
Hypertrophy and Heart Failure in Mice Overexpressing the Cardiac Sodium-Calcium Exchanger
12
Hypertrophy and Heart Failure in Mice Overexpressing the Cardiac Sodium-Calcium Exchanger KENNETH P. ROOS, PhD, 1 MARIA C. JORDAN, MD, 1 MICHAEL C. FISHBEIN, MD, 2 MATTHEW R. RITTER, PhD, 4 MARTIN FRIEDLANDER, PhD, 4 HELEN C. CHANG, BS, 1 PAYMON RAHGOZAR, BS, 1 TIEYAN HAN, MD, 1 ALEJANDRO J. GARCIA, BA, 3 W. ROBB MACLELLAN, MD, 3 ROBERT S. ROSS, MD, 5 AND KENNETH D. PHILIPSON, PhD 1,3 Los Angeles, California; La Jolla, California; San Diego, California ABSTRACT Background: The cardiac sodiumecalcium exchanger (NCX1) is a key sarcolemmal protein for the maintenance of calcium homeostasis in the heart. Because heart failure is associated with increased expression of NCX1, heterozygous (HET) and homozygous (HOM) transgenic mice overexpressing NCX1 were developed and evaluated. Methods and Results: The NCX1 transgenic mice display 2.3-fold (HET) and 3.1-fold (HOM) increases in exchanger activity from wild-type (WT) mice. Functional information was obtained by echocardiog- raphy and catheterizations before and after hemodynamic stress from pregnancy, treadmill exercise or transaortic constriction (TAC). HET and HOM mice exhibited hypertrophy and blunted responses with b-adrenergic stimulation. Postpartum mice from all groups were hypertrophied, but only the HOM mice exhibited premature death from heart failure. HOM mice became exercise intolerant after 6 weeks of daily treadmill running. After 21 days TAC, HET, and HOM mice exhibited significant contractile dysfunction and 15% to 40% mortality with clinical evidence of heart failure. Conclusions: Hemodynamic stress results in a compensated hypertrophy in WT mice, but NCX1 trans- genic mice exhibit decreased contractile function and heart failure in proportion to their level of NCX1 expression. Thus exchanger overexpression in mice leads to abnormal calcium handling and a decompen- satory transition to heart failure with stress. (J Cardiac Fail 2007;13:318e329) Key Words: Heart failure, hypertrophy, Na/Ca exchanger, E-C coupling, transgenic mice. Heart failure is a complex disease process that leads to severely depressed cardiac function and high mortality. Detrimental alterations in calcium homeostasis are gener- ally recognized as a universal factor in the development of heart failure. 1e5 Sodium-calcium (Na þ -Ca 2þ ) exchange is a key mechanism in maintaining calcium (Ca 2þ ) homeo- stasis during cardiac excitation-contraction coupling. 6e9 Ca 2þ entering cardiomyocytes via L-type Ca 2þ channels is primarily extruded by the Na þ -Ca 2þ exchanger (forward mode exchange). However, the net direction in which the exchanger will transport Ca 2þ is dependent on ionic condi- tions and membrane potential. Thus ‘‘reverse mode ex- change’’ is a possible Ca 2þ influx mechanism. 4,10e13 Though the magnitude of Ca 2þ influx through the ex- changer is probably modest under normal physiologic con- ditions, the exchanger is upregulated during hypertrophy and heart failure in most human and animal models when reverse mode exchange may become more promi- nent. 3,4,10,11,14e20 Predicting the effects of upregulated exchange activity and its relationship to heart failure is not straightforward. Increased levels of exchanger may compensate for defec- tive excitation-contraction coupling mechanisms such as the depressed function of the sarcoplasmic reticulum (SR) Ca 2þ pump (SERCA) that often accompanies hypertrophy and heart failure. 21e23 Alternatively, increased exchanger ac- tivity may act to decrease SR Ca 2þ stores and hence diminish contractility. Understanding the role of increased Na þ -Ca 2þ exchanger activity during heart failure is further complicated From the 1 Departments of Physiology; 2 Pathology; 3 Medicine, The Car- diovascular Research Laboratory, David Geffen School of Medicine at UCLA, Los Angeles, California; 4 Department of Cell Biology, The Scripps Research Institute, La Jolla, California and 5 The Department of Medicine, UCSD School of Medicine and Veterans Administration San Diego Health- care System, San Diego, California. Manuscript received June 24, 2006; revised manuscript received No- vember 3, 2006; revised manuscript accepted January 15, 2007. Reprint requests: Kenneth P. Roos, PhD, David Geffen School of Med- icine at UCLA, Physiology, 10833 LeConte Avenue, Los Angeles, CA 90095-1751. Supported by an American Heart Association Grant-in-Aid (KPR), American Heart Association (Western States) Undergraduate Fellowship (HCC, PR), Laubisch Endowment Funds (KPR, RSR), the Piansky Family Trust (MCF), and NIH HL-48509 (KDP). 1071-9164/$ - see front matter Ó 2007 Elsevier Inc. All rights reserved. doi:10.1016/j.cardfail.2007.01.004 318 Journal of Cardiac Failure Vol. 13 No. 4 2007
Transcript of Hypertrophy and Heart Failure in Mice Overexpressing the Cardiac Sodium-Calcium Exchanger

Journal of Cardiac Failure Vol. 13 No. 4 2007
Hypertrophy and Heart Failure in Mice Overexpressing theCardiac Sodium-Calcium Exchanger
KENNETH P. ROOS, PhD,1 MARIA C. JORDAN, MD,1 MICHAEL C. FISHBEIN, MD,2 MATTHEW R. RITTER, PhD,4 MARTINFRIEDLANDER, PhD,4 HELEN C. CHANG, BS,1 PAYMON RAHGOZAR, BS,1 TIEYAN HAN, MD,1 ALEJANDRO J. GARCIA, BA,3
W. ROBB MACLELLAN, MD,3 ROBERT S. ROSS, MD,5 AND KENNETH D. PHILIPSON, PhD1,3
Los Angeles, California; La Jolla, California; San Diego, California
ABSTRACT
Background: The cardiac sodiumecalcium exchanger (NCX1) is a key sarcolemmal protein for themaintenance of calcium homeostasis in the heart. Because heart failure is associated with increasedexpression of NCX1, heterozygous (HET) and homozygous (HOM) transgenic mice overexpressingNCX1 were developed and evaluated.Methods and Results: The NCX1 transgenic mice display 2.3-fold (HET) and 3.1-fold (HOM) increasesin exchanger activity from wild-type (WT) mice. Functional information was obtained by echocardiog-raphy and catheterizations before and after hemodynamic stress from pregnancy, treadmill exercise ortransaortic constriction (TAC). HET and HOM mice exhibited hypertrophy and blunted responses withb-adrenergic stimulation. Postpartum mice from all groups were hypertrophied, but only the HOMmice exhibited premature death from heart failure. HOM mice became exercise intolerant after 6 weeksof daily treadmill running. After 21 days TAC, HET, and HOM mice exhibited significant contractiledysfunction and 15% to 40% mortality with clinical evidence of heart failure.Conclusions: Hemodynamic stress results in a compensated hypertrophy in WT mice, but NCX1 trans-genic mice exhibit decreased contractile function and heart failure in proportion to their level of NCX1expression. Thus exchanger overexpression in mice leads to abnormal calcium handling and a decompen-satory transition to heart failure with stress. (J Cardiac Fail 2007;13:318e329)Key Words: Heart failure, hypertrophy, Na/Ca exchanger, E-C coupling, transgenic mice.
Heart failure is a complex disease process that leads toseverely depressed cardiac function and high mortality.Detrimental alterations in calcium homeostasis are gener-ally recognized as a universal factor in the developmentof heart failure.1e5 Sodium-calcium (Naþ-Ca2þ) exchangeis a key mechanism in maintaining calcium (Ca2þ) homeo-stasis during cardiac excitation-contraction coupling.6e9
From the 1Departments of Physiology; 2Pathology; 3Medicine, The Car-diovascular Research Laboratory, David Geffen School of Medicine atUCLA, Los Angeles, California; 4Department of Cell Biology, The ScrippsResearch Institute, La Jolla, California and 5The Department of Medicine,UCSD School of Medicine and Veterans Administration San Diego Health-care System, San Diego, California.
Manuscript received June 24, 2006; revised manuscript received No-vember 3, 2006; revised manuscript accepted January 15, 2007.
Reprint requests: Kenneth P. Roos, PhD, David Geffen School of Med-icine at UCLA, Physiology, 10833 LeConte Avenue, Los Angeles, CA90095-1751.
Supported by an American Heart Association Grant-in-Aid (KPR),American Heart Association (Western States) Undergraduate Fellowship(HCC, PR), Laubisch Endowment Funds (KPR, RSR), the Piansky FamilyTrust (MCF), and NIH HL-48509 (KDP).
