
Disturbed vitamin A metabolism in chronic liver disease and ...
267
University of Groningen Disturbed vitamin A metabolism in chronic liver disease and relevance for therapy Saeed, Ali IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it. Please check the document version below. Document Version Publisher's PDF, also known as Version of record Publication date: 2019 Link to publication in University of Groningen/UMCG research database Citation for published version (APA): Saeed, A. (2019). Disturbed vitamin A metabolism in chronic liver disease and relevance for therapy. University of Groningen. Copyright Other than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons). The publication may also be distributed here under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license. More information can be found on the University of Groningen website: https://www.rug.nl/library/open-access/self-archiving-pure/taverne- amendment. Take-down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons the number of authors shown on this cover page is limited to 10 maximum. Download date: 07-09-2022
-
Upload
khangminh22 -
Category
Documents
-
view
4 -
download
0
Transcript of Disturbed vitamin A metabolism in chronic liver disease and ...

University of Groningen
Disturbed vitamin A metabolism in chronic liver disease and relevance for therapySaeed, Ali
IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite fromit. Please check the document version below.
Document VersionPublisher's PDF, also known as Version of record
Publication date:2019
Link to publication in University of Groningen/UMCG research database
Citation for published version (APA):Saeed, A. (2019). Disturbed vitamin A metabolism in chronic liver disease and relevance for therapy.University of Groningen.
CopyrightOther than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of theauthor(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons).
The publication may also be distributed here under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license.More information can be found on the University of Groningen website: https://www.rug.nl/library/open-access/self-archiving-pure/taverne-amendment.
Take-down policyIf you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediatelyand investigate your claim.
Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons thenumber of authors shown on this cover page is limited to 10 maximum.
Download date: 07-09-2022

Disturbed vitamin A metabolism in chronic
liver disease and relevance for therapy
Ali Saeed

This work was supported by the Graduate School of Medical Sciences
(GSMS), University Medical Center Groningen, University of Groningen,
Groningen, The Netherlands. This work was also supported by
Bahauddin Zakariya University, Multan, Pakistan and Higher education
commission of Pakistan under Yousuf Raza Gilani faculty development
Ph.D. scholarships.
The printing of this thesis was financially supported by the following
organizations:
Groningen University Institute for Drug Explorations
Nederlandse vereniging voor Hepatologie (NVH)
Their contribution is greatly acknowledged!
© copyright 2019 A. Saeed
All rights reserved. No part of this book may be reproduced, stored in a
retrieval system, or transmitted in any form or by any means without written
permission of the author and the publisher holding the copyright of the
published articles.
ISBN / EAN: 978-94-034-1686-1 (Printed book)
ISBN: 978-94-034-1685-4 (Digital book)
Cover: Designed by A. Saeed
Lay-out by: A. Saeed
Printed by: PRINTSUPPORT4U

Disturbed vitamin A metabolism in chronic liver disease and relevance for therapy
PhD thesis
to obtain the degree of PhD at the University of Groningen on the authority of the
Rector Magnificus prof. E. Sterken and in accordance with
the decision by the College of Deans.
This thesis will be defended in public on
Tuesday 2 July 2019 at 9.00 hours
by
Ali Saeed
born on 6 January 1984 in Faisalabad, Pakistan

Supervisor Prof. K.N. Faber
Co-supervisor Prof. J. Blokzijl
Assessment Committee Prof. J.B. Helms
Prof. R.K. Weersma
Prof. P. Olinga

Paranymphs Janette Heegsma Manon Buist-Homan

Dedicated to my parents, my wife and my kids

Table of contents
Chapter 1: Introduction and aim of the thesis 9
Chapter 2: The interrelationship between bile acid and vitamin A homeostasis 17
Saeed A, Hoekstra M, Hoeke MO, Heegsma, Faber KN (2017). BBA-
Molecular and Cell Biology of Lipids, 1862(5):496-512.
Chapter 3: Hormone-sensitive lipase is a retinyl ester hydrolase in human and rat
quiescent hepatic stellate cells
75
Shajari S*, Saeed A*, Smith-Cortinez NF, Heegsma J, Sydor S, Faber
KN (2019) BBA-molecular and cell biology of lipids, In press.
*equal authors
Chapter 4: Disturbed Vitamin A Metabolism in Non-Alcoholic Fatty Liver Disease
(NAFLD)
105
Saeed A, Dullaart RPF, Schreuder TCMA, Blokzijl H, Faber KN (2017)
Nutrients 2017 Dec 29;10(1). pii: E29. doi: 10.3390/nu10010029
Chapter 5: Hepatic vitamin A metabolism is disturbed in mice with Non-Alcoholic
Fatty Liver Disease leading to vitamin A accumulation in hepatocytes
143
Submitted
Chapter 6: Glycogen storage disease type 1a is associated with disturbed vitamin
A metabolism and elevated serum retinol levels
176
Submitted
Chapter 7: Farnesoid X receptor (FXR) and bile acids regulate vitamin A storage 201
In preparation
Chapter 8: Discussion 229
Summary & Samenvatting 251
Acknowledgements 259
Curriculum Vitae & Publications 261


Chapter 1
Introduction and aim of the thesis
Ali Saeed1,2, Klaas Nico Faber1,3
1Department of Gastroenterology and Hepatology, 3Laboratory Medicine, Center for
Liver, Digestive, and Metabolic Diseases, University Medical Center Groningen,
University of Groningen, Groningen, The Netherlands.
2Institute of Molecular Biology and Biotechnology, Bahauddin Zakariya University
Multan, Pakistan

Introduction and aim of the thesis
10
1.1. INTRODUCTION
Vitamin A is an essential nutrient, principally meaning that humans and animals fully
depend on dietary sources to supply key physiological processes in the body with this
vitamin. Plants synthesize carotenoids, like alpha- and beta-carotene, that give the
characteristic color to carrot and corn and are the primary source of vitamin A.
Carotenoids absorb light energy for photosynthesis, but also protect chlorophyll from
light-inflicted photo damage. The typical color change of leaves in fall is a result of
degradation of chlorophyll (green pigment) into colorless tetrapyrroles, at the same
time revealing the color of other pigments of which carotenoids (from yellow to
orange) are often abundantly present [1,2]. Animals eat the plants and roots
containing the carotenoids and most of it this “pro-vitamin A” is absorbed in the small
intestine. As carotenoids are lipophilic compounds, efficient intestinal absorption
depends on the presence of sufficient bile acids, which are produced in the liver and
are released in the small intestine for that purpose: keeping fat-soluble compounds
“in solution”, either for uptake or effective excretion [3]. Most of the carotenoids are
converted to retinol in the intestine, which is subsequently esterified to long chain
fatty acids (LCFAs), such as palmitate and stearate, and released to the bloodstream
for distribution to peripheral tissues that need vitamin A [3,4]. As quite a bit of vitamin
A accumulates in fat tissue, many meat- and/or organ-based foods are also an
important nutritional source of vitamin A for humans [5,6]. Most of the “dietary”
vitamin A is, however, routed to the liver where excess vitamin A is stored and
typically contains over 80% of the total vitamin A pool in a healthy individual [5]. The
liver is “in control” of supplying the body with sufficient vitamin A in times when
dietary intake is low, and it has an impressive capacity to maintain normal vitamin A
homeostasis. Even in the absence of any vitamin A intake, it may take months to
years before humans experience vitamin A deficiency, also called hypovitaminosis A.
In the meantime, stable blood levels of retinol are maintained at around 2 µmol/L
independent of the vitamin A pool size in the liver [7]. The typical symptom of vitamin
A deficiency is night blindness, which is a result of the impaired production of
rhodopsin that depends on specific vitamin A metabolites [8]. Still, vitamin A
deficiency leads to many more problems as it also impairs skin and tissue
regeneration, immune control, metabolic control, fertility as well as predisposes for
cancer [9]. Retinol is secreted from the liver to the blood circulation bound to retinol
binding protein 4 (RBP4). Retinol is taken up by tissues requiring vitamin A and

Chapter 1
11
converted to retinoic acids, which are the active metabolites of vitamin A [10]. Most of
the processes that require vitamin A are actually controlled by retinoic acids that
activate ligand-dependent transcription factors, e.g. retinoic acid-activated receptors
(RARs) and retinoid X receptors (RXRs). Retinol is the mother compound that is
converted to different forms of retinoic acids, which in the end largely determines the
ultimate effect in physiological processes as all-trans retinoic acid (atRA) is a high-
affinity ligand for RARs, while 9-cis retinoic acid (9cRA) and 9-cis-13,14
dihydroretinoic acid (9cDHRA) activate RXRs [10,11]. Hepatic vitamin A uptake,
storage and release depends on a complex interplay between different cell types in
the liver, in particular the hepatocytes and hepatic stellate cells [5]. Up to 80-85% of
the total liver cell mass is taken by hepatocytes and they are considered to determine
most of the liver functions, e.g. controlling glucose and lipid metabolism, production
of blood proteins and detoxification [11,12]. Vitamin A absorbed in the intestine,
mostly retinyl esters in chylomicron remnants, first arrives in hepatocytes where it is
converted to retinol and secreted to the blood bound to RBP4. Excess retinol is taken
up by HSC and converted to retinyl esters again for long term storage [10]. Many
details of how hepatocytes and HSC communicate to maintain the stable retinol
levels in blood are, however, still unclear. Chronic liver diseases, including viral,
metabolic, immune-mediated and obstructive forms, are often associated with
impaired bile acid metabolism and/or bile flow, which as a result affects intestinal
absorption of fat-soluble nutrients, including vitamin A [13]. Moreover, the
progression of liver diseases also leads to fibrosis, which may progress to cirrhosis
and liver cancer. HSC are considered to be the main liver cell type causing fibrosis.
The vitamin A-containing “quiescent” HSC that are characteristic for the healthy liver
undergo a dramatic phenotypic and functional transdifferentiation in the chronically-
injured liver [12]. They transform to highly proliferative, migratory and extracellular
matrix-producing “activated’ HSC that lose their vitamin A content in this process [14].
Both pathological processes contribute to a reduction in the hepatic vitamin A pool,
that may even progress to systemic vitamin A deficiency (VAD), which is defined as
serum retinol levels below 0.7 mol/L [15]. The prevalence of VAD in chronic liver
diseases varies between studies, which may lie in differences in disease etiology,
group size, patient age and also the used definition of “vitamin A deficiency”. Some
studies the strict cut-off of serum retinol of 0.7 µM, while others also include the
range of 0.7-1.05 µM as “vitamin A inadequate”. Still, vitamin A deficiency has been

Introduction and aim of the thesis
12
reported for 20-40% of patients with primary biliary cholangitis (PBC) or primary
sclerosis cholangitis (PSC) [16–19], while up to 70% of pediatric patients with biliary
atresia may develop VAD [20]. Moreover, non-alcoholic fatty liver disease (NAFLD)
and related pathologies like obesity, type 2 diabetes and metabolic syndrome, have
repeatedly been shown to be associated with impaired systemic vitamin A status,
including lowered serum retinol levels and/or elevated serum RBP4 levels [21]. Still, it
remains largely unknown whether 1) this is due to loss of vitamin A from the liver or a
change in hepatic vitamin A metabolism and 2) whether impaired vitamin A status
actually contributes to disease progression. Recent observations indicate that
impaired vitamin A metabolism may contribute to chronic liver disease, including
NAFLD. One is that genome-wide association studies have now identified 2 genes
associated with NAFLD that encode enzymes involved in vitamin A metabolism, e.g.
the retinyl ester hydrolase patatin-like phospholipase domain-containing protein 3
(PNPLA3) and the retinol dehydrogenase hydroxysteroid 17-beta dehydrogenase 13
(HSD17B13) [22–24]. Secondly, vitamin A metabolites, especially atRA and retinyl
aldehyde as well as synthetic ligands for RARs, have therapeutic effects in animal
models of NAFLD [21].
Thus, there are ample indications that chronic liver diseases are linked to impaired
vitamin A status, but we know very little mechanistic details. Such knowledge is
required to evaluate the therapeutic potential of vitamin A and/or specific vitamin A
metabolites in these diseases.
1.2. THE AIM OF THE THESIS
The overall aim of this thesis is the delineate molecular mechanism that are involved
in hepatic vitamin A metabolism in chronic liver diseases and how this may be
affected by drugs currently studied for the treatment of NAFLD. In Chapter 2, we first
provide a comprehensive overview of the interrelationship between bile acid and
vitamin A homeostasis and how this is related to various chronic liver diseases. In
Chapter 3, we analyzed the role of the hormone sensitive lipase (HSL) as a retinyl
ester hydrolase and HSC and how this role may change during HSC
transdifferentiation. In Chapter 4, we summarize the current knowledge about
vitamin A metabolism in NAFLD and its putative role in disease progression, as well
as the therapeutic potential of vitamin A metabolites. From this critical review, we
conclude that there is actually quite a bit of controversy about what is going on with
vitamin A metabolism in the fatty liver. Thus, in Chapter 5 we analyzed vitamin A

Chapter 1
13
metabolism in 2 animal models of NAFLD, e.g. the high fat-high cholesterol diet
model, as well as the Leptinob mutant (ob/ob) genetic model. We quantified retinol
and retinol ester levels in the liver and found that technical aspects of the analytical
procedures for these compounds heavily affect the experimental outcome. Using
complementary approaches we obtained consistent results that reveal that NAFLD
does not lead to true vitamin A deficiency, but to cell type-specific rearrangements in
vitamin A metabolism. In Chapter 6, we analysed hepatic vitamin A metabolism in
glycogen storage disease type 1a (GSD Ia), a syndrome caused by mutations in the
catalytic subunit of glucose-6-phosphatase (G6PC). Increased liver fat is a typical
symptom of GSD Ia and we wanted to know whether this similarly affects vitamin A
metabolism as in NAFLD. Serum retinol levels were determined of GSD Ia patients,
as well as hepatic vitamin A metabolism in liver-specific G6pc knock-out mice.
Indeed, vitamin A metabolism is impaired in the absence of G6PC, but in a clear
different manner than observed for the NAFLD models in chapter 5. In Chapter 7, we
analyzed whether the farnesoid X receptor (FXR), which is the bile acid sensor and
therapeutic target in NAFLD, affects hepatic vitamin A metabolism. We quantified
retinol and retinyl esters in whole body- and intestine-specific FXR-null mice, as well
as after reintroduction of hepatic FXR in whole body FXR-null mice. Moreover, wild
type animals were treated with obeticholic acid (OCA), a high-affinity ligand of FXR
and currently under investigation for the treatment of NAFLD, and the normal bile
acid cholic acid (CA) to determine their effect on hepatic vitamin A metabolism.
Again, hepatic vitamin A metabolism was strongly affected by the absence of FXR as
well as the ligand-activation of FXR. Finally, in Chapter 8, we summarize the results
obtained in the experimental studies in this thesis and present and provide an outlook
for future directions to target vitamin A metabolism in the treatment of chronic liver
diseases.

Introduction and aim of the thesis
14
REFERENCES
[1] M. Shumskaya, E.T. Wurtzel, The carotenoid biosynthetic pathway: thinking in all dimensions, Plant Sci. Int. J. Exp. Plant Biol. 208 (2013) 58–63. doi:10.1016/j.plantsci.2013.03.012.
[2] J.-H. Park, S. Jung, Perturbations of carotenoid and tetrapyrrole biosynthetic pathways result in differential alterations in chloroplast function and plastid signaling, Biochem. Biophys. Res. Commun. 482 (2017) 672–677. doi:10.1016/j.bbrc.2016.11.092.
[3] E. Reboul, Absorption of vitamin A and carotenoids by the enterocyte: focus on transport proteins, Nutrients. 5 (2013) 3563–3581. doi:10.3390/nu5093563.
[4] T. Bohn, C. Desmarchelier, S.N. El, J. Keijer, E. van Schothorst, R. Rühl, P. Borel, β-Carotene in the human body: metabolic bioactivation pathways - from digestion to tissue distribution and excretion, Proc. Nutr. Soc. 78 (2019) 68–87. doi:10.1017/S0029665118002641.
[5] S.M. O’Byrne, W.S. Blaner, Retinol and retinyl esters: biochemistry and physiology, J. Lipid Res. 54 (2013) 1731–1743. doi:10.1194/jlr.R037648.
[6] R. Schreiber, U. Taschler, K. Preiss-Landl, N. Wongsiriroj, R. Zimmermann, A. Lass, Retinyl ester hydrolases and their roles in vitamin A homeostasis, Biochim. Biophys. Acta. 1821 (2012) 113–123. doi:10.1016/j.bbalip.2011.05.001.
[7] R. Blomhoff, M.H. Green, T. Berg, K.R. Norum, Transport and storage of vitamin A, Science. 250 (1990) 399–404.
[8] C.M. Kemp, S.G. Jacobson, D.J. Faulkner, R.W. Walt, Visual function and rhodopsin levels in humans with vitamin A deficiency, Exp. Eye Res. 46 (1988) 185–197.
[9] E.M. Wiseman, S. Bar-El Dadon, R. Reifen, The vicious cycle of vitamin a deficiency: A review, Crit. Rev. Food Sci. Nutr. 57 (2017) 3703–3714. doi:10.1080/10408398.2016.1160362.
[10] W.S. Blaner, Y. Li, P.-J. Brun, J.J. Yuen, S.-A. Lee, R.D. Clugston, Vitamin A Absorption, Storage and Mobilization, Subcell. Biochem. 81 (2016) 95–125. doi:10.1007/978-94-024-0945-1_4.
[11] A. Saeed, M. Hoekstra, M.O. Hoeke, J. Heegsma, K.N. Faber, The interrelationship between bile acid and vitamin A homeostasis, Biochim. Biophys. Acta. 1862 (2017) 496–512. doi:10.1016/j.bbalip.2017.01.007.
[12] Z. Kmieć, Cooperation of liver cells in health and disease, Adv. Anat. Embryol. Cell Biol. 161 (2001) III–XIII, 1–151.
[13] C. Freund, D.N. Gotthardt, Vitamin A deficiency in chronic cholestatic liver disease -is vitamin A therapy beneficial ?, Liver Int. Off. J. Int. Assoc. Study Liver. (2017). doi:10.1111/liv.13433.
[14] J.X. Jiang, N.J. Török, Liver Injury and the Activation of the Hepatic Myofibroblasts, Curr. Pathobiol. Rep. 1 (2013) 215–223. doi:10.1007/s40139-013-0019-6.
[15] World Health Organization, Indicators for assessing vitamin A deficiency and their application in monitoring and evaluating intervention programmes, Eileen Brown, James Akré, World Health Organization: Geneva, Switzerland, 1996. http://www.who.int/nutrition/publications/micronutrients/vitamin_a_deficiency/WHO_NUT_96.10/en/.
[16] J.R. Phillips, P. Angulo, T. Petterson, K.D. Lindor, Fat-soluble vitamin levels in patients with primary biliary cirrhosis, Am. J. Gastroenterol. 96 (2001) 2745–2750. doi:10.1111/j.1572-0241.2001.04134.x.

Chapter 1
15
[17] S.J. Muñoz, J.E. Heubi, W.F. Balistreri, W.C. Maddrey, Vitamin E deficiency in primary biliary cirrhosis: gastrointestinal malabsorption, frequency and relationship to other lipid-soluble vitamins, Hepatol. Baltim. Md. 9 (1989) 525–531.
[18] B.L. Shneider, J.C. Magee, J.A. Bezerra, B. Haber, S.J. Karpen, T. Raghunathan, P. Rosenthal, K. Schwarz, F.J. Suchy, N. Kerkar, Y. Turmelle, P.F. Whitington, P.R. Robuck, R.J. Sokol, Childhood Liver Disease Research Education Network (ChiLDREN), Efficacy of fat-soluble vitamin supplementation in infants with biliary atresia, Pediatrics. 130 (2012) e607-614. doi:10.1542/peds.2011-1423.
[19] R.A. Jorgensen, K.D. Lindor, J.S. Sartin, N.F. LaRusso, R.H. Wiesner, Serum lipid and fat-soluble vitamin levels in primary sclerosing cholangitis, J. Clin. Gastroenterol. 20 (1995) 215–219.
[20] Y.-M. Shen, J.-F. Wu, H.-Y. Hsu, Y.-H. Ni, M.-H. Chang, Y.-W. Liu, H.-S. Lai, W.-M. Hsu, H.-L. Weng, H.-L. Chen, Oral absorbable fat-soluble vitamin formulation in pediatric patients with cholestasis, J. Pediatr. Gastroenterol. Nutr. 55 (2012) 587–591. doi:10.1097/MPG.0b013e31825c9732.
[21] A. Saeed, R.P.F. Dullaart, T.C.M.A. Schreuder, H. Blokzijl, K.N. Faber, Disturbed Vitamin A Metabolism in Non-Alcoholic Fatty Liver Disease (NAFLD), Nutrients. 10 (2017). doi:10.3390/nu10010029.
[22] Y. Ma, O.V. Belyaeva, P.M. Brown, K. Fujita, K. Valles, S. Karki, Y.S. de Boer, C. Koh, Y. Chen, X. Du, S.K. Handelman, V. Chen, E.K. Speliotes, C. Nestlerode, E. Thomas, D.E. Kleiner, J.M. Zmuda, A.J. Sanyal, NASH CRN, N.Y. Kedishvili, T.J. Liang, Y. Rotman, HSD17B13 is a Hepatic Retinol Dehydrogenase Associated with Histological Features of Non-Alcoholic Fatty Liver Disease, Hepatol. Baltim. Md. (2018). doi:10.1002/hep.30350.
[23] J.A. Del Campo, R. Gallego-Durán, P. Gallego, L. Grande, Genetic and Epigenetic Regulation in Nonalcoholic Fatty Liver Disease (NAFLD), Int. J. Mol. Sci. 19 (2018). doi:10.3390/ijms19030911.
[24] A.A. Ashla, Y. Hoshikawa, H. Tsuchiya, K. Hashiguchi, M. Enjoji, M. Nakamuta, A. Taketomi, Y. Maehara, K. Shomori, A. Kurimasa, I. Hisatome, H. Ito, G. Shiota, Genetic analysis of expression profile involved in retinoid metabolism in non-alcoholic fatty liver disease, Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 40 (2010) 594–604. doi:10.1111/j.1872-034X.2010.00646.x.

Introduction and aim of the thesis
16

Chapter 2
The interrelationship between bile
acid and vitamin A homeostasis
Ali Saeed1, 3, Mark Hoekstra1, Martijn Oscar Hoeke1, Janette
Heegsma1, 2, Klaas Nico Faber1*
1Department of Gastroenterology and Hepatology, 2Laboratory Medicine, Center for
Liver, Digestive, and Metabolic Diseases, University Medical Center Groningen,
University of Groningen, Groningen, The Netherlands
3Institute of Molecular biology & Bio-technology, Bahauddin Zakariya University,
Multan, Pakistan
Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids; 1862,
(5), 2017, 496–512

Bile acid and Vitamin A homeostasis intertwined
18
ABSTRACT
Vitamin A is a fat-soluble vitamin important for vision, reproduction, embryonic
development, cell differentiation, epithelial barrier function and adequate immune
responses. Efficient absorption of dietary vitamin A depends on the fat-solubilizing
properties of bile acids. Bile acids are synthesized in the liver and maintained in an
enterohepatic circulation. The liver is also the main storage site for vitamin A in the
mammalian body, where an intimate collaboration between hepatocytes and hepatic
stellate cells leads to the accumulation of retinyl esters in large cytoplasmic lipid
droplet hepatic stellate cells. Chronic liver diseases are often characterized by
disturbed bile acid and vitamin A homeostasis, where bile production is impaired and
hepatic stellate cells lose their vitamin A in a transdifferentiation process to
myofibroblasts, cells that produce excessive extracellular matrix proteins leading to
fibrosis. Chronic liver diseases thus may lead to vitamin A deficiency. Recent data
reveal an intricate crosstalk between vitamin A metabolites and bile acids, in part via
the Retinoic Acid Receptor (RAR), Retinoid X Receptor (RXR) and the Farnesoid X
Receptor (FXR), in maintaining vitamin A and bile acid homeostasis. Here, we
provide an overview of the various levels of “communication” between vitamin A
metabolites and bile acids and its relevance for the treatment of chronic liver
diseases.

Chapter 2
19
2.1. INTRODUCTION
Efficient absorption of fat-soluble nutrients in the intestine requires the action of bile
acids. Bile acids are synthesized in the liver and actively secreted into bile. Bile is
collected in the gallbladder, which contracts upon intake of a meal and releases bile
in the proximal small intestine. Bile acids form mixed micelles with phospholipids and
these structures incorporate fat-soluble nutrients to allow their absorption in the
intestine. Simultaneously, bile acid-phospholipid micelles also carry fat-soluble
metabolites, like cholesterol, and toxins that need to be secreted from the body. One
group of nutrients that depend on bile acids for their efficient absorption are the fat-
soluble vitamins A, D, E and K. In contrast to water-soluble vitamins (B, C), the fat-
soluble vitamins can be stored in various tissues to buffer periods of low intake.
Vitamin A is predominantly stored in the liver and humans can maintain adequate
levels of serum retinol for months to years even if intake is minimal. Still vitamin A
deficiency is the most common micronutrient deficiency in the world, particularly in
many third-world countries because intake is too low. Vitamin A deficiency is also a
common condition in patients with liver disease, especially if this includes impairment
in bile flow, e.g. cholestasis. Not only is vitamin A uptake affected under cholestatic
conditions, but chronic liver injury also leads to rapid loss of hepatic vitamin A stores
that disappear from hepatic stellate cells when they transdifferentiate to
myofibroblasts that leads to liver fibrosis. Bile acids and vitamin A-metabolites, in
particular retinoic acids, are high-affinity ligands for the transcription factors
Farnesoid X Receptor (FXR), Retinoid X Receptor (RXR) and Retinoic Acid Receptor
(RAR), which act, in part as obligate partners in regulating bile acid, lipid and glucose
metabolism. There is a wealth of information and excellent reviews that specifically
focus on the function, metabolism and signaling functions of vitamin A-metabolites,
e.g. retinoic acids, on the one hand or on bile acids on the other hand [1–3]. By no
means, this review can cover all the details of the physiological actions of these
molecules. Here, we aim to provide an overview of how bile acid and vitamin A
metabolism are interrelated and may have implications for the treatment of (chronic)
liver diseases.
2.2. Function of vitamin A and its active metabolites
The term “vitamin A” is a generic descriptor for compounds that have the biological
activity of retinol or its metabolic products. Vitamin A-derivatives fulfil numerous
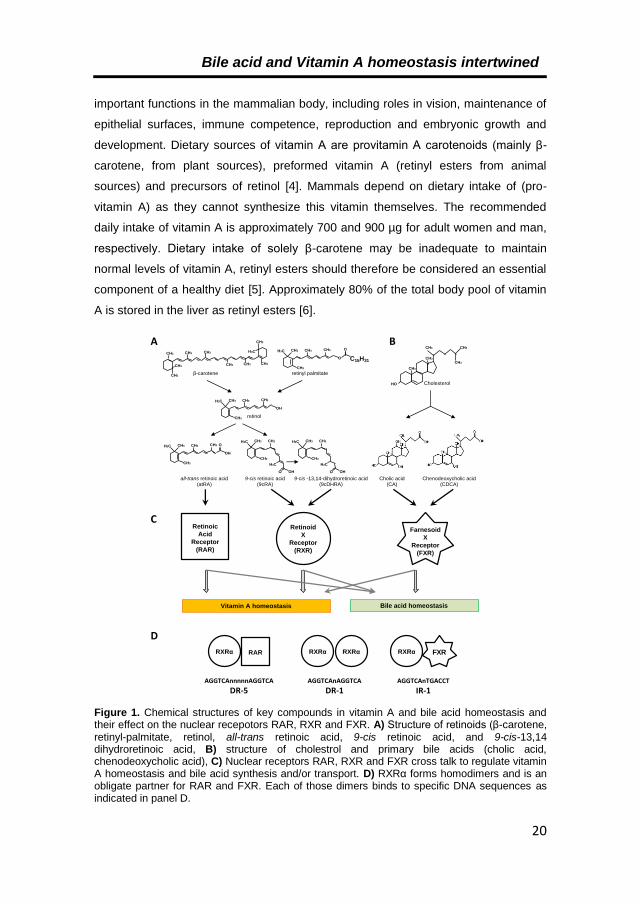
Bile acid and Vitamin A homeostasis intertwined
20
important functions in the mammalian body, including roles in vision, maintenance of
epithelial surfaces, immune competence, reproduction and embryonic growth and
development. Dietary sources of vitamin A are provitamin A carotenoids (mainly β-
carotene, from plant sources), preformed vitamin A (retinyl esters from animal
sources) and precursors of retinol [4]. Mammals depend on dietary intake of (pro-
vitamin A) as they cannot synthesize this vitamin themselves. The recommended
daily intake of vitamin A is approximately 700 and 900 µg for adult women and man,
respectively. Dietary intake of solely β-carotene may be inadequate to maintain
normal levels of vitamin A, retinyl esters should therefore be considered an essential
component of a healthy diet [5]. Approximately 80% of the total body pool of vitamin
A is stored in the liver as retinyl esters [6].
Figure 1. Chemical structures of key compounds in vitamin A and bile acid homeostasis and their effect on the nuclear recepotors RAR, RXR and FXR. A) Structure of retinoids (β-carotene, retinyl-palmitate, retinol, all-trans retinoic acid, 9-cis retinoic acid, and 9-cis-13,14 dihydroretinoic acid, B) structure of cholestrol and primary bile acids (cholic acid, chenodeoxycholic acid), C) Nuclear receptors RAR, RXR and FXR cross talk to regulate vitamin A homeostasis and bile acid synthesis and/or transport. D) RXRα forms homodimers and is an obligate partner for RAR and FXR. Each of those dimers binds to specific DNA sequences as indicated in panel D.
all-trans retinoic acid(atRA)
9-cis retinoic acid(9cRA)
Chenodeoxycholic acid(CDCA)
Cholic acid(CA)
Cholesterol
β-carotene retinyl palmitate
Bile acid homeostasisVitamin A homeostasis
retinol
A B
C
D
CH3
HO
CH3
CH3 CH3
CH3
CH3 CH3
CH3 CH3 CH3
H3C
CH3
CH3
CH3
CH3
CH3 CH3
CH3
H3C CH3
OH
C15H31
CH3 CH3
CH3
H3C CH3
O
O
CH3 CH3
CH3
H3C CH3
OH
OCH3
CH3
H3C CH3
H3C
OH
Retinoic
Acid
Receptor
(RAR)
Retinoid
X
Receptor
(RXR)
Farnesoid
X
Receptor
(FXR)
RARRXRα
AGGTCAnnnnnAGGTCA
DR-5
RXRα RXRα
AGGTCAnAGGTCA
DR-1
FXRRXRα
AGGTCAnTGACCT
IR-1
CH3
CH3
H3C CH3
H3C
OH
9-cis -13,14-dihydroretinoic acid(9cDHRA)

Chapter 2
21
Provitamin A carotenoids like -carotene, as well as non-provitamin A carotenoids
lycopene are potent antioxidants [7], but vitamin A itself is not. A well-known function
of vitamin A is its role in the visual cycle by photoisomerization. Rhodopsin, the visual
pigment of the rod photoreceptor cell, contains 11-cis retinal as its light-sensitive
cofactor. Light activation is achieved by 11-cis to all-trans isomerization, followed by
the release of all-trans retinaldehyde [8].
Most biological functions of vitamin A, however, involve the activation of ligand-
dependent transcription factors. This hormonal function of vitamin A gained
tremendous scientific interest with the discovery of two vitamin A receptors that are
members of the nuclear receptor superfamily [9]. All-trans retinoic acid (atRA) is a
high-affinity ligand for RAR, while 9-cis retinoic acid (9cRA) and 9-cis-13,14
dihydroretinoic acid (9cDHRA) are high-affinity ligands for RXR (Figure 1A). These
nuclear receptors will be discussed later.

Bile acid and Vitamin A homeostasis intertwined
22
Figure 2. A simplified scheme of the biosynthesis and enterohepatic cycling of bile acids. Only the proteins relevant for this review are included. Bile acid synthesis starts at the top of the figure by the conversion of cholesterol to the primary bile acids cholic acid and chenodeoxycholic acid, both either Glycine- or Taurine-conjugated. These bile acids are transported to the bile and small intestine where they maintain fat-soluble compounds, including (pro-)vitamin A, in solution. At the terminal ileum, bile acids are absorbed and transported back to the liver. Thick arrows show the principle pathway (and involved proteins) for bile salt synthesis and transport under normal conditions. Dashed lines indicate transporters that are involved in bile acid transport under cholestatic conditons. For each gene involved in bile acid synthesis or transport it is indicated whether its expression is regulated (directly or indirectly) by RAR, RXR and/or FXR. FXR-mediated suppression of transcription is regulated either via SHP or FGF15/19, as detailed in the main text. Abbreviations can be found seperately in the “list of abbreviations”. More details about the regulations of the individual genes, including references, can be found in Supplementary Table 2S.
Bile salts
Gut Lumen
Hepatocyte
Blood
Enterocyte
ASBT
Bile salts
Bile salts-IBABP
Bile salts
Bile salts
Cholesterol
Bile salts
G/T-Cholic acid G/T-Chenodeoxycholic acid
CYP8B1
BSEP
CYP7A1
MRP2
MDR1
MDR3
CYP27A1
7α-hydroxycholesterol
Bile salts
MRP3 OSTα OSTβ
Bile salts
Bile salts
OATPsNTCP
7α-hydroxy-4-cholesten-3-one
27α-hydroxycholesterol
Alternative pathwayAcidic pathway
OSTβOSTαMRP3
Bile Duct
Bile salts
BACS
BAAT
InduceSuppress
RXR
RAR
FXR
Legend
Phospholipids
BACS
BAAT

Chapter 2
23
2.3. Bile acid synthesis and enterohepatic circulation
Bile aids in the digestion and absorption of nutrients, and is a main route for the
excretion of waste products via the intestine. Major components of bile are bile salts,
phospholipids, cholesterol, bilirubin, alkaline hydrogen carbonate ions (HCO3-) and
water. Bile salts are the components in the bile that give it its fat-emulsifying
properties. Bile salts are synthesized by hepatocytes using cholesterol as substrate
[10,11]. Humans convert approximately 500 mg of cholesterol into primary bile acids
every day, which is an important way to eliminate cholesterol from the body (see for a
review [12]). The liver secretes bile via the bile ducts and the gall bladder into the
proximal small intestinal lumen (duodenum). Here, bile fulfils its function in the
digestion and absorption of fats and fat-soluble dietary compounds. In the terminal
ileum, bile salts are reabsorbed and transported back to the liver via the portal vein.
In human, bile salts shuttle between liver and intestine, a process called
enterohepatic circulation, about 6 to 10 times per day [13]. Enterohepatic cycling of
bile salts is a very efficient process as only 5% of the bile salts are lost in the faeces.
De novo synthesis of bile salts in the liver compensates for this loss and maintains a
balanced amount of bile salts in the enterohepatic cycle [10,11] (Figure 2).
Bile acid synthesis produces two main types of primary bile acids: cholic acid (CA)
and chenodeoxycholic acid (CDCA) (Figure 1B). These are conjugated to either
glycine or taurine, yielding the bile salts taurocholic acid (TCA), glycocholic acid
(GCA), taurochenodeoxycholic acid (TCDCA) and glycochenodeoxycholic acid
(GCDCA) [13].
Cholesterol is oxidized to bile acids through a cascade of enzymatic conversions
involving at least 13 different enzymes (see for a review [13]). Although the first steps
of this conversion may take place at extra-hepatic locations, the production of the end
products, e.g. bile salts, is restricted to the hepatocytes in the liver. Two enzymes,
cholesterol 7-alpha-hydroxylase (CYP7A1) and sterol 12-alpha-hydrolase (CYP8B1)
are of particular relevance. CYP7A1 is considered the rate-limiting enzyme in bile
acid synthesis [14], while CYP8B1 drives bile acid synthesis towards CA instead of
CDCA. CDCA is considered a hydrophobic bile acid while CA is at the hydrophilic
end and CYP8B1 thus has a major impact on the relative hydrophobicity of the BA
pool [15]. The final step in bile acid synthesis is the conjugation of taurine or glycine
and is exclusively performed by the peroxisomal enzyme bile acid coenzyme A:
amino acid N-acyltransferase (BAAT) [16].

Bile acid and Vitamin A homeostasis intertwined
24
Lipid membranes are impermeable for conjugated bile acids and have low
permeability for unconjugated bile acids. Therefore, the efficient uptake and secretion
of bile acids across cellular membranes is mainly dependent on bile salt transporters.
Excretion of bile salts into the bile canaliculus occurs against a steep concentration
gradient. Therefore, bile salt excretion is dependent on active transport. Bile salts are
excreted from the hepatocytes at the apical membrane by the ATP Binding Cassette
(ABC)-transporter Bile Salt Export Pump (BSEP/ABCB11). BSEP is the main bile
acid exporter in hepatocytes and excretes monovalent conjugated bile salts only.
Sulphoronidated or glucoronidated bile salts are excreted into the bile ducts by the
Multidrug Resistance-associated Protein 2 (MRP2/ABCC2). MRP2 is not a dedicated
bile acid transporter like BSEP as its primary substrate is bilirubin and also transports
organic anions, organic cations, glutathione and glutathione conjugates.
Phospholipids are excreted by MDR3 (ABCB4; the rodent homolog is called MDR2)
[17,18]. Bile acid and phospholipids form mixed micelles that are the actual carriers
of hydrophobic compounds through the digestive tract, including fat-soluble vitamins,
like vitamin A.
At the terminal ileum, bile salts are absorbed into enterocytes by the apical sodium-
dependent bile salt transporter (ASBT/SLC10A2) [19]. Inside enterocytes, bile salts
are bound to the Ileal Bile Acid Binding Protein (I-BABP) and transported to the
basolateral membrane where they are excreted to the circulation by the organic
solute transporter dimer α/β (OSTα/β). Some amount of bile acids “spill” into the
colon, where most of them are deconjugated by resident bacteria and converted into
secondary bile acids, including lithocholic acid (LCA) and deoxycholic acid (DCA)
[20]. A significantly amount of secondary bile salts are absorbed to the circulation by
processes that are not well-characterized. The mixture of conjugated and
unconjugated primary and secondary bile acids cycles back to the liver via the portal
track. The conjugated bile salts are taken up by the hepatocytes through basolateral
bile salts transporters, in particular by the sodium/taurocholate co-transporting
polypeptide (NTCP/SLC10A1) and organic anion transporting polypeptides OATPs
(OATP1, OATP4) [12]. Mouse Oatp1b2 (homologue of human OATP1B1/1B3) was
shown to specifically transport unconjugated bile salts into hepatocytes [21]. Hepatic
absorption completes the enterohepatic circulation of bile salts, which can enter a
next round of cycling either directly (for conjugated bile acids) or after reconjugation
(for unconjugated bile acids) to glycine or taurine.

Chapter 2
25
2.4. Vitamin A uptake, transport, storage and metabolism.
A simplified scheme showing the various cell types, intracellular-, extracellular-,
transmembrane-transporters and enzymes involved in (pro)vitamin A uptake,
transport, storage and metabolism that are relevant for this review is depicted in
Figure 3. Factors that are under transcriptional control of RAR, RXR or FXR and/or
their ligands are also indicated. The reader is referred to excellent reviews for more
detailed information about specific pathways in this scheme [22–29].
Uptake: Various forms of –precursors of- vitamin A are entering the digestive tract
depending on the composition of the diet and are predominantly absorbed in the
proximal part of the small intestine. Plant carotenoids were thought to be absorbed
into the intestinal epithelium by passive diffusion after being incorporated into
micelles that mainly consist of bile salts and dietary fats. However, recent studies
suggest that several receptors may facilitate uptake of carotenoids, most prominently
scavenger receptor class B member 1 (SR-BI), but also including Cluster
Determinant 36 (CD36) and Niemann-Pick C1-Like 1 (NPC1L1) [30–34]. All-trans
retinoic acid supplementation induced the expression of the intestinal transcription
factor ISX, which suppressed the expression of SR-BI, and was shown to reduce the
absorption of β-carotene. Conversely, SR-BI expression is enhanced in vitamin A
deficient conditions to promote β-carotene absorption from the intestine [31]. CD36 is
a ubiquitous scavenger receptor with broad substrate specificity and is present in the
brush boarder of the duodenum and jejunum. CD36 deficiency impairs lymph
secretion in mice. CD36 facilitates intestinal uptake of different carotenoids, including
β-carotene, lycopene and lutein. Further, CD36 localizes with caveolin-1 in lipid rafts,
which suggests its possible involvement into lipid micronutrient uptake (see review
[32]). Additionally, NPC1L1 is a cholesterol transporter that can also facilitate the
uptake of α/β carotene, β-cryptoxanthin, lycopene, lutein and zeaxanthin [33,34].
Retinyl esters from animal sources are first converted to retinol by retinyl ester
hydrolases (REHs) within the intestinal lumen, after which they are absorbed by
enterocytes [35]. Several REHs are implicated in the luminal hydrolysis of retinyl
esters, including pancreatic triglyceride lipase (PTL), carboxyl ester lipase (CEL) and
the intestinal brush border membrane enzyme phospholipase B (PLB) [36], where
PTL seems the most important REH in the intestinal lumen [37]. The enzymatic
activity of both PTL and CEL is enhanced by bile acids. Administration of bile salt
sequestering agents to humans lowers the total carotenoid levels in serum [38], while

Bile acid and Vitamin A homeostasis intertwined
26
administration of taurocholic acid enhanced vitamin A absorption in rats [39], further
underscoring the role of bile salts in vitamin A absorption. Absorption of retinol into
the enterocyte is still a largely uncharacterized process. While passive diffusion into
enterocytes is assumed, this seems only sufficient at supra-physiological
concentrations. It is likely that a saturable carrier-mediated process is involved, but
the identity of such intestinal retinol carrier is not established yet.
Intracellular transport: Beta-carotene can be converted to retinoids inside the
enterocyte [35]. Symmetric cleavage of one molecule β-carotene by beta-carotene
15,15'-monooxygenase 1 (BCMO1) yields two molecules of retinaldehyde [40].
Subsequently, retinaldehyde is reduced to retinol by retinaldehyde reductases (RRD).
Several enzymes are capable of catalyzing this conversion, including members of the
short- and medium-chain alcohol dehydrogenase/reductase superfamily that will be
discussed later. Inside enterocytes, free retinol is bound by the abundantly present
cellular retinol-binding protein type II (CRBP2). Next, most of the retinol is re-
esterified to saturated long-chain fatty acids, mainly palmitic acid. Binding of retinol to
CRBP2 facilitates the esterification of retinol by lecithin:retinol acyl tranferase (LRAT)
or diacylglycerol O-acyltransferase 1 (DGAT1) (also called acyl CoA:retinol acyl
transferase; ARAT). Uncleaved carotenoids and newly-synthesized retinyl esters are
packaged into chylomicrons (CMs) and secreted to the lymphatic system [30,41].
Chylomicrons are heterogeneously-sized particles that consist of a core of
triglycerides and cholesterol-esters and a monolayer of phospholipids, cholesterol
and proteins. CMs are formed in the Golgi and are excreted via exocytotic vesicles
from the enterocyte. CM excretion is impaired in the absence of bile salts [42]. In a
mouse model for CM retention disease it was observed that the absorption of fat,
vitamin A and E was severely impaired and significantly reduced growth rates [41],
underscoring the importance of CM in the efficient absorption of fat and fat-soluble
vitamins, such as vitamin A.
Although most retinoids leave the enterocyte as retinyl esters, but retinol can also be
released directly into the portal circulation [43], which may be facilitated by ABCA1
[30].
Storage and distribution: CMs distribute nutrients to peripheral tissues and the CM
remnants, which still contain most of the retinyl esters, are subsequently cleared by
the hepatocyte. CM remnants uptake is a complex process. Low-Density Lipoprotein
Receptor (LDLR) has a high affinity for apoE-rich CM remnants and mediates their

Chapter 2
27
internalization. Syndecan-1 (SDC1) is a heparin sulfate proteoglycan that also
facilitates apoE binding and may be a back-up system LDLR levels are low. Hepatic
lipases also facilitate the sequestration of CMs for uptake, and apoB in CM remnants
can increase this process. In addition, also an LDLR-related protein (LRP) may be
involved in CM remnant uptake. The relative contributions of all these proteins for
uptake of CM remnants, and thereby vitamin A uptake in the liver, requires further
research [44–46].
Within the hepatocyte, retinyl esters are again hydrolyzed to retinol by REHs, which
includes carboxylesterase ES-10 that is highly expressed in rat liver [47], but likely
also other REHs. Retinol is efficiently bound by apo-CRBP1 which is present in molar
excess of retinol in hepatocytes [48,49]. Transfer of retinol to endoplasmic reticulum-
localized RBP4 induces complex formation with transthyretin (TTR) and secretion of
holo-RBP4-TTR to the circulation [50,51]. Retinol-binding is crucial for efficient
secretion of RBP4, as serum levels are significantly reduced and strong accumulation
in the ER is observed in hepatocytes under vitamin A-deficient conditions [52].
Supplementation of retinol tot VAD rats induces release of RBP4 from hepatocytes
within minutes [52]. Approximately 95 % of plasma retinol-RBP4 is complexed with
TTR in a 1:1 ratio. This interaction reduces glomerular filtration of retinol [4,53].
Uptake of retinol in extrahepatic tissues with a high retinoid demand is facilitated by
“Stimulated by Retinoic Acid gene 6 homolog” (STRA6), an integral membrane
protein containing an extracellular RBP4-binding domain and 9 transmembrane
domains that form a channel for retinol to enter the cell [54]. The tissue localization of
STRA6 has been studied previously. STRA6 is expressed during embryonic
development and in the adult mouse brain, testis, female genital tract, kidney, and at
lower quantities in spleen, heart and lung [55,56]. Fitting its function, STRA6 is also
highly expressed in the retinal pigment epithelium (RPE) of the eye. The retinol-
RBP4-TTR complex dissociates at STRA6 and retinol is taken up by the cell, while
free RBP4 in the circulation is catabolized in the kidney. STRA6 is virtually absent in
the liver. Recently, though, a second receptor for RBP4 has been identified, RBP4
receptor 2 (RBPR2) that is highly expressed in the liver, as well as in the small
intestine, colon and spleen [57]. RBPR2 is structurally related to STRA6 and shows
highly similar retinol uptake characteristics, which is stimulated by RBP4 and TTR.
RBPR2 expression is inversely correlated with hepatic retinoid stores. Inline, retinol
and atRA strongly suppress its expression. Though it is suggested by the authors

Bile acid and Vitamin A homeostasis intertwined
28
that RBPR2 is most likely expressed in hepatocytes, this still requires confirmation
through cell type-specific analyses.
For storage in the liver, retinol is directed to the hepatic stellate cell. Remarkably,
how retinol is getting into the stellate cells is still unknown, but upon entering it is
immediately captured by CRBP1 and subsequently esterified to long chain fatty
acids, again predominantly palmitate, for storage in cytoplasmic lipid droplets.
Retinyl-esters make up 30-50% of the lipid content of the lipid droplets in stellate cells
[6]. Although the mechanism for retinol storage in, and mobilization from, stellate
cells has not been fully elucidated yet, it is clear that CRBP1 is required for shuttling
retinol between different liver cell types. The involvement and recycling of RBP4 has
been ruled out, since mice lacking Rbp4 accumulate excess of vitamin A in the liver
[58–61]. While still able to store vitamin A, these mice were unable to mobilize it to
plasma. Moreover, extrahepatic expression of RBP4 does not restore vitamin A
mobilization in these animals, indicating that circulating RBP4 is not re-used by the
liver. STRA6 is not expressed in the liver, ruling out a role of this receptor in hepatic
storage of retinoids [59,61]. Once inside hepatic stellate cells, retinol is esterified to
retinyl esters by LRAT and possibly also DGAT1, although the latter is controversial
as Lrat-/- mice do not store retinyl esters in the liver [62], even though stellate cells
from Lrat-/- mice are still able to synthesize retinyl esters [63]. Retinyl ester
hydrolases (REHs) are required to mobilize retinol from the lipid droplets in hepatic
stellate cells, a process that is essential to supply retinol to extrahepatic tissues [24].
Several enzymes have been shown to contain REH activity in hepatic stellate cells,
including ES-10, LpL, PLRP2, hormone sensitive lipase (HSL) [64,65], adipose
triglyceride lipase (ATGL) and patatin-like phospholipase domain-containing 3
(PNPLA3) [66–68], but their relative contribution to retinol production in HSC remains
to be determined. Pharmacological inhibition of ATGL leads to an increase of retinyl
esters in cultured mouse HSC [66], but hepatic retinyl ester stores and serum retinol
levels were not changed in Atgl-ko mice, implying a role for (an)other REH(s) in HSC
also [66]. Interestingly, PNPLA3 was recently shown to contain retinyl-palmitate
lipase activity in HSC [67] and the PNPLA3-I148M variant, which is the most
pronounced genetic factor associated with alcoholic liver disease (ALD) [69] and
nonalcoholic fatty liver disease (NAFLD) [70], is associated with reduced levels of
circulating retinol levels [68] and increased hepatic retinyl-palmitate storage [71]. In
line with these observations, the retinyl-palmitate lipase activity of the I148M variant

Chapter 2
29
appears markedly reduced [67]. While this suggests a prominent role of PNPLA3 in
mobilizing retinol from hepatic stores, a direct role of impaired retinol mobilization
from hepatic stellate cells in NAFLD remains elusive though, since PNPLA3 is also
expressed in hepatocytes and the loss of function (I148M variant) also leads to
triglyceride accumulation in hepatocytes [72]. A major unsolved issue is how retinol
that is liberated from the retinyl ester-stores actually is released from the HSC and
transferred to circulating RBP4.
Under normal (vitamin A sufficient) conditions, most of the retinyl esters absorbed
from the chylomicron remnants are transferred as retinol to the stellate cells, where
up to 80% of the body supply of vitamin A is stored [4]. Smaller amounts are also
stored in lipid droplets in the hepatocytes as well as in extrahepatic organs and
tissues, such as the eye, lung, adipose tissue, kidneys, small intestine, adrenal gland,
lung, testis, uterus, bone marrow, thymus, skin and spleen [6,48,73–76]. Extrahepatic
storage sites of retinyl esters may provide a local supply of vitamin A to tissues with a
high demand, such as the retina. The importance of extrahepatic vitamin A pools is
demonstrated by the observation that storage of retinyl esters in retinal pigment
epithelial cells is prerequisite for normal visual function [75]. The stores of vitamin A
are sufficient to maintain a steady physiological concentration above 2 µM retinol in
plasma in humans (and 1-2 µM in rodents), despite strong fluctuations in daily intake
of vitamin A [75].

Bile acid and Vitamin A homeostasis intertwined
30
Figure 3. A simplified scheme of vitamin A uptake, transport, storage and metabolism. Only the proteins relevant for this review are included. Absorption of dietary (pro-)vitamin A starts at the botttom of the figure in the small intestine. After inclusion into chylomicrons most of the retinyl-esters are transported to the liver for storage and/or redistribution. After uptake by the hepatocytes, retinol esters are hydrolyzed and retinol is transported to hepatic stellate cells that store most of the body reserves of vitamin A as retinyl-esters in large cytoplasmic lipid droplets. Controled release of retinol from those hepatic vitamin A stores maintains stable blood retinol levels even when dietary vitamin A intake is variable. Retinol binding protein 4 (RBP4) is the main factor in the circulation to transports retinol to peripheral tissues where it is metaboliized to high affinity ligands for RAR and RXR and ultimately catabolized. For each gene involved in vitamin A uptake, transport, storage and metabolism it is indicated whether its expression is regulated (directly or indirectly) by ligands of RAR, RXR and/or FXR. RDH10 and DHRS3 activate each other through a physical interaction. RDH10 is considered the key retinol dehydrogenase, at least in embryogenesis. Other enzymes that may act also as retinol dehydrogenases are included in a smaller letter type. Abbreviations can be found seperately in the “list of abbreviations”. More details about the regulations of the individual genes, including references, can be found in Supplementary Table S1.
α/β-carotene Retinyl-ester RetinolPTL,CEL
PLB
Gut Lumen
TTR-RBP4-Retinol
RBP4TTR
Cellsin target tissue
RARα/β/g
CYP26A1,26B1CYP2C8,2C9,26C1
CYP3A4/5
CRABP1/2-all trans-retinoic acid 9cis-retinoic acid-CRABP1/2
RALDH2RALDH3
RDH1
Hepatocyte Stellate cell
Retinyl-ester
Retinol Retinol-CRBP1
Retinyl-ester-CRBP1
LRAT
DGAT1
REH
ATGL
PNPLA3
Blood
Chylomicrons
CM-remnants
Lymph
Retinol-CRBP2Retinyl-ester
Enterocyte
BCMO1β-carotene Retinaldehyde-CRBP2
RRDREH
LRAT
Lipid droplet
apo-CRBP1
LDLR SDC1
REH,CEL
NPC1L1CD36 SR-BI
ISX
ABCA1
Lipases
DGAT1
ER/
Golgi
exocyt?
?
InduceSuppress
RXR
RAR
FXR
Legend
RXRα/β/γ
RALDH1RALDH4
Retinol-CRBP1
Retinaldehyde-CRBP1
CYP2A6,2B6
9cDHRA?
CRBP1-Retinol
apo-CRBP1
apo-CRBP1STRA6
DHRS3RDH10
TTR-RBP4-Retinol
Chylomicrons
ER/Golgi
exocytosis
RDH16
ADH1B
ADH2
ADH4
CYP1A1
CYP1B1
CYP3A4
RBPR2
RBP4TTR
TTR-RBP4-RetinolRBP4TTR
RBPR2

Chapter 2
31
Retinoic acid synthesis and degradation: Retinol is the main circulating form of
vitamin A, which is locally converted to the bioactive metabolites retinaldehyde and
retinoic acid in a two-step process [4]. First, retinol is oxidized to retinaldehyde, which
is considered to be the rate-limiting step for retinoic acid synthesis, but is also
reversible. The second step in which retinaldehyde is oxidized to retinoic acid is
irreversible. It is important to note that various isomers of retinol, including all trans-
retinol and 9 cis-retinol, co-exist in circulation and that they are the specific
precursors for atRA and 9cRA. As several enzymes involved in the RA synthesis
show selectivity towards at-Retinol or 9c-Retinol, these 2 pathways are separately
shown in Figure 3.
Many different enzymes have been shown to be able to convert retinol to
retinaldehyde, at least in in vitro assays. These enzymes belong to 3 different
enzyme families: 1) the (membrane-associated) short chain
dehydrogenases/reductases (SDR) [4,77] 2) the cytosolic medium-chain alcohol
dehydrogenases (ADH), and 3) various cytochrome P450s. However, recent studies
have identified RDH10, a member of the SDR16C subfamily, as an essential factor in
retinol-to-retinaldehyde conversion, at least during embryogenesis. Knock-out of the
gene encoding RDH10 leads to embryonic lethality and causes severe
developmental malformations due to insufficient RA production [78–80], a phenotype
that could be rescued by supplementation with at-Retinaldehyde or RA [79]. While
underscoring the central role of RDH10 during embryogenesis, a similar vital role for
RDH10 in retinylaldehyde production in adult tissues remains to be established.
Limited information about tissue-specific expression suggests that RDH10 has a very
restricted tissue distribution in adult mammals, where it was found predominantly in
bovine RPE, with much lower levels in retina and liver and was virtually undetectable
in other tissues, like brain, lung, kidney, pancreas, and skeletal muscle [81,82]. Thus,
other retinol dehydrogenases could still play a physiological role in adult tissues. For
instance, Rdh1-ko mice are viable, but when maintained on a vitamin A-restricted diet
show enhanced retinol levels in liver and kidney, suggesting a role for RDH1 in
retinol-to-retinaldehyde conversion in adult mice [83]. The human orthologue from the
SDR9C subfamily, RDH16, exhibits a liver-specific expression and is able to convert
retinol to retinaldehyde as well, although it shows higher catalytic activity towards 3α-
hydroxysteroids [84]. Human ADH1B, ADH2 and ADH4 show retinol dehydrogenase
activity in vitro, but both ADH inhibitory studies and ADH-null mice have questioned

Bile acid and Vitamin A homeostasis intertwined
32
their relevance in vivo [85]. Similarly, CYP1A1 and 3A4 were shown to convert retinol
to retinaldehyde in human liver, whereas CYP1A1 and CYP1B1 mainly catalyzed this
conversion in extrahepatic tissues. Moreover, CYP2C subfamily members and
CYP2E1 also exhibited some retinol dehydrogenase activity [86]. Thus, although
RDH10 has taken center stage as a key retinol dehydrogenase during
embryogenesis, other SRD family members, ADHs or CYPs may contribute to
retinaldehyde production in specific tissues after birth.
As indicated above, retinol-to-retinaldehyde conversion is reversible. The main
enzyme involved in the reduction of retinaldehyde to retinol is Dehydrogenase
Reductase 3 (DHRS3). DHRS3 has a broad tissue distribution, including human
heart, placenta, lung, liver, kidney and pancreas [87]. Remarkably, DHRS3 requires
the presence of RDH10 to acquire full enzymatic activity. Vice versa, RDH10 is
activated when physically interacting with DHRS3 [88]. Indeed, the transcripts of
RDH10 and DHRS3 colocalized in some of the tissues during embryogenesis, but
this needs to be established in adult tissue still.
In the second step, retinaldehyde is converted to RA in an irreversible reaction.
Multiple enzymes contain the retinaldehyde dehydrogenase (RALDH) activity. Best
characterized are the RALDH1 to 4 that have been shown to be involved in RA
synthesis in vivo [89]. RALDH1 and RALDH2 convert both at-retinaldehyde and 9c-
retinaldehyde to the respective retinoic acids, while RALDH3 appears selective for at-
retinaldehyde and RALDH4 selective for 9c-retinaldehyde [90–92]. RALDH2 and
RALDH3 appear particularly relevant during respectively early and late
embryogenesis [93,94]. RALDH1 is not essential for embryogenesis and is likely to
have role in RA biosynthesis during adulthood. In line, Aldh1-/- mice treated with
retinol show reduced hepatic RA biosynthesis and enhanced serum retinaldehyde
levels [95,96]. Also various enzymes belonging to the human cytochrome P-450
family can convert retinaldehydes to retinoic acids, including CYP1A1, CYP1A2 and
CYP3A4. CYP1A2 shows the highest activity for 9c-retinaldehyde [97].
Although produced locally, RA may act everywhere in the organism. The circulation
contains low levels of atRA (0.2 to 0.7 % of plasma retinol) and contributes to
variable extend to the tissue pool of RA, depending on the tissue/organ. In liver and
brain, the retinoic acid pool originates primarily from the circulation rather than from
local synthesis [98]. RA concentrations in tissue are very low, on average 3-15 µg per
kilogram [4]. As will be described in detail below, 9cRA levels are only detectable in

Chapter 2
33
the pancreas where it stimulates RXRα–mediated transcription [99]. 9cRA may be
further processed to 9-cis-13,14-dihydroretinoic acid (9cDHRA) that is readily
detectable in mouse serum and tissue and also acts as a RXRα ligand [100].
Local levels of RA are the result of an interplay between RA synthesizing, RA binding
and RA catabolizing enzymes. Cellular retinoic acid binding proteins (CRABP1 and
CRABP2) bind to newly synthesize retinoic acid, increase RA metabolism and protect
cells from excessive RA [77]. Overexpression of CRABP1 reduces the sensitivity of
retinoic acid [101]. Retinoic acids are catabolized in a two-step process. In the first
step, phase I enzymes of the CYP450 superfamily catabolize the different isomers of
RA. Retinoic acid-inactivating cytochrome P450s CYP26A1 and 26B1 predominantly
catabolize atRA and hardly cis-RA isomers (9cRA and 13cRA). CYP26A1 is the
predominant form in liver and lung, while other human adult tissues contain more
CYP26B1 [102,103]. AtRA induces expression of these CYPs, thereby controlling its
own degradation [104–107]. A third CYP26C1 has also been identified that can
metabolize both atRA and 9cRA [108]. Additionally, numerous CYP enzymes have
been identified that catabolize 9cRA. Of the CYPs that are dominantly expressed in
the adult human liver, CYP2C8, -2C9 and -3A4 are the major ones involved in 9cRA
catabolism. The efficiency of these CYPs to metabolize 9cRA is higher than that for
atRA, which may explain why the concentrations of 9cRA in vivo are lower compared
to atRA. In the second step, phase II enzymes facilitate the conjugation of phase I
metabolites. All trans-RA and its phase I metabolites 4-oxo-RA, 5,6-epoxy-RA, and 4-
OH-RA were found to be glucuronidated by the human glucuronyl transferase
UGT2B7 [77,109].
2.5. Nuclear receptors activated by retinoic acids and/or bile acids
Bile acids and retinoic acids are potent ligands for specific members of the nuclear
receptor (NR) family of transcription factors. Bile acids, and in particular CDCA,
activate the Farnesoid X Receptor (FXR). AtRA activates the Retinoic Acid Receptor
(RAR), and 9cRA activates the Retinoid X Receptor (RXR) (Figure 1C). While atRA
is readily detectable in rodent and human serum and various tissues and thereby
physiologically relevant for controlling RAR-mediated transcription, 9cRA is not and
its role as endogenous ligand remains therefore controversial [110]. However, the
presence of 9cRA has been firmly established in the mouse pancreas where its
concentration (~20 pmol/g tissue) surpluses that of atRA (~7 pmol/g tissue) and

Bile acid and Vitamin A homeostasis intertwined
34
regulates glucose-stimulated insulin secretion [99]. In the same study, 9cRA
remained undetectable in mouse pancreas, liver and serum using a sensitive
LC/MS/MS assay (levels below the detection limit of 0.05 pmol/g). Thus, 9cRA can
act as an endogenous ligand for RXR at least locally. Moreover, 9cRA may be
present in a specific subset of cells and is diluted out below detection limits when
whole tissue is processed for analysis. It should also be emphasized that retinoic
acids are highly susceptible to light-induced isomerization, temperature-induced
degradation and non-specific oxidation, putting restrictions on sample processing
preceding the quantification. More recently, however, an alternative endogenous
RXR ligand has been identified in the form of the 9cRA metabolite 9-cis-13,14-
dihydroretinoic acid (9cDHRA) [100]. 9cDHRA binds and transactivates RXR, albeit
at lower affinity as compared to 9cRA. 9cDHRA is readily detectable in serum (~120
ng/ml = ~400 nM), liver (135 ng/g = ~450 pmol/g) and brain of wild type mice (~7
ng/g = ~23 pmol/g), while levels of atRA in serum (0.3 ng/ml) were approximately
400-fold lower and 9cRA remained undetectable. As far as we know, it has not been
established yet whether 9cDHCA is also detectable in human serum and/or tissue,
thus it remains to be established whether this 9cRA metabolite could also act as a
genuine endogenous ligand for RXR in human.
The NR superfamily contains 49 members that are divided into seven subfamilies
(NR0 to NR6) based on sequence homology. The nomenclature of nuclear receptors
has been standardized per subfamily [111] and an in depth overview of the seven
subclasses and their members is given by Aranda et al. [112]. RXR-alpha (RXRα)
takes a special place in the NR superfamily as it is an obligate partner of several
NRs, including FXR and RAR (Figure 1D). Those transcription factors become active
as a NR/RXRα heterodimer. NRs have a modular structure consisting of multiple
functional domains. A typical nuclear receptor consists of a variable N-terminal region
(region A/B), a conserved DNA-binding domain (DBD) (region C), a linker (region D),
and a conserved E region that contains the ligand binding domain (LBD). Some
receptors contain also a C-terminal region (F) with unknown function. Isotypes
(alpha, beta, gamma) originate from homologous genes and isoforms (alpha 1, alpha
2) of nuclear receptors are generated via alternative translation initiation sites and/or
alternative mRNA splicing. The ligand-independent transcriptional activation domain
(AF-1) is contained within the A/B region, and the ligand-dependent transactivation
domain (AF-2) is located within the C-terminal portion of the LBD [112].

Chapter 2
35
Nuclear receptors regulate gene transcription by binding to specific DNA sequences,
so-called hormone responsive elements (HREs), in the promoter region of specific
genes. NR/RXRα heterodimers bind to a variety of tandem repeats of the hexamers
AGGTCA or AGTTCA typically spaced by 1 or more base pairs [113,114]. The
orientation of these hexamers, or so-called half-sites, may vary giving rise to a direct
repeat (DR) (AGGTCAnAGGTCA), a palindromic everted repeat (ER)
(TGACCTnAGGTCA) or a palindromic inverted repeat (IR) (AGGTCAnTGACCT). It
should be noted that these HREs are consensus sequences and that the actual
genomic HREs may slightly differ from the consensus.
Gene regulation by NRs is, however, far more complex than just receptor binding to a
responsive element in a promoter. Competition between agonists and antagonists,
RXRα availability, heterodimerization efficiency, cofactor recruitment and NR protein
modification, together determine the ultimate transcriptional efficiency [115].
The Retinoid X Receptor (RXR) was first described in 1990 [116] and later three
isotypes (RXRα/NR2B1, RXRβ/NR2B2, RXRγ/NR2B3 and two isoforms for each
isotype (RXRα1 and RXRα2; RXRβ1 and RXRβ2; RXRγ1 and RXRγ2) were
identified [117]. RXRα is predominantly expressed in liver and to lesser extend in
spleen, muscle, kidney, heart and adrenal gland. RXRγ is mainly found in kidney,
heart, spleen, intestine and adrenal gland. RXRβ is expressed ubiquitously, but
relatively low in intestine and liver [118]. 9cRA is a high-affinity ligand for RXRs, but
so far it has only been detected in the mouse pancreas keeping the question alive
whether other endogenous for RXRs exist [99,119]. 9cDHRA appears to be a good
candidate as physiological RXR ligand, but also unsaturated fatty acids (PUFAs),
including linoleic, oleic acid, linolenic, arachidonic acid, and docosahexaenoic acids,
have also been shown to activate RXRα [119]. Moreover, atRA was originally
reported to be a ligand for RXRα, although nowadays it is considered to be the
natural ligand for RAR [116] (see below). Visa versa, 9cRA has also been reported to
be a ligand for RAR [120]. Homodimeric RXRα interacts with DR-1 sequences [121]
(Figure 1D). More importantly though, RXRα is the obligate heterodimer partner of
most nuclear receptors belonging to the NR1 subfamily, including RARs and FXR. As
such, RXRα is a key factor in a multitude of metabolic processes, including bile salt
homeostasis. In NR/RXRα heterodimers, 2 different ligands play a role in modulating
the transcriptional activity of the protein complex. The presence of an RXRα ligand
may have different effects on the NR/RXRα activity depending on the specific NR

Bile acid and Vitamin A homeostasis intertwined
36
and/or target gene studied and have been subclassified as “permissive” and “non-
permissive” NR/RXRα complexes [112,122]. In permissive NR/RXRα heterodimers,
RXRα is both a structural and functional component, meaning that RXRα ligands
enhance signaling through such NR/RXRα heterodimers. Ligands of RXRα and its
NR partner can independently and synergistically activate gene transcription. This
has been described for PPARs [123,124], LXR [125] and also FXR [126]. In non-
permissive NR/RXRα heterodimers RXRα is merely a structural component of the
heterodimer required for DNA-binding, but not necessarily acting as a receptor.
Moreover, RXRα agonists may even repress expression of the target genes [122].
RXRα is non-permissive in heterodimers with RAR [127], TR [128] and VDR [129].
However, a given NR/RXRα combination is not strictly permissive or non-permissive,
as this also depends on the specific target gene. The FXR/RXRα heterodimer was
originally described as permissive, based on the regulation of IBABP and PLTP
[126,130]. We and others have shown that RXRα acts as a non-permissive partner to
FXR in the regulation of human and mouse BSEP (see below in “Vitamin A regulates
bile acid homeostasis”).
As for RXR, also three isotypes of the retinoic acid receptor exist, -alpha
(RARα/NR1B1), -beta (RARβ/NR1B2) and -gamma (RARγ/NR1B3) [120]. Each
isotype has several isoforms, of which the expression has been extensively studied in
mouse embryonic development. RARα is ubiquitously expressed, while RARβ and
RARγ show a more tissue- and cell type-specific distribution. RARβ is present in the
liver capsule as well as in the epithelium and outer mesenchyme of the intestine.
RARγ is largely absent from the gastrointestinal tract, with the exception of the
squamous epithelium of the stomach [131].
RAR forms heterodimers with RXRα [132]. RAR/RXRα typically interacts with retinoic
acid response elements (RAREs) consisting of a direct repeat of AGGTCA
interspaced by 5 nucleotides (DR-5) [133] (Figure 1D). The main natural ligand of
RAR is atRA. RARs also bind 9cRA, but with lower affinity than atRA [134,135].
FXR (NR1H4) was originally identified in 1995 as a retinoid receptor-interacting
protein (RIP14, [136]) and found to be activated by farnesol [137], retinoic acid and
TTNPB (4-[E-2-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)-1-propenyl]
benzoic acid) [138]. FXR was named after its first identified ligand, although a direct
interaction with any of the above compounds was never established.

Chapter 2
37
A few years later, it was found that actually bile acids are potent physiological
activators of FXR. The strongest activating bile acid is (unconjugated) CDCA [139–
141]. With the discovery of its endogenous ligands FXR was renamed bile acid
receptor (BAR) [139], but the name FXR is still commonly used today. Other
endogenous ligands of FXR include androsterone [142], PUFAs [143] and oxysterol
22(R)-hydroxycholesterol [144]. Synthetic ligands include GW4064 [145] and 6-ethyl-
CDCA (6-ECDCA) [146]. Plant-derived guggulsterone can function as an FXR
agonist [147], but also as an FXR antagonist [148].
FXR is profoundly expressed in tissues that are exposed to bile salts, such as liver
and intestine, but also in kidney and adrenal glands [137,149]. Four isoforms of FXR
have been identified in rodents and humans, designated FXRα1-4, all arising from
one gene. Due to differential translation initiation sites, FXRα3 and FXRα4 contain
an N-terminal extension in comparison to FXRα1 and 2. As a result of differential
transcript splicing, FXRα1 and FXRα3 contain a four amino acid-insert in the hinge
region that is not present in FXRα2 and FXRα4 [150]. The FXR isoforms are
differentially expressed. In adult humans, FXRα1/2 mRNA is predominant in liver and
adrenal gland. Expression of FXRα3/4 mRNA is most abundant in colon, duodenum,
and kidney. FXRα3/4 mRNA levels are generally lower than that of FXRα1/2 [151].
For most transcriptional targets, FXR needs to form a heterodimer with RXRα to be
transcriptionally active [138]. The typical FXR responsive element (FXRE) is an
inverted repeat spaced by one base pair (IR-1), with the consensus sequence
AGGTCAnTGACCT [137] (Figure 1D).
FXR functions go far beyond regulating bile acid synthesis and transport alone, as it
has also been shown to control expression of genes involved in glucose metabolism,
triglyceride metabolism, inflammation, coagulation and the list is still growing [152],
For this review we, however, restrict ourselves to its function in bile acid and vitamin
A metabolism. Also, FXR is not the only NR that is activated by bile acids. The other
two bile acid-sensing NRs are PXR and VDR. These are mainly involved in the
adaptive response to prevent bile acid hepatotoxicity. In addition, CAR is also
activated by increased levels of bile salts. Although CAR is not a bile acid sensor by
itself, bile salts and bilirubin induces nuclear translocation of CAR, thereby enhancing
transcription of CAR target genes. Like PXR and VDR, CAR is involved in the
detoxification of bile salts. These receptors are extensively reviewed by Moore et al.
2006 [150].

