
Photochemical internalization (PCI) in cancer therapy: From bench towards bedside medicine
36
Accepted Manuscript Photochemical internalization (PCI) in cancer therapy; from bench towards bedside medicine Ole-Jacob Norum, Pål Kristian Selbo, Anette Weyergang, Karl-Erik Giercksky, Kristian Berg PII: S1011-1344(09)00067-0 DOI: 10.1016/j.jphotobiol.2009.04.012 Reference: JPB 8917 To appear in: Journal of Photochemistry and Photobiology B: Bi‐ ology Received Date: 12 February 2009 Revised Date: 20 April 2009 Accepted Date: 23 April 2009 Please cite this article as: O-J. Norum, l.K. Selbo, A. Weyergang, K-E. Giercksky, K. Berg, Photochemical internalization (PCI) in cancer therapy; from bench towards bedside medicine, Journal of Photochemistry and Photobiology B: Biology (2009), doi: 10.1016/j.jphotobiol.2009.04.012 This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
-
Upload
rr-research -
Category
Documents
-
view
3 -
download
0
Transcript of Photochemical internalization (PCI) in cancer therapy: From bench towards bedside medicine

Accepted Manuscript
Photochemical internalization (PCI) in cancer therapy; from bench towards
bedside medicine
Ole-Jacob Norum, Pål Kristian Selbo, Anette Weyergang, Karl-Erik Giercksky,
Kristian Berg
PII: S1011-1344(09)00067-0
DOI: 10.1016/j.jphotobiol.2009.04.012
Reference: JPB 8917
To appear in: Journal of Photochemistry and Photobiology B: Bi‐
ology
Received Date: 12 February 2009
Revised Date: 20 April 2009
Accepted Date: 23 April 2009
Please cite this article as: O-J. Norum, l.K. Selbo, A. Weyergang, K-E. Giercksky, K. Berg, Photochemical
internalization (PCI) in cancer therapy; from bench towards bedside medicine, Journal of Photochemistry and
Photobiology B: Biology (2009), doi: 10.1016/j.jphotobiol.2009.04.012
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers
we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and
review of the resulting proof before it is published in its final form. Please note that during the production process
errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.

ACCEPTED MANUSCRIPT
1
Title Photochemical internalization (PCI) in cancer therapy; from bench towards bedside medicine. Authors Ole-Jacob Norum1+2
Pål Kristian Selbo1
Anette Weyergang1
Karl-Erik Giercksky 2 Kristian Berg 1
1Department of Radiation Biology, Institute for Cancer Research, Norwegian Radium Hospital, Montebello, N-0310 Oslo, Norway. 2Department of Surgical Oncology, Norwegian Radium Hospital, and University of Oslo Corresponding author Kristian Berg, Department of Radiation Biology, The Norwegian Radium Hospital, N-0310 Oslo, Norway E-mail: [email protected] Phone: +47 40873549 / +47 22934260 Fax: +47 22934270
Abstract
PDT in cancer therapy has been reviewed several times recently and many published reports
have been showing promising results. The clinical approvals for PDT include curative
treatment of early or superficial cancers and palliative treatment of more advanced disease.
Still PDT has yet to become a widely used cancer treatment. This may partly be due to
limitations in current PDT regimens and partly due to effective alternative treatment
modalities. If the specificity and selectivity of PDT could be improved, PDT would probably
make substantial progress and comprise an even more competitive alternative in cancer
treatment.
The PCI technology is based on the same principles as PDT, the activation of a
photosensitizer by light and subsequently followed by formation of reactive oxygen species.
Unlike PDT, the photosensitizer used in PCI has to be located in the endocytic vesicles of the
targeted cells and will, upon activation of light, induce a release of endocytosed therapeutic
agents after a photochemically induced rupture of the endocytic vesicles. The endocytosed

ACCEPTED MANUSCRIPT
2
therapeutic agent will then be released and may reach their intracellular target of action before
being degraded in lysosomes. This site-specific drug delivery induced by PCI will take place
in addition to the well described cytotoxic, vascular and immunostimulatory effects of PDT.
PCI has been shown to facilitate intracellular delivery of a large variety of macromolecules
that do not otherwise readily penetrate the plasma membrane, including type I ribosome-
inactivating proteins (RIPs), RIP-based immunotoxins, genes and some chemotherapeutic
agents.
Several animal models have been used for in vivo documentation of the PCI principle and
more animal models of clinical relevance have recently been utilized for addressing clinical
issues. This review will focus on the possibilities and limitations offered by PCI to overcome
some of the challenges recognized in current PDT regimens in cancer treatment.
Key words Photochemical internalization, photodynamic therapy, phthalocyanine, bleomycin, cancer therapy. Abbreviations AlPcS2a Disulfonated aluminum phthalocyanine with the sulfonate groups on adjacent
phthalate rings AlPcS4 Tetrasulfonated aluminum phthalocyanine ALA 5-aminolevulinic acid BLM Bleomycin BPD-MA Benzoporphyrin derivate monoacid EGF(R) Epidermal growth factor (reseptor) CE-MRI Contrast-enhanced magnetic resonance imaging HpD Hematoporphyrin derivate m-THPC Tetra (m-hydroxyphenyl)chlorine PCI Photochemical internalization PDT Photodynamic therapy RIP Ribosome inactivating protein

ACCEPTED MANUSCRIPT
3
1. Introduction
Photodynamic therapy (PDT) is a treatment modality that takes advantage of the cytotoxic
effects induced by a photosensitizer and light in the presence of oxygen [1]. The therapeutic
effect of PDT may be due to direct cytotoxicity (i.e. damage of cell membranes), vascular
damage, and inflammatory and immunological responses [1,2]. The relative importance of
these pathways to therapeutic effects depends on the tissue oxygenation, photosensitizer
formulation, distribution and light dosimetry. In this review we will discuss the therapeutic
possibilities and limitations of PDT and the potentials in the photochemical internalization
(PCI) technology to overcome some of the limitations of PDT.
1.1. PDT in cancer treatment
PDT in cancer treatment has been presented in several recent reviews [1,3-6]. PDT has the
advantage of good cosmetic results [3], low mutagenic potential [7] low systemic toxicity
except for skin photosensitivity, no cumulative or long term toxicity [8] and not inducing
resistance to chemotherapy or radiotherapy [9]. PDT is used on a regular basis in treating non-
melanotic skin cancers like superficial basal cell carcinoma, actinic keratosis and Bowen’s
disease (squamous cell carcinoma in situ) using the photosensitizer precursors 5-
aminolevulinic acid and methyl aminolevulinate (Levulan® / Metvix®). The cure rate is high
and the side effects are favorable compared to alternative treatments (e.g. surgery) [3]. These
lesions are ideal for clinical PDT treatment since they are available for topical administration
of the photosensitizing compounds, resulting in no systemic phototoxicity, and they are early
cancers or precancerous lesions with little or no metastatic potential. Another superficial and
early cancer that may be suitable for standard PDT treatment is carcinoma in situ of the
esophagus with a reported 89% complete remission rate after PDT [10]. The clinical outcome
seems independent of the photosensitizer used (m-THPC, HpD and Photofrin II). Similar rates

ACCEPTED MANUSCRIPT
4
of complete remission were obtained with ALA-PDT [11], with no complications observed.
Fistulas, perforations and stenosis are reported with chemically synthesized photosensitizers
exposed to red light, although, this is mainly prevented by using green light [10]. The
treatment of Barrett’s esophagus and early esophageal cancers will have to compete with the
therapeutic safety and costs benefit analysis of endoscopic mucosal resection and
radiofrequency ablation reporting even higher cure rates than PDT [12]. There is a need for
randomized trials to define the preferred treatment method, or combination of methods, of the
patient population with these lesions. Similarly, there are unanswered questions concerning
the optimal treatment of early lung cancer [13]. Local treatment options like laser therapy,
electrocautery, brachytherapy and cryotherapy and PDT will all need to withstand the
comparison with surgery. PDT has the advantage of better specificity and lower complications
rates compared to the other modalities. PDT is a good alternative in patients not eligible to
surgery due to co-morbidity or previous contralateral lung resection. Currently there are
clinical PDT approvals for treatment of early lung and esophageal cancer, superficial gastric
cancer, carcinoma in situ of cervix and vulva and recurrent superficial bladder cancer based
on Porfimer Sodium (Photofrin®) as photosensitizer [3]. One of the drawbacks of utilizing
Photofrin, however, is the long lasting skin photosensitivity. PDT is clearly suitable for
precancerous and early non-invasive cancers where PDT may be expected to result in high
cure rates. However, accurate staging is a prerequisite for predicting the clinical outcome of
PDT as more advanced disease yields lower complete remission rates [10,11].
PDT has also proven its suitability in palliative cancer therapy, especially in recanalization of
near obliterated tubular anatomical structures accessible via endoscope like the bile ducts
[14], bronchi [15] and esophagus [4] and for palliative treatment of head and neck cancer with
temoporfin (m-THPC / Foscan®) [16]. PDT is also an alternative when the patient does not

