65.8, 68.2, 114.6, 129.2, 134.6, 157.5; IR (NaCl, neat) 3406 cm-1;
HRMS (ESI-MS): m/z calcd for C20H36NO2 [M+H]+ 322.2746, found 322.2746.
(2S)-N-(4-(4’-Heptyloxyphenyl)-1-hydroxy-2-methylbutan-2-yl)acetamide (3.63)
Acetyl chloride (10 μL, 0.14 mmol) was added dropwise to a solution of AAL(S) 1.37 (42 mg, 0.14
mmol) and triethylamine (60 μL, 0.43 mmol) in dichloromethane (3 mL) at 0°C. The solution was
stirred at 0°C for 2 h. before being quenched with saturated aqueous sodium bicarbonate
solution. The mixture was extracted with dichloromethane (× 3). The organic extracts were
combined and washed with saturated aqueous sodium bicarbonate solution, wwater and brine,
then dried (Na2SO4). The solvent was removed under reduced pressure and the crude material
purified by flash chromatography on silica gel, eluting with ethyl acetate, to afford the product
3.63 as a clear colourless gum (17 mg, 35 %). [𝛼]𝐷26.9 = - 10 (0.5, CHCl3); 1H NMR (300 MHz;
CDCl3) δ 0.89 (t, J = 6.8 Hz, 3H), 1.26 (s, 3H), 1.28 – 1.46 (m, 8H), 1.69 – 1.80 (m, 2H), 1.84 –
1.97 (m, 1H), 1.91 (s, 3H), 2.01 – 2.11 (m, 1H), 2.47 – 2.58 (m, 1H), 2.61 -2.71 (m, 1H), 3.61 –
3.70 (m, 2H), 3.91 (t, J = 6.6 Hz, 2H), 4.93 (br s, 1H), 5.50 (br s, 1H), 6.80 – 6.84 (m, 2H), 7.06 –
7.11 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 14.2, 22.7, 23.0, 24.1, 26.1, 29.2, 29.3, 29.4, 31.9,
38.4, 59.1, 68.2, 69.7, 114.8, 129.3, 133.6, 157.6, 171.4; IR (NaCl, neat) 1742, 3089, 3191, 3288
cm-1; HRMS (ESI-MS): m/z calcd for C20H33NO3Na [M+Na]+ 358.2358, found 358.2336.
(2S)-t-Butyl(1-hydroxy-4-(4’-heptyloxyphenyl)-2-methylbutan-2-yl)carbamate (3.66)[34]
Di-t-butyl dicarbonate (0.46 g, 2.10 mmol) was added as a solid in one portion to a mixture of
AAL(S) 1.37 (0.45 g, 1.54 mmol) in saturated aqueous sodium bicarbonate solution (25 mL) and
ethyl acetate (20 mL). The mixture was heated at 60°C for 6.5 h then allowed to cool to room
temperature. The solution was diluted with water and extracted with ethyl acetate (× 3). The
organic extracts were combined and washed with brine, then dried (Na2SO4). The solvent was
removed under reduce pressure and the crude material purified by flash chromatography on silica
gel, eluting with 30 % ethyl acetate/n-hexane, to afford the product 3.66 as a white solid (0.51 g,

Chapter 5: Experimental
224
83 %). [𝛼]𝐷25.6 = +2 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.88 (t, J = 6.8 Hz, 3H), 1.22 (s,
3H), 1.26 – 1.47 (m, 8H), 1.43 (s, 9H), 1.71 – 1.89 (m, 3H), 2.02 (td, J = 12.8, 5.2 Hz, 1H), 2.46 –
2.67 (m, 2H), 3.61 – 3.73 (m, 2H), 3.92 (t, J = 6.6 Hz, 2H), 4.21 (br s, 1H), 4.63 (br s, 1H), 6.77 –
6.84 (m, 2H), 7.087 – 7.11 (m, 2H); 13C NMR (100 MHz; CDCl3) δ 14.2, 22.7, 23.0, 26.2, 28.5,
29.2, 29.3, 29.5, 31.9, 38.7, 57.1, 68.2, 69.8, 80.0, 114.7, 129.3, 133.9, 156.3, 157.5; IR (NaCl,
neat) 1678, 3077, 3278 cm-1; HRMS (ESI-MS): m/z calcd for C23H39NO4Na [M+Na]+ 416.2777,
found 416.2776. Spectroscopic data matched those reported in the literature.[34]
(4S)-4-(4’-Heptyloxyphenethyl)-4-methyloxazolidin-2-one (3.67)
Boc-AAL(S) 3.66 (22 mg, 49 μmol), potassium hydroxide (11 mg, 0.20 mmol) and tosyl chloride
(11 mg, 58 μmol) in diethyl ether (2 mL) was heated at reflux for 15 h. The solution was allowed
to cool to room temperature and poured onto ice water. The mixture was extracted with diethyl
ether (× 3). The organic extracts were combined and washed with water and brine, then dried
(Na2SO4). The solvent was removed under reduced pressure and the crude material purified by
flash chromatography on silica gel, eluting with 20 % ethyl acetate/n-hexane, to afford the product
3.67 as a clear colourless gum (14 mg, 90 %).[𝛼]𝐷26.7 = + 12 (0.5, CHCl3); 1H NMR (300 MHz;
CDCl3) δ 0.89 (t, J = 6.8 Hz, 3H), 1.26 – 1.47 (m, 8H), 1.39 (s, 3H), 1.71 – 1.81 (m, 2H), 1.83 –
1.89 (m, 2H), 2.58 – 2.63 (m, 2H), 3.91 (t, J = 6.6 Hz, 2H), 4.04 (d, J = 8.5 Hz, 1H), 4.15 (d, J =
8.5 Hz, 1H), 6.48 (br s, 1H), 6.78 – 6.83 (m, 2H), 7.04 – 7.08 (m, 2H); 13C NMR (75 MHz; CDCl3)
δ 14.2, 22.7, 25.9, 26.1, 29.2, 29.4, 29.5, 31.9, 42.6, 57.8, 68.1, 75.8, 114.7, 129.2, 132.7, 157.7,
159.6; IR (NaCl, neat) 1729, 1764, 3161, 3248 cm-1; HRMS (ESI-MS): m/z calcd for
C19H29NO3Na [M+Na]+ 342.2045, found 342.2045.
(4S)-4-(4’-Heptyloxyphenethyl)-3,4-dimethyloxazolidin-2-one (3.68)
Boc-AAL(S) 3.66 (50 mg, 0.11 mmol) was added portion-wise to a solution of sodium hydride (5
mg, 0.13 mmol) in DMF (0.5 mL) at room temperature. The solution was stirred at room
temperature for 1 h where it had turned dark orange. Methyl iodide (8 μL, 0.13 mmol) was added
dropwise and the solution stirred at room temperature for a further 10 h. water was added and the

Chapter 5: Experimental
225
mixture was extracted with ethyl acetate (× 4). The organic extracts were combined and washed
with water and brine, then dried (Na2SO4). The solvent was removed under reduced pressure and
the crude material purified by flash chromatography on silica gel, eluting with 1 % methanol/5 %
triethylamine/dichloromethane, to afford the product 3.68 as a light yellow oil (30 mg, 88 %).
[𝛼]𝐷24.0 = + 14 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.88 (t, J = 6.8 Hz, 3H), 1.25 – 1.46 (m,
8H), 1.31 (s, 3H), 1.71 – 1.93 (m, 4H), 2.36 – 2.46 (m, 1H), 2.52 – 2.63 (m, 1H), 2.76 (s, 3H), 3.92
(t, J = 6.6 Hz, 2H), 3.95 (d, J = 8.6 Hz, 1H), 4.19 (d, J = 8.6 Hz, 1H), 6.79 – 6.84 (m, 2H), 7.03 –
7.07 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 14.2, 22.7, 23.8, 25.5, 26.1, 29.1, 29.2, 29.4, 31.9,
39.4, 60.1, 68.2, 72.3, 114.8, 129.1, 132.5, 157.8, 158.0; IR (NaCl, neat) 1755 cm-1; HRMS (ESI-
MS): m/z calcd for C20H31NO3Na [M+Na]+ 356.2202, found 356.2192.
(4S)-t-Butyl-4-(4’-heptyloxyphenylethyl)-4-methyl-1,2,3-oxathiazolidine-3-carboxylate-2-
oxide (5.44 and 5.45)[35]
Thionyl chloride (0.12 mL, 1.67 mmol) and pyridine (0.27 mL, 3.34 mmol) were added
successively, dropwise to a solution of Boc-AAL(S) 3.66 (0.26 g, 0.67 mmol) in acetonitrile (15
mL) at -40°C (dry ice/acetonitrile). The solution was allowed to warm slowly, in the cold bath, to -
10°C over 2.5 h. The reaction mixture was poured onto 1M aqueous hydrochloric acid and
extracted with ethyl acetate (×3). The organic extracts were combined and washed with saturate
aqueous sodium bicarbonate, water and brine, the dried (Na2SO4). The solvent was removed
under reduced pressure to afford a 1:1 mixture of diastereomers 5.44 and 5.45 which were
purified by flash chromatography on silica gel, eluting with 5 % ethyl acetate/n-hexane, to afford
one diastereomer as a clear colourless oil (0.14 g, 48 %). [𝛼]𝐷27.2 = - 36 (0.5, CHCl3); 1H NMR
(300 MHz; CDCl3) δ 0.89 (t, J = 6.8 Hz, 3H), 1.26 – 1.47 (m, 8H), 1.45 (s, 3H), 1.54 (s, 9H), 1.71
– 1.81 (m, 2H), 2.00 – 2.10 (m, 1H), 2.28 – 2.39 (m, 1H), 2.44 – 2.55 (m, 1H), 2.59 – 2.69 (m,
1H), 3.92 (t, J = 6.6 Hz, 2H), 4.28 (d, J = 8.9 Hz, 1H), 4.96 (br d, J = 8.9 Hz, 1H), 6.79 – 6.84 (m,
2H), 7.04 – 7.10 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 14.2, 22.7, 26.2, 28.4, 29.2, 29.5, 30.2,
31.9, 40.0, 68.2, 83.8, 114.7, 129.3, 133.0, 157.7; IR (NaCl, neat) 1722 cm-1; HRMS (ESI-MS):
m/z calcd for C23H37NO5SNa [M+Na]+ 462.2290, found 462.2277.
Further elution afforded the other diastereomer as a clear colourless oil (0.15 g, 51 %). [𝛼]𝐷27.2 =
+ 40 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.89 (t, J = 6.8 Hz, 3H), 1.26 – 1.47 (m, 8H), 1.53

Chapter 5: Experimental
226
(s, 9H), 1.64 (s, 3H), 1.72 – 1.81 (m, 2H), 1.87 (td, J = 12.8, 5.2 Hz, 1H), 2.21 (td, J = 12.8, 4.6
Hz, 1H), 2.41 (td, J = 12.8, 4.8 Hz, 1H), 2.58 (td, J = 12.8, 5.2 Hz, 1H), 3.92 (t, J = 6.6 Hz, 2H),
4.59 (d, J = 9.2 Hz, 1H), 4.84 (br d, J = 9.2 Hz, 1H), 6.79 – 6.84 (m, 2H), 7.03 – 7.08 (m, 2H); 13C
NMR (75 MHz; CDCl3) δ 14.2, 22.7, 26.2, 28.4, 29.2, 29.4, 30.3, 31.9, 68.2, 83.9, 114.7, 129.2,
132.9, 157.7; IR (NaCl, neat) 1724 cm-1; HRMS (ESI-MS): m/z calcd for C23H37NO5SNa [M+Na]+
462.2290, found 462.2279.
(4S)-t-Butyl-4-(4’-heptyloxyphenylethyl)-4-methyl-1,2,3-oxathiazolidine-3-carboxylate-2,2-
dioxide (3.69)[35]
Ruthenium(III) chloride (17 mg, 65 μmol) and sodium periodate (0.21 g, 0.99 mmol) were added
successively to a solution of cyclic sulfamidite 5.44 and 5.45 (0.29 g, 0.66 mmol, 50:50 mixture of
diastereomers) in 3:1 acetonitrile:water (40 mL) at 0°C. The solution was stirred at 0°C for 1 h
then, the reaction solution was diluted with water and extracted with diethyl ether (× 3). The
organic extracts were combined and washed with saturated aqueous sodium bicarbonate, water
and brine, then dried (Na2SO4). The solvent was removed under reduced pressure and the crud
material purified by flash chromatography on silica gel, eluting with 10 % ethyl acetate/n-hexane,
to afford the product 3.69 as a clear colourless oil (0. 26 g, 85 %). [𝛼]𝐷27.2 = + 22 (0.5, CHCl3); 1H
NMR (300 MHz; CDCl3) δ 0.89 (t, J = 6.8 Hz, 3H), 1.26 – 1.47 (m, 8H), 1.57 (s, 9H), 1.61 (s, 3H),
1.72 – 1.81 (m, 2H), 1.96 (m, 1H), 2.33 – 2.44 (m, 1H), 2.49 – 2.68 (m, 2H), 3.92 (t, J = 6.6 Hz,
2H), 4.16 (d, J = 9.2 Hz, 1H), 4.41 (d, J = 9.2 Hz, 1H), 6.80 – 6.85 (m, 2H), 7.04 – 7.09 (m, 2H);
13C NMR (75 MHz; CDCl3) δ 14.2, 22.7, 23.4, 26.2, 28.1, 29.2, 29.37, 29.43, 31.9, 38.7, 65.0,
68.2, 73.9, 85.4, 114.8, 129.3, 132.2, 148.4, 157.9; IR (NaCl, neat) 1734 cm-1; HRMS (ESI-MS):
m/z calcd for C23H37NO6SNa [M+Na]+ 478.2239, found 478.2228.
(2S)-1-Fluoro-4-(4’-heptyloxyphenyl)-2-methylbutan-2-amine (3.64)
A solution of tetra-n-butylammonium fluoride in THF (0.28 mL, 1.0 M, 0.28 mmol) was added
dropwise to a solution of cyclic sulfamidate 3.69 (51 mg, 0.11 mmol) in acetonitrile (3 mL) at room
temperature. The solution was heated at reflux for 3 h. then, the solution was allowed to cool to

