
Effect of coenzyme Q10 and vitamin E on brain energy metabolism in the animal model of Huntington's...
7
Effect of coenzyme Q 10 and vitamin E on brain energy metabolism in the animal model of Huntington’s disease Svatava Kas ˇparova ´ a, * , Zuzana Sumbalova ´ b , Peter Bystricky ´ a , Jarmila Kucharska ´ b , Tibor Liptaj a , Vladimı ´r Mlyna ´rik c , Anna Gvozdja ´kova ´ b a Central Laboratory of NMR Spectroscopy, Faculty of Chemical and Food Technology, Slovak University of Technology, Bratislava, Radlinske ´ho 9, SK-812 37 Bratislava, Slovakia b Pharmacobiochemical Laboratory of the 3rd Department of Internal Medicine, School of Medicine, Comenius University, Bratislava, Slovakia c Laboratory for Functional and Metabolic Imaging, EPFL, Lausanne, Switzerland Received 12 July 2005; accepted 15 September 2005 Available online 14 November 2005 Abstract The neuropathological and clinical symptoms of Huntington’s disease (HD) can be simulated in animal model with systemic administration of 3-nitropropionic acid (3-NP). Energy defects in HD could be ameliorated by administration of coenzyme Q 10 (CoQ 10 ), creatine, or nicotinamid. We studied the activity of creatine kinase (CK) and the function of mitochondrial respiratory chain in the brain of aged rats administered with 3- NP with and without previous application of antioxidants CoQ 10 + vitamin E. We used dynamic and steady-state methods of in vivo phosphorus magnetic resonance spectroscopy ( 31 P MRS) for determination of the pseudo-first order rate constant (k for ) of the forward CK reaction, the phosphocreatine (PCr) to adenosinetriphosphate (ATP) ratio, intracellular pH i and Mg i 2+ content in the brain. The respiratory chain function of isolated mitochondria was assessed polarographically; the concentration of CoQ 10 and a-tocopherol by HPLC. We found significant elevation of k for in brains of 3-NP rats, reflecting increased rate of CK reaction in cytosol. The function of respiratory chain in the presence of succinate was severely diminished. The activity of cytochromeoxidase and mitochondrial concentration of CoQ 10 was unaltered; tissue content of CoQ 10 was decreased in 3-NP rats. Antioxidants CoQ 10 + vitamin E prevented increase of k for and the decrease of CoQ 10 content in brain tissue, but were ineffective to prevent the decline of respiratory chain function. We suppose that increased activity of CK system could be compensatory to decreased mitochondrial ATP production, and CoQ 10 + vitamin E could prevent the increase of k for after 3-NP treatment likely by activity of CoQ 10 outside the mitochondria. Results of our experiments contributed to elucidation of mechanism of beneficial effect of CoQ 10 administration in HD and showed that the rate constant of CK is a sensitive indicator of brain energy disorder reflecting therapeutic effect of drugs that could be used as a new in vivo biomarker of neurodegenerative diseases. # 2005 Elsevier Ltd. All rights reserved. Keywords: Creatine kinase; In vivo saturation transfer 31 P NMR; Rats; 3-Nitropropionic acid; Oxidative phosphorylation; Antioxidants 1. Introduction Huntington’s disease (HD) is a hereditary neurodegenerative disorder, which is characterized by psychiatric symptoms, movement disorder and progressive dementia (Browne et al., 1999). The mutant gene, located on the short arm of chromosome 4, results in an expanded CAG trinucleotide repeat (Beal, 1995). The function of the huntingtin protein is unknown. Several different etiological processes may play roles, and strong evidence from studies in both humans and animal models suggest the involvement of energy metabolism dysfunction, excitotoxic processes, and oxidative stress. The gene defect may cause a subtle impairment of energy metabolism that ultimately leads to neuronal degeneration, initially in the striatum and later in other brain regions (Jenkins et al., 1998). Impaired energy production may lead to increases in intracellular calcium, and generation of free radicals but exact mechanism of energy impairment in HD is unclear. Accidental human ingestion of 3-nitropropionic acid (3-NP), a potent blocker of succinate dehydrogenase (SDH), complex II of mitochondrial electron transport chain, results in chorea and dystonia and in basal ganglia degeneration (Ludolph et al., 1992). www.elsevier.com/locate/neuint Neurochemistry International 48 (2006) 93–99 * Corresponding author. Tel.: +421 2 5292 6018; fax: +421 2 5292 6018. E-mail address: [email protected] (S. Kas ˇparova ´). 0197-0186/$ – see front matter # 2005 Elsevier Ltd. All rights reserved. doi:10.1016/j.neuint.2005.09.002
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Effect of coenzyme Q10 and vitamin E on brain energy metabolism in the animal model of Huntington's...

Effect of coenzyme Q10 and vitamin E on brain energy metabolism
in the animal model of Huntington’s disease
Svatava Kasparova a,*, Zuzana Sumbalova b, Peter Bystricky a, Jarmila Kucharska b,Tibor Liptaj a, Vladimır Mlynarik c, Anna Gvozdjakova b
a Central Laboratory of NMR Spectroscopy, Faculty of Chemical and Food Technology, Slovak University of Technology,
Bratislava, Radlinskeho 9, SK-812 37 Bratislava, Slovakiab Pharmacobiochemical Laboratory of the 3rd Department of Internal Medicine, School of Medicine,
Comenius University, Bratislava, Slovakiac Laboratory for Functional and Metabolic Imaging, EPFL, Lausanne, Switzerland
Received 12 July 2005; accepted 15 September 2005
Available online 14 November 2005
Abstract
The neuropathological and clinical symptoms of Huntington’s disease (HD) can be simulated in animal model with systemic administration of
3-nitropropionic acid (3-NP). Energy defects in HD could be ameliorated by administration of coenzyme Q10 (CoQ10), creatine, or nicotinamid.
We studied the activity of creatine kinase (CK) and the function of mitochondrial respiratory chain in the brain of aged rats administered with 3-
NP with and without previous application of antioxidants CoQ10 + vitamin E. We used dynamic and steady-state methods of in vivo phosphorus
magnetic resonance spectroscopy (31P MRS) for determination of the pseudo-first order rate constant (kfor) of the forward CK reaction, the
phosphocreatine (PCr) to adenosinetriphosphate (ATP) ratio, intracellular pHi and Mgi2+ content in the brain. The respiratory chain function of
isolated mitochondria was assessed polarographically; the concentration of CoQ10 and a-tocopherol by HPLC.
We found significant elevation of kfor in brains of 3-NP rats, reflecting increased rate of CK reaction in cytosol. The function of respiratory chain
in the presence of succinate was severely diminished. The activity of cytochromeoxidase and mitochondrial concentration of CoQ10 was unaltered;
tissue content of CoQ10 was decreased in 3-NP rats. Antioxidants CoQ10 + vitamin E prevented increase of kfor and the decrease of CoQ10 content
in brain tissue, but were ineffective to prevent the decline of respiratory chain function.
We suppose that increased activity of CK system could be compensatory to decreased mitochondrial ATP production, and CoQ10 + vitamin E
could prevent the increase of kfor after 3-NP treatment likely by activity of CoQ10 outside the mitochondria. Results of our experiments contributed
to elucidation of mechanism of beneficial effect of CoQ10 administration in HD and showed that the rate constant of CK is a sensitive indicator of
brain energy disorder reflecting therapeutic effect of drugs that could be used as a new in vivo biomarker of neurodegenerative diseases.