ACCEPTED MANUSCRIPT
5
accept or tolerate surgery, ionizing radiation or chemotherapy due to anatomical
considerations, cumulative toxicity, comorbidity or when existing treatment modalities are
associated with mutilation or very high complication rates (e.g. surgery for head and neck
cancers or in case of esophagectomy). PDT has received attention as a potential treatment
modality for achieving local control in a variety of advanced malignant tumors of the
esophagus, bile ducts, pancreas, colon, rectum, carcinomatosis, bronchi, lung, pleura, head &
neck, connective tissue (sarcomas) and brain among others [17-24]. Many patients have been
treated in numerous small nonrandomized trials, but more data from trials with sufficient
statistical power demonstrating superiority to currently available treatment methods, is needed
to change clinical practice in larger scale, although there are some promising results from
smaller clinical studies [25,26]. The reason for PDT not being standard treatment in several
cancers may however be multifactorial; lack of coordinated randomized controlled trials, lack
of photosensitizer specificity and potency and practical challenges in optimizing the PDT
regime. The PDT regimens consist of a sequence of drug and light of which the individual
doses, administration form, irradiance and timing will have influence on the treatment effect
and hence the clinical outcome. Optimizing these parameters is difficult, expensive and time
consuming. PDT may also be limited by reduced penetration of light through the target tissue
and local acute phototoxicity to normal surrounding tissue.
PDT has been explored as an adjuvant treatment after debulking surgery with promising, but
not convincing results. Multiple Photofrin- and Foscan-based phase I and phase II clinical
trials with PDT after cytoreductive surgery [18-20,27-29] have been reported. In general,
maximum tolerated dose in these clinical trials is limited by treatment related side effects on
normal tissue. A problem using PDT after debulking surgery of peritoneal carcinomatosis has
been excessive fluid shift with volume overload has [19]. Moreover, PDT after surgical

ACCEPTED MANUSCRIPT
6
debulking of non-small cell lung cancer and malignant pleural mesothelioma in thoracic
surgery has resulted in esophageal perforations and bronchopulmonal fistula [17,18,29].
Regarding neurosurgery, the post treatment edema after PDT of brain tumors is potentially
limiting and has been challenging [20]. These clinical side effects have been tentatively
overcome by giving fluid resuscitation, anti-inflammatory drugs, adjusting the drug light
interval, and most of all by reducing the light and drug doses. In general, these clinical trials
have not been successful in achieving complete local responses. Many studies conclude that
there is a need for a more specific treatment effect either by adjusting current treatment
regimes, modifying the existing photosensitizers or developing new and more specific
photosensitizers. The reasons for treatment failure of PDT as an adjunct to surgery are
considered to be of critical importance and will be further discussed (see 4.2).
In summary, PDT is ideal for treating superficial and early cancers with little or no metastatic
potential, accurate staging being a matter of utmost importance and in many cases the
therapeutic ratio needs to be increased. The aims of palliative treatment are relief of symptom
and local control, however sustained local tumor control has not been achieved for most of the
suggested PDT indications. We hypothesize that the PCI technology may improve the results
of many already approved indications for PDT and possibly be able to obtain significant
clinical results where PDT has been undeceive. In addition to being a “fortified PDT” version,
PCI is also functioning as a system for site-specific drug delivery, opening up for new and yet
unmet needs in targeted cancer therapy not optimally addressed today (e.g. delivery of
immunotoxins and nucleic acids for gene therapy).

ACCEPTED MANUSCRIPT
7
2. Photochemical internalization (PCI)
Photochemical internalization (PCI) is a technology based on the same principles as PDT and
sharing many fundamental characteristics [30]. With PCI we have found the same three main
effect mechanisms to eradicate tumor cell as reported for PDT: direct cytotoxic [31], vascular
starvation [32] and a possible activation of the immune system (OJ Norum, K Berg
unpublished results). In addition PCI is introducing a fourth novel effect [30]; photochemical
release of the content of the endocytic vesicles into the cytosol in a functionally active form as
described in more detail below.
2.1. Endocytosis of macromolecules and photosensitizers
Mammalian cells have several different ways of taking up and internalize extracellular
substances, one of them being endocytosis. Most mature cells exert endocytic activity (except
mature erythrocytes), but the rate of endocytosis varies. Endocytosis involves internalization
of the extracellular substances through invagination of the cell membrane, forming an
intracellular bud, resulting in an endocytic vesicle containing the originally extracellular
substances inside the vesicles. In this way, cells can take up substances that do not readily
pass the plasma membrane. In general, molecules taken up by endocytosis are rapidly
processed and directed to other organelles, if not destined otherwise they are subjected to
enzymatic degradation [33]. Therapeutic agents that target intracellular sites have to penetrate
the plasma membrane or escape from the endocytic vesicles after endocytosis to accomplish
their therapeutic effect. Membrane impermeable molecules of therapeutic interest without any
alternative escape mechanism are consequently trapped in the endocytic vesicles (endosomal /
lysosomal compartments) and degraded [34]. There is probably a number of compounds that
have been excluded from further clinical development due to inadequate penetration through

ACCEPTED MANUSCRIPT
8
the plasma membrane and consequently low cytosolic concentration, in early pharmaceutical
screening experiments.
Some photosensitizers are for various reasons unable to penetrate the plasma membrane and
will enter the cells through endocytosis (Fig. 1). The most hydrophilic photosensitizers, such
as AlPcS4 and uroporphyrin do not efficiently intrude into the plasma membrane and will be
taken up into the cells by pinocytosis, which is an uptake mechanism of low efficacy.
Amphiphilic photosensitizers intruding into the plasma membrane, but unable to penetrate
through the plasma membrane will enter the cells via adsorptive endocytosis, which is a
substantially more efficient uptake mechanism than pinocytosis. These photosensitizers may
be incorporated into the outer leaflet of the plasma membrane and consequently ending up in
the inner leaflet of the endocytic vesicles as indicated by the localization of the disulfonated
photosensitizer in Fig.2. Photosensitizers may also be taken up by endocytosis through
receptor-mediated endocytosis when linked to targeting moieties such as antibodies or
exogene ligands. In all these cases the photosensitizer may reach the endosomes and
lysosomes. An alternative route for photosensitizers to be localized in endocytic vesicles is
directly through the plasma membrane and the endocytic membranes if the photosensitizer
contains lysosomotropic weak base properties, such as acridine orange, that cause the
photosensitizer to be trapped in the acidic compartments of late endosomes and lysosomes
(Fig. 1).
2.2. Activation of photosensitizers in PCI
The most efficient photosensitizers for use in PCI have an amphiphilic structure (Fig. 2) with
a hydrophilic part inhibiting penetration through cellular membranes as illustrated in Fig. 3.
As described above the amphiphilic photosensitizers will accumulate in the inner leaflet of the

ACCEPTED MANUSCRIPT
9
endocytic vesicles. As for PDT, reactive oxygen species are formed when the photosensitizer
is exposed to light (Fig. 3). Singlet oxygen (1O2), assumed to be most important reactive
oxygen species in PDT, has a short range of action (20-100 nm,[35-38]) and is more efficient
in the membranes than in an aqueous environment. It should be noted that hydrophilic
photosensitizers located in the matrix of the endocytic vesicles are not able to induce any PCI-
effect, pointing to the importance of the intravesicular localization of the photosensitizer [39].
The chemical reactions induced by 1O2 leads to disruption of the endocytic vesicle and release
of its contents (Fig. 4). Although the exact structure of the damaged vesicles has not been
revealed, results from previous studies indicate that relatively large particles can escape the
vesicles after PCI [40]. It was initially thought that the photosensitizer and macromolecules
to be released from the endocytic compartments had to be located in the same compartments
at the time of light exposure. However, it has been demonstrated that the photochemical
treatment can be performed up to 6-8 hours prior to the delivery without reducing the
synergistic effect of the combined treatment [40]. The assumed mechanistic basis for such an
observation is that newly formed vesicles are able to fuse with photochemically damaged
vesicles.
3. Possibilities and limitations of photochemical delivery of therapeutic agents
Therapeutic agents for photochemical delivery may be divided into four main groups: (I)
proteins, (II) nucleic acids, (III) synthetic polymers for delivery of chemotherapeutic agents
and (IV) other molecules that do not easily penetrate a cell membrane and therefore are
inhibited from entering the cellular cytosol. A prerequisite for PCI is that the molecule of
interest is accumulated in endocytic vesicles at some stage in the process. The environment of
the endocytic vesicles may set limits to the therapeutic effect of PCI in that the hydrolytic
enzymes in the endocytic vesicles may degrade the therapeutic agent. Furthermore, the