1071-9164/$ - see front matter� 2007 Elsevier Inc. All rights reserved.doi:10.1016/j.cardfail.2007.01.004
3
Ca2þ entering cardiomyocytes via L-type Ca2þ channelsis primarily extruded by the Naþ-Ca2þ exchanger (forwardmode exchange). However, the net direction in which theexchanger will transport Ca2þ is dependent on ionic condi-tions and membrane potential. Thus ‘‘reverse mode ex-change’’ is a possible Ca2þ influx mechanism.4,10e13
Though the magnitude of Ca2þ influx through the ex-changer is probably modest under normal physiologic con-ditions, the exchanger is upregulated during hypertrophyand heart failure in most human and animal models whenreverse mode exchange may become more promi-nent.3,4,10,11,14e20
Predicting the effects of upregulated exchange activityand its relationship to heart failure is not straightforward.Increased levels of exchanger may compensate for defec-tive excitation-contraction coupling mechanisms such asthe depressed function of the sarcoplasmic reticulum (SR)Ca2þ pump (SERCA) that often accompanies hypertrophyand heart failure.21e23 Alternatively, increased exchanger ac-tivity may act to decrease SR Ca2þ stores and hence diminishcontractility. Understanding the role of increased Naþ-Ca2þ
exchanger activity during heart failure is further complicated
18

Heart Failure in NCX Mice � Roos et al 319
by the many adaptive modifications of Ca2þ-related andcontractile proteins that simultaneously occur.5,11
To help reveal the contributions of discrete componentsassociated with the disease processes, we have used geneticmanipulations to alter the level of the cardiac Naþ-Ca2þ ex-changer (NCX1) in the myocardium.24e26 Numerous studieswith heterozygous (HET) transgenic a-myosin heavy chain-NCX1 overexpressing mice have reported alterations in Ca2þ
handling and contractile function in myocytes.12,14,27e34 Noadaptations in expression levels of other calcium handlingproteins (L-type Ca2þ channels, SR calcium pump, calse-questrin, or phospholamban) were reported to occur in themyocardium of these HET mice.12,28,29
Based on these HET data and the evidence of increasedexchanger levels in other models of heart failure, we hy-pothesized that further increases in NCX1 expression inour model would alter baseline function and predisposethe myocardium to heart failure after hemodynamic stress.Thus homozygous overexpressing mice (HOM) were devel-oped and found to have a 33% increase in NCX1 activityabove that of the HET mice examined in the previous pub-lications. Recently, we reported on significant defects in theexcitation-contraction coupling mechanism in myocytesfrom these HOM mice.9,25,26,35 We now examine the invivo phenotype of the HOM NCX1 mice and contrastthem with the HET and wild-type (WT) mice. Because noalterations in the expression of other Ca2þ handling pro-teins were also found in the HOM mice, the effects of ex-changer overexpression can be studied in a normalbackground of other excitation-contraction coupling pro-teins. The increased exchanger activity in the HET andHOM mice induced a compensated cardiac hypertrophyat baseline and a blunted contractile response to b-adrener-gic stimulation whose magnitude was proportional toNCX1 overexpression levels. Furthermore, mice chroni-cally stressed by pregnancy, exercise, or pressure overloadmanifest an accelerated hypertrophic response and transi-tion to decompensated heart failure in proportion to theirexpression levels. Thus the pathophysiology observed inHOM mice is associated with abnormalities in Ca2þ han-dling directly tied to the greater expression of the Naþ-Ca2þ exchanger as compared to the HET and WT mice.
Methods
Generation and Characterization of HOM TransgenicMice
The a-myosin heavy chain (MHC) Naþ-Ca2þ exchanger(NCX1.1) transgenic mice were generated previously described.24
These HET mice were mated to homozygosity. Genotypes wereconfirmed by Southern blot and polymerase chain reaction, andhomozygosity was confirmed genetically. All groups of mice(HOM, HET, and WT) are on the same C57Bl/6 x C3HF1 back-ground. Naþ-Ca2þ exchange activity was determined in a crudefraction of cardiac membrane vesicles by measuring the Naþ gra-dientedependent 45Ca2þ uptake.24 ANF and SERCA2 expressionwere respectively determined by Northern and Western blot
analyses as previously described.24 DNA microarray analysismethods were similar to those from Ritter et al.36
Unless specifically stated otherwise, mice from all groups andunder all conditions were evaluated at approximately 4 monthsof age. This investigation conforms to the Guide for the Careand Use of Laboratory Animals published the US National Insti-tutes for Health (NIH publication # 85-23, revised 1996) andwas approved by the University of California Los Angeles Officefor Protection of Research Subjects.
Echocardiography
Mice were sedated with Avertin (2,2,2, tribromo-ethanol, 2.5%solution, 0.016 mL/gm body mass, Aldrich Chemical Corp) andultrasonically imaged with either an ATL Interspec ApogeeCX200 instrument37,38 or with a Siemens Acuson Sequoia C256instrument (Siemens Medical Solutions, Mountain View, Califor-nia).39 Digitized 2-dimensionally guided M-mode images wereanalyzed with SigmaScan (Systat Software Inc, Richmond, Cali-fornia) or AccessPoint (Freeland Systems LLC, Santa Fe, NewMexico) software for dimensions of the left ventricular cavity(end-diastolic dimension [EDD] and end-systolic dimension[ESD]) and wall thickness (posterior wall thickness [PWT] andventricular septal thickness [VST]) during systole and diastole.Ejection times and heart rates were determined from Doppler im-ages. Left ventricular mass was calculated from the EDD, PWT,and VST values according to Tanaka et al.38 Left ventricular frac-tional shortening (%LVFS) and velocity of circumferential fibershortening (Vcf) were calculated as indices of contractility.38
Mouse electrocardiograms were obtained in the standard lead IIarrangement with subcutaneous Pt electrodes (Grass Instruments,Warwick, Rhode Island).
Exercise Testing
WT and HOM mice (n 5 6 each) were run daily on a Rotarodtreadmill (Ugo Basile, Italy) for 8 weeks. The mice were condi-tioned to the device during the first 2 weeks at increasing speeds.From the third through the eighth weeks, the mice were run twicedaily (30 minutes each) at a more strenuous pace about 10% lessthan their individual maximum determined at the end of the firstweek (3e4 m/min). All exercise sessions were performed duringthe mouse’s normal active (dark) circadian period in a room litby only two 15 W red bulbs. Exercise tolerance was scored bythe maximum speed and the number falls per hour for eachmouse.40
Transaortic Constriction
A fixed pressure overload was obtained by surgically constrict-ing (banding) the aorta between the right and left carotid arteriesto the diameter of a 27 gauge needle in an anesthetized, ventilatedmouse.41,42 At the end of the study period (7 or 21 days), the trans-aortic constriction (TAC) mice were evaluated a second time viaechocardiography before the terminal physiologic and morpho-metric assessment. For confirmation of the TAC pressure gradient,the right carotid and femoral arteries were catheterized withflame-stretched PE-50 tubing and pressures were simultaneouslymeasured.
Physiologic Assessment
Mice were anesthetized with a mixture of ketamine (100 mg/kgbody mass), xylazine (2 mg/kg), and buprenorphine (0.1 mg/kg),intubated, and hemodynamic data obtained. For assessment of

320 Journal of Cardiac Failure Vol. 13 No. 4 May 2007
contractility, the right carotid artery was catheterized with a 1.4 Frcatheter (Millar Instruments, Houston, Texas) advanced into theleft ventricle. After stabilization of baseline LV pressure and 6
dP/dT recordings, sequential 10e150 mg/kg dose (0.01-mL bolusintravenously) infusions of dobutamine were administered at 10-minute intervals. Hemodynamic measurements were acquired,digitized, displayed, and analyzed with HEM V3.3 software (No-tocord Systems, Croissy sur Seine, France). All hemodynamicdata were recorded continuously for at least 30 minutes to ensurephysiologic levels of pressures and heart rates.
Morphometric and Histologic Assessment
At the end of the study, standard morphometric measures wereobtained including body, heart, and lung weights as well as tibialength. In some cases, hearts were immediately fixed in formalinfor histologic assessment. Routinely processed paraffin-embeddedtissue sections were stained with hematoxylin and eosin and Mas-son’s trichrome stains. For morphometric analysis, sections wereimaged and evaluated using an automated image analysis system(Image Pro software, Media Cybernetics Inc, Silver Springs,Maryland).
Statistical Methods
Data were compiled and are shown as means and standard er-rors. Data were evaluated using a student’s 2-tailed t-test or anal-ysis of variance with Instat V3.05 statistical software (GraphPadInc, San Diego, California). In all analyses, P ! .05 was takento be statistically significant.
Results
Characterization of Baseline NCX1 Transgenic Mice
After producing homozygous offspring of our transgenicmice, we assessed the level of NCX1 activity in the HOM,HET, and WT mice. The NCX1 activity in membrane ves-icles was increased 2.34-fold in HET and 3.12 fold in HOMvs. WT controls. HET activity was significantly greater (P! .001) than WT controls and HOM activity was signifi-cantly (P ! .05) greater than both WT and HET values. Be-cause NCX1 activity has been suggested to decline withage,43 it was evaluated in both young mice (2 to 4 monthsof age) as well as elderly mice (16 to 19 months of age). Nosignificant differences were noted with age or gender.
Previously published studies have clearly established thatthere were no adaptations to expression levels of other cal-cium handling proteins in young adult HET mice.12,28,29
New analyses were performed to reveal whether the addi-tional NCX1 activity in HOM mice elicited any changes.Western blot analysis for SERCA2a revealed no differencesin expression in ventricles from HOM and WT mice fromboth genders (Fig. 1A; WT 0.96 6 0.13; HOM, 1.00 6
0.11; mean 6 SEM in arbitrary units; n 5 3 each). RNAexpression levels determined by microarray analysis formany other Ca2þ handling proteins (calsequestrin, ryano-dine receptor, L-type Ca2þ channel, the Naþ/Kþ-ATPase,and SERCA2a) as well as markers of hypertrophy (ANFand a-skeletal actin) were not significantly different be-tween any of the groups examined (WT, HET, and
HOM). Because the 4-month-old transgenic mice (HETand HOM) exhibited baseline hypertrophy (documentedin the following section), younger, 2-month-old micewere specifically chosen for this gene chip analysis to re-move any confounding variables from the hypertrophic re-sponses associated with aging.
