
Differential oxidation of apolipoprotein E isoforms and interaction with phospholipids
12
Original Contribution DIFFERENTIAL OXIDATION OF APOLIPOPROTEIN E ISOFORMS AND INTERACTION WITH PHOSPHOLIPIDS CORINNE JOLIVALT,* BRIGITTE LEININGER-MULLER,* PHILIPPE BERTRAND,* R ´ EGINE HERBER,* YVES CHRISTEN, ² and G ´ ERARD SIEST* From the *Centre du Me ´dicament, UPRES, Faculte ´ de Pharmacie, Universite ´ Henri Poincare ´ Nancy 1, Nancy, and ² Institut Ipsen, Paris, France (Received 19 February 1999; Revised 25 October 1999; Accepted 25 October 1999) Abstract—Accumulation of oxidized proteins has been demonstrated in the brain of patients suffering from Alzhei- mer’s disease (AD). Among the proteins found in cerebral amyloid deposits, apolipoprotein (apo) E is a polymorphic protein which one specific isoform, apo E4, has been widely associated with AD. Apo E may be linked with AD by its isoform-specific interaction with lipids or other proteins in amyloid plaques. Using the myeloperoxidase oxidative system, we report that oxidation of the three recombinant apo E isoforms is differential (as estimated using immunoblot and high-performance liquid chromatography analysis), with apo E4 being more susceptible than apo E3, which in turn is much more susceptible than apo E2. In addition, susceptibility to thrombin proteolysis is reduced when apo E is oxidized, and oxidation of apo E decreases its incorporation into phospholipid discs by approximately 50%. Oxidation of apo E may contribute to inefficient lipid recycling in the brain, particularly regarding apo E4 and E3. Our results link and strengthen both the e4 allele linkage with AD and the role of protein oxidation in AD. The cerebral mechanisms underlying apo E oxidation and/or myeloperoxidase functions in vivo remain to be assessed. © 2000 Elsevier Science Inc. Keywords—Apolipoprotein E, Oxidation, Myeloperoxidase, Alzheimer’s disease, Thrombin proteolysis, Phospholip- ids, Free radicals INTRODUCTION Free radicals are inevitable byproducts of biological re- dox reactions. The formation of free-oxygen species in the central nervous system is widely accepted as being one of the pathogenic mechanisms leading to neurode- generative diseases [1,2]. The brain is particularly vulnerable to oxidative dam- age because of its low levels of antioxidants and its high level of oxygen consumption. Several forms of protein oxidation may occur, including formation of carbonyls, attack on particular amino acid residues, and oxidation of sulfhydryl groups. Oxidation may lead to cross-linking of proteins, thus increasing protein susceptibility to pro- teolysis, conformational changes, and loss of biological function [3]. An increase of the cerebral content of oxidized proteins has been reported during normal aging, and this increase is more pronounced in neurodegenera- tive disorders such as Parkinson’s and Alzheimer’s dis- ease (AD) [4,5]. To our knowledge, no study regarding the in vitro oxidation of the specific target proteins implicated in AD has been reported to date. Apolipoprotein (apoE) is one of the major proteins colocalized with amyloid-b peptide (Ab) in cerebral senile plaques of patients with AD. Human apo E is a polymorphic protein which three main isoforms (E2, E3, and E4) are the products of three alleles (e2, e3, and e4) that differ from each other by a single mutation [6]. Apo E has been implicated in lipid homeostasis, and its main function in the brain is to locally recycle cholesterol and phospholipids in response to injury [7]. Apolipoprotein E received particular consideration in the field of AD research after demonstration of the link- age of the e4 allele with AD [7,8]. In a complementary fashion, apo E2 seems to be protective against AD [9]. Address correspondence to: G. Siest, Centre du Me ´dicament, Uni- versite ´ Henri Poincare ´ Nancy 1, 30 rue Lionnois, 54000 Nancy, France; Tel: 133 (0) 3-83-17-88-34; Fax: 133 (0) 3-83-32-13-22; E-Mail: [email protected]. Free Radical Biology & Medicine, Vol. 28, No. 1, pp. 129 –140, 2000 Copyright © 2000 Elsevier Science Inc. Printed in the USA. All rights reserved 0891-5849/00/$–see front matter PII S0891-5849(99)00232-4 129
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Differential oxidation of apolipoprotein E isoforms and interaction with phospholipids

Original Contribution
DIFFERENTIAL OXIDATION OF APOLIPOPROTEIN E ISOFORMS ANDINTERACTION WITH PHOSPHOLIPIDS
CORINNE JOLIVALT ,* BRIGITTE LEININGER-MULLER,* PHILIPPE BERTRAND,* REGINE HERBER,* Y VES CHRISTEN,†
and GERARD SIEST*From the *Centre du Me´dicament, UPRES, Faculte´ de Pharmacie, Universite´ Henri Poincare´ Nancy 1, Nancy, and
†Institut Ipsen, Paris, France
(Received19 February1999;Revised25 October1999;Accepted25 October1999)
Abstract—Accumulation of oxidized proteins has been demonstrated in the brain of patients suffering from Alzhei-mer’s disease (AD). Among the proteins found in cerebral amyloid deposits, apolipoprotein (apo) E is a polymorphicprotein which one specific isoform, apo E4, has been widely associated with AD. Apo E may be linked with AD by itsisoform-specific interaction with lipids or other proteins in amyloid plaques. Using the myeloperoxidase oxidativesystem, we report that oxidation of the three recombinant apo E isoforms is differential (as estimated using immunoblotand high-performance liquid chromatography analysis), with apo E4 being more susceptible than apo E3, which in turnis much more susceptible than apo E2. In addition, susceptibility to thrombin proteolysis is reduced when apo E isoxidized, and oxidation of apo E decreases its incorporation into phospholipid discs by approximately 50%. Oxidationof apo E may contribute to inefficient lipid recycling in the brain, particularly regarding apo E4 and E3. Our results linkand strengthen both thee4 allele linkage with AD and the role of protein oxidation in AD. The cerebral mechanismsunderlying apo E oxidation and/or myeloperoxidase functions in vivo remain to be assessed. © 2000 Elsevier ScienceInc.
Keywords—Apolipoprotein E, Oxidation, Myeloperoxidase, Alzheimer’s disease, Thrombin proteolysis, Phospholip-ids, Free radicals
INTRODUCTION
Free radicals are inevitable byproducts of biological re-dox reactions. The formation of free-oxygen species inthe central nervous system is widely accepted as beingone of the pathogenic mechanisms leading to neurode-generative diseases [1,2].
The brain is particularly vulnerable to oxidative dam-age because of its low levels of antioxidants and its highlevel of oxygen consumption. Several forms of proteinoxidation may occur, including formation of carbonyls,attack on particular amino acid residues, and oxidation ofsulfhydryl groups. Oxidation may lead to cross-linkingof proteins, thus increasing protein susceptibility to pro-teolysis, conformational changes, and loss of biologicalfunction [3]. An increase of the cerebral content of
oxidized proteins has been reported during normal aging,and this increase is more pronounced in neurodegenera-tive disorders such as Parkinson’s and Alzheimer’s dis-ease (AD) [4,5].
To our knowledge, no study regarding the in vitrooxidation of the specific target proteins implicated in ADhas been reported to date. Apolipoprotein (apoE) is oneof the major proteins colocalized with amyloid-b peptide(Ab) in cerebral senile plaques of patients with AD.Human apo E is a polymorphic protein which three mainisoforms (E2, E3, and E4) are the products of threealleles (e2, e3, ande4) that differ from each other by asingle mutation [6]. Apo E has been implicated in lipidhomeostasis, and its main function in the brain is tolocally recycle cholesterol and phospholipids in responseto injury [7].
Apolipoprotein E received particular consideration inthe field of AD research after demonstration of the link-age of thee4 allele with AD [7,8]. In a complementaryfashion, apo E2 seems to be protective against AD [9].
Address correspondence to: G. Siest, Centre du Me´dicament, Uni-versiteHenri Poincare´ Nancy 1, 30 rue Lionnois, 54000 Nancy, France;Tel: 133 (0) 3-83-17-88-34; Fax:133 (0) 3-83-32-13-22; E-Mail:[email protected].
