
Targeted homozygous deletion of M-band titin in cardiomyocytes prevents sarcomere formation
Upload
independentCategory
view
4download
0
Lipopolysaccharide Internalization ActivatesEndotoxin-Dependent Signal Transduction
in CardiomyocytesDouglas B. Cowan, Sabrena Noria, Christof Stamm, Lina M. Garcia, Dimitrios N. Poutias,
Pedro J. del Nido, Francis X. McGowan, Jr
Abstract—We tested the hypothesis that bacterial lipopolysaccharide (LPS) must be internalized to facilitate endotoxin-dependent signal activation in cardiac myocytes. Fluorescently labeled LPS was used to treat primary cardiomyocytecultures, perfused heart preparations, and the RAW264.7 macrophage cell line. Using confocal microscopy andspectrofluorometry, we found that LPS was rapidly internalized in cardiomyocyte cultures and Langendorff-perfusedhearts. Although LPS uptake was also observed in macrophages, only a fraction of these cells were found to internalizeendotoxin to the extent seen in cardiomyocytes. Colocalization experiments with organelle or structure-specificfluorophores showed that LPS was concentrated in the Golgi apparatus, lysosomes, and sarcomeres. Similar intracellularlocalization was demonstrated in cardiomyocytes by transmission electron microscopy using gold-labeled LPS. Theinternalization of LPS was dependent on endosomal trafficking, because an inhibitor of microfilament reorganizationprevented uptake in both cardiomyocytes and whole hearts. Inhibition of endocytosis specifically restricted earlyactivation of extracellular signal–regulated kinase proteins and nuclear factor-kB as well as later tumor necrosis factor-aproduction and inducible nitric oxide synthase expression. In conclusion, we have demonstrated that bacterial endotoxinis internalized and transported to specific intracellular sites in heart cells and that these events are obligatory foractivation of LPS-dependent signal transduction.(Circ Res. 2001;88:491-498.)
Key Words: Golgi apparatusn microfilamentsn endocytosisn lysosomesn signal transduction
Endotoxin or lipopolysaccharide (LPS) from the outermembrane of Gram-negative bacteria is the primary
trigger of the systemic inflammatory response in sepsis.1
Sepsis or septic shock develops in.500 000 patients eachyear in the United States alone, of which nearly half die.2 Theclinical manifestations of sepsis are in large part attributableto LPS-induced signal transduction and gene expression inboth myeloid cells (eg, macrophages) and nonmyeloid cells(eg, endothelial cells and cardiomyocytes). These events arethe primary cause of myocardial dysfunction in sepsis, whichis an important determinant of patient outcome.
In the heart, substantial evidence has been collected thatindicates LPS exerts its effects in 2 overlapping phases.Reduced systolic function and contractile reserve occurwithin minutes to hours of LPS exposure.3 These phenomenaoccur in the absence of systemic acidosis, hypotension, ordecreased coronary perfusion. Early myocardial dysfunctionhas been related to direct LPS effects and rapid LPS-stimulated production of proinflammatory cytokines, such astumor necrosis factor-a (TNF-a).4,5 Additional elaboration ofproinflammatory cytokines and other mediators in response
to the LPS signal results in injury from a variety of mecha-nisms that include free radical production, nitric oxide gen-eration, and arachidonic acid metabolite release. These eventsresult in progressive contractile dysfunction, diminishedb-adrenergic responsiveness, impaired oxidative metabolism,and cell death. Therefore, defining how the LPS signal istransduced in the heart is relevant to understanding thepathophysiology of myocardial dysfunction during sepsis.
The delayed effects of LPS in the heart have been wellstudied; however, little is known about the process of earlyLPS signaling or injury. Our laboratory has previously shownthat LPS rapidly activates members of the extracellularsignal–regulated kinase (ERK), signal transducer and activa-tor of transcription (STAT), and nuclear factor-kB (NF-kB)signal transduction cascades in cardiomyocytes.6 Unlike cellsof reticuloendothelial origin, signaling through these path-ways seems to be receptor-mediated but independent of theglycosyl phosphatidylinositol-linked receptor CD14 andlipopolysaccharide-binding protein.6 The signaling eventsthat precede activation of these pathways in cardiac cells,however, have not been thoroughly studied.
Original received October 3, 2000; revision received December 22, 2000; accepted January 11, 2001.From the Departments of Anesthesia (D.B.C., D.N.P., F.X.M.) and Cardiac Surgery (L.M.G., C.S., P.J.D.), Children’s Hospital and Harvard Medical
School, Boston, Mass, and the Department of Laboratory Medicine and Pathobiology (S.N.), University of Toronto, Toronto, Ontario, Canada.Correspondence to Douglas B. Cowan, PhD, Department of Anesthesia, Enders Room 1355, Children’s Hospital, 300 Longwood Ave, Boston, MA
02115. E-mail [email protected]© 2001 American Heart Association, Inc.
Circulation Researchis available at http://www.circresaha.org
491 by guest on February 17, 2015http://circres.ahajournals.org/Downloaded from

Recently, a number of laboratories have shown that LPS isinternalized by several cell types,7–11 but the role of LPSuptake in activating signal transduction remains controver-sial.12–15 Polymorphonuclear leukocytes and HeLa cells en-docytically transport monomeric LPS to the Golgi apparatus.8
Although CD14 may be important in monomeric LPS recog-nition and transfer to either a coreceptor or directly into theplasma membrane in myeloid cells, the intracellular traffick-ing of LPS to the Golgi appears independent of this recep-tor.11 On the other hand, LPS aggregates are likely trans-ported in macrophages to lysosomes in conjunction withCD14, where acyloxyacyl hydrolase deacylates LPS.7,10,11,16
The latter function is ostensibly more relevant to the detox-ification and clearance of endotoxin rather than the initiationof signaling.11,17
LPS-mediated signal transduction is presently believed tobe governed by the TLR4 transmembrane receptor in bothcardiac and myeloid cells.1,18–22 Because endocytosis ofligand-activated cell-surface receptors often regulates signaltransduction and gene expression,23 we have studied therelationship between LPS intracellular trafficking and signalactivation in the heart. Given the complexity and diversity oflater LPS-induced effects in this organ, it is evident thatidentifying the early mechanisms of endotoxin signaling inthe myocardium is essential for developing novel and specificstrategies to prevent cardiac dysfunction during sepsis.
