
On the lack of polymorphism in Aβ-peptide aggregates derived from patient brains
Upload
independentCategory
view
0download
0
A. Doevendans and Jeffery D. MolkentinJ. Glascock, Raisa Klevitsky, Timothy E. Hewett, Thomas R. Kimball, Bruce J. Aronow, Pieter Orlando F. Bueno, Daniel J. Lips, Robert A. Kaiser, Benjamin J. Wilkins, Yan-Shan Dai, Betty
Apoptosis and Dysfunction Gene Targeting Predisposes the Myocardium to Acute Ischemia-InducedβCalcineurin A
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2003 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/01.RES.0000107197.99679.772004;94:91-99; originally published online November 13, 2003;Circ Res.
http://circres.ahajournals.org/content/94/1/91World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on May 30, 2013http://circres.ahajournals.org/Downloaded from

Calcineurin A� Gene Targeting Predisposes theMyocardium to Acute Ischemia-Induced Apoptosis
and DysfunctionOrlando F. Bueno, Daniel J. Lips, Robert A. Kaiser, Benjamin, J. Wilkins, Yan-Shan Dai,
Betty J. Glascock, Raisa Klevitsky, Timothy E. Hewett, Thomas R. Kimball, Bruce J. Aronow,Pieter A. Doevendans, Jeffery D. Molkentin
Abstract—Cardiovascular disease is the leading cause of mortality and morbidity within the industrialized nations of theworld, with coronary heart disease (CHD) accounting for as much as 66% of these deaths. Acute myocardial infarctionis a typical sequelae associated with long-standing coronary heart disease resulting in large scale loss of ventricularmyocardium through both apoptotic and necrotic cell death. In this study, we investigated the role that the calciumcalmodulin-activated protein phosphatase calcineurin (PP2B) plays in modulating cardiac apoptosis after acuteischemia-reperfusion injury to the heart. Calcineurin A� gene–targeted mice showed a greater loss of viablemyocardium, enhanced DNA laddering and TUNEL, and a greater loss in functional performance compared withstrain-matched wild-type control mice after ischemia-reperfusion injury. RNA expression profiling was performed touncover potential mechanisms associated with this loss of cardioprotection. Interestingly, calcineurin A��/� hearts werecharacterized by a generalized downregulation in gene expression representing approximately 6% of all genes surveyed.Consistent with this observation, nuclear factor of activated T cells (NFAT)-luciferase reporter transgenic mice showedreduced expression in calcineurin A��/� hearts at baseline and after ischemia-reperfusion injury. Finally, expression ofan activated NFAT mutant protected cardiac myocytes from apoptotic stimuli, whereas directed inhibition of NFATaugmented cell death. These results represent the first genetic loss-of-function data showing a prosurvival role forcalcineurin-NFAT signaling in the heart. (Circ Res. 2004;94:91-99.)
Key Words: signaling � calcineurin � apoptosis � heart failure � transcription
Cardiovascular disease is the leading cause of mortality inadults in the United States where it is thought to account
for approximately 12 million deaths annually.1,2 Myocardialinfarction, as well as nonischemic forms of heart failure, arethought to involve an irreversible loss of cardiac myocytesthrough apoptosis or programmed cell death.2–4 In recentyears, a great body of research has focused on identifying thesignaling molecules responsible for myocardial cell death orthose molecules that might protect the myocardium fromischemic insults.5 For example, numerous studies have iden-tified signaling molecules that enhance cardiomyocyte apo-ptosis such as p38�, c-Jun N-terminal kinase, tumor necrosisfactor-�, p53, �-adrenergic receptors, and nitric oxide.5–7 Incontrast, other signaling pathways have been shown to protectthe heart from apoptosis such as cardiotrophin-1 through thegp130 receptor, p38�, insulin-like growth factor-1 (IGF-1),
Akt/protein kinase B, protein kinase C�, nitric oxide, andextracellular signal-regulated kinases 1/2.5,6,8–12
Another potential regulator of cardiac apoptosis is thecalcium-calmodulin–activated protein phosphatase cal-cineurin (or PP2B). Calcineurin was identified as an impor-tant regulator of cardiomyocyte hypertrophy in vivo and invitro, while more recent investigation has suggested a role forcalcineurin in the modulation of cardiomyocyte apopto-sis.13,14 Studies conducted in neurons, lymphocytes, andtumor cell lines have demonstrated either pro- or antiapoptot-ic effects associated with calcineurin activation.15–22 Simi-larly, both pro- and antiapoptotic modulatory roles have beenascribed to calcineurin in cardiomyocytes. For example,calcineurin activation in cultured cardiac myocytes confersprotection to H2O2- or 2-deoxyglucose–induced apoptosis,suggesting that calcineurin is cytoprotective.14,23 Moreover,
Original received May 28, 2003; revision received November 3, 2003; accepted November 6, 2003.From the Department of Pediatrics, Division of Molecular Cardiovascular Biology (O.F.B., R.A.K., B.J.W., Y.-S.D., R.K., T.E.H., J.D.M.), Children’s
Hospital Medical Center, Ohio; Department of Cardiology (D.J.L., P.A.D.), Cardiovascular Research Institute Maastricht, University Hospital Maastricht,the Netherlands, and Department of Cardiology, Heart, Lung Center Utrecht, Utrecht, the Netherlands; Department of Pediatrics, Division ofCardiovascular Imaging (B.J.G., T.R.K.), Children’s Hospital Medical Center, Ohio; and the Department of Pediatrics, Division of Pediatric Informatics(B.J.A.), Children’s Hospital Medical Center, Ohio.
Correspondence to Jeffery D. Molkentin, Department of Pediatrics, Division of Molecular Cardiovascular Biology, Children’s Hospital Medical Center,3333 Burnet Ave, Cincinnati, OH 45229-3039. E-mail [email protected]
© 2004 American Heart Association, Inc.
