
treatmentof benign prostatic hypertrophy in homoeopathy with ...
Upload
universityofvicCategory
view
1download
0
Left Ventricular Hypertrophy in Rats With BiliaryCirrhosis
Javier Inserte,1 Antonia Perello,2 Luis Agullo,1 Marisol Ruiz-Meana,1 Klaus-Dieter Schluter,3 Noelia Escalona,1
Mariona Graupera,2 Jaume Bosch,2 and David Garcia-Dorado1
Portal hypertension induces neuroendocrine activation and a hyperkinetic circulation state. Thisstudy investigated the consequences of portal hypertension on heart structure and function.Intrahepatic portal hypertension was induced in male Sprague-Dawley rats by chronic bile ductligation (CBDL). Six weeks later, CBDL rats showed higher plasma angiotensin-II and endothe-lin-1 (P < .01), 56% reduction in peripheral resistance and 73% reduction in pulmonaryresistance (P < .01), 87% increase in cardiac index and 30% increase in heart weight (P < .01),and increased myocardial nitric oxide (NO) synthesis. In CBDL rats, macroscopic analysisdemonstrated a 30% (P < .01) increase in cross-sectional area of the left ventricular (LV) wallwithout changes in the LV cavity or in the right ventricle (RV). Histomorphometric analysisrevealed increased cell width (12%, P < .01) of cardiomyocytes from the LV of CBDL rats, butno differences in myocardial collagen content. Myocytes isolated from the LV were wider (12%)and longer (8%) than right ventricular myocytes (P < .01) in CBDL rats but not in controls.CBDL rats showed an increased expression of ANF and CK-B genes (P < .01). Isolated perfusedCBDL hearts showed pressure/end-diastolic pressure curves and response to isoproterenol iden-tical to sham hearts, although generated wall tension was reduced because of the increased wallthickness. Coronary resistance was markedly reduced. This reduction was abolished by inhibi-tion of NO synthesis with N-nitro-L-arginine. Expression of eNOS was increased in CBDL hearts. Inconclusion, portal hypertension associated to biliary cirrhosis induces marked LV hypertrophy andincreased myocardial NO synthesis without detectable fibrosis or functional impairment. Thisobservation could be relevant to patients with cirrhosis. (HEPATOLOGY 2003;38:589-598.)
Hyperdynamic circulation is a common feature inhuman and experimental portal hypertension,with or without cirrhosis. This is also called hy-
perdynamic circulatory syndrome and is the consequence
of splanchnic and systemic vasodilatation, manifested byincreased heart rate, cardiac output, and regional bloodflow with decreased mean arterial pressure and systemicvascular resistance (SVR).1
The mechanism of systemic and splanchnic vasodila-tation is multifactorial. Many studies have suggested aprominent role for increased biosynthesis of nitric oxide(NO),2 which is due to increased endothelial nitric oxidesynthase (eNOS) activity.3,4 It has been shown that NOinhibition attenuates the hyperdynamic circulation ob-served in portal hypertensive rats.5,6 Other studies haveshown an increased splanchnic production of carbonmonoxide in portal hypertension7 as well as an increasedrelease of splanchnic vasodilatory peptides, such as gluca-gon.8,9 Vasoconstrictor systems are also up-regulated,probably as a counter-regulatory response to the hyperdy-namic state.10 Sympathetic nervous and renin-angioten-sin aldosterone systems are enhanced, and the plasmalevels of vasopressin and endothelin-1 are elevated.11,12
Both hemodynamic overload and neuroendocrineactivation occurring during portal hypertension are po-tentially able to induce cardiac hypertrophy and dysfunc-tion.13,14 However, although an echocardiographic studydescribed increased left ventricular (LV) wall thickness inpatients with advanced cirrhosis,15 there is no direct evi-
Abbreviations: SVR, systemic vascular resistance; NO, nitric oxide; eNOS, endothe-lial nitric oxide synthase; LV, left ventricle; CBDL, common bile duct ligated rat; MAP,mean arterial pressure; CI, cardiac index; PAP, pulmonary artery pressure; PVR, pul-monary vascular resistance; CWI, cardiac weight index; RV, right ventricle; LVEDP,left ventricular end-diastolic pressure; LVdevP, left ventricular developed pressure; L-NNA, N-nitro-L-arginine; SNAP, S-nitroso-N-acetyl-D,L-penicillamine; cGMP, cy-clic guanosine monophosphate; ANF, atrial natriuretic factor; CK-M, creatin kinaseisoform M; CK-B, creatin kinase isoform B; TGF-�1, tumor growth factor �1; iNOS,inducible nitric oxide synthase; nNOS, neuronal nitric oxide synthase.
From the 1Servicio de Cardiologıa, Hospital Universitari Vall d’Hebron, Bar-celona; 2Hepatic Hemodynamic Laboratory, Liver Unit, Institut de Malalties Di-gestives, Hospital Clınic, Institut d’Investigacions Biomediques August Pi i Sunyer,University of Barcelona, Barcelona, Spain; and 3Physiologisches Institut, Justus-Liebig Universitat, Giessen, Germany.
Received September 6, 2002; accepted June 4, 2003.Supported in part by CICYT grant (SAF 99/102) from Ministerio de Ciencia y Tecno-
logia, Spain, and by grants from Instituto de Salud Carlos III (PI02739 and C03/02).J.I. and A.P. contributed equally to this paper.Address reprint requests to: David Garcia-Dorado, M.D., Servicio de Cardiologıa, Hos-
pital Universitari Vall d’Hebron, Paseo Vall d’Hebron 119-129, 08035 Barcelona, Spain.E-mail: [email protected]; fax: (34) 93-489 4032. Jaume Bosch, M.D., Labo-ratori d’Hemodinamica Hepatica, Escala 5-7 3 planta, Hospital Clinico Barcelona,Villarroel 170, 08036 Barcelona, Spain. E-mail: [email protected].
Copyright © 2003 by the American Association for the Study of Liver Diseases.0270-9139/03/3803-0009$30.00/0doi:10.1053/jhep.2003.50369
589

dence of the induction of myocardial hypertrophy by por-tal hypertension. In patients with portal hypertensionand/or cirrhosis, LV function has been documented to benormal or hyperdynamic at baseline,16,17 whereas numer-ous studies demonstrate an altered function under condi-tions of physiologic18,19 or pharmacologic stress inducedby exercise or �-adrenergic agonist infusion.20,21
The aim of this study was to investigate the conse-quences of portal hypertension on the structure and func-tion of the heart. For this purpose, we induced portalhypertension in rats by common bile duct ligation, whichresults in a well-characterized, secondary biliary cirrhosiswith intrahepatic portal hypertension.