Evaluation of Baseline Phenotypes
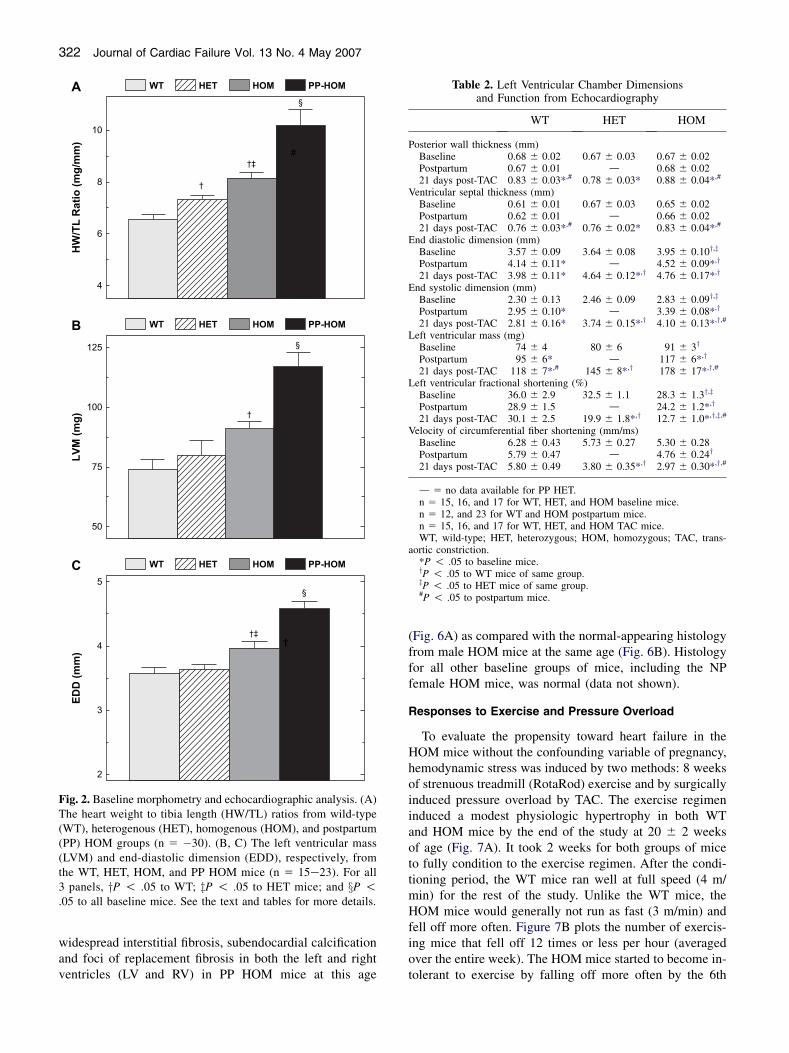
Baseline phenotypes were evaluated in WT, HET, andHOM mice at 4 months of age. Each group of mice con-sisted of approximately equal numbers of males and nullip-arous (never pregnant) females. Unlike at 2 months of age,ANF expression from Northern analysis is 20.7 timesgreater in HOM mice than in WT mice at 4 months ofage (Fig. 1B). Postmortem morphometry confirms the sug-gested hypertrophic response from the ANF data. Heartweights (HW) significantly increase from 119 6 4 mg inWT mice to 133 6 4 mg in HET and 146 6 4 mg inHOM mice (mean 6 SEM; P ! .001; n 5 30, 23, 30).When normalized to both tibia length (HW/TL) or bodyweights (HW/BW), the hypertrophic response was greatestin the HOM mice and at intermediate values in the HET inparallel to the increased expression level of the NCX1transgene (Table 1, baseline; Fig. 2A). Though both mea-sures revealed significant hypertrophy in HOM mice, wefeel that the HW/TL index is a better index of hypertrophythan HW/BW in these mice because the HET and HOMmice were, respectively, 10% and 13% heavier than theirWT counterparts, whereas there were no differences in tibialength. Morphometric data obtained from older (12 to 15months) nonfailing baseline mice are consistent with thedata from younger mice in all groups (data not shown).There were no significant gender differences in these data.
Ventricular remodeling and changes in function associ-ated with hypertrophy were assessed by echocardiographyat physiologic heart rates (513 6 19 beats/min; Table 2,
A SERCA2a Western Blot
HOM WT HOM WT HOM WT
B ANF Northern Blot
110 kDA
ANF GAPDH
WT HOM WT HOM
Fig. 1. Baseline sarcoplasmic reticulum (SR) Ca2þ pump (SERCAand ANF expression. (A) A Western blot analysis for SERCA2aprotein expression in wild-type (WT) and homogenous (HOM)mice. (B) A representative Northern blot analysis for baselineANF in WT and HOM mice at 4 months of age. See text fordetails.

Heart Failure in NCX Mice � Roos et al 321
Table 1. Morphometric Measurements
WT HET HOM
Heart weight/body weight (mg/g)Baseline 4.11 6 0.10 4.15 6 0.12 4.48 6 0.13y
Postpartum 3.52 6 0.19 5.66 6 0.38*,y 6.19 6 0.46*,y
21 days post-TAC 7.40 6 0.38*,# 8.77 6 0.41*,y,# 9.59 6 0.59*,y,#
Heart weight/tibia length (mg/mm)Baseline 6.54 6 0.19 7.32 6 0.16y 8.15 6 0.22y,z
Postpartum 7.38 6 0.20* 8.76 6 0.38*,y 10.18 6 0.62*,y
21 days post-TAC 10.79 6 0.43*,# 12.05 6 0.41*,y,# 13.04 6 0.59*,y,#
Lung weight/tibia length (mg/mm)Baseline 8.28 6 0.26 9.62 6 0.20y 9.54 6 0.18y
Postpartum 9.55 6 0.32* 9.80 6 0.41 10.71 6 0.31*,y
21 days post-TAC 13.14 6 1.60*,# 21.85 6 1.46*,y,# 20.90 6 1.54*,y,#
n 5 30, 23, and 30 for WT, HET, and HOM baseline mice.n 5 19, 8, and 9 for WT, HET, and HOM postpartum mice.n 5 13, 17, and 16 for WT, HET, and HOM TAC mice.WT, wild-type; HET, heterozygous; HOM, homozygous; TAC, trans-aortic constriction.*P ! .05 to baseline mice.yP ! .05 to WT mice of same group.zP ! .05 to HET mice of same group.#P ! .05 to postpartum mice.
baseline). Similar to the mice evaluated for Table 1, no an-imals in these groups showed overt clinical evidence ofheart failure. Figure 2B shows the significant left ventricu-lar mass increase in the HOM mice consistent with thepostmortem data. This appears to be an eccentric type ofhypertrophy because left ventricular chamber enlargementoccurred (increased EDD; Fig. 2C) without any significantchanges in wall thickness (PWT and VST; Table 2, base-line). Echo-based indices of ventricular function (%LVFSand Vcf) were not significantly different between WT andHET mice. However, 1 index of function, %LVFS, was sig-nificantly reduced in HOM mice suggesting some decom-pensation before failure (Table 2, baseline).
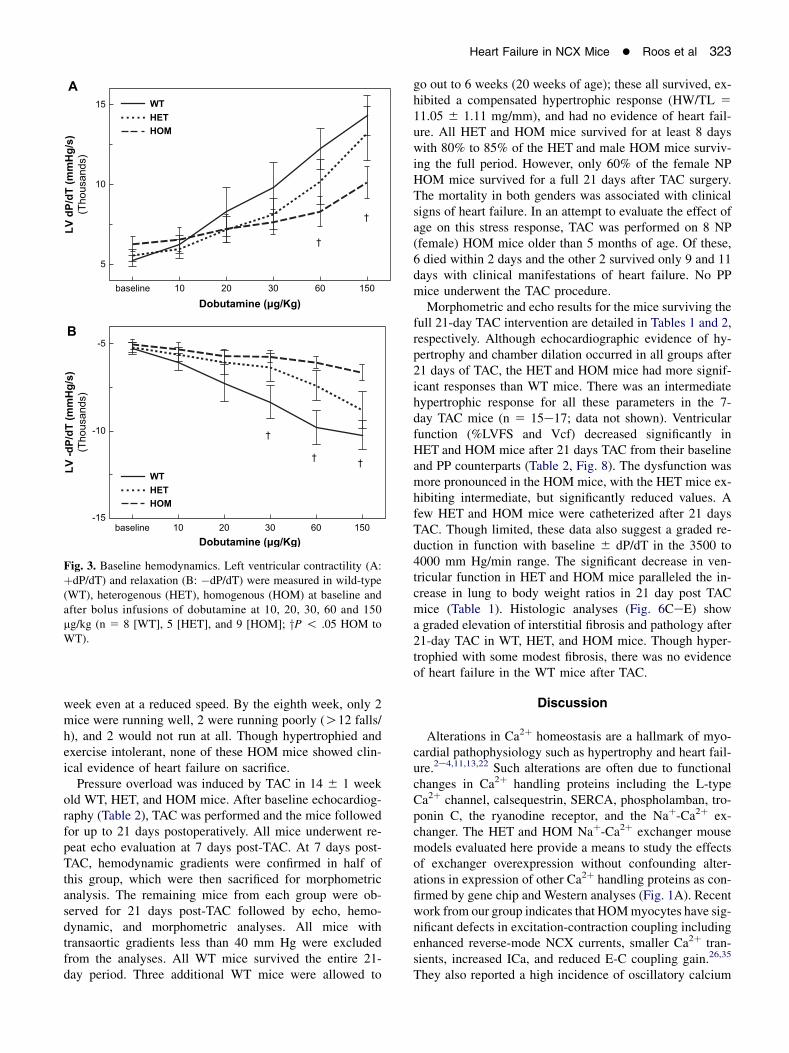
To further evaluate the ventricular function in vivo, weused LV catheterization. The LV pressures and heart rateswere in the physiological range with no statistical differ-ences between any of the groups. Contractility (þdP/dT)and relaxation (�dP/dT) were not different between thethree groups (Fig. 3, baseline). Subsequently, sequentialbolus infusions of dobutamine were given to evaluate theresponse of NCX1-overexpressing mice to b-adrenergicstimuli. HOM mice exhibited significantly blunted re-sponses (Fig. 3). Interestingly, HET mice generallyexhibited an intermediate response at higher doses.