Circulation Research is available at http://www.circresaha.org DOI: 10.1161/01.RES.0000107197.99679.77
91 by guest on May 30, 2013http://circres.ahajournals.org/Downloaded from

transgenic mice expressing an activated form of calcineurinin the heart are largely protected from ischemia-reperfusion–induced DNA laddering, further suggesting that calcineurinactivation antagonizes cardiomyocyte apoptosis in vivo.14 Incontrast, isoproterenol stimulation of cardiac �-adrenergicreceptors promoted myocyte apoptosis, in part, by stimulatingcalcineurin activity.24 Part of this disparity potentially stemsfrom the dichotomous actions of the often-employed inhibi-tory agent cyclosporine A. For example, cyclosporine A notonly inhibits calcineurin enzymatic activity, but also inhibitsmitochondrial permeability pore transition (MPTP) and apo-ptosis through its association with cyclophilin D in the innermitochondrial membrane.25 However, use of cyclosporine-independent strategies have also revealed pro- and antiapo-ptotic regulatory roles for calcineurin, suggesting multiplelevels of complexity underlying the biology of calcineurinand cell death. Given these issues, we pursued a geneticapproach to evaluate the functional significance of cal-cineurin as a potential modulator of ischemia-reperfusion–induced apoptosis in vivo.
Materials and MethodsSurgical Procedure and Animal ModelsMice were randomized to receive either ischemia-reperfusion injuryor a sham procedure. Mice were anesthetized and placed on awarming pad maintained at 37°C. The trachea was cannulated witha polyethylene tube connected to a respirator (Harvard Apparatus)with a tidal volume of 0.6 mL (110/min). A left lateral thoracotomywas performed between the fourth and fifth ribs, pericardial tissuewas removed, and the left anterior descending artery (LAD) wasvisualized and ligated with a slipknot of 8-0 silk. Reperfusion wasinitiated 60 minutes after the occlusion. The Calcineurin A�(CnA�)�/� mouse and the NFAT-dependent luciferase reportertransgenic line (containing nine copies of an NFAT binding siteupstream of a minimal promoter) were described previously.26,27 Allprocedures performed in animals were approved by the InstitutionalAnimal Care and Use Committee.
Measurements of Ischemic Area at Risk andInfarct SizeTwenty-four hours after reperfusion, mice were anesthetized, theLADs were reoccluded, and 1 mL of 1.0% Evans blue was injectedinto the apex of each heart to stain nonischemic tissue. The heartswere then excised, washed with PBS, embedded in agarose, and cutinto five transverse slices for 15 minutes of incubation at roomtemperature with 1.5% 2,3,5-triphenyltetrazolium chloride (TTC) tomeasure viable myocardium (red staining). Slices were photo-graphed (each side) under a microscope and left ventricular area,area at risk (AAR), and infarct area were determined by digitalplanimetry.
Echocardiography and Working Heart AnalysesCnA��/� or wild-type mice were anesthetized with 2% isofluraneand hearts were visualized using a Hewlett Packard Sonos 5500instrument and a 15-MHZ transducer. Cardiac ventricular dimen-sions were measured on M-mode three times for the number ofanimals indicated. The isolated ejecting mouse heart preparation hasbeen described in detail previously.28
DNA Laddering and TUNELCnA��/� mice or wild-type controls were subjected to 60 minutesLAD occlusion and 24 hours of reperfusion to induce cardiacapoptosis for DNA laddering and 3 weeks of reperfusion for terminaldeoxyribonucleotide transferase (TdT)–mediated dUTP nick-endlabeling (TUNEL) assessment as previously described.14
Assessment of Apoptosis in CulturedCardiac MyocytesConditions for generating and culturing neonatal cardiac myocyteswere described previously.14 The Ad�CnA and Ad�NFATc4 adeno-viruses were described previously,14 whereas the AdVIVIT NFATinhibitory adenovirus was composed of the green florescent protein(GFP) fused to the VIVIT sequence as described previously29 (gift ofDr Susan D. Kraner and Chris Norris, University of Kentucky,Lexington). Cardiac myocytes were treated with staurosporine (500nmol/L) for 18 hours, and all manipulations were performed 24hours after adenoviral infection to allow adequate protein expres-sion.14 DNA laddering and measurement of TUNEL were describedpreviously.14
Affymetrix Gene Expression Profilingand BioinformaticsTotal RNA samples were prepared from four individual hearts from8-week-old CnA��/� or wild-type control mice. Biotin-labeled targetcRNA was prepared from T7-transcribed cDNA made from 10 �g ofthe total RNA using the Affymetrix-recommended protocol30,31 andhybridized for expression analysis to the Affymetrix U74Av2 Gene-Chip using antibody-based fluorescence signal amplification. Datavalues used for filtering and clustering were Signal, Signal Confi-dence, Absolute Call (Absent/Present), and Change (Increase, De-crease, and Unchanged) as implemented in MicroArray Suite 5.0.
Statistical AnalysisStatistical analyses between the experimental groups were performedusing a Student’s t test or one-way ANOVA when comparingmultiple groups. Data were reported as mean�SEM. Values ofP�0.05 were considered significant.
ResultsIncreased Myocardial Infarct Size inCnA��/� MiceAlthough conclusive evidence has emerged implicatingcalcineurin as a pivotal regulator of cardiac myocytehypertrophy, its role in regulating cardiac myocyte sur-vival or apoptosis after ischemic or stress-related injuryhas engendered some controversy. To more thoroughlyevaluate the association between calcineurin signal trans-duction and cardiac survival/apoptosis, we analyzedCnA��/� mice for susceptibility to ischemia-reperfusioninjury. Null mice and strain-matched wild-type (WT)controls were subjected to 60 minutes of LAD coronaryartery occlusion followed by 24 hours of reperfusion. Afterthis 24-hour reperfusion period, hearts were removed andperfused with Evans blue dye, sectioned, and incubated in1.5% TTC to quantify area at risk and area of infarctnormalized to total left ventricular area (AAR/LV). Thearea at risk normalized to ventricular area was not differentbetween the two groups (WT, 79.8�3.3%; CnA��/� mice,76.5�2.9%; Figure 1A). These results indicated that therewere no genotype-dependent differences in the perfusedareas between the two groups. In contrast, loss of viablemyocardium was increased by approximately 30% inCnA��/� mice compared with WT mice (WT, 31.9�3.6%;CnA��/� mice, 46.19�3.6%, n�8; P�0.05, Figures 1Band 2C).