Materials and MethodsAnimal Models
This study was performed on adult male Sprague-Dawley rats weighing between 175 and 200 g. All exper-iments were performed according to the criteria outlinedin the “Guide for the Care and Use of Laboratory Ani-mals” prepared by the National Academy of Sciences andpublished by the National Institutes of Health (NIH pub-lication 86-23 revised 1985), after approval of the proto-col by the Ethical Committee and fulfilling all localregulations for research involving experimental animals.
The bile duct-ligated cirrhotic rat model (CBDL) haspreviously been described in detail.22 Briefly, under ket-amine anesthesia (100 mg/kg intramuscularly), cirrhosisand portal hypertension were induced by the doubleligature and section of the common bile duct. Benza-thine benzylpenicillin was administered postoperatively(50,000 U, intramuscularly) for prophylaxis of infection.Vitamin K (8 mg/kg, intramuscularly) was given aftersurgery at weekly intervals. The animals were studied 6weeks after surgery. In the control group of sham-oper-ated animals of the same age, the common bile duct wereisolated but not ligated.
Hemodynamic MeasurementsThese were done as previously described.23 The rats
were anesthetized by the intraperitoneal injection of thio-butabarbital sodium (150 mg/kg, Inactin; Research Bio-chemical International, Natick, MA). The left femoralartery was catheterized for mean arterial pressure (MAP)and heart-rate measurements. Right atrium was catheter-ized, and a thermistor (Columbus Instruments, Colum-bus, Ohio) was positioned in the aortic arch. Cardiacoutput was determined by thermodilution (Cardiotherm500-AC-R; Columbus Instruments). Cardiac index (CI)was calculated as cardiac output (mL/min) per 100 g ofbody weight. SVR (mm Hg/mL/min/100 g) was calcu-
lated as electronically averaged mean arterial pressure(MAP/CI).
Pulmonary artery pressure (PAP) was measured inCBDL rats by a catheter introduced through the rightjugular vein and advanced under continuous intravascularpressure monitoring into the right ventricle until its tipentered into the pulmonary artery. Pulmonary vascularresistance (PVR) (mm Hg/mL/min/100 g) was calculatedas PAP/CI. After completion of the systemic hemody-namic measurements, pulmonary pressure was measuredthrough a fluid-filled 20-gauge needle inserted into thesplecnic parenchyma.
Heart WeightAfter obtaining the hemodynamic data, the animals
were killed by intravenous injection of saturated potas-sium chloride solution. Hearts were immediately re-moved, dried with filter paper, and weighed on a precisionbalance. Cardiac weight index (CWI) was calculated asthe ratio of heart to body weight.
Morphometric Analysis of Rat HeartsMorphometric analyses were performed in the CBDL
model. Animals were anesthetized with thiopental so-dium (150 mg/kg), and the heart was removed and per-fused retrogradely through the aorta with cardioplegicsolution (Abboplegisol; Abbott Laboratories, Chicago,IL) and then fixed with 4% paraformaldehyde. A 6- to10-mm coronal slice of the ventricle was taken at theequator of the heart and embedded in paraffin, and 5-�msections were obtained and stained with Masson’strichrome (MicroStain). Digital images (�5) were ob-tained (Olympus Digital Camera C-1400L; OlympusOptical Co., Tokyo, Japan), and total ventricle area, cav-ity area, and average thickness of the ventricle wall werecalculated by means of commercially available software(Micro Image; Olympus Optical Co).
Histomorphometric AnalysisDigital microphotographs of adjacent optical fields
(Olympus DP10 camera; Olympus Optical Co.) wereobtained from the LV for subsequent analysis (Micro Im-age; Olympus Optical Co.). Average cardiomyocytewideness and nuclear length were determined in eachheart from 30 to 60 measurements made at a magnifica-tion of �400 in longitudinally oriented myocytes. Aver-age number of nuclei per area was obtained counting thetotal number of myocyte nuclear profile in a measuredarea (14 to 25 fields in each heart) in which cardiac musclefibers were sectioned transversely at a magnification of�600. Average area occupied by collagen was measuredin Masson’s trichrome-stained sections from the left ven-
590 INSERTE ET AL. HEPATOLOGY, September 2003

tricle at a magnification of �300. Interstitial collagen wascalculated as the percentage of collagen surface area overmyocardial surface area. Perivascular collagen was mea-sured separately around every coronary artery visible inthe analyzed field and was calculated as the ratio betweenthe area occupied by collagen in contact with microvesselsand the perimeter of coronary lumen.
Morphometric Analysis of Isolated CardiomyocytesCardiomyocytes from the LV and right ventricle (RV)
corresponding to CBDL and sham rat hearts were isolatedseparately as previously described.24 Rod-shaped cellswere selected by gravity sedimentation in a 4% bovineserum albumin gradient (fraction V, Boehringer Mann-heim) and plated with medium 199/HEPES (Sigma) and4% fetal calf serum (Gibco) for 2 hours. After plating,cells were fixed in 2% paraformaldehyde buffer (inmmol/L, NaF 100, KCl 50, MgCl2 2, EGTA 1, KH2PO4
10, DTT 0.2, sucrose 1,000, pH 6.8). Ten digital micro-photographs at a magnification of �40 per plate wereobtained. Long-axis section, length, and width of eachmyocyte was automatically calculated. A minimum of 30myocytes per heart was examined.
Functional Studies
Isolated Heart Model. Six weeks after surgery,CBDL and sham rats were anesthetized with thiopentalsodium (150 mg/kg, intraperitoneal). As previously de-scribed,24 hearts were mounted on a Langendorff appara-tus and perfused with Krebs-Henseleit bicarbonate bufferat a constant pressure of 60 mm Hg. LV pressure wasmonitored by means of a water-filled latex balloon placedin the LV. The variables measured included heart rate, LVend-diastolic pressure (LVEDP), and LV-developed pres-sure (LVdevP), calculated as the difference between LVpeak systolic pressure and LVEDP. Coronary flow wasmeasured by collecting coronary effluent at the end ofequilibration using a calibrated tube and expressed in mL/min.
Functional Study Protocol. Isolated hearts fromsham and CBDL rats were subjected to 2 different proto-cols. In the first protocol (n � 6 per group), after 15minutes of equilibration, LVEDP was steeply increasedby inflating the intraventricular balloon. After achievingthe maximal LVdevP, the balloon was deinflated to a finalvalue of LVEDP between 8 and 10 mm Hg. The secondprotocol assessed the response of hearts (n � 5 per group)to stimulation with isoproterenol (10�10 to 3 � 10�6
mol/L). Before stimulation, the area of sinus node wasdamaged by clamping to achieve atrioventricular block,and the right ventricle was paced at 5 Hz to avoid chro-notropic interference of isoproterenol.
