ORL1 receptor–mediated internalization of N-type calcium channels
10
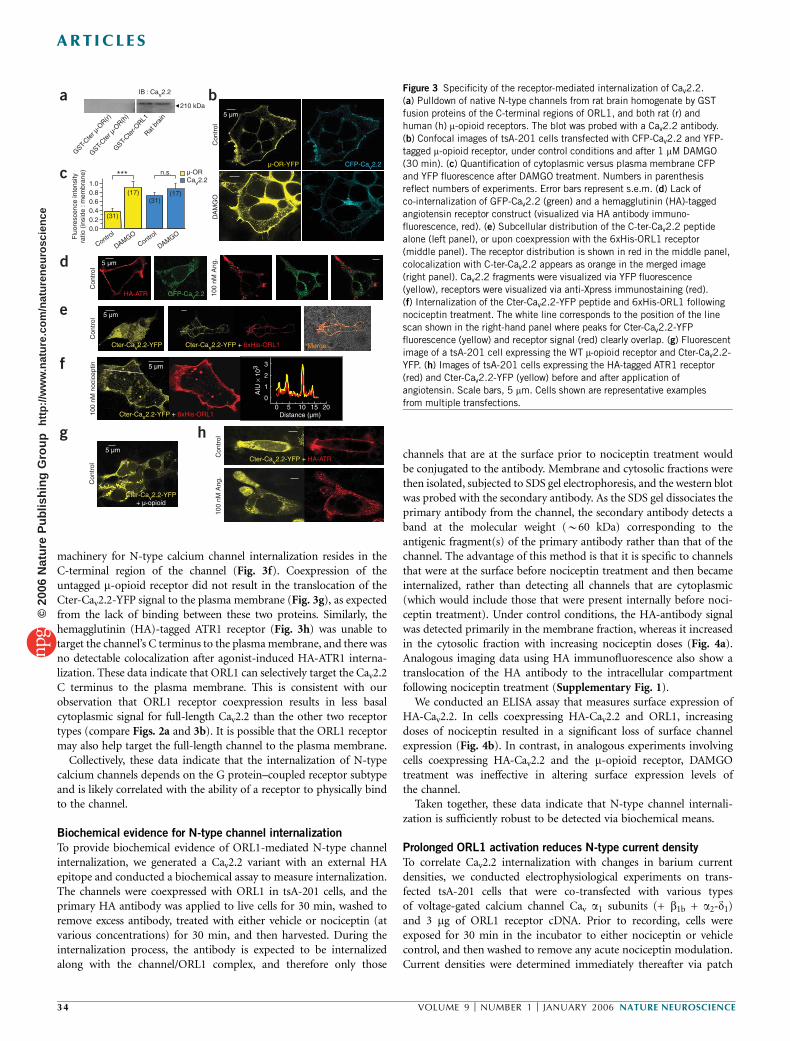
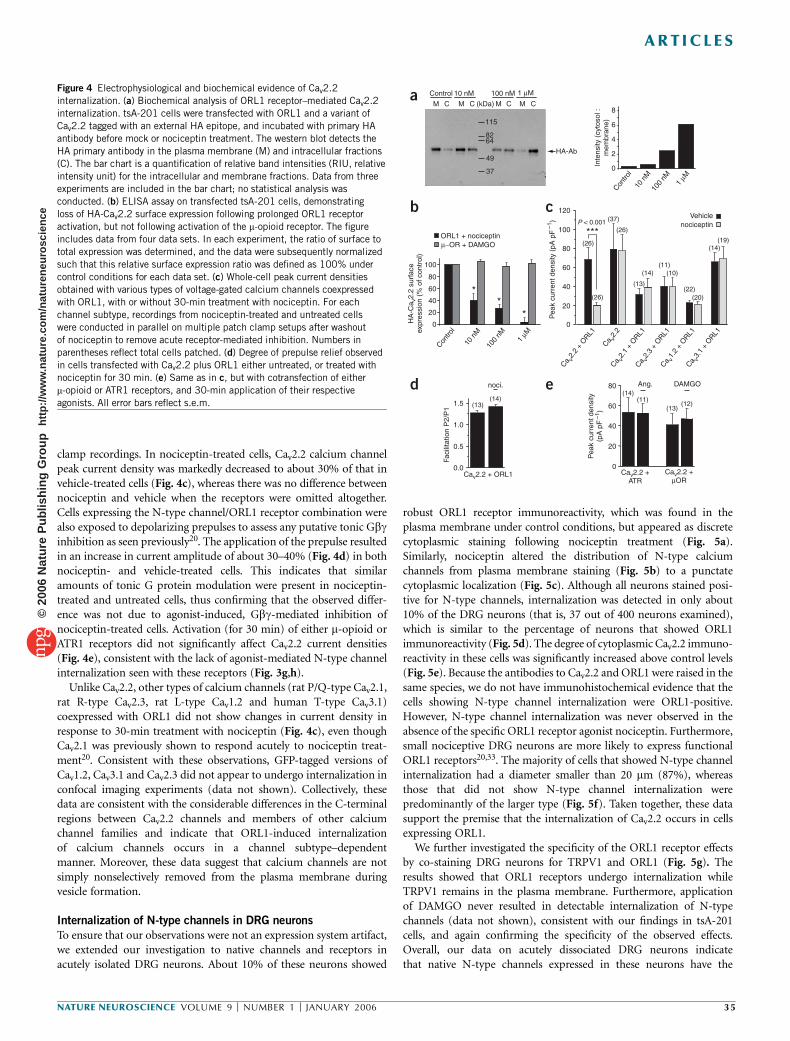
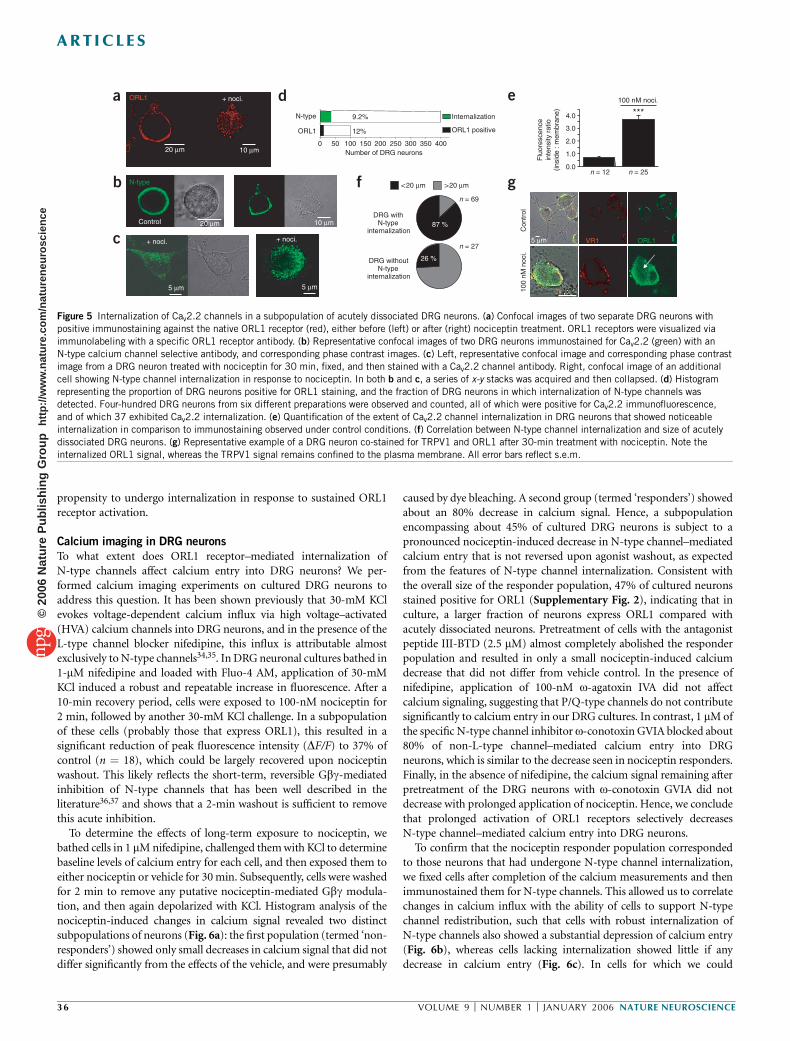
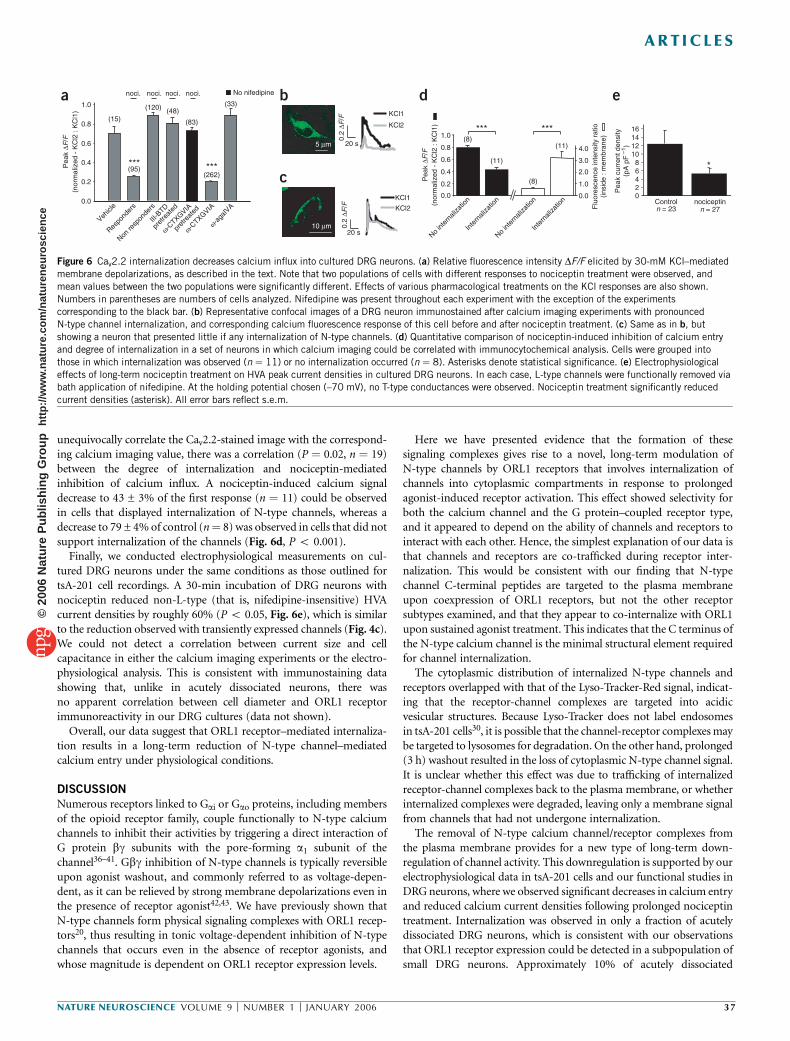
ORL1 receptor–mediated internalization of N-type calcium channels Christophe Altier 1 , Houman Khosravani 1 , Rhian M Evans 1 , Shahid Hameed 1 , Jean B Peloquin 1 , Brian A Vartian 1 , Lina Chen 1 , Aaron M Beedle 1 , Stephen S G Ferguson 2 , Alexandre Mezghrani 3 , Stefan J Dubel 3 , Emmanuel Bourinet 3 , John E McRory 1 & Gerald W Zamponi 1 The inhibition of N-type calcium channels by opioid receptor like receptor 1 (ORL1) is a key mechanism for controlling the transmission of nociceptive signals. We recently reported that signaling complexes consisting of ORL1 receptors and N-type channels mediate a tonic inhibition of calcium entry. Here we show that prolonged (~30 min) exposure of ORL1 receptors to their agonist nociceptin triggers an internalization of these signaling complexes into vesicular compartments. This effect is dependent on protein kinase C activation, occurs selectively for N-type channels and cannot be observed with l-opioid or angiotensin receptors. In expression systems and in rat dorsal root ganglion neurons, the nociceptin-mediated internalization of the channels is accompanied by a significant downregulation of calcium entry, which parallels the selective removal of N-type calcium channels