
Advanced glycation of apolipoprotein AI impairs its anti-atherogenic properties
Upload
independentCategory
view
0download
0
Apolipoprotein C-I Is Crucially Involved inLipopolysaccharide-Induced Atherosclerosis Development in
Apolipoprotein E–Knockout MiceMarit Westerterp, PhD*; Jimmy F.P. Berbée, PhD*; Nuno M.M. Pires, PhD;
Geertje J.D. van Mierlo, PhD; Robert Kleemann, PhD; Johannes A. Romijn, MD, PhD;Louis M. Havekes, PhD; Patrick C.N. Rensen, PhD
Background—Lipopolysaccharide (LPS), which is released from Gram-negative bacteria on multiplication or lysis,aggravates atherosclerosis in humans and rodents by inducing inflammation via toll-like receptors. Because apolipopro-tein C-I (apoCI) enhances the LPS-induced inflammatory response in macrophages in vitro and in mice, we investigatedthe effect of endogenous apoCI expression on LPS-induced atherosclerosis in mice.
Methods and Results—Twelve-week-old apoe�/�apoc1�/� and apoe�/�apoc1�/� mice received weekly intraperitonealinjections of LPS (50 �g) or vehicle for a period of 10 weeks, and atherosclerosis development was assessed in the aorticroot. LPS administration did not affect atherosclerotic lesion area in apoe�/�apoc1�/� mice but increased it inapoe�/�apoc1�/� mice. In fact, apoCI expression increased the LPS-induced atherosclerotic lesion area by 60%(P�0.05), concomitant with an increase in LPS-induced plasma levels of fibrinogen and E-selectin. This indicated thatapoCI increased the LPS-induced inflammatory state, both systemically (ie, fibrinogen) and at the level of the vesselwall (ie, E-selectin). In addition, both macrophage-derived apoCI and HDL-associated apoCI increased the LPS-inducedtumor necrosis factor-� response by macrophages in vitro.
Conclusions—We conclude that apoCI is crucially involved in LPS-induced atherosclerosis in apoe�/� mice, which mainlyrelates to an increased inflammatory response toward LPS. We anticipate that apoCI plasma levels contribute toaccelerated atherosclerosis development in individuals who have chronic infection. (Circulation. 2007;116:2173-2181.)
Key Words: apolipoproteins � atherosclerosis � inflammation � lipoproteins � hypercholesterolemia � leukocytes
Cardiovascular disease (CVD) is the principal cause ofdeath in Europe, the United States, and much of Asia.
The main cause of CVD is atherosclerosis.1–3
Clinical Perspective p 2181
Although hypertension and hyperlipidemia are commonrisk factors for atherosclerosis, it is also evident that bacterialinfections worsen the outcome of atherosclerosis by main-taining a heightened state of inflammatory response, thuspropagating this inflammatory disease.1–4 Lipopolysaccha-ride (LPS) is a highly inflammatory constituent of the outermembrane of Gram-negative bacteria. When these bacteriamultiply or lyse, LPS is released and then activates theMD-2/toll-like receptor 4 (TLR4) receptor complex on endo-thelial cells, neutrophils, and macrophages, which results in a
proinflammatory response through activation of the nuclearfactor–�B pathway.5 TLR4 is expressed in macrophages andendothelial cells within atherosclerotic lesions,6,7 and indeed,impaired LPS signaling in human carriers of the TLR4polymorphisms Asp299Gly or Thr399Ile is associated withLPS hyporesponsiveness,8 and the Asp299Gly mutation isassociated with decreased CVD.4 Conversely, infection withthe Gram-negative bacterium Chlamydia pneumoniae hasbeen associated with increased CVD in humans.9–11
In rodents, bacterial infection and LPS also promoteatherogenesis. For example, local delivery of C pneumoniaein the vessel wall of carotid arteries increases the develop-ment of atherosclerosis in low-density lipoprotein (LDL)receptor gene–deficient (ldlr�/�) mice,12 and infection with Cpneumoniae accelerates the development of atherosclerosis in
Received January 30, 2007; accepted August 31, 2007.From The Netherlands Organization for Applied Scientific Research–Quality of Life (M.W., J.F.P.B., N.M.M.P., R.K., L.M.H., P.C.N.R.), Department
of Biomedical Research, Gaubius Laboratory, Leiden, the Netherlands; and Departments of General Internal Medicine, Endocrinology, and MetabolicDiseases (M.W., J.F.P.B., J.A.R., L.M.H., P.C.N.R.), Cardiology (N.M.M.P., L.M.H.), Rheumatology (G.J.D.v.M.), and Vascular Surgery (R.K.), LeidenUniversity Medical Center, Leiden, the Netherlands.
*Drs Westerterp and Berbée contributed equally to this article.The online-only Data Supplement, consisting of an expanded Methods section, is available with this article at http://circ.ahajournals.org/cgi/content/
full/CIRCULATIONAHA.107.693382/DC1.Correspondence to Marit Westerterp, Leiden University Medical Center, Department of Endocrinology and Metabolic Diseases, C4-R, PO Box 9600,
2300 RC Leiden, the Netherlands. E-mail [email protected]© 2007 American Heart Association, Inc.
Circulation is available at http://circ.ahajournals.org DOI: 10.1161/CIRCULATIONAHA.107.693382
2173 by guest on February 17, 2015http://circ.ahajournals.org/Downloaded from

apolipoprotein (apo) E gene–deficient (apoe�/�), ldlr�/�, andAPOE*3-Leiden transgenic mice.13–15 In addition, LPS accel-erates intimal lesion development in a periadventitial cuffmodel in wild-type mice, which is largely decreased whenTLR4 expression is absent.16 Furthermore, repeated intrave-nous and intraperitoneal administration of LPS acceleratesatherosclerosis in rabbits and apoe�/� mice, respectively.17,18
Until recently, apolipoprotein C-I (apoCI) was knownprimarily for its role in lipoprotein metabolism. ApoCIcirculates in plasma with a concentration of 6 mg/dL and ismainly bound to the lipoproteins very-low-density lipoprotein(VLDL), chylomicrons, and high-density lipoprotein(HDL).19 ApoCI inhibits lipoprotein lipase (LPL),20,21 andendogenous apoCI expression was associated with modesthyperlipidemia in apoe�/� mice.21 Recently, we have discov-ered that apoCI strongly binds to LPS, thereby augmentingthe inflammatory response to LPS and the Gram-negativebacterium Klebsiella pneumoniae in mice in vivo and to LPSin macrophages in vitro.22
In the present study, we investigated the effect of endog-enous apoCI on the development of LPS-induced atheroscle-rosis in apoe�/�apoc1�/� versus apoe�/�apoc1�/� mice. Wefound that LPS-induced atherosclerosis is enhanced in apoCI-expressing mice, in association with a higher inflammatorystate achieved with LPS.