Coronary ResistanceCoronary resistance (perfusion pressure/coronary
flow) in CBDL hearts was studied in additional experi-ments. Perfused-isolated hearts from sham or CBDL ratswere allocated to receive, after 30 minutes of equilibra-tion, no drug (n � 8), 200 �mol/L N-nitro-L-arginine(L-NNA; n � 8), or 100 �mol/L S-nitroso-N-acetyl-D,L-penicillamine (SNAP; n � 8) for 15 minutes. At thispoint, half of the hearts of each group were immediatelyfrozen in liquid nitrogen for cyclic guanosine monophos-phate (cGMP) quantification. The rest of the hearts wereallowed to perfuse without drugs for another 30 minutes.
Determination of Endothelin-1 and Angiotensin IIConcentrations
Plasma concentration of immunoreactive endothe-lin-1 was measured in blood samples of sham and CBDLrats (n � 7 per group) by radioimmunoassay (NicholsInstitute Diagnostics B.V., Wijchen, The Netherlands)after endothelin-1 extraction on Sep-Pak C18 cartridges(Waters Associates, Milford, MA).25 Angiotensin II wasextracted from plasma with cold ethanol and measured byradioimmunoassay (Nichols Institute Diagnostics B.V.)as previously described.26
RNA AnalysisThe expression of atrial natriuretic factor (ANF),
creatin kinase (CK), and tumor growth factor �1
(TGF-�1) was measured by reverse-transcriptase poly-merase chain reaction methods as described.27 Oligo-nucleotide primers were synthesized by Gibco-BRLand had the following sequences: �-actin sense:5�-GAAGTGTGACGTTGACATCCG-3� and anti-sense: 5�-AGTAGATGTTTGGCCTCTCTGG-3�; ANFsense: 5�-ATGGGCTCCTTCTCTCCATCAC-3� andantisense: 5�-TCTTCGGTACCGGAAGCT-3�; CK-Bsense: 5�-GCCGCCATGCCCTTCCTC-3� and antisense:5�-CTTAGACTTGAAGAGAGTTTC-3�; CK-M sense:5�-CCACAACAAGTTCAAGCTGAA-3� and antisense:AAGATCTCCTCAATCTTCTGC3�; and TGF-�1
sense: 5�-AATACGTCAGACATTCGGGAA-3� and anti-sense: 5�-GTGGAGTACATTATCTTTGCT-3�. Afteramplification, reaction products were separated on 5% poly-acrylamide gels, stained with ethidium bromide, and photo-graphed under UV illumination. Density of the DNAfragments was determined by Image Quant (Molecular Dy-namics, Krefeld, Germany). The results were normalized forequal loading by �-actin amplification.
cGMP ContentcGMP content was measured in the whole heart, in
isolated myocytes, and endothelial cells. Endothelial cells
HEPATOLOGY, Vol. 38, No. 3, 2003 INSERTE ET AL. 591
were obtained as previously described in detail.28 To an-alyze cGMP in the whole hearts, hearts were pulverizedunder liquid nitrogen and homogenized in cold trichlo-roacetic acid at 7.5% (wt/vol). After centrifugation(14,000g, 15 minutes, 4°C), the supernatant was col-lected and washed with water-saturated diethylether. Toanalyze cGMP synthesis in isolated cells, cGMP degrada-tion was inhibited by adding 3-isobutyl-1-methylxan-thine at 1 mmol/L. cGMP content in myocytes andendothelial cells was assessed in ethanol extracts from thecorresponding cell cultures, dried, and resuspended inacetate buffer (5 mmol/L, pH 4.8). cGMP was measuredin the extracts by radioimmunoassay using acetylated[3H]-cGMP as previously described,28 and results wereexpressed as femtomoles of cGMP per milligram of pro-tein.
NOS Protein ContentProtein content of eNOS, inducible nitric oxide syn-
thase (iNOS), and neuronal nitric oxide synthase (nNOS)was determined in ventricular tissue from sham andCBDL hearts frozen with liquid nitrogen and pulverizedand homogenized in lysis buffer (20 mmol/L Tris-HCl,pH 7.4, 140 mmol/L NaCl, 10 mmol/L EDTA, 10%glycerol, 1% Nonidet P-40, and 1% protease inhibitorcocktail [Sigma]). The homogenate was centrifuged at15,000g for 15 minutes and supernatant diluted with anequal volume of Laemmli sample buffer (Sigma). Proteins(20 �g sample/line) and the corresponding positive con-trol were separated by electrophoresis on 7.5% sodiumdodecyl sulfate gel and transferred onto nitrocellulosemembrane (Hybond ECL, Amersham) by wet electro-blotting. At this point, membranes were stained withPonceau solution (Sigma) to test gel loading and transfer.After blocking, membranes were incubated with mouseantibodies to iNOS, eNOS, and nNOS (1:2,500,Trans-duction Laboratories, Lexington, KY). An anti-mouseIgG peroxidase conjugate (Sigma), at dilution 1:10,000,was used as secondary antibody. Protein bands were de-tected using an enhanced chemiluminescence detection
kit (SuperSignal West Dura Extended Duration Sub-strate, Pierce).
Data AnalysisAll morphometric data were collected blindly, and the
treatment code was broken at the end of the experiment.Statistics were done with the SPSS package of statisticalprograms (SPSS Inc., Chicago, IL). Results are expressedas mean � SEM. Student’s t test was used for the com-parison of quantitative variables. Pearson correlation co-efficient was used to test the relation between quantitativevariables. Statistical significance was established at P �.05.
ResultsHemodynamics and Heart Weight
Hemodynamics and heart weight data are summarizedin Table 1. CBDL rats developed portal hypertension andhyperdynamic circulation, as shown by higher pulmonarypressure and CI and lower SVR than their respective shamcontrols. PAP and PVR in CBDL rats were lower than insham animals (1.57 � 0.55 mm Hg vs. 3.80 � 0.95 mmHg, P � .048, and 2.29 � 0.91 mm Hg/mL/min/g vs.8.48 � 1.64 mm Hg/mL/min/g, P � .016, respectively).