ACCEPTED MANUSCRIPT
10
reactive oxygen species formed by the photochemical treatment may inactivate the therapeutic
agent by an oxidizing process. The quantum yield for inhibition of the therapeutic activity of
the molecule will depend on its 3-D structure, i.e. the access of reactive oxygen species to
amino acids or guanine of importance for the biologic activity. In addition, the distance
between the photosensitizer and the molecule to be delivered, will have impact on the rate of
inactivation. Finally, the rate of endocytosis, and hence the uptake of the desired therapeutic
agent, varies between different cell types. It should be noted however that 60% horseradish
peroxidase taken up into endocytic vesicles has been released into the cytosol in a functionally
active form after PCI [41]. This indicates that photochemical inactivation of therapeutic
macromolecules may be of marginal importance.
In summary: the success of PCI in inducing release of functionally active remedies from an
endocytic vesicle may depend on (I) the hydrolytic activity of the endocytic vesicles, (II) the
sensitivity of the molecule to the photochemical treatment and (III) the endocytic activity of
the target cells. The present status on the use of PCI for increasing the biological activity of
various substances and the PCI effect observed in different cell lines will be reviewed.
3.1. PCI of proteins
Ribosome-inactivating protein toxins (RIPs) have extensively been evaluated for use in cancer
therapy and other diseases [42], and has been used in the PCI technology because of its
general therapeutic potential. RIPs kill cells by inhibiting protein synthesis through
inactivation of 28S ribosome. The 28S RIPs we have been using are plant toxins taken up by
endocytosis. PCI has been shown to release the type I RIPs (like saporin and gelonin, small
proteins of about 30 kDa) from endocytic vesicles and to activate their cytotoxic effect
[31,43,44]. Type I RIPs de novo has minimal therapeutic effect due to their low escape rate

ACCEPTED MANUSCRIPT
11
from the endocytic vesicles after endocytosis. With PCI of type I RIPs, a curative rate of 60-
80% has been obtained in animal models, while similar photochemical treatment without
protein toxin injection (i.e. PDT) induced only 0-10% complete response [31,45]. However,
these studies have not been optimized with respect to maximum therapeutic effect and even
higher cure rates may be obtained.
3.2. PCI of targeted proteins, increasing the specificity and cytotoxicity
Side effects of therapeutic agents often limit the treatment dose. It might be feasible to
minimize the adverse effects through drug delivery modalities that reduce the drug uptake by
non-target cells and selectively deliver the drugs to the cancer cells at low extracellular
concentrations. A current approach to site-specific drug delivery is to conjugate the
therapeutic agent to a carrier recognized only by the cells where the pharmacological action is
desired. This will provide additional specificity to the already exciting dual specificity of PDT
and PCI arising from the tumor cell retention of photosensitizer and their biological activities
restricted to illuminated areas.
The uptake and specificity of protein toxins, like type I RIPs, may be enhanced by means of
targeting moieties such as monoclonal antibodies (i.e. immunotoxin) or growth factors (e.g
epidermal growth factor, EGF). On the cellular level, enzymatic degradation of such targeting
toxins in the endocytic vesicles is a major hindrance for their therapeutic effect [46]. We have
used gelonin conjugated to the monoclonal antibody MOC31, which targets an epithelial
antigen (EpCAM/EGP-2) that is expressed on nearly all types of carcinoma cells. After PCI-
induced release of the immunotoxin MOC31-gelonin, a >10-fold increase in cytotoxicity as
compared to PCI of gelonin was observed [47]. PCI of MOC31-gelonin was efficacious
against several different carcinoma cell lines (lung (H-146), breast (T47D) and colon (WiDr
and KM20L2)), demonstrating that PCI of immunotoxins has the potential to become a potent

ACCEPTED MANUSCRIPT
12
anti-cancer application. However, it was later found, by means of confocal microscopy, that
most of Alexa488-labelled MOC31-gelonin was bound to the cell surface and was not
endocytosed (data not published) after overnight incubation. Due to the low penetration rate
of immunotoxins through solid tumours, it is of outermost importance to select targeting
ligands, which are efficiently endocytosed by the tumour cells. Epidermal growth factor
receptor (EGFR) is overexpressed in many different types of solid tumours and is associated
with metastasis and poor prognosis [48]. Thus, both antibodies and tyrosine kinase inhibitors
against EGFR are used in treatment of several cancers [49]. After activation by a ligand such
as EGF, the EGFR-complex is rapidly endocytosed and may therefore be a good candidate
target for efficient endocytosis of therapeutic agents. EGF linked to the type I RIP saporin,
has been evaluated for its therapeutic potential as an affinity toxin in combination with PCI.
The results indicate that EGF-saporin is efficiently endocytosed and that PCI of EGF-saporin
improves both efficacy and specificity as compared to PCI of saporin [50]. Improvement of
efficacy has also been obtained by linking Cetuximab to saporin. Cetuximab is a monoclonal
antibody recognizing the ligand binding extracellular domain of the EGFR [51].
Immunotoxins and affinity toxins like MOC31-gelonin, EGF-saporin and Cetuximab-saporin
represents highly toxic targeted molecules with good therapeutic potential in combination
with the PCI technology.
3.3. PCI of nucleic acids
PCI in gene delivery has recently been reviewed in detail [52] and will only briefly be
described here. Gene therapy is recognized as having the treatment potential for a wide range
of diseases [53]. With most gene delivery vectors, the therapeutic gene is taken into the cell
by endocytosis, and for many of these vectors the efficient mechanisms for translocating the
gene out of the endocytic vesicles turns out to constitute a major hindrance for realization of

ACCEPTED MANUSCRIPT
13
the therapeutic potential. Photochemical internalization has been studied as a gene delivery
technology mainly by using reporter genes, but has also been shown to induce the delivery of
therapeutic genes [54]. The photochemical translocation of nucleic acids into cytosol has been
shown to enhance the transgene activity both when non-viral, adenoviral and adeno-assisiated
viral vectors are used. PCI has also been shown to enhance gene silencing by means of
peptide-nucleic acids and siRNA [20,55,56]. As shown for PCI of protein toxins, PCI of
targeted gene delivery vectors improves the specificity and efficacy of the transgene delivery
[57].
3.4. PCI of chemotherapeutic agents
Some chemotherapeutic agents accumulate in endocytic vesicles probably due to their size
and/or charge. Bleomycin (molecular weight about 1,4 kDa) is a widely used
chemotherapeutic agent for several types of cancer (e.g. testicular cancers, lymphomas and
germinal cell tumor of children), and known to accumulate in endocytic vesicles [58]. PCI has
been shown to improve its cytotoxic effect both in vitro and in vivo [59], indicating that not
only large macromolecules like proteins and genes may profit from a PCI-based treatment, but
also smaller molecules unable to penetrate the biological membranes efficiently. PCI has the
potential to reduce the administered dose of bleomycin, and hence the dose dependent side
effects, without losing clinical efficacy. These results warrant evaluation of PCI of BLM in
future clinical PCI protocols. PCI may also reverse resistance to weak base chemotherapeutics
such as doxorubicin when resistance is caused by lowered pH in endocytic vesicles [60]. The
specificity of small molecular chemotherapeutic agents may be improved by linking to
polymers. PCI has been shown to enhance the therapeutic efficacy of such complexes [61,62].
3.5. The PCI effect on different cell lines

ACCEPTED MANUSCRIPT
14
In addition to the ability of activating a plethora of therapeutic molecules accumulating in the
endocytic vesicles, it should be noted that the PCI technology induces biological effects in a
variety of cells and tissue types – both malignant and non-malignant cells. So far PCI of
gelonin, saporin, genes and bleomycin has been demonstrated in more than 30 cell lines and
documented in vivo with the same macromolecules in xenografted nude mice and allografted
immuno-competent mice using 6 different cell lines (WiDr, TAX-1, HT1080, CT26, THX ,
DU145) [61,63,64]. Since all mammalian cells, except mature erythrocytes, exert endocytotic
activity, the potential of the PCI technology is not restricted to specific indications, but may
be utilized in all cancers suitable for PDT as well as non-malignant diseases.
4. PCI in translational research
The PCI technology and its diversity have been described above. PCI is currently approaching
bedside medicine and we will review recent studies of assumed clinical relevance and the
possible clinical implications in the following paragraphs.
4.1. PCI for treatment of invasive cancers and as adjunct to surgery
The infiltrative growth of many cancers frequently results in local recurrences and is therefore
a major challenge in cancer treatment. All the in vivo PCI reports reviewed above are based
on subcutaneously located tumors in mice. However, PCI was found equally efficient on a
orthotopic, deep and invasive HT1080 fibrosarcoma model relevant for evaluating the clinical
potential of the PCI technology [32]. As in the subcutaneous tumor models PCI of BLM was
found to retard tumor growth significantly more than PDT (Fig. 5) and acting in a synergistic
manner as compared to the expected sum of BLM and PDT. Contrast enhanced magnetic
resonance images (CE-MRI) obtained two hours after PDT and histological sections seven
days after PDT both indicated that the tumor periphery responds differently from the central