Free Radical Biology & Medicine, Vol. 28, No. 1, pp. 129–140, 2000Copyright © 2000 Elsevier Science Inc.Printed in the USA. All rights reserved
0891-5849/00/$–see front matter
PII S0891-5849(99)00232-4
129

The relationship between AD and apo E is thus isoformdependent. It may derive from an apo E isoform-specificinteraction with Ab, the principal constituent of senileplaques [10]; with tau, the major constituent of tangles[11]; with lipids [12]; or from its decreased levels in ADbrain [13]. The differential implication of apo E isoformsin AD may be reinforced or correlated with a differentialdegradation of apo E isoforms resulting from oxidativemodifications. Such modifications could generate apo Efragments, as has been observed with several proteins[14]. In addition, the 10 kDa, thrombin-cleavage C-terminal fragment of apo E has been detected in senileplaques and shown to form fibrils [15]. The 22-kDaN-terminal fragment of apo E4 is more neurotoxic thanthe apo E3 fragment [16]. Moreover, apo E/Ab interac-tions have been proposed to be favored when apo E isoxidized [10], and we recently showed that apo E4oxidized by the myeloperoxidase (MPO; EC 1.11.1.7)enzymatic system significantly enhances Ab fibrillogen-esis, whereas nonoxidized apo E4 does not modify Abfibrillogenesis. This characteristic was specific for theapo E4 isoform, because we did not observe any differ-ence between oxidized and nonoxidized apo E3 [17].
It now is generally accepted that Ab aggregates pro-duce free radicals [18] and, therefore, that the colocal-ized apo E could be a target for these radicals and mayeven function as a local antioxidant [19]. Moreover,peroxidation of lipid gives rise to reactive byproductssuch as malondialdehyde and 4-hydroxynonenal, whichseem to covalently modify and cross-link apo E, thusaltering its metabolism [20]. Direct apolipoprotein oxi-dation, however, occurs independently of lipid oxidation[21,22] or earlier than lipid peroxidation during acutepancreatitis [23] or after oxidation of low-density li-poprotein (LDL) by the MPO system [24]. We previ-ously investigated the susceptibility of both recombinantapo E3 and apo E4 to free radicals produced in vitro bya strong vanadyl/hydrogen peroxide oxidizing system.Under these conditions, both apo E isoforms were frag-mented and/or polymerized [25].
Such a strong oxidation system, however, does notcorrespond to the physiologic conditions representativeof the progressive accumulation of free radical speciesduring aging. Thus, we studied apo E oxidation in a morephysiologic manner, with a milder MPO oxidizing sys-tem [26]. This enzyme, which is present in both poly-morphonuclear neutrophils and monocytes, is involvedin atherosclerosis via its implication in inflammatoryprocesses [27]. Formation of AD senile plaques alsodepends on inflammation [28], and we recently reportedthe presence of MPO in human brain [29]. In the pres-ence of H2O2 and Cl2, MPO catalyzes peroxidation ofchloride to hypochloride, which then rapidly interactswith the oxidizable moieties of protein [26]. Using this in
vitro oxidizing system, lipid-free apo E3 and E4 wereboth oxidized to a greater extent than albumin [29]. Mostphysiologic apo E, however, is complexed with lipids toform high-density lipoprotein (HDL)–like particles incerebrospinal fluid [30], and interaction of apo E withcellular lipoprotein receptors can only be achieved if theapo E is embedded in an adequate lipid environment [6].Thus, we prepared associated apo E–phospholipid com-plexes to mimic physiologic conditions.
The aim of this study was to compare the susceptibil-ity of the three major apo E isoforms to MPO oxidation.We also studied the consequences of apo E oxidation onits susceptibility to thrombin proteolysis, its interactionwith phospholipid, and the effect of the lipid environ-ment on apo E oxidation.
MATERIALS AND METHODS
Materials
The three recombinant human apo E (E2, E3, E4)cDNAs were cloned in a modified pARHS-2 vector andproduced inEscherichia coli BL21 (DE3) as fusionproteins with an apparent molecular weight (MW) ofapproximately 43 kDa. The 43 kDa apo E results fromthe fusion of apo E with a fusion peptide (4.5 kDa)containing a polyhistidine sequence on the N-terminalend of the apo E. This polyhistidine tag allows purifica-tion of the recombinant proteins by a single-step affinitychromatography on nickel gel [31] without modifyingthe major properties of the apo E isoforms [32]. Thethree recombinant apo Es were lyophilized and dena-tured for 2 h at room temperature in 4 M HCl-guanidine,100 mM Tris-HCl (pH, 8.0), 1 mM ethylenediaminetet-raacetic acid (EDTA), 2%b-mercaptoethanol. The sam-ples were then dialyzed against 0.1 M NH4HCO3 (pH,8.8) and stored at 4°C. The initial apo E2 preparation waspartly aggregated (Fig. 1).
Enterokinase was from Invitrogen (Groningen, TheNetherlands), thrombin from Boehringer Mannheim(Mannheim, Germany), and MPO from Calbiochem (LaJolla, CA, USA). Acrylamide and bis-acrylamide werefrom BDH, and the gel gradient (4–20%) was fromNovex. All other chemicals were from Sigma Chemical(St. Louis, MO, USA). Plasma apo AI was kindly pro-vided by Dr V. V. Shuvaev [33] and plasma apo E by DrA. Dergunov.
Hydrolysis assays
Cleavage of the fusion peptide was performed byincubating 20mg of apo E with 1 unit of enterokinase in50 mM Tris-HCl buffer (pH, 8.0), 1 mM CaCl2, 0.1%Tween 20 (Merck, Germany), and 1 Mb-mercaptoetha-
130 C. JOLIVALT et al.
nol for 12 h at room temperature. The reaction wasstopped by placing the sample on ice. The sample wasthen concentrated and washed on Centricon 30 (Amicon)to eliminate both fusion peptide andb-mercaptoethanol.
Thrombin cleavage occurred after introduction of ar-ginine residues to produce two major fragments (30 and10 kDa) for recombinant apo E. Twenty micrograms ofapo E was incubated with thrombin (120 U/mg) at aprotein:enzyme ratio of 50:1 (w:w) in 100 mM bicarbon-ate buffer for 24 h at room temperature. The reaction wasstopped by placing the sample on ice.
For cyanogen bromide (CNBr) cleavage, 20mg of apoE was incubated overnight at room temperature with thesame volume of CNBr diluted in 70% HCOOH. The
reaction was stopped by diluting the sample 10-fold withwater, and it was then lyophilized. After lyophilization,50 ml of 4 M HCl-guanidine was added for denaturation,and then 150ml of 100 mM bicarbonate buffer wasadded for reverse-phase high-performance liquid chro-matography (RP-HPLC) analysis.
Oxidation assays
In vitro oxidation assays of the recombinant proteinswith MPO isolated from polymorphonuclear neutrophils[34] were performed as described elsewhere [29], withslight modifications of pH adapted to the formation ofapo E–phospholipid discoidal complexes. Briefly, chlo-ramines were formed by the reaction of MPO at a finalconcentration of 10mg/ml in 55 mM phosphate buffer(pH, 4.5), 100 mM NaCl, and 10 mM leucine withdifferent concentrations of H2O2 (0, 0.625, 1.25, 3, 7.6,and 12.5 mM). Then, 1ml of the solution containing theextemporaneously formed chloramines was added to 0.5mg of native apo E at physiologic pH for 30 min in a finalvolume of 5ml at room temperature [35]. Proteins weresubjected to 12% sodium dodecyl sulfate–polyacrylam-ide gel-electrophoresis (SDS-PAGE) under nonreducingconditions and then transferred to PVDF (polyvinylidenefluoride, Millipore, Bedford, MA, USA) membranes.
The protein carbonyl content was evaluated by form-ing labeled protein hydrazone derivatives using 2,4-di-nitrophenyl hydrazine (DNPH) [36]. The dinitrophenylmoieties formed on apo E were revealed using enhancedchemiluminescence (Pierce, Rockford, IL, USA) afterincubation of the blot with rabbit anti-DNP antibodies(Sigma), which was then followed by incubation withhorseradish peroxidase–labeled antirabbit antibodies(Sigma). Alternatively, blots were incubated with mono-clonal antiapo E antibodies (kind gift from Dr. Y. Mar-cel, Montreal, Canada), which was followed by incuba-tion with secondary antimouse antibodies labeled withHRP (Sigma) and then revealed using enhanced chemi-luminescence.
Analysis of apparent molecular weight was performedusing Biomax 1D software (Kodak. Rochester, NY,USA). Apparent molecular weights of the bands werecalculated according to low-molecular-weight proteinstandards from 14 to 97 kDa (Biorad; Hercules, CA,USA).
To strip off the bound antibodies, the membrane wasincubated for 30 min at 50°C in 100 mMb-mercapto-ethanol, 2% sodium dodecyl sulfate, and 62.5 mM Tris-HCl (pH, 6.7) buffer. The membrane was then washedwith TBST-buffer (20 mM Tris-HCl [pH, 7.6], 137 mMNaCl, 0.1 % Tween 20).