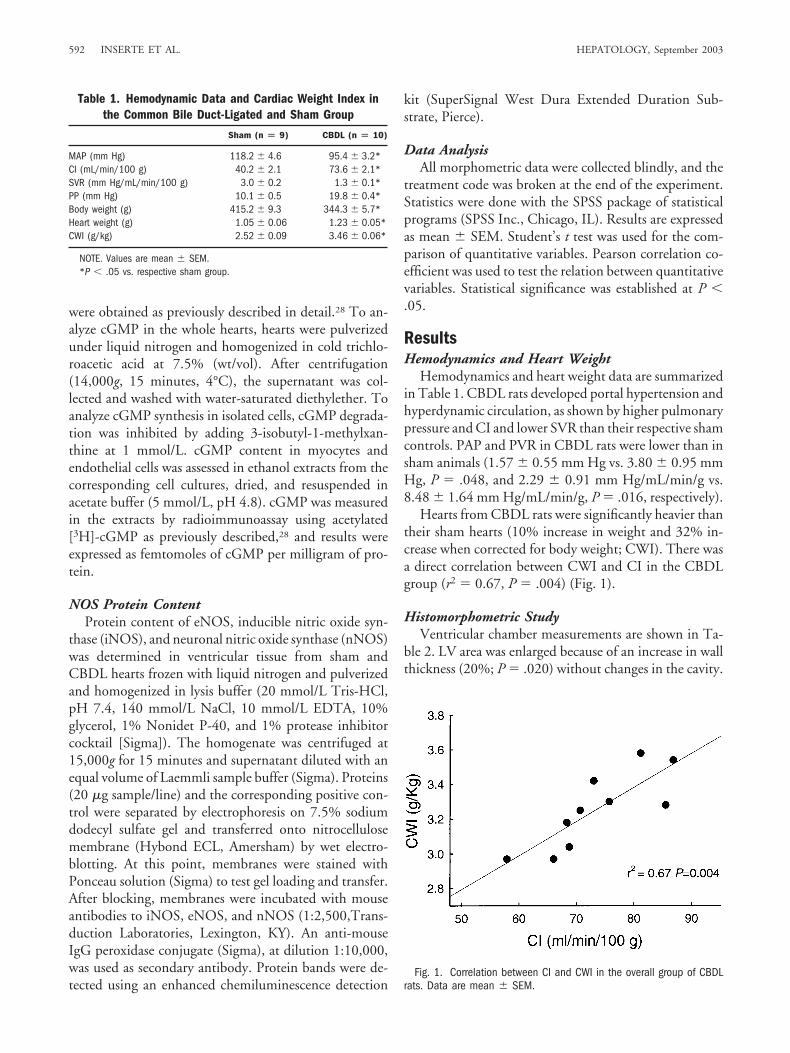
Hearts from CBDL rats were significantly heavier thantheir sham hearts (10% increase in weight and 32% in-crease when corrected for body weight; CWI). There wasa direct correlation between CWI and CI in the CBDLgroup (r2 � 0.67, P � .004) (Fig. 1).
Histomorphometric StudyVentricular chamber measurements are shown in Ta-
ble 2. LV area was enlarged because of an increase in wallthickness (20%; P � .020) without changes in the cavity.
Fig. 1. Correlation between CI and CWI in the overall group of CBDLrats. Data are mean � SEM.
Table 1. Hemodynamic Data and Cardiac Weight Index inthe Common Bile Duct-Ligated and Sham Group
Sham (n � 9) CBDL (n � 10)
MAP (mm Hg) 118.2 � 4.6 95.4 � 3.2*CI (mL/min/100 g) 40.2 � 2.1 73.6 � 2.1*SVR (mm Hg/mL/min/100 g) 3.0 � 0.2 1.3 � 0.1*PP (mm Hg) 10.1 � 0.5 19.8 � 0.4*Body weight (g) 415.2 � 9.3 344.3 � 5.7*Heart weight (g) 1.05 � 0.06 1.23 � 0.05*CWI (g/kg) 2.52 � 0.09 3.46 � 0.06*
NOTE. Values are mean � SEM.*P � .05 vs. respective sham group.
592 INSERTE ET AL. HEPATOLOGY, September 2003

In contrast, no significant differences were observed in theRV between CBDL and sham rats. Measurements per-formed in the LV (Table 2) disclosed an increase of 11%in average cardiomyocyte width (P � .002) and a less thanaverage number of myocyte nuclei per area unit, withoutdifferences in average nuclear length.
The percentage of area occupied by interstitial collagenand the ratio of perivascular collagen measured in the LVwere similar in both groups (3.12% � 0.34% and 3.05%� 0.22% of interstitial collagen in CBDL and shamgroups, respectively, and a ratio of perivascular collagenarea/perimeter of vascular lumen of 13.6 � 1.9 and12.5 � 2.4 for CBDL and sham group, respectively).
LV-isolated myocytes from CBDL hearts showed a sig-nificant increase in cell length (8%), cell width (12%),and long-axis section (21%) with respect to myocytes iso-lated from the RV (Table 3). No differences between LVand RV were observed in sham rats.
Functional Studies in Perfused Rat HeartsLV-developed pressure increased in the sham group in
response to the progressive rise in LVEDP and reached aplateau with an LVEDP of 16.4 � 2.1 mm Hg. No dif-ferences in LVdevP between sham and CBDL hearts wereobserved (Fig. 2A). In the second protocol, isolated heartswere paced at 5 Hz. Baseline LVdevP was similar in bothgroups (87.7 � 4.0 mm Hg in sham group vs. 84.5 � 8.4mm Hg in CBDL group). Stimulation with isoproterenolinduced an inotropic dose-response effect without differ-
ences between groups regarding the maximum responsecalculated as percentage with respect to basal values or inthe concentration inducing half-maximum response(EC50). Sham and CBDL maximum responses were248.9% � 13.7% and 250.8% � 15.6% and EC50
10.9 � 4.1 nmol/L and 13.1 � 3.2 nmol/L, respectively(Fig. 2B).
Coronary flow per gram of heart weight was signifi-cantly higher in the CBDL group (15.6 � 1.4 mL/g vs.11.6 � 0.8 mL/g in the sham group, P � .021). Basalcoronary resistance in CBDL-isolated hearts was reducedby 32% with respect to sham hearts. Addition of 200�mol/L L-NNA to the perfusion buffer was followed by aclear increase in resistance in CBDL hearts that was lesspronounced in the sham hearts (40.3% and 24.7%, re-spectively, after 15 minutes of perfusion with L-NNA,P � .01). After addition of L-NNA, the significant dif-ferences in coronary resistance between both groups wereno longer observed. Coronary resistance of CBDL heartswas not altered with perfusion with 100 �mol/L SNAP,whereas it was reduced in sham hearts (by 21.3% after 15minutes of perfusion with SNAP) (Fig. 3).
Contractility was not altered with the addition of L-NNA. However, perfusion with SNAP was associated to asignificant reduction of LVdevP compared with basal val-ues without differences between groups (from 116.2 �11.3 mm Hg to 75.4 � 15.2 mm Hg in sham hearts andfrom 105.0 � 5.2 mm Hg to 79.7 � 8.6 mm Hg inCBDL hearts).
