
Age-related dendritic hypertrophy and sexual dimorphism in rat basolateral amygdala

DOI: 10.1161/CIRCULATIONAHA.114.006185
1
The Cardiac CaMKII Genes and Contribute Redundantly to Adverse
Remodeling but Inhibit Calcineurin-Induced Myocardial Hypertrophy
Running title: Kreusser et al.; CaMKII isoforms and calcineurin
Michael M. Kreusser, MD1; Lorenz H. Lehmann, MD1; Stanislav Keranov, MD1; Marc-Oscar Hoting1; Michael Kohlhaas, PhD2; Jan-Christian Reil, MD2; Kay Neumann, MD3;
Michael D. Schneider, MD4; Joseph A. Hill, MD, PhD5; Dobromir Dobrev, MD6; Christoph Maack, MD2; Lars S. Maier, MD3; Hermann-Josef Gröne, MD7; Hugo A. Katus, MD8;
Eric N. Olson, PhD9; Johannes Backs, MD1
1Research Unit Cardiac Epigenetics, Dept of Cardiology, University of Heidelberg, Heidelberg, Germany; DZHK (German Centre for Cardiovascular Research), partner site Heidelberg/
Mannheim, Germany; 2Dept of Cardiology, Saarland University, Homburg, Germany; 3Dept of Internal Medicine II, University of Regensburg, Regensburg, Germany; 4British Heart Foundation Centre of Research Excellence, National Heart and Lung Institute, Faculty of Medicine, Imperial College London, United Kingdom; 5Dept of Internal Medicine, University of Southwestern Texas
Medical Center, Dallas, TX; 6Institute of Pharmacology, University of Duisburg-Essen, Essen, Germany; 7Dept of Molecular Pathology, German Cancer Research Center, Heidelberg, Germany; 8Dept of Cardiology, University of Heidelberg, Heidelberg, Germany; DZHK (German Centre for
Cardiovascular Research), partner site Heidelberg, Germany; 9Dept of Molecular Biology, University of Southwestern Texas Medical Center, Dallas, TX
Address for Correspondence:
Johannes Backs, MD
Research Unit Cardiac Epigenetics, Department of Cardiology
Heidelberg University
Im Neuenheimer Feld 410
69120 Heidelberg, Germany
Tel: +49-6221-56-37714
Fax: +49-6221-56-5573
E-mail: [email protected]
Journal Subject Codes: Heart failure:[110] Congestive, Myocardial biology:[155] Physiological and pathological control of gene expression, Basic science research:[130] Animal models of human disease
p g , p gy, y gg, gg,Germany; DZHK (German Centre for Cardiovascular Research), partner sitee HHHeeieiddedelblblberererg/g/g/
Mannheim, Germany; 2Dept of Cardiology, Saarland University, Homburg, Geermrmmananyy;y; 33DeDeDeptptpt oooff Internal Medicine II, University of Regensburg, Regensburg, Germany; 4British Heart FoundationCentre of Research Excellence, National Heart and Lung Institute, Faculty of Medicine, ImperialCoollllegegegeee LoLoLondndndonnn, UnUnU ited Kingdom; 5Dept of Inteernrnrnalaa Medicine, Univerersisisitytyty of Southwestern Texas
MeMeMedidicaall CeCeCenterer,, DaD lllasa ,, TXT ; 6InI sttiti utute ofo Phaarmmacaccologgy,y, UUnivererrsiis tyty oof f DDuisbuburgrg-Esssenn,, EsE sen, GGGerrmrmany; 7DeDeD ptptpt ooff MoMoMoleelecucuc lalalarr r PaPaPathththololologogo yy,y, GGGerere mmamann CCCancnccererer RRReseseseaeaarcrr h h h CeCeCenntnteeer, HeHeHeidididelelelbebebergrgg,,, GeGeGermrmrmanana y;rrr88Deeept of Cardiolllogggy, UnUnnivererrsisiityt oofff HHeHeidideelbeeerggg, Heeiiddelbebebergrgrg,,, GGGermmmaaany; DDDZZZHHKHK ((GeG rrmrmanan CCCeentrrre fof r
CaCaC rdiovavavascscs uululaaar RReeeseeaearchhh), papap rtrtnenen r r siitete HHHeiiidedelblbbeerrg,g,, GGGerermamamanyny;; 99DeDeptptt oof f MoMoMolelel cucullalarr r Biiollogogyyy, UnUnUnivivivererersisis ty oooff f SoSoSoututhwhwwesesesteteternrnn TTTexexexasasas MMMedededicicicalalal CCCennnteteterr,r, DDDalalallalalas,s TTTXX X
Address for rr CoCoCorrrrrresesspopop ndndn enenenccce:
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.114.006185
2
Abstract
Background—Ca2+-dependent signaling through CaMKII and calcineurin was suggested to
contribute to adverse cardiac remodeling. However, the relative importance of CaMKII versus
calcineurin for adverse cardiac remodeling remained unclear.
Methods and Results—We generated double-knockout mice (DKO) lacking the two cardiac
CaMKII genes and specifically in cardiomyocytes. We show that both CaMKII isoforms
contribute redundantly to phosphorylation not only of phospholamban, ryanodine receptor 2,
histone deacetylase 4 but also calcineurin. Under baseline conditions, DKO mice are viable and
display neither abnormal Ca2+ handling nor functional and structural changes. Upon pathological
pressure overload and beta-adrenergic stimulation, DKO mice are protected against cardiac
dysfunction and interstitial fibrosis. But surprisingly and paradoxically, DKO mice develop
cardiac hypertrophy driven by excessive activation of endogenous calcineurin, which is
associated with a lack of phosphorylation at the auto-inhibitory calcineurin A site Ser411.
Likewise, calcineurin inhibition prevents cardiac hypertrophy in DKO. Upon exercise
performance, DKO mice show an exaggeration of cardiac hypertrophy with increased expression
of the calcineurin target gene RCAN1-4 but no signs of adverse cardiac remodeling.
Conclusions—We established a mouse model in which CaMKII’s activity is specifically and
completely abolished. By the use of this model we show that CaMKII induces maladaptive
cardiac remodeling while it inhibits calcineurin-dependent hypertrophy. These data suggest
inhibition of CaMKII but not calcineurin as a promising approach to attenuate the progression of
heart failure.
Key words: heart failure, signal transduction, calcium/calmodulin-dependent protein kinase II, calcineurin, cardiac hypertrophy
display neither abnormal Ca2+ handling nor functional and structural changes. UpUppononon pppatatathohohololologigigicac lt
pressure overload and beta-adrenergic stimulation, DKO mice are protected against cardiac
dysfsfunununctctctioioion n n anana d d inininteterstitial fibrosis. But surprisiingngnglylyl and paradoxicallylyy, DKDD O mice develop
cacaardddiai c hypeertrtroroophphphy y drdrdrivvivenene bbyy y exexexcececessssssiviviveee acacctititivvavattioonn of enenendododogegegenonoousus cccalallcicicinnneururininin, , whwhwhicicich h h isisis
asassosos cicc atated wwwititith h aaa llaackck off phphooosphphphororylylylatatioionn atat thhhee aauuttoo-i-iinhnhnhibibiiitoorory y cacalclcineeueuririn n A A A sisitete SSeeer44411.1.
LiLiLikekekewiwiwisesese, cacacalclclcininineueueuriririnnn inininhihihibibibitititiononon ppprerereveveventntntsss cccararardididiacacac hhhypypypererertrtrtropopophyhyhy iiinnn DKDKDKOOO. UUUpopoponnn exexexererercicicisesese
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.114.006185
3
Introduction
Heart failure is the leading cause of death in developed countries.1 It results from adverse cardiac
remodeling upon pathological stress situations such as arterial hypertension, ischemic injuries or
genetic causes. Adverse cardiac remodeling is usually described by a combined appearance of
myocardial hypertrophy, activation of a fetal gene program, cell death and interstitial fibrosis.2
Ca2+-dependent signaling pathways including CaMKII and calcineurin were both proposed to
play pivotal roles in adverse cardiac remodeling.3
The protein kinase CaMKII consists of four different isoforms with distinct expression
patterns. CaMKII and CaMKII are enriched in the brain, but CaMKII and CaMKII are
expressed ubiquitously. CaMKII is the predominant cardiac CaMKII isoform but CaMKII is
also expressed in the heart.4 In human and experimental heart failure, CaMKII expression and
activity is enhanced.4, 5 Phosphorylation of the ryanodine receptor RyR2 by CaMKII has been
reported to cause sarcoplasmic reticulum (SR) Ca2+ leak, which in turn seems to drive heart
failure development.6, 7 At the epigenetic level, CaMKII inactivates the negative regulator of
adverse cardiac remodeling histone deacetylase 4 (HDAC4), leading to transcriptional activation
of the myocyte enhancer factor 2 (MEF2), and phosphorylates histone 3.8-14 Transgenic
overexpression of the splice variants CaMKII B (localizes to the nucleus) and CaMKII C
(localizes to the cytosol) promote cardiac hypertrophy and dilated cardiomyopathy,
respectively.15, 16 CaMKII inhibitory peptides prevent structural heart disease.17, 18 Mice with a
global deletion of CaMKII were protected against adverse cardiac remodeling.7, 8 However in
all of these models, substantial phosphorylation of the target phospholamban (PLB) was still
detectable, indicating an incomplete loss-of-function.
The protein phosphatase calcineurin consists of two subunits: calcineurin A (CnA), which
expressed ubiquitously. CaMKII is the predominant cardiac CaMKII isoform bubuut CaCaCaMKMKMKIIIIII iis s
also expressed in the heart.4 In human and experimental heart failure, CaMKII expression and
acctitiivivivitytyty iiiss enene hhhancncceeded.4, 5 Phosphorylation of the ryryyanannodine receptororr RyRyRR22 2 byb CaMKII has been
eepooortr ed to caaususeee sssarcrcopopplalalasmsmsmicicic rretetticicicululumumum (SSRR))) Caaa2+++ leaaakk,k, wwwhihiichch iinn tuturnrnn sseeeemsmsms tto o drdrd ivivive e heeearart tt
faailililururureee dedevevevelololopmpmmennnt.t.6,6, 7 AAAt t thhhe ee epepepigigigeeneneetticcc lllevevevelell,, CCaaMKMKMKIIII iiinnnacactititivavaateeesss ththt eee nnenegagaatitiivevev rrregegegullaaatooror ooff
adverse carddiaiaaccc rereemomomodeded lill ngngg hhhisi tototonenen dddeaaacececetytytylalalasesese 444 (((HDHDH ACACAC4)4)4), , leleleadadadinnng gg tototo tttrararansnsn crcrripipptititionononalaa activationnn
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.114.006185
4
contains the catalytic site and calcineurin B (CnB), the small regulatory Ca2+-binding subunit.19
Transgenic CnA overexpression in mice induces massive cardiac hypertrophy by
dephosphorylation-dependent translocation of the transcription factor NFAT from the cytosol to
the nucleus.20, 21 The CnA- and CnA- isoforms are expressed in the myocardium but only
CnA was demonstrated to be stress responsive.22 Moreover, mice lacking CnA were protected
against cardiac hypertrophy.23 Inhibition of CnA has been suggested as a treatment option for
cardiac hypertrophy because studies with the calcineurin inhibitory agent cyclosporine A (CyA)
have been successful in rodents.24 On the other hand, the splicing variant CnA- 1 seems to
activate Akt-dependent cardioprotective pathways without inducing NFAT-dependent
maladaptive hypertrophy.25
We developed a cardiomyocyte-specific double knockout model, in which the two
cardiac isoforms, CaMKII and CaMKII , were both deleted. This model identifies CaMKII as
the critical regulator of maladaptive cardiac remodeling and reveals that CaMKII serves as a
negative regulator of calcineurin activity in vivo. Unexpectedly, we provide evidence that cardiac
hypertrophy due to over-activation of endogenous calcineurin does not cause systolic cardiac
dysfunction.
Methods
Mouse experiments
We generated conditional knockout mice containing cardiomyocyte-specific deletions of
CaMKII ( -CKO), CaMKII ( -CKO) or both isoforms (DKO) by crossing previously
described conditional knockout mice8, 26 to transgenic mice expressing Cre recombinase under
the control of the -MHC promoter ( MHC-Cre) or by using the mER-Cre-mER fusion protein
maladaptive hypertrophy.25
We developed a cardiomyocyte-specific double knockout model, in which the two
caardrddiaiaiaccc iisisofofofororrmss,, CCaCaMKII and CaMKII , wereee bbbootth deleted. Thihiis s momodededel l identifies CaMKII as
hhhe crc itical reggulullatatoror ooof mamamalalaladadadaptptivivvee e cacaardddiac reeemoodedeelingngng aandndd rrreveveeaealsls tthahahat CaCaaMKMKM IIIII sererervevees aasas aaa
nenegagagatititivevev rregegeguluulatatooror oof f ccacallclcinineueuuririinnn acacacttitiviviitytyy ininin vvivivvooo.. UnUnUnexxxpepeectctctededdlylyly,, wewee ppproroovvividedee eeviviv dedeencncnce tththaatat ccaaarddidiac
hypertrophy y dududue e e tototo ooovevev r-rr accctitit vavv tititiononon ooof f enenendododogegegenononoususu cacaalcl ininneueueuririin n n dododoesss nnnototo cccauauauseses sssysysystototolililiccc cardiac
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.114.006185
5
under the control of a cardiomyocyte-specific -MHC-promoter.27, 28 As controls, littermates
homozygous for the conditional CaMKII and CaMKII alleles without MHC-Cre transgene
( -FF, -FF and FFFF) were used. For some experiments, C57BL/6 mice were obtained from
Charles River (wild type mice). A detailed description of the surgical methods, the exercise
protocol and in vivo imaging as well as hemodynamic measurement techniques can be found in
Supplemental Methods. All experimental procedures were reviewed and approved by the
Institutional Animal Care and Use Committee at the Regierungspräsidium Karlsruhe, Germany.
Histology
The protocols for hematoxylin and eosin (H&E), Masson´s trichrome staining and CD31
immunostaining can be found in the Supplemental Methods.
Western Blotting, GST-pulldown and immunocytochemistry
The immunoblotting, GST-pulldown and immunocytochemistry protocols as well as the
antibodies used in this study can be found in the Supplemental Methods.
CaMKII kinase activity
CaMKII kinase activity was measured by detecting the amount of endogenous CaMKII that
associates to GST-HDAC4 419-670 or by radioactive kinase assay using 32P-ATP.10 A detailed
description can be found in the Supplemental Methods.
Gene expression analysis
The RNA isolation and quantitative RT-PCR as well as the primer sequences used in this study
can be found in the Supplemental Methods.
Culture of primary cardiomyocytes
Adenoviruses for gene transfer into cardiomyocytes were produced as described in the
Supplemental Methods. Neonatal mouse ventricular myocytes (NMVMs) and adult mouse
mmunostaining can be found in the Supplemental Methods.
Western Blotting, GST-pulldown and immunocytochemistry
ThThee e imimimmumunonon bbblototttititinngng, GST-pulldown and immunnococo yytochemistry pppror toocococolls as well as the
anntiiibbodies useed d iini thihisss ststtuddudyy y ccacan n bebebe ffoouunnnd innn tthhe SuSuSuppplelelemememenntntalal MMMeeethohoddds..
CaCaMKMKMKIIIIII kkkinininasaseee acacactitivvvitytyy
CaMKII kinnasasase e e acacactititiviviv tytyty wwasasas mmmeaeaeasusus rerered d bybyby dddetetetececectititingngng tttheheh aaamomomounununt t t ofoo eeendndndogogogenenenououo ss s CaCaCaMKMKMKII that
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.114.006185
6
ventricular myocytes (AMVMs) were isolated from wild type and mutant mice, neonatal rat
ventricular myocytes (NRVMs) were isolated from Wistar rats (Charles River). A detailed
description is included in the Supplemental Methods.