MethodsFor an extended version of the Methods section, see the online-onlyData Supplement.
AnimalsApoe�/�apoc1�/�23 and apoe�/�24 mice were back-crossed at least 8times to the C57Bl/6 background. Apoe�/�apoc1�/� and apoe�/�
apoc1�/� littermates were generated, and female mice were used forexperiments, housed under standard conditions with a 12-hour lightcycle (7 AM to 7 PM), and fed ad libitum with regular chow.
Analysis of LPS Response in TimeTwelve-week-old apoe�/�apoc1�/� and apoe�/�apoc1�/� littermates(n�8 per genotype) received an intraperitoneal injection of LPS (50�g; Escherichia coli serotype 055:B5, Sigma, St Louis, Mo) in 200�L of PBS. Twenty-four hours before LPS injection (t�0) and every24 hours thereafter for a period of 1 week, 35 �L of blood wascollected after a 4-hour fast for measurement of total cholesterol(TC), fibrinogen, and E-selectin. At t�0, 24 hours after LPSinjection, and 1 week after the injection, triglyceride (TG) levelswere also measured.
Atherosclerosis StudyTwelve-week-old apoe�/�apoc1�/� and apoe�/�apoc1�/� littermatesreceived weekly intraperitoneal injections of LPS (50 �g) in 200 �Lof PBS or PBS alone (vehicle; n�8 to 11 per genotype per group),exactly as described previously.18,25 Plasma lipid and inflammatoryparameters of these mice were analyzed 1 week before the start of theinjections and 24 hours after the first, fifth, and tenth injections.Twenty-four hours after the first injection, lipid distribution overlipoproteins was assessed.
Plasma Lipid and Lipoprotein AnalysisBlood was collected, and plasma TC and TG, as well as TC and TGdistribution over lipoproteins, were determined.26 For analysis of theapolipoprotein distribution over lipoproteins, 2 adjacent fractions of50 �L were pooled, and apolipoproteins were separated.27
HDL AnalysisTo determine the apoCI level on HDL relative to HDL lipidconcentrations, HDL from apoe�/�apoc1�/� mice that received eithervehicle or LPS was isolated via precipitation of apoB-containinglipoproteins,27 and HDL phospholipids, HDL cholesterol, andapoCI21 were determined.
Plasma Analysis of Fibrinogen and E-SelectinPlasma fibrinogen levels were determined with a homemadeELISA,28 and plasma E-selectin levels were ascertained via acommercially available ELISA (R&D Systems Europe, Abingdon,UK).
Incubation of Macrophages With LPS In VitroApoe�/�apoc1�/� and apoe�/�apoc1�/� peritoneal macrophages wereisolated and cultured27 and then incubated with LPS (0.1 to 100ng/mL) in Dulbecco’s Modified Eagle Medium supplemented with0.01% human serum albumin (4 hours at 37°C). Tumor necrosisfactor (TNF)-� in the medium was determined.22 Murine RAW264.7 macrophages were cultured22 and incubated with LPS (100ng/mL) with or without HDL isolated from apoe�/�apoc1�/� andapoe�/�apoc1�/� littermates (24 hours at 37°C), and TNF-� wasdetermined in the medium.22
Atherosclerotic Lesion AnalysisFor determination of atherosclerosis development, mice were eutha-nized at 24 hours after the tenth injection. Hearts were fixed andembedded in paraffin, and sections were made.27 Lesion area andcomposition with regard to adherent monocytes, macrophages,collagen, smooth muscle cells (SMCs), and T cells weredetermined.29
Statistical AnalysisThe Mann-Whitney nonparametric test for 2 independent sampleswas used to define differences between data sets from experimentalgroups. The criterion for significance was set at P�0.05. Linearregression analysis was used to evaluate correlations between HDLapoCI levels versus HDL lipid parameters. Statistical analyses wereperformed with SPSS version 11.5 (SPSS Inc, Chicago, Ill).
The authors had full access to the data and take full responsibility forits integrity. All authors have read and agree to the manuscript aswritten.
ResultsEffect of ApoCI on Plasma Lipid Levels AfterLPS InjectionTo examine the effect of LPS on plasma lipid levels, wemeasured plasma levels of TC and TG before and during a
Figure 1. Effect of apoCI on plasma cholesterol levels after LPSinjection. Twelve-week-old apoe�/�apoc1�/� (●) and apoe�/�
apoc1�/� (�) mice received an injection of LPS (50 �g). Twenty-four hours before LPS injection (t�0) and every 24 hours there-after for a period of 7 days, plasma samples were taken after a4-hour fast. TC was measured, and the values shown indicatethe increase compared with t�0. Basal levels of TC were7.0�0.5 and 9.3�1.8 mmol/L in apoe�/�apoc1�/� and apoe�/�
apoc1�/� mice, respectively. Values are mean�SD; n�8.
2174 Circulation November 6, 2007
by guest on February 17, 2015http://circ.ahajournals.org/Downloaded from
period of 7 days after a single LPS injection (Figure 1). LPSinjection increased TC levels in both apoe�/�apoc1�/� andapoe�/�apoc1�/� mice at 24 hours after LPS injection by�2.5 mmol/L. The level of TC decreased thereafter to returnto its starting value after 7 days (Figure 1). LPS onlymodestly increased levels of TG, by �0.1 mmol/L, and thisalso returned to starting values within 7 days (results notshown). ApoCI expression apparently did not affect the totalLPS-induced increase in TC (Figure 1) and TG levels (resultsnot shown).