Table 2. Histomorphometric Data From Common Bile Duct-Ligated and Sham-Operated Heart Sections
LV RV
Sham CBDL P Value Sham CBDL P Value
Macroscopic analysisMid-ventricular wall thickness (mm) 2.0 � 0.1 2.4 � 0.1 .020 0.9 � 0.1 0.9 � 0.1 nsMid-ventricular cavity area (mm2) 18.3 � 2.8 18.8 � 2.2 ns 10.5 � 1.2 12.8 � 2.3 ns
Microscopic analysisCardiomyocyte thickness (�m) 10.6 � 0.3 11.8 � 0.2 .002 10.4 � 0.2 10.6 � 0.3 nsNuclear length (�m) 16.2 � 0.5 16.2 � 0.4 ns 16.0 � 0.3 15.9 � 0.4 nsNumber of nuclei per area (nn/mm2) 1,190 � 82 964 � 67 .038 1,251 � 74 1,098 � 90 ns
NOTE. Values are mean � SEM. P value vs. respective sham group.
Table 3. Dimensions of Cardiomyocytes Isolated From the Right and Left Ventricles of Hearts From CommonBile Duct-Ligated and Sham-Operated Rats
RV LV
Sham CBDL P Value Sham CBDL P Value
Length (�m) 112.6 � 2.1 111.3 � 1.5 ns 113.0 � 2.1 119.8 � 1.5 �.001Width (�m) 20.5 � 0.5 18.8 � 0.5 ns 18.8 � 0.5 21.1 � 0.5 �.001Area (�m2) 2,272 � 100 2,069 � 100 ns 2,201 � 150 2,501 � 100 �.001
NOTE. Values are mean � SEM. P value with respect to sham-operated group.
HEPATOLOGY, Vol. 38, No. 3, 2003 INSERTE ET AL. 593

Myocardial cGMP ContentWhole heart myocardial cGMP concentration was in-
creased in CBDL hearts under basal conditions by 90%with respect to sham hearts. The perfusion with 200�mol/L L-NNA reduced cGMP in both groups (37% inCBDL hearts and 43% in sham hearts with respect totheir respective basal values), whereas the perfusion with100 �mol/L SNAP induced a large increase in cGMPconcentration and abolished the differences betweengroups (Fig. 4).
No significant differences in cGMP content betweensham and CBDL isolated myocytes were observed(187.8 � 31.1 fmols/mg protein vs. 120.3 � 28.7fmols/mg protein, respectively), whereas CBDL micro-vascular endothelial cells showed a near significant trendtoward a higher cGMP concentration with respect to
sham endothelial cells (136.9 � 21.5 fmols/mg proteinvs. 556.1 � 240.6 fmols/mg protein, respectively, P �.078).
Angiotensin II and Endothelin-1 DeterminationsThe plasma content in angiotensin II and endothelin-1
was markedly increased in the CBDL group with respectto its sham group (52.9 � 3.2 pg/mL vs. 20.9 � 7.4pg/mL, respectively, P � .001, for angiotensin II and22.9 � 5.7 pg/mL vs. 4.6 � 0.4 pg/mL, respectively, P �.006, for endothelin-1).
RNA AnalysisIn CBDL rats, the myocardial expression of CK-B was
increased 186.0% � 28.1% with respect to sham hearts
Fig. 3. Effect of addition of 200 �mol/L L-NNA and 100 �mol/LSNAP during 15 minutes (dashed area) in isolated rat hearts on coronaryresistance, expressed as percentage of sham basal values (n � 4 heartsper group). Data are mean � SEM.
Fig. 2. Functional results of sham and CBDL isolated hearts. (A)Changes in LVdevP in response to progressive increase in LVEDP. (B)Dose-response curves to isoproterenol stimulation in hearts paced at 5Hz. LVdevP is expressed as percentage of basal values (n � 5 pergroup). Data are mean � SEM.
594 INSERTE ET AL. HEPATOLOGY, September 2003
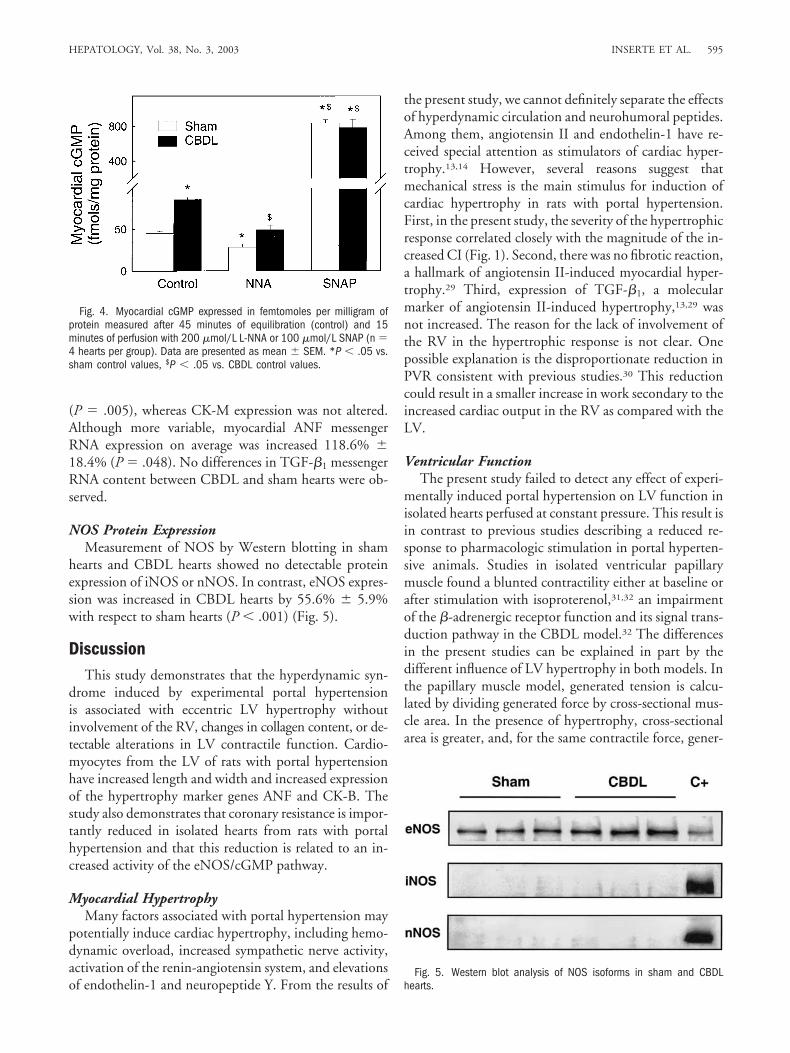
(P � .005), whereas CK-M expression was not altered.Although more variable, myocardial ANF messengerRNA expression on average was increased 118.6% �18.4% (P � .048). No differences in TGF-�1 messengerRNA content between CBDL and sham hearts were ob-served.
NOS Protein ExpressionMeasurement of NOS by Western blotting in sham
hearts and CBDL hearts showed no detectable proteinexpression of iNOS or nNOS. In contrast, eNOS expres-sion was increased in CBDL hearts by 55.6% � 5.9%with respect to sham hearts (P � .001) (Fig. 5).