# 2005 Elsevier Ltd. All rights reserved.
Keywords: Creatine kinase; In vivo saturation transfer 31P NMR; Rats; 3-Nitropropionic acid; Oxidative phosphorylation; Antioxidants
www.elsevier.com/locate/neuint
Neurochemistry International 48 (2006) 93–99
1. Introduction
Huntington’s disease (HD) is a hereditary neurodegenerative
disorder, which is characterized by psychiatric symptoms,
movement disorder and progressive dementia (Browne et al.,
1999). The mutant gene, located on the short arm of
chromosome 4, results in an expanded CAG trinucleotide
repeat (Beal, 1995). The function of the huntingtin protein is
unknown. Several different etiological processes may play
* Corresponding author. Tel.: +421 2 5292 6018; fax: +421 2 5292 6018.
E-mail address: [email protected] (S. Kasparova).
0197-0186/$ – see front matter # 2005 Elsevier Ltd. All rights reserved.
doi:10.1016/j.neuint.2005.09.002
roles, and strong evidence from studies in both humans and
animal models suggest the involvement of energy metabolism
dysfunction, excitotoxic processes, and oxidative stress. The
gene defect may cause a subtle impairment of energy
metabolism that ultimately leads to neuronal degeneration,
initially in the striatum and later in other brain regions (Jenkins
et al., 1998). Impaired energy production may lead to increases
in intracellular calcium, and generation of free radicals but
exact mechanism of energy impairment in HD is unclear.
Accidental human ingestion of 3-nitropropionic acid (3-NP),
a potent blocker of succinate dehydrogenase (SDH), complex II
of mitochondrial electron transport chain, results in chorea and
dystonia and in basal ganglia degeneration (Ludolph et al., 1992).

S. Kasparova et al. / Neurochemistry International 48 (2006) 93–9994
Systemic administration of 3-NP to both rats and primates
produces age-dependent striatal lesions, which are strikingly
similar to those seen in HD (Brouillet et al., 1995). Chronic 3-NP
treatment reproduces selective loss of spiny projection neurons
containing the inhibitory neurotransmitter g-aminobutyric acid
(GABA) and sparing of cholinergic neurons that are observed
typically in the HD brain (Beal et al., 1993). It is well known that
GABA is an important source of energy in the brain
(GABA = 1.6 mmol/g, glucose = 1–1.5 mmol/g), thus we sup-
pose that an impairment of GABAergic neurons by 3-NP could
cause changes in the brain energy metabolism leading to increase
of BB-CK activity. SDH inhibitor causes a rapid depletion of
intracellular ATP in neurons, leading to impairment of Ca2+-
ATPase and Na+/K+-ATPase and to progressive membrane
depolarization as a result of intracellular sodium overload
(Brouillet et al., 1999). BB-CK has been found to be functionally
coupled with the plasma membrane-bound Na+/K+-ATPase (Lim
et al., 1983; Wallimann et al., 1985) thus the creatine/
phosphocreatine (Cr/PCr) shuttle is supposed to be a major
component of the neuronal Ca2+ reconstitution mechanism
(Hemmer and Wallimann, 1993; Wallimann and Hemmer, 1994).
Although the main steps leading to 3-NP toxicity have been
recognized, the reasons for the vulnerability of striatal
GABAergic neurons in contrast to the resistance of cholinergic
interneurons to chronic 3-NP application remain not fully
understood.
3-NP lesions formation in rats that have been reported to be
associated with elevated lactate levels (Brouillet et al., 1995)
could be blocked by removal of glutamatergic excitatory
striatal inputs by decortication, by glutamate release inhibitors,
or by glutamate receptor antagonists (Jenkins et al., 1996).
Quantitative localized proton magnetic resonance spectro-
scopy (1H MRS) and proton decoupled 31P MRS are sensitive to
�10% alterations in key cerebral metabolites, and may be of
value in noninvasive monitoring of appropriate therapies in
neurological diseases (Hoang et al., 1998). 1H MRS revealed an
increase of lactate concentration in the occipital cortex of HD
patients that correlated with duration of illness (Jenkins et al.,
1993). A decrease in the phosphocreatine to inorganic
phosphate ratio (PCr/Pi) in HD muscle suggests that the defect
in energy metabolism extends also to non-neural tissue
(Koroshetz et al., 1997). However, measurements of steady-
state high-energy phosphate levels do not reflect energy
metabolism of brain tissue in chronic pathological states
satisfactorily (Kasparova et al., 2000, 2005; Sumbalova et al.,
2002). On the other hand, the rates of synthesis and/or
consumption of high-energy phosphate compounds, i.e.
adenosinetriphosphate (ATP) and phosphocreatine (PCr) could
be more reliable indicators of brain function under these
conditions. Creatine kinase (CK) is an important enzyme that
catalyses the reversible exchange of phosphate group between
PCr and ATP according to the equation:
PCr2� þMgADP� þHþ , MgATP2� þCr (1)
CK in brain exists in multiple forms, and together with Cr and
PCr constitute a system that seems to be critical in regulation of
energy homeostasis in the brain and other organs with high and
fluctuating energy demands (Hemmer and Wallimann, 1993).
Over the last years, the important role of creatine and CK
reaction in various pathological states have been highlighted
(Kasparova et al., 1998, 2000, 2005; Sumbalova et al., 2002;
Wyss and Schultze, 2002). Changes of CK activity may reflect
the energy disorder of the brain as well as therapeutic effect of
drugs (Wyss and Schultze, 2002). Novel therapeutic strategy to
ameliorate mitochondrial-induced dysfunction is to increase
intracellular energy stores. The administration of Cr, the sub-
strate of CK reaction, stimulates mitochondrial respiration and
PCr synthesis, which may help to sustain ATP levels under
oxidative stress (Matthews et al., 1998). There is evidence that
impaired energy metabolism contributes to neuronal death in
HD and the most rapid event after addition of 3-NP is a decrease
in PCr/Cr ratio (Erecinska and Nelson, 1994).
Measurements of lactate production in brains patients with
HD and animals with modeled HD by 1H MRS revealed that
creatine, cyclocreatine, coenzyme Q10 (CoQ10) and nicotina-
mide—compounds increasing energy metabolism could exert
neuroprotective effect in this disease (Koroshetz et al., 1997;
Matthews et al., 1998; Beal, 1999). Antioxidants vitamin E and
CoQ10 form an essential redox-couple (Kagan et al., 2000).
Pretreatment with a-tocopherol had no neuroprotective effect in
animal model of HD (Beal et al., 1988); and treatment with high
doses of a-tocopherol was effective only in patients in early
course of the disease (Peyser et al., 1995). On the other hand,
pretreatment with CoQ10 exerted neuroprotective effect in a
variety of animal models of HD and oral administration of CoQ10
significantly decreased elevated lactate levels in patients with
Huntington’s disease (Beal, 1999). CoQ10 serum levels of HD
patients were significantly lower than that of healthy controls and
HD patients treated with CoQ10 (Andrich et al., 2004). It was also
proposed that supplementation of HD patients with CoQ10, an
essential cofactor of respiratory chain, could reduce their
impaired mitochondrial function (Andrich et al., 2004).