Bile acid and Vitamin A homeostasis intertwined
38
2.6. Vitamin A regulates vitamin A homeostasis
Transcription of hundreds of genes is controlled by vitamin A-metabolites, in
particularly through retinoic acids [153]. This list is so long because those ligands act
via RXRs and RARs, but also through the nuclear receptor partners of which RXRα is
an obligate partner [122]. Also the expression of many genes involved in vitamin A
uptake, transport, storage and metabolism are controlled by retinoic acids, though for
many the physical binding of RXR and/or RAR in the promoter regions and/or
identification of the specific RXR or RAR response elements has not been
established yet. In Figure 3, we provide an as complete as possible overview of the
current knowledge of the RXR- and/or RAR-mediated regulation of genes in vitamin
A metabolism. More details and references are given in Supplementary Table S1. In
general terms it can stated that retinoic acids induce the expression of genes
involved in vitamin A uptake (like SR-BI), (cellular) transport (like CRBP1 and 2,
CRABP1 and 2), metabolism (RALDH), storage (LRAT) and catabolism (CYP26A1
and 26B1), the latter self-controlling retinoic acid breakdown. Effects of retinoic acids
may also be tissue-specific, as they have been shown to induce expression of RBP4
in human liver cells [154], but suppress RBP4 expression in mouse adipose tissue
[155]. Similarly, the effect of retinoic acids on circulating levels of RBP4 may be
context dependent as it has been reported that atRA enhances circulating RBP4
levels in normal mice [155], while it reduces circulating RBP4 levels in diabetic mice
[156]. Retinoic acids regulate LRAT expression, which involves RARα, RARβ, RARγ
and RXRα even though no typical DNA binding elements for these transcription
factors were found in the rat Lrat promotor region [157]. In vitamin A-deficient (VAD)
mice, hepatic Lrat expression is strongly suppressed, but not in testis and the
intestine. Hepatic Lrat expression was rapidly induced in VAD mice treated with
retinoic acid, allowing the buildup of vitamin A stores [158]. Activation of hepatic
stellate cells is associated with sharp reductions in RARβ and RXRα levels and
thereby inhibits vitamin A storage via Lrat [159]. Uptake of retinol is facilitated by
STRA6. The STRA6 gene encodes two distinct transcripts (STRA6L and STRA6S),
both of which are induced by all-trans retinoic acid. Levels of both transcripts were
decreased in the brain of VAD mice. In contrast, only STRA6S levels are reduced in
VAD mouse kidneys, while STRA6L levels were highly increased by differential use
of STRA6 promoter [160], also indicating tissue-specific differences in vitamin A-

Chapter 2
39
mediated gene expression as described for RPB4. Very little is known about the
retinoic acid-mediated regulation of retinyl ester hydrolases (REHs).
2.7. Bile acids regulate bile acid homeostasis
Bile acid-activated FXR suppresses bile acid synthesis and cellular uptake of bile
acids, while cellular excretion of bile acids is enhanced. An overview of regulatory
effects of (the ligands of) FXR in bile acid synthesis, transport and absorption are
included in Figure 2. More details and references are given in Supplementary Table
S2. Transcription of ABCB11 (encoding BSEP) and the genes encoding the 2
proteins forming Ostα/β contain IR-1 consensus sequences that bind FXR/RXRα
[161,162]. FXR-mediated induction of these efflux systems prevents potential toxic
accumulation of bile acids in either hepatocytes or enterocytes. Also bile acid
biosynthetic enzymes that are involved in uptake of unconjugated bile acids
(OATP1B3; (216) (re-)conjugation of bile acids (BACS and BAAT; [163], as well is
intracellular transport of bile acids (IBABP) [130] are under positive transcriptional
control of FXR. Suppression of bile acid synthesis and cellular uptake of conjugated
bile acids is an indirect process where the FXR-induced expression of the small
heterodimer partner (SHP/NR0B2) and fibroblast growth factor 19 (FGF19; the rodent
orthologue is called Fgf15) play central roles [164].
The small heterodimer partner (SHP/NR0B2) is an atypical member of the NR-family
lacking a DNA-binding domain. Also a natural ligand for SHP has not been identified.
SHP is a general negative transcription factor of signaling pathways involving nuclear
receptors. SHP represses gene transcription by directly interacting with other NRs,
cofactors or chromatin-modifying enzymes [165]. Bile acid-activated FXR/RXRα
enhances the expression of SHP [166], which in turn represses the expression of
hepatic CYP7A1 [166], CYP8B1 [167], NTCP [168] and ileal ASBT [169], thereby
reducing biosynthesis and cellular import of bile salts.
An alternative way to suppress bile acid synthesis and cellular uptake is mediated by
FGF19/Fgf15 [164]. Bile acids in the intestinal lumen activate ileal FXR/RXRα and
increase expression of FGF19/Fgf15. Ileal FGF15/19 is released in the blood and
signals to the liver via the hepatic fibroblast growth factor receptor 4 (FGFR4), which
leads to phosphorylation of JNK. Activated JNK represses CYP7A1 expression [164].
Additionally, FGF19/FGF15 has also been shown to inhibit ASBT expression [170],
expanding FXR-FGF19/FGF15 signaling to bile acid transport. Thus, the FXR-SHP

Bile acid and Vitamin A homeostasis intertwined
40
and the FXR-FGF19 signaling cascades are both involved in feedback mechanisms
that control bile salt synthesis and transport.
2.8. Vitamin A regulates bile acid homeostasis
Vitamin A metabolites regulate expression of genes involved in bile acid homeostasis
via at least 2 mechanisms; 1) direct control of expression by RXR and/or RAR and 2)
modulating the transcriptional activity of FXR/RXRα in a target gene-specific manner
(Figure 1C). An overview of regulatory effects of (the ligands of) RAR and RXR in
bile acid synthesis, transport and absorption are schematically summarized in Figure
2. More details and references are given in Supplementary Table S2. Functional
RAR/RXRα binding sites that have been identified in the genes encoding the rate-
limiting step in bile salt synthesis CYP7A1 [171], as well as in hepatic (NTCP
[168,172]) and ileal bile acid transporters (ASBT [169], OSTβ [173]) that suggest that
vitamin A promotes the expression of bile acid synthesis and cellular export.
However, both mechanisms that suppress bile acid synthesis, e.g. SHP and
FGF19/Fgf15 are also induced by vitamin A. Direct exposure of human hepatocytes
to atRA strongly suppressed CYP7A1 expression, which was accompanied by a
strong increase in SHP and FGF19 (FGF19 may also be synthesized in the human
liver, in contrast to mouse Fgf15 that is only produced in the intestine). AtRA was
found to activate FXR/RXRα transcriptional activity via RXRα. FXR/RXRα-stimulated
SHP and FGF19 subsequently suppress CYP7A1 expression. Still, also FXR/RXRα-
independent mechanism seem to be involved, as the atRA-mediated suppression of
CYP7A1 persistent in the absence of these factors and a role for atRA-induced
expression of PCG-1 was proposed [174]. In line with these observation, oral intake
of retinyl-palmitate in mice strongly increased ileal Fgf15 (4-fold) and hepatic Shp
expression (6-fold) and was accompanied by almost complete suppression of hepatic
Cyp7A1 expression [175]. Selective ligands for RAR (TTNPB) or RXRα (LG268)
revealed that both RAR and RXRα induce SHP [176], while expression of Fgf15 was
only controlled by RXRα. The vitamin A-mediated induction of mouse Fgf15 was
dependent on the presence of FXR, suggesting that it is mediated through the
FXR/RXRα heterodimer independently of bile acids, a similar observation was made
for human FXR/RXRα [174]. The vitamin-A mediated suppression of Cyp7A1 was so
strong that it could completely reverse the derepression of Cyp7A1 in
cholestyramine-treated mice, which suggest that vitamin A may be of therapeutic use

Chapter 2
41
in diseases characterized by severe bile acid malabsorption. Recently, the vitamin A-
mediated regulation of human FGF19 was also established, however, mechanistically
it appears different from mouse Fgf15 [177]. Within a few hours, 9cRA or atRA
strongly induced human FGF19 expression in intestinal cell lines. This result was
mostly reproduced by selective ligands for RAR (TTNPB), but not RXRα ligands
(methoprene acid), and was independent of FXR. A DR-5 was identified in the
second intron of FGF19 that binds RARγ [177]. Since it was reported earlier that
RARy is almost absent in the gastrointestinal tract [131], a central role for RARy in
controlling FGF19 expression and bile salt homeostasis requires further studies.
Also, the stimulation of FGF19 may not be completely independent of FXR, as 9cRA
also strongly induced FXR mRNA and protein levels and thereby indirectly promotes
FGF19 expression through bile acid-stimulated activity of FXR. Several studies
reported the transcriptional effects of supplementing mice with atRA and consistently
report that this suppresses hepatic Cyp7A1 and SHP, as well as ileal Fgf15
expression [178–180]. Additionally, suppressive effects on the expression of other
genes involved in bile acid synthesis (Cyp8B1, Akr1d1, Baat) as well as transport
(Ntcp/Slc10A1, Bsep/Abcb11) were found, but not studied in detail mechanistically.
While ligands for RXRα may stimulate expression of FXR/RXRα target genes in the
absence of bile acids, as described above for FGF19, and thus considered to be a
permissive partner for FXR, it may also show the opposite effect and act as a non-
permissive partner for FXR. Bile acid-induced expression of human BSEP is strongly
suppressed by 9cRA and synthetic ligands of RXRs [181,182]. Ligand-activation of
RXRα prevents the binding of FXR/RXRα to the typical FXRE, IR-1 in the BSEP
promotor region, as well as the recruitment of FXR/RXRα coactivators. Thus, RXRα-
agonists appear to dampen the response of FXR/RXRα to changing bile acid
concentrations. In line, hepatic expression of Bsep/Abcb11 was enhanced in vitamin
A-deficient mice exposed to cholic acid-feeding [182]. In contrast to bile acid/9cRA-
mediated expression of BSEP/ABCB11, human SHP/NR0B2 is maximally induced
when both ligands are present [176]. Here, the interaction between 9cRA/RXRα and
bile acid/FXR-mediated regulation becomes even more complex. Bile acid/FXR-
mediated activation of the SHP promoter appeared not to occur at the previously
identified IR-1 [176], but at a DNA region that conforms to an LRH-1 binding site.
Surprisingly, the 9cRA-mediated induction of SHP was dependent on the IR-1 and
was independent of bile acids. It remains unclear whether FXR is part of the binding

Bile acid and Vitamin A homeostasis intertwined
42
complex or whether other RXRα-containing heterodimers or homodimers are
involved in the vitamin A-induced expression of human SHP.
Collectively, the regulatory effects of vitamin A on bile acid synthesis and transport
can be interpreted as a control mechanism for dietary uptake of this vitamin: e.g.
sufficient vitamin A metabolite in the gastrointestinal tract suppress bile salt
synthesis, while vitamin A deficiency stimulates bile acid synthesis and export from
the liver to maximize intestinal absorption of vitamin A.
2.9. Bile acids regulate vitamin A homeostasis
So far, little is known about the possible role of FXR/RXRα in the regulation of
vitamin A metabolism. None of the factors involved in vitamin A uptake, transport,
storage, redistribution, retinoic acid synthesis or degradation has been identified or
studied as possible FXR target gene. Recently, however, “retinol metabolism” was
identified as one of the most significantly changed pathways by RNA-Seq analysis of
primary human hepatocytes exposed to the FXR ligand GW4064 [183]. Amongst
others, GW4064-induced changes were observed for expression of RDH16,
CYP1A1, ADH1B (retinol-to-retinaldehyde) and CYP26A1, CYP26B1 (atRA
catabolism). FXR-mediated regulation of ADH1B by endogenous (CDCA) and
synthetic (GW4064) FXR ligands in primary human hepatocytes and HepG2 cells
had been shown before [184]. Especially the effect on CYP26A1 and 26B1 may have
profound effects on RA-mediated signaling in the human liver. Moreover, the
simultaneous effect on several enzymes that harbor retinol dehydrogenase activity,
though not considered as rate-limiting factors individually, may contribute to an
unbalanced RA production in hepatocytes.
Apart from transcriptional effects, bile acids may also control vitamin A metabolism in
a more direct way. The enzyme activity of several retinyl ester hydrolases (REHs) is
activated by bile acids, which promotes the release of retinol from retinyl palmitate in
the small intestine [24]. For instance, carboxyl ester lipase (CEL) is a key enzyme to
facilitate retinol uptake and exhibits a broad substrate specificity, including retinyl
esters, but also cholesterol esters and phospholipids [185–188]. CEL activity is most
potently stimulated by 3 alpha, 7 alpha dihydroxylated bile salts [189]. However, CEL
deficiency does not affect vitamin A uptake, metabolism and distribution in mice as
other gut luminal retinyl ester hydrolases may compensate the loss of this enzyme
[190]. Also PLB can hydrolyze retinyl palmitate and its activity is induced by

Chapter 2
43
deoxycholate and taurocholate [191]. Bile acid-induced retinyl ester hydrolase activity
is not restricted to the intestinal lumen, but also found in the liver and is important for
hepatic uptake and mobilization of vitamin A [186]. Hepatic retinyl palmitate
hydrolase activity is also stimulated by bile acids (sodium cholate) and appeared to
be inhibited by α-tocopherol [186]. Still, there is a lot of redundancy in the total
intestinal and hepatic retinyl ester hydrolase activity as also several bile salt-
independent REHs have been characterized [192].
2.10. Vitamin A deficiency in liver disease, including role of cholestasis and
fibrosis
Liver diseases are often subdivided in acute and chronic forms. Acute liver diseases
are characterized by a rapid loss of functional liver tissue, mainly hepatocytes, as a
result of a toxic insult, such as acute viral hepatitis (A, B, E), toxins (mushroom
Amanita phalloides) or an acetaminophen overdose. Acute treatment is required to
prevent acute liver failure and the ultimate treatment option is liver transplantation to
save the patient. Though the inflammatory conditions may affect the serum retinol
levels in these patients, vitamin A deficiency per se is not associated with these
diseases. A far larger group of liver disease patients that are at risk for developing
vitamin A deficiency is suffering from chronic liver disease, where the liver is
constantly exposed to cell damaging factors, like viruses (HCV), alcohol, lipid
overload or auto-immune reactions. Examples of chronic liver diseases are primary
biliary cholangitis (formerly called primary biliary cirrhosis; PBC), primary sclerosing
cholangitis (PSC), biliary atresia, alcoholic liver disease (ALD), non-alcoholic
steatohepatitis (NASH) and autoimmune hepatitis (AIH). These diseases are
commonly associated with the development of fibrosis, the excessive deposition of
scar tissue in the liver as a result of a continuous healing process in response to the
chronic liver injury. Moreover, most chronic liver diseases are characterized by
chronic or transient episodes of cholestasis, an impairment in bile flow. These 2
processes, cholestasis and fibrosis, make these patients susceptible for developing
fat-soluble vitamin deficiencies, in particular hypovitaminosis A. First, impaired bile
flow from the liver limits the level of bile acids in the intestine and thereby hampers
efficient absorption of vitamin A. Second, the vitamin A-storing hepatic stellate cells
become activated in the injured liver and transdifferentiate from quiescent cells to
highly proliferative myofibroblasts that produce excessive extracellular matrix

Bile acid and Vitamin A homeostasis intertwined
44
proteins, such as collagens and fibronectins, e.g. they are main contributors to the
development of fibrosis in chronic liver diseases. Importantly, hepatic stellate cells
lose their vitamin A-containing lipid droplets in the transdifferentiation process. The
molecular mechanisms leading to the loss of retinoids in activating HSC are not
exactly known. Expression of the main enzyme involved in retinyl ester biosynthesis,
LRAT, is rapidly lost in the transdifferentiation process [193]. Culture-activated rat
HSC selectively loose retinyl esters (>80% during 7 days culture) from lipid droplets
and are replaced by polyunsaturated triacylglycerides (PUFA-TAG) while levels of
cholesterol esters remain the same during the first 7 days in culture [194]. Mouse
HSC were shown to produce and secrete atRA in the first days of culture-activation,
concomitant with a progressive decline in cellular retinol levels and a transient
induction of RALDH2 at day 4 and 7 [195]. Quiescent mouse HSC express already
high levels of RALDH1, which is hardly affected during the culture-activation process
up to 21 days. Similarly, quiescent HSC contain high levels of CRBP1 and CRABP as
well as RA-catabolic enzymes, like Cyp26B1 and Cyp2S1 [196–198]. Still, it is
unclear whether the retinyl-ester stores during HSC transdifferentiation to
myofibroblastst are quantitatively converted to retinol and retinoic acids followed by
RA catabolism or that significant amounts of retinol are transported to the circulation.
Taken together, chronic (cholestatic) liver diseases are characterized by impaired
intestinal absorption and active loss of hepatic pools of vitamin A. Indeed, vitamin A
deficiency is reported for all chronic liver diseases mentioned above. Vitamin A
deficiency is defined as serum retinol levels below 0.7 g/ml (where levels >2 g/ml
are normal for human). The prevalence of vitamin A deficiency varies between
studies because of differences in disease etiology, group size, patient age and the
definition of “deficiency”. Still, vitamin A deficiency is observed in 20-40% of PBC and
PSC patients [199–202] and up to approximately 70% of biliary atresia patients [203].
The latter group suffers from chronic cholestasis and development of fibrosis directly
after birth. Those patients did not get the opportunity to build up hepatic stores of
vitamin A and are therefore extra vulnerable for developing vitamin A deficiency. Also
diseases that are not typically associated with persistent cholestasis, e.g. ALD (20-
95%) [204,205], NASH (10-35%) [206–208], HCV (40-55%) [209,210], AIH (2-100%)
[202,211,212], show a high prevalence (shown between brackets) of patients with
vitamin A deficiency. Moreover, vitamin A deficiency is also associated with
increasing Child-Pugh score which is an indicator of liver disease progression,

Chapter 2
45
especially cirrhosis. In most diseases, vitamin A deficiency co-exists with deficiencies
in other fat-soluble vitamins (D, E, K). Taken together, these studies suggest that
vitamin A deficiency is the most frequent deficiency of all fat-soluble vitamins in
chronic liver diseases.
2.11. Bile acid therapy in liver disease
The only current treatment for cholestatic liver disease is the supplementation of a
relative hydrophilic bile acid, ursodeoxycholic acid (UDCA), originally isolated from
bear bile. However, the therapeutic potential of several new bile acid-derivatives are
under investigation in clinical trials. UDCA may slow down disease progression in
PBC and PSC, but the long-term therapeutic benefits remain uncertain (see for
recent review [213]). Many mechanistic actions have been assigned to UDCA,
including protection against apoptosis, enhancement of the secretory capacity of
hepatocytes and cholangiocytes, strengthening of the biliary HCO3- umbrella and
alleviation of endoplasmic reticulum stress. Also, the original proposal that this
hydrophilic bile acid simply dilutes out toxic hydrophobic bile acids in cholestatic
disease may still contribute to its therapeutic action. UDCA has no proven therapeutic
effect in other chronic (cholestatic) liver diseases. 24-norursodeoxycholic acid
(norUDCA) harbors a shorter side chain compared to UDCA and appears to be rather
resistant to conjugation to glycine and taurine [214]. Unconjugated norUDCA
undergoes “cholehepatic shunting”, e.g. after secretion by hepatocytes it is passively
absorbed by cholangiocytes and directly shuttles back to hepatocytes. This leads to
protective hypersecretion of HCO3-. NorUDCA has been extensively tested in
(animal) models resembling PSC and obstructive cholestasis and showed
remarkable therapeutic effects [215]. No final results from clinical trials with norUDCA
have been reported yet. Several FXR agonists are currently in clinical trials to test
their therapeutic potential in PBC [216], NAFLD/NASH [217] and bile acid diarrhea
[218], including obeticholic acid (OCA, INT-747, 6-ethyl-chenodeoxycholic acid,
OcalivaTM) and PX-102, a non-bile acid compound. In a 12-month phase III trial, daily
treatment with OCA significantly decreased alkaline phosphatase and total bilirubin
levels compared to placebo-treated patients [219]. No effects on (non-invasive
measures of) liver fibrosis were observed. Moreover, pruritus was more common in
OCA-treated patients, which also increased with the OCA dose. In a 72-week phase
III trial, daily OCA treatment improved histological features of NASH compared to

Bile acid and Vitamin A homeostasis intertwined
46
placebo-treated patients [217]. Also in this trial, pruritus was more frequent in the
OCA-treated patients compared to placebo, indicating that this may be directly OCA-
related. Thus, though FXR-ligands show promising effects on liver damage markers
and histology, their long-term benefit and safety need further study.
2.12. Vitamin A therapy in liver disease
Remarkably, monitoring vitamin A status is not generally implicated in the treatment
of chronic cholestatic patients in contrast to the other fat-soluble vitamins, like vitamin
D and K. Patients with PBC, PSC and BA are prone to develop vitamin A deficiency
which can cause severe (secondary) complications. The controversy about vitamin A
therapy in liver disease might be due to complicating factors in determining hepatic
vitamin A status and its cellular toxicity at high concentrations. Plasma vitamin A
(retinol) levels are not always a reliable indicator for hepatic vitamin A status,
because plasma retinol levels are very stable despite fluctuating hepatic retinoid
stores. Only complete exhaustion of hepatic retinol stores will finally lead to declining
retinol levels in plasma. Moreover, inflammation affects hepatic RBP4 synthesis and
the excretion of retinol:RBP4 complexes into plasma causing a drop in plasma retinol
levels [220,221]. Plasma retinol concentrations and RBP4 are inversely correlated
with acute phase response markers like C-Reactive protein [222].
Guidelines for supplementation of fat-soluble vitamins to patients suffering from
chronic liver diseases differ per country. Fat-soluble vitamin supplementation is
recommended in patients suffering from cholestatic liver disease, especially in
neonates and infants, as stores of fat-soluble vitamins are low at birth [223,224].
Vitamin A supplementation is recommended as a single 50.000 IU dose every 15
days for adults and every month for children. However, no clear guidelines exist for
vitamin A supplementation for patients with liver disease in the Netherlands.
Administration of vitamin E, 100-200-400 IU/day, is recommended for all patients with
cholestasis and neurological symptoms of unknown etiology. Vitamin K
malabsorption is rapidly corrected by subcutaneous administering of vitamin K, 10
mg/day for 3 days, followed by long-term oral supplements of vitamin K, 5-10 mg/day
or 10 mg/month subcutaneously [225,226].
When fat-soluble vitamin supplements are given attention should be paid to the route
of administration. Oral supplementation of fat-soluble vitamins to (obstructive)
cholestatic patients may not be as efficient as subcutaneous supplementation, since

Chapter 2
47
fat absorption is disturbed in these patients because of limited bile flow to the
intestine.
Given the multiple functions of vitamin A and it’s metabolites it may very well be that
impaired metabolism and/or depletion of this vitamin may aggravate liver injury in
patients with chronic liver disease, especially with respect to compromised immune
responses. Vitamin A deficiency leads to impaired mucosal barrier function by
reduced mucus production and regenerating capacity of the mucosal epithelial
barriers. Moreover, it is associated with impaired phagocytic activity and respiratory
burst of macrophages [227,228]. Natural killer cells were found to be reduced and
less active upon inflammation in vitamin A deficient rats [229]. Interestingly, vitamin A
deficiency may promote excessive inflammation by reducing IL10 synthesis and
increase production of TNFα and IL12 [230,231]. Also, lymphocyte development is
dependent on the activation of retinoic acid receptors [232]. Vitamin A maintains the
humoral mediated Th2 response by suppressing IL12, TNFα and IFNγ production of
Th1 lymphocytes. As a consequence, vitamin A deficiency leads to an exaggerated
pro inflammatory Th1 response with reduced anti-inflammatory Th2 cytokine
production (IL4, IL10, IL13), which stimulates B-cells.
Recently, studies have been initiated to study the therapeutic effect of atRA in
cholestatic liver disease, as sole therapy or in combination with UDCA [233–235].
Rats underwent bile duct ligation to induce obstructive cholestasis and were treated
daily with atRA (5 mg/kg) and/or UDCA (15 mg/kg) for 14 days by oral gavage. AtRA
treatment alone reduced the bile acid pool size, necrotic liver damage and markers of
inflammation and fibrosis. Overall it was superior to UDCA therapy. Remarkably,
UDCA appeared to enhance the therapeutic effect of atRA [233]. Similar therapeutic
effects of atRA, with or without the cotreatment with UDCA, were also observed in 2
additional rodent models of chronic cholestasis, a-naphthylisothiocyanate (ANIT)-
treated rats and the Mdr2/Abcb4 (-/-) mice [234]. These results formed the basis for a
human pilot study where combination therapy of atRA and UDCA was given to
patients with PSC [235]. Although this was a small study with 15 patients, preliminary
results are promising as serum ALT levels and a serum marker of bile acid
biosynthesis (7a-hydroxy-4-cholesten-3-one; C4) were significantly reduced by atRA
when given as co-treatment with UDCA. Though not significant in this small cohort,
also the primary endpoint, serum ALP levels, showed a trend towards decreased
levels upon atRA treatment. Thus, this first human trial shows promising therapeutic

Bile acid and Vitamin A homeostasis intertwined
48
effects of atRA in PSC even without detailed knowledge about the serum or hepatic
vitamin A status of these patients.
2.13. CONCLUDING REMARKS
Bile acid and vitamin A metabolism are heavily intertwined, extending far beyond the
“simple” requirement of bile acids for the efficient absorption of (pro-)vitamin A in the
intestine. Via nuclear receptors, vitamin A-metabolites directly regulate bile acid
synthesis and transport, while bile acids control expression of genes involved in
vitamin A metabolism. Bile acids in the intestinal lumen enhance the activity of retinyl
ester hydrolases to supply the circulation and peripheral tissues with retinol. The liver
is the central organ controlling bile acid metabolism as well as vitamin A metabolism.
It stores vitamin A in hepatic stellate cells that may maintain stable blood retinol
levels for months to years even when dietary intake is very low. Chronic liver
diseases are associated with vitamin A deficiency. Given the important role of vitamin
A metabolites in immunity and tissue differentiation/regeneration it is very likely that
insufficient levels of vitamin A may aggravate liver damage in chronic liver diseases,
an issue that needs further studies. Bile acids derivatives, being ligands for FXR or
not, as well as retinoic acids show promising therapeutic effects in chronic
(cholestatic) liver diseases, including PBC, PSC and NASH, which may ultimately
lead to a most effective therapeutic outcome using both compounds.

Chapter 2
49
REFERENCES
[1] T. Li, U. Apte, Bile Acid Metabolism and Signaling in Cholestasis, Inflammation, and Cancer, Adv. Pharmacol. San Diego Calif. 74 (2015) 263–302. doi:10.1016/bs.apha.2015.04.003.
[2] S.A. Kliewer, D.J. Mangelsdorf, Bile Acids as Hormones: The FXR-FGF15/19 Pathway, Dig. Dis. Basel Switz. 33 (2015) 327–331. doi:10.1159/000371670.
[3] T. Li, J.Y.L. Chiang, Bile acids as metabolic regulators, Curr. Opin. Gastroenterol. 31 (2015) 159–165. doi:10.1097/MOG.0000000000000156.
[4] R. Blomhoff, H.K. Blomhoff, Overview of retinoid metabolism and function, J. Neurobiol. 66 (2006) 606–630. doi:10.1002/neu.20242.
[5] D. Weber, T. Grune, The contribution of β-carotene to vitamin A supply of humans, Mol. Nutr. Food Res. 56 (2012) 251–258. doi:10.1002/mnfr.201100230.
[6] H. Senoo, Structure and function of hepatic stellate cells, Med. Electron Microsc. Off. J. Clin. Electron Microsc. Soc. Jpn. 37 (2004) 3–15. doi:10.1007/s00795-003-0230-3.
[7] L.M. Hix, S.F. Lockwood, J.S. Bertram, Bioactive carotenoids: potent antioxidants and regulators of gene expression, Redox Rep. Commun. Free Radic. Res. 9 (2004) 181–191. doi:10.1179/135100004225005967.
[8] E. Ritter, M. Elgeti, F.J. Bartl, Activity switches of rhodopsin, Photochem. Photobiol. 84 (2008) 911–920. doi:10.1111/j.1751-1097.2008.00324.x.
[9] M.L. Bonet, J. Ribot, F. Felipe, A. Palou, Vitamin A and the regulation of fat reserves, Cell. Mol. Life Sci. CMLS. 60 (2003) 1311–1321.
[10] M.O. Hoeke, The role of vitamin A in bile acid synthesis and transport and the relevance for cholestatic liver disease, Doctoral Thesis, University of Groningen, The Netherland, 2013. https://www.rug.nl/research/portal/publications/the-role-of-vitamin-a-in-bile-acid-synthesis-and-transport-and-the-relevance-for-cholestatic-liver-disease(5c3550c6-2741-4a82-8ee1-b24ca98b607b)/export.html.
[11] The Textbook of Hepatology: From Basic Science to Clinical Practice, 3rd Edition - Juan Rodes, Jean-Pierre Benhamou, Andres Blei, et al, (2008). http://eu.wiley.com/WileyCDA/WileyTitle/productCd-1405127414.html (accessed January 6, 2016).
[12] C. Thomas, R. Pellicciari, M. Pruzanski, J. Auwerx, K. Schoonjans, Targeting bile-acid signalling for metabolic diseases, Nat. Rev. Drug Discov. 7 (2008) 678–693. doi:10.1038/nrd2619.
[13] A. Pellicoro, K.N. Faber, The function and regulation of proteins involved in bile salt biosynthesis and transport, Aliment. Pharmacol. Ther. 26 Suppl 2 (2007) 149–160. doi:10.1111/j.1365-2036.2007.03522.x.
[14] S. Shefer, S. Hauser, I. Bekersky, E.H. Mosbach, Biochemical site of regulation of bile acid biosynthesis in the rat, J. Lipid Res. 11 (1970) 404–411.
[15] D.W. Russell, The enzymes, regulation, and genetics of bile acid synthesis, Annu. Rev. Biochem. 72 (2003) 137–174. doi:10.1146/annurev.biochem.72.121801.161712.
[16] A. Pellicoro, F.A.J. van den Heuvel, M. Geuken, H. Moshage, P.L.M. Jansen, K.N. Faber, Human and rat bile acid-CoA:amino acid N-acyltransferase are liver-specific peroxisomal enzymes: implications for intracellular bile salt transport, Hepatol. Baltim. Md. 45 (2007) 340–348. doi:10.1002/hep.21528.
[17] S. Ruetz, P. Gros, Phosphatidylcholine translocase: a physiological role for the mdr2 gene, Cell. 77 (1994) 1071–1081.

Bile acid and Vitamin A homeostasis intertwined
50
[18] T.H. Mauad, C.M. van Nieuwkerk, K.P. Dingemans, J.J. Smit, A.H. Schinkel, R.G. Notenboom, M.A. van den Bergh Weerman, R.P. Verkruisen, A.K. Groen, R.P. Oude Elferink, Mice with homozygous disruption of the mdr2 P-glycoprotein gene. A novel animal model for studies of nonsuppurative inflammatory cholangitis and hepatocarcinogenesis, Am. J. Pathol. 145 (1994) 1237–1245.
[19] G. Alpini, S.S. Glaser, R. Rodgers, J.L. Phinizy, W.E. Robertson, J. Lasater, A. Caligiuri, Z. Tretjak, G.D. LeSage, Functional expression of the apical Na+-dependent bile acid transporter in large but not small rat cholangiocytes, Gastroenterology. 113 (1997) 1734–1740.
[20] J.M. Ridlon, D.-J. Kang, P.B. Hylemon, Bile salt biotransformations by human intestinal bacteria, J. Lipid Res. 47 (2006) 241–259. doi:10.1194/jlr.R500013-JLR200.
[21] I.L. Csanaky, H. Lu, Y. Zhang, K. Ogura, S. Choudhuri, C.D. Klaassen, Organic anion-transporting polypeptide 1b2 (Oatp1b2) is important for the hepatic uptake of unconjugated bile acids: Studies in Oatp1b2-null mice, Hepatol. Baltim. Md. 53 (2011) 272–281. doi:10.1002/hep.23984.
[22] J.L. Napoli, Physiological insights into all-trans-retinoic acid biosynthesis, Biochim. Biophys. Acta. 1821 (2012) 152–167. doi:10.1016/j.bbalip.2011.05.004.
[23] M.A. Kane, Analysis, occurrence, and function of 9-cis-retinoic acid, Biochim. Biophys. Acta. 1821 (2012) 10–20. doi:10.1016/j.bbalip.2011.09.012.
[24] R. Schreiber, U. Taschler, K. Preiss-Landl, N. Wongsiriroj, R. Zimmermann, A. Lass, Retinyl ester hydrolases and their roles in vitamin A homeostasis, Biochim. Biophys. Acta. 1821 (2012) 113–123. doi:10.1016/j.bbalip.2011.05.001.
[25] S.M. O’Byrne, W.S. Blaner, Retinol and retinyl esters: biochemistry and physiology, J. Lipid Res. 54 (2013) 1731–1743. doi:10.1194/jlr.R037648.
[26] Y. Li, N. Wongsiriroj, W.S. Blaner, The multifaceted nature of retinoid transport and metabolism, Hepatobiliary Surg. Nutr. 3 (2014) 126–139. doi:10.3978/j.issn.2304-3881.2014.05.04.
[27] N. Kono, H. Arai, Intracellular transport of fat-soluble vitamins A and E, Traffic Cph. Den. 16 (2015) 19–34. doi:10.1111/tra.12231.
[28] W.S. Blaner, Y. Li, P.-J. Brun, J.J. Yuen, S.-A. Lee, R.D. Clugston, Vitamin A Absorption, Storage and Mobilization, Subcell. Biochem. 81 (2016) 95–125. doi:10.1007/978-94-024-0945-1_4.
[29] N.Y. Kedishvili, Retinoic Acid Synthesis and Degradation, Subcell. Biochem. 81 (2016) 127–161. doi:10.1007/978-94-024-0945-1_5.
[30] A. During, E.H. Harrison, Mechanisms of provitamin A (carotenoid) and vitamin A (retinol) transport into and out of intestinal Caco-2 cells, J. Lipid Res. 48 (2007) 2283–2294. doi:10.1194/jlr.M700263-JLR200.
[31] G.P. Lobo, S. Hessel, A. Eichinger, N. Noy, A.R. Moise, A. Wyss, K. Palczewski, J. von Lintig, ISX is a retinoic acid-sensitive gatekeeper that controls intestinal beta,beta-carotene absorption and vitamin A production, FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 24 (2010) 1656–1666. doi:10.1096/fj.09-150995.
[32] E. Reboul, Absorption of vitamin A and carotenoids by the enterocyte: focus on transport proteins, Nutrients. 5 (2013) 3563–3581. doi:10.3390/nu5093563.
[33] A. During, H.D. Dawson, E.H. Harrison, Carotenoid transport is decreased and expression of the lipid transporters SR-BI, NPC1L1, and ABCA1 is

Chapter 2
51
downregulated in Caco-2 cells treated with ezetimibe, J. Nutr. 135 (2005) 2305–2312.
[34] Y. Sato, R. Suzuki, M. Kobayashi, S. Itagaki, T. Hirano, T. Noda, S. Mizuno, M. Sugawara, K. Iseki, Involvement of cholesterol membrane transporter Niemann-Pick C1-like 1 in the intestinal absorption of lutein, J. Pharm. Pharm. Sci. Publ. Can. Soc. Pharm. Sci. Société Can. Sci. Pharm. 15 (2012) 256–264.
[35] T. Moore, Vitamin A and carotene: The absence of the liver oil vitamin A from carotene. VI. The conversion of carotene to vitamin A in vivo, Biochem. J. 24 (1930) 692–702.
[36] K.M. Rigtrup, L.R. McEwen, H.M. Said, D.E. Ong, Retinyl ester hydrolytic activity associated with human intestinal brush border membranes, Am. J. Clin. Nutr. 60 (1994) 111–116.
[37] A.M. van Bennekum, E.A. Fisher, W.S. Blaner, E.H. Harrison, Hydrolysis of retinyl esters by pancreatic triglyceride lipase, Biochemistry. 39 (2000) 4900–4906.
[38] J.L. Probstfield, T.L. Lin, J. Peters, D.B. Hunninghake, Carotenoids and vitamin A: the effect of hypocholesterolemic agents on serum levels, Metabolism. 34 (1985) 88–91.
[39] D. Hollander, Retinol lymphatic and portal transport: influence of pH, bile, and fatty acids, Am. J. Physiol. 239 (1980) G210-214.
[40] J. von Lintig, A. Wyss, Molecular analysis of vitamin A formation: cloning and characterization of beta-carotene 15,15’-dioxygenases, Arch. Biochem. Biophys. 385 (2001) 47–52. doi:10.1006/abbi.2000.2096.
[41] M.M. Hussain, A proposed model for the assembly of chylomicrons, Atherosclerosis. 148 (2000) 1–15.
[42] N.O. Davidson, M.E. Kollmer, R.M. Glickman, Apolipoprotein B synthesis in rat small intestine: regulation by dietary triglyceride and biliary lipid, J. Lipid Res. 27 (1986) 30–39.
[43] E.H. Harrison, Mechanisms of digestion and absorption of dietary vitamin A, Annu. Rev. Nutr. 25 (2005) 87–103. doi:10.1146/annurev.nutr.25.050304.092614.
[44] B.J. Zeng, B.C. Mortimer, I.J. Martins, U. Seydel, T.G. Redgrave, Chylomicron remnant uptake is regulated by the expression and function of heparan sulfate proteoglycan in hepatocytes, J. Lipid Res. 39 (1998) 845–860.
[45] K.I. Stanford, J.R. Bishop, E.M. Foley, J.C. Gonzales, I.R. Niesman, J.L. Witztum, J.D. Esko, Syndecan-1 is the primary heparan sulfate proteoglycan mediating hepatic clearance of triglyceride-rich lipoproteins in mice, J. Clin. Invest. 119 (2009) 3236–3245. doi:10.1172/JCI38251.
[46] T. Mondal, H. Wang, G.T. DeKoster, B. Baban, M.L. Gross, C. Frieden, ApoE: In Vitro Studies of a Small Molecule Effector, Biochemistry. 55 (2016) 2613–2621. doi:10.1021/acs.biochem.6b00324.
[47] T. Linke, H. Dawson, E.H. Harrison, Isolation and characterization of a microsomal acid retinyl ester hydrolase, J. Biol. Chem. 280 (2005) 23287–23294. doi:10.1074/jbc.M413585200.
[48] W.S. Blaner, J.A. Olson, Retinol and retinoic acid metabolism, Retin. Biol. Chem. Med. Sporn MB Roberts AB Goodman Eds Raven Press N. Y. 2 (1994) 229–256.
[49] E.H. Harrison, W.S. Blaner, D.S. Goodman, A.C. Ross, Subcellular localization of retinoids, retinoid-binding proteins, and acyl-CoA:retinol acyltransferase in rat liver, J. Lipid Res. 28 (1987) 973–981.

Bile acid and Vitamin A homeostasis intertwined
52
[50] H. Ronne, C. Ocklind, K. Wiman, L. Rask, B. Obrink, P.A. Peterson, Ligand-dependent regulation of intracellular protein transport: effect of vitamin a on the secretion of the retinol-binding protein, J. Cell Biol. 96 (1983) 907–910.
[51] D. Bellovino, T. Morimoto, F. Tosetti, S. Gaetani, Retinol binding protein and transthyretin are secreted as a complex formed in the endoplasmic reticulum in HepG2 human hepatocarcinoma cells, Exp. Cell Res. 222 (1996) 77–83. doi:10.1006/excr.1996.0010.
[52] A. Suhara, M. Kato, M. Kanai, Ultrastructural localization of plasma retinol-binding protein in rat liver, J. Lipid Res. 31 (1990) 1669–1681.
[53] P.A. Peterson, Studies on the interaction between prealbumin, retinol-binding protein, and vitamin A, J. Biol. Chem. 246 (1971) 44–49.
[54] H. Sun, R. Kawaguchi, The membrane receptor for plasma retinol-binding protein, a new type of cell-surface receptor, Int. Rev. Cell Mol. Biol. 288 (2011) 1–41. doi:10.1016/B978-0-12-386041-5.00001-7.
[55] P. Bouillet, V. Sapin, C. Chazaud, N. Messaddeq, D. Décimo, P. Dollé, P. Chambon, Developmental expression pattern of Stra6, a retinoic acid-responsive gene encoding a new type of membrane protein, Mech. Dev. 63 (1997) 173–186.
[56] J. Amengual, N. Zhang, M. Kemerer, T. Maeda, K. Palczewski, J. Von Lintig, STRA6 is critical for cellular vitamin A uptake and homeostasis, Hum. Mol. Genet. 23 (2014) 5402–5417. doi:10.1093/hmg/ddu258.
[57] P. Alapatt, F. Guo, S.M. Komanetsky, S. Wang, J. Cai, A. Sargsyan, E. Rodríguez Díaz, B.T. Bacon, P. Aryal, T.E. Graham, Liver retinol transporter and receptor for serum retinol-binding protein (RBP4), J. Biol. Chem. 288 (2013) 1250–1265. doi:10.1074/jbc.M112.369132.
[58] K.R. Norum, R. Blomhoff, McCollum Award Lecture, 1992: vitamin A absorption, transport, cellular uptake, and storage, Am. J. Clin. Nutr. 56 (1992) 735–744.
[59] L. Quadro, W.S. Blaner, L. Hamberger, P.M. Novikoff, S. Vogel, R. Piantedosi, M.E. Gottesman, V. Colantuoni, The role of extrahepatic retinol binding protein in the mobilization of retinoid stores, J. Lipid Res. 45 (2004) 1975–1982. doi:10.1194/jlr.M400137-JLR200.
[60] J. Paik, S. Vogel, L. Quadro, R. Piantedosi, M. Gottesman, K. Lai, L. Hamberger, M. de M. Vieira, W.S. Blaner, Vitamin A: overlapping delivery pathways to tissues from the circulation, J. Nutr. 134 (2004) 276S-280S.
[61] L. Quadro, W.S. Blaner, D.J. Salchow, S. Vogel, R. Piantedosi, P. Gouras, S. Freeman, M.P. Cosma, V. Colantuoni, M.E. Gottesman, Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein, EMBO J. 18 (1999) 4633–4644. doi:10.1093/emboj/18.17.4633.
[62] N. Wongsiriroj, H. Jiang, R. Piantedosi, K.J.Z. Yang, J. Kluwe, R.F. Schwabe, H. Ginsberg, I.J. Goldberg, W.S. Blaner, Genetic dissection of retinoid esterification and accumulation in the liver and adipose tissue, J. Lipid Res. 55 (2014) 104–114. doi:10.1194/jlr.M043844.
[63] M. Ajat, M. Molenaar, J.F.H.M. Brouwers, A.B. Vaandrager, M. Houweling, J.B. Helms, Hepatic stellate cells retain the capacity to synthesize retinyl esters and to store neutral lipids in small lipid droplets in the absence of LRAT, Biochim. Biophys. Acta. 1862 (2016) 176–187. doi:10.1016/j.bbalip.2016.10.013.
[64] T. Mello, A. Nakatsuka, S. Fears, W. Davis, H. Tsukamoto, W.F. Bosron, S.P. Sanghani, Expression of carboxylesterase and lipase genes in rat liver cell-types, Biochem. Biophys. Res. Commun. 374 (2008) 460–464. doi:10.1016/j.bbrc.2008.07.024.

Chapter 2
53
[65] W. Pang, Y. Zhang, S. Wang, A. Jia, W. Dong, C. Cai, Z. Hua, J. Zhang, The mPlrp2 and mClps genes are involved in the hydrolysis of retinyl esters in the mouse liver, J. Lipid Res. 52 (2011) 934–941. doi:10.1194/jlr.M010082.
[66] U. Taschler, R. Schreiber, C. Chitraju, G.F. Grabner, M. Romauch, H. Wolinski, G. Haemmerle, R. Breinbauer, R. Zechner, A. Lass, R. Zimmermann, Adipose triglyceride lipase is involved in the mobilization of triglyceride and retinoid stores of hepatic stellate cells, Biochim. Biophys. Acta. 1851 (2015) 937–945. doi:10.1016/j.bbalip.2015.02.017.
[67] C. Pirazzi, L. Valenti, B.M. Motta, P. Pingitore, K. Hedfalk, R.M. Mancina, M.A. Burza, C. Indiveri, Y. Ferro, T. Montalcini, C. Maglio, P. Dongiovanni, S. Fargion, R. Rametta, A. Pujia, L. Andersson, S. Ghosal, M. Levin, O. Wiklund, M. Iacovino, J. Borén, S. Romeo, PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells, Hum. Mol. Genet. 23 (2014) 4077–4085. doi:10.1093/hmg/ddu121.
[68] A. Mondul, R.M. Mancina, A. Merlo, P. Dongiovanni, R. Rametta, T. Montalcini, L. Valenti, D. Albanes, S. Romeo, PNPLA3 I148M Variant Influences Circulating Retinol in Adults with Nonalcoholic Fatty Liver Disease or Obesity, J. Nutr. 145 (2015) 1687–1691. doi:10.3945/jn.115.210633.
[69] C. Tian, R.P. Stokowski, D. Kershenobich, D.G. Ballinger, D.A. Hinds, Variant in PNPLA3 is associated with alcoholic liver disease, Nat. Genet. 42 (2010) 21–23. doi:10.1038/ng.488.
[70] S. Romeo, J. Kozlitina, C. Xing, A. Pertsemlidis, D. Cox, L.A. Pennacchio, E. Boerwinkle, J.C. Cohen, H.H. Hobbs, Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease, Nat. Genet. 40 (2008) 1461–1465. doi:10.1038/ng.257.
[71] M. Kovarova, I. Königsrainer, A. Königsrainer, F. Machicao, H.-U. Häring, E. Schleicher, A. Peter, The Genetic Variant I148M in PNPLA3 Is Associated With Increased Hepatic Retinyl-Palmitate Storage in Humans, J. Clin. Endocrinol. Metab. 100 (2015) E1568-1574. doi:10.1210/jc.2015-2978.
[72] E. Trépo, S. Romeo, J. Zucman-Rossi, P. Nahon, PNPLA3 gene in liver diseases, J. Hepatol. 65 (2016) 399–412. doi:10.1016/j.jhep.2016.03.011.
[73] J.M. Erickson, A.R. Mawson, Possible role of endogenous retinoid (Vitamin A) toxicity in the pathophysiology of primary biliary cirrhosis, J. Theor. Biol. 206 (2000) 47–54. doi:10.1006/jtbi.2000.2102.
[74] N.E. Nagy, K.B. Holven, N. Roos, H. Senoo, N. Kojima, K.R. Norum, R. Blomhoff, Storage of vitamin A in extrahepatic stellate cells in normal rats, J. Lipid Res. 38 (1997) 645–658.
[75] R. Blomhoff, M.H. Green, T. Berg, K.R. Norum, Transport and storage of vitamin A, Science. 250 (1990) 399–404.
[76] S. Vogel, M.V. Gamble, W.S. Blaner, Biosynthesis, Absorption, Metabolism and Transport of Retinoids, in: P.D.D. h c H. Nau, W.S. Blaner (Eds.), Retinoids, Springer Berlin Heidelberg, 1999: pp. 31–95. doi:10.1007/978-3-642-58483-1_2.
[77] J. Marill, N. Idres, C.C. Capron, E. Nguyen, G.G. Chabot, Retinoic acid metabolism and mechanism of action: a review, Curr. Drug Metab. 4 (2003) 1–10.
[78] L.L. Sandell, B.W. Sanderson, G. Moiseyev, T. Johnson, A. Mushegian, K. Young, J.-P. Rey, J. Ma, K. Staehling-Hampton, P.A. Trainor, RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development, Genes Dev. 21 (2007) 1113–1124. doi:10.1101/gad.1533407.

Bile acid and Vitamin A homeostasis intertwined
54
[79] M. Rhinn, B. Schuhbaur, K. Niederreither, P. Dollé, Involvement of retinol dehydrogenase 10 in embryonic patterning and rescue of its loss of function by maternal retinaldehyde treatment, Proc. Natl. Acad. Sci. U. S. A. 108 (2011) 16687–16692. doi:10.1073/pnas.1103877108.
[80] A.M. Ashique, S.R. May, M.A. Kane, A.E. Folias, K. Phamluong, Y. Choe, J.L. Napoli, A.S. Peterson, Morphological defects in a novel Rdh10 mutant that has reduced retinoic acid biosynthesis and signaling, Genes. N. Y. N 2000. 50 (2012) 415–423. doi:10.1002/dvg.22002.
[81] B.X. Wu, Y. Chen, Y. Chen, J. Fan, B. Rohrer, R.K. Crouch, J.-X. Ma, Cloning and characterization of a novel all-trans retinol short-chain dehydrogenase/reductase from the RPE, Invest. Ophthalmol. Vis. Sci. 43 (2002) 3365–3372.
[82] B.X. Wu, G. Moiseyev, Y. Chen, B. Rohrer, R.K. Crouch, J.-X. Ma, Identification of RDH10, an All-trans Retinol Dehydrogenase, in Retinal Muller Cells, Invest. Ophthalmol. Vis. Sci. 45 (2004) 3857–3862. doi:10.1167/iovs.03-1302.
[83] M. Zhang, P. Hu, C.R. Krois, M.A. Kane, J.L. Napoli, Altered vitamin A homeostasis and increased size and adiposity in the rdh1-null mouse, FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 21 (2007) 2886–2896. doi:10.1096/fj.06-7964com.
[84] W.H. Gough, S. VanOoteghem, T. Sint, N.Y. Kedishvili, cDNA cloning and characterization of a new human microsomal NAD+-dependent dehydrogenase that oxidizes all-trans-retinol and 3alpha-hydroxysteroids, J. Biol. Chem. 273 (1998) 19778–19785.
[85] M.A. Leo, C.S. Lieber, Normal testicular structure and reproductive function in deermice lacking retinol and alcohol dehydrogenase activity, J. Clin. Invest. 73 (1984) 593–596. doi:10.1172/JCI111248.
[86] H. Chen, W.N. Howald, M.R. Juchau, Biosynthesis of all-trans-retinoic acid from all-trans-retinol: catalysis of all-trans-retinol oxidation by human P-450 cytochromes, Drug Metab. Dispos. Biol. Fate Chem. 28 (2000) 315–322.
[87] F. Haeseleer, J. Huang, L. Lebioda, J.C. Saari, K. Palczewski, Molecular characterization of a novel short-chain dehydrogenase/reductase that reduces all-trans-retinal, J. Biol. Chem. 273 (1998) 21790–21799.
[88] M.K. Adams, O.V. Belyaeva, L. Wu, N.Y. Kedishvili, The retinaldehyde reductase activity of DHRS3 is reciprocally activated by retinol dehydrogenase 10 to control retinoid homeostasis, J. Biol. Chem. 289 (2014) 14868–14880. doi:10.1074/jbc.M114.552257.
[89] G. Duester, F.A. Mic, A. Molotkov, Cytosolic retinoid dehydrogenases govern ubiquitous metabolism of retinol to retinaldehyde followed by tissue-specific metabolism to retinoic acid, Chem. Biol. Interact. 143–144 (2003) 201–210.
[90] I. Gagnon, G. Duester, P.V. Bhat, Kinetic analysis of mouse retinal dehydrogenase type-2 (RALDH2) for retinal substrates, Biochim. Biophys. Acta. 1596 (2002) 156–162.
[91] M. Lin, M. Zhang, M. Abraham, S.M. Smith, J.L. Napoli, Mouse retinal dehydrogenase 4 (RALDH4), molecular cloning, cellular expression, and activity in 9-cis-retinoic acid biosynthesis in intact cells, J. Biol. Chem. 278 (2003) 9856–9861. doi:10.1074/jbc.M211417200.
[92] A. Sima, M. Parisotto, S. Mader, P.V. Bhat, Kinetic characterization of recombinant mouse retinal dehydrogenase types 3 and 4 for retinal substrates, Biochim. Biophys. Acta. 1790 (2009) 1660–1664. doi:10.1016/j.bbagen.2009.09.004.

Chapter 2
55
[93] K. Niederreither, V. Subbarayan, P. Dollé, P. Chambon, Embryonic retinoic acid synthesis is essential for early mouse post-implantation development, Nat. Genet. 21 (1999) 444–448. doi:10.1038/7788.
[94] V. Dupé, N. Matt, J.-M. Garnier, P. Chambon, M. Mark, N.B. Ghyselinck, A newborn lethal defect due to inactivation of retinaldehyde dehydrogenase type 3 is prevented by maternal retinoic acid treatment, Proc. Natl. Acad. Sci. U. S. A. 100 (2003) 14036–14041. doi:10.1073/pnas.2336223100.
[95] X. Fan, A. Molotkov, S.-I. Manabe, C.M. Donmoyer, L. Deltour, M.H. Foglio, A.E. Cuenca, W.S. Blaner, S.A. Lipton, G. Duester, Targeted disruption of Aldh1a1 (Raldh1) provides evidence for a complex mechanism of retinoic acid synthesis in the developing retina, Mol. Cell. Biol. 23 (2003) 4637–4648.
[96] A. Molotkov, G. Duester, Genetic evidence that retinaldehyde dehydrogenase Raldh1 (Aldh1a1) functions downstream of alcohol dehydrogenase Adh1 in metabolism of retinol to retinoic acid, J. Biol. Chem. 278 (2003) 36085–36090. doi:10.1074/jbc.M303709200.
[97] Q.Y. Zhang, D. Dunbar, L. Kaminsky, Human cytochrome P-450 metabolism of retinals to retinoic acids, Drug Metab. Dispos. Biol. Fate Chem. 28 (2000) 292–297.
[98] S.B. Kurlandsky, M.V. Gamble, R. Ramakrishnan, W.S. Blaner, Plasma delivery of retinoic acid to tissues in the rat, J. Biol. Chem. 270 (1995) 17850–17857.
[99] M.A. Kane, A.E. Folias, A. Pingitore, M. Perri, K.M. Obrochta, C.R. Krois, E. Cione, J.Y. Ryu, J.L. Napoli, Identification of 9-cis-retinoic acid as a pancreas-specific autacoid that attenuates glucose-stimulated insulin secretion, Proc. Natl. Acad. Sci. U. S. A. 107 (2010) 21884–21889. doi:10.1073/pnas.1008859107.
[100] R. Rühl, A. Krzyżosiak, A. Niewiadomska-Cimicka, N. Rochel, L. Szeles, B. Vaz, M. Wietrzych-Schindler, S. Álvarez, M. Szklenar, L. Nagy, A.R. de Lera, W. Krężel, 9-cis-13,14-Dihydroretinoic Acid Is an Endogenous Retinoid Acting as RXR Ligand in Mice, PLoS Genet. 11 (2015) e1005213. doi:10.1371/journal.pgen.1005213.
[101] J.F. Boylan, L.J. Gudas, The level of CRABP-I expression influences the amounts and types of all-trans-retinoic acid metabolites in F9 teratocarcinoma stem cells, J. Biol. Chem. 267 (1992) 21486–21491.
[102] A.R. Topletz, J.E. Thatcher, A. Zelter, J.D. Lutz, S. Tay, W.L. Nelson, N. Isoherranen, Comparison of the function and expression of CYP26A1 and CYP26B1, the two retinoic acid hydroxylases, Biochem. Pharmacol. 83 (2012) 149–163. doi:10.1016/j.bcp.2011.10.007.
[103] J. Xi, Z. Yang, Expression of RALDHs (ALDH1As) and CYP26s in human tissues and during the neural differentiation of P19 embryonal carcinoma stem cell, Gene Expr. Patterns GEP. 8 (2008) 438–442. doi:10.1016/j.gep.2008.04.003.
[104] P.M. Petkovich, Retinoic acid metabolism, J. Am. Acad. Dermatol. 45 (2001) S136-142. doi:10.1067/mjd.2001.113715.
[105] N. Idres, J. Marill, G.G. Chabot, Regulation of CYP26A1 expression by selective RAR and RXR agonists in human NB4 promyelocytic leukemia cells, Biochem. Pharmacol. 69 (2005) 1595–1601. doi:10.1016/j.bcp.2005.02.024.
[106] J.A. White, B. Beckett-Jones, Y.D. Guo, F.J. Dilworth, J. Bonasoro, G. Jones, M. Petkovich, cDNA cloning of human retinoic acid-metabolizing enzyme (hP450RAI) identifies a novel family of cytochromes P450, J. Biol. Chem. 272 (1997) 18538–18541.

Bile acid and Vitamin A homeostasis intertwined
56
[107] J.A. White, H. Ramshaw, M. Taimi, W. Stangle, A. Zhang, S. Everingham, S. Creighton, S.P. Tam, G. Jones, M. Petkovich, Identification of the human cytochrome P450, P450RAI-2, which is predominantly expressed in the adult cerebellum and is responsible for all-trans-retinoic acid metabolism, Proc. Natl. Acad. Sci. U. S. A. 97 (2000) 6403–6408. doi:10.1073/pnas.120161397.
[108] M. Taimi, C. Helvig, J. Wisniewski, H. Ramshaw, J. White, M. Amad, B. Korczak, M. Petkovich, A novel human cytochrome P450, CYP26C1, involved in metabolism of 9-cis and all-trans isomers of retinoic acid, J. Biol. Chem. 279 (2004) 77–85. doi:10.1074/jbc.M308337200.
[109] J. Marill, C.C. Capron, N. Idres, G.G. Chabot, Human cytochrome P450s involved in the metabolism of 9-cis- and 13-cis-retinoic acids, Biochem. Pharmacol. 63 (2002) 933–943.
[110] J.W. Jones, K. Pierzchalski, J. Yu, M.A. Kane, Use of fast HPLC multiple reaction monitoring cubed for endogenous retinoic acid quantification in complex matrices, Anal. Chem. 87 (2015) 3222–3230. doi:10.1021/ac504597q.
[111] P. Germain, B. Staels, C. Dacquet, M. Spedding, V. Laudet, Overview of nomenclature of nuclear receptors, Pharmacol. Rev. 58 (2006) 685–704. doi:10.1124/pr.58.4.2.
[112] A. Aranda, A. Pascual, Nuclear hormone receptors and gene expression, Physiol. Rev. 81 (2001) 1269–1304.
[113] M. Beato, P. Herrlich, G. Schütz, Steroid hormone receptors: many actors in search of a plot, Cell. 83 (1995) 851–857.
[114] B.M. Forman, R.M. Evans, Nuclear hormone receptors activate direct, inverted, and everted repeats, Ann. N. Y. Acad. Sci. 761 (1995) 29–37.
[115] S.J. Karpen, Nuclear receptor regulation of hepatic function, J. Hepatol. 36 (2002) 832–850.
[116] D.J. Mangelsdorf, E.S. Ong, J.A. Dyck, R.M. Evans, Nuclear receptor that identifies a novel retinoic acid response pathway, Nature. 345 (1990) 224–229. doi:10.1038/345224a0.
[117] P. Germain, P. Chambon, G. Eichele, R.M. Evans, M.A. Lazar, M. Leid, A.R. De Lera, R. Lotan, D.J. Mangelsdorf, H. Gronemeyer, International Union of Pharmacology. LXIII. Retinoid X receptors, Pharmacol. Rev. 58 (2006) 760–772. doi:10.1124/pr.58.4.7.
[118] D.J. Mangelsdorf, U. Borgmeyer, R.A. Heyman, J.Y. Zhou, E.S. Ong, A.E. Oro, A. Kakizuka, R.M. Evans, Characterization of three RXR genes that mediate the action of 9-cis retinoic acid, Genes Dev. 6 (1992) 329–344.
[119] G. Wolf, Is 9-cis-retinoic acid the endogenous ligand for the retinoic acid-X receptor?, Nutr. Rev. 64 (2006) 532–538.
[120] P. Germain, P. Chambon, G. Eichele, R.M. Evans, M.A. Lazar, M. Leid, A.R. De Lera, R. Lotan, D.J. Mangelsdorf, H. Gronemeyer, International Union of Pharmacology. LX. Retinoic acid receptors, Pharmacol. Rev. 58 (2006) 712–725. doi:10.1124/pr.58.4.4.
[121] X.K. Zhang, J. Lehmann, B. Hoffmann, M.I. Dawson, J. Cameron, G. Graupner, T. Hermann, P. Tran, M. Pfahl, Homodimer formation of retinoid X receptor induced by 9-cis retinoic acid, Nature. 358 (1992) 587–591. doi:10.1038/358587a0.
[122] B. Desvergne, RXR: from partnership to leadership in metabolic regulations, Vitam. Horm. 75 (2007) 1–32. doi:10.1016/S0083-6729(06)75001-4.
[123] I. Issemann, R.A. Prince, J.D. Tugwood, S. Green, The retinoid X receptor enhances the function of the peroxisome proliferator activated receptor, Biochimie. 75 (1993) 251–256.

Chapter 2
57
[124] S.A. Kliewer, K. Umesono, D.J. Noonan, R.A. Heyman, R.M. Evans, Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors, Nature. 358 (1992) 771–774. doi:10.1038/358771a0.
[125] P.J. Willy, K. Umesono, E.S. Ong, R.M. Evans, R.A. Heyman, D.J. Mangelsdorf, LXR, a nuclear receptor that defines a distinct retinoid response pathway, Genes Dev. 9 (1995) 1033–1045.
[126] N.L. Urizar, D.H. Dowhan, D.D. Moore, The farnesoid X-activated receptor mediates bile acid activation of phospholipid transfer protein gene expression, J. Biol. Chem. 275 (2000) 39313–39317. doi:10.1074/jbc.M007998200.
[127] J. DiRenzo, M. Söderstrom, R. Kurokawa, M.H. Ogliastro, M. Ricote, S. Ingrey, A. Hörlein, M.G. Rosenfeld, C.K. Glass, Peroxisome proliferator-activated receptors and retinoic acid receptors differentially control the interactions of retinoid X receptor heterodimers with ligands, coactivators, and corepressors, Mol. Cell. Biol. 17 (1997) 2166–2176.
[128] B.M. Forman, K. Umesono, J. Chen, R.M. Evans, Unique response pathways are established by allosteric interactions among nuclear hormone receptors, Cell. 81 (1995) 541–550.
[129] P.N. MacDonald, D.R. Dowd, S. Nakajima, M.A. Galligan, M.C. Reeder, C.A. Haussler, K. Ozato, M.R. Haussler, Retinoid X receptors stimulate and 9-cis retinoic acid inhibits 1,25-dihydroxyvitamin D3-activated expression of the rat osteocalcin gene, Mol. Cell. Biol. 13 (1993) 5907–5917.
[130] J. Grober, I. Zaghini, H. Fujii, S.A. Jones, S.A. Kliewer, T.M. Willson, T. Ono, P. Besnard, Identification of a bile acid-responsive element in the human ileal bile acid-binding protein gene. Involvement of the farnesoid X receptor/9-cis-retinoic acid receptor heterodimer, J. Biol. Chem. 274 (1999) 29749–29754.
[131] P. Dollé, Developmental expression of retinoic acid receptors (RARs), Nucl. Recept. Signal. 7 (2009) e006. doi:10.1621/nrs.07006.
[132] S.A. Kliewer, K. Umesono, D.J. Mangelsdorf, R.M. Evans, Retinoid X receptor interacts with nuclear receptors in retinoic acid, thyroid hormone and vitamin D3 signalling, Nature. 355 (1992) 446–449. doi:10.1038/355446a0.
[133] K. Umesono, K.K. Murakami, C.C. Thompson, R.M. Evans, Direct repeats as selective response elements for the thyroid hormone, retinoic acid, and vitamin D3 receptors, Cell. 65 (1991) 1255–1266.
[134] G. Allenby, R. Janocha, S. Kazmer, J. Speck, J.F. Grippo, A.A. Levin, Binding of 9-cis-retinoic acid and all-trans-retinoic acid to retinoic acid receptors alpha, beta, and gamma. Retinoic acid receptor gamma binds all-trans-retinoic acid preferentially over 9-cis-retinoic acid, J. Biol. Chem. 269 (1994) 16689–16695.
[135] J.J. Repa, K.K. Hanson, M. Clagett-Dame, All-trans-retinol is a ligand for the retinoic acid receptors, Proc. Natl. Acad. Sci. U. S. A. 90 (1993) 7293–7297.
[136] W. Seol, H.S. Choi, D.D. Moore, Isolation of proteins that interact specifically with the retinoid X receptor: two novel orphan receptors, Mol. Endocrinol. Baltim. Md. 9 (1995) 72–85. doi:10.1210/mend.9.1.7760852.
[137] B.M. Forman, E. Goode, J. Chen, A.E. Oro, D.J. Bradley, T. Perlmann, D.J. Noonan, L.T. Burka, T. McMorris, W.W. Lamph, R.M. Evans, C. Weinberger, Identification of a nuclear receptor that is activated by farnesol metabolites, Cell. 81 (1995) 687–693.
[138] A.M. Zavacki, J.M. Lehmann, W. Seol, T.M. Willson, S.A. Kliewer, D.D. Moore, Activation of the orphan receptor RIP14 by retinoids, Proc. Natl. Acad. Sci. U. S. A. 94 (1997) 7909–7914.

Bile acid and Vitamin A homeostasis intertwined
58
[139] H. Wang, J. Chen, K. Hollister, L.C. Sowers, B.M. Forman, Endogenous bile acids are ligands for the nuclear receptor FXR/BAR, Mol. Cell. 3 (1999) 543–553.
[140] D.J. Parks, S.G. Blanchard, R.K. Bledsoe, G. Chandra, T.G. Consler, S.A. Kliewer, J.B. Stimmel, T.M. Willson, A.M. Zavacki, D.D. Moore, J.M. Lehmann, Bile acids: natural ligands for an orphan nuclear receptor, Science. 284 (1999) 1365–1368.
[141] M. Makishima, A.Y. Okamoto, J.J. Repa, H. Tu, R.M. Learned, A. Luk, M.V. Hull, K.D. Lustig, D.J. Mangelsdorf, B. Shan, Identification of a nuclear receptor for bile acids, Science. 284 (1999) 1362–1365.
[142] S. Wang, K. Lai, F.J. Moy, A. Bhat, H.B. Hartman, M.J. Evans, The nuclear hormone receptor farnesoid X receptor (FXR) is activated by androsterone, Endocrinology. 147 (2006) 4025–4033. doi:10.1210/en.2005-1485.
[143] A. Zhao, J. Yu, J.-L. Lew, L. Huang, S.D. Wright, J. Cui, Polyunsaturated fatty acids are FXR ligands and differentially regulate expression of FXR targets, DNA Cell Biol. 23 (2004) 519–526. doi:10.1089/1044549041562267.
[144] R. Deng, D. Yang, J. Yang, B. Yan, Oxysterol 22(R)-hydroxycholesterol induces the expression of the bile salt export pump through nuclear receptor farsenoid X receptor but not liver X receptor, J. Pharmacol. Exp. Ther. 317 (2006) 317–325. doi:10.1124/jpet.105.097758.
[145] P.R. Maloney, D.J. Parks, C.D. Haffner, A.M. Fivush, G. Chandra, K.D. Plunket, K.L. Creech, L.B. Moore, J.G. Wilson, M.C. Lewis, S.A. Jones, T.M. Willson, Identification of a chemical tool for the orphan nuclear receptor FXR, J. Med. Chem. 43 (2000) 2971–2974.
[146] R. Pellicciari, S. Fiorucci, E. Camaioni, C. Clerici, G. Costantino, P.R. Maloney, A. Morelli, D.J. Parks, T.M. Willson, 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity, J. Med. Chem. 45 (2002) 3569–3572.
[147] J. Cui, L. Huang, A. Zhao, J.-L. Lew, J. Yu, S. Sahoo, P.T. Meinke, I. Royo, F. Pelaez, S.D. Wright, Guggulsterone is a farnesoid X receptor antagonist in coactivator association assays but acts to enhance transcription of bile salt export pump, J. Biol. Chem. 278 (2003) 10214–10220. doi:10.1074/jbc.M209323200.
[148] N.L. Urizar, A.B. Liverman, D.T. Dodds, F.V. Silva, P. Ordentlich, Y. Yan, F.J. Gonzalez, R.A. Heyman, D.J. Mangelsdorf, D.D. Moore, A natural product that lowers cholesterol as an antagonist ligand for FXR, Science. 296 (2002) 1703–1706. doi:10.1126/science.1072891.
[149] Y. Zhang, H.R. Kast-Woelbern, P.A. Edwards, Natural structural variants of the nuclear receptor farnesoid X receptor affect transcriptional activation, J. Biol. Chem. 278 (2003) 104–110. doi:10.1074/jbc.M209505200.
[150] D.D. Moore, S. Kato, W. Xie, D.J. Mangelsdorf, D.R. Schmidt, R. Xiao, S.A. Kliewer, International Union of Pharmacology. LXII. The NR1H and NR1I receptors: constitutive androstane receptor, pregnene X receptor, farnesoid X receptor alpha, farnesoid X receptor beta, liver X receptor alpha, liver X receptor beta, and vitamin D receptor, Pharmacol. Rev. 58 (2006) 742–759. doi:10.1124/pr.58.4.6.
[151] R.M. Huber, K. Murphy, B. Miao, J.R. Link, M.R. Cunningham, M.J. Rupar, P.L. Gunyuzlu, T.F. Haws, A. Kassam, F. Powell, G.F. Hollis, P.R. Young, R. Mukherjee, T.C. Burn, Generation of multiple farnesoid-X-receptor isoforms through the use of alternative promoters, Gene. 290 (2002) 35–43.

Chapter 2
59
[152] F.Y. Lee, H. Lee, M.L. Hubbert, P.A. Edwards, Y. Zhang, FXR, a multipurpose nuclear receptor, Trends Biochem. Sci. 31 (2006) 572–580. doi:10.1016/j.tibs.2006.08.002.
[153] J.E. Balmer, R. Blomhoff, Gene expression regulation by retinoic acid, J. Lipid Res. 43 (2002) 1773–1808.
[154] L. Panariello, L. Quadro, S. Trematerra, V. Colantuoni, Identification of a novel retinoic acid response element in the promoter region of the retinol-binding protein gene, J. Biol. Chem. 271 (1996) 25524–25532.
[155] J. Mercader, N. Granados, M.L. Bonet, A. Palou, All-trans retinoic acid decreases murine adipose retinol binding protein 4 production, Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 22 (2008) 363–372. doi:10.1159/000149815.
[156] D.-C. Manolescu, A. Sima, P.V. Bhat, All-trans retinoic acid lowers serum retinol-binding protein 4 concentrations and increases insulin sensitivity in diabetic mice, J. Nutr. 140 (2010) 311–316. doi:10.3945/jn.109.115147.
[157] R. Zolfaghari, A.C. Ross, An essential set of basic DNA response elements is required for receptor-dependent transcription of the lecithin:retinol acyltransferase (Lrat) gene, Arch. Biochem. Biophys. 489 (2009) 1–9. doi:10.1016/j.abb.2009.08.001.
[158] R. Zolfaghari, A.C. Ross, Lecithin:retinol acyltransferase from mouse and rat liver. CDNA cloning and liver-specific regulation by dietary vitamin a and retinoic acid, J. Lipid Res. 41 (2000) 2024–2034.
[159] S.-H. Yi, Y. Zhang, D. Tang, L. Zhu, Mechanical force and tensile strain activated hepatic stellate cells and inhibited retinol metabolism, Biotechnol. Lett. 37 (2015) 1141–1152. doi:10.1007/s10529-015-1785-5.
[160] K.B. Laursen, V. Kashyap, J. Scandura, L.J. Gudas, An alternative retinoic acid-responsive Stra6 promoter regulated in response to retinol deficiency, J. Biol. Chem. 290 (2015) 4356–4366. doi:10.1074/jbc.M114.613968.
[161] J.R.M. Plass, O. Mol, J. Heegsma, M. Geuken, K.N. Faber, P.L.M. Jansen, M. Müller, Farnesoid X receptor and bile salts are involved in transcriptional regulation of the gene encoding the human bile salt export pump, Hepatol. Baltim. Md. 35 (2002) 589–596. doi:10.1053/jhep.2002.31724.
[162] J.-F. Landrier, J.J. Eloranta, S.R. Vavricka, G.A. Kullak-Ublick, The nuclear receptor for bile acids, FXR, transactivates human organic solute transporter-alpha and -beta genes, Am. J. Physiol. Gastrointest. Liver Physiol. 290 (2006) G476-485. doi:10.1152/ajpgi.00430.2005.
[163] P.C. Pircher, J.L. Kitto, M.L. Petrowski, R.K. Tangirala, E.D. Bischoff, I.G. Schulman, S.K. Westin, Farnesoid X receptor regulates bile acid-amino acid conjugation, J. Biol. Chem. 278 (2003) 27703–27711. doi:10.1074/jbc.M302128200.
[164] J.A. Holt, G. Luo, A.N. Billin, J. Bisi, Y.Y. McNeill, K.F. Kozarsky, M. Donahee, D.Y. Wang, T.A. Mansfield, S.A. Kliewer, B. Goodwin, S.A. Jones, Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis, Genes Dev. 17 (2003) 1581–1591. doi:10.1101/gad.1083503.
[165] H.K. Lee, Y.K. Lee, S.H. Park, Y.S. Kim, S.H. Park, J.W. Lee, H.B. Kwon, J. Soh, D.D. Moore, H.S. Choi, Structure and expression of the orphan nuclear receptor SHP gene, J. Biol. Chem. 273 (1998) 14398–14402.
[166] B. Goodwin, S.A. Jones, R.R. Price, M.A. Watson, D.D. McKee, L.B. Moore, C. Galardi, J.G. Wilson, M.C. Lewis, M.E. Roth, P.R. Maloney, T.M. Willson, S.A.

Bile acid and Vitamin A homeostasis intertwined
60
Kliewer, A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis, Mol. Cell. 6 (2000) 517–526.
[167] A. del Castillo-Olivares, G. Gil, Suppression of sterol 12alpha-hydroxylase transcription by the short heterodimer partner: insights into the repression mechanism, Nucleic Acids Res. 29 (2001) 4035–4042.
[168] L.A. Denson, E. Sturm, W. Echevarria, T.L. Zimmerman, M. Makishima, D.J. Mangelsdorf, S.J. Karpen, The orphan nuclear receptor, shp, mediates bile acid-induced inhibition of the rat bile acid transporter, ntcp, Gastroenterology. 121 (2001) 140–147.
[169] E. Neimark, F. Chen, X. Li, B.L. Shneider, Bile acid-induced negative feedback regulation of the human ileal bile acid transporter, Hepatol. Baltim. Md. 40 (2004) 149–156. doi:10.1002/hep.20295.
[170] J. Sinha, F. Chen, T. Miloh, R.C. Burns, Z. Yu, B.L. Shneider, beta-Klotho and FGF-15/19 inhibit the apical sodium-dependent bile acid transporter in enterocytes and cholangiocytes, Am. J. Physiol. Gastrointest. Liver Physiol. 295 (2008) G996–G1003. doi:10.1152/ajpgi.90343.2008.
[171] M. Crestani, A. Sadeghpour, D. Stroup, G. Galli, J.Y. Chiang, Transcriptional activation of the cholesterol 7alpha-hydroxylase gene (CYP7A) by nuclear hormone receptors, J. Lipid Res. 39 (1998) 2192–2200.
[172] L.A. Denson, K.L. Auld, D.S. Schiek, M.H. McClure, D.J. Mangelsdorf, S.J. Karpen, Interleukin-1beta suppresses retinoid transactivation of two hepatic transporter genes involved in bile formation, J. Biol. Chem. 275 (2000) 8835–8843.
[173] S. Xu, A.-Q. Sun, F.J. Suchy, A novel RARα/CAR-mediated mechanism for regulation of human organic solute transporter-β gene expression, Am. J. Physiol. Gastrointest. Liver Physiol. 306 (2014) G154-162. doi:10.1152/ajpgi.00138.2013.
[174] S.-Y. Cai, H. He, T. Nguyen, A. Mennone, J.L. Boyer, Retinoic acid represses CYP7A1 expression in human hepatocytes and HepG2 cells by FXR/RXR-dependent and independent mechanisms, J. Lipid Res. 51 (2010) 2265–2274. doi:10.1194/jlr.M005546.
[175] D.R. Schmidt, S.R. Holmstrom, K. Fon Tacer, A.L. Bookout, S.A. Kliewer, D.J. Mangelsdorf, Regulation of bile acid synthesis by fat-soluble vitamins A and D, J. Biol. Chem. 285 (2010) 14486–14494. doi:10.1074/jbc.M110.116004.
[176] M.O. Hoeke, J. Heegsma, M. Hoekstra, H. Moshage, K.N. Faber, Human FXR regulates SHP expression through direct binding to an LRH-1 binding site, independent of an IR-1 and LRH-1, PloS One. 9 (2014) e88011. doi:10.1371/journal.pone.0088011.
[177] D. Jahn, D. Sutor, D. Dorbath, J. Weiß, O. Götze, J. Schmitt, H.M. Hermanns, A. Geier, Farnesoid X receptor-dependent and -independent pathways mediate the transcriptional control of human fibroblast growth factor 19 by vitamin A, Biochim. Biophys. Acta. 1859 (2016) 381–392. doi:10.1016/j.bbagrm.2015.12.007.
[178] Y. He, L. Gong, Y. Fang, Q. Zhan, H.-X. Liu, Y. Lu, G.L. Guo, L. Lehman-McKeeman, J. Fang, Y.-J.Y. Wan, The role of retinoic acid in hepatic lipid homeostasis defined by genomic binding and transcriptome profiling, BMC Genomics. 14 (2013) 575. doi:10.1186/1471-2164-14-575.
[179] F. Yang, Y. He, H.-X. Liu, J. Tsuei, X. Jiang, L. Yang, Z.-T. Wang, Y.-J.Y. Wan, All-trans retinoic acid regulates hepatic bile acid homeostasis, Biochem. Pharmacol. 91 (2014) 483–489. doi:10.1016/j.bcp.2014.08.018.

Chapter 2
61
[180] A. Mamoon, A. Subauste, M.C. Subauste, J. Subauste, Retinoic acid regulates several genes in bile acid and lipid metabolism via upregulation of small heterodimer partner in hepatocytes, Gene. 550 (2014) 165–170. doi:10.1016/j.gene.2014.07.017.
[181] A. Kassam, B. Miao, P.R. Young, R. Mukherjee, Retinoid X receptor (RXR) agonist-induced antagonism of farnesoid X receptor (FXR) activity due to absence of coactivator recruitment and decreased DNA binding, J. Biol. Chem. 278 (2003) 10028–10032. doi:10.1074/jbc.M208312200.
[182] M.O. Hoeke, J.R.M. Plass, J. Heegsma, M. Geuken, D. van Rijsbergen, J.F.W. Baller, F. Kuipers, H. Moshage, P.L.M. Jansen, K.N. Faber, Low retinol levels differentially modulate bile salt-induced expression of human and mouse hepatic bile salt transporters, Hepatol. Baltim. Md. 49 (2009) 151–159. doi:10.1002/hep.22661.
[183] L. Zhan, H.-X. Liu, Y. Fang, B. Kong, Y. He, X.-B. Zhong, J. Fang, Y.-J.Y. Wan, G.L. Guo, Genome-wide binding and transcriptome analysis of human farnesoid X receptor in primary human hepatocytes, PloS One. 9 (2014) e105930. doi:10.1371/journal.pone.0105930.
[184] C. Langhi, E. Pedraz-Cuesta, D. Haro, P.F. Marrero, J.C. Rodríguez, Regulation of human class I alcohol dehydrogenases by bile acids, J. Lipid Res. 54 (2013) 2475–2484. doi:10.1194/jlr.M039404.
[185] E.H. Harrison, Bile salt-dependent, neutral cholesteryl ester hydrolase of rat liver: possible relationship with pancreatic cholesteryl ester hydrolase, Biochim. Biophys. Acta. 963 (1988) 28–34.
[186] J.H. Prystowsky, J.E. Smith, D.S. Goodman, Retinyl palmitate hydrolase activity in normal rat liver., J. Biol. Chem. 256 (1981) 4498–4503.
[187] E.H. Harrison, M.Z. Gad, Hydrolysis of retinyl palmitate by enzymes of rat pancreas and liver. Differentiation of bile salt-dependent and bile salt-independent, neutral retinyl ester hydrolases in rat liver, J. Biol. Chem. 264 (1989) 17142–17147.
[188] W.S. Blaner, J.H. Prystowsky, J.E. Smith, D.S. Goodman, Rat liver retinyl palmitate hydrolase activity. Relationship to cholesteryl oleate and triolein hydrolase activities, Biochim. Biophys. Acta. 794 (1984) 419–427.
[189] D. Lombardo, O. Guy, Studies on the substrate specificity of a carboxyl ester hydrolase from human pancreatic juice. II. Action on cholesterol esters and lipid-soluble vitamin esters, Biochim. Biophys. Acta. 611 (1980) 147–155.
[190] A.M. van Bennekum, L. Li, R. Piantedosi, R. Shamir, S. Vogel, E.A. Fisher, W.S. Blaner, E.H. Harrison, Carboxyl ester lipase overexpression in rat hepatoma cells and CEL deficiency in mice have no impact on hepatic uptake or metabolism of chylomicron-retinyl ester, Biochemistry. 38 (1999) 4150–4156. doi:10.1021/bi981680+.
[191] K.M. Rigtrup, B. Kakkad, D.E. Ong, Purification and partial characterization of a retinyl ester hydrolase from the brush border of rat small intestine mucosa: probable identity with brush border phospholipase B, Biochemistry. 33 (1994) 2661–2666.
[192] T. Matsuura, M.Z. Gad, E.H. Harrison, A.C. Ross, Lecithin:retinol acyltransferase and retinyl ester hydrolase activities are differentially regulated by retinoids and have distinct distributions between hepatocyte and nonparenchymal cell fractions of rat liver, J. Nutr. 127 (1997) 218–224.
[193] J. Kluwe, N. Wongsiriroj, J.S. Troeger, G.-Y. Gwak, D.H. Dapito, J.-P. Pradere, H. Jiang, M. Siddiqi, R. Piantedosi, S.M. O’Byrne, W.S. Blaner, R.F. Schwabe, Absence of hepatic stellate cell retinoid lipid droplets does not enhance hepatic

Bile acid and Vitamin A homeostasis intertwined
62
fibrosis but decreases hepatic carcinogenesis, Gut. 60 (2011) 1260–1268. doi:10.1136/gut.2010.209551.
[194] N. Testerink, M. Ajat, M. Houweling, J.F. Brouwers, V.V. Pully, H.-J. van Manen, C. Otto, J.B. Helms, A.B. Vaandrager, Replacement of retinyl esters by polyunsaturated triacylglycerol species in lipid droplets of hepatic stellate cells during activation, PloS One. 7 (2012) e34945. doi:10.1371/journal.pone.0034945.
[195] B. Gao, S. Radaeva, W.-I. Jeong, Activation of natural killer cells inhibits liver fibrosis: a novel strategy to treat liver fibrosis, Expert Rev. Gastroenterol. Hepatol. 1 (2007) 173–180. doi:10.1586/17474124.1.1.173.
[196] R. Blomhoff, M. Rasmussen, A. Nilsson, K.R. Norum, T. Berg, W.S. Blaner, M. Kato, J.R. Mertz, D.S. Goodman, U. Eriksson, Hepatic retinol metabolism. Distribution of retinoids, enzymes, and binding proteins in isolated rat liver cells, J. Biol. Chem. 260 (1985) 13560–13565.
[197] W.S. Blaner, H.F. Hendriks, A. Brouwer, A.M. de Leeuw, D.L. Knook, D.S. Goodman, Retinoids, retinoid-binding proteins, and retinyl palmitate hydrolase distributions in different types of rat liver cells, J. Lipid Res. 26 (1985) 1241–1251.
[198] D.N. D’Ambrosio, J.L. Walewski, R.D. Clugston, P.D. Berk, R.A. Rippe, W.S. Blaner, Distinct populations of hepatic stellate cells in the mouse liver have different capacities for retinoid and lipid storage, PloS One. 6 (2011) e24993. doi:10.1371/journal.pone.0024993.
[199] J.R. Phillips, P. Angulo, T. Petterson, K.D. Lindor, Fat-soluble vitamin levels in patients with primary biliary cirrhosis, Am. J. Gastroenterol. 96 (2001) 2745–2750. doi:10.1111/j.1572-0241.2001.04134.x.
[200] S.J. Muñoz, J.E. Heubi, W.F. Balistreri, W.C. Maddrey, Vitamin E deficiency in primary biliary cirrhosis: gastrointestinal malabsorption, frequency and relationship to other lipid-soluble vitamins, Hepatol. Baltim. Md. 9 (1989) 525–531.
[201] B.L. Shneider, J.C. Magee, J.A. Bezerra, B. Haber, S.J. Karpen, T. Raghunathan, P. Rosenthal, K. Schwarz, F.J. Suchy, N. Kerkar, Y. Turmelle, P.F. Whitington, P.R. Robuck, R.J. Sokol, Childhood Liver Disease Research Education Network (ChiLDREN), Efficacy of fat-soluble vitamin supplementation in infants with biliary atresia, Pediatrics. 130 (2012) e607-614. doi:10.1542/peds.2011-1423.
[202] R.A. Jorgensen, K.D. Lindor, J.S. Sartin, N.F. LaRusso, R.H. Wiesner, Serum lipid and fat-soluble vitamin levels in primary sclerosing cholangitis, J. Clin. Gastroenterol. 20 (1995) 215–219.
[203] Y.-M. Shen, J.-F. Wu, H.-Y. Hsu, Y.-H. Ni, M.-H. Chang, Y.-W. Liu, H.-S. Lai, W.-M. Hsu, H.-L. Weng, H.-L. Chen, Oral absorbable fat-soluble vitamin formulation in pediatric patients with cholestasis, J. Pediatr. Gastroenterol. Nutr. 55 (2012) 587–591. doi:10.1097/MPG.0b013e31825c9732.
[204] M.A. Leo, C.S. Lieber, Hepatic vitamin A depletion in alcoholic liver injury, N. Engl. J. Med. 307 (1982) 597–601. doi:10.1056/NEJM198209023071006.
[205] M.A. Leo, C.S. Lieber, Alcohol, vitamin A, and beta-carotene: adverse interactions, including hepatotoxicity and carcinogenicity, Am. J. Clin. Nutr. 69 (1999) 1071–1085.
[206] G.V. Chaves, S.E. Pereira, C.J. Saboya, D. Spitz, C.S. Rodrigues, A. Ramalho, Association between liver vitamin A reserves and severity of nonalcoholic fatty liver disease in the class III obese following bariatric surgery, Obes. Surg. 24 (2014) 219–224. doi:10.1007/s11695-013-1087-8.

Chapter 2
63
[207] L. de Souza Valente da Silva, G. Valeria da Veiga, R.A. Ramalho, Association of serum concentrations of retinol and carotenoids with overweight in children and adolescents, Nutr. Burbank Los Angel. Cty. Calif. 23 (2007) 392–397. doi:10.1016/j.nut.2007.02.009.
[208] M.L. Neuhouser, C.L. Rock, A.L. Eldridge, A.R. Kristal, R.E. Patterson, D.A. Cooper, D. Neumark-Sztainer, L.J. Cheskin, M.D. Thornquist, Serum concentrations of retinol, alpha-tocopherol and the carotenoids are influenced by diet, race and obesity in a sample of healthy adolescents, J. Nutr. 131 (2001) 2184–2191.
[209] D. Bitetto, N. Bortolotti, E. Falleti, S. Vescovo, C. Fabris, G. Fattovich, A. Cussigh, S. Cmet, E. Fornasiere, E. Ceriani, M. Pirisi, P. Toniutto, Vitamin A deficiency is associated with hepatitis C virus chronic infection and with unresponsiveness to interferon-based antiviral therapy, Hepatol. Baltim. Md. 57 (2013) 925–933. doi:10.1002/hep.26186.
[210] W. a. F. Peres, G.V. Chaves, J.C.S. Gonçalves, A. Ramalho, H.S.M. Coelho, Vitamin A deficiency in patients with hepatitis C virus-related chronic liver disease, Br. J. Nutr. 106 (2011) 1724–1731. doi:10.1017/S0007114511002145.
[211] M.L.G. Saron, H.T. Godoy, G. Hessel, Nutritional status of patients with biliary atresia and autoimmune hepatitis related to serum levels of vitamins A, D and E, Arq. Gastroenterol. 46 (2009) 62–68.
[212] C.C. Chow, W.F. Mieler, Vitamin A deficiency and xerophthalmic fundus in autoimmune hepatitis and cirrhosis, Retin. Cases Brief Rep. 8 (2014) 164–166. doi:10.1097/ICB.0000000000000031.
[213] U. Beuers, M. Trauner, P. Jansen, R. Poupon, New paradigms in the treatment of hepatic cholestasis: from UDCA to FXR, PXR and beyond, J. Hepatol. 62 (2015) S25-37. doi:10.1016/j.jhep.2015.02.023.
[214] C.D. Schteingart, A.F. Hofmann, Synthesis of 24-nor-5 beta-cholan-23-oic acid derivatives: a convenient and efficient one-carbon degradation of the side chain of natural bile acids, J. Lipid Res. 29 (1988) 1387–1395.
[215] P. Fickert, M. Wagner, H.-U. Marschall, A. Fuchsbichler, G. Zollner, O. Tsybrovskyy, K. Zatloukal, J. Liu, M.P. Waalkes, C. Cover, H. Denk, A.F. Hofmann, H. Jaeschke, M. Trauner, 24-norUrsodeoxycholic acid is superior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice, Gastroenterology. 130 (2006) 465–481. doi:10.1053/j.gastro.2005.10.018.
[216] S. Mudaliar, R.R. Henry, A.J. Sanyal, L. Morrow, H.-U. Marschall, M. Kipnes, L. Adorini, C.I. Sciacca, P. Clopton, E. Castelloe, P. Dillon, M. Pruzanski, D. Shapiro, Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease, Gastroenterology. 145 (2013) 574-582.e1. doi:10.1053/j.gastro.2013.05.042.
[217] B.A. Neuschwander-Tetri, R. Loomba, A.J. Sanyal, J.E. Lavine, M.L. Van Natta, M.F. Abdelmalek, N. Chalasani, S. Dasarathy, A.M. Diehl, B. Hameed, K.V. Kowdley, A. McCullough, N. Terrault, J.M. Clark, J. Tonascia, E.M. Brunt, D.E. Kleiner, E. Doo, NASH Clinical Research Network, Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial, Lancet Lond. Engl. 385 (2015) 956–965. doi:10.1016/S0140-6736(14)61933-4.
[218] J.R.F. Walters, I.M. Johnston, J.D. Nolan, C. Vassie, M.E. Pruzanski, D.A. Shapiro, The response of patients with bile acid diarrhoea to the farnesoid X

Bile acid and Vitamin A homeostasis intertwined
64
receptor agonist obeticholic acid, Aliment. Pharmacol. Ther. 41 (2015) 54–64. doi:10.1111/apt.12999.
[219] F. Nevens, P. Andreone, G. Mazzella, S.I. Strasser, C. Bowlus, P. Invernizzi, J.P.H. Drenth, P.J. Pockros, J. Regula, U. Beuers, M. Trauner, D.E. Jones, A. Floreani, S. Hohenester, V. Luketic, M. Shiffman, K.J. van Erpecum, V. Vargas, C. Vincent, G.M. Hirschfield, H. Shah, B. Hansen, K.D. Lindor, H.-U. Marschall, K.V. Kowdley, R. Hooshmand-Rad, T. Marmon, S. Sheeron, R. Pencek, L. MacConell, M. Pruzanski, D. Shapiro, POISE Study Group, A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis, N. Engl. J. Med. 375 (2016) 631–643. doi:10.1056/NEJMoa1509840.
[220] F.J. Rosales, S.J. Ritter, R. Zolfaghari, J.E. Smith, A.C. Ross, Effects of acute inflammation on plasma retinol, retinol-binding protein, and its mRNA in the liver and kidneys of vitamin A-sufficient rats, J. Lipid Res. 37 (1996) 962–971.
[221] A. Tomkins, Assessing micronutrient status in the presence of inflammation, J. Nutr. 133 (2003) 1649S-1655S.
[222] J.A. Louw, A. Werbeck, M.E. Louw, T.J. Kotze, R. Cooper, D. Labadarios, Blood vitamin concentrations during the acute-phase response, Crit. Care Med. 20 (1992) 934–941.
[223] European Association for the Study of the Liver, EASL Clinical Practice Guidelines: management of cholestatic liver diseases, J. Hepatol. 51 (2009) 237–267. doi:10.1016/j.jhep.2009.04.009.
[224] R. Chapman, J. Fevery, A. Kalloo, D.M. Nagorney, K.M. Boberg, B. Shneider, G.J. Gores, American Association for the Study of Liver Diseases, Diagnosis and management of primary sclerosing cholangitis, Hepatol. Baltim. Md. 51 (2010) 660–678. doi:10.1002/hep.23294.
[225] T. Pérez Fernández, P. López Serrano, E. Tomás, M.L. Gutiérrez, J.L. Lledó, G. Cacho, C. Santander, C.M. Fernández Rodríguez, Diagnostic and therapeutic approach to cholestatic liver disease, Rev. Esp. Enfermedades Dig. Organo Of. Soc. Esp. Patol. Dig. 96 (2004) 60–73.
[226] O. Amédée-Manesme, M.S. Mourey, J. Therasse, M. Couturier, F. Alvarez, A. Hanck, O. Bernard, Short- and long-term vitamin A treatment in children with cholestasis, Am. J. Clin. Nutr. 47 (1988) 690–693.
[227] S.R. Sijtsma, J.H. Rombout, M.J. Dohmen, C.E. West, A.J. van der Zijpp, Effect of vitamin A deficiency on the activity of macrophages in Newcastle disease virus-infected chickens, Vet. Immunol. Immunopathol. 28 (1991) 17–27.
[228] M. Ongsakul, S. Sirisinha, A.J. Lamb, Impaired blood clearance of bacteria and phagocytic activity in vitamin A-deficient rats, Proc. Soc. Exp. Biol. Med. Soc. Exp. Biol. Med. N. Y. N. 178 (1985) 204–208.
[229] Z. Zhao, D.M. Murasko, A.C. Ross, The role of vitamin A in natural killer cell cytotoxicity, number and activation in the rat, Nat. Immun. 13 (1994) 29–41.
[230] P. Aukrust, F. Müller, T. Ueland, A.M. Svardal, R.K. Berge, S.S. Frøland, Decreased vitamin A levels in common variable immunodeficiency: vitamin A supplementation in vivo enhances immunoglobulin production and downregulates inflammatory responses, Eur. J. Clin. Invest. 30 (2000) 252–259.
[231] X. Wang, C. Allen, M. Ballow, Retinoic acid enhances the production of IL-10 while reducing the synthesis of IL-12 and TNF-alpha from LPS-stimulated monocytes/macrophages, J. Clin. Immunol. 27 (2007) 193–200. doi:10.1007/s10875-006-9068-5.