Epifluorescence microscopy
Freshly isolated AMVMs were incubated with Fura-2 AM or Indo-1 AM and Ca2+ transients as
well as SR Ca2+ load were measured. AMVMs were transilluminated by red light (>650 nm) to
visualize sarcomeres, and fractional shortening was measured. A detailed description is included
in the Supplemental Methods.
Statistical analysis
Data are summarized as mean±SEM. Statistical analysis was performed with the Graph-Pad
Prism Software Package Version 5.0 (GraphPad, Inc.). When two groups were compared, we
used Wilcoxon-Mann-Whitney U tests. When more than two groups were compared, a Kruskal-
Wallis test was applied. When we obtained a significant p-value we continued with pair-wise
comparisons using Wilcoxon-Mann-Whitney U tests according to the closed testing principle. A
value of P<0.05 was considered statistically significant.
Results
CaMKII and CaMKII contribute redundantly to cardiac CaMKII activity
Gene-expression analysis of the four CaMKII genes (CaMKII , CaMKII , CaMKII and
CaMKII ) in RNA samples from healthy and diseased wild type mouse hearts was performed in
relation to the expression levels in the brain. In accordance with others,4 we revealed that besides
CaMKII , CaMKII is also expressed in the heart, whereas CaMKII and CaMKII are not (Fig.
1A). Moreover, both genes were up-regulated in diseased hypertrophic hearts after transverse
Data are summarized as mean±SEM. Statistical analysis was performed with the e GrGrrapapph-hh-PaPaPad d d
Prism Software Package Version 5.0 (GraphPad, Inc.). When two groups were compared, we
ussededd WWWilililcococoxoxoxon-MaMaMannn-Whitney U tests. When mmmoroo eee than two grououupsp wwwererere e compared, a Kruskal-
WaWalllllis test waas s aaappplplieiei d.d. WWWhehehen nn wewee ooobtbtaaainnned aa ssignnnifffiicananntt t p-p-vavavaluluee wwewe ccooonttinunuuededed wwititith hh papairi -w-w-wissise
coompmpmparararisisononns ss ususininng g g WWiilcccoxoxonnn-M-MMananann-nn-WhWhWhitititneneney U UU teteeststtss aaaccococordrrdininng g g ttoto tttheheh ccclooosesed dd teteteststininingg g prprrinnnciciplplle.. AA
value of P<00.0.00555 wawawass s cococ nsnn ididderererededd ssstatatttisii titiicacacalllll y y y sisisigngngnififificicicanana t.t..
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.114.006185
7
aortic constriction (TAC)-induced pressure overload of the left ventricle. Thus, we took
advantage of two conditional CaMKII knockout models that we developed previously,8, 26 and
generated cardiomyocyte-specific CaMKII ( -CKO), CaMKII ( -CKO) and
CaMKII /CaMKII double knockout mice (DKO) (Fig. 1B). As controls, MHC-Cre negative
littermates ( -FF, -FF and FFFF) were used. Western blot analysis using an antibody, that was
shown to recognize at least CaMKII and CaMKII ,8, 26 confirmed a marked loss of CaMKII in
cardiac extracts of -CKO and DKO (Fig. 1C). With this antibody, CaMKII was almost not
detectable in -CKO, suggesting (under the assumption that the antibody has the same affinity to
CaMKII as to CaMKII ) that CaMKII is largely more abundant than CaMKII in the heart
(Fig. 1C, see also Suppl. Fig. 1A). A CaMKII -specific antibody could detect CaMKII in -
CKO but not in -CKO and DKO mice, suggesting that CaMKII could compensate for the loss
of CaMKII in -CKO mice. Notably, phosphorylation of the CaMKII target site of PLB Thr17
was only slightly reduced in -CKO mice, whereas it was completely abolished in DKO mice,
confirming that CaMKII and CaMKII exert redundant roles and largely compensate for each
other. As a control, phosphorylation at the protein kinase A (PKA) site PLB-Ser16 was not
reduced (Fig. 1C). Thus, DKO represents a so far unique model for a complete loss of CaMKII
activity in cardiomyocytes in vivo. We also analyzed the phosphorylation level of targets of
interest. Consistent with PLB, phosphorylation of RyR2-Ser2814 and HDAC4-Ser632 was
decreased moderately in -CKO but clearly in DKO animals. Again, phosphorylation at the PKA
site RyR2-Ser2808 was not affected by CaMKII deletion (Fig. 1C).
CaMKII DKO mice are viable and do not display cardiac abnormalities
DKO mice developed normally and showed no increase in mortality or apparent morphological
abnormalities. Cardiac size as indicated by heart weight/body weight ratios, ventricular chamber
Fig. 1C, see also Suppl. Fig. 1A). A CaMKII -specific antibody could detect CaCaaMKMKMKIIIII iiin n --
CKO but not in -CKO and DKO mice, suggesting that CaMKII could compensate for the loss
off CCCaMaMaMKIKIIIII iiin C-C-CKKO mice. Notably, phosphorrylyly aattion of the CaaMKMKM IIIII tttaararget site of PLB Thr17
wwasss ono ly slighhtltlyyy rrreduduuceceedd inin -C-CKOKOKO mmmiccce, wwwhhhereaasas it wawawass cccommpmpllletttelyl aaabboolilishshsheded iinnn DDKDKOO mimiccce,,
coconfnfnfiririrmimimingngng ttthhahat t t CaCaCaMKMKMKIIIII andndd CCCaMaMaMKIKIIII eeexexexertt rrredededunnndadadantntt rrroloolesess aanndnd lllaaargrgrgelely y y cocoompmpmpenenenssasatetee fororor eeeaacach h h
other. As a cococontntntrororol,l,l ppphohoh spsps hohohoryryrylaaatititiononon at t thththe e e prprp otototeieiein n n kikikinanan sesese AAA (((PKPKPKA)A)A sssititte e e PLPLPLB-B-B SeSeSer1r1r16 6 6 wawaw s not
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.114.006185
8
dimensions or fractional shortening, assessed by echocardiography were unchanged in mice at
age of 12 weeks (Suppl. Fig. 1B). To study functional parameters in more detail we used a
working heart preparation to standardize pre- and afterload (Suppl. Fig. 1C). We detected slight
improvements in ESPVR (which was not significant in all experimental groups used in this
study, see also Fig. 3A) as a measure for maximal pressure that can be developed by the ventricle
at any given LV volume. Based on the observation that the CaMKII sites on PLB and RyR2 were
hypo-phosphorylated, we measured global parameters of Ca2+ handling (Suppl. Fig. 1D-E).
However, we did not detect any change in Ca2+ transients, SR Ca2+ content, diastolic Ca2+
concentration and single-cell fractional shortening, surprisingly indicating that – at least under
basal conditions – CaMKII activity is dispensable for intracellular Ca2+ handling and
cardiomyocyte contractility.
Calcineurin precedes CaMKII activation upon pathological pressure overload
We analyzed the time course of CaMKII and calcineurin activation after TAC (Fig. 2A-B).
Starting at 24 hours after TAC, CaMKII expression (CaMKII C > CaMKII B) and CaMKII
activity, as judged by its activity-dependent binding affinity to HDAC4,10 increased (~2-fold)
(Fig. 2A). We did not use the usual antibodies recognizing CaMKII autophosphorylation to
determine CaMKII activity because in DKO one of these antibodies still showed a signal
although CaMKII protein was gone (Suppl. Fig. 2A and Suppl. Fig. 1A). As another indication
for CaMKII activation, phosphorylation of HDAC4 at its CaMKII phosphorylation site Ser632
also started to increase three days after TAC. In contrast, expression of RCAN1-4, a specific
target gene of the calcineurin-NFAT pathway, which is considered as an endogenous calcineurin
reporter,29 was dramatically overexpressed (~17-fold) one day after TAC (Fig. 2B). These data
indicate that CaMKII activation and HDAC4 phosphorylation depend on upstream signals
basal conditions – CaMKII activity is dispensable for intracellular Ca2+ handlingg aandndnd
cardiomyocyte contractility.
CaCalclclcininineeueuriririn n pprecececeededes CaMKII activation upononon pppathological prprpressusuurerere overload
WeWe aanalyzed ththhee tiiimeme cououoursrsrsee e ofoof CCCaMaMaMKKKIIII anndd ccalccinnneurrrininn aaccctiivvatatioionn afafftteterr TATAACCC ((FiFiig.g. 222A-A-A BBB).).
StStararartitiingngng aatt 242424 hhoouurrsrs aafffteerr TTACACAC, CaCaCaMKMKMKIIII eeexpxpxpreeesssss ioioonn n (C(CCaMaMMKKKIIII CC >> CCCaMaMMKKIKIII BBB))) anandd d CCaCaMKMKMKIIII
activity, as jjududdgegeged d d bybyby iiitstss accctititivivv tytyy-d-ddepepepennndededentntnt bbbininindidiingngng aaffffinininititity y y totoo HHHDADAAC4C4C4,,,101010 iiincncn rerereasasasededd (((~2~ -fold)
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.114.006185
9
distinct from the upstream signals of calcineurin activation.
Dissociation of cardiac dysfunction and interstitial fibrosis from myocardial hypertrophy
Next, we performed TAC surgeries in DKO and FFFF mice. Strikingly, DKO were protected
against cardiac fibrosis, increased caspase 3/7 activity as marker for apoptosis and systolic
dysfunction and display an attenuated rarefication of capillarization (Fig. 3A, see also Suppl.
Fig. 3A). This protective effect was not associated with changes in intracellular Ca2+ handling or
cellular contractility (Suppl. Fig. 2C+D). Surprisingly, TAC still induced cardiac and
cardiomyocyte hypertrophy in DKO. Moreover, the gene expression changes of typical fetal
genes (ANP, BNP, MHC) and remodeling marker ( MHC) were similar in DKO and FFFF
(Fig. 3B). These data indicate a dissociation of cardiac hypertrophy and re-activation of the fetal
gene program on one hand from maladaptive cardiac remodeling including interstitial fibrosis,
apoptosis, reduced capillarization and cardiac dysfunction on the other hand. To confirm these
data in an independent model, we applied neurohormonal stress to DKO. We injected
isoproterenol (Iso) to DKO and FFFF mice. Iso led to cardiac hypertrophy (Suppl. Fig. 4A) and
fetal gene activation in DKO as observed in FFFF mice (Suppl. Fig. 4B). But again and
consistent with the TAC data, we observed less cardiac fibrosis, apoptotic markers and collagen
expression in DKO (Suppl. Fig. 4A+B, see also Suppl. Fig. 3A). These observations were
especially surprising in view of our previous finding that global deletion of one isoform,
CaMKII , attenuated cardiac hypertrophy and fetal gene activation.8 However, in our previous
model we could still detect substantial phosphorylation of the typical CaMKII target PLB-Thr17
in the myocardium because CaMKII was not deleted (see also Fig. 1C). Thus, we hypothesized
that complete CaMKII inhibition might exert dual effects on anti- and pro-hypertrophic
pathways. To substantiate this hypothesis we performed a gene dosage experiment (Fig. 3C).
Fig. 3B). These data indicate a dissociation of cardiac hypertrophy and re-activavaatitionono oof f f thththe e e feffettal
gene program on one hand from maladaptive cardiac remodeling including interstitial fibrosis,
appopopoptototosisisis,s, rrredededuccededed ccapillarization and cardiac dyyyssfs uuunction on thee ooothererr hhhaand. To confirm these
ddadataaa in an indepepeenendedentnt mmmodododelelel,,, wewe aapppppllieeed neeeuuurohororormomoonanaal l sststreessss too DKDKKOO.. WWWe e ininjejeectctctededed
ssopopoprorootetet rerenononolll (I(Issoo) ) toto DDKKOKO aaandndnd FFFFFFFFFFF mimimicecece... Isssooo leleed d tot ccararardididiacacc hhhyypypererertrtrt ooophhyhy (((SuSuSuppppppll.l. FFigigg. 44A4A)) ) annnd d
fetal gene actctivivivatata ioioion n n ininn DDDKOKOKO aas ss obobobseseservrvvededed iiin n n FFFFFFFFFFFF mmmicicee (((SuSuSuppppppl.l.l. FFigigg.. 4B4B4B).).). BBBututt aaagagagaininin aaand
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.114.006185
10
Consistent with our previous findings, the homozygous deletion of either CaMKII or CaMKII
resulted in a reduction of cardiac hypertrophy upon TAC. Paradoxically, the additional
heterozygous deletion of the other CaMKII isoform resulted in a less pronounced reduction of
cardiac hypertrophy upon TAC. Furthermore, the complete cardiac-specific deletion of all four
CaMKII and CaMKII gene copies (DKO) did not prevent cardiac hypertrophy. We also
analyzed the effects of the single deletion of CaMKII and CaMKII in isolated adult
cardiomyocytes and could confirm the anti-hypertrophic effects of CaMKII and CaMKII
(Suppl. Fig. 3C). We were also interested whether CaMKII or CaMKII play a specific role for
apoptosis because our recent data suggested a specific role of CaMKII for apoptosis at least in
macrophages.30 However, TUNEL assays indicated that CaMKII deletion more than CaMKII
deletion attenuates cardiomyocyte apoptosis and that the combined deletion is even more
effective (Suppl. Fig. 4B).
CaMKII controls the calcineurin-NFAT pathway
Thus, we hypothesized that complete CaMKII deletion may result in an activation of a pro-
hypertrophic pathway. Therefore, we searched for candidate signaling molecules that are
activated by TAC or Iso. Whereas, protein kinase D (PKD) was not activated in DKO after TAC
(Suppl. Fig. 2A), we found the calcineurin reporter gene RCAN1-4 to be strongly up-regulated
in DKO mice after TAC and Iso (Fig. 4A, Suppl. Fig. 2E). This could not be observed after
deletion of only one CaMKII gene, in particular CaMKII (Suppl. Fig. 2E), indicating that
complete CaMKII deletion in the heart results in calcineurin activation. Consistently, Iso induced
a dramatic increase in nuclear NFAT translocation and NFAT activity in NMVMs from DKO
when compared to NMVMs from FFFF (Fig. 4B+C). In this regard, it was reported that CaMKII
directly phosphorylates CnA-Ser411 in vitro but it remained unclear whether this mechanism
macrophages.30 However, TUNEL assays indicated that CaMKII deletion moree thahaan CaCaCaMKMKMKIIII
deletion attenuates cardiomyocyte apoptosis and that the combined deletion is even more
efffefeectctctivivivee (((SuSuSuppppl.l. FFFigi . 4B).
CCCaMMMKII contrtroolo ss thheee ccacalclclcininneeueuririin-n-n-NFNFFAAAT pppaaathwwwaayy
ThThhususu , wewew hhhypypypotothhehessisizezedd tththata cccomommplplpletetetee CaCaCaMKMKMKIIII dddeelletettioionn n mamamayyy rereresusuultt iiin n n aanan aaactctivivivatatatioionnn ooof aaa pproro--
hypertrophicc pppatatathwhwhwayayay.. ThTT erererefefeforrre,e,e, wwweee seseseararrchchchededed ffororo ccanana didididadadatetete sisisigngngnalllinining g g momomolelelecucuuleleles ss ththhatatat are
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.114.006185
11
was of biological significance in vivo.30, 31 Thus, using a newly synthesized specific antibody
against CnA-Ser411 (Fig. 4D) we aimed to detect endogenous CnA phosphorylation. Indeed,
overexpression of active CaMKII (T287D) in NRVMs resulted in hyper-phosphorylation of
CnA-Ser411 (Fig. 4E). In accordance, we found that CnA was strongly hypo-phosphorylated in
cardiac lysates from sham- and TAC-operated DKO hearts, which was associated with RCAN1-
4 upregulation (Fig. 4F). Moreover, as compared to DKO CnA-Ser411 hypo-phosphorylation
was less pronounced in cardiac extracts of -CKO, and not found in -CKO, explaining why