Chapter 5: Experimental
227
room temperature and 6 M aqueous hydrochloric acid solution was added dropwise. The reaction
mixture was heated at reflux for a further 3 h. after which, it was allowed to cool to room
temperature. The acetonitrile was removed under reduced pressure. The residue was neutralised
with solid sodium bicarbonate, diluted with water and extracted with ethyl acetate (× 3). The
organic extracts were combined and washed with water and brine, then dried (Na2SO4). The
solvent was removed under reduced pressure and the crude material purified by flash
chromatography on silica gel, eluting with 1 % triethylamine/ethyl acetate, to afford the product
3.64 as a clear colourless oil (29 mg, 88 %) [𝛼]𝐷26.2 = + 4 (0.5, CHCl3); 1H NMR (600 MHz;
CDCl3) δ 0.89 (t, J = 6.8 Hz, 3H), 1.14 (d, JH-F = 1.9 Hz, 3H), 1.28 – 1.47 (m, 10H), 1.67 – 1.73
(m, 2H), 1.74 – 1.79 (m, 2H), 2.56 – 2.66 (m, 2H), 3.92 (t, J = 6.6 Hz, 2H), 4.12 (dd, J = 17.7, 8.8
Hz, 1H), 4.20 (dd, J = 17.7, 8.8 Hz, 1H), 6.81 – 6.83 (m, 2H), 7.08 – 7.10 (m, 2H); 13C NMR (150
MHz; CDCl3) δ 14.2, 22.7, 23.8 (d, JC-F = 4.2 Hz), 26.1, 29.2, 29.3, 29.4, 31.9, 41.2 (d, JC-F = 2.9
Hz), 52.2 (d, JC-F = 17.5 Hz), 68.2, 90.9 (d, JC-F = 173.3 Hz), 114.6, 129.2, 134.1, 157.5; IR (NaCl,
neat) 3294, 3370 cm-1; HRMS (ESI-MS): m/z calcd for C18H31FNO [M+H]+ 296.2390, found
296.2384.
(2S)-4-(4’-Heptyloxyphenyl)-1-methoxy-2-methylbutan-2-amine (3.65)
Methyl iodide (11 μL, 0.18 mmol) was added dropwise to a solution Boc-AAL(S) 3.66 (14 mg, 34
μmol) and tetra-n-butylammonium sulphate (2 mg, 5.9 μmol) in 50 % aqueous sodium hydroxide
solution (0.3 mL) and THF (0.3 mL) at room temperature. The solution was stirred at room
temperature for 72 h. the reaction solution was diluted with water and extracted with ethyl acetate
(× 3). The organic extracts were combined and washed with water and brine, then dried
(Na2SO4). The solvent was removed under reduced pressure. The crude material was dissolved
in acetonitrile (1 mL) and 2 M aqueous hydrochloric acid solution (2 mL) was added. The
suspension was heated at reflux for 9 h. The solution was cooled and the acetonitrile was
removed under reduced pressure. The residue was diluted with water and neutralised with solid
sodium bicarbonate before being extracted with ethyl acetate (× 3). The organic extracts were
combined and washed with water and brine, then dried (Na2SO4). The solvent was removed
under reduced pressure and the crude material purified by flash chromatography on silica gel,
eluting with 1 % triethylamine/ethyl acetate, to afford the product 3.65 as a colourless gum (9 mg,
82 %). [𝛼]𝐷24.4 = + 4 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.89 (t, J = 6.8 Hz, 3H), 1.11 (s,

Chapter 5: Experimental
228
3H), 1.26 – 1.44 (m, 12H), 1.63 – 1.81 (m, 4H), 2.50 – 2.65 (m, 2H), 2.74 (br s, 1H), 3.15 (d, J =
8.7 Hz, 1H), 3.20 (d, J = 8.7 Hz, 1H), 3.36 (s, 3H), 3.92 (t, J = 6.6 Hz, 2H), 6.78 – 6.83 (m, 2H),
7.07 – 7.12 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 14.2, 22.8, 25.1, 26.2, 29.2, 29.5, 29.6, 31.9,
42.5, 52.3, 59.4, 68.2, 81.9, 114.6, 129.3, 134.7, 157.4; IR (NaCl, neat) 3305, 3368 cm-1; HRMS
(ESI-MS): m/z calcd for C19H34NO2 [M+H]+ 308.2589, found 308.2587.
4-(4’-(Heptyloxy)phenyl)butane-2-one (5.46)
Phenol 3.70 (1.33 g, 8.12 mmol) was added as a solid in one portion to a solution of cesium
carbonate (2.91 g, 8.25 mmol) in dry DMF (12 mL) at room temperature. The solution was stirred
for 15 min., where it had turned bright yellow, then 1-bromoheptane (1.3 mL, 8.27 mmol) was
added dropwise. The solution was stirred at room temperature for 24 h. The suspension was
poured onto 2M aqueous hydrochloric acid solution and extracted with ethyl acetate (× 3). The
organic extracts were combined and washed with water (× 2) and brine, then dried (Na2SO4). The
solvent was removed under reduced pressure and the crude material purified by flash
chromatography on silica gel, eluting with 10 % ethyl acetate/n-hexane, to afford the product 5.46
as a clear colourless oil (1.94 g, 91 %). 1H NMR (300 MHz; CDCl3) δ 0.89 (t, J = 6.8 Hz, 3H), 1.31
– 1.49 (m, 8H), 1.72 – 1.81 (m, 2H), 2.12 (s, 3H), 2.69 – 2.74 (m, 2H), 2.81 – 2.86 (m, 2H), 3.92
(t, J = 6.6 Hz, 2H), 6.79 – 6.83 (m, 2H), 7.06 – 7.10 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 14.2,
22.7, 26.1, 29.0, 29.2, 29.4, 30.2, 31.9, 45.6, 68.1, 114.6, 129.3, 132.9, 157.7, 208.3; IR (NaCl,
neat) 1717 cm-1; HRMS (ESI-MS): m/z calcd for C17H26NO2Na [M+Na]+ 285.1831, found
285.1827.
1-(Heptyloxy)-4-(3’-methylbut-3’-enyl)benzene (3.61)
A solution of n-butyllithium in hexanes (4.00 mL, 1.4 M, 5.40 mmol) was added dropwise to a
solution of methyltriphenylphosphonium bromide (2.06 g, 5.76 mmol) in freshly distilled THF (50
mL) at room temperature. The solution was stirred at room temperature for 15 min. where it had
turned dark orange. A solution of ketone 5.46 (1.01 g, 3.84 mmol) in freshly distilled THF (15 mL)
was added dropwise after which, the reaction solution was stirred at room temperature for an
additional 5 h. The reaction was quenched with water and the mixture extracted with n-pentane (×

Chapter 5: Experimental
229
3). The organic extracts were combined and washed with water and brine, then dried (Na2SO4).
The solvent was removed under reduced pressure and the crude material purified by flash
chromatography on silica gel, eluting with n-hexane, to afford the product 3.61 as a clear
colourless oil (0.83 g, 83 %). 1H NMR (300 MHz; CDCl3) δ 0.90 (t, J = 6.8 Hz, 3H), 1.26 – 1.50
(m, 8H), 1.72 – 1.82 (m, 2H), 1.77 (s, 3H), 2.26 – 2.31 (m, 2H), 2.67 – 2.72 (m, 2H), 3.93 (t, J =
6.6 Hz, 2H), 4.70 – 4.71 (m, 1H), 4.73 – 4.74 (m, 1H), 6.79 – 6.84 (m, 2H), 7.07 – 7.12 (m, 2H);
13C NMR (75 MHz; CDCl3) δ 14.2, 22.8, 26.2, 29.2, 29.5, 31.9, 33.5, 40.0, 68.2, 110.3, 114.5,
129.3, 134.2, 145.7, 157.4.
(2S)-1-Amino-4-(4’-heptyloxyphenyl)-2-methylbutan-2-ol (3.21)
(a) A solution of aqueous sodium hydroxide (3.5 mL, 0.4 M, 1.40 mmol) was added
dropwise to a solution of 2-(trimethylsilyl)ethyl carbamate (3.26) (0.23 g, 1.40 mmol) in n-propanol
(1 mL) at room temperature. 1,3-Dichloro-5,5-dimethylhydantoin (0.18 g, 0.93 mmol) was added
as a solid in one portion, followed by a solution of (DHQD)2AQN (3.27) (20 mg, 23.3 μmol) in n-
propanol (2.1 mL) and a solution of alkene 3.61 (0.12 g, 0.47 mmol) in n-propanol (0.4 mL). The
solution was stirred vigorously at room temperature until it became homogeneous, then
potassium osmate dihydrate (7 mg, 19.0 μmol) was added as a solid in one portion. The solution
immediately turned dark green and was stirred at room temperature for 24 h after which, it had
turned light yellow. Sodium sulphite (0.59 g, 4.70 mmol) was added and the suspension was
stirred for a further 10 min. The mixture was diluted with water and extracted with ethyl acetate (×
3). The organic extracts were combined and washed with brine, then dried (Na2SO4). The solvent
was removed under reduced pressure and the crude material was purified by flash
chromatography on silica gel, eluting with dichloromethane, to remove any residual 2-
(trimethylsilyl)ethyl carbamate, then 30 % ethyl acetate/n-hexane, to afford the product 5.47 as a
clear colourless oil (0.16 g, 77 %). [𝛼]𝐷21.0 = -2 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.04
(s, 9H), 0.89 (t, J = 6.8 Hz, 3H), 0.95 – 1.01 (m, 2H), 1.23 (s, 3H), 1.26 – 1.47 (m, 10H), 1.72 –
1.81 (m, 2H), 2.23 (br s, 1H), 2.61 – 2.67 (m, 2H), 3.15 – 3.28 (m, 2H), 3.92 (t, J = 6.6 Hz, 2H),
4.14 – 4.19 (m, 2H), 5.01 (br s, 1H), 6.80 – 6.83 (m, 2H), 7.08 – 7.10 (m, 2H); 13C NMR (75 MHz;
CDCl3) δ -1.34, 14.2, 17.9, 22.7, 24.7, 26.2, 29.2, 29.3, 29.5, 31.9, 42.1, 50.6, 63.5, 68.2, 77.4,
114.7, 129.3, 134.1, 157.5, 157.9; IR (NaCl, neat) 1699, 3417 cm-1; HRMS (ESI-MS): m/z calcd
for C24H43NO4SiNa [M+Na]+ 460.2859, found 460.2860.

Chapter 5: Experimental
230
(b) A solution of hydrochloric acid in diethyl ether (0.53 mL, 2 M, 1.06 mmol) was
added dropwise to a solution Teoc-protected aminoalcohol 5.47 (92 mg, 0.21 mmol) in methanol
(5 mL) at room temperature. The solution was stirred at room temperature for 5 h. then the
solvent was removed under reduced pressure. The residue was dissolved in 1M aqueous
hydrochloric acid and extracted with diethyl ether (× 3). The remaining aqueous layer was
collected and neutralised with solid sodium bicarbonate and extracted with dichloromethane (× 3).
The organic extracts were combined and washed with brine, then dried (Na2SO4). The solvent
was removed under reduced pressure and the crude material purified by flash chromatography
on silica gel, eluting with 1 % triethylamine/5 % methanol/ dichloromethane, to afford the product
3.21 as a clear colourless gum (28 mg, 45 %). [𝛼]𝐷25.6 = -2 (0.5, CHCl3); 1H NMR (300 MHz;
CDCl3) δ 0.89 (t, J = 6.5 Hz, 3H), 1.20 (s, 3H), 1.26 – 1.44 (m, 8H), 1.71 – 1.80 (m, 4H), 2.61 –
2.71 (m, 4H), 2.95 (br s, 3H), 3.91 (t, J = 6.5 Hz, 2H), 6.79 – 6.82 (m, 2H), 7.08 – 7.11 (m, 2H);
13C NMR (75 MHz; CDCl3) δ 14.2, 22.7, 24.3, 26.2, 29.2, 29.3, 29.5, 31.9, 42.0, 50.7, 68.2, 71.4,
114.6, 129.3, 134.4, 157.4; IR (NaCl, neat) 3298, 3362 cm-1; HRMS (ESI-MS): m/z calcd for
C18H32NO2 [M+H]+ 294.2433, found 294.2432.
(2S)-4-(4’-Heptyloxyphenyl)-2-methylbutane-1,2-diol (3.71)
Potassium osmate dihydrate (1 mg, 2.71 μmol) was added as a solid in one portion to a
suspension of alkene 3.61 (0.13 g, 0.48 mmol), potassium carbonate (0.20 g, 1.45 mmol),
potassium ferricyanide (0.48 g, 1.45 mmol) and (DHQD)2PHAL (3.25) (20 mg, 24 μmol) in 1:1 t-
butanol:water (8 mL) at 0°C. The solution was allowed to slowly warm to room temperature, in
the cold bath, over 20 h. Sodium sulfite (0.55 g, 4.35 mmol) was added and the solution allowed
to stir for a further 15 min. The mixture was diluted with water and extracted with ethyl acetate (×
3). The organic extracts were combined and washed with brine, then dried (Na2SO4). The solvent
was removed under reduced pressure and the crude material purified by flash chromatography
on silica gel, eluting with 60 % ethyl acetate/n-hexane, to afford the product 3.71 as a clear
colourless oil (0.12 g, 83 %). [𝛼]𝐷21.0 = -4 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.91 (t, J =
6.8 Hz, 3H), 1.22 (s, 3H), 1.32 – 1.48 (m, 8H), 1.73 – 1.82 (m, 4H), 2.58 – 2.67 (m, 2H), 2.94 (br
s, 1H), 3.29 (br s, 1H), 3.42 (d, J = 11.0 Hz, 1H), 3.50 (d, J = 11.0 Hz, 1H), 3.91 (t, J = 6.6 Hz,
2H), 6.79 – 6.83 (m, 2H), 7.08 – 7.11 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 14.2, 22.7, 23.2, 26.1,