The purpose of the present study was to study the CK
reaction in 3-NP model of HD by kinetic parameter of in vivo31P MR spectroscopy and to elucidate the mechanism of
beneficial effect of the pretreatment with CoQ10 + vitamin E on
brain energy metabolism in 3-NP model of HD.
2. Materials
Male Wistar rats 20–24 months old were injected with 3-NP (10 mg/kg/
every 12 h, i.p.) for 11 days to develop chronic model of HD (Matthews et al.,
1998). Group QE + 3-NP received coenzyme Q10 and vitamin E by the use of
gastric tube (hydrosoluble Q-GEL1, Tishcon Corp., USA) (250 mg CoQ10
+ 530 mg vit. E/kg/day) during 10 days before application of 3-NP. The control
group (C) received corresponding vehicle. At the end of the experiment all the
animals were measured by 31P MRS. The last dose of 3-NP was given to animals
in the evening before their sacrifying.
3. Experimental procedures
3.1. In vivo 31P MRS technique
In vivo 31P MRS experiments were performed at 4.7 T on a SISCO 200/300
imaging spectrometer equipped with horizontal bore magnet for measurements
on animals. 31P MR spectra were collected using 16 mm surface coil. The static

S. Kasparova et al. / Neurochemistry International 48 (2006) 93–99 95
Graph 1. Unidirectional rate constant of CK reaction, kfor, calculated from 31P
MRS saturation transfer experiment. C, control aged rats; 3-NP, aged rats after
administration of 3-nitropropionic acid; QE + 3-NP, rats pretreated with
CoQ10 + vitamin E. Number of animals in groups was 4–6. *p < 0.05 vs. C.
magnetic field was shimmed using the proton signal of water that showed a
typical line width of 20–35 Hz. Relative concentrations of phosphate metabo-
lites were determined from integrals of their signals in 31P MR spectra using
program MESTRE-C 1.5.1. Values of intracellular pHi and Mgi2+ concentration
were calculated using equations given by Jelicks and Gupta (1992).
Time-dependent 31P MRS saturation transfer was applied to determine the
pseudo-first order rate constant of forward CK reaction (kfor) as described
previously (Clark et al., 1991; Mlynarik et al., 1998). Time-dependent satura-
tion transfer allows one to measure simultaneously two parameters, T1 and kfor.
Saturation of the g-ATP resonance for increasing time periods induces an
exponential decay of the PCr resonance to a new steady state. The evolution of
longitudinal magnetization as a function of time is described by the equation
(Forsen and Hoffman, 1963):
dMPCr
dt¼ �½kfor þ ðT1PCrÞ�1�ðMPCr �M0
PCrÞ þ krevðMATP �M0ATPÞ (2)
The dependence of the longitudinal magnetization of the PCr signal, MPCr, on
the time t of the g-ATP signal saturation is given by the equation:
MPCr ¼ M0PCr
�1 � kforT1sPCr
�1 � exp
��t
T1sPCr
���(3)
where M0PCr is the magnetization of PCr in the absence of g-ATP saturation, kfor
the forward CK reaction rate constant, T�11sPCr ¼ kfor þ T�1
1PCr the apparent
longitudinal relaxation rate in the presence of g-ATP saturation, and t is the
irradiation time. The T1sPCr value was calculated as a slope of the semiloga-
rithmic plot of MPCr �M1PCr against t where M1
PCr ¼ M0PCrT
�11PCr=ðkfor þ T�1
1PCrÞ is
the steady-state magnetization of PCr after a long-term irradiation of the g-ATP
signal. The pseudo first-order rate constant kfor was calculated according to the
equation:
kfor ¼1 �M1
PCr=M0PCr
T1sPCr
(4)
In place of the magnetization values, corresponding PCr signal intensities were
used in these calculations. M1PCr was read from the spectrum with 10-s
irradiation of the g-ATP resonance, and M0PCr was obtained from the reference
spectrum measured with the irradiation offset in the mirror position relative to
the PCr resonance and with the irradiation time of 1 s. The saturation was
accomplished by an on-resonance DANTE sequence consisting of a series of
10 ms radio frequency pulses with interpulse delays of 400 ms. The time of
irradiation of the g-ATP resonance was varied from 0.3 to 10 s (Clark et al.,
1991). As a check of validity of the results, the T1PCr values were calculated
using the following equation:
T1PCr ¼ T1sPCr
M0PCr
M1PCr
(5)
3.2. Biochemical analyses
The animals were sacrificed by decapitation. Brain mitochondria were
isolated by differential centrifugation (Sarma et al., 1976). The functions of
respiratory chain in the presence of glutamate as a NAD-substrate or succinate/
rotenone as a FAD-substrate were assessed in isolated mitochondria by means
of Oxygraph Gilson 5/6H (USA) using an oxygen Clark-type electrode. The
activity of cytochromeoxidase (COX) was determined from oxygen consump-
tion after addition of cytochrome c to mitochondria pre-incubated with 0.1%
Triton X-100 (Muscatello and Carafoli, 1969). Mitochondrial proteins were
determined by the method of Lowry et al. (1951) using bovine serum albumin as
a standard. The concentrations of a-tocopherol, CoQ9 and CoQ10 in brain tissue
and mitochondria were determined by the use of high-performance liquid
chromatography (Lang et al., 1986; Kucharska et al., 1998).
3.3. In vitro CK determination
The brain tissue was homogenized with Ultra Turrax in 14 volumes of the
medium containing: 10 mM HEPES, 137 mM NaCl, 4.6 mM KCl, 1.1 mM
KH2PO4, 0.6 mM MgSO4, and protease inhibitors pepstatin (0.7 mg/ml) and
phenyl-methylsulphonyl fluoride (PMSF, 40 mg/ml) (pH 7.4) (Horakova et al.,
2000). The samples were frozen and stored at �20 8C. The activity of CK was
determined using the Creatine Phosphokinase kit (Procedure no. 661) from
Sigma Diagnostic.
3.4. Statistics
The differences between groups were evaluated using Student’s t-test,
p < 0.05 was considered statistically significant. Values in tables are mean
� S.E.M.
4. Results
Using in vivo 31P MRS saturation transfer experiment, we
observed a significant elevation of the rate constant, kfor in brain
of 3-NP rats versus control values (Graph 1). The increase of
kfor was prevented by 10-day pretreatment of rats with high
doses of the antioxidants CoQ10 and vitamin E. On the other
hand, no significant differences between groups were found by
steady-state 31P MR spectroscopy in the ratio of high-energy
metabolites concentrations, intracellular pHi or Mgi2+ content
(Table 1). Also the activities of CK measured by biochemical
method in brain homogenates were similar in all experimental
groups (Table 1).