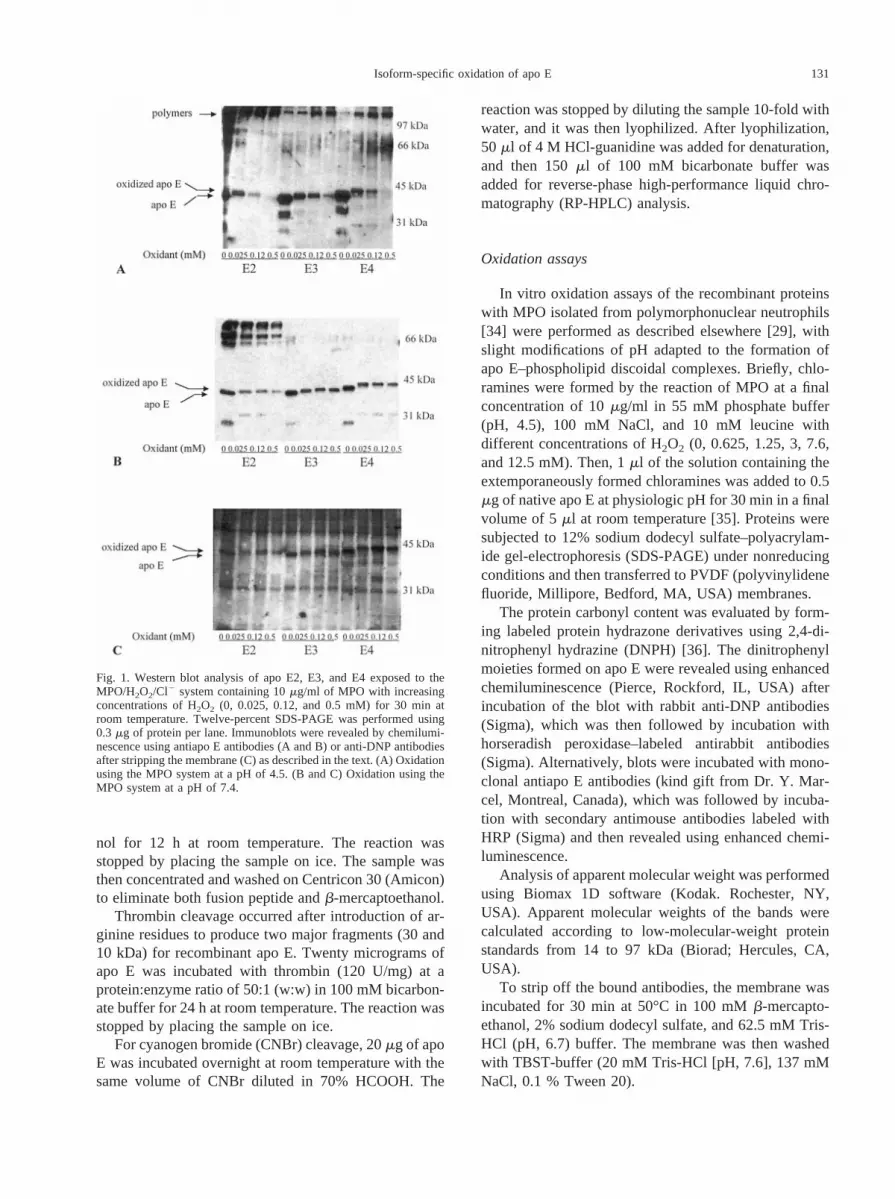
Fig. 1. Western blot analysis of apo E2, E3, and E4 exposed to theMPO/H2O2/Cl2 system containing 10mg/ml of MPO with increasingconcentrations of H2O2 (0, 0.025, 0.12, and 0.5 mM) for 30 min atroom temperature. Twelve-percent SDS-PAGE was performed using0.3 mg of protein per lane. Immunoblots were revealed by chemilumi-nescence using antiapo E antibodies (A and B) or anti-DNP antibodiesafter stripping the membrane (C) as described in the text. (A) Oxidationusing the MPO system at a pH of 4.5. (B and C) Oxidation using theMPO system at a pH of 7.4.
131Isoform-specific oxidation of apo E

Separation of apo E by RP-HPLC
Both oxidized and nonoxidized apo E were resolvedusing RP-HPLC, which was performed on a RPC-4column (5 m, 4.6 3 150 mm; Vydac, Hesperia, CA,USA) using a gradient of acetonitrile in 0.1% trifluoro-acetic acid at a flow rate of 1 ml/min, from 20 to 40% for10 min, and followed by a gradient from 40 to 60% for30 min on an HPLC pump 422 system (Kontron, Everett,MA, USA). Peaks were detected at 220 nm using aspectrophotometer (Waters, Milford, MA, USA).
Preparation of discoidal complexes of apo E
Discoidal complexes were prepared using a detergentreconstitution method adapted from that of Jonas [37].The phospholipid used, dipalmitoyl phosphatidyl-choline(DPPC), was dispersed at a concentration of 6 mg/ml ina 50 mM Tris (pH, 7.4), 150 mM NaCl, 0.1 mM EDTA,and 0.02% NaN3 buffer containing 7.5 mM sodiumcholate. The phospholipid was incubated with apo Eaccording to a 3:1 (w:w) DPPC:protein ratio. The spon-taneously formed discs were isolated by adsorption ofthe cholate on Bio Beads (Biorad). The solution contain-ing the complexes was then subjected to gel filtration ona Sephacryl HR300 column (1.63 55 cm, PharmaciaLKB Biotechnology; Piscataway, NJ, USA) to separatethe apo E–DPPC discs from free apo E and free phos-pholipid. Fractions of 2 ml were collected. Materialeluted up to fraction 10 corresponded to the apo E–DPPCcomplexes. Subsequent fractions contained free apo E.The apo E–DPPC complexes were concentrated usingcentrifugation on a Centriplus 100 (Amicon, Beverly,MA, USA) and were examined at 280 nm for theirprotein content. The lipid content of the complexes was
measured using an enzymatic colorimetric assay (Boehr-inger Mannheim). The same protocol was followed forplasma apo AI, which was used as a control. The com-plexes formed next underwent nondenaturing gel gradi-ent electrophoresis for 5 h at 120 V andwere then stainedwith Coomassie blue reagent.
Statistical analysis
Each experiment was conducted at least three times.When adequate, results were compared using Student’st-test.
RESULTS
Differential oxidation of the apo E isoforms
The electrophoretic profiles of the apo E isoformsafter oxidation by the MPO system are shown in Fig. 1.On oxidation at a pH of 7.4, the apo E–specific bandmigrated, with a higher apparent molecular weight.These shifts in apparent molecular weight increased to aplateau when the oxidant concentration was increased(Fig. 2). These shifts were observed for the three apo Eisoforms after detection with anti-DNP as well as withanti-apo E antibodies (Figs. 1B and 1C), and their levelsdiffered from one isoform to another. These shifts in theapparent molecular weight of apo E were observed evenin the absence of DNPH, thus suggesting they did notresult from the formation of DNP adducts. Because thebackground was less important on anti–apo E than onanti-DNP blots, after assays were investigated using an-tiapo E hybridization. At the highest concentration ofoxidant, the difference in the migration level observedbetween the main apo E band before and after oxidation
Fig. 2. Variations in the apparent molecular weight of the three apo E isoforms after oxidation at a pH of 7.4 plotted as a function ofoxidant concentration. Statistical analysis was performed using a Student’st-test against the corresponding values for apo E4. Resultsrepresent the mean6 SEM of four determinations. *p , .05; **p , .01.
132 C. JOLIVALT et al.

was similar for both apo E4 (DkDa 5 2.0) and apo E3(DkDa 5 1.8) but much lower for apo E2 (DkDa50.5)(Fig. 2). In addition, using a lower concentration ofoxidant (0.05 mM), it seemed that apo E4 was morereadily oxidized than both apo E3 and apo E2 (Fig. 2).After oxidation at the lower pH of 4.5, these shifts in theapparent molecular weight of apo E were even morepronounced (DkDa5 2.2 for apo E4, 2.0 for apo E3, and0.8 for apo E2) (Fig. 1A). Concomitantly, the intensity ofthe major apo E band decreased after oxidation, andhigher–molecular weight bands appeared.
The global decrease in intensity of the apo E bandafter oxidation at a pH of 4.5 as well as at a pH of 7.4could have resulted from technical limitations or de-creased apo E binding to PVDF membrane and/or tooxidative modification of epitopes leading to a lowerrecognition by antibodies, because we did not observethis decrease after Coomassie blue staining (data notshown). Using plasma apo E as well as recombinant apoE with the fusion peptide removed through enterokinasehydrolysis, similar shifts in the apparent molecularweight of apo E were observed after oxidation (data notshown). The results obtained for recombinant apo Eoxidation may reflect oxidation of plasma apo E, thusvalidating the use of recombinant protein during oxida-tive studies. The nonoxidized apo E isoforms (Fig. 1C)as well as the nonoxidized plasma apo E, however, wereall labeled using anti-DNP antibodies, thus suggestingthat recombinant apo E already was partly oxidized toform carbonyl groups. This initial oxidation could haveresulted from the bacterial production and/or the purifi-cation step on nickel gel, but the oxidation of apo E by
the MPO/H2O2/Cl2 system produces additional modifi-cations, such as the shift in apparent molecular weight orthe detection of higher–molecular weight bands. This lastmodification, however, does not seem to support theformation of carbonyl groups. Oxidation by Ni11 is wellaccepted, but Zhuang et al. [38] showed that NiCl2 (0.2mM) produced oxidation of albumin only at a rather highconcentration of H2O2 (5 mM) compared with that usedin our system (0.5 mM). In addition, their results showedthat the carbonyl content after albumin was exposed toNiCl2 was much lower than that when albumin wasexposed to copper. In our study, incubation of humanapo E as well as recombinant apo E with NiCl2 atconcentrations of 0.3 M or lower in the presence orabsence of H2O2 (0.5 mM) did not modify the DNPdetection or the apparent molecular weight of apo E (datanot shown).