Chapter 5: Experimental
231
29.16, 29.21, 29.4, 31.9, 40.7, 68.1, 69.8, 73.1, 114.6, 129.2, 134.2, 157.4; IR (NaCl, neat) 3352
cm-1; HRMS (ESI-MS): m/z calcd for C18H30O3Na [M+Na]+ 317.2093, found 317.2093.
4-(4’-Heptyloxyphenyl)-2-methylbutan-2-amine (3.72)
(a) Under a static atmosphere, a solution of concentrated sulfuric acid (0.2 mL, 1.11
mmol) in acetic acid (1 mL) was added dropwise to a solution of alkene 3.61 (0.19 g, 0.73 mmol)
and potassium cyanide (48 mg, 0.74 mmol) in acetic acid (1 mL) at 0°C. The solution was
allowed to slowly warm to room temperature, in the cold bath, over 24 h. The reaction solution
was basified to pH 12 with 2M aqueous sodium hydroxide solution and extracted with diethyl
ether (× 3). The organic extracts were combined and dried (Na2SO4). The solvent was removed
under reduced pressure and the crude material purified by flash chromatography on silica gel,
eluting with 50 % ethyl acetate/n-hexane, to afford the product 5.48 as a 50:50 mixture rotamers,
as a white solid (0.16 g, 97 %). 1H NMR (400 MHz; CDCl3) δ 0.89 (t, J = 6.9 Hz, 3H, both
rotamers), 1.28 – 1.46 (m, 20H), 1.36 (s, 6H, one rotamer), 1.40 (s, 6H, one rotamer), 1.72 – 1.82
(m, 8H, both rotamers), 1.99 – 2.03 (m, 2H, one rotamer), 2.52 – 2.60 (m, 4H), 3.91 (t, J = 6.6 Hz,
2H, one rotamer), 3.92 (t, J = 6.6 Hz, 2H, one rotamer), 5.25 (br s, 1H, one rotamer), 5.98 (br d, J
= 12.0 Hz, one rotamer), 6.79 – 6.83 (m, 4H), 7.05 – 7.09 (m, 4H), 8.05 (d, J = 2.0 Hz, 1H, one
rotamer), 8.28 (d, J = 12.0 Hz, 1H, one rotamer); 13C NMR (100 MHz; CDCl3) δ 14.2, 22.7, 26.1,
27.3, 28.8, 29.18, 29.19, 29.43, 29.45, 29.51, 29.8, 31.9, 42.6, 46.1, 52.9, 54.1, 68.16, 68.17,
114.6, 114.7, 129.2, 129.3, 133.2, 133.9, 157.5, 157.7, 160.6, 163.2; IR (NaCl, neat) 1683, 3290
cm-1; HRMS (ESI-MS): m/z calcd for C19H31O2Na [M+Na]+ 328.2252, found 328.2247.
(b) Formamide 5.48 (56 mg, 0.18 mmol) in 6 M aqueous hydrochloric acid solution
(9 mL) was heated at reflux for 16 h. The solution was cooled to room temperature and basified to
pH 14 using 2 M aqueous sodium hydroxide solution. The suspension was extracted with
dichloromethane (× 3). The organic extracts were combined and dried (Na2SO4). The solvent was
removed under reduced pressure to afford the product 3.72 as a white solid (49 mg, 96 %). 1H
NMR (300 MHz; CDCl3) δ 0.89 (t, J = 6.8 Hz, 3H), 1.16 (s, 6H), 1.29 – 1.46 (m, 10H), 1.61 – 1.67
(m, 2H), 1.71 – 1.81 (m, 2H) 2.56 – 2.61 (m, 2H), 3.92 (t, J = 6.6 Hz, 2H), 6.79 – 6.84 (m, 2H),
7.07 – 7.12 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 14.2, 22.7, 26.2, 29.2, 29.5, 30.3, 30.5, 31.9,
47.5, 49.6, 68.2, 114.6, 129.2, 134.7, 157.4; IR (NaCl, neat) 3189, 3284, 3353 cm-1; HRMS (ESI-
MS): m/z calcd for C18H32NO [M+H]+ 278.2484, found 278.2480.

Chapter 5: Experimental
232
(2S,5S)-5-Isopropyl-3,6-dimethoxy-2-(4’-t-butyldimethylsilyloxyphenethyl)-2,5-
dihydropyrazine (3.77)
A solution of n-butyllithium in hexanes (0.17 mL, 2.1 M, 0.36 mmol) was added dropwise to a
solution of bis-lactim ether 3.75 (0.15 g, 0.35 mmol) in freshly distilled THF (2 mL) at -78°C (dry
ice/acetone). The solution was stirred at -78°C for 15 min after which, it had turned dark yellow.
Methanol (28 μL, 0.69 mmol) was added dropwise and the solution allowed to warm slowly, in the
cold bath, to -40°C over 3 h. The reaction was quenched with saturated aqueous sodium
bicarbonate solution and allowed to warm to room temperature. The THF was removed under
reduced pressure and the residue extracted with dichloromethane (× 4). The organic extracts
were combined and dried (Na2SO4). The solvent was removed under reduced pressure to afford
a yellow oil. 1HNMR analysis showed the diastereometric ratio to be 70:30, in favour of the
starting material, through integration of the benzylic multiplets at δ 2.46 – 2.62 and δ 2.65 – 2.81
respectively. The mixture was purified by flash chromatography on silica gel, eluting with 3 %
ethyl acetate/n-hexane, to afford the starting material 3.75 as a clear colourless oil (89 mg, 59 %).
All spectroscopic data matched those reported previously.
Further elution afforded the minor diastereomer 3.77 as a clear colourless oil (41 mg, 27 %).
[𝛼]𝐷25.0 = - 6 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.18 (s, 6H), 0.75 (d, J = 6.8 Hz, 3H),
0.98 (s, 9H), 1.08 (d, J = 6.8 Hz, 3H), 1.68 – 1.80 (m, 1H), 2.12 – 2.29 (m, 2H), 2.65 – 2.81 (m,
2H), 3.68 (s, 3H), 3.71 (s, 3H), 3.94 – 3.03 (m, 2H), 6.73 – 6.78 (m, 2H), 7.05 – 7.10 (m, 2H); 13C
NMR (75 MHz; CDCl3) δ -4.3, 17.7, 18.3, 19.7, 25.9, 31.4, 31.6, 37.6, 52.38, 52.41, 55.2, 61.1,
120.0, 129.5, 135.0, 153.8, 163.2, 163.9; IR (NaCl, neat) 1697 cm-1; HRMS (ESI-MS): m/z calcd
for C23H39N2O3Si [M+Na]+ 419.2729, found 419.2714.
(2S,5S)-5-Isopropyl-3,6-dimethoxy-2-(4’-heptyloxyphenethyl)-2,5-dihydropyrazine (3.43)
Cesium fluoride (0.20 g, 1.33 mmol) was added as a solid in one portion to a solution of bis-lactim
ether 3.77 (0.28 g, 0.67 mmol) in dry DMF (5 mL) at room temperature. The solution was stirred

Chapter 5: Experimental
233
for 15 min, where it had turned dark orange, before 1-bromoheptane (0.20 mL, 1.27 mmol) was
added dropwise. The solution was stirred at room temperature for 17 h. Water was added and the
mixture was extracted with ethyl acetate (× 3). The organic extracts were combined and washed
with water and brine, then dried (Na2SO4). The solvent was removed under reduced pressure and
the crude material purified by flash chromatography on silica gel, eluting with 2 % ethyl acetate/n-
hexane, to afford the product as a clear colourless oil (0.19 g, 72 %). All spectroscopic data
matched those reported previously for 3.43.
Methyl (2S)-2-amino-4-(4’-heptyloxyphenyl)butanoate (5.49)
A solution of TFA (2.5 mL) in water (5 mL) was added dropwise to a solution of bis-lactim ether
3.43 (0.19 g, 0.48 mmol) in acetonitrile (12 mL). The solution was stirred at room temperature for
4 h after which the acetonitrile was removed under reduced pressure. The residue was diluted
with water and neutralised with portions of solid sodium bicarbonate, then extracted with
dichloromethane (× 4). The organic extracts were combined and dried (Na2SO4). The solvent was
removed under reduced pressure and the crude material purified by flash chromatography on
silica gel, eluting with ethyl acetate, to afford the product 5.49 as a clear colourless oil (0.15 g, 99
%). [𝛼]𝐷22.3 = + 14 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.88 (t, J = 6.8 Hz, 3H), 1.26 – 1.46
(m, 8H), 1.53 (br s, 2H), 1.70 – 1.86 (m, 3H), 1.97 – 2.08 (m, 1H), 2.57 – 2.73 (m, 2H), 3.43 (dd, J
= 7.9, 5.2 Hz, 1H) 3.69 (s, 3H), 3.91 (t, J = 6.6 Hz, 2H), 6.78 – 6.83 (m, 2H), 7.06 – 7.11 (m, 2H);
13C NMR (75 MHz; CDCl3) δ 14.1, 22.7, 26.1, 29.1, 29.4, 31.1, 31.9, 36.7, 52.0, 53.9, 68.1, 114.5,
129.4, 133.1, 157.6, 176.6; IR (NaCl, neat) 1738, 3317, 3382 cm-1; HRMS (ESI-MS): m/z calcd
for C18H29NO3Na [M+Na]+ 330.2045, found 330.2031.
(2S)-2-Amino-4-(4’-heptyloxyphenyl)butan-1-ol (3.73)
Lithium aluminium hydride (37 mg, 0.98 mmol) was added as a solid in one portion to a solution
of aminoester 5.49 (0.15 g, 0.48 mmol) in freshly distilled THF (4 mL) at 0°C. The solution was
stirred at 0°C for 20 min then the cold bath was removed and the solution stirred at room
temperature for 2 h. The reaction was quenched with saturated aqueous sodium sulfate solution

Chapter 5: Experimental
234
and the mixture was extracted with ethyl acetate (× 4). The organic extracts were combined and
washed with saturated aqueous sodium bicarbonate solution, water and brine, then dried
(Na2SO4). The solvent was removed under reduced pressure and the crude material purified by
flash chromatography on silica gel, eluting with 3 % methanol/3 % triethylamine/dichloromethane,
to afford the product 3.73 as a clear gum (59 mg, 44 %). [𝛼]𝐷26.4 = - 2 (0.5, CHCl3); 1H NMR (300
MHz; CDCl3) δ 0.89 (t, J = 6.8 Hz, 3H), 1.26 – 1.46 (m, 8H), 1.51 – 1.63 (m, 1H), 1.69 – 1.81 (m,
3H), 2.07 (br s, 1H), 2.17 (br s, 2H), 2.54 – 2.73 (m, 2H), 2.83 – 2.91 (m, 1H), 3.32 (dd, J = 10.6,
7.8 Hz, 1H), 3.61 (dd, J = 10.6, 3.5 Hz, 1H), 3.91 (t, J = 6.6 Hz, 2H), 6.69 – 6.83 (m, 2H), 7.06 –
7.09 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 14.2, 22.8, 26.2, 29.2, 29.5, 31.6, 31.9, 36.3, 52.47,
52.48, 66.6, 68.2, 114.7, 129.3, 133.6, 157.6; IR (NaCl, neat) 3120, 3280, 3339 cm-1; HRMS
(ESI-MS): m/z calcd for C17H30NO2 [M+H]+ 280.2277, found 280.2265.
(2S,5S)-5-Isopropyl-3,6-dimethoxy-2-(4’-t-butyldimethylsilyloxyphenethyl)-2-ethyl-2,5-
dihydropyrazine (3.76)
A solution of n-butyllithium in hexanes (0.47 mL, 2.1 M, 0.99 mmol) was added dropwise to a
solution of bis-lactim ether 3.75 (0.39 g, 0.94 mmol) in freshly distilled THF (6 mL) at -78°C (dry
ice/acetone). The solution was stirred at -78°C for 15 min after which, it had turned dark yellow.
Ethyliodide (83 μL, 1.03 mmol) was added dropwise and the solution allowed to warm slowly, in
the cold bath, to -10°C over 5 h. The reaction was quenched with saturated aqueous sodium
bicarbonate solution and allowed to warm to room temperature. The THF was removed under
reduced pressure and the residue extracted with dichloromethane (× 4). The organic extracts
were combined and dried (Na2SO4). The solvent was removed under reduced pressure and the
crude material purified by flash chromatography on silica gel, eluting with 3 % ethyl acetate/n-
hexane, to afford the product 3.76 as a clear colourless oil (0.37 g, 87 %). [𝛼]𝐷24.3 = + 36 (0.5,
CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.17 (s, 6H), 0.66 (t, J = 7.4 Hz, 3H), 0.71 (d, J = 6.8 Hz,
3H), 0.97 (s, 9H), 1.12 (d, J = 6.8 Hz, 3H), 1.51 – 1.63 (m, 1H), 1.75 – 1.90 (m, 2H), 2.09 (td, J =
12.8, 4.2 Hz, 1H), 2.21 (td, J = 12.8, 4.2 Hz, 1H), 2.32 – 2.46 (m, 2H), 3.71 (s, 3H), 3.72 (s, 3H),
3.93 (d, J = 3.3 Hz, 1H), 6.71 – 6.75 (m, 2H), 6.98 – 7.02 (m, 2H); 13C NMR (75 MHz; CDCl3) δ -
4.3, 8.4, 17.2, 18.4, 19.7, 25.9, 30.78, 30.81, 34.0, 42.4, 52.3, 52.4, 60.9, 62.7, 119.9, 129.3,
135.5, 153.6, 162.9, 164.1; IR (NaCl, neat) 1691 cm-1; HRMS (ESI-MS): m/z calcd for
C25H42N2O3SiNa [M+Na]+ 469.2862, found 469.2845.