In the present study, we found that LPS was rapidlyinternalized in both cardiomyocytes and perfused wholehearts. In cardiomyocytes, LPS was sorted through an endo-somal pathway to the Golgi complex, lysosomes, and con-tractile apparatus. This process was linked with the immedi-ate activation of ERK and NF-kB signaling pathways and thelater production of TNF-a and nitric oxide. Our findingsindicate that endocytic membrane trafficking and retrogradetransport of LPS regulates endotoxin-dependent signal trans-duction in cardiac muscle.
Materials and MethodsCell Culture and Isolated PerfusedHeart PreparationsAnimal procedures received institutional approval and were con-ducted according to National Institutes of Health guidelines (publi-cation No. 85-23, 1985). Wistar rat (Charles River Laboratories,Portage, Manitoba, Canada) cardiomyocytes and the RAW264.7mouse macrophage cell line (American Type Culture Collection)were cultured as described earlier.6 Adult rat hearts wereLangendorff-perfused with Krebs-Henseleit buffer at 10 mL/minconstant flow essentially as described.3 A spectrofluorometer (SLM-Aminco) measured real-time emission light from hearts at 550 to 650nm after excitation at 524 nm.24
Fluorescent and Gold Labeling of LPSSalmonella typhosaLPS (Sigma) was labeled with BODIPY FL(4,4-difluoro-4-bora-3a, 4a-diaza-s-indacene fluorescein), OregonGreen 488, or Texas Red X succinimidyl ester derivatives (Molec-ular Probes), as described earlier.25 LPS was also labeled with1.4-nm-diameter mono-Sulfo-N-hydroxysuccinimide ester Nanogoldparticles (Nanoprobes) and purified by gel filtration. The labelingefficiency for each of the succinimidyl ester compounds wascalculated to be 82.6% to 93.5% on thebasis of absorbance measure-ments and known extinction coefficients. In addition, both fluorophoreand gold-labeled LPS (LPS-Au) stimulated TNF-a secretion and nitrite
production in cardiomyocytes and RAW264.7 cells to the same extentas unlabeled LPS.
Treatments and StainingCells were treated with 0.01 to 1mg/mL labeled LPS. Some cellswere treated with 50 nmol/L LysoTracker Red DND-99, 50 nmol/LBODIPY TR C5-ceramide, or 500 nmol/L MitoTracker CMX Ros(Molecular Probes) along with BODIPY FL–labeled LPS (BODIPYFL-LPS). Others were pretreated for 30 minutes with 1, 10, or 100mmol/L cytochalasin D (Sigma) before LPS or 50 and 500mmol/LH2O2 treatment in combination with cytochalasin. For staining,treated cultures were fixed and mounted directly or stained witheither TRITC-phalloidin (Sigma) or Texas Red X-phalloidin (Mo-lecular Probes). Perfused hearts were treated with Texas RedX-LPS610 mmol/L cytochalasin D and then fixed, paraffin-embedded, sectioned, and mounted for visualization.
Confocal Laser MicroscopySlides were visualized using a BioRad MRC1024 confocal micro-scope with a Nikon360 oil immersion objective, NA5160/0.17.BODIPY FL was excited at 488 nm and detected between 506 to 538nm. TRITC and Texas Red X were excited at 568 nm and detectedbetween 589 and 621 nm. Optical sections (0.5mm) were mergedand projected with the BioRad software.
Transmission Electron MicroscopyLPS-Au–treated cells were fixed in 2.5% grade I glutaraldehyde(Sigma) and silver (Ag)-enhanced (Nanoprobes) before staining with0.25% uranyl acetate and 0.5% osmium tetroxide (Electron Micros-copy Sciences). Sections (60 nm) were cut with a Reichert Ultracut-Sultramicrotome and mounted on copper grids (200 mesh) for electronmicroscopy.
Immunoblot AnalysesImmunoblotting was performed as described earlier.6 The anti-ERK1(K-23) antibody (Santa Cruz) was used at 0.1mg/mL to detectERK1/2, whereas the antiphospho-p44/42 mitogen-activated proteinkinase (MAPK) E10 antibody (New England Biolabs) was diluted to0.05mg/mL to detect phosphorylated (active) forms of ERK1/ERK2.The anti-inducible nitric oxide synthase (iNOS; NOS type II)antibody (Transduction Laboratories) was diluted to 0.1mg/mL.
Electrophoretic Mobility Shift AssaysNuclear extracts were isolated from LPS-treated cardiomyocytesusing the method of Andrews and Faller,26 and gel shift assayreactions were carried out as described previously.6
Measurement of TNF-a and Nitrite LevelsRat TNF-a levels were determined in culture media samples usingthe Quantikine (R&D Systems) sandwich ELISA. Nitric oxideproduction was determined in culture media using Greiss reagent.27
Comparisons of TNF-a and nitrite production were made using anANOVA followed by the Tukey-Kramer test.
ResultsLPS Is Internalized in Cardiomyocyte Culturesand Whole HeartsTo determine whether BODIPY FL-LPS was internalized incardiomyocyte cultures, we incubated cells with 1mg/mLlabeled LPS for various times (Figures 1A through 1D). Fixedcardiomyocytes were stained for filamentous actin (F-actin)(red). A diffuse distribution of LPS (green) was apparent incardiomyocytes by 30 minutes, with more intense globularstaining at perinuclear locations at 60 minutes and 24 hours.Labeled LPS was also observed to localize to a regionconsistent with either the A- or H-band of the sarcomere.