ACCEPTED MANUSCRIPT
15
region after PDT in accordance with recent report on BPD-MA-based PDT [65]. The CE-MRI
indicated that PDT induced inhibition of perfusion throughout the tumor, except in the tumor
periphery where the tumor regrowth was confirmed by histology. The link between these
observations has not been unequivocally confirmed, but surgical removal of the central tumor
indicates low sensitivity of the tumor periphery to PDT (OJ Norum, K berg, unpublished
results). In contrast, PCI of BLM in the cavity after inadequate (R1) surgical resection of the
tumor induced a strong delay in tumor growth (Fig.6). Our results indicate that the limited
success of PDT as an adjuvant to surgery may be due to lower PDT sensitivity of the
infiltrative transitional zone left after cytoreductive surgery. We therefore anticipate that the
synergistic effect of AlPcS2a-PDT and BLM (i.e PCI of BLM) after cytoreductive surgery is
due to the internalization of BLM and that PCI in this study acts as an effective drug delivery
system in which BLM is released to the cytosol where it can reach its target and inhibit tumor
growth. The results also indicate that the photosensitizer is taken up by the remaining tumor
cells after surgery, but for reasons that are not fully understood is not able to induce detectable
photocytotoxicity. It is however not clear whether the enhanced treatment effect observed by
PCI is due to targeting of the parenchyma or endothelial cells of the tumor. The results may
indicate that PCI offers a new possibility as an adjuvant to surgery more effective than PDT.
4.2. PCI as adjunct to radiotherapy
Ionizing radiation has continuously been refined through many decades and is a generally
accepted standard in both curative and palliative therapy for a variety of cancers. In general
the therapeutic effect of ionizing radiation is via DNA damage while PDT mainly affects the
cellular membranes [8]. Previously published in vitro and in vivo studies on the interaction of
ionizing radiation and PDT have shown contradictory results, ranging from synergistic to
antagonistic effects [66-71]. In an athymic mouse model using a transplantable fibrosarcoma

ACCEPTED MANUSCRIPT
16
TAX-1, PDT and PCI were found to interact in an additive to antagonistic manner when
combined with ionizing radiation (OJ Norum, ØS Bruland, K Berg unpublished results).
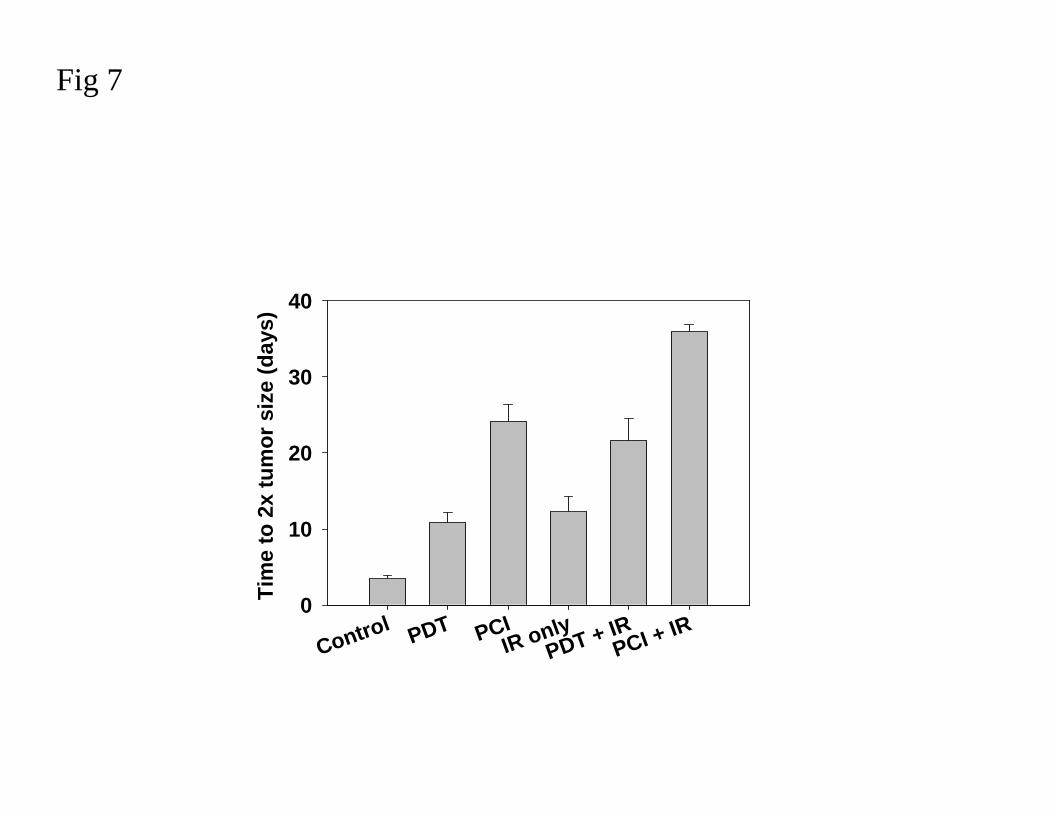
However, PCI as neoadjuvant treatment to ionizing radiation induced a prolonged time to
progression as compared to PDT (Fig 7). The effect of BLM in combination with ionizing
radiation would be increased by higher cumulative doses of both fractionated BLM and
ionizing radiation [72,73]. However, there are animal and clinical reports showing that
increasing the BLM or ionizing radiation doses in radiochemotherapy will increase the side
effects to unacceptable levels [72-75]. PCI of BLM will increase the therapeutic effect of
BLM and hence lower the needed drug dose. When the PCI of BLM and ionizing radiation
were separated in time by one week, no side effects were observed. Thus, PCI of BLM
appears as a safe neoadjuvant to ionizing radiation exerting therapeutic advantages and may
be considered as a treatment modality to preceed a standard radiotherapy regimen.
4.3. PCI and the immunological response
While radiotherapy, chemotherapy and major surgery [76] predominantly evoke an
immunosuppressive response in the patients, PDT is reported to cause a strong local
inflammation leading to activation of the systemic immune system in the host [77]. The
immune response seems important for both the immediate tumor cell eradication and for the
long term tumor control [78]. Knowledge on PDT and immunity is manly obtained from
preclinical studies, however a few clinical reports [79,80] provide indications that the
immunostimulatory effects initiated by PDT may also influence on the clinical outcome of
PDT. Still much need to be done to close in on the optimal PDT regimen [81] and the
development of a clinically useful PDT tumor specific whole-cell cancer vaccine. One
possible great advantage of the potential of a PDT generated vaccine is that no immuno-
adjuvants are needed and that the vaccine may be developed in situ [82].

ACCEPTED MANUSCRIPT
17
In experimental models PDT has been proven to evoke an acute inflammation and activating
the immune system in immuno-competent mice to a degree that facilitates tumor specific
resistance if a re-challenge with the same cell line is administrated [82-84]. The vaccination
ability of PDT appears independent of the photosensitizer [83]. Accordingly, AlPcS2a-PDT
has been found to induce a similar inhibition of growth of CT26.CL25 mouse colon
carsinoma cells when immuno-competent BALB/c mice previously had been subjected to
curative PDT. PCI of BLM appears equally efficient in inhibiting regrowth of CT26.CL25
tumor cells as PDT (Fig. 8). Thus, PCI does not seem to be a hindrance for the immunological
activation and development of a cancer vaccine (unpublished, OJ Norum, K. Berg). A
possible vaccinating response to PDT and PCI would develop these treatment modalities from
aids to local cancer control to a curative endpoint. However, clinical documentation is still
sparse and in the case of PCI the influence of the macromolecule to be delivered on the
immune response among others need further evaluation.
5. Future perspectives
In this review, we focused on PCI as a potential treatment modality in cancer care. PDT is
today approved for treating superficial, precancerous or early malignancies with little or no
metastatic potential, but despite promising and impressive results from many clinical trials
PDT is only regarded as standard therapy within a limited number of malignancies.
PDT is challenged by alternative treatment methods (open or endoscopic surgery,
radiofrequency ablation, cryotherapy, radiotherapy, chemotherapy among others) and for PDT
to succeed, PCI may contribute with increased therapeutic efficacy and specificity to the
photochemical treatment. It has been demonstrated that PCI is able to increase the depth of
necrosis and that the tumor periphery seems to be more susceptible to PCI treatment than
PDT. Thus, PCI may be more effective than PDT and by combination with appropriately