CnA��/� Mice Show Greater Cardiac Cell DeathAfter Ischemia-Reperfusion InjuryDNA laddering was examined by agarose gel electrophoresisusing hearts from sham operated animals and mice subjected
92 Circulation Research January 9/23, 2004
by guest on May 30, 2013http://circres.ahajournals.org/Downloaded from

to ischemia-reperfusion injury. CnA��/� mice showed aqualitative increase in the degree of DNA laddering com-pared with hearts from WT mice, suggestive of more apo-ptotic damage (n�6 hearts in each group, although only twoeach are shown) (Figure 2A). These data were extendedthrough independent assessment of DNA fragmentation byligation-mediated polymerase chain reaction, which alsoshowed significantly greater signal in the CnA��/� heartsafter ischemia-reperfusion injury (Figure 2B). Histologicalanalysis was performed using Masson’s trichrome staining(blue color shows fibrosis and scar) on myocardial tissuefrom WT and CnA��/� mice subjected to ischemia and 3weeks of reperfusion. Analysis of six individual hearts ineach group revealed greater myocardial damage and in-creased interstitial fibrosis in CnA��/� mice compared withWT animals (two representative hearts are shown for eachgroup) (Figure 2C). As a more quantifiable measure of celldeath, TUNEL was performed from histological sections, 3weeks after ischemia-reperfusion injury. Rates of TUNEL incardiac myocytes were assessed within the remaining regionof viable left ventricle and septum, which demonstratedapproximately twice the rate in CnA��/� hearts comparedwith WT controls (Figure 2D). Basal TUNEL rates did notvary between WT and CnA��/� hearts from sham animals(data not shown). Collectively, these results indicate thatCnA��/� mice undergo enhanced cell death after ischemia-reperfusion injury, suggesting that calcineurin is normallycytoprotective in the heart.
CnA��/� mice have a partially compromised immuneresponse,26 an effect that might secondarily lessen the degree
of myocardial injury, ventricular remodeling, and the inflam-matory response after ischemia-reperfusion injury. In attemptto control for such variables, cardiac histological sectionswere subjected to immunohistochemical analysis for CD45
Figure 1. CnA��/� mice show greater myocardial injury afterischemia-reperfusion. A, Area at risk (AAR) to total left ventricu-lar area ratios. B, Infarcted area (IA) normalized to AAR (n�8;P�0.05). C, Representative injured hearts from wild-type (Wt)and CnA��/� mice stained with Evans blue and TTC.
Figure 2. CnA��/� mice show greater myocyte death after is-chemia-reperfusion. A, Representative DNA ladder as an indica-tion of apoptosis from wild-type (WT) and CnA��/� hearts afterischemia-reperfusion injury (n�6). B, Representative DNA frag-mentation detection assays by ligation-mediated PCR. C, Rep-resentative histological sections showing greater scarring inCnA��/� hearts after ischemia-reperfusion injury (n�6). D,TUNEL quantification of cardiomyocytes in 2 CnA��/� and wild-type control hearts 3 weeks after ischemic injury expressed as apercentage of total myocytes in the remaining viable myocardi-um. E, Hematoxylin and eosin– and chloroacetate esterase–stained cardiac histological sections from the indicated mice (24hours of reperfusion; arrows show mast cells).
Bueno et al Calcineurin Regulates Cardiac Apoptosis In Vivo 93
by guest on May 30, 2013http://circres.ahajournals.org/Downloaded from

(total leukocytes), CD3 (T lymphocytes), and B220 (Blymphocytes), which showed no differences in cellular re-cruitment 24 hours after ischemia-reperfusion in either group(data not shown). Histological sections were also stained withhematoxylin and eosin (H&E) and chloroacetate esterase (formast cells) (Figure 2E). The data show similar degrees ofcellularity augmentation in both groups 24 hours after ische-mia-reperfusion injury (H&E sections), as well as equivalentlevels of mast cells in the pericardium (Figure 2E).
CnA��/� Mice Display Worse Cardiac FunctionAfter Ischemia-ReperfusionAt baseline, hearts from CnA��/� mice functioned equivalentto WT control hearts with respect to �dP/dt, maximal leftventricular pressure developed, and time to peak pressure,each assessed by working heart preparation (Table). Theseresults indicate that loss of calcineurin A� does not obviouslyalter cardiac function in the unstimulated state. To furtherevaluate the functional ramifications of enhanced cell deathafter ischemia-reperfusion injury, transthoracic echocardiog-raphy was performed in CnA��/� and WT mice. Echocardi-ography was performed at baseline and 1, 2, and 3 weeks afterischemia-reperfusion injury to serially evaluate function non-invasively. As shown in Figures 3A and 3B, left ventricularend-diastolic dimensions (LVED) and left ventricular end-systolic dimensions (LVES) increased to a larger extent inCnA��/� mice compared with WT mice (P�0.05). Theseresults indicate greater left ventricular dilation in the heart,presumably due to enhanced cell death in the absence ofCnA�. More importantly, fractional shortening, which ap-proximates cardiac contractile performance, was reduced to agreater extent in CnA��/� mice compared with WT mice 2and 3 weeks after the ischemic event (P�0.05, n�9; Figure3C). These results indicate that CnA��/� mice had reducedcardiac function after ischemia-reperfusion injury comparedwith WT controls, consistent with the notion that CnA��/�
hearts are more susceptible to a cell death–promoting stimu-lus. However, it is possible that CnA��/� mice could alsoshow alterations in function due to differences in ventricularremodeling that typifies postinfarction injury. To address thispossibility, heart weight to body weight ratio assessmentswere performed after the final 3-week echocardiographicmeasurement in both groups. Neither WT nor CnA��/� miceunderwent significant cardiac hypertrophy over this timeperiod under the conditions used, suggesting that the differ-ence in functional performance likely reflects the degree ofinjury (data not shown).