Chapter 5: Experimental
235
(2S,5S)-5-Isopropyl-3,6-dimethoxy-2-(4’-heptyloxyphenethyl)-2-ethyl-2,5-dihydropyrazine
(5.50)
Cesium fluoride (0.12 g, 0.77 mmol) was added as a solid in one portion to a solution of bis-lactim
ether 3.76 (0.17 g, 0.39 mmol) in dry DMF (3.5 mL) at room temperature. The solution was stirred
for 15 min, where it had turned dark orange, before 1-bromoheptane (90 μL, 0.44 mmol) was
added dropwise. The solution was stirred at room temperature for 17 h. Water was added and the
mixture was extracted with ethyl acetate (× 3). The organic extracts were combined and washed
with water and brine, then dried (Na2SO4). The solvent was removed under reduced pressure and
the crude material purified by flash chromatography on silica gel, eluting with 3 % ethyl acetate/n-
hexane, to afford the product 5.50 as a clear colourless oil (0.14 g, 84 %). [𝛼]𝐷24.8 = + 30 (0.5,
CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.66 (t, J = 7.4 Hz, 3H), 0.71 (d, J = 6.8 Hz, 3H), 0.89 (t, J =
6.8 Hz, 3H), 1.12 (d, J = 6.8 Hz, 3H), 1.31 – 1.47 (m, 8H), 1.51 – 1.63 (m, 1H), 1.71 – 1.90 (m,
4H), 2.09 (td, J = 12.8, 4.2 Hz, 1H), 2.22 (td, J = 12.8, 4.2 Hz, 1H), 2.33 – 2.47 (m, 2H), 3.71 (s,
3H), 3.72 (s, 3H), 3.90 – 3.94 (m, 3H), 6.79 – 6.82 (m, 2H), 7.04 – 7.07 (m, 2H); 13C NMR (75
MHz; CDCl3) δ 8.4, 14.2, 17.2, 19.7, 22.8, 26.2, 29.2, 29.5, 30.7, 30.8, 32.0, 34.0, 42.6, 52.3,
52.4, 60.9, 62.7, 68.2, 114.5, 129.3, 134.8, 157.3, 162.9, 164.2; IR (NaCl, neat) 1691 cm-1;
HRMS (ESI-MS): m/z calcd for C26H43N2O3 [M+H]+ 431.3274, found 431.3261.
Methyl (2S)-2-amino-2-ethyl-4-(4’-heptyloxyphenyl)butanoate (5.51)
A solution of TFA (3 mL) in water (6 mL) was added dropwise to a solution of bis-lactim ether
5.50 (0.14 g, 0.33 mmol) in acetonitrile (6 mL). The solution was stirred at room temperature for
76 h after which the acetonitrile was removed under reduced pressure. The residue was diluted
with water and neutralised with portions of solid sodium bicarbonate, then extracted with
dichloromethane (× 4). The organic extracts were combined and dried (Na2SO4). The solvent was
removed under reduced pressure and the crude material purified by flash chromatography on
silica gel, eluting with 50 % ethyl acetate/n-hexane, to afford the product 5.51 as a clear
colourless oil (27 mg, 25 %). [𝛼]𝐷27.8 = +20 (0.5, CHCl3); 1H NMR (400 MHz; CDCl3) 0.84 – 0.90

Chapter 5: Experimental
236
(m, 6H), 1.25 – 1.37 (m, 6H), 1.40 – 1.47 (m, 2H), 1.56 -1.65 (m, 1H), 1.69 (br s, 2H), 1.71 – 1.86
(m, 4H), 1.99 – 2.06 (m, 1H), 2.39 (td, J = 12.6, 4.8 Hz, 1H), 2.62 (td, J = 12.6, 5.3 Hz, 1H), 3.71
(s, 3H), 3.91 (t, J = 6.6 Hz, 2H), 6.79 – 6.82 (m, 2H), 7.05 – 7.08 (m, 2H); 13C NMR (100 MHz;
CDCl3) δ 8.4, 14.2, 22.7, 26.1, 29.2, 29.4, 29.8, 31.9, 33.2, 42.0, 52.2, 61.7, 68.1, 114.6, 129.3,
133.6, 157.5, 177.6; IR (NaCl, neat) 1732, 3325, 3385 cm-1; HRMS (ESI-MS): m/z calcd for
C20H34NO3 [M+H]+ 336.2539, found 336.22530.
(2S)-2-Amino-2-ethyl-4-(4’-heptyloxyphenyl)butan-1-ol (3.74)
Lithium aluminium hydride (5 mg, 0.13 mmol) was added as a solid in one portion to a solution of
aminoester 5.51 (27 mg, 80.5 μmol) in freshly distilled THF (0.5 mL) at 0°C. The solution was
stirred at 0°C for 20 min then the cold bath was removed and the solution stirred at room
temperature for 1 h. The reaction was quenched with saturated aqueous sodium sulfate solution
and the mixture was extracted with ethyl acetate (× 4). The organic extracts were combined and
washed with saturated aqueous sodium bicarbonate solution, water and brine, then dried
(Na2SO4). The solvent was removed under reduced pressure and the crude material purified by
flash chromatography on silica gel, eluting with 1 % methanol/3 % triethylamine/dichloromethane,
to afford the product 3.74 as a clear gum (15 mg, 60 %). [𝛼]𝐷26.9 = - 2 (0.5, CHCl3); 1H NMR (300
MHz; CDCl3) 0.86 0.93 (m, 6H), 1.25 – 1.60 (m, 12H), 1.64 – 1.81 (m, 3H), 1.89 (br s, 2H), 2.47
– 2.58 (m, 2H), 2.37 (s, 2H), 3.92 (t, J = 6.6 Hz, 2H), 6.79 – 6.83 (m, 2H), 7.06 – 7.11 (m, 2H); 13C
NMR (75 MHz; CDCl3) δ 7.9, 14.2, 22.7, 26.2, 29.1, 29.17, 29.20, 29.5, 31.9, 38.7, 55.3, 68.0,
68.2, 114.6, 129.2, 134.3, 157.5; IR (NaCl, neat) 3355 cm-1; HRMS (ESI-MS): m/z calcd for
C19H34NO2 [M+H]+ 308.2589, found 308.2572.
2-(4’-Benzyloxyphenyl)ethanol (5.52)[36]
Benzyl bromide (3.2 mL, 26.90 mmol) was added dropwise to a suspension of tyrosol 3.41 (3.33
g, 24.07 mmol) and potassium carbonate (5.00 g, 36.17 mmol) in methanol (28 mL) at room
temperature. The reaction mixture was stirred at room temperature for 18 h. The methanol was
removed under reduced pressure. The residue was dissolved in water and extracted with ethyl
acetate (× 3). The organic extracts were combined and washed with water and brine, then dried
(Na2SO4). The solvent was removed under reduced pressure and the crude material was

Chapter 5: Experimental
237
recystallised (n-hexane) to afford the product 5.52 as a white solid (5.33 g, 97 %) with all
analytical data matching that reported in the literature.[36] 1H NMR (300 MHz; CDCl3) δ, 1.61 (br s,
1H), 2.82 (t, J = 6.6 Hz, 2H), 3.83 (t, J = 6.6 Hz, 2H), 5.06 (s, 2H), 6.91 – 6.96 (m, 2H), 7.13 –
7.17 (m, 2H), 7.30 – 7.46 (m, 5H).
2-(4’-Benzyloxyphenyl)-1-iodoethane (3.83)[37]
(a) Methanesulfonyl chloride (1.9 mL, 24.55 mmol) was added dropwise to a solution of
alcohol 5.52 (5.00 g, 21.90 mmol) and triethylamine (9.2 mL, 66.01 mmol) in dichloromethane
(220 mL) at 0°C. The solution was stirred at 0°C for 15 min. then the cold bath was removed and
the solution stirred at room temperature for 3 h. The reaction mixture was poured onto brine and
extracted with dichloromethane (× 2). The organic extracts were combined and dried (Na2SO4).
The solvent was removed under reduced pressure to afford a light orange residue which was
used in the next step without further purification.
(b) The crude material was dissolved in acetone (110 mL) and sodium iodide (32.84 g, 0.22
mol) was added. The solution was stirred at room temperature, protected from light for 17 h. The
acetone was removed under reduced pressure. Water was added to the residue which was
extracted with dichloromethane (× 3). The organic extracts were combined and washed with
water and brine, then dried (Na2SO4). The solvent was removed under reduced pressure and the
crude material was recrystallised (diethyl ether) to afford the product 3.83 as a white solid (5.03 g,
68 %) with all the analytical data matching that reported in the literature.[37] 1H NMR (300 MHz;
CDCl3) δ 3.12 (t, J = 7.7 Hz, 2H), 3.32 (t, J = 7.7 Hz, 2H), 5.05 (s, 2H), 6.91 – 6.95 (m, 2H), 7.09
– 7.14 (m, 2H), 7.30 – 7.45 (m, 5H).
(2R,5S)-5-isopropyl-3,6-dimethoxy-2-(4’-benzyloxyphenethyl)-2,5-dihydropyrazine (5.54)
A solution of n-butyllithium in hexanes (3.0 mL, 1.4 M, 4.20 mmol) was added dropwise to a
solution of freshly distilled (S)-Schöllkopf’s reagent (S)-3.7 (0.74 g, 4.24 mmol) in freshly distilled
THF (8.5 mL) at -78°C (dry ice/acetone). The solution was stirred at -78°C for 15 min, where it
had turned dark yellow. A solution of iodide 3.83 (1.36 g, 4.02 mmol) in freshly distilled THF (8
mL) at -78°C was added dropwise. The solution was stirred for a further 30 min at -78°C then

Chapter 5: Experimental
238
allowed to slowly warm to -15°C over 4 h. The reaction mixture was quenched with saturated
aqueous sodium bicarbonate solution and allowed to warm to room temperature. The THF was
removed under reduced pressure and the residue extracted with dichloromethane (× 4). The
organic extracts were combined and dried (Na2SO4). The solvent was removed under reduced
pressure and the crude material was purified by flash chromatography on silica gel, eluting with 3
% ethyl acetate/n-hexane, to afford the product 5.54 as a clear colourless oil (1.47 g, 92 %).
[𝛼]𝐷25.0 = - 6 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3) δ 0.70 (d, J = 6.8 Hz, 3H), 1.06 (d, J = 6.8
Hz, 3H), 1.90 – 2.02 (m, 1H), 2.07 – 2.19 (m, 1H), 2.28 (septd, J = 6.8, 3.3 Hz, 1H), 2.48 – 2.65
(m, 1H), 3.69 (s, 3H), 3.70 (s, 3H), 3.97 (t, J = 3.3 Hz, 1H), 4.02 – 4.07 (m, 1H), 5.04 (s, 2H), 6.87
– 6.92 (m, 2H), 7.09 – 7.14 (m, 2H), 7.29 – 7.45 (m, 5H); 13C NMR (75 MHz; CDCl3) δ 16.8, 19.2,
30.2, 32.0, 36.2, 52.5, 55.1, 60.9, 70.2, 114.8, 127.6, 128.0, 128.7, 129.5, 134.7, 137.4, 157.1,
163.7, 163.9; IR (NaCl, neat) 1694 cm-1; HRMS (ESI-MS): m/z calcd for C24H31N2O3 [M+H]+
395.2334, found 395.2320.
(2S,5S)-5-Isopropyl-3,6-dimethoxy-2-(4’-benzyloxyphenethyl)-2-methyl-2,5-dihydropyrazine
(5.32)
A solution of n-butyllithium in hexanes (2.8 mL, 1.4 M, 3.92 mmol) was added dropwise to a
solution of bis-lactim ether 5.54 (1.47 g, 3.72 mmol) in freshly distilled THF (18.5 mL) at -78°C
(dry ice/acetone). The solution was stirred at -78°C for 15 min, where it had turned dark yellow.
Methyl iodide (0.46 mL, 7.39 mmol) was added dropwise. The solution was stirred for a further 30
min at -78°C then allowed to slowly warm to -15°C over 4 h. The reaction mixture was quenched
with saturated aqueous sodium bicarbonate solution and allowed to warm to room temperature.
The THF was removed under reduced pressure and the residue extracted with dichloromethane
(× 4). The organic extracts were combined and dried (Na2SO4). The solvent was removed under
reduced pressure and the crude material was purified by flash chromatography on silica gel,
eluting with 2 % ethyl acetate/n-hexane, to afford the title compound as a clear colourless oil
(1.01 g, 67 %) with all the analytical data matching that reported previously for 5.32.