Chapter 2
65
[232] Y. Yang, M.S. Vacchio, J.D. Ashwell, 9-cis-retinoic acid inhibits activation-driven T-cell apoptosis: implications for retinoid X receptor involvement in thymocyte development, Proc. Natl. Acad. Sci. U. S. A. 90 (1993) 6170–6174.
[233] H. He, A. Mennone, J.L. Boyer, S.-Y. Cai, Combination of retinoic acid and ursodeoxycholic acid attenuates liver injury in bile duct-ligated rats and human hepatic cells, Hepatol. Baltim. Md. 53 (2011) 548–557. doi:10.1002/hep.24047.
[234] S.-Y. Cai, A. Mennone, C.J. Soroka, J.L. Boyer, All-trans-retinoic acid improves cholestasis in α-naphthylisothiocyanate-treated rats and Mdr2-/- mice, J. Pharmacol. Exp. Ther. 349 (2014) 94–98. doi:10.1124/jpet.113.209353.
[235] D.N. Assis, O. Abdelghany, S.-Y. Cai, A.A. Gossard, J.E. Eaton, J.C. Keach, Y. Deng, K.D.R. Setchell, M. Ciarleglio, K.D. Lindor, J.L. Boyer, Combination Therapy of All-Trans Retinoic Acid With Ursodeoxycholic Acid in Patients With Primary Sclerosing Cholangitis: A Human Pilot Study, J. Clin. Gastroenterol. (2016). doi:10.1097/MCG.0000000000000591.
[236] T. Shimada, A.C. Ross, D.D. Muccio, W.J. Brouillette, Y.F. Shealy, Regulation of hepatic lecithin:retinol acyltransferase activity by retinoic acid receptor-selective retinoids, Arch. Biochem. Biophys. 344 (1997) 220–227. doi:10.1006/abbi.1997.0209.
[237] A. Bosch, S.P. Bertran, Y. Lu, A. Garcia, A.M. Jones, M.I. Dawson, E.F. Farias, Reversal by RARα agonist Am580 of c-Myc-induced imbalance in RARα/RARγ expression during MMTV-Myc tumorigenesis, Breast Cancer Res. BCR. 14 (2012) R121. doi:10.1186/bcr3247.
[238] L.B. Todaro, M.J. Veloso, P.B. Campodónico, L.I. Puricelli, E.F. Farías, E.D. Bal de Kier Joffé, A clinically relevant bi-cellular murine mammary tumor model as a useful tool for evaluating the effect of retinoic acid signaling on tumor progression, Breast Cancer Tokyo Jpn. 20 (2013) 342–356. doi:10.1007/s12282-012-0342-5.
[239] K. Kohlstedt, C. Gershome, C. Trouvain, W.-K. Hofmann, S. Fichtlscherer, I. Fleming, Angiotensin-converting enzyme (ACE) inhibitors modulate cellular retinol-binding protein 1 and adiponectin expression in adipocytes via the ACE-dependent signaling cascade, Mol. Pharmacol. 75 (2009) 685–692. doi:10.1124/mol.108.051631.
[240] O. Ziouzenkova, G. Orasanu, M. Sharlach, T.E. Akiyama, J.P. Berger, J. Viereck, J.A. Hamilton, G. Tang, G.G. Dolnikowski, S. Vogel, G. Duester, J. Plutzky, Retinaldehyde represses adipogenesis and diet-induced obesity, Nat. Med. 13 (2007) 695–702. doi:10.1038/nm1587.
[241] D.J. Mangelsdorf, K. Umesono, S.A. Kliewer, U. Borgmeyer, E.S. Ong, R.M. Evans, A direct repeat in the cellular retinol-binding protein type II gene confers differential regulation by RXR and RAR, Cell. 66 (1991) 555–561.
[242] H. Nakshatri, P. Chambon, The directly repeated RG(G/T)TCA motifs of the rat and mouse cellular retinol-binding protein II genes are promiscuous binding sites for RAR, RXR, HNF-4, and ARP-1 homo- and heterodimers, J. Biol. Chem. 269 (1994) 890–902.
[243] A.W. Ross, C.A. Webster, J.G. Mercer, K.M. Moar, F.J. Ebling, S. Schuhler, P. Barrett, P.J. Morgan, Photoperiodic regulation of hypothalamic retinoid signaling: association of retinoid X receptor gamma with body weight, Endocrinology. 145 (2004) 13–20. doi:10.1210/en.2003-0838.
[244] M.S. Levin, A.E. Davis, Retinoic acid increases cellular retinol binding protein II mRNA and retinol uptake in the human intestinal Caco-2 cell line, J. Nutr. 127 (1997) 13–17.

Bile acid and Vitamin A homeostasis intertwined
66
[245] L.N. Wei, W.S. Blaner, D.S. Goodman, M.C. Nguyen-Huu, Regulation of the cellular retinoid-binding proteins and their messenger ribonucleic acids during P19 embryonal carcinoma cell differentiation induced by retinoic acid, Mol. Endocrinol. Baltim. Md. 3 (1989) 454–463. doi:10.1210/mend-3-3-454.
[246] L.N. Wei, C.H. Lee, Demethylation in the 5’-flanking region of mouse cellular retinoic acid binding protein-I gene is associated with its high level of expression in mouse embryos and facilitates its induction by retinoic acid in P19 embryonal carcinoma cells, Dev. Dyn. Off. Publ. Am. Assoc. Anat. 201 (1994) 1–10. doi:10.1002/aja.1002010102.
[247] A.C. Chen, L.J. Gudas, An analysis of retinoic acid-induced gene expression and metabolism in AB1 embryonic stem cells, J. Biol. Chem. 271 (1996) 14971–14980.
[248] D.A. Kleinjan, S. Dekker, M.J. Vaessen, F. Grosveld, Regulation of the CRABP-I gene during mouse embryogenesis, Mech. Dev. 67 (1997) 157–169.
[249] R. Faraonio, M. Galdieri, V. Colantuoni, Cellular retinoic-acid-binding-protein and retinol-binding-protein mRNA expression in the cells of the rat seminiferous tubules and their regulation by retinoids, Eur. J. Biochem. FEBS. 211 (1993) 835–842.
[250] L.N. Wei, C.H. Lee, P. Filipcik, L. Chang, Regulation of the mouse cellular retinoic acid-binding protein-I gene by thyroid hormone and retinoids in transgenic mouse embryos and P19 cells, J. Endocrinol. 155 (1997) 35–46.
[251] A.L. Means, J.R. Thompson, L.J. Gudas, Transcriptional regulation of the cellular retinoic acid binding protein I gene in F9 teratocarcinoma cells, Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 11 (2000) 71–82.
[252] R. Taneja, P. Bouillet, J.F. Boylan, M.P. Gaub, B. Roy, L.J. Gudas, P. Chambon, Reexpression of retinoic acid receptor (RAR) gamma or overexpression of RAR alpha or RAR beta in RAR gamma-null F9 cells reveals a partial functional redundancy between the three RAR types, Proc. Natl. Acad. Sci. U. S. A. 92 (1995) 7854–7858.
[253] P.M. Amann, C. Hofmann, M. Freudenberger, S. Holland-Cunz, S.B. Eichmüller, A.V. Bazhin, Expression and activity of alcohol and aldehyde dehydrogenases in melanoma cells and in melanocytes, J. Cell. Biochem. 113 (2012) 792–799. doi:10.1002/jcb.23406.
[254] O. Ziouzenkova, J. Plutzky, Retinoid metabolism and nuclear receptor responses: New insights into coordinated regulation of the PPAR-RXR complex, FEBS Lett. 582 (2008) 32–38. doi:10.1016/j.febslet.2007.11.081.
[255] Y. Ohoka, A. Yokota-Nakatsuma, N. Maeda, H. Takeuchi, M. Iwata, Retinoic acid and GM-CSF coordinately induce retinal dehydrogenase 2 (RALDH2) expression through cooperation between the RAR/RXR complex and Sp1 in dendritic cells, PloS One. 9 (2014) e96512. doi:10.1371/journal.pone.0096512.
[256] G. Duester, M.L. Shean, M.S. McBride, M.J. Stewart, Retinoic acid response element in the human alcohol dehydrogenase gene ADH3: implications for regulation of retinoic acid synthesis, Mol. Cell. Biol. 11 (1991) 1638–1646.
[257] P.P. Harding, G. Duester, Retinoic acid activation and thyroid hormone repression of the human alcohol dehydrogenase gene ADH3, J. Biol. Chem. 267 (1992) 14145–14150.
[258] M. Zgombić, G. Duester, DNA elements mediating retinoid and thyroid hormone regulation of alcohol dehydrogenase gene expression, Adv. Exp. Med. Biol. 328 (1993) 571–580.

Chapter 2
67
[259] M.A. Gray, E.J. Squires, Effects of nuclear receptor transactivation on steroid hormone synthesis and gene expression in porcine Leydig cells, J. Steroid Biochem. Mol. Biol. 133 (2013) 93–100. doi:10.1016/j.jsbmb.2012.09.014.
[260] E. Pavez Loriè, J.C. Chamcheu, A. Vahlquist, H. Törmä, Both all-trans retinoic acid and cytochrome P450 (CYP26) inhibitors affect the expression of vitamin A metabolizing enzymes and retinoid biomarkers in organotypic epidermis, Arch. Dermatol. Res. 301 (2009) 475–485. doi:10.1007/s00403-009-0937-7.
[261] C.M. Soref, Y.P. Di, L. Hayden, Y.H. Zhao, M.A. Satre, R. Wu, Characterization of a novel airway epithelial cell-specific short chain alcohol dehydrogenase/reductase gene whose expression is up-regulated by retinoids and is involved in the metabolism of retinol, J. Biol. Chem. 276 (2001) 24194–24202. doi:10.1074/jbc.M100332200.
[262] Y. Li, Y. Zhang, J. Hill, H.-T. Kim, Q. Shen, R.P. Bissonnette, W.W. Lamph, P.H. Brown, The rexinoid, bexarotene, prevents the development of premalignant lesions in MMTV-erbB2 mice, Br. J. Cancer. 98 (2008) 1380–1388. doi:10.1038/sj.bjc.6604320.
[263] F. Cerignoli, X. Guo, B. Cardinali, C. Rinaldi, J. Casaletto, L. Frati, I. Screpanti, L.J. Gudas, A. Gulino, C.J. Thiele, G. Giannini, retSDR1, a short-chain retinol dehydrogenase/reductase, is retinoic acid-inducible and frequently deleted in human neuroblastoma cell lines, Cancer Res. 62 (2002) 1196–1204.
[264] L. Feng, R.E. Hernandez, J.S. Waxman, D. Yelon, C.B. Moens, Dhrs3a regulates retinoic acid biosynthesis through a feedback inhibition mechanism, Dev. Biol. 338 (2010) 1–14. doi:10.1016/j.ydbio.2009.10.029.
[265] Y.F. Wong, P.D. Wilson, R.J. Unwin, J.T. Norman, M. Arno, B.M. Hendry, Q. Xu, Retinoic acid receptor-dependent, cell-autonomous, endogenous retinoic acid signaling and its target genes in mouse collecting duct cells, PloS One. 7 (2012) e45725. doi:10.1371/journal.pone.0045725.
[266] X.Y. Li, A. Aström, E.A. Duell, L. Qin, C.E. Griffiths, J.J. Voorhees, Retinoic acid antagonizes basal as well as coal tar and glucocorticoid-induced cytochrome P4501A1 expression in human skin, Carcinogenesis. 16 (1995) 519–524.
[267] K. Inouye, T. Mae, S. Kondo, H. Ohkawa, Inhibitory effects of vitamin A and vitamin K on rat cytochrome P4501A1-dependent monooxygenase activity, Biochem. Biophys. Res. Commun. 262 (1999) 565–569. doi:10.1006/bbrc.1999.1240.
[268] S.R. Howell, M.A. Shirley, E.H. Ulm, Effects of retinoid treatment of rats on hepatic microsomal metabolism and cytochromes P450. Correlation between retinoic acid receptor/retinoid x receptor selectivity and effects on metabolic enzymes, Drug Metab. Dispos. Biol. Fate Chem. 26 (1998) 234–239.
[269] P.T. Vedell, Y. Lu, C.J. Grubbs, Y. Yin, H. Jiang, K.I. Bland, D.D. Muccio, D. Cvetkovic, M. You, R. Lubet, Effects on gene expression in rat liver after administration of RXR agonists: UAB30, 4-methyl-UAB30, and Targretin (Bexarotene), Mol. Pharmacol. 83 (2013) 698–708. doi:10.1124/mol.112.082404.
[270] P.M. Amann, K. Czaja, A.V. Bazhin, R. Rühl, S.B. Eichmüller, H.F. Merk, J.M. Baron, LRAT overexpression diminishes intracellular levels of biologically active retinoids and reduces retinoid antitumor efficacy in the murine melanoma B16F10 cell line, Skin Pharmacol. Physiol. 28 (2015) 205–212. doi:10.1159/000368806.
[271] R. Zolfaghari, A.C. Ross, Hepatocyte nuclear factor 4α (HNF4α) in coordination with retinoic acid receptors increases all-trans-retinoic acid-

Bile acid and Vitamin A homeostasis intertwined
68
dependent CYP26A1 gene expression in HepG2 human hepatocytes, J. Cell. Biochem. 115 (2014) 1740–1751. doi:10.1002/jcb.24839.
[272] S. Tay, L. Dickmann, V. Dixit, N. Isoherranen, A comparison of the roles of peroxisome proliferator-activated receptor and retinoic acid receptor on CYP26 regulation, Mol. Pharmacol. 77 (2010) 218–227. doi:10.1124/mol.109.059071.
[273] A.M. Anisfeld, H.R. Kast-Woelbern, M.E. Meyer, S.A. Jones, Y. Zhang, K.J. Williams, T. Willson, P.A. Edwards, Syndecan-1 expression is regulated in an isoform-specific manner by the farnesoid-X receptor, J. Biol. Chem. 278 (2003) 20420–20428. doi:10.1074/jbc.M302505200.
[274] C. Röhrl, K. Eigner, S. Fruhwürth, H. Stangl, Bile acids reduce endocytosis of high-density lipoprotein (HDL) in HepG2 cells, PloS One. 9 (2014) e102026. doi:10.1371/journal.pone.0102026.
[275] Y. Ma, Y. Huang, L. Yan, M. Gao, D. Liu, Synthetic FXR agonist GW4064 prevents diet-induced hepatic steatosis and insulin resistance, Pharm. Res. 30 (2013) 1447–1457. doi:10.1007/s11095-013-0986-7.
[276] S. Han, N. Sidell, Peroxisome-proliferator-activated-receptor gamma (PPARgamma) independent induction of CD36 in THP-1 monocytes by retinoic acid, Immunology. 106 (2002) 53–59.
[277] G.D. Norata, M. Ongari, P. Uboldi, F. Pellegatta, A.L. Catapano, Liver X receptor and retinoic X receptor agonists modulate the expression of genes involved in lipid metabolism in human endothelial cells, Int. J. Mol. Med. 16 (2005) 717–722.
[278] L. Malerød, M. Sporstøl, L.K. Juvet, S.A. Mousavi, T. Gjøen, T. Berg, N. Roos, W. Eskild, Bile acids reduce SR-BI expression in hepatocytes by a pathway involving FXR/RXR, SHP, and LRH-1, Biochem. Biophys. Res. Commun. 336 (2005) 1096–1105. doi:10.1016/j.bbrc.2005.08.237.
[279] A. Mencarelli, B. Renga, E. Distrutti, S. Fiorucci, Antiatherosclerotic effect of farnesoid X receptor, Am. J. Physiol. Heart Circ. Physiol. 296 (2009) H272-281. doi:10.1152/ajpheart.01075.2008.
[280] P.R. Manna, Retinoid regulated macrophage cholesterol efflux involves the steroidogenic acute regulatory protein, Data Brief. 7 (2016) 940–945. doi:10.1016/j.dib.2016.03.055.
[281] M. Ayaori, E. Yakushiji, M. Ogura, K. Nakaya, T. Hisada, H. Uto-Kondo, S. Takiguchi, Y. Terao, M. Sasaki, T. Komatsu, M. Iizuka, M. Yogo, Y. Uehara, H. Kagechika, T. Nakanishi, K. Ikewaki, Retinoic acid receptor agonists regulate expression of ATP-binding cassette transporter G1 in macrophages, Biochim. Biophys. Acta. 1821 (2012) 561–572. doi:10.1016/j.bbalip.2012.02.004.
[282] P. Costet, F. Lalanne, M.C. Gerbod-Giannone, J.R. Molina, X. Fu, E.G. Lund, L.J. Gudas, A.R. Tall, Retinoic acid receptor-mediated induction of ABCA1 in macrophages, Mol. Cell. Biol. 23 (2003) 7756–7766.
[283] M. Tachibana, M. Shinohara, Y. Yamazaki, C.-C. Liu, J. Rogers, G. Bu, T. Kanekiyo, Rescuing effects of RXR agonist bexarotene on aging-related synapse loss depend on neuronal LRP1, Exp. Neurol. 277 (2016) 1–9. doi:10.1016/j.expneurol.2015.12.003.
[284] F. Lalloyer, C. Fiévet, S. Lestavel, G. Torpier, J. van der Veen, V. Touche, S. Bultel, S. Yous, F. Kuipers, R. Paumelle, J.-C. Fruchart, B. Staels, A. Tailleux, The RXR agonist bexarotene improves cholesterol homeostasis and inhibits atherosclerosis progression in a mouse model of mixed dyslipidemia, Arterioscler. Thromb. Vasc. Biol. 26 (2006) 2731–2737. doi:10.1161/01.ATV.0000248101.93488.84.

Chapter 2
69
[285] K.-H. Song, T. Li, E. Owsley, S. Strom, J.Y.L. Chiang, Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha-hydroxylase gene expression, Hepatol. Baltim. Md. 49 (2009) 297–305. doi:10.1002/hep.22627.
[286] J. Liu, H. Lu, Y.-F. Lu, X. Lei, J.Y. Cui, E. Ellis, S.C. Strom, C.D. Klaassen, Potency of individual bile acids to regulate bile acid synthesis and transport genes in primary human hepatocyte cultures, Toxicol. Sci. Off. J. Soc. Toxicol. 141 (2014) 538–546. doi:10.1093/toxsci/kfu151.
[287] A. Szanto, S. Benko, I. Szatmari, B.L. Balint, I. Furtos, R. Rühl, S. Molnar, L. Csiba, R. Garuti, S. Calandra, H. Larsson, U. Diczfalusy, L. Nagy, Transcriptional regulation of human CYP27 integrates retinoid, peroxisome proliferator-activated receptor, and liver X receptor signaling in macrophages, Mol. Cell. Biol. 24 (2004) 8154–8166. doi:10.1128/MCB.24.18.8154-8166.2004.
[288] D. Jung, J.P. York, L. Wang, C. Yang, A. Zhang, H.L. Francis, P. Webb, W.L. McKeehan, G. Alpini, G.D. Lesage, D.D. Moore, X. Xia, FXR-induced secretion of FGF15/19 inhibits CYP27 expression in cholangiocytes through p38 kinase pathway, Pflugers Arch. 466 (2014) 1011–1019. doi:10.1007/s00424-013-1364-3.
[289] M. Ananthanarayanan, N. Balasubramanian, M. Makishima, D.J. Mangelsdorf, F.J. Suchy, Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor, J. Biol. Chem. 276 (2001) 28857–28865. doi:10.1074/jbc.M011610200.
[290] M. Le Vee, E. Jouan, B. Stieger, O. Fardel, Differential regulation of drug transporter expression by all-trans retinoic acid in hepatoma HepaRG cells and human hepatocytes, Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 48 (2013) 767–774. doi:10.1016/j.ejps.2013.01.005.
[291] Y. Liu, J. Binz, M.J. Numerick, S. Dennis, G. Luo, B. Desai, K.I. MacKenzie, T.A. Mansfield, S.A. Kliewer, B. Goodwin, S.A. Jones, Hepatoprotection by the farnesoid X receptor agonist GW4064 in rat models of intra- and extrahepatic cholestasis, J. Clin. Invest. 112 (2003) 1678–1687. doi:10.1172/JCI18945.
[292] D. Jung, M. Podvinec, U.A. Meyer, D.J. Mangelsdorf, M. Fried, P.J. Meier, G.A. Kullak-Ublick, Human organic anion transporting polypeptide 8 promoter is transactivated by the farnesoid X receptor/bile acid receptor, Gastroenterology. 122 (2002) 1954–1966.
[293] Q. Li, Y. Sai, Y. Kato, I. Tamai, A. Tsuji, Influence of drugs and nutrients on transporter gene expression levels in Caco-2 and LS180 intestinal epithelial cell lines, Pharm. Res. 20 (2003) 1119–1124.
[294] H.R. Kast, B. Goodwin, P.T. Tarr, S.A. Jones, A.M. Anisfeld, C.M. Stoltz, P. Tontonoz, S. Kliewer, T.M. Willson, P.A. Edwards, Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X-activated receptor, and constitutive androstane receptor, J. Biol. Chem. 277 (2002) 2908–2915. doi:10.1074/jbc.M109326200.
[295] S.E. Bates, L.A. Mickley, Y.N. Chen, N. Richert, J. Rudick, J.L. Biedler, A.T. Fojo, Expression of a drug resistance gene in human neuroblastoma cell lines: modulation by retinoic acid-induced differentiation, Mol. Cell. Biol. 9 (1989) 4337–4344.
[296] L.D. Teeter, T. Eckersberg, Y. Tsai, M.T. Kuo, Analysis of the Chinese hamster P-glycoprotein/multidrug resistance gene pgp1 reveals that the AP-1 site is essential for full promoter activity, Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 2 (1991) 429–437.

Bile acid and Vitamin A homeostasis intertwined
70
[297] T.P. Stromskaya, E.Y. Rybalkina, A.A. Shtil, T.N. Zabotina, N.A. Filippova, A.A. Stavrovskaya, Influence of exogenous RAR alpha gene on MDR1 expression and P-glycoprotein function in human and rodent cell lines, Br. J. Cancer. 77 (1998) 1718–1725.
[298] W. Chen, S.-Y. Cai, S. Xu, L.A. Denson, C.J. Soroka, J.L. Boyer, Nuclear receptors RXRalpha:RARalpha are repressors for human MRP3 expression, Am. J. Physiol. Gastrointest. Liver Physiol. 292 (2007) G1221-1227. doi:10.1152/ajpgi.00191.2006.
[299] H. Lee, Y. Zhang, F.Y. Lee, S.F. Nelson, F.J. Gonzalez, P.A. Edwards, FXR regulates organic solute transporters alpha and beta in the adrenal gland, kidney, and intestine, J. Lipid Res. 47 (2006) 201–214. doi:10.1194/jlr.M500417-JLR200.
[300] W. Chen, E. Owsley, Y. Yang, D. Stroup, J.Y. Chiang, Nuclear receptor-mediated repression of human cholesterol 7alpha-hydroxylase gene transcription by bile acids, J. Lipid Res. 42 (2001) 1402–1412.
[301] T.T. Lu, M. Makishima, J.J. Repa, K. Schoonjans, T.A. Kerr, J. Auwerx, D.J. Mangelsdorf, Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors, Mol. Cell. 6 (2000) 507–515.
[302] J. Li, P.C. Pircher, I.G. Schulman, S.K. Westin, Regulation of complement C3 expression by the bile acid receptor FXR, J. Biol. Chem. 280 (2005) 7427–7434. doi:10.1074/jbc.M411473200.

Chapter 2
71
Supplementary tables.
Table 1S.RXR/RAR/FXR targets genes involve in Vitamin A uptake, transport
and metabolism
Function Gene NRs Ligands (Ant)agoni-
st /
(De)activat
or
Expressio
n Up (↑),
down (↓),
no (→)
Refere-
nce
Vitamin A
Storage
LRAT
RAR all-trans-RA,
AM580, all-
trans-UAB8
Agonist ↑ [236]
RXR 9-cis-RA Agonist ↑
Vitamin A
transport
CRBP1 RARα Am580 Agonist ↑ [237]
RAR all-trans-RA Agonist ↑ [238]
RXR 9-cis-RA Agonist ↑ [239]
RAR all-trans-RA Agonist ↑
RAR/RXR ACE
inhibitors
Activator ↑
RXRα/PPA
Rγ
Retinaldehyd
e
Antagonist - [240]
CRBPII RAR/RXR Retinoic acid Agonist ↑ [241–
243]
RAR all-trans-RA Agonist ↑ [244]
RXR 9-cis-RA Agonist
CRABP
1
RAR/RXR Retinoic acid Agonist ↑ [245–
248]
RAR/RXR Retinoic
acid, retinol
Agonist → [249]
RAR/RXR Retinoic acid Agonist ↓ [250,25
1]
RBP4
RAR/RXR Retinoic acid Agonist ↑ [154]
RXRα/PPA
Rγ
Retinaldehyd
e
Antagonist - [240]
RAR all-trans-RA Agonist ↓ [155]
RAR/RXR Retinaldehyd
e,
Agonist ↓
RAR TTNPB Agonist ↓
RXR Methoprene Agonist ↓

Bile acid and Vitamin A homeostasis intertwined
72
RBPR2 RAR all-trans-RA Agonist ↓ [57]
STRA6 RAR Retinoic acid Agonist ↑ [252]
Vitamin A
metabolis
-m
RALDH
1/
ALDH1
RXR/PPAR
γ
All-trans RAL Antagonist ↑ [253,25
4]
RALDH
3/
ALDH6
RXR/PPAR
γ
All-trans RAL Antagonist ↑
RALDH
2
RAR/RXR Retinoic acid Agonist ↑ [255]
ADH1A FXR/RXR CDCA Agonist ↑ [184]
FXR/RXR GW4064 Agonist ↑
ADH1B FXR/RXR CDCA Agonist ↑
FXR/RXR GW4064 Agonist ↑
ADH1C
/ADH3
RARβ Retinoic acid Agonist ↑ [256–
258]
FXR GW4064 Agonist ↑ [183]
RDH12 FXR CDCA Agonist ↑ [259]
RDH16 RAR all-trans-RA Agonist ↓ [260]
RDH RAR all-trans-RA Agonist ↑ [261]
RXR 9-cis-RA Agonist ↑
DHRS3 RXR Bexarotene Agonist ↑ [262]
RAR all-trans-RA Agonist ↑ [263–
265]
CYP1A
1
RAR Retinoic acid Agonist ↓ [266,26
7]
CYP1A
2
RAR ALRT1550 Agonist ↓ [268]
RXR LG100268,
LGD 1069
Agonist ↓ [268]
CYP1B
1
RXR TRG, 4-Me-
UAB30,UAB
30
Agonist ↑ [269]
CYP3A RAR ALRT1550 Agonist ↓ [268]
RXR LG100268,
LGD 1069
Agonist ↑ [268]
CYP4A RAR ALRT1550 Agonist ↑ [268]
RXR LG100268, Agonist ↑ [268]

Chapter 2
73
LGD 1069
CYP26
A1
RARα Am580 Agonist ↑ [237]
RAR All-trans RA Agonist ↑ [270]
RAR All-trans RA Agonist ↑ [271]
RAR All-trans RA Agonist ↑ [272]
RXR+
RAR
BMS649+
BMS753
Agonist ↑ [105]
Agonist ↑
CYP26
B1
RAR All-trans RA Agonist ↑ [272]
RARα AM580 Agonist ↑
Others Syndec
an-1
(SDC-1)
FXR GW4064 Agonist ↑ [273]
ISX RAR/RXR Retinoic acid Agonist ↑ [31]
RAR TTNPB Agonist ↑
CD36 FXR CDCA Agonist ↓ [274]
FXR GW4064 Agonist ↓ [274,27
5]
RAR TTNPB, RA Agonist ↑ [276]
RXR LG153, RA Agonist ↑
SR-B1 RXR 9-cis-RA Agonist ↑ [277]
FXR GW4064,
CDCA
Agonist ↓ [278]
ABCA1 FXR INT-747 Agonist ↑ [279]
RAR All-trans RA Agonist ↑ [280,28
1]
RAR TTNPB Agonist ↑ [281,28
2]
RARγ/RXR All-trans RA Agonist ↑ [282]
RXR 9-cis-RA, Agonist ↑ [280],
RXR Bexarotein Agonist ↑ [283]
NPC1L
1
RXR Bexarotene Agonist ↓ [284]

Bile acid and Vitamin A homeostasis intertwined
74
Table 2S: RXR/RAR targets gene involve in bile acid synthesis and transport.
Function Target genes RXR or
RAR
Expression Up
(↑), down (↓), no
(→)
Reference
Bile acid
synthesis
CYP7A1 RAR/RXR ↓ (via FXR/RXR-
SHP/FGF19)
[174]
FXR/RXR ↓ (via
SHP/FGF19)
[166,285,286]
CYP27A1 RAR/RXR ↑ [287]
FXR/RXR ↓ (via FGF19) [288]
CYP8B1 RAR/RXR ↓ (via
SHP/FGF19)
[179,180]
FXR/RXR ↓ (via
SHP/FGF19)
[286]
BACS FXR/RXR ↑ [163]
BAAT FXR/RXR ↑ [163]
RAR/RXR ↓ (via SHP) [180]
Bile acid
transport
BSEP FXR/RXR ↑ [161,289]
RAR/RXR ↓ [290]
mMdr2/hMDR3 FXR ↑ [291]
NTCP RAR/RXR ↑ [168,172]
OATP1B3/
OATP8
FXR/RXR ↑ [292]
RAR/RXR ↓ [290]
MRP2 RAR/RXR ↑ [172,293]
FXR/RXR ↑ [294]
MDR1 RAR/RXR ↑ [290,295–297]
MRP3 RAR/RXR ↓ [298]
ASBT RAR/RXR ↓ [169,170]
I-BABP FXR/RXR ↑ [130,141]
OSTα/β RAR/RXR ↑ [173]
FXR/RXR ↑ [162,299]
Bile acid
homeostasis
regulators
SHP FXR/RXR ↑ [164,168,174,300,301]
RAR/
RXR
↑ [179,180]
FGF15/19 FXR/RXR ↑ [164,302]
RAR/RXR ↑ [177]

Chapter 3
Hormone-sensitive lipase is a
retinyl ester hydrolase in human
and rat quiescent hepatic stellate
cells
Shiva Shajari1*., Ali Saeed1,4*., Natalia F. Smith-Cortinez1,
Janette Heegsma1,2., Svenja Sydor3, Klaas Nico Faber1,2
1Department of Gastroenterology and Hepatology, University of Groningen,
University Medical Center Groningen, Groningen, The Netherlands.
2Department of Laboratory Medicine, University of Groningen, University Medical
Center Groningen, Groningen, The Netherlands.
3Department of Gastroenterology, Hepatology and Infectious Diseases, Otto von
Guericke University Magdeburg, Magdeburg, Germany.
4Institute of Molecular Biology & Biotechnology, Bahauddin Zakariya University,
Multan, Pakistan
Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids
(In press, *equal authors)

HSL hydrolyzes retinyl esters in qHSC
76
ABSTRACT
Hepatic stellate cells (HSC) store vitamin A as retinyl esters and control circulating
retinol levels. Upon liver injury, quiescent (q)HSC lose their vitamin A and
transdifferentiate to myofibroblasts, e.g. activated (a)HSC, that promote fibrosis by
producing excessive extracellular matrix. Adipose triglyceride lipase/patatin-like
phospholipase domain-containing protein 2 (ATGL/PNPLA2) and adiponutrin
(ADPN/PNPLA3) have so far been shown to mobilize retinol from retinyl esters in
HSC. Here, we studied the putative role of hormone-sensitive lipase (HSL/LIPE) in
HSC, as it is the major retinyl ester hydrolase (REH) in adipose tissue.
Lipe/HSL expression was analyzed in rat liver and primary human and rat qHSC and
culture-activated aHSC. Retinyl hydrolysis was analyzed after Isoproterenol-mediated
phosphorylation/activation of HSL.
Primary human HSC contain 2.5-fold higher LIPE mRNA levels compared to
hepatocytes. Healthy rat liver contains significant mRNA and protein levels of
HSL/Lipe, which predominates in qHSC and cells of the portal tree. Q-PCR
comparison indicates that Lipe mRNA levels in qHSC are dominant over Pnpla2 and
Pnpla3. HSL is mostly phosphorylated/activated in qHSC and partly colocalizes with
vitamin A-containing lipid droplets. Lipe/HSL and Pnpla3 expression is rapidly lost
during HSC culture-activation, while Pnpla2 expression is maintained. HSL super-
activation by isoproterenol accelerates loss of lipid droplets and retinyl palmitate from
HSC, which coincided with a small, but significant reduction in HSC proliferation and
suppression of Collagen1A1 mRNA and protein levels.
In conclusion, HSL participates in vitamin A metabolism in qHSC. Equivalent
activities of ATGL and ADPN provide the healthy liver with multiple routes to control
circulating retinol levels.

Chapter 3
77
3.1. INTRODUCTION
Vitamin A is an essential nutrient important for many physiological functions,
including vision, embryogenesis, reproduction, cell differentiation and immune
regulation [1]. Animals and humans need to acquire vitamin A from the diet.
Carotenes in fruits and vegetables and retinyl esters in meat, fish, eggs and milk are
important sources of vitamin A. The liver is the main storage site for vitamin A and
controls stable circulating levels of retinol to supply peripheral tissues with vitamin A
[1,2].
Dietary carotenes are converted in the intestinal epithelium to retinyl esters and
together with animal-originated retinyl esters packed in chylomicrons and transported
to the circulation [3]. Chylomicron remnants still contain most of the vitamin A content
and are taken up by hepatocytes in the liver. Retinyl esters are hydrolyzed to retinol,
which subsequently is exported from the hepatocyte to the circulation bound to retinol
binding protein 4 (RBP4). For storage in the liver, retinol is taken up by hepatic
stellate cells (HSC) that convert it back to retinyl esters and store it in large
cytoplasmic lipid droplets that also contain cholesterol, cholesterol esters, and
phospholipids [4,5]. Controlled hydrolysis of retinyl esters to retinol in HSC is believed
to maintain stable circulating retinol levels in blood around 2 µmol/L in humans (1-1.5
µmol/L in rodents) [5,6]. Liver injury leads to activation of HSC, which
transdifferentiate to migratory and proliferative myofibroblasts that produce matrix
proteins to promote wound healing. In chronic liver disease, this often leads to scar
tissue formation and fibrosis. Importantly, HSC rapidly lose their vitamin A stores in
the differentiation process and many chronic liver diseases are associated with
vitamin A deficiency as a consequence.
The vitamin A pool in HSC is a resultant of enzymes that esterify retinol and enzymes
that hydrolyse retinyl esters. Lecithin retinol acyltransferase (LRAT) and acyl-
CoA:diacylglycerol acyltransferase 1 (DGAT1) are the main enzymes involved in
retinol esterification [7,8]. LRAT appears to be the most relevant retinol esterase in
the liver as its absence leads to severe depletion of hepatic vitamin A pools [7]. In
line, LRAT expression is rapidly lost in transdifferentiating HSC. Retinyl ester
hydrolases (REH) in HSC include adipose triglyceride lipase/patatin-like
phospholipase domain containing 2 (ATGL/PNPLA2) and adiponutrin
(ADPN/PNPLA3) [9–12]. A single nucleotide polymorphism (SNP) in the PNPLA3
gene (PNPLA3-I148M), the most prominent genetic risk factor associated with non-

HSL hydrolyzes retinyl esters in qHSC
78
alcoholic liver disease (NAFLD), leads to accumulation of retinyl esters in the liver in
conjunction with reduced serum retinol levels. Atgl-/- mice, on the other hand, contain
normal hepatic vitamin A levels suggesting that redundancy in HSC-associated REH
activity likely exists. Besides the earlier-mentioned retinyl esterases, hormone-
sensitive lipase (HSL/LIPE) is also expressed in the liver and harbors retinyl ester
hydrolase activity [13,14]. A role of HSL in vitamin A metabolism is so far, however,
only established in adipose tissue [14,15]. Adipose tissue contains the largest
extrahepatic store of vitamin A accounting for up to 15-20% of the total body pool
[16]. REH activity is absent in adipose tissue of Hsl/Lipe-/- mice and accompanied by
a strong increase in retinyl esters and reduction in retinol in this tissue [14]. Hepatic
HSL activity has been assigned to hepatocytes where its major substrates are
cholesteryl esters [13], while hepatic retinyl ester levels are normal in Hsl/Lipe-/-
mice, like in the Atgl-/- mice [15]. Moreover, Lipe mRNA was reported to be absent in
freshly-isolated, as well as culture-activated rat HSC [17]. These findings lead to the
general assumption that HSL is not involved in hepatic vitamin A metabolism.
Here, we re-evaluated the cell-type specific expression of hepatic HSL in rat and
human. We found that HSL is expressed in quiescent rat and human HSC and is
rapidly lost when HSC transdifferentiate to myofibroblasts. HSL partly colocalizes
with HSC lipid droplets and pharmacological activation of HSL promotes retinyl ester
loss from HSC, concomitant with lipid droplet loss. These data support a role for HSL
in hepatic vitamin A metabolism, particularly in the healthy liver.
3.2. MATERIALS AND METHODS
3.2.1. Animals
Specified pathogen-free male Wistar rats (350-400 g; Charles River Laboratories
Inc., Wilmington, MA, USA) were housed under standard laboratory conditions with
free access to standard laboratory chow and water. All experiments were carried out
according to the Dutch law on the welfare of laboratory animals and guidelines of the
ethics committee of University of Groningen for care and use of laboratory animals.
3.2.2. Hepatic stellate cell isolation and culture
Primary rat hepatic stellate cells were isolated after pronase (Merck; Amsterdam, The
Netherlands) and collagenase-P (Roche; Almere, The Netherlands) perfusion of the
liver, followed by Nycodenz (Axis-Shield POC; Oslo, Norway) density gradient

Chapter 3
79
centrifugation, as described previously [18]. HSC were cultured in Iscove’s Modified
Dulbecco’s Medium (IMDM) with Glutamax (GibcoTM by Life technologies,
Thermofisher Scientific, Bleiswijk, The Netherlands) supplemented with 20% heat-
inactivated fetal calf serum (FCS), 1 mmol/L sodium-pyruvate, 1× MEM non-essential
amino acids, 50 µg/mL gentamycin, 100 U/mL Antibiotic-Antimycotic (all GibcoTM) in
a humidified incubator at 37°C with 5% CO2. After 7 to 10 days of culture-activation
cells were harvested for the experiments.
3.2.3. Fluorescence-Activated Cell Sorting
Following Nycodenz gradient purification, freshly-isolated qHSC were further purified
by Side Scatter-Activated Cell Sorting as described before [19] using MoFlo™ XDP
(Beckman Coulter, Woerden, The Netherlands). Briefly, two cell populations were
sorted with high side scatter (SSC), one with low forward scatter (FSC; R1) and one
with high FSC (R2) (Supplementary Figure S1A). R1 cells were highly pure (>98%
based on vitamin A autofluorescence) single cell HSC. R2 represents clusters of cells
containing HSC together with Kupffer cells and/or endothelial cells (Supplementary
Figure S1B). The R1 population was used as highly purified fractions of primary
quiescent HSC.
3.2.4. Hepatocyte isolation and culture
Primary rat hepatocytes were isolated by collagenase (Sigma Aldrich, The
Netherlands) perfusion as described before [20]. Cell viability was greater than 85%
as determined by Trypan Blue (Sigma Aldrich). Hepatocytes were cultured on
collagen-coated plates in William’s E medium (GibcoTM) supplemented with 10%
FCS, 50 µg/mL gentamycin and 100 U/mL penicillin/strep/amphotericin B. During the
4 h of attachment, 50 nmol/L dexamethasone (Department of Pharmacy UMCG,
Groningen, The Netherlands) was supplemented to the medium. Cells were
harvested for analysis 4h or 24h after isolation. Cells were cultured in a humidified
incubator at 37°C and 5% CO2.
3.2.5. Portal myofibroblasts (PMF) isolation and culture
Portal myofibroblasts (PMF) were isolated from the portal tree fraction obtained
during the hepatocyte isolation as previously described [21]. Briefly, the portal tree
was minced and incubated in a digestion solution containing collagenase, pronase,
DNase (Roche, The Netherlands) and hyaluronidase (Sigma Aldrich, The

HSL hydrolyzes retinyl esters in qHSC
80
Netherlands) for 1 h at 37°C. PMF were cultured in IMDM medium supplemented
supplemented with 20% heat-inactivated fetal calf serum (FCS), 1 mmol/L sodium-
pyruvate, 1× MEM non-essential amino acids, 50 µg/mL gentamycin, 100 U/mL
Antibiotic-Antimycotic (all GibcoTM) in a humidified incubator at 37°C and 5% CO2 for
7 days and harvested for analysis.
3.2.6. Kupffer cell isolation
Kupffer cells were isolated from the non-parenchymal cell fraction obtained during the
hepatocyte isolation. Kupffer cells were purified using a Percoll density cushion at
1800 g for 15 min at 4°C, as previously described [22]. Kupffer cells were allowed to
adhere in culture plates at 37°C for 30 min in Mg2+- and Ca2+-containing HBSS
(GibcoTM) supplemented with 10% FCS. Next, remaining hepatocytes were washed
away and attached Kupffer cells were further cultured for 24 h in RPMI (GibcoTM)
supplemented with 10% FCS and Antibiotic-Antimycotic (GibcoTM) (100 U/mL) and
harvested after 24 h for analysis.
3.2.7. Tissue specimens
Rat adipose tissue, portal tree and liver specimens were harvested and snap-frozen
in liquid nitrogen and stored at -80°C for further use. The frozen tissue was crushed
and dissolved in Tri-reagent (Sigma-Aldrich) for RNA isolation and qPCR analyses.
3.2.8. The human hepatic stellate cell line (LX-2)
LX-2 cells were kindly provided by Scott Friedman [23]. Cells were cultured in
Dulbecco's Modified Eagle Medium (DMEM) (GibcoTM) supplemented with 10% FCS
and Antibiotic-Antimycotic (GibcoTM) (100 U/mL).
3.2.9. Isolation and culture of primary human hepatocytes and HSC.
Freshly-isolated primary human hepatocytes and HSC were isolated from
macroscopically normal liver specimens obtained from fresh tumor resections by an
“all-in-one” liver cell purification procedure as described previously [24]. Primary
human hepatocytes were harvested after 3 day in vitro culturing. Cultured primary
human HSC were harvested approximately 5 days after isolation when they show an
intermediate phenotype between quiescent (vitamin A storage) and activation
(collagen production) [24].
3.2.10. Quantitative real-time reverse transcription polymerase chain reaction

Chapter 3
81
Quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR)
was performed as previously described [25]. Shortly, total RNA was isolated from
tissue samples using TRIzol® reagent according to supplier’s instruction
(ThermoFisher Scientific, The Netherlands). RNA quality and quantity were
determined using a Nanodrop 2000c UV-vis spectrophotometer (Thermo Fisher
Scientific). cDNA was synthesized from 2.5 µg RNA using random nonamers and M-
MLV reverse transcriptase (Invitrogen, USA). Taqman primers and probes were
designed using Primer Express 3.0.1 and are shown in Supplementary Table S1. All
target genes were amplified using the Q-PCR core kit master mix (Eurogentec, The
Netherlands) on a 7900HT Fast Real-Time PCR system (Applied Biosystems
Europe, The Netherlands). SDSV2.4.1 (Applied Biosystems Europe, The
Netherlands) software was used to analyze the data. Expression of genes is
presented in 2-delta CT and normalized to 18S.
3.2.11. Western blot analysis
Protein samples were prepared for Western blot analysis as described previously
[26]. Protein concentrations were quantified using the Bio-Rad protein assay (Bio-
Rad, Hercules, CA, USA) with bovine serum albumin (BSA) as a standard. Equal
amounts of protein (5-20 µg) were separated on Mini-PROTEAN® TGX™ precast 4-
15% gradient gels (Bio-Rad, Hercules, CA, USA) and transferred to nitrocellulose
membranes using the Trans-Blot Turbo transfer system, (Bio-Rad). Primary
antibodies (against HSL, pHSL, αSMA, Collagen1A1, LRAT, GAPDH and PEX-14;
details and dilutions are listed in Supplementary Table S2) and appropriate
horseradish peroxidase (HRP)-conjugated secondary antibodies (1:2,000; P0448,
DAKO) were used for detection. Proteins were detected using the Pierce ECL
Western blotting kit (Thermo Scientific). Images were captured using the chemidoc
XRS system and Image Lab version 3.0 (Bio-Rad). The intensity of bands was
quantified using ImageJ version 1.51 (NIH, USA).
3.2.12. Real-time monitoring of cell proliferation
The proliferation of freshly-isolated rat hepatic stellate cells was monitored for 5 days
in presence or absence of Isoproterenol using Real Time Cell Analyzer (RTCA,
xCELLigence RTCA DP, ACEA Biosciences, Inc.). Freshly-isolated rat qHSC were
plated in E plates having interdigitated gold microelectrodes to constantly record cell
proliferation, according to manufacturer’s instructions. Isoproterenol treatment was

HSL hydrolyzes retinyl esters in qHSC
82
started at 4 h after the attachment of cells. Results were recorded and analyzed by
RTCA Software.
3.2.13. Oil Red O Staining
Rat HSC and LX-2 cells were seeded on coverslips and cultured for the indicated
periods, after which they were fixed with 4% paraformaldehyde (Merck Millipore, The
Netherlands). LX-2 cells were additionally exposed to BSA-conjugated oleic acid (0.1
mmol/L; Sigma-Aldrich) for 24 h, as previously described [27]. HSC and LX-2 were
treated with different concentrations (5, 10, 50 µmol/L) of Isoproterenol (Iso).
Intracellular lipids were stained with Oil Red O solution (Sigma-Aldrich, The
Netherlands) and Hematoxylin (Sigma-Aldrich) were stained the nuclei as described
before [28]. Slides were scanned using a Nonozoomer 2.0HT (Hamamatsu
Photonics, Hamamatsu, Japan) and analyzed with Aperio ImageScope (version 11.1,
Leica Microsystems, Amsterdam, The Netherlands).
3.2.14. Immunohistochemistry
Immunohistochemistry for phosphorylated HSL (anti-pHSL; supplementary Table S2)
was performed on paraffin-embedded rat liver sections as previously described [29].
Briefly, after deparaffinization antigen retrieval was performed by microwave
irradiation in citrate buffer (10 mmol/L), pH 6.0 and blocking of endogenous
peroxidase with 0.3% H2O2 for 30 min. After blocking (1% BSA for 30 min) tissue was
incubated with polyclonal-rabbit-phospho-HSL (Ser660) primary antibody (1:50
dilution in 1% BSA) overnight at 4°C in a humidity chamber. Horseradish peroxidase
(HRP)-conjugated goat anti-rabbit antibodies (1:50; #170-6515, Bio-Rad) was used
as secondary antibody. Slides were stained with ImmPact NovaRED Peroxidase
(HRP) substrate (cat# SK-4805, Vector Laboratories, The Netherlands) for
11 minutes, and Haematoxylin was used as a counter nuclear stain (2 min at RT).
Finally, slides were dehydrated and mounted with Eukitt®, (Sigma-Aldrich). Slides
were scanned using a nanozoomer 2.0 HT digital slide scanner (C9600-12,
Hamamatsu Photonics, Hamamatsu, Japan) and analyzed with Aperio ImageScope
(version 11.1, Leica Microsystems, Amsterdam, The Netherlands).
3.2.15. Fluorescence microscopy
Freshly isolated primary rat HSC or LX-2 were seeded in 12-well plates (1*105

Chapter 3
83
cells/well) containing 18 mm glass coverslips and cultured for the indicated periods.
Coverslips were washed with PBS three times, fixed with 4% PFA in PBS at room
temperature (RT) for 10 minutes and permeabilized with 0.1% triton X-100 in PBS for
10 minutes at RT. Non-specific antibody binding was blocked with 2% BSA in PBS
for 30 min. After blocking, coverslips were incubated with anti-pHSL antibody (1:200,
see supplementary Table S2) for 1 h at RT. Coverslips were then washed 3 times
with blocking solution and incubated 30 min with secondary antibody (goat-anti-rabbit
488, 1:500 in blocking solution) at RT and covered from light. Next, coverslips were
washed 3 times and then mounted with VECTASHIELD Antifade Mounting Medium
with DAPI (Vector Laboratories). Coverslips were air dried, sealed using nail polish,
stored at 4°C and covered from light until further use. Images were obtained using a
Zeiss LSM 780 NLO two photon Confocal Laser Scan Microscope (Carl Zeiss, Jena,
Germany). After double-photon simultaneous excitation, image was obtained for each
fluorophore sequentially according to their individual unique emission spectra
(emission of autofluoresce at 410 nm, DAPI at 460 nm, pHSL at 530 nm). Images
were then processed and analyzed using ImageJ version 1.51 (NIH, Maryland, USA).
3.2.16. Retinyl palmitate and retinol measurement
Vitamin A content was analyzed by reverse phase HPLC as previously described
[30]. Retinol and retinyl esters were extracted and deproteinized twice with n-hexane
from tissue and serum with retinol acetate as the internal standard. Samples were
diluted in 200 µL ethanol and 50 µL was injected for phase separation (150 x 3.0 mm
of 5 µM column) and/or measurements (Photo Diode Array Detector at 325 nm) by
HPLC.
3.2.17. Statistical analysis
Data is presented as Mean ± SEM by using the GraphPad Prism 7 software package
(GraphPad Software, San Diego, CA, USA). Statistical significance was determined
by the unpaired t-test. P-values ≤0.05*, ≤0.01**, ≤0.001*** were considered
significant.

HSL hydrolyzes retinyl esters in qHSC
84
3.3. RESULTS
First, we compared mRNA levels of Lipe (encoding HSL) in purified cell types from
healthy rat livers and related it to its expression in adipose tissue (Figure 1A). Lipe
mRNA levels were well-detectable in total rat liver, though were 38-fold lower
compared to adipose tissue. Comparing purified hepatic cell fractions, the highest
Lipe levels were found in freshly-isolated FACS-sorted quiescent hepatic stellate
cells (qHSC; marker Lrat), with drastically lower levels in purified hepatocytes
(marker: Abcb11), Kupffer cells (marker: CD68) and cultured portal myofibroblasts
(marker: Acta2) (Figure 1A inset). In addition, high Lipe mRNA levels were detected
in the portal tree fractions, that mainly contain cholangiocytes (marker: Slc10a2) and
portal myofibroblasts. Primary human HSC also contained readily detectable mRNA
levels of LIPE, which were significantly higher compared to primary human
hepatocytes (Figure 1B). In contrast to quiescent HSC from rat liver, the purification
protocol for human HSC included a short in vitro cultivation period, in which these
cells show early signs of activation (COL1A1 and ACTA2), while a typical marker for
quiescent HSC (LRAT) is still significantly detected.

Chapter 3
85
A
B
Figure 1. Rat and human quiescent hepatic stellate cells express hormone sensitive lipase. (A) Quiescent hepatic stellate cells (qHSC), Kupffer cells (KC), Portal myofibroblasts (PMF), hepatocytes and the portal tree fraction were purified form healthy rat liver and mRNA levels of Lipe and cell type-specific marker genes (Lrat, Cd68, Acta2, Abcb11, Slc10a2, respectively) were quantified by Q-PCR and compared to whole liver and adipose tissue. Hepatic Lipe mRNA was most abundant in qHSC and in the portal tree that mostly consists of cholangiocytes. (The inset in the left panel shows a zoom of Lipe mRNA levels in rat qHSC, KC, PMF and hepatocytes). (B) Primary human HSC and hepatocytes were purified from healthy tissue in surgical resections. hHSC purification requires short in vitro culture to remove contaminating cells, resulting in co-expression of LRAT, COL1A1 and ACTA2. LIPE mRNA levels were 2.5-fold higher in the hHSC compared to purified human hepatocytes (expressing ALB).

HSL hydrolyzes retinyl esters in qHSC
86
An earlier study reported the absence of HSL/Lipe in (partially activated) rat HSC
[17]. Therefore, we next studied whether culture-activation of rat HSC affects Lipe
expression. Highly-pure FACS-sorted rat HSC were either processed directly (qHSC)
or culture-activated for 7-10 days (aHSC). Lipe mRNA (Figure 2A) and HSL protein
(Figure 2B) were readily detected in purified qHSC from healthy rat livers, but were
strongly decreased –to undetectable levels- in culture-activated aHSC. A similar
profile was observed for the qHSC marker Lrat/LRAT, while Acta2/αSMA showed the
expected low levels in qHSC and a sharp increase in aHSC. In fact, HSL protein had
already largely disappeared in 3-day cultured HSC (Supplementary Figure S2).
Thus, HSL expression is rapidly lost during HSC transdifferentiation.
Figure 2. HSL expression is lost during culture-activation of hepatic stellate cells. Freshly-isolated and FACS-purified rat HSC were either processed directly (qHSC) or culture-activated for 7-10 days (aHSC) and Lipe mRNA and HSL protein levels were analyzed by Q-PCR (A) and Western blotting (B), respectively. qHSC show high expression of Lipe/HSL and Lrat/LRAT which are lost in aHSC. Instead, aHSH show high expression of Acta2/αSMA.

Chapter 3
87
Two enzymes have been characterized to play a role in hydrolyzing retinyl esters in
mouse and/or human HSC, e.g. ATGL/PNPLA2 and ADPN/PNPLA3 [9,10]. HSL
could be a third enzyme involved in controlling hepatic vitamin A metabolism, as such
role for HSL is already well-established in adipose tissue [14,31,32]. We next
analyzed the relative abundance of mRNA levels of these 3 hydrolases in freshly-
isolated and purified qHSC from healthy rat liver and after culture-activation (Figure
3). Quiescent HSC contained high mRNA levels of Lipe, with clearly lower levels of
Atgl/Pnpla2 and even lower levels of Adpn/Pnpla3. Transcript levels of Lipe and
Adpn/Pnpla3 were sharply decreased in aHSC (>100-fold and >10-fold, respectively),
while Atgl/Pnpla2 mRNA levels were comparable between aHSC and qHSC.
Figure 3. Quiescent hepatic stellate cells contain high Lipe mRNA levels in comparison to other retinyl ester hydrolase-encoding genes. Freshly-isolated and FACS-purified rat HSC were either processed directly (qHSC) or culture-activated for 7-10 days (aHSC) and the mRNA levels of three retinyl ester hydrolases, Lipe, Pnpla2 and Pnpla3 was quantified by q-PCR qHSC contained high mRNA levels of Lipe, with lower levels of Atgl/Pnpla2 and even lower levels of Adpn/Pnpla3. Transcript levels of Lipe and Adpn/Pnpla3 sharply decreased with activation of HSC, while Atgl/Pnpla2 mRNA levels were similar in aHSC and qHSC.
HSL enzyme activity is activated by phosphorylation (at serine 563, 565 and 660) and
we next analyzed the presence of pHSL in rat liver tissue and purified qHSC by
immunohistochemical staining (Figure 4). pHSL-specific staining was sparsely
distributed throughout the liver parenchyma and detected in small cells located close
to hepatocytes and sinusoids (Figure 4A), the typical location for qHSC. In addition,
strong pHSL staining was detected in portal areas (Figure 4B), in line with the high
Lipe mRNA levels detected in these cellular fractions (Figure 1).
Immunofluorescence microscopy analysis of pHSL in purified rat qHSC revealed

HSL hydrolyzes retinyl esters in qHSC
88
pronounced cytoplasmic staining that partly co-localized with lipid droplets that
showed strong vitamin A autofluorescence (Figure 4C). Together, these data
indicate that HSL/Lipe is a prominent lipase/hydrolase in rat qHSC.
Figure 4. Quiescent hepatic stellate cells in healthy liver contain phosphorylated, thus active, HSL. Paraffin-sections of healthy rat liver (A, B) and freshly-isolated quiescent hepatic stellate cells (qHSC; C) were stained for phosphorylated-HSL (pHSL) by immunohistochemistry and immunofluorescence microscopy, respectively. pHSL-specific staining was detected in small cells located close to hepatocytes and sinusoids, the typical location for qHSC (indicated by red arrows in A). Additional pHSL-specific staining was detected in the portal areas (yellow arrows in B). Freshly-isolated qHSC showed a pronounced cytoplasmic location of pHSL that partly co-localized with vitamin A-containing lipid droplets, as visualized by the autofluorescence of vitamin A (C).

Chapter 3
89
HSL is best known for its hydrolase activity towards triglycerides and cholesteryl
esters. This activity can be enhanced by isoproterenol (Iso) that stimulates HSL
phosphorylation at serine 563, 565 and 660 [33–35]. Basel levels of pHSLS660 in LX-2
cells (model for human stellate cells) were low, but Iso (5 µmol/L) quickly and
strongly induced HSLS660 phosphorylation and promoted clearance of lipid droplets
from LX-2 cells (Supplementary Figure S3A-C). LX-2 cells do not store retinyl
esters, so a potential role of HSL in hydrolyzing retinyl palmitate was analyzed in
primary rat HSC. In contrast to LX-2 cells, primary rat qHSC showed high basal
levels of pHSL, which could be further induced by Iso in the early phase of culture-
activation of rat HSC (Figure 5A). Like in LX-2 cells, Iso dose-dependently
accelerated lipid loss from primary rat HSC in the first 3 days of culture activation
(Figure 5B, quantification in D: after 24 h with and without 50 µmol/L Iso), which was
accompanied by a dose-dependent decrease in vitamin A-autofluorescence in HSC
(Figure 5C, quantification in D: after 24 h with and without 50 µmol/L Iso). In line, Iso
treatment (50 µmol/L) lead to a significant reduction in cellular retinyl palmitate levels
in 1 day-cultured primary rat HSC, while cellular retinol levels were unchanged
(Figure 5D). These data support a role for HSL in the hydrolysis of retinyl esters, as
well as other esterified lipids in HSC.

HSL hydrolyzes retinyl esters in qHSC
90
Figure 5. Hormone-sensitive lipase activation promotes retinyl ester hydrolysis in HSC. Freshly-isolated rat qHSC were allowed to attach for 4 h and subsequently exposed to Isoproterenol (5 and 50 µmol/L) to enhance HSL phosphorylation and thus its enzyme activity. Untreated qHSL contain significant levels of phosphorylated HSL, which could be further increased by Isoproterenol treatment (A), total-HSL and GAPDH are included as controls.
Isoproterenol treatment for 1 and 3 days dose-dependently accelerated lipid loss from primary rat qHSC (as shown by oil red O (ORO) staining) (B), which was accompanied by a dose-dependent decrease in vitamin A-autofluorescence (C). ORO staining, vitamin A-autofluorescence and cellular retinyl palmitate and retinol levels were quantified after 24 h exposure with and without 24 h Isoproterenol (D).

Chapter 3
91
As “super”-activation of HSL promoted retinoid loss from cultured rat HSC, we
analyzed whether this may also affect the activation process of HSC. Freshly-isolated
qHSC were seeded in a Real-Time Cell Analyzer (RCTA; xCELLigence) and after the
initial 4h-attachment phase cells were treated with 0, 5, 10 and 50 µmol/L Iso (Figure
6A). Iso treatment delayed the initial increase in cell index (reflecting cell stretching
and/or proliferation) in the first 24 h of culture, after which the cell index increased
with similar kinetics as the untreated HSC. A 3-day Iso treatment caused a small, but
significant reduction in Collagen 1a1 levels already at 5 umol/L, both at mRNA and
protein level, while only trends in lowering Acta2/aSma, Tgf-β and Lrat levels were
detected at that concentration. (Figure 6B and C).

HSL hydrolyzes retinyl esters in qHSC
92
Figure 6. Super-activation of hormone-sensitive lipase reduces hepatic stellate cell proliferation and activation. Freshly-isolated rat qHSC were allowed to attached for 4 h and subsequently exposed to Isoproterenol (5, 10 and 50 µmol/L). Cell proliferation was monitored for 5 days in a Real-Time Cell Analyzer (RCTA; xCELLigence; A). Markers of HSC activation were quantified by Q-PCR after 3 days (B) and Western blotting after 30 min, 1 day and 3 days (C). Isoproterenol treatment delayed the initial increase in cell index (reflecting cell stretching and/or proliferation) in the first 24 h of culture, after which the cell index increased with similar
kinetics as the untreated HSC (A). mRNA levels of Col1a1 and Tgf-β were significantly reduced
after 3 days Isoproterenol treatment, while mRNA levels of Acta2, Timp-1 and Lrat were not affected (B). Protein levels of COL1A1 were reduced after 3 days of Isoproterenol treatment, while it did not change the aSMA protein level.
Taken together, our data show that quiescent rat and human HSC contain significant
amounts of (phosphorylated, thus active) HSL and promotes retinyl ester hydrolysis
in these cells. HSL expression is rapidly lost during transdifferentiation of HSC and
thus does not seem to play a role in vitamin A metabolism in aHSC.

Chapter 3
93
3.4. DISCUSSION
In this study, we show for the first time that hormone sensitive lipase (HSL) is
expressed in human and rat quiescent hepatic stellate cells (qHSC) where it plays a
role in retinyl ester hydrolysis. HSL is mostly present in it is active phosphorylated
state and partly colocalizes with vitamin A-containing lipid droplets in qHSC. HSL
expression is rapidly lost when HSC transdifferentiate to myofibroblasts.
Pharmacological HSL super-activation accelerated retinyl ester loss from HSC and
lead to transient suppression of HSC activation in vitro. Thus, HSL is a third retinyl
ester hydrolase in HSC, where it controls vitamin A metabolism together with
ATGL/PNPLA2 and ADPN/PNPLA3.
The fact that HSL contains retinyl ester hydrolase activity is well-known from its role
in adipose tissue [14], but it was deemed absent from HSC based on earlier studies
[15,17]. The experimental data supporting this is, however, limited. When comparing
mRNA levels of various carboxylesterase and lipase genes in different rat liver cell
types, no significant mRNA levels of Lipe were detected in any of the cell types
analyzed, e.g. hepatocytes, endothelial cells, Kupffer cells, qHSC and aHSC [17].
Still, HSL is present in the liver based on other studies [13]. Furthermore, Taschler et
al.[15] only tested HSL protein expression in αSMA-positive aHSC of mice and found
it to be absent, which is in line with our results with rat aHSC. It remains puzzling to
us why no Lipe mRNA was detected in any of the rat hepatic cell types analyzed by
others [17], in particular in the highly-pure fractions of qHSC, since mRNA levels of
Lipe in rat qHSC appeared even dominant over Pnpla2 and Pnpla3 in our study.
Moreover, Western blotting, immunohistochemical staining and immunofluorescence
microscopy further confirmed that rat qHSC do express HSL and appears to be
mostly in the activate phosphorylated state (pHSL). In addition, and for the first time,
we also show that Lipe/HSL is expressed in primary human HSC, even more so than
in human hepatocytes, as well as in the human HSC cell line LX-2. LX-2 cells have
an intermediate transdifferentiation state and most of the HSL appears non-
phosphorylated. Upon pharmacological induction of phosphorylation (and activation)
by Iso, pHSL immunefluoresce staining becomes apparent and shows a punctate
staining pattern, indicative of co-localization with lipid droplets in LX-2 cells, similar as
we observed in rat qHSC and as described earlier for pHSL in 3T3L1 adipocytes [36].
Iso treatment accelerated retinyl palmitate loss from rat HSC, implying a role of HSL
in retinyl ester hydrolysis in these cells.

HSL hydrolyzes retinyl esters in qHSC
94
So far, 2 enzymes were implicated in retinyl ester hydrolysis in HSC, ATGL/PNPLA2
and ADPN/PNPLA3, so HSL/LIPE is a third one. Whole body knock-out mice for
either Pnpla2 or Lipe do not show altered levels of hepatic retinyl esters [15]. Pnpla3
knockout mice have not been analyzed for hepatic retinyl ester content yet, but do
not show global alterations in lipid homeostasis compared to wild type animals, either
on chow or fatty liver-inducing diets [37,38]. This does not necessarily mean that
vitamin A metabolism is not disturbed in these mice and thus requires a focused
analysis of hepatic vitamin A metabolites in Pnpla3-/- mice. Still, it may very well be
that like Pnpla2-/- and Lipe-/- mice also Pnpla3-/- mice may not have altered hepatic
retinyl ester levels and that these genes can fully compensate each other for retinyl
ester hydrolysis in HSC. Both Lipe and Pnpla3 expression are strongly suppressed
during HSC transdifferentiation, while Pnpla2 levels remain stable. This is in line with
earlier observations that pharmacological inhibition of ATGL (encoded by Pnpla2)
leads to accumulation of retinyl esters in activated mouse HSC when exposed to
retinol-containing media for 10 days in vitro, while this was not observed with HSL
inhibitors [15]. Thus, while HSL, ATGL and ADPN have overlapping roles in retinyl
ester hydrolysis in qHSC in the healthy liver, ATGL is probably the most important for
retinyl ester hydrolysis in aHSC in the chronically injured liver.
Notably, transcriptional regulation of both HSL/Lipe and ATGL/Pnpla2 is under
control of PPARγ, which is a well-known marker for qHSC and is lost during
transdifferentiation. Lipe mRNA levels follow Pparγ levels during HSC
transdifferentiation [39,40], while Pnpla2 expression is not affected (our data). This
indicates that undefined compensatory mechanisms exist to maintain Pnpla2
expression in aHSC to secure retinyl ester hydrolase activity in aHSC.
Our work may also have relevance to hepatic HSL functions beyond vitamin A
metabolism. HSL is an intracellular neutral lipase that besides retinyl esters
hydrolyses cholesterol esters, triglycerides, mono- and di-acylglycerol and other lipids
[13]. Hepatic expression was most dominant in the portal tree fractions and purified
qHSC, while much lower levels were observed in hepatocytes, both in rat and human
liver cells.
The physiological relevance of hepatic HSL is evident from the whole body Lipe-/-
mouse that is characterized by exaggerated accumulation of cholesteryl esters in in
vivo models of fatty liver disease [13,41]. Given our data, it will be interesting to
analyze which cells in the liver actually accumulate cholesteryl esters in Lipe-/- mice.

Chapter 3
95
Cells other than hepatocytes may very well be good candidates as hepatocyte-
specific deletion of Lipe did not replicate the fatty liver phenotype in mice [42]. While
these authors concluded that HSL has little involvement in hepatic lipid metabolism, it
may actually be other cell types than hepatocytes that play a crucial role here. It is
therefore interesting to determine how much HSC contribute to the accumulation of
cholesteryl esters in Lipe-/- mice.
Taken together, our data show that HSL is a prominent retinyl ester hydrolase in
quiescent rat and human HSC. Retinol release from HSC in the healthy liver is
therefore a fine-tuned collaboration between HSL, ATGL/PNAPL2 and
ADPN/PNPLA3 to maintain whole body vitamin A homeostasis. HSL expression in
qHSC is significantly higher than in hepatocytes, which may point to a broader role of
HSC-located HSL in hepatic lipid metabolism. Future work should take the cell type-
specific location of hepatic HSL into account when analyzing its role in lipid
metabolism.

HSL hydrolyzes retinyl esters in qHSC
96
REFERENCES
[1] A. Cañete, E. Cano, R. Muñoz-Chápuli, R. Carmona, Role of Vitamin A/Retinoic Acid in Regulation of Embryonic and Adult Hematopoiesis, Nutrients. 9 (2017). doi:10.3390/nu9020159.
[2] R. Blomhoff, H.K. Blomhoff, Overview of retinoid metabolism and function, J. Neurobiol. 66 (2006) 606–630. doi:10.1002/neu.20242.
[3] E. Reboul, Absorption of vitamin A and carotenoids by the enterocyte: focus on transport proteins, Nutrients. 5 (2013) 3563–3581. doi:10.3390/nu5093563.
[4] H. Senoo, Structure and function of hepatic stellate cells, Med. Electron Microsc. Off. J. Clin. Electron Microsc. Soc. Jpn. 37 (2004) 3–15. doi:10.1007/s00795-003-0230-3.
[5] A. Saeed, M. Hoekstra, M.O. Hoeke, J. Heegsma, K.N. Faber, The interrelationship between bile acid and vitamin A homeostasis, Biochim. Biophys. Acta. 1862 (2017) 496–512. doi:10.1016/j.bbalip.2017.01.007.
[6] R. Blomhoff, M.H. Green, T. Berg, K.R. Norum, Transport and storage of vitamin A, Science. 250 (1990) 399–404.
[7] S.M. O’Byrne, N. Wongsiriroj, J. Libien, S. Vogel, I.J. Goldberg, W. Baehr, K. Palczewski, W.S. Blaner, Retinoid absorption and storage is impaired in mice lacking lecithin:retinol acyltransferase (LRAT), J. Biol. Chem. 280 (2005) 35647–35657. doi:10.1074/jbc.M507924200.
[8] M. Ajat, M. Molenaar, J.F.H.M. Brouwers, A.B. Vaandrager, M. Houweling, J.B. Helms, Hepatic stellate cells retain the capacity to synthesize retinyl esters and to store neutral lipids in small lipid droplets in the absence of LRAT, Biochim. Biophys. Acta - Mol. Cell Biol. Lipids. 1862 (2017) 176–187. doi:10.1016/j.bbalip.2016.10.013.
[9] U. Taschler, R. Schreiber, C. Chitraju, G.F. Grabner, M. Romauch, H. Wolinski, G. Haemmerle, R. Breinbauer, R. Zechner, A. Lass, R. Zimmermann, Adipose triglyceride lipase is involved in the mobilization of triglyceride and retinoid stores of hepatic stellate cells, Biochim. Biophys. Acta BBA - Mol. Cell Biol. Lipids. 1851 (2015) 937–945. doi:10.1016/j.bbalip.2015.02.017.
[10] C. Pirazzi, L. Valenti, B.M. Motta, P. Pingitore, K. Hedfalk, R.M. Mancina, M.A. Burza, C. Indiveri, Y. Ferro, T. Montalcini, C. Maglio, P. Dongiovanni, S. Fargion, R. Rametta, A. Pujia, L. Andersson, S. Ghosal, M. Levin, O. Wiklund, M. Iacovino, J. Borén, S. Romeo, PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells, Hum. Mol. Genet. 23 (2014) 4077–4085. doi:10.1093/hmg/ddu121.
[11] T.O. Eichmann, L. Grumet, U. Taschler, J. Hartler, C. Heier, A. Woblistin, L. Pajed, M. Kollroser, G. Rechberger, G.G. Thallinger, R. Zechner, G. Haemmerle, R. Zimmermann, A. Lass, ATGL and CGI-58 are lipid droplet proteins of the hepatic stellate cell line HSC-T6, J. Lipid Res. 56 (2015) 1972–1984. doi:10.1194/jlr.M062372.
[12] M. Tuohetahuntila, M.R. Molenaar, B. Spee, J.F. Brouwers, M. Houweling, A.B. Vaandrager, J.B. Helms, ATGL and DGAT1 are involved in the turnover of newly synthesized triacylglycerols in hepatic stellate cells., J. Lipid Res. (2016). doi:10.1194/jlr.M066415.
[13] M. Sekiya, J.-I. Osuga, N. Yahagi, H. Okazaki, Y. Tamura, M. Igarashi, S. Takase, K. Harada, S. Okazaki, Y. Iizuka, K. Ohashi, H. Yagyu, M. Okazaki, T. Gotoda, R. Nagai, T. Kadowaki, H. Shimano, N. Yamada, S. Ishibashi, Hormone-sensitive lipase is involved in hepatic cholesteryl ester hydrolysis., J. Lipid Res. 49 (2008) 1829–38. doi:10.1194/jlr.M800198-JLR200.

Chapter 3
97
[14] K. Ström, T.E. Gundersen, O. Hansson, S. Lucas, C. Fernandez, R. Blomhoff, C. Holm, Hormone-sensitive lipase (HSL) is also a retinyl ester hydrolase: evidence from mice lacking HSL., FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 23 (2009) 2307–2316. doi:10.1096/fj.08-120923.
[15] U. Taschler, R. Schreiber, C. Chitraju, G.F. Grabner, M. Romauch, H. Wolinski, G. Haemmerle, R. Breinbauer, R. Zechner, A. Lass, R. Zimmermann, Adipose triglyceride lipase is involved in the mobilization of triglyceride and retinoid stores of hepatic stellate cells, Biochim. Biophys. Acta. 1851 (2015) 937–945. doi:10.1016/j.bbalip.2015.02.017.
[16] C. Tsutsumi, M. Okuno, L. Tannous, R. Piantedosi, M. Allan, D.S. Goodman, W.S. Blaner, Retinoids and retinoid-binding protein expression in rat adipocytes., J. Biol. Chem. 267 (1992) 1805–1810.
[17] T. Mello, A. Nakatsuka, S. Fears, W. Davis, H. Tsukamoto, W.F. Bosron, S.P. Sanghani, Expression of carboxylesterase and lipase genes in rat liver cell-types, Biochem. Biophys. Res. Commun. 374 (2008) 460–464. doi:10.1016/j.bbrc.2008.07.024.
[18] H.A.N. Moshace, A. Casini, S. Lieber, Acetaldehyde Selectively Stimulates Collagen Production in Cultured Rat Liver Fat-storing Cells but not in Hepatocytes, (n.d.) 511–518.
[19] A. Geerts, T. Niki, K. Hellemans, D. De Craemer, K. Van Den Berg, J.M. Lazou, G. Stange, M. Van De Winkel, P. De Bleser, Purification of rat hepatic stellate cells by side scatter-activated cell sorting, Hepatol. Baltim. Md. 27 (1998) 590–598. doi:10.1002/hep.510270238.
[20] T.E. Woudenberg-Vrenken, M. Buist-Homan, L. Conde de la Rosa, K.N. Faber, H. Moshage, Anti-oxidants do not prevent bile acid-induced cell death in rat hepatocytes., Liver Int. Off. J. Int. Assoc. Study Liver. 30 (2010) 1511–21. doi:10.1111/j.1478-3231.2010.02325.x.
[21] H. El Mourabit, E. Loeuillard, S. Lemoinne, A. Cadoret, C. Housset, Culture model of rat portal myofibroblasts, Front. Physiol. 7 (2016) 1–9. doi:10.3389/fphys.2016.00120.
[22] O. Teufelhofer, W. Parzefall, E. Kainzbauer, F. Ferk, C. Freiler, S. Knasm??ller, L. Elbling, R. Thurman, R. Schulte-Hermann, Superoxide generation from Kupffer cells contributes to hepatocarcinogenesis: Studies on NADPH oxidase knockout mice, Carcinogenesis. 26 (2005) 319–329. doi:10.1093/carcin/bgh320.
[23] L. Xu, a Y. Hui, E. Albanis, M.J. Arthur, S.M. O’Byrne, W.S. Blaner, P. Mukherjee, S.L. Friedman, F.J. Eng, Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis., Gut. 54 (2005) 142–151. doi:10.1136/gut.2004.042127.
[24] M. Werner, S. Driftmann, K. Kleinehr, G.M. Kaiser, Z. Mathé, J.W. Treckmann, A. Paul, K. Skibbe, J. Timm, A. Canbay, G. Gerken, J.F. Schlaak, R. Broering, All-in-one: Advanced preparation of human parenchymal and non-parenchymal liver cells, PLoS ONE. (2015). doi:10.1371/journal.pone.0138655.
[25] H. Blokzijl, S. Vander Borght, L.I.H. Bok, L. Libbrecht, M. Geuken, F.A.J. van den Heuvel, G. Dijkstra, T.A.D. Roskams, H. Moshage, P.L.M. Jansen, K.N. Faber, Decreased P-glycoprotein (P-gp/MDR1) expression in inflamed human intestinal epithelium is independent of PXR protein levels, Inflamm. Bowel Dis. 13 (2007) 710–720. doi:10.1002/ibd.20088.
[26] A. Pellicoro, F.A.J. van den Heuvel, M. Geuken, H. Moshage, P.L.M. Jansen, K.N. Faber, Human and rat bile acid-CoA:amino acid N-acyltransferase are

HSL hydrolyzes retinyl esters in qHSC
98
liver-specific peroxisomal enzymes: implications for intracellular bile salt transport, Hepatol. Baltim. Md. 45 (2007) 340–348. doi:10.1002/hep.21528.
[27] Y.M. Song, S.-O. Song, Y.-K. Jung, E.-S. Kang, B.S. Cha, H.C. Lee, B.-W. Lee, Dimethyl sulfoxide reduces hepatocellular lipid accumulation through autophagy induction, Autophagy. 8 (2012) 1085–1097. doi:10.4161/auto.20260.
[28] S. Shajari, A. Laliena, J. Heegsma, M.J. Tu????n, H. Moshage, K.N. Faber, Melatonin suppresses activation of hepatic stellate cells through ROR??-mediated inhibition of 5-lipoxygenase, J. Pineal Res. 59 (2015) 391–404. doi:10.1111/jpi.12271.
[29] M. Aparicio-Vergara, P.P.H. Hommelberg, M. Schreurs, N. Gruben, R. Stienstra, R. Shiri-Sverdlov, N.J. Kloosterhuis, A. de Bruin, B. van de Sluis, D.P.Y. Koonen, M.H. Hofker, Tumor necrosis factor receptor 1 gain-of-function mutation aggravates nonalcoholic fatty liver disease but does not cause insulin resistance in a murine model, Hepatol. Baltim. Md. 57 (2013) 566–576. doi:10.1002/hep.26046.
[30] Z. Zaman, P. Fielden, P.G. Frost, Simultaneous determination of vitamins A and E and carotenoids in plasma by reversed-phase HPLC in elderly and younger subjects, Clin. Chem. 39 (1993) 2229–2234.
[31] S. Wei, K. Lai, S. Patel, R. Piantedosi, H. Shen, V. Colantuoni, F.B. Kraemer, W.S. Blaner, Retinyl ester hydrolysis and retinol efflux from BFC-1beta adipocytes, J. Biol. Chem. 272 (1997) 14159–14165.
[32] R. Zimmermann, J.G. Strauss, G. Haemmerle, G. Schoiswohl, R. Birner-Gruenberger, M. Riederer, A. Lass, G. Neuberger, F. Eisenhaber, A. Hermetter, R. Zechner, Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase., Science. 306 (2004) 1383–1386. doi:10.1126/science.1100747.
[33] S. Kim, T. Tang, M. Abbott, J.A. Viscarra, Y. Wang, H.S. Sul, Lipase To Regulate Lipolysis and Fatty Acid Oxidation within, 36 (2016) 1961–1976. doi:10.1128/MCB.00244-16.Address.
[34] S.M. Choi, D.F. Tucker, D.N. Gross, R.M. Easton, L.M. DiPilato, A.S. Dean, B.R. Monks, M.J. Birnbaum, Insulin regulates adipocyte lipolysis via an Akt-independent signaling pathway., Mol. Cell. Biol. 30 (2010) 5009–5020. doi:10.1128/MCB.00797-10.
[35] G.P.C. Martins, C.O. Souza, S. Marques, T.F. Luciano, B.L. DA Silva Pieri, J.C. Rosa, A.S.R. DA Silva, J.R. Pauli, D.E. Cintra, E.R. Ropelle, B. Rodrigues, F.S. DE Lira, C.T. DE Souza, Topiramate effects lipolysis in 3T3-L1 adipocytes., Biomed. Rep. 3 (2015) 827–830. doi:10.3892/br.2015.514.
[36] P.M. McDonough, R.S. Ingermanson, P.A. Loy, E.D. Koon, R. Whittaker, C.A. Laris, J.M. Hilton, J.B. Nicoll, B.M. Buehrer, J.H. Price, Quantification of hormone sensitive lipase phosphorylation and colocalization with lipid droplets in murine 3T3L1 and human subcutaneous adipocytes via automated digital microscopy and high-content analysis, Assay Drug Dev. Technol. 9 (2011) 262–280. doi:10.1089/adt.2010.0302.
[37] W. Chen, B. Chang, L. Li, L. Chan, Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease, Hepatol. Baltim. Md. 52 (2010) 1134–1142. doi:10.1002/hep.23812.
[38] M.K. Basantani, M.T. Sitnick, L. Cai, D.S. Brenner, N.P. Gardner, J.Z. Li, G. Schoiswohl, K. Yang, M. Kumari, R.W. Gross, R. Zechner, E.E. Kershaw, Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease

Chapter 3
99
or metabolic syndrome, J. Lipid Res. 52 (2011) 318–329. doi:10.1194/jlr.M011205.
[39] T. Deng, S. Shan, P.-P. Li, Z.-F. Shen, X.-P. Lu, J. Cheng, Z.-Q. Ning, Peroxisome proliferator-activated receptor-gamma transcriptionally up-regulates hormone-sensitive lipase via the involvement of specificity protein-1, Endocrinology. 147 (2006) 875–884. doi:10.1210/en.2005-0623.
[40] E.E. Kershaw, M. Schupp, H.-P. Guan, N.P. Gardner, M.A. Lazar, J.S. Flier, PPARgamma regulates adipose triglyceride lipase in adipocytes in vitro and in vivo, Am. J. Physiol. Endocrinol. Metab. 293 (2007) E1736-1745. doi:10.1152/ajpendo.00122.2007.
[41] C. Fernandez, M. Lindholm, M. Krogh, S. Lucas, S. Larsson, P. Osmark, K. Berger, J. Borén, B. Fielding, K. Frayn, C. Holm, Disturbed cholesterol homeostasis in hormone-sensitive lipase-null mice, Am. J. Physiol. Endocrinol. Metab. 295 (2008) E820-831. doi:10.1152/ajpendo.90206.2008.
[42] B. Xia, G.H. Cai, H. Yang, S.P. Wang, G.A. Mitchell, J.W. Wu, Adipose tissue deficiency of hormone-sensitive lipase causes fatty liver in mice, PLoS Genet. 13 (2017) e1007110. doi:10.1371/journal.pgen.1007110.
[43] M. Fransen, S.R. Terlecky, S. Subramani, Identification of a human PTS1 receptor docking protein directly required for peroxisomal protein import, Proc. Natl. Acad. Sci. U. S. A. 95 (1998) 8087–8092.

HSL hydrolyzes retinyl esters in qHSC
100
SUPPLEMENTARY FIGURES and TABLES
Supplementary figure S1. Isolation of highly-pure rat hepatic stellate cells by side scatter-activated cell sorting. Following Nycodenz gradient purification, freshly-isolated qHSC were further purified by Side Scatter-Activated Cell Sorting as described before [19] using MoFlo™ XDP (Beckman Coulter, Woerden, The Netherlands). A) As described before, two cell populations were detected with high side scatter (SSC), one with low forward scatter (FSC; R1) and one with high FSC (R2). B) Highly pure single cell HSC in fraction R1 (red bars) were characterized by high Lrat mRNA levels and low Cd68 (marker for Kupffer cells) and Enos (marker for endothelial cells) mRNA levels. Cells from fraction R2 (green bars), still contains high mRNA levels of Cd68 and Enos, which suggests that the R2 cell fraction contain (clusters of HSC with) Kupffer and/or endothelial cells. Supplementary Figure S2.

Chapter 3
101
Supplementary figure S2. HSL expression is rapidly lost during HSC differentiation. Freshly-isolated rat HSC were allowed to attach to culture disks for 4h and either harvested directly (D0) or cultured for 1, 3 or 7 days. Lipe mRNA levels were analyzed by Q-PCR (left panel) and HSL protein levels were analyzed by Western blotting (right panel). mRNA levels in qHSC were set to 1. Ponceau-S staining of total protein was included as loading control, while aSMA was analyzed to confirm activation of HSC. Lipe and HSL levels rapidly decreased upon culture-activation of HSC.