DiscussionThis study demonstrates that the hyperdynamic syn-
drome induced by experimental portal hypertensionis associated with eccentric LV hypertrophy withoutinvolvement of the RV, changes in collagen content, or de-tectable alterations in LV contractile function. Cardio-myocytes from the LV of rats with portal hypertensionhave increased length and width and increased expressionof the hypertrophy marker genes ANF and CK-B. Thestudy also demonstrates that coronary resistance is impor-tantly reduced in isolated hearts from rats with portalhypertension and that this reduction is related to an in-creased activity of the eNOS/cGMP pathway.
Myocardial HypertrophyMany factors associated with portal hypertension may
potentially induce cardiac hypertrophy, including hemo-dynamic overload, increased sympathetic nerve activity,activation of the renin-angiotensin system, and elevationsof endothelin-1 and neuropeptide Y. From the results of
the present study, we cannot definitely separate the effectsof hyperdynamic circulation and neurohumoral peptides.Among them, angiotensin II and endothelin-1 have re-ceived special attention as stimulators of cardiac hyper-trophy.13,14 However, several reasons suggest thatmechanical stress is the main stimulus for induction ofcardiac hypertrophy in rats with portal hypertension.First, in the present study, the severity of the hypertrophicresponse correlated closely with the magnitude of the in-creased CI (Fig. 1). Second, there was no fibrotic reaction,a hallmark of angiotensin II-induced myocardial hyper-trophy.29 Third, expression of TGF-�1, a molecularmarker of angiotensin II-induced hypertrophy,13,29 wasnot increased. The reason for the lack of involvement ofthe RV in the hypertrophic response is not clear. Onepossible explanation is the disproportionate reduction inPVR consistent with previous studies.30 This reductioncould result in a smaller increase in work secondary to theincreased cardiac output in the RV as compared with theLV.
Ventricular FunctionThe present study failed to detect any effect of experi-
mentally induced portal hypertension on LV function inisolated hearts perfused at constant pressure. This result isin contrast to previous studies describing a reduced re-sponse to pharmacologic stimulation in portal hyperten-sive animals. Studies in isolated ventricular papillarymuscle found a blunted contractility either at baseline orafter stimulation with isoproterenol,31,32 an impairmentof the �-adrenergic receptor function and its signal trans-duction pathway in the CBDL model.32 The differencesin the present studies can be explained in part by thedifferent influence of LV hypertrophy in both models. Inthe papillary muscle model, generated tension is calcu-lated by dividing generated force by cross-sectional mus-cle area. In the presence of hypertrophy, cross-sectionalarea is greater, and, for the same contractile force, gener-
Fig. 5. Western blot analysis of NOS isoforms in sham and CBDLhearts.
Fig. 4. Myocardial cGMP expressed in femtomoles per milligram ofprotein measured after 45 minutes of equilibration (control) and 15minutes of perfusion with 200 �mol/L L-NNA or 100 �mol/L SNAP (n �4 hearts per group). Data are presented as mean � SEM. *P � .05 vs.sham control values, $P � .05 vs. CBDL control values.
HEPATOLOGY, Vol. 38, No. 3, 2003 INSERTE ET AL. 595

ated tension will be smaller. In contrast, in the isolatedheart model generated pressure, instead of tension, is an-alyzed. According to the Laplace law, wall tension in theintact heart is calculated as the product of pressure bycavity radius divided by wall thickness. Because cavityradius in CBDL and control hearts are equal, by applyingthe Laplace formula, generated wall tension in our CBDLmodel is reduced by 16.7% with respect to hearts fromsham-operated rats. In other words, because, in CBDLrats, higher ventricular mass produces the same totalwork, work produced by unit of myocardial mass issmaller.
To our knowledge, only one study assessed cardiacfunction in isolated CBDL rat hearts perfused at constantflow,33 and it found impaired baseline cardiac contractil-ity. This discrepancy can be explained by methodologicdifferences in both studies. Under constant flow perfu-sion, it can be predicted that the perfusion pressure shouldbe much lower in the maximally vasodilated CBDL heartsthan in controls. In fact, from the data shown by theauthors, it can be calculated that a substantial proportionof hearts in the CBDL group should have been perfused atvery low pressure (below 35 mm Hg).
Coronary VasodilationIn hearts with portal hypertension, coronary resistance
was markedly diminished. The abnormally low coronaryresistance observed in hearts from CBDL rats cannot beexplained by an increased work demand in that coronaryflow adaptation to changes in demand are extremelyrapid, and, in our ex vivo studies, demand was the same orlower in CBDL hearts. The fact that the correlation be-tween systemic vascular resistance and cardiac output inCBDL hearts was not significant (r2 � 0.103, P � .897)is in agreement with this interpretation.
The reduction in coronary resistance was NO relatedbecause inhibition of NOS with the L-arginine analogueNNA increased coronary resistance and abolished the dif-ferences between CBDL and sham hearts. CBDL heartsshowed a 2-fold increase in myocardial cGMP as com-pared with hearts from sham rats, and addition of L-NNAabolished the differences between both groups. These re-sults are in agreement with previous studies demonstrat-ing a relationship between NO overproduction and thevascular modifications observed in portal hyperten-sion.2,5,6,33 The observation that the administration ofSNAP in CBDL rat hearts fails to further reduce coronaryresistance suggests that NO-dependent reduction of cor-onary resistance is maximum in CBDL hearts as furtherincrease in NO availability is not associated with furtherdecrease in resistance. The absence of differences betweenSNAP-treated CBDL and sham groups regarding the
concentration of cGMP also discards the possibility thatincreased cGMP was due to alterations in the guanylylcyclase activity. Measurement of cGMP production inisolated cells indicated that the rise in myocardial cGMPcontent observed in CBDL rats is mainly due to increasedcGMP production in endothelial cells rather than tochanges in myocytes. On the other hand, increasedcGMP content was due to enhanced eNOS expression.
Contradictory data have been published on the alter-ations of the NO pathway in the heart during experimen-tal portal hypertension. In the only study performed inisolated CBDL rat hearts, addition of NMMA increasedcoronary resistance.33 In hearts of 4-week CBDL models,Liu et al. recently showed cytokine activation of iNOSand improvement of papillary muscle contractility whenthe enzyme was inhibited.34 This result is in contrast withthe increase in eNOS without activation of iNOS ob-served in aortic rings by the same group35 and others36
and with the results in the partial portal vein ligation ratheart37 in which NOS expression and activity and end-products resulting from NO pathway did not differ fromthose in sham hearts. An intriguing aspect of the presentresults is the mechanism of the persistently increased syn-thesis of NO by eNOS in isolated hearts. Recent studieshave shown that stimulation of the G-protein-coupledCB1 receptor by the endogenous cannabinoid anandam-ide may be responsible, at least in part, for the increasedNO synthesis in cirrhosis. Monocytes from cirrhotic ratsstimulate NO overproduction in noncirrhotic rats, andthis effect is prevented in the presence of pharmacologicblockade of the CB1 receptor.38,39 It is thus conceivablethat anandamide secreted by myocardial histiocytes couldmaintain increased NO synthesis during ex vivo condi-tions.