calcineurin activity was only strongly increased in DKO but not in the single knockouts (Fig.
4G).
Cardiac hypertrophy, but not dysfunction is controlled by calcineurin during pathological
and physiological hypertrophy in DKO
To test whether cardiac hypertrophy after TAC in DKO depended on calcineurin activation, we
performed CnA knockdown experiments in isolated NMVMs of mutant mice. CnA knockdown,
which resulted in approximately 50% decrease of CnA protein (Suppl. Fig. 5A), prevented Iso-
induced cellular hypertrophy and activation of fetal genes in myocytes derived from DKO but
not FFFF (Fig. 5A-B). We then repeated the TAC experiment in DKO with the calcineurin
inhibitor cyclosporine A (CyA). CyA was shown to inhibit cardiac hypertrophy after TAC in
dosages from 12.5 - 50 mg/kg body weight per day.31 Using a dosage of 4 mg/kg body weight
per day, which did not affect TAC-induced cardiac hypertrophy in FFFF, we were able to inhibit
RCAN1-4 expression as well as cardiac and cardiomyocyte hypertrophy in DKO but not FFFF
(Fig. 5C and Suppl. Fig. 5B), indicating that TAC-dependent hypertrophy in DKO depends on
calcineurin. As CnA knockdown, CyA attenuated also the expression of fetal genes in DKO
(Fig. 5B and Suppl. Fig. 5B). In contrast to the anti-hypertrophic effects of CyA in DKO, CyA
Cardiac hypertrophy, but not dysfunction is controlled by calcineurin duriinngng pppattthohohololologiggicacal
and physiological hypertrophy in DKO
Too tttesesesttt whwhwhetetethehh r cacacardrdr iac hypertrophy after TAC ininin DDDKO depended d d on cccalalalccineurin activation, we
perffforo med CnnA AA knknknocckdkdk owowownn n exexxpeperirir mmementntss in issoolatteddd NNMVMVM MsMsMs ooff f mmumutatannnt mmiciccee.e. CCnAnAnA kkknonockckckdododowwwn,
whwhhicici hhh rerer susuultltltedeed iinnn apapapprprproxxximimatatatelellyyy 505050%%% dededecrcrcreaeaeaseee ooff f CCnCnAAA prprprotototeiiin n n ((SuSuSuppppp ll.. FFFigig. . 5A5A5A))),,, prprpreveveenentteted d IIsoo-o-
nduced cellulullararar hhhypypypererrtrtrropophyhyhy aandndnd acacctitit vavaatitiononon ooof f f fefefetatatal l gegeg nenenes s ininin mymymyocoo ytytyteseses ddderererivivivededd fffrororom m m DKD O but
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.114.006185
12
did not affect the protective effects of DKO on cardiac fibrosis or systolic function, providing
evidence that - at least in this model - calcineurin-dependent cardiac hypertrophy is not
maladaptive on the one hand but also not cardioprotective on the other hand. In line with the
latter, we could not accumulate evidence that the potential cardioprotective calcineurin-
dependent Akt pathway was activated (Suppl. Fig. 5C).
To clarify the role of the CaMKII-calcineurin pathway in physiological hypertrophy, we
performed endurance exercise training by a swimming protocol in DKO and FFFF mice (Fig.
5D-F). Interestingly, whereas CaMKII protein levels were not altered after swimming in control
mice (FFFF), CaMKII activity decreased about 30% (Fig. 5D). Remarkably, we found
exaggerated heart weight/body weight (HW/BW) ratios and increased myocyte size in DKO
mice after swimming, which was associated with increased expression of the calcineurin target
gene RCAN1-4 (Fig. 5E+F). Again, despite calcineurin activation and increased cardiac
hypertrophy no signs for systolic cardiac dysfunction were detected, indicating that activation of
endogenous calcineurin is not maladaptive.
Discussion
By generating mice lacking the two cardiac CaMKII isoforms and we introduce a new loss-
of-function model with the deletion of both cardiac CaMKII isoforms in cardiomyocytes, and
demonstrate that CaMKII - under unstressed conditions - is dispensable in cardiomyocytes with
regard to integrity, growth, cardiomyocyte contractility and Ca2+-handling. This is somewhat
surprising in light of innumerous reports about CaMKII functions. But from the drug developer’s
point of view, this indicates that CaMKII inhibition does not exert major unwanted side effects
on basal cardiac function. Furthermore, we show that - upon pathological stress conditions -
exaggerated heart weight/body weight (HW/BW) ratios and increased myocyte sisiizeee iinn n DKDKDKO OO
mice after swimming, which was associated with increased expression of the calcineurin target
gegenenene RRRCACACAN1N1N1-4 (((FFiFigg. 5E+F). Again, despite calclccinini eeeurin activationonn andndd iiinnncreased cardiac
hhyhyppepertrophy nono sssigggnsns ffororr sssysysystototolilic c cacacardrdiaiaccc dyysfffuuncttiooon wwweerere e dedeetetecccteeded, ininindiiicacatititinngng tthahahat t acactitt vavavatitiononon of
enndodod gegegenonoususus cccalalcciinneneururinin iis s nooot t mamamalalaladdadapptptivivve.ee.
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.114.006185
13
CaMKII plays a dual role in the regulation of adverse cardiac remodeling (Fig. 6). On the one
hand, CaMKII transduces maladaptive signaling. On the other hand, CaMKII negatively
regulates calcineurin-dependent cardiac hypertrophy in vivo.
Redundancy of CaMKII and CaMKII
Because of a lack of loss-of-function models with the deletion of both cardiac CaMKII isoforms,
it was so far not possible to study all essential roles of CaMKII in myocardial disease. Moreover,
most of the reported CaMKII studies used non-genetic approaches such as overexpression of
peptides or pharmacologic compounds such as KN-93. However, off-target effects, as e.g.
described on protein kinase D or ion channels (e.g. L-type Ca2+ channels),8, 32, 33 could have
complicated the conclusions. Therefore, we focused first on the development of a specific and
complete genetic mouse model, in which the two CaMKII genes that are expressed in
cardiomyocytes are deleted. We found that CaMKII and CaMKII compensate for each other
and act redundantly on the phosphorylation of PLB-Thr17, RyR2-Ser2814, CnA-Ser411 and
HDAC4-Ser632. In contrast to previously described approaches aiming at inhibiting CaMKII,7, 8,
17, 18, 34, 35 the new DKO model results in profound hypo-phosphorylation of these typical
CaMKII phosphorylation targets.
CaMKII controls calcineurin signaling
Previous data from our lab showed that homozygous CaMKII knockout mice are protected
against cardiac hypertrophy and interstitial fibrosis.8 Here we demonstrate that the loss of more
than two cardiac CaMKII gene copies unmasks a dual role of CaMKII in regulating cardiac
hypertrophy. DKO mice appear to develop a similar hypertrophic response as FFFF littermates
after TAC and Iso, but histological, gene expression and biochemical analyses revealed that
cardiac hypertrophy in DKO depends on activation of endogenous calcineurin. CaMKII
complicated the conclusions. Therefore, we focused first on the development of f aaa spppecccififificicic aaandnnd
complete genetic mouse model, in which the two CaMKII genes that are expressed int
caardrddioioiommymyocococytytytes aaarrere deleted. We found that CaMKMKMKIIII and CaMKIKIIIII ccomommppensate for each other
anndd d aca t redunddananntllyy ononn ttthehehe pphohohospsphohohorryryllattiion offf PLLBLB--Thhrhr1717,, RRyRyRR22--SSerer2822 1414,, CnCnCnA-A-SeSeSer4r4r411111 aandndnd
HDHDDACACAC4-4-SeSeer6r6r63232.. InInn cconoontrtrt aast t totot ppprerereviviv ououuslllyy y ddedescsccririr bbbeddd apapapprprproaoaoachhhesese aimimimiinngng aaat t ininnhihihibibib titiingngng CCCaMMMKKIII,,7,7 8
777, 181818 33, 3444, 353535 thhe e nenenew w w DKDKDKO OO momomodeded l rereresusuultltl s ininin ppprororofffououundndnd hhhypypy o-o-o phphphosososphphphororylylylatatatioioion n n ofofo tthehehesesese tttypypypical ffffff
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.114.006185
14
phosphorylates CnA-Ser411, which was shown to lead to a decrease in its phosphatase
activity.36, 37 Here, we show that this phosphorylation event occurs in vivo and in a CaMKII-
dependent manner. Moreover, we found CnA-Ser411 hypo-phosphorylation to be associated
with cardiac hypertrophy after pathological and physiological stress. Importantly and in contrast
to current paradigms, the data of this study suggest that anti-hypertrophic effects are not required
for an improved cardiac function. Moreover, a vast amount of literature suggested that
calcineurin induces pathological cardiac hypertrophy, ultimately leading to heart failure.20, 21, 38,
39 However, the latter studies used forced overexpression of a truncated CnA construct lacking
the regulatory domain that is phosphorylated by CaMKII, complicating the interpretation of
these findings. A recent CnA loss-of-function study seemed to prove the idea that calcineurin
mediates pathological cardiac hypertrophy but this study focused mainly on cardiac hypertrophy
but not on other aspects of pathological remodeling such as cardiac dysfunction.23 In fact,
histological analyses did not show a protection against cardiac fibrosis in CnA null mice.23 In
the present study we identify DKO mice as a model with activation of endogenous calcineurin.
We challenge current paradigms and conclude that - at least in the absence of CaMKII signals -
activation of endogenous calcineurin is not maladaptive. Support comes from a recent study,
demonstrating that calcineurin induces physiological hypertrophy in pregnant mice.40 Moreover,
Heineke et al. suggested that calcineurin exerts cardioprotective functions under certain
conditions: hearts of transgenic mice with mild overexpression of CnA were protected in a
murine model of dilated cardiomyopathy.41 Thus it is tempting to speculate that calcineurin
activation in DKO contributes to the observed cardioprotective effects upon TAC. Because it
was suggested that the splice variant CnA 1 exerts cardioprotection via Akt,25 we measured Akt
phosphorylation. But we found no changes. We cannot completely rule out that other
hese findings. A recent CnA loss-of-function study seemed to prove the idea thahat cccalclclcininineueueuririr n n
mediates pathological cardiac hypertrophy but this study focused mainly on cardiac hypertrophy
buutt t nononottt onn ooothththerr aaasspspects of pathological remodelililingngng such as cardiiacaca ddysyssfufufunction.23 In fact,
hhistttolo ogical ananallyysssess dddiddd nnotott ssshhohow w w aaa prproottectiiionnn aggagaiinst t t ccacardrddiaaacc fifiibrrosisis innn CnCnCnAAA nnululu l l l mmmiccece.23323 Innn
hhe e prprpresesesenenent t t stsstududdy y y wewewe iiidedeentntn ifi y y DKDKDKO O O mimicecec aaasss a a momomoddedel ll wiwiwiththh aaacctc ivivvatatioioi n n ofofof enenndodod gegegenononoususus ccalalalcicic neneneurururininn..
We challengegege cccururu rererentntn ppparara adaddigigigmsmss aaandndd cconononclclcludududeee thththatatat --- aaat leleleasasast t t ininin ttthehh aaabsbsbsenenencecece ooof f CaCaCaMKMKMKIIII signals -
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.114.006185
15
calcineurin-dependent pathways contribute to the cardioprotective effects but our data show that
CyA-treatment after TAC in DKO did not lead to cardiac dysfunction. Thus, we conclude that -
in the setting of the current study - CaMKII-dependent but calcineurin-independent processes
mediate pathological remodeling.
CaMKII is required for pathological remodeling but not cardiac hypertrophy and fetal
gene expression
The question arises of how CaMKII regulates pathological remodeling, especially cardiac
fibrosis. We have shown that HDAC4 transmits CaMKII signals to the cardiac genome via
MEF2.8, 10, 13 Because gene deletion and overexpression studies revealed that MEF2D primarily
drives cardiac fibrosis and dysfunction rather than cardiac hypertrophy,42 the CaMKII-HDAC4-
MEF2 pathway is a likely pathway leading to adverse cardiac remodeling (see model in Fig. 6).
Likewise, HDAC4 hypo-phosphorylation was associated to the protective situation in DKO
mice. But it was remarkable that DKO mice were not protected against re-activation of certain
fetal genes including ANP, BNP and MHC. It is important to note, that fetal gene activation is
suggested to be driven not only by MEF2 but also NFAT.43 Thus, we speculate that over-
activation of the calcineurin-NFAT pathway in DKO compensates for the attenuation of the
CaMKII-HDAC4-MEF2 pathway with regard to ANP, BNP and MHC but not for attenuation
of yet unidentified genes that cause cardiac fibrosis. Natriuretic peptides are actually known not
to be causative but rather to be adaptive to combat the hemodynamic load and oxidative capacity
of muscle cells.44, 45 On the other hand, MEF2-dependent genes that cause interstitial fibrosis are
not clearly described yet. Taken together, our data indicate that fetal genes associated with
calcineurin-dependent hypertrophy do not cause cardiac dysfunction. Likewise, in a model of
physiological hypertrophy we found no activation of CaMKII but activation of calcineurin-
drives cardiac fibrosis and dysfunction rather than cardiac hypertrophy,42 the CaMaMMKIKIK II-I-HDHDHDACACAC44-
MEF2 pathway is a likely pathway leading to adverse cardiac remodeling (see model in Fig. 6).
Liikekekewiwiwisese, HDHDHDACCC444 hyh po-phosphorylation was aassssssocciated to the prprroto ecctititivveve situation in DKO
mmiccece. But it wwasas reememaarrkakaablblble e thththatat DDDKOKOKO mmmicee wwweree nnot ppprroroteteectteded aaggaininstst rre-e-t acacactititivavatititiononn oof ff cececertrtaiaa nnn
feetatatal l l gegegenenen ss s inininclcluududininng g g ANANANP,P BBBNPNPNP aaandndn MHMHMHCCC. IIIt t t isiss impmpmpororortatatanntnt ttto o o nnootetete,,, ththhatt ffetettalala gggenenne e e aca ttitivvaatitioonon iis s
uggested too bbbe e e drdrdrivivivenenen nnnotot ooonlnln y y bybyby MMMEFEFEF2 2 2 bububut t t alalalsososo NNNFAFAFAT.T.T 43 TTThuhuhus,ss wwwe e e spspspecececululu atatte e e thththatatat ooovev r-
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.114.006185
16
NFAT signaling without signs of pathological remodeling.
CaMKII is dispensable for intracellular Ca2+ handling under unstressed conditions
Another puzzling observation of this study was the lack of effect on Ca2+ handling although
profound changes on phosphorylation on RyR2 and PLB were detected. We speculate that the
lack of CaMKII phosphorylation on Ca2+ handling proteins might be compensated by other
mechanisms such as PKA-mediated phosphorylation, although we could not detect significant
increases in phosphorylation at the PKA sites PLB-Ser16 or RyR2-Ser2808. Knockin mouse
models with a specific mutation of these sites would clarify the specific role of CaMKII