HSL hydrolyzes retinyl esters in qHSC
102
Supplementary figure S3. Isoproterenol treatment of human hepatic stellate cells (LX-2) induced HSL phosphorylation HSL activation and promotes lipid clearance. Basal levels of pHSL in LX-2 are low in human hepatic stellate cells (LX-2), which can be rapidly and strongly increased by Isoproterenol as shown by Western blotting (A) WB and immunofluorescence microscopy (B; pHSL is shown in green, dapi in blue). The secondary antibody control is shown for comparison. C) Oil red O (ORO) staining indicate that LX-2 accumulated intracellular lipids in presence of Oleate (0.1 mmol/L), while Iso (5µmol/L, 10µmol/L) decreased the intracellular lipid accumulation.

Chapter 3
103
Supplementary Table S1: Primers and Probes for Real-time Quantitative PCR Analysis
Gene Taqman primers and probes
18S Fwd: 5'-CGGCTACCACATCCAAGGA-3' Rev: 5'-CCAATTACAGGGCCTCGAAA-3' Probe: 5'-CGCGCAAATTACCCACTCCCGA-3'
Abcb11 Fwd: 5'-CCAAGCTGCCAAGGATGCTA-3' Rev: 5'-CCTTCTCCAACAAGGGTGTCA-3' Probe: 5'-CATTATGGCCCTGCCGCAGCA-3'
Acta2 Fwd: 5’-GCCAGTCGCCATCAGGAAC-3’ Rev: 5’-CACACCAGAGCTGTGCTGTCTT-3’ Probe:-CTTCACACATAGCTGGAGCAGCTTCTCGA-3’
ACTA2 Fwd: 5'-GGGACGACATGGAAAAGATCTG-3' Rev: 5'-CAGGGTGGGATGCTCTTCA-3' Probe: 5'-CACTCTTTCTACAATGAGCTTCGTGTTGCCC-3'
ALB Fwd: 5’-GCTGCCATGGAGATCTGCTT-3’ Rev: 5'-TTCCTTCAGTTTACTGGAGATCGAA-3’ Probe: 5'-CTTGGCAAGGTCCGCCCTGTCATC-3’
Cd68 Fwd: 5’-ACAGTGCCCATCCCCACTT-3’ Rev: 5'-GCTTGTGGGAAGGACACATTGT-3’ Probe: 5'-TTCAAACAGGACCGACATCAGAGCCAC-3’
Col1α1 Fwd: 5’-CGGCTCCTGCTCCTCTTAGG-3’ Rev: 5’-CTGACTTCAGGGATGTCTTCTTGG-3’ Probe: 5’-CCACTGCCCTCCTGACGCATGG-3’
COL1A1 Fwd: 5’-GGCCCAGAAGAACTGGTACATC-3’ Rev: 5'-CCGCCATACTCGAACTGGAA-3’ Probe: 5'-CCCCAAGGACAAGAGGCATGTCTG-3’
Lipe Fwd: 5’-GAGGCCTTTGAGATGCCACT-3’ Rev: 5’- AGATGAGCCTGGCTAGCACAG-3’ Probe: 5’-CCATCTCACCTCCCTTGGCACACAC-3’
LIPE Fwd: 5’-CTGCATAAGGGATGCTTCTATGG-3’ Rev: 5’-GCCTGTCTCGTTGCGTTTG-3’ Probe: CTGCCTGGGCTTCCAGTTCACGC-3’
Lpl Fwd: 5’-AAGGTCAGAGCCAAGAGAAGCA-3’ Rev: 5’-CCAGAAAAGTGAATCTTGACTTGGT-3’ Probe: 5’-CCTGAAGACTCGCTCTCAGATGCCCTACA-3’
Lrat Fwd: 5’-ACTGTGGAACAACTGCGAACAC-3’ Rev: 5’-AGGCCTGTGTAGATAATAGACACTAATCC-3’ Probe: 5'-AGGCCTGTGTAGATAATAGACACTAATCC-3’
LRAT Fwd: 5’-TGCGAGCACTTCGTGACCTA-3’ Rev: 5’-TTATCTTCACAGTCTCACAAAACTTGTC-3’ Probe: 5'-TGCAGATATGGCACCCCGATCAGTC-3’
Pnpla2 Fwd: 5'-AGCATCTGCCAGTATCTGGTGAT-3' Rev: 5'-CACCTGCTCAGACAGTCTGGAA-3' Probe: 5’-ATGGTCACCCAATTTCCTCTTGGCCC-3'
Pnpla3 Fwd: 5'-GTAGCCACTGGATATCTTCATGGA-3' Rev: 5'-TCTTGCTGCCCTGCACTCT-3' Probe: 5’-CACCAGCCTGTGGACTGCAGCG-3'
PPARγ Fwd: 5'-GATGTCTCATAATGCCATCAGGTT-3' Rev: 5'-GGATTCAGCTGGTCGATATCACT-3' Probe: 5’-CCAACAGCTTCTCCTTCTCGGCCTG-3'
Slc10a2 Fwd: 5'-ACCACTTGCTCCACACTGCTT-3' Rev: 5'-CGTTCCTGAGTCAACCCACAT-3' Probe: 5’-CTTGGAATGATGCCCCTTTGCCTCT-3'
Tgfβ Fwd: 5'-GGGCTACCATGCCAACTTCTG-3' Rev: 5'-GAGGGCAAGGACCTTGCTGTA-3' Probe: 5'-CCTGCCCCTACATTTGGAGCCTGGA-3'

HSL hydrolyzes retinyl esters in qHSC
104
Timp1 Fwd: 5'-GCAGGCAGTGATGTGCAAAT-3' Rev: 5'-ACCGCAGCGAGGAGTTTCT-3' Probe: 5'-CGTTCCTTAAACGGCCCGCGAT-3'
Supplementary Table S2: Antibodies used for Immunofluorescence and Western blotting
Antibody Dilution Catalog
number Company
Poly rabbit-anti HSL 1:1,000 4107 Cell Signaling Technology, Leiden, The Netherlands
Poly rabbit-anti LRAT 1:2,000 28075 Sigma Aldrich, The Netherlands
Mono mouse-anti GAPDH
1:2,000 CB1001 Calbiochem, San Diego, CA, United States
Mono mouse-anti αSMA
1:2,000 A5228 Sigma-Aldrich, The Netherlands
Poly-rabbit-phospho-HSL (Ser660)
1:1,000 4126 Cell Signaling Technology, The Netherlands
Poly goat-anti Collagen type 1
1:2,000 1310-01
Southern Biotech, The Netherlands

Chapter 4
Disturbed Vitamin A Metabolism in
Non-Alcoholic Fatty Liver Disease
(NAFLD)
Ali Saeed1,2, Robin P.F. Dullaart3, Tim C.M.A. Schreuder1, Hans
Blokzijl1, Klaas Nico Faber1,4*
1Department of Gastroenterology and Hepatology, 3Department of Endocrinology,
University Medical Center Groningen, 4Department of Laboratory Medicine, University
of Groningen, Groningen, The Netherlands
2Institute of Molecular Biology & Bio-technology, Bahauddin Zakariya University,
Multan, Pakistan
Nutrients 2017, 29;10 (1).

Disturbed vitamin A metabolism in NAFLD
106
ABSTRACT
Vitamin A is required for important physiological processes, including embryogenesis,
vision, cell proliferation and differentiation, immune regulation, as well as glucose and
lipid metabolism. Many of vitamin A’s functions are executed through retinoic acids that
activate transcriptional networks controlled by retinoic acid receptors (RARs) and
retinoid X receptors (RXRs).The liver plays a central role in vitamin A metabolism; 1) it
produces bile supporting efficient intestinal absorption of fat-soluble nutrients like
vitamin A, 2) it produces retinol binding protein 4 (RBP4) that distributes vitamin A, as
retinol, to peripheral tissues and 3) it harbors the largest body supply of vitamin A,
mostly as retinyl esters, in hepatic stellate cells (HSC). In times of inadequate dietary
intake, the liver maintains stable circulating retinol levels of approximately 2 µmol/L,
sufficient to provide the body for months with this vitamin. Liver diseases, in particular
those leading to fibrosis and cirrhosis, are associated with impaired vitamin A
homeostasis and may lead to vitamin A deficiency. Liver injury triggers HSC to
transdifferentiate to myofibroblasts that produce excessive amounts of extracellular
matrix leading to fibrosis. HSC lose the retinyl ester stores in this process ultimately
leading to vitamin A deficiency. Non-alcoholic fatty liver disease (NAFLD) is the hepatic
manifestation of the metabolic syndrome and is a spectrum of conditions ranging from
benign hepatic steatosis to non-alcoholic steatohepatitis (NASH) and may progress to
cirrhosis and liver cancer. NASH is projected to be the main cause of liver failure in the
near future. Retinoic acids are key regulators of glucose and lipid metabolism in liver
and adipose tissue, but it is unknown whether impaired vitamin A homeostasis
contributes to or suppresses the development of NAFLD. A genetic variant of patatin-
like phospholipase domain-containing 3, (PNPLA3-I148M) is the most prominent
heritable factor associated with NAFLD. Interestingly, PNPLA3 harbors retinyl ester
hydrolase activity and PNPLA3-I148M associates with low serum retinol levels, but
enhanced retinyl esters in the liver of NAFLD patients. Low circulating retinol in NAFLD
may therefore not reflect true “vitamin A deficiency”, but rather disturbed vitamin A
metabolism. Here, we summarize current knowledge about vitamin A metabolism in
NAFLD and its putative role in progression of liver disease, as well as the therapeutic
potential of vitamin A metabolites.
Keywords: Non-alcoholic fatty liver disease; Vitamin A deficiency; Hepatic stellate
cells

Chapter 4
107
4.1. Vitamin A, its active metabolites and dietary causes of vitamin A
deficiency
The generic term "Vitamin A" is used for compounds having the biological activity of
retinol or its metabolic products, such as all-trans retinoic acid (atRA), 9-cis retinoic
acid (9cRA) and 9-cis-13,14 dihydroretinoic acid (9cDHRA). Mammals cannot
synthesize retinol themselves, so depend on the diet to acquire sufficient amounts of
this micronutrient. Dietary sources of vitamin A are carotenoids, such as β-carotene
(rich plant sources are sweet potato, carrots and dark green leafy vegetables, like
spinach) and retinyl esters (rich animal sources are liver, eggs and fish) [1]. Vitamin A
is an essential fat-soluble vitamin and adequate daily intake (~700-900 µg for humans)
and hepatic storage (~80% in a healthy individual) are required to maintain plasma
retinol levels around 2 µmol/L in humans (1-2 µmol/L in rodents) [2,3]. Vitamin A plays
important physiological roles in vision, reproduction, growth, development, immunity
and metabolic programs [4–10]. Most functions of vitamin A are mediated through
activation of ligand-activated transcription factors. For example, atRA is a high affinity
ligand for retinoic acid receptors (RARα, β and ), while 9cRA and 9cDHRA activate
retinoid X receptors (RXRα, β and ). The ligand-dependent activation and the
physiological roles of RAR and RXR have been extensively reviewed over the past
years [10–12].
Vitamin A deficiency (VAD) is common in low-income countries. According to the World
Health Organization (WHO) an estimated 250 million preschool children in these
countries have VAD and are predisposed to developmental and immune disorders,
preventable (night) blindness and infection-associated morbidity and mortality.
Moreover, VAD in pregnant women increases the risk for maternal mortality, though
high doses of vitamin A supplementation in pregnant women are contraindicated as
they are associated with teratogenicity for the fetus [6,13]. The liver plays a central
role in controlling vitamin A metabolism and chronic liver diseases, like biliary atresia
[14,15], primary biliary cholangitis (PBC) [16–18], primary sclerosing cholangitis (PSC)
[19], viral hepatitis [20] [20], alcoholic liver disease (ALD) [21,22], and non-alcoholic
fatty liver disease (NAFLD, NASH) [23–25] are associated with VAD. Moreover, VAD
in hepatitis C virus (HCV) is associated with lack of response to interferon-based
antiviral therapy [20]. Thus, VAD is also a highly relevant condition in high-income
countries given the increased incidence of liver diseases associated with a Western
life style, e.g. 20-30% of the population of industrialized countries is suspected of

Disturbed vitamin A metabolism in NAFLD
108
having NAFLD [26–28]. Many epidemiological studies have reported VAD in liver
diseases, including NAFLD, where the golden standard for assessing the vitamin A
status is measuring serum retinol levels (< 0.7 µmol/L is considered VAD) [29].
Moreover, serum retinol levels are inversely associated with disease progression
[11,24,25,30,31]. Oral vitamin A supplements are often not very effective in restoring
adequate serum retinol levels in NAFLD patients, indicating a fundamentally impaired
vitamin A metabolism. Animal studies have shown beneficial effects of therapeutic use
of vitamin-A derivatives, e.g. retinoic acids or pharmacological modulators of RARs
and RXRs. Relatively little is known, however, about changes in vitamin A metabolism
in liver disease, including possible changes in storage (retinyl esters) vs. circulating
(retinol, RA) forms and possible redirection of vitamin A pools within the liver
(hepatocytes vs. hepatic stellate cells) and/or to extrahepatic tissues. Insight in these
processes is needed to get a better view on the possible therapeutic application of
vitamin A and/or its derivatives (retinoids acting on RARs and rexinoids acting on
RXRs) in liver diseases. This review aims to give an overview of vitamin A metabolism
in patients and animal models of NAFLD and associated obesity, as well as the
possible therapeutic value of vitamin-A and its metabolites in NAFLD.
4.2. Vitamin A uptake, storage and metabolism
A schematic representation of vitamin A uptake, transport, storage and metabolism is
given in Figure 1. Plant-derived carotenes and animal-derived retinyl esters are the
main sources of vitamin A in the diet. Bile salts produced by the liver are important to
keep these fat-soluble compounds in solution in the digestive tract and make them
available for absorption in the proximal part of the small intestine [32,33]. Retinyl ester
hydrolases (REH) in the gut lumen release retinol from retinyl esters, after which it is
absorbed by enterocytes by a yet to-be-characterized mechanism. Carotenes are
taken up by membrane-bound transporters, including fatty acid translocase
(FAT/CD36), Niemann-Pick C1-Like 1 (NPC1L1) and scavenger receptor class B
member 1 (SR-B1), and metabolized to retinol inside enterocytes. Next, retinol is re-
esterified to retinyl esters, mainly by lecithin:retinol acyl transferase (LRAT) and
diacylglycerol O-acyltransferase (DGAT1), sequestered into chylomicrons (CM) and
secreted to the circulation [34,35]. CM remnants that still contain most of the retinyl
esters are taken up by hepatocytes mostly through the LDL receptor (LDLR) [36]. The
retinyl esters are then hydrolyzed to retinol through the action of REHs [37] followed

Chapter 4
109
by transfer of retinol to RBP4 and transthyretin (TTR), which stimulates its secretion
into the circulation [38–40]. From here, retinol is directed to either of 2 main
destinations: 1) peripheral tissues in demand of retinol(-metabolites). Here, the RBP4-
TTR-retinol complex binds to “stimulated by retinoic acid gene 6 homolog” (STRA6),
which facilitates uptake of retinol [41] and makes it available for conversion to bioactive
retinoic acids; or 2) hepatic stellate cells (HSC) that convert it back again to retinyl
esters (by LRAT and DGAT1 [42,43]) and store it in large cytoplasmic lipid droplets. It
is estimated that 60-95% of the whole body reservoir of vitamin A resides in the liver
of a healthy individual, but considerable amounts may also reside in adipose tissue,
pancreas, intestines and the eyes. Still, the liver is considered to be the central player
in providing retinol to peripheral tissues in times of insufficient dietary intake. This is a
tightly controlled process maintaining well-balanced levels of approximately 2 µmol/L
retinol in blood. However, very little mechanistic details are known how this is achieved,
e.g. it is unknown how retinol gets in and out of HSC and which (molecular) signals
control this. Mobilization of retinol from lipid droplets in HSC is catalyzed by REHs, and
several enzymes are implicated in this process, including adipose triglyceride lipase
(ATGL/PNPLA2) [44], patatin-like phospholipase domain-containing 3 (PNPLA3) [44–
46] and hormone-sensitive lipase (HSL) [47,48]. Interestingly, genetic studies have
identified the PNPLA3-I148M variant as a prominent genetic factor associated with
NAFLD, and even more prominently with disease progression within NAFLD to NASH
and NASH-associated cirrhosis [49–51]. PNPLA3 affects vitamin A homeostasis, as
will be discussed in more detail in following sections. Liver diseases, in particular
chronic liver diseases that lead to liver fibrosis, have major impact on hepatic vitamin
A metabolism and storage. Quiescent vitamin A-storing HSC (qHSC) become
activated as a result of liver injury, and transdifferentiate to migratory and highly-
proliferative myofibroblasts (aHSC) that produce excessive amounts of extracellular
matrix proteins (ECM), mainly collagens and fibronectins, leading to fibrosis. HSC lose
their vitamin A stores during this transdifferentiation process. Consequently, chronic
liver diseases, including NAFLD, may lead to vitamin A inadequacy.
Given the various forms and storage sites of vitamin A in the body it is largely unclear
whether the reported vitamin A deficiency (VAD) in NAFLD is truly a reflection of
complete depletion of vitamin A from the body, or whether it is rather a reflection of
impaired vitamin A metabolism. Clinical examination of vitamin A status is typically
performed by determining serum retinol levels, where levels below 0.7 µmol/L are

Disturbed vitamin A metabolism in NAFLD
110
considered deficient. Retinol is also present in the liver, but most vitamin A is esterified,
predominantly as retinyl palmitate. Both forms reside in hepatocytes (initial site of
absorption from the circulation) and HSC (final destination and controller of serum
retinol levels). In addition, a significant amount of vitamin A is present in adipose tissue
and the pancreas, both as retinol and retinyl esters. Thus, for thorough evaluation of
the vitamin A status, both retinol and retinyl esters need to be quantified, in blood, liver
and preferably also adipose tissue. Hepatic retinol levels are sometimes analyzed in
liver biopsies or explant livers [25,52] but quantification of retinyl ester is rarely
performed [52]. Therefore, when scanning literature for the vitamin A status in liver
disease it is important to take into account what exactly is measured and keep in mind
that the limited data on hepatic retinyl ester levels prevents us from establishing the
true VAD prevalence in humans.
Figure 1. Schematic representation of intestinal vitamin A absorption, transport to and storage in the liver and redistribution to peripheral tissues. (see main text for details). Carotenes (from plants) and retinyl esters (from animals) are the main dietary sources of vitamin A (lower left corner). They are absorbed in the proximal small intestine and transported as retinyl esters in chylomicrons (CM) to the liver. Chylomicron remnants are taken up by hepatocytes and retinyl esters are hydrolyzed to form retinol. Hepatocytes produce retinol binding protein 4 (RBP4) and retinol binding to RBP4 stimulates the secretion of retinol-carrying holo-RBP4 to the circulation. Retinol is then either transported to hepatic stellate cells for storage as retinyl esters or transported to peripheral tissues where it is converted to retinoic acids that activate the transcription factors retinoic acid receptor (RAR) or retinoid X receptor (RXR). In times of inadequate vitamin A intake, retinol is released from the HSC stores to maintain stable levels of circulating retinol (~2 µM in human).

Chapter 4
111
4.3. Vitamin A and RBP4 in the clinical course of NAFLD and metabolic
syndrome
NAFLD is characterized by the accumulation of fat in the liver, in particular non-
esterified fatty acids (NEFA), triglycerides and non-esterified cholesterol. NAFLD is
regarded as a consequence of excessive dietary intake of fat and/or sugars (glucose
and fructose) typical for the “Western” life style, in combination with limited physical
activity. Benign steatosis may progress to an inflammatory state in the liver leading to
chronic liver injury, e.g. non-alcoholic steatohepatitis (NASH). In turn, this causes a
chronic healing response leading to liver fibrosis that may progress to cirrhosis that
predisposes for liver cancer. NAFLD is commonly appreciated to be the hepatic
manifestation of the metabolic syndrome (MetS). Numerous studies have reported on
vitamin A status in MetS, while reports specifically focusing on NAFLD are limited.
Almost all studies documenting vitamin A status in MetS report reductions in serum
retinol, retinoic acid and/or β-carotene that inversely correlate with MetS features,
including obesity, insulin resistance, glucose intolerance and hypertriglyceridemia
[53–62]. In line with these observations, inadequate serum retinol levels (<1.05 µmol/L)
were found in 11-36% of morbidly obese adults with ultrasonography-proven NAFLD
and a significant association between low retinol levels and insulin resistance (IR) was
found [25,53]. A similar trend was observed in obese children with NAFLD [23].
Moreover, serum retinol levels were inversely associated with body mass and serum
transaminases in patients with NAFLD, suggesting a link between retinol inadequacy
and development of disease [24]. In one study, both serum and hepatic retinol levels
were analyzed in NAFLD patients and the latter was found more frequently inadequate
(36% vs. 68% for serum and hepatic retinol inadequacy, respectively) in these patients
[25]. Hepatic retinol levels showed a strong inverse correlation with the histological
classification of the disease (sub-classified in mild and severe steatosis, NASH and
hepatocyte necrosis), which was not observed for serum retinol levels. These
observations were confirmed by Trasino et al. [63] who reported an inverse correlation
between the level of steatosis and hepatic retinol and retinyl palmitate concentrations.
However, the cause of hepatic steatosis was unknown in this study, as liver tissue was
obtained from deceased individuals after motor vehicle accidents or head trauma [63].
More recently, also serum retinoic acid levels were shown to be inversely associated
with hepatic steatosis and liver injury in NAFLD. While reference values for retinol are
approximately 2 µmol/L in human blood, atRA concentrations are approximately 200-

Disturbed vitamin A metabolism in NAFLD
112
fold lower (~10 pmol/L). Still, also for circulating atRA, levels were markedly reduced
in NAFLD (-47%) and even more pronounced in NASH (-58%) patients compared to
control subjects [64]. Moreover, atRA concentration and RXRα levels were inversely
correlated to liver triglyceride content, grade of hepatic steatosis and severity of liver
disease [64]. In comparing the expression of 51 genes involved in RA signaling in
control, simple steatotic and NASH livers, Ashla et al. [65] observed a hyper-dynamic
state of RA metabolism and degradation in the liver of NAFLD patients, which further
increased when it included NASH. Hepatic expression of genes involved in vitamin A
storage (LRAT and DGAT1) as well as RA production retinaldehyde dehydrogenase 1
and 3 (RALDH1 and 3) and degradation (Cyp26A) were all significantly increased in
NAFLD patients. This expression profile may indeed lead to low hepatic retinol and
retinoic acids levels, but not necessarily to VAD, as it suggests that also production of
retinyl esters is enhanced. Low retinol levels appear predictive for the development of
hepatocellular carcinoma (HCC) in cirrhotic patients [66,67], though this has not been
studied yet specifically for NAFLD-associated HCC. Impaired RAR- and RXR-
mediated signaling is assumed to promote HCC. Hepatocyte-specific overexpressing
of a dominant negative form of RAR induces hepatic tumor development, which is
suppressed by a diet containing retinoic acid [68].
In contrast to reduced retinol, serum levels of RBP4 are typically elevated in MetS
patients and obese animals. A landmark paper by Yang et al. [69] revealed a direct
role of serum RBP4 in the development of insulin resistance in obesity and type 2
diabetes [69]. RBP4 expression was selectively enhanced in adipose tissue in animal
models of type 2 diabetes, but not in liver. Transgenic overexpression of human RBP4
in mice or injection with recombinant RBP4 in normal mice lead to insulin resistance.
These findings suggest that adipose-derived RBP4 increases serum levels of RBP4
and plays a pathological role in type 2 diabetes (T2D). Many papers have confirmed
the elevated serum RBP4 levels in obese patients with or without T2D (for example
see [69–73]). However, also a significant number of similar studies did not replicate
the enhanced levels of RBP4 in serum of these patients (for example see [74–78]).
Thus, the specific correlation between serum RBP4 levels and components of the MetS
spectrum remain an active debate today. Since serum retinol levels are typically
reduced in MetS, this feature may be an important factor to consider together with
RBP4. Low serum retinol in combination with stable or enhanced RBP4 levels infers
the presence of more retinol-free (apo-)RBP4 in circulation. The few studies that

Chapter 4
113
quantified both retinol and RBP4 in serum indeed confirm that a low retinol-to-RBP4
ratio is a better predictor for obesity, T2D and other components of metabolic syndrome
in children and adults then RBP4 alone [79–81].
In addition to the debate on the relevance of serum RBP4 levels as indicator for MetS
components, also the origin of the increased levels of RBP4 is an ongoing puzzle.
Enhanced RBP4 production by adipose tissue was the original hypothesis, because of
the selective increase of RBP4 mRNA levels in this tissue in T2D mice and not in liver
[69]. More recent data indicate that elevated serum RBP4 levels are specifically
associated with obesity- and T2D-associated reduced kidney function, suggesting that
impaired renal clearance of RBP4 is an important contributing factor [82–86]. Recently,
it was found that hepatocyte-specific deletion of Rbp4 in mice completely abolishes
RBP4 from circulation, both in lean and obese animals, providing strong evidence that
the liver, more specifically the hepatocytes, is the primary –if not sole- source of serum
RBP4 [87]. In line with this finding is that adipocyte-specific overexpression of human
RBP4 did not increase circulating RBP4, but did cause hepatic steatosis in mice [88].
These findings suggest that adipocyte-produced RBP4 acts locally to activate signaling
cascades that cause fat-accumulation in the liver, but is not a circulating adipokine
itself. Secretion of RBP4 by hepatocytes is strongly stimulated by retinol [38–40] and
this may be impaired in NAFLD patients where both hepatic and serum retinol levels
are reduced. RBP4 indeed accumulates in livers of NAFLD patients as determined by
immunohistochemistry [89]. Hepatic RBP4 retention in low-retinol NAFLD livers
suggests that impaired renal clearance might even be a more prominent factor in
enhancing serum RBP4 levels.
Bariatric surgery is nowadays a common approach to treat morbid obesity, where
adjustable gastric band (AGB), Roux-en-Y gastric bypass (RYGB), biliopancreatic
diversion with a duodenal switch (BPD-DS) and vertical sleeve gastrectomy (VSG) are
the 4 most applied procedures. Remarkably, serum retinol levels do not return to
normal after bariatric surgery and in most studies even further decline in the 6-12
months post-surgery, also under impressive weight loss and improvement of MetS
components [90–94]. Ocular problems related to low vitamin A status, such as night
blindness, are commonly reported in patients who underwent bariatric surgery. BPD-
DS appears to induce a stronger decline in serum retinol compared to RYGB [91,95]
and vitamin A deficiency was still observed in 23-69% patients 4-10 years post-PBD-
DS [96–98]. Impaired serum retinol levels were also observed in neonates from

Disturbed vitamin A metabolism in NAFLD
114
mothers who had underwent RYGB 11-69 months before pregnancy onset, which may
cause irreversible eye problems in the child [99]. In contrast to retinol, circulating levels
of vitamin D and E were higher in neonates of mothers who had undergone bariatric
surgery as compared to neonates from healthy mothers [100]. This argues against a
general deficiency in fat-soluble vitamins in these children. Inadequate serum retinol
levels before and/or after bariatric surgery are often linked to inadequate dietary intake,
as well as the anatomical changes resulting from bariatric surgery. Large parts of the
jejunum do not receive dietary input post-surgery and this is exactly the site where
most vitamin A is normally absorbed. However, intramuscular [101] or dietary vitamin
A supplementation (alone or in multivitamins) does not effectively elevate serum retinol
levels and/or prevent the decline in serum retinol post-surgery [91,92,94,102]. Few
case reports have shown that only very high doses of vitamin A relieve signs of severe
VAD, like night blindness [103]. Little is currently known about the effect of diet-induced
weight loss by calorie restriction and its effect of serum retinol levels. Serum RBP4
decline after this treatment, but this is not consistently associated with improvement in
insulin sensitivity [104–107].
4.4. PNPLA3 variant I148M regulates vitamin A in NAFLD
Several genomic loci have been identified to increase the susceptibility for NAFLD.
The most prominent NAFLD-associated genetic risk factor is a specific variant of
PNPLA3, PNPLA3-I148M, which also predisposes for disease progression and
NAFLD-associated hepatocellular carcinoma [108–111]. PNPLA3 is a multifunctional
enzyme acting as a triglyceride hydrolase, an acetyl-CoA-independent transacylase
and a retinyl esterase [112]. Originally, it was found to contain hydrolase activity
towards triglycerides, in particular those containing mono- and poly-unsaturated fatty
acids. PNPLA3-I148M shows reduced hydrolase activity and promotes hepatic
triglyceride accumulation, all in line with the primary phenotype of NAFLD: fat
accumulation in hepatocytes. In line, chronic overexpression of PNPLA3-I148M (but
not PNPLA3-I148) in mice leads to hepatic steatosis [113]. Notably, while the variant
is associated with increased liver fat content, PNPLA3-I148M appears not to be
associated with other features of the metabolic syndrome, like insulin resistance
[114,115]. Serum triglycerides (TG) levels are either the same or lower in PNPLA3-
I148M carriers compared to non-carriers, consistent with a lack of association with
insulin resistance [115].

Chapter 4
115
A recent paper, however, showed that PNPLA3 mRNA and protein levels are
significantly higher in HSC compared to hepatocytes. PNPLA3 was found to contain
retinyl-esterase activity and promotes the release of retinol from lipid droplets [45].
Carriers of the PNPLA3-I148M allele with either NAFLD or obesity alone have reduced
fasting circulating retinol and RBP4 levels. No association was found between this
genotype and β-carotene, indicating a specific association with retinol [46]. On the
other hand, hepatic retinyl palmitate levels are increased in individuals homozygous
for PNPLA3-I148M [116], supporting a role for PNPLA3 in hepatic retinoid metabolism
[116]. PNPLA3 expression in HSC is regulated by retinol and insulin and is inversely
related to lipid droplet content. Retinol suppresses PNPLA3 expression in HSC, while
it is induced upon retinol depletion. Moreover, PNPLA3 expression is induced upon
HSC activation and the PNPLA3-I148M variant further promotes fibrogenic features of
HSC, including enhanced proliferation, migration and expression of collagen type 1
alpha 1 (COL1A1), pro-inflammatory cytokines and chemokines alongside lower
cellular retinol levels. Remarkably, PNPLA3-I148M-carrying HSC contain more lipid
droplets, which is a typical characteristic of HSC quiescence [117]. These features are
in line with a higher risk for progressive liver disease in PNPLA3-I148M carriers, but
are seemingly in contrast to the increased hepatic retinyl palmitate levels in those
patients. Thus, these NAFLD patients did not have VAD, although the low circulating
retinol levels are interpreted as such. It remains to be determined whether this is a
more general phenomenon in NAFLD. Since 1) dietary retinyl esters are first delivered
to -and hydrolyzed in- hepatocytes before they move as retinol to HSC to become
esterified again, and since 2) PNPLA3 is expressed both in hepatocytes and HSC, it
remains an open question in which hepatic cell type retinyl esters accumulate in
NAFLD and specifically in PNPLA3-I148M patients.
4.5. Vitamin A and hepatic lipid metabolism.
The hepatic lipid content is a resultant of 1) de novo lipogenesis (DNL) in the liver, 2)
influx of dietary lipids (delivered as non-esterified free fatty acids (NEFAs) or as TG in
chylomicrons), 3) influx of NEFAs produced by adipose tissue (primarily from white
adipose tissue [WAT]), 4) esterification of lipids (mainly to TG) and packaging into lipid
droplets, 5) influx of TG carried in CM remnants and low density lipoproteins (LDL), 6)
efflux of TG carried in very low density lipoprotein (VLDL)-particles, 7) TG hydrolysis
producing NEFAs and 8) catabolism of NEFAs through mitochondrial and peroxisomal

Disturbed vitamin A metabolism in NAFLD
116
β-oxidation (Figure 2). NAFLD is characterized by hepatic TG accumulation, enhanced
VLDL production and secretion leading to hypertriglyceridemia. Vitamin A-metabolites,
especially retinoic acids, are involved in regulatory networks that affect all these
processes either directly or indirectly, as described below and schematically depicted
in Figure 2.
Hepatic DNL (#1 in Figure 2) is under primary transcriptional control of the sterol
response element binding protein-1c (SREBP-1c) and carbohydrate response element
binding protein (ChREBP) that induce expression of key glycolytic enzymes
(glucokinase (GCK), pyruvate kinase isozymes R/L (PKLR), ATP citrate lyase (ACL),
acetyl-CoA synthetase (ACS) and lipogenic enzymes (acetyl-CoA carboxylase 1
(ACC1), fatty acid synthase (FASN), ELOVL fatty acid elongase 6 (ELOVL6), stearoyl-
CoA desaturase-1 (SCD), glycerol-3-phosphate acyltransferase, mitochondrial
(GPAT)) in the liver. Absence or inhibition of SREBP-1c or ChREBP impairs lipid
synthesis and reduces hepatic steatosis [118–120]. Insulin, glucose and fructose
stimulate SREBP-1c and ChREBP expression to promote hepatic DNL. Retinoic acids,
as well as synthetic ligands of RXRα (e.g. bexarotene) enhance hepatic DNL and
plasma TG levels by activating Liver X receptor (LXR)/RXR and peroxisome
proliferator-activated receptor γ (PPARγ)/RXR that, in turn, enhance expression of
SREBP-1c and ChREBP [121]. Although LXR and PPAR are typically activated by
oxycholesterols and NEFAs, respectively, RXRα is a permissive dimerization partner,
meaning that RXR ligands enhance DNL through those heterodimers independently of
the co-presence of ligands for LXR or PPARγ. LXR/RXR also directly induces FAS
expression, thereby promoting DNL and enhancing plasma triglyceride levels in mice
[122]. In contrast, atRA suppresses DNL by activating RARα that, via induction of Hes
family BHLH transcription factor 6 (HES6) and subsequent inhibition of hepatocyte
nuclear factor 4α (HNF4α), thereby reducing PPAR expression and downstream
SREBP-1c activity. In a counteracting mechanism, 9cRA-activated RXRα induces
expression of small heterodimer partner (SHP), which inhibits HES6 expression
promoting DNL via the PPAR-SREBP-1c axis [123]. However, SHP also inhibits
LXR/RXR transcriptional activity, thereby simultaneously inhibiting SREBP-1c-
mediated DNL [124]. This highlights the delicate position of SHP in the development
of hepatic steatosis, inhibiting RXR/LXR-ChREBP/SREBP-1c-mediated DNL, while at
the same time promoting DNL via the HES6-HNF4α-PPAR pathway. Absence of SHP

Chapter 4
117
protects mice against diet-induced hepatic steatosis, suggesting a most prominent role
for the HES6-HNF4α-PPAR axis, which is activated by atRA and RARα [123].
Figure 2. Regulation of hepatic lipid metabolism by vitamin A metabolites. Triglyceride synthesis and breakdown is subdivided in 8 steps: 1) de novo lipogenesis (DNL) in the liver, 2) influx of dietary lipids (delivered as non-esterified free fatty acids (NEFAs) or as triglycerides (TG) in chylomicrons), 3) influx of NEFAs produced by adipose tissue (primarily from white adipose tissue [WAT]), 4) esterification of lipids (mainly to TG) and packaging into lipid droplets, 5) influx of TG carried in CM remnants and low density lipoproteins (LDL), 6) efflux of TG carried in very low density lipoprotein (VLDL)-particles, 7) TG hydrolysis producing NEFAs and 8) catabolism of NEFAs through mitochondrial and peroxisomal β-oxidation. Direct transcriptional regulation of lipogenic/lipolytic genes is shown in the inner (light grey) ring. Indirect transcriptional regulation is shown in or outside the outer (dark grey) ring. Vitamin A-related factors are indicated in blue. Factors that promote lipogenesis are shown in red, factors promoting lipolysis in green. Relevant regulation and factors in adipose tissue and the intestine are also included. (see main text for details about the specific genes that are regulated in each step).
Influx of NEFA, either from dietary sources or adipose tissue (#’s 2 and 3 in Figure 2)
is facilitated by fatty acid translocase (FAT/CD36). Hepatocyte-specific deletion of
CD36 protects against HFD-induced lipid accumulation in mouse liver, underscoring
its role in NAFLD pathogenesis. Expression of CD36 is controlled by SREBP-1c,
RXR/PPAR and RXR/PPARα, transcription factors that are regulated directly or
indirectly by RA (see above). Moreover, RAR/RXR may also directly enhance

Disturbed vitamin A metabolism in NAFLD
118
expression of CD36, though this regulatory pathway has only been studied in human
THP-1 monocytes so far [125,126].
Hepatic TG synthesis (#4 in Figure 2) is catalyzed by GPAT, mannosyl (alpha-1,6-)-
glycoprotein beta-1,2-N-acetylglucosaminyltransferase (MGAT2) and diacylglycerol O-
acyltransferase 2 (DGAT2) all of which are under transcriptional control of ChREBP.
In addition, GPAT is also controlled by SREBP-1c. Though no specific data is available
on RA-mediated expression of those genes, it is likely that they are co-regulated with
key genes in DNL as a resultant of RXR/LXR and RAR-mediated effects on ChREBP
and SREBP-1c.
Hepatic uptake of TG-containing CM remnants or LDL particles (#5 in Figure 2) is
controlled by the LDL receptor (LDLR). Expression of human LDLR is controlled by
LXR/RXR [127], but co-regulation by retinoic acids has not yet been studied in detail.
However, given the potent effects of RXR-ligands on LXR/RXR-mediated regulation of
SREBP-1c and ChREBP (described above) it is likely that retinoic acids may also
promote LDLR-mediated uptake of TG in the liver.
Hepatic VLDL particle formation and secretion (#6 in Figure 2) is facilitated, amongst
others, by apo-CIII [128]. Apo-CIII null mice fail to stimulate VLDL production upon
HFD-feeding, while Apo-CIII overexpressing mice show enhanced diet-induced
triglyceride accumulation in the liver [129]. A genetic variant of Apo-CIII leads to
enhanced circulating Apo-CIII in humans and is associated with NAFLD [130]. Hepatic
Apo-CIII expression is suppressed by RARα via a pathway involving SHP and HNF4α,
thereby reducing hepatic and plasma triglyceride levels [131]. Earlier studies have
shown that RXR ligands sort the opposite effect and enhance hepatic apo-CIII
expression, either via RXR homodimers or RXR/PPARα, and thereby promote
hypertriglyceridemia, a well-known adverse effect of pharmacological ligands of RXR
and a risk factor for cardiovascular disease (CVD) [132]. This emphasizes the opposite
roles of RAR-ligands (e.g. atRA) and RXR-ligands (e.g. 9cRA) on VLDL particle
production and secretion by the liver. In addition, Apo-CIII expression is controlled by
ChREBP adding an additional layer of indirect RA-responsiveness as described above
[133].
Key enzymes involved in TG lipolysis (#7 in Figure 2) in hepatocytes are adipose
triglyceride lipase (ATGL/PNPLA2), hormone-sensitive lipase (HSL) and PNPLA3.
Those are exactly the same enzymes responsible for retinyl ester hydrolysis that
release retinol from cellular stores, though that activity is believed to occur

Chapter 4
119
predominantly in HSC (see above). Absence of either ATGL or HSL aggravate diet-
induced hepatic TG accumulation and steatosis in mice [134–136]. In line, hepatic
overexpression of ATGL and/or HSL reduced TG levels by 40-60% in ob/ob mice,
enhanced fatty acid oxidation and ameliorated hepatic steatosis, while fasting plasma
TG and NEFA were not affected [137]. Expression of both ATGL and HSL is controlled
by PPARα and PPARα-agonist-mediated decrease in hepatic lipids in HFD-fed mice
was paralleled by a strong increases in expression of these genes [138]. PPARα
ligands also induce hepatic ATGL expression in rats and reduce hepatic TG levels
[139]. It is interesting to note that hepatic lipolysis produces ligands for PPARα to
further enhance catabolism via mitochondrial and/or peroxisomal β-oxidation.
Moreover, HSL expression is under (positive) control of PPAR [140]. LXRα-agonists,
on the other hand, were found to suppress HSL expression, but so far, this has only
been analyzed in adipocytes [141]. There is no information available on the effect of
RA on PPARα- and/or PPAR-mediated regulation of ATGL and HSL, but these vitamin
A–metabolites likely have modulatory functions on their expression given the
collaborative actions of those factors with RXRα. Interestingly, PNPLA3 expression is
under direct control of SREBP-1c and ChREBP [142,143], which was linked to its role
in conversion of TG to NEFA. Given the recent data that PNPLA3 also contains retinyl
esterase activity and is highly expressed in HSC, this may imply a direct role of these
transcription factors in vitamin A metabolism.
Finally, hepatic fatty acid β-oxidation (#8 in Figure 2) is largely controlled by
PPARα/RXR. There is a wealth of information about PPARα agonists and how they
protect against and/or relieve fat accumulation in the liver (see for recent reviews
[144,145]). There is, however, limited information on whether vitamin A-metabolites
have a modulatory action on RXR/PPARα-mediated fatty acid catabolism. atRA
treatment does enhance hepatic PPARα and RXRα levels in mice, as well as key target
genes uncoupling-protein-2 (UCP2), carnitine-palmitoyltransferase 1A (CPT1) and
carnitine/acylcarnitine carrier, while suppressing SREBP-1c and FAS levels [146]. Both
9cRA and atRA were shown to induce CPT1 in vitro most likely via RXR/PPARα [147].
PPARα also induces expression of Fibroblast Growth Factor 21 (FGF21), a
hepatocyte-derived hormone suppressing obesity-induced fatty liver. FGF21 controls
glucose and lipid metabolism and induces PPAR-coactivator 1-alpha (PGC-1α)
signaling resulting in enhanced fatty acid oxidation and suppression of lipid synthesis
(reviewed in [148]). atRA induces expression of FGF21 through RARα and RARβ.

Disturbed vitamin A metabolism in NAFLD
120
Adenoviral overexpression of RARβ enhances hepatic production and secretion of
FGF21 and promotes hepatic fatty acid β-oxidation [149]. FGF21 expression is also
controlled by RXR/Farnesoid X receptor (FXR), where RXR acts as a permissive
partner. Thus, FGF21 expression is enhanced by 9cRA via FXR/RXR [150]. Hepatic
lipid metabolism is also modulated by another FGF, e.g. FGF19 (the ortholog of FGF15
in rodents). Murine FGF15 is produced in the intestine, while human FGF19 is
produced in the intestine as well as in the liver [151–153]. FGF19 suppresses
lipogenesis by blocking SREBP-1c signaling and simultaneously inducing fatty acid β-
oxidation by blocking ACC2 (reviewed in [154]). Mouse FGF15 expression is under
control of RXR/FXR where ligands of RXR induce its expression independently from
bile acid-induced FXR activation. A similar effect of retinoic acids was found for human
FGF19 expression, however, mechanistically a more prominent role was observed for
RXR/RAR-mediated regulation of FGF19 [155].
Taken together, it is evident that vitamin A-metabolites are key (co-)regulators of
hepatic lipid metabolism. RAR-mediated signaling (via atRA or synthetic ligands)
shows most consistently a suppression of hepatic NEFA and TG accumulation. RXR-
mediated signaling (via 9cRA, synthetic ligands or other NR, like PPARs, LXR and
FXR) may cause opposing effects at different levels in the metabolic pathway leading
to hepatic lipid accumulation. However, the liver is not the only tissue involved in
obesity-induced hepatic steatosis that is heavily regulated by vitamin A metabolites.
The most important –and intensively studied- being adipose tissue, which is discussed
next.
4.6. Vitamin A and fat metabolism in adipose tissue
Besides controlling lipid metabolism in the liver, vitamin-A metabolites also play key
roles in differentiation, maturation and function of adipose tissue. In obesity-associated
NAFLD, there is an increase in NEFA flowing from adipose tissue to the liver, in part
as a result of insulin resistance. Adipogenesis is a tightly regulated cellular
differentiation process, in which preadipocytes are transformed into lipid-storing
adipocytes with enhanced expression of lipogenic genes. Insulin promotes glucose
transport to adipocytes and PPAR-dependent DNL leads to lipid accumulation.
atRA has a dual role in adipocyte differentiation and functionality: 1) it suppresses
adipogenesis, while 2) it promotes lipolysis in differentiated adipocytes. Two RA-
binding proteins, cellular retinoic acid binding protein 2 (CRABP2) and Fatty Acid

Chapter 4
121
Binding Protein 5 (FABP5), play a key role in directing atRA to either RXR/RAR or
RXR/PPARβ/δ, activation of which differentiates between the two pathways. atRA
inhibits adipogenesis through the CRABP2-RXR/RAR pathway that induces
expression of inhibitors of adipocyte differentiation, amongst others SOX9 that blocks
CCAAT/enhancer binding protein beta (C/EBPβ) and CCAAT/enhancer binding protein
gamma (C/EBP)-mediated differentiation and Kruppel-like factor 2 (KLF2) that blocks
CCAAT/enhancer binding protein alpha (C/EBPα)-, SCREBP-1c- and PPAR-
mediated adipogenesis. KLF2 also induces RAR and CRABP2, providing a positive
feedback loop to suppress adipogenesis [156,157]. Adipocyte differentiation (for
instance induced by insulin) is initiated by lowering CRABP2 levels and redirection of
atRA-mediated signaling to FABP5-RXR/PPARβ/δ that induces lipolysis (via HSL
upregulation), mitochondrial activity (via uncoupling proteins/UCP1) and fatty acid β-
oxidation, as well as enhancing the insulin-responsive glucose transporter type 4
(GLUT4). In vivo, atRA raises body temperature, decreases body weight and reduces
plasma triglycerides and insulin levels in obese mice, and might do so more potently
than selective PPARβ/δ ligands [156,157]. Obviously, this is not a stand-alone effect
on adipose tissue, but heavily intertwined with effects of hepatic lipid metabolism
described above. Interestingly, impaired supply of retinols to (pre)adipocytes may also
stimulate adipogenesis, as excess of (retinol-free) apoRBP4 (as observed in obese
individuals with a low retinol:RBP4 ratio) promotes retinol-efflux via STRA6, thereby
reducing RAR activity leading to enhanced adipogenesis [158]. As stated earlier,
adipocytes are the main extrahepatic cells that express RBP4, but this does not
contribute to circulating RBP4. Adipocyte-specific overexpression of (human) RBP4
aggravated diet-induced obesity, glucose intolerance and hepatic TG levels. RBP4-
induced inflammation in adipose tissue stimulated lipolysis in adipocytes leading to
enhanced circulating NEFA leading to elevated triglycerides in the liver [88].
In addition to atRA-signaling, synthetic ligands for RXR promote adipogenesis in 3T3-
L1 cells, a commonly used model to study adipocyte differentiation, by activation of
RXR/PPAR-mediated adipogenesis [159]. On the contrary, retinaldehyde, the
precursor for retinoic acid, inhibits 9cRA-mediated activation of RXR/PPAR thereby
suppressing adipogenesis and lipid accumulation in adipose tissue. Moreover,
retinaldehyde activates RAR, thereby recruiting PGC-1α and inducing UCP1
expression leading to enhanced mitochondrial respiration and adaptive
thermogenesis, which promotes “browning” of white adipose tissue [160]. While

Disturbed vitamin A metabolism in NAFLD
122
endogenous levels of retinoic acids are generally undetectable in tissues,
retinaldehyde levels in mouse adipose tissue are ~1 nmol/g [161]. Intraperitoneal
administration of retinaldehyde significantly suppressed adipogenesis and diet-
induced obesity in mice, while such effect was not observed after oral delivery of
retinaldehyde [161,162]. Genetic ablation of Aldh1a1 (encoding RALDH1) and
administration of RALDH inhibitors increase tissue levels of retinaldehyde and protect
against diet-induced obesity and diabetes [160,161].
Finally, glucose may be an important modulator of the effect of RA on adipocytes as
recent data show that RA suppresses lipid accumulation under normal glucose levels,
while this effect shifts to lipid accumulation at high glucose conditions, a metabolic
switch controlled by SREBP-1c [163].
4.7. Insulin and vitamin A cross talk in NAFLD
Vitamin A is required for normal development and endocrine functions of the pancreas,
including the insulin-producing β-cells and the glucagon-producing α-cells in the islets
of Langerhans. The pancreas stores retinoids in pancreatic stellate cells that are
essential for normal islet function. Similar to the liver, retinoid storage in the pancreas
is impaired during development of pancreatic diseases. VAD reduces β-cell mass and
increases α-cell mass. Consequently, VAD in mice leads to aberrant pancreatic
endocrine function due to lower insulin secretion and promotes hyperglycemia [164].
atRA-activated RARα induces pancreatic glucose transporter type 2 (Glut2) and Gck
expression and is required in the adult pancreas for maintaining β-cell mass and
function [165,166]. Similarly, RARβ2 agonists improved insulin sensitivity, and lowered
serum glucose and insulin levels and reduces triglycerides and steatosis in liver,
pancreas and kidneys of obese and diabetic mice [167].
In liver, insulin promotes activation of HSC via phosphorylation of forkhead box gene,
group O1 (FoxO1). Active (non-phosphorylated) FoxO1 suppresses HSC activation.
Insulin signals via the PI3/AKT pathway to phosphorylate FoxO1, thereby allowing
activation of HSC, characterized by enhanced proliferation and expression of fibrotic
markers and aggravation of bile duct ligation-induced fibrosis in FoxO1+/-
heterozygous mice compared to FoxO1+/+ wild types [168–170]. A recent paper
suggests though that the effect of insulin may turn around in the presence of vitamin
A. Co-treatment with insulin potentiated the vitamin A-mediated suppression of HSC
activation markers through stimulating Janus kinase 2/ Signal transducer and activator

Chapter 4
123
of transcription 5A (JAK2/STAT5) signaling and SREBP1 expression [171]. Thus, VAD
and hyperinsulinemia may synergize to activate HSC and promote fibrosis in NAFLD.
4.8. Vitamin A therapy in NAFLD
Despite extensive historical and recent evidence that 1) vitamin A metabolism is
disturbed in obesity and NAFLD and 2) vitamin A-metabolites, especially atRA and
synthetic RAR ligands, have beneficial effects on hepatic lipid metabolism and obesity-
induced NAFLD in animal models, no clinical trials are ongoing to evaluate their
therapeutic potential in patients. Instead, multiple trials are being performed to test the
therapeutic value of synthetic ligands that modulate the activity of other nuclear
receptors that control hepatic glucose and lipid metabolism, such as PPARα, PPARβ/δ,
PPAR, and FXR, all dimerization partners of RXRα. Also, variants of RAR-controlled
FGF19 and FGF21 are in phase II clinical trials for the treatment of NAFLD [172]. As
outlined above, vitamin A metabolism is heavily disturbed in NAFLD and will affect the
activation status of RXRs and RARs. As an additional result, it will also modulate the
activity of PPARs and FXR via the heterodimer partner RXR. Thus, the synthetic
ligands for these receptors that are currently in clinical trials for the treatment of NAFLD
and/or NASH may only reside partial effects because of impaired activation of the
heterodimer partner RXR. Re-establishing proper levels of vitamin A metabolites,
either systemically or directed to the liver, may have therapeutic value on its own, or
potentiate the therapeutic effect of ligands of PPARs and/or FXR. Moreover, vitamin
A-metabolites also regulate bile acid synthesis directly via RXR- and RAR-mediated
regulation of SHP and FGF15/19, as well as indirectly via RXR/FXR [155,173–176],
which may feedback into FXR-mediated regulation of lipid and glucose metabolism.
In addition, retinaldehyde was identified about 10 years ago as promising compound
to treat diet-induced obesity and diabetes in animal models. Raldh1-/- mice, which
accumulate retinaldehyde in tissues, showed reduced hepatic lipid accumulation
compared to wild type (WT) mice when fed a high fat diet (HFD) [161]. However, no
application of retinaldehyde or RALDH inhibitors in NAFLD patients has been reported
so far or is in early phase of clinical testing.
Fenretinide (4-hydroxy(phenyl)retinamide; 4-HPR) is a synthetic retinoid that has been
extensively studied for its potential therapeutic action against cancers, especially
breast cancer, non-small cell lung cancer, neuroblastoma and prostate cancer. Both
RXR- and RAR-dependent and independent mechanism have been proposed to

Disturbed vitamin A metabolism in NAFLD
124
underlie the therapeutic action of fenretinide that includes inhibition of cell proliferation
and the induction of apoptosis in cancer cells [177–179]. Fenretinide is well tolerated
with limited side effects in daily treatment regimens for 5 years or more [180,181].
Fenretinide also improves symptoms of diet-induced obesity, insulin resistance and
NAFLD. In line, fenretinide improved insulin sensitivity and decreased serum leptin
levels in a clinical trial in overweight women [182]. Fenretinide reduces circulating
RBP4 levels, but the key importance of this effect in the therapeutic action of fenretinide
is controversial, as it also prevents and/or reverses obesity, insulin resistance, and
hepatic steatosis in Rbp4 knockout mice on HFD [183]. Indeed, also additional RAR-
dependent and -independent mechanisms have been identified that may contribute to
the therapeutic effect of Fenretinide in obesity-related pathologies, including enhanced
mitochondrial and peroxisomal β-oxidation [184–188], ER-stress-mediated
degradation of SCD1 [189], inhibition of ceramide synthesis, enhanced reactive
oxygen species (ROS) production [184,190], enhanced retinoid signaling [191] and
inhibition of hepatic FGF21 expression [192]. The many clinical trials aimed at
evaluating fenretinide therapy in cancer have, however, shown that it dose-
dependently reduces plasma retinol levels up to 90% compared to baseline pre-
therapy leading to vitamin A deficiency and impaired dark-adaptation was a regular
observed adverse effect [193–212]. The drop in circulating retinol cannot be prevented
by vitamin A supplementation [196]. The retinol-lowering effects of fenretinide may be
particular relevant for patients with metabolic syndrome and diabetes as plasma retinol
levels are already low in those patents. A phase 2 clinical trial to evaluate the insulin-
sensitizing effect of fenretinide in subjects with insulin resistance and NAFLD has been
initiated in 2007, but no results have been reported yet [213].
In summary, both natural and synthetic retinoids show important potential for the
treatment of NAFLD and associated syndromes, like diabetes. However, these
compounds act also on many other biological processes. Thus, tissue-specific
targeting and/or characterization of derivatives that selectively modulate specific
pathways may be required to come to safe and effective treatment in NAFLD.
4.9. Conclusions
It is evident that hepatic glucose and lipid metabolism are regulated by vitamin A
metabolites at many different levels. Moreover, disease progression within the NAFLD
spectrum to NASH, cirrhosis and cancer is associated with declining circulating and

Chapter 4
125
hepatic retinol levels. This is not necessarily true for hepatic retinyl esters as individuals
homozygous for the PNPLA3-I148M risk allele are predisposed for NAFLD and
disease progression, while their hepatic retinyl palmitate levels are increased
compared to NAFLD-I148 (protective allele) carriers. It is unknown whether this shift
from retinol to retinyl esters is more common in NALFD patients. Thus, we still lack
important knowledge on hepatic vitamin A metabolism and the true meaning of VAD in
NAFLD. To specify a few open questions: 1) are retinyl ester stores depleted in NAFLD
or is retinol release from such stores impaired?; 2) how can PNPLA3-I148M
predispose for fibrosis while it leads to increased hepatic retinyl ester levels?; 3) what
is the contribution of hepatocytes and stellate cells to impaired retinol metabolism in
NAFLD?; 4) what is the absolute contribution of adipose-derived lipids and de novo
lipogenesis in NAFLD and how is this controlled by vitamin A metabolites?; 5) why do
circulating retinol levels stay low or even further decline after bariatric surgery, even
under impressive weight loss and/or vitamin A supplementation therapy? 6) is the
therapeutic efficacy of nuclear receptor ligands that are currently under investigation
for NAFLD limited by impaired vitamin A metabolism in the liver?; 7) does VAD actually
contribute to the development of fatty liver?
With respect to the latter: Severe VAD in lean rats decreases serum triacylglycerol,
cholesterol and HDL-cholesterol levels as well as hepatic phospholipids compared to
VA-sufficient animals [214]. Expression of acetyl-CoA carboxylase was decreased,
suggesting impaired fatty acid synthesis, while mitochondrial fatty acid β-oxidation was
enhanced. However, (free) cholesterol levels were enhanced in the hearts of VAD rats
[215], as well as concentrations of triglycerides, total cholesterol, free and esterified
cholesterol, and phospholipids in the aorta [216]. In contrast, VAD in mice appears to
have the opposite effect. Hepatic TG levels are enhanced in VAD mice compared to
control animals, which is associated with strongly reduced expression of PPARα and
genes involved in mitochondrial and peroxisomal β-oxidation [217]. Thus, it also
remains unclear whether VAD contributes to hepatic steatosis, and human data are so
far lacking on this topic.
Thus, there is still a lot to learn about vitamin A metabolism in the liver in healthy and
pathological conditions, which hopefully will reveal novel therapeutic targets for the
treatment of NAFLD and in particular to prevent pathological conditions caused by
NASH, cirrhosis and hepatocellular carcinoma.

Disturbed vitamin A metabolism in NAFLD
126
REFERENCES
[1] R. Blomhoff, H.K. Blomhoff, Overview of retinoid metabolism and function, J. Neurobiol. 66 (2006) 606–630. doi:10.1002/neu.20242.
[2] C.S. Lo, M.L. Wahlqvist, Y. Horie, Determination of retinoic acid and retinol at physiological concentration by HPLC in Caucasians and Japanese women, Asia Pac. J. Clin. Nutr. 5 (1996) 173–174.
[3] D. Weber, T. Grune, The contribution of β-carotene to vitamin A supply of humans, Mol. Nutr. Food Res. 56 (2012) 251–258. doi:10.1002/mnfr.201100230.
[4] S. Bar-El Dadon, R. Reifen, Vitamin A and the epigenome, Crit. Rev. Food Sci. Nutr. 57 (2017) 2404–2411. doi:10.1080/10408398.2015.1060940.
[5] M. Zhong, R. Kawaguchi, M. Kassai, H. Sun, Retina, retinol, retinal and the natural history of vitamin A as a light sensor, Nutrients. 4 (2012) 2069–2096. doi:10.3390/nu4122069.
[6] A. Comptour, M. Rouzaire, C. Belville, D. Bouvier, D. Gallot, L. Blanchon, V. Sapin, Nuclear retinoid receptors and pregnancy: placental transfer, functions, and pharmacological aspects, Cell. Mol. Life Sci. CMLS. 73 (2016) 3823–3837. doi:10.1007/s00018-016-2332-9.
[7] S.A. Tanumihardjo, R.M. Russell, C.B. Stephensen, B.M. Gannon, N.E. Craft, M.J. Haskell, G. Lietz, K. Schulze, D.J. Raiten, Biomarkers of Nutrition for Development (BOND)-Vitamin A Review, J. Nutr. 146 (2016) 1816S–48S. doi:10.3945/jn.115.229708.
[8] T.J. Cunningham, G. Duester, Mechanisms of retinoic acid signalling and its roles in organ and limb development, Nat. Rev. Mol. Cell Biol. 16 (2015) 110–123. doi:10.1038/nrm3932.
[9] M.R. Bono, G. Tejon, F. Flores-Santibañez, D. Fernandez, M. Rosemblatt, D. Sauma, Retinoic Acid as a Modulator of T Cell Immunity, Nutrients. 8 (2016). doi:10.3390/nu8060349.
[10] P. Huang, V. Chandra, F. Rastinejad, Retinoic acid actions through mammalian nuclear receptors, Chem. Rev. 114 (2014) 233–254. doi:10.1021/cr400161b.
[11] A. Saeed, M. Hoekstra, M.O. Hoeke, J. Heegsma, K.N. Faber, The interrelationship between bile acid and vitamin A homeostasis, Biochim. Biophys. Acta. 1862 (2017) 496–512. doi:10.1016/j.bbalip.2017.01.007.
[12] R. Zhang, Y. Wang, R. Li, G. Chen, Transcriptional Factors Mediating Retinoic Acid Signals in the Control of Energy Metabolism, Int. J. Mol. Sci. 16 (2015) 14210–14244. doi:10.3390/ijms160614210.
[13] V. Azaïs-Braesco, G. Pascal, Vitamin A in pregnancy: requirements and safety limits, Am. J. Clin. Nutr. 71 (2000) 1325S–33S.
[14] W.S. Andrews, C.M. Pau, H.P. Chase, L.C. Foley, J.R. Lilly, Fat soluble vitamin deficiency in biliary atresia, J. Pediatr. Surg. 16 (1981) 284–290.
[15] M.S. Mourey, G. Siegenthaler, O. Amédée-Manesme, Regulation of metabolism of retinol-binding protein by vitamin A status in children with biliary atresia, Am. J. Clin. Nutr. 51 (1990) 638–643.
[16] H.F. Herlong, R.M. Russell, W.C. Maddrey, Vitamin A and zinc therapy in primary biliary cirrhosis, Hepatol. Baltim. Md. 1 (1981) 348–351.
[17] R.P. Walt, C.M. Kemp, L. Lyness, A.C. Bird, S. Sherlock, Vitamin A treatment for night blindness in primary biliary cirrhosis, Br. Med. J. Clin. Res. Ed. 288 (1984) 1030–1031.
[18] J.R. Phillips, P. Angulo, T. Petterson, K.D. Lindor, Fat-soluble vitamin levels in patients with primary biliary cirrhosis, Am. J. Gastroenterol. 96 (2001) 2745–2750. doi:10.1111/j.1572-0241.2001.04134.x.

Chapter 4
127
[19] R.A. Jorgensen, K.D. Lindor, J.S. Sartin, N.F. LaRusso, R.H. Wiesner, Serum lipid and fat-soluble vitamin levels in primary sclerosing cholangitis, J. Clin. Gastroenterol. 20 (1995) 215–219.
[20] D. Bitetto, N. Bortolotti, E. Falleti, S. Vescovo, C. Fabris, G. Fattovich, A. Cussigh, S. Cmet, E. Fornasiere, E. Ceriani, M. Pirisi, P. Toniutto, Vitamin A deficiency is associated with hepatitis C virus chronic infection and with unresponsiveness to interferon-based antiviral therapy, Hepatol. Baltim. Md. 57 (2013) 925–933. doi:10.1002/hep.26186.
[21] S.K. Majumdar, G.K. Shaw, A.D. Thomson, Vitamin A utilization status in chronic alcoholic patients, Int. J. Vitam. Nutr. Res. Int. Z. Vitam.- Ernahrungsforschung J. Int. Vitaminol. Nutr. 53 (1983) 273–279.
[22] M.B. Ray, C.L. Mendenhall, S.W. French, P.S. Gartside, Serum vitamin A deficiency and increased intrahepatic expression of cytokeratin antigen in alcoholic liver disease, Hepatol. Baltim. Md. 8 (1988) 1019–1026.
[23] F.I. Suano de Souza, O.M. Silverio Amancio, R.O. Saccardo Sarni, T. Sacchi Pitta, A.P. Fernandes, F.L. Affonso Fonseca, S. Hix, R.A. Ramalho, Non-alcoholic fatty liver disease in overweight children and its relationship with retinol serum levels, Int. J. Vitam. Nutr. Res. Int. Z. Vitam.- Ernahrungsforschung J. Int. Vitaminol. Nutr. 78 (2008) 27–32. doi:10.1024/0300-9831.78.1.27.
[24] J.I. Botella-Carretero, J.A. Balsa, C. Vázquez, R. Peromingo, M. Díaz-Enriquez, H.F. Escobar-Morreale, Retinol and alpha-tocopherol in morbid obesity and nonalcoholic fatty liver disease, Obes. Surg. 20 (2010) 69–76. doi:10.1007/s11695-008-9686-5.
[25] G.V. Chaves, S.E. Pereira, C.J. Saboya, D. Spitz, C.S. Rodrigues, A. Ramalho, Association between liver vitamin A reserves and severity of nonalcoholic fatty liver disease in the class III obese following bariatric surgery, Obes. Surg. 24 (2014) 219–224. doi:10.1007/s11695-013-1087-8.
[26] E.H. van den Berg, M. Amini, T.C.M.A. Schreuder, R.P.F. Dullaart, K.N. Faber, B.Z. Alizadeh, H. Blokzijl, Prevalence and determinants of non-alcoholic fatty liver disease in lifelines: A large Dutch population cohort, PloS One. 12 (2017) e0171502. doi:10.1371/journal.pone.0171502.
[27] W.N. Hannah, S.A. Harrison, Lifestyle and Dietary Interventions in the Management of Nonalcoholic Fatty Liver Disease, Dig. Dis. Sci. 61 (2016) 1365–1374. doi:10.1007/s10620-016-4153-y.
[28] V. Nguyen, J. George, Nonalcoholic Fatty Liver Disease Management: Dietary and Lifestyle Modifications, Semin. Liver Dis. 35 (2015) 318–337. doi:10.1055/s-0035-1562950.
[29] World Health Organization, Indicators for assessing vitamin A deficiency and their application in monitoring and evaluating intervention programmes, Eileen Brown, James Akré, World Health Organization: Geneva, Switzerland, 1996. http://www.who.int/nutrition/publications/micronutrients/vitamin_a_deficiency/WHO_NUT_96.10/en/.
[30] L. de Souza Valente da Silva, G. Valeria da Veiga, R.A. Ramalho, Association of serum concentrations of retinol and carotenoids with overweight in children and adolescents, Nutr. Burbank Los Angel. Cty. Calif. 23 (2007) 392–397. doi:10.1016/j.nut.2007.02.009.
[31] M.L. Neuhouser, C.L. Rock, A.L. Eldridge, A.R. Kristal, R.E. Patterson, D.A. Cooper, D. Neumark-Sztainer, L.J. Cheskin, M.D. Thornquist, Serum concentrations of retinol, alpha-tocopherol and the carotenoids are influenced by diet, race and obesity in a sample of healthy adolescents, J. Nutr. 131 (2001) 2184–2191.

Disturbed vitamin A metabolism in NAFLD
128
[32] N.A. Malik, Solubilization and Interaction Studies of Bile Salts with Surfactants and Drugs: a Review, Appl. Biochem. Biotechnol. 179 (2016) 179–201. doi:10.1007/s12010-016-1987-x.
[33] B.K. Nordskog, C.T. Phan, D.F. Nutting, P. Tso, An examination of the factors affecting intestinal lymphatic transport of dietary lipids, Adv. Drug Deliv. Rev. 50 (2001) 21–44.
[34] A. During, E.H. Harrison, Mechanisms of provitamin A (carotenoid) and vitamin A (retinol) transport into and out of intestinal Caco-2 cells, J. Lipid Res. 48 (2007) 2283–2294. doi:10.1194/jlr.M700263-JLR200.
[35] M.M. Hussain, A proposed model for the assembly of chylomicrons, Atherosclerosis. 148 (2000) 1–15.
[36] S. Ishibashi, S. Perrey, Z. Chen, J. i Osuga, M. Shimada, K. Ohashi, K. Harada, Y. Yazaki, N. Yamada, Role of the low density lipoprotein (LDL) receptor pathway in the metabolism of chylomicron remnants. A quantitative study in knockout mice lacking the LDL receptor, apolipoprotein E, or both, J. Biol. Chem. 271 (1996) 22422–22427.
[37] T. Linke, H. Dawson, E.H. Harrison, Isolation and characterization of a microsomal acid retinyl ester hydrolase, J. Biol. Chem. 280 (2005) 23287–23294. doi:10.1074/jbc.M413585200.
[38] H. Ronne, C. Ocklind, K. Wiman, L. Rask, B. Obrink, P.A. Peterson, Ligand-dependent regulation of intracellular protein transport: effect of vitamin a on the secretion of the retinol-binding protein, J. Cell Biol. 96 (1983) 907–910.
[39] J.L. Dixon, D.S. Goodman, Studies on the metabolism of retinol-binding protein by primary hepatocytes from retinol-deficient rats, J. Cell. Physiol. 130 (1987) 14–20. doi:10.1002/jcp.1041300104.
[40] D. Bellovino, Y. Lanyau, I. Garaguso, L. Amicone, C. Cavallari, M. Tripodi, S. Gaetani, MMH cells: An in vitro model for the study of retinol-binding protein secretion regulated by retinol, J. Cell. Physiol. 181 (1999) 24–32. doi:10.1002/(SICI)1097-4652(199910)181:1<24::AID-JCP3>3.0.CO;2-0.
[41] H. Sun, R. Kawaguchi, The membrane receptor for plasma retinol-binding protein, a new type of cell-surface receptor, Int. Rev. Cell Mol. Biol. 288 (2011) 1–41. doi:10.1016/B978-0-12-386041-5.00001-7.
[42] H. Senoo, Structure and function of hepatic stellate cells, Med. Electron Microsc. Off. J. Clin. Electron Microsc. Soc. Jpn. 37 (2004) 3–15. doi:10.1007/s00795-003-0230-3.
[43] N. Wongsiriroj, H. Jiang, R. Piantedosi, K.J.Z. Yang, J. Kluwe, R.F. Schwabe, H. Ginsberg, I.J. Goldberg, W.S. Blaner, Genetic dissection of retinoid esterification and accumulation in the liver and adipose tissue, J. Lipid Res. 55 (2014) 104–114. doi:10.1194/jlr.M043844.
[44] U. Taschler, R. Schreiber, C. Chitraju, G.F. Grabner, M. Romauch, H. Wolinski, G. Haemmerle, R. Breinbauer, R. Zechner, A. Lass, R. Zimmermann, Adipose triglyceride lipase is involved in the mobilization of triglyceride and retinoid stores of hepatic stellate cells, Biochim. Biophys. Acta. 1851 (2015) 937–945. doi:10.1016/j.bbalip.2015.02.017.
[45] C. Pirazzi, L. Valenti, B.M. Motta, P. Pingitore, K. Hedfalk, R.M. Mancina, M.A. Burza, C. Indiveri, Y. Ferro, T. Montalcini, C. Maglio, P. Dongiovanni, S. Fargion, R. Rametta, A. Pujia, L. Andersson, S. Ghosal, M. Levin, O. Wiklund, M. Iacovino, J. Borén, S. Romeo, PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells, Hum. Mol. Genet. 23 (2014) 4077–4085. doi:10.1093/hmg/ddu121.

Chapter 4
129
[46] A. Mondul, R.M. Mancina, A. Merlo, P. Dongiovanni, R. Rametta, T. Montalcini, L. Valenti, D. Albanes, S. Romeo, PNPLA3 I148M Variant Influences Circulating Retinol in Adults with Nonalcoholic Fatty Liver Disease or Obesity, J. Nutr. 145 (2015) 1687–1691. doi:10.3945/jn.115.210633.
[47] T. Mello, A. Nakatsuka, S. Fears, W. Davis, H. Tsukamoto, W.F. Bosron, S.P. Sanghani, Expression of carboxylesterase and lipase genes in rat liver cell-types, Biochem. Biophys. Res. Commun. 374 (2008) 460–464. doi:10.1016/j.bbrc.2008.07.024.
[48] W. Pang, Y. Zhang, S. Wang, A. Jia, W. Dong, C. Cai, Z. Hua, J. Zhang, The mPlrp2 and mClps genes are involved in the hydrolysis of retinyl esters in the mouse liver, J. Lipid Res. 52 (2011) 934–941. doi:10.1194/jlr.M010082.
[49] H. Kim, K.-W. Lee, K. Lee, S. Seo, M.-Y. Park, S.W. Ahn, S.K. Hong, K.C. Yoon, H.-S. Kim, Y. Choi, H.W. Lee, N.-J. Yi, K.-S. Suh, Effect of PNPLA3 I148M polymorphism on histologically proven non-alcoholic fatty liver disease in liver transplant recipients, Hepatol. Res. Off. J. Jpn. Soc. Hepatol. (2017). doi:10.1111/hepr.12940.
[50] M. Krawczyk, R. Jiménez-Agüero, J.M. Alustiza, J.I. Emparanza, M.J. Perugorria, L. Bujanda, F. Lammert, J.M. Banales, PNPLA3 p.I148M variant is associated with greater reduction of liver fat content after bariatric surgery, Surg. Obes. Relat. Dis. Off. J. Am. Soc. Bariatr. Surg. 12 (2016) 1838–1846. doi:10.1016/j.soard.2016.06.004.
[51] M.-F. Xia, Y. Ling, H. Bian, H.-D. Lin, H.-M. Yan, X.-X. Chang, X.-M. Li, H. Ma, D. Wang, L.-S. Zhang, S.-S. Wang, B.-J. Wu, W.-Y. He, N.-Q. Zhao, X. Gao, I148M variant of PNPLA3 increases the susceptibility to non-alcoholic fatty liver disease caused by obesity and metabolic disorders, Aliment. Pharmacol. Ther. 43 (2016) 631–642. doi:10.1111/apt.13521.
[52] H. Bell, A. Nilsson, K.R. Norum, L.B. Pedersen, N. Raknerud, M. Rasmussen, Retinol and retinyl esters in patients with alcoholic liver disease, J. Hepatol. 8 (1989) 26–31.
[53] G. Villaça Chaves, S.E. Pereira, C.J. Saboya, A. Ramalho, Non-alcoholic fatty liver disease and its relationship with the nutritional status of vitamin A in individuals with class III obesity, Obes. Surg. 18 (2008) 378–385. doi:10.1007/s11695-007-9361-2.
[54] M.A. Beydoun, M.R. Shroff, X. Chen, H.A. Beydoun, Y. Wang, A.B. Zonderman, Serum antioxidant status is associated with metabolic syndrome among U.S. adults in recent national surveys, J. Nutr. 141 (2011) 903–913. doi:10.3945/jn.110.136580.
[55] S.E. Pereira, C.J. Saboya, C. Saunders, A. Ramalho, Serum levels and liver store of retinol and their association with night blindness in individuals with class III obesity, Obes. Surg. 22 (2012) 602–608. doi:10.1007/s11695-011-0522-y.
[56] M.A. Beydoun, J.A. Canas, H.A. Beydoun, X. Chen, M.R. Shroff, A.B. Zonderman, Serum antioxidant concentrations and metabolic syndrome are associated among U.S. adolescents in recent national surveys, J. Nutr. 142 (2012) 1693–1704. doi:10.3945/jn.112.160416.
[57] P. Lefebvre, F. Letois, A. Sultan, D. Nocca, T. Mura, F. Galtier, Nutrient deficiencies in patients with obesity considering bariatric surgery: a cross-sectional study, Surg. Obes. Relat. Dis. Off. J. Am. Soc. Bariatr. Surg. 10 (2014) 540–546. doi:10.1016/j.soard.2013.10.003.
[58] M. Teske, A.P.B. Melges, F.I.S. de Souza, F.L.A. Fonseca, R.O.S. Sarni, Plasma concentrations of retinol in obese children and adolescents: relationship to

Disturbed vitamin A metabolism in NAFLD
130
metabolic syndrome components, Rev. Paul. Pediatr. Orgao Of. Soc. Pediatr. Sao Paulo. 32 (2014) 50–54.
[59] E. Wolf, M. Utech, P. Stehle, M. Büsing, B. Stoffel-Wagner, S. Ellinger, Preoperative micronutrient status in morbidly obese patients before undergoing bariatric surgery: results of a cross-sectional study, Surg. Obes. Relat. Dis. Off. J. Am. Soc. Bariatr. Surg. 11 (2015) 1157–1163. doi:10.1016/j.soard.2015.03.018.
[60] Y. Liu, H. Chen, D. Mu, J. Fan, J. Song, Y. Zhong, D. Li, M. Xia, Circulating Retinoic Acid Levels and the Development of Metabolic Syndrome, J. Clin. Endocrinol. Metab. 101 (2016) 1686–1692. doi:10.1210/jc.2015-4038.
[61] X. Wei, R. Peng, J. Cao, Y. Kang, P. Qu, Y. Liu, X. Xiao, T. Li, Serum vitamin A status is associated with obesity and the metabolic syndrome among school-age children in Chongqing, China, Asia Pac. J. Clin. Nutr. 25 (2016) 563–570.
[62] M.M. Godala, I. Materek-Kuśmierkiewicz, D. Moczulski, M. Rutkowski, F. Szatko, E. Gaszyńska, S. Tokarski, J. Kowalski, The risk of plasma vitamin A, C, E and D deficiency in patients with metabolic syndrome: a case-control study, Adv. Clin. Exp. Med. Off. Organ Wroclaw Med. Univ. (2017). doi:10.17219/acem/62453.
[63] S.E. Trasino, X.-H. Tang, J. Jessurun, L.J. Gudas, Obesity Leads to Tissue, but not Serum Vitamin A Deficiency, Sci. Rep. 5 (2015) 15893. doi:10.1038/srep15893.
[64] Y. Liu, H. Chen, J. Wang, W. Zhou, R. Sun, M. Xia, Association of serum retinoic acid with hepatic steatosis and liver injury in nonalcoholic fatty liver disease, Am. J. Clin. Nutr. 102 (2015) 130–137. doi:10.3945/ajcn.114.105155.
[65] A.A. Ashla, Y. Hoshikawa, H. Tsuchiya, K. Hashiguchi, M. Enjoji, M. Nakamuta, A. Taketomi, Y. Maehara, K. Shomori, A. Kurimasa, I. Hisatome, H. Ito, G. Shiota, Genetic analysis of expression profile involved in retinoid metabolism in non-alcoholic fatty liver disease, Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 40 (2010) 594–604. doi:10.1111/j.1872-034X.2010.00646.x.
[66] C. Clemente, S. Elba, G. Buongiorno, P. Berloco, V. Guerra, A. Di Leo, Serum retinol and risk of hepatocellular carcinoma in patients with child-Pugh class A cirrhosis, Cancer Lett. 178 (2002) 123–129.
[67] P.N. Newsome, I. Beldon, Y. Moussa, T.E. Delahooke, G. Poulopoulos, P.C. Hayes, J.N. Plevris, Low serum retinol levels are associated with hepatocellular carcinoma in patients with chronic liver disease, Aliment. Pharmacol. Ther. 14 (2000) 1295–1301.
[68] A. Yanagitani, S. Yamada, S. Yasui, T. Shimomura, R. Murai, Y. Murawaki, K. Hashiguchi, T. Kanbe, T. Saeki, M. Ichiba, Y. Tanabe, Y. Yoshida, S.-I. Morino, A. Kurimasa, N. Usuda, H. Yamazaki, T. Kunisada, H. Ito, Y. Murawaki, G. Shiota, Retinoic acid receptor alpha dominant negative form causes steatohepatitis and liver tumors in transgenic mice, Hepatol. Baltim. Md. 40 (2004) 366–375. doi:10.1002/hep.20335.
[69] Q. Yang, T.E. Graham, N. Mody, F. Preitner, O.D. Peroni, J.M. Zabolotny, K. Kotani, L. Quadro, B.B. Kahn, Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes, Nature. 436 (2005) 356–362. doi:10.1038/nature03711.
[70] D.G. Haider, K. Schindler, G. Prager, A. Bohdjalian, A. Luger, M. Wolzt, B. Ludvik, Serum retinol-binding protein 4 is reduced after weight loss in morbidly obese subjects, J. Clin. Endocrinol. Metab. 92 (2007) 1168–1171. doi:10.1210/jc.2006-1839.