492 Circulation Research March 16, 2001
by guest on February 17, 2015http://circres.ahajournals.org/Downloaded from

Serial 0.5-mm mid (Z)-plane optical sections of cardiomy-ocytes treated with labeled LPS for 60 minutes are shown inFigure 2 (top). An intense punctate staining pattern wasobserved in the perinuclear region in addition to staining inthe contractile apparatus. For comparison, BODIPY FL-LPSstaining of RAW264.7 macrophages indicated that'15% ofthe macrophages avidly internalized LPS whereas the remain-ing cells exhibited relatively low levels of uptake (bottom).Despite variability in the degree of internalization, all mac-rophages were found to contain labeled LPS.
To establish that LPS internalization was not attributable tothe BODIPY FL compound, we labeled endotoxin with 2structurally different fluorophores. LPS labeled with eitherOregon Green 488 or Texas Red X succinimidyl estersexhibited a similar pattern of staining as demonstrated forBODIPY FL-LPS in both cardiac myocytes and RAW264.7cells (not shown). Furthermore, unreacted BODIPY FL iso-lated from the gel filtration column used to purify labeledLPS exhibited no internalization. Similar levels of internal-ization were seen in both cell types at LPS concentrationsranging from 0.1 to 10mg/mL; however, detection of fluo-rescent LPS at concentrations#0.01 mg/mL was moredifficult than that observed at higher concentrations.
Langendorff-perfused whole hearts were also found tointernalize LPS. Perfused hearts were fixed and sectioned toconfirm the intracellular localization of Texas Red X-LPS(Figures 3A through 3C). Figure 3A shows the level ofautofluorescence and background fluorescence in a controlheart perfused with unreacted fluorophore alone, whereas
Figures 3B and 3C demonstrate endotoxin uptake in heartsperfused with Texas Red X-LPS. Figure 3D shows the rate ofTexas Red X-LPS uptake in a working heart model. TexasRed X-LPS uptake occurred within 10 minutes. To demon-strate that LPS was localized to an intracellular space, weperfused hearts for 8.5 minutes with labeled LPS followed bya washout period using perfusate alone (Figure 3E). A drop influorescent emission was seen between 8.5 and 10 minutes,indicating that labeled LPS was eliminated from the vascularlumen. After the initial decline in signal intensity, cardiacfluorescent emission stabilized (10 to 30 minutes), demon-strating that Texas Red X-LPS remained within heart cells.
LPS Is Transported to the Golgi Complexand LysosomesThe intracellular location of LPS was investigated using 2strategies. In the first, cardiomyocyte cultures were treatedwith 0.1 mg/mL BODIPY FL-LPS for 60 minutes. Simulta-neously, the cells were exposed to 3 different organelle-specific stains (BODIPY TR C5-ceramide, Golgi apparatus;LysoTracker Red DND-99, lysosomes; and MitoTrackerCMX Ros, mitochondria) and then visualized on a confocalmicroscope. The second approach used an electron micro-scope and LPS labeled with 1.4-nm-diameter gold particles.
Figure 4 shows typical results from the experiments usingconfocal microscopy. For simplicity, cardiomyocytes with asingle nuclei are depicted; however, binucleated cells exhib-ited comparable staining patterns. The left column of thephotomicrographs (panels A, D, and G) shows the perinuclear
Figure 1. Confocal photomicrographs of cardiomyocytesexposed to BODIPY FL-LPS for various times. Cells were incu-bated with 1 mg/mL fluorescently labeled S. typhosa LPS (green)for 0 (A), 30 (B), and 60 minutes (C) and 24 hours (D). After fixa-tion, cells were cross-stained for F-actin with Texas RedX-phalloidin (red) and examined. The micrographs represent aseries projection of 0.5-mm optical sections from the basal tothe apical surface of the cells. Scale bars510 mm. Experimentswere repeated 8 times.
Figure 2. Photomicrographs of cardiomyocyte cultures (CMCs)and RAW264.7 macrophages treated with 1 mg/mL BODIPYFL-LPS for 60 minutes. Cells were stained for F-actin andexamined on a confocal microscope. LPS appears green andF-actin appears red. A through D, 0.5-mm (top) or 1-mm (bot-tom) optical sections midway between the apical and basal sur-faces of the cells and the projected series of these sections (E).Scale bars550 mm. Experiments were repeated 6 times.
Cowan et al LPS Uptake and Signaling in Cardiomyocytes 493
by guest on February 17, 2015http://circres.ahajournals.org/Downloaded from

staining by LPS, as noted in Figures 1 and 2. The centercolumn (panels B, E, and H) shows the organelle-specificstains for the Golgi complex (top), lysosomes (middle), andmitochondria (bottom). The right column (panels C, F, and I)reveals the merged images from the corresponding left andcenter columns. Any overlap in the areas stained with LPS(shown in green) and the organelle-specific stains (shown inred) appear as yellow in the right column. As demonstrated inpanels C and F, there is considerable overlap between LPSand Golgi or lysosome-specific stains. The yellow stainingseen in panel I is bleed-through between red and greenfluorescent channels and is not considered colocalization.This assertion was confirmed when individual 0.5-mm opticalsections were examined rather than images of the projectedseries shown in Figure 4 (not shown). The greatest degree ofLPS colocalization was seen with the Golgi-specific stain,
with nearly all of the spherical bodies observed in panel Cappearing yellow. The merged image in panel F shows that amajority of the lysosomes colocalize with LPS; however,individual green and red areas of staining are also apparent(particularly when discrete optical sections were examined),indicative of a less-uniform distribution of LPS in thelysosomes compared with that of the Golgi.