phosphorylation at PLB-Thr17 and RyR2-Ser2814. Indeed, Respress et al. described RyR2-
2814-S/A knockin mice.6 Consistent with our data, they also observed an unaltered SR Ca2+
content, Ca2+ transient decay and Ca2+ sparks at baseline, but they discovered that in the situation
of heart failure, mutant mice were protected. Thus, further experiments are warranted to
investigate DKO mice and Ca2+ handling in models of severe heart failure. However, it appears
unlikely that an attenuation of disturbed Ca2+ handling contributes to the improved cardiac
function as observed in DKO after TAC in the present study.
Summary
Taken together, we show that CaMKII is a key driver of maladaptive cardiac remodeling. This
study describes maladaptive cardiac remodeling as a process of interstitial fibrosis and cardiac
dysfunction but not cardiac hypertrophy and activation of commonly measured fetal genes such
as ANP and BNP. Under certain conditions, CaMKII inhibits cardiac hypertrophy by a
previously underestimated cross talk mechanism to calcineurin. In the absence of CaMKII
signals, calcineurin does not seem to contribute to maladaptive cardiac remodeling, highlighting
CaMKII and not calcineurin as a promising drug target to combat heart failure.
2814-S/A knockin mice.6 Consistent with our data, they also observed an unalteerereed d SRSRR CCCaaa2+ 2+2+
content, Ca2+ transient decay and Ca2+ sparks at baseline, but they discovered that in the situation
off hhheaeaeartrtrt fffaiaiailululurrre, mumumutat nt mice were protected. Thuhuhus,s further experimimmenntstss aaarer warranted to
nnveeests igate DKKOO O mmimicecee aaandndnd CCCaaa2+ 2+2+ hahaanndndliingng in mmmodeeelss of f seseevevereree hheeaarrtt ffaiaiilull rrere. HoHoHowewevevev r,r,r iiitt apapappepeearrrs
ununlililikekekelylyly tthahahat t t anan aaattttenenuuuattitionon ooof f dididistststuruurbebeed d CaCaCa222+ hhhananandldliingg g cococontntntriibububuttees s tototo tthehehe iimpmpmprorooveveedd d ccarrdrdiiaac c
function as obobbseseservrvvededed iiin n n DKDKKO O O afffteteterr TATAT C CC ininin ttthehehe ppprerer sesesentnn ssstututudydydy..
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.114.006185
17
Acknowledgments: The authors thank Ulrike Oehl, Lonny Jürgensen, Jutta Krebs, Sylvia Katz,
Claudia Heft, Michaela Oestringer and Claudia Liebetrau for their excellent technical assistance.
We thank David Stanmore for editing the manuscript. We thank Lorenz Uhlmann for statistical
advice.
Funding Sources: J.B., H.A.K., D.D. and L.S.M. were supported by the DZHK (Deutsches
Zentrum für Herz-Kreislauf-Forschung - German Centre for Cardiovascular Research) and by
the BMBF (German Ministry of Education and Research). J.B. was supported by the Deutsche
Forschungsgemeinschaft (BA 2258/2-1, SFB 1118) and by the European Commission (FP7-
Health-2010; MEDIA-261409). L.S.M. was supported by the Deutsche Forschungsgemeinschaft
(SFB 1002, IRTG 1816) and the Fondation Leducq. L.H.L. is recipient of a HRCMM
(Heidelberg Research Center for Molecular Medicine) career development fellowship. M.M.K.
was supported by a research grant from the Ernst-und-Berta-Grimmke foundation and by a
Young Investigator grant of the University of Heidelberg. D.D. was supported by a grant from
Foundation Leducq (07CVD03).
Conflict of Interest Disclosures: None.
References: 1. Vitali E, Colombo T, Bruschi G, Garatti A, Russo C, Lanfranconi M and Frigerio M. Different clinical scenarios for circulatory mechanical support in acute and chronic heart failure. Am J Cardiol. 2005;96:34L-41L.
2. Backs J and Olson EN. Control of cardiac growth by histone acetylation/deacetylation. CircRes. 2006;98:15-24. 3. Zarain-Herzberg A, Fragoso-Medina J and Estrada-Aviles R. Calcium-regulated transcriptional pathways in the normal and pathologic heart. IUBMB life. 2011;63:847-855. 4. Colomer JM, Mao L, Rockman HA and Means AR. Pressure overload selectively up-regulates Ca2+/calmodulin-dependent protein kinase II in vivo. Mol Endocrinol. 2003;17:183-192. 5. Hoch B, Meyer R, Hetzer R, Krause EG and Karczewski P. Identification and expression of delta-isoforms of the multifunctional Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circ Res. 1999;84:713-721.
g ) p pp
was supported by a research grant from the Ernst-und-Berta-Grimmke foundationonn aaandndnd bbby y y a aa
Young Investigator grant of the University of Heidelberg. D.D. was supported by a grant from
Foundation Leducq q (07CVD03).
CCConnfnflict of Innteterererestt DDDisssclclcloososururureses:: NNoNonnene..
Refefererencceses:
11 VVititalalii EE CCololomombobo TT BrBrususchchii GG GGararatattiti AA RuRussssoo CC LLananfrfranancoconini MM aandnd FrFrigigererioio MM DiDiffffererenenn
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.114.006185
18
6. Loneragan K, Cheah TB, Bunn SJ and Marley PD. The role of protein kinase C in nicotinic responses of bovine chromaffin cells. Eur J Pharmacol. 1996;311:87-94.
7. Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, Dalton ND, Peterson KL, Chen J, Bers D and Brown JH. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009;119:1230-1240. 8. Backs J, Backs T, Neef S, Kreusser MM, Lehmann LH, Patrick DM, Grueter CE, Qi X, Richardson JA, Hill JA, Katus HA, Bassel-Duby R, Maier LS and Olson EN. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc Natl Acad Sci U S A. 2009;106:2342-2347. 9. Zhang T, Kohlhaas M, Backs J, Mishra S, Phillips W, Dybkova N, Chang S, Ling H, Bers DM, Maier LS, Olson EN and Brown JH. CaMKIIdelta isoforms differentially affect calcium handling but similarly regulate HDAC/MEF2 transcriptional responses. J Biol Chem. 2007;282:35078-35087. 10. Backs J, Song K, Bezprozvannaya S, Chang S and Olson EN. CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J Clin Invest. 2006;116:1853-1864. 11. Awad S, Kunhi M, Little GH, Bai Y, An W, Bers D, Kedes L and Poizat C. Nuclear CaMKII enhances histone H3 phosphorylation and remodels chromatin during cardiac hypertrophy. Nucleic Acids Res. 2013;41:7656-7672. 12. Little GH, Bai Y, Williams T and Poizat C. Nuclear calcium/calmodulin-dependent protein kinase IIdelta preferentially transmits signals to histone deacetylase 4 in cardiac cells. J Biol Chem. 2007;282:7219-7231. 13. Hohl M, Wagner M, Reil JC, Muller SA, Tauchnitz M, Zimmer AM, Lehmann LH, Thiel G, Bohm M, Backs J and Maack C. HDAC4 controls histone methylation in response to elevated cardiac load. J Clin Invest. 2013;123:1359-1370. 14. Kreusser MM and Backs J. Integrated mechanisms of CaMKII-dependent ventricular remodeling. Front Pharmacol. 2014;5:36. 15. Zhang T, Johnson EN, Gu Y, Morissette MR, Sah VP, Gigena MS, Belke DD, Dillmann WH, Rogers TB, Schulman H, Ross J, Jr. and Brown JH. The cardiac-specific nuclear delta(B) isoform of Ca2+/calmodulin-dependent protein kinase II induces hypertrophy and dilated cardiomyopathy associated with increased protein phosphatase 2A activity. J Biol Chem. 2002;277:1261-1267.
16. Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J, Jr., Bers DM and Brown JH. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res. 2003;92:912-919.
10. Backs J, Song K, Bezprozvannaya S, Chang S and Olson EN. CaM kinase III sselellecectitiivevevelylyly ignals to histone deacetylase 4 during cardiomyocyte hypertrophy. J Clin Investt.
2006;116:1853-1864.
111. . AwAwAwadadad SSS,, KKuKunhnhnhii M, Little GH, Bai Y, An W,, BeBeBers D, Kedes L L ana d d PoPoPoizi at C. Nuclear CaMKIIenennhahaances hhisstttonene HHH33 phphphosossphphp ororylylylatatatioionn anand d reremmododeeelsss chroroomamamatit n dududurriringng ccarara diacac hhhypypyperertrtropopophyhyhy.. NNucclcleic Acids s ReReRes.. 2201010 3;3;3;414141:7:77656656-6--76767672722.
1222. LiLiLittttttlele GGGH,H,H BBaai YY, , WiWWilllliaiammsms TTT aaandndn PPPoioioizazazattt C.C.C NNNucucclelearar cccalalalciciumumum/c/calalalmomomoduduulilinn-n-dededepepependndndennnt pprprototteieinnn kinanasese IIIdIdelelta pprer fefererentn ialllly y trtranansmsmits siigngnalals s toto hhistoonene ddeeacacetetylasasee 44 ini cacarddiaiacc cellls.s. J BiBiolol Chem. 2007;2;2;2828282:7:772121219-9-9 7277 313131.
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.114.006185
19
17. Joiner ML, Koval OM, Li J, He BJ, Allamargot C, Gao Z, Luczak ED, Hall DD, Fink BD, Chen B, Yang J, Moore SA, Scholz TD, Strack S, Mohler PJ, Sivitz WI, Song LS and Anderson ME. CaMKII determines mitochondrial stress responses in heart. Nature. 2012;491:269-273. 18. Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Jr., Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ and Anderson ME. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005;11:409-417. 19. Heineke J and Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589-600. 20. Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR and Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215-228. 21. Berry JM, Le V, Rotter D, Battiprolu PK, Grinsfelder B, Tannous P, Burchfield JS, Czubryt M, Backs J, Olson EN, Rothermel BA and Hill JA. Reversibility of adverse, calcineurin-dependent cardiac remodeling. Circ Res. 2011;109:407-417. 22. Taigen T, De Windt LJ, Lim HW and Molkentin JD. Targeted inhibition of calcineurin prevents agonist-induced cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. 2000;97:1196-1201. 23. Zhou XF, Marley PD and Livett BG. Substance P modulates the time course of nicotinic but not muscarinic catecholamine secretion from perfused adrenal glands of rat. Br J Pharmacol. 1991;104:159-165. 24. Sussman MA, Lim HW, Gude N, Taigen T, Olson EN, Robbins J, Colbert MC, Gualberto A, Wieczorek DF and Molkentin JD. Prevention of cardiac hypertrophy in mice by calcineurin inhibition. Science. 1998;281:1690-1693. 25. Felkin LE, Narita T, Germack R, Shintani Y, Takahashi K, Sarathchandra P, Lopez-Olaneta MM, Gomez-Salinero JM, Suzuki K, Barton PJ, Rosenthal N and Lara-Pezzi E. Calcineurin splicing variant calcineurin Abeta1 improves cardiac function after myocardial infarction without inducing hypertrophy. Circulation. 2011;123:2838-2847. 26. Backs J, Stein P, Backs T, Duncan FE, Grueter CE, McAnally J, Qi X, Schultz RM and Olson EN. The gamma isoform of CaM kinase II controls mouse egg activation by regulating cell cycle resumption. Proc Natl Acad Sci U S A. 2010;107:81-86. 27. Marley PD and Thomson KA. The Ca++/calmodulin-dependent protein kinase II inhibitors KN62 and KN93, and their inactive analogues KN04 and KN92, inhibit nicotinic activation of tyrosine hydroxylase in bovine chromaffin cells. Biochem Biophys Res Commun. 1996;221:15-18.
M, Backs J, Olson EN, Rothermel BA and Hill JA. Reversibility of adverse, calcinineueueuririn-n-dependent cardiac remodeling. Circ Res. 2011;109:407-417.
22. Taigen T, De Windt LJ, Lim HW and Molkentin JD. Targeted inhibition of calcineurin prevents agoonist-induced cardiomyocyte hypertropphy. Proc Natl Acad SSci U S A. 2000;97:1196-122010101..
2233. ZZhou XF, MMMararleley y PDPDD aaandndnd LLLivivetete tt t BBGG.. Suubsbsstanccee P momom duduulalaatetesss thhhe e titiimememe ccououoursrsee ofofof nnnicicicotttinininiccic bbbut nonoot mmum scarinnici ccatateechhoolaamiminnene seccreetitit oonn ffrommm pperffufusesed d adadadrererenaaal ggllaannds ooof f rarattt. BBrBr J PPPhhaharrmrmaacooll.199991919 ;1;1;10404:1:11595959-1166565..
24. Sussman n MAMAMA, , LiLiL m m m HWHWHW, , , GuGuGudedede NNN,, TaTaTaigigigenenen TTT,,, OlOlOlsosoon n ENENEN,, RoRoRobbbbbbininss s J,J,J, CCCololo bebeb rtrtt MMMC,C,C, GGGualberto AA,WiWiececzozorerekk DFDF aandnd MMololkekentntinin JJDD PPrereveventntioionn ofof ccarardidiacac hhypyperertrtropophyhy iinn mimicece bbyy cacalclcinineueuririnn
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.114.006185
20
28. Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA and Schneider MD. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J Clin Invest. 1997;100:169-179. 29. Frey N, Frank D, Lippl S, Kuhn C, Kogler H, Barrientos T, Rohr C, Will R, Muller OJ, Weiler H, Bassel-Duby R, Katus HA and Olson EN. Calsarcin-2 deficiency increases exercise capacity in mice through calcineurin/NFAT activation. J Clin Invest. 2008;118:3598-3608. 30. Timmins JM, Ozcan L, Seimon TA, Li G, Malagelada C, Backs J, Backs T, Bassel-Duby R, Olson EN, Anderson ME and Tabas I. Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J Clin Invest. 2009;119:2925-2941.
31. Hill JA, Karimi M, Kutschke W, Davisson RL, Zimmerman K, Wang Z, Kerber RE and Weiss RM. Cardiac hypertrophy is not a required compensatory response to short-term pressure overload. Circulation. 2000;101:2863-2869. 32. Rezazadeh S, Claydon TW and Fedida D. KN-93 (2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)]amino-N-(4-chlorocinnamyl)-N -methylbenzylamine), a calcium/calmodulin-dependent protein kinase II inhibitor, is a direct extracellular blocker of voltage-gated potassium channels. J Pharmacol Exp Ther. 2006;317:292-299. 33. Gao L, Blair LA and Marshall J. CaMKII-independent effects of KN93 and its inactive analog KN92: reversible inhibition of L-type calcium channels. Biochem Biophys Res Commun. 2006;345:1606-1610.
34. Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, Dalton ND, Peterson KL, Chen J, Bers D and Heller Brown J. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009;119:1230-1240. 35. Yang Y, Zhu WZ, Joiner ML, Zhang R, Oddis CV, Hou Y, Yang J, Price EE, Gleaves L, Eren M, Ni G, Vaughan DE, Xiao RP and Anderson ME. Calmodulin kinase II inhibition protects against myocardial cell apoptosis in vivo. Am J Physiol Heart Circ Physiol. 2006;291:H3065-3075. 36. Hashimoto Y, King MM and Soderling TR. Regulatory interactions of calmodulin-binding proteins: phosphorylation of calcineurin by autophosphorylated Ca2+/calmodulin-dependent protein kinase II. Proc Natl Acad Sci U S A. 1988;85:7001-7005. 37. MacDonnell SM, Weisser-Thomas J, Kubo H, Hanscome M, Liu Q, Jaleel N, Berretta R, Chen X, Brown JH, Sabri AK, Molkentin JD and Houser SR. CaMKII negatively regulates calcineurin-NFAT signaling in cardiac myocytes. Circ Res. 2009;105:316-325. 38. Wilkins BJ and Molkentin JD. Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem Biophys Res Commun. 2004;322:1178-1191.
32. Rezazadeh S, Claydon TW and Fedida D. KN-93 (2-[N-(2-hydroxyethyl)]-N-((4-4-4-methoxybenzenesulfonyl)]amino-N-(4-chlorocinnamyl)-N -methylbenzylamine)e),, , a calcium/calmodulin-dependent protein kinase II inhibitor, is a direct extracellularar bbblololockckkererer ooofff voltage-gated potassium channels. J Pharmacol Exp Ther. 2006;317:292-299.