The measurement of respiratory activities of isolated brain
mitochondria revealed that 3-NP administration induced
pronounced decrease of the rate of ATP production
(�75.3%) as well as of ADP-stimulated (�71.4%) and basal
respiration (�68.8%) in the presence of succinate, i.e. through
complex II of mitochondrial respiratory chain (Table 2). The
rate of ATP production as well as state 3 respiration in the
presence of glutamate were decreased in 3-NP brain
mitochondria (�28.2 and �25.1% versus C), however, these
changes were not statistically significant (Table 2). Antiox-
idants CoQ10 + vitamin E given to rats before induction of HD
were not effective in preventing of 3-NP induced changes in
function of respiratory chain in the presence of succinate and
resulted in the decrease of parameters of oxidative phosphor-
ylation in the presence of glutamate (Table 2). Concentrations
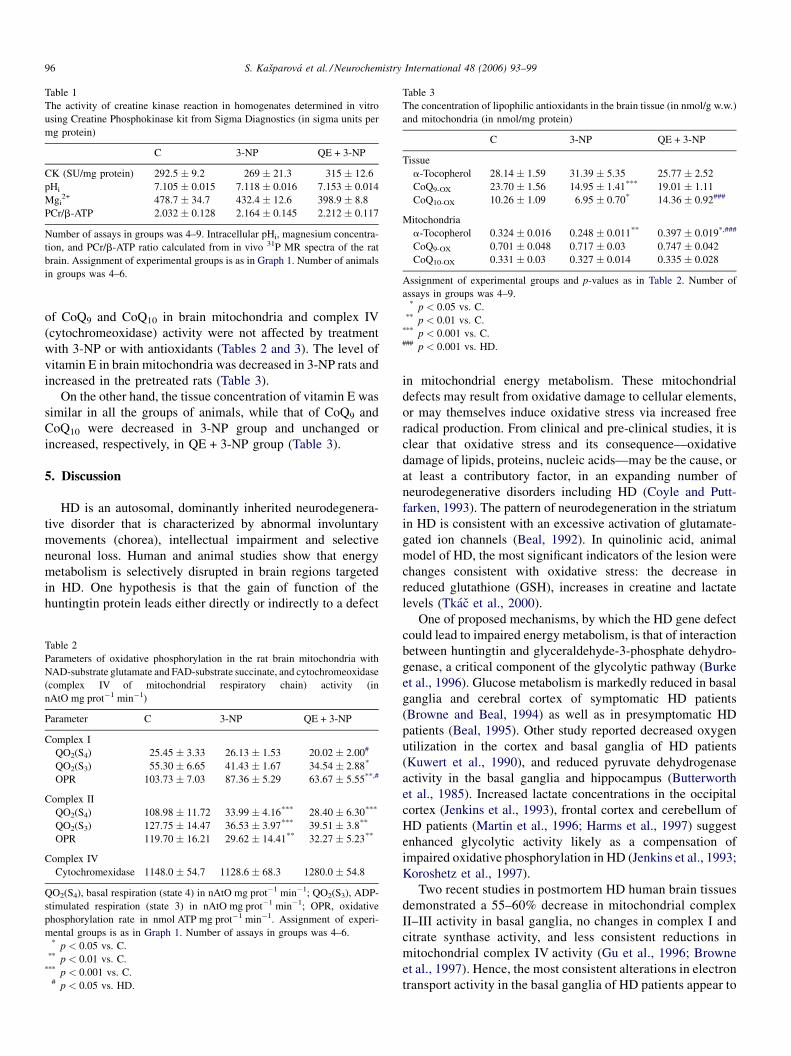
S. Kasparova et al. / Neurochemistry International 48 (2006) 93–9996
Table 1
The activity of creatine kinase reaction in homogenates determined in vitro
using Creatine Phosphokinase kit from Sigma Diagnostics (in sigma units per
mg protein)
C 3-NP QE + 3-NP
CK (SU/mg protein) 292.5 � 9.2 269 � 21.3 315 � 12.6
pHi 7.105 � 0.015 7.118 � 0.016 7.153 � 0.014
Mgi2+ 478.7 � 34.7 432.4 � 12.6 398.9 � 8.8
PCr/b-ATP 2.032 � 0.128 2.164 � 0.145 2.212 � 0.117
Number of assays in groups was 4–9. Intracellular pHi, magnesium concentra-
tion, and PCr/b-ATP ratio calculated from in vivo 31P MR spectra of the rat
brain. Assignment of experimental groups is as in Graph 1. Number of animals
in groups was 4–6.
Table 3
The concentration of lipophilic antioxidants in the brain tissue (in nmol/g w.w.)
and mitochondria (in nmol/mg protein)
C 3-NP QE + 3-NP
Tissue
a-Tocopherol 28.14 � 1.59 31.39 � 5.35 25.77 � 2.52
CoQ9-OX 23.70 � 1.56 14.95 � 1.41*** 19.01 � 1.11
CoQ10-OX 10.26 � 1.09 6.95 � 0.70* 14.36 � 0.92###
Mitochondria
a-Tocopherol 0.324 � 0.016 0.248 � 0.011** 0.397 � 0.019*,###
CoQ9-OX 0.701 � 0.048 0.717 � 0.03 0.747 � 0.042
CoQ10-OX 0.331 � 0.03 0.327 � 0.014 0.335 � 0.028
Assignment of experimental groups and p-values as in Table 2. Number of
assays in groups was 4–9.* p < 0.05 vs. C.
** p < 0.01 vs. C.*** p < 0.001 vs. C.### p < 0.001 vs. HD.
of CoQ9 and CoQ10 in brain mitochondria and complex IV
(cytochromeoxidase) activity were not affected by treatment
with 3-NP or with antioxidants (Tables 2 and 3). The level of
vitamin E in brain mitochondria was decreased in 3-NP rats and
increased in the pretreated rats (Table 3).
On the other hand, the tissue concentration of vitamin E was
similar in all the groups of animals, while that of CoQ9 and
CoQ10 were decreased in 3-NP group and unchanged or
increased, respectively, in QE + 3-NP group (Table 3).
5. Discussion
HD is an autosomal, dominantly inherited neurodegenera-
tive disorder that is characterized by abnormal involuntary
movements (chorea), intellectual impairment and selective
neuronal loss. Human and animal studies show that energy
metabolism is selectively disrupted in brain regions targeted
in HD. One hypothesis is that the gain of function of the
huntingtin protein leads either directly or indirectly to a defect
Table 2
Parameters of oxidative phosphorylation in the rat brain mitochondria with
NAD-substrate glutamate and FAD-substrate succinate, and cytochromeoxidase
(complex IV of mitochondrial respiratory chain) activity (in
nAtO mg prot�1 min�1)
Parameter C 3-NP QE + 3-NP
Complex I
QO2(S4) 25.45 � 3.33 26.13 � 1.53 20.02 � 2.00#
QO2(S3) 55.30 � 6.65 41.43 � 1.67 34.54 � 2.88*
OPR 103.73 � 7.03 87.36 � 5.29 63.67 � 5.55**,#
Complex II
QO2(S4) 108.98 � 11.72 33.99 � 4.16*** 28.40 � 6.30***
QO2(S3) 127.75 � 14.47 36.53 � 3.97*** 39.51 � 3.8**
OPR 119.70 � 16.21 29.62 � 14.41** 32.27 � 5.23**
Complex IV
Cytochromexidase 1148.0 � 54.7 1128.6 � 68.3 1280.0 � 54.8
QO2(S4), basal respiration (state 4) in nAtO mg prot�1 min�1; QO2(S3), ADP-
stimulated respiration (state 3) in nAtO mg prot�1 min�1; OPR, oxidative
phosphorylation rate in nmol ATP mg prot�1 min�1. Assignment of experi-
mental groups is as in Graph 1. Number of assays in groups was 4–6.* p < 0.05 vs. C.