Our Findings and Other Models of PortalHypertension
The current study was performed in rats with advancedsecondary biliary cirrhosis, 6 weeks after CBDL. This is awidely used model of cirrhosis that has been previouslyshown, and confirmed by the present study, to develop allthe main complications of human cirrhosis, including in-trahepatic portal hypertension, hyperkinetic circulation,portal-systemic collaterals, liver failure, ascites, splanchnicvasodilatation, activation of endogenous vasoactive sys-tems, and hepatopulmonary syndrome.22,29,30,40,41 Never-theless, the model is also associated with markedcholestasis. Because of this, the findings in this model maynot be extrapolated to other models of hepatic cirrhosis orto other human diseases. It is important to note that manyprevious experimental studies on the cirrhotic cardiomy-opathy have been performed in the CBDL model, al-
596 INSERTE ET AL. HEPATOLOGY, September 2003

though in less advanced stages. It is of interest that, in astudy using CCl4-induced cirrhotic rats, heart weight wasincreased without histologic abnormalities.42 The conclu-sions of a study of Ma et al.31 performed in hearts ofCBDL rats were similar. Few human studies have de-scribed the influence of portal hypertension on heartstructure.43 A previous study reported an increase in LVwall thickness measured by echocardiography in cirrhoticpatients,15 whereas another group, using the samemethod, found enlarged diameters in right and left atriumand RV without changes in the LV.44 More recently, in-creased septal thickness with normal LV ejection fractionwas described in cirrhotic patients without cardiovascularsymptoms.17
In conclusion, our results show that portal hyperten-sion in rats with biliary cirrhosis is associated with reducedsystemic, pulmonary, and coronary vascular resistanceand to LV hypertrophy, in which magnitude correlateswith the degree of hyperdynamic circulation. The lack offibrosis and contractile abnormalities and the absence ofup-regulation of TGF-�1 in myocytes suggest that thehypertrophic response is mainly caused by mechanicaloverload with less important participation of the associ-ated neuroendocrine activity. The observed reduction ofcoronary resistance was secondary to increased synthesisof NO by eNOS.
References1. Bosch J, Pizcueta P, Feu F, Fernandez M, Garcıa-Pagan JC. Pathophysi-
ology of portal hypertension. Gastroenterol Clin North Am 1992;21:1-14.2. Pizcueta MP, Pique JM, Fernandez M, Bosch J, Rodes J, Whittle BJ,
Moncada S. Modulation of the hyperdynamic circulation of cirrhotic ratsby nitric oxide inhibition. Gastroenterology 1992;103:1909-1915.
3. Fernandez M, Garcıa-Pagan JC, Casadevall M, Bernadich C, Piera C,Whittle BJR, Pique JM, et al. Evidence against a role for inducible nitricoxide synthase in the hyperdynamic circulation of portal-hypertensive rats.Gastroenterology 1995;108:1487-1495.
4. Shah V, Wiest R, Garcia-Cardena G, Cadelina G, Groszmann RJ, SessaWC. Hsp90 regulation of endothelial nitric oxide synthase contributes tovascular control in portal hypertension. Am J Physiol 1999;277:G463-G468.
5. Pizcueta MP, Pique JM, Bosch J, Whittle BJ, Moncada S. Effects of inhib-iting nitric oxide biosynthesis on the systemic and splanchnic circulation ofrats with portal hypertension. Br J Pharmacol 1992;105:184-190.
6. Lee FY, Albillos A, Colombato LA, Groszmann RJ. The role of nitric oxidein the vascular hyporesponsiveness to methoxamine in portal hypertensiverats. HEPATOLOGY 1992;16:1043-1048.
7. Fernandez M, Bonkovsky HL. Increased heme oxygenase-1 gene expres-sion in liver cells and splanchnic organs from portal hypertensive rats.HEPATOLOGY 1999;29:1672-1679.
8. Pizcueta MP, Garcıa-Pagan JC, Fernandez M, Casamitjana R, Bosch J,Rodes J. Glucagon hinders the effects of somatostatin on portal hyperten-sion. A study in rats with partial portal vein ligation. Gastroenterology1991;101:1710-1715.
9. Benoit JN, Zimmerman B, Premen AJ, Go VL, Granger DN. Role ofglucagon in splanchnic hyperemia of chronic portal hypertension. Am JPhysiol 1986;251:G674-G677.
10. Schrier RW, Arroyo V, Bernardi M, Epstein M, Henriksen JH, Rodes J.Peripheral arterial vasodilation hypothesis: a proposal for the initiation of
renal sodium and water retention in cirrhosis. HEPATOLOGY 1988;8:1151-1157.
11. Luo B, Abrams GA, Fallon MB. Endothelin-1 in the rat bile duct ligationmodel of hepatopulmonary syndrome: correlation with pulmonary dys-function. J Hepatol 1998;29:571-578.
12. Moller S, Bendtsen F, Henriksen JH. Splanichnic and systemic hemody-namic derangement in decompensated cirrhosis. Can J Gastroenterol2001;15:94-106.
13. Sadoshima J, Izumo S. Molecular characterization of angiotensin II-in-duced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibro-blasts: a critical role of the AT1 receptor subtype. Circ Res 1993;73:413-423.
14. Ito H, Hirata Y, Hiroe M. Endothelin-1 induces hypertrophy with en-hanced expression of muscle-specific genes in cultured neonatal rat cardi-omyocytes. Circ Res 1991;69:209-215.
15. Pozzi M, Carugo S, Boari G, Pecci V, de Ceglia S, Maggiolini S, Bolla GB,et al. Evidence of functional and structural cardiac abnormalities in cir-rhotic patients with and without ascites. HEPATOLOGY 1997;26:1131-1137.
16. Lewis FL, Adair O, Rector WG. Arterial vasodilation is not the cause ofincreased cardiac output in cirrhosis. Gastroenterology 1992;102:1024-1029.
17. Wong F, Liu P, Tobe S, Morali G, Blendis L. Central blood volume incirrhosis: measurement with radionuclide angiography. HEPATOLOGY
1994;19:312-321.18. Bernardi M, Rubboli A, Trevisani F, Cancellieri C, Ligabue A, Baraldini
M, Gasbarrini G. Reduced cardiovascular responsiveness to exercise in-duced sympathoadrenergic stimulation in patients with cirrhosis. J Hepa-tol 1991;12:207-216.