Altered Gene Expression in CnA��/� MiceThe enhanced myocardial damage observed after ischemia-reperfusion injury in CnA��/� mice suggested an alteration inone or more molecular pathway(s) that influences cell sur-vival. Given the vast number of effectors that might poten-tially influence the survival versus apoptotic decision ofmyocardial cells after ischemia-reperfusion injury, we per-formed a large scale, unbiased screen for altered geneexpression. Specifically, hearts from 8-week-old CnA��/�
and strain-matched WT mice (2 each) were harvested andRNA was purified for gene expression profiling using theAffymetrix U74Av2 array. This array contains all genes inthe murine Unigene database that have been functionallycharacterized (approximately 6000) as well as an additional6000 expressed sequence tags (ESTs). Duplicate heart sam-ples were cross-compared between WT and CnA��/� miceresulting in 5735 genes being significantly detected in one ormore groups after internal normalization. Of these genes, 437were significantly altered in expression between CnA��/� andWT hearts (P�0.05). The raw data shown in Figure 4represent the 2n relationship of expression between Wt andCnA��/� hearts. For example, a WT value of 1.0 and a nullvalue of 0.33 or 3.0 translates into an 8-fold change in geneexpression either way. The data are also depicted on acolorimetric scale for simplicity (Figure 4A). Remarkably,383 genes showed downregulated expression in CnA��/�
hearts, whereas only 54 genes showed upregulated expres-
Cardiac Functional Analysis Using an Ex Vivo Working HeartPreparation in CnA��/� and Wild-Type Controls
WT (n�2) CnA��/� (n�2) P
Heart rate, bpm 401�1 397�5 Paced
�dP/dt, mm Hg/ms 6595�282 7399�321 NS
LVP, mm Hg 109�3 107�2 NS
TPP, ms 47�1 49�3 NS
�dP/dt represents a measure of contractility based on the derivative ofpressure change versus time. LVP indicates left ventricular pressure developed;TPP, time to peak pressure; and NS, not significant.
Figure 3. Cardiac morphology and function as determined byechocardiography. A and B, Left ventricular end-diastolicdimensions (LVED) and left ventricular end-systolic dimensions(LVES) in CnA��/� mice compared with wild-type (WT) mice(*P�0.05 vs baseline; #P�0.05 vs WT after ischemia-reperfusion). Data are expressed in millimeters, average�SEM,n�9. C, Fractional shortening assessed by echocardiography inCnA��/� and WT mice (*P�0.05 vs baseline; #P�0.05 vs WTafter ischemia-reperfusion, n�9). Fractional shortening was cal-culated as (LVED�LVES)/LVED�100 and expressed as percent-age, average�SEM, n�9.
94 Circulation Research January 9/23, 2004
by guest on May 30, 2013http://circres.ahajournals.org/Downloaded from

Figure 4. A, Relative expression ratios of selected functional gene groups assessed from microarray screening. Cardiac RNA was col-lected from 2 CnA��/� mice (KO) and 2 wild-type mice (WT) and subjected to expression profiling using the Affymetrix U74Av2 array.Left panels show a color scale representation of gene expression levels, with yellow equal to 1 (no change), blue equal to 0.2 (reducedexpression), and red equal to 5 (increased expression). Absolute normalized expression data are shown for each sample in the right-hand columns along with the GenBank accession number. B, Select group of genes showing altered expression in the array screenwas subject to a confirmatory RT-PCR analysis of expression levels at varying cycles (to show the most linear point in amplification).
Bueno et al Calcineurin Regulates Cardiac Apoptosis In Vivo 95
by guest on May 30, 2013http://circres.ahajournals.org/Downloaded from
sion. This general profile suggests that the loss of CnA�reduces the expression of a subset of cardiac genes, implicat-ing calcineurin as an important positive transcriptional effec-tor pathway in the heart.
Expression signatures were further analyzed in CnA��/�
hearts to potentially implicate individual genes as phenotypicmodulators. The most significantly altered genes within sevenfunctional categories were assembled and annotated; any oneof which might affect the viability of cardiac myocytes afterinjury. An interesting alteration was identified in a selectgroup of genes that regulate sterol/cholesterol metabolismsuch as ATP-binding cassette transporter ABC1, the low-density lipoprotein receptor-related protein 10, and thestearoyl-coenzyme A desaturase 1 (Figure 4A). Also ofinterest, four cell cycle regulatory genes were significantlyaltered in CnA��/� hearts, as well as five genes encodingmitochondrial-localized proteins (Figure 4A). Within thetranscription category, the DNA binding factors Nkx2.5,Hand2, YB-1, and ATF-3 were each downregulated inCnA��/� hearts, whereas ATF4 and DBP were upregulated.The observed reduction in Nkx2.5 and Hand2 expression,which themselves are important regulators of cardiac geneexpression, is consistent with the proposed paradigm wherebycalcineurin-NFAT signaling directly and indirectly augmentsexpression of a subset of cardiac genes. Another intriguingpathway alteration was observed in structural genes thatfacilitate cellular adhesion and/or intracellular support. Forexample, four and a half LIM domains 1, �-sarcoglycan,�-sarcoglycan, desmoglein-2, �-6 integrin, and �-4 tubulineach showed altered expression in the CnA��/� heart (Figure4A). A large number of genes that participate in cellularsignaling, apoptosis signaling, or stress-responsive signalingwere also identified as significantly altered in the absence ofCnA�. Finally, a group of genes showing altered expressionin the array screen were selected for reverse transcriptasepolymerase chain reaction (RT-PCR) analysis at varyingcycles to confirm the observed relationships (Figure 4B). Thedata obtained by RT-PCR showed the same profile ofincreased or decrease expression, confirming the reliability ofthe array screen (normalized to L7 expression) (Figure 4B).