Thrombolytic digestion
In general, oxidized proteins seem to be more suscep-tible than nonoxidized proteins to proteolytic degrada-tion [39]. Therefore, we studied the susceptibility of apoE to thrombin proteolysis both before and after oxida-tion. After thrombin digestion and Coomassie blue stain-ing, the three isoforms of apo E were resolved as twobands that corresponded to nonhydrolyzed apo E and toa 30 kDa band that corresponded to the N-terminalfragment, as shown for apo E4 in Figure 3. After 7 h,hydrolysis was not complete. When apo E4 was oxidizedunder both pH conditions (7.4 and 4.5), hydrolysis was
Fig. 3. Apo E4 was exposed to the MPO/H2O2/Cl2 system with 0.5 mM H2O2 at a pH of 4.5 for 30 min at room temperature. Thesamples were then incubated with thrombin according to a protein:thrombin ratio of 50:1 (w:w) for 7 h atroom temperature. Next, 15%SDS-PAGE was performed using 1mg of protein per lane, and the gel was stained with Coomassie blue. Similar profiles were obtainedfor the three isoforms of apo E. Lane 1: apo E4; lane 2: apo E4 hydrolyzed by thrombin; lane 3: oxidized apo E4; lane 4: oxidizedapo E4 hydrolyzed by thrombin.
133Isoform-specific oxidation of apo E
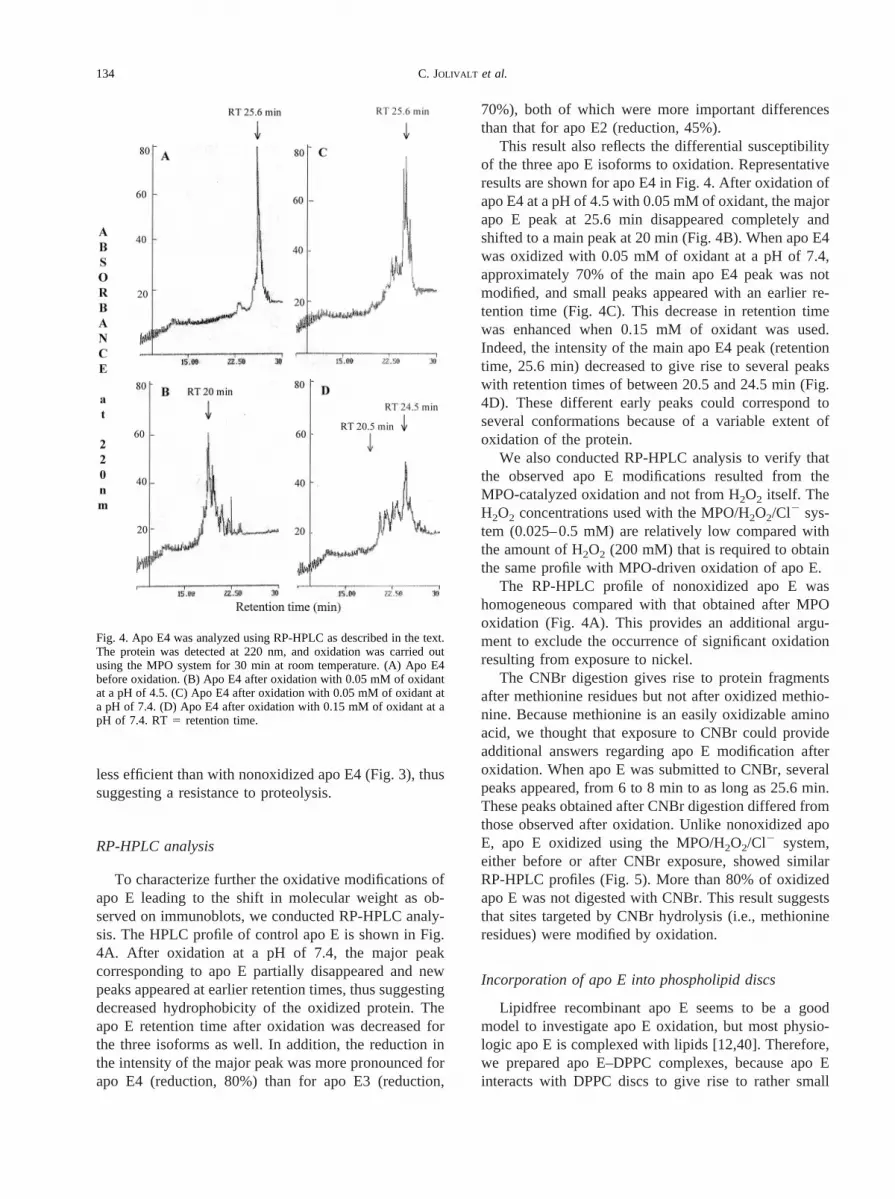
less efficient than with nonoxidized apo E4 (Fig. 3), thussuggesting a resistance to proteolysis.
RP-HPLC analysis
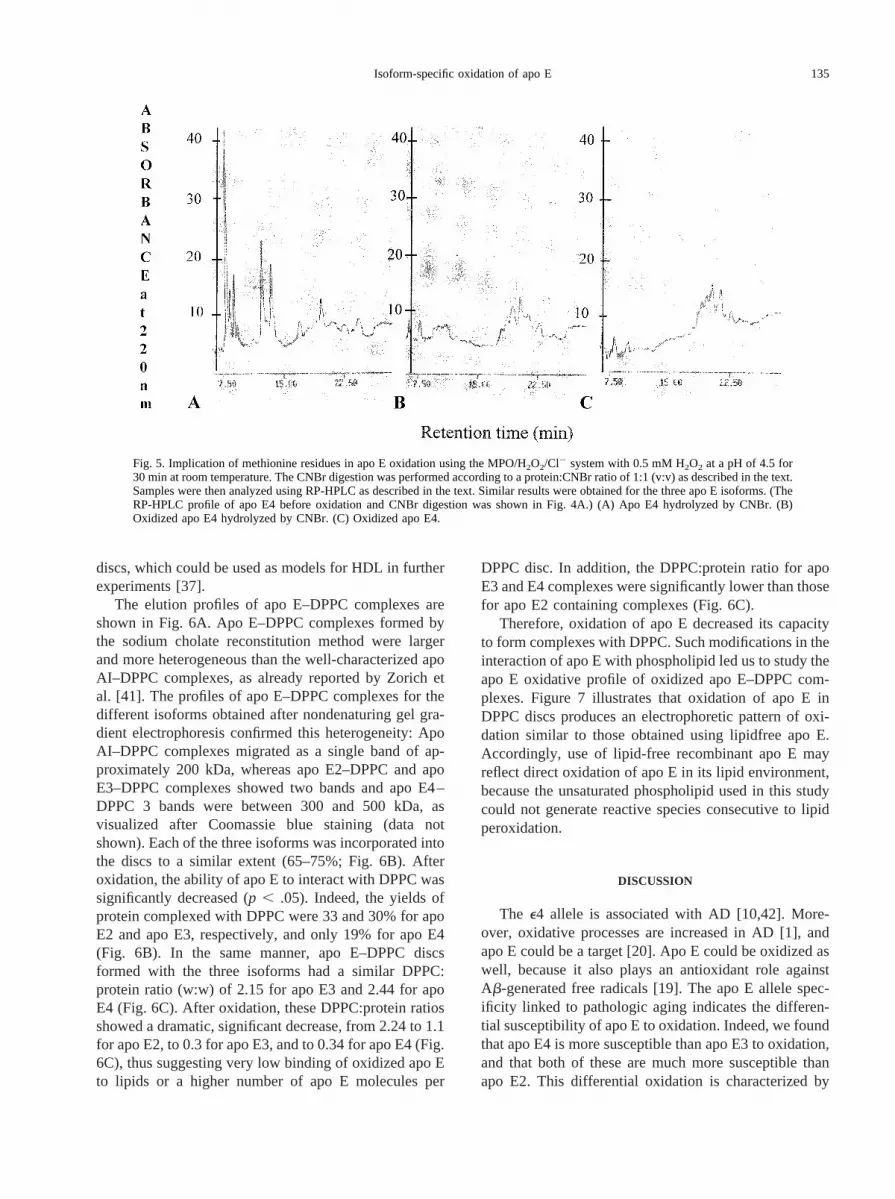
To characterize further the oxidative modifications ofapo E leading to the shift in molecular weight as ob-served on immunoblots, we conducted RP-HPLC analy-sis. The HPLC profile of control apo E is shown in Fig.4A. After oxidation at a pH of 7.4, the major peakcorresponding to apo E partially disappeared and newpeaks appeared at earlier retention times, thus suggestingdecreased hydrophobicity of the oxidized protein. Theapo E retention time after oxidation was decreased forthe three isoforms as well. In addition, the reduction inthe intensity of the major peak was more pronounced forapo E4 (reduction, 80%) than for apo E3 (reduction,
70%), both of which were more important differencesthan that for apo E2 (reduction, 45%).