ACCEPTED MANUSCRIPT
18
targeted therapeutic agents it should be possible to enhance specificity and lowering the total
PDT dose, but still increasing the therapeutic efficacy.
In palliative cancer care aiming at prolonged local control, PDT has achieved some
impressive results (e.g. cholangiocarcinoma). PCI should be applicable for the same
indications assumed for PDT and warrant further studies. In case of PCI in combination with
surgery, our study demonstrates that the potential of synergistic achievements by means of
PCI may be superior to PDT.
It is important to emphasize that the current phase I study on PCI in humans is carried out
with bleomycin. Although PCI of BLM has shown promising synergistic effects, it is to be
expected that PCI of targeted therapeutics will further enhance the specificity and efficacy of
this treatment modality. The PCI technology may be regarded as a “fortified PDT”, but
perhaps more important is that the PCI technology is able to activate a plethora of potentially
biological active molecules that are today not in clinical use because they have intracellular
targets and do not readily pass the plasma membrane. With an ideal therapeutic agent, having
low biological activity without PCI, and high toxicity once released in cytosol, the PCI
technology may function as an efficient drug delivery system. In addition to the well-known
PDT-related cytotoxic mechanisms, this may be utilized to obtain better clinical results.
Acknowledgements
This study has been financially supported by the Norwegian Radium Hospital Research
Foundation, Norwegian Research Council and the Norwegian Cancer Society.

ACCEPTED MANUSCRIPT
19
References
1 D.E.Dolmans, D.Fukumura, and R.K.Jain, Photodynamic therapy for cancer, Nat. Rev. Cancer. 3 (2003) 380-387.
2 A.P.Castano, P.Mroz, and M.R.Hamblin, Photodynamic therapy and anti-tumour immunity, Nat. Rev. Cancer. 6 (2006) 535-545.
3 S.B.Brown, E.A.Brown, and I.Walker, The present and future role of photodynamic therapy in cancer treatment, Lancet Oncol. 5 (2004) 497-508.
4 C.Hopper, Photodynamic therapy: a clinical reality in the treatment of cancer, Lancet Oncol. 1:212-9. (2000) 212-219.
5 M.Triesscheijn, P.Baas, J.H.Schellens, and F.A.Stewart, Photodynamic therapy in oncology, Oncologist. 11 (2006) 1034-1044.
6 B.C.Wilson and M.S.Patterson, The physics, biophysics and technology of photodynamic therapy, Phys. Med. Biol. 53 (2008) R61-109.
7 A.Juzeniene, Q.Peng, and J.Moan, Milestones in the development of photodynamic therapy and fluorescence diagnosis, Photochem. Photobiol. Sci. 6 (2007) 1234-1245.
8 F.Stewart, P.Baas, and W.Star, What does photodynamic therapy have to offer radiation oncologists (or their cancer patients)?, Radiother. Oncol. 48 (1998) 233-248.
9 R.Hornung, H.Walt, N.E.Crompton, K.A.Keefe, B.Jentsch, G.Perewusnyk, U.Haller, and O.R.Kochli, m-THPC-mediated photodynamic therapy (PDT) does not induce resistance to chemotherapy, radiotherapy or PDT on human breast cancer cells in vitro, Photochem. Photobiol. 68 (1998) 569-574.
10 A.Radu, P.Grosjean, C.Fontolliet, G.Wagnieres, A.Woodtli, H.V.Bergh, and P.Monnier, Photodynamic therapy for 101 early cancers of the upper aerodigestive tract, the esophagus, and the bronchi: a single-institution experience, Diagn. Ther. Endosc. 5 (1999) 145-154.
11 G.D.Mackenzie, N.F.Jamieson, M.R.Novelli, C.A.Mosse, B.R.Clark, S.M.Thorpe, S.G.Bown, and L.B.Lovat, How light dosimetry influences the efficacy of photodynamic therapy with 5-aminolaevulinic acid for ablation of high-grade dysplasia in Barrett's esophagus, Lasers Med. Sci. 23 (2008) 203-210.
12 R.E.Pouw, J.J.Gondrie, C.M.Sondermeijer, F.J.ten Kate, T.M.van Gulik, K.K.Krishnadath, P.Fockens, B.L.Weusten, and J.J.Bergman, Eradication of Barrett esophagus with early neoplasia by radiofrequency ablation, with or without endoscopic resection, J. Gastrointest. Surg. 12 (2008) 1627-1636.
13 F.D.Sheski and P.N.Mathur, Endoscopic treatment of early-stage lung cancer, Cancer Control. 7 (2000) 35-44.
14 T.Zoepf, Photodynamic therapy of cholangiocarcinoma, HPB (Oxford). 10 (2008) 161-163.
15 K.Moghissi, K.Dixon, M.Stringer, T.Freeman, A.Thorpe, and S.Brown, The place of bronchoscopic photodynamic therapy in advanced unresectable lung cancer: experience of 100 cases, Eur. J. Cardiothorac. Surg. 15 (1999) 1-6.
16 P.J.Lou, H.R.Jager, L.Jones, T.Theodossy, S.G.Bown, and C.Hopper, Interstitial photodynamic therapy as salvage treatment for recurrent head and neck cancer, Br. J. Cancer. 91 (2004) 441-446.

ACCEPTED MANUSCRIPT
20
17 J.S.Friedberg, R.Mick, J.Stevenson, J.Metz, T.Zhu, J.Buyske, D.H.Sterman, H.I.Pass, E.Glatstein, and S.M.Hahn, A phase I study of Foscan-mediated photodynamic therapy and surgery in patients with mesothelioma, Ann. Thorac. Surg. 75 (2003) 952-959.
18 J.S.Friedberg, R.Mick, J.P.Stevenson, T.Zhu, T.M.Busch, D.Shin, D.Smith, M.Culligan, A.Dimofte, E.Glatstein, and S.M.Hahn, Phase II trial of pleural photodynamic therapy and surgery for patients with non-small-cell lung cancer with pleural spread, J. Clin. Oncol. 22 (2004) 2192-2201.
19 S.M.Hahn, D.L.Fraker, R.Mick, J.Metz, T.M.Busch, D.Smith, T.Zhu, C.Rodriguez, A.Dimofte, F.Spitz, M.Putt, S.C.Rubin, C.Menon, H.W.Wang, D.Shin, A.Yodh, and E.Glatstein, A phase II trial of intraperitoneal photodynamic therapy for patients with peritoneal carcinomatosis and sarcomatosis, Clin. Cancer Res. 12 (2006) 2517-2525.
20 S.S.Stylli, A.H.Kaye, L.MacGregor, M.Howes, and P.Rajendra, Photodynamic therapy of high grade glioma - long term survival, J. Clin. Neurosci. 12 (2005) 389-398.
21 J.Usuda, H.Kato, T.Okunaka, K.Furukawa, H.Tsutsui, K.Yamada, Y.Suga, H.Honda, Y.Nagatsuka, T.Ohira, M.Tsuboi, and T.Hirano, Photodynamic therapy (PDT) for lung cancers, J. Thorac. Oncol. 1 (2006) 489-493.
22 J.B.Wang and L.X.Liu, Use of photodynamic therapy in malignant lesions of stomach, bile duct, pancreas, colon and rectum, Hepatogastroenterology. 54 (2007) 718-724.
23 T.Nakamura, K.Kusuzaki, T.Matsubara, A.Matsumine, H.Murata, and A.Uchida, A new limb salvage surgery in cases of high-grade soft tissue sarcoma using photodynamic surgery, followed by photo- and radiodynamic therapy with acridine orange, J. Surg. Oncol. 97 (2008) 523-528.
24 W.Jerjes, T.Upile, C.S.Betz, M.M.El, S.Abbas, A.Wright, and C.Hopper, The application of photodynamic therapy in the head and neck, Dent. Update. 34 (2007) 478-4, 486.
25 M.E.Ortner, K.Caca, F.Berr, J.Liebetruth, U.Mansmann, D.Huster, W.Voderholzer, G.Schachschal, J.Mossner, and H.Lochs, Successful photodynamic therapy for nonresectable cholangiocarcinoma: a randomized prospective study, Gastroenterology. 125 (2003) 1355-1363.
26 R.E.Cuenca, R.R.Allison, C.Sibata, and G.H.Downie, Breast cancer with chest wall progression: treatment with photodynamic therapy, Ann. Surg. Oncol. 11 (2004) 322-327.
27 J.S.Friedberg, R.Mick, J.Stevenson, J.Metz, T.Zhu, J.Buyske, D.H.Sterman, H.I.Pass, E.Glatstein, and S.M.Hahn, A phase I study of Foscan-mediated photodynamic therapy and surgery in patients with mesothelioma, Ann. Thorac. Surg. 75 (2003) 952-959.
28 H.I.Pass, B.K.Temeck, K.Kranda, G.Thomas, A.Russo, P.Smith, W.Friauf, and S.M.Steinberg, Phase III randomized trial of surgery with or without intraoperative photodynamic therapy and postoperative immunochemotherapy for malignant pleural mesothelioma, Ann. Surg. Oncol. 4 (1997) 628-633.
29 H.B.Ris, Photodynamic therapy as an adjunct to surgery for malignant pleural mesothelioma, Lung Cancer. 49 Suppl 1:S65-8. (2005) S65-S68.
30 K.Berg, P.K.Selbo, L.Prasmickaite, T.E.Tjelle, K.Sandvig, J.Moan, G.Gaudernack, O.Fodstad, S.Kjolsrud, H.Anholt, G.H.Rodal, S.K.Rodal, and A.Hogset, Photochemical internalization: a novel technology for delivery of macromolecules into cytosol, Cancer Res. 59 (1999) 1180-1183.
31 P.K.Selbo, G.Sivam, O.Fodstad, K.Sandvig, and K.Berg, In vivo documentation of photochemical internalization, a novel approach to site specific cancer therapy, Int. J. Cancer 92 (2001) 761-766.
32 O.J.Norum, J.-V.Gaustad, E.Angell-Pettersen, E.K.Rofstad, Q.Peng, K.-E.Giercksky, and K.Berg, Photochemical Internalization of Bleomycin is Superior to Photodynamic Therapy due to the Therapeutic Effect in the Tumor Periphery, Photochem. Photobiol. In Press (2008).
33 S.Mukherjee, R.N.Ghosh, and F.R.Maxfield, Endocytosis, Physiol Rev. 77 (1997) 759-803.