Chapter 4
131
[71] T. Reinehr, B. Stoffel-Wagner, C.L. Roth, Retinol-binding protein 4 and its relation to insulin resistance in obese children before and after weight loss, J. Clin. Endocrinol. Metab. 93 (2008) 2287–2293. doi:10.1210/jc.2007-2745.
[72] M. Tajtáková, Z. Semanová, G. Ivancová, J. Petrovicová, V. Donicová, E. Zemberová, [Serum level of retinol-binding protein 4 in obese patients with insulin resistance and in patients with type 2 diabetes treated with metformin], Vnitr. Lek. 53 (2007) 960–963.
[73] F. Ulgen, C. Herder, M.C. Kühn, H.S. Willenberg, M. Schott, W.A. Scherbaum, S. Schinner, Association of serum levels of retinol-binding protein 4 with male sex but not with insulin resistance in obese patients, Arch. Physiol. Biochem. 116 (2010) 57–62. doi:10.3109/13813451003631421.
[74] M. Broch, J. Vendrell, W. Ricart, C. Richart, J.-M. Fernández-Real, Circulating retinol-binding protein-4, insulin sensitivity, insulin secretion, and insulin disposition index in obese and nonobese subjects, Diabetes Care. 30 (2007) 1802–1806. doi:10.2337/dc06-2034.
[75] J. Gómez-Ambrosi, A. Rodríguez, V. Catalán, B. Ramírez, C. Silva, F. Rotellar, M.J. Gil, J. Salvador, G. Frühbeck, Serum retinol-binding protein 4 is not increased in obesity or obesity-associated type 2 diabetes mellitus, but is reduced after relevant reductions in body fat following gastric bypass, Clin. Endocrinol. (Oxf.). 69 (2008) 208–215. doi:10.1111/j.1365-2265.2007.03156.x.
[76] C. Kanaka-Gantenbein, A. Margeli, P. Pervanidou, S. Sakka, G. Mastorakos, G.P. Chrousos, I. Papassotiriou, Retinol-binding protein 4 and lipocalin-2 in childhood and adolescent obesity: when children are not just “small adults,” Clin. Chem. 54 (2008) 1176–1182. doi:10.1373/clinchem.2007.099002.
[77] N. Alkhouri, R. Lopez, M. Berk, A.E. Feldstein, Serum retinol-binding protein 4 levels in patients with nonalcoholic fatty liver disease, J. Clin. Gastroenterol. 43 (2009) 985–989. doi:10.1097/MCG.0b013e3181a0998d.
[78] K.B. Comerford, W. Buchan, S.E. Karakas, The effects of weight loss on FABP4 and RBP4 in obese women with metabolic syndrome, Horm. Metab. Res. Horm. Stoffwechselforschung Horm. Metab. 46 (2014) 224–231. doi:10.1055/s-0033-1353204.
[79] I. Aeberli, R. Biebinger, R. Lehmann, D. L’allemand, G.A. Spinas, M.B. Zimmermann, Serum retinol-binding protein 4 concentration and its ratio to serum retinol are associated with obesity and metabolic syndrome components in children, J. Clin. Endocrinol. Metab. 92 (2007) 4359–4365. doi:10.1210/jc.2007-0468.
[80] J.P. Mills, H.C. Furr, S.A. Tanumihardjo, Retinol to retinol-binding protein (RBP) is low in obese adults due to elevated apo-RBP, Exp. Biol. Med. Maywood NJ. 233 (2008) 1255–1261. doi:10.3181/0803-RM-94.
[81] C. Erikstrup, O.H. Mortensen, A.R. Nielsen, C.P. Fischer, P. Plomgaard, A.M. Petersen, R. Krogh-Madsen, B. Lindegaard, J.G. Erhardt, H. Ullum, C.S. Benn, B.K. Pedersen, RBP-to-retinol ratio, but not total RBP, is elevated in patients with type 2 diabetes, Diabetes Obes. Metab. 11 (2009) 204–212. doi:10.1111/j.1463-1326.2008.00901.x.
[82] A. Henze, S.K. Frey, J. Raila, M. Tepel, A. Scholze, A.F.H. Pfeiffer, M.O. Weickert, J. Spranger, F.J. Schweigert, Evidence that kidney function but not type 2 diabetes determines retinol-binding protein 4 serum levels, Diabetes. 57 (2008) 3323–3326. doi:10.2337/db08-0866.
[83] A. Zwolak, A. Szuster-Ciesielska, J. Daniluk, J. Semeniuk, M. Kandefer-Szerszen, Chemerin, retinol binding protein-4, cytokeratin-18 and transgelin-2

Disturbed vitamin A metabolism in NAFLD
132
presence in sera of patients with non-alcoholic liver fatty disease, Ann. Hepatol. 15 (2016) 862–869. doi:10.5604/16652681.1222102.
[84] M. Rahimlou, K. Mirzaei, S.A. Keshavarz, A. Hossein-Nezhad, Association of circulating adipokines with metabolic dyslipidemia in obese versus non-obese individuals, Diabetes Metab. Syndr. 10 (2016) S60-65. doi:10.1016/j.dsx.2015.09.015.
[85] C.-H. Chu, H.-C. Lam, J.-K. Lee, C.-C. Lu, C.-C. Sun, H.-J. Cheng, M.-C. Wang, M.-J. Chuang, Elevated serum retinol-binding protein 4 concentrations are associated with chronic kidney disease but not with the higher carotid intima-media thickness in type 2 diabetic subjects, Endocr. J. 58 (2011) 841–847.
[86] H. Wu, W. Jia, Y. Bao, J. Lu, J. Zhu, R. Wang, Y. Chen, K. Xiang, Serum retinol binding protein 4 and nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus, Diabetes Res. Clin. Pract. 79 (2008) 185–190. doi:10.1016/j.diabres.2007.08.016.
[87] S.J. Thompson, A. Sargsyan, S.-A. Lee, J.J. Yuen, J. Cai, R. Smalling, N. Ghyselinck, M. Mark, W.S. Blaner, T.E. Graham, Hepatocytes Are the Principal Source of Circulating RBP4 in Mice, Diabetes. 66 (2017) 58–63. doi:10.2337/db16-0286.
[88] S.-A. Lee, J.J. Yuen, H. Jiang, B.B. Kahn, W.S. Blaner, Adipocyte-specific overexpression of retinol-binding protein 4 causes hepatic steatosis in mice, Hepatol. Baltim. Md. 64 (2016) 1534–1546. doi:10.1002/hep.28659.
[89] M. Schina, J. Koskinas, D. Tiniakos, E. Hadziyannis, S. Savvas, B. Karamanos, E. Manesis, A. Archimandritis, Circulating and liver tissue levels of retinol-binding protein-4 in non-alcoholic fatty liver disease, Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 39 (2009) 972–978. doi:10.1111/j.1872-034X.2009.00534.x.
[90] D.A. de Luis, D. Pacheco, O. Izaola, M.C. Terroba, L. Cuellar, T. Martin, Clinical results and nutritional consequences of biliopancreatic diversion: three years of follow-up, Ann. Nutr. Metab. 53 (2008) 234–239. doi:10.1159/000185641.
[91] E.T. Aasheim, S. Björkman, T.T. Søvik, M. Engström, S.E. Hanvold, T. Mala, T. Olbers, T. Bøhmer, Vitamin status after bariatric surgery: a randomized study of gastric bypass and duodenal switch, Am. J. Clin. Nutr. 90 (2009) 15–22. doi:10.3945/ajcn.2009.27583.
[92] S. Pereira, C. Saboya, G. Chaves, A. Ramalho, Class III obesity and its relationship with the nutritional status of vitamin A in pre- and postoperative gastric bypass, Obes. Surg. 19 (2009) 738–744. doi:10.1007/s11695-008-9478-y.
[93] M. Cuesta, L. Pelaz, C. Pérez, M.J. Torrejón, L. Cabrerizo, P. Matía, N. Pérez-Ferre, A. Sánchez-Pernaute, A. Torres, M.A. Rubio, Fat-soluble vitamin deficiencies after bariatric surgery could be misleading if they are not appropriately adjusted, Nutr. Hosp. 30 (2014) 118–123. doi:10.3305/nh.2014.30.1.7471.
[94] J.S. Silva, G.V. Chaves, A.P. Stenzel, S.E. Pereira, C.J. Saboya, A. Ramalho, Improvement of anthropometric and biochemical, but not of vitamin A, status in adolescents who undergo Roux-en-Y gastric bypass: a 1-year follow up study, Surg. Obes. Relat. Dis. Off. J. Am. Soc. Bariatr. Surg. 13 (2017) 227–233. doi:10.1016/j.soard.2016.09.002.
[95] T.T. Søvik, E.T. Aasheim, O. Taha, M. Engström, M.W. Fagerland, S. Björkman, J. Kristinsson, K.I. Birkeland, T. Mala, T. Olbers, Weight loss, cardiovascular risk factors, and quality of life after gastric bypass and duodenal switch: a randomized trial, Ann. Intern. Med. 155 (2011) 281–291. doi:10.7326/0003-4819-155-5-201109060-00005.

Chapter 4
133
[96] G.H. Slater, C.J. Ren, N. Siegel, T. Williams, D. Barr, B. Wolfe, K. Dolan, G.A. Fielding, Serum fat-soluble vitamin deficiency and abnormal calcium metabolism after malabsorptive bariatric surgery, J. Gastrointest. Surg. Off. J. Soc. Surg. Aliment. Tract. 8 (2004) 48-55-55.
[97] P. Nett, Y. Borbély, D. Kröll, Micronutrient Supplementation after Biliopancreatic Diversion with Duodenal Switch in the Long Term, Obes. Surg. 26 (2016) 2469–2474. doi:10.1007/s11695-016-2132-1.
[98] R. Bolckmans, J. Himpens, Long-term (>10 Yrs) Outcome of the Laparoscopic Biliopancreatic Diversion With Duodenal Switch, Ann. Surg. 264 (2016) 1029–1037. doi:10.1097/SLA.0000000000001622.
[99] H. Gilchrist, D.A. Taranath, G.A. Gole, Ocular malformation in a newborn secondary to maternal hypovitaminosis A, J. AAPOS Off. Publ. Am. Assoc. Pediatr. Ophthalmol. Strabismus. 14 (2010) 274–276. doi:10.1016/j.jaapos.2010.01.015.
[100] G. Gascoin, M. Gerard, A. Sallé, G. Becouarn, S. Rouleau, L. Sentilhes, R. Coutant, Risk of low birth weight and micronutrient deficiencies in neonates from mothers after gastric bypass: a case control study, Surg. Obes. Relat. Dis. Off. J. Am. Soc. Bariatr. Surg. 13 (2017) 1384–1391. doi:10.1016/j.soard.2017.03.017.
[101] S. Pereira, C. Saboya, A. Ramalho, Impact of different protocols of nutritional supplements on the status of vitamin A in class III obese patients after Roux-en-Y gastric bypass, Obes. Surg. 23 (2013) 1244–1251. doi:10.1007/s11695-013-0885-3.
[102] P. Topart, G. Becouarn, A. Sallé, P. Ritz, Biliopancreatic diversion requires multiple vitamin and micronutrient adjustments within 2 years of surgery, Surg. Obes. Relat. Dis. Off. J. Am. Soc. Bariatr. Surg. 10 (2014) 936–941. doi:10.1016/j.soard.2014.02.007.
[103] J.S. Fok, J.Y.Z. Li, T.Y. Yong, Visual deterioration caused by vitamin A deficiency in patients after bariatric surgery, Eat. Weight Disord. EWD. 17 (2012) e144-146.
[104] M. Vitkova, E. Klimcakova, M. Kovacikova, C. Valle, C. Moro, J. Polak, J. Hanacek, F. Capel, N. Viguerie, B. Richterova, M. Bajzova, J. Hejnova, V. Stich, D. Langin, Plasma levels and adipose tissue messenger ribonucleic acid expression of retinol-binding protein 4 are reduced during calorie restriction in obese subjects but are not related to diet-induced changes in insulin sensitivity, J. Clin. Endocrinol. Metab. 92 (2007) 2330–2335. doi:10.1210/jc.2006-2668.
[105] M.C. Mitterberger, M. Mattesich, E. Klaver, S. Lechner, T. Engelhardt, L. Larcher, G. Pierer, H. Piza-Katzer, W. Zwerschke, Adipokine profile and insulin sensitivity in formerly obese women subjected to bariatric surgery or diet-induced long-term caloric restriction, J. Gerontol. A. Biol. Sci. Med. Sci. 65 (2010) 915–923. doi:10.1093/gerona/glq107.
[106] M.C. Klempel, K.A. Varady, Reliability of leptin, but not adiponectin, as a biomarker for diet-induced weight loss in humans, Nutr. Rev. 69 (2011) 145–154. doi:10.1111/j.1753-4887.2011.00373.x.
[107] P. Wang, R.-Y. Zhang, J. Song, Y.-F. Guan, T.-Y. Xu, H. Du, B. Viollet, C.-Y. Miao, Loss of AMP-activated protein kinase-α2 impairs the insulin-sensitizing effect of calorie restriction in skeletal muscle, Diabetes. 61 (2012) 1051–1061. doi:10.2337/db11-1180.
[108] S. Romeo, J. Kozlitina, C. Xing, A. Pertsemlidis, D. Cox, L.A. Pennacchio, E. Boerwinkle, J.C. Cohen, H.H. Hobbs, Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease, Nat. Genet. 40 (2008) 1461–1465. doi:10.1038/ng.257.

Disturbed vitamin A metabolism in NAFLD
134
[109] L. Valenti, A. Al-Serri, A.K. Daly, E. Galmozzi, R. Rametta, P. Dongiovanni, V. Nobili, E. Mozzi, G. Roviaro, E. Vanni, E. Bugianesi, M. Maggioni, A.L. Fracanzani, S. Fargion, C.P. Day, Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease, Hepatol. Baltim. Md. 51 (2010) 1209–1217. doi:10.1002/hep.23622.
[110] S. Sookoian, C.J. Pirola, Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease, Hepatol. Baltim. Md. 53 (2011) 1883–1894. doi:10.1002/hep.24283.
[111] Y.-L. Liu, G.L. Patman, J.B.S. Leathart, A.-C. Piguet, A.D. Burt, J.-F. Dufour, C.P. Day, A.K. Daly, H.L. Reeves, Q.M. Anstee, Carriage of the PNPLA3 rs738409 C >G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma, J. Hepatol. 61 (2014) 75–81. doi:10.1016/j.jhep.2014.02.030.
[112] T.J. Severson, S. Besur, H.L. Bonkovsky, Genetic factors that affect nonalcoholic fatty liver disease: A systematic clinical review, World J. Gastroenterol. 22 (2016) 6742–6756. doi:10.3748/wjg.v22.i29.6742.
[113] J.Z. Li, Y. Huang, R. Karaman, P.T. Ivanova, H.A. Brown, T. Roddy, J. Castro-Perez, J.C. Cohen, H.H. Hobbs, Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis, J. Clin. Invest. 122 (2012) 4130–4144. doi:10.1172/JCI65179.
[114] S. Lallukka, H. Yki-Järvinen, Non-alcoholic fatty liver disease and risk of type 2 diabetes, Best Pract. Res. Clin. Endocrinol. Metab. 30 (2016) 385–395. doi:10.1016/j.beem.2016.06.006.
[115] E.M. Petäjä, H. Yki-Järvinen, Definitions of Normal Liver Fat and the Association of Insulin Sensitivity with Acquired and Genetic NAFLD-A Systematic Review, Int. J. Mol. Sci. 17 (2016). doi:10.3390/ijms17050633.
[116] M. Kovarova, I. Königsrainer, A. Königsrainer, F. Machicao, H.-U. Häring, E. Schleicher, A. Peter, The Genetic Variant I148M in PNPLA3 Is Associated With Increased Hepatic Retinyl-Palmitate Storage in Humans, J. Clin. Endocrinol. Metab. 100 (2015) E1568-1574. doi:10.1210/jc.2015-2978.
[117] F.V. Bruschi, T. Claudel, M. Tardelli, A. Caligiuri, T.M. Stulnig, F. Marra, M. Trauner, The PNPLA3 I148M variant modulates the fibrogenic phenotype of human hepatic stellate cells, Hepatol. Baltim. Md. 65 (2017) 1875–1890. doi:10.1002/hep.29041.
[118] R. Papazyan, Z. Sun, Y.H. Kim, P.M. Titchenell, D.A. Hill, W. Lu, M. Damle, M. Wan, Y. Zhang, E.R. Briggs, J.D. Rabinowitz, M.A. Lazar, Physiological Suppression of Lipotoxic Liver Damage by Complementary Actions of HDAC3 and SCAP/SREBP, Cell Metab. 24 (2016) 863–874. doi:10.1016/j.cmet.2016.10.012.
[119] S. Softic, D.E. Cohen, C.R. Kahn, Role of Dietary Fructose and Hepatic de novo Lipogenesis in Fatty Liver Disease, Dig. Dis. Sci. 61 (2016) 1282–1293. doi:10.1007/s10620-016-4054-0.
[120] K. Iizuka, Y. Horikawa, ChREBP: a glucose-activated transcription factor involved in the development of metabolic syndrome, Endocr. J. 55 (2008) 617–624.
[121] F. Lalloyer, T.A. Pedersen, B. Gross, S. Lestavel, S. Yous, E. Vallez, J.-A. Gustafsson, S. Mandrup, C. Fiévet, B. Staels, A. Tailleux, Rexinoid bexarotene modulates triglyceride but not cholesterol metabolism via gene-specific permissivity of the RXR/LXR heterodimer in the liver, Arterioscler. Thromb. Vasc. Biol. 29 (2009) 1488–1495. doi:10.1161/ATVBAHA.109.189506.

Chapter 4
135
[122] S.B. Joseph, B.A. Laffitte, P.H. Patel, M.A. Watson, K.E. Matsukuma, R. Walczak, J.L. Collins, T.F. Osborne, P. Tontonoz, Direct and indirect mechanisms for regulation of fatty acid synthase gene expression by liver X receptors, J. Biol. Chem. 277 (2002) 11019–11025. doi:10.1074/jbc.M111041200.
[123] S.C. Kim, C.-K. Kim, D. Axe, A. Cook, M. Lee, T. Li, N. Smallwood, J.Y.L. Chiang, J.P. Hardwick, D.D. Moore, Y.K. Lee, All-trans-retinoic acid ameliorates hepatic steatosis in mice by a novel transcriptional cascade, Hepatol. Baltim. Md. 59 (2014) 1750–1760. doi:10.1002/hep.26699.
[124] M. Watanabe, S.M. Houten, L. Wang, A. Moschetta, D.J. Mangelsdorf, R.A. Heyman, D.D. Moore, J. Auwerx, Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c, J. Clin. Invest. 113 (2004) 1408–1418. doi:10.1172/JCI21025.
[125] D.M. Wuttge, A. Romert, U. Eriksson, H. Törmä, G.K. Hansson, A. Sirsjö, Induction of CD36 by all-trans retinoic acid: retinoic acid receptor signaling in the pathogenesis of atherosclerosis, FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 15 (2001) 1221–1223.
[126] S. Han, N. Sidell, Peroxisome-proliferator-activated-receptor gamma (PPARgamma) independent induction of CD36 in THP-1 monocytes by retinoic acid, Immunology. 106 (2002) 53–59.
[127] K. Ishimoto, K. Tachibana, M. Sumitomo, S. Omote, I. Hanano, D. Yamasaki, Y. Watanabe, T. Tanaka, T. Hamakubo, J. Sakai, T. Kodama, T. Doi, Identification of human low-density lipoprotein receptor as a novel target gene regulated by liver X receptor alpha, FEBS Lett. 580 (2006) 4929–4933. doi:10.1016/j.febslet.2006.08.010.
[128] Z. Yao, Human apolipoprotein C-III - a new intrahepatic protein factor promoting assembly and secretion of very low density lipoproteins, Cardiovasc. Hematol. Disord. Drug Targets. 12 (2012) 133–140.
[129] H.-Y. Lee, A.L. Birkenfeld, F.R. Jornayvaz, M.J. Jurczak, S. Kanda, V. Popov, D.W. Frederick, D. Zhang, B. Guigni, K.G. Bharadwaj, C.S. Choi, I.J. Goldberg, J.-H. Park, K.F. Petersen, V.T. Samuel, G.I. Shulman, Apolipoprotein CIII overexpressing mice are predisposed to diet-induced hepatic steatosis and hepatic insulin resistance, Hepatol. Baltim. Md. 54 (2011) 1650–1660. doi:10.1002/hep.24571.
[130] R.-N. Zhang, R.-D. Zheng, Y.-Q. Mi, D. Zhou, F. Shen, G.-Y. Chen, C.-Y. Zhu, Q. Pan, J.-G. Fan, APOC3 rs2070666 Is Associated with the Hepatic Steatosis Independently of PNPLA3 rs738409 in Chinese Han Patients with Nonalcoholic Fatty Liver Diseases, Dig. Dis. Sci. 61 (2016) 2284–2293. doi:10.1007/s10620-016-4120-7.
[131] S.J. Lee, M. Mahankali, A. Bitar, H. Zou, E. Chao, H. Nguyen, J. Gonzalez, D. Caballero, M. Hull, D. Wang, P.G. Schultz, W. Shen, A Novel Role for RARα Agonists as Apolipoprotein CIII Inhibitors Identified from High Throughput Screening, Sci. Rep. 7 (2017) 5824. doi:10.1038/s41598-017-05163-w.
[132] N. Vu-Dac, P. Gervois, I.P. Torra, J.C. Fruchart, V. Kosykh, T. Kooistra, H.M. Princen, J. Dallongeville, B. Staels, Retinoids increase human apo C-III expression at the transcriptional level via the retinoid X receptor. Contribution to the hypertriglyceridemic action of retinoids, J. Clin. Invest. 102 (1998) 625–632. doi:10.1172/JCI1581.
[133] S. Caron, A. Verrijken, I. Mertens, C.H. Samanez, G. Mautino, J.T. Haas, D. Duran-Sandoval, J. Prawitt, S. Francque, E. Vallez, A. Muhr-Tailleux, I. Berard, F. Kuipers, J.A. Kuivenhoven, S.B. Biddinger, M.-R. Taskinen, L. Van Gaal, B. Staels, Transcriptional activation of apolipoprotein CIII expression by glucose

Disturbed vitamin A metabolism in NAFLD
136
may contribute to diabetic dyslipidemia, Arterioscler. Thromb. Vasc. Biol. 31 (2011) 513–519. doi:10.1161/ATVBAHA.110.220723.
[134] K.T. Ong, M.T. Mashek, S.Y. Bu, A.S. Greenberg, D.G. Mashek, Adipose triglyceride lipase is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid signaling and partitioning, Hepatol. Baltim. Md. 53 (2011) 116–126. doi:10.1002/hep.24006.
[135] P. Jha, T. Claudel, A. Baghdasaryan, M. Mueller, E. Halilbasic, S.K. Das, A. Lass, R. Zimmermann, R. Zechner, G. Hoefler, M. Trauner, Role of adipose triglyceride lipase (PNPLA2) in protection from hepatic inflammation in mouse models of steatohepatitis and endotoxemia, Hepatol. Baltim. Md. 59 (2014) 858–869. doi:10.1002/hep.26732.
[136] K. Harada, W.-J. Shen, S. Patel, V. Natu, J. Wang, J. Osuga, S. Ishibashi, F.B. Kraemer, Resistance to high-fat diet-induced obesity and altered expression of adipose-specific genes in HSL-deficient mice, Am. J. Physiol. Endocrinol. Metab. 285 (2003) E1182-1195. doi:10.1152/ajpendo.00259.2003.
[137] B.N. Reid, G.P. Ables, O.A. Otlivanchik, G. Schoiswohl, R. Zechner, W.S. Blaner, I.J. Goldberg, R.F. Schwabe, S.C. Chua, L.-S. Huang, Hepatic overexpression of hormone-sensitive lipase and adipose triglyceride lipase promotes fatty acid oxidation, stimulates direct release of free fatty acids, and ameliorates steatosis, J. Biol. Chem. 283 (2008) 13087–13099. doi:10.1074/jbc.M800533200.
[138] M. Rakhshandehroo, L.M. Sanderson, M. Matilainen, R. Stienstra, C. Carlberg, P.J. de Groot, M. Müller, S. Kersten, Comprehensive analysis of PPARalpha-dependent regulation of hepatic lipid metabolism by expression profiling, PPAR Res. 2007 (2007) 26839. doi:10.1155/2007/26839.
[139] M. Karahashi, M. Hoshina, T. Yamazaki, T. Sakamoto, A. Mitsumoto, Y. Kawashima, N. Kudo, Fibrates reduce triacylglycerol content by upregulating adipose triglyceride lipase in the liver of rats, J. Pharmacol. Sci. 123 (2013) 356–370.
[140] T. Deng, S. Shan, P.-P. Li, Z.-F. Shen, X.-P. Lu, J. Cheng, Z.-Q. Ning, Peroxisome proliferator-activated receptor-gamma transcriptionally up-regulates hormone-sensitive lipase via the involvement of specificity protein-1, Endocrinology. 147 (2006) 875–884. doi:10.1210/en.2005-0623.
[141] B.M. Stenson, M. Rydén, N. Venteclef, I. Dahlman, A.M.L. Pettersson, A. Mairal, G. Aström, L. Blomqvist, V. Wang, J.W.E. Jocken, K. Clément, D. Langin, P. Arner, J. Laurencikiene, Liver X receptor (LXR) regulates human adipocyte lipolysis, J. Biol. Chem. 286 (2011) 370–379. doi:10.1074/jbc.M110.179499.
[142] C. Dubuquoy, C. Robichon, F. Lasnier, C. Langlois, I. Dugail, F. Foufelle, J. Girard, A.-F. Burnol, C. Postic, M. Moldes, Distinct regulation of adiponutrin/PNPLA3 gene expression by the transcription factors ChREBP and SREBP1c in mouse and human hepatocytes, J. Hepatol. 55 (2011) 145–153. doi:10.1016/j.jhep.2010.10.024.
[143] J. Perttilä, C. Huaman-Samanez, S. Caron, K. Tanhuanpää, B. Staels, H. Yki-Järvinen, V.M. Olkkonen, PNPLA3 is regulated by glucose in human hepatocytes, and its I148M mutant slows down triglyceride hydrolysis, Am. J. Physiol. Endocrinol. Metab. 302 (2012) E1063-1069. doi:10.1152/ajpendo.00125.2011.
[144] S. Kersten, R. Stienstra, The role and regulation of the peroxisome proliferator activated receptor alpha in human liver, Biochimie. 136 (2017) 75–84. doi:10.1016/j.biochi.2016.12.019.
[145] M. Pawlak, P. Lefebvre, B. Staels, Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease, J. Hepatol. 62 (2015) 720–733. doi:10.1016/j.jhep.2014.10.039.

Chapter 4
137
[146] J. Amengual, J. Ribot, M.L. Bonet, A. Palou, Retinoic acid treatment enhances lipid oxidation and inhibits lipid biosynthesis capacities in the liver of mice, Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 25 (2010) 657–666. doi:10.1159/000315085.
[147] J. Amengual, P. Petrov, M.L. Bonet, J. Ribot, A. Palou, Induction of carnitine palmitoyl transferase 1 and fatty acid oxidation by retinoic acid in HepG2 cells, Int. J. Biochem. Cell Biol. 44 (2012) 2019–2027. doi:10.1016/j.biocel.2012.07.026.
[148] A. Patton, F.H. Khan, R. Kohli, Impact of Fibroblast Growth Factors 19 and 21 in Bariatric Metabolism, Dig. Dis. Basel Switz. 35 (2017) 191–196. doi:10.1159/000450910.
[149] Y. Li, K. Wong, K. Walsh, B. Gao, M. Zang, Retinoic acid receptor β stimulates hepatic induction of fibroblast growth factor 21 to promote fatty acid oxidation and control whole-body energy homeostasis in mice, J. Biol. Chem. 288 (2013) 10490–10504. doi:10.1074/jbc.M112.429852.
[150] H.A. Cyphert, X. Ge, A.B. Kohan, L.M. Salati, Y. Zhang, F.B. Hillgartner, Activation of the farnesoid X receptor induces hepatic expression and secretion of fibroblast growth factor 21, J. Biol. Chem. 287 (2012) 25123–25138. doi:10.1074/jbc.M112.375907.
[151] J.A. Holt, G. Luo, A.N. Billin, J. Bisi, Y.Y. McNeill, K.F. Kozarsky, M. Donahee, D.Y. Wang, T.A. Mansfield, S.A. Kliewer, B. Goodwin, S.A. Jones, Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis, Genes Dev. 17 (2003) 1581–1591. doi:10.1101/gad.1083503.
[152] T. Inagaki, M. Choi, A. Moschetta, L. Peng, C.L. Cummins, J.G. McDonald, G. Luo, S.A. Jones, B. Goodwin, J.A. Richardson, R.D. Gerard, J.J. Repa, D.J. Mangelsdorf, S.A. Kliewer, Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis, Cell Metab. 2 (2005) 217–225. doi:10.1016/j.cmet.2005.09.001.
[153] G. Alvarez-Sola, I. Uriarte, M.U. Latasa, R. Urtasun, M. Bárcena-Varela, M. Elizalde, M. Jiménez, C.M. Rodriguez-Ortigosa, F.J. Corrales, M.G. Fernández-Barrena, C. Berasain, M.A. Avila, Fibroblast Growth Factor 15/19 in Hepatocarcinogenesis, Dig. Dis. Basel Switz. 35 (2017) 158–165. doi:10.1159/000450905.
[154] C. Degirolamo, C. Sabbà, A. Moschetta, Therapeutic potential of the endocrine fibroblast growth factors FGF19, FGF21 and FGF23, Nat. Rev. Drug Discov. 15 (2016) 51–69. doi:10.1038/nrd.2015.9.
[155] D. Jahn, D. Sutor, D. Dorbath, J. Weiß, O. Götze, J. Schmitt, H.M. Hermanns, A. Geier, Farnesoid X receptor-dependent and -independent pathways mediate the transcriptional control of human fibroblast growth factor 19 by vitamin A, Biochim. Biophys. Acta. 1859 (2016) 381–392. doi:10.1016/j.bbagrm.2015.12.007.
[156] N. Noy, The one-two punch, Adipocyte. 2 (2013) 184–187. doi:10.4161/adip.23489.
[157] N. Noy, Vitamin A in regulation of insulin responsiveness: mini review, Proc. Nutr. Soc. 75 (2016) 212–215. doi:10.1017/S0029665115004322.
[158] M. Muenzner, N. Tuvia, C. Deutschmann, N. Witte, A. Tolkachov, A. Valai, A. Henze, L.E. Sander, J. Raila, M. Schupp, Retinol-binding protein 4 and its membrane receptor STRA6 control adipogenesis by regulating cellular retinoid homeostasis and retinoic acid receptor α activity, Mol. Cell. Biol. 33 (2013) 4068–4082. doi:10.1128/MCB.00221-13.
[159] S.S. Canan Koch, L.J. Dardashti, R.M. Cesario, G.E. Croston, M.F. Boehm, R.A. Heyman, A.M. Nadzan, Synthesis of retinoid X receptor-specific ligands that are

Disturbed vitamin A metabolism in NAFLD
138
potent inducers of adipogenesis in 3T3-L1 cells, J. Med. Chem. 42 (1999) 742–750. doi:10.1021/jm980621r.
[160] F.W. Kiefer, C. Vernochet, P. O’Brien, S. Spoerl, J.D. Brown, S. Nallamshetty, M. Zeyda, T.M. Stulnig, D.E. Cohen, C.R. Kahn, J. Plutzky, Retinaldehyde dehydrogenase 1 regulates a thermogenic program in white adipose tissue, Nat. Med. 18 (2012) 918–925. doi:10.1038/nm.2757.
[161] O. Ziouzenkova, G. Orasanu, M. Sharlach, T.E. Akiyama, J.P. Berger, J. Viereck, J.A. Hamilton, G. Tang, G.G. Dolnikowski, S. Vogel, G. Duester, J. Plutzky, Retinaldehyde represses adipogenesis and diet-induced obesity, Nat. Med. 13 (2007) 695–702. doi:10.1038/nm1587.
[162] K. Ito, L. Hao, A.E. Wray, A.C. Ross, Lipid emulsion administered intravenously or orally attenuates triglyceride accumulation and expression of inflammatory markers in the liver of nonobese mice fed parenteral nutrition formula, J. Nutr. 143 (2013) 253–259. doi:10.3945/jn.112.169797.
[163] M.A. Abd Eldaim, S. Matsuoka, Y. Okamatsu-Ogura, A. Kamikawa, M.M. Ahmed, A. Terao, K.-I. Nakajima, K. Kimura, Retinoic acid modulates lipid accumulation glucose concentration dependently through inverse regulation of SREBP-1 expression in 3T3L1 adipocytes, Genes Cells Devoted Mol. Cell. Mech. 22 (2017) 568–582. doi:10.1111/gtc.12498.
[164] S.E. Trasino, Y.D. Benoit, L.J. Gudas, Vitamin A Deficiency Causes Hyperglycemia and Loss of Pancreatic β-Cell Mass, J. Biol. Chem. 290 (2015) 1456–1473. doi:10.1074/jbc.M114.616763.
[165] P.-J. Brun, A. Grijalva, R. Rausch, E. Watson, J.J. Yuen, B.C. Das, K. Shudo, H. Kagechika, R.L. Leibel, W.S. Blaner, Retinoic acid receptor signaling is required to maintain glucose-stimulated insulin secretion and β-cell mass, FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 29 (2015) 671–683. doi:10.1096/fj.14-256743.
[166] P.-J. Brun, N. Wongsiriroj, W.S. Blaner, Retinoids in the pancreas, Hepatobiliary Surg. Nutr. 5 (2016) 1–14. doi:10.3978/j.issn.2304-3881.2015.09.03.
[167] S.E. Trasino, X.-H. Tang, J. Jessurun, L.J. Gudas, Retinoic acid receptor β2 agonists restore glycaemic control in diabetes and reduce steatosis, Diabetes Obes. Metab. 18 (2016) 142–151. doi:10.1111/dom.12590.
[168] G. Svegliati-Baroni, F. Ridolfi, A. Di Sario, A. Casini, L. Marucci, G. Gaggiotti, P. Orlandoni, G. Macarri, L. Perego, A. Benedetti, F. Folli, Insulin and insulin-like growth factor-1 stimulate proliferation and type I collagen accumulation by human hepatic stellate cells: differential effects on signal transduction pathways, Hepatol. Baltim. Md. 29 (1999) 1743–1751. doi:10.1002/hep.510290632.
[169] M. Adachi, Y. Osawa, H. Uchinami, T. Kitamura, D. Accili, D.A. Brenner, The forkhead transcription factor FoxO1 regulates proliferation and transdifferentiation of hepatic stellate cells, Gastroenterology. 132 (2007) 1434–1446. doi:10.1053/j.gastro.2007.01.033.
[170] J. Zhang, S. Li, J. Li, C. Han, Z. Wang, C. Li, X. Wang, Z. Liu, J. Wen, L. Zheng, Expression and significance of fat mass and obesity associated gene and forkhead transcription factor O1 in non-alcoholic fatty liver disease, Chin. Med. J. (Engl.). 127 (2014) 3771–3776.
[171] A. Yoneda, K. Sakai-Sawada, Y. Niitsu, Y. Tamura, Vitamin A and insulin are required for the maintenance of hepatic stellate cell quiescence, Exp. Cell Res. 341 (2016) 8–17. doi:10.1016/j.yexcr.2016.01.012.
[172] Y. Rotman, A.J. Sanyal, Current and upcoming pharmacotherapy for non-alcoholic fatty liver disease, Gut. 66 (2017) 180–190. doi:10.1136/gutjnl-2016-312431.

Chapter 4
139
[173] D.R. Schmidt, S.R. Holmstrom, K. Fon Tacer, A.L. Bookout, S.A. Kliewer, D.J. Mangelsdorf, Regulation of bile acid synthesis by fat-soluble vitamins A and D, J. Biol. Chem. 285 (2010) 14486–14494. doi:10.1074/jbc.M110.116004.
[174] M.O. Hoeke, J. Heegsma, M. Hoekstra, H. Moshage, K.N. Faber, Human FXR regulates SHP expression through direct binding to an LRH-1 binding site, independent of an IR-1 and LRH-1, PloS One. 9 (2014) e88011. doi:10.1371/journal.pone.0088011.
[175] F. Yang, Y. He, H.-X. Liu, J. Tsuei, X. Jiang, L. Yang, Z.-T. Wang, Y.-J.Y. Wan, All-trans retinoic acid regulates hepatic bile acid homeostasis, Biochem. Pharmacol. 91 (2014) 483–489. doi:10.1016/j.bcp.2014.08.018.
[176] A. Mamoon, A. Subauste, M.C. Subauste, J. Subauste, Retinoic acid regulates several genes in bile acid and lipid metabolism via upregulation of small heterodimer partner in hepatocytes, Gene. 550 (2014) 165–170. doi:10.1016/j.gene.2014.07.017.
[177] N. Hail, H.J. Kim, R. Lotan, Mechanisms of fenretinide-induced apoptosis, Apoptosis Int. J. Program. Cell Death. 11 (2006) 1677–1694. doi:10.1007/s10495-006-9289-3.
[178] J. Brtko, Role of retinoids and their cognate nuclear receptors in breast cancer chemoprevention, Cent. Eur. J. Public Health. 15 (2007) 3–6.
[179] D. Macis, S. Gandini, A. Guerrieri-Gonzaga, H. Johansson, P. Magni, M. Ruscica, M. Lazzeroni, D. Serrano, M. Cazzaniga, S. Mora, I. Feroce, M. Pizzamiglio, M.T. Sandri, M. Gulisano, B. Bonanni, A. Decensi, Prognostic effect of circulating adiponectin in a randomized 2 x 2 trial of low-dose tamoxifen and fenretinide in premenopausal women at risk for breast cancer, J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 30 (2012) 151–157. doi:10.1200/JCO.2011.35.2237.
[180] F. Formelli, M. Clerici, T. Campa, M.G. Di Mauro, A. Magni, G. Mascotti, D. Moglia, G. De Palo, A. Costa, U. Veronesi, Five-year administration of fenretinide: pharmacokinetics and effects on plasma retinol concentrations, J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 11 (1993) 2036–2042. doi:10.1200/JCO.1993.11.10.2036.
[181] A. Decensi, H. Johansson, R. Miceli, L. Mariani, T. Camerini, E. Cavadini, M.G. Di Mauro, A. Barreca, A.G. Gonzaga, S. Diani, M.T. Sandri, G. De Palo, F. Formelli, Long-term effects of fenretinide, a retinoic acid derivative, on the insulin-like growth factor system in women with early breast cancer, Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 10 (2001) 1047–1053.
[182] H. Johansson, S. Gandini, A. Guerrieri-Gonzaga, S. Iodice, M. Ruscica, B. Bonanni, M. Gulisano, P. Magni, F. Formelli, A. Decensi, Effect of fenretinide and low-dose tamoxifen on insulin sensitivity in premenopausal women at high risk for breast cancer, Cancer Res. 68 (2008) 9512–9518. doi:10.1158/0008-5472.CAN-08-0553.
[183] F. Preitner, N. Mody, T.E. Graham, O.D. Peroni, B.B. Kahn, Long-term Fenretinide treatment prevents high-fat diet-induced obesity, insulin resistance, and hepatic steatosis, Am. J. Physiol. Endocrinol. Metab. 297 (2009) E1420-1429. doi:10.1152/ajpendo.00362.2009.
[184] G.D. Mcilroy, S.R. Tammireddy, B.H. Maskrey, L. Grant, M.K. Doherty, D.G. Watson, M. Delibegović, P.D. Whitfield, N. Mody, Fenretinide mediated retinoic acid receptor signalling and inhibition of ceramide biosynthesis regulates adipogenesis, lipid accumulation, mitochondrial function and nutrient stress signalling in adipocytes and adipose tissue, Biochem. Pharmacol. 100 (2016) 86–97. doi:10.1016/j.bcp.2015.11.017.

Disturbed vitamin A metabolism in NAFLD
140
[185] M. Rahmaniyan, R.W. Curley, L.M. Obeid, Y.A. Hannun, J.M. Kraveka, Identification of dihydroceramide desaturase as a direct in vitro target for fenretinide, J. Biol. Chem. 286 (2011) 24754–24764. doi:10.1074/jbc.M111.250779.
[186] M. Valsecchi, M. Aureli, L. Mauri, G. Illuzzi, V. Chigorno, A. Prinetti, S. Sonnino, Sphingolipidomics of A2780 human ovarian carcinoma cells treated with synthetic retinoids, J. Lipid Res. 51 (2010) 1832–1840. doi:10.1194/jlr.M004010.
[187] A.L. Sabichi, H. Xu, S. Fischer, C. Zou, X. Yang, V.E. Steele, G.J. Kelloff, R. Lotan, J.L. Clifford, Retinoid receptor-dependent and independent biological activities of novel fenretinide analogues and metabolites, Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 9 (2003) 4606–4613.
[188] I. Koh, H.-S. Jun, J.S. Choi, J.H. Lim, W.H. Kim, J.B. Yoon, J. Song, Fenretinide ameliorates insulin resistance and fatty liver in obese mice, Biol. Pharm. Bull. 35 (2012) 369–375.
[189] W. Samuel, R.K. Kutty, T. Duncan, C. Vijayasarathy, B.C. Kuo, K.M. Chapa, T.M. Redmond, Fenretinide induces ubiquitin-dependent proteasomal degradation of stearoyl-CoA desaturase in human retinal pigment epithelial cells, J. Cell. Physiol. 229 (2014) 1028–1038. doi:10.1002/jcp.24527.
[190] N. Mody, G.D. Mcilroy, The mechanisms of Fenretinide-mediated anti-cancer activity and prevention of obesity and type-2 diabetes, Biochem. Pharmacol. 91 (2014) 277–286. doi:10.1016/j.bcp.2014.07.012.
[191] K.D. Shearer, N. Morrice, C. Henderson, J. Reekie, G.D. Mcilroy, P.J. McCaffery, M. Delibegovic, N. Mody, Fenretinide prevents obesity in aged female mice in association with increased retinoid and estrogen signaling, Obes. Silver Spring Md. 23 (2015) 1655–1662. doi:10.1002/oby.21164.
[192] N. Morrice, G.D. Mcilroy, S.R. Tammireddy, J. Reekie, K.D. Shearer, M.K. Doherty, M. Delibegović, P.D. Whitfield, N. Mody, Elevated Fibroblast growth factor 21 (FGF21) in obese, insulin resistant states is normalised by the synthetic retinoid Fenretinide in mice, Sci. Rep. 7 (2017) 43782. doi:10.1038/srep43782.
[193] Y.M. Peng, W.S. Dalton, D.S. Alberts, M.J. Xu, H. Lim, F.L. Meyskens, Pharmacokinetics of N-4-hydroxyphenyl-retinamide and the effect of its oral administration on plasma retinol concentrations in cancer patients, Int. J. Cancer. 43 (1989) 22–26.
[194] A. Costa, W. Malone, M. Perloff, F. Buranelli, T. Campa, G. Dossena, A. Magni, M. Pizzichetta, C. Andreoli, M. Del Vecchio, Tolerability of the synthetic retinoid Fenretinide (HPR), Eur. J. Cancer Clin. Oncol. 25 (1989) 805–808.
[195] F. Formelli, R. Carsana, A. Costa, F. Buranelli, T. Campa, G. Dossena, A. Magni, M. Pizzichetta, Plasma retinol level reduction by the synthetic retinoid fenretinide: a one year follow-up study of breast cancer patients, Cancer Res. 49 (1989) 6149–6152.
[196] N.V. Dimitrov, C.J. Meyer, M. Perloff, M.M. Ruppenthal, M.J. Phillipich, D. Gilliland, W. Malone, F.L. Minn, Alteration of retinol-binding-protein concentrations by the synthetic retinoid fenretinide in healthy human subjects, Am. J. Clin. Nutr. 51 (1990) 1082–1087.
[197] A. Decensi, R. Torrisi, A. Polizzi, R. Gesi, V. Brezzo, M. Rolando, G. Rondanina, M.A. Orengo, F. Formelli, A. Costa, Effect of the synthetic retinoid fenretinide on dark adaptation and the ocular surface, J. Natl. Cancer Inst. 86 (1994) 105–110.
[198] A. Decensi, V. Fontana, M. Fioretto, G. Rondanina, R. Torrisi, M.A. Orengo, A. Costa, Long-term effects of fenretinide on retinal function, Eur. J. Cancer Oxf. Engl. 1990. 33 (1997) 80–84.

Chapter 4
141
[199] R.C. Caruso, J. Zujewski, F. Iwata, M.J. Podgor, B.A. Conley, L.M. Ayres, M.I. Kaiser-Kupfer, Effects of fenretinide (4-HPR) on dark adaptation, Arch. Ophthalmol. Chic. Ill 1960. 116 (1998) 759–763.
[200] B. Conley, J. O’Shaughnessy, S. Prindiville, J. Lawrence, C. Chow, E. Jones, M.J. Merino, M.I. Kaiser-Kupfer, R.C. Caruso, M. Podgor, B. Goldspiel, D. Venzon, D. Danforth, S. Wu, M. Noone, J. Goldstein, K.H. Cowan, J. Zujewski, Pilot trial of the safety, tolerability, and retinoid levels of N-(4-hydroxyphenyl) retinamide in combination with tamoxifen in patients at high risk for developing invasive breast cancer, J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 18 (2000) 275–283. doi:10.1200/JCO.2000.18.2.275.
[201] J.M. Kurie, J.S. Lee, F.R. Khuri, L. Mao, R.C. Morice, J.J. Lee, G.L. Walsh, A. Broxson, S.M. Lippman, J.Y. Ro, B.L. Kemp, D. Liu, H.A. Fritsche, X. Xu, R. Lotan, W.K. Hong, N-(4-hydroxyphenyl)retinamide in the chemoprevention of squamous metaplasia and dysplasia of the bronchial epithelium, Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 6 (2000) 2973–2979.
[202] C. Thaller, M. Shalev, A. Frolov, G. Eichele, T.C. Thompson, R.H. Williams, O. Dillioglugil, D. Kadmon, Fenretinide therapy in prostate cancer: effects on tissue and serum retinoid concentration, J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 18 (2000) 3804–3808. doi:10.1200/JCO.2000.18.22.3804.
[203] L. Baglietto, R. Torrisi, G. Arena, F. Tosetti, A.G. Gonzaga, W. Pasquetti, C. Robertson, A. Decensi, Ocular effects of fenretinide, a vitamin A analog, in a chemoprevention trial of bladder cancer, Cancer Detect. Prev. 24 (2000) 369–375.
[204] H. Abou-Issa, R.W. Curley, G.A. Alshafie, K.L. Weiss, M. Clagett-Dame, J.S. Chapman, S.M. Mershon, Chemotherapeutic evaluation of 4-hydroxybenzylretinone (4-HBR), a nonhydrolyzable C-linked analog of N-(4-hydroxyphenyl) retinamide (4-HPR) against mammary carcinogenesis, Anticancer Res. 21 (2001) 3839–3844.
[205] F. Formelli, T. Camerini, E. Cavadini, V. Appierto, M.G. Villani, A. Costa, G. De Palo, M.G. Di Mauro, U. Veronesi, Fenretinide breast cancer prevention trial: drug and retinol plasma levels in relation to age and disease outcome, Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 12 (2003) 34–41.
[206] A.L. Sabichi, M.R. Modiano, J.J. Lee, Y.-M. Peng, M.-J. Xu, H. Villar, W.S. Dalton, S.M. Lippman, Breast tissue accumulation of retinamides in a randomized short-term study of fenretinide, Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 9 (2003) 2400–2405.
[207] A. Garaventa, R. Luksch, M.S. Lo Piccolo, E. Cavadini, P.G. Montaldo, M.R. Pizzitola, L. Boni, M. Ponzoni, A. Decensi, B. De Bernardi, F.F. Bellani, F. Formelli, Phase I trial and pharmacokinetics of fenretinide in children with neuroblastoma, Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 9 (2003) 2032–2039.
[208] R.A. Radu, Y. Han, T.V. Bui, S. Nusinowitz, D. Bok, J. Lichter, K. Widder, G.H. Travis, N.L. Mata, Reductions in serum vitamin A arrest accumulation of toxic retinal fluorophores: a potential therapy for treatment of lipofuscin-based retinal diseases, Invest. Ophthalmol. Vis. Sci. 46 (2005) 4393–4401. doi:10.1167/iovs.05-0820.
[209] S.M. Lippman, J.J. Lee, J.W. Martin, A.K. El-Naggar, X. Xu, D.M. Shin, M. Thomas, L. Mao, H.A. Fritsche, X. Zhou, V. Papadimitrakopoulou, F.R. Khuri, H. Tran, G.L. Clayman, W.N. Hittelman, W.K. Hong, R. Lotan, Fenretinide activity in

Disturbed vitamin A metabolism in NAFLD
142
retinoid-resistant oral leukoplakia, Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 12 (2006) 3109–3114. doi:10.1158/1078-0432.CCR-05-2636.
[210] N. Colombo, F. Formelli, M.G. Cantù, G. Parma, M. Gasco, A. Argusti, A. Santinelli, R. Montironi, E. Cavadini, L. Baglietto, A. Guerrieri-Gonzaga, G. Viale, A. Decensi, A phase I-II preoperative biomarker trial of fenretinide in ascitic ovarian cancer, Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 15 (2006) 1914–1919. doi:10.1158/1055-9965.EPI-06-0183.
[211] B.J. Schneider, F.P. Worden, S.M. Gadgeel, R.E. Parchment, C.M. Hodges, J. Zwiebel, R.L. Dunn, A.J. Wozniak, M.J. Kraut, G.P. Kalemkerian, Phase II trial of fenretinide (NSC 374551) in patients with recurrent small cell lung cancer, Invest. New Drugs. 27 (2009) 571–578. doi:10.1007/s10637-009-9228-6.
[212] F. Formelli, E. Cavadini, R. Luksch, A. Garaventa, V. Appierto, S. Persiani, Relationship among pharmacokinetics and pharmacodynamics of fenretinide and plasma retinol reduction in neuroblastoma patients, Cancer Chemother. Pharmacol. 66 (2010) 993–998. doi:10.1007/s00280-010-1370-5.
[213] M. Chojkier, A Randomized, Double-blind Study of the Effects of Fenretinide Administered in Subjects With Obesity - Full Text View - ClinicalTrials.gov, (n.d.). https://clinicaltrials.gov/ct2/show/NCT00546455 (accessed September 8, 2017).
[214] L.B. Oliveros, M.A. Domeniconi, V.A. Vega, L.V. Gatica, A.M. Brigada, M.S. Gimenez, Vitamin A deficiency modifies lipid metabolism in rat liver, Br. J. Nutr. 97 (2007) 263–272. doi:10.1017/S0007114507182659.
[215] V.A. Vega, A.C. Anzulovich, S.M. Varas, M.R. Bonomi, M.S. Giménez, L.B. Oliveros, Effect of nutritional vitamin A deficiency on lipid metabolism in the rat heart: Its relation to PPAR gene expression, Nutr. Burbank Los Angel. Cty. Calif. 25 (2009) 828–838. doi:10.1016/j.nut.2009.01.008.
[216] L.V. Gatica, V.A. Vega, F. Zirulnik, L.B. Oliveros, M.S. Gimenez, Alterations in the lipid metabolism of rat aorta: effects of vitamin a deficiency, J. Vasc. Res. 43 (2006) 602–610. doi:10.1159/000096247.
[217] H.W. Kang, G.R. Bhimidi, D.P. Odom, P.-J. Brun, M.-L. Fernandez, M.M. McGrane, Altered lipid catabolism in the vitamin A deficient liver, Mol. Cell. Endocrinol. 271 (2007) 18–27. doi:10.1016/j.mce.2007.03.002.

Chapter 5
Impaired hepatic vitamin A
metabolism in NAFLD mice leading
to vitamin A accumulation in
hepatocytes
Ali Saeed1,2, Paulina Bartuzi3, Janette Heegsma1,4, Daphne
Dekker3, Niels Kloosterhuis3, Alain de Bruin3,5, Johan W.
Jonker6, Bart van de Sluis3, Klaas Nico Faber1,4$
1Department of Gastroenterology and Hepatology, 4Laboratory Medicine, Department
of Pediatrics, 3Section of Molecular Genetics and 4Section of Molecular Metabolism
and Nutrition, University Medical Center Groningen, University of Groningen,
Groningen, The Netherlands. 5Dutch Molecular Pathology Center, Department of
Pathobiology, Faculty of Veterinary Medicine, Utrecht University, Utrecht, The
Netherlands.
2Institute of Molecular Biology and Biotechnology, Bahauddin Zakariya University
Multan, Pakistan.
(Submitted)

Impaired hepatic vitamin A metabolism in NAFLD
144
ABSTRACT
Vitamin A homeostasis is centrally controlled by the liver, involving close
collaboration between hepatocytes and hepatic stellate cells (HSC). Non-alcoholic
fatty liver disease (NAFLD), as well as other chronic liver diseases, is associated with
low hepatic and serum retinol levels, suggestive of systemic vitamin A deficiency. In
this study we show that, in contrast to retinol, hepatic levels of retinyl palmitate (the
main vitamin A storage form) are strongly enhanced in 2 in vivo mouse models of
NAFLD. Transcriptome analysis supports the simultaneous enhancement of retinol
conversion to retinyl esters and retinoic acids in NAFLD livers. Vitamin A
accumulates in hepatocytes in NAFLD, while this occurs in HSC in control mice,
findings corroborated by palmitic acid-treated hepatocytes and HSC in vitro. Thus,
NAFLD does not lead to true vitamin A deficiency, but to cell type-specific
rearrangements in vitamin A metabolism leading to hepatic retinyl ester accumulation
and retinol depletion.

Chapter 5
145
5.1. INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is the most common liver disease
worldwide. The prevalence of NAFLD is estimated at 20-30% in the general
population in Western countries. Starting with benign steatosis, patients are at risk to
develop non-alcoholic steatohepatitis (NASH), progress to cirrhosis and
hepatocellular carcinoma [1]. NALFD prevalence is particularly high in obese
individuals (80-90%), diabetes (30-50%) and/or hyperlipidemia (90%) [1,2]. Hepatic
fat accumulation is a combined result of a high fat- and high carbohydrate-containing
diet and insufficient catabolism of these energy sources, in part due to low physical
activity [3]. A tipping point NAFLD pathology is when simple steatosis is being
accompanied by hepatic inflammation and fibrosis. Inflammation leads to the
activation of hepatic stellate cells(HSC) that transdifferentiate to proliferative and
migratory myofibroblasts, e.g. activated HSC, that produce excessive amounts of
extracellular matrix proteins, like collagens and fibronectins, the typical feature of
fibrosis [4].
HSC is considered “quiescent” in the healthy liver, though they play a key role in
controlling vitamin A metabolism [5]. Vitamin A is important for many physiological
processes, including reproduction, embryogenesis, glucose- and lipid metabolism
and vision, most of which are controlled by the retinoic acid-activated transcription
factors retinoid X receptor (RXR) and retinoic acid receptor (RAR). Quiescent HSC
contains the main body reserve of vitamin A, which is stored as retinyl esters in large
cytoplasmic lipid droplets [6,7]. Controlled conversion to retinol in qHSC is believed to
maintain stable circulating serum retinol levels around 2.0 µmol/L in humans (1.0-1.5
µmol/L in rodents). The specific (control) mechanisms involved are, however, largely
unknown. Dietary vitamin A follows a complex route for final storage in HSC [7]. In
the small intestine, retinyl esters are incorporated in chylomicrons and after transport
through the circulation taken up by hepatocytes. In hepatocytes, they are converted
to retinol and subsequent binding to retinol binding protein 4 (RBP4) stimulates the
release of the retinol-RBP4 complex back to the circulation. Via an unknown
mechanism, retinol is transferred to HSC, re-esterified and stored in lipid droplets.
Liver injury-induced activation of HSC leads to the rapid loss of vitamin A-containing
lipid droplets and, as a consequence, chronic liver diseases are associated with
vitamin A deficiency (VAD), including viral hepatitis, primary biliary cholangitis (PBC),
primary sclerosing cholangitis (PSC), biliary atresia, alcoholic hepatitis and also

Impaired hepatic vitamin A metabolism in NAFLD
146
NAFLD [8]. VAD is defined as serum retinol levels below 0.7 mmol/L [9]. Not only
serum, but also hepatic retinol levels were found to be reduced in class-III (BMI≥40
kg/m2) obese patients and negatively correlated with histological severity of hepatic
steatosis [10,11]. These analyses, however, do not take retinyl esters into account,
which form the largest pool of vitamin A. It thus remains unclear whether the low
serum and hepatic retinol levels truly reflect complete depletion of vitamin A stores
or, alternatively, point to aberrant vitamin A metabolism in the fatty liver. Notably,
individuals carrying the NAFLD risk variant of patatin-like phospholipase domain-
containing protein 3 (PNPLA3, encoding adiponutrin) show low serum retinol levels,
while hepatic retinyl ester levels are increased, pointing to impaired hepatic retinyl
ester-to-retinol conversion [12]. Moreover, transcriptome analyses revealed a
hyperdynamic state, of hepatic retinol metabolism in NAFLD patients, e.g. enhanced
expression of genes involved in vitamin A storage, hydrolysis and transport, though
with unknown effects on the various vitamin A pools [13].
Thus, in this study we analyzed serum and hepatic retinol, retinyl ester and RBP4
levels in 2 NAFLD mouse models, as well as expression of genes/proteins involved in
vitamin A metabolism. Our data show that NAFLD in mice does not lead to plain
vitamin A deficiency, but enhances vitamin A storage in the liver while retinol levels
are reduced. Importantly, the bulk of the vitamin A accumulates in hepatocytes,
rather than in HSC.
5.2. MATERIALS AND METHODS
Animals
All animal experiments were approved by the Institutional Animal Care and Use
Committee, University of Groningen, The Netherlands. Age- and sex-matched (8-10
weeks old) C57BL/6J mice, (Charles River, Saint-Germain-Nuelles, France) were fed
either regular chow or a high fat, high cholesterol (HFC) diet (containing 21% fat, with
45% calories from butter-fat, 0.2% cholesterol and per gram of diet) (Scientific Animal
Food and Engineering (SAFE), Villemoisson-Sur-Orge, France) for period of 12 or 20
weeks (n=8). Both diets contained 20 IU of retinyl acetate/g as a source of vitamin A.
Leptinob mutant (JAX™ ob/ob) mice (10-12 weeks; Charles River) on a C57BL/6J
genetic background and appropriate controls (C57BL/6J) were also analyzed and
kept on chow diet. All animals were kept in a pathogen-free environment with

Chapter 5
147
alternating dark-light cycles of 12 hours, and controlled temperature (20-24 ºC) and
relative humidity (55%±15%). Animals received food and water ad libitum. All animals
were fasted 4 hours before sacrifice. Tissues were snap-frozen in liquid nitrogen or
fixed in paraformaldehyde. Blood was collected by heart puncture.
Primary hepatocyte increased the accumulation of vitamin A in high
fat In vitro
Primary rat hepatocytes and hepatic stellate cells (HSC) were isolated from Wister rat
(Charles River) and cultured in William’s E medium (Thermo Fisher Scientific, Breda,
The Netherlands) and Iscove’s Modified Dulbecco’s Medium with Glutamax
(ThermoFisher, Scientific), respectively, in a humidified incubator at 37ºC and 5%
CO2, as previously described [14–16]. Primary hepatocytes and HSC were exposed
to BSA-conjugated palmitic acid (Sigma-Aldrich, St Louis, USA) (0.25 or 0.5 mmol/L),
with or without retinol (Sigma-Aldrich) (5 µM) for 24-48 hours as previously described
[17].
Quantitative real-time reverse transcription polymerase chain
reaction (qRT-PCR)
Quantitative real-time reverse transcription polymerase chain reaction was performed
as previously described [18]. Shortly, total RNA was isolated from tissue samples
using TRIzol® reagent according to the supplier’s instructions (ThermoFisher
Scientific). The RNA quality and quantity were determined using a Nanodrop 2000c
UV-vis spectrophotometer (Thermo Fisher Scientific). cDNA was synthesized from
2.5 µg of RNA by using random nonamers and M-MLV reverse transcriptase
(Thermo Fisher Scientific). Taqman primers and probes were designed using Primer
Express 3.0.1 (ThermoFisher, Scientific) and are shown in Supplementary Table
S1. All target genes were amplified using the Q-PCR core kit master mix
(Eurogentec, Maastricht, The Netherlands) on a 7900HT Fast Real-Time PCR
system (ThermoFisher, Scientific). SDSV2.4.1 (ThermoFisher, Scientific) was used to
analyze the data. Expression of genes is presented in 2-delta CT and normalized to
36B4.

Impaired hepatic vitamin A metabolism in NAFLD
148
Western blot analysis
Protein samples were prepared for Western blot analysis as described previously
[19]. Protein concentrations were quantified using the Bio-Rad protein assay (Bio-
Rad, Hercules, CA, USA) with bovine serum albumin (BSA) as standard. Equal
amounts of protein were separated on Mini-PROTEAN® TGX™ precast 4-15%
gradient gels (Bio-Rad) and transferred to nitrocellulose membranes using the Trans-
Blot turbo transfer system, (Bio-Rad). Primary antibodies (anti-RBP4, 1:2,000;
#ab109193, Abcam, Cambridge, UK and anti-GAPDH, 1:40,000 #CB1001,
Calbiochem, Merck-Millipore Amsterdam-Zuidoost, The Netherland) and horseradish
peroxidase (HRP)-conjugated goat anti-rabbit secondary antibodies (1:2,000; Agilent
DAKO, Amstelveen, The Netherlands) were used for detection. Proteins were
detected using the Pierce ECL Western blotting kit (Thermo Fisher Scientific).
Images were captured using the chemidoc XRS system and Image Lab version 3.0
(Bio-Rad). The intensity of bands was quantified using ImageJ version 1.51 (NIH,
Maryland, USA).
Histology and pathological scoring
Hematoxylin and Eosin (H&E) staining’s on liver sections (4 µm) were performed.
Snap-frozen liver sections (5 µm) were stained using Oil Red O (ORO) [20,21]. A
pathological score was calculated as previously described [22] to determine the level
of steatosis, as well as the grade of lobular inflammation.
CD68 staining
Immunohistochemistry for CD68 (1:300; rabbit anti-CD68; #137002, Biolegio,
Nijmegen, the Netherlands) was performed on snap-frozen liver sections as
previously described [23]. Antigen retrieval was performed by microwave irradiation
in citrate buffer, pH 6.0 and blocking of endogenous peroxidase with 0.3% H2O2 for
30 min. Horseradish peroxidase (HRP)-conjugated goat anti-rabbit antibodies (1:50;
#170-6515, Bio-Rad) was used secondary antibody. Slides were stained with 3,3'-
Diaminobenzidine (DAB) for 10 min and Haematoxylin was used as a counter nuclear
stain (2 min at RT). Finally, slides were dehydrated and mounted with Eukitt®,
(Sigma-Aldrich). Slides were scanned using a nanozoomer 2.0 HT digital slide
scanner (C9600-12, Hamamatsu Photonics, Hamamatsu, Japan) and analyzed with
Aperio ImageScope (version 11.1, Leica Microsystems, Amsterdam, The

Chapter 5
149
Netherlands). CD68-positive cells were counted to assess the expansion of
macrophages in the liver.
Lipid analysis
Lipids were extracted from 15% (w/v) liver homogenate in PBS by using the Bligh &
Dyer method [24]. A colorimetric assay was used to determine total cholesterol
(11489232, Roche Molecular Biochemicals, Almere, the Netherlands) and free
cholesterol (113609910930, DiaSys Diagnostic Systems Gmbh, Holzheim,
Germany). Cholesterol standards (DiaSys Diagnostic Systems Gmbh) were used as
a reference. Triglycerides were quantified using the Trig/GB kit (1187771, Roche
Molecular Biochemicals) and Roche Precimat Glycerol standards (16658800) were
used as a reference.
Serum and hepatic vitamin A analysis
Serum and tissue vitamin A content was analyzed by reverse phase HPLC as
previously described [25]. Briefly, tissue (30-50 mg) was homogenized in PBS to
create a 15% (w/v) tissue homogenate. Then, tissue homogenate (66.70 µL equal to
10 mg of tissue) or serum (50 µL) were added in antioxidant mix (2.75 mL, containing
1153.39 mmol/L pyrogallol, 66.01 mmol/L butylated hydroxytoluene, 311.08 mmol/L
ethylenediaminetetraacetic acid and 2064.71 mmol/L ascorbic acid, dissolved in
methanol (2.67):dH20 (1), pH 5.4) and vortexed thoroughly for 1 min.
Retinol and retinyl esters were extracted and deproteinized twice with n-hexane in
the presence of retinol acetate (100 µL, concentration 4 µmol/L) as an external
standard to assess the level of recovery after the extraction procedure. Standard
curves created from a range of concentrations of retinol and retinyl palmitate were
used to determine absolute tissue and serum concentrations of these compounds.
Additionally, two negative controls (only containing internal standard) and two
positive controls (low and high concentrations of retinol plus internal standard) were
included in each series of extractions. Samples were evaporated under N2 and
diluted in 300 µL 100% ultrapure ethanol. Then, 50 µL was injected in HPLC (Waters
2795 Alliance HT Separations Module, Connecticut, USA) for phase separation on a
C18 column (Waters Symmetry C18, dimension 150 x 3.0 mm, particle size 5 µm,
Waters Corporation, Milford, MA, USA) and measurement (UV-VIS, dual wavelength,
UV-4075 Jasco, Tokyo, Japan). Retinoids in samples were identified by the exact

Impaired hepatic vitamin A metabolism in NAFLD
150
retention time of known standards in ultraviolet absorption at 325 nm by HPLC.
Finally, retinol and retinyl palmitate concentrations were calculated and normalized to
final volume or tissue weight.
Vitamin A autofluorescence in liver tissue
Autofluorescence analysis was performed on unstained cryostatic liver sections using
a Zeiss LSM 780 NLO two-photon CLSM (Carl Zeiss, Jena, Germany) as previously
described [26,27]. Briefly, cryostat liver sections were illuminated with an excitation
filter of 366 nm band-pass interference, and spectra were recorded in the range of
400-680 nm with spectrum acquisition from 0.2 to 3 seconds.
Statistical analysis
Data is presented in group Mean±SEM and statistical analysis was performed using
the GraphPad Prism 6 software package (GraphPad Software, San Diego, CA, USA).
Statistical significance was determined by Mann-Whitney test (two groups), One-way
ANOVA or Kruskal-Wallis (more than two groups) followed by post-hoc Dunns
(compare all pairs of columns). P-values with ≤0.05*, ≤0.01**, ≤0.001*** were
considered significant.
5.3. RESULTS
HFC diet increases body weight and liver fat content.
In order to study the effect of NAFLD on vitamin A metabolism, we fed mice a high-fat
and high-cholesterol (HFC)-containing diet for 12 or 20 weeks and first confirmed the
development and progression of fatty liver disease. As described by us [28,29] and
other [30,31], the HFC diet induced significant body and liver weight gain after 20
weeks (Figure 1A, B). Hepatic total and free cholesterol, as well as total
triglycerides, were significantly increased after 12 weeks of HFC-feeding and
remained enhanced after 20 weeks (Figure 1C-E). Similarly, plasma total and free
cholesterol, as well as insulin levels were significantly elevated HFC-fed mice (Figure
1F-H). H&E and Oil-Red-O (ORO) staining confirmed excessive fat accumulation in
the livers of HFC-fed mice and was associated with accumulation of CD68-positive
inflammatory cells (Figure 1I).

Chapter 5
151
Figure 1. Hepatic fat accumulation and steatohepatitis in HFC-fed mice. Mice fed chow or HFC-diet for 12 or 20 weeks were analyzed for A) body weight, B) liver weight, C) liver total cholesterol levels, D) liver triglyceride levels, E) liver free cholesterol levels, F) plasma cholesterol levels, G) plasma free cholesterol levels, H) plasma insulin levels, I) H&E staining, Oil red O staining (ORO) and CD68 immunohistochemistry. Quantification of (immuno)histochemistry is described in Supplementary Methods and Materials.

Impaired hepatic vitamin A metabolism in NAFLD
152
HFC diet induces markers of hepatic lipid uptake and synthesis,
inflammation and fibrosis in mice
Hepatic expression of genes involved in lipid uptake (Srb1, Cd36) and synthesis
(Scd1, Fasn, Acc1, Srebp1c) were strongly induced by HFC-fed mice (Figure 2A).
Moreover, the HFC diet lead to an early progression to steatohepatitis, with
enhanced expression of inflammatory markers, including Cd68, Tnf-α, Nos2, Ccl2, Il-
1β and Il-6 (Figure 2B) as well as the progressive induction of markers of fibrosis
(Coll1a1, Acta2, Tgf-β, and Timp1) (Figure 2C).

Chapter 5
153
Figure 2. Expression of hepatic genes involved in lipid uptake, lipid synthesis, inflammation and fibrosis in HFC-fed mice. Mice fed chow or HFC-diet for 12 or 20 weeks were analyzed by Q-PCR for hepatic expression of genes involved in A) hepatic lipid uptake (Srb1, Cd36) and synthesis (Scd1, Fasn, Acc1), B) hepatic inflammation (Cd68, Tnf-α, Nos2, Ccl2, Il-1β, Il-6) and C) liver fibrosis (Coll1a1, Acta2, Tgf-β and Timp1). Hepatic lipid uptake and synthesis, hepatic inflammation and hepatic fibrosis were strongly increased in HFC-fed mice.

Impaired hepatic vitamin A metabolism in NAFLD
154
Reduced retinol, but elevated retinyl palmitate in livers of HFC-fed
mice
Next, we determined the effect of the HFC diet on the levels of retinol in liver and
plasma, as well as hepatic retinyl palmitate, the major storage form of vitamin A.
Hepatic retinol levels were comparable in 12- and 20-week chow-fed mice (24.0 ± 2.4
and 24.0 ± 2.5 µg/g liver, respectively; Figure 3A). In contrast, hepatic retinol levels
were progressively decreased in HFC-fed mice (9.1 ± 0.8 and 5.7 ± 0.8 µg/g liver
after 12 and 20 weeks, respectively; Figure 3A). On the other hand, hepatic retinyl
palmitate levels progressively increased in HFC-fed mice (785 ± 34 and 1,060 ± 182
µg/g liver after 12 and 20 weeks, respectively) and were significantly higher than in
chow-fed mice (331 ± 50 and 413 ± 58 µg/g liver after 12 and 20 weeks, respectively;
Figure 3B). The disturbed hepatic retinol/retinyl palmitate balance was not
accompanied by changes in plasma levels of retinol after 12 weeks HFC diet (1.4 ±
0.1 and 1.6 ± 0.1 µmol/L for chow and HFC diet, respectively), while it was increased
in HFC-fed mice after 20 weeks (1.1 ± 0.1 and 2.1 ± 0.1 µmol/L for chow and HFC
diet, respectively; Figure 3C). Hepatic RBP4 protein levels were decreased in HFC-
fed mice compared to chow-fed mice (Figure 3D), while hepatic mRNA expression of
Rbp4 was comparable in all animal groups (Figure 4A). In contrast, serum RBP4
levels were elevated in HFC-fed mice, which was particularly evident after 20 weeks
(Figure 3E). These results show that a drastic reduction in hepatic retinol levels in
HFC-fed mice is accompanied by a significant increase in hepatic retinyl esters.

Chapter 5
155
Figure 3. A HFC diet leads to impaired hepatic vitamin A metabolism in mice. Mice fed chow or HFC-diet for 12 or 20 weeks were analyzed for hepatic levels of A) retinol B) retinyl palmitate, and C) plasma levels of retinol. Hepatic retinol levels were strongly reduced in HFC-fed mice, while retinyl palmitate levels were significantly increased compared to control mice. 12-week HFC-feeding did not alter plasma retinol levels, which were slightly elevated after 20 weeks. D) Hepatic RBP4 protein levels were decreased in 12 week HFC-fed mice, while this decrease was no longer significant after 20 week HFC feeding. E) Plasma RBP4 progressively increased in HFC- fed mice.

Impaired hepatic vitamin A metabolism in NAFLD
156
Hepatic expression of genes involved in vitamin A storage, transport,
hydrolysis, as well as retinoic acid targets are elevated in HFC-fed
mice
Hepatic mRNA levels of Lrat, coding for the main enzyme producing retinyl esters in
the liver, increased with time in HFC-fed mice (Figure 4A). No or minor changes
were detected in mRNA levels for alternative retinol-esterifying enzymes, like Dgat1
and Dgat2. Of the various retinyl ester hydrolases (REHs) that mobilize retinol from
hepatic retinyl ester stores, only Adpn/Pnpla3 mRNA levels were strongly induced in
HFC-fed mice, while Atgl/Pnpla2 and Hsl/Lipe mRNA levels were not altered
compared to chow-fed mice. Raldh2 mRNA levels were significantly enhanced in
livers of HFC-fed mice, while levels of other members of the retinol-to-retinoic-acid-
converting enzymes [32–34] were not changed (Raldh1, Raldh3 and Raldh4) (Figure
4B). Hepatic expression of retinoic acid-responsive genes (RAR-β, Cyp26a1,
Hsd17b13, Ucp2, Cpt1a, Fgf21) was enhanced in the mice fed the HFC diet (Figure
4C). Thus, the HFC diet leads to a hyperdynamic state of retinol metabolism in mice,
confirming earlier observations in NAFLD patients [13].

Chapter 5
157
Figure 4. A HFC diet strongly affects the hepatic expression of genes involved in vitamin A homeostasis. Mice fed chow or HFC-diet for 12 or 20 weeks were analyzed by Q-PCR for hepatic expression of genes and transcription factors involved in A) vitamin A storage (Lrat, Dgat1, Dgat2) and transport (Rbp4), B) retinyl ester hydrolysis (Atgl/Pnpla2, Pnpla3, Lipe) and retinol-to-retinoic acid conversion (Raldh1, Raldh2, Raldh3, Raldh4) and C) retinoic acid target genes (Rar-β, Cyp26a1, Hsd17b13, Ucp2, Cpt1a, Fgf21). Hepatic expression of vitamin A storage and hydrolysis were increased in HFC-fed mice, in conjunction with enhanced expression of retinoic acid responsive genes.

Impaired hepatic vitamin A metabolism in NAFLD
158
Reduced retinol and enhanced retinyl palmitate in livers of ob/ob
mice
To validate our findings, we next analyzed hepatic vitamin A metabolism in another
model of fatty liver disease, e.g. the leptin-deficient ob/ob mouse. In line with the
observations described above for HFC-fed mice, hepatic retinol levels were
significantly lower in ob/ob mice compared to age-matched wild-type littermates (2.8
± 0.1 versus 16.9 ± 5.1 µg/g liver, respectively; Figure 5A), in conjunction with
strongly enhanced levels of retinyl palmitate (359 ± 28 versus 160 ± 26 µg/g liver,
respectively; Figure 5B). Moreover, plasma retinol and RBP4 levels were
significantly higher in ob/ob mice as compared to wild-type littermates (2.5 ± 0.2
versus 0.9 ± 0.2 µM retinol, respectively; Figure 5C and D). Finally, also hepatic
RBP4 protein levels were reduced in ob/ob mice independently of (unchanged) Rbp4
mRNA levels as compared to wild-type mice (Figure 5D and 6A), all features being
comparable between HFC-fed wild type mice and chow-fed ob/ob mice.
Figure 5. Ob/ob mice show disturbed hepatic vitamin A metabolism. Ob/ob mice and age-matched wild-type littermates were sacrificed and analyzed for A) hepatic retinol, B) hepatic retinyl palmitate, C) plasma retinol and hepatic (D) and plasma (E) RBP4 levels. Hepatic retinol levels were strongly reduced, while retinyl palmitate levels were significantly increased in ob/ob mice. Plasma retinol and RBP4 protein levels were significantly elevated in ob/ob mice. In contrast, hepatic RBP4 protein levels were reduced. Note that hepatic RBP4 mRNA levels were not changed in ob/ob mice (see Figure 6A).

Chapter 5
159
Hepatic expression of genes involved in vitamin A storage, transport,
hydrolysis and retinoic acid-responsive targets are elevated in ob/ob
mice
Similar to HFC-fed mice, hepatic expression of Lrat and Dgat1 were significantly
enhanced in ob/ob mice as compared to controls, while expression of Dgat2 and
Rbp4 was not different between both animal groups (Figure 6A). Pnpla3 was
strongly enhanced in ob/ob mice compared to control mice, while Pnpla2 and Lipe
levels were unchanged. In contrast to HFC-fed mice, Raldh2 mRNA levels were
reduced in ob/ob mice, while Raldh1 and Raldh3 levels were significantly elevated
(Figure 6B). Finally, hepatic expression of various retinoic acid-responsive genes
was also enhanced in ob/ob mice livers as compared to controls (Figure 6C). Taken
together, both NAFLD mouse models present comparable impairments in hepatic
vitamin A homeostasis.

Impaired hepatic vitamin A metabolism in NAFLD
160
Figure 6. Hepatic expression of genes involved in vitamin A homeostasis is strongly affected in ob/ob mice. Ob/ob mice and age-matched wild-type littermates were sacrificed and analyzed by Q-PCR for hepatic expression of genes and transcription factors involved in A) vitamin A storage (Lrat, Dgat1, Dgat2), transport (Rbp4), B) vitamin A hydrolysis (Atgl/Pnpla2, Pnpla3, Lipe) and retinol-to-retinoic acid conversion (Raldh1, Raldh2, Raldh3, Raldh4) and C) retinoic acid target genes (Rar-β, Cyp26a1, Hsd17b13, Ucp2, Cpt1a, Fgf21). Hepatic expression of vitamin A storage and hydrolysis was increased in ob/ob, in conjunction with enhanced expression of retinoic acid responsive genes.

Chapter 5
161
Palmitic acid increases Lrat expression and vitamin A accumulation
in primary rat hepatocytes
Dietary vitamin A is absorbed by hepatocytes through retinyl ester-carrying
chylomicron remnants, after which it is redistributed to HSC for storage (see
introduction). Thus, both hepatocytes and HSC are possible sites for retinyl ester
accumulation and earlier work has suggested vitamin A accumulation in hepatocytes
of human and rodent fatty livers [26,35]. Microscopical analysis of liver tissue for
vitamin A-specific autofluorescence revealed few bright (auto)fluorescent dots in the
hepatic parenchyma of chow-fed mice (Figure 7A, top right panel) reminiscent of the
distribution of quiescent HSC and the pattern observed by others in healthy rat and
human liver [26,35]. The autofluorescence signal strongly increased in livers of HFC-
fed mice, showing a completely altered staining pattern, which now appeared in large
vesicular structures (“lipid droplets”) that localize predominantly in hepatocytes
(Figure 7A, bottom right panel).
In order to study cell-type specific effects of vitamin A storage in an in vitro model of
fatty liver disease, we exposed primary rat hepatocytes, as well as quiescent and
activated primary rat HSC, to palmitate and found that it enhanced Lrat mRNA levels
only in hepatocytes and not in quiescent nor in activated HSC (Figure 7B).
Conversely, PNPLA3 levels were elevated by palmitate exposure specifically in HSC.
Moreover, cellular retinyl palmitate levels significantly increased only in primary rat
hepatocytes exposed to palmitate together with retinol for 48 h (Figure 7C). These
results suggest that NAFLD promotes vitamin A loss in HSC while vitamin A storage
is induced in hepatocytes.

Impaired hepatic vitamin A metabolism in NAFLD
162
Figure 7. Autofluorescence in liver increases with fatty liver and palmitic acid induces Lrat expression and cellular retinyl palmitate accumulation in primary rat hepatocytes. Oil-Red-O staining (left panels) and vitamin A-specific autofluorescence (right panels of liver sections of chow-fed (top panels) and HFC-fed (bottom panels) mice. Vitamin A-specific autofluorescence was strongly increased in livers of HFC-fed mice and was located predominantly in hepatocytes compared to its location in sparsely-present HSC in livers of chow-fed mice. B) Freshly-isolated and 4 h-attached primary rat hepatocytes and quiescent primary HSC were and immediately treated with and without palmitate for 24 h. Cells were harvested and analyzed by Q-PCR analysis for Lrat and Pnpla3 expression. The gene expression is presented in 2-∆∆CT and normalized to 18S. C) Freshly-isolated and 4 h-attached primary rat hepatocytes were treated for 48 h with and without palmitate and retinol, followed by an analysis of cellular retinyl palmitate and retinol levels.

Chapter 5
163
5.4. DISCUSSION
In this study, we show that steatohepatitis in mice heavily affects hepatic vitamin A
metabolism leading to reduced retinol and enhanced retinyl ester levels in the liver.
Notably, retinyl esters accumulate in hepatocytes rather than in hepatic stellate cells,
cells that store vitamin A esters in a healthy liver. Following retinol levels, hepatic
RBP4 levels are also reduced, while increased in circulation. Thus, steatohepatitis
does not lead to true vitamin A deficiency as suggested by earlier studies, but rather
leads to hepatic retinol deficiency due to metabolic changes. Vitamin A
supplementation in the form of retinyl esters may, therefore, be counterproductive in
NAFLD, as it will likely accumulate in the already overloaded hepatocytes. Also,
aberrant vitamin A metabolism may directly affect the activity of nuclear receptors,
such as RAR, PPARs, LXR and FXR, as they require RXR for most of their actions.
Chronic liver diseases, including NAFLD, are associated with low serum and hepatic
retinol levels, which is generally considered to be a sign of systemic vitamin A
deficiency [10,11,36–38]. Moreover, serum and hepatic retinol levels are negatively
correlated with liver disease progression [10,36,39,40]. However, systemic retinol
levels are only a small fraction of the total pool of vitamin A, which mostly consists of
retinyl esters stored in liver (≥80%) and adipose tissue (10-20%) [41,42]. Thus,
systemic retinol levels alone are not a reliable marker for hypovitaminosis A per se.
Earlier studies have actually suggested that NAFLD is associated with hepatic
vitamin A accumulation in humans and rodents [11,43].
We show that hepatic fat accumulation in mice is associated with a strong reduction
in hepatic retinol levels, but not in circulation. This is in line with earlier observations
[10,11,36–38] and indicates that the mouse NAFLD models used do show the
hepatic phenotype, but do not replicate the reduced circulatory retinol levels
observed in NAFLD patients [10,36,44]. NAFLD mouse models likely reflect only the
initiating phase of NAFLD and serum retinol levels are also hardly affected at that
stage in patients. Alternatively, the observed difference may be species related, but
this requires analyses of vitamin A metabolism in more severe mouse NAFLD
models. We did observe, however, that serum RBP4 levels are elevated in NAFLD
mice, which is also found in obese individuals with or without established NAFLD
[45–49]. In contrast, hepatic RBP4 levels are reduced in NAFLD mice. This implies
that, even though hepatic retinol levels are low, sufficient retinol is produced to
promote -and even enhance- RBP4 secretion from the liver. Efficient RBP4 secretion

Impaired hepatic vitamin A metabolism in NAFLD
164
from the liver strongly depends on retinol availability. Vitamin A deficiency leads to
pronounced hepatic accumulation of retinol-free apo-RBP4 under unchanged Rbp4
mRNA levels and is rapidly released into the circulation upon retinol or retinoic acid
treatment [50–52]. As hepatic retinol levels were low, enhanced RBP4 secretion may
also result from enhanced production of retinoic acids. Indeed, mRNA levels of
Raldh1, the main enzyme responsible for RA production in the liver, were significantly
elevated in ob/ob mice, with similar trends observed for 12- and 20-wk HFC-fed mice.
Moreover, expression of various RA-responsive genes, such as RAR-β, Cyp26a1,
Hsd17b13, Ucp2, Cpt1a, Fgf21, was also induced in NAFLD mice. This is in line with
the earlier reported hyper-metabolic state of hepatic vitamin A metabolism and
degradation in NAFLD patients [13]. That study also observed enhanced expression
of hepatic LRAT and DGAT1 in NAFLD patients, genes that encode enzymes that
esterify retinol, which we also detected in NAFLD mice. This coincides with a strong
increase in hepatic retinyl palmitate levels in NAFLD mice, which is the main retinyl
ester in both human and rodents [53,54]. Particularly relevant is the apparent
redistribution of vitamin A from HSC in control livers to lipid-loaded hepatocytes in
fatty livers. Retinyl esters are rapidly converted to retinol in the healthy liver.
Accumulation of retinyl esters in lipid-loaded hepatocytes may, therefore, result from
decreased hydrolysis and/or increased esterification. Expression of Atgl/Pnpla2
(hydrolysis) was not changed, while both Lrat and Dgat1 (esterification) were
enhanced in mouse fatty livers, indicating a shift to vitamin A storage in retinyl esters.
On the other hand, PNPLA3/ADPN is also able to hydrolyze retinyl esters [55] and its
expression is strongly increased in mouse fatty liver, like in human NAFLD patients
[56]. Still, PNPLA3’s main substrates are TG that also strongly accumulate in mouse
fatty liver. This implies that its activity cannot compensate for the build-up of TG in
hepatocytes, as observed for retinyl palmitate. Notably, Lrat expression was induced
in primary rat hepatocytes exposed to palmitate and they accumulate retinyl-
palmitate when co-exposed to retinol. These conditions did not regulate Lrat
expression in qHSC, but rather enhance retinyl ester hydrolysis by increasing
PNPLA3 expression. These findings are in line with the shift in the main location of
vitamin A to hepatocytes in fatty liver in mice (this study), rats [35] and human [26].
The redistribution of retinyl esters from HSC to hepatocytes may also promote
NAFLD-associated fibrosis, as differentiation of qHSC to myofibroblasts is
characterized by the loss of vitamin A-containing lipid droplets. In NAFLD, this

Chapter 5
165
apparently happens in conjunction with a pronounced accumulation of retinyl esters
in the liver, but then primarily in hepatocytes.
Our observation of retinyl palmitate accumulation in mouse fatty liver contrasts to
recent findings by others, who reported pronounced reductions in hepatic retinol and
retinyl palmitate in HFD-fed mice and ob/ob mice [11]. Several reasons may account
for this discrepancy; 1) HFD vs HFC-diets: A high-fat (only) diet may affect vitamin A
metabolism differently than the high-fat high-cholesterol diets (HFC) used in this
study. However, we did not detect a reduction in hepatic retinyl palmitate levels in
mice fed HFD diet for 12 weeks, while hepatic retinol levels were reduced in both
HFD- and HFC-fed mice (Supplementary Figure S1 and Figure 3A). 2) Amount of
vitamin A in the diets: In our study, both control chow and HFC diet contained equal
amounts of vitamin A, e.g. 20 IU/g. The earlier study [11] used a HFD that contained
significantly less vitamin A compared to the control chow (3.8 vs. 15 IU/g plus
additional β-carotene, respectively). Dietary intake of vitamin A, even above daily
recommendations, correlates directly with hepatic levels of retinyl palmitate and
retinol [57–60]. Thus, for establishing an effect of a (high-fat) diet on hepatic vitamin
A metabolism it is crucial to standardize the dietary vitamin A intake for control and
experimental diet. This is the case in the ob/ob mouse studies and cannot explain the
opposing results between our and the earlier study. 3) Retinol/retinyl ester extraction
procedure: extraction of retinol and retinyl esters from serum or tissue is typically
performed with either n-hexane [25,41] or acetonitrile [11], though this is not always
specified in methods sections. We compared both methods and found that
acetonitrile very inefficiently extracts retinyl palmitate specifically from fatty liver
tissue, while retinol extraction was similar with these solvents (Supplementary
Figure S2). This suggests that the extraction of retinyl esters by acetonitrile is
compromised when a lot of fat is present in the (liver) tissue, and this may (in part)
explain the controversial findings on retinyl ester levels in fatty liver.
Thus, our study shows that fatty liver disease is associated with hepatic accumulation
of retinyl esters and disturbed vitamin A metabolism. The latter condition likely
modulates disease progression, as it was recently shown that even moderate
changes in hepatic RA production significantly enhance hepatic lipid accumulation
[61]. Moreover, lipid metabolism is tightly controlled by various nuclear receptors
(NR), like PPARs, FXR, LXR and RAR, ligands of which are in advanced clinical trial
stages for the treatment of NAFLD [62]. All these factors require RXR as an obligate
partner and aberrant production of RXR-activating retinoids will affect NR/RXR

Impaired hepatic vitamin A metabolism in NAFLD
166
signaling. Further studies are needed to determine the impact of changed vitamin A
metabolism in NAFLD on the therapeutic efficacy of these drug targets.
ACKNOWLEDGEMENTS
The authors want to acknowledge J.W. (Hans) Jonker (group), Tim Van Zutphen and
Dicky Struik for providing ob/ob mice materials.