This result also reflects the differential susceptibilityof the three apo E isoforms to oxidation. Representativeresults are shown for apo E4 in Fig. 4. After oxidation ofapo E4 at a pH of 4.5 with 0.05 mM of oxidant, the majorapo E peak at 25.6 min disappeared completely andshifted to a main peak at 20 min (Fig. 4B). When apo E4was oxidized with 0.05 mM of oxidant at a pH of 7.4,approximately 70% of the main apo E4 peak was notmodified, and small peaks appeared with an earlier re-tention time (Fig. 4C). This decrease in retention timewas enhanced when 0.15 mM of oxidant was used.Indeed, the intensity of the main apo E4 peak (retentiontime, 25.6 min) decreased to give rise to several peakswith retention times of between 20.5 and 24.5 min (Fig.4D). These different early peaks could correspond toseveral conformations because of a variable extent ofoxidation of the protein.
We also conducted RP-HPLC analysis to verify thatthe observed apo E modifications resulted from theMPO-catalyzed oxidation and not from H2O2 itself. TheH2O2 concentrations used with the MPO/H2O2/Cl2 sys-tem (0.025–0.5 mM) are relatively low compared withthe amount of H2O2 (200 mM) that is required to obtainthe same profile with MPO-driven oxidation of apo E.
The RP-HPLC profile of nonoxidized apo E washomogeneous compared with that obtained after MPOoxidation (Fig. 4A). This provides an additional argu-ment to exclude the occurrence of significant oxidationresulting from exposure to nickel.
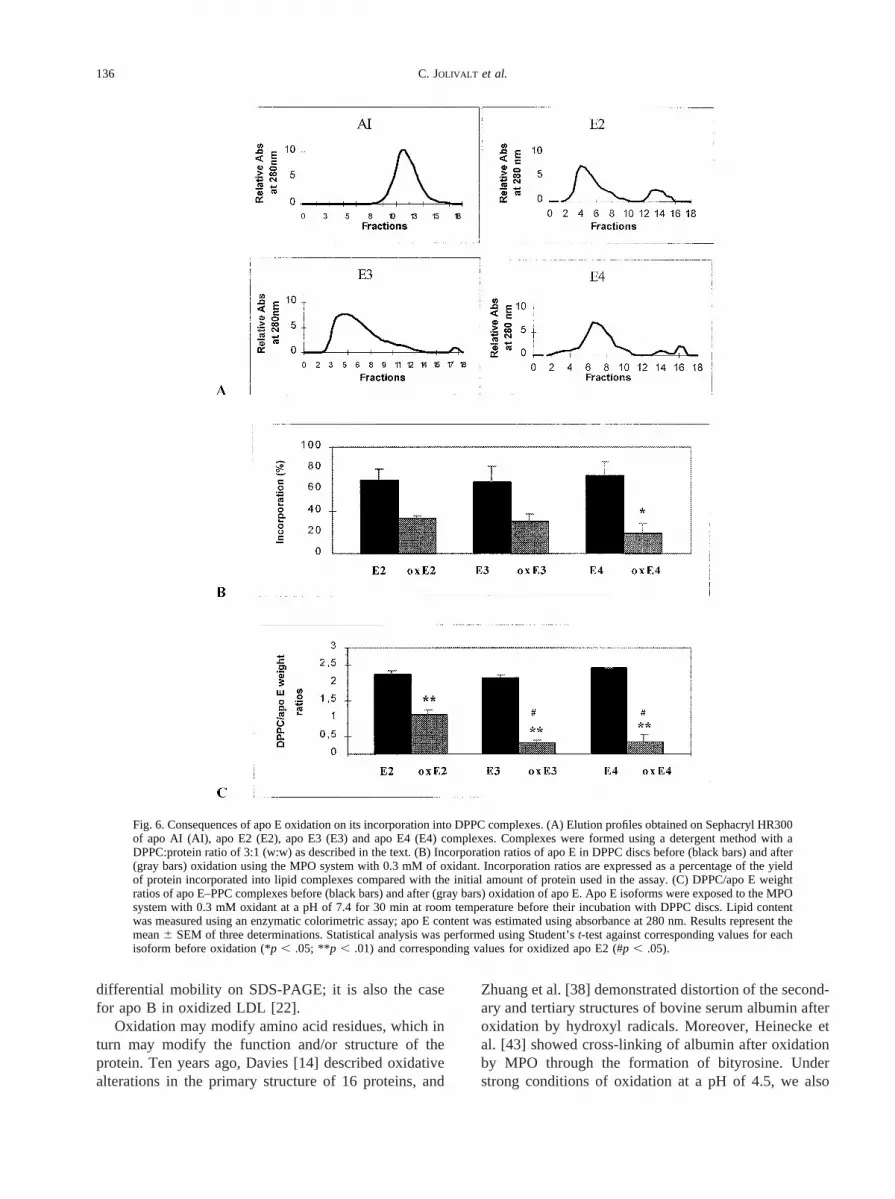
The CNBr digestion gives rise to protein fragmentsafter methionine residues but not after oxidized methio-nine. Because methionine is an easily oxidizable aminoacid, we thought that exposure to CNBr could provideadditional answers regarding apo E modification afteroxidation. When apo E was submitted to CNBr, severalpeaks appeared, from 6 to 8 min to as long as 25.6 min.These peaks obtained after CNBr digestion differed fromthose observed after oxidation. Unlike nonoxidized apoE, apo E oxidized using the MPO/H2O2/Cl2 system,either before or after CNBr exposure, showed similarRP-HPLC profiles (Fig. 5). More than 80% of oxidizedapo E was not digested with CNBr. This result suggeststhat sites targeted by CNBr hydrolysis (i.e., methionineresidues) were modified by oxidation.
Incorporation of apo E into phospholipid discs
Lipidfree recombinant apo E seems to be a goodmodel to investigate apo E oxidation, but most physio-logic apo E is complexed with lipids [12,40]. Therefore,we prepared apo E–DPPC complexes, because apo Einteracts with DPPC discs to give rise to rather small
Fig. 4. Apo E4 was analyzed using RP-HPLC as described in the text.The protein was detected at 220 nm, and oxidation was carried outusing the MPO system for 30 min at room temperature. (A) Apo E4before oxidation. (B) Apo E4 after oxidation with 0.05 mM of oxidantat a pH of 4.5. (C) Apo E4 after oxidation with 0.05 mM of oxidant ata pH of 7.4. (D) Apo E4 after oxidation with 0.15 mM of oxidant at apH of 7.4. RT5 retention time.
134 C. JOLIVALT et al.
discs, which could be used as models for HDL in furtherexperiments [37].
The elution profiles of apo E–DPPC complexes areshown in Fig. 6A. Apo E–DPPC complexes formed bythe sodium cholate reconstitution method were largerand more heterogeneous than the well-characterized apoAI–DPPC complexes, as already reported by Zorich etal. [41]. The profiles of apo E–DPPC complexes for thedifferent isoforms obtained after nondenaturing gel gra-dient electrophoresis confirmed this heterogeneity: ApoAI–DPPC complexes migrated as a single band of ap-proximately 200 kDa, whereas apo E2–DPPC and apoE3–DPPC complexes showed two bands and apo E4–DPPC 3 bands were between 300 and 500 kDa, asvisualized after Coomassie blue staining (data notshown). Each of the three isoforms was incorporated intothe discs to a similar extent (65–75%; Fig. 6B). Afteroxidation, the ability of apo E to interact with DPPC wassignificantly decreased (p , .05). Indeed, the yields ofprotein complexed with DPPC were 33 and 30% for apoE2 and apo E3, respectively, and only 19% for apo E4(Fig. 6B). In the same manner, apo E–DPPC discsformed with the three isoforms had a similar DPPC:protein ratio (w:w) of 2.15 for apo E3 and 2.44 for apoE4 (Fig. 6C). After oxidation, these DPPC:protein ratiosshowed a dramatic, significant decrease, from 2.24 to 1.1for apo E2, to 0.3 for apo E3, and to 0.34 for apo E4 (Fig.6C), thus suggesting very low binding of oxidized apo Eto lipids or a higher number of apo E molecules per
DPPC disc. In addition, the DPPC:protein ratio for apoE3 and E4 complexes were significantly lower than thosefor apo E2 containing complexes (Fig. 6C).