ACCEPTED MANUSCRIPT
21
34 J.B.Lloyd, Lysosome membrane permeability: implications for drug delivery, Adv. Drug Deliv. Rev. 41 (2000) 189-200.
35 S.Hatz, L.Poulsen, and P.R.Ogilby, Time-resolved singlet oxygen phosphorescence measurements from photosensitized experiments in single cells: effects of oxygen diffusion and oxygen concentration, Photochem. Photobiol. 84 (2008) 1284-1290.
36 J.Moan and K.Berg, The photodegradation of porphyrins in cells can be used to estimate the lifetime of singlet oxygen, Photochem. Photobiol. 53 (1991) 549-553.
37 M.J.Niedre, M.S.Patterson, A.Giles, and B.C.Wilson, Imaging of photodynamically generated singlet oxygen luminescence in vivo, Photochem. Photobiol. 81 (2005) 941-943.
38 A.Jimenez-Banzo, M.L.Sagrista, M.Mora, and S.Nonell, Kinetics of singlet oxygen photosensitization in human skin fibroblasts, Free Radic. Biol. Med. 44 (2008) 1926-1934.
39 L.Prasmickaite, A.Hogset, and K.Berg, Evaluation of different photosensitizers for use in photochemical gene transfection, Photochem. Photobiol. 73 (2001) 388-395.
40 L.Prasmickaite, A.Hogset, P.K.Selbo, B.O.Engesaeter, M.Hellum, and K.Berg, Photochemical disruption of endocytic vesicles before delivery of drugs: a new strategy for cancer therapy, Br. J. Cancer. 86 (2002) 652-657.
41 K.Berg and J.Moan, Lysosomes as photochemical targets, Int. J. Cancer. 59 (1994) 814-822.
42 G.R.Thrush, L.R.Lark, B.C.Clinchy, and E.S.Vitetta, Immunotoxins: an update, Annu. Rev. Immunol. 14:49-71. (1996) 49-71.
43 K.Berg, L.Prasmickaite, P.K.Selbo, M.Hellum, A.Bonsted, and A.Hogset, Photochemical internalization (PCI)--a novel technology for release of macromolecules from endocytic vesicles, Oftalmologia. 56 (2003) 67-71.
44 P.K.Selbo, K.Sandvig, V.Kirveliene, and K.Berg, Release of gelonin from endosomes and lysosomes to cytosol by photochemical internalization, Biochim. Biophys. Acta. 1475 (2000) 307-313.
45 A.Dietze, Q.Peng, P.K.Selbo, O.Kaalhus, C.Muller, S.Bown, and K.Berg, Enhanced photodynamic destruction of a transplantable fibrosarcoma using photochemical internalisation of gelonin, Br. J. Cancer. 92 (2005) 2004-2009.
46 M.Wu, Enhancement of immunotoxin activity using chemical and biological reagents, Br. J. Cancer. 75 (1997) 1347-1355.
47 P.K.Selbo, G.Sivam, O.Fodstad, K.Sandvig, and K.Berg, Photochemical internalisation increases the cytotoxic effect of the immunotoxin MOC31-gelonin, Int. J. Cancer. 87 (2000) 853-859.
48 J.Baselga, The EGFR as a target for anticancer therapy--focus on cetuximab, Eur. J. Cancer. 37 Suppl 4:S16-22. (2001) S16-S22.
49 L.Castillo, M.C.Etienne-Grimaldi, J.L.Fischel, P.Formento, N.Magne, and G.Milano, Pharmacological background of EGFR targeting, Ann. Oncol. 15 (2004) 1007-1012.
50 A.Weyergang, P.K.Selbo, and K.Berg, Photochemically stimulated drug delivery increases the cytotoxicity and specificity of EGF-saporin, J. Control Release. 111 (2006) 165-173.
51 W.L.Yip, A.Weyergang, K.Berg, H.H.Tonnesen, and P.K.Selbo, Targeted delivery and enhanced cytotoxicity of cetuximab-saporin by photochemical internalization in EGFR-positive cancer cells, Mol. Pharm. 4 (2007) 241-251.
52 A.Hogset, L.Prasmickaite, P.K.Selbo, M.Hellum, B.O.Engesaeter, A.Bonsted, and K.Berg, Photochemical internalisation in drug and gene delivery, Adv. Drug Deliv. Rev. 56 (2004) 95-115.

ACCEPTED MANUSCRIPT
22
53 N.Somia and I.M.Verma, Gene therapy: trials and tribulations, Nat. Rev. Genet. 1 (2000) 91-99.
54 L.Prasmickaite, A.Hogset, V.M.Olsen, O.Kaalhus, S.O.Mikalsen, and K.Berg, Photochemically enhanced gene transfection increases the cytotoxicity of the herpes simplex virus thymidine kinase gene combined with ganciclovir, Cancer Gene Ther. 11 (2004) 514-523.
55 S.Oliveira, M.M.Fretz, A.Hogset, G.Storm, and R.M.Schiffelers, Photochemical internalization enhances silencing of epidermal growth factor receptor through improved endosomal escape of siRNA, Biochim. Biophys. Acta. 1768 (2007) 1211-1217.
56 M.Folini, K.Berg, E.Millo, R.Villa, L.Prasmickaite, M.G.Daidone, U.Benatti, and N.Zaffaroni, Photochemical internalization of a peptide nucleic acid targeting the catalytic subunit of human telomerase, Cancer Res. 63 (2003) 3490-3494.
57 A.Bonsted, E.Wagner, L.Prasmickaite, A.Hogset, and K.Berg, Photochemical enhancement of DNA delivery by EGF receptor targeted polyplexes, Methods Mol. Biol. 434:171-81. (2008) 171-181.
58 G.Pron, N.Mahrour, S.Orlowski, O.Tounekti, B.Poddevin, J.Belehradek, Jr., and L.M.Mir, Internalisation of the bleomycin molecules responsible for bleomycin toxicity: a receptor-mediated endocytosis mechanism, Biochem. Pharmacol. 57 (1999) 45-56.
59 K.Berg, A.Dietze, O.Kaalhus, and A.Hogset, Site-specific drug delivery by photochemical internalization enhances the antitumor effect of bleomycin, Clin. Cancer Res. 11 (2005) 8476-8485.
60 P.J.Lou, P.S.Lai, M.J.Shieh, A.J.MacRobert, K.Berg, and S.G.Bown, Reversal of doxorubicin resistance in breast cancer cells by photochemical internalization, Int. J. Cancer. 119 (2006) 2692-2698.
61 H.Cabral, M.Nakanishi, M.Kumagai, W.D.Jang, N.Nishiyama, and K.Kataoka, A Photo-Activated Targeting Chemotherapy Using Glutathione Sensitive Camptothecin-Loaded Polymeric Micelles, Pharm. Res. (2008).
62 M.J.Shieh, C.L.Peng, P.J.Lou, C.H.Chiu, T.Y.Tsai, C.Y.Hsu, C.Y.Yeh, and P.S.Lai, Non-toxic phototriggered gene transfection by PAMAM-porphyrin conjugates, J. Control Release. 129 (2008) 200-206.
63 A.Ndoye, G.Dolivet, A.Hogset, A.Leroux, A.Fifre, P.Erbacher, K.Berg, J.P.Behr, F.Guillemin, and J.L.Merlin, Eradication of p53-mutated head and neck squamous cell carcinoma xenografts using nonviral p53 gene therapy and photochemical internalization, Mol. Ther. 13 (2006) 1156-1162.
64 A.Dietze, P.K.Selbo, L.Prasmickaite, A.Weyergang, A.Bonsted, B.Engesaeter, A.Hogset, and K.Berg, Photochemical internalization (PCI): a new modality for light activation of endocytosed therapeuticals, J. Environ. Pathol. Toxicol. Oncol. 25 (2006) 521-536.
65 B.Chen, C.Crane, C.He, D.Gondek, P.Agharkar, M.D.Savellano, P.J.Hoopes, and B.W.Pogue, Disparity between prostate tumor interior versus peripheral vasculature in response to verteporfin-mediated vascular-targeting therapy, Int. J. Cancer. 123 (2008) 695-701.
66 G.Graschew and M.Shopova, Photodynamic Therapy and gamma-irradiation of tumors:Effect on Tumor-Cell Reoxygenation, 1:193 1986 ed., 2008.
67 H.Kostron, M.R.Swartz, D.C.Miller, and R.L.Martuza, The interaction of hematoporphyrin derivative, light, and ionizing radiation in a rat glioma model, Cancer. 57 (1986) 964-970.
68 Z.Luksiene, A.Kalvelyte, and R.Supino, On the combination of photodynamic therapy with ionizing radiation, J. Photochem. Photobiol. B. 52 (1999) 35-42.
69 R.Allman, P.Cowburn, and M.Mason, Effect of photodynamic therapy in combination with ionizing radiation on human squamous cell carcinoma cell lines of the head and neck, Br. J. Cancer. 83 (2000) 655-661.