The large number of candidate genes showing alteredexpression made it difficult to mechanistically establishwhich factors or pathways might ultimately underlie theobserved profile of greater stress-induced cell death in theabsence of CnA�. Despite this qualification, the overallobservation that large subsets of genes are reduced in expres-sion suggests that calcineurin functions as a general transcrip-tional regulator in the heart, which might predispose the heartto cell death. Indeed, NFAT transcription factors are primaryeffectors of calcineurin signaling that could fulfill such a role(see next section).
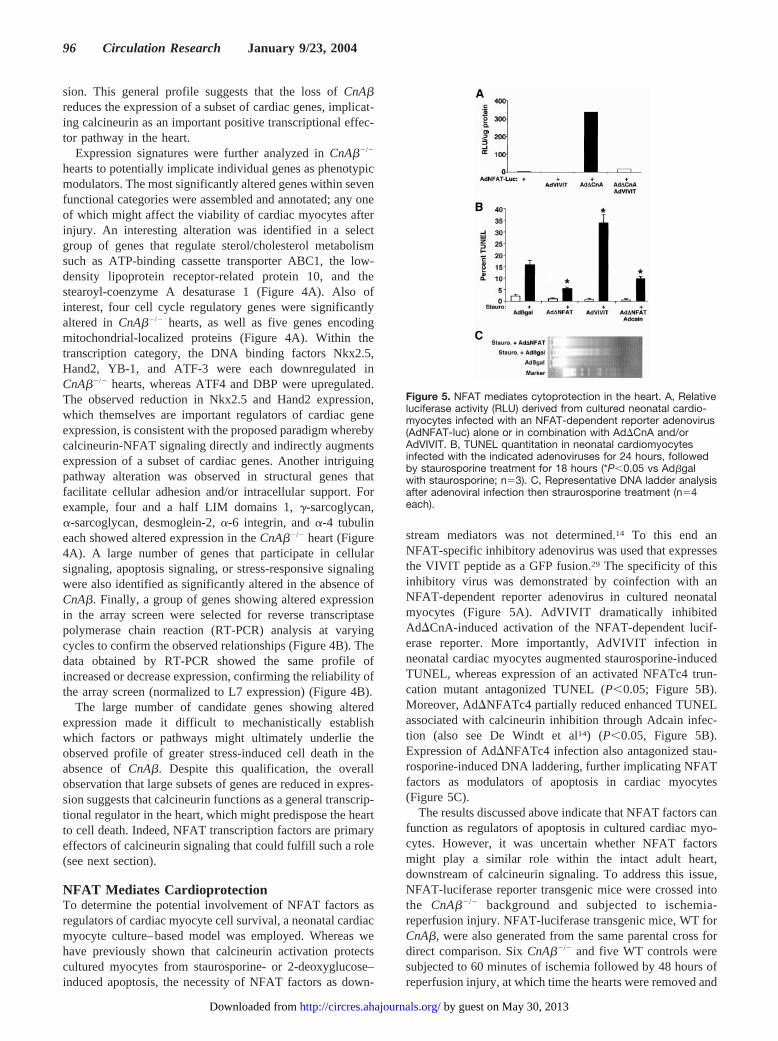
NFAT Mediates CardioprotectionTo determine the potential involvement of NFAT factors asregulators of cardiac myocyte cell survival, a neonatal cardiacmyocyte culture–based model was employed. Whereas wehave previously shown that calcineurin activation protectscultured myocytes from staurosporine- or 2-deoxyglucose–induced apoptosis, the necessity of NFAT factors as down-
stream mediators was not determined.14 To this end anNFAT-specific inhibitory adenovirus was used that expressesthe VIVIT peptide as a GFP fusion.29 The specificity of thisinhibitory virus was demonstrated by coinfection with anNFAT-dependent reporter adenovirus in cultured neonatalmyocytes (Figure 5A). AdVIVIT dramatically inhibitedAd�CnA-induced activation of the NFAT-dependent lucif-erase reporter. More importantly, AdVIVIT infection inneonatal cardiac myocytes augmented staurosporine-inducedTUNEL, whereas expression of an activated NFATc4 trun-cation mutant antagonized TUNEL (P�0.05; Figure 5B).Moreover, Ad�NFATc4 partially reduced enhanced TUNELassociated with calcineurin inhibition through Adcain infec-tion (also see De Windt et al14) (P�0.05, Figure 5B).Expression of Ad�NFATc4 infection also antagonized stau-rosporine-induced DNA laddering, further implicating NFATfactors as modulators of apoptosis in cardiac myocytes(Figure 5C).
The results discussed above indicate that NFAT factors canfunction as regulators of apoptosis in cultured cardiac myo-cytes. However, it was uncertain whether NFAT factorsmight play a similar role within the intact adult heart,downstream of calcineurin signaling. To address this issue,NFAT-luciferase reporter transgenic mice were crossed intothe CnA��/� background and subjected to ischemia-reperfusion injury. NFAT-luciferase transgenic mice, WT forCnA�, were also generated from the same parental cross fordirect comparison. Six CnA��/� and five WT controls weresubjected to 60 minutes of ischemia followed by 48 hours ofreperfusion injury, at which time the hearts were removed and
Figure 5. NFAT mediates cytoprotection in the heart. A, Relativeluciferase activity (RLU) derived from cultured neonatal cardio-myocytes infected with an NFAT-dependent reporter adenovirus(AdNFAT-luc) alone or in combination with Ad�CnA and/orAdVIVIT. B, TUNEL quantitation in neonatal cardiomyocytesinfected with the indicated adenoviruses for 24 hours, followedby staurosporine treatment for 18 hours (*P�0.05 vs Ad�galwith staurosporine; n�3). C, Representative DNA ladder analysisafter adenoviral infection then straurosporine treatment (n�4each).
96 Circulation Research January 9/23, 2004
by guest on May 30, 2013http://circres.ahajournals.org/Downloaded from

parsed into three components for measurement of luciferaseactivity: right ventricle (RV), nonischemic left ventricle andseptum (LV-non), and the injured area of the LV (LV-injured). The RV was analyzed to measure NFAT activity asa secondary consequence of ischemia-reperfusion injury–induced remodeling/hypertrophy. Ischemia-reperfusion in-jury dramatically enhanced NFAT luciferase activity in eachof the assayed ventricular compartments in WT controls(Figure 6). More importantly, CnA��/� mice consistentlyshowed less NFAT activation after injury (Figure 6). Theseresults suggest an association between reduced NFAT tran-scriptional activity and diminished cardioprotection inCnA��/� mice.