** p < 0.01 vs. C.*** p < 0.001 vs. C.
# p < 0.05 vs. HD.
in mitochondrial energy metabolism. These mitochondrial
defects may result from oxidative damage to cellular elements,
or may themselves induce oxidative stress via increased free
radical production. From clinical and pre-clinical studies, it is
clear that oxidative stress and its consequence—oxidative
damage of lipids, proteins, nucleic acids—may be the cause, or
at least a contributory factor, in an expanding number of
neurodegenerative disorders including HD (Coyle and Putt-
farken, 1993). The pattern of neurodegeneration in the striatum
in HD is consistent with an excessive activation of glutamate-
gated ion channels (Beal, 1992). In quinolinic acid, animal
model of HD, the most significant indicators of the lesion were
changes consistent with oxidative stress: the decrease in
reduced glutathione (GSH), increases in creatine and lactate
levels (Tkac et al., 2000).
One of proposed mechanisms, by which the HD gene defect
could lead to impaired energy metabolism, is that of interaction
between huntingtin and glyceraldehyde-3-phosphate dehydro-
genase, a critical component of the glycolytic pathway (Burke
et al., 1996). Glucose metabolism is markedly reduced in basal
ganglia and cerebral cortex of symptomatic HD patients
(Browne and Beal, 1994) as well as in presymptomatic HD
patients (Beal, 1995). Other study reported decreased oxygen
utilization in the cortex and basal ganglia of HD patients
(Kuwert et al., 1990), and reduced pyruvate dehydrogenase
activity in the basal ganglia and hippocampus (Butterworth
et al., 1985). Increased lactate concentrations in the occipital
cortex (Jenkins et al., 1993), frontal cortex and cerebellum of
HD patients (Martin et al., 1996; Harms et al., 1997) suggest
enhanced glycolytic activity likely as a compensation of
impaired oxidative phosphorylation in HD (Jenkins et al., 1993;
Koroshetz et al., 1997).
Two recent studies in postmortem HD human brain tissues
demonstrated a 55–60% decrease in mitochondrial complex
II–III activity in basal ganglia, no changes in complex I and
citrate synthase activity, and less consistent reductions in
mitochondrial complex IV activity (Gu et al., 1996; Browne
et al., 1997). Hence, the most consistent alterations in electron
transport activity in the basal ganglia of HD patients appear to

S. Kasparova et al. / Neurochemistry International 48 (2006) 93–99 97
be the decrease in complex II–III activity. Genetic defects
affecting complex II–III activity are associated with basal
ganglia degeneration (Bourgeron et al., 1995), comparable
defects in experimental animals result in striatal degeneration
(Brouillet et al., 1995).
3-NP toxicity produces an energy defect in vivo and has
gained acceptance as an animal model of HD. In the present
study, we confirmed that rats injected with 3-NP exhibit
significant disturbance in the brain energy metabolism. As
expected, 3-NP administration resulted in severe inhibition of
respiration and ATP production in brain mitochondria in the
presence of succinate. We observed also a slight decrease in
state 3 respiration and rate of oxidative phosphorylation in the
presence of glutamate. The activity of complex IV was not
affected. Klivenyi et al. (2004) reported approximately 30%
decrease of state 3 respiration supported with pyruvate plus
malate, and 60% decrease in state 3 respiration supported with
a-ketoglutarate in brain mitochondria as well as decreased
a-ketoglutarate dehydrogenase activity in brain tissue of 3-NP-
treated mice. Henry et al. (2002) reported decreased
tricarboxylic acid (TCA) cycle rate and increased exchange
rate between a-ketoglutarate and glutamate in the rat brain 24 h
after single dose of 3-NP. Our results of decreased parameters
of oxidative phosphorylation in the presence of glutamate are
consistent with works of Henry et al. (2002) and Klivenyi et al.
(2004) and could be a result of decreased activity of complex I
or of decreased activity of TCA cycle and particularly of
decreased a-ketoglutarate dehydrogenase activity. GABA is
formed from glutamate in ‘‘GABA shunt’’ bypassing the step of
oxidative decarboxylation of a-ketoglutarate in TCA cycle.
Intrasynaptosomal levels of glutamate and GABA increase
after addition of 3-NP to synaptosomes (Erecinska and Nelson,
1994). The neurotransmitter efflux from synaptosomes is
supposed to be an early step of 3-NP toxicity occurring at
terminal level (Marti et al., 2003).
The lactate–pyruvate ratio is a measure of the redox state of
the cell and it parallels the NADH/NAD+ ratio. On the basis of
oxidative stress, as a hypothesized mechanism for neuronal
vulnerability in HD, we carried out a study on the simultaneous
effect of antioxidants CoQ10 + vitamin E on brain energy
metabolism in 3-NP model of HD. Concerted action of vitamin
E and CoQ10 provides for an effective antioxidant protection of
membranes and lipoproteins (Kagan et al., 2000). Antioxidative
effectiveness of vitamin E is enhanced by CoQ10, which links
recycling process of vitamin E to major electron transport
systems. Treatment with high doses of D-a-tocopherol was not
effective in whole group of HD patients, however, it had
significant selective therapeutic effect on neurological symp-
toms of patients in the early course of the HD disorder (Peyser
et al., 1995). In quinolinic acid (QA) model of HD adm-
inistration of a-tocopherol for 3 days prior to QA had no effect
on striatal lesions (Beal et al., 1988); melatonin suppressed
changes induced by QA (Southgate et al., 1998). a-Lipoic acid,
a potent antioxidant and coenzyme for pyruvate dehydrogenase
and a-ketoglutarate dehydrogenase increased survival of rats in
two transgenic models of HD (Andreassen et al., 2001). The
lactate concentrations in occipital cortex decreased when
patients with HD were taken CoQ10 (Koroshetz et al., 1997).
Pretreatment with CoQ10 exerted neuroprotective effect in a
variety of HD animal models (Beal, 1999). Oral administration
of CoQ10 during 2 months increased its cerebral cortex
concentration in adult rats (Beyer, 1992).
Our steady-state in vivo 31P MRS measurements showed that
concentrations of high-energy phosphate compounds do not
reflect brain function with sufficient sensitivity under this
chronic pathological condition. However, our kinetic results of
in vivo 31P MRS saturation transfer experiment reveal a
significant elevation of kfor in brains of 3-NP rats and provide
evidence for an impairment of energy metabolism in this model
of HD. In vivo 31P NMR transfer measurements showed a
correlation between CK flux and brain 2-deoxyglucose uptake
leading to changes of the brain activity (Sauter and Rudin,
1993; Corbett and Laptook, 1994). Our result of increased CK
activity corresponds well with reported increased 2-deoxyglu-
cose uptake in basal ganglia of HD patients (Beal, 1998).
Furthermore, it has been shown that excitation of the brain
activity (measured by EEG) by bicuculline—a specific
antagonist of GABA effect on GABA-A receptor—induced a
pronounced increase of the rate constant of CK in the rat brain
(Sauter and Rudin, 1993). Thus, we suppose that chronic
administration of 3-NP resulting in selective loss of GABAer-
gic neurons could induce similar increase in kfor.