333. . GaGaGaooo LL,L, BBBlllairr LLLAA and Marshall J. CaMKII-indndndepeppendent effectstss of KNKNKN939 and its inactiveanannalalloog KN9N9222: rrevevverere sisiblblle e inininhihibibitititiononon ooff L-L-tytyypepep ccalalcciuuum cchahahannnnnnele s.s. BiBiBiocochehem m m Biiopopphyhyhysss ReRes s CoCoC mmmmmmunu . 220000606;345:16006-6-1616161000..
3444. LiLiLingngng HHH,,, ZhZZhananng g g T,T, PPeerereie raaa LLL, MeMeMeananss s CKCKCK,,, ChChChenenng g H,H,H, GGGuuu YY,Y, DDDaalaltooon n n NDNDND, , , PePeetetetersrsr ononn KKKLL,L, CCChehennn JJ,J, Bersrs DD andnd Helellel r BrBroown n JJ. RReqequiuirementnt fforor CaCa2+2 /ccalalmomodudulilin-n dedepependndennt t kikinan sese II inin thehe ransition froom m m prprp esesessususurerere oveveverlrlrloaoaad-d-d ininndududucecec dd d cacacarrrdididiacacac hhhypypy ererrtrtrtropopophyhyhy tttooo heheheararart t t fafafailillururu e e e ininin mmmicicce.e J Clin
InInvevestst 2200009;9;11119:9:12123030 1-1242400
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.114.006185
21
39. Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, Jones F, Kimball TR and Molkentin JD. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94:110-118.
40. Marley PD and Thomson KA. Inhibition of nicotinic responses of bovine adrenal chromaffin cells by the protein kinase C inhibitor, Ro 31-8220. Br J Pharmacol. 1996;119:416-422. 41. Heineke J, Wollert KC, Osinska H, Sargent MA, York AJ, Robbins J and Molkentin JD. Calcineurin protects the heart in a murine model of dilated cardiomyopathy. J Mol Cell Cardiol. 2010;48:1080-1087. 42. Kim Y, Phan D, van Rooij E, Wang DZ, McAnally J, Qi X, Richardson JA, Hill JA, Bassel-Duby R and Olson EN. The MEF2D transcription factor mediates stress-dependent cardiac remodeling in mice. J Clin Invest. 2008;118:124-132. 43. Kuwahara K and Nakao K. New molecular mechanisms for cardiovascular disease:transcriptional pathways and novel therapeutic targets in heart failure. J Pharmacol Sci. 2011;116:337-342. 44. Nishikimi T, Kuwahara K and Nakao K. Current biochemistry, molecular biology, and clinical relevance of natriuretic peptides. J Cardiol. 2011;57:131-140. 45. Engeli S, Birkenfeld AL, Badin PM, Bourlier V, Louche K, Viguerie N, Thalamas C, Montastier E, Larrouy D, Harant I, de Glisezinski I, Lieske S, Reinke J, Beckmann B, Langin D, Jordan J and Moro C. Natriuretic peptides enhance the oxidative capacity of human skeletal muscle. J Clin Invest. 2012;122:4675-4679. Figure Legends
Figure 1. CaMKII and CaMKII contribute redundantly to cardiac CaMKII activity. (A) Real-
time-PCR analysis of cardiac CaMKII isoforms three weeks after sham (SH) or transverse aortic
constriction (TAC) surgery in wild type mice. The relative expression levels were normalized to
CaMKII in brain tissue. All values are reported as mean±SEM (n 3, *p<0.05). (B) To generate
cardiomyocyte-specific conditional CaMKII and single knockout ( -CKO and -CKO) and
CaMKII /CaMKII double knockout mice (DKO), CaMKII loxP-exon1/2-loxP ( -FF), CaMKII loxP-
exon1/2-loxP ( -FF) and CaMKII loxP-exon1/2-loxP, CaMKII loxP-exon1/2-loxP (FFFF) mice were crossed to
2011;116:337-342.
44. Nishikimi T, Kuwahara K and Nakao K. Current biochemistry, molecular biiolollogogogyy, aaandndnd clinical relevance of natriuretic peptides. J Cardiol. 2011;57:131-140.
455. . EnEnEnggegelilili SSS, Biirkrkrkeenenfeld AL, Badin PM, Bourlieeerr r VVV, Louche K, VVVigi ueeriririee e N, Thalamas C, MMoMonntntastier EEE,,, LaLarrrrrrououy y y D,D,D, HHHararanannttt I,I,I, ddee GlGlisisezezininskskiii II,, Lieeskskskeee S,S ReReReiinnkeke JJ, , BeB ckckmamamannnnn BB,,, LaLaL ngngngininin D,Joordddana J and MMoorrooo C.C.C NNatatatriririuruureeteticic pppeepepttiiddees enhnhhanccce theee oooxixidadad titivevee ccappacaccittyy ofoff hhhumummanana sskekek leleetatal llmmmusscscle. J ClClinin Innvnveest. 200012;2;121222:464 75757 --4466679.
Figure Leggenenndsdsds
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.114.006185
22
transgenic mice harboring Cre-recombinase under the control of the MHC promoter. (C)
Western blot analysis using antibodies directed against CaMKII, CaMKII , total PLB, phospho-
PLB-Thr17, phospho-PLB-Ser16, total RyR2, phospho-RyR2-Ser2814, phospho-RyR2-Ser2808,
total HDAC4, phospho-HDAC4-Ser632 and GAPDH (as loading control). Left ventricular
extracts from -CKO, -CKO and DKO mice and their Cre-negative littermates ( -FF, -FF and
FFFF) were analyzed (n=4 per group). Quantitative analysis of the expression and
phosphorylation of the proteins is shown at right. All values are presented as mean±SEM.
*p<0.05. n.s., not significant.
Figure 2. Calcineurin precedes CaMKII activation upon pathological pressure overload. Western
blot analysis were performed in cardiac extracts from wild type mice. Organs were harvested at
different time points after TAC surgery as indicated. (A) Western blot analysis after GST-
HDAC4 pulldown using GST-HDAC4 419–670, which contains a CaMKII-activity dependent
binding site. GST-HDAC4 2-250, which lacks a CaMKII binding domain, served as a negative
control. As an additional negative control, protein lysates from global CaMKII mice versus wild
type mice were used. GST-HDAC4 input was visualized by GST-immunoblotting, associated
endogenous CaMKII is visualized by CaMKII-immunoblotting. The degree of CaMKII binding
to GST-HDAC4 419-670 is a specific measure for CaMKII activity.10 A quantification of
CaMKII binding normalized to the input of GST-HDAC4 is shown. (B) Western blot analysis
using antibodies directed against CaMKII, HDAC4, phospho-HDAC4-Ser632, RCAN1-4 and
GAPDH. Quantitative analysis of the expression and phosphorylation of the proteins is shown.
For all experiments n 3 per group were used. All values are presented as mean±SEM. *p<0.05.
n.s., not significant.
Figure 2. Calcineurin precedes CaMKII activation upon pathological pressure ovvoverrrlloadadad.. WeWeWeststerern
blot analysis were performed in cardiac extracts from wild type mice. Organs were harvested at
didifffffererereenentt tititimememe poioioinntnts s after TAC surgery as indicatatateddd. (A) Westernnn bbloot t anananala ysis after GST-
HDHDAAC4 pulldodownwnwn uusisingngg GGGSTSTST-H-HDADADAC4C44 44419––6–670, wwhhiccch hh coconntntaiainsnss aaa CCaMaMMKKIKII-I-I-aacactitivivivitytyy ddeepepeenendedeenntnt
bibindndndininnggg sisisitetete.. GSGSST-T-T-HDHDHDACACC4 4 2-2-2-2525250,0,0, wwwhihichchch lllacacacksss aa CCCaaaMKMKMKIIIIII bbbinindidid ngngg dddomomomaiin,n,n sserere veveveddd aasas aa negegegatattivive ee
control. As ananan aaadddddditititioioionananal l nenenegagagatiiveveve ccconono tttroorol,l,l, ppprorooteteeininin lllysysysatateseses fffrororom m m glglg obobbalalal CCCaMaMaMKIKIKIIII mmmicicice e e versus willd
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.114.006185
23
Figure 3. Dissociation of cardiac dysfunction and interstitial fibrosis from myocardial
hypertrophy. (A) DKO and FFFF mice were randomized to either TAC or SH surgery and
sacrificed after 3 weeks. Representative images of the total hearts, H&E, Masson´s trichrome and
CD31 staining, echocardiographic M-modes and quantification of heart weight/body weight
ratios, myocyte size, fibrosis area, capillary density and fractional shortening are shown (n 8 per
group). Values of left ventricular ejection fraction, isovolumetric relaxation time constant (Tau),
end systolic pressure volume relation (ESPVR) and stroke volume measured in a working heart
preparation are shown (n 7 per group). (B) Fold-changes in mRNA levels of the hypertrophic
markers ANP, BNP, MHC and MHC and fold-changes in collagen expression (n 5 per
group). (C) Heart weight/body weight ratios of SH- and TAC-operated mice with the indicated
genotypes (n 4 per group). All values are presented as mean±SEM. *p<0.05. n.s., not
significant.
Figure 4. CaMKII controls CnA activity. (A) Real-time-PCR analysis of RCAN1-4 in hearts
from animals with genotypes and treatments as indicated ( 5 per group). (B+C) NMVMs from
FFFF and DKO were used for cell-based experiments. (B) NFAT-GFP was adenovirally
expressed in NMVMs. Cells were stained for -actinin (shown in red) and with DAPI nuclear
stain (shown in blue). The percentage of cells in which NFAT-GFP was localized to the nucleus
is indicated (>100 cardiomyocytes per well; n 3 per condition). (C) NFAT-luciferase reporter
assay in NMVMs (n 3 per condition). (D) Western blot analysis was performed using lysates
from COS-cells that were transfected with CnA in the absence and presence of constitutive
active CaMKII (CaMKII-T287D). A schematic diagram of CnA indicates the position of the
CaMKII phosphorylation site at Ser411, which is located within the calmodulin-binding domain
group). (C) Heart weight/body weight ratios of SH- and TAC-operated mice withthh ttheheh iindndndicicicatatateded
genotypes (n 4 per group). All values are presented as mean±SEM. *p<0.05. n.s., not
iigngnnififificicicananntt.t.
FiFigugugurerere 44.. CaCaCaMKMKMKIIII cconontrtrrolols CnCnCnAAA acacacttitiviviitytyy.. (((AAA) RRReaeaal---tit mememe-P-P-PCRCRCR aannanalylylysisis ss offf RRRCACACAN1N11-4-44 inn n hheheararttts
from animalss wwwititi h h h gegegenononotytypepepes s s annnd d d trtrreaeae tmtmmenenentststs aaas s s ininndidid cacac teeed d d ((( 555 pepeper r r grgrrouououp)p)p). . (B(B(B+C+CC))) NNNMVMVMVMs from
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.114.006185
24
of CnA (CnB, calcineurin B-binding domain; CaM, calmodulin-binding domain; AID,
autoinhibitory domain). (E) Adenoviral overexpression of CaMKII-T287D (adCaMKII-T287D)
in NRVMs results in hyper-phosphorylation of CnA-Ser411 compared to non infected (control)
NRVMs and NRVMs infected with GFP. (F) Western blot analysis of total CaMKII, RCAN1-4,
total CnA and, CnA-Ser411 in cardiac extracts from FFFF and DKO mice 3 weeks after SH or
TAC surgery (n 3 per group). (G) Western blot analysis of total CnA and CnA-Ser411 in -
CKO, -CKO or DKO versus their Cre-negative littermates (n=4 per group). For all Western blot
experiments, GAPDH was used as loading control. Quantitative analysis of the expression and
phosphorylation of the proteins is shown at right (F and G). All values are presented as
mean±SEM. *p<0.05. n.s., not significant.
Figure 5. Cardiac hypertrophy, but not dysfunction is controlled by calcineurin during
pathological and physiological hypertrophy in DKO. (A-B) NMVMs from FFFF and DKO mice
were transfected with two siRNAs directed against CnA or with scrambled siRNA (knockdown
efficiency is shown in Suppl. Fig. 5A). NMVMs were then treated with Iso or vehicle. (A) ANP
and BNP gene expression in NMVMs after Iso-stimulation (n 3 per condition). (B) Cells were
stained for -actinin (shown in red) and cardiomyocyte size was determined after Iso-stimulation
(>100 cardiomyocytes per well; n 3 per condition). (C) FFFF and DKO mice underwent SH or
TAC surgery and were treated with cyclosporine A (CyA; 4 mg/Kg/d, 3 weeks i.p.; n 7) or
vehicle (n 3) as indicated. CyA treatment was started at the day of TAC or SH surgery. Three
weeks later the mice were sacrificed. HW/BW ratios, myocyte cross sectional areas,
quantification of cardiac fibrosis from Masson´s trichrome-stained myocardial sections and
fractional shortening assessed by echocardiography are shown. (D-F) FFFF and DKO mice were
mean±SEM. *p<0.05. n.s., not significant.
Fiigugugurerere 555. CaCaCarrrdiaaaccc hhypertrophy, but not dysfunctititiononon is controlled bybb ccalalalccicinen urin during
pathhholo ogical aandndnd pphyhysisis olollogoggicicicalalal hhypypypeerertrrropphy innn DKKKOOO. (A(AA-B-B))) NNNMMVVMsMs fffrorom m FFFFFFFFFF aaandndnd DDDKOKOKO mmmiici e
weweererer tttrarar nsnsfefefectccteded wwwitith h twtwwoo siiRRRNANANAsss diddirereectcttededed aagagagaininstst CCnAnAnA ooorr r wiwiwiththt scrcrcramamamblblleded sssiRiRRNANAA (((knknnoccckdkdowowown nRR
efficiency is shshshowowown n n ininin SSSupupplplpl... FiFiig.g.g. 55A)AA .. NMNMNMVMVMVMs s wewewererer tthehehen n n trtrtreaeaeateteted wiwiwiththth IIIsososo oor r r vevevehihihiclclcle.e (A) ANP
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.114.006185
25
randomly assigned to a 14-day swimming or resting group, respectively. (D) Cardiac CaMKII
activity was determined in heart lysates by GST-HDAC4 pulldown using GST-HDAC4 419–670
(GST-HDAC4 2-250 served as specificity control). A quantification of CaMKII-activity
dependent binding as measure for CaMKII activity is given (n 11 per group). (E) Representative
images of whole hearts as well as H&E and Masson´s trichrome staining of left ventricular wall
sections. Quantification of HW/BW ratios, myocyte size, fibrosis area and fractional shortening
(n 3 per group). (F) Western blot analysis using antibodies directed against CaMKII, total CnA,
phospho-CnA-Ser411, RCAN1-4 and GAPDH. Quantitative analysis of the expression and
phosphorylation of the proteins is shown (n 7 per group). All values are presented as
mean±SEM. *p<0.05. n.s., not significant.
Figure 6. Schematic model. Stimulation of -adrenergic receptors by norepinephrine (NE) leads
to an activation of the two Ca2+-dependent pathways calcineurin and CaMKII (mediated by the
isoforms CaMKII and CaMKII ). Activated CaMKII phosphorylates HDAC4, which leads to a
translocation of HDAC4 from the nucleus to the cytosol. Thereby, the repressive effect of
HDAC4 on transcription factors as MEF2 is interrupted and a maladaptive remodeling gene
program is initiated. As a second mechanism, CaMKII phosphorylates CnA, which leads to
repression of the calcineurin-NFAT pathway and thereby acts anti-hypertrophic. Unexpectedly,
the endogenous calcineurin-NFAT driven pro-hypertrophic pathway is not maladaptive.
mean±SEM. *p<0.05. n.s., not significant.
Fiigugugurerere 666. ScScSchhhemamamattiticc model. Stimulation of -adrdrdrenenenergic receptorrss s byy nnnoororepinephrine (NE) leads
oo aaan n activationon ooof f thhhe e twwwo oo CaCaCa2+2+2+-d-depepepenendedeent pppattthwayayays ccaallclcininneuuuririnnn aannd d CaCC MKMKMKIIIIII ((memeedididiatatedede bbby y ththhe
ssofofofororormsmsm CCCaMaMaMKIKIII aanndnd CCCaMaMMKIKIKIIII ).).). AAccttivvvatatateeed CCCaMaMMKKKII I I phphphosososphphhororo ylyly atatateeses HHDADAAC4C4C4,,, whwhwhiichh h leeeadadsss tooo a
ranslocatioonnn ofofof HHHDADADAC4C4C4 ffrororom mm ththhe e e nununuclleuueuss s tototo ttthhhe e e cycycytototososol.l.l. TTTheheherererebybyby, thththee e rerereprprpresese sisiiveveve eeffffffecece t of
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from
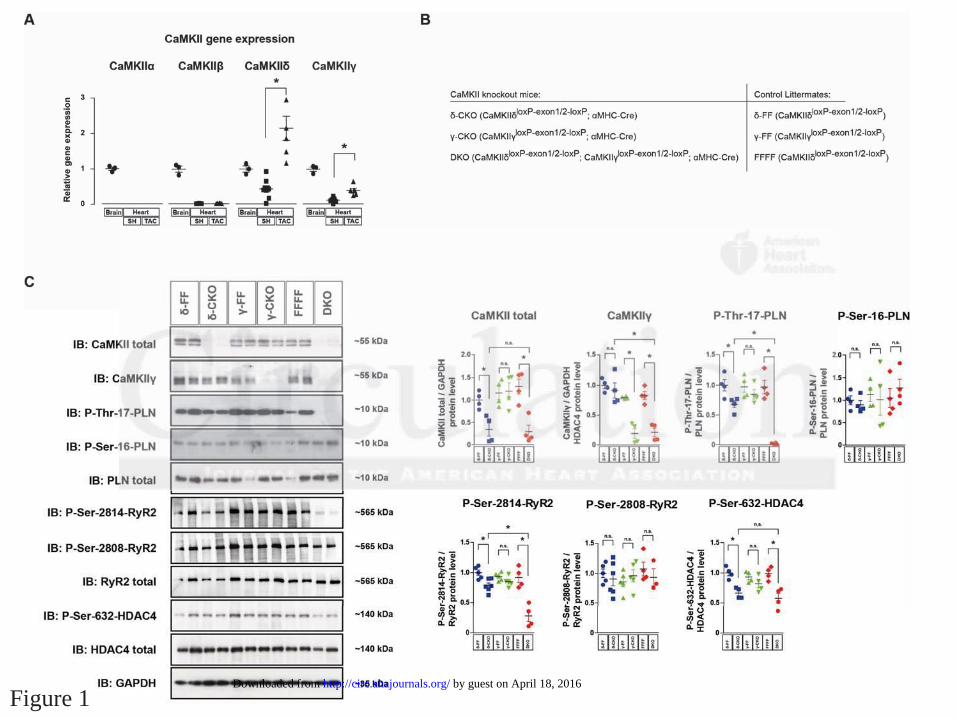
Figure 1 by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