For transmission electron microscopy, LPS was labeledwith NanoGold particles rather than a gold colloid to mini-mize the size of the attached label and ensure uniformity ofparticle size. LPS-Au-Ag associated with structures, consis-tent with the interior of endosomes or early lysosomes(Figures 5A through 5C). In most cases, LPS-Au-Ag wasassociated with membranous regions within these vesicles,similar to the findings of Kreigsmann et al.28 It was alsoobserved that some LPS-Au-Ag was associated with theplasma membrane (Figure 5A); however, surface stainingwas never seen at sites consistent with caveolae or clathrin-coated pits. A longer treatment (60 minutes) resulted inlocalization of LPS-Au-Ag in lysosomes (arrows in Figures5D and E) or compact vesicular structures surrounding theGolgi complex (Figure 5F). These vesicles were primarilylocated neartrans-face and alongside the Golgi. Labeled LPSwas not found associated with the cisternae of the Golgidespite the high level of colocalization demonstrated inFigure 4.
Figure 3. Photomicrographs of rat heart tissue perfused withTexas Red X-LPS for 30 minutes. Hearts were either perfused inthe absence (A) or presence (B and C) of 1 mg/mL fluorescentlylabeled LPS and examined by confocal microscopy. Micro-graphs represent 0.5-mm optical sections of tissue aligned ineither the longitudinal (A and B) or transverse (C) orientation.Arrowheads indicate blood vessels. Scale bars550 mm. D andE, Spectrofluorometric measurement of Texas Red X-LPSuptake in the left ventricle of perfused hearts. Fluorescence at614 nm in hearts either continuously perfused with LPS sus-pended in Krebs-Henseleit buffer for 30 minutes (D) or perfusedfor 8.5 minutes with LPS followed by 21.5 minutes perfusionwith buffer alone (E). Values are expressed as a percentage ofthe peak fluorescence and plotted as mean6SEM (n54). Allmeasurements have been adjusted for background autofluores-cence and intraluminal vascular fluorescence.
Figure 4. Colocalization of BODIPY FL-LPS with Golgi-,lysosomal-, or mitochondrial-specific fluorophores. Primary car-diomyocytes were treated with 0.1 mg/mL fluorescently labeledLPS and BODIPY TR C5-ceramide (A through C), LysoTrackerRed DND-99 (D through F), or MitoTracker CMX Ros (G throughI) for 60 minutes. BODIPY FL-LPS is shown in green (A, D, andG), and C5 ceramide (B), LysoTracker (E), or MitoTracker (H) isshown in red. Merged images of A and B, D and E, and G andH are depicted in C, F, and I, respectively. Colocalization of redand green pixels appears in yellow. A projection of 0.5-mm opti-cal sections is shown. Scale bars510 mm. Experiments wererepeated 6 times.
494 Circulation Research March 16, 2001
by guest on February 17, 2015http://circres.ahajournals.org/Downloaded from

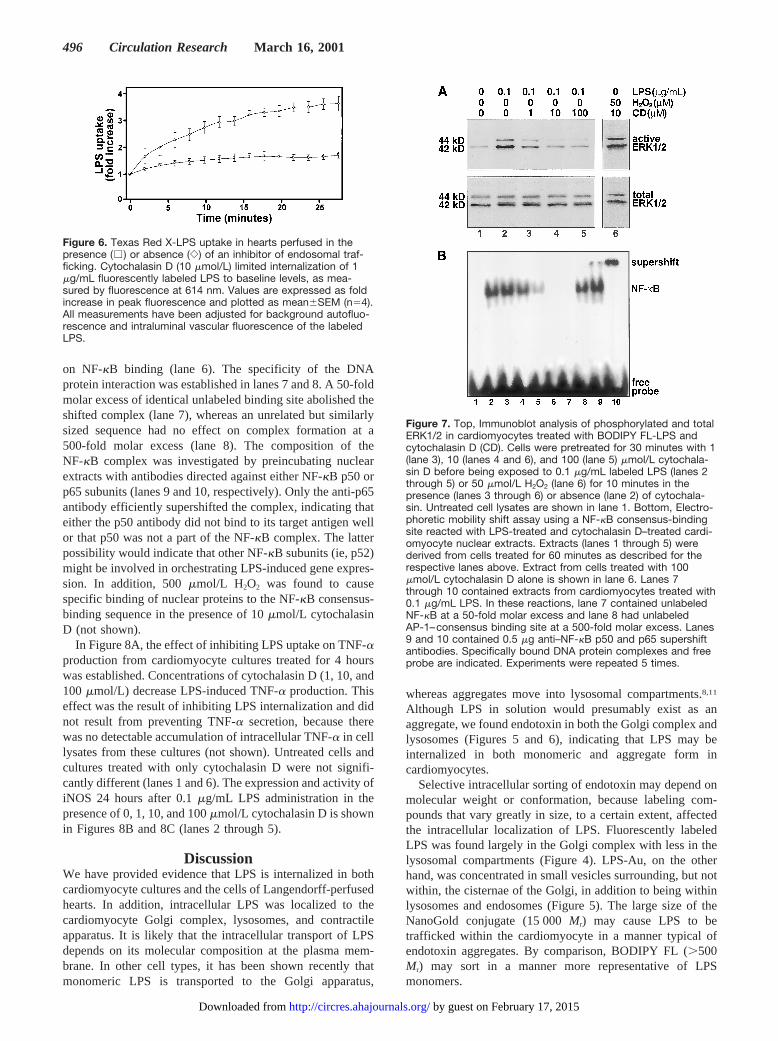
Activation of Signal Transduction byLipopolysaccharide Is Dependent on UptakeTo ascertain whether LPS internalization and intracellulartrafficking are required for activation of signal transduction incardiomyocytes, we inhibited endocytosis with cytochalasinD.29,30 Figure 6 demonstrates that in perfused whole hearts,10 mmol/L cytochalasin D completely blocked the internal-ization of Texas Red X-LPS. Similarly, BODIPY FL-LPSwas not internalized in cardiomyocyte cultures treated withcytochalasin D (not shown).