Chapter 5
167
REFERENCES
[1] S. Albhaisi, A. Sanyal, Recent advances in understanding and managing non-alcoholic fatty liver disease, F1000Research. 7 (2018). doi:10.12688/f1000research.14421.1.
[2] S. Bellentani, F. Scaglioni, M. Marino, G. Bedogni, Epidemiology of Non-Alcoholic Fatty Liver Disease, Dig. Dis. 28 (2010) 155–161. doi:10.1159/000282080.
[3] C.N. Katsagoni, M. Georgoulis, G.V. Papatheodoridis, D.B. Panagiotakos, M.D. Kontogianni, Effects of lifestyle interventions on clinical characteristics of patients with non-alcoholic fatty liver disease: A meta-analysis, Metabolism. 68 (2017) 119–132. doi:10.1016/j.metabol.2016.12.006.
[4] J.P. Iredale, A. Thompson, N.C. Henderson, Extracellular matrix degradation in liver fibrosis: Biochemistry and regulation, Biochim. Biophys. Acta. 1832 (2013) 876–883. doi:10.1016/j.bbadis.2012.11.002.
[5] R. Schreiber, U. Taschler, K. Preiss-Landl, N. Wongsiriroj, R. Zimmermann, A. Lass, Retinyl ester hydrolases and their roles in vitamin A homeostasis, Biochim. Biophys. Acta. 1821 (2012) 113–123. doi:10.1016/j.bbalip.2011.05.001.
[6] W.S. Blaner, S.M. O’Byrne, N. Wongsiriroj, J. Kluwe, D.M. D’Ambrosio, H. Jiang, R.F. Schwabe, E.M.C. Hillman, R. Piantedosi, J. Libien, Hepatic stellate cell lipid droplets: a specialized lipid droplet for retinoid storage, Biochim. Biophys. Acta. 1791 (2009) 467–473. doi:10.1016/j.bbalip.2008.11.001.
[7] H. Senoo, Y. Mezaki, M. Fujiwara, The stellate cell system (vitamin A-storing cell system), Anat. Sci. Int. 92 (2017) 387–455. doi:10.1007/s12565-017-0395-9.
[8] A. Saeed, M. Hoekstra, M.O. Hoeke, J. Heegsma, K.N. Faber, The interrelationship between bile acid and vitamin A homeostasis, Biochim. Biophys. Acta. 1862 (2017) 496–512. doi:10.1016/j.bbalip.2017.01.007.
[9] World Health Organization, Indicators for assessing vitamin A deficiency and their application in monitoring and evaluating intervention programmes, Eileen Brown, James Akré, World Health Organization: Geneva, Switzerland, 1996. http://www.who.int/nutrition/publications/micronutrients/vitamin_a_deficiency/WHO_NUT_96.10/en/.
[10] G.V. Chaves, S.E. Pereira, C.J. Saboya, D. Spitz, C.S. Rodrigues, A. Ramalho, Association between liver vitamin A reserves and severity of nonalcoholic fatty liver disease in the class III obese following bariatric surgery, Obes. Surg. 24 (2014) 219–224. doi:10.1007/s11695-013-1087-8.
[11] S.E. Trasino, X.-H. Tang, J. Jessurun, L.J. Gudas, Obesity Leads to Tissue, but not Serum Vitamin A Deficiency, Sci. Rep. 5 (2015) 15893. doi:10.1038/srep15893.
[12] M. Kovarova, I. Königsrainer, A. Königsrainer, F. Machicao, H.-U. Häring, E. Schleicher, A. Peter, The Genetic Variant I148M in PNPLA3 Is Associated With Increased Hepatic Retinyl-Palmitate Storage in Humans, J. Clin. Endocrinol. Metab. 100 (2015) E1568-1574. doi:10.1210/jc.2015-2978.
[13] A.A. Ashla, Y. Hoshikawa, H. Tsuchiya, K. Hashiguchi, M. Enjoji, M. Nakamuta, A. Taketomi, Y. Maehara, K. Shomori, A. Kurimasa, I. Hisatome, H. Ito, G. Shiota, Genetic analysis of expression profile involved in retinoid metabolism in non-alcoholic fatty liver disease, Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 40 (2010) 594–604. doi:10.1111/j.1872-034X.2010.00646.x.

Impaired hepatic vitamin A metabolism in NAFLD
168
[14] T.E. Vrenken, M. Buist-Homan, A.J. Kalsbeek, K.N. Faber, H. Moshage, The active metabolite of leflunomide, A77 1726, protects rat hepatocytes against bile acid-induced apoptosis, J. Hepatol. 49 (2008) 799–809. doi:10.1016/j.jhep.2008.07.019.
[15] H. Moshage, A. Casini, C.S. Lieber, Acetaldehyde selectively stimulates collagen production in cultured rat liver fat-storing cells but not in hepatocytes, Hepatol. Baltim. Md. 12 (1990) 511–518.
[16] S. Shajari, A. Laliena, J. Heegsma, M.J. Tuñón, H. Moshage, K.N. Faber, Melatonin suppresses activation of hepatic stellate cells through RORα-mediated inhibition of 5-lipoxygenase, J. Pineal Res. 59 (2015) 391–401. doi:10.1111/jpi.12271.
[17] Y.M. Song, S.-O. Song, Y.-K. Jung, E.-S. Kang, B.S. Cha, H.C. Lee, B.-W. Lee, Dimethyl sulfoxide reduces hepatocellular lipid accumulation through autophagy induction, Autophagy. 8 (2012) 1085–1097. doi:10.4161/auto.20260.
[18] H. Blokzijl, S. Vander Borght, L.I.H. Bok, L. Libbrecht, M. Geuken, F.A.J. van den Heuvel, G. Dijkstra, T.A.D. Roskams, H. Moshage, P.L.M. Jansen, K.N. Faber, Decreased P-glycoprotein (P-gp/MDR1) expression in inflamed human intestinal epithelium is independent of PXR protein levels, Inflamm. Bowel Dis. 13 (2007) 710–720. doi:10.1002/ibd.20088.
[19] A. Pellicoro, F.A.J. van den Heuvel, M. Geuken, H. Moshage, P.L.M. Jansen, K.N. Faber, Human and rat bile acid-CoA:amino acid N-acyltransferase are liver-specific peroxisomal enzymes: implications for intracellular bile salt transport, Hepatol. Baltim. Md. 45 (2007) 340–348. doi:10.1002/hep.21528.
[20] A.H. Fischer, K.A. Jacobson, J. Rose, R. Zeller, Hematoxylin and eosin staining of tissue and cell sections, CSH Protoc. 2008 (2008) pdb.prot4986.
[21] A. Mehlem, C.E. Hagberg, L. Muhl, U. Eriksson, A. Falkevall, Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease, Nat. Protoc. 8 (2013) 1149–1154. doi:10.1038/nprot.2013.055.
[22] D.E. Kleiner, E.M. Brunt, M. Van Natta, C. Behling, M.J. Contos, O.W. Cummings, L.D. Ferrell, Y.-C. Liu, M.S. Torbenson, A. Unalp-Arida, M. Yeh, A.J. McCullough, A.J. Sanyal, Nonalcoholic Steatohepatitis Clinical Research Network, Design and validation of a histological scoring system for nonalcoholic fatty liver disease, Hepatol. Baltim. Md. 41 (2005) 1313–1321. doi:10.1002/hep.20701.
[23] M. Aparicio-Vergara, P.P.H. Hommelberg, M. Schreurs, N. Gruben, R. Stienstra, R. Shiri-Sverdlov, N.J. Kloosterhuis, A. de Bruin, B. van de Sluis, D.P.Y. Koonen, M.H. Hofker, Tumor necrosis factor receptor 1 gain-of-function mutation aggravates nonalcoholic fatty liver disease but does not cause insulin resistance in a murine model, Hepatol. Baltim. Md. 57 (2013) 566–576. doi:10.1002/hep.26046.
[24] E.G. Bligh, W.J. Dyer, A rapid method of total lipid extraction and purification, Can. J. Biochem. Physiol. 37 (1959) 911–917. doi:10.1139/o59-099.
[25] Y.-K. Kim, L. Quadro, Reverse-phase high-performance liquid chromatography (HPLC) analysis of retinol and retinyl esters in mouse serum and tissues, Methods Mol. Biol. Clifton NJ. 652 (2010) 263–275. doi:10.1007/978-1-60327-325-1_15.
[26] A.C. Croce, U. De Simone, I. Freitas, E. Boncompagni, D. Neri, U. Cillo, G. Bottiroli, Human liver autofluorescence: an intrinsic tissue parameter discriminating normal and diseased conditions, Lasers Surg. Med. 42 (2010) 371–378. doi:10.1002/lsm.20923.

Chapter 5
169
[27] A.C. Croce, U. De Simone, M. Vairetti, A. Ferrigno, E. Boncompagni, I. Freitas, G. Bottiroli, Liver autofluorescence properties in animal model under altered nutritional conditions, Photochem. Photobiol. Sci. Off. J. Eur. Photochem. Assoc. Eur. Soc. Photobiol. 7 (2008) 1046–1053. doi:10.1039/b804836c.
[28] P. Bartuzi, T. Wijshake, D.C. Dekker, A. Fedoseienko, N.J. Kloosterhuis, S.A. Youssef, H. Li, R. Shiri-Sverdlov, J.-A. Kuivenhoven, A. de Bruin, E. Burstein, M.H. Hofker, B. van de Sluis, A cell-type-specific role for murine Commd1 in liver inflammation, Biochim. Biophys. Acta. 1842 (2014) 2257–2265. doi:10.1016/j.bbadis.2014.06.035.
[29] P. Bartuzi, D.D. Billadeau, R. Favier, S. Rong, D. Dekker, A. Fedoseienko, H. Fieten, M. Wijers, J.H. Levels, N. Huijkman, N. Kloosterhuis, H. van der Molen, G. Brufau, A.K. Groen, A.M. Elliott, J.A. Kuivenhoven, B. Plecko, G. Grangl, J. McGaughran, J.D. Horton, E. Burstein, M.H. Hofker, B. van de Sluis, CCC- and WASH-mediated endosomal sorting of LDLR is required for normal clearance of circulating LDL, Nat. Commun. 7 (2016) 10961. doi:10.1038/ncomms10961.
[30] R. Costa, I. Rodrigues, L. Guardão, S. Rocha-Rodrigues, C. Silva, J. Magalhães, M. Ferreira-de-Almeida, R. Negrão, R. Soares, Xanthohumol and 8-prenylnaringenin ameliorate diabetic-related metabolic dysfunctions in mice, J. Nutr. Biochem. 45 (2017) 39–47. doi:10.1016/j.jnutbio.2017.03.006.
[31] M.S. Kim, M.-S. Choi, S.N. Han, High fat diet-induced obesity leads to proinflammatory response associated with higher expression of NOD2 protein, Nutr. Res. Pract. 5 (2011) 219–223. doi:10.4162/nrp.2011.5.3.219.
[32] N.L. Makia, P. Bojang, K.C. Falkner, D.J. Conklin, R.A. Prough, Murine hepatic aldehyde dehydrogenase 1a1 is a major contributor to oxidation of aldehydes formed by lipid peroxidation, Chem. Biol. Interact. 191 (2011) 278–287. doi:10.1016/j.cbi.2011.01.013.
[33] E.C. Kathmann, S. Naylor, J.J. Lipsky, Rat liver constitutive and phenobarbital-inducible cytosolic aldehyde dehydrogenases are highly homologous proteins that function as distinct isozymes, Biochemistry. 39 (2000) 11170–11176.
[34] D.K. Bhatt, A. Gaedigk, R.E. Pearce, J.S. Leeder, B. Prasad, Age-dependent Protein Abundance of Cytosolic Alcohol and Aldehyde Dehydrogenases in Human Liver, Drug Metab. Dispos. Biol. Fate Chem. 45 (2017) 1044–1048. doi:10.1124/dmd.117.076463.
[35] A.C. Croce, A. Ferrigno, V.M. Piccolini, E. Tarantola, E. Boncompagni, V. Bertone, G. Milanesi, I. Freitas, M. Vairetti, G. Bottiroli, Integrated autofluorescence characterization of a modified-diet liver model with accumulation of lipids and oxidative stress, BioMed Res. Int. 2014 (2014) 803491. doi:10.1155/2014/803491.
[36] J.I. Botella-Carretero, J.A. Balsa, C. Vázquez, R. Peromingo, M. Díaz-Enriquez, H.F. Escobar-Morreale, Retinol and alpha-tocopherol in morbid obesity and nonalcoholic fatty liver disease, Obes. Surg. 20 (2010) 69–76. doi:10.1007/s11695-008-9686-5.
[37] P.V. Havaldar, V.D. Patel, B.M. Siddibhavi, Recurrent vitamin A deficiency and fatty liver, J. Trop. Pediatr. 37 (1991) 87–88.
[38] Y. Liu, H. Chen, J. Wang, W. Zhou, R. Sun, M. Xia, Association of serum retinoic acid with hepatic steatosis and liver injury in nonalcoholic fatty liver disease, Am. J. Clin. Nutr. 102 (2015) 130–137. doi:10.3945/ajcn.114.105155.
[39] L. de Souza Valente da Silva, G. Valeria da Veiga, R.A. Ramalho, Association of serum concentrations of retinol and carotenoids with overweight in children and adolescents, Nutr. Burbank Los Angel. Cty. Calif. 23 (2007) 392–397. doi:10.1016/j.nut.2007.02.009.

Impaired hepatic vitamin A metabolism in NAFLD
170
[40] M.L. Neuhouser, C.L. Rock, A.L. Eldridge, A.R. Kristal, R.E. Patterson, D.A. Cooper, D. Neumark-Sztainer, L.J. Cheskin, M.D. Thornquist, Serum concentrations of retinol, alpha-tocopherol and the carotenoids are influenced by diet, race and obesity in a sample of healthy adolescents, J. Nutr. 131 (2001) 2184–2191.
[41] M.A. Kane, A.E. Folias, J.L. Napoli, HPLC/UV quantitation of retinal, retinol, and retinyl esters in serum and tissues, Anal. Biochem. 378 (2008) 71–79. doi:10.1016/j.ab.2008.03.038.
[42] W.S. Blaner, Y. Li, P.-J. Brun, J.J. Yuen, S.-A. Lee, R.D. Clugston, Vitamin A Absorption, Storage and Mobilization, Subcell. Biochem. 81 (2016) 95–125. doi:10.1007/978-94-024-0945-1_4.
[43] S.E. Trasino, X.-H. Tang, J. Jessurun, L.J. Gudas, Retinoic acid receptor β2 agonists restore glycaemic control in diabetes and reduce steatosis, Diabetes Obes. Metab. 18 (2016) 142–151. doi:10.1111/dom.12590.
[44] F.I. Suano de Souza, O.M. Silverio Amancio, R.O. Saccardo Sarni, T. Sacchi Pitta, A.P. Fernandes, F.L. Affonso Fonseca, S. Hix, R.A. Ramalho, Non-alcoholic fatty liver disease in overweight children and its relationship with retinol serum levels, Int. J. Vitam. Nutr. Res. Int. Z. Vitam.- Ernahrungsforschung J. Int. Vitaminol. Nutr. 78 (2008) 27–32. doi:10.1024/0300-9831.78.1.27.
[45] Q. Yang, T.E. Graham, N. Mody, F. Preitner, O.D. Peroni, J.M. Zabolotny, K. Kotani, L. Quadro, B.B. Kahn, Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes, Nature. 436 (2005) 356–362. doi:10.1038/nature03711.
[46] D.G. Haider, K. Schindler, G. Prager, A. Bohdjalian, A. Luger, M. Wolzt, B. Ludvik, Serum retinol-binding protein 4 is reduced after weight loss in morbidly obese subjects, J. Clin. Endocrinol. Metab. 92 (2007) 1168–1171. doi:10.1210/jc.2006-1839.
[47] T. Reinehr, B. Stoffel-Wagner, C.L. Roth, Retinol-binding protein 4 and its relation to insulin resistance in obese children before and after weight loss, J. Clin. Endocrinol. Metab. 93 (2008) 2287–2293. doi:10.1210/jc.2007-2745.
[48] M. Tajtáková, Z. Semanová, G. Ivancová, J. Petrovicová, V. Donicová, E. Zemberová, [Serum level of retinol-binding protein 4 in obese patients with insulin resistance and in patients with type 2 diabetes treated with metformin], Vnitr. Lek. 53 (2007) 960–963.
[49] F. Ulgen, C. Herder, M.C. Kühn, H.S. Willenberg, M. Schott, W.A. Scherbaum, S. Schinner, Association of serum levels of retinol-binding protein 4 with male sex but not with insulin resistance in obese patients, Arch. Physiol. Biochem. 116 (2010) 57–62. doi:10.3109/13813451003631421.
[50] H. Ronne, C. Ocklind, K. Wiman, L. Rask, B. Obrink, P.A. Peterson, Ligand-dependent regulation of intracellular protein transport: effect of vitamin a on the secretion of the retinol-binding protein, J. Cell Biol. 96 (1983) 907–910.
[51] J.L. Dixon, D.S. Goodman, Studies on the metabolism of retinol-binding protein by primary hepatocytes from retinol-deficient rats, J. Cell. Physiol. 130 (1987) 14–20. doi:10.1002/jcp.1041300104.
[52] D. Bellovino, Y. Lanyau, I. Garaguso, L. Amicone, C. Cavallari, M. Tripodi, S. Gaetani, MMH cells: An in vitro model for the study of retinol-binding protein secretion regulated by retinol, J. Cell. Physiol. 181 (1999) 24–32. doi:10.1002/(SICI)1097-4652(199910)181:1<24::AID-JCP3>3.0.CO;2-0.

Chapter 5
171
[53] S. Futterman, J.S. Andrews, The Composition of Liver Vitamin A Ester and the Synthesis of Vitamin A Ester by Liver Microsomes, J. Biol. Chem. 239 (1964) 4077–4080.
[54] M.W. Schäffer, S.S. Roy, S. Mukherjee, D. Nohr, M. Wolter, H.K. Biesalski, D.E. Ong, S.K. Das, Qualitative and quantitative analysis of retinol, retinyl esters, tocopherols and selected carotenoids out of various internal organs form different species by HPLC, Anal. Methods Adv. Methods Appl. 2 (2010) 1320–1332. doi:10.1039/c0ay00288g.
[55] C. Pirazzi, L. Valenti, B.M. Motta, P. Pingitore, K. Hedfalk, R.M. Mancina, M.A. Burza, C. Indiveri, Y. Ferro, T. Montalcini, C. Maglio, P. Dongiovanni, S. Fargion, R. Rametta, A. Pujia, L. Andersson, S. Ghosal, M. Levin, O. Wiklund, M. Iacovino, J. Borén, S. Romeo, PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells, Hum. Mol. Genet. 23 (2014) 4077–4085. doi:10.1093/hmg/ddu121.
[56] G. Aragonès, T. Auguet, S. Armengol, A. Berlanga, E. Guiu-Jurado, C. Aguilar, S. Martínez, F. Sabench, J.A. Porras, M.D. Ruiz, M. Hernández, J.J. Sirvent, D. Del Castillo, C. Richart, PNPLA3 Expression Is Related to Liver Steatosis in Morbidly Obese Women with Non-Alcoholic Fatty Liver Disease, Int. J. Mol. Sci. 17 (2016). doi:10.3390/ijms17050630.
[57] P.R. Sundaresan, S.M. Kaup, P.W. Wiesenfeld, S.J. Chirtel, S.C. Hight, J.I. Rader, Interactions in indices of vitamin A, zinc and copper status when these nutrients are fed to rats at adequate and increased levels, Br. J. Nutr. 75 (1996) 915–928.
[58] I. Johansson, G. Hallmans, A. Wikman, C. Biessy, E. Riboli, R. Kaaks, Validation and calibration of food-frequency questionnaire measurements in the Northern Sweden Health and Disease cohort, Public Health Nutr. 5 (2002) 487–496. doi:10.1079/PHNPHN2001315.
[59] C.J. Cifelli, A.C. Ross, Chronic vitamin A status and acute repletion with retinyl palmitate are determinants of the distribution and catabolism of all-trans-retinoic acid in rats, J. Nutr. 137 (2007) 63–70.
[60] A. Olivares, A. Daza, A.I. Rey, C.J. López-Bote, Dietary vitamin A concentration alters fatty acid composition in pigs, Meat Sci. 81 (2009) 295–299. doi:10.1016/j.meatsci.2008.07.029.
[61] D. Yang, M.G. Vuckovic, C.P. Smullin, M. Kim, C. Pui-See Lo, E. Devericks, H.S. Yoo, M. Tintcheva, Y. Deng, J.L. Napoli, Modest Decreases in Endogenous All-Trans-Retinoic Acid Produced by a MouseRdh10Heterozygote Provoke Major Abnormalities in Adipogenesis and Lipid Metabolism, Diabetes. (2018). doi:10.2337/db17-0946.
[62] A. Saeed, R.P.F. Dullaart, T.C.M.A. Schreuder, H. Blokzijl, K.N. Faber, Disturbed Vitamin A Metabolism in Non-Alcoholic Fatty Liver Disease (NAFLD), Nutrients. 10 (2017). doi:10.3390/nu10010029.

Impaired hepatic vitamin A metabolism in NAFLD
172
Supplementary Figure 1: A high-fat diet (HFD) leads to impaired hepatic vitamin A metabolism in mice. Mice were fed a chow diet or HFD diet for 12 weeks and analyzed for hepatic levels of retinol hepatic levels of retinyl palmitate. Hepatic retinol levels were strongly reduced in HFD-fed mice as compared to control mice. However, hepatic retinyl palmitate levels did change in both groups.
Supplementary Figure 2: Comparison n-hexane and ACN extraction in the mice liver. Effect of two different methods of vitamin A extraction was analyzed with two different extraction methods 1) n-hexane (n-hex) (used in this study or 2) acetonitrile method (ACN) as previously described (Trasino et al. 2015. [11]) from same mice livers fed a chow diet or HFC diet for 20 weeks (n=3). Remarkably, both methods extracted a similar amount of retinol, but a significant less fraction of retinyl esters were extracted with ACN method.

Chapter 5
173
Supplementary Table S1: Primers and probes used in this study.
Gene / ID Taqman primers and probe
36B4 NM_022402
Fwd: 5'-GCTTCATTGTGGGAGCAGACA-3' Rev: 5'-CATGGTGTTCTTGCCCATCAG-3' Probe: 5'-TCCAAGCAGATGCAGCAGATCCGC-3'
Acaca / Acc1 NM_133360.1 / NM_022193.1
Fwd: 5'-GCCATTGGTATTGGGGCTTAC-3' Rev: 5'-CCCGACCAAGGACTTTGTTG-3' Probe: 5'-CTCAACCTGGATGGTTCTTTGTCCCAGC-3'
Act2 / α-Sma NM_007392
Fwd: 5'-TTCGTGTGGCCCCTGAAG-3' Rev: 5'-GGACAGCACAGCCTGAATAGC-3' Probe: 5'-TTGAGACCTTCAATGTCCCCGCCA-3'
Cd36 BC010262 / NM_031561
Fwd: 5'-GATCGGAACTGTGGGCTCAT-3' Rev: 5'-GGTTCCTTCTTCAAGGACAACTTC-3' Probe: 5'-AGAATGCCTCCAAACACAGCCAGGAC-3'
Cd68 NM_009853
Fwd: 5'-CACTTCGGGCCATGTTTCTC-3' Rev: 5'-AGGACCAGGCCAATGATGAG-3' Probe: 5'-CAACCGTGACCAGTCCCTCTTGCTG-3'
Ccl2 NM_031530.1
Fwd: 5'-TGTCTCAGCCAGATGCAGTTAAT-3' Rev: 5'-CCGACTCATTGGGATCATCTT-3' Probe: 5'-CCCCACTCACCTGCTGCTACTCATTCA-3'
Col1a1 NM_007742
Fwd: 5'-TGGTGAACGTGGTGTACAAGGT-3' Rev: 5'-CAGTATCACCCTTGGCACCAT-3' Probe: 5'-TCCTGCTGGTCCCCGAGGAAACA-3'
Cpt1a NM_013495.1
Fwd: 5'-CTCAGTGGGAGCGACTCTTCA-3' Rev: 5'-GGCCTCTGTGGTACACGACAA-3' Probe: 5'-CCTGGGGAGGAGACAGACACCATCCAAC-3'
Mlxipl / Chrebp NM_021455.3 / NM_133552.1
Fwd: 5'-GATGGTGCGAACAGCTCTTCT-3' Rev: 5'-CTGGGCTGTGTCATGGTGAA-3' Probe: 5'-CCAGGCTCCTCCTCGGAGCCC-3'
Cyp26a1 NM_007811.1
Fwd: 5'-GGAGACCCTGCGATTGAATC-3' Rev: 5'-GATCTGGTATCCATTCAGCTCAAA-3' Probe: 5'-TCTTCAGAGCAACCCGAAACCCTCC-3'
Dgat1 NM_010046.2 / NM_053437.1
Fwd: 5'-GGTGCCCTGACAGAGCAGAT-3' Rev: 5'-CAGTAAGGCCACAGCTGCTG-3' Probe: 5'-CTGCTGCTACATGTGGTTAACCTGGCCA-3'
Dgat2 NM_026384.2 / NM_001012345.1
Fwd: 5'-GGGTCCAGAAGAAGTTCCAGAAG-3' Rev: 5'-CCCAGGTGTCAGAGGAGAAGAG-3' Probe: 5'-CCCCTGCATCTTCCATGGCCG-3'
Fasn NM_007988 / NM_017332
Fwd: 5'-GGCATCATTGGGCACTCCTT-3' Rev: 5'-GCTGCAAGCACAGCCTCTCT-3' Probe: 5'-CCATCTGCATAGCCACAGGCAACCTC-3'
Fgf21 NM_020013.4 / NM_130752.1
Fwd: 5'-CCGCAGTCCAGAAAGTCTCC-3' Rev: 5'-TGACACCCAGGATTTGAATGAC-3' Probe: 5'-CCTGGCTTCAAGGCTTTGAGCTCC A-3'
Hsd17b13 NM_198030.2 / NM_001163486.1
Fwd: 5'- AAAGCAGAAAAGCAGACTGGTTCT-3' Rev: 5'- CCCCAGTTTCCTGCATTTGT-3' Probe: 5'-CGGTTTCCTCAACACCACGCTTATTGA-3'
Lipe / HSL NM_010719 / X51415
Fwd: 5'-GAGGCCTTTGAGATGCCACT-3' Rev: 5'-AGATGAGCCTGGCTAGCACAG-3' Probe: 5'-CCATCTCACCTCCCTTGGCACACAC-3'
IL-1β NM_008361
Fwd: 5'-ACCCTGCAGCTGGAGAGTGT-3' Rev: 5'-TTGACTTCTATCTTGTTGAAGACAAACC-3' Probe: 5'-CCCAAGCAATACCCAAAGAAGAAGATGGAA -3'
IL-6 NM_031168
Fwd: 5'-CCGGAGAGGAGACTTCACAGA-3' Rev: 5'-AGAATTGCCATTGCACAACTCTT-3'

Impaired hepatic vitamin A metabolism in NAFLD
174
Probe: 5'-ACCACTTCACAAGTCGGAGGCTTAATTACA-3'
iNos / Nos2 AF049656 / NM_010927 / NM_012611
Fwd: 5'- CTATCTCCATTCTACTACTACCAGATCGA-3' Rev: 5'- CCTGGGCCTCAGCTTCTCAT-3' Probe: 5'- CCCTGGAAGACCCACATCTGGCAG-3'
Ldlr NM_010700 / NM_175762
Fwd: 5'-GCATCAGCTTGGACAAGGTGT-3' Rev: 5'-GGGAACAGCCACCATTGTTG-3' Probe: 5'-CACTCCTTGATGGGCTCATCCGACC-3'
Lpl NM_008509 / NM_012598
Fwd: 5'-AAGGTCAGAGCCAAGAGAAGCA-3' Rev: 5'-CCAGAAAAGTGAATCTTGACTTGGT-3' Probe: 5'-CCTGAAGACTCGCTCTCAGATGCCCTACA-3'
Lrat NM_023624
Fwd: 5'-TCCATACAGCCTACTGTGGAACA-3' Rev: 5'-CTTCACGGTGTCATAGAACTTCTCA-3' Probe: 5'-ACTGCAGATATGGCTCTCGGATCAGTCC-3'
Pck1 NM_011044 / NM_198780
Fwd: 5'-GTGTCATCCGCAAGCTGAAG-3' Rev: 5'-CTTTCGATCCTGGCCACATC-3' Probe: 5'-CAACTGTTGGCTGGCTCTCACTGACCC-3'
Ppargc1 α / Pgc1α NM_008904 / NM_031347
Fwd: 5'-GACCCCAGAGTCACCAAATGA-3' Rev: 5'-GGCCTGCAGTTCCAGAGAGT-3' Probe: 5'-CCCCATTTGAGAACAAGACTATTGAGCGAACC-3'
Pnpla2 / Atgl NM_025802 / XM_347183
Fwd: 5'-AGCATCTGCCAGTATCTGGTGAT-3' Rev: 5'-CACCTGCTCAGACAGTCTGGAA-3' Probe: 5'-ATGGTCACCCAATTTCCTCTTGGCCC-3'
Pnpla3 NM_054088
Fwd: 5'-ATCATGCTGCCCTGCAGTCT-3' Rev: 5'-GCCACTGGATATCATCCTGGAT-3' Probe: 5'-CACCAGCCTGTGGACTGCAGCG-3'
RAR-β Assay on demand, Mm01319677_m1 (ThermoFisher)
Raldh1 Assay on demand, Mm00657317_m1 (ThermoFisher)
Raldh2 Assay on demand, Mm00501306_m1 (ThermoFisher)
Raldh3 Assay on demand, Mm00474049_m1 (ThermoFisher)
Raldh4 NM_178713.4
Fwd: 5'-TGGAGCAGTCTCTGGAGGAGTT-3' Rev: 5'- GAAGTTCAGAACAGACCGAGGAA-3' Probe: 5'- AATCTAAAGACCAAGGGAAAACCCTCACGC-3'
Rbp4 NM_011255.2 / XM_215285.3
Fwd: 5'-GGTGGGCACTTTCACAGACA-3' Rev: 5'-GATCCAGTGGTCATCGTTTCCT-3' Probe: 5'-CCCCAGTACTTCATCTTGAACTTGGCAGG-3'
Scd1 NM_009127.2
Fwd: 5'-ATGCTCCAAGAGATCTCCAGTTCT-3' Rev: 5'-CTTCACCTTCTCTCGTTCATTTCC-3' Probe: 5'-CCACCACCACCATCACTGCACCTC-3'
TGF-β1 NM_021578.1
Fwd: 5'-GGGCTACCATGCCAACTTCTG-3' Rev: 5'-GAGGGCAAGGACCTTGCTGTA-3' Probe: 5'-CCTGCCCCTACATTTGGAGCCTGGA-3'
TGF-β1 NM_021578.1
Fwd: 5'-GGGCTACCATGCCAACTTCTG-3' Rev: 5'-GAGGGCAAGGACCTTGCTGTA-3' Probe: 5'-CCTGCCCCTACATTTGGAGCCTGGA-3'
Timp1 NM_001044384.1 / NM_011593.2
Fwd: 5'-TCTGAGCCCTGCTCAGCAA-3' Rev: 5'-AACAGGGAAACACTGTGCACAC-3' Probe: 5'-CCACAGCCAGCACTATAGGTCTTTGAGAAAGC-3'
TNF-α NM_013693 / NM_012675
Fwd: 5'- GTAGCCCACGTCGTAGCAAAC-3' Rev: 5'- AGTTGGTTGTCTTTGAGATCCATG-3' Probe: 5'- CGCTGGCTCAGCCACTCCAGC-3'
Ucp2 NM_011671.2
Fwd: 5'-CGAAGCCTACAAGACCATTGC-3' Rev: 5'-ACCAGCTCAGCACAGTTGACA-3' Probe: 5'-CAGAGGCCCCGGATCCCTTCC-3'

Chapter 6
Glycogen storage disease type 1a
is associated with disturbed
vitamin A metabolism and elevated
serum retinol levels.
Ali Saeed1,2$, Joanne A. Hoogerland3, Janette Heegsma1,4,
Terry G.J. Derks5, Eveline van der Veer4, Gilles Mithieux6,7,8,
Fabienne Rajas6,7,8, Maaike H. Oosterveer3* and Klaas Nico
Faber1,4*$
1Department of Gastroenterology and Hepatology, 3Pediatrics and 4Laboratory
Medicine, 5Section of Metabolic Diseases, Beatrix Children's Hospital, Center for
Liver Digestive, and Metabolic Diseases, University Medical Center Groningen,
University of Groningen, Groningen, The Netherlands. 6Institut National de la Santé
et de la Recherche Médicale, U1213, Lyon, F-69008, 7Universite de Lyon, Lyon, F-
69008 and 8Université Lyon 1, Villeurbanne, F-69622, France. 2Institute of Molecular
Biology and Biotechnology, Bahauddin Zakariya University Multan, Pakistan.
(Submitted)

Impaired vitamin A metabolism in GSD Ia
176
ABSTRACT
Objective: Glycogen storage disease type 1a (GSD Ia or von Gierke disease) is an
inborn error of metabolism caused by mutations in the G6PC gene, encoding the
catalytic subunit of glucose-6-phosphatase. Early symptoms include severe fasting
intolerance, failure to thrive and hepatomegaly, biochemically associated with
nonketotic hypoglycemia, fasting hyperlactidemia, hyperuricemia and hyperlipidemia.
Dietary management is the cornerstone of treatment aiming at maintaining
euglycemia, prevention of secondary metabolic perturbations and long-term
complications, including liver (hepatocellular adenomas and carcinomas), kidney and
bone disease (hypovitaminosis D and osteoporosis).
Methods: As impaired vitamin A homeostasis also associates with similar symptoms
and is coordinated by the liver, we analysed vitamin A metabolism in GSD Ia patients
and liver-specific glucose-6-phosphatase (L-G6pc-/-) knockout mice.
Results: Serum levels of retinol and retinol binding protein 4 (RBP4) were
significantly increased in both GSD Ia patients and L-G6pc-/- mice. In contrast,
hepatic retinol levels were significantly reduced in L-G6pc-/- mice, while hepatic retinyl
palmitate (vitamin A storage form) and RBP4 levels were not altered. Transcript and
protein analyses indicate an enhanced production of retinol and reduced conversion
the retinoic acids (unchanged LRAT, Pnpla2/Atgl and Pnpla3 up, Cyp26a1 down) in
L-G6pc-/- mice. Aberrant expression of genes involved in vitamin A metabolism was
associated with reduced basal mRNA levels of markers of inflammation (Cd68, Tnfα,
Nos2, Il-6) and fibrosis (Col1a1, Acta2, Tgfβ, Timp1) in livers of L-G6pc-/- mice.
Conclusion: In conclusion, GSD Ia is associated with elevated serum retinol and
RBP4 levels, which may contribute to disease symptoms, including osteoporosis and
hepatic steatosis.

Chapter 6
177
6.1. INTRODUCTION
Glycogen storage disease type 1a (GSD Ia) is an autosomal recessive inherited
disorder of carbohydrate metabolism. Mutations in G6PC, encoding the catalytic
subunit of glucose-6-phosphatase (G6PC), limit the production of glucose from
glucose-6-phosphate (G6P) leading to hepatic glycogen accumulation and life-
threatening hypoglycemia in times of inadequate dietary carbohydrate intake. In
addition, GSD Ia is associated with hepatic steatosis, hyperlipidemia,
hyperlactacidaemia, hepatocellular tumor formation and intestinal and renal
impairments [1]. Untreated GSD Ia patients display a protruding abdomen,
hepatomegaly, wasted muscles, a bleeding tendency, truncal obesity, a rounded doll
face and short stature. No cure is available yet and prevention of hypoglycemia and
related metabolic dysfunctions are the main goals of dietary management and control
of GSD Ia [2]. Still, numerous additional nutritional and metabolic concerns are
associated with GSD Ia, including a frequently detected deficiency in vitamin D.
Suboptimal levels of serum 25-hydroxyvitamin-D (<30 ng/mL) are observed in most
patients, even under supplementation of vitamin D and calcium [3]. Restrictive dietary
plans, intestinal malabsorption, poor compliance to dietary plans and metabolic
derangements may cause hypovitaminosis D in GSD Ia patients [3].
Hypomagnesaemia, hypercalciuria and low tubular resorption of phosphate, along
with vitamin D deficiency may reduce bone mineral content and matrix formation in
GSD Ia and increase the risk of bone fractures and osteoporosis [4]. Besides vitamin
D, very limited information is available about other potential vitamins deficiencies in
GSD Ia. Vitamin A may be particularly relevant for GSD Ia as these patients develop
significant hepatic pathologies, such as hepatic steatosis, hyperlipidemia and
adenomas that may affect the liver’s role in regulating vitamin A homeostasis.
Vitamin A is an essential fat-soluble vitamin and approximately ~80% of the total
vitamin A pool is stored as retinyl esters, mainly retinyl palmitate, in the liver. White
adipose tissue (WAT) contains the second-largest pool of vitamin A (10-20%).
Adequate hepatic storage is required to maintain plasma retinol levels around 2
µmol/L in healthy humans (1-1.5 µmol/L in mice) [5]. Vitamin A plays important
physiological roles in vision, reproduction, growth, development, immunity and
metabolic programs [6]. Impaired triglyceride and/or cholesterol metabolism often
associates with impaired vitamin A metabolism and homeostasis [7,8]. Indeed,
reduced serum retinol levels are associated with hepatic steatosis [9,10],

Impaired vitamin A metabolism in GSD Ia
178
hypertriglyceridemia, glucose intolerance, insulin resistance and obesity [11,12]. On
the other hand, excess of vitamin A-metabolites may also cause hyperlipidemia by
modulating hepatic triglyceride synthesis and very-low density lipoprotein (VLDL)
production [13,14]. As steatosis and hypertriglyceridemia are prevalent metabolic
symptoms in GSD Ia patients [1], it is relevant to determine whether this also
associates with abnormal circulating vitamin A levels, as this may affect immune
regulation, tissue differentiation and metabolic pathways in these patients.
Indeed, we detected abnormal circulating retinol levels in GSD Ia patients, but rather
unexpectedly, they appeared elevated as compared to age- and sex-matched healthy
controls. Similar observations were made in transgenic mice that were studied 10
days after a hepatocyte-specific deletion of the G6pc gene (L-G6pc-/-). Our study
reveals that GSD Ia is characterized by hypervitaminosis A, which may contribute to
the pathology of this disease.
6.2. MATERIALS AND METHODS
6.2.1. Patients
The study was performed in accordance with the Declaration of Helsinki and the
institutional rules for studying biological rest materials. Retinol analysis was
performed in serum samples from 22 genetically confirmed GSD Ia patients (male
n=9 and female n=13), who visited the Beatrix Childrens’ Hospital, UMCG. Samples
were randomly obtained during the day. The controls included 20 healthy, age- and
sex-matched control subjects (male n=8 and female n=12) aged between 10 and 45
years.
6.2.2. Animal model
The tamoxifen-inducible hepatocyte-specific G6pc-knockout (L-G6pc-/-) mice were
used in this study as a model of the liver-specific pathologies of GSD Ia [15]. Briefly,
G6pc recombinant mice with two loxP sites flank G6pc exon 3 (B.G6pclox/w) were
crossed with transgenic mice expressing the tamoxifen-inducible recombinase
(CREERT2) under control of the serum albumin promoter to confer hepatocyte-specific
expression in B6.SAcreERT2/w mutant mice. Male B6.G6pcex3lox/ex3lox.SACreERT2/+ mice (8-
12 weeks old) were injected intraperitoneally once daily with 100 µL tamoxifen (10
mg/ml, Sigma–Aldrich) for five consecutive days to obtain L-G6pc-/- mice. All mice

Chapter 6
179
were sacrificed 10 days after the last tamoxifen injection. Animal experiments were
performed after approval of all procedures by the Institutional Animal Care and Use
Committee, University of Groningen, the Netherlands. All animals (n=6-7) were kept
in an environment with alternating dark and light cycles (07:00 PM-07:00 AM), with
controlled temperature (20-24 ºC) and relative humidity (55% ± 15%) and ad libitum
access to food and water. Prior to sacrifice, the mice were fasted from 10:00 PM until
08:00 AM the next day. Tissue and plasma samples were collected for further
analysis.
6.2.3. Cholesterol and triglyceride analysis in liver and plasma
A colorimetric assay was used to determine triglyceride, free or total cholesterol by
using commercial kits (Roche Diagnostics and Wako Chemicals, USA) after lipid
extraction according to the Bligh and Dyer method [16].
6.2.4. Vitamin A analysis
Serum and tissue vitamin A content was analyzed by reverse phase HPLC as
previously described [17]. Retinol and retinyl esters were extracted and deproteinized
twice with n-hexane from tissue and serum with retinol acetate as an internal
standard. Samples were diluted in 200 µL ethanol and 50 µL was injected for phase
separation (150 x 3.0 mm of 5 µM column) and/or measurements (UV-VIS, dual
wave length, UV-4075 Jasco, Tokyo, Japan) by HPLC.
6.2.5. Histology
Liver histology (Hematoxylin & Eosin stain and Oil Red O) was performed on paraffin
sections and/or snap-frozen liver sections as previously described [18].
6.2.6. Quantitative real-time reverse transcription polymerase chain
reaction (qRT-PCR)
Quantitative real-time reverse transcription polymerase chain reaction was performed
as previously described [19]. Shortly, total RNA was isolated from tissue samples
using TRIzol® reagent according to supplier’s instruction (ThermoFisher, Scientific,
The Netherlands). RNA quality and quantity were determined using a Nanodrop
2000c UV-vis spectrophotometer (ThermoFisher Scientific, The Netherlands). cDNA
was synthesized from 2.5 µg RNA using random nanomers and M-MLV reverse
transcriptase (Invitrogen, USA). Taqman primers and probes were designed on

Impaired vitamin A metabolism in GSD Ia
180
Primer Express 3.0.1 and are shown in Supplementary Table S1. All target genes
were amplified using the QPCR core kit master mix (Eurogentec, The Netherlands)
on a 7900HT Fast Real-Time PCR system (Applied Biosystems Europe, The
Netherlands). SDSV2.4.1 (Applied Biosystems Europe, The Netherlands) software
was used to analyze the data. Expression of genes is presented in 2-delta CT and
normalized to 36B4.
6.2.7. Western blot analysis
Protein samples were prepared for Western blot analysis as described previously
[20]. Protein concentrations were quantified using the Bio-Rad protein assay (Bio-
Rad, Hercules, CA, USA) with bovine serum albumin (BSA) as a standard. Equal
amounts of protein (5-20 µg) were separated on Mini-PROTEAN® TGX™ precast 4-
15% gradient gels (Bio-Rad, Hercules, CA, USA) and transferred to nitrocellulose
membranes using the Trans-Blot Turbo transfer system, (Bio-Rad, Hercules, CA,
USA). Primary antibodies (anti-RBP4, 1:2,000; #ab109193, Abcam, Cambridge, UK,
anti-β-ACTIN, 1:1000 #4979, Cell Signaling, Leiden, The Netherlands and anti-
GAPDH, 1:40,000 #CB1001, Calbiochem, Merck-Millipore Amsterdam-Zuidoost, The
Netherland and anti-LRAT, 1:1,000; with horseradish peroxidase (HRP)-conjugated
appropriate combination of secondary antibody (1:2,000; P0448, DAKO) were used
for detection. Proteins were detected using the Pierce ECL Western blotting kit
(ThermoFisher Scientific). Images were captured using the chemidoc XRS system
and Image Lab version 3.0, (Bio-Rad, Hercules, CA, USA). The intensity of bands
was quantified using ImageJ version 1.51 (NIH, USA).
6.2.8. Statistical analysis
Data is presented as Mean ± SEM and statistical analysis was performed using the
GraphPad Prism 7 software package (GraphPad Software, San Diego, CA, USA).
Statistical significance was determined by the Mann-Whitney test. P-values ≤0.05*,
≤0.01**, ≤0.001*** were considered significant.

Chapter 6
181
6.3. RESULTS
6.3.1. GSD Ia patients and L-G6pc-/- mice have elevated serum retinol
levels
Serum retinol levels were determined in 22 GSD Ia patients and 20 age- and sex-
matched healthy controls (age range 10-45 years) (Figure 1A). Circulating retinol
levels were significantly increased in GSD Ia patients as compared to the healthy
controls (2.96 ± 0.22 versus 2.31 ± 0.12 µM, respectively). No correlation was
observed between serum retinol levels and sex or age of the patients and controls
(Figure 1B). We next aimed to analyze whether the elevated serum retinol levels are
also observed in a mouse model of GSD Ia (L-G6pc-/- mice) [15], and if so, what
molecular mechanisms may be involved. The induction of the GSD Ia phenotype in L-
G6pc-/- mice was supported by: 1) increased liver weight (+47%); 2) elevated hepatic
triglyceride levels (both concentration (+56%) and total pool (+120%), 3) minor
effects on free cholesterol levels, but an increase in total hepatic pool of total
cholesterol; 4) fasting hypoglycemia, when compared to control mice
(Supplementary Figure S1A). Excessive lipid accumulation in the L-G6pc-/- mice
was also confirmed by H&E and ORO staining of the liver tissue (Supplementary
Figure S1B). Similar to GSD Ia patients, plasma retinol levels were significantly
increased in L-G6pc-/- mice (2.33 0.18 µM) as compared to control mice (1.23
0.08 µM) (Figure 2A).
Figure 1. Serum retinol levels are increased in GSD Ia patients. Serum retinol levels were determined in GSD Ia patients (n=22) and age- and sex-matched controls (n=20) (A). No correlation was observed between age and sex (male- Δ or Female- O) in GSD Ia patients (red symbols) nor in healthy controls (black symbols) (B).

Impaired vitamin A metabolism in GSD Ia
182
6.3.2. G6pc deficiency reduces hepatic retinol levels, while retinyl palmitate
is unchanged.
In contrast, hepatic retinol concentrations (6.59 0.66 versus 11.57 0.72 µg/g liver)
as well as the total hepatic pool of retinol were significantly lower in L-G6pc-/- mice
compared to control mice (Figure 2B and C). White adipose tissue (WAT) is a
second storage site of vitamin A and retinol levels were similarly reduced in WAT of
L-G6pc-/- mice as compared to control mice (0.38 0.02 versus 0.54 0.02 µg/g
WAT, respectively) (Figure 2D).
On the other hand, hepatic retinyl palmitate levels were not different between L-G6pc-
/- mice and control mice, not in concentration and not in the total pool (Figure 2E and
F) and the same was observed for retinyl palmitate concentrations in WAT (Figure
2G). These results indicate that hepatic vitamin A metabolism is affected in the
absence of G6pc, leading to increased levels of circulating retinol and reduced levels
in liver and WAT, while hepatic vitamin A storage is maintained.
Figure 2. G6pc deficiency in mice increases plasma retinol and reduces hepatic retinol, while retinyl palmitate remains unchanged. Ten (10) days after tamoxifen-induced deletion of the G6pc gene in hepatocytes, L-G6pc-/- and control mice were sacrificed and analyzed for A) plasma retinol levels, B) liver retinol concentrations, C) total liver retinol pool, D) retinol concentrations in white adipose tissue (WAT), E) liver retinyl palmitate concentrations, F) total liver retinyl palmitate pool; G) retinyl palmitate concentrations in WAT.

Chapter 6
183
6.3.3. Low expression of vitamin A storing enzymes, while higher
expression of vitamin A hydrolyzing enzymes in L-G6pc-/- fasted-
mouse liver
Hepatic retinol and retinyl ester levels are a resultant of enzymes that catalyze the
esterification of retinol (predominantly LRAT and to a lesser extent by DGAT1), 2)
enzymes that hydrolyze retinyl esters (ATGL/PNPLA2 and PNPLA3) and 3) enzymes
that convert retinol to retinoic acids (ADH and RALDH). Moreover, retinol promotes
its own release from the liver to the circulation by binding to retinol binding protein 4
(RBP4) in hepatocytes. Hepatic mRNA levels of Lrat were strongly reduced in L-
G6pc-/- mice as compared to control mice (Figure 3A). However, LRAT protein levels
appeared not different between L-G6pc-/- and control mice (Figure 3B). Moreover,
hepatic mRNA levels of alternative enzymes involved in retinol esterification (Dgat1
and Dgat2) were elevated in L-G6pc-/- mice (Figure 3A). DGAT1 and 2 are key
enzymes in triglyceride synthesis and, indeed, hepatic triglyceride accumulation was
observed in L-G6pc-/- mice (Supplementary Figure S1). Hepatic mRNA levels of
both Pnpla2 and Pnpla3 were strongly induced in L-G6pc-/- mice as compared to
control mice, which may contribute in hepatic retinol production (Figure 3C). Hepatic
mRNA levels of Rbp4 were similar in control and L-G6pc-/- mice (Figure 3D). It is
important to note, however, that hepatic and serum RBP4 protein levels are primarily
regulated by the availability of retinol in the liver, where retinol binding promotes the
secretion of RBP4 from hepatocytes [21]. Conversely, the absence of retinol leads to
strong hepatic accumulation of RBP4 even at stable Rbp4 mRNA levels, as observed
in vitamin A-deficient mice (see supplementary Figure S2) and also observed in rat
[22]. Thus RBP4 protein levels were analyzed next. RBP4 protein levels in livers and
WAT of L-G6pc-/- mice were similar to control mice (Figure 4A and B, notably,
hepatic RBP4 sometimes appears as a double band in western blot analyses, as
observed by others [23–25], but with unknown cause). In contrast, serum RBP4
levels in L-G6pc-/- mice were clearly elevated up to 3-fold compared to control mice
(Figure 4C). Sera of GSD Ia patients also contained significantly elevated levels of
RBP4 compared to age- and sex-matched healthy control (Figure 4D). Serum retinol
levels were largely in line with serum RBP4 levels in healthy controls, while such
association was less evident in GSD Ia patients (Figure 4D).

Impaired vitamin A metabolism in GSD Ia
184
Figure 3. Hepatic expression of genes involved in vitamin A storage, hydrolysis and export in L-G6pc-/- fasted-mice. Ten (10) days after tamoxifen-induced deletion of the G6pc gene in hepatocytes, L-G6pc-/- and control mice were sacrificed and analyzed by Q-PCR (A, C and D) and Western blot analysis (B) for hepatic expression of genes/proteins involved in A) retinyl ester formation (Lrat, Dgat1, and Dagt2), B) protein levels of LRAT and β-ACTIN (loading control), C) retinol synthesis (Pnpla2/Agtl and Pnpla3) and D) retinol export from the liver (Rbp4). Transcript analyses suggest that the balance between vitamin A storage/retinol synthesis in the liver shifts to retinol synthesis in L-G6pc-/- mice.

Chapter 6
185
Figure 4. Serum RBP4 levels are elevated in L-G6pc-/- mice and GSD Ia patients. A-C) Ten (10) days after tamoxifen-induced deletion of the G6pc gene in hepatocytes, L-G6pc-/- and control mice were sacrificed and analyzed by Western blotting for RBP4 protein levels in A) liver, B) WAT and C) plasma. D) Similarly, RBP4 protein levels were analyzed in sera of age- and sex-matched healthy controls and GSD Ia patients. β-ACTIN and Ponceau S stainings are included as loading controls. Protein signal intensities were quantified and are shown to the right.
6.3.4. G6pc deficiency suppresses expression of retinoic acid-responsive
Cyp26a1
Next, we aimed to analyze whether G6pc deficiency may affect the production of
retinoic acids in the liver. Hepatic mRNA levels of Hsd17b13, a recently identified
retinol dehydrogenase [26] and all 4 retinaldehyde dehydrogenases (Raldh1-4) were
hardly affected by the absence of GSD Ia in mice (Figure 5). Only a small significant
increase in Raldh2 and decrease in Raldh4 were observed in G6pc deficient mice.

Impaired vitamin A metabolism in GSD Ia
186
However, mRNA levels of the highly retinoic acid-sensitive Cyp26a1 were strongly
(89%) decreased (Figure 5) compared to control mice.
Figure 5. Gene expression of retinoic acid-responsive Cyp26a1 is strongly suppressed in the livers of L-G6pc-/- mice. Ten (10) days after tamoxifen-induced deletion of the G6pc gene in hepatocytes, L-G6pc-/- and control mice were sacrificed and analyzed by Q-PCR for hepatic mRNA levels of genes involved in the conversion of retinol to retinoic acids (Hsd1713, Raldh1, Raldh2, Raldh3, Raldh4) or catabolism of retinoic acids (Cyp26a1). Transcriptional regulation of Cyp26a1 is highly responsive to retinoic acids.
6.3.5. Liver-specific G6pc deficiency in mice does not cause hepatic
inflammation, nor fibrosis.
Finally, we analyzed whether the abnormal vitamin A metabolism in L-G6pc-/- mice
leads to hepatic inflammation and/or fibrosis. Hepatic mRNA levels of markers of
inflammation, e.g. Cd68, Tnfα, Nos2, Ccl2, Il6 were reduced in L-G6pc-/- mice as
compared to control mice (Figure 6A). A similar suppression of hepatic mRNA levels
of markers of fibrosis, e.g. Coll1a1, Acta2, Tgf-β and Timp1, was observed in L-G6pc-
/- mice as compared to control mice (Figure 6B).

Chapter 6
187
Figure 6. Hepatic G6pc-deficiency suppresses basal levels of inflammation and fibrosis in mice. Ten (10) days after tamoxifen-induced deletion of the G6pc gene in hepatocytes, L-G6pc-/- and
control mice were sacrificed and analyzed by Q-PCR for hepatic mRNA levels of markers of A)
inflammation (Cd68, Tnfα, Nos2, Ccl2 and Il6) or B) fibrosis (Col1a1, Acta2, Tgf- and Timp1).
Both markers of inflammatory and fibrosis were not increased in L-G6pc-/- livers. Instead, a
significant reduction was observed for hepatic expression of Cd68, Tnfα and Timp1 in L-G6pc-/-
mice compared to controls, while all other markers showed similar trends.
Taken together, our data show that G6pc deficiency leads to elevated serum retinol
and RBP4 levels in humans and in mice. In contrast, hepatic retinol levels are
reduced, most probably because of enhanced mobilization of retinol from retinyl ester
stores and subsequent RBP4-mediated release from hepatocytes.

Impaired vitamin A metabolism in GSD Ia
188
6.4. DISCUSSION
This study shows for the first time that G6pc deficiency in human and mouse is
associated with elevated levels of circulating retinol, concomitantly with an increase
of circulating RBP4 levels. On the contrary, retinol levels are reduced in the liver and
WAT. Tissue retinyl palmitate levels are not changed in liver-specific G6pc deficient
mice, at least not within 10 days after ablation of the gene in hepatocytes. Hepatic
expression profiling suggests that metabolism of retinyl ester to retinol that may
promote secretion of retinol-bound RBP4 to the circulation. A persistent increase in
circulatory retinol may contribute to symptoms of GSD Ia patients and aggravate
steatosis and osteoporosis.
Clinical management of GSD Ia is primarily aimed at maintaining steady circulating
glucose levels by strictly controlled intake of dietary carbohydrates during day and
night. Due to the impaired ability to produce glucose from glucose-6-phosphate
(G6P), cellular glycogen content increases, in conjugation with elevated triglyceride
storage leading to steatosis. GSD Ia is associated with hypovitaminosis D, which has
been linked to the development of osteoporosis in these patients [3,27]. As fatty liver
disease is associated with hypovitaminosis A [9,10], we were interested whether
GSD Ia patients may also show aberrant circulating vitamin A levels. To our surprise,
we found that circulating retinol revels, as well as RBP4 levels, were significantly
elevated, instead of being reduced, in GSD Ia patients and L-G6pc-/- mice. Apart from
cases of excessive dietary vitamin A intake, elevated circulating retinol levels are a
rare phenomenon. Impaired kidney function is described as a pathological condition
that leads to circulating hypervitaminosis A [28,29]. Moreover, idiopathic intracranial
hypertension (IIH) has been associated with elevated retinol levels in serum and
cerebrospinal fluid [30,31], although this is a controversial observation as circulating
retinol levels were not different in IIH patients compared to BMI-matched controls in
the recently reported “IIH Treatment Trial (IIHTT)” [32]. Still, vitamin A status is an
important factor affecting intracranial pressure as both high dietary vitamin A intake
as well as hypovitaminosis A may cause (benign) IH [33,34]. These conditions can be
reversed by normalizing vitamin A intake. Serum retinol levels are, however, not a
sensitive measure of sharp fluctuations in dietary intake of vitamin A as early work
showed that daily retinyl palmitate supplementation in a range of 0 to 36,000 IU
(equal to 0 to 12 times the RDA [Recommended Daily Allowance]) increased serum
retinol levels on average by only 2% (= 0.04 µM) per 10,000 IU vitamin A [35]. Even

Chapter 6
189
though GSD Ia patients are often prescribed multivitamin supplements, it is unlikely
to cause the increased circulating retinol levels in these patients. GSD Ia is
associated with impaired renal function [36], which may contribute to the elevated
circulating retinol levels. However, the liver-specific G6pc-/- mice also showed
markedly elevated retinol levels, while there is as yet no evidence that these animals
develop any kidney abnormalities, not even 15 months after ablation of the gene in
the liver [15]. Thus, also hepatic vitamin A metabolism may (primarily) contribute to
the elevated serum levels of retinol and RBP4 in GSD Ia appear. In contrast to blood,
tissue retinol levels in the liver and WAT were reduced in L-G6pc-/- mice compared to
controls, while retinyl palmitate, the main storage form of vitamin A in the liver, was
not changed. Indeed, protein levels of LRAT, the main hepatic enzyme catalyzing
esterification of retinol, were normal in L-G6pc-/- mice. Expression profiling revealed
an induction of retinyl ester-hydrolyzing activity (Pnpla2 and Pnpla3 up) and a
reduction of retinoic acid catabolism (Cyp26a1 down). De novo lipogenesis is
increased in G6PC deficiency and activation of the carbohydrate-response-element-
binding protein (ChREBP) likely contributes to this phenomenon [37,38]. Hepatic
Chrebp mRNA levels were indeed increased in L-G6pc-/- mice compared to controls
(Supplementary Figure S3). Notably, PNPLA3 expression is controlled by ChREBP
[39] and NAFLD patients carrying the PNPLA3-I148M variant show reduced
circulating retinol levels and enhanced hepatic retinyl palmitate contents [40,41].
Thus, ChREBP-mediated induction of Pnpla3, together with elevated levels of
Atgl/Pnpla2 may contribute to the enhanced conversion of hepatic retinyl esters to
retinol. The strong reduction in Cyp26a1 mRNA levels in L-G6pc-/- mice primarily
hints to reduced production of retinoic acids, as they are potent inducers of Cyp26a1
transcription (summarized in ref [5]. Enhanced hepatic retinyl ester-hydrolysis and
reduced retinoic acid catabolism is theoretically expected to lead to accumulation of
retinol. However, hepatic retinol levels were actually reduced, while an increase was
observed circulatory retinol and RBP4 in GDS Ia patients and L-G6pc-/- mice.
Enhanced retinol production in the liver promotes its own release from hepatocytes,
bound to RBP4, to the circulation and contributes to elevated plasma levels of retinol
and RBP4 [42–44]. Thus, we hypothesize that an enhanced production of retinol
pushes itself out of the liver contributes to the elevated serum levels of RBP4 and
retinol found in GSD Ia patients and L-G6pc-/- mice. The reduced hepatic retinol
levels may be an early response to the induced deletion of the G6pc gene in this

Impaired vitamin A metabolism in GSD Ia
190
mouse model (10 days gene deletion). Future studies may include L-G6pc mice after
long term gene deletion [15] to determine whether the low hepatic retinol levels
persist.
Chronically elevated retinol in circulation may contribute to clinical symptoms
associated with GSD Ia. Hypervitaminosis A promotes osteoclast formation, skeleton
fragility and osteoporosis due to decrease cortical bone mass and bone formation
[45]. Hypervitaminosis A-associated osteoporosis may already occur at twice the
recommended daily allowances (RDA) of vitamin A [45,46]. Osteoporosis is also
observed in GSD Ia patients and typically linked to hypovitaminosis D, which is
analyzed in routine surveillance [1,3]. Abnormal vitamin D and A levels may
synergize in aberrant bone homeostasis and may need to be monitored both to
prevent this complication in GSD Ia patients. In fact, there are quite a few additional
commonalities in symptoms in GSD Ia and hypervitaminosis A, like impaired growth,
dizziness and irritability [47–50]. Though these symptoms likely primarily result of
poorly controlled blood glucose levels, it could be that chronically elevated serum
retinol levels may also contributes to such symptoms. Hypervitaminosis A causes
hepatic steatosis in rats [51], while vitamin A deficiency reduces hepatic lipid
accumulation [52].
Increased circulating retinol has been found to reduce the risk for hepatocellular
carcinoma (HCC) [53,54]. GSD Ia patients are actually at risk for the development of
hepatic adenomas that may progress to HCC. The effects on retinol and tumor
development in GSD Ia therefore appear counterintuitive. However, hepatic retinol
levels are reduced and may promote adenoma development specifically in the liver.
Here also, it is of interest what the long-term effect is of the absence of hepatic G6PC
activity on vitamin A metabolism in the liver [15]. It may very well be that hepatic
vitamin A stores get depleted in the long-term and predispose to liver tumor
development in GSD Ia. One older patient indeed showed very low circulating retinol
levels (Figure 1B), which suggests extremely low hepatic vitamin A stores.
The hepatic pathologies and disturbed vitamin A metabolism did not induce an
inflammatory or fibrotic response in livers of L-G6pc-/- mice. In fact, all tested markers
for hepatic inflammation and fibrosis were suppressed to greater of lesser extent in L-
G6pc-/- mice. This may also be a result of changes in hepatic retinol metabolism as
vitamin A metabolites are potent controllers of hepatic inflammation and fibrosis
[55,56].

Chapter 6
191
Management of hypervitaminosis A is currently limited to controlling the dietary intake
of vitamin A. Given the “metabolic origin” of hypervitaminosis A in GSD Ia patients, it
is important to monitor circulating retinol levels and refrain from vitamin A
supplementation when plasma retinol levels are close to or above normal (~2 µM)
levels. Future studies need to establish the course of vitamin A levels in the absence
of G6PC activity in patients and/or mice in order to determine the necessity of
management of vitamin A levels in early and late stages of disease development.
Taken together, our study shows that vitamin A metabolism is disturbed in the
absence of G6PC activity in mice and GSD Ia patients, resulting in elevated
circulating retinol levels. This condition may contribute to various symptoms of GSD
Ia, in particular, osteoporosis, which has been linked to hypovitaminosis D in these
patients so far. Vitamin A is thus a second vitamin that needs attention in the
management of GSD Ia.
ACKNOWLEGEMENTS: The authors are thankful to Trijnie Bos, Brenda Hijmans
and Aycha Bleeker for providing the technical assistance in the execution of animal
experiments in mice (L-G6pc-/-).
Funding: MHO holds a Rosalind Franklin Fellowship from the University of
Groningen.

Impaired vitamin A metabolism in GSD Ia
192
REFERENCES
[1] D.S. Bali, Y.-T. Chen, S. Austin, J.L. Goldstein, Glycogen Storage Disease Type I, in: R.A. Pagon, M.P. Adam, H.H. Ardinger, S.E. Wallace, A. Amemiya, L.J. Bean, T.D. Bird, N. Ledbetter, H.C. Mefford, R.J. Smith, K. Stephens (Eds.), GeneReviews(®), University of Washington, Seattle, Seattle (WA), 1993. http://www.ncbi.nlm.nih.gov/books/NBK1312/ (accessed May 19, 2017).
[2] K.M. Ross, L.M. Brown, M.M. Corrado, T. Chengsupanimit, L.M. Curry, I.A. Ferrecchia, L.Y. Porras, J.T. Mathew, D.A. Weinstein, Safety and Efficacy of Chronic Extended Release Cornstarch Therapy for Glycogen Storage Disease Type I, JIMD Rep. 26 (2016) 85–90. doi:10.1007/8904_2015_488.
[3] S.G. Banugaria, S.L. Austin, A. Boney, T.J. Weber, P.S. Kishnani, Hypovitaminosis D in glycogen storage disease type I, Mol. Genet. Metab. 99 (2010) 434–437. doi:10.1016/j.ymgme.2009.12.012.
[4] J. Cabrera-Abreu, N.J. Crabtree, E. Elias, W. Fraser, R. Cramb, S. Alger, Bone mineral density and markers of bone turnover in patients with glycogen storage disease types I, III and IX, J. Inherit. Metab. Dis. 27 (2004) 1–9. doi:10.1023/B:BOLI.0000016632.13234.56.
[5] A. Saeed, M. Hoekstra, M.O. Hoeke, J. Heegsma, K.N. Faber, The interrelationship between bile acid and vitamin A homeostasis, Biochim. Biophys. Acta. 1862 (2017) 496–512. doi:10.1016/j.bbalip.2017.01.007.
[6] S. Bar-El Dadon, R. Reifen, Vitamin A and the epigenome, Crit. Rev. Food Sci. Nutr. 57 (2017) 2404–2411. doi:10.1080/10408398.2015.1060940.
[7] M. Zhang, C. Liu, M. Hu, J. Zhang, P. Xu, F. Li, Z. Zhong, L. Liu, X. Liu, High-fat diet enhanced retinal dehydrogenase activity, but suppressed retinol dehydrogenase activity in liver of rats, J. Pharmacol. Sci. 127 (2015) 430–438. doi:10.1016/j.jphs.2015.03.001.
[8] M.-J. Shin, S.-M. Kang, Y. Jang, J.H. Lee, J. Oh, J.H. Chung, N. Chung, Serum retinol binding protein 4 levels are associated with serum adiponectin levels in non-diabetic, non-obese subjects with hypercholesterolemia, Clin. Chim. Acta Int. J. Clin. Chem. 378 (2007) 227–229. doi:10.1016/j.cca.2006.11.026.
[9] J.I. Botella-Carretero, J.A. Balsa, C. Vázquez, R. Peromingo, M. Díaz-Enriquez, H.F. Escobar-Morreale, Retinol and alpha-tocopherol in morbid obesity and nonalcoholic fatty liver disease, Obes. Surg. 20 (2010) 69–76. doi:10.1007/s11695-008-9686-5.
[10] G. Villaça Chaves, S.E. Pereira, C.J. Saboya, A. Ramalho, Non-alcoholic fatty liver disease and its relationship with the nutritional status of vitamin A in individuals with class III obesity, Obes. Surg. 18 (2008) 378–385. doi:10.1007/s11695-007-9361-2.
[11] Y. Liu, H. Chen, D. Mu, J. Fan, J. Song, Y. Zhong, D. Li, M. Xia, Circulating Retinoic Acid Levels and the Development of Metabolic Syndrome, J. Clin. Endocrinol. Metab. 101 (2016) 1686–1692. doi:10.1210/jc.2015-4038.
[12] M.M. Godala, I. Materek-Kuśmierkiewicz, D. Moczulski, M. Rutkowski, F. Szatko, E. Gaszyńska, S. Tokarski, J. Kowalski, The risk of plasma vitamin A, C, E and D deficiency in patients with metabolic syndrome: a case-control study, Adv. Clin. Exp. Med. Off. Organ Wroclaw Med. Univ. (2017). doi:10.17219/acem/62453.
[13] M. Krupková, F. Liška, L. Šedová, D. Křenová, V. Křen, O. Šeda, Pharmacogenomic analysis of retinoic-acid induced dyslipidemia in congenic rat model, Lipids Health Dis. 13 (2014) 172. doi:10.1186/1476-511X-13-172.

Chapter 6
193
[14] N. Vu-Dac, P. Gervois, I.P. Torra, J.C. Fruchart, V. Kosykh, T. Kooistra, H.M. Princen, J. Dallongeville, B. Staels, Retinoids increase human apo C-III expression at the transcriptional level via the retinoid X receptor. Contribution to the hypertriglyceridemic action of retinoids, J. Clin. Invest. 102 (1998) 625–632. doi:10.1172/JCI1581.
[15] E. Mutel, A. Abdul-Wahed, N. Ramamonjisoa, A. Stefanutti, I. Houberdon, S. Cavassila, F. Pilleul, O. Beuf, A. Gautier-Stein, A. Penhoat, G. Mithieux, F. Rajas, Targeted deletion of liver glucose-6 phosphatase mimics glycogen storage disease type 1a including development of multiple adenomas, J. Hepatol. 54 (2011) 529–537. doi:10.1016/j.jhep.2010.08.014.
[16] E.G. Bligh, W.J. Dyer, A rapid method of total lipid extraction and purification, Can. J. Biochem. Physiol. 37 (1959) 911–917. doi:10.1139/o59-099.
[17] Y.-K. Kim, L. Quadro, Reverse-phase high-performance liquid chromatography (HPLC) analysis of retinol and retinyl esters in mouse serum and tissues, Methods Mol. Biol. Clifton NJ. 652 (2010) 263–275. doi:10.1007/978-1-60327-325-1_15.
[18] M. Aparicio-Vergara, P.P.H. Hommelberg, M. Schreurs, N. Gruben, R. Stienstra, R. Shiri-Sverdlov, N.J. Kloosterhuis, A. de Bruin, B. van de Sluis, D.P.Y. Koonen, M.H. Hofker, Tumor necrosis factor receptor 1 gain-of-function mutation aggravates nonalcoholic fatty liver disease but does not cause insulin resistance in a murine model, Hepatol. Baltim. Md. 57 (2013) 566–576. doi:10.1002/hep.26046.
[19] H. Blokzijl, S. Vander Borght, L.I.H. Bok, L. Libbrecht, M. Geuken, F.A.J. van den Heuvel, G. Dijkstra, T.A.D. Roskams, H. Moshage, P.L.M. Jansen, K.N. Faber, Decreased P-glycoprotein (P-gp/MDR1) expression in inflamed human intestinal epithelium is independent of PXR protein levels, Inflamm. Bowel Dis. 13 (2007) 710–720. doi:10.1002/ibd.20088.
[20] A. Pellicoro, F.A.J. van den Heuvel, M. Geuken, H. Moshage, P.L.M. Jansen, K.N. Faber, Human and rat bile acid-CoA:amino acid N-acyltransferase are liver-specific peroxisomal enzymes: implications for intracellular bile salt transport, Hepatol. Baltim. Md. 45 (2007) 340–348. doi:10.1002/hep.21528.
[21] M. Schina, J. Koskinas, D. Tiniakos, E. Hadziyannis, S. Savvas, B. Karamanos, E. Manesis, A. Archimandritis, Circulating and liver tissue levels of retinol-binding protein-4 in non-alcoholic fatty liver disease, Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 39 (2009) 972–978. doi:10.1111/j.1872-034X.2009.00534.x.
[22] Y. Muto, J.E. Smith, P.O. Milch, D.S. Goodman, Regulation of retinol-binding protein metabolism by vitamin A status in the rat, J. Biol. Chem. 247 (1972) 2542–2550.
[23] S.J. Thompson, A. Sargsyan, S.-A. Lee, J.J. Yuen, J. Cai, R. Smalling, N. Ghyselinck, M. Mark, W.S. Blaner, T.E. Graham, Hepatocytes Are the Principal Source of Circulating RBP4 in Mice, Diabetes. 66 (2017) 58–63. doi:10.2337/db16-0286.
[24] P. Alapatt, F. Guo, S.M. Komanetsky, S. Wang, J. Cai, A. Sargsyan, E. Rodríguez Díaz, B.T. Bacon, P. Aryal, T.E. Graham, Liver retinol transporter and receptor for serum retinol-binding protein (RBP4), J. Biol. Chem. 288 (2013) 1250–1265. doi:10.1074/jbc.M112.369132.
[25] C. Zhu, Y. Xiao, X. Liu, J. Han, J. Zhang, L. Wei, W. Jia, Pioglitazone lowers serum retinol binding protein 4 by suppressing its expression in adipose tissue of obese rats, Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 35 (2015) 778–788. doi:10.1159/000369737.

Impaired vitamin A metabolism in GSD Ia
194
[26] Y. Ma, O.V. Belyaeva, P.M. Brown, K. Fujita, K. Valles, S. Karki, Y.S. de Boer, C. Koh, Y. Chen, X. Du, S.K. Handelman, V. Chen, E.K. Speliotes, C. Nestlerode, E. Thomas, D.E. Kleiner, J.M. Zmuda, A.J. Sanyal, NASH CRN, N.Y. Kedishvili, T.J. Liang, Y. Rotman, HSD17B13 is a Hepatic Retinol Dehydrogenase Associated with Histological Features of Non-Alcoholic Fatty Liver Disease, Hepatol. Baltim. Md. (2018). doi:10.1002/hep.30350.
[27] K. Soejima, B.H. Landing, T.F. Roe, V.L. Swanson, Pathologic studies of the osteoporosis of Von Gierke’s disease (glycogenosis 1a), Pediatr. Pathol. 3 (1985) 307–319.
[28] B. Manickavasagar, A.J. McArdle, P. Yadav, V. Shaw, M. Dixon, R. Blomhoff, G.O. Connor, L. Rees, S. Ledermann, W. Van’t Hoff, R. Shroff, Hypervitaminosis A is prevalent in children with CKD and contributes to hypercalcemia, Pediatr. Nephrol. Berl. Ger. 30 (2015) 317–325. doi:10.1007/s00467-014-2916-2.
[29] M.T. Vannucchi, H. Vannucchi, M. Humphreys, Serum levels of vitamin A and retinol binding protein in chronic renal patients treated by continuous ambulatorial peritoneal dialysis, Int. J. Vitam. Nutr. Res. Int. Z. Vitam.- Ernahrungsforschung J. Int. Vitaminol. Nutr. 62 (1992) 107–112.
[30] D.M. Jacobson, R. Berg, M. Wall, K.B. Digre, J.J. Corbett, R.D. Ellefson, Serum vitamin A concentration is elevated in idiopathic intracranial hypertension, Neurology. 53 (1999) 1114–1118.
[31] A. Tabassi, A.H. Salmasi, M. Jalali, Serum and CSF vitamin A concentrations in idiopathic intracranial hypertension, Neurology. 64 (2005) 1893–1896. doi:10.1212/01.WNL.0000163556.31080.98.
[32] J. Libien, M.J. Kupersmith, W. Blaner, M.P. McDermott, S. Gao, Y. Liu, J. Corbett, M. Wall, NORDIC Idiopathic Intracranial Hypertension Study Group, Role of vitamin A metabolism in IIH: Results from the idiopathic intracranial hypertension treatment trial, J. Neurol. Sci. 372 (2017) 78–84. doi:10.1016/j.jns.2016.11.014.
[33] G.R. Aispuru, E.E. Pascual-Pablo, [Idiopathic intracranial hypertension caused by dietary hypervitaminosis A], Semergen. 41 (2015) e24-26. doi:10.1016/j.semerg.2014.05.011.
[34] M. Obeid, J. Price, L. Sun, M.H. Scantlebury, P. Overby, R. Sidhu, C.A. Chiriboga, L.M. Quittell, Facial palsy and idiopathic intracranial hypertension in twins with cystic fibrosis and hypovitaminosis A, Pediatr. Neurol. 44 (2011) 150–152. doi:10.1016/j.pediatrneurol.2010.10.002.
[35] N.J. Wald, H.S. Cuckle, R.D. Barlow, P. Thompson, K. Nanchahal, R.J. Blow, I. Brown, C.C. Harling, W.J. McCulloch, J. Morgan, The effect of vitamin A supplementation on serum retinol and retinol binding protein levels, Cancer Lett. 29 (1985) 203–213.
[36] M. Gjorgjieva, M. Raffin, A. Duchampt, A. Perry, A. Stefanutti, M. Brevet, A. Tortereau, L. Dubourg, A. Hubert-Buron, M. Mabille, C. Pelissou, L. Lassalle, P. Labrune, G. Mithieux, F. Rajas, Progressive development of renal cysts in glycogen storage disease type I, Hum. Mol. Genet. 25 (2016) 3784–3797. doi:10.1093/hmg/ddw224.
[37] R.H.J. Bandsma, B.H. Prinsen, M. de S. van Der Velden, J.-P. Rake, T. Boer, G.P.A. Smit, D.-J. Reijngoud, F. Kuipers, Increased de novo lipogenesis and delayed conversion of large VLDL into intermediate density lipoprotein particles contribute to hyperlipidemia in glycogen storage disease type 1a, Pediatr. Res. 63 (2008) 702–707. doi:10.1203/PDR.0b013e31816c9013.

Chapter 6
195
[38] A. Grefhorst, M. Schreurs, M.H. Oosterveer, V.A. Cortés, R. Havinga, A.W. Herling, D.-J. Reijngoud, A.K. Groen, F. Kuipers, Carbohydrate-response-element-binding protein (ChREBP) and not the liver X receptor α (LXRα) mediates elevated hepatic lipogenic gene expression in a mouse model of glycogen storage disease type 1, Biochem. J. 432 (2010) 249–254. doi:10.1042/BJ20101225.
[39] J. Perttilä, C. Huaman-Samanez, S. Caron, K. Tanhuanpää, B. Staels, H. Yki-Järvinen, V.M. Olkkonen, PNPLA3 is regulated by glucose in human hepatocytes, and its I148M mutant slows down triglyceride hydrolysis, Am. J. Physiol. Endocrinol. Metab. 302 (2012) E1063-1069. doi:10.1152/ajpendo.00125.2011.
[40] M. Kovarova, I. Königsrainer, A. Königsrainer, F. Machicao, H.-U. Häring, E. Schleicher, A. Peter, The Genetic Variant I148M in PNPLA3 Is Associated With Increased Hepatic Retinyl-Palmitate Storage in Humans, J. Clin. Endocrinol. Metab. 100 (2015) E1568-1574. doi:10.1210/jc.2015-2978.
[41] A. Mondul, R.M. Mancina, A. Merlo, P. Dongiovanni, R. Rametta, T. Montalcini, L. Valenti, D. Albanes, S. Romeo, PNPLA3 I148M Variant Influences Circulating Retinol in Adults with Nonalcoholic Fatty Liver Disease or Obesity, J. Nutr. 145 (2015) 1687–1691. doi:10.3945/jn.115.210633.
[42] A. Suhara, M. Kato, M. Kanai, Ultrastructural localization of plasma retinol-binding protein in rat liver, J. Lipid Res. 31 (1990) 1669–1681.
[43] H. Ronne, C. Ocklind, K. Wiman, L. Rask, B. Obrink, P.A. Peterson, Ligand-dependent regulation of intracellular protein transport: effect of vitamin a on the secretion of the retinol-binding protein, J. Cell Biol. 96 (1983) 907–910.
[44] J.L. Dixon, D.S. Goodman, Studies on the metabolism of retinol-binding protein by primary hepatocytes from retinol-deficient rats, J. Cell. Physiol. 130 (1987) 14–20. doi:10.1002/jcp.1041300104.
[45] K.L. Penniston, S.A. Tanumihardjo, The acute and chronic toxic effects of vitamin A, Am. J. Clin. Nutr. 83 (2006) 191–201.
[46] D. Feskanich, V. Singh, W.C. Willett, G.A. Colditz, Vitamin A intake and hip fractures among postmenopausal women, JAMA. 287 (2002) 47–54.
[47] C.P. Mahoney, M.T. Margolis, T.A. Knauss, R.F. Labbe, Chronic vitamin A intoxication in infants fed chicken liver, Pediatrics. 65 (1980) 893–897.
[48] J.A. Oliveira, C.R. Silva-Netto, M.A. Sala, R.A. Lopes, G. Maia Campos, [Experimental hypervitaminosis A in the rat. 14. Morphological and morphometric study of changes in the esophageal epithelium], Rev. Odontol. Univ. Sao Paulo. 4 (1990) 200–205.
[49] H. Özen, Glycogen storage diseases: New perspectives, World J. Gastroenterol. WJG. 13 (2007) 2541–2553. doi:10.3748/wjg.v13.i18.2541.
[50] D.D. Koeberl, P.S. Kishnani, D. Bali, Y.-T. Chen, Emerging therapies for glycogen storage disease type I, Trends Endocrinol. Metab. TEM. 20 (2009) 252–258. doi:10.1016/j.tem.2009.02.003.
[51] M. Singh, V.N. Singh, Fatty liver in hypervitaminosis A: synthesis and release of hepatic triglycerides, Am. J. Physiol. 234 (1978) E511-514.
[52] L.B. Oliveros, M.A. Domeniconi, V.A. Vega, L.V. Gatica, A.M. Brigada, M.S. Gimenez, Vitamin A deficiency modifies lipid metabolism in rat liver, Br. J. Nutr. 97 (2007) 263–272. doi:10.1017/S0007114507182659.
[53] J.-M. Yuan, Y.-T. Gao, C.-N. Ong, R.K. Ross, M.C. Yu, Prediagnostic level of serum retinol in relation to reduced risk of hepatocellular carcinoma, J. Natl. Cancer Inst. 98 (2006) 482–490. doi:10.1093/jnci/djj104.

Impaired vitamin A metabolism in GSD Ia
196
[54] C. Clemente, S. Elba, G. Buongiorno, P. Berloco, V. Guerra, A. Di Leo, Serum retinol and risk of hepatocellular carcinoma in patients with child-Pugh class A cirrhosis, Cancer Lett. 178 (2002) 123–129.
[55] R. Weiskirchen, F. Tacke, Cellular and molecular functions of hepatic stellate cells in inflammatory responses and liver immunology, Hepatobiliary Surg. Nutr. 3 (2014) 344–363. doi:10.3978/j.issn.2304-3881.2014.11.03.
[56] T.-B. Zhou, G.P.C. Drummen, Y.-H. Qin, The controversial role of retinoic acid in fibrotic diseases: analysis of involved signaling pathways, Int. J. Mol. Sci. 14 (2012) 226–243. doi:10.3390/ijms14010226.

Chapter 6
197
Supplementary Figure S1. Development of hepatic steatosis in L-G6pc-/- mice. Ten (10) days after tamoxifen-induced deletion of the G6pc gene in hepatocytes, L-G6pc-/- and control mice were sacrificed and analyzed for body weight, liver weight, hepatic triglyceride concentration and total hepatic triglyceride pool, hepatic free cholesterol concentration and total hepatic free cholesterol pool, hepatic total (free+esterified) cholesterol concentration and total hepatic total cholesterol pool, fasting glucose levels (A) and H&E and Oil red O staining of liver tissue (B). L-G6pc-/- showed significantly increased liver weight and hepatic triglyceride accumulation in conjunction with fasting hypoglycemia within 10 days after tamoxifen-induced gene deletion.