Figure 2 by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

Figure 3 by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

Figure 4 by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from
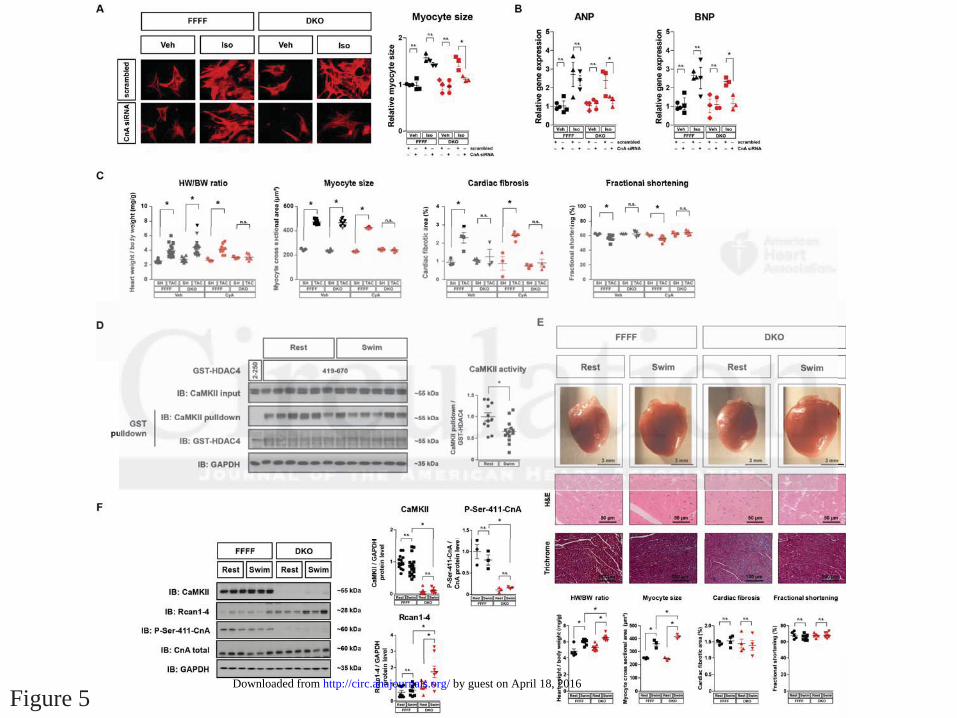
Figure 5 by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

Figure 6 by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

Johannes BacksDobrev, Christoph Maack, Lars S. Maier, Hermann-Josef Gröne, Hugo A. Katus, Eric N. Olson and
Kohlhaas, Jan-Christian Reil, Kay Neumann, Michael D. Schneider, Joseph A. Hill, Dobromir Michael M. Kreusser, Lorenz H. Lehmann, Stanislav Keranov, Marc-Oskar Hoting, Michael
Inhibit Calcineurin-Induced Myocardial Hypertrophy Contribute Redundantly to Adverse Remodeling butγ and δThe Cardiac CaMKII Genes
Print ISSN: 0009-7322. Online ISSN: 1524-4539 Copyright © 2014 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation published online August 14, 2014;Circulation.
http://circ.ahajournals.org/content/early/2014/08/14/CIRCULATIONAHA.114.006185World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circ.ahajournals.org/content/suppl/2014/08/14/CIRCULATIONAHA.114.006185.DC1.htmlData Supplement (unedited) at:
http://circ.ahajournals.org//subscriptions/
is online at: Circulation Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer available in the
Permissions in the middle column of the Web page under Services. Further information about this process isOnce the online version of the published article for which permission is being requested is located, click Request
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the Editorial Office.Circulation Requests for permissions to reproduce figures, tables, or portions of articles originally published inPermissions:
by guest on April 18, 2016http://circ.ahajournals.org/Downloaded from

SUPPLEMENTAL MATERIAL
The cardiac CaMKII genes δ and γ contribute redundantly to adverse
remodeling but inhibit calcineurin-induced myocardial hypertrophy
Michael M. Kreusser, MD1; Lorenz H. Lehmann, MD1; Stanislav Keranov, MD1; Marc-Oscar Hoting1;
Michael Kohlhaas, PhD2; Jan-Christian Reil, MD2; Kay Neumann, MD3; Michael D. Schneider, MD4;
Joseph A. Hill, MD, PhD5; Dobromir Dobrev, MD6; Christoph Maack, MD2; Lars S. Maier, MD3;
Hermann-Josef Gröne, MD7; Hugo A. Katus, MD8; Eric N. Olson, PhD9; Johannes Backs, MD1, 10
1 Research Unit Cardiac Epigenetics, Department of Cardiology, University of Heidelberg, Heidelberg,
Germany; DZHK (German Centre for Cardiovascular Research), partner site Heidelberg/Mannheim,
Germany.
2 Department of Cardiology, Saarland University, Homburg, Germany
3 Department of Internal Medicine II, University of Regensburg, Regensburg, Germany
4 British Heart Foundation Centre of Research Excellence, National Heart and Lung Institute, Faculty
of Medicine, Imperial College London, United Kingdom
5 Department of Internal Medicine, University of Southwestern Texas Medical Center, Dallas, TX, USA
6 Institute of Pharmacology, University of Duisburg-Essen, Essen, Germany
7 Department of Molecular Pathology, German Cancer Research Center, Heidelberg, Germany
8 Department of Cardiology, University of Heidelberg, Heidelberg, Germany; DZHK (German Centre
for Cardiovascular Research), partner site Heidelberg, Germany
9 Department of Molecular Biology, University of Southwestern Texas Medical Center, Dallas, TX, USA
10 Address for correspondence:
Johannes Backs, MD, Research Unit Cardiac Epigenetics / Department of Cardiology, Heidelberg
University, Im Neuenheimer Feld 410, 69120 Heidelberg, Germany. Phone: +49-6221-56-37714. Fax:
+49-6221-56-5573. e-mail: [email protected]

Supplemental Methods
Generation of DKO mice. All DKO animals (except the animals that were used for the
endurance exercise experiment) were generated by crossing FFFF mice with transgenic
mice expressing Cre-recombinase under the control of the a-MHC promoter (aMHC-Cre).1
DKO mice that were used for the endurance exercise experiment were generated by
crossing FFFF mice with transgenic mice expressing the mER-Cre-mER fusion protein under
control of a cardiomyocyte-specific αMHC-promoter.2 The latter animals were treated with
tamoxifen to achieve CaMKII deletion. Therefore, 10 mg/ml tamoxifen (Sigma) was dissolved
in a regular sunflower oil solution containing 6% ethanol. Tamoxifen was administered via
gavage for 10 consecutive days once per day in a dose of 80 mg/Kg body weight per day.
Efficient take-out of CaMKII was verified by Western blot. To avoid interferences with
transient Cre expression, experiments were conducted at least 3 weeks after the last
tamoxifen application.
Transverse aortic constriction. TAC to a 27 gauge stenosis was performed in 9-10 week-
old male DKO, δ-KO, γ-KO, CaMKII δ-/-/γ+/-, CaMKIIγ-/-/δ+/-, FFFF and wild type mice as
described previously.3, 4 A subgroup of the animals was treated twice daily by i.p. injection
with cyclosporin A (CyA; Sandimmun, Novartis) with 24 mg, 12 mg or 4 mg/kg body weight
per day for 7 days or 3 weeks, beginning at the day of surgery. CyA was diluted in 0.9%
saline solution with 12% ethanol, and the vehicle alone served as control.
Isoproterenol administration. Isoproterenol (Iso; Sigma) diluted in 0.1% ascorbic acid was
injected intraperitoneally (i.p.) once daily in 8-week-old male DKO mice and FFFF mice in a
given dose of 10 mg/kg body weight per day over 14 days. 0.1% ascorbic acid alone
(vehicle) served as control.