We next investigated the effect of repeated injections ofLPS or vehicle on plasma TC and TG levels in apoe�/�
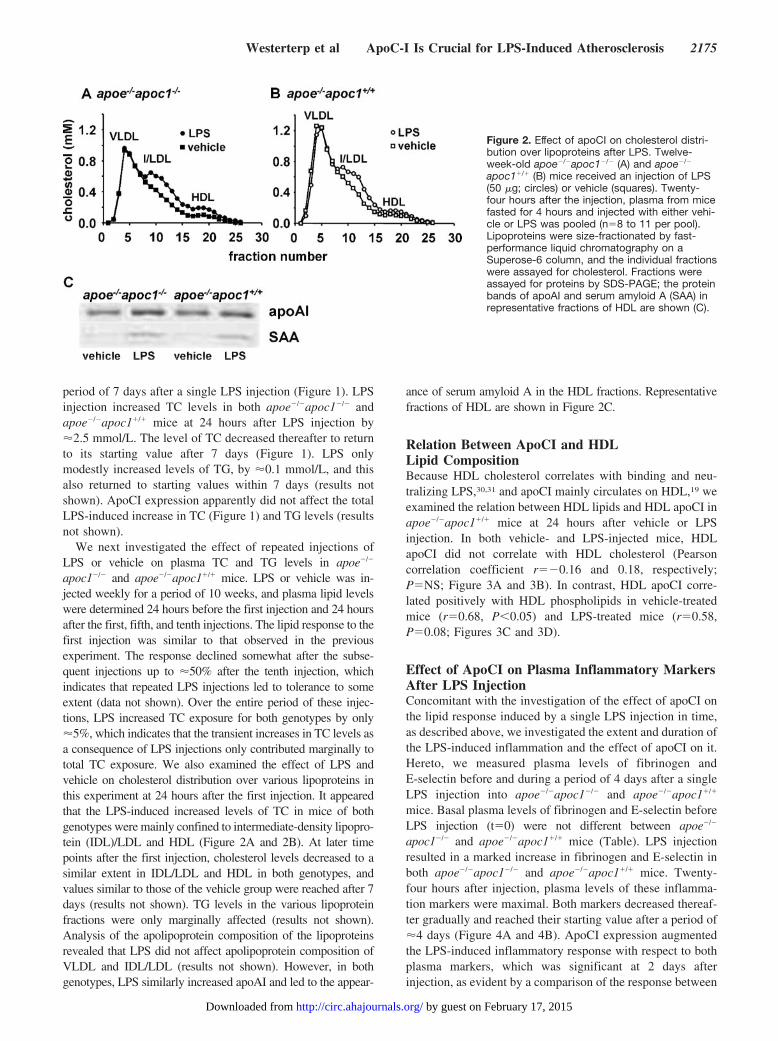
apoc1�/� and apoe�/�apoc1�/� mice. LPS or vehicle was in-jected weekly for a period of 10 weeks, and plasma lipid levelswere determined 24 hours before the first injection and 24 hoursafter the first, fifth, and tenth injections. The lipid response to thefirst injection was similar to that observed in the previousexperiment. The response declined somewhat after the subse-quent injections up to �50% after the tenth injection, whichindicates that repeated LPS injections led to tolerance to someextent (data not shown). Over the entire period of these injec-tions, LPS increased TC exposure for both genotypes by only�5%, which indicates that the transient increases in TC levels asa consequence of LPS injections only contributed marginally tototal TC exposure. We also examined the effect of LPS andvehicle on cholesterol distribution over various lipoproteins inthis experiment at 24 hours after the first injection. It appearedthat the LPS-induced increased levels of TC in mice of bothgenotypes were mainly confined to intermediate-density lipopro-tein (IDL)/LDL and HDL (Figure 2A and 2B). At later timepoints after the first injection, cholesterol levels decreased to asimilar extent in IDL/LDL and HDL in both genotypes, andvalues similar to those of the vehicle group were reached after 7days (results not shown). TG levels in the various lipoproteinfractions were only marginally affected (results not shown).Analysis of the apolipoprotein composition of the lipoproteinsrevealed that LPS did not affect apolipoprotein composition ofVLDL and IDL/LDL (results not shown). However, in bothgenotypes, LPS similarly increased apoAI and led to the appear-
ance of serum amyloid A in the HDL fractions. Representativefractions of HDL are shown in Figure 2C.
Relation Between ApoCI and HDLLipid CompositionBecause HDL cholesterol correlates with binding and neu-tralizing LPS,30,31 and apoCI mainly circulates on HDL,19 weexamined the relation between HDL lipids and HDL apoCI inapoe�/�apoc1�/� mice at 24 hours after vehicle or LPSinjection. In both vehicle- and LPS-injected mice, HDLapoCI did not correlate with HDL cholesterol (Pearsoncorrelation coefficient r��0.16 and 0.18, respectively;P�NS; Figure 3A and 3B). In contrast, HDL apoCI corre-lated positively with HDL phospholipids in vehicle-treatedmice (r�0.68, P�0.05) and LPS-treated mice (r�0.58,P�0.08; Figures 3C and 3D).
Effect of ApoCI on Plasma Inflammatory MarkersAfter LPS InjectionConcomitant with the investigation of the effect of apoCI onthe lipid response induced by a single LPS injection in time,as described above, we investigated the extent and duration ofthe LPS-induced inflammation and the effect of apoCI on it.Hereto, we measured plasma levels of fibrinogen andE-selectin before and during a period of 4 days after a singleLPS injection into apoe�/�apoc1�/� and apoe�/�apoc1�/�
mice. Basal plasma levels of fibrinogen and E-selectin beforeLPS injection (t�0) were not different between apoe�/�
apoc1�/� and apoe�/�apoc1�/� mice (Table). LPS injectionresulted in a marked increase in fibrinogen and E-selectin inboth apoe�/�apoc1�/� and apoe�/�apoc1�/� mice. Twenty-four hours after injection, plasma levels of these inflamma-tion markers were maximal. Both markers decreased thereaf-ter gradually and reached their starting value after a period of�4 days (Figure 4A and 4B). ApoCI expression augmentedthe LPS-induced inflammatory response with respect to bothplasma markers, which was significant at 2 days afterinjection, as evident by a comparison of the response between
Figure 2. Effect of apoCI on cholesterol distri-bution over lipoproteins after LPS. Twelve-week-old apoe�/�apoc1�/� (A) and apoe�/�
apoc1�/� (B) mice received an injection of LPS(50 �g; circles) or vehicle (squares). Twenty-four hours after the injection, plasma from micefasted for 4 hours and injected with either vehi-cle or LPS was pooled (n�8 to 11 per pool).Lipoproteins were size-fractionated by fast-performance liquid chromatography on aSuperose-6 column, and the individual fractionswere assayed for cholesterol. Fractions wereassayed for proteins by SDS-PAGE; the proteinbands of apoAI and serum amyloid A (SAA) inrepresentative fractions of HDL are shown (C).
Westerterp et al ApoC-I Is Crucial for LPS-Induced Atherosclerosis 2175
by guest on February 17, 2015http://circ.ahajournals.org/Downloaded from

apoe�/�apoc1�/� and apoe�/�apoc1�/� mice (P�0.05; Figures4A and 4B). Fibrinogen and E-selectin were also measuredafter repeated injections of LPS and vehicle. The response tothe first injection was similar to that described above anddeclined �15% after repeated LPS injections (data notshown), which indicates the development of some toleranceto LPS injections in both genotypes.