Impaired vitamin A metabolism in GSD Ia
198
Supplementary Figure S2. RBP4 protein accumulates in livers of vitamin A-depleted mice. Pregnant mice were fed either a vitamin A-sufficient (VAS) control-diet or a vitamin A-deficient (VAD)-diet during pregenacy. New born mice were fed VAS- or VAD-diet for additional 10 weeks and analyzed for hepatic mRNA and protein levels of RBP4. Hepatic mRNA expression of RBP4 did not change in mice fed a VAD-diet compared to VAS controls, while a prominent accumulation of RBP4 protein was detected in mice fed a VAD-diet compared to VAS controls.
Supplementary Figure S3. Hepatic Mlxipl(Chrebp) mRNA levels are elevated in L-G6pc-/- mice. Ten (10) days after tamoxifen-induced deletion of the G6pc gene in hepatocytes, L-G6pc-
/- and control mice were sacrificed and analyzed by Q-PCR for hepatic mRNA levels of Mlxipl(Chrebp). Hepatic mRNA expression of Mlxipl(Chrebp) was increased in L-G6pc-/- mice compared to controls.

Chapter 6
199
Supplementary Table S1: Primers and probes used in study for analysis of target genes
Gene / ID Taqman primers and probe
36B4 NM_022402
Fwd: 5'-GCTTCATTGTGGGAGCAGACA-3' Rev: 5'-CATGGTGTTCTTGCCCATCAG-3' Probe: 5'-TCCAAGCAGATGCAGCAGATCCGC-3'
Act2 / α-Sma NM_007392
Fwd: 5'-TTCGTGTGGCCCCTGAAG-3' Rev: 5'-GGACAGCACAGCCTGAATAGC-3' Probe: 5'-TTGAGACCTTCAATGTCCCCGCCA-3'
Cd68 NM_009853
Fwd: 5'-CACTTCGGGCCATGTTTCTC-3' Rev: 5'-AGGACCAGGCCAATGATGAG-3' Probe: 5'-CAACCGTGACCAGTCCCTCTTGCTG-3'
Ccl2 NM_031530.1
Fwd: 5'-TGTCTCAGCCAGATGCAGTTAAT-3' Rev: 5'-CCGACTCATTGGGATCATCTT-3' Probe: 5'-CCCCACTCACCTGCTGCTACTCATTCA-3'
Col1a1 NM_007742
Fwd: 5'-TGGTGAACGTGGTGTACAAGGT-3' Rev: 5'-CAGTATCACCCTTGGCACCAT-3' Probe: 5'-TCCTGCTGGTCCCCGAGGAAACA-3'
Cyp26a1 NM_007811.1
Fwd: 5'-GGAGACCCTGCGATTGAATC-3' Rev: 5'-GATCTGGTATCCATTCAGCTCAAA-3' Probe: 5'-TCTTCAGAGCAACCCGAAACCCTCC-3'
Mlxipl / Chrebp NM_021455.3 / NM_133552.1
Fwd: 5'-GATGGTGCGAACAGCTCTTCT-3' Rev: 5'-CTGGGCTGTGTCATGGTGAA-3' Probe: 5'-CCAGGCTCCTCCTCGGAGCCC-3'
Dgat1 NM_010046.2 / NM_053437.1
Fwd: 5'-GGTGCCCTGACAGAGCAGAT-3' Rev: 5'-CAGTAAGGCCACAGCTGCTG-3' Probe: 5'-CTGCTGCTACATGTGGTTAACCTGGCCA-3'
Dgat2 NM_026384.2 / NM_001012345.1
Fwd: 5'-GGGTCCAGAAGAAGTTCCAGAAG-3' Rev: 5'-CCCAGGTGTCAGAGGAGAAGAG-3' Probe: 5'-CCCCTGCATCTTCCATGGCCG-3'
Fasn NM_007988 / NM_017332
Fwd: 5'-GGCATCATTGGGCACTCCTT-3' Rev: 5'-GCTGCAAGCACAGCCTCTCT-3' Probe: 5'-CCATCTGCATAGCCACAGGCAACCTC-3'
Hsd17b13 NM_198030.2, NM_001163486.1
Fwd: 5'-AAAGCAGAAAAGCAGACTGGTTCT-3' Rev: 5'-CCCCAGTTTCCTGCATTTGT-3' Probe: 5'- CGGTTTCCTCAACACCACGCTTATTGA-3'
Lipe / HSL NM_010719 / X51415
Fwd: 5'-GAGGCCTTTGAGATGCCACT-3' Rev: 5'-AGATGAGCCTGGCTAGCACAG-3' Probe: 5'-CCATCTCACCTCCCTTGGCACACAC-3'
IL-1β NM_008361
Fwd: 5'-ACCCTGCAGCTGGAGAGTGT-3' Rev: 5'-TTGACTTCTATCTTGTTGAAGACAAACC-3' Probe: 5'-CCCAAGCAATACCCAAAGAAGAAGATGGAA -3'
IL-6 NM_031168
Fwd: 5'-CCGGAGAGGAGACTTCACAGA-3' Rev: 5'-AGAATTGCCATTGCACAACTCTT-3' Probe: 5'-ACCACTTCACAAGTCGGAGGCTTAATTACA-3'
iNos / Nos2 AF049656 / NM_010927 / NM_012611
Fwd: 5'- CTATCTCCATTCTACTACTACCAGATCGA-3' Rev: 5'- CCTGGGCCTCAGCTTCTCAT-3' Probe: 5'- CCCTGGAAGACCCACATCTGGCAG-3'
Lrat NM_023624
Fwd: 5'-TCCATACAGCCTACTGTGGAACA-3' Rev: 5'-CTTCACGGTGTCATAGAACTTCTCA-3' Probe: 5'-ACTGCAGATATGGCTCTCGGATCAGTCC-3'
Mlxipl/Chrebp NM_021455.3
Fwd: 5'-GATGGTGCGAACAGCTCTTCT-3' Rev: 5'-CTGGGCTGTGTCATGGTGAA-3' Probe: 5'-CCAGGCTCCTCCTCGGAGCCC-3'
Pck1 NM_011044 / NM_198780
Fwd: 5'-GTGTCATCCGCAAGCTGAAG-3' Rev: 5'-CTTTCGATCCTGGCCACATC-3' Probe: 5'-CAACTGTTGGCTGGCTCTCACTGACCC-3'

Impaired vitamin A metabolism in GSD Ia
200
Pnpla2 / Atgl NM_025802 / XM_347183
Fwd: 5'-AGCATCTGCCAGTATCTGGTGAT-3' Rev: 5'-CACCTGCTCAGACAGTCTGGAA-3' Probe: 5'-ATGGTCACCCAATTTCCTCTTGGCCC-3'
Pnpla3 NM_054088
Fwd: 5'-ATCATGCTGCCCTGCAGTCT-3' Rev: 5'-GCCACTGGATATCATCCTGGAT-3' Probe: 5'-CACCAGCCTGTGGACTGCAGCG-3'
Raldh1 Assay on demand, Mm00657317_m1 (ThermoFisher)
Raldh2 Assay on demand, Mm00501306_m1 (ThermoFisher)
Raldh3 Assay on demand, Mm00474049_m1 (ThermoFisher)
Raldh4 NM_178713.4
Fwd: 5'-TGGAGCAGTCTCTGGAGGAGTT-3' Rev: 5'- GAAGTTCAGAACAGACCGAGGAA-3' Probe: 5'- AATCTAAAGACCAAGGGAAAACCCTCACGC-3'
Rbp4 NM_011255.2 / XM_215285.3
Fwd: 5'-GGTGGGCACTTTCACAGACA-3' Rev: 5'-GATCCAGTGGTCATCGTTTCCT-3' Probe: 5'-CCCCAGTACTTCATCTTGAACTTGGCAGG-3'
TGF-β1 NM_021578.1
Fwd: 5'-GGGCTACCATGCCAACTTCTG-3' Rev: 5'-GAGGGCAAGGACCTTGCTGTA-3' Probe: 5'-CCTGCCCCTACATTTGGAGCCTGGA-3'
Timp1 NM_001044384.1 / NM_011593.2
Fwd: 5'-TCTGAGCCCTGCTCAGCAA-3' Rev: 5'-AACAGGGAAACACTGTGCACAC-3' Probe: 5'-CCACAGCCAGCACTATAGGTCTTTGAGAAAGC-3'
TNF-α NM_013693 / NM_012675
Fwd: 5'- GTAGCCCACGTCGTAGCAAAC-3' Rev: 5'- AGTTGGTTGTCTTTGAGATCCATG-3' Probe: 5'- CGCTGGCTCAGCCACTCCAGC-3'

Chapter 7
Farnesoid X receptor (FXR) and bile
acids regulate vitamin A storage
Ali Saeed1,7, Jing Yang1,6, Janette Heegsma1,2, Albert K. Groen3,
Saskia W.C. van Mil5, Coen C. Paulusma4, Lu Zhou6, Bangmao
Wang6, Klaas Nico Faber1, 2
1Department of Gastroenterology and Hepatology, 2Laboratory Medicine, 3Department
of Pediatrics, Center for Liver, Digestive, and Metabolic Diseases, University
Medical Center Groningen, University of Groningen, Groningen, The Netherlands.
4Amsterdam UMC, University of Amsterdam, Tytgat Institute for Liver and Intestinal
Research, Amsterdam Gastroenterology and Metabolism, Amsterdam, the
Netherlands.
5Department of Molecular Cancer Research, Center for Molecular Medicine, University
Medical Center Utrecht, Utrecht, The Netherlands.
6Tianjin Medical University General Hospital, Gastroenterology and Hepatology
Department, China.
7Institute of Molecular Biology and Biotechnology, Bahauddin Zakariya University
Multan, Pakistan.
(In preparation)

FXR and hepatic vitamin A
202
ABSTRACT
The nuclear receptor Farnesoid X Receptor (FXR) is an intracellular bile acid sensor.
The retinoid X receptor-alpha (RXRα) is an obligate partner of FXR and is activated by
retinoic acids, e.g. the active metabolites of vitamin A. FXR/RXRα play a crucial role in
bile acid and lipid metabolism. Earlier, we showed that RXR activation modulates FXR-
mediated regulation of bile acid synthesis and transport. Here, we analyzed whether
FXR in turn affects hepatic vitamin A metabolism, a process needed for proper
metabolic and immune control.
Hepatic retinol and retinyl ester levels, as well as mRNA and protein levels of vitamin
A metabolizing factors, were analyzed in 1) FXR-null mice without and with adenoviral
reintroduction of hepatic FXR; 2) intestine-specific FXR-null mice (iFXR-null) and 3)
obeticholic acid (OCA)- and cholic acid (CA)-treated wild type mice.
Hepatic retinol and retinyl palmitate levels were strongly reduced (>90%) in FXR-null
mice compared to wild type littermates. iFXR-null mice had normal levels of these
retinoids, while hepatic reintroduction of FXR in FXR-null mice increased hepatic
retinoid levels. 3-week OCA-treatment in mice strongly reduced (>60%) hepatic retinyl
palmitate levels, concurrently with strongly increased retinol levels (>5-fold) in the liver.
CA-treated animals only showed a mild reduction in hepatic retinyl palmitate after 12
weeks when compared to control animals. Remarkable inconsistent relationships were
observed between transcript and protein levels of key factors in hepatic vitamin A
metabolism in OCA-treated animals, which precluded the identification of molecular
mechanism causing the strongly impaired vitamin A metabolism.
In conclusion, hepatic FXR is required for efficient vitamin A storage in mouse liver,
while activation of FXR by OCA or CA also suppresses hepatic vitamin A storage.
These data suggest that FXR-targeted therapies may be prone to cause vitamin A-
related pathologies.

Chapter 7
203
7.1. INTRODUCTION
The Farnesoid X Receptor (FXR/NR1H) is a pleiotropic ligand-activated nuclear
receptor controlling a great variety of cellular processes, including energy metabolism,
immunomodulation and tissue regeneration. Bile acids are the natural ligands for FXR
and these molecules are increasingly recognized as key signaling molecules
controlling whole body homeostasis, much more than the original view of being just
detergents for lipid-soluble compounds [1]. FXR typically forms a heterodimer with the
Retinoid X Receptor-alpha (RXRα/NR2B1)), a common obligatory partner for many
nuclear receptors, including peroxisome proliferator-activated receptors (PPARs),
Liver X receptor (LXR), Retinoic acid receptor (RAR) and the vitamin D receptor (VDR).
RXRα is activated by retinoic acids, particularly 9-cis retinoic acid (9cRA), the active
metabolites of vitamin A [2]. RXRα is not a silent partner of FXR. Co-activation of
FXR/RXRα with 9cRA may have strong transcriptional effects of FXR-target genes.
This can go either way: e.g. further enhancing bile acid-induced expression, as for the
small heterodimer partner (SHP/NR0B2) [3], or strongly suppressing it, as for the bile
salt export pump (BSEP/ABCB11) [4,5]. Moreover, retinoic acids suppress Cyp7A1
expression, the rate limiting factor in hepatic bile acid synthesis, through direct
transcriptional effects in the liver, as well as by inducing expression of intestinal
fibroblast growth factor 15/19 (mouse FGF15/human FGF19), which in turn
suppresses hepatic bile acid synthesis [6–9]
Vitamin A deficiency is a common condition in chronic liver diseases. This is mainly
the result of 2 pathological processes, e.g. cholestasis and fibrosis. Vitamin A is a fat-
soluble vitamin and its intestinal absorption depends on bile acids [10]. Cholestasis is
characterized by insufficient bile flow from the liver and thereby impairs intestinal
vitamin A absorption. Absorbed vitamin A is efficiently transported to the liver, where it
is stored as retinyl esters, mainly retinyl palmitate, in hepatic stellate cells [11]. The
“quiescent” HSC (qHSC) in the healthy liver maintain stable circulating levels of retinol
at approximately 1.5-2.0 µmol/L to be delivered to peripheral tissues to support proper
function [12]. Liver injury in chronic liver diseases, however, induce a phenotypic
change in HSC that transdifferentiate to contractile, mobile and extracellular matrix-
producing myofibroblasts, so-called activated HSC (aHSC). The qHSC-to-aHSC
transdifferentiation process is the driving force for the development of fibrosis and
characterized by progressive loss of the retinyl ester stores from these cells, ultimately
leading to hepatic an systemic vitamin A deficiency [10,13].

FXR and hepatic vitamin A
204
While the vitamin A-mediated regulation of bile acid synthesis and transport is well
established [3,5–10,14], very little is known about a putative role of FXR in regulating
hepatic vitamin A metabolism. Key factors for hepatic vitamin A metabolism are 1) the
LDL receptor in hepatocytes, which absorbs retinyl ester-containing chylomicron
remnants coming from the gut; 2) Retinyl hydrolases (ATGL, PNPLA3, amongst
others) in hepatocytes that convert retinyl ester to retinol; 3) Retinol Binding Protein 4
(RBP4) that exports retinol from the hepatocytes to the circulation; 4) retinyl esterases
(LRAT and DGAT1) in hepatic stellate cells that convert retinol to retinyl esters for
storage; 5) Retinyl hydrolases (ATGL, PNPLA3, HSL) in HSC for controlled release of
retinol in times of insufficient dietary intake; 6) RDHs and RALDHs that convert retinol
to retinoic acids and 7) cytochrome P450s (particularly Cyp26A1) that catabolize
retinoic acids (see also Saeed 2017 BBA) [10]. Previous transcriptome analysis have
not hinted to a clear effect of FXR on hepatic vitamin A metabolism [15–17]. Such FXR-
mediated transcriptional effects have been well-established for regulation of hepatic
lipid and glucose metabolism [15–18]. The pharmacological FXR agonist obeticholic
acid (OCA; 6α-ethyl-chenodeoxycholic acid; INT-747) is used for the treatment of
Primary biliary cholangitis (PBC), either as mono therapy or in combination with
ursodeoxycholic acid (UDCA) [19–21](refs). In addition, OCA and several other
pharmacological FXR ligands are being evaluated in clinical trials for their therapeutic
value in other chronic liver diseases, including primary sclerosing cholangitis (PSC)
and non-alcoholic fatty liver disease (NAFLD). As vitamin A metabolism is typically
impaired in chronic liver diseases like PBC, PSC and NAFLD [22–27], FXR may also
play a role in this, either directly or indirectly.
In this study, we therefore analyzed vitamin A metabolism in total and tissue-specific
FXR-null mice, all on normal chow, as well as in WT mice treated with OCA, or cholic
acid (CA) to dissect the effects of FXR in vitamin A metabolism. To our surprise, we
found that both the absence of hepatic FXR, as well as the pharmacological activation
by OCA, lead to vitamin A depletion from the liver. Thus, proper control of vitamin A
metabolism in the liver depends on a tightly-balanced action of FXR.

Chapter 7
205
7.2. MATERIALS AND METHODS
7.2.1. Animal experiments
Animal experiments were approved by local ethics committees for animal testing (e.g.
Universities of Groningen, Amsterdam and Utrecht, The Netherlands and Tianjin
Medical University, China). All animals were kept in a pathogen-free control
environment with alternating dark and light cycles of 12 hours, temperature (20-24 ºC)
and relative humidity (55%±15%). Animals received food and water ad libitum.
FXR-null mice mice [28,29] or intestinal specific FXR-null mice (iFXR-null) mice [30]
and wild-type mice were kept on standard chow diet (RMHB; Hope Farms, The
Netherlands; contains 10 IU of retinyl acetate/g as source of vitamin A) for 8-10 weeks,
and then sacrificed for analysis. FXR2 and FXR4 isoforms were reintroduced in
livers of FXR-null mice as described previously [31] using self-complementary adeno-
associated virus (scAAV) serotype 8 vectors under control of the liver-specific LP1-
promoter. ScAAV expressing the Green Fluorescent Protein (GFP) were used as
control. FXR-null mice received 1x1011 AAV particles into the retro-orbital sinus with
vector genomes. All animals were sacrificed after one month of scAAV transfection for
analysis, as described previously [31]. Wild type C57BL/6 mice were fed a chow diet
diet (K4068.02; Arie Blok diervoeders, Woerden, The Netherlands; contains 10 IU of
retinyl acetate/g as source of vitamin A) mixed with either vehicle or obeticholic acid
(OCAl; 0.025% w/w) for 3 weeks. Alternatively, wild type C57BL/6 mice were fed a
chow diet (H10293G, Hua Fukang Biological, technology, Beijing, China; contains 4 IU
of retinyl acetate/g as source of vitamin A) mixed with either vehicle or cholic acid (0.1%
w/w) (Sigma-Aldrich, China, Inc) for 4, 8 or 12 weeks.
7.2.2. Serum and hepatic vitamin A analysis
Serum and tissue retinoid content (both retinol and retinyl palmitate) was analyzed by
reverse phase HPLC as previously described [32]. Briefly, tissue (30-50 mg) was
homogenized in PBS to create a 15% (w/v) tissue homogenate. Then, tissue
homogenate (66.7 µL equal to 10 mg of tissue) or serum (50 µL) were added in the
antioxidant mix (containing pyrogallol, butylated hydroxytoluene,
ethylenediaminetetraacetic acid and ascorbic acid) and vortexed thoroughly for 1 min.
Retinol and retinyl esters were extracted and deproteinized twice with n-hexane in the
presence of retinol acetate (100 µL, concentration 4 µmol/L) as an internal standard to
assess the recovery efficiency after the extraction procedure. Standard curves created

FXR and hepatic vitamin A
206
from a range of concentrations of retinol and retinyl palmitate were used to determine
absolute tissue and serum concentrations of these compounds. Additionally, two
negative controls (only containing internal standard) and two positive controls (low and
high concentrations of retinol plus internal standard) were included in each series of
extractions. Samples were evaporated under N2 and diluted in 300 µL 100% ultrapure
ethanol. Then, 50 µL was injected into HPLC (Waters 2795 Alliance HT Separations
Module, Connecticut, USA) for phase separation on a C18 column (Waters Symmetry
C18, dimension 150 x 3.0 mm, particle size 5 µm, Waters Corporation, Milford, MA,
USA) and measurement (UV-VIS, dual wavelength, UV-4075 Jasco, Tokyo, Japan).
Retinoids in samples were identified by exact retention time of known standards in
ultraviolet absorption at 325 nm by HPLC. Finally, retinol and retinyl palmitate
concentrations were calculated and normalized to final volume or tissue weight.
7.2.3. Quantitative real-time reverse transcription polymerase chain reaction
(qRT-PCR)
Quantitative real-time reverse transcription polymerase chain reaction was performed
as previously described [33]. Shortly, total RNA was isolated from tissue samples using
TRIzol® reagent according to supplier’s instructions (ThermoFisher Scientific, Breda,
The Netherlands). RNA quality and quantity were determined using a Nanodrop 2000c
UV-vis spectrophotometer (Thermo Fisher Scientific). cDNA was synthesized from 2.5
µg of RNA by using random nonamers and M-MLV reverse transcriptase
(ThermoFisher Scientific). Taqman primers and probes were designed using Primer
Express 3.0.1 (ThermoFisher Scientific) and are shown in Supplementary Table S1.
All target genes were amplified using the Q-PCR core kit master mix (Eurogentec,
Maastricht, The Netherlands) on a 7900HT Fast Real-Time PCR system
(ThermoFisher Scientific). SDSV2.4.1 (ThermoFisher Scientific) was used to analyze
the data. Expression of genes is presented in 2-delta CT and normalized to 36B4.
7.2.4. SDS-PAGE and Western Blotting
Protein samples are prepared for Western blot analysis as described previously [34].
Protein concentrations were quantified using the Bio-Rad protein assay (Bio-Rad,
Hercules, CA, USA) with bovine serum albumin (BSA) as standard. Equal amounts of
protein were separated on Mini-PROTEAN® TGX™ precast 4-15% gradient gels (Bio-
Rad) and transferred to nitrocellulose membranes using the Trans-Blot turbo transfer
system, (Bio-Rad). Primary antibodies (anti-LRAT, 1:500; # SAB4503589, Sigma-

Chapter 7
207
Aldrich), RBP4 (1:2,000; # ab109193, Abcam), ATGL (1:1,000; # 2138, Cell Signaling
Technology), PNPLA3 (1:1,000; #PA5-18901, ThermoFisher, scientific), pHSL
(1:1,000# 4126, Cell Signaling Technology), HSL (# 4107, Cell Signaling Technology),
PEPCK1 (1:500; 10004943, Cayman chem. Abcam), CYP7A1 (H-58): sc-25536,
SantaCruz), rabbit-NTCP (1:1,000; kind gift from Dr. B. Stieger, Zurich, Switzerland
[35]), anti-GAPDH, (1:40,000 #CB1001, Calbiochem, Merck-Millipore Amsterdam-
Zuidoost, The Netherland) and horseradish peroxidase (HRP)-conjugated secondary
antibody (1:2,000; DAKO, Amstelveen, The Netherlands) were used for detection.
Proteins were detected using the pierce ECL Western blotting kit (ThermoFisher
scientific). Images were captured using the chemidoc XRS system and Image Lab
version 3.0, (Bio-Rad). The intensity of bands was quantified using ImageJ version
1.51 (NIH, USA).
7.2.5. Microscopy
Hematoxylin and Eosin (H&E) staining on liver sections (4 µm) was performed on snap-
frozen liver sections as previously described [36]. Immunohistochemistry was
performed on the paraffin-embedded liver tissue. Briefly, after deparaffinization,
antigen retrieval was performed by using microwave irradiation in citrate buffer, pH 6.0
and blocking of endogenous peroxidase with 0.3% H2O2 for 30 min. Primary antibodies
used were rabbit anti-mouse LRAT (#28075 Takara, Japan). Horse peroxidase-
conjugated goat anti-rabbit secondary antibody and rabbit anti-goat tertiary antibodies
were used. Slides were stained with the Vector® NovaRED™ substrate Kit (# SK-4800,
Vector Laboratories, Inc., USA) for 10 min and Haematoxylin was used as a counter
nuclear stain for 2 min at room temperature. Finally, slides were dehydrated and
mounted with Eukitt® (Sigma-Aldrich). Slides were scanned on a nanozoomer 2.0
digital slide scanner (C9600-12, Hamamatsu Photonics, Hamamatsu, Japan) and
analyzed using Aperio ImageScope (version 11.1, Leica Microsystems, Amsterdam,
The Netherlands). Autofluorescence analysis was performed on unstained cryostatic
liver sections using a Leica CTR 6000 FS fluorescence microscope (Leica
Microsystems, Amsterdam, The Netherlands) as previously described [37,38]. Briefly,
cryostat liver sections were illuminated with an excitation filter of 366 nm band-pass
interference, and spectra were recorded in the range of 400-680 nm with spectrum
acquisition from 0.2 to 3 seconds.

FXR and hepatic vitamin A
208
7.2.6. Statistical analysis
Data are presented as Mean±SEM and statistical analysis was performed with
GraphPad Prism 6 software package (GraphPad Software, San Diego, CA, USA).
Statistical significance was determined by Mann-Whitney test (two groups), One-way
ANOVA or Kruskal-Wallis (more than two groups) followed by post-hoc Dunns:
Compare all pairs of columns. P-values ≤0.05*, ≤0.01**, ≤0.001*** were considered
significant.
7.3. RESULTS
7.3.1. Hepatic fat accumulation, but vitamin A depletion in FXR null mice
FXR-null mice have previously been shown to develop mild steatohepatitis, particularly
accumulating triglycerides, which may progress to NASH even when fed a chow diet
[28,39–42]. Indeed, Oil-Red-O staining of liver tissue of 10-12 week-old FXR-null mice
demonstrated fat accumulation compared to their wild type litter mates (Figure 1 A,
middle panels). Moreover, liver weight was slightly increased in FXR-null mice and
showed enhanced mRNA levels of genes involved in lipogenesis, such as Plin2, Fasn
and Acc1 (Figure 1 B). In sharp contrast and unexpectedly, hepatic retinyl palmitate
and retinol levels were strongly (>90%) reduced in FXR-null mice compared to their
WT littermates (Figure 1 C). Lower hepatic vitamin A levels in FXR-null mice were also
confirmed by reduced vitamin A-specific autofluorescence in liver tissue (Figure 1 A,
right panels). The sharp depletion of hepatic vitamin A was, however, not accompanied
by a reduction in plasma retinol levels in FXR-null mice (Figure 1 C). This is not
necessarily surprising as mice on a vitamin A-deficient diet are also able to maintain
normal serum retinol levels, even if the liver contains less than 5% retinyl esters
compared to animals on a vitamin A-containing diet (Supplementary Figure S1). Still,
these results show that hepatic retinoid levels are drastically decreased in mice lacking
FXR.

Chapter 7
209
Figure 1. FXR deficiency impairs hepatic retinoid storage. Whole body FXR-null mice and age-match wild-type control mice were analyzed for A) H&E and oil red O staining, which revealed fat accumulation in livers of FXR-null mice as compared to control mice, while autofluorescence analysis revealed reduced vitamin A levels in livers of FXR-null mouse. B) The body weight was not altered FXR-null mice, while the liver weight was increased in FXR-null mice. As expected, hepatic expression of Plin2, Fasn and Acc1 were increased in FXR-null mice vs wild-type mice. C) hepatic retinyl palmitate and retinol levels were reduced in FXR-null mice, while plasma retinol levels were equal in both groups.

FXR and hepatic vitamin A
210
7.3.2. Hepatic and not intestinal FXR is essential for efficient vitamin A storage
in the liver
As dietary vitamin A is absorbed in the intestine and transported to the liver for storage,
we next studied whether hepatic and/or intestinal FXR is controlling hepatic vitamin A
levels. Hepatic retinyl palmitate and retinol levels were not significantly different in
intestine-specific FXR-null mice (iFXR-null) as compared to WT control animals
(Figure 2 A), suggesting that vitamin A metabolism is normal in these mice. In contrast,
reintroduction of hepatic FXR2 or FXR4 in FXR-null mice through adenoviral
expression (4 weeks) elevated both retinyl palmitate and retinol levels in mouse livers
liver (Figure 2 B). As FXR expression was driven by the hepatocyte-specific Lp1
promoter [43,44], these data also suggest that particularly hepatocytes play a key role
in the FXR-mediated control of hepatic vitamin A levels.
Figure 2. Hepatic and not intestinal FXR is essential for normal retinoid levels in the liver. A) Hepatic retinyl palmitate and retinol were analyzed in intestinal specific FXR-null mice (iFXR-null) and wild-type littermates. Intestine-specific FXR-deficiency did not alter hepatic vitamin A levels. B) Whole body FXR-null mice were transduced with ScAAV-produced FXRα2 or FXRα4 or GFP controls and after 4 weeks analyzed for hepatic retinyl palmitate and retinol levels. Both FXR-isoform enhanced hepatic retinoid levels, suggesting that hepatic FXR plays a crucial role in vitamin A storage and metabolism.

Chapter 7
211
7.3.3. Post-transcriptional reduction of retinol esterifying LRAT in FXR-null
mice
Hepatic retinyl ester and retinol levels are a resultant of the activity of retinyl esterases
(LRAT and to a lesser extent by DGAT1 and 2) and retinyl ester hydrolases (ATGL,
PNPLA3 and HSL). mRNA levels of Dgat1 and 2, Pnpla2 (ATGL) and Lipe (HSL) were
comparable between FXR-null mice and WT littermates, while both Lrat and Pnpla3
levels were elevated in FXR-null mice (Figure 3 A). As the latter may hint to a more
dynamic vitamin A metabolism in the liver, it does not provide a clear explanation for
the depletion of hepatic retinyl esters and retinol in FXR-null mice. Detailed (re)analysis
of public transcriptome analysis comparing wild-type and FXR-null mice also did not
reveal a prominent effect on genes involved in hepatic vitamin A metabolism when
FXR is absent (Supplementary Figure S2). As LRAT plays a key role in retinyl ester
accumulation in the liver, we analyzed the protein level in more detail. In contrast to
the elevated Lrat mRNA levels, hepatic LRAT protein levels were significantly reduced
(>60%) in FXR-null mice compared to WT littermates, as quantified by Western blotting
(Figure 3 B). In line, LRAT-specific immunohistochemical staining was strongly
reduced in liver sections of FXR-null mice compared to WT animals, while a stellate
cell-specific staining was preserved (Figure 3 C). These data reveal a post-
transcriptional effect caused by the absence of FXR that may aid to the impaired
accumulation of hepatic retinoids.

FXR and hepatic vitamin A
212
Figure 3. Post-transcriptional reduction of LRAT in FXR-null mice. Whole body FXR-null and wild type mice were analyzed for A) hepatic mRNA expression of Lrat, Dgat1, Dgat2, Pnpla2 (Atgl), Pnpla3, Lipe (Hsl); Hepatic Lrat and Pnpla3 mRNA levels were increased in FXR-null mice, with no change in other vitamin A metabolizing enzymes as compared to control. Western blot analysis (B) and immunohistochemical staining (C) for LRAT revealed that hepatic protein levels of LRAT were significantly reduced in FXR-null mice as compared to wild-type control mice.
7.3.4. OCA-mediated activation of FXR reduces hepatic vitamin A levels
Given the pronounced reduction of hepatic retinoids in the absence of FXR in mouse
livers, we next analyzed the effect of pharmacological activation of FXR in WT mice.
Mice were treated for 3 weeks with obeticholic acid (OCA). As expected, mRNA levels
of the FXR target genes Nr0b2 (Shp) and Abcb11 (Bsep) were enhanced by OCA
treatment (Figure 4 A). In contrast to our expectation, OCA treatment also strongly
decreased hepatic retinyl palmitate levels (-64 %) compared to control animals, while
retinol concentrations were sharply elevated (+ 5-fold) in the liver (Figure 4 B). mRNA

Chapter 7
213
levels of Lrat and Rbp4 were not affected by OCA treatment, while a pronounced
induction was observed for the genes encoding the retinyl ester hydrolases PNPLA3
and HSL (Lipe), concomitant with a reduction in Pnpla2 (ATGL) mRNA levels (Figure
4 C). Surprisingly, however, Western blot analysis revealed that protein levels of all 3
hepatic retinyl ester hydrolases (ATGL, PNPLA3 and HSL, including its active,
phosphorylated form pHSL) were drastically decreased in OCA-treated animals
(Figure 4 D), while LRAT levels were not changed compared to control animals. The
high retinol levels in the livers of OCA-treated animals (Figure 4 B) are therefore
unlikely to come from hydrolysis of hepatic retinyl esters. Interestingly, hepatic RBP4
protein levels appeared lower in OCA-treated animals, while Rbp4 mRNA levels
tended to be increased, which may be a result of retinol-induced release from the liver
[45–47]. Importantly, OCA did lead to expected induction of PEPCK (encoded by Pck1)
and reductions in CYP7A1 and NTCP (Figure 4 D). To examine whether hepatic retinol
metabolism to retinoic acids was blocked, we analyzed expression of RALDHs and
found that mRNA levels of Raldh1, 2 and 4 were induced by OCA (Figure 4 E) and
was accompanied by increased levels of retinoic acid-responsive genes, like Cpt1a,
Pck1, Ppargc1a, Cyp26a1, Ucp2, Fgf21 (Figure 4 F). Taken together, these data show
that, even though the capacity to esterify retinol seems to be increased by OCA (LRAT
maintained, while retinyl hydrolases are strongly reduced), it enhances hepatic retinol
levels while retinyl ester stores go down. Moreover, the production of retinoic acids in
the liver seems not impaired or even enhanced in OCA-treated animals.

FXR and hepatic vitamin A
214
Figure 4. OCA treatment reduces hepatic retinyl palmitate, while increasing retinol levels in mouse livers. Control and OCA-treated mice were analyzed for hepatic vitamin A levels, mRNA and protein levels of FXR targets and vitamin A metabolizing factors. A) The mRNA levels of FXR target genes Nr0b2 (encoding SHP) and Abcb11 (encoding BSEP) were strongly enhanced in livers of OCA-treated mice. B) Hepatic retinyl palmitate levels were significantly reduced in OCA-treated mice, while hepatic retinol levels were significantly increased as compared to control mice. C) Hepatic mRNA levels of Lrat, Rbp4 and Pnpla3 were not changed, while levels of Pnpla2 (ATGL) were decreased and Lipe (HSL) were increased in OCA-treated mice. D) Protein levels of LRAT were not affected by OCA-treatment, while RBP4, ATGL, PNPLA3, HSL (and its active phosphorylated form pHSL), CYP7A1 and NTCP were all decreased and PEPCK was increased. GAPDH was used as loading control. E) mRNA levels of genes involved in the conversion of retinol into retinoic acid (Raldh1, Raldh2, Raldh4) and of F) retinoic acid-responsive genes (Cpt1a, Pck1, Ppargc1a, Cyp26a1, Ucp2, Fgf21) were enhanced in OCA-treated mice as compared to control animals.

Chapter 7
215
7.3.5. Cholic acid feeding also impairs retinyl ester accumulation in mouse liver
Given the remarkable observation that both the absence and pharmacological
activation of FXR leads to reduction of retinyl ester levels in the liver, we also analyzed
whether cholic acid treatment may affect hepatic vitamin A metabolism in mice. Mice
were fed a diet containing 0.1% CA for up to 12 weeks. Both retinyl palmitate and
retinol levels in the liver increase with age when animals are fed normal (vitamin A-
containing) chow, as has been reported before [48,49] No significant differences in
retinyl palmitate levels were detected after 4 or 8 week CA feeding compared to
animals on control chow, although the accumulation of hepatic retinyl palmitate
seemed delayed between week 4 and 8 in CA-fed mice. This indeed progressed to a
moderate (-29%), but significant reduction in retinyl palmitate after 12 weeks feeding a
CA-containing diet, compared to control animals (Figure 5 A). At none of the time
points an effect of the CA diet on hepatic and serum retinol levels was observed
(Figure 5 B). The CA-diet induced the hepatic expression of of Nr0b2 (Shp) and
Abcb11 (Bsep) (Figure 5 C), though the effect was less pronounced compared to OCA
(Figure 4 A).
Thus, all together, our data show that both the absence and activation of FXR impairs
hepatic vitamin A storage.
Figure 5. Cholic acid (0.1%) treatment delays the accumulation of hepatic retinoids in mice. Mice were fed chow or a CA (0.1%)-containing diet for 4, 8 and 12 weeks. A) Hepatic retinyl palmitate was significantly decreased CA-fed treated mice after 12 weeks as compared to control mice. B) Hepatic and serum retinol levels did not differ at any time point between both groups of mice C) Hepatic mRNA levels FXR-responsive genes Nr0b2 (SHP) and Abcb11 (BSEP) were moderately increased in CA-treated animals compared to control mice.

FXR and hepatic vitamin A
216
7.4. DISCUSSION
In this study, we report the remarkable observation that both the absence, as well as
ligand-activation of FXR leads to a reduction in hepatic retinyl ester stores in mice.
FXR presence in the liver appears essential for promoting retinyl ester accumulation,
while intestinal FXR is not. Pharmacological activation of FXR by OCA left hepatic
LRAT expression unaffected, while it strongly suppressed levels of hepatic retinyl ester
hydrolases (ATGL, PNPLA3 and HSL) by combined transcriptional and non-
transcriptional mechanisms. The accompanied retinyl ester depletion and retinol
accumulation appears therefore not (solely) a result of impaired vitamin A metabolism
inside the liver. FXR is an important therapeutic target for liver disease and is currently
evaluated in clinical trials for the treatment of chronic liver diseases, including PSC and
NASH [50–52]. Effects of FXR ligands on vitamin A metabolism therefore deserve
attention, as vitamin A metabolites serve many key physiological functions disturbed
in liver disease, such as in immune regulation, tissue differentiation, bile acid, glucose
and fat metabolism.
To the best of our knowledge, this is the first study that analyzed the putative role of
FXR in vitamin A metabolism. FXR has already been shown to control diverse
metabolic processes, varying from bile acid, triglyceride, cholesterol, glucose to amino
acid metabolism [53–56]. In addition, it regulates autophagy, inflammation and
extracellular matrix production [57–59]. A direct effect of FXR on transcriptional
regulation of genes involved in these processes has been confirmed [53–60]. In this
study, we have so far been unable to make such direct link with FXR-controlled gene
expression for vitamin A metabolism. We have re-evaluated public databases with
micro-array and RNAseq data on FXR-mediated effects, but have not discovered clear
overlooked genes involved in vitamin A metabolism. Both the absence of FXR and
OCA-treatment in this study lead to expected transcriptional effects on hepatic genes
involved in lipid (Figure 1B) and bile acid (Figure 4A) metabolism, respectively. With
respect to the genes involved in vitamin A metabolism, FXR absence enhanced mRNA
levels of Lrat while reducing Pnpla3 and leaving Pnpla2 and Lipe levels unchanged.
While this may suggest an enhanced metabolism back and forth between retinyl esters
and retinol, it does not push a direction to retinyl ester and/or retinol loss from the liver.
In contrast to the enhanced Lrat mRNA levels, LRAT protein levels were actually
decreased in FXR-null mice. LRAT-null mice are unable to store retinyl esters in the
liver [61], thus a significant reduction in LRAT protein may impair retinyl ester storage

Chapter 7
217
and aid to an explanation for the reduced retinyl ester levels in FXR-null mice.
However, this appears not a result of FXR-mediated transcriptional regulation as Lrat
mRNA levels were unaffected by the absence of FXR in the liver. Analysis of hepatic
mRNA levels after 3 week OCA treatment suggested some hints for the rapid change
in retinyl ester (> 60% down) and retinol (>5-fold up) levels in the liver, where enhanced
mRNA levels of Pnpla3 and Lipe were detected with stable Lrat mRNA levels.
Surprisingly, however, protein levels of enzymes known to hydrolyse retinyl esters
(ATGL, PNPLA3 and HSL) were all strongly reduced in the liver. Thus, as observed in
FXR-null mice, post-transcriptional effects associated with OCA treatment appear to
have pronounced effects on vitamin A metabolism. At present, we cannot explain the
high hepatic retinol levels induced by OCA with the transcriptional and post-
translational regulatory mechanisms in the liver. One hypothesis to be tested is
whether the high retinol levels in the liver may actually arise from the gut after it is
absorbed from the intestinal lumen. This requires a detailed analysis of vitamin A
metabolites in the intestine and circulation, samples that were unfortunately not
available for this study.
In contrast to a potential effect of OCA on intestinal vitamin A metabolism leading to a
strongly disturbed retinyl ester/retinol balance in the liver, hepatic FXR seems to be
critically important to maintain normal levels of hepatic retinyl esters and retinol.
Previously, adenoviral reintroduction of the FXR isoforms α2 and α4 in FXR-null mice
was shown to enhance mRNA levels of various lipases (incl. Pnpla2 and Lipe,
encoding ATGL and HSL respectively) and decrease hepatic triglyceride levels [62].
Interestingly, ATGL and HSL hydrolyse both triglycerides and retinyl esters. Still,
hepatic retinyl ester stores increase with reintroduction of hepatic FXR (this study)
while triglycerides decrease [62]. On the other hand, hepatic retinol levels go up after
reintroduction of Fxra2 and a4, which could be a result of the enhanced Pnpla2 and
Lipe levels. Given the fact that protein levels of these lipases may strongly deviate from
the corresponding mRNA levels, as we observed in the OCA-treated animals, it
remains to be determined what is the driving force of the triglyceride decline and
vitamin A increase in these animals.
Some of the hepatic effects observed earlier in the FXR-null mice after reintroduction
of FXR may actually be caused directly or indirectly by the changed hepatic vitamin A
metabolism. Retinoic acids regulate hepatic lipid metabolism at various levels by
activating RARs and RXRs, either directly or in complex with other nuclear receptors,

FXR and hepatic vitamin A
218
like PPARa, PPARg, LXR and also FXR. All-trans retinoic acids, typically activating
RARs, have been shown to suppress hepatic steatosis [63]. Activation of RXR by 9-
cis retinoic acid or pharmacological ligands has a less-predictable outcome, as it co-
activates nuclear receptors promoting lipolysis (PPARa, FXR) [64–66], as well as
those that promote lipogenesis (PPARg, LXR) [67,68]. It will therefore greatly depend
on local levels of the various nuclear receptors and the type of retinoic acid produced
which process will dominate, either lipolysis or lipogenesis. Thus, the therapeutic
action of FXR ligands, including OCA, may in part be due to effects on hepatic vitamin
A metabolism.
Not only OCA, but also CA-feeding impaired hepatic vitamin A storage in mice, which
was significantly reduced after 12 weeks on a diet containing 0.1% CA. This
concentration was chosen as it resembles the mouse-equivalent of the therapeutic
dose as FDA-approved bile acids for the treatment of several liver diseases, including
gallstones, inborn errors of bile acid synthesis and peroxisomal disorders (Zellweger
syndrome) [69–71]. Particularly, the age-dependent increase in hepatic retinyl
palmitate levels was suppressed in mice on a CA-containing diet although the absolute
effect was much less pronounced as compared to the 3 week OCA treatment. Still, it
may be recommended to regularly check patients on bile acid therapy for circulating
retinol levels. Although serum retinol levels do not accurately represent the hepatic
pool of vitamin A, they will drop at the stage when the liver gets depleted from vitamin
A. According to our results, hepatic vitamin A depletion is likely to develop more rapidly
under OCA treatment. It is important to note that the OCA therapy is already available
for PBC and in clinical trials for other chronic liver diseases that are known to be
associated with vitamin A deficiency, including PBC, PSC and NASH. OCA treatment
may therefore accelerate vitamin A depletion in these patients affecting proper immune
regulation and metabolic control. Future studies need to address whether patients
treated with therapeutic bile acids or FXR-ligands are truly at risk for conditions caused
by impaired vitamin A metabolism.
An important limitation of our study is the fact that the various animal experiments were
performed at 4 different laboratories with different suppliers of the chow. As hepatic
retinoid levels are strongly dependent on the composition of the diet and the age of the
animals, levels of hepatic retinyl palmitate and retinol varied significantly between the
various control groups. The amount of vitamin A in the chow was 10 IU of vitamin A
per gram chow in the genetic models and OCA-treatment experiment, while the CA-

Chapter 7
219
treatment experiment contained 4 IU vitamin A/g. It was therefore crucial that each
experiment had the proper animal control group included, where the hepatic retinyl
palmitate and retinol levels differed significantly between these control groups.
Importantly, all vitamn A measurements were performed in the same laboratory by the
same researchers to exclude analytical differences. On the other hand, the fact that a
clear effect of FXR and its ligands on hepatic retinoid levels was observed in all animal
experiments also underscores the fact that a clear relationship seems to exist between
FXR and vitamin A metabolism, although this may not be regulated primarily at the
transcriptional level. This, in fact, uncovers a completely unexplored research field of
the possible post-transcriptional effects associated with manipulation of FXR activity.
In conclusion, our data show that the absence of hepatic FXR, as well as the oral
supplementation of natural and synthetic bile acids impair vitamin A metabolism in the
liver. Vitamin A serves a great variety of physiological functions in and outside the liver,
including immune regulation and metabolic control. Future studies therefore need to
address whether the therapeutics targeting FXR may lead to vitamin A-related adverse
effects.

FXR and hepatic vitamin A
220
REFERENCES
[1] C.Y. Han, Update on FXR Biology: Promising Therapeutic Target?, Int. J. Mol. Sci. 19 (2018). doi:10.3390/ijms19072069.
[2] M.I. Dawson, Z. Xia, The retinoid X receptors and their ligands, Biochim. Biophys. Acta. 1821 (2012) 21–56. doi:10.1016/j.bbalip.2011.09.014.
[3] M.O. Hoeke, J. Heegsma, M. Hoekstra, H. Moshage, K.N. Faber, Human FXR regulates SHP expression through direct binding to an LRH-1 binding site, independent of an IR-1 and LRH-1, PloS One. 9 (2014) e88011. doi:10.1371/journal.pone.0088011.
[4] W. Zheng, Y. Lu, S. Tian, F. Ma, Y. Wei, S. Xu, Y. Li, Structural insights into the heterodimeric complex of the nuclear receptors FXR and RXR, J. Biol. Chem. 293 (2018) 12535–12541. doi:10.1074/jbc.RA118.004188.
[5] M.O. Hoeke, J.R.M. Plass, J. Heegsma, M. Geuken, D. van Rijsbergen, J.F.W. Baller, F. Kuipers, H. Moshage, P.L.M. Jansen, K.N. Faber, Low retinol levels differentially modulate bile salt-induced expression of human and mouse hepatic bile salt transporters, Hepatol. Baltim. Md. 49 (2009) 151–159. doi:10.1002/hep.22661.
[6] D.R. Schmidt, S.R. Holmstrom, K. Fon Tacer, A.L. Bookout, S.A. Kliewer, D.J. Mangelsdorf, Regulation of bile acid synthesis by fat-soluble vitamins A and D, J. Biol. Chem. 285 (2010) 14486–14494. doi:10.1074/jbc.M110.116004.
[7] S.-Y. Cai, H. He, T. Nguyen, A. Mennone, J.L. Boyer, Retinoic acid represses CYP7A1 expression in human hepatocytes and HepG2 cells by FXR/RXR-dependent and independent mechanisms, J. Lipid Res. 51 (2010) 2265–2274. doi:10.1194/jlr.M005546.
[8] F. Yang, Y. He, H.-X. Liu, J. Tsuei, X. Jiang, L. Yang, Z.-T. Wang, Y.-J.Y. Wan, All-trans retinoic acid regulates hepatic bile acid homeostasis, Biochem. Pharmacol. 91 (2014) 483–489. doi:10.1016/j.bcp.2014.08.018.
[9] D. Jahn, D. Sutor, D. Dorbath, J. Weiß, O. Götze, J. Schmitt, H.M. Hermanns, A. Geier, Farnesoid X receptor-dependent and -independent pathways mediate the transcriptional control of human fibroblast growth factor 19 by vitamin A, Biochim. Biophys. Acta. 1859 (2016) 381–392. doi:10.1016/j.bbagrm.2015.12.007.
[10] A. Saeed, M. Hoekstra, M.O. Hoeke, J. Heegsma, K.N. Faber, The interrelationship between bile acid and vitamin A homeostasis, Biochim. Biophys. Acta. 1862 (2017) 496–512. doi:10.1016/j.bbalip.2017.01.007.
[11] C. Freund, D.N. Gotthardt, Vitamin A deficiency in chronic cholestatic liver disease -is vitamin A therapy beneficial ?, Liver Int. Off. J. Int. Assoc. Study Liver. (2017). doi:10.1111/liv.13433.
[12] R. Blomhoff, M.H. Green, T. Berg, K.R. Norum, Transport and storage of vitamin A, Science. 250 (1990) 399–404.
[13] J.X. Jiang, N.J. Török, Liver Injury and the Activation of the Hepatic Myofibroblasts, Curr. Pathobiol. Rep. 1 (2013) 215–223. doi:10.1007/s40139-013-0019-6.
[14] Y. He, L. Gong, Y. Fang, Q. Zhan, H.-X. Liu, Y. Lu, G.L. Guo, L. Lehman-McKeeman, J. Fang, Y.-J.Y. Wan, The role of retinoic acid in hepatic lipid homeostasis defined by genomic binding and transcriptome profiling, BMC Genomics. 14 (2013) 575. doi:10.1186/1471-2164-14-575.
[15] N. Ijssennagger, A.W.F. Janssen, A. Milona, J.M. Ramos Pittol, D.A.A. Hollman, M. Mokry, B. Betzel, F.J. Berends, I.M. Janssen, S.W.C. van Mil, S. Kersten, Gene expression profiling in human precision cut liver slices in response to the

Chapter 7
221
FXR agonist obeticholic acid, J. Hepatol. 64 (2016) 1158–1166. doi:10.1016/j.jhep.2016.01.016.
[16] L. Zhan, H.-X. Liu, Y. Fang, B. Kong, Y. He, X.-B. Zhong, J. Fang, Y.-J.Y. Wan, G.L. Guo, Genome-wide binding and transcriptome analysis of human farnesoid X receptor in primary human hepatocytes, PloS One. 9 (2014) e105930. doi:10.1371/journal.pone.0105930.
[17] G. Porez, B. Gross, J. Prawitt, C. Gheeraert, W. Berrabah, J. Alexandre, B. Staels, P. Lefebvre, The hepatic orosomucoid/α1-acid glycoprotein gene cluster is regulated by the nuclear bile acid receptor FXR, Endocrinology. 154 (2013) 3690–3701. doi:10.1210/en.2013-1263.
[18] A. Sirvent, T. Claudel, G. Martin, J. Brozek, V. Kosykh, R. Darteil, D.W. Hum, J.-C. Fruchart, B. Staels, The farnesoid X receptor induces very low density lipoprotein receptor gene expression, FEBS Lett. 566 (2004) 173–177. doi:10.1016/j.febslet.2004.04.026.
[19] F. Nevens, P. Andreone, G. Mazzella, S.I. Strasser, C. Bowlus, P. Invernizzi, J.P.H. Drenth, P.J. Pockros, J. Regula, U. Beuers, M. Trauner, D.E. Jones, A. Floreani, S. Hohenester, V. Luketic, M. Shiffman, K.J. van Erpecum, V. Vargas, C. Vincent, G.M. Hirschfield, H. Shah, B. Hansen, K.D. Lindor, H.-U. Marschall, K.V. Kowdley, R. Hooshmand-Rad, T. Marmon, S. Sheeron, R. Pencek, L. MacConell, M. Pruzanski, D. Shapiro, POISE Study Group, A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis, N. Engl. J. Med. 375 (2016) 631–643. doi:10.1056/NEJMoa1509840.
[20] K.V. Kowdley, V. Luketic, R. Chapman, G.M. Hirschfield, R. Poupon, C. Schramm, C. Vincent, C. Rust, A. Parés, A. Mason, H.-U. Marschall, D. Shapiro, L. Adorini, C. Sciacca, T. Beecher-Jones, O. Böhm, R. Pencek, D. Jones, Obeticholic Acid PBC Monotherapy Study Group, A randomized trial of obeticholic acid monotherapy in patients with primary biliary cholangitis, Hepatol. Baltim. Md. 67 (2018) 1890–1902. doi:10.1002/hep.29569.
[21] G.M. Hirschfield, A. Mason, V. Luketic, K. Lindor, S.C. Gordon, M. Mayo, K.V. Kowdley, C. Vincent, H.C. Bodhenheimer, A. Parés, M. Trauner, H.-U. Marschall, L. Adorini, C. Sciacca, T. Beecher-Jones, E. Castelloe, O. Böhm, D. Shapiro, Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid, Gastroenterology. 148 (2015) 751-761.e8. doi:10.1053/j.gastro.2014.12.005.
[22] A.A. Ashla, Y. Hoshikawa, H. Tsuchiya, K. Hashiguchi, M. Enjoji, M. Nakamuta, A. Taketomi, Y. Maehara, K. Shomori, A. Kurimasa, I. Hisatome, H. Ito, G. Shiota, Genetic analysis of expression profile involved in retinoid metabolism in non-alcoholic fatty liver disease, Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 40 (2010) 594–604. doi:10.1111/j.1872-034X.2010.00646.x.
[23] G.V. Chaves, S.E. Pereira, C.J. Saboya, D. Spitz, C.S. Rodrigues, A. Ramalho, Association between liver vitamin A reserves and severity of nonalcoholic fatty liver disease in the class III obese following bariatric surgery, Obes. Surg. 24 (2014) 219–224. doi:10.1007/s11695-013-1087-8.
[24] P. Pettinelli, B.M. Arendt, A. Teterina, I. McGilvray, E.M. Comelli, S.K. Fung, S.E. Fischer, J.P. Allard, Altered hepatic genes related to retinol metabolism and plasma retinol in patients with non-alcoholic fatty liver disease, PloS One. 13 (2018) e0205747. doi:10.1371/journal.pone.0205747.
[25] J.R. Phillips, P. Angulo, T. Petterson, K.D. Lindor, Fat-soluble vitamin levels in patients with primary biliary cirrhosis, Am. J. Gastroenterol. 96 (2001) 2745–2750. doi:10.1111/j.1572-0241.2001.04134.x.

FXR and hepatic vitamin A
222
[26] J.M. Erickson, A.R. Mawson, Possible role of endogenous retinoid (Vitamin A) toxicity in the pathophysiology of primary biliary cirrhosis, J. Theor. Biol. 206 (2000) 47–54. doi:10.1006/jtbi.2000.2102.
[27] R.A. Jorgensen, K.D. Lindor, J.S. Sartin, N.F. LaRusso, R.H. Wiesner, Serum lipid and fat-soluble vitamin levels in primary sclerosing cholangitis, J. Clin. Gastroenterol. 20 (1995) 215–219.
[28] C.J. Sinal, M. Tohkin, M. Miyata, J.M. Ward, G. Lambert, F.J. Gonzalez, Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis, Cell. 102 (2000) 731–744.
[29] E. Hambruch, S. Miyazaki-Anzai, U. Hahn, S. Matysik, A. Boettcher, S. Perović-Ottstadt, T. Schlüter, O. Kinzel, H.D. Krol, U. Deuschle, M. Burnet, M. Levi, G. Schmitz, M. Miyazaki, C. Kremoser, Synthetic farnesoid X receptor agonists induce high-density lipoprotein-mediated transhepatic cholesterol efflux in mice and monkeys and prevent atherosclerosis in cholesteryl ester transfer protein transgenic low-density lipoprotein receptor (-/-) mice, J. Pharmacol. Exp. Ther. 343 (2012) 556–567. doi:10.1124/jpet.112.196519.
[30] B.B. Madison, L. Dunbar, X.T. Qiao, K. Braunstein, E. Braunstein, D.L. Gumucio, Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine, J. Biol. Chem. 277 (2002) 33275–33283. doi:10.1074/jbc.M204935200.
[31] M. Boesjes, V.W. Bloks, J. Hageman, T. Bos, T.H. van Dijk, R. Havinga, H. Wolters, J.W. Jonker, F. Kuipers, A.K. Groen, Hepatic farnesoid X-receptor isoforms α2 and α4 differentially modulate bile salt and lipoprotein metabolism in mice, PloS One. 9 (2014) e115028. doi:10.1371/journal.pone.0115028.
[32] Y.-K. Kim, L. Quadro, Reverse-phase high-performance liquid chromatography (HPLC) analysis of retinol and retinyl esters in mouse serum and tissues, Methods Mol. Biol. Clifton NJ. 652 (2010) 263–275. doi:10.1007/978-1-60327-325-1_15.
[33] H. Blokzijl, S. Vander Borght, L.I.H. Bok, L. Libbrecht, M. Geuken, F.A.J. van den Heuvel, G. Dijkstra, T.A.D. Roskams, H. Moshage, P.L.M. Jansen, K.N. Faber, Decreased P-glycoprotein (P-gp/MDR1) expression in inflamed human intestinal epithelium is independent of PXR protein levels, Inflamm. Bowel Dis. 13 (2007) 710–720. doi:10.1002/ibd.20088.
[34] A. Pellicoro, F.A.J. van den Heuvel, M. Geuken, H. Moshage, P.L.M. Jansen, K.N. Faber, Human and rat bile acid-CoA:amino acid N-acyltransferase are liver-specific peroxisomal enzymes: implications for intracellular bile salt transport, Hepatol. Baltim. Md. 45 (2007) 340–348. doi:10.1002/hep.21528.
[35] B. Stieger, B. Hagenbuch, L. Landmann, M. Höchli, A. Schroeder, P.J. Meier, In situ localization of the hepatocytic Na+/Taurocholate cotransporting polypeptide in rat liver, Gastroenterology. 107 (1994) 1781–1787.
[36] A.H. Fischer, K.A. Jacobson, J. Rose, R. Zeller, Hematoxylin and eosin staining of tissue and cell sections, CSH Protoc. 2008 (2008) pdb.prot4986.
[37] A.C. Croce, U. De Simone, I. Freitas, E. Boncompagni, D. Neri, U. Cillo, G. Bottiroli, Human liver autofluorescence: an intrinsic tissue parameter discriminating normal and diseased conditions, Lasers Surg. Med. 42 (2010) 371–378. doi:10.1002/lsm.20923.
[38] A.C. Croce, U. De Simone, M. Vairetti, A. Ferrigno, E. Boncompagni, I. Freitas, G. Bottiroli, Liver autofluorescence properties in animal model under altered nutritional conditions, Photochem. Photobiol. Sci. Off. J. Eur. Photochem. Assoc. Eur. Soc. Photobiol. 7 (2008) 1046–1053. doi:10.1039/b804836c.

Chapter 7
223
[39] M. Miyata, Y. Sakaida, H. Matsuzawa, K. Yoshinari, Y. Yamazoe, Fibroblast growth factor 19 treatment ameliorates disruption of hepatic lipid metabolism in farnesoid X receptor (Fxr)-null mice, Biol. Pharm. Bull. 34 (2011) 1885–1889.
[40] M. Bjursell, M. Wedin, T. Admyre, M. Hermansson, G. Böttcher, M. Göransson, D. Lindén, K. Bamberg, J. Oscarsson, M. Bohlooly-Y, Ageing Fxr deficient mice develop increased energy expenditure, improved glucose control and liver damage resembling NASH, PloS One. 8 (2013) e64721. doi:10.1371/journal.pone.0064721.
[41] J. Schmitt, B. Kong, B. Stieger, O. Tschopp, S.M. Schultze, M. Rau, A. Weber, B. Müllhaupt, G.L. Guo, A. Geier, Protective effects of farnesoid X receptor (FXR) on hepatic lipid accumulation are mediated by hepatic FXR and independent of intestinal FGF15 signal, Liver Int. Off. J. Int. Assoc. Study Liver. 35 (2015) 1133–1144. doi:10.1111/liv.12456.
[42] L. Sheng, P.K. Jena, H.-X. Liu, K.M. Kalanetra, F.J. Gonzalez, S.W. French, V.V. Krishnan, D.A. Mills, Y.-J.Y. Wan, Gender Differences in Bile Acids and Microbiota in Relationship with Gender Dissimilarity in Steatosis Induced by Diet and FXR Inactivation, Sci. Rep. 7 (2017) 1748. doi:10.1038/s41598-017-01576-9.
[43] A.C. Nathwani, J.T. Gray, C.Y.C. Ng, J. Zhou, Y. Spence, S.N. Waddington, E.G.D. Tuddenham, G. Kemball-Cook, J. McIntosh, M. Boon-Spijker, K. Mertens, A.M. Davidoff, Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver, Blood. 107 (2006) 2653–2661. doi:10.1182/blood-2005-10-4035.
[44] A.C. Nathwani, M. Cochrane, J. McIntosh, C.Y.C. Ng, J. Zhou, J.T. Gray, A.M. Davidoff, Enhancing transduction of the liver by adeno-associated viral vectors, Gene Ther. 16 (2009) 60–69. doi:10.1038/gt.2008.137.
[45] H. Ronne, C. Ocklind, K. Wiman, L. Rask, B. Obrink, P.A. Peterson, Ligand-dependent regulation of intracellular protein transport: effect of vitamin a on the secretion of the retinol-binding protein, J. Cell Biol. 96 (1983) 907–910.
[46] J.L. Dixon, D.S. Goodman, Studies on the metabolism of retinol-binding protein by primary hepatocytes from retinol-deficient rats, J. Cell. Physiol. 130 (1987) 14–20. doi:10.1002/jcp.1041300104.
[47] D. Bellovino, Y. Lanyau, I. Garaguso, L. Amicone, C. Cavallari, M. Tripodi, S. Gaetani, MMH cells: An in vitro model for the study of retinol-binding protein secretion regulated by retinol, J. Cell. Physiol. 181 (1999) 24–32. doi:10.1002/(SICI)1097-4652(199910)181:1<24::AID-JCP3>3.0.CO;2-0.
[48] H.D. Dawson, Y. Yamamoto, R. Zolfaghari, F.J. Rosales, J. Dietz, T. Shimada, N. Li, A.C. Ross, Regulation of hepatic vitamin A storage in a rat model of controlled vitamin A status during aging, J. Nutr. 130 (2000) 1280–1286. doi:10.1093/jn/130.5.1280.
[49] J. Sundboom, J.A. Olson, Effect of aging on the storage and catabolism of vitamin A in mice, Exp. Gerontol. 19 (1984) 257–265.
[50] P.J. Trivedi, G.M. Hirschfield, M.E. Gershwin, Obeticholic acid for the treatment of primary biliary cirrhosis, Expert Rev. Clin. Pharmacol. 9 (2016) 13–26. doi:10.1586/17512433.2015.1092381.
[51] S. Fiorucci, E. Distrutti, P. Ricci, V. Giuliano, A. Donini, F. Baldelli, Targeting FXR in cholestasis: hype or hope, Expert Opin. Ther. Targets. 18 (2014) 1449–1459. doi:10.1517/14728222.2014.956087.
[52] V. Sepe, C. Festa, B. Renga, A. Carino, S. Cipriani, C. Finamore, D. Masullo, F. del Gaudio, M.C. Monti, S. Fiorucci, A. Zampella, Insights on FXR selective

FXR and hepatic vitamin A
224
modulation. Speculation on bile acid chemical space in the discovery of potent and selective agonists, Sci. Rep. 6 (2016). doi:10.1038/srep19008.
[53] H. Tu, A.Y. Okamoto, B. Shan, FXR, a bile acid receptor and biological sensor, Trends Cardiovasc. Med. 10 (2000) 30–35.
[54] G. Lambert, M.J.A. Amar, G. Guo, H.B. Brewer, F.J. Gonzalez, C.J. Sinal, The farnesoid X-receptor is an essential regulator of cholesterol homeostasis, J. Biol. Chem. 278 (2003) 2563–2570. doi:10.1074/jbc.M209525200.
[55] B. Cariou, D. Duran-Sandoval, F. Kuipers, B. Staels, Farnesoid X receptor: a new player in glucose metabolism?, Endocrinology. 146 (2005) 981–983. doi:10.1210/en.2004-1595.
[56] V. Massafra, A. Milona, H.R. Vos, R.J.J. Ramos, J. Gerrits, E.C.L. Willemsen, J.M. Ramos Pittol, N. Ijssennagger, M. Houweling, H.C.M.T. Prinsen, N.M. Verhoeven-Duif, B.M.T. Burgering, S.W.C. van Mil, Farnesoid X Receptor Activation Promotes Hepatic Amino Acid Catabolism and Ammonium Clearance in Mice, Gastroenterology. 152 (2017) 1462-1476.e10. doi:10.1053/j.gastro.2017.01.014.
[57] S. Seok, T. Fu, S.-E. Choi, Y. Li, R. Zhu, S. Kumar, X. Sun, G. Yoon, Y. Kang, W. Zhong, J. Ma, B. Kemper, J.K. Kemper, Transcriptional regulation of autophagy by an FXR-CREB axis, Nature. 516 (2014) 108–111. doi:10.1038/nature13949.
[58] Y.-D. Wang, W.-D. Chen, M. Wang, D. Yu, B.M. Forman, W. Huang, Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response, Hepatol. Baltim. Md. 48 (2008) 1632–1643. doi:10.1002/hep.22519.
[59] L. Verbeke, I. Mannaerts, R. Schierwagen, O. Govaere, S. Klein, I. Vander Elst, P. Windmolders, R. Farre, M. Wenes, M. Mazzone, F. Nevens, L.A. van Grunsven, J. Trebicka, W. Laleman, FXR agonist obeticholic acid reduces hepatic inflammation and fibrosis in a rat model of toxic cirrhosis, Sci. Rep. 6 (2016) 33453. doi:10.1038/srep33453.
[60] J.K. Kemper, Regulation of FXR transcriptional activity in health and disease: Emerging roles of FXR cofactors and post-translational modifications, Biochim. Biophys. Acta. 1812 (2011) 842–850. doi:10.1016/j.bbadis.2010.11.011.
[61] L. Liu, L.J. Gudas, Disruption of the lecithin:retinol acyltransferase gene makes mice more susceptible to vitamin A deficiency, J. Biol. Chem. 280 (2005) 40226–40234. doi:10.1074/jbc.M509643200.
[62] J.C. Correia, J. Massart, J.F. de Boer, M. Porsmyr-Palmertz, V. Martínez-Redondo, L.Z. Agudelo, I. Sinha, D. Meierhofer, V. Ribeiro, M. Björnholm, S. Sauer, K. Dahlman-Wright, J.R. Zierath, A.K. Groen, J.L. Ruas, Bioenergetic cues shift FXR splicing towards FXRα2 to modulate hepatic lipolysis and fatty acid metabolism, Mol. Metab. 4 (2015) 891–902. doi:10.1016/j.molmet.2015.09.005.
[63] S.C. Kim, C.-K. Kim, D. Axe, A. Cook, M. Lee, T. Li, N. Smallwood, J.Y.L. Chiang, J.P. Hardwick, D.D. Moore, Y.K. Lee, All-trans-retinoic acid ameliorates hepatic steatosis in mice by a novel transcriptional cascade, Hepatol. Baltim. Md. 59 (2014) 1750–1760. doi:10.1002/hep.26699.
[64] S. Kersten, R. Stienstra, The role and regulation of the peroxisome proliferator activated receptor alpha in human liver, Biochimie. 136 (2017) 75–84. doi:10.1016/j.biochi.2016.12.019.
[65] M. Pawlak, P. Lefebvre, B. Staels, Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease, J. Hepatol. 62 (2015) 720–733. doi:10.1016/j.jhep.2014.10.039.

Chapter 7
225
[66] H.A. Cyphert, X. Ge, A.B. Kohan, L.M. Salati, Y. Zhang, F.B. Hillgartner, Activation of the farnesoid X receptor induces hepatic expression and secretion of fibroblast growth factor 21, J. Biol. Chem. 287 (2012) 25123–25138. doi:10.1074/jbc.M112.375907.
[67] F. Lalloyer, T.A. Pedersen, B. Gross, S. Lestavel, S. Yous, E. Vallez, J.-A. Gustafsson, S. Mandrup, C. Fiévet, B. Staels, A. Tailleux, Rexinoid bexarotene modulates triglyceride but not cholesterol metabolism via gene-specific permissivity of the RXR/LXR heterodimer in the liver, Arterioscler. Thromb. Vasc. Biol. 29 (2009) 1488–1495. doi:10.1161/ATVBAHA.109.189506.
[68] S.B. Joseph, B.A. Laffitte, P.H. Patel, M.A. Watson, K.E. Matsukuma, R. Walczak, J.L. Collins, T.F. Osborne, P. Tontonoz, Direct and indirect mechanisms for regulation of fatty acid synthase gene expression by liver X receptors, J. Biol. Chem. 277 (2002) 11019–11025. doi:10.1074/jbc.M111041200.
[69] D. Herebian, E. Mayatepek, Inborn errors of bile acid metabolism and their diagnostic confirmation by means of mass spectrometry, J. Pediatr. Sci. 3 (2011). doi:10.17334/jps.55342.
[70] S.S. Sundaram, K.E. Bove, M.A. Lovell, R.J. Sokol, Mechanisms of disease: Inborn errors of bile acid synthesis, Nat. Clin. Pract. Gastroenterol. Hepatol. 5 (2008) 456–468. doi:10.1038/ncpgasthep1179.
[71] R.D. Jones, J.J. Repa, D.W. Russell, J.M. Dietschy, S.D. Turley, Delineation of biochemical, molecular, and physiological changes accompanying bile acid pool size restoration in Cyp7a1−/− mice fed low levels of cholic acid, Am. J. Physiol. - Gastrointest. Liver Physiol. 303 (2012) G263–G274. doi:10.1152/ajpgi.00111.2012.

FXR and hepatic vitamin A
226
Supplementary Figure S1: Hepatic and serum vitamin A level in mice fed-vitamin A deficient diet. Four (4)-week old mice were fed either a chow diet (H10293G, Hua Fukang Biological, technology, Beijing, China) with 4 IU of retinyl acetate/g as a vitamin A source or a vitamin A deficient (VAD) diet (H10293G, Hua Fukang Biological, technology, Beijing, China). Hepatic Retinol and retinyl palmitate levels were analyzed after 4, 8 and 12 weeks diet. Hepatic retinyl palmitate and retinol levels were significantly reduced already after 4 weeks in mice fed a VAD diet while serum retinol levels were stable in both groups of mice during the whole course of the dietary intervention.
Supplementary Figure S2: A heat map of significantly altered KEGG pathway associated with liver-specific FXR deletion. KEGG analysis of liver-specific FXR-null mice vs wild type mice was performed using online available DAVID 6.7 software on differentially regulated genes in publicly available microarray data (E-MTAB-1722). A heat map was generated from the –log10 p-value of the significantly altered KEGG pathways associated with liver-specific FXR-deletion vs control. Fatty acid metabolism was the top hit in this analysis, however, retinol metabolism was not significantly associated with L-FXR deletion in the liver of mice.

Chapter 7
227
Supplementary Table S1: Primers and probes used in study for analysis of target genes
Gene / ID Taqman primers and probe
36B4 NM_022402
Fwd: 5'-GCTTCATTGTGGGAGCAGACA-3' Rev: 5'-CATGGTGTTCTTGCCCATCAG-3' Probe: 5'-TCCAAGCAGATGCAGCAGATCCGC-3'
Abcb11 / Bsep NM_021022.3
Fwd: 5'-CTGCCAAGGATGCTAATGCA -3' Rev: 5'-CGATGGCTACCCTTTGCTTCT-3' Probe: 5'-TGCCACAGCAATTTGACACCCTAGTTGG-3'
Acaca / Acc1 NM_133360.1 / NM_022193.1
Fwd: 5'-GCCATTGGTATTGGGGCTTAC-3' Rev: 5'-CCCGACCAAGGACTTTGTTG-3' Probe: 5'-CTCAACCTGGATGGTTCTTTGTCCCAGC-3'
Cpt1a NM_013495.1
Fwd: 5'-CTCAGTGGGAGCGACTCTTCA-3' Rev: 5'-GGCCTCTGTGGTACACGACAA-3' Probe: 5'-CCTGGGGAGGAGACAGACACCATCCAAC-3'
Cyp26a1 NM_007811.1
Fwd: 5'-GGAGACCCTGCGATTGAATC-3' Rev: 5'-GATCTGGTATCCATTCAGCTCAAA-3' Probe: 5'-TCTTCAGAGCAACCCGAAACCCTCC-3'
Dgat1 NM_010046.2 / NM_053437.1
Fwd: 5'-GGTGCCCTGACAGAGCAGAT-3' Rev: 5'-CAGTAAGGCCACAGCTGCTG-3' Probe: 5'-CTGCTGCTACATGTGGTTAACCTGGCCA-3'
Dgat2 NM_026384.2 / NM_001012345.1
Fwd: 5'-GGGTCCAGAAGAAGTTCCAGAAG-3' Rev: 5'-CCCAGGTGTCAGAGGAGAAGAG-3' Probe: 5'-CCCCTGCATCTTCCATGGCCG-3'
Fasn NM_007988 / NM_017332
Fwd: 5'-GGCATCATTGGGCACTCCTT-3' Rev: 5'-GCTGCAAGCACAGCCTCTCT-3' Probe: 5'-CCATCTGCATAGCCACAGGCAACCTC-3'
Fgf21 NM_020013.4 / NM_130752.1
Fwd: 5'-CCGCAGTCCAGAAAGTCTCC-3' Rev: 5'-TGACACCCAGGATTTGAATGAC-3' Probe: 5'-CCTGGCTTCAAGGCTTTGAGCTCC A-3'
Lipe / HSL NM_010719 / X51415
Fwd: 5'-GAGGCCTTTGAGATGCCACT-3' Rev: 5'-AGATGAGCCTGGCTAGCACAG-3' Probe: 5'-CCATCTCACCTCCCTTGGCACACAC-3'
Lrat NM_023624
Fwd: 5'-TCCATACAGCCTACTGTGGAACA-3' Rev: 5'-CTTCACGGTGTCATAGAACTTCTCA-3' Probe: 5'-ACTGCAGATATGGCTCTCGGATCAGTCC-3'
Nr0b2 / Shp
Fwd: 5'-CCTTCTGGAGCCTGGAGCTTA -3' Rev: 5'-CTGGCACATCGGGGTTGA-3' Probe: 5'-ATGGTCCCTTTCAGGCAGGCATATTCCTT-3'
Pck1 NM_011044 / NM_198780
Fwd: 5'-GTGTCATCCGCAAGCTGAAG-3' Rev: 5'-CTTTCGATCCTGGCCACATC-3' Probe: 5'-CAACTGTTGGCTGGCTCTCACTGACCC-3'
Plin2 NM_175640.2 / NM_001113471.1
Fwd: 5'-AGAACGTGCTCAGAGAGGTTACAG-3' Rev: 5'-GTGTTCTGCACGGTGTGTACC-3' Probe: 5-CCTGCCCAACCCGAGAGGCC-3'
Ppargc1 α / Pgc1α NM_008904 / NM_031347
Fwd: 5'-GACCCCAGAGTCACCAAATGA-3' Rev: 5'-GGCCTGCAGTTCCAGAGAGT-3' Probe: 5'-CCCCATTTGAGAACAAGACTATTGAGCGAACC-3'
Pnpla2 / Atgl NM_025802 / XM_347183
Fwd: 5'-AGCATCTGCCAGTATCTGGTGAT-3' Rev: 5'-CACCTGCTCAGACAGTCTGGAA-3' Probe: 5'-ATGGTCACCCAATTTCCTCTTGGCCC-3'
Pnpla3 NM_054088
Fwd: 5'-ATCATGCTGCCCTGCAGTCT-3' Rev: 5'-GCCACTGGATATCATCCTGGAT-3' Probe: 5'-CACCAGCCTGTGGACTGCAGCG-3'
Raldh1 Assay on demand, Mm00657317_m1 (ThermoFisher)
Raldh2 Assay on demand, Mm00501306_m1 (ThermoFisher)
Raldh4 Fwd: 5'-TGGAGCAGTCTCTGGAGGAGTT-3'

FXR and hepatic vitamin A
228
NM_178713.4 Rev: 5'- GAAGTTCAGAACAGACCGAGGAA-3' Probe: 5'- AATCTAAAGACCAAGGGAAAACCCTCACGC-3'
Ucp2 NM_011671.2
Fwd: 5'-CGAAGCCTACAAGACCATTGC-3' Rev: 5'-ACCAGCTCAGCACAGTTGACA-3' Probe: 5'-CAGAGGCCCCGGATCCCTTCC-3'

Chapter 8
General Discussion
Ali Saeed1,2, Klaas Nico Faber1,3
1Department of Gastroenterology and Hepatology, 3Laboratory Medicine, University
Medical Center Groningen, University of Groningen, Groningen, The Netherlands.
2Institute of Molecular Biology and Biotechnology, Bahauddin Zakariya University
Multan, Pakistan

Discussion
230
The liver produces bile for efficient hepatic secretion of fat-soluble waste products, as
well as effective absorption of fat-soluble nutrients in the intestine, including vitamin A.
Bile acids are essential components in bile for this physiologically important process.
Excess vitamin A is efficiently stored in the liver, from which it is released in a highly
controlled manner in times of insufficient dietary intake. However, the interdependence
between vitamin A and bile acids goes far beyond the simple requirement of bile acids
for intestinal absorption of (pro-)vitamin A. Vitamin A metabolites collaborate with bile
acids to control key physiological processes, like glucose and lipid metabolism as well
as immune regulation. Moreover, they control their own (bile acid and vitamin A)
homeostasis. The retinoic acid receptor (RAR), retinoid X receptor (RXR) and
farnesoid X receptor (FXR) are the central players that control these processes [1–3].
The liver plays a central role in controlling vitamin A metabolism and chronic liver
diseases, such as biliary atresia, primary biliary cholangitis (PBC), primary sclerosing
cholangitis (PSC), viral hepatitis, alcoholic liver disease (ALD), and non-alcoholic fatty
liver disease (NAFLD and the progressive form non-alcoholic steatohepatitis [NASH])
are associated with impaired vitamin A homeostasis that may lead to vitamin A
deficiency [1]. Animal studies have shown the beneficial effects of vitamin A
derivatives, e.g., retinoic acids or pharmacological modulators of RARs and RXRs, in
the treatment of chronic liver diseases. However, oral vitamin A supplements appear
not very effective in restoring adequate serum vitamin A (retinol) levels in NAFLD
patients, suggesting that vitamin A metabolism is intrinsically impaired. Relatively little
is known, however, about changes in vitamin A metabolism in the liver diseases,
including possible changes in storage (retinyl esters) vs. circulating (retinol, RA) forms
and possible redirection of vitamin A pools within the liver (hepatocytes vs. hepatic
stellate cells) and/or to extrahepatic tissues. Insight into these processes is needed to
get a better view of the possible therapeutic application of vitamin A and/or its
derivatives (retinoids acting on RARs and rexinoids acting on RXRs) in the liver
diseases. Therefore in this thesis, we studied vitamin A metabolism in patients, as well
as animal and cellular models of chronic liver diseases.
The key findings reported in this thesis are: 1) Quiescence hepatic stellate cells (qHSC)
expresses hormone-sensitive lipase (HSL), which harbors retinyl ester hydrolyzing
activity and contributes to hepatic vitamin A metabolism (chapter 3). 2) NAFLD is
associated with perturbed vitamin A metabolism characterized by low hepatic retinol
levels, while circulatory retinol and RBP4 levels increased in NAFLD. Moreover, retinyl

Chapter 8
231
palmitate (being the most dominant retinyl ester) levels are clearly enhanced in NAFLD
and accumulate in hepatocytes (rather than in HSC) (chapter 5). An important
technical issue addressed in this chapter is that the accurate quantification of retinyl
esters in fatty livers is highly dependent on the lipid extraction method used. 3) fatty
liver that is associated with glycogen storage disease type 1a (GSD Ia) is also
characterized by impaired vitamin A metabolism leading to increased serum retinol and
RBP4 levels (chapter 6). 4) Manipulation of FXR activity in mice (either by gene
deletion or pharmacological activation) effectively deplete hepatic vitamin A stores, in
which hepatic FXR plays a key role (chapter 7).
A main finding of these studies is that metabolic diseases, as well as drug therapies to
treat them, heavily affect vitamin A metabolism in the liver that is not reflected at all by
the serum retinol levels. This may be good for providing peripheral tissues with
necessary vitamin A, but may go at the expense of disease progression in the liver.
Thus, manipulation of vitamin A metabolism may hold great promise in the treatment
of chronic liver diseases, but requires more sophisticated approaches than simply
(oral) vitamin A supplementation when serum retinol levels are low.
8.1. Hormone-sensitive lipase (HSL) acts as a retinyl ester hydrolase in
quiescence hepatic stellate cells (qHSC)
Vitamin A homeostasis in the liver is a result of a fine-tuned balance between hepatic
retinol esterification (storage) and retinyl ester hydrolysis (release). Lecithin retinol
acyltransferase (LRAT) is the major enzyme involved in retinol esterification, while
adipose triglyceride lipase (ATGL) and patatin-like phospholipase domain-containing
protein 3 (PNPLA3) have been previously been identified as retinyl ester hydrolases
(REHs). In chapter 3, we identified HSL as a 3rd REH in qHSC that contributes to
hepatic vitamin A metabolism. HSL has a broad substrate-specificity and besides
acting as a REH, it also converts cholesterol esters and triglycerides to free cholesterol
and free fatty acids, respectively. We found that mRNA levels of Lipe, the gene
encoding HSL, were actually higher qHSC as compared to hepatocytes, which may
point to a broader role of qHSC-located HSL in hepatic lipid metabolism. Our study
shows that HSL is mostly in its active phosphorylated state in qHSL, but
pharmacological induction of HSL-phosphorylation further enhances retinyl ester
hydrolysis in qHSC.