We investigated whether cytochalasin D blocked early(#60 minutes), intermediate (4 hours), or late (24 hours)signaling in cardiomyocytes. Figure 7A shows the activity ofERK proteins at 10 minutes after LPS or H2O2 treatment inthe presence of 1, 10, and 100mmol/L cytochalasin D. ERKwas maximally phosphorylated 10 minutes after LPS expo-
sure, as shown in lanes 1 (untreated) and 2 (0.1mg/mL LPS),in agreement with our previous observations.6 Increasingdoses of cytochalasin D (lanes 3 through 5) attenuated ERKphosphorylation, whereas cytochalasin D alone had no effecton ERK activity (not shown). To demonstrate that cytocha-lasin was inhibiting ERK phosphorylation specificallythrough prevention of endocytosis and not via inhibition ofthe MAPK pathway, 50mmol/L hydrogen peroxide (H2O2)plus cytochalasin D was used to treat cardiomyocytes. ERKproteins were activated by H2O2 in the presence of 10mmol/Lcytochalasin D (lane 6).
Moreover, the ability of nuclear proteins to specificallybind a NF-kB consensus binding sequence after 60 minutesof LPS treatment (Figure 7B; compare lanes 1 and 2) wasreduced in the presence of escalating concentrations ofcytochalasin (lanes 3 through 5). Inhibitor alone had no effect
Figure 5. Transmission electron micros-copy of cardiomyocytes treated with 0.01mg/mL LPS-Au. NanoGold-labeled LPS wasapplied to cell cultures for 5 minutes (Athrough C) or 60 minutes (D through F)before fixation and processing. N indicatesnuclei. Arrows point toward endosomes (Athrough C), lysosomes (D and E), and vesi-cles surrounding the Golgi (F). Other areaslabeled with LPS-Au-Ag include the cellsurface (A), a residual body (below thearrow in B), and several lysosomes (C andD). Areas not found to contain LPS-Auinclude the nucleus, mitochondria, lipiddroplets, and endoplasmic reticulum. Scalebars5200 nm. Experiments were repeated4 times.
Cowan et al LPS Uptake and Signaling in Cardiomyocytes 495
by guest on February 17, 2015http://circres.ahajournals.org/Downloaded from
on NF-kB binding (lane 6). The specificity of the DNAprotein interaction was established in lanes 7 and 8. A 50-foldmolar excess of identical unlabeled binding site abolished theshifted complex (lane 7), whereas an unrelated but similarlysized sequence had no effect on complex formation at a500-fold molar excess (lane 8). The composition of theNF-kB complex was investigated by preincubating nuclearextracts with antibodies directed against either NF-kB p50 orp65 subunits (lanes 9 and 10, respectively). Only the anti-p65antibody efficiently supershifted the complex, indicating thateither the p50 antibody did not bind to its target antigen wellor that p50 was not a part of the NF-kB complex. The latterpossibility would indicate that other NF-kB subunits (ie, p52)might be involved in orchestrating LPS-induced gene expres-sion. In addition, 500mmol/L H2O2 was found to causespecific binding of nuclear proteins to the NF-kB consensus-binding sequence in the presence of 10mmol/L cytochalasinD (not shown).
In Figure 8A, the effect of inhibiting LPS uptake on TNF-aproduction from cardiomyocyte cultures treated for 4 hourswas established. Concentrations of cytochalasin D (1, 10, and100 mmol/L) decrease LPS-induced TNF-a production. Thiseffect was the result of inhibiting LPS internalization and didnot result from preventing TNF-a secretion, because therewas no detectable accumulation of intracellular TNF-a in celllysates from these cultures (not shown). Untreated cells andcultures treated with only cytochalasin D were not signifi-cantly different (lanes 1 and 6). The expression and activity ofiNOS 24 hours after 0.1mg/mL LPS administration in thepresence of 0, 1, 10, and 100mmol/L cytochalasin D is shownin Figures 8B and 8C (lanes 2 through 5).
DiscussionWe have provided evidence that LPS is internalized in bothcardiomyocyte cultures and the cells of Langendorff-perfusedhearts. In addition, intracellular LPS was localized to thecardiomyocyte Golgi complex, lysosomes, and contractileapparatus. It is likely that the intracellular transport of LPSdepends on its molecular composition at the plasma mem-brane. In other cell types, it has been shown recently thatmonomeric LPS is transported to the Golgi apparatus,
whereas aggregates move into lysosomal compartments.8,11
Although LPS in solution would presumably exist as anaggregate, we found endotoxin in both the Golgi complex andlysosomes (Figures 5 and 6), indicating that LPS may beinternalized in both monomeric and aggregate form incardiomyocytes.
Selective intracellular sorting of endotoxin may depend onmolecular weight or conformation, because labeling com-pounds that vary greatly in size, to a certain extent, affectedthe intracellular localization of LPS. Fluorescently labeledLPS was found largely in the Golgi complex with less in thelysosomal compartments (Figure 4). LPS-Au, on the otherhand, was concentrated in small vesicles surrounding, but notwithin, the cisternae of the Golgi, in addition to being withinlysosomes and endosomes (Figure 5). The large size of theNanoGold conjugate (15 000Mr) may cause LPS to betrafficked within the cardiomyocyte in a manner typical ofendotoxin aggregates. By comparison, BODIPY FL (.500Mr) may sort in a manner more representative of LPSmonomers.