The study of the brain ATP metabolism, including its
synthesis and consumption, is complicated by regional and
cellular heterogenity. However, it has been demonstrated that
cytosolic isoforms and only minority of mitochondrial isoforms
of CK are monitored in the brain tissue in our experimental
setup of in vivo 31P MRS saturation transfer (Jost et al., 2002).
Thus, the unidirectional rate constant of CK, kfor, is a parameter
predominantly reflecting activity of the cytosolic isoforms of
CK in the rat brain. Our in vitro biochemical study of CK in
brain homogenates showed no significant changes in 3-NP
model of HD. The discrepancy between our in vivo and in vitro
results will be subject of further studies.
Pretreatment of 3-NP-treated rats with high doses of the
antioxidants CoQ10 + vitamin E prevented the increase of kfor.
In our study, the time course of the pretreatment—10 days,
had stimulating effect on content of a-tocopherol predomi-
nantly in the brain mitochondria, without changes in its tissue
level. On the other hand, contents of CoQ9 and CoQ10 were not
changed in brain mitochondria, but were decreased in HD brain
tissue and restored to/over control values in brain tissue of rats
pretreated with antioxidants (Table 3). CoQ is present
ubiquitously in all types of membranes. As an essential
cofactor of mitochondrial electron transport chain CoQ is
inevitable for ATP production. Its antioxidant properties are
associated with major metabolic electron transport systems
(Kagan et al., 2000). Increased trans-plasma-membrane
electron transport has been reported to accompany increased
content of CoQ10 in plasma membrane (Villalba et al., 1995).
The trans-plasma-membrane electron transport chain (Morre,
2005) regenerating cytosolic NAD+ has been reported to play a
key role for viability cells lacking functional mitochondria
(Lawen et al., 1994). In our study, neither the content of CoQ10

S. Kasparova et al. / Neurochemistry International 48 (2006) 93–9998
in mitochondria, nor the mitochondrial function was improved
in rats pretreated with CoQ10 + vitamin E. Nevertheless, the
elevation of the activity of CK in cytosol was prevented.
Jenkins et al. (1996) reported decrease of lactate level in brains
of rats after intrastriatal injection with malonate, which were
pretreated with CoQ10 for 10 days in similar doses as we used in
our experiment. Interestingly, the lactate level was decreased
also when the animals were pretreated with nicotinamide—a
precursor of NAD (Jenkins et al., 1996). We suppose that the
positive effect of CoQ10 + vitamin E pretreatment on brain
energy metabolism of 3-NP rats in our experiment could be
attributed to non-mitochondrial activity of CoQ10 and could be
mediated by cytosolic NADH/NAD+ ratio, which could be
affected by activity of CoQ10 in trans-plasma-membrane
electron transport chain.
Literature data indicate that GABAergic neurons are
especially sensitive to energy deficit (Marti et al., 2003).
Mitochondrial defect produced by 3-NP could be ameliorated
by increasing intracellular energy stores. Administration of
creatine to cultured striatal tissue protected GABAergic
neurons against 3-NP toxicity (Andres et al., 2005). In our
experiment, the energy deficit was reduced by increasing
concentration of CoQ10 in the brain tissue. We suppose that
CoQ10 + vitamin E pretreatment improving brain energy
metabolism could protect GABAergic neurons against 3-NP
toxicity as well.
In summary, our investigation confirmed that systemic 3-NP
administration produces energy defects in brain mitochondria
and affects activity of the CK system as well. We suppose that
increased activity of CK system could be a compensatory
mechanism to decreased ATP production in brain mitochondria
of 3-NP-treated rats.
Results of our 31P MRS experiment confirm importance of
CK/PCr system in the regulation of brain energy metabolism
and contribute to elucidation of mechanisms of a beneficial
effect of CoQ10 administration in HD. Our findings may have
significant implications for clinical treatment of HD, because
we confirmed sensitivity of the method of in vivo 31P MRS
saturation transfer in measurement of CK activity. The
parameter kfor reflects changes in brain energy metabolism
in the 3-NP model of HD and its response to treatment with
antioxidants CoQ10 + vitamin E. We suppose that measurement
of the rate of CK reaction by 31P MR saturation transfer could
be useful for diagnostics of brain metabolic changes in
Huntington’s disease.
Acknowledgments
NMR part of this work was facilitated by the support of the
Slovak State Program of Research and Development no.
2003SP200280203. The financial support from Grant of
Ministry of Education, Slovak Republic and Slovak Grant
Agency for Science (Grants No 1/7547/20 and 1/0546/03) are
gratefully acknowledged. We thank very much for technical
assistance to: Ing. E. Benko, Ing. P. Zauskova, V. Jeskova, A.
Stetkova, and Ing. D. Ivanicka.
References
Andreassen, O.A., Ferrante, R.J., Dedeoglu, A., Beal, M.F., 2001. Lipoic acid
improves survival in transgenic mouse models of Huntington’s disease.
NeuroReport 12, 3371–3373.
Andres, R.H., Ducray, D., Huber, A.W., Perez-Bouza, A., Krebs, S.H., Schlatt-
ner, U., Seiler, R.W., Wallimann, T., Widmer, H.R., 2005. Effects of creatine
treatment on survival and differentiation of GABA-ergic neurons in cultured
striatal tissue. J. Neurochem. 95, 33–45.
Andrich, J., Saft, C., Gerlach, M., Schneider, B., Arz, A., Kuhn, W., Muller, T.,
2004. Coenzyme Q10 serum levels in Huntington’s disease. J. Neural.
Transm. Suppl. 68, 111–116.
Beal, M.F., 1992. Does impairment of energy metabolism result in excitotoxic
neuronal death in neurodegenerative illness? Ann. Neurol. 31, 168–171.
Beal, M.F., 1995. Aging, energy and oxidative stress in neurodegenerative
diseases. Ann. Neurol. 38, 357–366.
Beal, M.F., 1998. Mitochondrial dysfunction in neurodegenerative diseases.
Biochim. Biophys. Acta 1366, 211–223.
Beal, M.F., 1999. Coenzyme Q10 administration and its potential for treatment
of neurodegenerative diseases. Biofactors 9 (2–4), 261–266.
Beal, M.F., Brouillet, E., Jenkins, B.G., Ferrante, R.J., Kowall, N.W., Miller,
J.M., Storey, E., Srivastava, R., Rosen, B.R., Hyman, B.T., 1993. Neuro-
chemical and histologic characterization of striatal excitotoxic lesions
produced by the mitochondrial toxin 3-nitropropionic acid. J. Neurosci.
13, 4181–4192.
Beal, M.F., Kowall, N.W., Swartz, K.J., Ferrante, R.J., Martin, J.B., 1988.
Systemic approaches to modifying quinolinic acid striatal lesions in rats. J.
Neurosci. 10, 3901–3908.
Beyer, R.E., 1992. An analysis of the role of coenzyme Q in free radical
generation and as an antioxidant. Biochem. Cell. Biol. 70, 390–403.