DiscussionIn this study, we investigated the association between thecalcineurin-NFAT signaling pathway and cell death in themyocardium after stress stimulation. Loss of CnA� renderedthe myocardium more susceptible to ischemia-reperfusioninjury, suggesting that calcineurin signaling helps maintainmyocyte viability in vivo. Part of the mechanism responsiblefor increased ischemic damage in CnA��/� mice is likely asecondary consequence of altered gene expression due toreduced NFAT activity.
Role of Calcineurin as a Modulator ofCellular ApoptosisAlthough calcineurin has been recognized as a central regu-lator of the hypertrophic growth of the myocardium, its roleas an effector of myocardial cell death is more controversial.In other tissues or cell-types, calcineurin has been shown toeither agonize or antagonize apoptosis after stress stimula-tion. Studies conducted in neurons, lymphocytes, and tumorcell lines have shown both pro- or antiapoptotic effects ofcalcineurin activation.15–22 The exact decision of cytoprotec-tion versus apoptosis is likely regulated by coordinatedsignals from other costimulated signaling pathways or de-pends on cell-type specific calcineurin effector/docking pro-teins. Indeed, calcineurin activation was shown to eitherinduce apoptosis or to antagonize apoptosis depending on thestatus of p38 activation.32 More recently, calcineurin wasshown to localize to the mitochondria in fibroblasts throughdocking with the inhibitory protein FKBP38, resulting in
Bcl-2 and Bcl-xl redistribution.33 Calcineurin has also beenimplicated as a direct inducer of apoptosis in primary hip-pocampal neurons through dephosphorylating the proapoptot-ic factor Bad.15 In addition to cell-type specific regulatoryconsiderations, the pro- or antiapoptotic effects attributed tocalcineurin have been obscured by the use of cyclosporine A,which can modulate apoptosis independent of calcineurinthrough direct effects on mitochondrial permeability transi-tion pore (MPTP) formation within the inner mitochondrialmembrane.25 Given the calcineurin-independent effects asso-ciated with cyclosporine A and FK506, the use of calcineuringene-targeted mice and/or null cells derived from these miceshould help elucidate the direct role that calcineurin plays inmodulating cellular apoptosis in diverse tissues.
In the present study, we demonstrated that genetic disrup-tion of the CnA� gene in the mouse enhanced cardiac damageinduced by ischemia-reperfusion injury. This result indicatesthat calcineurin signaling imparts a degree of protectionagainst cell death in the heart. Consistent with this notion, wehave previously shown that transgenic mice expressing aconstitutively active mutant of calcineurin in the heart aresignificantly protected from ischemia-reperfusion-inducedcell death.14 In cultured cardiomyocytes, adenoviral-mediatedgene transfer of activated calcineurin reduced 2-deoxyglu-cose–induced TUNEL, whereas calcineurin inhibition with aCain-expressing adenovirus increased TUNEL.14 In contrast,a subsequent study reported that isoproterenol stimulation ofneonatal cardiomyocytes promoted apoptosis, in part, bystimulating calcineurin activity.24 These authors demon-strated that cyclosporine A and FK506 blocked the increasein cardiomyocyte apoptosis induced by isoproterenol stimu-lation and, more significantly, that transgenic mice express-ing dominant-negative calcineurin in the heart were refrac-tory to isoproterenol-induced TUNEL reactivity in vivo.These results suggest that calcineurin activation is associatedwith enhanced apoptosis in cardiomyocytes, in contrast to ouroriginal and subsequent observations. It is likely that thenature of the stimulus (isoproterenol) partially underlies thedisparity between the two studies discussed above. Forexample, isoproterenol induces a prominent elevation incAMP, which induces protein kinase A signaling and second-ary alterations in inotropy, events not typically associatedwith ischemic injury or straurosporine stimulation.
By comparison, Kakita et al23 recently identified an anti-apoptotic role for calcineurin activation in cardiomyocytesafter endothelin-1 stimulation. Specifically, endothelin-1stimulation protected cardiac myocytes in culture from H2O2-induced TUNEL reactivity, DNA laddering, caspase-3 cleav-age, and loss of mitochondrial membrane potential. Thisendothelin-1–mediated cytoprotection from H2O2-induced ap-optosis was blocked by inhibition of calcineurin with eithercyclosporine A or FK506. Collectively, these disparate ac-counts underscore the complexity of intracellular signalingnetworks within mammalian cells, such that seemingly re-lated stress stimuli can elicit fundamentally different re-sponses depending on the nature of the stimulus(mitochondrial- versus death receptor–mediated), the statusof other parallel signaling pathways, and the context ofcell-type specific calcineurin modulatory factors.
Figure 6. Relative luciferase activity from the hearts of NFAT-luciferase reporter transgenic mice at baseline or after ischemia-reperfusion injury. These reporter mice were crossed into theCnA��/� or CnA��/� background (n�6 and 5, respectively).Activity is shown from the whole heart (control), right ventricle(RV), nonischemic region of the LV and septum (LV-non), andthe LAD-perfused area of the LV subjected to ischemia(LV-injured).