from the plasma membrane. This may provide a new means for long-term regulation of calcium entry in the pain pathway. Calcium influx through N-type calcium channels into dorsal root ganglion (DRG) neurons is an essential step in the transmission of nociceptive signals at the spinal level 1 . Consequently, the inhibition of N-type channel activity by calcium channel blockers 2,3 , or through activation of opioid receptors, mediates analgesia in animals and humans. The regulation of N-type channels by members of the opioid receptor family has been extensively described. Like many other members of the Ga i/o -linked G protein–coupled receptors, opioid receptor activation results in membrane-delimited, voltage-dependent inhibition of N-type channels by Gbg subunits. This inhibition can be rapidly reversed upon agonist washout, or by the application of strong membrane depolarizations 4–8 . The clinical use of opiates in the treat- ment of pain is associated with the development of tolerance 9 , the underlying molecular basis of which is complex and remains incompletely understood. ORL1 receptors (also known as nociceptin, orphanin FQ or NOP receptors) are structurally related to m-, d- and k-opioid receptors 10–12 . ORL1 receptors are activated by the endogenous neuropeptide nociceptin 13 , but are resistant to classical opioid agonists such as morphine 14 . They are expressed in the CNS, as well as in centrally projecting small nociceptive DRG neurons 7,14–16 , and their activation via intrathecal injection of nociceptin diminishes pain sensation and spinal neurotransmission at the level of small nociceptive DRG neurons 17,18 . In DRG neurons and in expression systems, ORL1 receptors have been shown to inhibit N-type, T-type, P/Q-type and possibly R-type channels 7,19,20 , consistent with the analgesic effects of nociceptin. Increased mRNA expression of the endogenous nociceptin peptide and upregulation of nociceptin binding sites is observed following peripheral nerve inflammation 21,22 . ORL1 receptor gene expression is also increased after sciatic nerve ligation 23 and during development of morphine tolerance 24 . Conversely, knockout of the ORL1 receptor results in resistance to morphine tolerance without changing the acute effects of morphine 25 , indicating a complex inter- play between opioid and ORL1 receptors. We recently reported that ORL1 receptors can form signaling complexes with N-type channels, resulting in tonic, agonist- independent G protein inhibition of N-type channel activity 20 . This effect could be observed in expression systems and in DRG neurons, and involved a physical binding interaction between the C termini of the two proteins. Because ORL1 receptors have been shown to internalize in response to sustained agonist application 26–28 , we hypothesized that channels complexed with ORL1 might also be removed from the plasma membrane. As we show