Chapter 5: Experimental
239
(2S)-t-Butyl(1-hydroxy-4-(4’-benzyloxyphenyl)-2-methylbutan-2-yl)carbamate (5.55)[34]
Di-t-butyl dicarbonate (0.31 g, 1.08 mmol) was added as a solid in one portion to a mixture of
aminoalcohol 3.57 (0.35 g, 1.62 mmol) in saturated aqueous sodium bicarbonate solution (10.8
mL) and ethyl acetate (10.8 mL). The mixture was heated at 70°C for 16 h then allowed to cool to
room temperature. The solution was diluted with water and extracted with ethyl acetate (× 3). The
organic extracts were combined and washed with brine, then dried (Na2SO4). The solvent was
removed under reduce pressure and the crude material was purified by flash chromatography on
silica gel, eluting with 30 % ethyl acetate/n-hexane, to afford the product 5.55 as a white solid
(0.38 g, 91 %) with all analytical data matching that reported in the literature.[34] [𝛼]𝐷25.6 = +2 (0.5,
CHCl3); 1H NMR (400 MHz; CDCl3) δ 1.22 (s, 3H), 1.44 (s, 9H), 1.84 (td, J = 12.3, 5.3 Hz, 1H),
2.02 (td, J = 12.3, 5.3 Hz, 1H), 2.53 (td, J = 12.3, 5.2 Hz, 1H), 2.63 (td, J = 12.3, 5.2 Hz, 1H), 3.62
– 3.72 (m, 2H), 4.10 (br s, 1H), 4.63 (s, 1H), 5.04 (s, 2H), 6.88 – 6.92 (m, 2H), 7.09 – 7.13 (m,
2H), 7.30 – 7.44 (m, 5H); 13C NMR (100 MHz; CDCl3) δ 23.0, 28.5, 29.3, 38.6, 57.1, 69.8, 70.2,
80.1, 115.0, 127.6, 128.0, 128.7, 129.4, 134.5, 137.3, 156.3, 157.2; IR (NaCl, neat) 1674, 3275
cm-1; HRMS (ESI-MS): m/z calcd for C23H31NO4Na [M+Na]+ 408.2150, found 418.2148.
(2S)-t-Butyl(1-hydroxy-4-(4’-hydroxyphenyl)-2-methylbutan-2-yl)carbamate (3.82)[34]
Palladium on carbon (0.28 mg, 10 wt. %) was added to a solution of Boc-aminoalcohol 5.57 (0.23
g, 0.59 mmol) in dry methanol (18.4 mL) at room temperature. A hydrogen balloon was attached
to the flask and the flask was evacuated and purged with hydrogen (× 3). The solution was stirred
at room temperature for 18 h. The reaction mixture was diluted with dichloromethane and filtered
through a short pad of Celite, eluting with dichloromethane. The solvent was removed under
reduced pressure to afford the product 3.82 as a white solid (0.11 g, 63 %) with all analytical data
matching that reported in the literature.[34]. [𝛼]𝐷25.6 = +5 (0.5, CHCl3); 1H NMR (300 MHz; CDCl3)
δ 1.21 (s, 3H), 1.44 (s, 9H), 1.74 (br s, 1H), 1.82 (td, J = 12.6, 5.6 Hz, 1H), 2.05 (td, J = 12.6, 5.6
Hz, 1H), 2.44 – 2.64 (m, 2H), 3.65 – 3.73 (m, 2H), 4.56 (br s, 1H), 4.69 (s, 1H), 6.10 (br s, 1H),
6.72 – 6.77 (m, 2H), 6.99 – 7.02 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 23.0, 28.5, 29.3, 38.4,

Chapter 5: Experimental
240
57.0, 69.9, 80.3, 115.5, 129.4, 133.6, 154.4, 156.5; IR (NaCl, neat) 1743, 3333 cm-1; HRMS (ESI-
MS): m/z calcd for C16H25NO4Na [M+Na]+ 318.1681, found 318.1675
General Procedure E for the Alkylation of Phenol 3.82
(a) Substituted benzyl bromide (1 eq.) was added dropwise to a suspension of phenol 3.82
(1 eq.) and potassium carbonate (3 eq.) in dry DMF (0.1 M). The suspension was stirred at room
temperature for 15 h. The solution was diluted with water and extracted with ethyl acetate (× 3).
The organic extracts were combined and washed with water and brine, then dried (Na2SO4). The
solvent was removed under reduced pressure and the crude material was purified by flash
chromatography on silica gel, eluting with 30 % ethyl acetate/n-hexane, to afford the product.
(b) A solution of methanolic hydrochloric acid (3 eq) was added dropwise to a solution of N-
Boc compound (1 eq) in methanol (0.1 M). The solution was heated at reflux for 4 h then allowed
to cool to room temperature. The solvent was removed under reduced pressure. The residue was
dissolved in chloroform and washed with saturated aqueous sodium bicarbonate solution (× 2)
and brine, then dried (Na2SO4). The solvent was removed under reduced pressure to afford the
product 3.79 – 3.81.
(2S)-2-Amino-4-(4’-(4-chlorobenzyloxy)phenyl)-2-methylbutan-1-ol (3.79)
Prepared using general procedure E by Stephen Butler using phenol 3.82 (40 mg, 0.14 mmol), 4-
chlorobenzyl bromide (22 mg, 0.14 mmol), potassium carbonate (56 mg, 0.41 mmol) and DMF
(1.4 mL) then, methanolic hydrochloric acid (0.2 mL, 2 M, 0.4 mmol) and methanol (0.8 mL) to
afford the product 3.79 as a white solid (22 mg, 51 %). [𝛼]𝐷26.5 = - 2 (0.5, CHCl3); 1H NMR (300
MHz; CDCl3) δ 1.13 (s, 3H), 1.60 – 1.73 (m, 2H), 1.84 (br s, 3H), 2.58 (t, J = 8.5 Hz, 2H), 3.36 (br
s, 2H), 5.00 (s, 2H), 6.85 – 6.88 (m, 2H), 7.09 – 7.12 (m, 2H), 7.32 – 7.38 (m, 4H); 13C NMR (75
MHz; CDCl3) δ 29.5, 29.8, 69.4, 70.4, 77.4, 115.0, 128.86, 128.88, 129.4, 133.8, 135.0, 135.6,
135.8, 156.9; IR (NaCl, neat) 3500 cm-1; HRMS (ESI-MS): m/z calcd for C18H23NO2Cl [M+H]+
320.1417, found 320.1405.

Chapter 5: Experimental
241
(2S)-2-Amino-4-(4’-(4-methylbenzyloxy)phenyl)-2-methylbutan-1-ol (3.80)
Prepared using general procedure E by Stephen Butler using phenol 3.82 (40 mg, 0.14 mmol), 4-
methylbenzyl bromide (27 mg, 0.15 mmol), potassium carbonate (56 mg, 0.41 mmol) and DMF
(1.4 mL) then, methanolic hydrochloric acid (0.2 mL, 2 M, 0.4 mmol) and methanol (0.8 mL) to
afford the product 3.80 as a white solid (20 mg, 49 %). [𝛼]𝐷25.5 = - 2 (0.5, CHCl3); 1H NMR (300
MHz; CDCl3) δ 1.13 (s, 3H), 1.58 – 1.76 (m, 5H), 2.36 (s, 3H), 2.59 (t, J = 8.5 Hz, 2H), 3.33 (d, J
= 10.5 Hz, 1H), 3.38 (d, J = 10.5 Hz, 1H), 4.99 (s, 2H), 6.88 – 6.91 (m, 2H), 7.09 – 7.12 (m, 2H),
7.17 – 7.20 (m, 2H), 7.30 – 7.33 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 21.3, 29.5, 29.8, 70.1,
70.5, 77.4, 115.0, 127.7, 129.3, 129.4, 134.2, 134.7, 135.6, 137.8, 157.2; IR (NaCl, neat) 3335
cm-1; HRMS (ESI-MS): m/z calcd for C19H26NO2 [M+H]+ 300.1963, found 300.1946.
(2S)-2-Amino-4-(4’-(3,4-dichlorobenzyloxy)phenyl)-2-methylbutan-1-ol (3.81)
Prepared using general procedure E using phenol 3.82 (24 mg, 81.3 μmol), 3,4-dichlorobenzyl
bromide (20 mg, 81.3 μmol), potassium carbonate (34 mg, 0.25 mmol) and DMF (0.8 mL) then,
methanolic hydrochloric acid (0.12 mL, 2 M, 0.24 mmol) and methanol (0.5 mL) to afford the
product 3.81 as a white solid (16 mg, 57 %). [𝛼]𝐷25.5 = - 2 (0.5, CHCl3); 1H NMR (300 MHz;
CDCl3) δ 1.15 (s, 3H), 1.63 – 1.77 (m, 2H), 1.83 (br s, 3H), 2.60 (t, J = 8.5 Hz, 2H), 3.35 (d, J =
10.8 Hz, 1H), 3.41 (d, J = 10.8 Hz, 1H), 4.98 (s, 2H), 6.85 – 6.87 (m, 2H), 7.10 – 7.13 (m, 2H),
7.24 – 7.35 (m, 1H), 7.44 (d, J = 8.3 Hz, 1H), 7.53 (d, J = 1.6 Hz, 1H); 13C NMR (75 MHz; CDCl3)
δ 24.4, 29.5, 53.1, 68.8, 70.2, 77.4, 115.0, 126.7, 129.3, 129.5, 130.7, 135.2, 135.51, 135.54,
137.7, 156.6; IR (NaCl, neat) 3425 cm-1; HRMS (ESI-MS): m/z calcd for C18H22NO2Cl2 [M+H]+
354.1027, found 354.1017.
4-Nitrophenyl-5-azidopentanoate ((C5)-3.89)[38]
1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (0.35 g, 1.82 mmol) was added to a
solution of azide 5.56 (0.24 g, 1.66 mmol) and 4-nitrophenol (0.23 g, 1.65 mmol) in
dichloromethane (4 mL) at room temperature. The solution was stirred at room temperature for 17
h. Water was added and the mixture was extracted with dichloromethane (× 3). The organic
extracts were combined and washed with water, brine, then dried (Na2SO4). The solvent was
removed under reduced pressure and the crude material purified by flash chromatography on
silica gel, eluting with 15 % ethyl acetate/n-hexane, to afford the product (C5)-3.89 as a clear light
yellow oil (0.33 g, 75 %) with all the analytical data matching that reported in the literature. [38] 1H

Chapter 5: Experimental
242
NMR (300 MHz; CDCl3) δ 1.68 – 1.78 (m, 2H), 1.81 – 1.91 (m, 2H), 2.66 (t, J = 7.2 Hz, 2H), 3.36
(t, J = 6.6 Hz, 2H), 7.26 – 7.31 (m, 2H), 8.25 – 8.30 (m, 2H).
4-Nitrophenyl-1-azido-3,6,9,12-tetraoxapentadecanoate ((PEG4)-3.89)
1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (81 mg, 0.42 mmol) was added to a
solution of azide 5.59 (0.117 g, 0.40 mmol) and 4-nitrophenol (56 mg, 0.40 mmol) in
dichloromethane (2 mL) at room temperature. The solution was stirred at room temperature for 15
h. Water was added and the mixture was extracted with dichloromethane (× 3). The organic
extracts were combined and washed with water, brine, then dried (Na2SO4). The solvent was
removed under reduced pressure and the crude material purified by flash chromatography on
silica gel, eluting with 50 % ethyl acetate/n-hexane, to afford the product (PEG4)-3.89 as a clear
light yellow oil (0.107 g, 65 %). 1H NMR (300 MHz; CDCl3) δ 2.87 (t, J = 6.2 Hz, 2H), 3.37 (t, J =
5.1 Hz, 2H), 3.64 – 3.68 (m, 14H), 3.87 (t, J = 6.2 Hz, 2H), 7.27 – 7.32 (m, 2H), 8.24 – 8.29 (m,
2H); 13C NMR (75 MHz; CDCl3) δ 35.4, 50.8, 66.4, 70.2, 70.6, 70.7, 70.79, 70.81, 122.6, 125.3,
145.5, 155.5, 169.4; IR (NaCl, neat) 1763, 2107 cm-1; HRMS (ESI-MS): m/z calcd for
C17H24N4O8Na [M+Na]+ 435.1492, found 435.1474.
Attachment of Azide Linker to Affinity Chromatography bead[38]
A Thermo Scientific Carboxylink column (2mL, 16-20 μmol amine/mL, 32 – 40 μmol) was
clamped upright and the Carboxylink gel allowed to settle for 30 min. The column was flushed
with pH 4.7 aqueous MES buffer (5 × 2 mL, 0.1 M). The column was drained and a solution of p-
nitrophenylester 3.89 (10 eq) in DMF (2 mL) was added. The column was sealed and shaken
gently (160 rpm) for 50 h. pH 7 aqueous phosphate buffer (0.3 mL, 1.5 M) was added and the
column was shaken, as before, for a further 30 min. The column was drained and washed with
consecutive portions of 1 M aqueous sodium chloride solution (2.5 mL), DMF (2.5 mL), 1 M
aqueous sodium chloride solution (2.5 mL) and DMF (2.5 mL). The filtrates were combined and
the total volume was adjusted to 12 mL with DMF. An aliquot of 3 mL was removed and diluted
again, to a total volume of 12 mL with DMF. 3 mL of this solution was removed and centrifuged
(1000 rpm) for 1 min. The supernatant was transferred to a quartz cuvette and the absorbance
measured at 412 nm, against a standard of DMF. The amount of azide linker attached to the

Chapter 5: Experimental
243
bead was assumed to be proportional to the amount of p-nitrophenol detected (A = , ℓ = 1 cm, ε =
432 M-1 cm-1, dilution × 4 × 4). The above process was repeated once more to increase the
overall amount of azide on the bead. The azide-linker Carboxylink beads were stored under 1 M
aqueous sodium chloride solution (2 mL, 0.05 % sodium azide) at 4°C until required.
Linker Yield Yield Total Yield (%)
-(CH2)4- (C5) 25 23 48 (9.69 μmol/mL) -(CH2CH2O)4- (PEG4) 36 29 65 (12.85 μmol/mL)
7-Octyn-1-ol (3.92)[39]
Sodium hydride (3.52 g, 88.00 mmol) was added as a solid in one portion to a solution dry
ethylenediamine (28 mL) at room temperature. The mixture was stirred at 70°C for 1 h where the
solution had turned dark purple/brown. The solution was cooled to room temperature and 3-
octynol (3.91) (2.5 mL, 17.63 mmol) was added in one portion. The mixture was heated again at
70°C for a further 1 h. The dark green reaction mixture was diluted with diethyl ether and water
then cooled to 0°C and slowly acidified to pH 1 with conc. hydrochloric acid solution. The mixture
was extracted with diethyl ether (× 3). The organic extracts were combined and washed with
brine, then dried (Na2SO4). The solvent was removed under reduced pressure to afford an
orange oil which was purified by flash chromatography on silica gel, eluting with 30 % ethyl
acetate/n-hexane, to afford the product 3.92 as a clear colourless oil (1.96 g, 88 %) with all the
analytical data matching that reported in the literature.[40] 1H NMR (300 MHz; CDCl3) δ 1.34 –
1.47 (m, 4H), 1.48 – 1.62 (m, 4H). 1.65 (br s, 1H), 1.94 (t, J = 2.7 Hz, 1H), 2.19 (td, J = 6.9, 2.7
Hz, 2H), 3.64 (t, J = 6.6 Hz, 2H).
8-Iodo-1-octyne (3.93)[41]
(a) Methanesulfonyl chloride (1.26 mL, 16.27 mmol) was added dropwise to a solution of
alcohol 3.92 (1.96 g, 15.49 mmol) and triethylamine (4.32 mL, 30.99 mmol) in dichloromethane
(35 mL) at 0°C. The solution was stirred at 0°C for 15 min then the cold bath was removed and
the solution stirred at room temperature for 2 h. The reaction mixture was poured onto brine and
the organic layer was removed. The aqueous layer was extracted further with dichloromethane (×
2). The organic extracts were combined washed with brine, then dried (Na2SO4). The solvent was