In all, these data show that LPS transiently increasesboth plasma lipid levels and the inflammatory response inapoe�/�apoc1�/� and apoe�/�apoc1�/� mice. Whereas thelipid response after LPS injection was relatively small, LPSinjection led to a marked increase in fibrinogen and E-selectinlevels. The presence of apoCI had no effect on LPS-inducedchanges in plasma lipid concentrations but increased themagnitude of the LPS-induced inflammatory response.
Effect of ApoCI on LPS-Induced TNF-� Responseof MacrophagesBecause we found previously that exogenous apoCI increasedthe LPS-induced TNF-� response in macrophages,22 andapoCI mainly circulates on HDL,19 we next examined theeffect of HDL (0.1 and 1 �g protein/mL) from apoe�/�
apoc1�/� and apoe�/�apoc1�/� mice on the LPS-inducedTNF-� response by macrophages. ApoCI presence on HDLincreased the TNF-� response by 77% (P�0.05; HDL�0.1�g protein/mL) and 66% (P�0.05; HDL�1 �g protein/mL)as evident from a comparison of the response betweenapoe�/�apoc1�/� and apoe�/�apoc1�/� HDL (Figure 5A). Inaddition, we investigated the effect of apoCI expression bymacrophages on the LPS-induced TNF-� response usingapoe�/�apoc1�/� and apoe�/�apoc1�/� peritoneal macro-phages. Endogenous apoCI expression by macrophagesincreased the TNF-� response elicited by 1, 10, and 100ng/mL LPS, which reached significance with 100 ng/mLLPS (45%; P�0.05; Figure 5B). Collectively, both apoCIon HDL and macrophage-derived apoCI contributed toenhancement of the LPS-induced TNF-� response bymacrophages.
Effect of ApoCI on LPS-InducedAtherosclerosis DevelopmentWe next examined the effect of repeated LPS injections onatherosclerosis development and the effect of apoCI onLPS-induced atherosclerosis. For this purpose, mice wereeuthanized 24 hours after the tenth LPS injection, and thedevelopment of atherosclerosis was studied at the level of theaortic root. Representative lesions are shown in Figure 6Athrough 6D, and the data of all mice are summarized in Figure6E. LPS increased atherosclerotic lesion area by 53%(P�0.05) in apoe�/�apoc1�/� mice, as derived from thecomparison between vehicle- and LPS-treated apoe�/�
apoc1�/� mice (Figure 6C versus 6D). In contrast, LPS did notaffect the atherosclerotic lesion area in apoe�/�apoc1�/� mice(Figure 6A versus 6B). Furthermore, apoCI expression in-
Figure 3. Relation between HDL apoCI andHDL lipids. Twelve-week-old apoe�/�apoc1�/�
mice received injection of vehicle (A, C) or LPS(50 �g; B, D). Twenty-four hours after injection,HDL was isolated from plasma from micefasted for 4 hours and assayed for apoCI, cho-lesterol, and phospholipids. ApoCI is expressedin arbitrary units (AU) relative to a pool ofplasma from apoe�/�apoc1�/� mice. Lines indi-cate linear regression analysis between HDLapoCI and HDL cholesterol (A, B) or HDL phos-pholipids (C, D). n�9 to 10.
Table. Effect of Endogenous ApoCI Expression on BasalPlasma Levels of Inflammatory Markers in apoe�/� Mice
Mouse Genotype Fibrinogen, mg/mL E-Selectin, ng/mL
apoe�/�apoc1�/� 1.23�0.28 96.2�5.9
apoe�/�apoc1�/� 1.26�0.10 91.7�13.7
Blood was obtained from 4-hour-fasted apoe�/�apoc1�/� and apoe�/
�apoc1�/� mice. Plasma levels of fibrinogen and E-selectin were measuredwith ELISA. Values are mean�SD. n�8.
2176 Circulation November 6, 2007
by guest on February 17, 2015http://circ.ahajournals.org/Downloaded from

creased LPS-induced atherosclerosis by 60% (P�0.05), asevident from a comparison of LPS-injected apoe�/�apoc1�/�
and apoe�/�apoc1�/� mice (Figure 6B versus 6D). In contrast,apoCI expression did not affect atherosclerosis in mice
injected with vehicle (Figure 6A versus 6C). These data thusindicate that endogenous apoCI expression is a strong deter-minant of the LPS-induced increase in atherosclerotic lesionsize (Figure 6E).
Figure 4. Effect of apoCI on plasma inflamma-tory markers after LPS injection. Twelve-week-old apoe�/�apoc1�/� (●) and apoe�/�apoc1�/�
(�) mice received an injection of LPS (50 �g).Twenty-four hours before LPS injection (t�0)and every 24 hours thereafter for a period of 4days, plasma samples were taken after a4-hour fast. Fibrinogen (A) and E-selectin (B)were measured, and the values shown indicatethe increase compared with t�0. Values aremean�SD; n�8. *P�0.05, significant differencebetween genotypes.
Figure 5. Effect of apoCI on the LPS-induced TNF-� responsein macrophages. RAW 264.7 macrophages were incubated withLPS (100 ng/mL) in Dulbecco’s Modified Eagle Medium supple-mented with 0.01% human serum albumin, in the presence orabsence of HDL isolated from apoe�/�apoc1�/� or apoe�/�
apoc1�/� littermates (0.1 and 1 �g HDL protein/mL). After 24hours, cells were washed and lysed, and TNF-� was determinedin the medium (A). Peritoneal macrophages from apoe�/�
apoc1�/� and apoe�/�apoc1�/� littermates were incubated with-out or with LPS (0.1 to 100 ng/mL) in Dulbecco’s Modified EagleMedium supplemented with 0.01% human serum albumin. After4 hours, cells were washed and lysed, and TNF-� was deter-mined in the medium (B). Values are represented relative to cellprotein content as mean�SD; n�4. *P�0.05.
Figure 6. Effect of apoCI on LPS-induced atherosclerosis devel-opment in the aortic root. Twelve-week-old apoe�/�apoc1�/�
(solid symbols) and apoe�/�apoc1�/� (open symbols) micereceived weekly injections of LPS (50 �g; circles) or vehicle(squares) for a period of 10 weeks and were euthanized afterthe last injection. Hearts were isolated, cross-sectioned (5 �m)throughout the aortic root, and stained with hematoxylin-phloxin-saffron. Representative pictures are shown (A throughD). Atherosclerotic lesion area was measured in 4 sections permouse at 40-�m intervals. Each data point represents the meanper mouse. Each line indicates the mean of the data points (E).n�8 to 11. *P�0.05.