Heart Failure in Postpartum HOM Mice
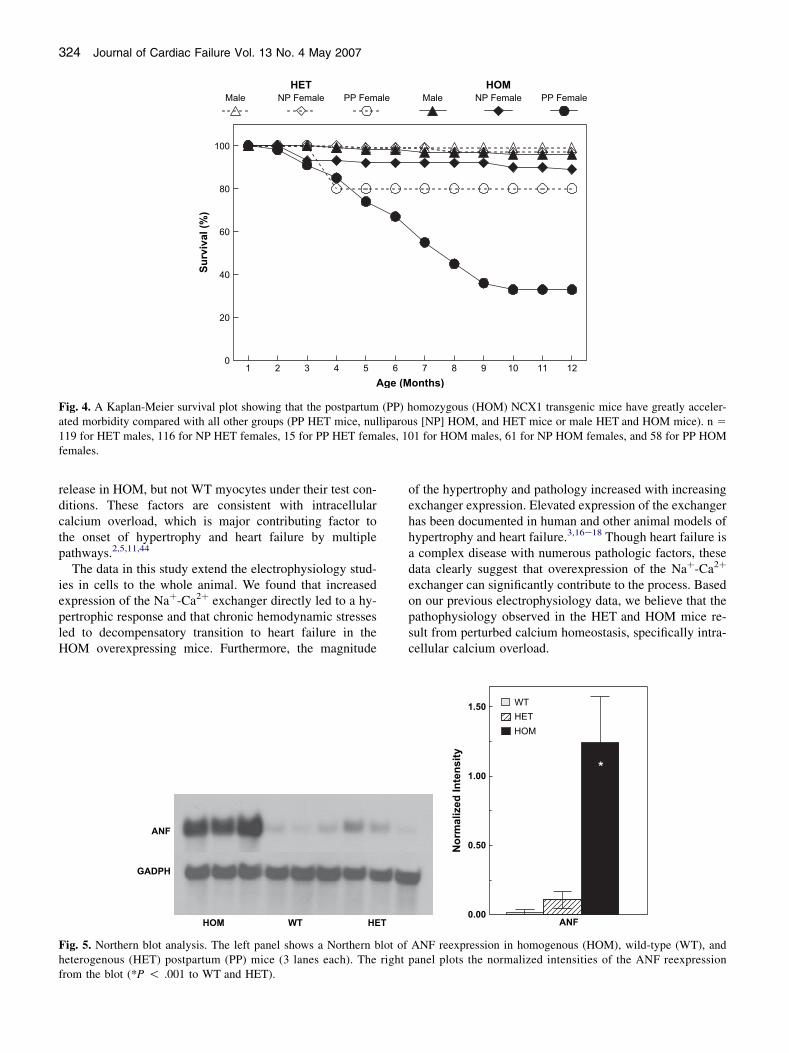
Our initial observation suggesting that increased levels ofNCX1 expression provoked cardiac dysfunction and heartfailure was noted as premature mortality in female HOMbreeders. Figure 4 shows a Kaplan-Meier survival curvegathered over a 12-month period for male, nulliparous(NP) female, and postpartum (PP) female HOM and HETmice. Only 33% of the PP HOM group survived past 12months of age, whereas 89% of the NP HOM femalesand 96% of the HOM males survived. In the HET groups,80% of the PP females, 97% of the NP females, and 99%
of the males survived to 1 year of age. All the PP HETdeaths occurred between 3 and 4 months of age and nonethereafter. Only 1 mouse died from all WT groups duringthis period (data not shown).
Postmortem examinations of PP HOM mice were indic-ative of congestive heart failure revealing severe edema,pleural effusions, and ascites as compared with the PPand baseline WT and HET mice. PP HOM mice sacrificedbefore the onset of heart failure (19 6 1 weeks of age) alsoexhibited a significantly increased lung weight to tibialength ratio consistent with heart failure (Table 1, postpar-tum). Additionally, both the PP HET and PP HOM mice ex-hibited a significant hypertrophic response (heart weight tobody weight or tibia length) compared with baseline and PPWT mice at the same age (Table 1, Fig. 2A). Interestingly,the magnitude of the hypertrophic response in the PP HETmice was at an intermediate level roughly in proportion tothe level of NCX1 overexpression.
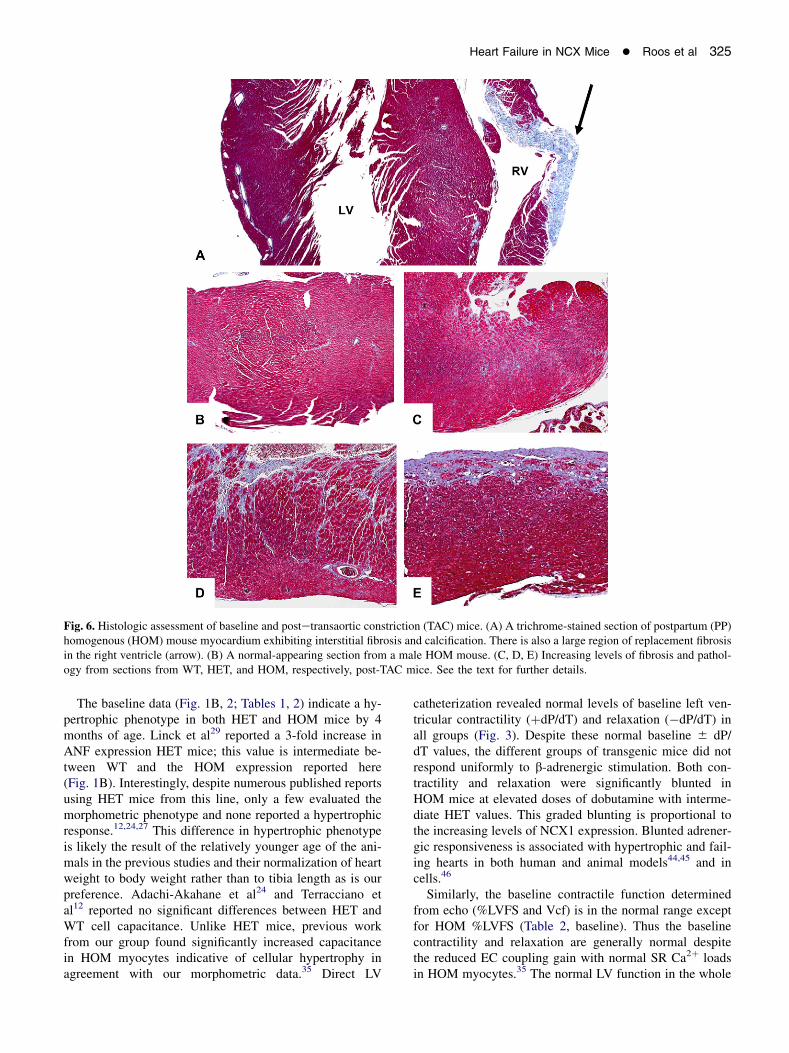
Echocardiographic evaluation in PP WT and PP HOMmice confirmed the hypertrophy measured from morphom-etry and revealed significantly reduced ventricular function(Table 2, postpartum; Fig. 2B, C). Specifically, the chamberdimensions (EDD and ESD) and left ventricular mass weresignificantly increased over both the PP WT and baselineHOM mice. Ventricular function in PP HOM mice was sig-nificantly reduced from PP WT mice, and, in the case of the%LVFS index, also to the baseline HOM mice. Two failingPP HOM mice were successfully catheterized, and had anaverage þdP/dT of 3180 6 552 mm Hg/min and a �dP/dT of 3054 6 595 mm Hg/min. These values are nearlyhalf of that of the nonfailing mice in Fig. 3. At the molec-ular level, Northern blot analysis (Fig. 5) revealed high-level expression of ANF in ventricular tissue of the PPHOM mice (WT, 0.1 6 0.2; HET, 1.1 6 0.6; HOM, 12.4 6
3.3; mean 6 SEM in arbitrary units; n 5 3 for each; P !.05 HOM to HET and WT). Histologic analysis revealed
322 Journal of Cardiac Failure Vol. 13 No. 4 May 2007
widespread interstitial fibrosis, subendocardial calcificationand foci of replacement fibrosis in both the left and rightventricles (LV and RV) in PP HOM mice at this age
4
6
8
10
HW
/T
L R
atio
(m
g/m
m)
†
†‡#
§
2
3
4
5
ED
D (m
m)
†‡†
§
50
75
100
125
LV
M (m
g)
WT HET HOM PP-HOM
†
§
A
WT HET HOM PP-HOMB
WT HET HOM PP-HOMC
Fig. 2. Baseline morphometry and echocardiographic analysis. (A)The heart weight to tibia length (HW/TL) ratios from wild-type(WT), heterogenous (HET), homogenous (HOM), and postpartum(PP) HOM groups (n 5 �30). (B, C) The left ventricular mass(LVM) and end-diastolic dimension (EDD), respectively, fromthe WT, HET, HOM, and PP HOM mice (n 5 15e23). For all3 panels, yP ! .05 to WT; zP ! .05 to HET mice; and xP !.05 to all baseline mice. See the text and tables for more details.