Chapter 5: Experimental
244
removed under reduced pressure to afford an orange residue (3.26 g), which was used in the
next step without further purification.
(b) The crude mesylate 5.58 (3.26 g) was dissolved in acetone (80 mL) and sodium
iodide (23.20 g, 154.78 mmol) was added in one portion. The solution was stirred at room
temperature protected from light for 15 h. The acetone was removed under reduced pressure.
Water was added to the residue which was extracted with dichloromethane (× 3). The organic
extracts were combined and washed with water, brine, then dried (Na2SO4). The solvent was
removed under reduced pressure to afford a dark orange residue which was purified by flash
chromatography on silica gel, eluting with 1 % ethyl acetate/n-hexane, to afford the product 3.93
as a clear colourless oil (2.54 g, 70 %) with all the analytical data matching that reported in the
literature.[41] 1H NMR (400 MHz; CDCl3) δ 1.40 – 1.46 (m, 4H), 1.51 – 1.56 (m, 4H), 1.81 – 1.88
(m, 2H), 1.94 (t, J = 2.8 Hz, 1H), 2.20 (td, J = 6.8, 2.8 Hz, 2H), 3.19 (t, J = 7.0 Hz, 2H).
(2S,5S)-5-Isopropyl-3,6-dimethoxy-2-(4’-(oct-7-ynyl-1-oxy)phenethyl)-2-methyl-2,5-
dihydropyrazine (5.59)
Cesium fluoride (0.33 g, 2.15 mmol) was added as a solid in one portion to a solution of bis-lactim
ether 3.49 (0.47 g, 1.08 mmol) in dry DMF (6 mL) at room temperature. The solution was stirred
for 15 min and 8-iodo-1-octyne (3.93) (0.33 g, 1.40 mmol) was added dropwise. The solution was
stirred at room temperature for 14 h. Water was added and the mixture was extracted with ethyl
acetate (× 3). The organic extracts were combined and washed with water, brine, then dried
(Na2SO4). The solvent was removed under reduced pressure and the crude material purified by
flash chromatography on silica gel, eluting with 2 % ethyl acetate/n-hexane, to afford the product
5.59 as a clear colourless oil (0.40 g, 87 %). [𝛼]𝐷27.2 = + 38 (0.5, CHCl3); 1H NMR (300 MHz;
CDCl3) δ 0.70 (d, J = 6.8 Hz, 3H), 1.12 (d, J = 6.8 Hz, 3H), 1.30 (s, 3H), 1.45 – 1.58 (m, 7H), 1.73
– 1.82 (m, 2H), 1.85 (td, J = 12.8, 5.0 Hz, 1H), 1.94 (t, J = 2.6 Hz, 1H), 2.08 (td, J = 12.9, 4.3 Hz,
1H), 2.20 (td, J = 6.9, 2.7 Hz, 2H), 2.23 (td, J = 12.9, 4.3 Hz, 1H), 2.32 – 2.47 (m, 2H), 3.70 (s,
3H), 3.71 (s, 3H), 3.93 (t, J = 6.5 Hz, 2H), 3.94 (d, J = 3.4 Hz, 1H), 6.77 – 6.82 (m, 2H), 7.03 –
7.08 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 17.1, 18.5, 19.7, 25.7, 28.5, 28.6, 29.4, 30.7, 30.8,
43.0, 52.4, 58.4, 60.5, 68.0, 68.4, 84.7, 114.5, 129.3, 134.8, 157.3, 162.1, 165.7; IR (NaCl, neat)
cm-1 1690, 2118; HRMS (ESI-MS): m/z calcd for C26H39N2O3 [M+H]+ 427.2961, found 427.2947.

Chapter 5: Experimental
245
Methyl (2S)-2-amino-4-(4’-(oct-7-ynyl-1-oxy)phenethyl)-2-methylbutanoate (5.60)
A solution of TFA (3 mL) in water (6 mL) was added dropwise to a solution of bis-lactim ether
5.59 (0.17 g, 0.41 mmol) in acetonitrile (9 mL). The solution was stirred at room temperature for 4
h after which the acetonitrile was removed under reduced pressure. The residue was diluted with
water and neutralised with portions of solid sodium bicarbonate, then extracted with
dichloromethane (× 4). The organic extracts were combined and dried (Na2SO4). The solvent was
removed under reduced pressure and the crude material purified by flash chromatography on
silica gel, eluting with ethyl acetate, to afford the product 5.60 as a clear colourless oil (0.11 g, 82
%). [𝛼]𝐷24.7 = + 18 (0.5, CHCl3); 1H NMR (400 MHz; CDCl3) δ 1.34 (s, 3H), 1.43 – 1.55 (m, 6H),
1.73 – 1.77 (m, 4H), 1.83 (td, J = 12.7, 5.4 Hz, 1H), 1.93 (t, J = 2.6 Hz, 1H), 1.98 (td, J = 12.7, 5.0
Hz, 1H), 2.17 (td, J = 6.9, 2.6 Hz, 2H), 2.43 (td, J = 12.7, 5.0 Hz, 1H), 2.57 (td, J = 12.7, 5.4 Hz,
1H), 3.68 (s, 3H), 3.90 (t, J = 6.5 Hz, 2H), 6.77 – 6.80 (m, 2H), 7.03 – 7.07 (m, 2H); 13C NMR
(100 MHz; CDCl3) δ 18.4, 25.6, 26.6, 28.4, 28.5, 29.2, 29.8, 43.1, 52.2, 57.8, 67.9, 68.3, 84.6,
114.5, 129.2, 133.5, 157.4, 178.0; IR (NaCl, neat) cm-1 1731, 2116, 3294, 3377; HRMS (ESI-MS):
m/z calcd for C20H30NO3 [M+H]+ 332.2226, found 332.2217.
(2S)-2-Amino-4-(4’-(oct-7-ynyl-1-oxy)phenethyl)-2-methyl-1-butanol (3.94)
Lithium aluminium hydride (28 mg, 0.74 mmol) was added as a solid in one portion to a solution
of aminoester 5.60 (0.19 g, 0.57 mmol) in freshly distilled THF (7 mL) at 0°C. The solution was
stirred at 0°C for 20 min then the cold bath was removed and the solution stirred at room
temperature for 1 h. The reaction was quenched with saturated aqueous sodium sulfate solution
and the mixture was extracted with ethyl acetate (× 4). The organic extracts were combined and
washed with saturated aqueous sodium bicarbonate solution, water, brine, then dried (Na2SO4).
The solvent was removed under reduced pressure and the crude material purified by flash
chromatography on deactivated silica gel, eluting with 2 % methanol/4 %
triethylamine/dichloromethane, to afford the product 3.94 as a clear colourless gum (0.13 g, 77
%). [𝛼]𝐷26.7 = + 4 (0.5 , CHCl3); 1H NMR (600 MHz; CDCl3) δ 1.13 (s, 3H), 1.45 – 1.48 (m, 4H),

Chapter 5: Experimental
246
1.53 – 1.58 (m, 2H), 1.62 – 1.73 (m, 2H), 1.75 – 1.79 (m, 2H), 1.94 (t, J = 2.6 Hz, 1H), 2.19 (td, J
= 7.0, 2.6 Hz, 2H), 2.44 (br s, 2H), 2.48 (br s, 1H), 2.57 (t, J = 8.6 Hz, 2H), 3.34 (d, J = 10.5 Hz,
1H), 3.39 (d, J = 10.5 Hz, 1H), 3.91 (t, J = 6.5 Hz, 2H), 6.79 – 6.81 (m, 2H), 7.07 – 7.09 (m, 2H);
13C NMR (150 MHz; CDCl3) δ 18.5, 24.3, 25.7, 28.5, 28.6, 29.3, 29.4, 42.0, 53.5, 68.0, 68.4, 69.9,
84.7, 114.6, 129.2, 134.2, 157.4; IR (NaCl, neat) cm-1 2115, 3176, 3266, 3290, 3332; HRMS
(ESI-MS): m/z calcd for C19H30NO2 [M+H]+ 304.2277, found 304.2268.
t-Butyl-(S)-(3-(2-(2-(4-(6-(4-(3’-amino-4’-hydroxy-3’-methylbutyl)phenoxy)hexyl)-1H-1,2,3-
triazol-1-yl)ethoxy)ethoxy)propyl)carbamate (3.95)
Copper(I) iodide (1 mg, 5.3 μmol) was added as a solid to a solution of alkyne 3.94 (11 mg, 36.3
μmol), azide 3.96 (11 mg, 40.1 μmol), N,N-diisopropylethylamine (21 μL, 0.12 μmol) and TBTA (2
mg, 3.8 μmol) in DMF (0.4 mL) at room temperature. The solution was stirred at room
temperature for 38 h. after which it was diluted with water and extracted with EtOAc (× 4). The
organic extracts were combined and washed with water and brine, then dried (Na2SO4). The
solvent was removed under reduced pressure and the crude material purified by flash
chromatography on silica gel, eluting with 2 % methanol/10 % triethylamine/dichloromethane, to
afford the product 3.95 as a clear colourless gum (11 mg, 53 %). [𝛼]𝐷26.2 = - 8 (0.5, CHCl3); ); 1H
NMR (300 MHz; CDCl3) δ 1.23 (s, 3H), 1.37 – 1.45 (m, 6H), 1.43 (s, 9H), 1.64 – 1.83 (m, 6H),
2.57 (t, J = 8.0 Hz, 2H), 2.71 (t, J = 7.7 Hz, 2H), 2.83 – 2.97 (m, 8H), 3.28 – 3.32 (m, 2H), 3.45 –
3.52 (m, 4H), 3.56 (s, 3H), 3.85 (t, J = 5.3 Hz, 2H), 3.89 (t, J = 6.6 Hz, 2H), 4.49 (t, J = 5.3 Hz,
2H), 4.92 (br s, 1H), 5.47 (br s, 1H), 6.76 – 6.79 (m, 2H), 7.08 – 7.11 (m, 2H), 7.42 (s, 1H); 13C
NMR (150 MHz; CDCl3) δ 8.1, 25.69, 25.9, 28.5, 29.1, 29.2, 29.3, 29.5, 40.2, 40.4, 50.2, 53.0,
55.8, 68.0, 69.8, 70.2, 70.4, 70.6, 77.4, 114.6, 121.9, 129.4, 133.8, 148.3, 156.1, 157.5; IR (NaCl,
neat) cm-1 1704, 3398; HRMS (ESI-MS): m/z calcd for C30H52N5O6 [M+H]+ 578.3917, found
578.3917.