Figure 6. Texas Red X-LPS uptake in hearts perfused in thepresence (e ) or absence (e) of an inhibitor of endosomal traf-ficking. Cytochalasin D (10 mmol/L) limited internalization of 1mg/mL fluorescently labeled LPS to baseline levels, as mea-sured by fluorescence at 614 nm. Values are expressed as foldincrease in peak fluorescence and plotted as mean6SEM (n54).All measurements have been adjusted for background autofluo-rescence and intraluminal vascular fluorescence of the labeledLPS.
Figure 7. Top, Immunoblot analysis of phosphorylated and totalERK1/2 in cardiomyocytes treated with BODIPY FL-LPS andcytochalasin D (CD). Cells were pretreated for 30 minutes with 1(lane 3), 10 (lanes 4 and 6), and 100 (lane 5) mmol/L cytochala-sin D before being exposed to 0.1 mg/mL labeled LPS (lanes 2through 5) or 50 mmol/L H2O2 (lane 6) for 10 minutes in thepresence (lanes 3 through 6) or absence (lane 2) of cytochala-sin. Untreated cell lysates are shown in lane 1. Bottom, Electro-phoretic mobility shift assay using a NF-kB consensus-bindingsite reacted with LPS-treated and cytochalasin D–treated cardi-omyocyte nuclear extracts. Extracts (lanes 1 through 5) werederived from cells treated for 60 minutes as described for therespective lanes above. Extract from cells treated with 100mmol/L cytochalasin D alone is shown in lane 6. Lanes 7through 10 contained extracts from cardiomyocytes treated with0.1 mg/mL LPS. In these reactions, lane 7 contained unlabeledNF-kB at a 50-fold molar excess and lane 8 had unlabeledAP-1–consensus binding site at a 500-fold molar excess. Lanes9 and 10 contained 0.5 mg anti–NF-kB p50 and p65 supershiftantibodies. Specifically bound DNA protein complexes and freeprobe are indicated. Experiments were repeated 5 times.
496 Circulation Research March 16, 2001
by guest on February 17, 2015http://circres.ahajournals.org/Downloaded from

We also found LPS associates with the contractile appara-tus. The lack of overlap between the phalloidin and BODIPYFL-LPS staining suggests that LPS associates with theH-band of sarcomeric thick filaments, which are composedprincipally of myosin. It is possible that some of the earlycardiac contractile effects of LPS may be attributed theaccumulation of endotoxin in the sarcomere (Figures 1 and2).3 In addition, because endotoxin was found to persist in theGolgi apparatus for at least 24 hours (Figure 1 and data notshown), LPS has the potential to obstruct Golgi and endo-plasmic reticular processing of cellular proteins destined formembrane compartments or the extracellular space. Because
LPS cannot be metabolized in the Golgi complex, a situationreminiscent of the endoplasmic reticulum (ER)-overloadresponse may be occurring.8,31 In the ER-stress response, theNF-kB signaling pathway is activated because of an accumu-lation of proteins in the ER. This could occur because of abacklog from the Golgi complex as the result of LPSdeposition in that organelle. Our present and earlier6 resultsare consistent with this phenomenon but do not exclude otherpossibilities.
LPS was also internalized in the cardiomyocytes andvascular cells of perfused whole hearts (Figure 3). Thisfinding represents the first demonstration of LPS internaliza-tion in a solid organ. The spectrofluorometer used for some ofthese studies measured output signal from a section of the leftventricular free wall. The effective excitation light penetra-tion from the 400-W xenon lamp used in these experimentswas'4 mm, and the resultant emission light was estimated tobe unaffected by tissue absorbance.24 Microscopic examina-tion of tissue sections from the perfused hearts verified thatfluorescent LPS was evenly distributed intracellularlythroughout the ventricular wall. A longer wavelength fluoro-phore (Texas Red X) was used to avoid the large amount ofautofluorescence attributable to myoglobin that is observednear the peak emission wavelength for BODIPY FL orOregon Green 488.
We observed that uptake of LPS can be completelyprevented by treating the perfused hearts with cytochalasin Djust before and during the administration of Texas RedX-LPS (Figure 6). There are several studies in the literaturethat support our finding that LPS internalization is dependenton microfilament reorganization.12,13 Although cytochalasinshave been used to block LPS uptake in several cell types, thismolecule has also been shown to prevent downstream signal-ing as a consequence of LPS exposure.32–34 Interestingly,Poussin et al12 showed that cytochalasin D did not preventLPS-dependent p38 MAPK and NF-kB activation in THP-1cells. In their study, cytochalasin D actually increasedinterleukin-8 secretion after LPS treatment.12
Additionally, we have demonstrated that cytochalasin Dtreatment of cardiomyocytes, in a dose-dependent manner,prevented the immediate activation of ERK and NF-kBsignaling pathways (Figure 7) and the delayed production ofTNF-a and nitric oxide (Figure 8). Activation of thesesignaling proteins was specifically attributable to the preven-tion of internalization of LPS and not the result of directinhibition of the ERK and NF-kB pathways, because H2O2
could stimulate these signaling cascades in the presence ofcytochalasin. Hydrogen peroxide has previously been provento stimulate both of these pathways in cardiomyocytes,35,36
likely through a receptor-independent means.On the basis of the assumption that in cardiac muscle cells
TNF-a secretion leads to inducible nitric oxide synthase geneexpression,37 it is not surprising that microfilament disruptionprevents nitric oxide production, because TNF-a is regulatedby the NF-kB and ERK pathways.6 Consequently, neitherpathway can be activated in response to LPS, when internal-ization is blocked (Figure 7).