here, transiently expressed and native N-type channels are internalized to vesicular compartments following prolonged (B30 min) exposure to nociceptin. This effect is paralleled by a decrease in N-type channel– mediated calcium entry, occurs selectively for N-type channels and does not take place with receptors that do not physically interact with these channels. Our findings indicate that internalization of receptor-channel complexes constitutes a previously unrecognized mechanism for long-term downregulation of N-type channel activity in the pain pathway. Received 20 September; accepted 27 October; published online 27 November 2005; doi:10.1038/nn1605 1 Department of Physiology and Biophysics, Hotchkiss Brain Institute, University of Calgary, Calgary, Alberta T2N 4N1, Canada. 2 Cell Biology Research Group, Robarts Research Institute, London, Ontario, N6A 5K8, Canada. 3 De ´partement de Physiologie, Institut de Genomique Fonctionnelle, Centre National de la Recherche Scientifique (CNRS) UMR5203, Institut National de la Sante ´ et de la Recherche Me ´dicale (INSERM) U661 Universite ´s UMI & UMII, 34094 Montpellier, France. Correspondence should be addressed to G.W.Z. ([email protected]). NATURE NEUROSCIENCE VOLUME 9 [ NUMBER 1 [ JANUARY 2006 31 ARTICLES © 2006 Nature Publishing Group http://www.nature.com/natureneuroscience
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of ORL1 receptor–mediated internalization of N-type calcium channels