Westerterp et al ApoC-I Is Crucial for LPS-Induced Atherosclerosis 2177
by guest on February 17, 2015http://circ.ahajournals.org/Downloaded from

Effect of ApoCI on LPS-Induced AtheroscleroticLesion CompositionTo study whether LPS treatment affected lesion composition,we characterized the atherosclerotic lesions with respect tomonocyte adhesion, the content of CD3� T cells, macro-phages, SMCs, and collagen. In mice of both genotypes, LPStreatment tended to stimulate monocyte adhesion (Figure 7A)and T-cell recruitment (Figure 7B), although the effects didnot reach statistical significance. These effects are consistentwith previous observations that LPS stimulates monocyteadhesion and T-cell recruitment.5,18 In mice deficient inapoCI, LPS treatment did not affect macrophage area (Figure7C), SMC area (Figure 7D), or collagen area (Figure 7E), asevident from a comparison of vehicle-treated and LPS-treatedapoe�/�apoc1�/� mice. In contrast, in mice expressing apoCI,LPS increased the macrophage area significantly (P�0.05;Figure 7C), tended to increase SMC area (Figure 7D), andsignificantly increased collagen area (P�0.05; Figure 7E), asevident from a comparison of vehicle-treated apoe�/�
apoc1�/� mice and LPS-treated apoe�/�apoc1�/� mice.In both the vehicle- and LPS-treated groups, apoCI expres-
sion did not affect monocyte adhesion (Figure 7A), yet itshowed a tendency to increase T-cell recruitment (Figure 7B)and macrophage area (Figure 7C), as evident from a compar-ison of the vehicle- or LPS-treated apoe�/�apoc1�/� mice andthe vehicle- or LPS-treated apoe�/�apoc1�/� mice. Further-more, in the LPS-treated groups, apoCI expression signifi-
cantly increased SMC (Figure 7D) and collagen (Figure 7E)area, as evident from a comparison of LPS-treated apoe�/�
apoc1�/� mice and LPS-treated apoe�/�apoc1�/� mice.Taken together, apoCI expression accelerated atheroscle-
rosis after treatment with LPS. As a result, the atheroscleroticlesions increased in size and concomitantly contained moreSMCs and collagen, which reflected the progression ofatherosclerosis.
DiscussionGram-negative bacteria such as C pneumoniae release LPSon multiplication or lysis during infections, which leads to achronic inflammatory state that accelerates atherosclerosis inhumans and rodents.9–18 We have shown previously thatapoCI binds to LPS, thereby augmenting the inflammatoryresponse to LPS and K pneumoniae in mice and in macro-phages in vitro.22 In the present study, we investigated thesignificance of these observations for a chronic inflammatorydisease, atherosclerosis, by assessing the effect of apoCIexpression on LPS-induced atherosclerosis in apoe�/� mice invivo. We found that endogenous apoCI increased atheroscle-rosis development in apoe�/� mice induced by chronic treat-ment with LPS.
We showed that injection of LPS similarly increased theTC content of IDL/LDL and HDL in both apoe�/�apoc1�/�
and apoe�/�apoc1�/� mice and marginally increased TGlevels. This response was transient, which indicates that the
Figure 7. Effect of apoCI on LPS-induced ath-erosclerotic lesion composition. In 4 cross sec-tions of the aortic root (5 �m), at 40-�m inter-vals, lesion composition was assessed byimmunohistochemistry with respect to mono-cyte adhesion (AIA-31240– and hematoxylin-positive; A), CD3� T cells (CD3�; B), macro-phages (AIA-31240–positive; C), collagen(Sirius-red–positive; D), and SMCs (�-actin–positive; E). Values are expressed as numberof cells that stained positive (A, B) or as totalarea (C through E). Each data point representsthe mean per mouse. Each line indicates themean of the data points. n�8 to 11. *P�0.05;**P�0.01.
2178 Circulation November 6, 2007
by guest on February 17, 2015http://circ.ahajournals.org/Downloaded from

effects were induced after LPS injection, returning to theirstarting value before the next LPS injection. It is tempting tospeculate about the mechanism underlying the lipid changesin the present study after LPS injection. Serum amyloid A andapoAI, which were both elevated after LPS injection, cancontribute to an increased HDL plasma level by stimulatingHDL generation.32,33 In addition, serum amyloid A can inhibitHDL clearance.34 In contrast to the present findings, HDLcholesterol decreases in humans upon challenge with LPS.35
We speculate that the increased HDL cholesterol in thepresent mouse model is mainly related to reduced clearanceof HDL as a consequence of the absence of apoE thatmediates the clearance of HDL, as reported previously.36 Theincreased cholesterol content in IDL/LDL might be second-ary to reduced LPL activity, because LPS injection decreasesLPL activity.37 The effects of LPS on lipid levels weretransient and similar in both mouse models, and because theincreased levels of plasma cholesterol contributed only �5%to the total cholesterol exposure over time until atheroscle-rosis assessment (not shown), it is unlikely that the transientincrease in plasma lipid levels after LPS injection was aprimary contributor to development of atherosclerosis.
The augmenting effect of apoCI on LPS-induced athero-sclerosis is related to a greater inflammatory status in LPS-treated apoe�/�apoc1�/� mice than in apoe�/�apoc1�/� mice.We showed that apoCI expression enhanced the effect of LPSon fibrinogen and E-selectin. Fibrinogen is an acute-phaseprotein that is secreted from the liver and that reflects thegeneral inflammatory status of mice and putatively partici-pates in atherosclerotic lesion development.38 Fibrinogen isprimarily regulated by interleukin-6.39 E-selectin is a targetgene of the nuclear factor–�B pathway in macrophages andreflects the inflammatory state of the vessel wall.40 ApoCIthus increased LPS-induced inflammatory responses bothsystemically and at the vascular level. Although the differ-ence in magnitude of the LPS-induced inflammatory responsewas relatively small, it has recently been stated in humansthat small effects on inflammatory responses contributesubstantially to CVD.41 ApoCI expression per se did notaffect these inflammatory markers in vehicle-treated mice, anobservation that is in agreement with the observed compara-ble atherosclerotic lesion area in vehicle-treated apoe�/�
apoc1�/� and apoe�/�apoc1�/� mice. In addition, apoCI ex-pression by apoe�/� macrophages per se increased the LPS-induced inflammatory response, and apoCI on HDL fromapoe�/� mice was crucial for increasing the inflammatoryresponse of macrophages toward LPS.
A recent human study showed that HDL cholesterolnegatively correlates with the response to a single adminis-tration of LPS.30 Because apoCI is mainly localized on HDL,it may have been expected that apoCI would be associatedwith a decreased LPS response in the present mouse study.However, although HDL apoCI correlated with HDL phos-pholipids, owing to the distribution of both components in theparticle shell, HDL apoCI did not correlate at all with HDLcholesterol. Therefore, the present study does not exclude thepossibility that apoCI also increases the LPS response inhumans.