Therefore, oxidation of apo E decreased its capacityto form complexes with DPPC. Such modifications in theinteraction of apo E with phospholipid led us to study theapo E oxidative profile of oxidized apo E–DPPC com-plexes. Figure 7 illustrates that oxidation of apo E inDPPC discs produces an electrophoretic pattern of oxi-dation similar to those obtained using lipidfree apo E.Accordingly, use of lipid-free recombinant apo E mayreflect direct oxidation of apo E in its lipid environment,because the unsaturated phospholipid used in this studycould not generate reactive species consecutive to lipidperoxidation.
DISCUSSION
The e4 allele is associated with AD [10,42]. More-over, oxidative processes are increased in AD [1], andapo E could be a target [20]. Apo E could be oxidized aswell, because it also plays an antioxidant role againstAb-generated free radicals [19]. The apo E allele spec-ificity linked to pathologic aging indicates the differen-tial susceptibility of apo E to oxidation. Indeed, we foundthat apo E4 is more susceptible than apo E3 to oxidation,and that both of these are much more susceptible thanapo E2. This differential oxidation is characterized by
Fig. 5. Implication of methionine residues in apo E oxidation using the MPO/H2O2/Cl2 system with 0.5 mM H2O2 at a pH of 4.5 for30 min at room temperature. The CNBr digestion was performed according to a protein:CNBr ratio of 1:1 (v:v) as described in the text.Samples were then analyzed using RP-HPLC as described in the text. Similar results were obtained for the three apo E isoforms. (TheRP-HPLC profile of apo E4 before oxidation and CNBr digestion was shown in Fig. 4A.) (A) Apo E4 hydrolyzed by CNBr. (B)Oxidized apo E4 hydrolyzed by CNBr. (C) Oxidized apo E4.
135Isoform-specific oxidation of apo E
differential mobility on SDS-PAGE; it is also the casefor apo B in oxidized LDL [22].
Oxidation may modify amino acid residues, which inturn may modify the function and/or structure of theprotein. Ten years ago, Davies [14] described oxidativealterations in the primary structure of 16 proteins, and
Zhuang et al. [38] demonstrated distortion of the second-ary and tertiary structures of bovine serum albumin afteroxidation by hydroxyl radicals. Moreover, Heinecke etal. [43] showed cross-linking of albumin after oxidationby MPO through the formation of bityrosine. Understrong conditions of oxidation at a pH of 4.5, we also
Fig. 6. Consequences of apo E oxidation on its incorporation into DPPC complexes. (A) Elution profiles obtained on Sephacryl HR300of apo AI (AI), apo E2 (E2), apo E3 (E3) and apo E4 (E4) complexes. Complexes were formed using a detergent method with aDPPC:protein ratio of 3:1 (w:w) as described in the text. (B) Incorporation ratios of apo E in DPPC discs before (black bars) and after(gray bars) oxidation using the MPO system with 0.3 mM of oxidant. Incorporation ratios are expressed as a percentage of the yieldof protein incorporated into lipid complexes compared with the initial amount of protein used in the assay. (C) DPPC/apo E weightratios of apo E–PPC complexes before (black bars) and after (gray bars) oxidation of apo E. Apo E isoforms were exposed to the MPOsystem with 0.3 mM oxidant at a pH of 7.4 for 30 min at room temperature before their incubation with DPPC discs. Lipid contentwas measured using an enzymatic colorimetric assay; apo E content was estimated using absorbance at 280 nm. Results represent themean6 SEM of three determinations. Statistical analysis was performed using Student’st-test against corresponding values for eachisoform before oxidation (*p , .05; **p , .01) and corresponding values for oxidized apo E2 (#p , .05).
136 C. JOLIVALT et al.

observed that oxidative modification of apo E can lead toproducts of polymerization [29]. As shown by Anan-tharamaiah et al. [44] through a combination of RP-HPLC and CNBr cleavage, oxidation of apo AI methio-nine residues alters the secondary structure of theprotein. In the present study, we demonstrated usingRP-HPLC that the modification of apo E induced byoxidation decreases its retention time, which in turnssuggests a decrease of its hydrophobicity and/or a mod-ification of its conformation. The noncleavage of oxi-dized apo E by CNBr indicated that apo E methionineswere oxidized. Methionine residues have recently beenproposed to act as internal antioxidants in proteins [45],and they could play a significant role in the reduction ofhydroperoxides of cholesteryl esters and phosphatidylcholine associated with human HDL, as shown for apoAI and AII methionine residues [46]. Depending on theexposure of the methionine residues (8 in apo E), wemight expect a differential capacity of apo E isoforms toresist attack by reactive species. Such properties could becorrelated with the differential antioxidant properties ofapo E isoforms [19]. The similarity of oxidative modifi-cations between recombinant apo E and the same proteinless most of its fusion peptide containing four additionalmethionines indicates that the fusion peptide does notinterfere with apo E oxidation by endogenous methio-nine residues.
The different susceptibilities to oxidation for the threeapo E isoforms also may be linked to their tertiarystructure because of a variable exposure of oxidizableamino acid residues. Discrete amino acid residues maymediate isoform-specific conformational changes that al-ter the interaction of apo E with lipids or receptors[12,47]. The primary structure of apo E could be in-volved, because the three isoforms differ in their cysteinecontent at positions 112 and 158. Cysteine residues also
may act as antioxidants [48]. Apo E4 is Arg112–Arg158,apo E3 is Arg112–Cys158, and apo E2 is Cys112–Cys158.
Modification of apo E after MPO-driven oxidationmay alter its susceptibility to proteases. Oxidized apo Eisoforms showed no greater susceptibility to thrombincleavage than the nonoxidized isoforms, but they wererather resistant to the cleavage. This result is similar tothe increased resistance of oxidized albumin to proteol-ysis compared with that of native albumin [49]. Thisproteolytic resistance of apo E after oxidation could notcorrelate with the presence of thrombolytic fragments ofapo E in senile plaques [15], but it might contribute to anincreased local concentration of full-length oxidized apoE, which would enhance Ab fibrillogenesis, especiallyfor apo E4 [17].
Oxidative alterations of apo E could modify some ofits many functional properties. The discoidal apo E–D-PPC complexes are a good model of lipid particles in thebrain. Indeed, the particles produced by nascent astro-cytes are discoidal and contain both apo E and apo J,which is in contrast with CSF lipoproteins, which arespherical and contain esterified cholesterol, apo E, J, AI,and AII [40]. The decreased incorporation of oxidizedapo E into DPPC is in agreement, however, with thereduction of apo AI incorporation into dimyristol phos-phatidycholine complexes after oxidation [44]. Theseresults suggest that the oxidative modifications of apo Eaffect its lipid-binding capacity, or that oxidized apo Ecan disorganize lipid micelles. It should be remembered,however, that apo E plays its role in lipid recyclingthrough binding to several specific membrane receptors[6]. This binding can only be achieved when apo E isembedded in an adequate lipid environment. Oxidationof apo E then may lead to alterations of its functionalproperties, which could dramatically impair phospho-
Fig. 7. Western blot analysis of apo E–DPPC complexes oxidized by the MPO/H2O2/Cl2 system. The different samples were exposedto increased concentrations of oxidant (0, 0.025, and 0.5 mM) at a pH of 7.4 for 30 min at room temperature. Twelve-percentSDS-PAGE was performed using 0.3mg of protein per lane. The immunoblot was revealed by chemiluminescence using antiapo Eantibodies as described in the text. E2: apo E2; E2/DPPC: apo E2 in DPPC discs; E3: apo E3; E3/DPPC: apo E3 in DPPC discs; E4:apo E4; E4/DPPC: apo E4 in DPPC discs.
137Isoform-specific oxidation of apo E

lipid recycling in the brain. This hypothesis is in agree-ment with the known loss of cerebral cholesterol andphospholipid in patients with AD [50], with the decreaseof neuronal plasticity in the brains of patients with AD[51], and with the decreased compensatory response ofapo E–deficient mice to entorhinal cortex lesions [52].