Swimming exercise protocol. Endurance exercise was carried out as described by others.5,
6 In brief, a ramp protocol was used starting at 10 minutes two times daily, with 10 minutes
increase each day until 90 minutes, two times per day was reached. The protocol ended after
14 days. The animals were closely observed at all times to avoid relative hypoxia. With this
protocol, there were no events of mice submerging under the water surface.
Transthoracic echocardiography and working heart preparation. Echocardiography and
studies on isolated hearts were carried out as previously described. 7, 8
Histology. Hematoxylin and eosin (H&E), Masson´s trichrome and CD31 stainings were
performed as previously described.9, 10 Cardiomyocyte size was assessed on H&E-stained
sections by using Image J software (http://rsb.info.nih.gov/ij/). More than 200 randomly
chosen cardiomyocytes from each group were analyzed to measure cross-sectional
cardiomyocyte area. To quantify cardiac fibrosis, 20 trichrome-stained sections
(magnification 20x) from the left ventricle were randomly selected, and morphometric
analysis by using Image J was performed. Capillary density was determined as the ratio of
capillaries to 100 cardiomyocytes in transversely sectioned left ventricular tissue
immunostained with CD31 and counterstained with DAPI. Photographs were acquired with
an Olympus SZH zoom stereo dissection scope with an Optronics DEI-750 CCD digital
camera. All data were analyzed by a single observer blinded to the mouse genotypes.
Western Blotting and GST-pulldown. Proteins from heart tissue and cultured
cardiomyocytes were isolated, and Western blot analysis was performed according to
protocols described previously.11 GST-pulldown experiments were performed as described.11
Besides GST-HDAC4 419-670 (which contains a CaMKII activity-dependent binding domain)
we used GST-HDAC4 2-250 (which does not contain a CaMKII binding domain) as a
negative control. Primary antibodies used were anti-CaMKII total (BD Bioscience), anti-
CaMKIIγ (Santa Cruz), anti-p-CaMKII (Ser287) (Affinity Bioreagents), anti-PKD (Cell

signaling), anti-p-PKD (Ser744) (Cell signaling), anti-CnA (Abcam), anti-p-CnA (Ser411)
(generated by Pineda antibodies), anti-GAPDH (Chemicon), anti-HDAC4 (Santa Cruz), anti-
p-HDAC4 (Ser632) (Abcam), anti-phospholamban (PLB) (Upstate), anti-p-PLB (Thr17)
(Santa Cruz), anti-p-PLB (Ser16) (Upstate), anti-RyR2 (Affinity BioReagents), anti-p-RyR2
(Ser2808) and anti-RyR2 (Ser2814) (kind gifts from Dr. Xander Wehrens, Houston, USA),
anti-Akt (Cell signaling), anti-p-Akt (Ser473) (Cell signaling), anti-RCAN1-4 (kind gift from Dr.
Timothy McKinsey, Denver, USA), and anti-α-actin (Sigma-Aldrich). Primary antibody
incubation was followed by corresponding horseradish peroxidase (HRP)-conjugated
secondary anti-mouse and anti-rabbit antibodies and ECL detection. Relative protein levels
were detected by densitometry using the Image J program.
CaMKII kinase activity. CaMKII kinase activity was measured by radioactive kinase assays
using GST-HDAC4 419-670 as a substrate and indirectly by detecting the amount of
endogenous CaMKII that associates to GST-HDAC4 419-670 (see also above under GST
pulldown). A detailed protocol has been described previously.3, 11
Caspase 3/7 activity measurements. The Caspase-Glo 3/7 Assay (Promega Corporation)
was used to measure caspase-3 and -7 activities according to the manufacturer´s
instructions. Heart lysates were mixed with reaction solution at equal volumes in a 96-well
plate. Luminescence was detected after 60 minutes of incubation at room temperature.
RNA analysis. Total RNA was isolated from ventricular tissue or from cultured
cardiomyocytes using TRIzol (Invitrogen). Total RNA was digested with DNase, and cDNA
synthesis from 500 ng of RNA was carried out using a SuperScript first-strand synthesis
system for RT-PCR (Invitrogen). Quantitative real-time PCR (qPCR) was performed with
Universal ProbeLibrary (Roche) by using TaqMan Universal PCR Mastermix (Applied
Biosystems) and detection on a 7500 Fast Cycler (Applied Biosystems). Primers and probes
were: For rat: ANP sense 5´-cccgacccagcatgg-3´ and antisense 5´-caactgctttctgaaaggggtg-

3´; For mouse: CaMKIIα sense 5´-gctgccaagattatcaacacc-3´ and antisense 5´-
cacgctccagcttctggt-3´; CaMKIIβ sense 5´-gccatcctcaccactatgct-3´ and antisense 5´-
ctccatctgctttcttgttgagt-3´; CaMKIIγ sense 5´-agttcacagggacctgaagc-3´ and antisense 5´-
cgccttgaacttctatggcta-3´; CaMKIIδ sense 5´-gtgccatcctcacaaccat-3´ and antisense 5´-
catctgacttcttgttcaataggc-3´; GAPDH sense 5´-gggttcctataaatacggactgc-3` and antisense 5´-
ccattttgtctacgggacga-3`; Col5a1 sense 5`-ctacatccgtgccctggt-3` and antisense 5`-
ccagcaccgtcttctggtag-3`; Col16a1 sense 5`-gcattgcaggagaaaatggt-3` and antisense 5`-
ccatcttgccataacctgga-3`; ANP sense 5`-cacagatctgatggatttcaaga-3` and antisense 5`-
cctcatcttctaccggcatc-3`; BNP sense 5´-gtctggccggacactcag-3` and antisense 5`-
tgcactggtgtcttcaacaac-3`; bMHC sense 5`-cgcatcaaggagctcacc-3` and antisense 5`-
ctgcagccgcagtaggtt; aMHC sense 5`-cgcatcaaggagctcacc-3` and antisense 5`-
cctgcagccgcattaagt-3`; Rcan1-4 sense 5`-actggaaggtggtgtccttgt-3` and antisense 5´-
tccagcttgggcttgactgag-3` .
Adenovirus production. Adenoviruses harboring CaMKII-T287D and GFP were generated
according to the manufacturer’s instructions (ViraPower Adenoviral Expression System;
Invitrogen). Nuclear factor of activated T-cells (NFAT) luciferase, and NFAT-GFP
adenoviruses were obtained from Seven Hills Bioreagents. After generation, the
adenoviruses were amplified, purified with the Adeno-X Purification Kit (BD Biosciences) and
its infectious units per µl were determined with the Adeno-X Rapid Titer Kit (BD Biosciences).
Cell culture of COS cells and transfection assays. COS cells were maintained in DMEM
with 10% FBS, 2 mM l-glutamine, and penicillin-streptomycin. Transfection was performed
with GeneJammer (Agilent Technologies) according to manufacturer’s instructions. The
expression constructs for CaMKII-T287D and CnA were described before.11, 12
Culture of neonatal rat ventricular cardiomyocytes (NRVMs). NRVMs were isolated from
1 to 2-day old Wistar rats as previously described.11 After isolation, NRVMs were maintained

in DMEM/199 medium (4:1) with 10% FBS, 2 mM l-glutamine, and penicillinstreptomycin.
NRVMs were infected 24 h after plating, grown 12 h later in serum-free media for another 4
h.
Culture of neonatal mouse ventricular cardiomyocytes (NMVMs). Neonatal
cardiomyocytes were isolated from 1- to 2-day-old DKO or FFFF mice according to
previously published methods.13 Cells grew for 24 h in the fresh complete medium. Some of
the cells were treated with CnA-siRNA or scrambled siRNA according to the manufacturer’s
instructions (Sigma-Aldrich). Medium was then replaced with serum-free medium 24 h prior
to adding isoproterenol (Sigma-Aldrich, 0.1 μM) for another 24 h. Cells were then harvested
for immunocytochemical staining or luciferase assay. For some of the immunostainings and
for luciferase assay, adenovirus harboring NFAT-GFP or NFAT luciferase reporter were
added, respectively.
Culture of adult mouse ventricular myocytes (AMVMs). For AMVM isolation, δ-CKO, γ-
CKO and DKO mice and their Cre-negative littermates (control), respectively, were
anesthetized with isoflurane and hearts were excised. Explanted hearts were retrogradely
perfused and digested as described before.14-16 Cells were plated onto regular cell culture
plates or superfusion chambers, with the glass bottoms treated with laminin. Cells were
immediately used for epifluorescence microscopy experiments or grew for 24 h in the fresh
complete medium. Medium was then replaced with serum-free medium 24 h prior to adding
isoproterenol (Sigma-Aldrich, 0.1 μM) for another 24 h.
Immunocytochemistry. NMVMs or AMVMs were fixed in 4 % paraformaldehyde (Sigma-
Aldrich) in PBS, permeabilized with 0.3 % Triton X-100 (Sigma-Aldrich) for 10 min at room
temperature and blocked for 30-60 min with 5 % goat serum (PAA) or BSA in PBS. Primary
antibody for α-actinin (Sigma) was applied in PBS containing 5 % goat serum or BSA and
0.05 % Triton X-100 for 1 h. Cells were treated with secondary antibody (Texas Red-coupled

anti-mouse-antibody) in PBS containing 5 % goat serum and 0.1 % Triton X-100 for 1 h.
Nuclei were labeled with DAPI (Invitrogen).
Apoptosis assay and cell size in AMVMs. TUNEL assays were performed using the in situ
cell death detection kit (Roche) according to the manufacturer’s protocol. To quantify the
number of apoptotic AMVMs, AMVMs were counterstained with sarcomeric α-actinin, and the
total numbers of AMVMs and TUNEL-positive nuclei were counted in 10 low power fields.
For each experimental condition, contiguous visual fields were counted to accumulate data
on 200 AMVMs per condition. Stained cells were normalized to total cell count as judged by
DAPI staining. More than 80% of cells were sarcomeric α-actinin positive. Cardiomyocyte
size was assessed by using Image J software (http://rsb.info.nih.gov/ij/). More than 100
randomly chosen cardiomyocytes from each group were analyzed to measure cross-
sectional cardiomyocyte area.
Epifluorescence microscopy. A Nikon Eclipse TE2000-U inverted microscope which was
provided with an IonOptix fluorescence detection system for assessment of Ca2+ handling
properties was used (IonOptix). For the studies on unstressed mice (was performed in the
Maier lab), AMVMs were loaded with Fura-2AM and analysed as described previously 14 For
the studies on sham and TAC-operated mice (was performed in the Maack lab), AMVMs ere
loaded with Indo-1AM and analysed as described previously. 15, 16

Supplemental References:
1. Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA and Schneider MD. Gene
recombination in postmitotic cells. Targeted expression of Cre recombinase provokes
cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J
Clin Invest. 1997;100:169-79.
2. Marley PD and Thomson KA. The Ca++/calmodulin-dependent protein kinase II
inhibitors KN62 and KN93, and their inactive analogues KN04 and KN92, inhibit
nicotinic activation of tyrosine hydroxylase in bovine chromaffin cells. Biochem
Biophys Res Commun. 1996;221:15-8.
3. Backs J, Backs T, Neef S, Kreusser MM, Lehmann LH, Patrick DM, Grueter CE, Qi
X, Richardson JA, Hill JA, Katus HA, Bassel-Duby R, Maier LS and Olson EN. The
delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and
remodeling after pressure overload. Proc Natl Acad Sci U S A. 2009;106:2342-7.
4. Hill JA, Karimi M, Kutschke W, Davisson RL, Zimmerman K, Wang Z, Kerber RE
and Weiss RM. Cardiac hypertrophy is not a required compensatory response to short-
term pressure overload. Circulation. 2000;101:2863-9.
5. Kirchhefer U, Schmitz W, Scholz H and Neumann J. Activity of cAMP-dependent
protein kinase and Ca2+/calmodulin-dependent protein kinase in failing and nonfailing
human hearts. Cardiovasc Res. 1999;42:254-61.
6. Toischer K, Rokita AG, Unsold B, Zhu W, Kararigas G, Sossalla S, Reuter SP, Becker
A, Teucher N, Seidler T, Grebe C, Preuss L, Gupta SN, Schmidt K, Lehnart SE,
Kruger M, Linke WA, Backs J, Regitz-Zagrosek V, Schafer K, Field LJ, Maier LS and
Hasenfuss G. Differential cardiac remodeling in preload versus afterload. Circulation.
2010;122:993-1003.
7. Malekar P, Hagenmueller M, Anyanwu A, Buss S, Streit MR, Weiss CS, Wolf D,
Riffel J, Bauer A, Katus HA and Hardt SE. Wnt signaling is critical for maladaptive
cardiac hypertrophy and accelerates myocardial remodeling. Hypertension.
2010;55:939-45.
8. Reil JC, Hohl M, Oberhofer M, Kazakov A, Kaestner L, Mueller P, Adam O, Maack
C, Lipp P, Mewis C, Allessie M, Laufs U, Bohm M and Neuberger HR. Cardiac Rac1
overexpression in mice creates a substrate for atrial arrhythmias characterized by
structural remodelling. Cardiovasc Res. 2010;87:485-93.
9. Song K, Backs J, McAnally J, Qi X, Gerard RD, Richardson JA, Hill JA, Bassel-Duby
R and Olson EN. The transcriptional coactivator CAMTA2 stimulates cardiac growth
by opposing class II histone deacetylases. Cell. 2006;125:453-66.
10. Hamzah J, Jugold M, Kiessling F, Rigby P, Manzur M, Marti HH, Rabie T, Kaden S,
Grone HJ, Hammerling GJ, Arnold B and Ganss R. Vascular normalization in Rgs5-
deficient tumours promotes immune destruction. Nature. 2008;453:410-4.
11. Backs J, Song K, Bezprozvannaya S, Chang S and Olson EN. CaM kinase II
selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J Clin
Invest. 2006;116:1853-64.
12. Frey N, Richardson JA and Olson EN. Calsarcins, a novel family of sarcomeric
calcineurin-binding proteins. Proc Natl Acad Sci U S A. 2000;97:14632-7.
13. Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, Hill JA,
Richardson JA and Olson EN. Histone deacetylases 1 and 2 redundantly regulate
cardiac morphogenesis, growth, and contractility. Genes Dev. 2007;21:1790-802.
14. Sag CM, Kohler AC, Anderson ME, Backs J and Maier LS. CaMKII-dependent SR Ca
leak contributes to doxorubicin-induced impaired Ca handling in isolated cardiac
myocytes. J Mol Cell Cardiol. 2011;51:749-59.

15. Kohlhaas M and Maack C. Adverse bioenergetic consequences of Na+-Ca2+
exchanger-mediated Ca2+ influx in cardiac myocytes. Circulation. 2010;122:2273-80.
16. Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, Bohm M, O'Rourke B and Maack C.
Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species
in failing cardiac myocytes. Circulation. 2010;121:1606-13.