There is presently insufficient evidence to conclude thatLPS signaling occurs as a direct result of concentration in the
Figure 8. A, TNF-a production in cardiomyocytes treated withBODIPY FL-LPS and cytochalasin D (CD). Cells were pretreatedfor 30 minutes with 1, 10, and 100 mmol/L cytochalasin D (bars3 through 5, respectively) before being exposed to 0.1 mg/mLlabeled LPS for 4 hours (bars 2 through 5) in the presence (bars3 through 5) or absence (bar 2) of cytochalasin. After treat-ments, culture media were analyzed for TNF-a, and untreatedcardiomyocyte culture media were used as a negative control(bar 1). Results are presented as pg/mL TNF-a (mean6SD),where n54 and all experiments had an equal number of cells.There was a significant increase (*P,0.001) in TNF-a secretionin cells treated with LPS alone (bar 2) compared with untreatedcardiomyocytes (bar 1), whereas increasing concentrations ofcytochalasin D (bars 3 through 5) significantly reduced(**P,0.001) TNF-a production. Cytochalasin D alone (bar 6) didnot significantly alter TNF-a secretion compared with the control(bar 1). B, Immunoblot analysis of iNOS. C, Nitrite production incardiomyocyte cultures treated with BODIPY FL-LPS andcytochalasin for 24 hours. Samples were run in the same order(ie, 1 to 6) as described for TNF-a. A significant increase(*P,0.001) was demonstrated in nitrite accumulation in cellstreated with LPS alone (bar 2) compared with untreated cardio-myocytes (bar 1). Increasing concentrations of cytochalasin D(bars 3 through 5) significantly reduced (**P,0.001) nitric oxideproduction. Results are presented as mmol/L nitrite (mean6SD),where n54 and values have been adjusted for total cardiomyo-cyte protein.
Cowan et al LPS Uptake and Signaling in Cardiomyocytes 497
by guest on February 17, 2015http://circres.ahajournals.org/Downloaded from

Golgi apparatus or other cellular compartments. On the basisof the rapidity of some responses, it is probable that signalsare generated from an intermediate structure like the endo-some. This supposition is supported by the observation thatalthough LPS-Au was biologically active (as measured by theability to stimulate TNF-a secretion and nitric oxide produc-tion), it was not localized within the Golgi complex. WhetherLPS is receptor associated (eg, TLR4) within cellular com-partments or integrated into an intracellular membrane alsoremains to be elucidated. Internalization of ligand-activatedreceptors is a well-recognized means of modulating signaltransduction, with most examples of signaling from withinendosomes indicating that ligand receptor complexes areassociated with caveolae or clathrin-coated pits.23 We did notobserve LPS associated with either type of structure (Figure5), suggesting that other mechanisms may be involved.
In conclusion, we have shown that LPS is internalized andsorted to specific locations in cardiomyocytes and that theseevents are required for endotoxin-dependent signal activation.A complete understanding of the initial events that result inproduction of deleterious gene products or directly interferewith contractile function may lead to the development oftherapies designed to protect the heart from endotoxinexposure.
AcknowledgmentsThis work was supported by the Charles H. Hood Foundation ofBoston, the National Institutes of Health (grants HL52589 andHL46207), and the Children’s Hospital Anesthesia Foundation.
References1. Beutler B. Tlr4: central component of the sole mammalian LPS sensor.
Curr Opin Immunol. 2000;12:20–26.2. Wheeler AP, Bernard GR. Treating patients with severe sepsis.N Engl
J Med. 1999;340:207–214.3. Takeuchi K, del Nido PJ, Ibraham AE, Poutias DN, Glynn P, Cao-Danh
H, Cowan DB, McGowan FX Jr. Increased myocardial calcium cyclingand reduced myofilament calcium sensitivity in early endotoxemia.Surgery. 1999;126:231–238.
4. Yasuda S, Lew WYW. Lipopolysaccharide depresses cardiac contractilityand b-adrenergic contractile response by decreasing myofilamentresponse to Ca21 in cardiac myocytes.Circ Res. 1997;81:1011–1020.
5. Kapadia S, Lee J, Torre-Amione G, Birdsall HH, Ma TS, Mann DL.Tumor necrosis factor-a gene and protein expression in adult felinemyocardium after endotoxin administration.J Clin Invest. 1995;96:1042–1052.
6. Cowan DB, Poutias DN, del Nido PJ, McGowan FX Jr. CD14-independent activation of cardiomyocyte signal transduction by bacterialendotoxin.Am J Physiol. 2000;279:H619–H629.
7. Kitchens RL, Wang P, Munford RS. Bacterial lipopolysaccharide canenter monocytes via two CD14-dependent pathways.J Immunol. 1998;161:5534–5545.
8. Thieblemont N, Wright SD. Transport of bacterial lipopolysaccharide tothe Golgi apparatus.J Exp Med. 1999;190:523–534.
9. Beatty WL, Meresse S, Gounon P, Davoust J, Mounier J, Sansonetti PJ,Gorvel JP. Trafficking ofShigella lipopolysaccharide in polarizedintestinal epithelial cells.J Cell Biol. 1999;145:689–698.
10. Forestier C, Moreno E, Pizarro-Cerda J, Gorvel JP. Lysosomal accumu-lation and recycling of lipopolysaccharide to the cell surface of murinemacrophages, an in vitro and in vivo study.J Immunol. 1999;162:6784–6791.
11. Vasselon T, Hailman E, Thieringer R, Detmers PA. Internalization ofmonomeric lipopolysaccharide occurs after transfer out of cell surfaceCD14.J Exp Med. 1999;190:509–521.
12. Poussin C, Foti M, Carpentier JL, Pugin J. CD14-dependent endotoxininternalization via a macropinocytic pathway.J Biol Chem. 1998;273:20285–20291.
13. Detmers PA, Thieblemont N, Vasselon T, Pironkova R, Miller DS,Wright SD. Potential role of membrane internalization and vesicle fusionin adhesion of neutrophils in response to lipopolysaccharide and TNF.J Immunol. 1996;157:5589–5596.