(Fig. 6A) as compared with the normal-appearing histologyfrom male HOM mice at the same age (Fig. 6B). Histologyfor all other baseline groups of mice, including the NPfemale HOM mice, was normal (data not shown).
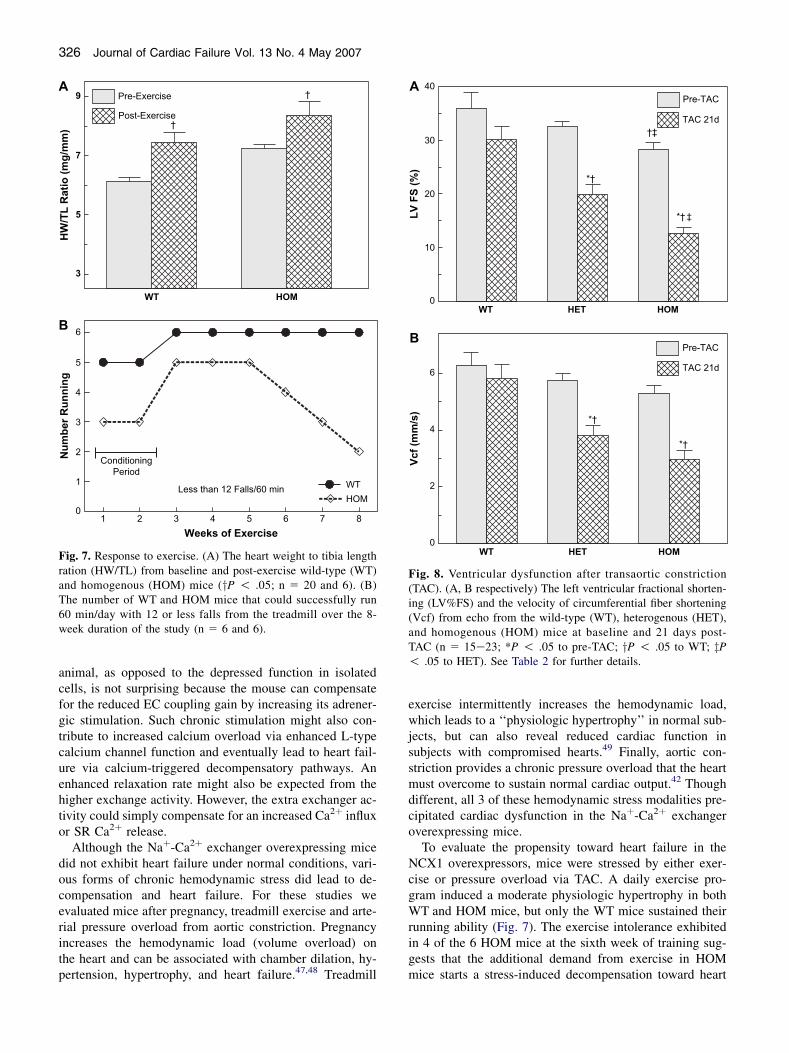
Responses to Exercise and Pressure Overload
To evaluate the propensity toward heart failure in theHOM mice without the confounding variable of pregnancy,hemodynamic stress was induced by two methods: 8 weeksof strenuous treadmill (RotaRod) exercise and by surgicallyinduced pressure overload by TAC. The exercise regimeninduced a modest physiologic hypertrophy in both WTand HOM mice by the end of the study at 20 6 2 weeksof age (Fig. 7A). It took 2 weeks for both groups of miceto fully condition to the exercise regimen. After the condi-tioning period, the WT mice ran well at full speed (4 m/min) for the rest of the study. Unlike the WT mice, theHOM mice would generally not run as fast (3 m/min) andfell off more often. Figure 7B plots the number of exercis-ing mice that fell off 12 times or less per hour (averagedover the entire week). The HOM mice started to become in-tolerant to exercise by falling off more often by the 6th
Table 2. Left Ventricular Chamber Dimensionsand Function from Echocardiography
WT HET HOM
Posterior wall thickness (mm)Baseline 0.68 6 0.02 0.67 6 0.03 0.67 6 0.02Postpartum 0.67 6 0.01 d 0.68 6 0.0221 days post-TAC 0.83 6 0.03*,# 0.78 6 0.03* 0.88 6 0.04*,#
Ventricular septal thickness (mm)Baseline 0.61 6 0.01 0.67 6 0.03 0.65 6 0.02Postpartum 0.62 6 0.01 d 0.66 6 0.0221 days post-TAC 0.76 6 0.03*,# 0.76 6 0.02* 0.83 6 0.04*,#
End diastolic dimension (mm)Baseline 3.57 6 0.09 3.64 6 0.08 3.95 6 0.10y,z
Postpartum 4.14 6 0.11* d 4.52 6 0.09*,y
21 days post-TAC 3.98 6 0.11* 4.64 6 0.12*,y 4.76 6 0.17*,y
End systolic dimension (mm)Baseline 2.30 6 0.13 2.46 6 0.09 2.83 6 0.09y,z
Postpartum 2.95 6 0.10* d 3.39 6 0.08*,y
21 days post-TAC 2.81 6 0.16* 3.74 6 0.15*,y 4.10 6 0.13*,y,#
Left ventricular mass (mg)Baseline 74 6 4 80 6 6 91 6 3y
Postpartum 95 6 6* d 117 6 6*,y
21 days post-TAC 118 6 7*,# 145 6 8*,y 178 6 17*,y,#
Left ventricular fractional shortening (%)Baseline 36.0 6 2.9 32.5 6 1.1 28.3 6 1.3y,z
Postpartum 28.9 6 1.5 d 24.2 6 1.2*,y
21 days post-TAC 30.1 6 2.5 19.9 6 1.8*,y 12.7 6 1.0*,y,z,#
Velocity of circumferential fiber shortening (mm/ms)Baseline 6.28 6 0.43 5.73 6 0.27 5.30 6 0.28Postpartum 5.79 6 0.47 d 4.76 6 0.24y
21 days post-TAC 5.80 6 0.49 3.80 6 0.35*,y 2.97 6 0.30*,y,#
d 5 no data available for PP HET.n 5 15, 16, and 17 for WT, HET, and HOM baseline mice.n 5 12, and 23 for WT and HOM postpartum mice.n 5 15, 16, and 17 for WT, HET, and HOM TAC mice.WT, wild-type; HET, heterozygous; HOM, homozygous; TAC, trans-
aortic constriction.*P ! .05 to baseline mice.yP ! .05 to WT mice of same group.zP ! .05 to HET mice of same group.#P ! .05 to postpartum mice.
Heart Failure in NCX Mice � Roos et al 323
week even at a reduced speed. By the eighth week, only 2mice were running well, 2 were running poorly (O12 falls/h), and 2 would not run at all. Though hypertrophied andexercise intolerant, none of these HOM mice showed clin-ical evidence of heart failure on sacrifice.
Pressure overload was induced by TAC in 14 6 1 weekold WT, HET, and HOM mice. After baseline echocardiog-raphy (Table 2), TAC was performed and the mice followedfor up to 21 days postoperatively. All mice underwent re-peat echo evaluation at 7 days post-TAC. At 7 days post-TAC, hemodynamic gradients were confirmed in half ofthis group, which were then sacrificed for morphometricanalysis. The remaining mice from each group were ob-served for 21 days post-TAC followed by echo, hemo-dynamic, and morphometric analyses. All mice withtransaortic gradients less than 40 mm Hg were excludedfrom the analyses. All WT mice survived the entire 21-day period. Three additional WT mice were allowed to
baseline 10 20 30 60 150
Dobutamine (µg/Kg)
5
10
15
LV
d
P/d
T (m
mH
g/s)
(Tho
usan
ds)
WT
HET
HOM
WT
HET
HOM
†
†
baseline 10 20 30 60 150Dobutamine (µg/Kg)
-15
-10
-5
LV
-d
P/d
T (m
mH
g/s)
(Tho
usan
ds)
†
† †
A
B
Fig. 3. Baseline hemodynamics. Left ventricular contractility (A:þdP/dT) and relaxation (B: �dP/dT) were measured in wild-type(WT), heterogenous (HET), homogenous (HOM) at baseline andafter bolus infusions of dobutamine at 10, 20, 30, 60 and 150mg/kg (n 5 8 [WT], 5 [HET], and 9 [HOM]; yP ! .05 HOM toWT).
go out to 6 weeks (20 weeks of age); these all survived, ex-hibited a compensated hypertrophic response (HW/TL 5
11.05 6 1.11 mg/mm), and had no evidence of heart fail-ure. All HET and HOM mice survived for at least 8 dayswith 80% to 85% of the HET and male HOM mice surviv-ing the full period. However, only 60% of the female NPHOM mice survived for a full 21 days after TAC surgery.The mortality in both genders was associated with clinicalsigns of heart failure. In an attempt to evaluate the effect ofage on this stress response, TAC was performed on 8 NP(female) HOM mice older than 5 months of age. Of these,6 died within 2 days and the other 2 survived only 9 and 11days with clinical manifestations of heart failure. No PPmice underwent the TAC procedure.