19. Grose RD, Nolan J, Dillon JF, Errington M, Hannan WJ, Bouchier IAD,Hayes PC. Exercise-induced left ventricular dysfunction in alcoholic andnon-alcoholic cirrhosis. J Hepatol 1995;26:326-332.
20. Lunzer MR, Manghani KK, Newman SP, Sherlock SP, Bernard AG, Gins-burg J. Impaired cardiovascular responsiveness in liver disease. Lancet1975;2:382-385.
21. Donovan CL, Marcovitz PA, Punch JD, Bach DS, Brown KA, Lucey MR,Armstrong WF. Two-dimensional and dobutamine stress echocardiogra-phy in the preoperative assessment of patients with end-stage liver diseaseprior to orthotopic liver transplantation. Transplantation 1996;61:1180-1188.
22. Fernandez M, Pizcueta P, Garcıa-Pagan JC, Feu F, Cirera I, Bosch J, RodesJ. Effects of ritanserin, a selective and specific S2-serotoninergic antagonist,on portal pressure and splanchnic hemodynamics in rats with long-termbile duct ligation. HEPATOLOGY 1993;18:389-393.
23. Castaneda B, Debernardi-Venon W, Bandi JC, Andreu V, Perez-del-Pulgar S, Moitinho E, Pizcueta P, et al. The role of portal pressure in theseverity of bleeding in portal hypertensive rats. HEPATOLOGY 2000;31:581-586.
24. Garcia-Dorado D, Inserte J, Ruiz-Meana M, Gonzalez MA, Solares J, JuliaM, Barrabes JA, et al. Gap junction uncoupler heptanol prevents cell-to-cell progression of hypercontracture and limits necrosis during myocardialreperfusion. Circulation 1997;96:3579-3586.
25. Poo JL, Jimenez W, Maria MR, Bosch-Marce M, Bordas N, Morales-RuizM, Perez M, et al. Chronic blockade of endothelin receptors in cirrhoticrats: hepatic and hemodynamic effects. Gastroenterology 1999;116:161-167.
26. Castro A, Jimenez W, Claria J, Ros J, Martinez JM, Bosch M, Arroyo V, etal. Impaired responsiveness to angiotensin II in experimental cirrhosis: roleof nitric oxide. HEPATOLOGY 1993;18:367-372.
27. Schluter KD, Frischkopf K, Flesch M, Rosenkranz S, Taimor G, PiperHM. Central role for ornithine decarboxylase in �-adrenoceptor mediatedhypertrophy. Cardiovasc Res 2000;45:410-417.
28. Agullo L, Garcia-Dorado G, Escalona N, Inserte J, Ruiz-Meana M, Bar-rabes JA, Mirabet M, et al. Hypoxia and acidosis impair cyclic GMPsynthesis in coronary enthothelial cells. Am J Physiol 2002;283:H917-H925.
HEPATOLOGY, Vol. 38, No. 3, 2003 INSERTE ET AL. 597

29. Kim S, Iwao H. Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacol Rev 2000;52:11-34.
30. Fallon MB, Abrams GA, McGrath JW, Hou Z, Luo B. Common bile ductligation in the rat: a model of intrapulmonary vasodilatation and hepato-pulmonary syndrome. Am J Physiol 1997;272:G779-G784.
31. Ma Z, Miyamoto A, Lee SS. Role of altered �-adrenoceptor signal trans-duction in the pathogenesis of cirrhotic cardiomyopathy in rats. Gastroen-terology 1996;110:1191-1198.
32. Battarbee HD, Zavecz JH. Cardiac performance in the portal vein-ste-nosed rat. Am J Physiol 1992;263:G181-G185.
33. Van Obbergh L, Vallieres Y, Blaise G. Cardiac modifications occurring inthe ascitic rat with biliary cirrhosis are nitric oxide related. J Hepatol1996;24:747-752.
34. Liu H, Ma Z, Lee SS. Contribution of nitric oxide to the pathogenesis ofcirrhotic caridomyopathy in bile duct-ligated rats. Gastroenterology 2000;118:937-944.
35. Liu H, Song D, Lee SS. Increased nitric oxide synthase expression in aortaof cirrhotic rats. Life Sci 1999;64:1753-1759.
36. Gadano AC, Sogni P, Heller J, Moreau R, Bories PN, Lebrec D. Vascularnitric oxide production during the development of two experimental mod-els of portal hypertension. J Hepatol 1999;30:896-903.
37. Battarbee HD, Zavecz JH, Grisham MB, Maloney RE, Chandler LJ,Mercer JW, Cady FM. Cardiac impairment and nitric oxide synthaseactivity in the chronic portal vein-stenosed rat. Am J Physiol 1999;276:G363-G372.
38. Batkai S, Jarai Z, Wagner JA, Goparaju SK, Varga K, Liu J, Wang L,et al. Endocannabinoids acting at vascular CB1 receptors mediatethe vasodilated state in advanced liver cirrhosis. Nat Med 2001;7:827-832.
39. Ros J, Claria J, To-Figueras J, Planaguma A, Cejudo-Martin P, Fernandez-Varo G, Martin-Ruiz R, et al. Endogenous cannabinoids: a new systeminvolved in the homeostasis of arterial pressure in experimental cirrhosis inthe rat. Gastroenterology 2002;122:85-93.
40. Ma Z, Lee SS. Cirrhotic cardiomyopathy: getting to the heart of the mat-ter. HEPATOLOGY 1996;24:451-459.
41. Liu L, Zhang M, Luo B, Abrams GA, Fallon MB. Biliary cyst fluid fromcommon bile duct-ligated rats stimulates endothelial nitric oxide synthasein pulmonary artery endothelial cells: a potential role in hepatopulmonarysyndrome. HEPATOLOGY 2001;33:722-727.
42. Fort J, Pilette C, Oberti F, Veal N, Gallois Y, Douay O, Cales P. Long-term administration of PGE1 increases liver fibrosis and collateral bloodflow in bile-duct-ligated rats. J Hepatol 1999;30:70-76.
43. Caramelo C, Fernandez-Munoz D, Santos JC, Blanchart A, Rodriguea-Puyol D, Lopez-Novoa JM, Hernando L. Effect of volume expansionhemodynamics, capillary permeability and renal function in concious, cir-rhotic rats. HEPATOLOGY 1986;6:129-134.
44. Ruwhof C, van der Laarse A. Mechanical stress-induced cardiac hypertro-phy: mechanisms and signal trasnduction pathways. Cardiovasc Res 2000;47:23-37.
45. Valeriano V, Funaro S, Lionetti R, Riggio O, Pulcinelli G, Fiore P, MasiniA, et al. Modification of cardiac function in cirrhotic patients with andwithout ascites. Am J Gastroenterol 2000;95:3200-3205.
598 INSERTE ET AL. HEPATOLOGY, September 2003
Copyright © 2022 FDOKUMEN




