14. Thieblemont N, Wright SD. Mice genetically hyporesponsive to lipopoly-saccharide (LPS) exhibit a defect in endocytic uptake of LPS andceramide.J Exp Med. 1997;185:2095–2100.
15. Thieblemont N, Thieringer R, Wright SD. Innate immune recognition ofbacterial lipopolysaccharide: dependence on interactions with membranelipids and endocytic movement.Immunity. 1998;8:771–777.
16. Luchi M, Munford RS. Binding, internalization, and deacylation of bac-terial lipopolysaccharide by human neutrophils.J Immunol. 1993;151:959–969.
17. Kitchens RL, Munford RS. CD14-dependent internalization of bacteriallipopolysaccharide (LPS) is strongly influenced by LPS aggregation butnot by cellular responses to LPS.J Immunol. 1998;160:1920–1928.
18. Frantz S, Kobzik L, Kim YD, Fukazawa R, Medzhitov R, Lee RT, KelleyRA. Toll4 (TLR4) expression in cardiac myocytes in normal and failingmyocardium.J Clin Invest. 1999;104:271–280.
19. Qureshi ST, Gros P, Malo D. Thelps locus: genetic regulation of hostresponses to bacterial lipopolysaccharide.Inflamm Res. 1999;48:613–620.
20. Anderson KV. Toll signaling pathways in the innate immune response.Curr Opin Immunol. 2000;12:13–19.
21. Beutler B. Endotoxin, toll-like receptor 4, and the afferent limb of innateimmunity. Curr Opin Immunol.2000;3:23–28.
22. Imler JL, Hoffmann JA. Signaling mechanisms in the antimicrobial hostdefense ofDrosophila.Curr Opin Immunol. 2000;3:16–22.
23. Ceresa BP, Schmid SL. Regulation of signal transduction by endocytosis.Curr Opin Cell Biol. 2000;12:204–210.
24. del Nido PJ, Glynn P, Buenaventura P, Salama G, Koretsky AP. Fluo-rescence measurement of calcium transients in perfused rabbit heart usingRhod-2.Am J Physiol. 1998;274:H728–H741.
25. Yu B, Wright SD. Catalytic properties of lipopolysaccharide (LPS)binding protein: transfer of LPS to soluble CD14.J Biol Chem. 1996;271:4100–4105.
26. Andrews NC, Faller DV. A rapid micropreparation technique forextraction of DNA-binding proteins from limiting numbers of mam-malian cells.Nucleic Acids Res. 1991;19:2499.
27. Luss H, Watkins SC, Freeswick PD, Imro AK, Nussler AK, Billiar TR,Simmons RL, del Nido PJ, McGowan FX Jr. Characterization ofinducible nitric oxide synthase expression in endotoxemic rat cardiomy-ocytes in vivo and following cytokine exposure in vitro.J Mol CellCardiol. 1995;27:2015–2029.
28. Kriegsmann J, Gay S, Brauer R. Endocytosis of lipopolysaccharide inmouse macrophages.Cell Mol Biol. 1993;39:791–800.
29. Cooper JA. Effects of cytochalasin and phalloidin on actin.J Cell Biol.1987;105:1473–1478.
30. Isowa N, Xavier AM, Dziak E, Opas M, McRitchie DI, Slutsky AS,Keshavjee SH, Lui MY. LPS-induced depolymerization of cytoskeletonand its role in TNFa production by rat pneumocytes.Am J Physiol.1999;277:L606–L615.
31. Pahl HL, Baeuerle PA. The ER-overload response: activation of NF-kB.Trends Biochem Sci. 1997;22:63–67.
32. Shinji H, Akagawa KS, Yoshida T. Cytochalasin D inhibitslipopolysaccharide-induced tumor necrosis factor production in macro-phages.J Leukoc Biol. 1993;54:336–342.
33. Fernandes PD, Araujo HM, Riveros-Moreno V, Aussreuy J. Depolymer-ization of macrophage microfilaments prevents induction and inhibitsactivity of nitric oxide synthase.Eur J Cell Biol. 1996;71:356–362.
34. Lichtman SN, Wang J, Lemasters JJ. Lipopolysaccharide-stimulatedTNFa release from cultured rat Kupffer cells: sequence of intracellularevents.J Leukoc Biol. 1998;64:368–372.
35. Aikawa R, Komuro I, Yamazaki T, Zou Y, Kudoh S, Tanaka M, ShiojimaI, Hiroi Y, Yazaki Y. Oxidative stress activates extracellular signal-regulated kinases through Src and Ras in cultured cardiac myocytes ofneonatal rats.J Clin Invest. 1997;100:1813–1821.
36. Adderley SR, Fitzgerald DJ. Oxidative damage of cardiomyocytes islimited by extracellular signal-regulated kinases 1/2-mediated inductionof cyclooxygenase-2.J Biol Chem. 1999;274:5038–5046.
37. Cain BS, Harken AH, Meldrum DR. Therapeutic strategies to reduceTNFa mediated cardiac contractile depression following ischemia andreperfusion.J Mol Cell Cardiol. 1999;31:931–947.
498 Circulation Research March 16, 2001
by guest on February 17, 2015http://circres.ahajournals.org/Downloaded from

Pedro J. del Nido and Francis X. McGowan, JrDouglas B. Cowan, Sabrena Noria, Christof Stamm, Lina M. Garcia, Dimitrios N. Poutias,
in CardiomyocytesLipopolysaccharide Internalization Activates Endotoxin-Dependent Signal Transduction
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2001 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/01.RES.88.5.4912001;88:491-498Circ Res.
http://circres.ahajournals.org/content/88/5/491World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on February 17, 2015http://circres.ahajournals.org/Downloaded from
Copyright © 2022 FDOKUMEN



