Bourgeron, T., Rustin, P., Chretien, D., Birch-Machin, M., Bourgeois, M.,
Viegas-Pequignot, E., Munnich, A., Rotig, A., 1995. Mutation of a nuclear
succinate dehydrogenase gene results in mitochondrial respiratory chain
deficiency. Nat. Genet. 11, 144–149.
Brouillet, E., Conde, E., Beal, M.F., 1999. Replicating Huntington’s disease
phenotype in experimental animals. Prog. Neurobiol. 59, 427–468.
Brouillet, E., Hantraye, P., Ferrante, R.J., Dolan, R., Leroy-Willig, A., Kowall,
N.W., Beal, M.F., 1995. Chronic mitochondrial energy impairment pro-
duces selective striatal degeneration and abnormal choreiform movements
in primates. Proc. Natl. Acad. Sci. U.S.A. 92, 7105–7109.
Browne, S.E., Beal, M.F., 1994. Oxidative damage and mitochondrial dysfunc-
tion in neuro-degenerative diseases. Biochem. Soc. Trans. 22, 1002–
1006.
Browne, S.E., Bowling, A.C., MacGavey, U., Baik, M.J., Berger, S.C., Muqit,
M.M.K., Bird, E.D., Beal, M.F., 1997. Oxidative damage and metabolic
dysfunction in Huntington’s disease: selective vulnerability of the basal
ganglia. Ann. Neurol. 41, 646–653.
Browne, S.E., Ferrante, R.J., Beal, M.F., 1999. Oxidative stress in Huntington’s
disease. Proceedings of the Symposium on Oxidative Stress in Neurological
Disease, Brain Pathol. 9, 147–163.
Burke, J.R., Enghild, J.J., Martin, M.E., Jou, Y.-S., Myers, R.M., Roses, A.D.,
Vance, J.M., Strittmatter, W.J., 1996. Huntington and DRPLA proteins
selectively interact with the enzyme GAPDH. Nat. Med. 2, 347–350.
Butterworth, R.F., Giguere, J.F., Besnard, A.M., 1985. Activities of thiamine-
dependent enzymes in two experimental models of thiamine-deficiency
encephalopathy: 1. The pyruvate dehydrogenase complex. Neurochem. Res.
10, 1417–1428.
Clark, J.F., Harris, G.I., Dillon, P.F., 1991. Multisite saturation transfer using
DANTE and continuous wave. Magn. Res. Med. 17, 274–278.
Corbett, R.J.T., Laptook, A.R., 1994. Age-related changes in swine brain
creatine kinase katalyzed 31P exchange measured in vivo using 31P NMR
magnetization transfer. J. Cereb. Blood Flow Metab. 14, 1070–1077.
Coyle, J.T., Puttfarken, P., 1993. Oxidative stress, glutamate and neurodegen-
erative disorders. Science 262, 689–695.
Erecinska, M., Nelson, D., 1994. Effects of 3-nitropropionic acid on synapto-
somal energy and transmitter metabolism: relevance to neurodegenerative
brain diseases. J. Neurochem. 63, 1033–1041.

S. Kasparova et al. / Neurochemistry International 48 (2006) 93–99 99
Forsen, F., Hoffman, R.A., 1963. Study of moderately rapid chemical exchange
reactions by means of nuclear magnetic double resonance. J. Chem. Phys.
39, 2892–2901.
Gu, M., Gash, M.T., Mann, V.M., Javoy-Agid, F., Cooper, J.M., Schapira,
A.H.V., 1996. Mitochondrial defect in Huntington’s disease caudate
nucleus. Ann. Neurol. 39, 385–389.
Harms, L., Meiercord, H., Timm, G., Pfeiffer, L., Ludolph, A.C., 1997.
Decreased N-acetyl-aspartate/choline ratio and increased lactate in the
frontal lobe of patients with Huntington’s disease: a proton magnetic
resonance study. J. Neurol. Neurosurg. Psychiat. 62, 27–30.
Hemmer, W., Wallimann, T., 1993. Functional Aspects of creatine kinase in
brain. Dev. Neurosci. 15, 249–260.
Henry, P.G., Lebon, V., Vaufrey, F., Brouillet, E., Hantraye, P., Bloch, G., 2002.
Decreased TCA cycle rate in the rat brain after acute 3-NP treatment measured
by in vivo 1H-(13C) NMR spectroscopy. J. Neurochem. 82, 857–866.
Hoang, T.Q., Bluml, S., Dubowitz, D.J., Moats, R., Kopyov, O., Jacques, D.,
Ross, B.D., 1998. Quantitative proton-decoupled 31P MRS and 1H MRS in
the evaluation of Huntington’s and Parkinson’s diseases. Neurology 50 (4),
1033–1040.
Horakova, L’., Ondrejickova, O., Bachrata, K., Vajdova, M., 2000. Preventive
effect of several antioxidants after oxidative stress on rat brain homoge-
nates. Gen. Physiol. Biophys. 19, 195–205.
Jelicks, L.A., Gupta, R.K., 1992. 31P-NMR of high energy phosphates in perfused
rat heart during metabolic acidosis. Am. J. Physiol. 263, H903–H909.
Jenkins, B.G., Koroshetz, W.J., Bael, M.F., Rosem, B.R., 1993. Evidence for
impairment of energy metabolism in vivo in HD using localized 1H NMR
spectroscopy. Neurology 43 (12), 2689–2695.
Jenkins, B.G., Brouillet, E., Chen, Y.-Ch.I., Storey, E., Schultz, J.B., Kirschner,
P., Beal, M.F., Rosen, B.R., 1996. Non-invasive neurochemical analysis of
focal excitotoxic lesions in models of neurodegenerative illness using
spectroscopic imaging. J. Cereb. Blood Flow Metab. 16, 450–461.
Jenkins, B.G., Rosas, H.D., Chen, Y.C., Makabe, T., Myers, R., MacDonald, M.,
Rosen, B.R., Beal, M.F., Koroshetz, W.J., 1998. 1H NMR spectroscopy
studies of Huntington’s disease: correlations with CAG repeat numbers.
Neurology 50 (5), 1357–1365.
Jost, C.R., Van der Zee, C.E.E.M., in’t Zandt, H.J.A., Oerlemans, F., Verheij,
M., Streijger, F., Fransen, J., Heerschap, A., Cools, A.R., Wieringa, B.,
2002. Creatine kinase B-driven energy transfer in the brain is important for
habituation, and spatial learning behaviour, mossy fibre field size and
determination of seizure susceptibility. Eur. J. Neurosci. 15, 1692–1706.
Kagan, V.E., Fabisiak, J.P., Tyurina, Y.Y., 2000. Independent and concerted
antioxidant functions of coenzyme Q. In: Kagan, V.E., Quinn, P.J. (Eds.),
Coenzyme Q: Molecular Mechanisms in Health and Disease. CRC Press
LLC, pp. 119–129.
Kasparova, S., Mlynarik, V., Liptaj, T., Dobrota, D., Horecky, J., 1998. A study
of creatine kinase reaction in the rat brain during chronic ischemia. In:
Proceedings of the 15th ESMRMB. p. 572.
Kasparova, S., Dobrota, D., Mlynarik, V., Pham, T.N., Liptaj, T., Horecky, J.,
Braunova, Z., Gvozdjakova, A., 2000. A study of creatine kinase reaction in
rat brain under chronic pathological conditions—chronic ischemia and
ethanol intoxication. Brain Res. Bull. 53, 431–435.