Discussion
232
So far, limited data is available on the expression and function of HSL in the liver, while
its retinyl ester hydrolase activity has been well-documented for adipose tissue [4].
HSL also hydrolyses cholesterol esters, triglycerides, mono- and di-acylglycerol and
other lipids [5]. However, in vitro studies using recombinant HSL indicate that retinyl
esters are the preferred substrate for HSL as compared to diacylglycerols, which was
considered the best substrate for HSL until that report [4]. Earlier studies analyzed the
putative presence of HSL in HSC, but were unable to confirm this. [6,7]. We cannot
fully explain this apparent discrepancy at this point, other than that these earlier studies
may have used (partly) activated HSC [6]. Moreover, Lipe expression was not detected
in any type of liver cell [7], while it has been detected by many others in total liver tissue
[5,8–10]. Our data suggest that HSL is not only dominantly present in qHSC, but also
likely regulates retinyl ester hydrolysis along with lipases (ATGL and PNPLA3) to
control the intracellular lipid turnover in HSC, especially in the early phase of HSC
activation. However, the relative contribution of each lipase in retinyl esters hydrolysis
in qHSC still needs to be determined.
Pharmacological inhibitors of HSL display great therapeutic potential in various
metabolic complications, including dyslipidemia, elevated blood glucose levels and
insulin resistance [11–13]. HSL null mice accumulate retinyl esters in adipose tissue
[4], while the hepatic retinyl ester pool is comparable to WT control animal [6]. This
may be explained by the fact that HSC expresses 3 different REH, ATGL, PNPLA3
and HSL, that can compensate for each other absence. Indeed, also ATGL-null mice
do not have altered hepatic retinyl ester levels [6]. It may actually require a triple knock
of Pnpla2 (ATGL), Pnpla3 and Lipe (HSL) to sufficiently eliminate REH activity in the
liver. Cholesteryl esters accumulated in whole body HSL-null mice [5,14], but this
phenotype was not replicated in hepatocyte-specific HSL-null mice [15]. This indicates
the possible involvement of another cell type in the accumulation of cholesterol esters,
which, taken our data in account, most likely are HSC. It is well-known that
accumulation of cholesterol and retinyl esters in HSC also disturbs the intrinsic vitamin
A metabolism. For example, Lxrαβ-/- mice accumulate cholesterol and retinyl esters
and enhance intrinsic retinoic acid receptor signaling [16]. Primary HSC isolated from
Lxrαβ-/- mice rapidly lose vitamin A stores and quickly acquire a more fibrogenic
phenotype during culture activation. Moreover, Lxrαβ-/- mice show increased
susceptibility to CCL4-induced liver injury as compared to control animals, and is –in
part- the result of enhanced intrinsic RAR-signaling [16]. Inversely, our results with the

Chapter 8
233
pharmacological activation of HSL indicate that enhanced hydrolysis of intracellular
retinyl esters in HSC combined with reduced retinoic acid production (in thereby
decreases intrinsic RAR-signaling) is associated with reduced the proliferation and
activation of in vitro-cultured HSC. Collectively, these data support the notion that
intrinsic retinoic acid metabolism directly impacts on HSC activation.
It needs to be noted that HSL is also expressed in other liver cells types, such as
Kupffer cells, cholangiocytes and possibly other cells in the biliary tree (Chapter 3).
Future studies need to address its role in these cell types in liver health and disease.
8.2. Redundancy of retinyl ester hydrolases in the liver/stellate cell (e.g. ATGL.
PNPLA3 and HSL)
Together with our results presented in chapter 3, there are now three enzymes
identified that are involved in retinyl ester hydrolysis in HSC, e.g. ATGL/PNPLA2,
ADPN/PNPLA3, and HSL/LIPE. Their relative contribution to vitamin A metabolism,
however, will strongly depend on the activation state of HSC, as we show that
HSL/LIPE and ADPN/PNPLA3 expression is rapidly lost when qHSC transdifferentiate
to aHSC, while ATGL/PNPLA2 levels remain the same. These gene-specific
expression characteristics also explain earlier observations that pharmacological
inhibition of ATGL leads to accumulation of retinyl esters in in vitro-cultured activated
mouse HSC, while this was not observed with HSL inhibitors [6]. Assuming that the
regulation of Pnpla2 and Lipe is the same in mouse and rat HSC, Lipe expression is
extremely low in mouse aHSC and thus blocking its activity does not resort in a
measurable effect on vitamin A metabolism in these cells. Whole body knock-out mice
for either Pnpla2 or Lipe did not show altered levels of hepatic retinyl esters [6]. As
these animals were not exposed to a fibrosis-inducing liver disease model, it is
expected that all three different REHs are expressed in the quiescent HSC in the
healthy livers, and can largely compensate for each other’s absence. Interestingly,
though, levels of retinyl esters in both human liver [17] and isolated primary HSC [18]
are elevated in patients/cells homozygous for the PNPLA3 I148M allele. The enzyme
activity of this variant of ADPN is markedly decreased. This observation may imply that
ADPN/PNPLA3 may be the most dominant REH in the liver. Still, it cannot be excluded
that the impaired function of ADPN may also affect the expression and/or activity of
the other REHs, as for instance the absence of ATGL causes hyper-
phosphorylation/activation of HSL [6]. Evidently, HSC harbor multiple and

Discussion
234
interconnected mechanisms to secure efficient hydrolysis of retinyl esters, most likely
in order to maintain release of retinol to the circulation in times where dietary input of
vitamin A is insufficient. This seems in contrast to the mechanism of hepatic vitamin A
storage, is this (almost) completely depends on the enzyme LRAT [19]. Future studied
using single, double and triple knock-out models of the different REHs need to
establish the individual contribution of these enzymes in regulating hepatic vitamin A
stores.
8.3. Impaired vitamin A metabolism is associated with NAFLD
NAFLD is the most prevalent chronic liver disease and its association with impaired
vitamin A status is well-documented. In chapter 4, we reviewed the current knowledge
about vitamin A metabolism in NAFLD, as well as the overlap with Type 2 diabetes
(T2D) and metabolic syndrome (MetS). Most studies show that NAFLD is associated
with vitamin A insufficiency or even vitamin A deficiency (VAD). This is primarily based
on analysis of circulating retinol, where serum levels between 0.7 and 1.05 µmol/L are
considered insufficient and below 0.7 µmol/L as deficient. Only a few anecdotal reports
analyzed hepatic retinol and/or retinyl esters, which also seem reduced in fatty livers,
but controversy exists on this topic.
This prompted us to analyze hepatic vitamin A metabolism in more detail in chapter
5. We indeed confirmed reduced retinol levels in fatty livers in mice. In sharp contrast,
however, hepatic levels of retinyl palmitate (being the most dominant retinyl ester) were
significantly increased. We noticed that the method of processing of the liver tissue for
vitamin A quantification strongly influenced the ultimate levels of retinyl palmitate
obtained, which may underlie the controversy between previous reports (discussed
later). mRNA levels of retinoic acid-responsive genes were increased suggesting that
production of retinoic acids is also enhanced in NAFLD. Interestingly, we found that
vitamin A accumulated preferentially in hepatocytes in NAFLD (rather than in stellate
cells), which may be an important additional factor contributing to disturbed vitamin A
metabolism in NAFLD.
Limited data are available in which hepatic vitamin A storage (retinyl ester levels) were
analyzed in NAFLD. However, three different studies by the same scientific group
evaluated hepatic retinyl palmitate levels in NAFLD and reported strongly reduced
retinyl esters levels in mice with NAFLD [20–22]. These contradicting results compared
to our findings have long been a puzzling issue to us, as we consistently detected
enhanced hepatic retinyl palmitate levels in the liver in different models of NAFLD mice.

Chapter 8
235
Still, circulatory and hepatic retinol levels in these studies were in line with our findings.
A critical evaluation of methods used in these and other previous studies [20,21],
revealed that this may be caused by different protocols to extract lipophilic compounds
out of the liver tissue (discussed in next section). The enhanced hepatic retinyl
palmitate levels we detected in mouse NAFLD livers, were in line with studies that
analyzed vitamin A autofluorescence in liver tissue of NAFLD patients and mice, which
also indicated a redistribution of vitamin A pools to hepatocytes [23–25]. In addition to
this, we also detected a hyperdynamic state of vitamin A metabolism in the liver of
mice with NAFLD, similar to previous patient studies [26,27]. This hypermetabolic state
was characterized by enhanced hepatic expression of the genes involved in vitamin A
storage as well as conversion to retinoic acids.
In contrast to the reduced serum retinol levels, serum RBP4 levels are typically
elevated in MetS patients and NAFLD with or without T2D [28–32]. In line, we also
found elevated serum RBP4 levels in mice with NAFLD. Hepatocytes are the primary
source of circulatory RBP4 [28], and secretion of hepatic RBP4 is strongly stimulated
by the availability of retinol [33–35] as well as retinoic acids [34]. The combination of
1) reduced RBP4 protein levels in the liver, 2) unchanged Rbp4 mRNA levels in the
liver and 3) increased mRNA levels of retinoic acid-responsive genes led us to
speculate that enhanced RBP4 release from the liver may contribute to accumulation
of RBP4 in the serum. However, impaired kidney function may also lead to elevated
circulatory RBP4 levels [36–40] as this is the organ where RBP4 is cleared. Indeed,
Western type high-fat diets have been shown to cause kidney injury as well [41–43],
thus, it remains to be determined what the organ-specific (liver versus kidney)
contributions are to the enhanced serum RBP4 levels in NAFLD. Unfortunately, we did
not collect data on kidney function in our studies analyzing NAFLD mice.
Figure 1 shows our current working model to explain the biochemical effects we
observed with respect to impaired vitamin A metabolism in NAFLD; the low hepatic
retinol levels in NAFLD are likely a cumulative effect of elevated hepatic retinyl esters
formation, excessive release of retinol and RBP4 from the liver, and enhanced retinol
metabolism to retinoic acids.

Discussion
236
Figure 1. A schematic diagram presenting the effect NAFLD on vitamin A homeostasis. 1) Retinyl esters increase and accumulate in the hepatocytes in NAFLD. 2) Elevated excretion of retinol and RBP4, increased circulatory levels in NAFLD mice, 3) but decreases hepatic levels of retinol in NAFLD. 4) Hyper-dynamic state of vitamin A metabolism increased the conversion of retinol metabolism into retinoic acid and 5) increased hepatic expression of retinoic acid-responsive genes, inflammatory markers and fibrotic markers in NAFLD. (↑)upregulated, (↓)downregulated. “Green” arrow marks increased flow and “red” arrow marks decreased flow. “Black” arrow marks normal flow.
8.4. Technical issues related to hepatic retinyl ester quantification.
Typically serum retinol levels are analyzed to assess the individuals vitamin A status.
It is, however, well-known that serum retinol levels are maintained at a very stable level
irrespective of the size of hepatic pool. Only when the hepatic pools are depleted, this
will also be reflected in circulating retinol levels. Moreover, the systemic retinol levels
are only a small fraction of the total body pool of vitamin A, which mostly consists of
retinyl esters stored in the liver (≥80%) and adipose tissue (10-20%) [44–46]. Since it
requires liver biopsies or resection material, hepatic vitamin A levels cannot easily be
analyzed in NAFLD patients. Still, a few recent studies [20–22] analyzed both hepatic
retinol and retinyl palmitate levels in human and mouse NAFLD and reported that
hepatic levels of both retinol and retinyl palmitate were strongly reduced compared to
healthy controls [20]. In chapter 5, we also detected the low hepatic retinol levels in
NAFLD mice, but, in contrast found that hepatic retinyl palmitate levels were
significantly increased. How to explain this apparent contradiction?

Chapter 8
237
A critical and thorough comparison of the protocols used for retinol and retinyl palmitate
quantification used by us and others revealed that there are different solvents in use
to extract lipophilic compounds, like retinol and retinyl esters, from serum and/or liver
tissue. We used n-hexane in our protocol, while others use acetonitrile-butanol for
retinol and retinyl palmitate extraction from liver tissue and plasma. Remarkably, in an
ono-to-one comparison of both methods using the same liver samples, we found that
these solvents show very different capacities to extract retinyl palmitate from fatty liver
tissue. Retinyl palmitate recovery was similar with both methods using healthy control,
but in sharp contrast, recovery of retinyl palmitate from fatty liver tissue was
significantly lower when using acetonitrile-butanol compared to n-hexane-based
procedures. To complex matters, the recovery of retinol from control and fatty livers
was not different between both procedures. This indicates that the extraction efficiency
of retinyl esters by acetonitrile-butanol is compromised when a lot of fat is present in
the (liver) tissue, while this is not the case for retinol. As these extraction procedures
are likely optimized for retinol extraction (mostly from plasma or serum), such a
differentiating effect on retinyl esters from fatty tissue has remained unnoticed so far.
Evidently, this technical issue heavily affects the interpretation of the results reported
so far. As we have performed the methods side-by-side in an attempt to exactly copy
the procedures used by others (using the same quantities of tissue, extraction volumes
and buffer compositions, including the use of anti-oxidants), we feel that using n-
hexane as extraction solution gives the most reliable quantification of retinyl esters.
Thus, we conclude that mouse NALFD livers are not prone to develop vitamin A
deficiency, but rather show impaired vitamin A metabolism leading to retinyl ester
accumulation, but a reduction in hepatic retinol levels. Importantly, the increased
autofluorescence of vitamin A in NAFLD livers, as observed by us (Chapter 5) and
others [23,25], further supports this interpretation.
8.5. Not vitamin A deficiency, but vitamin A metabolism should be targeted in
NAFLD
Our data indicate that NAFLD is not associated with “plain” VAD, as hepatic storage of
vitamin A in retinyl esters is even increased. Evidently, hepatic vitamin A metabolism
is disturbed in NAFLD, with cell type-specific redistribution of vitamin A pools within the
liver and reduced hepatic and serum retinol levels, possibly with increased serum
RBP4. These effects may be a result of lipid accumulation in the liver, but may also

Discussion
238
further contribute to disease progression. Accordingly, restoring proper vitamin A
metabolism in the liver is a relevant therapeutic target for the treatment of NAFLD.
Natural and synthetic ligands for RAR and RXR target cellular mechanisms that are
impaired in NAFLD, including lipogenesis, lipolysis, fatty acid β-oxidation,
adipogenesis, production of reactive oxygen species, cell proliferation and cell death
(Chapter 4 [47]). However, this may require highly-selective therapeutic targeting of
these pathways in NAFLD. Indiscriminate supplementation of vitamin A and/or its
metabolites may lead to toxicity or adverse effects. A recent study emphasized the
significance of a selective targeting approach in NAFLD and exemplified how adverse
consequences in NAFLD may arise with alternative approaches are used to target the
RAR-controlled transcription program. Selective targeting of RAR-β2 using the
pharmacological ligand AC261066 improved hyperglycemia and hepatic steatosis in
several animal models of NAFLD and type 2 diabetes, while targeting of RAR-α with
AM80 exacerbated hyperglycemia and NAFLD in such animal models [21,22].
Moreover, the natural ligand for RARs, all-trans-retinoic acid; ATRA) stimulates the
insulin secretion, while 9 cis-retinoic acid (9cRA), the ligand for RXR, suppressed
insulin secretion from pancreatic β-cells [48]. These findings support the notion that
vitamin A-responsive transcription factors are important therapeutic targets for NAFLD,
but this requires highly selective modulation of specific pathways.
Still, manipulation of endogenous intrahepatic retinoic acid synthesis and metabolism
may be another therapeutic target in NAFLD. Retinoic acid production from retinol is a
2 step process. First, retinol is converted into retinaldehyde by retinol dehydrogenases
(RDH), followed by the production of retinoic acid by retinaldehyde dehydrogenase
(RALDH) [1]. Inhibition of these enzymes suppresses the production of retinoic acids
and subsequent RAR/RXR-signaling. Some studies have already identified possible
targets relevant to the treatment of NAFLD. For example, heterozygous mice
containing only one genomic copy of the RDH10 gene give rise to a moderate (-25%)
reduction in hepatic ATRA levels, which was associated with a significant increase in
hepatic steatosis and glucose intolerance in Western diet-fed mice, while these
metabolic effects were corrected by pharmacological application of ATRA [49]. On the
other hand, inhibition of Raldh1 has also beneficial effects in animal models of NAFLD,
as it improved genetic- and diet-induced obesity, insulin resistance and energy
dissipation [50]. This therapeutic effect was primarily assigned to the accumulation of
retinyl aldehyde.

Chapter 8
239
Our data indicate that sufficient vitamin is present in the fatty liver and that vitamin A
supplementation, which could be indicated clinically due to the low serum retinol levels,
may actually work counterproductive and cause vitamin A hepatotoxicity. Instead,
supplementation or inhibition of the synthesis of particular metabolites may achieve
more desirable therapeutic effects. For example, retinaldehyde supplementation or
inhibition of retinaldehyde dehydrogenase show therapeutic potential in animal models
of NAFLD [50]. Moreover, FXR ligands, like obeticholic acid (OCA), may reduce
hepatic retinyl ester accumulation (Chapter 7) in NAFLD, and contribute to the
therapeutic effect by normalizing vitamin A metabolism in the liver.
8.6. Genetic association of two novel genes (PNPLA3 and HSD17B13) of
vitamin A metabolism with NAFLD
Interestingly, also genome-wide associations studies (GWAS) have clearly linked
impaired vitamin A metabolism to NAFLD [26,51,52]. The first one is ADPN/PNPLA3.
ADPN/PNPLA3 is a lipase that hydrolyses triglycerides into free fatty acids. A specific
SNP was identified in this gene, PNPLA3 I148M, which is strongly associated with
NAFLD and disease progression. The enzymatic activity of the PNPLA3 I148M variant
is markedly reduced, leading to hepatic steatosis [18,53]. However, PNPLA3 also
contains high REH activity and is highly expressed both in hepatocytes and HSC. As
described before, PNPLA3 I148M carriers show increased levels of retinyl ester in the
liver, in conjunction with reduced serum retinol levels [17]. So far, it is unknown how
much the impaired function of PNPLA3 I148M in vitamin A metabolism contributes to
the disease phenotype, as its role in triglyceride and retinyl ester hydrolysis go hand-
in-hand. More recently, several SNPs in the gene encoding 17β-Hydroxysteroid
dehydrogenase type 13 (HSD17B13) were found associated with specific histological
features of NAFLD such as increased steatosis, but reduced inflammation, hepatocyte
ballooning and cirrhosis. HSD17B13 proteins associates with lipid droplets in
hepatocytes and harbors retinol dehydrogenase (RDH) activity. Overexpression of
HSD17B13 markedly increased hepatic lipid accumulation [54]. Hepatic expression of
both PNPLA3 and HSD17B13 is strongly increased in NAFLD [51,55].
Collectively we can conclude that genetic, biochemical and pharmaceutical
approaches all point to a clear relationship between impaired vitamin A metabolism
and NAFLD. However, despite all the compelling evidence, no clinical studies or trials

Discussion
240
are ongoing to evaluate the therapeutic effect of selective targeting of vitamin A
metabolism in NAFLD [56].
8.7. Perturbed vitamin A metabolism is also associated with GSD Ia
Patients with glycogen storage disease Ia (GSD Ia) are at risk to develop a fatty liver.
Given our observations in chapter 5, we got interested in studying vitamin A
metabolism in this patient group as well, as fatty liver is a common pathology in GSD
Ia. Accordingly, in chapter 6 we analyzed whether GSD Ia patients and the liver-
specific glucose-6-phosphatase knockout mouse (L-G6pc-/-) mouse are at risk for
impaired vitamin A metabolism. To our surprise, we found that serum retinol and RBP4
levels were significantly enhanced in GSD Ia patients and L-G6pc-/- mice, whereas
hepatic retinol levels were significantly reduced. No change in retinyl ester levels was
observed. These results indicate that GSD Ia is a rare example of a disease
characterized by chronically elevated circulating retinol levels (a condition we termed
“metabolic hypervitaminosis A”), which may contribute to various disease symptoms,
including osteoporosis and hepatic steatosis.
GSD Ia and NAFLD share a number of common pathologies including hyperlipidemia,
hyperinsulinemia and hepatic steatosis. With respect to vitamin A metabolism, both
conditions are characterized by strongly reduced retinol levels in the liver, in
conjunction with enhanced serum RBP levels. Other than that, many features were
clearly different. Hepatic retinyl ester levels were clearly increased in the NAFLD
animal models, while they were not affected –and even showed a trend towards
reduction, in L-G6pc-/- mice. Mechanistically, this may be related to the fact that LRAT
levels, the enzyme responsible for retinol esterification, was strongly increased in
NAFLD mice, while they were not changed (at protein level) in L-G6pc-/- mice. A
schematic representation of vitamin A metabolism in GSD Ia is presented in Figure 2.

Chapter 8
241
Figure 2: A schematic diagram presenting the effect GSD Ia on vitamin A homeostasis. 1) Retinol-RBP4-TTR release from liver enhances in GSD Ia and 2) cause metabolic circulatory hypervitaminosis A and 3) reduced the tissue levels of retinol. 4) Retinoic acid conversion reduced while 5) hydrolysis of retinyl esters (vitamin A storage) was enhanced likely compensate the reduced hepatic retinol and retinoic acid levels. 6) No significant effect on retinyl esters synthesis was observed. (↑) upregulated, (↓) downregulated. “Green” arrow marks increased flow and “red” arrow marks decreased flow and “Black” arrow marks normal flow.
Interestingly, mice fat a high-fat diet (without the addition of cholesterol), also did not
further accumulate retinyl esters in the liver. This may suggest that particularly the
addition of cholesterol to the diet promotes hepatic retinyl ester accumulation. Indeed,
HFC-diets lead to a significant increase in concentrations of total and free cholesterol
in the liver features that are absent in the livers of L-G6pc-/- mice. Another clear
difference between fatty livers in the NAFLD mice and the L-G6pc-/- mice is the
differential expression of Cyp26a1, encoding an enzyme that catabolizes retinoic acids
[57]. Cyp26a1 transcription is under the strict control of retinoic acids [58], so may
serve as an indirect measure of these metabolites in the liver. Cyp26a1 mRNA levels
are evidently elevated in NAFLD mice, while the opposite (strongly decreased) was
observed in L-G6pc-/- mice. This indicated that the hepatic production of retinoic acids
is enhanced in NAFLD, while this is impaired in L-G6pc-/- mice. So far, the
mechanisms involved remain unclear. Interestingly, livers from NAFLD mice and L-
G6pc-/- mice also showed opposite features of markers of inflammation and fibrosis.
Where livers from HFC fed mice showed clear signs of inflammation and fibrosis, all
such markers were suppressed in livers of the L-G6pc-/- mice. Future research should
determine whether this might be caused by the differential effects on hepatic retinoic
acid production in these two mouse models. Alternatively, such effects may also be

Discussion
242
caused by the differences in hepatic cholesterol accumulation in these mouse models,
as described above.
Thus, much remains to be studied to delineate the molecular mechanism that leads to
impaired vitamin A metabolism in GSD Ia patients. Still, the ultimate effect seems to
be that this disease is associated with chronically elevated circulating retinol levels.
The absolute increase in serum retinol level appears relatively small, but the fact that
this is a chronic condition may mean that it does contribute to symptoms, like
osteoporosis, skin problems and steatosis. These symptoms are now typically linked
to insufficient vitamin D levels in these patients. Unfortunately, there is no easy way
the reduce serum retinol levels. In clinical management of GSD Ia, it is at least
important to be extra careful with vitamin A-containing supplements.
8.8. Farnesoid X receptor (FXR) and bile acids regulate vitamin A storage
Finally, in chapter 7, we analyzed the role of FXR and FXR agonists in vitamin A
metabolism. As eluded to in chapter 2, vitamin A metabolites are well-known to
(co)regulate bile acid metabolism. In contrast, nothing was known about the regulatory
role of FXR(agonists) on vitamin A metabolism. Moreover, FXR is an important
therapeutic target in chronic liver diseases, including NAFLD, making such information
even more relevant. Our primary observation was that FXR null mice contain very low
levels of vitamin A (both retinyl esters and retinol) in the liver, suggesting a crucial role
for this transcription factor in hepatic vitamin A storage and metabolism. Liver-specific
deletion revealed a similar phenotype, whereas intestine-specific FXR null mice had
normal hepatic vitamin A levels. In contrast to what then might be expected, treatment
of mice with the FXR ligands obeticholic acid (OCA) or cholic acid (CA) also lead to
depletion of hepatic retinyl esters. So far, we are unable to trace these effects to
transcriptional effects of OCA-activated FXR. Instead, OCA showed pronounced post-
transcriptional effects in the liver, in particular on retinyl hydrolases, e.g. ATGL,
PNPLA3 and HSL. Further studies are needed to delineate the mechanisms that are
involved in the FXR-mediated effects on vitamin A metabolism.
FXR controls highly diverse metabolic processes, varying from bile acid, triglyceride,
cholesterol, glucose to amino acid metabolism [59–62]. In addition, it regulates
autophagy, inflammation and extracellular matrix production [63–65]. Our study was
the first in which the putative role of FXR in hepatic vitamin A metabolism is analyzed
(chapter 7). FXR-null mice have previously been shown to develop mild
steatohepatitis, particularly accumulating triglycerides, which may progress to NASH

Chapter 8
243
even when fed a chow diet [66–70]. Dietary vitamin A uptake is accompanied by the
intestinal fat absorption and any impairment in the intestinal absorption of dietary fat
also impairs the uptake of dietary vitamin A [10,11]. Intestinal FXR is pivotal for the
absorption of fats and regulates lipid homeostasis. Intestinal-specific FXR-deletion in
mice suppresses diet-induced obesity [12], while hepatic FXR-deficiency increases
diet-induced lipid accumulation in the liver independent to FGF-15 signaling [13]. In
sharp contrast to the enhanced hepatic lipid content, our data show that the hepatic
vitamin A pool (retinyl palmitate and retinol) was strongly reduced (>90%) in total FXR-
null mice as compared to the age and sex-matched controls. Serum retinol levels were
not decreased in these animals. Moreover, intestine-specific FXR-null mice had normal
vitamin A reserves as compared to the control mice. So, normal circulatory retinol level,
in the presence of extremely low vitamin A reserves, once again reveal that little
hepatic reserves (5% in FXR-null mice) are sufficient to maintain normal circulatory
retinol levels and do not mirror what might be happening with vitamin A metabolism in
the liver.
The analysis of hepatic vitamin A levels in OCA-fed mice (3 weeks) surprised us with
a sharp reduction in retinyl ester (> 60% down) and retinol (>5-fold up) levels in the
liver. At present, we are unable to explain the underlying mechanism even after
intensive transcriptional and post-translational analysis of regulatory pathways in the
liver. Although we detected increased mRNA levels of hepatic lipases (Pnpla3 and
Lipe), the corresponding protein levels were sharply reduced. One hypothesis still to
be tested is whether this effect may possibly develop from the gut. OCA is a high-
affinity ligand for FXR, but may impair bile acid-driven vitamin A absorption in the
intestine and thereby prevent efficient hepatic storage. As a consequence, vitamin A
would be mobilized from the liver to provide peripheral tissues with this essential
vitamin. Hepatic retinyl ester and retinol levels rapidly build up in early life, so “the
reduction” caused by 3-week OCA treatment in ~10 week old mice may be caused by
a block in retinyl ester storage, enhanced retinyl ester hydrolysis or a combination of
both. A detailed analysis of vitamin A metabolites in the intestine and circulation is
required to shed light on this issue, samples that were unfortunately not available for
this study.
Importantly, our data suggest that patients under OCA or bile acid therapy may be at
risk to develop hepatic vitamin A deficiency, which may compromise metabolic,
immunologic and regenerative control in the liver. This is particularly relevant for

Discussion
244
patients with chronic liver disease, as they already show impaired vitamin A
metabolism. However, this could go either way: enhanced metabolism of vitamin A
could actually be part of the therapeutic effect in NAFLD as it helps to mobilize hepatic
retinol. In contrast, OCA treatment may further deplete hepatic vitamin A stores in other
chronic liver diseases that are associated with true hepatic and circulatory VAD, such
as PBC and PSC.
8.9. Conclusion and future perspective
The liver is the central organ controlling bile acid and vitamin A metabolism. It stores
vitamin A in hepatic stellate cells that are capable of maintaining stable blood retinol
levels for months to years even when dietary intake is insufficient. This is a very
important function of the liver as vitamin A metabolites are required for many different
physiological processes, including growth, vision, immunity and tissue
differentiation/regeneration. Vitamin A metabolism is impaired in chronic liver diseases,
but we need to redefine our clinical view that low serum retinol levels are a sign of
systemic and hepatic vitamin A deficiency. Hepatic retinol levels rare indeed typically
reduced in fatty liver, but hepatic vitamin A storage in retinyl esters may be actually
increased in these conditions. So, dietary vitamin A supplementation is unlikely to
provide any therapeutic effect in NAFLD. In contrast, it may even aggravate disease
progression. Specific retionic acids have already shown promising therapeutic effects
in animal models of NASH and cholestatic liver disease and this needs further
exploration. Not only as a single therapy but maybe also combined with FXR agonists,
as they also affect hepatic vitamin A metabolism.
With respect to vitamin A metabolism in the liver, many fundamental questions remain
unanswered: 1) How and in what form is vitamin A transported between hepatocytes,
HSC and the circulation? 2) What is the relative contribution of different retinyl ester
hydrolases (HSL, ATGL, PNPLA3) to the release of retinol form HSC? 3) 3) Does
impaired vitamin A metabolism actually contribute to the progression of liver diseases?
4) How do glucose and/or glycogen interact with vitamin A metabolism? 5) Is the
therapeutic effect of FXR agonists, like OCA, linked to its effect on vitamin A
metabolism? In conclusion, there is still a lot to learn about vitamin A metabolism in
the liver in healthy- and pathological conditions, which will hopefully reveal novel
therapeutic targets for the treatment of chronic liver disorders, and in particular to
prevent the development of advanced hepatic pathologies, such as fibrosis, cirrhosis
and hepatocellular carcinoma.

Chapter 8
245
REFERENCES
[1] A. Saeed, M. Hoekstra, M.O. Hoeke, J. Heegsma, K.N. Faber, The interrelationship between bile acid and vitamin A homeostasis, Biochim. Biophys. Acta. 1862 (2017) 496–512. doi:10.1016/j.bbalip.2017.01.007.
[2] P. Huang, V. Chandra, F. Rastinejad, Retinoic acid actions through mammalian nuclear receptors, Chem. Rev. 114 (2014) 233–254. doi:10.1021/cr400161b.
[3] R. Zhang, Y. Wang, R. Li, G. Chen, Transcriptional Factors Mediating Retinoic Acid Signals in the Control of Energy Metabolism, Int. J. Mol. Sci. 16 (2015) 14210–14244. doi:10.3390/ijms160614210.
[4] K. Ström, T.E. Gundersen, O. Hansson, S. Lucas, C. Fernandez, R. Blomhoff, C. Holm, Hormone-sensitive lipase (HSL) is also a retinyl ester hydrolase: evidence from mice lacking HSL., FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 23 (2009) 2307–2316. doi:10.1096/fj.08-120923.
[5] M. Sekiya, J.-I. Osuga, N. Yahagi, H. Okazaki, Y. Tamura, M. Igarashi, S. Takase, K. Harada, S. Okazaki, Y. Iizuka, K. Ohashi, H. Yagyu, M. Okazaki, T. Gotoda, R. Nagai, T. Kadowaki, H. Shimano, N. Yamada, S. Ishibashi, Hormone-sensitive lipase is involved in hepatic cholesteryl ester hydrolysis., J. Lipid Res. 49 (2008) 1829–38. doi:10.1194/jlr.M800198-JLR200.
[6] U. Taschler, R. Schreiber, C. Chitraju, G.F. Grabner, M. Romauch, H. Wolinski, G. Haemmerle, R. Breinbauer, R. Zechner, A. Lass, R. Zimmermann, Adipose triglyceride lipase is involved in the mobilization of triglyceride and retinoid stores of hepatic stellate cells, Biochim. Biophys. Acta. 1851 (2015) 937–945. doi:10.1016/j.bbalip.2015.02.017.
[7] T. Mello, A. Nakatsuka, S. Fears, W. Davis, H. Tsukamoto, W.F. Bosron, S.P. Sanghani, Expression of carboxylesterase and lipase genes in rat liver cell-types, Biochem. Biophys. Res. Commun. 374 (2008) 460–464. doi:10.1016/j.bbrc.2008.07.024.
[8] L. Yang, Y. Zhao, Y. Pan, D. Li, G. Zheng, Dietary supplement of Smilax china L. ethanol extract alleviates the lipid accumulation by activating AMPK pathways in high-fat diet fed mice, Nutr. Metab. 16 (2019) 6. doi:10.1186/s12986-019-0333-z.
[9] S. Ahmed El-Shazly, M.M. Ahmed, Z.S. Ibrahim, M.S. Refat, Synthesis, characterization, and efficacy evaluation of a new anti-diabetic vanadyl(II) thiamine hydrochloride complex in streptozotocin-induced diabetic rats, Int. J. Immunopathol. Pharmacol. 28 (2015) 227–239. doi:10.1177/0394632015576036.
[10] D.F. Jiang, W.T. Li, H.L. Yang, Z.Z. Zhang, D. Chen, C. Sun, Long-term effects of evodiamine on expressions of lipogenesis and lipolysis genes in mouse adipose and liver tissues, Genet. Mol. Res. GMR. 13 (2014) 1038–1046. doi:10.4238/2014.February.20.5.
[11] T. Ogiyama, M. Yamaguchi, N. Kurikawa, S. Honzumi, Y. Yamamoto, D. Sugiyama, S. Inoue, Identification of a novel boronic acid as a potent, selective, and orally active hormone sensitive lipase inhibitor, Bioorg. Med. Chem. 24 (2016) 3801–3807. doi:10.1016/j.bmc.2016.06.022.
[12] T. Ogiyama, M. Yamaguchi, N. Kurikawa, S. Honzumi, Y. Yamamoto, D. Sugiyama, H. Takakusa, S. ichi Inoue, Identification of a novel hormone sensitive lipase inhibitor with a reduced potential of reactive metabolites formation, Bioorg. Med. Chem. 25 (2017) 2234–2243. doi:10.1016/j.bmc.2017.02.045.
[13] T.H. Claus, D.B. Lowe, Y. Liang, A.I. Salhanick, C.K. Lubeski, L. Yang, L. Lemoine, J. Zhu, K.B. Clairmont, Specific inhibition of hormone-sensitive lipase

Discussion
246
improves lipid profile while reducing plasma glucose, J. Pharmacol. Exp. Ther. 315 (2005) 1396–1402. doi:10.1124/jpet.105.086926.
[14] C. Fernandez, M. Lindholm, M. Krogh, S. Lucas, S. Larsson, P. Osmark, K. Berger, J. Borén, B. Fielding, K. Frayn, C. Holm, Disturbed cholesterol homeostasis in hormone-sensitive lipase-null mice, Am. J. Physiol. Endocrinol. Metab. 295 (2008) E820-831. doi:10.1152/ajpendo.90206.2008.
[15] B. Xia, G.H. Cai, H. Yang, S.P. Wang, G.A. Mitchell, J.W. Wu, Adipose tissue deficiency of hormone-sensitive lipase causes fatty liver in mice, PLoS Genet. 13 (2017) e1007110. doi:10.1371/journal.pgen.1007110.
[16] F. O’Mahony, K. Wroblewski, S.M. O’Byrne, H. Jiang, K. Clerkin, J. Benhammou, W.S. Blaner, S.W. Beaven, Liver X receptors balance lipid stores in hepatic stellate cells through Rab18, a retinoid responsive lipid droplet protein, Hepatol. Baltim. Md. 62 (2015) 615–626. doi:10.1002/hep.27645.
[17] M. Kovarova, I. Königsrainer, A. Königsrainer, F. Machicao, H.-U. Häring, E. Schleicher, A. Peter, The Genetic Variant I148M in PNPLA3 Is Associated With Increased Hepatic Retinyl-Palmitate Storage in Humans, J. Clin. Endocrinol. Metab. 100 (2015) E1568-1574. doi:10.1210/jc.2015-2978.
[18] C. Pirazzi, L. Valenti, B.M. Motta, P. Pingitore, K. Hedfalk, R.M. Mancina, M.A. Burza, C. Indiveri, Y. Ferro, T. Montalcini, C. Maglio, P. Dongiovanni, S. Fargion, R. Rametta, A. Pujia, L. Andersson, S. Ghosal, M. Levin, O. Wiklund, M. Iacovino, J. Borén, S. Romeo, PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells, Hum. Mol. Genet. 23 (2014) 4077–4085. doi:10.1093/hmg/ddu121.
[19] S.M. O’Byrne, N. Wongsiriroj, J. Libien, S. Vogel, I.J. Goldberg, W. Baehr, K. Palczewski, W.S. Blaner, Retinoid absorption and storage is impaired in mice lacking lecithin:retinol acyltransferase (LRAT), J. Biol. Chem. 280 (2005) 35647–35657. doi:10.1074/jbc.M507924200.
[20] S.E. Trasino, X.-H. Tang, J. Jessurun, L.J. Gudas, Obesity Leads to Tissue, but not Serum Vitamin A Deficiency, Sci. Rep. 5 (2015) 15893. doi:10.1038/srep15893.
[21] S.E. Trasino, X.-H. Tang, J. Jessurun, L.J. Gudas, Retinoic acid receptor β2 agonists restore glycaemic control in diabetes and reduce steatosis, Diabetes Obes. Metab. 18 (2016) 142–151. doi:10.1111/dom.12590.
[22] M. Melis, X.-H. Tang, S.E. Trasino, V.M. Patel, D.J. Stummer, J. Jessurun, L.J. Gudas, Effects of AM80 compared to AC261066 in a high fat diet mouse model of liver disease, PloS One. 14 (2019) e0211071. doi:10.1371/journal.pone.0211071.
[23] A.C. Croce, U. De Simone, I. Freitas, E. Boncompagni, D. Neri, U. Cillo, G. Bottiroli, Human liver autofluorescence: an intrinsic tissue parameter discriminating normal and diseased conditions, Lasers Surg. Med. 42 (2010) 371–378. doi:10.1002/lsm.20923.
[24] A.C. Croce, U. De Simone, M. Vairetti, A. Ferrigno, E. Boncompagni, I. Freitas, G. Bottiroli, Liver autofluorescence properties in animal model under altered nutritional conditions, Photochem. Photobiol. Sci. Off. J. Eur. Photochem. Assoc. Eur. Soc. Photobiol. 7 (2008) 1046–1053. doi:10.1039/b804836c.
[25] A.C. Croce, A. Ferrigno, V.M. Piccolini, E. Tarantola, E. Boncompagni, V. Bertone, G. Milanesi, I. Freitas, M. Vairetti, G. Bottiroli, Integrated autofluorescence characterization of a modified-diet liver model with accumulation of lipids and oxidative stress, BioMed Res. Int. 2014 (2014) 803491. doi:10.1155/2014/803491.

Chapter 8
247
[26] A.A. Ashla, Y. Hoshikawa, H. Tsuchiya, K. Hashiguchi, M. Enjoji, M. Nakamuta, A. Taketomi, Y. Maehara, K. Shomori, A. Kurimasa, I. Hisatome, H. Ito, G. Shiota, Genetic analysis of expression profile involved in retinoid metabolism in non-alcoholic fatty liver disease, Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 40 (2010) 594–604. doi:10.1111/j.1872-034X.2010.00646.x.
[27] P. Pettinelli, B.M. Arendt, A. Teterina, I. McGilvray, E.M. Comelli, S.K. Fung, S.E. Fischer, J.P. Allard, Altered hepatic genes related to retinol metabolism and plasma retinol in patients with non-alcoholic fatty liver disease, PloS One. 13 (2018) e0205747. doi:10.1371/journal.pone.0205747.
[28] Q. Yang, T.E. Graham, N. Mody, F. Preitner, O.D. Peroni, J.M. Zabolotny, K. Kotani, L. Quadro, B.B. Kahn, Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes, Nature. 436 (2005) 356–362. doi:10.1038/nature03711.
[29] D.G. Haider, K. Schindler, G. Prager, A. Bohdjalian, A. Luger, M. Wolzt, B. Ludvik, Serum retinol-binding protein 4 is reduced after weight loss in morbidly obese subjects, J. Clin. Endocrinol. Metab. 92 (2007) 1168–1171. doi:10.1210/jc.2006-1839.
[30] T. Reinehr, B. Stoffel-Wagner, C.L. Roth, Retinol-binding protein 4 and its relation to insulin resistance in obese children before and after weight loss, J. Clin. Endocrinol. Metab. 93 (2008) 2287–2293. doi:10.1210/jc.2007-2745.
[31] M. Tajtáková, Z. Semanová, G. Ivancová, J. Petrovicová, V. Donicová, E. Zemberová, [Serum level of retinol-binding protein 4 in obese patients with insulin resistance and in patients with type 2 diabetes treated with metformin], Vnitr. Lek. 53 (2007) 960–963.
[32] F. Ulgen, C. Herder, M.C. Kühn, H.S. Willenberg, M. Schott, W.A. Scherbaum, S. Schinner, Association of serum levels of retinol-binding protein 4 with male sex but not with insulin resistance in obese patients, Arch. Physiol. Biochem. 116 (2010) 57–62. doi:10.3109/13813451003631421.
[33] H. Ronne, C. Ocklind, K. Wiman, L. Rask, B. Obrink, P.A. Peterson, Ligand-dependent regulation of intracellular protein transport: effect of vitamin a on the secretion of the retinol-binding protein, J. Cell Biol. 96 (1983) 907–910.
[34] J.L. Dixon, D.S. Goodman, Studies on the metabolism of retinol-binding protein by primary hepatocytes from retinol-deficient rats, J. Cell. Physiol. 130 (1987) 14–20. doi:10.1002/jcp.1041300104.
[35] D. Bellovino, Y. Lanyau, I. Garaguso, L. Amicone, C. Cavallari, M. Tripodi, S. Gaetani, MMH cells: An in vitro model for the study of retinol-binding protein secretion regulated by retinol, J. Cell. Physiol. 181 (1999) 24–32. doi:10.1002/(SICI)1097-4652(199910)181:1<24::AID-JCP3>3.0.CO;2-0.
[36] A. Henze, S.K. Frey, J. Raila, M. Tepel, A. Scholze, A.F.H. Pfeiffer, M.O. Weickert, J. Spranger, F.J. Schweigert, Evidence that kidney function but not type 2 diabetes determines retinol-binding protein 4 serum levels, Diabetes. 57 (2008) 3323–3326. doi:10.2337/db08-0866.
[37] A. Zwolak, A. Szuster-Ciesielska, J. Daniluk, J. Semeniuk, M. Kandefer-Szerszen, Chemerin, retinol binding protein-4, cytokeratin-18 and transgelin-2 presence in sera of patients with non-alcoholic liver fatty disease, Ann. Hepatol. 15 (2016) 862–869. doi:10.5604/16652681.1222102.
[38] M. Rahimlou, K. Mirzaei, S.A. Keshavarz, A. Hossein-Nezhad, Association of circulating adipokines with metabolic dyslipidemia in obese versus non-obese individuals, Diabetes Metab. Syndr. 10 (2016) S60-65. doi:10.1016/j.dsx.2015.09.015.

Discussion
248
[39] C.-H. Chu, H.-C. Lam, J.-K. Lee, C.-C. Lu, C.-C. Sun, H.-J. Cheng, M.-C. Wang, M.-J. Chuang, Elevated serum retinol-binding protein 4 concentrations are associated with chronic kidney disease but not with the higher carotid intima-media thickness in type 2 diabetic subjects, Endocr. J. 58 (2011) 841–847.
[40] H. Wu, W. Jia, Y. Bao, J. Lu, J. Zhu, R. Wang, Y. Chen, K. Xiang, Serum retinol binding protein 4 and nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus, Diabetes Res. Clin. Pract. 79 (2008) 185–190. doi:10.1016/j.diabres.2007.08.016.
[41] Y. Liu, L. Wang, M. Luo, N. Chen, X. Deng, J. He, L. Zhang, P. Luo, J. Wu, Inhibition of PAI-1 attenuates perirenal fat inflammation and the associated nephropathy in high-fat diet-induced obese mice, Am. J. Physiol. Endocrinol. Metab. 316 (2019) E260–E267. doi:10.1152/ajpendo.00387.2018.
[42] H. Xu, Z. Ma, S. Lu, R. Li, L. Lyu, L. Ding, Q. Lu, Renal Resistive Index as a Novel Indicator for Renal Complications in High-Fat Diet-Fed Mice, Kidney Blood Press. Res. 42 (2017) 1128–1140. doi:10.1159/000485781.
[43] A. Odermatt, The Western-style diet: a major risk factor for impaired kidney function and chronic kidney disease, Am. J. Physiol. Renal Physiol. 301 (2011) F919-931. doi:10.1152/ajprenal.00068.2011.
[44] H. Senoo, Structure and function of hepatic stellate cells, Med. Electron Microsc. Off. J. Clin. Electron Microsc. Soc. Jpn. 37 (2004) 3–15. doi:10.1007/s00795-003-0230-3.
[45] C.S. Lo, M.L. Wahlqvist, Y. Horie, Determination of retinoic acid and retinol at physiological concentration by HPLC in Caucasians and Japanese women, Asia Pac. J. Clin. Nutr. 5 (1996) 173–174.
[46] D. Weber, T. Grune, The contribution of β-carotene to vitamin A supply of humans, Mol. Nutr. Food Res. 56 (2012) 251–258. doi:10.1002/mnfr.201100230.
[47] A. Saeed, R.P.F. Dullaart, T.C.M.A. Schreuder, H. Blokzijl, K.N. Faber, Disturbed Vitamin A Metabolism in Non-Alcoholic Fatty Liver Disease (NAFLD), Nutrients. 10 (2017). doi:10.3390/nu10010029.
[48] P.-J. Brun, K.J.Z. Yang, S.-A. Lee, J.J. Yuen, W.S. Blaner, Retinoids: Potent regulators of metabolism, BioFactors Oxf. Engl. 39 (2013) 151–163. doi:10.1002/biof.1056.
[49] D. Yang, M.G. Vuckovic, C.P. Smullin, M. Kim, C. Pui-See Lo, E. Devericks, H.S. Yoo, M. Tintcheva, Y. Deng, J.L. Napoli, Modest Decreases in Endogenous All-Trans-Retinoic Acid Produced by a MouseRdh10Heterozygote Provoke Major Abnormalities in Adipogenesis and Lipid Metabolism, Diabetes. (2018). doi:10.2337/db17-0946.
[50] O. Ziouzenkova, G. Orasanu, M. Sharlach, T.E. Akiyama, J.P. Berger, J. Viereck, J.A. Hamilton, G. Tang, G.G. Dolnikowski, S. Vogel, G. Duester, J. Plutzky, Retinaldehyde represses adipogenesis and diet-induced obesity, Nat. Med. 13 (2007) 695–702. doi:10.1038/nm1587.
[51] Y. Ma, O.V. Belyaeva, P.M. Brown, K. Fujita, K. Valles, S. Karki, Y.S. de Boer, C. Koh, Y. Chen, X. Du, S.K. Handelman, V. Chen, E.K. Speliotes, C. Nestlerode, E. Thomas, D.E. Kleiner, J.M. Zmuda, A.J. Sanyal, NASH CRN, N.Y. Kedishvili, T.J. Liang, Y. Rotman, HSD17B13 is a Hepatic Retinol Dehydrogenase Associated with Histological Features of Non-Alcoholic Fatty Liver Disease, Hepatol. Baltim. Md. (2018). doi:10.1002/hep.30350.
[52] J.A. Del Campo, R. Gallego-Durán, P. Gallego, L. Grande, Genetic and Epigenetic Regulation in Nonalcoholic Fatty Liver Disease (NAFLD), Int. J. Mol. Sci. 19 (2018). doi:10.3390/ijms19030911.

Chapter 8
249
[53] E. Trépo, S. Romeo, J. Zucman-Rossi, P. Nahon, PNPLA3 gene in liver diseases, J. Hepatol. 65 (2016) 399–412. doi:10.1016/j.jhep.2016.03.011.
[54] W. Su, Z. Mao, Y. Liu, X. Zhang, W. Zhang, J.-A. Gustafsson, Y. Guan, Role of HSD17B13 in the liver physiology and pathophysiology, Mol. Cell. Endocrinol. (2018). doi:10.1016/j.mce.2018.10.014.
[55] G. Aragonès, T. Auguet, S. Armengol, A. Berlanga, E. Guiu-Jurado, C. Aguilar, S. Martínez, F. Sabench, J.A. Porras, M.D. Ruiz, M. Hernández, J.J. Sirvent, D. Del Castillo, C. Richart, PNPLA3 Expression Is Related to Liver Steatosis in Morbidly Obese Women with Non-Alcoholic Fatty Liver Disease, Int. J. Mol. Sci. 17 (2016). doi:10.3390/ijms17050630.
[56] ClinicalTrials.gov, (n.d.). https://clinicaltrials.gov/ (accessed March 5, 2019). [57] J.E. Thatcher, A. Zelter, N. Isoherranen, The relative importance of CYP26A1 in
hepatic clearance of all-trans retinoic acid, Biochem. Pharmacol. 80 (2010) 903–912. doi:10.1016/j.bcp.2010.05.023.
[58] S. Tay, L. Dickmann, V. Dixit, N. Isoherranen, A comparison of the roles of peroxisome proliferator-activated receptor and retinoic acid receptor on CYP26 regulation, Mol. Pharmacol. 77 (2010) 218–227. doi:10.1124/mol.109.059071.
[59] H. Tu, A.Y. Okamoto, B. Shan, FXR, a bile acid receptor and biological sensor, Trends Cardiovasc. Med. 10 (2000) 30–35.
[60] G. Lambert, M.J.A. Amar, G. Guo, H.B. Brewer, F.J. Gonzalez, C.J. Sinal, The farnesoid X-receptor is an essential regulator of cholesterol homeostasis, J. Biol. Chem. 278 (2003) 2563–2570. doi:10.1074/jbc.M209525200.
[61] B. Cariou, D. Duran-Sandoval, F. Kuipers, B. Staels, Farnesoid X receptor: a new player in glucose metabolism?, Endocrinology. 146 (2005) 981–983. doi:10.1210/en.2004-1595.
[62] V. Massafra, A. Milona, H.R. Vos, R.J.J. Ramos, J. Gerrits, E.C.L. Willemsen, J.M. Ramos Pittol, N. Ijssennagger, M. Houweling, H.C.M.T. Prinsen, N.M. Verhoeven-Duif, B.M.T. Burgering, S.W.C. van Mil, Farnesoid X Receptor Activation Promotes Hepatic Amino Acid Catabolism and Ammonium Clearance in Mice, Gastroenterology. 152 (2017) 1462-1476.e10. doi:10.1053/j.gastro.2017.01.014.
[63] S. Seok, T. Fu, S.-E. Choi, Y. Li, R. Zhu, S. Kumar, X. Sun, G. Yoon, Y. Kang, W. Zhong, J. Ma, B. Kemper, J.K. Kemper, Transcriptional regulation of autophagy by an FXR-CREB axis, Nature. 516 (2014) 108–111. doi:10.1038/nature13949.
[64] Y.-D. Wang, W.-D. Chen, M. Wang, D. Yu, B.M. Forman, W. Huang, Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response, Hepatol. Baltim. Md. 48 (2008) 1632–1643. doi:10.1002/hep.22519.
[65] L. Verbeke, I. Mannaerts, R. Schierwagen, O. Govaere, S. Klein, I. Vander Elst, P. Windmolders, R. Farre, M. Wenes, M. Mazzone, F. Nevens, L.A. van Grunsven, J. Trebicka, W. Laleman, FXR agonist obeticholic acid reduces hepatic inflammation and fibrosis in a rat model of toxic cirrhosis, Sci. Rep. 6 (2016) 33453. doi:10.1038/srep33453.
[66] C.J. Sinal, M. Tohkin, M. Miyata, J.M. Ward, G. Lambert, F.J. Gonzalez, Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis, Cell. 102 (2000) 731–744.
[67] M. Miyata, Y. Sakaida, H. Matsuzawa, K. Yoshinari, Y. Yamazoe, Fibroblast growth factor 19 treatment ameliorates disruption of hepatic lipid metabolism in farnesoid X receptor (Fxr)-null mice, Biol. Pharm. Bull. 34 (2011) 1885–1889.
[68] M. Bjursell, M. Wedin, T. Admyre, M. Hermansson, G. Böttcher, M. Göransson, D. Lindén, K. Bamberg, J. Oscarsson, M. Bohlooly-Y, Ageing Fxr deficient mice

Discussion
250
develop increased energy expenditure, improved glucose control and liver damage resembling NASH, PloS One. 8 (2013) e64721. doi:10.1371/journal.pone.0064721.
[69] J. Schmitt, B. Kong, B. Stieger, O. Tschopp, S.M. Schultze, M. Rau, A. Weber, B. Müllhaupt, G.L. Guo, A. Geier, Protective effects of farnesoid X receptor (FXR) on hepatic lipid accumulation are mediated by hepatic FXR and independent of intestinal FGF15 signal, Liver Int. Off. J. Int. Assoc. Study Liver. 35 (2015) 1133–1144. doi:10.1111/liv.12456.
[70] L. Sheng, P.K. Jena, H.-X. Liu, K.M. Kalanetra, F.J. Gonzalez, S.W. French, V.V. Krishnan, D.A. Mills, Y.-J.Y. Wan, Gender Differences in Bile Acids and Microbiota in Relationship with Gender Dissimilarity in Steatosis Induced by Diet and FXR Inactivation, Sci. Rep. 7 (2017) 1748. doi:10.1038/s41598-017-01576-9.

Summary

Summary
252
Vitamin A is an essential nutrient for healthy reproduction, vision, organ (re)generation,
immune control and energy metabolism. Fortunately, our body very efficiently
manages dietary uptake, storage and distribution of vitamin A to all tissues. By far the
most vitamin A (>80%) is stored (as retinyl esters) in the liver, which also regulates
controlled release to the bloodstream (as retinol) to distribute to vitamin A-demanding
tissues. Even in the complete absence of vitamin A intake, a healthy liver can support
normal circulating retinol levels for many months before vitamin A deficiency (VAD)
kicks in, which typically reveals itself in night blindness and uncontrolled infections.
Patients with chronic liver disease are believed to be at increased risk for developing
VAD as their sick liver is unable to support efficient bile production for intestinal
absorption of vitamin A. Moreover, the development of fibrosis leads to active release
of vitamin A from the liver. However, there is still a lot of controversy about the effect
of liver disease on vitamin A metabolism.
In this thesis, we report a few remarkable observations that may change our view on
the association between liver disease and VAD. Chapter 2 provides a detailed
overview of the body vitamin A metabolism and its mutual interaction with bile acid
metabolism. Hepatic vitamin A storage is a resultant of vitamin A esterification (via
LRAT) and hydrolysis (via ATGL and PNPLA3). In chapter 3, we identified another
novel enzyme, hormone-sensitive lipase (HSL), predominantly present in vitamin A
storing hepatic stellate cells that contribute in the release of vitamin A from the healthy
liver along with other enzymes (ATGL and PNPLA3). This indicates the presence of
alternative pathways for the mobilization and release of vitamin A from the liver for
distribution to extra-hepatic tissues.
There are many reports that show that chronic liver diseases are associated with
impaired vitamin A status. In chapter 4, we reviewed the current knowledge about
vitamin A metabolism in non-alcoholic fatty liver disease (NAFLD) and associated Type
2 diabetes and metabolic syndrome. Most studies report that NAFLD is associated with
systemic VAD. This is primarily based on levels of circulating retinol. Only few
anecdotal reports analyzed hepatic vitamin A reserves (both retinol and retinyl esters).
This prompted us to analyze hepatic vitamin A metabolism in more detail in chapter
5. We indeed confirmed reduced hepatic retinol levels in 2 mouse models of NAFLD.
In sharp contrast, hepatic retinyl palmitate (vitamin A storage) was increased and
accumulated preferentially in hepatocytes in NAFLD as compared to the normal
vitamin A-storing hepatic stellate cells in the healthy liver. This leads to retinol

Summary
253
deficiency in liver, but not true VAD. Remarkably, efficient extraction of retinyl esters
from fatty liver tissue was strongly influenced by the lipid extraction procedure applied,
which may underlie the controversy between earlier previous reports that used different
extraction protocols.
Glycogen Storage Disease Ia (GSD Ia) patients are at risk to develop fatty liver and in
chapter 6, we analyzed vitamin A metabolism in patients and mice to determine
whether such patients may also be at risk for impaired vitamin A metabolism. Similar
to NAFLD, hepatic retinol levels were reduced, while circulatory retinol levels were
elevated in GSD Ia. In contrast to NAFLD, vitamin A reserves did not change in GSD
Ia mice. These results indicate that GSD Ia patients are prone to develop circulatory
hypervitaminosis A that may cause vitamin A-toxicity and contribute to disease
symptoms including hepatic steatosis, hyperlipidemia and osteoporosis.
Production of bile is one of the main functions of the liver. Bile acids are the major
components of bile and they facilitate the absorption of lipid and lipid-soluble vitamins,
including vitamin A, in the proximal part of the small intestine. Bile acids are
endogenous ligands for the nuclear receptor Farnesoid X receptor (FXR). Ligand-
activated FXR regulates various vital functions in the liver related to lipid, glucose and
protein metabolism. FXR-targeted therapies have shown therapeutic potential in
NAFLD in several clinical trials. Vitamin A metabolites are well-known to (co)regulate
bile acid metabolism. However, the possible role of FXR-activation on vitamin A
metabolism was not studied before. In chapter 7, we show that FXR plays a vital role
in maintaining effective storage of vitamin A in the liver. An impairment in FXR function
may, therefore, increase the risk for developing VAD. Secondly, clinical available FXR-
targeted therapies also impaired hepatic vitamin A storage, which may be a desirable
effect in NAFLD, but this could also further aggravate the disease symptoms with
increased risks to develop VAD.
In conclusion, hepatic vitamin A metabolism is heavily disturbed in chronic liver
diseases but does not necessarily lead to true VAD. The clinically available therapies
such as bile acids or FXR-agonists may fix or aggravate this metabolic impairment,
though this still remains to be established in future studies. However, it seems clear
that oral supplementation of vitamin A is unlikely to be of any therapeutic value for
NAFLD patients. Thus, vitamin A metabolite-specific therapies may achieve more
desirable outcomes in chronic liver disease. This thesis provides an outlook for future
directions to target vitamin A metabolism in the treatment of chronic liver diseases.

Summary
254

Samenvatting

Samenvatting
256
In onze voeding is vitamine A mede essentieel voor een gezonde voortplanting, goed
zicht, orgaan (re)generatie, regulatie van het immuunsysteem en energiemetabolisme.
Op zeer efficiënte wijze wordt in ons lichaam de opname, opslag en transport van
vitamine A naar alle weefsels gereguleerd. Verreweg de meeste vitamine A (> 80%)
wordt opgeslagen (als retinylesters) in de lever. Ook reguleert de lever de afgifte aan
bloed (als retinol), waarna het verder getransporteerd wordt naar weefsels die vitamine
A nodig hebben. Zelfs wanneer er geen vitamine A wordt ingenomen kan een gezonde
lever maandenlang het retinolgehalte in bloed op peil te houden voordat er vitamine A-
deficiëntie (VAD) optreedt, wat zich gewoonlijk uit in nachtblindheid en
ongecontroleerde infecties. Men denkt echter dat patiënten met chronische
leveraandoeningen een verhoogd risico hebben op het ontwikkelen van VAD. Dit
omdat door hun aangedane lever de galproductie niet optimaal genoeg is voor de
intestinale absorptie van vitamine A. Bovendien leidt de ontwikkeling van fibrose tot
het actief vrijkomen van vitamine A uit de lever. Toch zijn de meningen over het effect
van leverziekte op vitamine A-metabolisme nog verdeeld.
In dit proefschrift rapporteren we enkele opmerkelijke observaties die ons inzicht op
de associatie tussen leverziekte en VAD kunnen veranderen. Hoofdstuk 2 geeft een
gedetailleerd overzicht van het vitamine A-metabolisme in ons lichaam en de
onderlinge interactie ervan met het galzuurmetabolisme. Opslag van vitamine A in de
lever is het gevolg van de balans tussen verestering (via LRAT) en hydrolyse (via
ATGL en PNPLA3) van vitamine A. In hoofdstuk 3 hebben we een ander nieuw enzym
geïdentificeerd, hormone-sensitive lipase (HSL), welke voornamelijk aanwezig is in de
hepatische stellaatcel waar vitamine A wordt opgeslagen. Samen met andere
enzymen (ATGL en PNPLA3) draagt het bij aan de afgifte van vitamine A uit de
gezonde lever. Dit wijst op de aanwezigheid van alternatieve routes voor de mobilisatie
en afgifte van vitamine A vanuit de lever voor transport naar extrahepatische weefsels.
Er zijn veel studies die aantonen dat chronische leverziekten geassocieerd zijn met
een verminderde vitamine A-status. In hoofdstuk 4 hebben we de huidige kennis over
vitamine A-metabolisme bij niet-alcoholische leververvetting (NAFLD) en de daarbij
geassocieerde diabetes type 2 en metabool syndroom besproken. De meeste studies
tonen aan dat NAFLD is geassocieerd met systemische VAD. Dit is voornamelijk
gebaseerd op de concentratie retinol dat in het bloed gemeten wordt. Slechts enkele
anekdotische rapporten analyseerden vitamine A-reserves in de lever (zowel retinol
als retinylesters).

Samenvatting
257
Dit heeft ons ertoe aangezet om het vitamine A-metabolisme in de lever in hoofdstuk
5 gedetailleerder te analyseren. We vonden inderdaad ook lagere retinol concentraties
retinol in muizenlevers in 2 diermodellen van NAFLD. In tegenstelling hiermee was dat
de concentraties retinylpalmitaat (vitamine A-opslag) in de lever sterk verhoogd waren
en bleek voornamelijk in hepatocyten aanwezig te zijn in deze NAFLD levers, in plaats
van in de hepatische stellaatcellen waar vitamine A normaliter in de gezonde lever
wordt opgeslagen. Dit leidt tot retinoldeficiëntie in de lever, maar niet tot echte VAD.
Opmerkelijk was dat een efficiënte isolatie van retinylpalmitaat uit vettig leverweefsel
sterk werd beïnvloed door de gebruikte lipide-extractieprocedure. Dit kan mogelijk ook
de oorzaak zijn van de controverse tussen eerdere studies die deze verschillende
extractieprotocollen gebruikten.
Patiënten met glycogeenstapelingsziekte type Ia (GSD Ia) hebben een verhoogd risico
op het ontwikkelen van een vette lever. In hoofdstuk 6 hebben we het metabolisme
van vitamine A bij GSD Ia patiënten en muizen geanalyseerd om te bepalen of deze
ziekte mogelijk ook een verhoogd risico hebben op een ontregeld vitamine A-
metabolisme. Net als bij NAFLD waren ook in GSD Ia de retinol concentraties in de
lever verlaagd, terwijl de concentraties in het bloed juist verhoogd waren. In
tegenstelling tot NAFLD veranderden de vitamine A-reserves in de lever niet in GSD
Ia muizen. Deze resultaten tonen aan dat GSD Ia-patiënten vatbaar zijn voor het
ontwikkelen van hypervitaminose A (te hoge vitamine A waarden) in het bloed dat
mogelijk zelfs vitamine A-toxiciteit kan veroorzaken en kan bijdragen aan
ziektesymptomen, waaronder leververvetting (steatose), te veel vetten in het bloed
(hyperlipidemie) en botontkalking (osteoporose).
De productie van gal is een van de belangrijkste functies van de lever. Galzuren zijn
belangrijke componenten van gal en ze vergemakkelijken de absorptie van vetten en
vet-oplosbare vitaminen, waaronder vitamine A, in het eerste deel van de dunne darm.
Galzuren zijn lichaamseigen liganden die de nucleaire receptor Farnesoid X-receptor
(FXR) kunnen activeren. Geactiveerd FXR reguleert verschillende vitale functies in de
lever die verband houden met het metabolisme van vetten, suikers en eiwitten. Op dit
moment wordt er veel onderzoek gedaan naar medicijnen die FXR activeren, omdat
deze mogelijk een therapeutische werking kunnen hebben in patiënten met NAFLD.
Vitamine A-metabolieten zijn in staat om het metabolisme van galzuren (mede) te
reguleren. De mogelijke rol van FXR op het vitamine A-metabolisme is echter niet
eerder geanalyseerd. In hoofdstuk 7 laten we zien dat FXR een essentiële rol speelt

Samenvatting
258
in de effectieve opslag van vitamine A in de lever. Een afgenomen functie van FXR
zou daarom het risico op het krijgen van VAD kunnen verhogen. Opmerkelijk was dat
een experimenteel medicijn dat FXR activeert ook de opslag van vitamine A in de lever
sterk verminderd. dit zou een positief effect kunnen zijn in patiënten met vette lever
(NAFLD), maar het zou ook symptomen kunnen verergeren met daarbij verhoogde
risico’s op de ontwikkeling van VAD.
Als conclusie van het werk beschreven in mijn proefschrift kan ik zeggen dat
chronische leveraandoeningen gepaard gaan met een sterk ontregeld vitamine A
metabolisme in de lever, maar dat dit niet noodzakelijkerwijs ook leidt tot echte
vitamine A deficiëntie. Beschikbare (experimentele) medicijnen, zoals galzuren of
FXR-agonisten, kunnen deze metabole stoornis verhelpen of juist verergeren, maar dit
zal nog moeten worden geanalyseerd in toekomstige studies. Het is echter wel
duidelijk dat orale vitamine A supplementen waarschijnlijk niet van enige
therapeutische waarde zal zijn voor NAFLD-patiënten. Hiervoor zullen therapieën
ontwikkeld moeten worden die zich richten op specifieke vitamine A metabolieten.
Hiermee is dus zeker nog geen einde gekomen aan het belangrijke onderzoek naar
de rol van vitamine A in chronische leverziekten.

Acknowledgements
First of all, thanks to Almighty for giving me the strength to achieve this goal. I would
like to express my appreciation to those who contributed to this journey. The first
person I want to acknowledge is my great father (Saeed Iqbal Anjum). His presence
in my life is a great blessing. He gave me all type of support, courage and motivation
throughout my life and in my Ph.D. I have no words to say thanks to my respected
mother (Ishrat) and parents of my wife (M. Arshad and Sajida Parveen). I have
achieved this milestone due to their support, love and prayers. This is impossible to
achieve without the sacrifices, love and support of my beloved wife (Memoona
Arshad). She gave tremendous support and care to me and to my kids (M. Rafay Ali
and Anaaya). My whole family (Umer, Hasan, Rehman, Shoaib, Awais, Rimsha,
Zonra, Farhan , Iqra, Hina, Alvina, Naila and my kids) was with me at each step of
my life and in this journey.
My research in master degree really motivates me towards science. So, I cannot
forget to acknowledge my first Scientific supervisor Dr. Qasier Mahemood Khan, my
first mentor in science, he provided me very first opportunity to work in a scientific
laboratory to express myself. This was also impossible without the funding
opportunity provided by BZU, HEC-Pakistan in a great collaboration with UMCG-
RUG. I am grateful to my wonderful colleagues in BZU (Dr. Muhammad Ali, Dr. M.
Baber, Dr. Rehan, Dr. Shehzad, Dr. Hammad, Dr. Sumaira, Dr. R. Khalid, Dr.
Tahir, Dr. Assad, Dr. Habib, Dr. Samra, Dr. Farhan, Dr. Muzaffar).
Now I want to express my heartfelt thanks of gratitude to my respected supervisor,
mentor and a great scientist and real soul of this project, Prof. Dr. Klaas Nico Faber.
I am very grateful to him for his intensive, kind and efficient supervision. I am also
grateful to him for his highly proficient input in my thesis and publications. He wrote
almost several letters of progress report in Pakistan (after every 6-month). I
appreciate all his support. Now working as Postdoc in his lab for next 2.5 more years
is a great honor for me. Thanks once again for all support, help and supervision!
I am extremely thankful to Prof. Dr. Han Moshage. I found him very kind and very
nice personality with a great scientific knowledge of this field. His input was also very
valuable and precious in my Ph.D. I am indebted to Janette Heegsma, she gave me
a great scientific and social support throughout my Ph.D. I appreciate her help and
support in my experimental work and all kind of technical assistance. I am also

Acknowledgements
260
grateful to Manon Buist-Homan and Tjasso Blokzijl for their support and all kind of
technical assistance. I learned a lot from these senior technicians.
This achievement is completely a team effort. So, I want to say now special thanks to
my team including Mark, Martijn, Floris, Shiva, Joanne, Natalia for their
contribution in different projects. I highly appreciated the contribution of Yang. I want
to say my affectionate and profound thanks to him for conducting some animal
experiments in China for us. I want to say thanks to my MDL-colleagues including
Dianne, Alfredo, Herson, Archie, Turu, Yana Geng, Fabio, Ying Cui, Natalia,
Mengfan, Guido, Danial, Raphael, Werna, Arnu.
A heartfelt thanks to some other friends those gave me tremendous support during
my Ph.D. including Dicky, Tim, Jan Freark, Brenda, Marleen Schonewille,
Marcella, Ivo, Marije, Federico, Onne, Sandra, Anne Claire, Twan, Vincent,
Ingrid, Marleen Dommerholt, Maaike Blankestijn, Sarah Stolle, Fan, Niels,
Aycha, Renze, Trijnie, Henk, Hermie, Gaby, Hannah, Hilde, Dolf.
My heartfelt gratitude is also reserved for a few clinicians and scientists including
Maaike H. Oosterveer, Hans Blokzijl, Tim CMA Schreuder, Robin P.F. Dullaart,
Henkjan Verkade, Albert Groen, Saskia van Mil, Coen Paulusma, Lu Zhou,
Bangmao Wang, Daphne Dekker, Niels Kloosterhuis, Alain de Bruin, Hans
Jonker, Bart van de Sluis, Terry G.J. Derks, Eveline Van der Veer, Giles
Mithieux, Fabienne Rajas, for collaborations and significant contribution.
I want to say thanks to my Pakistani friends and community including Rana Irfan,
Kashif Bilal, Saleem, Imran Fazal and his family, Ishtiaq Ahmad and his family,
Zaheer and his family, Asif, Atta ur Rehman, Ayaz and his family, Kashan
Arshad and his family, Sherjeel Aslam, Naveed (late) for their great support. I
would like to say thanks to all my friends in ZCC-Groningen, those provided me the
opportunity to play Cricket in The Netherlands.
While writing this section, I was afraid not to miss someone’s name. If I missed
someone’s name please accept my apology in advance. This was not my intention. I
am also still here for the next couple of years. I still need your support, love and care.

Curriculum Vitae
Ali Saeed was born on January 6, 1984 in Faisalabad, Pakistan. He finished his
bachelor degree (Doctor of Veterinary Medicine) in 2005 and joined his Master
degree in the area of Health Biotechnology in Quaid-I-Azam University, Islamabad,
Pakistan and finished his degree with successful thesis defense on “Molecular
Epidemiology of FMDV in Province Punjab, Pakistan” best public sector institute
(NIBGE). This challenging research environment gave him the opportunity to learn
advanced scientific methods and to publish his research in well-reputed journals. He
has research and teaching experience before the start of Ph.D. He worked as
Veterinary Officer (Health) (1.10 years) and lecturer/researcher in Institute of
Molecular biology & Biotechnology, Bahauddin Zakariya University, Multan, Pakistan
(3.7 years). He got faculty development Ph.D. scholarship from Bahauddin Zakariya
University, Multan, Pakistan-funded by higher education commission of Pakistan.
April 2013, He joined the group of Prof. Dr. Klaas Nico Faber, in the department of
Gastroenterology and Hepatology, University Medical Center Groningen, University
of Groningen, Groningen, The Netherlands as a Ph.D. student. His research topic
was “Disturbed vitamin A metabolism in chronic liver disease and relevance for
therapy”. Since June 1, 2018, he was appointed as a post-doctoral fellow in the same
department and supervision. Now his research topic is “Modulating gut host-microbe
interactions in a human in vitro (HoxBan) system to develop pre/pro-biotic therapies
for Inflammatory Bowel Disease”.
LIST OF PUBLICATIONS
1. Ali Saeed, Mark Hoekstra, Martijn Oscar Hoeke, Janette Heegsma, Klaas Nico
Faber. The interrelationship between bile acid and vitamin A homeostasis,
Biochim. Biophys. Acta. (Molecular and cell biology of lipids). 1862 (2017) 496–
512.
2. Shiva Shajari*, Ali Saeed*, Janette Heegsma, Natalia F. Smith-Cortinez,
Svenja Sydor, Klaas Nico Faber. Hormone-sensitive lipase is a retinyl ester
hydrolase in human and rat quiescent hepatic stellate cells. (Conditionally
accepted).

Curriculum Vitae
262
3. Ali Saeed, Robin P.F. Dullaart, Tim C.M.A. Schreuder, Hans Blokzijl, Klaas Nico
Faber. Disturbed Vitamin A Metabolism in Non-Alcoholic Fatty Liver Disease
(NAFLD). Nutrients 10, 29 (2017).
4. Ali Saeed, Paulina Bartuzi, Janette Heegsma., Daphne Dekker., J.W. (Hans)
Jonker., A.J.A. (Bart) van der Sluis., Klaas Nico Faber. Non-alcoholic fatty liver
disease disturbs vitamin A metabolism leading to enhanced storage in the liver,
but normal circulatory retinol (Submitted).
5. Ali Saeed, Joanne A. Hoogerland, Janette Heegsma, Terry G.J. Derks, Eveline
van der Veer, Gilles Mithieux, Fabienne Rajas, Maaike H. Oosterveer, Klaas
Nico Faber. Impaired vitamin A homeostasis in glycogen storage disease type
1a (Submitted).
6. Ali Saeed, Jing Yang, Janette Heegsma, Albert K. Groen, Saskia W.C. van Mil,
Coen C. Paulusma, Lu Zhou, Bangmao Wang, Klaas Nico Faber. Farnesoid X
receptor (FXR) and bile acids regulate vitamin A storage (In preparation).
7. Muhammad Munir, Ali Saeed, Muhammad Abubakar, Sehrish Kanwal and
Mikael Berg. Molecular Characterization
of Peste des Petits Ruminants Viruses From Outbreaks Caused by Unrestricted
Movements of Small Ruminants in Pakistan, Transboundary and Emerging
Diseases. 62 (1), 108-114 (2015).
8. Ali Saeed, Sehrish Kanwal, Memoona Arshad, Rehan Sadiq Shaikh,
Muhammad Ali, Muhammad Abubakar. Foot-and-mouth disease: overview of
motives of disease spread and efficacy of available vaccines. Journal of Animal
Science and Technology. 57 (1), 10 (2015).
9. Sehrish Kanwal, Ali Saeed, Muhammad Munir, Memoona Arshad.
Phylogenetics of Foot and Mouth Disease Virus in Punjab, Pakistan. British
Journal of Virology. 1(1): 21-28 (2014) .
10. Usman Waheed, Muhammad Siddique, Muhammad Arshad, Muhammad Ali, Ali
Saeed. Preparation of OE Newcastle disease vaccine from VG/GA strain and its
evaluation in commercial broiler chicks. Pakistan Journal of Zoology. 45(2), 339-
344 (2013).
11. Muhammad Munir, Siamak Zohari, Ali Saeed, Qaiser Mahmood Khan,
Muhammad Abubakar, Neil LeBlanc, Mikael Berg. Detection and phylogenetic
analysis of Peste des Petits Ruminants Virus isolated from outbreaks in Punjab,
Pakistan. Transboundary and Emerging Diseases. 59 85–93 (2012).

Curriculum Vitae
263
12. Samreen Zulfiqar, Sadia Shahnawaz, Muhammad Ali, Arif Mahmood Bhutta,
Shahid Iqbal, Sikandar Hayat, Shazia Qadir, Muhammad Latif, Nazia Kiran, Ali
Saeed, Muhammad Ali, Furhan Iqbal. Detection of Babesia bovis in blood
samples and its effect on the hematological serum biochemical profile in large
ruminants from southern Punjab. Asian Pacific Journal of Tropical Biomedicine.
104-108 (2012).
13. Farhan Afzal, Ali Saeed, Muhammad Azhar Sharif, Najma Ayub, Shamsul
Hassan. Pathogenicity of avian influenza virus H5N1 2007 isolates from
Pakistan. Asian Pacific Journal of Tropical Biomedicine. S380-S382, (2012).
14. Ali Saeed, Farhan Afzal, Memoona Arshad, Shamsul Hassan and Muhmmad
Abuabakar “Detection of avian Influenza virus H5N1 serotype in backyard
poultry, wild and zoo birds in Pakistan”. Revue de Médecine Vétérinaire.
163(11), 552-557 (2012).
15. Muhammad Abubakar , Sehrish Kanwal, Ali Saeed. Persistence, Emergence
and Distribution of Foot and Mouth Disease Virus (FMDV); Global and Pakistan
Perspectives. Pakistan Journal of Life and Social Sciences. 10(2): 84-90 (2012).
(Review Article)
16. Ali Saeed, Qaiser Mahmood Khan, Usman Waheed, Memoona Arshad,
Muhammad Asif, Muhammad Farooq. RT-PCR evaluation for identification and
sequence analysis of foot-and-mouth disease serotype O from 2006 to 2007 in
Punjab, Pakistan. Comparative Immunology, Microbiology and Infectious
Diseases. 34, pp. 95-101 (2011).
17. Ali Saeed, Usman Waheed, Memoona Arshad, Qaiser Mahmood Khan,
Muhammad Ali, Muhammad Baber. RT-PCR evaluation of foot-and-mouth
disease serotype O in saliva, tracheal and vesicular samples of goats in Punjab,
Pakistan. Pakistan Journal of Zoology. 43(4), 808-811 (2011).
18. Usman Waheed, Ali Saeed, Ameena Mobeen, Qaiser Mahmood Khan. The VP1
(Capsid Protein) Gene Based DNA Sequencing for Epidemiological Analysis of
FMDV, Isolated from Buffaloes in Pakistan. Pakistan Journal of Zoology.
Supplementary series, No.9 pp.333-339 (2009).
CONFERENCES
1. Ali Saeed, Paulina Bartuzi, Janette Heegsma, Daphne Dekker, Niels
Kloosterhuis, Alain de Bruin, Johan W. Jonker, Bart van der Sluis, Klaas Nico

Curriculum Vitae
264
Faber. Hepatic vitamin A metabolism is disturbed in mice with Non-Alcoholic
Fatty Liver Disease leading to vitamin A accumulation in hepatocytes. American
Association for the Study of Liver Diseases (AASLD-2018), 09–13 November
2018, San Francisco, USA.
2. Ali Saeed, Paulina Bartuzi, Janette Heegsma, Daphne Dekker, Niels
Kloosterhuis, Alain de Bruin, Johan W. Jonker, Bart van der Sluis, Klaas Nico
Faber. Hepatic vitamin A metabolism is disturbed in mice with Non-Alcoholic
Fatty Liver Disease leading to vitamin A accumulation in hepatocytes. European
Club for Liver Cell Biology (ECLCB-8), 04–06 October 2018, Bonn, Germany.
3. Ali Saeed, Paulina Bartuzi, Janette Heegsma, Daphne Dekker, A.J.A. (Bart)
van der Sluis, Klaas Nico Faber. Hepatic vitamin A metabolism is disturbed in
mice with Non-Alcoholic Fatty Liver Disease leading to vitamin A accumulation
in hepatocytes. 2nd European Fatty Liver Conference (2nd EFLC 2018), 28
February–2 March Maastricht, 2018, The Netherlands.
4. Ali Saeed, Joanne A. Hoogerland, Janette Heegsma, Terry G.J. Derks, Eveline
van der Veer, Gilles Mithieux, Fabienne Rajas, Maaike H. Oosterveer* and
Klaas Nico Faber. Retinol levels are elevated in circulation, but reduced in liver
in Glycogen storage disease type Ia. NVL-Dutch Liver Retreat 2018 (DLR2018),
1-2 February 2018, Spier, The Netherlands. (Oral presentation).
5. Ali Saeed, Martijn Oscar Hoeke, Mark Hoekstra, Janette Heegsma, Han
Moshage, Klaas Nico Faber. Vitamin A deficiency leads to mild cholestasis and
a “humanized” bile acid profile in rats. Symposium 203, XXIV International Bile
Acid Meeting: Bile Acids in Health and Disease. 17-18 June 2016, Düsseldorf,
Germany. (Poster wins 3rd position in the conference)
6. Ali Saeed, Mark Hoekstra, Martijn Oscar Hoeke, Janette Heegsma, Han
Moshage, Klaas Nico Faber. Vitamin A deficiency promotes hepatic expansion
of resident (Kupffer cells) and peripheral (Grhi) macrophages leading to
excessive liver damage in rats with obstructive cholestasis. Annual 66th Liver
Meeting®, AASLD, 13-17 November 2015, Moscone West Convention Center,
San Francisco, California, USA. (Poster)
7. Ali Saeed, Mark Hoekstra, Martijn Oscar Hoeke, Janette Heegsma, Han
Moshage, Klaas Nico Faber. Vitamin A controls the expression of inflammatory
cytokines and macrophage numbers in obstructive cholestasis in rat. NVL-

Curriculum Vitae
265
Dutch Liver Retreat (5th DLR2014), 30th and 31th October 2014, Spier, The
Netherlands. (Oral presentation)
8. Mark Hoekstra, Martijn Oscar Hoeke, Janette Heegsma, Renze Boverhof, Ali
Saeed, V.W. (Vincent) Bloks, Han Moshage, and Klaas Nico Faber. Vitamin A
promotes the synthesis of biliary beta-muricholic acid in rat. NVL-Dutch Liver
Retreat (4th DLR2013), 26th and 27th September, 2013, Spier, The Netherlands.
(Oral presentation)
9. Ali Saeed, Uman Waheed, Muhammad Ali, Muhammad Ali, Muhammad Qaiser
Khan. Foot-and-mouth disease detection by RT-PCR from original field
materials from Multan district of Punjab, Pakistan; Abstract is published in
National Conference Bahauddin Zakariya University, Multan. 13 October, 2010,
Multan, Pakistan. (Abstract)
10. Uman Waheed, Ali Saeed, Ehtisham-ul-Haque, Qaiser Mahmood Khan.
Diagnosis of recent outbreak of FMD in different districts of Punjab, Pakistan
through RT-PCR; National Conference Rawlakot 21-23, July 2010, Rawlakot,
Pakistan. (Abstract)
11. Qaiser Mahmood Khan, Uman Waheed, Sabir Farooq, Ali Saeed, Muhammad
Farooq. PCR based detection and epidemiology of FMDV and PPRV in field
samples from Punjab, Pakistan. FAO/IAEA international symposium on
sustainable improvement of animal production and health. Synopses, Vienna
(Austria); 8-11 Jun 2009; IAEA-CN--174/93.
12. Uman Waheed, Ali Saeed, Muhammad Asif, Qaiser Mahmood Khan.
Phylogenetic Analysis on the Basis of Amplified VP1 (Capsid Protein) Gene of
FMDV in Clinical Samples from Punjab Areas. Plenary talk - Prof. M.I.D.
Chughtai Memorial Lecture. 9th Biennial Conference Pakistan Society for
Biochemistry and Molecular Biology, Arid Agriculture University, Rawalpindi,
Dec. 17-18, 2008. Page 51. Rawalpindi, Pakistan. (Oral presentation)
13. Usman Waheed, Ali Saeed, Muhammad Asif, Qaiser Mahmood Khan.
Amplification of VP1 (capsid protein) gene of FMDV in clinical samples from
Punjab, Pakistan and its phylogeny. 35th All Pakistan Science Conference,
December 22-23 (2008), Karachi-Pakistan. (Abstract)

Curriculum Vitae
266
BOOK CHAPTERS
1) Ali Saeed, Muhammad Abubakar and Oguz Kul. Modification of Animal
Products for Fat and Other Characteristics. Modification of Animal Products for
Fat and Other Characteristics, pp 55-89. ISBN 978-3-662-46789-3, (2015)
(eBook).
2) Ali Saeed, Ali H. Sayyed, Sohail Safdar and Shumaila Manzoor. Reduction in
Animal Waste, pp 111-134. ISBN 978-3-662-46789-3, (2015) (eBook).
3) Ali Saeed, Muhammad Abubakar and Sehrish Kanwal. Future Challenges
Related to Animal Biotechnology. Modification of Animal Products for Fat and
Other Characteristics, pp 135-147. ISBN 978-3-662-46789-3, (2015) (eBook).
BOOKS
1) Muhammad Abubakar, Ali Saeed, Oguz Kul. The Role of Biotechnology in
Improvement of livestock. Animal health and biotechnology, pp 1-147. ISBN
978-3-662-46788-6; ISBN 978-3-662-46789-3 (eBook). (DOI 10.1007/978-3-
662-46789-3) (2015). (Editor of book).
2) Ali Saeed, Muhammad Abubakar, Qaiser Mahmood Khan. Foot and Mouth
Disease Virus (FMDV); Molecular Epidemiology of FMDV in Province Punjab,
Pakistan. LAP LAMBERT Academic Publishing (2011-09-05).




