Incorporation of recombinant apo E into discoidalcomplexes did not affect its susceptibility to oxidation,thus indicating that the changes in conformation apo Eundergoes on unfolding in a phospholipid phase do notseem to modify the exposure of oxidizable amino acidmoieties to free radicals. Thus, our results suggest thatlipid-free recombinant apo E represents a good modelfor use in further studies involving oxidation. As theconsequences of oxidation at a pH of 4.5 are moremarked than those at a pH of 7.4, free radicals prob-ably are the major factor responsible for apo E mod-ification in the present work, but we cannot excludethe coexistence of both aldehyde adduct formation anddirect radical attack of amino acids at a pH of 7.4.Further analysis of apo E oxidation should include thesearch for aldehyde adducts using specific antibodiesand the variations of oxidized apo E profile afterincorporation into various kinds of lipid complexes.Because MPO is likely to mediate preferential andfaster oxidation of an apo E moiety of lipoproteins, asshown for apo B-100 after LDL oxidation [24], thegeneral promotion of lipid peroxidation in neuropatho-logic aging will contribute further to modifying apo Ethrough reactive aldehyde adduct formation [20].
CONCLUSION
In conclusion, in vitro MPO-driven oxidation of apo Eleads to isoform-dependent susceptibility, with apo E4being the most and apo E2 the least oxidizable of thethree isoforms studied. In addition to carbonyl contentmeasurement in pathologic conditions [23,53,54], ourresults highlight that not only carbonyl content but alsosulfhydryl content or additional amino acid oxidativemodification of protein should be sought, in particularregarding AD. Modifications of apo E induced by oxi-dation render the protein resistant to thrombin proteoly-sis and alter its interaction with phospholipid. The lowerincorporation of oxidized apo E, and more particularlyapo E4, into phospholipids may induce a less efficientbinding to lipoprotein receptors and modify the lipidmetabolism, ultimately rendering as inefficient the lipidrecycling and neuronal plasticity in the brains of patientswith AD. Considered together, our results link andstrengthen both thee4 allele linkage with, and the role ofoxidation in, AD. At least two antioxidant treatments,such as selegiline and vitamin E [55] or Ginkgo bilobaextract (EGb 761) [56], improve the cognitive functions
of patients with AD. Because cerebral apo E function isa crucial determinant of lipid metabolism and, likely, ofantioxidant balance as well, such antioxidant treatmentmay help to prevent apo E oxidation and to improve lipidrecycling in AD brain.
The rate of oxidative modifications should further beassessed by quantification of oxidized amino acid resi-dues using mass spectrometry as well as specific aminoacid modification analysis, such as lysine acetylation andformation of chlorotyrosine. It now is important to assessthe cerebral mechanisms underlying apo E oxidationand/or MPO functions as well.
Acknowledgments— This work was supported by the Ipsen Institute(Paris), the European Community MICAD research program (EN E105200), the Region Lorrainem and Parke Davis. We thank Dr. A.Dergunov for helping to set up the apo E–phospholipid associationassay and Dr. V. Shuvaev for providing the apo AI. We also thank Dr.Y. Marcel for the kind gift of monoclonal anti–apo E antibodies and Dr.L. Aggerbeck for critical reading of the manuscript.
REFERENCES
[1] Halliwell, B. Reactive oxygen species and the central nervoussystem.J. Neurochem.59:1609–1623; 1992.
[2] Frolich, L.; Riederer, P. Free radical mechanisms in dementia ofAlzheimer type and the potential for antioxidative treatment.Drug. Res.45:443–446; 1995.
[3] Berlett, B. S.; Stadtman, E. R. Protein oxidation in aging, diseaseand oxidative stress.J. Biol. Chem.272:20313–20316; 1997.
[4] Smith, C. D.; Carney, J. M.; Starke-Reed, P. E.; Oliver, C. N.;Stadtman, E. R.; Floyd, R. A.; Markesbery, W. R. Excess brainprotein oxidation and enzyme dysfunction in normal aging and inAlzheimer’s disease.Proc. Natl. Acad. Sci. USA88:10540–10543;1991.
[5] Carney, J. M.; Carney, A. M. Role of protein oxidation in agingand in age-associated neurodegenerative diseases.Life Sci. 55:2097–2103; 1994.
[6] Mahley, R. W. Apolipoprotein E: cholesterol transport proteinwith expanding role in cell biology.Science240:622–630; 1988.
[7] Poirier, J. Apolipoprotein E in animal models of brain injury andin Alzheimer’s disease.Trends Neurosci.12:525–530; 1994.
[8] Siest, G.; Pillot, T.; Re´gis-Bailly, A.; Leininger Muller, B.; Stein-metz, J.; Galteau, M. M.; Visvikis, S. Apolipoprotein E: animportant gene and protein to follow in laboratory medicine.Clin.Chem.41:1068–086; 1995.
[9] West, H. L.; Rebeck, G. W.; Hyman, B. T. Frequency of theapolipoprotein E epsilon 2 allele is diminished in sporadic Alz-heimer disease.Neurosci. Lett.175:46–48; 1994.
[10] Strittmatter, W. J.; Saunders, A.; Schmechel, D.; Pericakvance,M.; Englhild, J.; Salvesen, O. S.; Roses, A. D. Apolipoprotein E:high affinity bindingb-amyloid and increased frequency of type4 allele in late onset Alzheimer’s disease.Proc. Natl. Acad. Sci.USA90:1977–1981; 1993.
[11] Strittmatter, W. J.; Saunders, A. M.; Goedert, M.; Weisgraber,K. H.; Dong, L.; Jakes, R.; Huang, D. Y.; Pericak-Vance, M.;Schmechel, D.; Roses, A. D. Isoform-specific interactions ofapolipoprotein E with microtubule-associated protein tau: impli-cations for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA91:11183–11186; 1994.
[12] Weisgraber, K. H. Apolipoprotein E: structure-function relation-ships.Adv. Protein Chem.45:249–302; 1994.
[13] Bertrand, P.; Poirier, J.; Oda, T.; Finch, C. E.; Pasinetti, G. M.Association of apolipoprotein E genotype with brain levels ofapolipoprotein E and apolipoprotein J (clusterin) in Alzheimerdisease.Mol. Brain Res.33:174–178; 1995.
138 C. JOLIVALT et al.

[14] Davies, K. J. A. Protein damage and degradation by oxygenradicals. II. Modification of amino acids.J. Biol. Chem.262:9895–9901; 1987.
[15] Castano, E. M.; Prelli, F.; Pras, M.; Frangione, B. ApolipoproteinE carboxyl-terminal fragments are complexed to amyloids A andL. Implications for amyloidogenesis and Alzheimer’s disease.J. Biol. Chem.270:17610–17615; 1995.
[16] Marques, M. A.; Tolar, M.; Harmony, J. A. K.; Crutcher, K. A. Athrombin cleavage fragment of apolipoprotein E exhibits isoform-specific neurotoxicity.Neuroreport7:2529–2532; 1996.
[17] Leininger Muller, B.; Jolivalt, C.; Bertrand, P.; Siest, G. Oxida-tion of human apolipoprotein E and cerebral enzymatic systems.Alzheimer’s Rep.1:95–99; 1998.
[18] Hensley, K.; Carney, J. M.; Mattson, M. P.; Aksenova, M.; Harris,M.; Wu, J. F.; Floyd, R. A.; Butterfield, D. A. A model forb-amyloid aggregation and neurotoxicity based on free radicalgeneration by the peptide: relevance to Alzheimer’s disease.Proc.Natl. Acad. Soc. USA91:3270–3274; 1994.
[19] Miyata, M.; Smith, J. D. Apolipoprotein E allele-specific antiox-idant activity and effects on cytotoxicity by oxidative insults andb-amyloid peptides.Nature Genet.14:55–61; 1996.
[20] Montine, T. J.; Huang, D. Y.; Valentine, W. M.; Amarnath, V.;Saunders, A.; Weisgraber, K. H.; Graham, D. G.; Strittmatter,W. J. Crosslinking of apolipoprotein E by products of lipidperoxidation. J. Neuropathol. Exp. Neurol.55:202–210; 1996.
[21] Hunt, J. V.; Bailey, J. R.; Schultz, D. L.; McKay, A. G.; Michin-son, M. J. Apolipoprotein oxidation in the absence of lipid per-oxidation enhances LDL uptake by macrophage.FEBS Lett.349:375–379; 1994.
[22] Noguchi, N.; Gotoh, N.; Niki, E. Effects of ebselen and probucolon oxidative modifications of lipid and protein of low densitylipoprotein induced by free radicals.Biochim. Biophys. Acta1213:176–182; 1994.
[23] Reinheckel, T.; Nedelev, B.; Prause, J.; Augustin, W.; Schulz,H. U.; Lippert, H.; Halangk, W. Occurrence of oxidatively mod-ified proteins: an early event in experimental acute pancreatitis.Free Radic. Biol. Med.24:393–400; 1998.
[24] Jerlich, A.; Fabjan, J. S.; Tschabushing, S.; Smirnova, A. V.;Horakova, L.; Hayn, M.; Auer, H.; Guttenberger, H.; Leis, H. J.;Tatzber, F.; Waeg, G.; Schaur, R. J. Human low density lipopro-tein as a target of hypochloride generated by myeloperoxidase.Free Radic. Biol. Med.24:1139–1148; 1998.