Morphometric and echo results for the mice surviving thefull 21-day TAC intervention are detailed in Tables 1 and 2,respectively. Although echocardiographic evidence of hy-pertrophy and chamber dilation occurred in all groups after21 days of TAC, the HET and HOM mice had more signif-icant responses than WT mice. There was an intermediatehypertrophic response for all these parameters in the 7-day TAC mice (n 5 15e17; data not shown). Ventricularfunction (%LVFS and Vcf) decreased significantly inHET and HOM mice after 21 days TAC from their baselineand PP counterparts (Table 2, Fig. 8). The dysfunction wasmore pronounced in the HOM mice, with the HET mice ex-hibiting intermediate, but significantly reduced values. Afew HET and HOM mice were catheterized after 21 daysTAC. Though limited, these data also suggest a graded re-duction in function with baseline 6 dP/dT in the 3500 to4000 mm Hg/min range. The significant decrease in ven-tricular function in HET and HOM mice paralleled the in-crease in lung to body weight ratios in 21 day post TACmice (Table 1). Histologic analyses (Fig. 6CeE) showa graded elevation of interstitial fibrosis and pathology after21-day TAC in WT, HET, and HOM mice. Though hyper-trophied with some modest fibrosis, there was no evidenceof heart failure in the WT mice after TAC.
Discussion
Alterations in Ca2þ homeostasis are a hallmark of myo-cardial pathophysiology such as hypertrophy and heart fail-ure.2e4,11,13,22 Such alterations are often due to functionalchanges in Ca2þ handling proteins including the L-typeCa2þ channel, calsequestrin, SERCA, phospholamban, tro-ponin C, the ryanodine receptor, and the Naþ-Ca2þ ex-changer. The HET and HOM Naþ-Ca2þ exchanger mousemodels evaluated here provide a means to study the effectsof exchanger overexpression without confounding alter-ations in expression of other Ca2þ handling proteins as con-firmed by gene chip and Western analyses (Fig. 1A). Recentwork from our group indicates that HOM myocytes have sig-nificant defects in excitation-contraction coupling includingenhanced reverse-mode NCX currents, smaller Ca2þ tran-sients, increased ICa, and reduced E-C coupling gain.26,35
They also reported a high incidence of oscillatory calcium
324 Journal of Cardiac Failure Vol. 13 No. 4 May 2007
1 2 3 4 5 6 7 8 9 10 11 12Age (Months)
0
20
40
60
80
100
Su
rvival (%
)
HET HOM
Male NP Female PP Female Male NP Female PP Female
Fig. 4. A Kaplan-Meier survival plot showing that the postpartum (PP) homozygous (HOM) NCX1 transgenic mice have greatly acceler-ated morbidity compared with all other groups (PP HET mice, nulliparous [NP] HOM, and HET mice or male HET and HOM mice). n 5
119 for HET males, 116 for NP HET females, 15 for PP HET females, 101 for HOM males, 61 for NP HOM females, and 58 for PP HOMfemales.
release in HOM, but not WT myocytes under their test con-ditions. These factors are consistent with intracellularcalcium overload, which is major contributing factor tothe onset of hypertrophy and heart failure by multiplepathways.2,5,11,44
The data in this study extend the electrophysiology stud-ies in cells to the whole animal. We found that increasedexpression of the Naþ-Ca2þ exchanger directly led to a hy-pertrophic response and that chronic hemodynamic stressesled to decompensatory transition to heart failure in theHOM overexpressing mice. Furthermore, the magnitude
of the hypertrophy and pathology increased with increasingexchanger expression. Elevated expression of the exchangerhas been documented in human and other animal models ofhypertrophy and heart failure.3,16e18 Though heart failure isa complex disease with numerous pathologic factors, thesedata clearly suggest that overexpression of the Naþ-Ca2þ
exchanger can significantly contribute to the process. Basedon our previous electrophysiology data, we believe that thepathophysiology observed in the HET and HOM mice re-sult from perturbed calcium homeostasis, specifically intra-cellular calcium overload.
ANF
0.00
0.50
1.00
1.50
No
rm
alized
In
ten
sity
WTHETHOM
*
ANF
GADPH
HOM WT HET
Fig. 5. Northern blot analysis. The left panel shows a Northern blot of ANF reexpression in homogenous (HOM), wild-type (WT), andheterogenous (HET) postpartum (PP) mice (3 lanes each). The right panel plots the normalized intensities of the ANF reexpressionfrom the blot (*P ! .001 to WT and HET).
Heart Failure in NCX Mice � Roos et al 325
Fig. 6. Histologic assessment of baseline and postetransaortic constriction (TAC) mice. (A) A trichrome-stained section of postpartum (PP)homogenous (HOM) mouse myocardium exhibiting interstitial fibrosis and calcification. There is also a large region of replacement fibrosisin the right ventricle (arrow). (B) A normal-appearing section from a male HOM mouse. (C, D, E) Increasing levels of fibrosis and pathol-ogy from sections from WT, HET, and HOM, respectively, post-TAC mice. See the text for further details.
The baseline data (Fig. 1B, 2; Tables 1, 2) indicate a hy-pertrophic phenotype in both HET and HOM mice by 4months of age. Linck et al29 reported a 3-fold increase inANF expression HET mice; this value is intermediate be-tween WT and the HOM expression reported here(Fig. 1B). Interestingly, despite numerous published reportsusing HET mice from this line, only a few evaluated themorphometric phenotype and none reported a hypertrophicresponse.12,24,27 This difference in hypertrophic phenotypeis likely the result of the relatively younger age of the ani-mals in the previous studies and their normalization of heartweight to body weight rather than to tibia length as is ourpreference. Adachi-Akahane et al24 and Terracciano etal12 reported no significant differences between HET andWT cell capacitance. Unlike HET mice, previous workfrom our group found significantly increased capacitancein HOM myocytes indicative of cellular hypertrophy inagreement with our morphometric data.35 Direct LV
catheterization revealed normal levels of baseline left ven-tricular contractility (þdP/dT) and relaxation (�dP/dT) inall groups (Fig. 3). Despite these normal baseline 6 dP/dT values, the different groups of transgenic mice did notrespond uniformly to b-adrenergic stimulation. Both con-tractility and relaxation were significantly blunted inHOM mice at elevated doses of dobutamine with interme-diate HET values. This graded blunting is proportional tothe increasing levels of NCX1 expression. Blunted adrener-gic responsiveness is associated with hypertrophic and fail-ing hearts in both human and animal models44,45 and incells.46
Similarly, the baseline contractile function determinedfrom echo (%LVFS and Vcf) is in the normal range exceptfor HOM %LVFS (Table 2, baseline). Thus the baselinecontractility and relaxation are generally normal despitethe reduced EC coupling gain with normal SR Ca2þ loadsin HOM myocytes.35 The normal LV function in the whole
326 Journal of Cardiac Failure Vol. 13 No. 4 May 2007
animal, as opposed to the depressed function in isolatedcells, is not surprising because the mouse can compensatefor the reduced EC coupling gain by increasing its adrener-gic stimulation. Such chronic stimulation might also con-tribute to increased calcium overload via enhanced L-typecalcium channel function and eventually lead to heart fail-ure via calcium-triggered decompensatory pathways. Anenhanced relaxation rate might also be expected from thehigher exchange activity. However, the extra exchanger ac-tivity could simply compensate for an increased Ca2þ influxor SR Ca2þ release.
Although the Naþ-Ca2þ exchanger overexpressing micedid not exhibit heart failure under normal conditions, vari-ous forms of chronic hemodynamic stress did lead to de-compensation and heart failure. For these studies weevaluated mice after pregnancy, treadmill exercise and arte-rial pressure overload from aortic constriction. Pregnancyincreases the hemodynamic load (volume overload) onthe heart and can be associated with chamber dilation, hy-pertension, hypertrophy, and heart failure.47,48 Treadmill
WT HOM
3
5
7
9
HW
/T
L R
atio
(m
g/m
m)
Pre-Exercise
Post-Exercise
†
†
1 2 3 4 5 6 7 8Weeks of Exercise
0
1
2
3
4
5
6
Nu
mb
er R
un
nin
g
WTHOM
Less than 12 Falls/60 min
ConditioningPeriod
A
B
Fig. 7. Response to exercise. (A) The heart weight to tibia lengthration (HW/TL) from baseline and post-exercise wild-type (WT)and homogenous (HOM) mice (yP ! .05; n 5 20 and 6). (B)The number of WT and HOM mice that could successfully run60 min/day with 12 or less falls from the treadmill over the 8-week duration of the study (n 5 6 and 6).
exercise intermittently increases the hemodynamic load,which leads to a ‘‘physiologic hypertrophy’’ in normal sub-jects, but can also reveal reduced cardiac function insubjects with compromised hearts.49 Finally, aortic con-striction provides a chronic pressure overload that the heartmust overcome to sustain normal cardiac output.42 Thoughdifferent, all 3 of these hemodynamic stress modalities pre-cipitated cardiac dysfunction in the Naþ-Ca2þ exchangeroverexpressing mice.