ACCEPTED MANUSCRIPT
23
70 K.Berg, Z.Luksiene, J.Moan, and L.Ma, Combined treatment of ionizing radiation and photosensitization by 5-aminolevulinic acid-induced protoporphyrin IX, Radiat. Res. 142 (1995) 340-346.
71 S.J.Madsen, C.H.Sun, B.J.Tromberg, A.T.Yeh, R.Sanchez, and H.Hirschberg, Effects of combined photodynamic therapy and ionizing radiation on human glioma spheroids, Photochem. Photobiol. 76 (2002) 411-416.
72 J.Molin, P.E.Sogaard, and J.Overgaard, Experimental studies on the radiation-modifying effect of bleomycin in malignant and normal mouse tissue in vivo, Cancer Treat. Rep. 65 (1981) 583-589.
73 P.Lelieveld, M.A.Scoles, J.M.Brown, and R.F.Kallman, The effect of treatment in fractionated schedules with the combination of X-irradiation and six cytotoxic drugs on the RIF-1 tumor and normal mouse skin, Int. J. Radiat. Oncol. Biol. Phys. 11 (1985) 111-121.
74 K.K.Fu, T.L.Phillips, I.J.Silverberg, C.Jacobs, D.R.Goffinet, C.Chun, M.A.Friedman, M.Kohler, K.McWhirter, and S.K.Carter, Combined radiotherapy and chemotherapy with bleomycin and methotrexate for advanced inoperable head and neck cancer: update of a Northern California Oncology Group randomized trial, J. Clin. Oncol. 5 (1987) 1410-1418.
75 H.Vermund, O.Kaalhus, F.Winther, J.Trausjo, E.Thorud, and R.Harang, Bleomycin and radiation therapy in squamous cell carcinoma of the upper aero-digestive tract: a phase III clinical trial, Int. J. Radiat. Oncol. Biol. Phys. 11 (1985) 1877-1886.
76 C.S.Ng, T.W.Lee, S.Wan, I.Y.Wan, A.D.Sihoe, A.A.Arifi, and A.P.Yim, Thoracotomy is associated with significantly more profound suppression in lymphocytes and natural killer cells than video-assisted thoracic surgery following major lung resections for cancer, J. Invest Surg. 18 (2005) 81-88.
77 M.Korbelik, PDT-associated host response and its role in the therapy outcome, Lasers Surg. Med. 38 (2006) 500-508.
78 A.P.Castano, P.Mroz, and M.R.Hamblin, Photodynamic therapy and anti-tumour immunity, Nat. Rev. Cancer. 6 (2006) 535-545.
79 E.S.bdel-Hady, P.Martin-Hirsch, M.Duggan-Keen, P.L.Stern, J.V.Moore, G.Corbitt, H.C.Kitchener, and I.N.Hampson, Immunological and viral factors associated with the response of vulval intraepithelial neoplasia to photodynamic therapy, Cancer Res. 61 (2001) 192-196.
80 H.Yanai, Y.Kuroiwa, N.Shimizu, Y.Matsubara, T.Okamoto, A.Hirano, Y.Nakamura, K.Okita, and T.Sekine, The pilot experience of immunotherapy-combined photodynamic therapy for advanced gastric cancer in elderly patients, Int. J. Gastrointest. Cancer. 32 (2002) 139-142.
81 B.W.Henderson, S.O.Gollnick, J.W.Snyder, T.M.Busch, P.C.Kousis, R.T.Cheney, and J.Morgan, Choice of oxygen-conserving treatment regimen determines the inflammatory response and outcome of photodynamic therapy of tumors, Cancer Res. 64 (2004) 2120-2126.
82 S.O.Gollnick, L.Vaughan, and B.W.Henderson, Generation of effective antitumor vaccines using photodynamic therapy, Cancer Res. 62 (2002) 1604-1608.
83 M.Korbelik, G.Krosl, J.Krosl, and G.J.Dougherty, The role of host lymphoid populations in the response of mouse EMT6 tumor to photodynamic therapy, Cancer Res. 56 (1996) 5647-5652.
84 G.Canti, D.Lattuada, A.Nicolin, P.Taroni, G.Valentini, and R.Cubeddu, Antitumor immunity induced by photodynamic therapy with aluminum disulfonated phthalocyanines and laser light, Anticancer Drugs. 5 (1994) 443-447.

ACCEPTED MANUSCRIPT
24
Figure legends
Fig 1. Intracellular transport pathways for photosensitizers to endocytic vesicles.
Photosensitizers (P) may enter endocytic vesicles through 1) pinocytosis (Pp), e.g. in case of
hydrophilic photosensitizers and photosensitizers linked to hydrophilic moieties; 2) adsorptive
endocytosis (Pa), e.g. in case of amphiphilic photosensitizers; 3) receptor-mediated
endocytosis (P-λ) when the photosensitizer is linked to a ligand with affinity for a plasma
membrane receptor or 4) trapped in acidic vesicles when the photosensitizer exerts
lysosomotropic weak base properties (P-H+).
Fig 2. Proposed localization of sulphonated photosensitizers used in the endocytic
vesicle.
The figure shows a schematic drawing of an endocytic vesicle and the main localization of
tetrasulfonated (tetra/4-sulfonatophenyl)phorphine, TPPS4) and two amphiphilic disulfonated
photosensitizers (tetraphenylporphine disulfonate, TPPS2a) and aluminium phthalocyanine
disulfonate (AlPcS2a). The drawings are not in scale. AlPcS2a and TPPS2a are amphifilic
photosensitizers and are first adsorbed into the plasma membrane. By endocytosis the
photosensitizer is then transported into the cell and kept localized in the membrane of the
endocytic vesicle. TPPS4 is not localized in the membrane, but in the matrix and taken up by
pinocytosis.
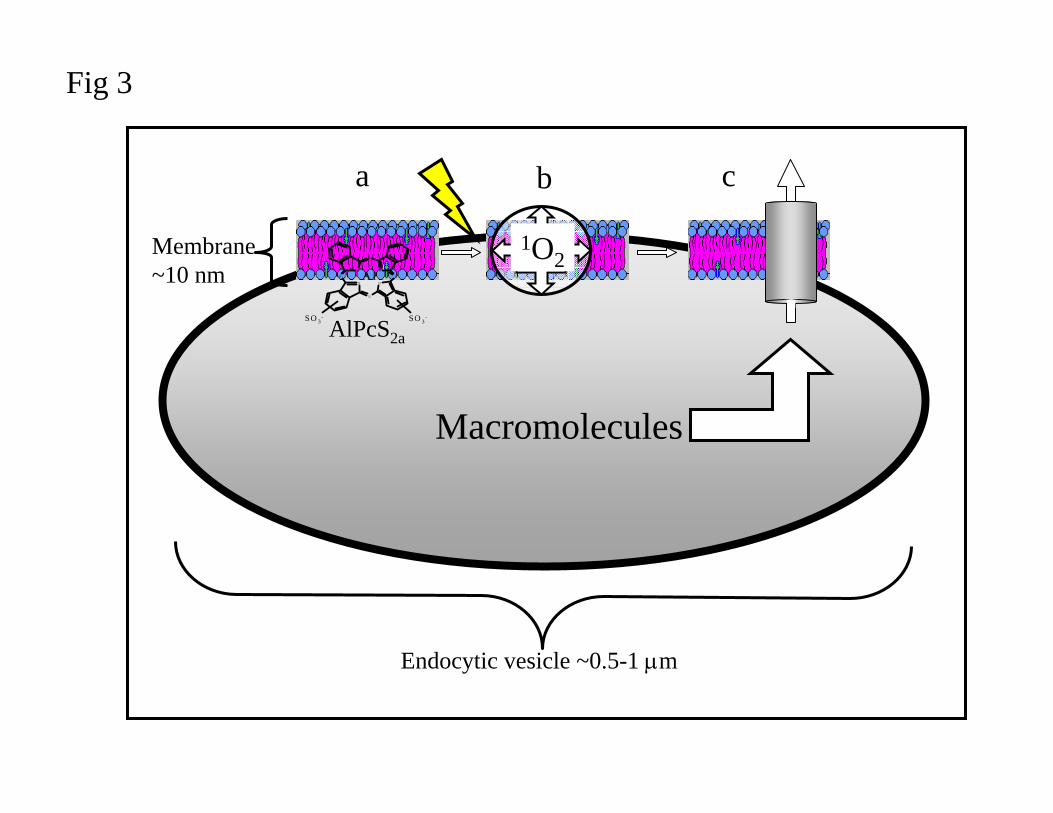
Fig 3. Schematic representation of photochemical internalization (PCI).
The figure illustrates an endocytic vesicle. In photochemical internalization (PCI),
photosensitizer molecules and a therapeutic agent (a macromolecule) accumulate in the
endocytic vesicles as indicated. The most efficient photosensitizers for PCI are mainly located