Kasparova, S., Brezova, V., Valko, M., Horecky, J., Mlynarik, V., Liptaj, T.,
Vancova, O., Ulicna, O., Dobrota, D., 2005. Study of the oxidative stress in a
rat model of chronic brain hypoperfusion. Neurochem. Int. 46 (8), 601–611.
Klivenyi, P., Starkov, A.A., Calingasan, N.Y., Gardian, G., Browne, S.E., Yang, L.,
Bubber, P., Gibson, G.E., Patel, M.S., Beal, M.F., 2004. Mice deficient in di-
hydrolipoamide dehydrogenase show increased vulnerability to MPTP, mal-
onate and 3-nitropropionic acid neurotoxicity. J. Neurochem. 88, 1352–1360.
Koroshetz, W.J., Jenkins, B.G., Rosen, B.R., Beal, M.F., 1997. Energy meta-
bolism defects in Huntington’s disease and effects of coenzyme Q10. Ann.
Neurol. 41, 160–165.
Kucharska, J., Gvozdjakova, A., Mizera, S., Braunova, Z., Schramekova, E.,
Schreinerova, Z., Pechan, I., Fabian, J., 1998. Participation of coenzyme Q10
in the rejection development of the transplanted heart: a clinical study.
Physiol. Res. 47 (6), 399–404.
Kuwert, T., Lange, H.W., Langen, K.J., Herzog, H., Aulich, A., Feinendegen,
L.E., 1990. Cortical and subcortical glucose consumption measured by PET
in patients with Huntington’s disease. Brain 113 (Pt. 5), 1405–1423.
Lang, J.K., Gohil, K., Packer, L., 1986. Simultaneous determination of toco-
pherols, ubiquinols, and ubiquinones in blood, plasma, tissue homogenates,
and subcellular fractions. Anal. Biochem. 157, 106–116.
Lawen, A., Martinus, R.D., McMullen, G.L., Nagley, P., Vaillant, F., Wolvetang,
E.J., Linnane, A.W., 1994. The universality of bioenergetic disease: the role
of mitochondrial mutation and the putative inter-relationship between
mitochondria and plasma membrane NADH oxidoreductase. Mol. Aspects
Med. 15, S13–S27.
Lim, L., Hall, C., Leung, T., Mahadevan, L., Whatley, S., 1983. Neurone-
specific enolase and creatine phosphokinase are protein components of rat
brain synaptic plasma membranes. J. Neurochem. 41, 1177–1182.
Lowry, D.H., Rosenbrough, N.Y., Farr, A.L., Randall, R.J., 1951. Protein
measurement with the Folin phenol reagent. J. Biol. Chem. 193, 265–276.
Ludolph, A., Seeling, M., Ludolph, A., Novitt, P., Allen, C.N., Spencer, P.S.,
Sabri, M.I., 1992. 3-Nitropropionic acid: exogenous animal neurotoxin and
possible human striatal toxin. Can. J. Neuro. Sci. 18, 492–498.
Marti, M., Mela, F., Ulazzi, L., Hanau, S., Stocchi, S., Paganini, F., Beani, L.,
Bianchi, C., Morari, M., 2003. Differential responsiveness of rat striatal
nerve endings to the mitochondrial toxin 3-nitropropionic acid: implications
for Huntington’s disease. Eur. J. Neurosci. 18, 759–767.
Martin, W.R.W., Hanstock, C., Hodder, J., Allen, P.S., 1996. Brain energy
metabolism in Huntington’s disease measured with in vivo proton magnetic
resonance spectroscopy. Ann. Neurol. 40, 538.
Matthews, R.T., Yang, L., Jenkins, B.G., Ferrante, R.J., Rosen, B.R., Kaddurah-
Daouk, R., Beal, M.F., 1998. Neuroprotective effects of creatine and
cyclocreatine in animal models of Huntington’s disease. J. Neurosci. 18,
156–163.
Mlynarik, V., Kasparova, S., Liptaj, T., Dobrota, D., Horecky, J., Belan, V.,
1998. Creatine kinase reaction rates in rat brain during chronic ischemia.
MAGMA 7, 162–165.
Morre, D.J., 2005. Quinone oxidoreductases of the plasma membrane. Methods
Enzymol. 378, 179–199.
Muscatello, U., Carafoli, E., 1969. The oxidation of exogenous and endogenous
cytochrome C in mitochondria. A biochemical and ultrastructural study. J.
Cell Biol. 40 (3), 602–621.
Peyser, C.E., Folstein, M., Chase, G.A., Starkstein, S., Brandt, J., Cockrell, J.R.,
Blysma, F., Coyle, J.T., McHugh, P.R., Folstein, S.E., 1995. Trial of D-a-
tocopherol in Huntington’s disease. Am. J. Psychiat. 152 (12), 1771–1775.
Sauter, A., Rudin, M., 1993. Determination of creatine kinase kinetic para-
meters in rat brain by NMR magnetization transfer. J. Biol. Chem. 682,
13166–13171.
Sarma, J.S., Ikeda, S., Fischer, R., Maruyama, Y., Weishaar, R., Bing, R.J.,
1976. Biochemical and contractile properties of heart muscle after pro-
longed alcohol administration. J. Mol. Cell. Cardiol. 8, 951–972.
Southgate, G.S., Daya, S., Potgeiter, B., 1998. Melatonin plays protective role in
quinolinic acid-induced neurotoxicity in the rat hippocampus. J. Chem.
Neuroanat. 14, 151–156.
Sumbalova, Z., Kasparova, S., Bystricky, P., Kucharska, J., Mlynarik, V.,
Gvozdjakova, A., 2002. Effect of coenzyme Q10 and vitamin E on brain
and skeletal muscle energy metabolism in animal model of Huntington’s
disease. In: Proceedings of the Third Conference of the International
Coenzyme Q10 Association, London 2002, pp. 193–195.
Tkac, I., Keene, C.D., Pfeuffer, J., Low, W.C., Gruetter, R., 2000. Metabolic
changes in quinolinic acid-lesioned rat striatum detected non-invasively by
in vivo 1H NMR spectroscopy. J. Neurosci. Res. 66 (5), 891–898.
Villalba, J.M., Navarro, F., Cordoba, F., Serrano, A., Arroyo, A., Crane, F.L.,
Navas, P., 1995. Coenzyme Q reductase from liver plasma membrane:
Purification and role in trans-plasma-membrane electron transport. Proc.
Natl. Acad. Sci. U.S.A. 92, 4887–4891.
Wallimann, T., Hemmer, W., 1994. Creatine kinase in non-muscle tissues and
cells. Mol. Cell. Biochem. 133–134, 193–220.
Wallimann, T., Walzthony, D., Wegmann, G., Moser, H., Eppenberger, H.M.,
Barrantes, F.J., 1985. Subcellular localization of creatine kinase in Torpedo
electrocytes: association with acetylcholine receptor-rich membranes. J.
Cell Biol. 100, 1063–1072.
Wyss, M., Schultze, A., 2002. Health implications of creatine: can oral creatine
supplementation protect against neurological and atherosclerotic disease?
Neuroscience 112 (2), 243–260.




