To evaluate the propensity toward heart failure in theNCX1 overexpressors, mice were stressed by either exer-cise or pressure overload via TAC. A daily exercise pro-gram induced a moderate physiologic hypertrophy in bothWT and HOM mice, but only the WT mice sustained theirrunning ability (Fig. 7). The exercise intolerance exhibitedin 4 of the 6 HOM mice at the sixth week of training sug-gests that the additional demand from exercise in HOMmice starts a stress-induced decompensation toward heart
WT HET HOM
0
10
20
30
40Pre-TAC
TAC 21d
*†
*† ‡
†‡
WT HET HOM
0
2
4
6
Vcf (m
m/s)
Pre-TAC
TAC 21d
*†
*†
A
B
LV
F
S (%
)
Fig. 8. Ventricular dysfunction after transaortic constriction(TAC). (A, B respectively) The left ventricular fractional shorten-ing (LV%FS) and the velocity of circumferential fiber shortening(Vcf) from echo from the wild-type (WT), heterogenous (HET),and homogenous (HOM) mice at baseline and 21 days post-TAC (n 5 15e23; *P ! .05 to pre-TAC; yP ! .05 to WT; zP! .05 to HET). See Table 2 for further details.

Heart Failure in NCX Mice � Roos et al 327
failure. With exercise, increased adrenergic stimulationleads to higher heart rates and contractility. These wouldlikely trigger higher intracellular calcium levels in an al-ready calcium overloaded heart.
Unlike exercise, a substantial hypertrophic response wasevident in TAC-induced pressure overload in all groups by7 days and continued to increase until the end of the 21-daystudy (Tables 1, 2). As with exercise, WT mice were ableto sustain their function and had no postsurgical mortality.But the HET and HOM mice had a graded reduction in func-tion, increased fibrosis, and increased mortality before 21days post-TAC (Table 2; Fig. 6 CeE, Fig. 8). Interestingly,the NP (female) HOM mice appeared to be more severely ef-fected by TAC with a somewhat greater mortality than themales. The source or this gender difference in mortality is un-clear and is in contrast to increased male mortality followingischemia/reperfusion injury in HET mice.28 Older HOMmice were completely intolerant of the TAC procedure.Taken as a whole, these data suggest that the WT, HET, andHOM mice have increasing levels of dysfunction and pathol-ogy after pressure overload in proportion to their increasedexpression of NCX1. Thus the level of NCX1 expressionlikely modulates the intracellular calcium levels which, incombination with pressure overload, accelerate the hypertro-phic response sufficiently to decompensate into heart failure.
The high rate of premature death due to heart failure inPP HOM mice was unexpected (Fig. 4, 6A; Tables 1, 2).The improved survival, graded reduction in hypertrophic re-sponse (PP ANF; Fig. 5, morphometry and echo), the lack ofheart failure in the PP WTand PP HET mice supports the con-cept of a direct relationship between NCX1 activity and path-ophysiologic consequences. The hemodynamic stressassociated with pregnancy appears sufficient to induce a de-compensatory transition to heart failure accompanied by tis-sue calcification and fibrosis in at least two thirds of the PPHOM mice by 9 months of age. As with TAC, the added stressof the volume overload commonly associated with pregnancytriggers this accelerated decompensation.
Why does overexpression of NCX1 lead to a hypertrophicphenotype? Hypertrophy, with or without fibrosis and heartfailure, is a complex process resulting from a combinationof multiple factors: genetic, structural (remodeling), auto-crine and paracrine.2,4,44 Indeed, a large number of changesin gene product expression have been elucidated in severalexperimental models. It is clear that multiple biochemicalsignaling events are involved in the pathogenesis andprogression of hypertrophy and eventual failure. As hyper-trophy progresses, it is almost always accompanied by fi-brous tissue proliferation and loss of myocytes (apoptosisor necrosis), that play a role in the ultimate decompensationof the hypertrophied heart. Additionally, an alteration in cal-cium homeostasis is a major contributing factor to the cas-cade of pathways leading to hypertrophy and failure.3,5,11,22
In our NCX-overexpressing mouse model, one might ex-pect intracellular Ca2þ levels to be lower with more ex-changer available for extrusion, but this is not necessarilythe case. Rodent cardiomyocytes have elevated internal
Naþ levels compared with cardiomyocytes of rabbit andother larger mammals.34,50,51 Increased Naþ levels inmice favor increased Ca2þ influx or decreased Ca2þ effluxduring diastole via the exchanger leading to chronic cal-cium overload.12,13,52 Similar to previous studies on HETmyocytes,12,24,31e34 the HOM myocytes appear to havean enhanced reverse-mode exchange and also a reducedgain of excitation-contraction coupling,35 which maymake them more susceptible to the stresses of pregnancy,exercise, or TAC.
In summary, overexpression of the cardiac Naþ-Ca2þ ex-changer in HET and HOM transgenic mice leads to 2- to 3-fold increases in NCX1 activity without alterations in otherCa2þ handling proteins. Increased NCX1 activity provokesa dilated ventricular phenotype, induces exercise intoler-ance, and leads to premature mortality and heart failurein mice after pregnancy or pressure overload. Increasinglevels of exchanger expression lead to a graded develop-ment of hypertrophy and susceptibility to a decompensatorytransition to heart failure. The NCX overexpressing modeldeveloped and evaluated in our study should also be usefulin future studies designed to elucidate the numerous stepsin the progression from hypertrophy to heart failure.
Acknowledgments
We wish to thank Dr. Scott Henderson for his helpful dis-cussions. We also thank Jeanne Kim, Liyan Lu, and YujuanLu for their technical assistance.
References
1. Houser SR. When does spontaneous sarcoplasmic reticulum Ca2þ re-
lease cause a triggered arrhythmia? Cellular versus tissue require-
ments. Circ Res 2000;87:725e7.
2. Mann DL, Bristow MR. Mechanisms and models in heart failure: the
biomechanical model and beyond. Circulation 2005;111:2837e49.
3. Pogwizd SM, Schlotthauer K, Li L, Yuan WL, Bers DM. Arrhythmo-
genesis and contractile dysfunction in heart failuredroles of sodium-
calcium exchange, inward rectifier potassium current, and residual
beta-adrenergic responsiveness. Circ Res 2001;88:1159e67.
4. Sipido KR, Volders PGA, Vos MA, Verdonck F. Altered Na/Ca ex-
change activity in cardiac hypertrophy and heart failure: a new target
for therapy? Cardiovasc Res 2002;53:782e805.
5. Yano M, Ikeda Y, Matsuzaki M. Altered intracellular Ca2þ handling
in heart failure. J Clin Invest 2005;115:556e64.
6. Bers DM. Cardiac excitation-contraction coupling. Nature 2002;415:
198e205.
7. Hilgemann DW. New insights into the molecular and cellular work-
ings of the cardiac Naþ/Ca2þ exchanger. Am J Physiol 2004;287:
C1167e72.
8. Philipson KD, Nicoll DA. Sodium-calcium exchange: a molecular
perspective. Annu Rev Physiol 2000;62:111e33.
9. Reuter H, Pott C, Goldhaber JI, Henderson SA, Philipson KD,
Schwinger RHG. Naþ-Ca2þexchange in the regulation of cardiac
excitation-contraction coupling. Cardiovasc Res 2005;67:198e207.
10. Gaughan JP, Furukawa S, Jeevanandam V, Hefner CA, Kubo H,
Margulies KB, et al. Sodium/calcium exchange contributes to contrac-
tion and relaxation in failed human ventricular myocytes. Amer J
Physiol 1999;277:H714e24.

328 Journal of Cardiac Failure Vol. 13 No. 4 May 2007
11. Houser SR, Piacentino V, Weisser J. Abnormalities of calcium cycling
in the hypertrophied and failing heart. J Mol Cell Cardiol 2000;32:
1595e607.
12. Terracciano CMN, DeSouza AI, Philipson KD, MacLeod KT. Naþ-
Ca2þ exchange and sarcoplasmic reticular Ca2þ regulation in ven-
tricular myocytes overexpressing the Naþ-Ca2þ exchanger. J Physiol
London 1998;512:651e67.
13. Weisser-Thomas J, Piacentino V, Gaughan JP, Margulies K,
Houser SR. Calcium entry via Na/Ca exchange during the action po-
tential directly contributes to contraction of failing human ventricular
myocytes. Cardiovasc Res 2003;57:974e85.
14. Hampton TG, Wang JF, DeAngelis J, Amende I, Philipson KD,
Morgan JP. Enhanced gene expression of Naþ/Ca2þ exchanger atten-
uates ischemic and hypoxic contractile dysfunction. Amer J Physiol
2000;279:H2846e54.
15. Hobai IA, ORourke B. Enhanced Ca2þ-activated Naþ-Ca2þexchange activity in canine pacing-induced heart failure. Circ Res
2000;87:690e8.