ACCEPTED MANUSCRIPT
25
in the membranes (a) of the vesicle as illustrated. Upon illumination (b) reactive oxygen
species, mainly singlet oxygen, are formed. Constituents of the membranes are destroyed and
the passage of the therapeutic agent to cytosol becomes possible (c).
Fig 4. Schematic illustration of the PCI process.
The figure illustrates the photosensitizer (S) and the therapeutic agent (D) is endocytosed by
the cell. Endocytosis is illustrated with an invagination of the plasma membrane where both
compounds end up in the endocytic vesicle named endosome. In absences of light the
endocytic vesicle will degrade or recycle its content in the lysosomes. In the presence of light,
the membranes will rupture and release the therapeutic agent to cytosol where it can reach its
target.
Fig 5. Growth curves of HT1080 fibrosarcoma xenograft in mice subjected to PDT or
PCI of bleomycin.
The tumor was located intramuscular in the lateral gastrocnemius muscle. The various
treatments are indicated on the figure; i.e. BLM, bleomycin only; PDT, AlPcS2a and light;
PCI, AlPcS2a, BLM and light. The photosensitizer AlPcS2a (10 mg/kg) and bleomycin (1500
IU) was administrated intraperitoneally 48 hours and 30 minutes prior to light exposure
respectively. The tumors were illuminated with a diode laser emitting at 670 nm (90 mW/cm2
and 30 J/cm2). The tumors were 80-150 mm3 at the day of illumination. The tumor size was
measured five times per week and the tumor volume (V) was calculated using the following
formula: V = (W · W · L) ⁄ 2 where W (width) is the shorter and L (length) is the longer of two
perpendicular diameters, measured with a caliper. The figure shows the synergistic effect of
PCI on tumor growth as compared to the individual treatments, i.e. BLM and PDT.
Fig 6. The treatment effect of HT1080 Fibrosarcoma xenograft in mice subjected to PDT
or PCI of bleomycin in combination with surgery. The tumor was located intramuscular in

ACCEPTED MANUSCRIPT
26
the lateral aspect of the femoral quadriceps muscle and the various treatments are indicated on
the figure. The photosensitizer AlPcS2a and bleomycin was administrated intraperitoneally
respectively 48 hours and 30 minutes prior to light exposure. The tumor bed was illuminated
with a diode laser emitting at 670 nm (90 mW/cm2 and 15 J/cm2) immediately after marginal
surgical resection of the tumor. The bars represent mean tumor volume and standard error on
day 20 after treatment. The tumors were 80-150 mm3 at the day of illumination. The tumor
size was measured five times per week and the tumor volume (V) was calculated using the
following formula: V = (W · W · L) ⁄ 2 where W (width) is the shorter and L (length) is the
longer of two perpendicular diameters, measured with a caliper. The figure shows the lack of
effect of PDT as an adjuvant to surgery in contrast to PCI resulting in a significant growth
delay in animals subjected to surgery.
Fig 7. The treatment effect of combining PDT and PCI with ionizing radiation.
The effect of combining PDT and PCI with ionizing radiation was assessed in TAX-1
sarcoma xenograft by determining the time for the tumors to double in volume after treatment.
The individual treatments are indicated on the figure. IR indicates ionizing radiation.
Photochemical treatment was carried out with AlPcS2a (10 mg/kg) and a diode laser emitting
at 670 nm, (90 mW/cm2 and 20 J/cm2) with or without administration of 1500 IU of
bleomycin 30 min prior to the light exposure. PCI + IR and PDT + IR indicate ionizing
radiation 7 days after the photochemical treatment. In all treatment regimens, except for IR
only, the cure rate was 10-20%. These animals are excluded from the figure. The figure
illustrates the additional treatment effect gained with neoadjuvant PCI before ionizing
radiation. The bars, with standard error, represent mean time to reach double tumor size.
Fig 8. The treatment effect of PDT and PCI of bleomycin on the CT26.CL25 tumors in
thymic and athymic mice.
PDT and PCI were performed in Balb/c nude/nude (athymic) and Balb/c (thymic) mice and
the treatment effects assessed 20 days after light exposure. The individual treatments are

ACCEPTED MANUSCRIPT
27
indicated on the figure. The bars represent the number of animals that did not reach the
endpoint of 1000 mm3 at day 20. PDT 30 indicates a treatment regimen with photodynamic
therapy using the photosensitizer AlPcS2a (10 mg/kg) and a diode laser emitting at 670 nm,
(90 mW/cm2 and 30 J/cm2). PDT 15 similarly represents photodynamic therapy with a total
light dose of 15 J/cm2. The figure illustrated the enhanced therapeutic effect of both PDT and
PCI in thymic mice compared to the same treatment performed in athymic mice.

ACCEPTED MANUSCRIPT
Endosomes
Lysosomes
Trans Golgi network
Golgi
ER
Secretoryvesicles
P-H+
P
P P-H+
P
P P-H+
PLDL
P
Pp
Pa
PaUncoated pit
Clathrin coated pit
H+Pa
Pp
Fig 1

ACCEPTED MANUSCRIPT
Fig 2
InsideSO3- SO3
-
Endosomes/lysosomes
SO3-
SO3-
N
NN
N
SO3-
SO3-
N
NN
NSO3
-
SO3-
SO3-
SO3-
N
NN
NNN
N
N
SO3-
SO3-
Al
TPPS2a AlPcS2a
TPPS4SO3
-
SO3-
ACCEPTED MANUSCRIPT
Fig 3
N
NN
N
NN
N
N
SO 3-
A l
SO 3-
1O2
Macromolecules
Endocytic vesicle ~0.5-1 µm
Membrane~10 nm
AlPcS2a
a b c

ACCEPTED MANUSCRIPT
Extracellularspace
Cytosol
D
DD
DDDD
DD
D
DD D
DD
DDDD
D
Lysosome
Endosome
Drug
PhotosensitizerS
S
S S
SSS
SS S
S SS
D DD
DDDS
S
S
SSLight
DD
D
Fig 4
S

ACCEPTED MANUSCRIPT
Fig 5
Time after light exposure (days)0 5 10 15 20 25
Rel
ativ
e tu
mor
siz
e
0
2
4
6
8
10
12Control BLM PDT PCI

ACCEPTED MANUSCRIPT
Control PDT PCI
Surgery alone
PDT + Surgery
PCI + Surgery
Rel
ativ
e tu
mor
siz
e da
y 5
0
2
4
6
8
10
Fig 6
ACCEPTED MANUSCRIPT
Fig 7
Control PDT PCIIR only
PDT + IRPCI + IR
Tim
e to
2x
tum
or s
ize
(day
s)
0
10
20
30
40

ACCEPTED MANUSCRIPT
Fig 8
PDT 30 athymic
PCI 30 athymic
PDT 30 thymic
PDT 15 thymic
PCI 15 thymicTum
ors
< 10
00 m
m3
(%) o
n da
y 20
00
20
40
60
80
100




