Chapter 5: Experimental
247
(2S)-(9H-Fluoren-9-yl)methyl-(1-hydroxy-2-methyl-4-(4’-oct-7-ynyloxyphenyl)butan-2-
yl)carbamate (3.90)
9-Fluorenylmethyl chloroformate (0.13 g, 0.49 mmol) was added in one portion to a solution of
alkyne (0.13 g, 0.44 mmol) in a mixture of ethyl acetate (2.5 mL) and saturated aqueous sodium
bicarbonate solution (2.5 mL). The solution was stirred vigorously and heated at reflux for 16 h.
The solution was allowed to cool to room temperature and and the mixture was extracted with
ethyl acetate (× 3). The organic extracts were combined and washed with water, brine, then dried
(Na2SO4). The solvent was removed under reduced pressure and the crude material was purified
by flash chromatography on silica gel, eluting with 30 % ethyl acetate/n-hexane, to afford the
product 3.90 as a white gum (0.15 g, 63 %). [𝛼]𝐷22.2 = + 4 (0.5 , CHCl3); 1H NMR (600 MHz;
CDCl3) δ 1.26 (s, 3H), 1.46 – 1.51 (m, 4H), 1.55 – 1.60 (m, 2H), 1.76 – 1.80 (m, 2H), 1.87 (br s,
1H), 1.98 (t, J = 2.6 Hz, 1H), 2.03 (br s, 1H), 2.22 (td, J = 6.8, 2.6 Hz, 2H), 2.50 – 2.53 (m, 1H),
2.58 – 2.62 (m, 1H), 3.63 (br s, 1H), 3.72 (br s, 1H), 3.92 (t, J = 6.5 Hz, 2H), 4.20 (t, J = 6.5 Hz,
1H), 4.43 (br s, 2H), 4.98 (br s, 1H), 6.82 – 6.84 (m, 2H), 7.09 – 7.10 (m, 2H), 7.31 – 7.34 (m,
2H), 7.39 – 7.42 (m, 2H), 7.59 – 7.61 (m, 2H), 7.76 – 7.78 (m, 2H); 13C NMR (150 MHz; CDCl3) δ
18.4, 22.5, 28.4, 28.5, 29.1, 29.2, 38.3, 47.4, 57.1, 66.3, 67.9, 68.36, 68.38, 68.8, 84.6, 114.5,
120.0, 125.0, 127.1, 127.8, 129.2, 133.7, 141.36, 141.37, 143.9, 156.1, 157.4; IR (NaCl, neat)
1699, 2115, 3300, 3407 cm-1; HRMS (ESI-MS): m/z calcd for C34H39NO4Na [M+Na]+ 548.2777
found 548.2762.
(Oct-7-ynyloxy)benzene (3.98)
8-Iodooctyne 3.93 (0.50 g, 1.95 mmol) was added dropwise to a suspension of phenol 5.61 (0.17
g, 1.78 mmol) and potassium carbonate (0.74 g, 5.33 mmol) in DMF (2 mL). The reaction mixture
was stirred at room temperature for 14 h. Water was added and the mixture was extracted with
ethyl acetate (× 3). The organic extracts were combined and washed with water, brine, then dried
(Na2SO4). The solvent was removed under reduced pressure and the crude material purified by
flash chromatography on silica gel, eluting with 2 % dichloromethane/n-hexane, to afford the
product 3.98 as a clear colourless oil (0.24 g, 66 %). 1H NMR (300 MHz; CDCl3) δ 1.42 – 1.56 (m,
6H), 1.72 - 1.81 (m, 2H), 1.91 (t, J = 2.6 Hz, 1H), 2.17 (td, 6.8, 2.6 Hz, 2H), 3.92 (t, J = 6.5 Hz,
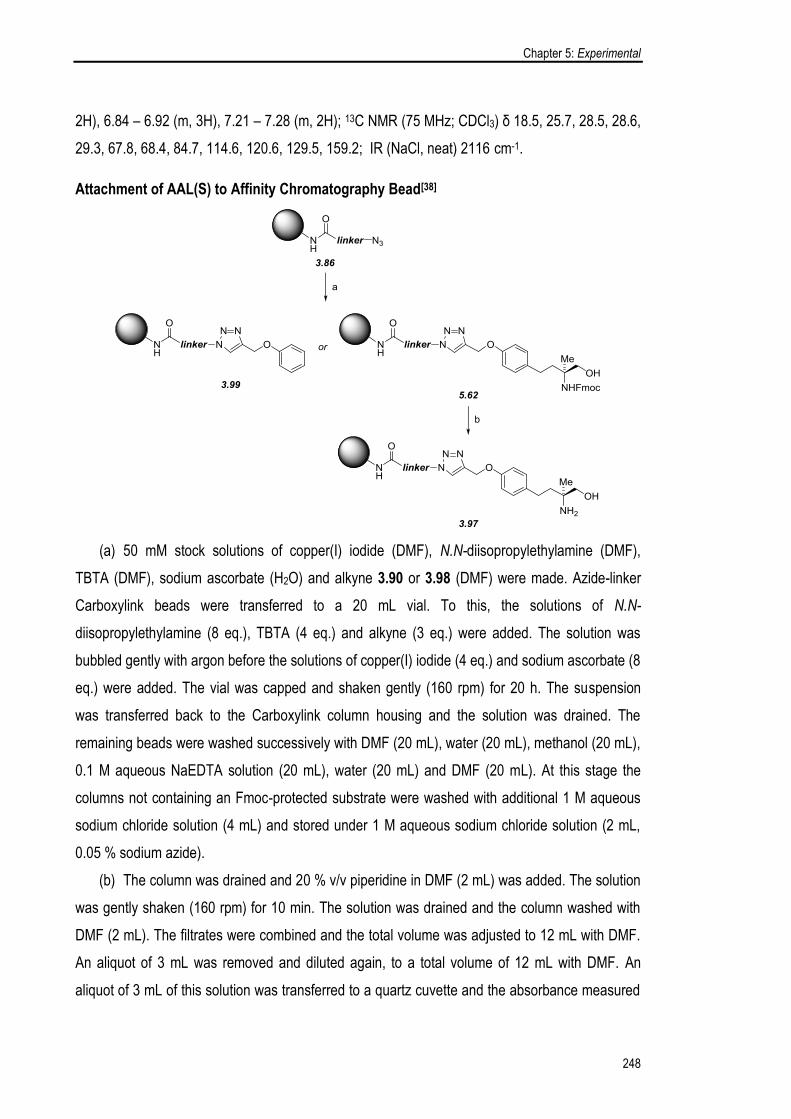
Chapter 5: Experimental
248
2H), 6.84 – 6.92 (m, 3H), 7.21 – 7.28 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 18.5, 25.7, 28.5, 28.6,
29.3, 67.8, 68.4, 84.7, 114.6, 120.6, 129.5, 159.2; IR (NaCl, neat) 2116 cm-1.
Attachment of AAL(S) to Affinity Chromatography Bead[38]
(a) 50 mM stock solutions of copper(I) iodide (DMF), N.N-diisopropylethylamine (DMF),
TBTA (DMF), sodium ascorbate (H2O) and alkyne 3.90 or 3.98 (DMF) were made. Azide-linker
Carboxylink beads were transferred to a 20 mL vial. To this, the solutions of N.N-
diisopropylethylamine (8 eq.), TBTA (4 eq.) and alkyne (3 eq.) were added. The solution was
bubbled gently with argon before the solutions of copper(I) iodide (4 eq.) and sodium ascorbate (8
eq.) were added. The vial was capped and shaken gently (160 rpm) for 20 h. The suspension
was transferred back to the Carboxylink column housing and the solution was drained. The
remaining beads were washed successively with DMF (20 mL), water (20 mL), methanol (20 mL),
0.1 M aqueous NaEDTA solution (20 mL), water (20 mL) and DMF (20 mL). At this stage the
columns not containing an Fmoc-protected substrate were washed with additional 1 M aqueous
sodium chloride solution (4 mL) and stored under 1 M aqueous sodium chloride solution (2 mL,
0.05 % sodium azide).
(b) The column was drained and 20 % v/v piperidine in DMF (2 mL) was added. The solution
was gently shaken (160 rpm) for 10 min. The solution was drained and the column washed with
DMF (2 mL). The filtrates were combined and the total volume was adjusted to 12 mL with DMF.
An aliquot of 3 mL was removed and diluted again, to a total volume of 12 mL with DMF. An
aliquot of 3 mL of this solution was transferred to a quartz cuvette and the absorbance measured

Chapter 5: Experimental
249
at 300 nm, against a standard of DMF. The amount of alkyne attached to the bead was assumed
to be proportional to the amount of released fluorene.
Linker Click Precurser Yield
-(CH2)4- AAL(S)-alkyne 3.90 24 (2.6 μmol/mL) -(CH2CH2O)4- AAL(S)-alkyne 3.90 16 (2.1 μmol/mL) -(CH2)4- Alkyne 3.98 ND -(CH2CH2O)4- Alkyne 3.98 ND
ND: not determined
Oct-1-yn-1-ylbenzene (3.104)[42]
Tetrakis(triphenylphosphine)palladium(0) (55 mg, 47.6 μmol) and copper(I) iodide (85 mg, 0.45
mmol) were added as solids successively to a Youngs tube containing iodobenzene (3.104) (0.5
mL, 4.49 mmo) and 1-octyne (1 mL, 6.78 mmol) in triethylamine (22 mL) at room temperature.
The solution was degassed using a standard freeze-pump-thaw technique (× 3) and backfilled
with argon. The reaction solution was stirred at room temperature for 16 h. The solution was
diluted with water and extracted with ethyl acetate (× 3). The organic extracts were combined and
washed with saturated aqueous sodium bicarbonate solution, water and brine, then dried
(Na2SO4). The solvent was removed under reduced pressure and the crude material was purified
by bulb-to-bulb distillation (85 -88°C at 1 mm/Hg) to afford the product 3.104 as a clear colourless
oil (0.73 g, 88 %) with all the analytical data matching that reported in the literature.[42] 1H NMR
(300 MHz; CDCl3) δ 0.91 (t, J = 6.9 Hz, 3H) 1.26 – 1.37 (m, 4H), 1.40 – 1.52 (m, 2H), 1.54 – 1.65
(m, 2H), 2.40 (t, J = 7.1 Hz, 2H), 7.24 – 7.31 (m, 3H), 7.37 – 7.42 (m, 2H).
Diethyl 2-acetamido-2-(4’-(t-butyldimethylsilyloxy)phenethyl)malonate (3.108)[43]
A solution of diethylacetamidomalonate (3.107) (2.75 g, 12.7 mmol) in DMF (20 mL) was added
dropwise to a solution of sodium hydride (0.31 g, 12.8 mmol) in DMF (20 mL) at room
temperature. The solution was stirred at room temperature for 1.5 h then a solution of iodide 3.48
(4.17 g, 11.5 mmol) in DMF (30 mL) was added dropwise. The solution was heated at 90°C for
36 h. The solution was cooled to room temperature and poured onto water. The mixture was
extracted with ethyl acetate (× 3). The organic extracts were combined and washed with water
and brine, then dried (Na2SO4). The solvent was removed under reduced pressure and the crude

Chapter 5: Experimental
250
material purified by flash chromatography on silica gel, eluting with 30 % ethyl acetate/n-hexane,
to afford the product 3.108 as a white solid (3.63 g, 70 %) with all the analytical data matching
that reported in the literature.[43] Mp 83-84°C; 1H NMR (300 MHz; CDCl3) δ 0.16 (s, 6H), 0.97 (s,
9H), 1.24 (t, J = 7.1 Hz, 6H), 1.99 (s, 3H), 1.38 – 2.43 (m, 2H), 2.62 – 2.68 (m, 2H), 4.15 – 4.25
(m, 4H), 6.71 – 6.75 (m, 2H), 6.76 (br s, 1H), 6.97 – 7.00 (m, 2H); 13C NMR (75 MHz; CDCl3) δ -
4.3, 14.1, 18.3, 23.2, 25.8, 29.4, 33.6, 62.7, 66.5, 120.0, 129.4, 133.3, 154.0, 168.2, 169.1; IR
(NaCl, neat) 1664, 1737, 1757, 3367 cm-1; HRMS (ESI-MS): m/z calcd for C23H37NO6SiNa
[M+Na]+ 474.2287 found 474.2287.
Diethyl 2-acetamido-2-(4’-heptyloxyphenethyl)malonate (5.63)[30]
Cesium fluoride (0.32 g, 2.09 mmol) was added as a solid in one portion to a solution of malonate
3.108 (0.63 g, 1.39 mmol) in dry DMF (14 mL) at room temperature. The solution was stirred for
15 min, whereupon it turned dark orange, before 1-bromoheptane (0.24 mL, 1.53 mmol) was
added dropwise. The solution was stirred at room temperature for 17 h. Water was added and the
mixture was extracted with ethyl acetate (× 3). The organic extracts were combined and washed
with water and brine, then dried (Na2SO4). The solvent was removed under reduced pressure and
the crude material purified by flash chromatography on silica gel, eluting with 50 % ethyl
acetate/n-hexane, to afford the product 5.63 as a white solid (0.57 g, 94 %) with all the analytical
data matching that reported in the literature.[30] Mp 79-81°C; 1H NMR (300 MHz; CDCl3) δ 0.89 (t,
J = 6.8 Hz, 3H), 1.24 (t, J = 7.3 Hz, 6H), 1.28 – 1.48 (m, 8H), 1.71 – 1.80 (m, 2H), 1.99 (s, 3H),
2.39 – 2.44 (m, 2H), 2.62 – 2.68 (m, 2H), 3.90 (t, J = 6.6 Hz, 2H), 4.16 – 4.24 (m, 4H), 6.76 (br s,
1H), 6.78 – 6.82 (m, 2H), 7.01 -7.06 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 14.1, 14.2, 22.8, 23.2,
26.1, 29.2, 29.37, 29.44, 31.9, 33.7, 62.7, 66.5, 68.2, 114.6, 129.4, 132.5, 157.7, 168.2, 169.1.
2-Amino-2-(4’-heptyloxyphenethyl)propane-1,3-diol (3.106)[30]
(a) Lithium aluminium hydride (78 mg, .2.06 mmol) was added as a solid in one
portion to a solution of malonate 5.63 (0.36 g, 0.84 mmol) in freshly distilled THF (4 mL) at 0°C.
The solution was stirred at 0°C for 20 min then the cold bath was removed and the solution
stirred at room temperature for 1 h. The reaction was quenched with saturated aqueous sodium

Chapter 5: Experimental
251
sulfate solution and the mixture was extracted with ethyl acetate (× 4). The organic extracts were
combined and washed with saturated aqueous sodium bicarbonate solution, water and brine,
then dried (Na2SO4). The solvent was removed under reduced pressure and the crude material
used in the next step without any further purification.
(b) The crude material was dissolved in ethanol (4 mL) and concentrated
hydrochloric acid solution (32 %, 1 mL) was added dropwise. The mixture was heated at reflux for
16 h. The solution was allowed to cool to room temperature and the ethanol was removed under
reduced pressure. The residue was neutralised with solid sodium bicarbonate and extracted with
ethyl acetate (× 4). The organic extracts were combined and washed with brine, then dried
(Na2SO4). The solvent was removed under reduced pressure to afford a brown gum which was
recystallised from ethanol to afford the product 3.106 as a white solid (0.16 g, 62 %) with all the
analytical data matching that reported in the literature.[30] 1H NMR (300 MHz; MeOD) 0.91 (t, J =
6.7 Hz, 3H), 1.33 – 1.50 (m, 8H), 1.65 – 1.76 (m, 4H), 2.55 – 2.61 (m, 2H), 3.48 (d, J = 11.0 Hz,
2H), 3.54 (d, J = 11.0 Hz, 2H), 3.91 (t, J = 6.4 Hz, 2H), 6.78 – 6.81 (m, 2H), 7.10 – 7.13 (m, 2H);
13C NMR (75 MHz; MeOD) δ 19.0, 26.7, 29.49, 29.53, 29.6, 30.3, 66.4, 68.9, 69.5, 85.0, 115.47,
115.51, 130.2, 135.9, 158.9.
Diethyl 2-acetamido-2-(4’-(oct-7-ynyloxy)phenethyl)malonate (5.64)
Cesium fluoride (0.47 g, 3.08 mmol) was added as a solid in one portion to a solution of malonate
3.108 (0.70 g, 1.54 mmol) in dry DMF (15 mL) at room temperature. The solution was stirred for
15 min, where it had turned dark orange, before 8-iodo-1-octyne (0.55 g, 2.31 mmol) was added
dropwise. The solution was stirred at room temperature for 17 h. Water was added and the
mixture was extracted with ethyl acetate (× 3). The organic extracts were combined and washed
with water and brine, then dried (Na2SO4). The solvent was removed under reduced pressure and
the crude material purified by flash chromatography on silica gel, eluting with 50 % ethyl
acetate/n-hexane, to afford the product 5.64 as a white solid (0.56 g, 82 %). Mp 72 – 74°C; 1H
NMR (300 MHz; CDCl3) δ 1.24 (t, J = 7.1 Hz, 6H), 1.44 – 1.58 (m, 6H), 1.72 – 1.81 (m, 2H), 1.94
(t, J = 2.6 Hz, 1H), 1.99 (s, 3H), 2.20 (td, J = 6.9, 2.6 Hz, 2H), 2.39 – 2.44 (m, 2H), 2.62 – 2.68
(m, 2H), 3.92 (t, J = 6.5 Hz, 2H), 4.16 – 4.24 (m, 4H), 6.76 (br s, 1H), 6.77 – 6.81 (m, 2H), 7.01 –
7.06 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 14.1, 18.5, 23.2, 25.7, 28.5, 28.6, 29.3, 29.4, 33.7,
62.7, 66.5, 68.0, 68.4, 84.7, 114.6, 129.5, 132.6, 157.6, 168.2, 169.1; IR (NaCl, neat) 1681, 1739,