16. Kent RL, Rozich JD, McCollam PL, McDermott DE, Thacker UF,
Menick DR, et al. Rapid expression of the Naþ-Ca2þ exchanger in
response to cardiac pressure overload - rapid communication. Am J
Physiol 1993;265:H1024e9.
17. Schillinger W, Fiolet JW, Schlotthauer K, Hasenfuss G. Relevance of
Naþ-Ca2þ exchange in heart failure. Cardiovasc Res 2003;57:
921e33.
18. Studer R, Reinecke H, Bilger J, Eschenhagen T, Bohm M,
Hasenfuss G, et al. Gene expression of the cardiac Naþ-Ca2þ ex-
changer in end-stage human heart failure. Circ Res 1994;75:443e53.
19. Wang ZY, Nolan B, Kutschke W, Hill JA. Naþ-Ca2þ exchanger re-
modeling in pressure overload cardiac hypertrophy. J Biol Chem
2001;276:17706e11.
20. Zhang XQ, Song JL, Rothblum LI, Lun MY, Wang XJ, Ding F, et al.
Overexpression of Naþ/Ca2þ exchanger alters contractility and SR
Ca2þ content in adult rat myocytes. Amer J Physiol 2001;281:
H2079e88.
21. Gomez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF,
Cannell MB, et al. Defective excitation-contraction coupling in exper-
imental cardiac hypertrophy and heart failure. Science 1997;276:
800e6.
22. Schultz JEJ, Glascock BJ, Witt SA, Nieman ML, Nattamai KJ,
Liu LH, et al. Accelerated onset of heart failure in mice during pres-
sure overload with chronically decreased SERCA2 calcium pump
activity. Am J Physiol 2004;286:H1146e53.
23. Weisser-Thomas J, Kubo H, Hefner CA, Gaughan JP, McGowan BS,
Ross R, et al. The Naþ/Ca2þ exchanger/SR Ca2þ ATPase transport
capacity regulates the contractility of normal and hypertrophied feline
ventricular myocytes. J Card Failure 2005;11:380e7.
24. Adachi-Akahane S, Lu LY, Li ZP, Frank JS, Philipson KD, Morad M.
Calcium signaling in transgenic mice overexpressing cardiac Naþ-
Ca2þ exchanger. J Gen Physiol 1997;109:717e29.
25. Goldhaber JI, Henderson SA, Reuter H, Pott C, Philipson KD. Effects
of Naþ-Ca2þ exchange expression on excitation-contraction cou-
pling in genetically modified mice. Annals N Y Acad Sci 2005;
1047:122e6.
26. Pott C, Goldhaber JI, Philipson KD. Genetic manipulation of cardiac
Naþ/Ca2þ exchange expression. Biochem Biophys Res Commun
2004;322:1336e40.
27. Baumer AT, Flesch M, Kilter H, Philipson KD, Bohm M. Overexpres-
sion of the Naþ-Ca2þ exchanger leads to enhanced inotropic respon-
siveness to Naþ-channel agonist without sarcoplasmic reticulum
protein changes in transgenic mice. Biochem Biophys Res Commun
1998;249:786e90.
28. Cross HR, Lu LY, Steenbergen C, Philipson KD, Murphy E. Overex-
pression of the cardiac Naþ/Ca2þ exchanger increases susceptibility
to ischemia/reperfusion injury in male, but not female, transgenic
mice. Circ Res 1998;83:1215e23.
29. Linck B, Boknik P, Huke S, Kirchhefer U, Knapp J, Luss H, et al.
Functional properties of transgenic mouse hearts overexpressing
both calsequestrin and the Naþ-Ca2þ exchanger. J Pharmacol Exp
Ther 2000;294:648e57.
30. Stagg MA, Malik AH, MacLeod KT, Terracciano CMN. The effects
of overexpression of the Naþ/Ca2þ exchanger on calcium regulation
in hypertrophied mouse cardiac myocytes. Cell Calcium 2004;36:
111e8.
31. Su Z, Bridge JHB, Philipson KD, Spitzer KW, Barry WH. Quantita-
tion of Na Ca exchanger function in single ventricular myocytes.
J Mol Cell Cardiol 1999;31:1125e35.
32. Sugishita K, Su Z, Li FH, Philipson KD, Barry WH. Gender influences
[Ca2þ](I) during metabolic inhibition in myocytes overexpressing the
Naþ-Ca2þ exchanger. Circulation 2001;104:2101e6.
33. Terracciano CMN, Philipson KD, MacLeod KT. Overexpression of the
Naþ/Ca2þ exchanger and inhibition of the sarcoplasmic reticulum
Ca2þ-ATPase in ventricular myocytes from transgenic mice. Cardio-
vasc Res 2001;49:38e47.
34. Yao AS, Su Z, Nonaka A, Zubair I, Lu LY, Philipson KD, et al. Effects
of overexpression of the Naþ-Ca2þ exchanger on [Ca2þ]i transients
in murine ventricular myocytes. Circ Res 1998;82:657e65.
35. Reuter H, Han T, Motter C, Philipson KD, Goldhaber JI. Mice
overexpressing the cardiac sodium-calcium exchanger: Defects in
excitation-contraction coupling. J Physiol London 2004;544:779e89.
36. Ritter MR, Dorrell MI, Edmonds J, Friedlander SF, Friedlander M.
Insulin-like growth factor 2 and potential regulators of hemangioma
growth and involution identified by large-scale expression analysis.
Proc Nat Acad Sci U S A 2002;99:7455e60.
37. Shai SY, Harpf AE, Babbitt CJ, Jordan MC, Fishbein MC, Chen J,
et al. Cardiac myocyte-specific excision of the beta 1 integrin gene
results in myocardial fibrosis and cardiac failure. Circ Res 2002;90:
458e64.
38. Tanaka N, Dalton N, Mao L, Rockman HA, Peterson KL,
Gottshall KR, et al. Transthoracic echocardiography in models of
cardiac disease in the mouse. Circulation 1996;94:1109e17.
39. Henderson SA, Goldhaber JI, So JM, Han T, Motter C, Ngo A, et al.
Functional adult myocardium in the absence of Naþ-Ca2þ exchange.
Cardiac-specific knockout of NCX1. Circ Res 2004;95:604e11.
40. Huynh L, Scremin OU, Jordan MC, Roos KP. Rota-rod treadmill ex-
ercise enhances murine ventricular mass:[abstract]. FASEB J 2001;15:
A793.
41. Keller RS, Shai SY, Babbitt CJ, Pham CG, Solaro RJ, Valencik ML,
et al. Disruption of integrin function in the murine myocardium leads
to perinatal lethality, fibrosis, and abnormal cardiac performance. Am
J Pathol 2001;158:1079e90.
42. Rockman HA, Ross RS, Harris AN, Knowlton KU, Steinhelper ME,
Field LJ, et al. Segregation of atrial-specific and inducible expres-
sion of an atrial natriutetic factor transgene and in vivo murine
model of cardiac hypertrophy. Proc Natl Acad Sci U S A 1991;88:
8277e81.
43. Lim CC, Liao RL, Varma N, Apstein CS. Impaired lusitropy-fre-
quency in the aging mouse: role of Ca2þ-handling proteins and effects
of isoproterenol. Amer J Physiol 1999;277:H2083e90.
44. Brodde OE, Michel MC, Zerkowski HR. Signal transduction mecha-
nisms controlling cardiac contractility and their alterations in chronic
heart failure. Cardiovasc Res 1995;30:570e84.
45. Feldman AM, Tena RG, Kessler PD, Weisman HF, Schulman SP,
Blumenthal RS, et al. Diminished b-andrenergic receptor responsive-
ness and cardiac dilation in hearts of myopathic Syrian hamsters (BIO
53.58) are associated with a functional abnormality of the G stimula-
tory protein. Circ Res 1990;81:1341e52.
46. Sato M, Gong H, Terracciano CMN, Ranu H, Harding SE. Loss of
b-adrenoceptor response in myocytes overexpressing the Naþ/Ca2þ-
exchanger. J Molec Cell Cardiol 2004;36:43e8.
47. Eghbali M, Deva R, Alioua A, Minosyan TY, Ruan H, Wang Y, et al.
Molecular and functional signature of heart hypertrophy during preg-
nancy. Circ Res 2005;96:1208e16.
48. Wong AYH, Kulandavelu S, Whiteley KJ, Qu D, Langille BL,
Adamson SL. Maternal cardiovascular changes during pregnancy
and postpartum in mice. Am J Physiol 2002;282:H918e25.

Heart Failure in NCX Mice � Roos et al 329
49. Haubold KW, Allen DL, Capetanaki Y, Leinwand LA. Loss of desmin
leads to impaired voluntary wheel running and treadmill exercise
performance. J Appl Physiol 2003;95:1617e22.
50. Despa S, Islam MA, Pogwizd SM, Bers DM. Intracellular [Naþ] and
Naþ pump rate in rat and rabbit ventricular myocytes. J Physiol
London 2002;539:133e43.
51. Pogwizd SM, Sipido KR, Verdonck F, Bers DM. Intracellular Na in
animal models of hypertrophy and heart failure: contractile function
and arrhythmogenesis. Cardiovasc Res 2003;57:887e96.
52. Baartscheer A, Schumacher CA, Belterman CNW, Coronel R,
Fiolet JWT. [Naþ](I) and the driving force of the Naþ/Ca2þ-
exchanger in heart failure. Cardiovasc Res 2003;57:986e95.




