FFFF DKO
5 mm
5 mm
5 mm
5 mm
Fractional shortening
80
20
0
40
SH TAC SH
Fra
cti
on
al sh
ort
en
ing
(%
)
FFFF DKO
n.s.
60
HW/BW ratio
6
2
0
4
SH TAC SH
Heart
weig
ht
/ b
od
y w
eig
ht
mg
/g)
FFFF DKO
n.s.
B
Ca2+ transient amplitude
1.02
0.96
0.94
1.00
F
(340 n
m /
380 n
m)
0.98
FFFF
DKO
0 200
time (ms)
SR Ca2+ content
1.40
1.55
1.45
F
(340 n
m / 3
80 n
m)
1.50
0 200
time (ms)
400
FFFF
DKO
D
Fractional Ca2+ release
80
20
0
40
SH TAC SH
F
racti
on
al re
lease
Ca
2+ tr
an
sie
nts
/ C
a2+ c
on
ten
t (%
)
FFFF DKO
n.s.
60
SR Ca2+ content
1.5
0.5
0FFFF DKO
S
R C
a2+
co
nte
nt
F (
340 n
m / 3
80 n
m)
n.s.
1.0
C
Ca2+ transient amplitude
0.8
0.2
0
0.6
Ca
2+ tr
an
sie
nt
am
plitu
de
F
(3
40 n
m /
380 n
m)
0.4
FFFF
DKO
n.s.
n.s.
n.s.
1 Hz 2 Hz 3 Hz
Ca2+ transient decay 80%
0.4
0.1
0
0.3
Ca
2+
tran
sie
nt
decay 8
0%
F
(3
40 n
m /
380 n
m)
0.2
FFFF
DKO
n.s. n.s.
n.s.
1 Hz 2 Hz 3 Hz
Diastolic Ca2+
1.6
KO NT
n.s.
n.s.
n.s.
1 Hz 2 Hz 3 Hz
1.2
1.0
0.8
1.4
D
iasto
lic C
a2+
F (
340 n
m / 3
80 n
m)
FFFF
DKO
Fractional shortening
4
3
KO NT
FFFF
DKO
n.s.n.s.
n.s.
1 Hz 2 Hz 3 Hz
2
1
0
F
racti
on
al sh
ort
en
ing
(% o
f re
sti
ng
cell len
gh
t)
E
Fig. S1
Ejection fraction
80
20
0
40
SH TAC SH
Eje
cti
on
fra
cti
on
(%
)
FFFF DKO
n.s.
60
ESPVR
8
2
0FFFF DKO
ES
PV
R
4
*
Stroke volume
50
10
0
20
SH TAC SH
Str
oke v
olu
me (
µl)
FFFF DKO
n.s.
30
Tau
15
5
0FFFF DKO
Iso
vo
lum
etr
ic r
ela
xati
on
t
ime c
on
sta
nt
(tau
)
10
n.s.
6
40
IB: CaMKII total
IB: CaMKII
IB: GAPDH
Co
ntr
ol
KO
DK
O
~35 kDa
~55 kDa
~55 kDa
~55 kDa
A

Fig. S1. Baseline characterization of DKO mice. (A) Western blot analysis of total CaMKII,
CaMKII�, phospho-CaMKII-Thr287, and GAPDH as loading control in adult ventricular
myocytes (AMVMs) from !-CKO, �-CKO and DKO mice and Cre-negative littermates (Control).
(B) Representative images of whole hearts, sections and M-mode echocardiograms from DKO
mice and floxed Cre-negative littermates (FFFF). Heart weight/body weight (HW/BW) ratios
and fractional shortening for both groups are also indicated. Values are presented as
mean±SEM (n"18). (C) Hemodynamic measurements in isolated Langendorff-perfused hearts
(n"10). Ejection fraction, end systolic pressure volume relations (ESPVR), stroke volume and
isovolumetric relaxation time constant (tau) are shown. (D) Representative single cell
measurements for Ca2+ transient amplitude, fractional Ca2+ release and sarcoplasmatic
reticulum (SR) Ca2+ content in isolated ventricular cardiomyocytes from DKO and FFFF mice,
loaded with fluo-4. (E) Ca2+ transient decay after field stimulation with 1, 2 and 3 Hz.
Measurements for intracellular diastolic Ca2+ and fractional shortening (FS) given as % of
resting cell length after stimulation with 1, 2 and 3 Hz. All measurements were conducted in
n"30 single cardiomyocytes from n"3 mice. Values are reported as mean±SEM. *p<0.05. n.s.,
not significant.

IB: P-Thr-287 CaMKII
IB: CaMKII total
IB: P-Thr-17 PLN
FFFF DKO
SH TAC SH TAC
~55 kDa
~55 kDa
~10 kDa
IB: P-Ser-744 PKD ~110 kDa
CaMKII activity
³²P-autoradiogram
Coomassie
B
A
Fractional
Ca2+ release
150
0
50
SH TAC SH TAC
Fra
cti
on
al
rele
as
e
Ca
2+
tra
ns
ien
ts /
Ca
2+
co
nte
nt
(%)
FFFF DKO
100
n.s. n.s.
SR Ca2+ content
0.8
0.2
0
0.6
SH TAC SH TAC
SR
Ca2+
c
on
ten
t
F (
40
5 n
m /
48
5 n
m)
FFFF DKO
0.4
n.s. n.s.
Tau (s)
4
2
0SH TAC SH TAC
Ta
u (
s)
FFFF DKO
3
n.s. n.s.
1 Hz
SH TAC SH TAC
FFFF DKO
n.s. n.s.
Caffeine
D
Fig. S2
CaMKII activity
0
2
4
6
³²P
acti
vit
y /
to
tal
pro
tein
*
SH TAC SHSH TAC
~110 kDa
IB: GAPDH ~35 kDa
IB: PKD total
SH TAC
C
E
CaMKII
0
0.5
1.0
1.5
CaM
KII
/ G
AP
DH
pro
tein
level
SH TAC SH TAC
FFFF DKO
n.s.
*
*
P-Thr17-PLN
0
0.5
1.0
1.5
P-T
hr-
17-P
LN
/
GA
PD
H p
rote
in l
evel
SH TAC SH TAC
FFFF DKO
n.s.
n.s.
*
P-Thr282-CaMKII
0
0.5
1.0
1.5
2.0
2.5
P-T
hr-
282-C
aM
KII
/
GA
PD
H p
rote
in l
evel
SH TAC SH TAC
FFFF DKO
**
n.s.
P-Ser-744-PKD
0
1
2
3
5
P-S
er-
744-P
DK
/
PK
D p
rote
in l
evel
n.s.
*n.s.
SH TAC SH TAC
FFFF DKO
0
2
4
6
8
MHC
Fo
ldch
an
ge T
AC
vs.
SH
n.s.
*
WT FFFF DKO
SH2
4
6
8
10
ANP
Fo
ldch
an
ge T
AC
vs.
SH
n.s.
*
0WT FFFF DKO
SH0
10
20
30
Rcan1-4
Fo
ldch
an
ge T
AC
vs.
SH
*
n.s.
SHWT FFFF DKO
SH
4
1
Ca2+ transient amplitude
0.3
0.1
02 Hz
Ca
2+
tr
an
sie
nt
am
pli
tud
e
F
(40
5 n
m /
48
5 n
m)
0.2
FFFF SH
FFFF TAC
n.s.
n.s.n.s.
0.5 1 3 2 4 5 Hz
n.s. n.s.
DKO SH
DKO TAC
Fractional shortening
8
2
0
6
F
rac
tio
na
l s
ho
rte
nin
g
(%
of
res
tin
g c
ell
le
ng
ht)
4
FFFF SH
FFFF TAC
n.s.
n.s.
n.s.n.s.
n.s.
DKO SH
DKO TAC
2 Hz 0.5 1 3 2 4 5 Hz
n.s.
n.s.

Fig. S2. Ca2+-handling after TAC in DKO. (A) Western blot analysis of total CaMKII, phospho-
CaMKII-Thr287, phospho-PLB-Thr17, phospho-PKD-Ser744, total PKD and GAPDH as
loading control in cardiac extracts from FFFF and DKO mice three weeks after SH or TAC
surgery. Quantitative analysis is shown (n�3 per group). (B) Radioactive CaMKII kinase activity
measured by in vitro kinase assay using GST-HDAC4 419-670 as a substrate. Coomassie
staining was used to demonstrate equivalent GST-HDAC4 input. Representative blots and
quantification (n=4) are shown. (C+D) Intracellular Ca2+ handling and cell shortening after TAC.
Measurements were conducted in adult mouse ventricular myocytes (AMVMs) from FFFF and
DKO mice three weeks after SH or TAC surgery. AMVMs were loaded with indo-1. Averaged
data for intracellular Ca2+ transient amplitudes and fractional shortening given as % of resting
cell length after field stimulation with 0.5, 1, 2, 3, 4 and 5 Hz. Fractional Ca2+ release and
averaged sarcoplasmatic (SR) Ca2+ content were estimated from the caffeine-induced peak in
Ca2+ transients. Time constant (tau) of intracellular Ca2+ decline after field stimulation and
under caffeine. All measurements were conducted in n�25 single AMVMs from n�3 mice. (E)
Gene expression in wild type and !-KO as well as DKO and FFFF mice after TAC or SH
surgery. Animals were sacrificed after 3 weeks. Fold-changes in mRNA levels of ANP, "MHC
and Rcan1-4 (n�3 per group). All values are reported as mean±SEM. *p<0.05. n.s., not
significant.

KO KO
Iso
DKO
+ + + +
a diomyocy e apop osis
0
2
4
6
8
Tu
ep
osi
ive c
ad
iom
yo
cy
es (
%)
KO
KO
DK
O
Iso � + + + +
B
DA
PI
KO DKOKO
Ve
hIs
o
Myocy e size
0
0.5
1.0
1.5
2.0
Re
aiv
e m
yo
cy
e s
ize
KO
KO
KO
DK
O
FF
FF
Iso � + + ++
DK
O
DK
OKO
� � �
*
*
*
**
.s.2.0
0.5
0
1.0
1.5
Veh Iso Veh Iso
*
FFFF DKO
.s.
2.5
0.5
0
1.0
1.5
*
FFFF DKO
A
2.0

Fig. S3. Redundant roles for CaMKII� and � for cardiomyocyte apoptosis and
hypertrophy. (A) Quantification of cardiac caspase 3/7 activity in DKO and FFFF mice as
measure for apoptosis induced by 3 weeks after TAC or 14 days of isoproterenol (Iso)
treatment (n�3 per group). (B-C) Experiments were conducted in adult ventricular myocytes
(AMVMs) from !-CKO, "-CKO and DKO mice and their Cre-negative littermates (Control).
Cells were stimulated with Iso or treated with vehicle. (B) Cells were stained for #-actinin
(shown in red) and with DAPI nuclear stain (shown in blue). TUNEL-positive cells display green
nuclei (more than 200 myocytes were analyzed per well). Shown are selected myocytes with
TUNEL-positive and negative staining. The percentage TUNEL-positive cells is indicated (n=4
per condition). (C) Myocyte size was determined by measuring >100 cardiomyocytes per well
(n=4 per condition). All values are presented as mean±SEM. *p<0.05. n.s., not significant.

Veh
FFFF DKO
Iso Veh Iso
3 mm 3 mm 3 mm 3 mm
BA
3 mm 3 mm 3 mm 3 mm
H&
ETri
ch
rom
e
50 µm 50 µm 50 µm 50 µm
100 µm 100 µm 100 µm 100 µm
ANP
4
1
0
3
Veh Iso Veh IsoRe
lati
ve
ge
ne
ex
pre
ss
ion *
FFFF DKO
*
n.s.
MHC
1.5
0.5
0
1.0
Veh Iso Veh Iso
*
FFFF DKO
*
n.s.
Re
lati
ve
ge
ne
ex
pre
ss
ion
BNP
6
2
0Veh Iso Veh Iso
*
FFFF DKO
*
n.s.
Re
lati
ve
ge
ne
ex
pre
ss
ion
MHC
4
2
0
3
Veh Iso Veh Iso
*
FFFF DKO
*
n.s.
Re
lati
ve
ge
ne
ex
pre
ss
ion
Col5a1
6
0
4
Veh Iso Veh Iso
FFFF DKO
2
**
Re
lati
ve
ge
ne
ex
pre
ss
ion
Col16a1
Veh Iso Veh Iso
FFFF DKO
* 10
2
0
8
6
4
*
n.s.
Re
lati
ve
ge
ne
ex
pre
ss
ion
Cardiac fibrosis
Veh Iso Veh Iso
Ca
rdia
c f
ibro
tic
are
a (
%)
*
FFFF DKO
15
10
0
5
*
My
oc
yte
cro
ss
se
cti
on
al
are
a (
µm
²)
Myocyte size
500
200
0
400
Veh Iso Veh Iso
*
FFFF DKO
*
100
300
n.s.
HW/BW ratio
10
2
0Veh Iso Veh Iso
He
art
we
igh
t /
bo
dy
we
igh
t (m
g/g
)
*
FFFF DKO
*
6
4
n.s.
Fig. S4
n.s.
n.s.
2
4
1
8

Fig. S4. CaMKII-dependent dissociation of cardiac fibrosis and dysfunction from cardiac
hypertrophy and fetal gene activation after isoproterenol. (A) Representative images of
whole hearts as well as H&E and Masson´s trichrome staining of left ventricular wall sections.
Quantification of HW/BW ratios, myocyte size and fibrosis area (n>6 per group). FFFF and
DKO mice were treated i.p. with isoproterenol (Iso) 10 mg/Kg/d or vehicle for 14 days,
respectively (B) Fold-changes in mRNA levels of the hypertrophic markers ANP, BNP, �-MHC
and !-MHC and fold-changes in collagen expression (n"4 per group). All values are presented
as mean±SEM. *p<0.05. n.s., not significant.

IB: CaMKII
IB: P-Ser-473-Akt
FFFF DKO
SH TAC SH TAC
IB: Akt total
Veh
FFFF DKO
SH TAC SH TAC
~55 kDa
~60 kDa
~60 kDa
CyA
IB: GAPDH ~35 kDa
C
Ve
h
Cy
A
Figure S5
CaMKII
0
0.5
1.0
1.5
2.0
2.5
CaM
KII
/ G
AP
DH
pro
tein
level
SH TAC SH TAC
FFFF DKO
n.s.
**
CaMKII
0
1
2
CaM
KII
/G
AP
DH
pro
tein
level
SH TAC SH TAC
FFFF DKO
n.s.
**
P-Ser-473-Akt
0
1
2
P-S
er-
473-A
kt
/
Akt
pro
tein
level
SH TAC SH TAC
FFFF DKO
n.s.
n.s.
P-Ser-473-Akt
0
1
2
P-S
er-
473-A
kt
/
Akt
pro
tein
level
SH TAC SH TAC
FFFF DKO
n.s.n.s.
SH TACTACFFFF DKO
ANP
0
5
10
15
Fo
ldch
an
ge T
AC
vs.
SH
BNP
0
1
2
3
5
Fo
ldch
an
ge T
AC
vs.
SH
Rcan1-4
0
5
10
15
Fo
ldch
an
ge T
AC
vs.
SH
MHC
0
1
2
Fo
ldch
an
ge T
AC
vs.
SH
MHC
0
2
4
6
8
Fo
ldch
an
ge T
AC
vs.
SH
B
FFFF DKO
Veh CyA
SH TACTACFFFF DKO FFFF DKO
Veh CyA
SH TACTACFFFF DKO FFFF DKO
Veh CyA
SH TACTACFFFF DKO FFFF DKO
Veh CyA
SH TACTACFFFF DKO FFFF DKO
Veh CyA
SH
SH
SH
SH
SH*
*
*
n.s.
n.s.
n.s.
n.s.
*
n.s.
*
IB: GAPDH ~35 kDa
IB: CnA ~60 kDa
non trea
ted
scra
mble
dC
nA s
iRN
A
A
*
4

Fig. S5. Normalization of fetal gene program and unaltered Akt phosphorylation after
CnA inhibition in DKO mice. (A) For data presented in Fig. 5A+B, neonatal mouse ventricular
myocytes (NMVMs) were transfected with CnA-siRNA or scrambled siRNAs, or they were not
transfected (non treated). Western blot analysis demonstrates efficient CnA knockdown.
GAPDH shows equal loading. (B+C) Analog to the experiment shown in Fig. 5C, DKO and
FFFF mice were randomized to either TAC or SH surgery and were treated with cyclosporine
A (CyA; 4 mg/Kg/d i.p.) or vehicle (n�3). Mice were sacrificed 7 days after TAC surgery. CyA-
treated DKO mice were protected from TAC-induced hypertrophy (data not shown) as
described in Fig. 5C (3 weeks after TAC). (B) Fold-changes in cardiac mRNA levels of the
hypertrophic markers ANP, BNP, !MHC and "MHC and fold-changes of RCAN 1-4 expression
(n�3 per group). (C) Western blots for CaMKII, Akt total, phospho-Akt-Ser473, and GAPDH as
loading control were performed in lysates from left ventricle. Quantitative analysis of the
expression and phosphorylation of the proteins is shown (n=3). All values are presented as
mean±SEM. *p<0.05. n.s., not significant.
Copyright © 2022 FDOKUMEN




