We thus conclude that apoCI accelerates LPS-inducedatherosclerosis progression in apoe�/� mice mainly as aconsequence of increasing inflammation. This is in line withthe finding that LPS-treated apoe�/� mice expressing humanapoA-IV show reduced production of proinflammatory cyto-kines and reduced atherosclerosis compared with theirapoe�/� littermates.25
LPS treatment did not affect lesion composition of micedeficient in apoCI, yet it increased macrophage and collagenarea in mice expressing apoCI. The increased macrophagecontent might have been the consequence of the increasedinflammation in macrophages after apoCI expression, as weshowed in vitro. However, comparison of lesions from bothvehicle groups and LPS-injected groups with the sameatherosclerotic lesion area revealed no differences regardingatherosclerotic lesion composition (not shown). Also, inadvanced lesions, we observed adventitial infiltrates of acti-vated lymphocytes, which have been reported to be caused byLPS injections in apoe�/� mice.18 Because we only observedthese infiltrates in advanced atherosclerotic lesions, we sug-gest that these infiltrates were the consequence of the severityof the lesion rather than being caused by LPS specifically. Inaddition, it has been reported that apoCI increases apoptosisin human aortic SMCs by recruiting neutral sphingomyeli-nase.42 It is unlikely that this effect contributed to atheroscle-rosis in the aortic root in the present study, because apoCI didnot affect SMCs in vehicle-treated mice.
At first glance, the finding that apoCI expression did notsignificantly affect atherosclerosis in the vehicle-treated miceappears to be in contrast to our previous data showing thatapoCI accelerates atherosclerosis as related to increasedVLDL lipid levels in mice of 26 weeks of age.26 However, inthe present study, VLDL lipid levels were somewhat in-creased on apoCI expression, yet the absolute values in bothmouse genotypes were �20% lower than in our previousstudy. In fact, the relatively low lipid levels in the presentstudy led to a relatively slow progression of atherosclerosisthat was not different between mice of both genotypes, andthis enabled us to investigate the effect of apoCI on LPS-induced atherosclerosis specifically. Indeed, mice need toexpress a certain level of hyperlipidemia for LPS to have aneffect on atherosclerosis development, as was demonstratedin former studies using C pneumoniae, which mainly exertsits effects via LPS.15,43,44 We speculate that the effects ofapoCI on LPS-induced atherosclerosis are mediated viaTLR4 in macrophages, because TLR4 is primarily activatedby LPS45 and has been demonstrated to be involved inLPS-induced atherosclerosis development in mice.16 Themechanism behind the augmenting effect of apoCI on theresponse toward LPS is currently under investigation.
We conclude that apoCI is crucially involved in LPS-inducedatherosclerosis in apoe�/� mice, mainly as a consequence ofenhancing the inflammatory response. We anticipate that inhumans who suffer from chronic inflammation, plasma apoCImay enhance atherosclerosis development and CVD.
AcknowledgmentsWe thank Erik Offerman and Karin Toet for excellent technicalassistance.
Westerterp et al ApoC-I Is Crucial for LPS-Induced Atherosclerosis 2179
by guest on February 17, 2015http://circ.ahajournals.org/Downloaded from

Sources of FundingThis work was performed within the framework of the “LeidenCenter for Cardiovascular Research LUMC-TNO” and was sup-ported by the Netherlands Organization for Scientific Research(NWO-VIDI grant 917.36.351 to Dr Rensen, program grant903.39.291 to Dr Havekes, and NWO-VENI grant 016.036.061 to DrKleemann), the Netherlands Heart Foundation (NHS grant2005B226 to Dr Rensen), and the LUMC (Gisela Thier Fellowshipto Dr Rensen).
DisclosuresNone.
References1. Glass CK, Witztum JL. Atherosclerosis: the road ahead. Cell. 2001;104:
503–516.2. Hansson GK. Mechanisms of disease: inflammation, atherosclerosis, and
coronary artery disease. N Engl J Med. 2005;352:1685–1695.3. Ross R. Mechanisms of disease: atherosclerosis: an inflammatory disease.
N Engl J Med. 1999;340:115–126.4. Kiechl S, Wiedermann CJ, Willeit J. Toll-like receptor 4 and athero-
genesis. Ann Med. 2003;35:164–171.5. Zhang FX, Kirschning CJ, Mancinelli R, Xu XP, Jin YP, Faure E,
Mantovani A, Rothe M, Muzio M, Arditi M. Bacterial lipopolysaccharideactivates nuclear factor-kappa B through interleukin-1 signalingmediators in cultured human dermal endothelial cells and mononuclearphagocytes. J Biol Chem. 1999;274:7611–7614.
6. Edfeldt K, Swedenborg J, Hansson GK, Yan ZQ. Expression of toll-likereceptors in human atherosclerotic lesions: a possible pathway for plaqueactivation. Circulation. 2002;105:1158–1161.
7. Xu XH, Shah PK, Faure E, Equils O, Thomas L, Fishbein MC, LuthringerD, Xu XP, Rajavashisth TB, Yano J, Kaul S, Arditi M. Toll-likereceptor-4 is expressed by macrophages in murine and human lipid-richatherosclerotic plaques and upregulated by oxidized LDL. Circulation.2001;104:3103–3108.
8. Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, Frees K,Watt JL, Schwartz DA. TLR4 mutations are associated with endotoxinhyporesponsiveness in humans. Nat Genet. 2000;25:187–191.
9. Campbell LA, Kuo CC. Chlamydia pneumoniae: an infectious risk factorfor atherosclerosis? Nat Rev Microbiol. 2004;2:23–32.
10. Mussa FF, Chai H, Wang X, Yao Q, Lumsden AB, Chen C. Chlamydiapneumoniae and vascular disease: an update. J Vasc Surg. 2006;43:1301–1307.
11. Nabipour I, Vahdat K, Jafari SM, Pazoki R, Sanjdideh Z. The associationof metabolic syndrome and Chlamydia pneumoniae, Helicobacter pylori,cytomegalovirus, and herpes simplex virus type 1: the Persian GulfHealthy Heart Study. Cardiovasc Diabetol. 2006;5:25.
12. Hauer AD, de Vos P, Peterse N, ten Cate H, Van Berkel TJ, Stassen FR,Kuiper J. Delivery of Chlamydia pneumoniae to the vessel wallaggravates atherosclerosis in LDLr�/� mice. Cardiovasc Res. 2006;69:280–288.