Bueno et al Calcineurin Regulates Cardiac Apoptosis In Vivo 97
by guest on May 30, 2013http://circres.ahajournals.org/Downloaded from

Mechanism Whereby Calcineurin SignalingProtects the MyocardiumAlthough a large number of studies have implicated cal-cineurin as a modulator of cell death in varied cell types, onlya few downstream mechanisms responsible for death orprotection have been identified. As discussed above, cal-cineurin was reported to directly dephosphorylate Bad inneurons, thus enhancing apoptosis.15 Careful evaluation ofBad phosphorylation at serine 112, 136, and 128 from thehearts of calcineurin transgenic mice or CnA��/� mice failedto show any difference from controls (data not shown).Kakita et al23 reported that calcineurin-mediated protectionfrom H2O2-induced apoptosis was associated with an increasein Bcl-2 expression in cultured neonatal cardiac myocytes.However, expression levels of Bcl-2 did not change in thehearts of either calcineurin TG mice or CnA��/� mice, norwas expression altered for Bcl-xl, Bad, caspase-1, -3, -8, -9,or FKBP38 (data not shown). Previously, we identified aminor but significant increase in Akt phosphorylation in thehearts of calcineurin transgenic mice.14 However, CnA��/�
mice showed no difference in basal or stimulated Aktphosphorylation in the heart, ruling out this potential mech-anism (data not shown). Collectively, these negative findingssuggest that the classical effectors of the apoptotic responseare not directly linked to calcineurin signaling in the heart.
To investigate other potential mechanisms, an unbiasedarray screen was performed from the hearts of CnA��/� andmatched WT mice. Although a number of potential regulatoryproteins were altered in expression, the most significantobservation was the dramatic downregulation in expressionof large subsets of unrelated genes (about 6% of detectablegenes in the heart). This observation suggested a defect in thetranscriptional potency underlying a group of genes in thehearts of CnA��/� mice. This interpretation is consistent withknown role of NFAT transcription factors as importanteffectors of calcineurin-regulated gene expression in mostcell types.34 Indeed, the full potency of calcineurin-inducedhypertrophy in the heart was shown to require NFATc3 usinggene-targeted mice.35
With respect to apoptosis signaling, we previously ob-served that overexpression of an activated NFATc4 in cul-tured neonatal cardiomyocytes partially antagonized2-deoxyglucose-induced apoptosis.14 Furthermore, Kakita etal23 showed that endothelin-1–mediated protection fromH2O2-induced apoptosis also promoted NFAT dephosphory-lation. In this study, we showed that specific inhibition ofNFAT with AdVIVIT augmented cardiomyocyte cell deathafter staurosporine treatment. More importantly, CnA��/�
mice showed reduced NFAT transcriptional activity in vivoafter ischemia-reperfusion injury. These results are alsoconsistent with a recent report by Izumo and colleagues36 inwhich NFAT inhibition augmented cardiac myocyte apopto-sis after phenylephrine stimulation in culture.
Collectively, these results discussed above suggest that thehomeostatic transcriptional activity of NFAT provides thenecessary framework of basal gene expression that affordscardiac “health” and resistance to apoptotic stimuli. However,the exact array of downstream effectors that are regulated byNFAT factors in providing protection is not known. Despite
this, the results of this study provide the first genetic dataindicating that calcineurin signaling is necessary for cardiacprotection and suggests that strategies to acutely agonizecalcineurin might be of therapeutic benefit during or aftermyocardial infarction. However, this notion is in dramaticcontrast to the function of calcineurin as a mediator of cardiachypertrophy, which itself can lead to cardiomyopathy andheart failure if constitutively activated (although it is apopto-sis-independent). These observations suggest that calcineurinmay function as a “double-edge sword,” such that transientactivation antagonizes myocyte apoptosis, but long-standingactivation induces cardiac hypertrophy and deleterious ven-tricular remodeling associated with heart failure.
AcknowledgmentsThis work was supported by the NIH (to J.D.M. and B.J.A.) and thePew Charitable Trusts (J.D.M). O.F.B was supported by Post-Doctoral Fellowships from the NIH (HL10336). B.J.W. was sup-ported by an MD/PhD scholar award from the University ofCincinnati Physician Scientist Training Program and the Albert J.Ryan Foundation.
References1. Kannel WB. Overview of atherosclerosis. Clin Ther. 1998;20:B2–B17.2. Gill C, Menstril R, Samali A. Losing heart: the role of apoptosis in heart
disease: a novel therapeutic target? FASEB J. 2002;16:135–146.3. Guerra S, Leri A, Wang X, Finato N, Di Loreto C, Beltrami CA, Katstura
J, Anversa P. Myocyte death in the failing human heart is genderdependent. Circ Res. 1999;85:856–866.
4. Kang PM, Izumo S. Apoptosis and heart failure: a critical review of theliterature. Circ Res. 2000;86:1107–1113.
5. Bishopric NH, Andreka P, Slepak T, Webster KA. Molecular mech-anisms of apoptosis in the cardiac myocyte. Curr Opin Pharmacol.2001;1:141–150.
6. Dawn B, Bolli R. Role of nitric oxide in myocardial preconditioning. AnnN Y Acad Sci. 2002;962:18–41.
7. Aoki H, Kang PM, Hampe J, Yoshimura K, Noma T, Matsuzaki M,Izumo S. Direct activation of mitochondrial apoptosis machinery by c-JunN-terminal kinase in adult cardiac myocytes. J Biol Chem. 2002;277:10244–10250.
8. Bueno OF, Molkentin JD. Involvement of extracellular signal-regulatedkinases 1/2 in cardiac hypertrophy and cell death. Circ Res. 2002;91:776–781.
9. Matsui T, Li L, Wu JC, Cook SA, Nagoshi T, Picard MH, Liao R,Rosenzweig A. Phenotypic spectrum caused by transgenic overexpressionof activated Akt in the heart. J Biol Chem. 2002;277:22896–22901.
10. Miao W, Luo Z, Kitsis RN, Walsh K. Intracoronary, adenovirus-mediatedAkt gene transfer in heart limits infarct size following ischemia-reperfusion injury in vivo. J Mol Cell Cardiol. 2000;32:2397–2402.
11. Yamashita K, Kajstura J, Discher DJ, Wasserlauf BJ, Bishopric NH,Anversa P, Webster KA. Reperfusion-activated Akt kinase prevents apo-ptosis in transgenic mouse hearts overexpressing insulin-like growthfactor-1. Circ Res. 2001;88:609–614.
12. Brar BK, Stephanou A, Pennica D, Latchman DS. CT-1 mediated car-dioprotection against ischemic re-oxygenation injury is mediated by PI3kinase, Akt and MEK1/2 pathways. Cytokine. 2001;16:93–96.
13. Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J,Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway forcardiac hypertrophy. Cell. 1998;93:215–228.
14. De Windt LJ, Lim HW, Taigen T, Wencker D, Condorelli G, Dorn GWII, Kitsis RN, Molkentin JD. Calcineurin-mediated hypertrophy protectscardiomyocytes from apoptosis in vitro and in vivo: an apoptosis-independent model of dilated heart failure. Circ Res. 2000;86:255–263.
15. Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F,McKeon F, Bobo T, Franke TF, Reed JC. Ca2�-induced apoptosis throughcalcineurin dephosphorylation of BAD. Science. 1999;284:339–343.
16. Tombal B, Weeraratna AT, Denmeade SR, Isaacs JT. Thapsigargininduces a calmodulin/calcineurin-dependent apoptotic cascaderesponsible for the death of prostatic cancer cells. Prostate. 2000;43:303–317.
98 Circulation Research January 9/23, 2004
by guest on May 30, 2013http://circres.ahajournals.org/Downloaded from

17. Jayaraman T, Marks AR. Calcineurin is downstream of the inositol1,4,5-trisphosphate receptor in the apoptotic and cell growth pathways.J Biol Chem. 2000;275:6417–6420.
18. Shibasaki F, McKeon F. Calcineurin functions in Ca2�-activated celldeath in mammalian cells. J Cell Biol. 1995;131:735–743.
19. Zhao Y, Tozawa Y, Iseki R, Mukai M, Iwata M. Calcineurin activationprotects T cells from glucocorticoid-induced apoptosis. J Immunol. 1995;154:6346–6354.
20. Asada A, Zhao Y, Kondo S, Iwata M. Induction of thymocyte apoptosisby Ca2�-independent protein kinase C (nPKC) activation and its regu-lation by calcineurin activation. J Biol Chem. 1998;273:28392–28398.
21. Ankarcrona M, Dypbukt JM, Orrenius S, Nicotera P. Calcineurin andmitochondrial function in glutamate-induced neuronal cell death. FEBSLett. 1996;394:321–324.
22. Wood AM, Bristow DR. N-methyl-D-aspartate receptor desensitisation isneuroprotective by inhibiting glutamate-induced apoptotic-like death.J Neurochem. 1998;70:677–687.
23. Kakita T, Hasegawa K, Iwai-Kanai E, Adachi S, Morimoto T, Wada H,Kawamura T, Yanazume T, Sasayama S. Calcineurin pathway is requiredfor endothelin-1-mediated protection against oxidant stress-induced apo-ptosis in cardiac myocytes. Circ Res. 2001;88:1239–1246.
24. Saito S, Hiroi Y, Zou Y, Aikawa R, Toko H, Shibasaki F, Yazaki Y,Nagai R, Komuro I. �-Adrenergic pathway induces apoptosis throughcalcineurin activation in cardiac myocytes. J Biol Chem. 2000;275:34528–34533.
25. Crompton M. Mitochondrial intermembrane junctional complexes andtheir role in cell death. J Physiol. 2000;529:11–21.
26. Bueno OF, Brandt EB, Rothenberg ME, Molkentin JD. Defective T celldevelopment and function in calcineurin A�-deficient mice. Proc NatlAcad Sci U S A. 2002;99:9398–9403.
27. Braz JC, Bueno OF, Liang Q, Wilkins BJ, Dai Y-S, Parsons S, BraunwartJ, Glascock BJ, Klevitsky R, Kimball TF, Hewett TE. Molkentin JD.
Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopa-thy through upregulation of calcineurin NFAT signaling. J Clin Invest.2003;111:1475–1486.
28. Gulick J, Hewett TE, Klevitsky R, Buck SH, Moss RL, Robbins J.Transgenic remodeling of the regulatory myosin light chains in themammalian heart. Circ Res. 1997;80:655–664.
29. Aramburu J, Yaffe MB, Lopez-Rodriguez C, Cantley LC, Hogan PG, RaoA. Affinity driven peptide selection of an NFAT inhibitor more selectivethan cyclosporin A. Science. 1999;285:2129–2133.
30. Cho RJ, Huang M, Campbell MJ, Dong H, Steinmetz L, Sapinoso L,Hampton G, Elledge SJ, Davis RW, Lockhart DJ. Transcriptional regu-lation and function during the human cell cycle. Nat Genet. 2001;27:48–54.
31. Notterman DA, Alon U, Sierk AJ, Levine AJ. Transcriptional geneexpression profiles of colorectal adenoma, adenocarcinoma, and normaltissue examined by oligonucleotide arrays. Cancer Res. 2001;61:3124–3130.
32. Lotem J, Kama R, Sachs L. Suppression or induction of apoptosis byopposing pathways downstream from calcium-activated calcineurin. ProcNatl Acad Sci U S A. 1999;96:12016–12020.
33. Shirane M, Nakayama KI. Inherent calcineurin inhibitor FKBP38 targetsBcl-2 to mitochondria and inhibits apoptosis. Nat Cell Biol. 2003;5:28–37.
34. Crabtree GR, Olson EN. NFAT signaling: choreographing the social livesof cells. Cell. 2002;109:S67–S79.
35. Wilkins BJ, De Windt LJ, Bueno OF, Braz JC, Glascock BJ, Kimball TF,Molkentin JD. Targeted disruption of NFATc3, but not NFATc4 revealsan intrinsic defect in calcineurin-mediated cardiac hypertrophic growth.Mol Cell Biol. 2002;22:7603–7613.
36. Pu WT, Ma Q, Izumo S. NFAT transcription factors are critical survivalfactors that inhibit cardiomyocyte apoptosis during phenylephrine stim-ulation in vitro. Circ Res. 2003;92:725–731.
Bueno et al Calcineurin Regulates Cardiac Apoptosis In Vivo 99
by guest on May 30, 2013http://circres.ahajournals.org/Downloaded from
Copyright © 2022 FDOKUMEN




