[25] Leininger Muller, B.; Jolivalt, C.; Pillot, T.; Lagrange, P.; Liver-toux, M. H.; Grassiot, M. C.; Minn, A.; Siest, G. . ApolipoproteinE and Alzheimer’s disease. In: Roses A. D.; Weisgraber K. H.;Christen Y., eds.Research and perspectives in Alzheimer’s dis-ease.Paris: Springer-Verlag; 1995: 161–69.
[26] Drozdz, R.; Naskalski, J. W. Action of myeloperoxidase-hydro-gen peroxide-chloride system on the egg white lysozyme.ActaBiochim. Pol.35:277–286; 1988.
[27] Heineke, J. W. Mechanisms of oxidative damage of low densitylipoprotein in human atherosclerosis.Curr. Opin. Lipidol.8:268–274; 1997.
[28] Kisilevsky, R. Inflammation-associated amyloidogenesis. Les-sons for Alzheimer’s amyloidogenesis.Mol. Neurobiol.8:65–66;1993.
[29] Jolivalt, C.; Leininger Muller, B.; Drozdz, R.; Naskalski, J. W.;Siest, G. Apolipoprotein E is highly susceptible to oxidation bymyeloperoxidase, an enzyme present in the brain.Neurosci. Lett.210:61–64; 1996.
[30] Pitas, R. E.; Boyles, J. K.; Lee, S. H.; Hui, D.; Weisgraber, K. H.Lipoproteins and their receptors in the central nervous system.Characterization of the lipoproteins in cerebrospinal fluid andidentification of apolipoprotein B, E (LDL) receptors in the brain.J. Biol. Chem.262:14352–14360; 1987.
[31] Janknecht, R.; de Martynoff, G.; Lou, J.; Hpiskind, R. A.; Nor-dheim, A.; Stunnenberg, H. G. Rapid and efficient purification ofnative histidine-tagged protein expressed by recombinant vacciniavirus. Proc. Natl. Acad. Sci. USA88:8972–8976; 1991.
[32] Barbier, A.; Visvikis, A.; Mathieu, F.; Diez, L.; Havekes, L.;Siest, G. Characterization of three human apolipoprotein E iso-
forms (E2, E3 and E4) expressed inEscherichia coli. Eur. J. Clin.Chem. Clin. Biochem.35:581–589; 1997.
[33] Sigalov, A.; Alexandrovitch, O.; Strizevskaya, E. Large-scaleisolation and purification of human apolipoproteins A-I and A-II.J. Chromatogr.537:464–468; 1991.
[34] Naskalski, J. W. Simple procedure of isolation of myeloperoxi-dase from leukocytes, pus and bone marrow cells.Acta Med. Pol.21:129–137; 1980.
[35] Zcliczynski, J. M.; Stelmaszynska, T.; Domanski, J.; Ostrowski,W. Chloramines as intermediates of oxidation reaction of aminoacids by myeloperoxidase.Biochim. Biophys. Acta235:419–424;1971.
[36] Keller, R. J.; Halmes, N. C.; Hinson, J. A.; Pumford, N. R.Immunochemical detection of oxidized proteins.Chem. Res.Toxicol.6:430–433; 1993.
[37] Jonas, A. A review of plasma apolipoprotein A-I interaction withphosphatidylcholines.Exp. Lung Res.6:255–270; 1984.
[38] Zhuang, Z.; Huang, X.; Costa, M. Paradoxical enhancement ofatherosclerosis by probucol treatment in apolipoprotein E-defi-cient mice.Toxicol. Appl. Pharmacol.126:319–325; 1994.
[39] Davies, K. J. A.; Delsignore, M. E. Protein damage and degra-dation by oxygen radicals.J. Biol. Chem.262:9908–9913; 1987.
[40] Ladu, M. J.; Gilligan, S. M.; Kukens, J. R.; Gabana, V. G.;Reardon, C. A.; Van Eldik, L. J.; Holtzman, D. M. Nascentastrocyte particles differ from lipoproteins in CSF.J. Neurochem.70:2070–2081; 1998.
[41] Zorich, N.; Jonas, A.; Pownall, H. J. Activation of lecithin cho-lesterol acyltransferase by human apolipoprotein E in discoidalcomplexes with lipids.J. Biol. Chem.260:8831–8837; 1985.
[42] Poirier, J.; Davignon, J.; Bouthillier, D.; Kogan, S.; Bertrand, P.;Gauthier, S. Apolipoprotein E polymorphism and Alzheimer’sdisease.Lancet342:697–699; 1993.
[43] Heinecke, J. W.; Li, W.; Francis, G. A.; Golstein, J. A. Tyrosylradical generated by myeloperoxidase catalyzes the oxidativecross-linking of proteins.J. Clin. Invest.91:2866–2872; 1993.
[44] Anantharamaiah, G. M.; Hughes, T. A.; Iqbal, M.; Gawish, A.;Neame, P. J.; Medley, M. F.; Segrest, J. P. Effect of oxidation onthe properties of apolipoproteins AI and AII.J. Lipid Res.29:309–318; 1988.
[45] Levine, R. L.; Mosoni, L.; Berlett, B. S.; Stadtman, E. R. Methi-onine residues as endogenous antioxidants in proteins.Proc. Natl.Acad. Sci. USA93:15036–15040; 1996.
[46] Garner, B.; Waldeck, A. R.; Witting, P. K.; Rye, K. A.; Stocker,R. Oxidation of high density lipoproteins. II. Evidence for directreduction of lipid hydroperoxides by methionine residues of apo-lipoproteins AI and AII.J. Biol. Chem.273:6088–6095; 1998.
[47] Dong, L. M.; Parkin, S.; Trakhanov, S. D.; Rupp, B.; Simmons,T.; Arnold, K. S.; Newhouse, Y. M.; Innerarity, T. L.; Weisgra-ber, K. H. Novel mechanism for defective receptor binding ofapolipoprotein E2 in type III hyperlipoproteinemia.Nat. Struct.Biol. 3:718–722; 1996.
[48] Droge, W.; Schulze-Osthoff, K.; Mihm, S.; Galter, D.; Schenk,H.; Eck, H. P.; Roth, S.; Gmunder, H. Functions of glutathioneand glutathione disulfide in immunology and immunopathology.FASEB J.8:1131–1138; 1994.
[49] Grant, A. J.; Jessup, W.; Dean, R. T. Inefficient degradation ofoxidized regions of protein molecules.Free Radic. Res. Commun.18:259–267; 1993.
[50] Wurtman, R. J. Choline metabolism as a basis for selectivevulnerability of cholinergique neurons.Trends Neurosci.15:117–122; 1992.
[51] Terry, R. D. Regeneration in Alzheimer disease and aging.Adv.Neurol.59:1–4; 1993.
[52] Masliah, E.; Mallory, M.; Ge, N.; Alford, M.; Veinberg, I.; Roses,A. D. Neurodegeneration in the central nervous system of apoE-deficient mice.Exp. Neurol.136:107–122; 1995.
[53] Reynolds, W. F.; Rhees, J.; Maciejewski, D.; Paladino, T.;Sieburg, H.; Maki, R. A.; Masliah, E. Myeloperoxidase polymor-phism is associated with gender specific risk for Alzheimer’sdisease.Exp. Neurol.155:31–41; 1999.
139Isoform-specific oxidation of apo E

[54] Lyras, L.; Evans, P. J.; Shaw, P. J.; Ince, P. G.; Halliwell, B.Oxidative damage and motor neurone disease difficulties in themeasurement of protein carbonyls in human brain tissue.FreeRadic. Res. Commun.24:397–406; 1996.
[55] Alam, Z. I.; Daniel, S. E.; Lees, A. J.; Marsden, D. C.; Jenner, P.;Halliwell, B. A generalised increase in protein carbonyls in thebrain in Parkinson’s but not incidental Lewy body disease.J. Neu-rochem.69:1326–1329; 1997.
[56] Sano, M.; Ernesto, C.; Thomas, R. G.; Klauber, M. R.; Schafer,
K.; Grundman, M.; Woodbuy, P.; Grewdon, J.; Cotman, C. W.;Pfeiffer, E.; Schneider, L. S.; Thal, L. J. A controlled trial ofselegiline, alpha-tocopherol, or both as treatment for Alzheimer’sdisease. The Alzheimer’s disease cooperative study.N. Engl.J. Med.336:1216–1222; 1997.
[57] Le Bars, P. L.; Katz, M. M.; Berman, N.; Itil, T. M.; Freedman,A. M.; Schatzberg, A. F. A placebo-controlled, double-bind,randomized trial of an extract of Ginkgo biloba for dementia.JAMA 278:1327–1332; 1997.
140 C. JOLIVALT et al.




