13. Ezzahiri R, Nelissen-Vrancken HJ, Kurvers HA, Stassen FR, Vliegen I,Grauls GE, van Pul MM, Kitslaar PJ, Bruggeman CA. Chlamydophilapneumoniae (Chlamydia pneumoniae) accelerates the formation ofcomplex atherosclerotic lesions in Apo E3-Leiden mice. Cardiovasc Res.2002;56:269–276.
14. Liu L, Hu H, Ji H, Murdin AD, Pierce GN, Zhong G. Chlamydiapneumoniae infection significantly exacerbates aortic atherosclerosis inan LDLR�/� mouse model within six months. Mol Cell Biochem.2000;215:123–128.
15. Moazed TC, Campbell LA, Rosenfeld ME, Grayston JT, Kuo CC.Chlamydia pneumoniae infection accelerates the progression of athero-sclerosis in apolipoprotein E-deficient mice. J Infect Dis. 1999;180:238–241.
16. Vink A, Schoneveld AH, van der Meer JJ, van Middelaar BJ, Sluijter JP,Smeets MB, Quax PH, Lim SK, Borst C, Pasterkamp G, de Kleijn DP. Invivo evidence for a role of toll-like receptor 4 in the development ofintimal lesions. Circulation. 2002;106:1985–1990.
17. Lehr HA, Sagban TA, Ihling C, Zahringer U, Hungerer KD, Blumrich M,Reifenberg K, Bhakdi S. Immunopathogenesis of atherosclerosis:endotoxin accelerates atherosclerosis in rabbits on hypercholesterolemicdiet. Circulation. 2001;104:914–920.
18. Ostos MA, Recalde D, Zakin MM, Scott-Algara D. Implication of naturalkiller T cells in atherosclerosis development during a LPS-inducedchronic inflammation. FEBS Lett. 2002;519:23–29.
19. Jong MC, Hofker MH, Havekes LM. Role of ApoCs in lipoproteinmetabolism: functional differences between ApoC1, ApoC2, and ApoC3.Arterioscler Thromb Vasc Biol. 1999;19:472–484.
20. Berbee JFP, van der Hoogt CC, Sundararaman D, Havekes LM, RensenPCN. Severe hypertriglyceridemia in human APOC1 transgenic mice iscaused by apoC-I-induced inhibition of LPL. J Lipid Res. 2005;46:297–306.
21. Westerterp M, de Haan W, Berbee JF, Havekes LM, Rensen PC. Endog-enous apoC-I increases hyperlipidemia in apoE-knockout mice by stim-ulating VLDL production and inhibiting LPL. J Lipid Res. 2006;47:1203–1211.
22. Berbee JF, van der Hoogt CC, Kleemann R, Schippers EF, Kitchens RL,van Dissel JT, Bakker-Woudenberg IA, Havekes LM, Rensen PC. Apo-lipoprotein CI stimulates the response to lipopolysaccharide and reducesmortality in Gram-negative sepsis. FASEB J. 2006;20:2162–2164.
23. vanRee JH, Vandenbroek WJJA, vanderZee A, Dahlmans VEH,Wieringa B, Frants RR, Havekes LM, Hofker MH. Inactivation of Apoeand Apoc1 by 2 consecutive rounds of gene targeting: effects onmessenger-RNA expression levels of gene-cluster members. Hum MolGenet. 1995;4:1403–1409.
24. Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hyper-cholesterolemia and arterial lesions in mice lacking apolipoprotein-E.Science. 1992;258:468–471.
25. Recalde D, Ostos MA, Badell E, Garcia-Otin AL, Pidoux J, Castro G,Zakin MM, Scott-Algara D. Human apolipoprotein A-IV reducessecretion of proinflammatory cytokines and atherosclerotic effects of achronic infection mimicked by lipopolysaccharide. Arterioscler ThrombVasc Biol. 2004;24:756–761.
26. Westerterp M, vanEck M, de Haan W, Offerman EH, Van Berkel TJ,Havekes LM, Rensen PCN. Apolipoprotein CI aggravates atherosclerosisdevelopment in ApoE-knockout mice despite mediating cholesterol effluxfrom macrophages. Atherosclerosis. 2007 Feb 22 [E-pub ahead of print].
27. Westerterp M, van der Hoogt CC, de Haan W, Offerman EH,Dallinga-Thie GM, Jukema JW, Havekes LM, Rensen PC. Cholesterylester transfer protein decreases high-density lipoprotein and severelyaggravates atherosclerosis in APOE*3-Leiden mice. Arterioscler ThrombVasc Biol. 2006;26:2552–2559.
28. Kockx M, Gervois PP, Poulain P, Derudas B, Peters JM, Gonzalez FJ,Princen HM, Kooistra T, Staels B. Fibrates suppress fibrinogen geneexpression in rodents via activation of the peroxisome proliferator-acti-vated receptor-alpha. Blood. 1999;93:2991–2998.
29. Hu L, Boesten LS, May P, Herz J, Bovenschen N, Huisman MV, BerbeeJF, Havekes LM, van Vlijmen BJ, Tamsma JT. Macrophage low-densitylipoprotein receptor-related protein deficiency enhances atherosclerosis inApoE/LDLR double knockout mice. Arterioscler Thromb Vasc Biol.2006;26:2710–2715.
30. Birjmohun RS, van Leuven SI, Levels JH, van’t Veer C, Kuivenhoven JA,Meijers JC, Levi M, Kastelein JJ, van der PT, Stroes ES. High-densitylipoprotein attenuates inflammation and coagulation response on endotoxinchallenge in humans. Arterioscler Thromb Vasc Biol. 2007;27:1153–1158.
31. Levels JH, Abraham PR, van den EA, van Deventer SJ. Distribution andkinetics of lipoprotein-bound endotoxin. Infect Immun. 2001;69:2821–2828.
32. Abe-Dohmae S, Kato KH, Kumon Y, Hu W, Ishigami H, Iwamoto N,Okazaki M, Wu CA, Tsujita M, Ueda K, Yokoyama S. Serum amyloid Agenerates high density lipoprotein with cellular lipid in an ABCA-1 orABCA7-dependent manner. J Lipid Res. 2006;47:1542–1550.
33. Timmins JM, Lee JY, Boudyguina E, Kluckman KD, Brunham LR,Mulya A, Gebre AK, Coutinho JM, Colvin PL, Smith TL, Hayden MR,Maeda N, Parks JS. Targeted inactivation of hepatic Abca1 causesprofound hypoalphalipoproteinemia and kidney hypercatabolism ofapoA-I. J Clin Invest. 2005;115:1333–1342.