Chapter 5: Experimental
252
2116, 3292, 3391 cm-1; HRMS (ESI-MS): m/z calcd for C25H35NO6Na [M+Na]+ 468.2362 found
468.2349.
2-Amino-2-(4-(oct-7-ynyloxy)phenethyl)propane-1,3-diol (5.65)
(a) Lithium aluminium hydride (0.22 g, 5.90 mmol) was added in one portion to a
solution of malonate 5.64 (1.05 g, 2.36 mmol) in freshly distilled THF (24 mL) at 0°C. The solution
was stirred at 0°C for 20 min then the cold bath was removed and the solution stirred at room
temperature for 1 h. The reaction was quenched with saturated aqueous sodium sulfate solution
and the mixture was extracted with ethyl acetate (× 4). The organic extracts were combined and
washed with saturated aqueous sodium bicarbonate solution, water and brine, then dried
(Na2SO4). The solvent was removed under reduced pressure and the crude material used in the
next step without any further purification.
(b) The crude material was dissolved in ethanol (24 mL) and concentrated
hydrochloric acid solution (32 %, 8 mL) was added dropwise. The mixture was heated at reflux for
16 h. The solution was allowed to cool to room temperature and the ethanol was removed under
reduced pressure. The residue was neutralised with sodium bicarbonate and extracted with ethyl
acetate (× 4). The organic extracts were combined and washed with brine, then dried (Na2SO4).
The solvent was removed under reduced pressure to afford a brown gum which was recystallised
(ethanol) to afford the product 5.65 as a white solid (0.34 g, 47 %). 1H NMR (300 MHz; MeOD) δ
1.36 – 1.47 (m, 8H), 1.59 – 1.68 (m, 2H), 2.11 (td, J = 6.5, 2.2 Hz, 2H), 2.43 – 2.49 (m, 2H), 2.69
(t, J = 2.2 Hz, 1H), 3.18 (d, J = 10.5 Hz, 2H), 3.23 (d, J = 10.5, 2H), 3.85 (t, J = 6.4 Hz, 2H), 6.75
– 6.77 (m, 2H), 7.01 – 7.04 (m, 2H); 13C NMR (75 MHz; MeOD) δ 27.08, 27.12, 34.5, 37.40,
37.44, 38.1, 46.2, 65.4, 74.5, 76.7, 80.6, 94.0, 123.7, 138.5, 144.4, 168.1; IR (NaCl, neat) 2115,
3288, 3333 cm-1; HRMS (ESI-MS): m/z calcd for C19H30NO3 [M+H]+ 320.2225 found 320.2209.
(9H-Fluoren-9-yl)methyl-(1-hydroxy-2-(hydroxymethyl)-4-(4-oct-7-ynyloxyphenyl)butan-2-
yl)carbamate (3.109)
Fmoc-Cl (44 mg, 0.17 mmol) was added as a solid in one portion to a solution of amino diol 5.65
(60 mg, 0.17 mmol) in ethyl acetate (7 mL) and saturated aqueous sodium bicarbonate solution

Chapter 5: Experimental
253
(3.5 mL). The solution was stirred at room temperature for 17 h. The mixture was diluted with
water and extracted with dichloromethane (× 3). The organic extracts were combined and
washed with water and brine, then dried (Na2SO4). The solvent was removed under reduced
pressure and the crude material purified by flash chromatography on silica gel, eluting with 80 %
diethyl ether/n-hexane, to afford the product 3.109 as a white solid (40 mg, 44 %). 1H NMR (300
MHz; CDCl3) δ 1.39 – 1.64 (m, 6H), 1.74 – 1.79 (m, 2H), 1.84 (br s, 1H), 1.94 (t, J = 2.6 Hz, 1H),
2.20 (td, J = 6.8, 2.6 Hz, 2H), 2.34 (t, J = 7.2 Hz, 2H), 2.49 – 2.54 (m, 2H), 3.17 (br s, 2H), 3.63
(d, J = 10.0 Hz, 2H), 3.86 (J = 10.0 Hz, 2H) 3.91 (t, J = 6.5 Hz, 2H), 4.20 (t, J = 6.5 Hz, 1H), 4.46
(br s, 2H), 5.24 (br s, 1H), 6.79 – 6.82 (m, 2H), 7.05 – 7.07 (m, 2H), 7.29 – 7.34 (m, 2H), 7.38 –
7.43 (m, 2H), 7.58 – 7.60 (m, 2H), 7.76 – 7.78 (m, 2H); 13C NMR (75 MHz; CDCl3) δ 18.5, 25.7,
28.5, 28.6, 28.7, 29.3, 47.5, 66.6, 66.7, 68.0, 68.4, 114.7, 121.2, 125.1, 127.2, 127.9, 129.3,
133.5, 135.5, 141.5, 143.9, 156.5, 157.6; IR (NaCl, neat) 1699, 2115, 3301, 3400 cm-1; HRMS
(ESI-MS): m/z calcd for C34H39NO5Na [M+Na]+ 564.2725 found 564.2709.
Attachment of O-FTY720 to Affinity Chromatography Bead[38]
(a) 50 mM stock solutions of copper(I) iodide (DMF), N,N-diisopropylethylamine (DMF),
TBTA (DMF), sodium ascorbate (H2O) and alkyne 3.109 (DMF) were made. Azide-linker
Carboxylink beads were transferred to a 20 mL vial. To this, the solutions of N,N-
diisopropylethylamine (8 eq.), TBTA (4 eq.) and Fmoc-O-FTY720-alkyne 3.109 (3 eq.) were
added. The solution was bubbled gently with argon before the solutions of copper(I) iodide (4 eq.)
and sodium ascorbate (8 eq.) were added. The vial was capped and shaken gently (160 rpm) for
20 h. The suspension was transferred back to the Carboxylink column housing and the solution
was drained. The remaining beads were washed successively with DMF (20 mL), water (20 mL),
methanol (20 mL), 0.1 M aqueous NaEDTA solution (20 mL), water (20 mL) and DMF (20 mL). At
this stage the columns not containing an Fmoc-protected substrate were washed with additional 1

Chapter 5: Experimental
254
M aqueous sodium chloride solution (4 mL) and stored under 1 M aqueous sodium chloride
solution (2 mL, 0.05 % sodium azide).
(b) The column was drained and 20 % v/v piperidine in DMF (2 mL) was added. The solution
was gently shaken (160 rpm) for 10 min. The solution was drained and the column washed with
DMF (2 mL). The filtrates were combined and the total volume was adjusted to 12 mL with DMF.
An aliquot of 3 mL was removed and diluted again, to a total volume of 12 mL with DMF. An
aliquot of 3 mL of this solution was transferred to a quartz cuvette and the absorbance measured
at 300 nm, against a standard of DMF. The amount of alkyne attached to the bead was assumed
to be proportional to the amount of released fluorene which was found to generate O-FTY720
affinity chromatography beads (PEG4)-3.108 in 45 % (3.9 μmol/mL).

Chapter 5: Experimental
255
5.4. References for Chapter 5
[1] G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw, K. I. Goldberg, Organometallics 2010, 29, 2176-2179.
[2] D. D. Perrin, W. L. F. Armarego, Purification of Laboratory Chemicals, 3rd Edn. ed., Pergamon Press Ltd., Great Britain, 1988.
[3] S. C. Watson, J. F. Eastham, J. Organomet. Chem. 1967, 9, 165-168. [4] T. R. Hoye, A. W. Aspaas, B. M. Eklov, T. D. Ryba, Org. Lett. 2005, 7, 2205-2208. [5] N. Uematsu, A. Fujii, S. Hashiguchi, T. Ikariya, R. Noyori, J. Am. Chem. Soc. 1996, 118,
4916-4917. [6] D. R. Coulson, L. C. Satek, S. O. Grim, in Inorg. Synth., John Wiley & Sons, Inc., 2007,
pp. 121-124. [7] R. J. Linderman, M. Jaber, B. D. Griedel, J. Org. Chem. 1994, 59, 6499-6500. [8] K. L. Reddy, K. R. Dress, K. B. Sharpless, Tetrahedron Lett. 1998, 39, 3667-3670. [9] C. Z. Ding, R. B. Silverman, Synth. Commun. 1993, 23, 1467-1471. [10] T. R. Chan, R. Hilgraf, K. B. Sharpless, V. V. Fokin, Org. Lett. 2004, 6, 2853-2855. [11] W. C. Still, M. Kahn, A. Mitra, J. Org. Chem. 1978, 43, 2923-2925. [12] L. E. Fisher, J. M. Muchowski, R. D. Clark, J. Org. Chem. 1992, 57, 2700-2705. [13] T. Schubert, M.-R. Kula, M. Müller, Synthesis 1999, 1999, 2045-2048. [14] W. Li, P. Sun, J. Org. Chem. 2012, 77, 8362-8366. [15] T. H. Jones, M. S. Blum, H. M. Fales, Synth. Commun. 1981, 11, 889-894. [16] C. J. Bungard, PhD Thesis, The University of Canterbury 2001. [17] C. J. Bungard, J. C. Morris, Synthesis 2001, 2001, 0741-0744. [18] M. Watanabe, S. Hisamatsu, H. Hotokezaka, S. Furukawa, Chem. Pharm. Bull. 1986, 34,
2810-2820. [19] G. Bringmann, G. Zhang, A. Hager, M. Moos, A. Irmer, R. Bargou, M. Chatterjee, Eur. J.
Med. Chem. 2011, 46, 5778-5789. [20] K. W. Kells, J. M. Chong, Org. Lett. 2003, 5, 4215-4218. [21] G. Bringmann, K. Messer, K. Wolf, J. Mühlbacher, M. Grüne, R. Brun, A. M. Louis,
Phytochemistry 2002, 60, 389-397. [22] F. Ferreira, M. Audouin, F. Chemla, Chem. -Eur. J. 2005, 11, 5269-5278. [23] J. Brusnahan, Unpublished Results 2009. [24] F. A. Davis, P. K. Mohanty, D. M. Burns, Y. W. Andemichael, Org. Lett. 2000, 2, 3901-
3903. [25] J. S. Brusnahan, PhD Thesis, The University of Adelaide (Adelaide, South Australia),
2009. [26] M. Amat, F. Subrizi, V. Elias, N. Llor, E. Molins, J. Bosch, Eur. J. Org. Chem. 2012, 2012,
5491-5497. [27] H. E. Zimmerman, R. L. Swafford, J. Org. Chem. 1984, 49, 3069-3083. [28] S. Datta, C.-L. Chang, K.-L. Yeh, R.-S. Liu, J. Am. Chem. Soc. 2003, 125, 9294-9295. [29] C.-L. Chen, S. M. Sparks, S. F. Martin, J. Am. Chem. Soc. 2006, 128, 13696-13697. [30] M. Kiuchi, K. Adachi, T. Kohara, M. Minoguchi, T. Hanano, Y. Aoki, T. Mishina, M. Arita,
N. Nakao, M. Ohtsuki, Y. Hoshino, K. Teshima, K. Chiba, S. Sasaki, T. Fujita, J. Med. Chem. 2000, 43, 2946-2961.
[31] A. B. Smith, J. B. Sperry, Q. Han, J. Org. Chem. 2007, 72, 6891-6900. [32] P. C. Kearney, M. W. Nowak, W. Zhong, S. K. Silverman, H. A. Lester, D. A. Dougherty,
Mol. Pharmacol. 1996, 50, 1401-1412. [33] A. Gissibl, C. Padié, M. Hager, F. Jaroschik, R. Rasappan, E. Cuevas-Yañez, C.-O.
Turrin, A.-M. Caminade, J.-P. Majoral, O. Reiser, Org. Lett. 2007, 9, 2895-2898.

Chapter 5: Experimental
256
[34] T. Nakamura, T. Tsuji, Y. Iio, S. Miyazaki, T. Takemoto, T. Nishi, Tetrahedron: Asymmetry 2006, 17, 2781-2792.
[35] K. Högenauer, K. Hinterding, P. Nussbaumer, Bioorg. Med. Chem. Lett. 2010, 20, 1485-1487.
[36] H. Tokuyama, K. Okano, H. Fujiwara, T. Noji, T. Fukuyama, Chemistry – An Asian Journal 2011, 6, 560-572.
[37] C. Cativiela, J. L. Serrano, M. M. Zurbano, J. Org. Chem. 1995, 60, 3074-3083. [38] S. Punna, E. Kaltgrad, M. G. Finn, Bioconjugate Chem. 2005, 16, 1536-1541. [39] R. E. Beveridge, R. A. Batey, Org. Lett. 2013, 15, 3086-3089. [40] H. Hopf, A. Krüger, Chem. Eur. J. 2001, 7, 4378-4385. [41] L. Peng, J. DeSousa, Z. Su, B. M. Novak, A. A. Nevzorov, E. R. Garland, C. Melander,
Chem. Commun. 2011, 47, 4896-4898. [42] T. Fukuyama, M. Shinmen, S. Nishitani, M. Sato, I. Ryu, Org. Lett. 2002, 4, 1691-1694. [43] S. Nakayama, Y. Uto, K. Tanimoto, Y. Okuno, Y. Sasaki, H. Nagasawa, E. Nakata, K.
Arai, K. Momose, T. Fujita, T. Hashimoto, Y. Okamoto, Y. Asakawa, S. Goto, H. Hori, Bioorg. Med. Chem. 2008, 16, 7705-7714.




