34. Cai L, de Beer MC, de Beer FC, van der Westhuyzen DR. Serum amyloidA is a ligand for scavenger receptor class B type I and inhibits highdensity lipoprotein binding and selective lipid uptake. J Biol Chem.2005;280:2954–2961.
35. Hudgins LC, Parker TS, Levine DM, Gordon BR, Saal SD, Jiang XC,Seidman CE, Tremaroli JD, Lai J, Rubin AL. A single intravenous doseof endotoxin rapidly alters serum lipoproteins and lipid transfer proteinsin normal volunteers. J Lipid Res. 2003;44:1489–1498.
36. Arai T, Rinninger F, Varban L, Fairchild-Huntress V, Liang CP, Chen W,Seo T, Deckelbaum R, Huszar D, Tall AR. Decreased selective uptake of
2180 Circulation November 6, 2007
by guest on February 17, 2015http://circ.ahajournals.org/Downloaded from

high density lipoprotein cholesteryl esters in apolipoprotein E knock-outmice. Proc Natl Acad Sci U S A. 1999;96:12050–12055.
37. Feingold KR, Staprans I, Memon RA, Moser AH, Shigenaga JK, DoerrlerW, Dinarello CA, Grunfeld C. Endotoxin rapidly induces changes in lipidmetabolism that produce hypertriglyceridemia: low doses stimulatehepatic triglyceride production while high doses inhibit clearance. J LipidRes. 1992;33:1765–1776.
38. Kooistra T, Verschuren L, de Vries-van der Weij, Koenig W, Toet K,Princen HM, Kleemann R. Fenofibrate reduces atherogenesis inApoE*3Leiden mice: evidence for multiple antiatherogenic effectsbesides lowering plasma cholesterol. Arterioscler Thromb Vasc Biol.2006;26:2322–2330.
39. Haziot A, Lin XY, Zhang F, Goyert SM. The induction of acute phaseproteins by lipopolysaccharide uses a novel pathway that is CD14-independent. J Immunol. 1998;160:2570–2572.
40. Michelsen KS, Doherty TM, Shah PK, Arditi M. TLR signaling: anemerging bridge from innate immunity to atherogenesis. J Immunol.2004;173:5901–5907.
41. Pussinen PJ, Tuomisto K, Jousilahti P, Havulinna AS, Sundvall J,Salomaa V. Endotoxemia, immune response to periodontal pathogens,and systemic inflammation associate with incident cardiovascular diseaseevents. Arterioscler Thromb Vasc Biol. 2007;27:1433–1439.
42. Kolmakova A, Kwiterovich P, Virgil D, Alaupovic P, Knight-Gibson C,Martin SF, Chatterjee S. Apolipoprotein C-I induces apoptosis in humanaortic smooth muscle cells via recruiting neutral sphingomyelinase. Arte-rioscler Thromb Vasc Biol. 2004;24:264–269.
43. Blessing E, Campbell LA, Rosenfeld ME, Kuo CC. Chlamydia pneu-moniae and hyperlipidemia are co-risk factors for atherosclerosis:infection before induction of hyperlipidemia does not accelerate devel-opment of atherosclerotic lesions in C57BL/6J mice. Infect Immun. 2002;70:5332–5334.
44. Hu H, Pierce GN, Zhong G. The atherogenic effects of chlamydia aredependent on serum cholesterol and specific to Chlamydia pneumoniae.J Clin Invest. 1999;103:747–753.
45. Michelsen KS, Doherty TM, Shah PK, Arditi M. Role of Toll-likereceptors in atherosclerosis. Circ Res. 2004;95:e96–e97.
CLINICAL PERSPECTIVEAccumulating evidence indicates that Gram-negative bacteria such as Chlamydia pneumoniae and Porphyromonasgingivalis are involved in cardiovascular disease. Infection with these pathogens may cause a chronic inflammatory stateas a result of the release of endotoxins such as lipopolysaccharide (LPS), which accelerates the development ofatherosclerosis in humans and rodents. It has been generally assumed that high-density lipoprotein (HDL) binds LPS,thereby attenuating the inflammatory response, which may contribute to the antiatherosclerotic potential of HDL. However,we have shown previously that apolipoprotein C-I (apoCI), a small and highly positively charged surface apolipoproteinthat is mainly HDL-associated, binds LPS and augments the inflammatory response toward LPS and Gram-negativebacteria in mice and macrophages. In the present study, we investigated the significance of these observations foratherogenesis. We assessed the effect of endogenous apoCI expression on atherosclerosis in apoE-knockout mice that weretreated chronically with LPS. Although the present study confirmed that LPS itself aggravates atherosclerosis inapoE-knockout mice, apoCI appeared to be a crucial determinant of LPS-induced aggravation of atherosclerosis.Endogenous apoCI expression enhanced inflammation both systemically and at the level of the vessel wall. In addition,HDL-associated apoCI increased the inflammatory response toward LPS in macrophages in vitro. These data thus indicatethat plasma apoCI may increase the cardiovascular disease risk associated with Gram-negative bacterial infections.Furthermore, the present study underscores the importance of the apolipoprotein composition of HDL for theantiatherosclerotic potential of HDL, which should be considered in the design and analysis of HDL-raising strategiesaimed at reducing cardiovascular disease risk.
Westerterp et al ApoC-I Is Crucial for LPS-Induced Atherosclerosis 2181
by guest on February 17, 2015http://circ.ahajournals.org/Downloaded from

Kleemann, Johannes A. Romijn, Louis M. Havekes and Patrick C.N. RensenMarit Westerterp, Jimmy F.P. Berbée, Nuno M.M. Pires, Geertje J.D. van Mierlo, Robert
Knockout Mice−Development in Apolipoprotein E Apolipoprotein C-I Is Crucially Involved in Lipopolysaccharide-Induced Atherosclerosis
Print ISSN: 0009-7322. Online ISSN: 1524-4539 Copyright © 2007 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation doi: 10.1161/CIRCULATIONAHA.107.693382
2007;116:2173-2181; originally published online October 22, 2007;Circulation.
http://circ.ahajournals.org/content/116/19/2173World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circ.ahajournals.org/content/suppl/2007/10/17/CIRCULATIONAHA.107.693382.DC1.htmlData Supplement (unedited) at:
http://circ.ahajournals.org//subscriptions/
is online at: Circulation Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer this process is available in the
click Request Permissions in the middle column of the Web page under Services. Further information aboutOffice. Once the online version of the published article for which permission is being requested is located,
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the EditorialCirculationin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on February 17, 2015http://circ.ahajournals.org/Downloaded from
Copyright © 2022 FDOKUMEN




















